Submitted:
17 April 2023
Posted:
18 April 2023
Read the latest preprint version here
Preprints on COVID-19 and SARS-CoV-2
Abstract
ORF7b-2 is an accessory protein of the SARS-CoV-2 virus of only 43 amino acids. It has been implicated in various functional hypotheses, some of which predict its involvement as a trans-membrane protein. In this study, ORF7b-2 has often been compared to ORF7b-1 of the SARS virus to highlight differences and similarities with a protein that should have a similar biological role. Structural analysis of ORF7b-2 and its electrostatic characteristics show a polypeptide with both ends negatively charged and a diffuse negative charge over the entire structure. Therefore, its behavior in solution is like that of a weak negative polyelectrolyte, more precisely a polyanion with a net charge of – 4 at neutral pH. Its structure was modeled with two different modeling systems, one of which was ab initio. The two best models are similar, as confirmed by the Ramachandran plot, and show a central alpha-helical structure with two disordered and mobile ends. A normal mode analysis characterized the low-frequency dynamic aspects of the protein. The analysis of the structural shows a rigid central segment with mobile and fluctuating extremities, involved in a conformational equilibrium of the helix ↔ coil type. The calculation of the dipole moment shows its vector is not aligned with the main axis of the structure with an outward tilt of 24°. Molecular dynamics simulations were also conducted and the one in water is in good agreement with the previous results. While, the simulation performed by inserting a pre-oriented dimer (OMP) into a solvated lipid showed the low tendency of the protein to solvate in the apolar environment of the membrane. ORF7b-2 also shows a widespread distribution of negative surfaces that dynamically adjust to changes in structural organization. The BioGrid platform's [Biological General Repository for Interaction Datasets] through the BioGrid COVID-19 Coronavirus Curation Project shows a very large number of experimentally proven physical interactions unique to ORF7b-2, some of them with cytoplasmic proteins. These features of ORF7b-2, evaluated together, suggest a remarkable propensity of ORF7b-2 to interact with multiple molecular partners on both an electrostatic and hydrophobic basis. All this makes it unreasonable that the only biological function of this protein should be that exerted as an intramembrane protein.
Keywords:
ORF7b
; SARS-CoV-2
; COVID-19
; 3D-structure
; molecular dynamics
; dipole vector
; Electrostatic properties of alpha-helix.
1. Introduction
ORF7b, an accessory protein of SARS-CoV-2 [UniProtKB Accession: P0DTD8-1], is made from 43 amino acid residues [1], one residue less than the orthologous protein of SARS-CoV [Table 1]. Both proteins [described here as ORF7b-1 and ORF7b-2, from SARS and SARS-CoV-2, respectively] share 85.4% identity and 97.2% sequence similarity, but they show different charged amino acid compositions [1,2,3] [Table 2]. Accessory proteins are considered not essential for viral replication but involved in pathogenesis, thus, many structural and functional hypotheses for ORF7b-2 have been made by in vitro studies on model cell systems or by invoking a structural similarity with ORF7b-1 [3,4,5]. One study found that ORF7b-2 is related to the endoplasmic reticulum [ER] region [6], others have located ORF7b-1 in the Golgi compartment [7,8,9] and identified a leucine zipper sequence within the trans-membrane segment [7]. On this basis, a report has hypothesized that ORF7b-2 too is a trans-membrane protein localized in the Golgi apparatus [10].
The biological success of the virus is based on its exceptional ability to neutralize the host organism's defenses through its set of proteins. In particular, many of them counteract cellular defensive responses, such as interferon production or immune suppression. The author of an atlas on SARS-CoV-2 proteins [11] suggests that 21 viral proteins concur in blocking the interferon immune response and among them inserts ORF7b based on the information in a preprint [12]. In this preprint, Forgeon et al. hypothesized that ORF7b-2 might interfere with cellular processes. The protein, by interacting through a leucine-zipper, should form multimers. The protein was treated in the various steps of preparation, also with surfactants. The pellets show an M.W. of about 6 KD [M.W. from the sequence is 5.175] while in the presence of various surfactants it appears as a dimer of about 11-12 KD. But it shows high M.W. complexes when switched to cyclodextrin. Cyclodextrins are a well-known complexing agent for proteins and show a large internal cavity [diameter 0.60–0.65 nm and height 0.78 nm] with a rigid toroidal-shaped structure, which makes them ideal neutral complexing agents [13]. The effect of increasing cyclodextrin concentrations is to increase the number of complexed protein molecules, but it is difficult to establish without specific experiments whether the multimers are artifacts of the cyclodextrin or aggregates or the natural self-association form of ORF7b2. NMR experiments on ORF7b2 [3] with the protein in surfactants showed weak and uninterpretable spectra because of the interference of the detergent. These authors found a strict dependency on detergents for its follow-up. Thus, because of the necessity of the solubilizing agent and its tendency to produce oligomers, they stated that structure determination is challenging with the need to undertake other forms of investigation.
Therefore, there is still no clear experimental evidence on the protein structure or its possible multimeric organization. However, the most likely data on the protein's structural organization is functional and indirect.
Toft-Bertelsen et al., [14] identified ORF7b-2 as a novel viroporin. They used electrophysiological techniques on the Xenopus laevis [X. laevis] oocyte expression system to look for potential ion channel activity for the protein and found that a viroporin inhibitor drug dampened the electrical signal. This observation suggested that ORF7b-2 might act as an ion channel. Summing up, while some authors have shown that ORF7b-1 is a membrane protein localized in the Golgi complex, only indirect clues exist that ORF7b-2 is related to the ER. This also means that the influence of SARS-CoV-2 on host metabolism has yet to be fully understood.
An AP-MS analysis identified 332 high-confidence protein interactions between SARS-CoV-2 proteins and human proteins [98]. This means that each viral protein could
interact with at least 11 human proteins on average. Therefore, to get an effective physical interaction in a highly crowded environment such as the cell, we need that the two interacting molecules to have not only a great affinity and good quantitative ratios but also similar space-time characteristics. The picture that can be perceived is that every single viral protein might interact with many molecular partners, therefore, at least a part of them should have structural characteristics such as to satisfy these multiple needs.
ORF7b-2 should also be subject to this rule. Unfortunately, we know very little about its structure and nothing about its function. Thus, understanding the physic-chemical properties of sequence and the structural determinants of this protein should be helpful because the architecture of a protein drives the function. However, the structural properties of mini-proteins such as ORF7b-2 are often elusive, so it will be mainly its physicochemical properties to play a fundamental role in driving its structure-function behavior. The aim of this study is to understand what potential structural roles we can attribute to ORF7b-2 from an analysis of its sequence, physicochemical and electrostatic properties, corroborated by an analysis of low-frequency normal structure modes and molecular dynamics simulations, all performed on a complete structural model of the protein.
2. Results
2.1. Sequence
The 3D structure of ORF7b-2 has not yet been experimentally determined, nor is its function fully known. Data analysis, when there are deficiencies in the knowledge and complexity of the system, is often useful by sharing protein data of other coronaviruses.. This has often prompted various researchers to compare ORF7b-2 to ORF7b-1 [15] assuming a localization of the two proteins in similar cell environments to perform corresponding functions. However, despite the remarkable similarity in composition and primary structure [Table 1 and Table 2], the two proteins show some differences that change their respective physicochemical properties and, therefore, the structure-function relationships.
The residues in red have a significant statistical propensity for the alpha-helix, those in green for the coil, and those in blue for the extended beta structure [105]. The 20 underlined residues (from 9 to 29) are those supposed to be transmembrane. In the whole molecule only 46% of the residues have a helical propensity and, in the helix, shown as trans-membrane, out of 20 residues only 9 have a helical propensity. The two proteins show an identical 9-29 sequence. A visual analysis of the N-terminal sequences of both shows the lack of any signal sequences [translocon sequence]. Signal sequences are N-terminal extensions of the nascent polypeptides [pre-proteins] of secretory and membrane proteins. They are of about 15-30 amino acid residues and comprised a positively charged N-terminal region with the cleavage site for signal peptidase [Ala-X-Ala motif at the C-terminal end of the signal peptide]. Both proteins do not show these features [101], essential for entering the ER.
A comparison between the two proteins is useful to understand how similar they really are structurally and, ultimately, how similar their functional behavior may also be. Comparing compositions and sequences, we note the lack of positively charged residues and proline in ORF7b-2, while ORF7b-1 possesses both proline and a positive charge. In both tails of ORF7b-2 we found residues with high propensity for disorder [16], such as T, A, D, H, Q, S, E in C-term and E, S, D in N-term, while D is known helix disruption residue [17]. These are not weightless differences, as the particular compositions of the tails could affect the structural organization. Everything leads us to predict that the terminal segments should be very mobile and, with an improbable or reduced local helical organization. For instance, the instability index reported in Table 1 is an estimate of the overall stability of a protein [18,19]. It is 50.96 and 39.77 for ORF7b-2 and ORF7b-1, respectively, predicting ORF7b-2 more stable than ORF7b-1.
2.2. Electrostatic properties
Before any three-dimensional consideration, it is important to evaluate the physico-chemical properties of both proteins, among which the electrostatic effects are of particular interest when hypothesizing interactions with membranes. Rohit Pappu has developed [20,21] an analysis for small peptides and proteins that provides a series of parameters to help evaluate, with a good basic approximation, the conformations that molecules can have in solution and, among the calculated parameters, there is also an evaluation of the electrostatic properties [22].
The analysis of the charge distribution of the two proteins [Table 3, Figure 1 and Figure 2] shows rather similar negative values of the net-charge distribution per residue [NCPR], but different values of the charged residue fractions [FCR] with a more asymmetrical distribution for ORF7b-1. These values allow to characterize the organizational tendency of a polypeptide in solution by classifying it in a State Diagram. The diagram [Figure 1] shows that both proteins are in region 1, characterized by globular or extended structural organizations [globule/tadpole conformation], thus in solution, they behave as globule-like.
The charge distribution of the protein was evaluated according to Das and Pappu [20,21]. In particular, the fraction of charged residues, as FCR = |f+ + f-|, and the net charge per residue [NCPR] = |f+ - f-|, are calculated. In this context, f+ and f- represent the fraction of positive and negative charges, respectively. Sigma, σ = [f+- f-]/[f++ f-], where f- and f+ refer to the fraction of negative and positive residues across the entire sequence and sigma their distribution symmetry.
K is specific to a sequence, ranging from 0 to 1. At 0, the charges are randomly distributed, while at 1 they are totally separated into two distinct groups. It describes, in a non-linear way, the extent of mixing of charged amino acid clusters along the sequence. Values between 0.2 and 0.3 are significant of a diffuse distribution. These calculated values allow the classification of the behavioral tendency in solution of the segmental sequences of protein into distinct regions of the Diagram of States for IDPs. pI is attributed according to Lukasz et al., [99]. AH according to Kyte and Doolitle [100].
According to the model used in this analysis [20,22], the globular organization is preferred when there are electrostatic attractions between opposite charged residues, while the extended structure is favored by the free hydration energies of the similarly charged residues which repel each other. While, a low net charge per residue with high fractions of positively and negatively charged residues characterizes polyampholytes [23]. Therefore, the behavior of ORF7b-2 in solution should be that of a negative weak polyelectrolyte [FCR <0.3] and should behave as an extended-like system with negative charges asymmetrically arranged in both terminal segments [Figure 2]. The negative charge is significant and distributed over the entire protein (K = 0.25372) with an average net-charge per residue of -0.1163 and, in solution, the protein shows a negative net-charge [Figure 3] strongly dependent on pH, between 4.3 and 10. Instead, ORF7b-1 behaves like a weak polyampholyte because of the positive charge, with a more asymmetrical charge distribution than ORF7b-2 [Table 3] but a similar mean value of NCPR. This characteristic drives a dependence of the net charge on the pH similar to that of ORF7b2.
This result opens to careful consideration of the trans-membrane hypothesis of ORF7b-2 because both sides of the phospholipid bilayer, where lipid heads are located, show a negative charge at physiological pH, similar to that shown by protein tails. [24,25,26]. Unfortunately, these important electrostatic characteristics were rarely highlighted in the structural papers on ORF7b-2, as well as in those on ORF7b-1, because only the central part of the protein is discussed as a trans-membrane helix [7,10,12,14]. In doing so, any structure-function consideration of these proteins could be distorted.
2.3. 3D structure
The 3D structure of ORF7b-2 and ORF7b1 are available only through modeling. A typical model of ORF7b2 is that from ModBase [University of California San Francisco–UCSF]. [Fig 1S]. The model, like others, shows only the 3D structure of the region between Leu4 and His37, predicted as a helix, but all terminal residues are missing.
In Figure 4, we can see the complete models of the two proteins got through two different platforms, PHYRE2 [27] and PEP-FOLD3 [28], with similar results. Each platform produced several dozen models and the overall reliability of the best models is 88% for both proteins. The central helical residues were modeled referring to specific templates [Table 1S and 2S], while the outer segments, C- and N-terminal [in green], were modeled ab initio. The charge distribution analysis (Figure 2) demonstrated an asymmetric distribution of the negative electric charge on proteins. Three-dimensional models reflect these effects. Both proteins show terminal segments with a three-dimensional organization detached from the compact one of the central helices. In particular, the C-terminal extremes have many more differently organized residues than the N-terminal extremes. The C-terminals are very long, between 14 and 16 residues, and should protrude into the cytoplasm, while the N-terminals of 5 and 7 residues should be luminal. If we observe [Fig 2S] the length of the central helices [TM] we can see that while that of ORF7b-1 appears adequate to the standard thickness of the membrane [about 32 Å], that of ORF7b-2 is overall 44 Å, therefore longer. The two platforms, although they reach similar general conclusions, are differently affected by the algorithms. For example, in ORF7b-2, both PHYRE2 and PEP-FOLD3 predict 12 differently organized terminal residues, where PHYRE2 predicts a small helix and PEP-FOLD3 only disorganized residues. A similar situation occurs for N-terminal residues.
Ramachandran plots support this view [29]. The diagram shows which dihedral angles are best suited for a α-helix and possible steric conflicts. Both models have many terminal residues with angles Φ and Ψ not suited for an alpha helix. Figure 5A,B, shows the residues in the anomalous areas. We can see that many of these residues are involved in the terminal segments of both proteins. This justifies the non-helical organization of the tails but does not explain why one algorithm sees some in the helix while the other sees the same residues in an extended conformation. From Table 3S to Table 6S we can see the Ramachandran statistics for the PHYRE2 and PEP-FOLD3 protein models.
Summing up, from the charge distribution analysis, we do not expect the tails of both proteins to be helical and immersed in the membrane. Even modeling systems confirm a not helical organization, likely mobile and fluctuating. The Φ and Ψ angles of the Ramachandran graphs support this general view with non-helical residuals. It remains to understand the different modeling of the terminal segments.
An explanation can be got if we analyze the weight of the conformational probabilities of each residue in the two proteins. This analysis, performed by PEP-FOLD3, is based on the concept of structural alphabet [30] and determines the mean weight of each elemental conformation that each residue uses in determining the conformation of the protein. The Figure 6 shows the set of conformational information by residue, namely the weighted population of all conformations for residue [30,31], for both proteins. From the conformational point of view, the two proteins have a compact helical core of 11 - 12 residues, insufficient for the structural needs of a trans-membrane helix, which are of about twenty residues.
A last, but no less important observation, derives from the set of conformations per residue that characterizes the terminal segments of the two helices. We can observe the weighted composition of the conformations for both N-terminus. The elongated and spiral conformations [green and blue in the figure] together have a considerable percentage weight, with the greatest weight for the extended one. Also, the two C-terminals show a similar condition, but with a different conformational incidence of the coil and of the extended structure. Roughly from the residue 26 to 33, we have a preponderance of extended conformation (green), and from 33 to the end coil (blue). Both tails degrade into an increasingly less organized and flexible segment, with a probable coil/extended dynamic inter-conversion. The N-terminal segment seems also flexible but with a greater propensity for extended organization. In fact, the terminal segments experience non-helical organizations, where the residues are likely to undergo continuous conformational changes.
The picture that is taking shape is that ORF7b2, regardless of the characteristics possessed by ORF7b-1 with which we have compared it until now, has many structural aspects that are not always compatible with those of a trans-membrane protein. The perplexity increases when one considers that both terminal segments of ORF7b2 are disorganized, charged, and suggested rather mobile. C-terminal instability is consistent with the previously reported result from generalized ensemble simulations of poly-Ala peptides [32,33].
2.4. Dynamic properties of ORF7b-2
Most of the functional activities of a protein fall within a wide temporal scale of movements. There are the very rapid ones of intra-molecular vibrations to the slower ones of conformational transitions along the structural hinges. Thus, proteins can sample many conformations [or also equilibrium fluctuations] in the neighborhood of their native conformation [34].
Normal mode analysis [NMA] is a helpful method for characterizing these various dynamic aspects of proteins [35]. In particular, NMA is very useful in evaluating the dynamic properties of helical peptides. In small molecules, we frequently evaluate only the Ca atoms because the backbone motions are all that is necessary for characterizing the lowest-frequency normal modes [36]. NMA is based on elastic network models and ORF7b2 was analyzed to calculate atomic fluctuations and displacements and superposition and correlation, showing the correlations between the motions of the C-alpha atoms of the backbones. Two Web-servers, elNémo, Network Elastic Model [37,38] and HINGE-Prot [39], were used for the automated computational analysis of the low-frequency normal structure mode.
NMA is performed through low-frequency movements with simplified mechanical models and provides a detailed description of the dynamics of small polypeptides by allowing the localization of relatively rigid segments and more flexible regions [40]. It is a suitable method for calculating vibrational modes and protein flexibility as the independent motion of atoms in a molecule relative to any of the other modes.
Table 4 reports hinge residues with the best score, calculated from the conformational models that describe the fluctuations of residues from the average structure in the principal directions of motion. Models were calculated with the Gaussian network model [GNM] and anisotropic network model (ANM) [41] by HINGE-Prot.
The table shows the best hinge residues in the ORF7-b2 structure and the reliability of the result as calculated by HINGE-Prot [the score varies between 0 and 1]. These residues represent twist angles or points of rigidity around which the movement of the entire structure is organized (see also Figure 7).
The movement of the hinge is characterized by large changes in the twist angles of the main-chain that occur at residues 9, 20, and 32. The conformational fluctuations that drive the twisting of each residue generate these movements. A close view of ORF7b-2 motions is shown in Figure 7. The figure shows some motion sequences around the 3 hinge residues of ORF7b-2. As seen from different views, the protein is mobile and flexible [see also fig 5S and fig 6S].
An overview is presented in Figure 8, where the 9 normal modes calculated by elNémo for ORF7b-2 are collectively represented. In the supplements [Figure 3S and 4S], we have calculated the calculated values of ORF7b-2 fluctuations and displacements. All these data show that the protein has significant segmental motions at both ends. Greater stability of the α-helical conformation is found in the middle of the polypeptide chain than in the terms. However, the surprising lack of gly in the helix induces a loosely packed backbone with a strain amplitude of about 12 Å, which is shared between bending and torsion. Its overall flexibility can be explained in terms of collective flexible movements of the structure that can be resolved in individual strain modes, such as physical, flexural and torsional strains along the principal axis and with torsional strains at both segmental α-helix terms. These additional degrees of freedom increase the entropy of the protein, favoring the decrease in free energy towards greater stability. However, the dynamic patterns of normal mode analysis [NMA] on the behavior of α-helices as deformable bodies appear similar between transmembrane, extra-membrane and soluble protein α-helices [42], because the deformations of the α-helix are independent of cell location [102]. Thus, this approach describes the dynamic of the local deformabilities of the helix structure but does not show which protein category the helix belongs to. It should only be noted that a transmembrane helix should be stiff on average.
Finally, the knowledge of the magnitude of the helix macro-dipole is also of fundamental interest in understanding ORF7b-2 structure. The helix macro-dipole has been often implicated in function and in stabilizing structural motifs containing helices. The strength of the helix dipole is given by the sum of the microscopic dipole moments [43,44], arising from the individual peptide bonds. They vary with the charge, orientation and position of residue; therefore, it is necessary to have also a satisfactory dipolar model of ORF7b-2.
The three-dimensional structure got by PHYRE2 was used for the calculation on the server at http://bip.weizmann.ac.il/dipol [45]. The server calculates the dipole moment and displays the dipole vector superimposed on a protein ribbon backbone [Figure 9]. There is no obvious relationship between a protein's dipole moment and its function, but, in this case, we may gain more insight into the ability of ORF7b-2 to be incorporated into a membrane, because of the peculiar electrostatic properties of this protein. The calculated dipole for ORF7b-2 is 488 Debye, lower regarding the average value for similar helical proteins [45] that is 542.66 D. In the figure, we can see the ribbon diagram of the protein with its dipole and mass moment vectors displayed, thus allowing the dipole moment to be appreciated in relation to the overall protein structure. The server also calculated a Radius of gyration [Rg] of 10.91 Å. Rg is one measure of the size of the shape which polymers adopt in solution and an indicator of protein structure compactness. It describes the equilibrium conformation of the total system. An ideal a-helix of 43 aa should have a Rg around 19-20 Å [103]. The lower value of Rg [10.91 Å = 50% less] found for ORF7b-2 means that this helix behaves in solution as a globule-like or more compact helix than the ideal reference helix, because more flexible. Thus, the shape of ORF7b-2 should be a prolate ellipsoid with the electric moment not parallel to the major axis. In fact, the calculated dipole vector points outward, as shown by the angle between helix and vector. This angle is 24°.
This means that this helix, inserted in a membrane, will be distorted or inclined, both because it is longer [39.7 Å] than the distance between the outer sheets of the membrane [about 32 Å], and because the dipole will seek its orientation regarding the helix body.
To have more details on the insertion of the protein into the membrane, we performed molecular dynamics experiments in water, as a single molecule, and in the membrane, as a dimer.
2.5. Molecular dynamics of ORF7b-2 in explicit water.
The best model of ORF7b-2 was subjected to minimizations and molecular dynamics simulations in explicit water at neutral pH and 300 °K (details in Materials and Methods). The protein, being a small peptide, rapidly reached equilibrium in about 25 ns. (Figure 10). The trend of various molecular parameters over time (Hydrogen bonds, Radius of gyration, Percentage of helicity, RMS fluctuation, Solvent accessible surface, and Area per residue over the trajectory) are reported in the Supplements (Figure 10S). Figure 10 shows the trend of the root-mean-square deviation (RMDS) of atomic positions over time. The RMSD value of about 1 nm (10 Å) at equilibrium agrees with the values of low-frequency molecular vibrations found in the normal mode analysis. Also, the other molecular parameters found in the dynamics show a good agreement with what has already been seen in silico. ORF7b-2 is a small molecule whose behavior in solution appears driven mainly by its physicochemical characteristics. During the dynamics, the protein is subjected to conformational changes which, even without generating unfolding phenomena, involve structural variations in which parts of the protein rearrange regarding others (see, for example, the trends of percentage of helicity, hydrogen bonds, area per residue, and gyration radius). Since it is a rather mobile small protein, the variation of the distribution of its electrostatic surfaces is an interesting parameter. Figure 10 shows the variations of the surface electrostatic distribution during the simulation for every 10 ns. The surface electrostatic potentials were calculated with the DelPhi program, which also incorporates the effects of ionic strength to evaluate the Poisson-Boltzmann equation (details in Material and Methods).
During the simulation, the 3D electrostatic surfaces shown above are always centered on the same side view of ORF7b-2. As we can see, the protein shows how the charge distribution on its surface varies continuously, even for relatively small conformational changes, as shown by the changes in helicity (Figure 10S). The equilibrium model at 40 ns shows a diffuse positive charge on an entire side. This suggests that the protein is also stable in aqueous solution and the response to its conformational changes is expressed through variations in the distribution of its surface charge, which drives its interactions with the solvent. This electrostatic behavior could allow it to guide the search for different molecular partners with electrostatically based interactions. A more detailed view of the conformation at 40 ns is shown in Figure 11. In the cartoon model (in green), the evidence shows that from L17 to W21, the protein is rigid and this segment is the pivot for slight bending of the surrounding parts. But if we look at the distribution of electrostatic potentials on the protein surface, in the top right model, one whole side of the protein surface is negatively charged (in red), while with a 180° rotation, the other side shows the charges positioned on the two tails and an uncharged, i.e., apolar surface, appears. Obviously, this is a static view but useful to get an idea of the position of the lysines of which the protein is rich. A lysine zipper was used to support the transmembrane localization of ORF7b-1 [7] from which to hypothesize that of ORF7b-2 as similar in behavior [12]. As regards ORF7b-2, the strip proposed (Lys 4, 11, 18, 25) by Forgeon et al. [12] is ideal and does not consider the structural and chemical-physical characteristics of the protein. These lysines do not form a zip on the same apolar side of the protein, but are dispersed along the structure even on electrostatically charged surfaces. For example, Lys 4 is in the N-terminus, in a charged region highly mobile by helix-coil inter-conversion, while 11 and 18 are on the other side of the molecule, embedded in a large molecular surface with diffuse negative charge. Figure 11S clearly illustrates these discrepancies. These results should not surprise because ORF7b-2 is a polyanion and, being small, the surface charge density is quite high. Thus, the electrostatic properties associated with its intrinsic mobility heavily influence its structural behavior.
2.6. Molecular dynamics of ORF7b-2 in membrane.
The ability of ORF7b-2 to form multimers in the membrane remains poorly understood. Thus, one way to test this experimentally is through the molecular dynamics of a dimeric structure of ORF7b-2 in a lipid bilayer surrounded by water. The dimer represents the minimal structural organization of ORF7b-2 that should exist stable in a membrane. To reduce the equilibration times, a dimer was simulated by HDOCK and its best model (fig 12S, left side) was then pre-oriented in a Golgi membrane using the Orientations of Proteins in Membranes (OPM) database (fig 12S, right side). This new model was used for molecular dynamics in POPC lipid bilayers for a 100 ns long simulation (details in Methods). The system reaches equilibrium in about 60 ns. Figure 12 shows the key features of this simulation. As we can see, the two components of the dimer change their mutual positions during the simulation. Between 35 and 50 ns, the dimer exhibits structural relaxation, as shown by the increase in RMSD and decrease in total helicity, with a concomitant change in the relative positions between the monomers. At 100 ns, the complex seems stable but the monomers are distorted, partially unwound, with an overall decrease in alpha helical organization. In another experiment, the simulation time was brought to 200 ns with no appreciable variation (result not shown). This result should not surprise, because, in a lipid bilayer, the formation of a dimer between two similar molecules can occur both by interaction through apolar surfaces and through surfaces with opposite charges. ORF7b-2 has a limited apolar surface on structurally similar sides of the molecule (see Figure 11). The rest of the molecular surface has a broad distribution of negative charge, which does not favor any interaction with an apolar medium. Indeed, if the molecules interacted with the apolar patches, the entire external surface of the resulting system would be diffusely negative and with no possibility of existence in an environment with a dielectric constant around 2. We must not forget that the peptide is a polyanion. The pH dependence on water and in the membrane is obviously different. While in water, each acid-base residue can be easily protonated-deprotonated as the pH changes, in a non-aqueous environment with a low dielectric constant, the protonable/deprotonable groups have no possibility of exchanging or transferring protons. This crystallizes the charges already present and induces distortions in the molecule.
Ultimately, the most favored structural organization in an apolar environment should be the one that is energetically constrained to expose as many apolar residues as possible. But this solution seems to involve a rather destructive reorganization of the system. If even a biological activity could be associated with this reorganization, it is difficult to establish in this context of studies, in which the structural characterization aims to highlight the most important chemical-physical properties that guide the behavior of ORF7b-2. In the supplements, the various structural organizations of the dimer in the membrane at different simulation times are also shown (fig 14S). Also, in these cases, the dominant influence on the behavior of the molecules in the apolar bilayer is exerted by the electrostatic characteristics of the molecular system which, attracted towards the more polar zones of the membrane, undergoes structural deformations.
All the results tell us that ORF7b-2 is a small alpha-helical macromolecular polyanion with a prolate ellipsoidal shape and endowed with high structural mobility, in particular at the ends. A strong net charge of - 4 at neutral pH, distributed over a relatively small surface, and an electric moment not parallel to the major axis of the molecule, give a peculiar behavior to its electrostatic surfaces, very sensitive even to small conformational changes caused by perturbations environmental. These perturbations result in significant changes in the surface electrostatic distribution. This peculiarity also suggests a potential electrostatic interaction with different molecular partners. The molecular dynamics experiments, in excellent agreement with the chemical-physical and structural data, show that these characteristics are probably inadequate to produce self-association effects such as the formation of multimers in an apolar environment. These conclusions, while not supporting the formation of ORF7b-2 oligomeric systems in membranes, do not exclude that ORF7b-2 may have hitherto unconsidered molecular partners.
3. Discussion
ORF7b-2, although tiny and essentially believed to be a trans-membrane helix, is a protein with a unique fold. Baruah et al., [46] noted that, in matching 2413 structures to the sequence of ORF7b-2 to predict its 3D structure, the most similar structural folds showed between 11 and 16% structural identity, endorsing the structural uniqueness of this protein. Other authors have also reached similar conclusions, i.e., that ORF7b-2 has no matching structures [72]. The protein was compared to the ORF7b-1 as far as possible, but there are various differences. The main one is that ORF7b-1 is a polyampholytes, while ORF7b-2 is a tiny macromolecular polyanion with a central helix. ORF7b-2 has also an asymmetrical electrostatic distribution. The entire structure is characterized by a diffuse negative charge [NCPR = - 0.1163] also on the residues of the central backbone but with a greater weight on both end segments [FCR = 0.2 and 0.4 for N- and C-terms, respectively]. These electrostatic characteristics are reflected in the entire system inducing a strong net negative charge of - 3.90 at pH 7.0 and a pI of 4.32, very low and uncommon among the proteins. Electrostatic surface analysis also supported these results. All of this generates a behavior similar to that of a polyelectrolyte, more precisely, a polyanion. [fig 3]. In the cell, various macromolecular entities possess polyanionic character, such as proteoglycans, lipid bilayer surfaces, tubulin and its microtubules, actin and its filaments. Nor are polycationic macromolecular entities lacking. Because of the specific functional properties of these natural polyelectrolytes, they have attracted much interest from the pharmacological and biotechnology industries.
Macromolecular polyanions are involved in protein structure stabilization and destabilization [47]. It has been experimentally showed on cationic model proteins, such as chymotrypsinogen A, ribonuclease A, cytochrome c, and lysozyme, that polyanions with negatively charged groups on their chain induce relatively minor structural destabilization, while the hydrophobic part of the polyanion handles protein unfolding. In these protein-polyanion complexes, protein stability inversely depends on charge-related electrostatic properties of proteins, such as isoelectric point and surface charge density, while hydrophobic polyanions have perturbed both tertiary and secondary structures of cytochrome c, even at neutral pH and at room temperature [48,49]. The full ability of polyanions to interact with proteins depends on their minimum length associated with a high net charge of the protein surface at neutral pH, which leads to a high diffuse charge density on the structure [50]. It is important to emphasize that the complexation between proteins and polyanions is modulated by various other factors, such as the surface charge and hydrophobicity distribution and the flexibility/stiffness of their structure [51]. These proteins form protein-polyelectrolyte complexes through a 1:1 stoichiometric binding between oppositely charged groups [52,53]. At acidic pH, the net charge zeros with a gradual lack of electrostatic repulsion and promoting hydrophobic interactions. This could also lead to the hypothesis that ORF7b-2 may work in a rather acidic, aqueous environment. However, whatever our structural view of ORF7b-2, it is a small biological object, a nano-particle if we want, with the properties of a polyanion. The results support this view. What functional roles it may play is cannot be defined with these data. The data show only a great potential of ORF7b-2 to interact.
The results also show that the ORF7b-2 sequence has the central segment 9 – 29 quite fluctuating in amplitude (10.2 Å), with a high helical tendency, but which corresponds to less than 50% of the total residues. This rather mobile, and diffusely negatively charged, helical core should be the major contributor to ORF7b-2 as a transmembrane protein. Many experimental observations have revealed that the central apolar nucleus of membranes has the tendency to become overpopulated with non-polar side chains, and to be under-populated with polar side chains and even more with charged side chains [54]. This implies that, in an apolar environment, these domains are oriented to maximize the sum of the distances of the charged residues from the central plane of the membrane [55] and to minimize the sum of the inclination angles of the helices [54]. As a result, charged residues, both basic and acidic, are often found around the lipid surface of the extracellular side than on the periplasmic side. This charge asymmetry is thought to be related to better partitioning of charged residues in the more hydrophilic outer leaflet of the outer membranes [57]. Thus, besides driving the orientation of proteins in the membrane, because of their strong preference for the carbonyl and glycerol regions of the lipid head groups, charged residues of the chain are also important for the positioning of membrane proteins along the membrane. In this activity, they are helped by Trp and Tyr, who prefer the same regions [57]. It has also been observed that the helix changes its angle of inclination to favor the movement of charges out of the non-polar nucleus [58]. Consequently, TM proteins must exist in the highly anisotropic environment of the lipid bilayer, hydrophilic at the edges and highly hydrophobic at the core. This requires both that the TM proteins are structurally adequate, but also that the non-polar environment is carefully balanced to accommodate them. In conclusion, if the charged side chains react with the lipid head groups, this favors all those movements of the helix and of the extended structures towards the membrane-water interface to adapt energetically to the environment. Evidently the electrostatic attraction plays a predominant role which, in the physical reality of the movement through the membrane, is transformed into structural distortions of the protein.
In conclusion, if the charged side chains react with the lipid head groups, this should favor all those movements of the helix and of the extended structures towards the membrane-water interface to adapt energetically to the environment. Here, the electrostatic attraction should play a role which, in the physical reality of the movement through the membrane, is transformed into structural distortions of the protein. This also means that the TM sequences and their physical properties must be optimized for insertion and function in the residence membrane [59].
Although ORF7b-2 possesses a helical core that might appear to be suitable for insertion into an ER membrane, the protein lacks a key feature that distinguishes proteins that must move into the Golgi membrane, or ER. The primary constraint on all TM domains that enter the ER is that they must be partitioned out of the translocon into the ER membrane during synthesis [60,61]. ORF7b-2, as well as ORF7b-1, do not show this feature.
We know that approximately 40% of nascent proteins in humans are translated by ribosomes on the surface of the ER [62]. The ER is the entry site to the endomembrane system of the Golgi apparatus, also for peroxisomes and lipid droplets [63,64,65]. To be inserted into the membrane, both ORF7b-1 and ORF7b-2 should interact in the inner channel of the translocon with a segment of at least 15-30 residues. Translocon is a hetero-trimeric protein complex [Sec61] that transports nascent polypeptides, through a targeting signal sequence, into the interior space of the ER from the cytosol, or to integrate nascent proteins into the membrane itself. This process is necessary for the protein to cross the lipid bilayer. If the transfer is co-translational, the protein should have the signal sequence that targets the proteins to the mammalian ER. But, both ORF7b-2 and ORF7b-1 are too small to cross the ER membrane co-translationally, via translocon, nor have a suitable signal sequence, which seems missing in their Opening Reading Frames, nor positive charges at N-terms.
A sequence signature is the most conserved region in a sequence pattern that occurs repeatedly in a group of related or homologous sequences [66]. According to this definition, a sequence signature could be a functional site, or any conserved region, even if of unknown function, but always in a protein family or group. Therefore, although the actual physical manifestation of a signature may vary [66], if ORF7b-2 does not possess homologous proteins [46] or does not belong to a protein group, it means that the signal-sequence is missing.
However, the translocation is not obligatorily co-translational, it may also be post-translational through a gate or pore that enables insertion of the substrate into the lumen or membrane of the desired organelle [67].
Ribosomes must ensure that the native states of viral proteins are generated thermodynamically stable. The early stages of folding during the release of the nascent chain are critical to stability and function. But these steps can also produce aggregates or poorly folded shapes, because during ribosomal translation [68], at least 30-40 residues of the nascent proteins must act as a signal. They interact with the ribosomal channel, to undergo co-translational compaction.
Therefore, this process involves a consistent part of the nascent protein. But with very short proteins, such as ORF7b-2, a substantial portion of the chain must fold post-translationally. ORF7b-2, because of the limited length of its sequence, cannot process the number of residues needed as a co-transactional signal, because the signal is longer than the entire ORF7b-2 sequence. So, it should be prone to aggregation upon release from the ribosome. However, the ribosome, the nascent protein, and molecular chaperones, such as Hsp70, should work together to overcome aggregation propensities and achieve a well-folded and stable native state [69].
In particular, the molecular chaperone Hsp70 [70] controls the solubility and structural accuracy of newly synthesized protein chains, assisting their protein folding, refolding of misfolded proteins, and protein trafficking. With ORF7b-2, the role of Hsp70 seems crucial because it has to promote ORF7b-2 solubility by preventing the formation of soluble and insoluble aggregates. 99% of the "Hsp70-cured-proteins" were found to contain at least one Hsp70 binding site, where the number of Hsp70 binding sites is directly proportional to the size of the bound protein [71,72]. Hsp70 chaperones have been implicated in assisting protein folding of newly synthesized polypeptide chains, refolding of misfolded proteins, and protein trafficking. For these functions, the chaperones need to exhibit a significant promiscuity in binding to different sequences of hydrophobic peptide stretches. For these purposes, the chaperone has developed the ability to bind heptad sequences of peptides in both orientations in the substrate binding cleft with comparable energy without rearrangements of the protein [71].
Thorough tests on ORF7b-2 by Limbo-Switch-lab Server [72] for "Best sequence", "High specificity prediction" (few past positive), and High sensitivity prediction (more true binders) demonstrated (Figure 15S) that ORF7b-2 had, at position 24-30, a statistically significant binding site for Hsp70 (with a high score of 23.17), a canonical heptad sequence (MLIIFWF) for binding to the chaperone.
This suggests that the protein is not directly inserted into the membrane through the translocon, but, aided by Hsp70, does not aggregate and is soluble in the cytoplasm.
Molecular dynamics in water showed the structural stability of the protein in a medium with a high dielectric constant, at neutral pH and 300 °K. The trans-bilayer helices possess many glycines and large hydrophobic amino acids for a better packing [73]. ORF7b-2 does not possess glycine and the mean HV product value (Figure 6S) is rather low, suggesting a widespread presence of small-volume residues and low hydrophobicity in the central segment of ∼20 amino acids, which instead should be strongly hydrophobic for a TM.
If we use the values of the hydrophobicity scale reported by Hessa [60,61], for transferring protein residues from the cytoplasm into the ER membrane, we can calculate the change as free energy of transfer. Even if the calculation is crude without some minor corrections, transferring hydrophobic/apolar residues and polar/charged residues from the cytoplasm to the membrane, respectively, can be estimated at - 3.17 kcal/mole and +6.66 kcal/mole. From which, the total free energy of transfer is + 3.49 kcal/mole. Basically, the "solubilization" of the ORF7b-2 sequence in the apolar environment of the membrane is not thermodynamically favored.
The second constraint is the "internal positive rule" reported by von Heijne [74,75], which postulates the preferential presence of positively charged residues as a topological determinant of TMs (lysine and arginine). This rule is valid for almost all helical membrane proteins, regardless of the organism and the membrane system. It is a powerful determinant of membrane protein topology, where "topology" refers to how one helical chain intertwines or interacts with similar helical chains in the membrane.
Here we have that the positive inside rule is not applicable both because of the mere presence of aspartic acid and glutamic acid residues which disfavor protein-lipid interactions, and because ORF7b-2 is diffusely negative.
This generates an additional constraint. In our case, an ORF7b-2 self-association should form multimers with the ability to create pores for the substrate. The dimerization test performed by molecular dynamics in the bilayer suggested that ORF7b-2, under the experimental conditions used, does not self-associate, most likely because of electrostatic repulsion. ORF7b-2 is not an ampholyte, it has no positive charges; it is a negative polyelectrolyte, a polyanion, with the charge spread over the structure. A negatively charged TM protein in a membrane is thermodynamically disadvantaged because of the very low dielectric constant of the membrane environment. Whatever the structural organization of ORF7b-2, reciprocal remodeling of the membrane and protein is difficult. Molecular dynamics simulations have highlighted these difficulties, showing a disorganization of the system with no dimer formation.
ORF7b-2 dimerization should be the side-to-side self-association of two similar molecules. In the membrane, oligomerization is understood as the lateral association of domains that are hydrophobically coupled to the bilayer [76]. It is likely that these specific associations giving rise to unique structures should show a rather detailed steric adaptation of the surfaces. Therefore, the side-to-side association of helices is a crucial event in the stability of many membrane complexes [77]. In our case, molecular dynamics simulation revealed that we have no lateral association between the two trans-bilayer helices. Nor we have observed specific associations giving rise to unique structures by a detailed steric fit of surfaces. Rather, we have observed steric clashes at helix-helix interfaces that are expected to act against interactions between helices [77]. The presence of so many constraints does not seem to support a function of ORF7b-2 as a transmembrane protein. The same structural organization with the terminal parts both negatively charged and fluctuating, with a dipole moment not aligned with the main structural axis, makes it difficult to conceive its involvement in a membrane.
Apparently, both the composition and structure of this tiny helical protein by itself contain little or no information about its ability to penetrate and associate in the membrane. Its behavior is mainly determined by the modification of the negative charge distribution on the surface of ORF7b-2. The ability of the protein to adapt its surface electrostatic distribution to environmental changes or even to small conformational variations is remarkable. This suggests a peptide with great possibilities of interaction with different molecular partners.
It is well known that viruses employ an evolutionary strategy to keep their genome size small [78,79]. This results in viruses that often encode tiny proteins, less than 50 amino acids long and not homologous to cellular proteins [80]. Because these proteins are very short, their open reading frames are often overlooked or considered very late. However, some of them are short transmembrane proteins, but many others bind variously to peripheral proteins on the outer surface of the membrane, changing their activity [81]. This second group of proteins is characterized by much more basic residues, which are prone to interact to the peripheral membrane proteins [82].
To date, membrane channel activities have not been conclusively associated with characteristic biological functions [83,84]. While protein-protein and protein-phospholipid interactions have been associated with specific biological activities [84]. Indeed, it should be noted that as research progresses, less and less interest is being paid to the involvement of these tiny proteins in membrane channels and more to the many other biological functions they perform [84]. On this basis, we could hypothesize that ORF7b-2 may belong to a protein class that provides a versatile mechanism for regulating a broad range of cellular activities through interactions.
There is evidence that many macromolecular polyanions and polycations act in the body and control important biological functions. For example, there is growing evidence that several polyanions have a procoagulant nature in blood [85]. This has aroused many pharmacological interests. Polyanionic biopolymers have also been discovered that can hinder blood clotting, for example, heparin and heparin mimics. They are antithrombotic, but with many side effects. But in the body, there are also many cationic proteins, very well studied (e.g., protamines), which neutralize the biological activity of heparins to balance their hemorrhagic effects [86]. With viral infections, the interest is greater because it is associated with the eosinophilic antiviral response against respiratory viruses, in vitro and in vivo [87,88], involving these cationic proteins. Eosinophil tissue cells can release some cationic proteins, such as major basic protein 1 (MBP1; also known as MBP and PRG2], eosinophil cationic protein (ECP; also known as RNase3), eosinophil-derived neurotoxin (EDN; also known as RNase2) and eosinophil peroxidase (EPX; also known as EPO). The great interest in these proteins lies in their biological properties and activities in the cellular functions of eosinophils. Eosinophils are strongly involved as effector cells in host defense and, in particular, in the mediation of inflammatory responses in human viral diseases and cationic proteins are part of these defensive mechanisms [110]. Some authors [11,12] reported that ORF7b-2 is involved in counteracting the immune defense mechanisms of the host cell. These contrasts are very often implemented through direct protein-protein or protein-complex molecular interactions, both in the cytoplasm and in the peripheral areas of the membrane. The chemical-physical and above all polyanionic characteristics of ORF7b-2 are very specific precisely in the management of interactions. The presence of cationic proteins in the defense against viruses suggests multiple roles for ORF7b-2.
Although all these results do not give us the opportunity to show a physical insertion of ORF7b-2 into membranes, as hypothesized by others, the protein has clearly shown to possess remarkable aptitudes to interact electrostatically with other molecular partners. This study alone does not support which functional activities can be accounted for by the remarkable structural properties and capabilities shown.
Some molecular partners of ORF7b-2 are known, albeit with different reliability and statistical significance, regardless of the knowledge of the spatiotemporal characteristics of where, how and when they perform their interactions and functions [111]. The project by BioGrid curators could shed some light.
BioGRID, the Biological General Repository for Interaction Datasets, is the platform that collects and takes care of all the physical interactions of the proteins of the human genome, with each other and with those of other organisms [89]. As part of the BioGRID COVID-19 Coronavirus Curation Project, the physical interactions between SARS-CoV-2 proteins with those of humans were experimentally detected and biophysically defined. The techniques used are eminently physical, such as Affinity Capture-MS or Proximity Label-MS. Of course, not all the physical interactions identified for ORF7b-2 develop into functional interactions that express a specific function, some are random. However, these data represent an important and concrete starting point. The BioGRID's curated set of data for SARS-CoV-2 has been updated to include interactions, chemical associations, and post-translational modifications [PTM], and in these protein sets we also find the physical interactions related to ORF7b-2 with the human proteome [https://thebiogrid.org/4383871/summary/severe-acute-respiratory-syndrome-coronavirus-2/ORF7b.html]. BioGrid reports 1599 unique physical interactors for ORF7b-2, who are involved in 2458 interactions. BioGrid curators classified the interactions into 5 levels of significance. Physical interaction means that two proteins meet and react by binding to each other, more or less strongly. We are considering a huge number of interactions that are scattered in different places of the complex human cellular organization. Many of these proteins are cytoplasmic. Briefly summarized, the protein has the central alpha-helix residues in common with membrane proteins. But all its chemical-physical characteristics seriously question this hypothesis, also supported by the vast number of cytoplasmic proteins with which it interacts experimentally, as reported by BioGrid.
Overall, the results, and the considerations made, show that the structural characterization of ORF7b-2 is an absolutely necessary prerequisite to understand its behavior, both in solution and in membrane, but also to understand the functional potential that this protein can exert. However, ORF7b-2 is also a protein with too many molecular partners, although the functions resulting from these interactions are still unknown. Thus, we need to find its targets to discover its many functional aspects and, eventually, to characterize them (manuscript in preparation). One way to get answers capable of untangling this functional dilemma is through interactomics analysis [90,91]. If we possess the human molecular interactors of this protein, as we have them in BioGrid, we can also extract the hidden functional relationships in the human proteome through a PPI network. This is an analysis indispensable for ORF7b of SARS-CoV-2.
4. Materials and Methods
Electrostatic properties - The charge distribution of the proteins was evaluated in agreement with Das and Pappu [20,21,22,23]. Particularly, we calculated the fraction of charged residues, as FCR = |f+ + f_|, and the net charge per residue, as NCPR = |f+ - f_|. In this context, f+ and f_ represent the fraction of positive and negative charges, respectively. These calculated values allow one to classify the protein sequences into distinct regions of the Diagram of States for IDPs: [22] (i) weak polyampholytes and polyelectrolytes named as Region 1 with values of FCR<0.25 and NCPR<0.25 and propensity for ensembles of Globule and Tadpole; (ii) a boundary region or Region 2 between 1 and 3 characterized by 0.25 ≤ FCR ≤ 0.35 and NCPR ≤ 0.35 values; (iii) strong polyampholytes (Region 3) with FCR > 0.35 and NCPR ≤ 0.35, and propensity for ensembles of Coils, Hairpins, and Chimeras; and (iv) strong polyelectrolytes (Region 4) where FCR > 0.35 and NCPR > 0.35, with a propensity for ensembles of Swollen Coils.
Finally, we have calculated the parameter k to distinguish between different sequence variants based on the linear sequence distributions of oppositely charged residues [20-22). The overall charge asymmetry was calculated as σ = (f+ - f_)2/(f+ + f_). For each sequence variant, we calculated k by partitioning the sequence into Nblob overlapping segments of size g. For each g residue segment, we calculated σί = (f+ - f_)2ί/(f+ + f_)ί , which is the charge asymmetry for the sequence of interest. We quantified the squared deviation from σ as
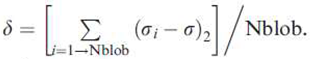
In particular, has been used g = 5 and hypothesized different sequence variants on which were evaluated different values of δ. Hence, the maximal value δmax for an amino acid composition was used to define k = (δ/δmax).
Net Charge Calculation - The net charges of proteins at a given pH are based on the formula below:
Z = ∑i Ni [10pKai/(10pH + 10pKai)] - ∑j Nj [10pH/(10pH + 10pKaj)]
Where Z is the Net charge of the peptide sequence. Ni: Number of arginine, lysine, and histidine residues and the N-terminus; pKai, pKa values of the N-terminus and the arginine, lysine, and histidine residues; Nj, Number of aspartic acid, glutamic acid, cysteine, and tyrosine residues and the C-terminus; pKaj, pKa values of the C-terminus and the aspartic acid, glutamic acid, cysteine and tyrosine residues pH: pH values. The pKa values used for: cysteine (pKa = 8.33), aspartic acid (pKa = 3.86), glutamic acid (pKa = 4.25), histidine (pKa = 6.0), lysine (pKa = 10.53), arginine (pKa = 12.48), tyrosine (pKa = 10.07), the N-terminal (pKa = 9.69) and C-terminal (pKa = 2.34). The isoelectric point is the pH at which the peptide Z shows zero value. Formulas and pKa value from Biochemistry text books.
Dipole moment - The dipole moment, in Debyes, is calculated as the magnitude of the dipole vector D = 4.803*Σriqi, summing over all atoms 'i ', where 4.803 converts from Angstrom-electron-charge units to Debyes. The mass moment vector of the protein is calculated as Rx =Σxi2, Ry=Σyi2, and Rz=Σzi2, and the associated mean radius RM = [(Rx + Ry + Rz)/3]1/2 is a measure of the overall protein size. The server at the following address was also used for the calculations: http://bip.weizmann.ac.il/dipol
CIDER (Classification of Intrinsically Disordered Ensemble Regions) is a web-server developed by the Pappu lab [22], at Washington University in St. Louis. CIDER allows for the calculation of many parameters associated with any protein sequences. It is very specific for small proteins. The server is at the address, http://pappulab.wustl.edu/CIDER/analysis/ The calculation of the average hydrophilicity of a peptide is based on the data from Hopp&Woods [91].
PHYRE2, Protein Homology/analogY Recognition Engine V 2.0, is a web portal for protein modeling, prediction and analysis at Structural Bioinformatics Group, Imperial College, London, UK.. (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index )
PEP-FOLD3 is a de novo approach aimed at predicting peptide structures from amino acid sequences through a series of 100 simulations. Each simulation samples a different region of the conformational space (prediction is limited to an amino acid sequence between 5 and 50 residues in FASTA format). It returns an archive of all the models generated by the detail of the clusters and the best conformation of the 5 best clusters. Once complete, a Monte Carlo procedure refines the peptide structure. (https://bioserv.rpbs.univ-paris-diderot.fr/services/PEP-FOLD3/ )
MEMEMBED 1.15 (Bioinformatics Group – University College London) Membrane Protein Orientation Predictor
(https://mybiosoftware.com/memembed-1-15-membrane-protein-orientation-predictor.html ) accurately orientates and refines both alpha-helical and beta-barrel membrane proteins within the lipid bilayer using a genetic algorithm and knowledge-based statistical potential. The Workbench provides a range of protein structure prediction methods. The site can be used interactively via a web browser or programmatically via our REST API.
PEPPI (Pipeline for the Extraction of Predicted Protein-protein Interactions) (https://seq2fun.dcmb.med.umich.edu/PEPPI/) is a computational program for protein-protein interaction (PPI) prediction. It is a whole-proteome protein-protein interaction prediction through structure and sequence similarity, functional association, and machine learning.
HINGEProt (http://bioinfo3d.cs.tau.ac.il/HingeProt/hingeprot.html ) is an Algorithm For Protein Hinge Prediction Using Elastic Network Models [95]. HingeProt makes use of both Gaussian Network Model (GNM) decomposes the fluctuations of N residues of a structure into a series of N-1 nonzero modes, given the Cartesian coordinates of Ca atoms [96]. The eigenvectors corresponding to the slowest first and second modes are extracted. The square of these vectors describes the mean-square fluctuations (the autocorrelations) of residues from equilibrium positions along the principal coordinates (first and second modes here). Minima of mean square fluctuations at a mode describe the flexible joints of the structure, i.e. the hinge regions, which connect the rigid units and mobile loops. The hinge regions are the mechanistically informative regions of the structure and are of importance in mediating cooperative motions that have functional importance. GNM calculates the mean-square fluctuations and the correlation between the fluctuations of residues in the most dominant (slowest two) modes, which were shown to overlap with known protein motions. These suggest hinge regions and the cooperation between them. ANM provides the direction of the fluctuations in the corresponding modes. GNM fluctuations being isotropic by definition, the directions of fluctuations are characterized by ANM. ANM predicts the fluctuations of N residues in the x, y and z directions from the average structure (X-ray or NMR) in 3N-6 ANM nonzero modes [97]. After mapping the ANM modes to GNM modes by comparing the square fluctuations between the resulting modes in the two models, the directions of the fluctuations of residues in the slowest first and second modes of GNM are got by ANM analysis. As the fluctuations are symmetric regarding the equilibrium positions, ANM predicted deformed structures could be got by adding and subtracting the fluctuations of each residue to/from its equilibrium position.
Molecular Dynamics - The best model of ORF7b-2 was subjected to molecular dynamics (MD) simulations by the GROMACS software (v4.5.6) [92,93] using GROMOS43a1 all-atom force field at neutral pH. In a recent paper of ours [94], this force field has been evaluated among the most suitable to simulate the folding of short peptides. In particular, the model was put into a cubic box with sides of 86.2 Å and covered with 21329 SPC216 water molecules. Initially, 2000 steps of energy minimization and 25000 steps of position restrains were executed to equilibrate the protein and to balance the water molecules around the protein, respectively. The complete 3D structure of ORF7b-2 was subjected to MD simulations for 40 ns in explicit water setting the time step at 2 fs, the temperature at 300 K and the time constant at 0.1 ps, pH 7.0.
A second set of experiments was performed in a solvated lipid bilayer under similar experimental conditions in the presence of a dimeric 3D structure of ORF7b-2. This structure was modeled using HDOCK. For this purpose, we prepared a lipid bilayer composed of 130 POPC (phosphatidylcholine) molecules by 'VMD membrane builder' plugin and placed the dimeric model of ORF7b-2 in the membrane based on not only the hydrophobicity of its residues but using the model pre-oriented by the 'Orientations of Protein in Membranes (OPM)' database (http://opm.phar.umich.edu) to provide a rigorous way, based on energetic and thermodynamic properties, of how the helix may be embedded in the membrane.. The OPM model is shown in Supplements (Figure 12S). When the correctly oriented helix was inserted into the membrane, all the system was solvated in a box of 10985 water molecules, also ionized using VMD and processed according to three steps: (i) equilibration and melting of lipid tails, (ii) minimization and equilibration with constrained protein and (iii) equilibration with released protein. After these three steps, the entire system was subjected to MD simulation for 100 ns, at 300 K and neutral pH.
Molecular dynamics (MD) analysis - The trajectories, which contain the information about the coordinates in terms of time evolution of all the atoms making up the system, were analyzed using different GROMACS routine utilities, which include the root-mean-square deviation (RMSD), gyration radius (Rg), root-mean-square fluctuations (RMSF), helicity, total solvent accessible area, ASA and so forth. Relevant functional motions were calculated by Principal Components Analysis (PCA). The number of H-bonds, and interactions with their closest atoms (IAC) were calculated using the Protein Interactions Calculator (PIC),55 HBPLUS56 and COCOMAPS57 tools.
(ORF7b-2-ORF7b-2) Docking - HDOCK server (http://hdock.phys.hust.edu.cn/), a web server for protein-protein docking based on a hybrid strategy [106], was used to model ORF7b-2 dimerization in silico. The information entered for receptor and ligand molecules was the best ORF7b-2 Phyre2-model. The server automatically predicts their interaction through a hybrid algorithm of template-based and template-free docking. Data input that accepts both sequence and structure is the first step of the process. The second step of the workflow is a sequence similarity search. Given the sequences from input or converted from structures, a sequence similarity search is conducted against the PDB sequence database to find the homologous sequences for both receptor and ligand molecules. The third step compares PDB codes and a common template will be selected for both receptor and ligand. If there is no overlap between two sets of homologous templates, the best templates will be selected for the receptor protein and/or the ligand protein from two sets of templates, respectively. If multiple templates are available, the one with the highest sequence coverage, the highest sequence similarity and the highest resolution will be selected. With the selected templates, models are built using MODELLER, in which the sequence alignment is conducted using ClustalW. The last step is traditional global docking. Here, HDOCKlite, a hierarchical FFT-based docking program, is used to sampling putative binding orientations. The top 10 docking models are interactively provided to users through a web page.
Orientations of Proteins in Membranes (OPM) database - OPM provides spatial arrangements of membrane proteins regarding the core of the lipid bilayer [107]. OPM provides preliminary results of a computational analysis of transmembrane α-helix binding in experimental structures for dimeric proteins. On PPM3 server, proteins are positioned in a bilayer of adjustable thickness and curvature to minimize their transfer energy from water to the membrane. Each protein is considered as a rigid body floating in a hydrophobic slab of adjustable thickness. In our experiment, a membrane with a Golgi-like composition with a thickness of 29.4 ± 2.7 Å was settled. Orientation of the proteins was determined by minimizing its overall transfer energy to – 28.8 kcal/mole regarding variables in a coordinate system whose axis Z coincides with the bilayer normal. Longitudinal axes of TM proteins are calculated as vector averages of TM segment vectors. The resulting tilt angles were 13 ± 2°, and 15 ± 2.5° for the two monomers.
The reason for using the OPM server is to pre-orient probable transmembrane proteins in a lipid sheet system. In this way, a reduction of the equilibration times is got in the simulations of molecular dynamics in the membrane. The results of the orientation are shown in Figure 12S.
Charge distributions and electrostatic potential calculations. Charge distributions and electrostatic potential were calculated by DelPhi [108] using a finite difference solution to the Poisson-Boltzmann equation. DelPhi is an electrostatics simulation program that can investigate electrostatic fields in a variety of molecular systems, including proteins. It is possible for DelPhi to take as input a coordinate file. DelPhi includes solutions to the nonlinear form of the Poisson-Boltzmann equation, which provides more accurate solutions for highly charged proteic systems. Many other features enhance the speed and versatility of DelPhi to handle complicated systems and finite difference lattices of extremely high dimension. We ran the DelPhi executable on a server with Fortran and C compilers. The program can be downloaded at this address:
https://honiglab.c2b2.columbia.edu/software/cgi-bin/software.pl?input=DelPhi. at Columbia University. The input pdb file should be in PQR format, which includes atomic radii and atomic charges. For this purpose, it was used PDB2PQR [109], a Python software package that automates many of the common tasks of preparing structures for continuum electrostatics calculations, providing a platform-independent utility for converting protein files in PDB format to PQR format. For the result, analysis are required to read out and display the potentials. The options were to output a potential map that could be read and contoured into the PyMol (or even Biosym) program. A utility has been provided to facilitate this.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
"This research received no external funding”.
Acknowledgments
I thank Dr. Vincenzo Saviano and Rosario Della Santa (AOU L. Vanvitelli – University of Campania, Naples, Italy) for their help in implementing some software platforms.
Conflicts of Interest
I declare that the content of this manuscript has no conflict of interest.
References
- Redondo N., Zaldivar-Lopez S., Garrido J.J., Montoya M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immun., [2021] VOL 12. ISSN 1664-3224. [CrossRef]
- Shang J, Han N, et al., Compositional diversity and evolutionary pattern of coronavirus accessory proteins. Brief Bioinform. [2021] Mar 22;22[2]:1267-1278. PMID: 33126244; PMCID: PMC7665327. [CrossRef]
- Altincekic N., Korn S.M., et al., Large-Scale Recombinant Production of the SARS-CoV-2 Proteome for High-Thoughput and structural Biology Applications. Front. Mol. Biosci. [2021], Vol 8, Article 653148.
- Yang R, Zhao Q, et al., SARS-CoV-2 Accessory protein ORF7b Mediates Tumor Necrosis Factor-α-Induced Apoptosis in Cells. Front. Microbiol. [2021] 12:654709. [CrossRef]
- Ramasamy S, Subbian S. Critical determinants of cytokine storm and type I interferon response in COVID-19 pathogenesis. Clin. Microbiol. Rev. [2021]: 34:e00299-20. [CrossRef]
- Zhang, J., Cruz-cosme, R., et al.. A systemic and molecular study of subcellular localization of SARSCoV-2 proteins. Signal Transduct. Target. Ther. [2020] 5:269. [CrossRef]
- Schaecher SR, Diamond MS, Pekosz A. The transmembrane domain of the severe acute respiratory syndrome coronavirus ORF7b protein is necessary and sufficient for its retention in the Golgi complex. J Virol. [2008] Oct;82[19]:9477-91. Epub 2008 Jul 16. PMID: 18632859; PMCID: PMC2546951. [CrossRef]
- Schaecher, S. R., J. M. Mackenzie, and A. Pekosz.. The ORF7b protein of severe acute respiratory syndrome coronavirus [SARS-CoV] is expressed in virus-infected cells and incorporated into SARS-CoV particles. J. Virol. [2007] 81718-731. [CrossRef]
- Liu DX, Fung TS, Chong KK, Shukla A, Hilgenfeld R. Accessory Proteins of SARS-CoV and Other Coronaviruses. Antiviral Res [2014] 109:97–109. [CrossRef]
- Debnath P, Khan U, Khan MS. Characterization and Structural Prediction of Proteins in SARS-CoV-2 Bangladeshi Variant Through Bioinformatics. Microbiol Insights. [2022] Aug 9; 15:11786361221115595. PMID: 35966939; PMCID: PMC9373114. [CrossRef]
- Mitch Leslie. A viral arsenal. SARS-CoV-2 wields versatile proteins to foil our immune system's counterattack. Science, [2022], vol 378, 6616, 128-131. [CrossRef]
- Marie-Laure Fogeron, Roland Montserret, et al., SARS-CoV-2 ORF7b: is a bat virus protein homologue a major cause of COVID-19 symptoms? - bioRxiv prep. [2021]. [CrossRef]
- Szente L, Singhal A, Domokos A, Song B. Cyclodextrins: Assessing the Impact of Cavity Size, Occupancy, and Substitutions on Cytotoxicity and Cholesterol Homeostasis. Molecules. [2018] May 20;23[5]:1228. PMID: 29783784; PMCID: PMC6100472. [CrossRef]
- Toft-Bertelsen TL, Jeppesen MG, et al., Amantadine has potential for the treatment of COVID-19 because it inhibits known and novel ion channels encoded by SARS-CoV-2. Commun Biol. [2021] Dec 1;4[1]:1347. Erratum in: Commun Biol. 2021 Dec 10;4[1]:1402. PMID: 34853399; PMCID: PMC8636635. [CrossRef]
- Scott R. Schaecher, Jason M. Mackenzie, Andrew Pekosz. The ORF7b Protein of Severe Acute Respiratory Syndrome Coronavirus [SARS-CoV] Is Expressed in Virus-Infected Cells and Incorporated into SARS-CoV Particles. J. Virol. [2007], Vol. 81, No. 2. [CrossRef]
- Campen, A., Williams, RM, Brown, C.J., Meng, J., Uversky, V.N., Dunker, A.K. TOP-IDP-scale: a new amino acid scale measuring propensity for intrinsic disorder. [2008] Protein Pept Lett. 15[9] pp 956 – 963.
- Huyghues-Despointes BM, Scholtz JM, Baldwin RL. Effect of a single aspartate on helix stability at different positions in a neutral alanine-based peptide. Protein Sci. [1993] Oct;2[10]:1604-11. PMID: 8251935; PMCID: PMC2142265. [CrossRef]
- Guruprasad, K., Reddy, B.V.B. and Pandit, M.W. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. [1990] 4,155-161. [PubMed: 2075190].
- Deller MC, Kong L, Rupp B. Protein stability: a crystallographer's perspective. Acta Crystallogr F Struct Biol Commun. [2016] Feb;72[Pt 2]:72-95. Epub 2016 Jan 26. PMID: 26841758; PMCID: PMC4741188. [CrossRef]
- Das RK, Pappu RV. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. PNAS [2013] Aug 13;110[33]:13392-7. Epub 2013 Jul 30. PMID: 23901099; PMCID: PMC3746876. [CrossRef]
- Lyle N, Das RK, Pappu RV. A quantitative measure for protein conformational heterogeneity. J Chem Phys. [2013] Sep 28;139[12]:121907. PMID: 24089719; PMCID: PMC3724800. [CrossRef]
- Holehouse AS, Das RK, Ahad JN, Richardson MO, Pappu RV. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys J. [2017] Jan 10;112[1]:16-21. PMID: 28076807; PMCID: PMC5232785. [CrossRef]
- Zeng X, Ruff KM, Pappu RV. Competing interactions give rise to two-state behavior and switch-like transitions in charge-rich intrinsically disordered proteins. Proc Natl Acad Sci U S A. [2022] May 10;119[19]:e2200559119. Epub 2022 May 5. PMID: 35512095; PMCID: PMC9171777. [CrossRef]
- Gurtovenko AA, Vattulainen I. Membrane potential and electrostatics of phospholipid bilayers with asymmetric transmembrane distribution of anionic lipids. J Phys Chem B. [2008] Apr 17;112[15]:4629-34. Epub 2008 Mar 26. PMID: 18363402. [CrossRef]
- Nordlund JR, Schmidt CF, Thompson TE. Transbilayer distribution in small unilamellar phosphatidylglycerol-phosphatidylcholine vesicles. Biochemistry. [1981] Oct 27;20[22]:6415-20. PMID: 7197988. [CrossRef]
- M.R.Moncelli, L.Becucci, R.Guidelli. The intrinsic pKa values for phosphatidylcholine, phosphatidylethanolamine, and phosphatidylserine in monolayers deposited on mercury electrodes. Biophys. J., [1994], Vol: 66, Issue: 6, Page: 1969-1980. ISSN: 0006-3495. PMCIDPMC1275922 PMID8075331. [CrossRef]
- Kelley LA et al. The Phyre2 web portal for protein modeling, prediction and analysis Nature Protocols [2015] 10, 845-858.
- Lamiable A, Thévenet P, et al., PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. [2016] Jul 8;44[W1]:W449-54.
- G.N. Ramachandran, C. Ramakrishnan & V. Sasisekharan: Stereochemistry of polypeptide chain configurations. J. Mol. Biol. [1963] vol. 7, p. 95-99. PMID 13990617.
- Camproux AC, Gautier R, Tuffery P. A hidden markov model derived structural alphabet for proteins. J Mol Biol. [2004] Jun 4;339[3]:591-605.
- Shen Y, Maupetit J, Derreumaux P, Tufféry P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction J. Chem. Theor. Comput. [2014]; 10:4745-4758.
- William S. Young, Charles L. Brooks III, A Microscopic View of Helix Propagation: N and C-terminal Helix Growth in Alanine Helices, J Mol Biol, [1996] Volume 259, Issue 3, Pages 560-572, ISSN 0022-2836. [CrossRef]
- Wieczorek R, Dannenberg JJ. H-bonding cooperativity and energetics of alpha-helix formation of five 17-amino acid peptides. J Am Chem Soc. [2003] Jul 9;125[27]:8124-9. PMID: 12837081. [CrossRef]
- H. Frauenfelder, S.G. Sligar, P.G. Wolynes The energy landscapes and motions of proteins Science, [1991] 254, pp. 1598-1603.
- Bauer JA, Pavlovič J, Bauerová-Hlinková V. Normal Mode Analysis as a Routine Part of a Structural Investigation. Molecules. [2019] ;24:3293.
- Levy RM, Karplus M. Vibrational Approach to the Dynamics of an α-helix. Biopolymers. [1979] ;18:2465–2495.
- K. Suhre & Y.H. Sanejouand, ElNemo: a normal mode web-server for protein movement analysis and the generation of templates for molecular replacement. N Acid Res, [2004] 32, W610-W614.
- K. Suhre & Y.H. Sanejouand, On the potential of normal mode analysis for solving difficult molecular replacement problems. Acta Cryst. D [2004] vol.60, p796-799, International Union of Crystallography.
- Emekli U, Schneidman-Duhovny D, Wolfson HJ, Nussinov R, Haliloglu T. HingeProt: Automated Prediction of Hinges in Protein Structures. Proteins, [2008] 70[4]:1219-27.
- López-Blanco JR, Chacón P. New generation of elastic network models. Curr Opin Struct Biol. [2016]; 37:46–53.
- Atilgan, A. R., Durell, A. R., et al., Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys J, [2001] 80, 505-515.
- Eldon G. Emberly, Ranjan Mukhopadhyay, Ned S. Wingreen, Chao Tang, Flexibility of alpha-Helices: Results of a Statistical Analysis of Database Protein Structures, JMB, [2003] Volume 327, Issue 1, Pages 229-237,ISSN 0022-2836. [CrossRef]
- T.E. Creighton, Proteins: Structures and Molecular Properties, WH Freeman and Co., New York [1993].
- W.G.J. Hol, P.T. van Duijen, H.J.C. Berendsen The α-helix dipole and the properties of proteins Nature, [1978] 273, pp.443-446.
- Clifford E. Felder, Jaime Prilusky, Israel Silman, and Joel L. Sussman, " A server and database for dipole moments of proteins", Nu Ac Res, [2007] 35, special Web Servers Issue. https://academic.oup.com/nar/article/35/suppl_2/W512/2922221.
- Baruah C, Mahanta S, Devi P, Sharma DK. In Silico Proteome Analysis of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2). J Nanotechnol Nanomaterials. [2021]; 2[1]: 1-19.
- Sedla E., Fedunova D., et al., Polyanion Hydrophobicity and Protein Basicity Affect Protein Stability in Protein-Polyanion Complexes. Biomacromolecules (2009), 10, 2533–2538.
- Sedlák E., Antalı́k M, et al., Interaction of ferricytochrome c with polyanion Nafion, Biochimica et Biophysica Acta (BBA) - Bioenergetics, 1997, Volume 1319, Issues 2–3, Pages 258-266, ISSN 0005-2728. (https://www.sciencedirect.com/science/article/pii/S0005272896001703). [CrossRef]
- Erik Sedlák, Marian Antalík. Coulombic and noncoulombic effect of polyanions on cytochrome c structure. Biopolymers (1998) Vol46, No.3 Pag 145-154. [CrossRef]
- Antalı M., Bágelová J., et al., Effect of varying polyglutamate chain length on the structure and stability of ferricytochrome c. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, (2003) Vol 1646, Issues 1–2, Pages 11-20, ISSN 1570-9639. (https://www.sciencedirect.com/science/article/pii/S1570963902005435). [CrossRef]
- Gong J., Yao, P., et al., Structural Transformation of Cytochrome c and Apo Cytochrome c Induced by Sulfonated Polystyrene. Biomacromolecules (2003), Vol 4 Is 5, pg. 1293-1300. [CrossRef]
- Kokufuta, E., Shimizu, H., et al.,Salt linkage formation of poly(diallyldimethylammonium chloride) with acidic groups in the polyion complex between human carboxyhemoglobin and potassium poly(vinyl alcohol) sulfate. Macromolecules (1981), Vol 14, Is 5, 1178-1180 https://doi.org/10.1021/ma50006a008 ACS . [CrossRef]
- Tsuboi, A.; Izumi, T.; et al., Complexation of Proteins with a Strong Polyanion in an Aqueous Salt-free System, Langmuir 1996, 12, 6295–6303. ACS. [CrossRef]
- Ulmschneider MB, Sansom MS, Di Nola A. Properties of Integral Membrane Protein Structures: Derivation of an Implicit Membrane Potential. Proteins: Struct, Funct Genet. 2005; 59: 252–265. [CrossRef]
- Dong H, Sharma M, Zhou HX, Cross TA. Glycines: Role in Alpha-Helical Membrane Protein Structures and a Potential Indicator of Native Conformation. Biochemistry. 2012;51:4779–4789. ACS. [CrossRef]
- Slusky JS, Dunbrack RL., Jr, Charge Asymmetry in the Proteins of the Outer Membrane. Bioinformatics. 2013; 29: 2122–2128. [CrossRef]
- Monne M, Nilsson I, et al., Positively and Negatively Charged Residues Have Different Effects on the Position in the Membrane of a Model Transmembrane Helix. J Mol Biol. 1998; 284: 1177–1183. [CrossRef]
- Segrest JP, De Loof H, et al., Amphipathic Helix Motif: Classes and Properties. Proteins: Struct, Funct Genet. 1990;8:103–117. [CrossRef]
- Von Heijne G. Recent advances in the understanding of membrane protein assembly and structure. Quart Rev Biophys 2000; 32: 285–307.
- Hessa, T., Kim, H., Bihlmaier, K. et al. Recognition of transmembrane helices by the endoplasmic reticu-lum translocon. Nature 433, 377–381 (2005). [CrossRef]
- Hessa, T., Meindl-Beinker, N., Bernsel, A. et al. Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 (2007). [CrossRef]
- Liezel A. Lumangtad, Thomas W. Bell, The signal peptide as a new target for drug design, Bioorg&Med Chem Lett, [2020] Volume 30, Issue 10, 127115, ISSN 0960-894X. [CrossRef]
- M. Uhlén, L. Fagerberg, B.M. Hallström, et al. Tissue-based map of the human proteome. Science, [2015] 347, p. 1260419.
- J. Dudek, S. Pfeffer, P.-H. Lee, et al. Protein transport into the human endoplasmic reticulum J Mol Biol, [2015] 427, pp. 1159-1175.
- Simon SM, Blobel G. Signal peptides open protein-conducting channels in E. coli. Cell. [1992] May 15;69[4]:677-84. PMID: 1375130. [CrossRef]
- Fang J., Haasl R.J. Dong Y. Lushington G.H. Discover protein sequence signatures from protein-protein interaction data. BMC Bioinformatics. 2005; 6: 277-284.
- Naama Aviram and Maya Schuldiner, Targeting and translocation of proteins to the endoplasmic reticulum at a glance. J Cell Sci [2017] 130, 4079-4085. [CrossRef]
- Harry F. Noller, Ribosomal RNA and Translation, Annual Review of Biochemistry (1991) Vol. 60: 191-227. [CrossRef]
- Miranda F. Mecha*, Rachel B. Hutchinson*, Jung Ho Lee, and Silvia Cavagnero Protein folding in vitro and in the cell: From a solitary journey to a team effort. Biophysical Chemistry, 2022, Volume 287,106821, ISSN 0301-4622. [CrossRef]
- Matthias P. Mayer Lila M. Gierasch. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones, JBC REVIEWS, (2019), Vol 294, Iss 6, Pgg 2085-2097. [CrossRef]
- Zahn M, Berthold N, Kieslich B, Knappe D, Hoffmann R, Sträter N. Structural studies on the forward and reverse binding modes of peptides to the chaperone DnaK. J Mol Biol. 2013 Jul 24;425(14):2463-79. Epub 2013 Apr 2. PMID: 23562829. [CrossRef]
- Van Durme J, Maurer-Stroh S, Gallardo R, Wilkinson H, Rousseau F, Schymkowitz J. Accurate prediction of DnaK-peptide binding via homology modelling and experimental data. PLoS Comput Biol. 2009 Aug;5(8):e1000475. Epub 2009 Aug 21. PMID: 19696878; PMCID: PMC2717214. [CrossRef]
- Baeza-Delgado C, Marti-Renom MA, Mingarro I. Structure-based statistical analysis of transmembrane helices. Eur Biophys J. 2013;42(2–3):199–207. pmid:22588483.
- Killian J.A., von Heijne G. How proteins adapt to a membrane-water interface. Trends Biochem. Sci. 2000; 25: 429-434.
- Lundin C., Kim H., Nilsson I., White S.H., von Heijne G. Molecular code for protein insertion in the endoplasmic reticulum membrane is similar for N(in)-C(out) and N(out)-C(in) transmembrane helices. Proc. Natl. Acad. Sci. USA. 2008; 105: 15702-15707.
- Wang S., Munro R., et al., Paramagnetic Relaxation Enhancement Reveals Oligomerization Interface of a Membrane Protein J. Am. Chem. Soc. 2012, 134, 41, 16995–16998. [CrossRef]
- Gupta, K., Donlan, J., Hopper, J. et al. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature 541, 421–424 (2017). [CrossRef]
- Keren Limor-Waisberg, Asaf Carmi, Avigdor Scherz, Yitzhak Pilpel, Itay Furman, Specialization versus adaptation: two strategies employed by cyanophages to enhance their translation efficiencies, Nucleic Acids Research, Volume 39, Issue 14, 1 August 2011, Pages 6016–6028. [CrossRef]
- Rampersad S, Tennant P. Replication and Expression Strategies of Viruses. Viruses. 2018:55–82. Epub 2018 Mar 30. PMCID: PMC7158166. [CrossRef]
- Stanley J Opella, Relating structure and function of viral membrane-spanning miniproteins, Current Opinion in Virology, Volume 12,2015, Pages 121-125, ISSN 1879-6257. [CrossRef]
- DiMaio D. Viral miniproteins. Annu Rev Microbiol. 2014;68:21-43. Epub 2014 Apr 10. PMID: 24742054; PMCID: PMC4430842. [CrossRef]
- Zhou HX, Pang X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem Rev. 2018 Feb 28;118(4):1691-1741. Epub 2018 Jan 10. PMID: 29319301; PMCID: PMC5831536. [CrossRef]
- Duarte, J. M., Biyani, N., Baskaran, K. & Capitani, G. An analysis of oligomerization interfaces in transmembrane proteins. BMC Struct. Biol. 13, 21 (2013).
- Stanley J Opella, Relating structure and function of viral membrane-spanning miniproteins, Current Opinion in Virology, Volume 12,2015, Pages 121-125, ISSN 1879-6257. [CrossRef]
- Chanel C. La, Lily E. Takeuchi, Srinivas Abbina, Sreeparna Vappala, Usama Abbasi, and Jayachandran N. Kizhakkedathu. Targeting Biological Polyanions in Blood: Strategies toward the Design of Therapeutics. Biomacromolecules 2020 21 (7), 2595-2621. [CrossRef]
- Alphonse DeLucia, Thomas W. Wakefield, et al., Efficacy and toxicity of differently charged polycationic protamine-like peptides for heparin anticoagulation reversal, Journal of Vascular Surgery, Volume 18, Issue 1, 1993, Pages 49-60, ISSN 0741-5214. (https://www.sciencedirect.com/science/article/pii/074152149370014P). [CrossRef]
- Armando S. Flores-Torres, Mario C. Salinas-Carmona, Eva Salinas, and Adrian G. Rosas-Taraco Eosinophils and Respiratory Viruses, Viral Immunology VOL. 32, NO. 5 Published Online: 4 Jun 2019. [CrossRef]
- Helene F. Rosenberg, Kimberly D. Dyer, Joseph B. Domachowske, Respiratory viruses and eosinophils: Exploring the connections, Antiviral Research, Volume 83, Issue 1, 2009, Pages 1-9, ISSN 0166-3542. (https://www.sciencedirect.com/science/article/pii/S0166354209002988). [CrossRef]
- Oughtred R, Rust J, et al., The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. [2020] Oct 18.
- Helisa H. Wippel, Juan D. Chavez, Xiaoting Tang, James E. Bruce, Quantitative interactome analysis with chemical cross-linking and mass spectrometry, CuOpChemBiol, [2022] Volume 66, 102076, ISSN 1367-5931. [CrossRef]
- Braun, P., Interactome mapping for analysis of complex phenotypes: Insights from benchmarking binary interaction assays. Proteomics, [2012] 12: 1499-1518. https://doi.org/10.1002/pmic.201100598. Hopp T.P., and Woods K.R. Amino acid scale: Hydrophilicity. [1981] Proc. Natl. Acad. Sci. U.S.A. 78:3824-3828.
- B. Hess, C. Kutzner, D. van der Spoel, E. Lindahl, J. Chem. Theory Comput. 2008, 4, 435.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. Shirts, J. Smith, P. Kasson, D. van der Spoel, B. Hess, E. Lindahl, Bioinformatics 2013, 29, 845.
- Raucci, R., Colonna, G., Castello, G. et al. Peptide Folding Problem: A Molecular Dynamics Study on Polyalanines Using Different Force Fields. Int J Pept Res Ther 19, 117–123 (2013). [CrossRef]
- Emekli U, Schneidman-Duhovny D, Wolfson HJ, Nussinov R, Haliloglu T. (2008) HingeProt: Automated Prediction of Hinges in Protein Structures. Proteins, 70(4):1219-27.
- Bahar, I., Atilgan A. R., Erman, B. (1997) Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Folding and Design, (1997), 2, 173-181.
- Atilgan, A. R., Durell, A. R., Jernigan, R. L., Demirel, M. C. , Keskin, O. , Bahar, I. (2001), Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophysical Journal, 80, 505-515.
- Gordon, D.E., Jang, G.M., et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature [2020] 583, 459–468. [CrossRef]
- Lukasz P. Kozlowski, Proteome-pI: proteome isoelectric point database, in Nu Ac Res, [2017] vol. 45, D1, pp. D1112–D1116, The UniProt Consortium: a hub for protein information. [CrossRef]
- Kyte J., Doolittle R., A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. [1982] 157, pp 105– 132.
- Simon SM, Blobel G. Signal peptides open protein-conducting channels in E. coli. Cell. [1992] May 15;69[4]:677-84. PMID: 1375130. [CrossRef]
- Bevacqua A, Bakshi S, Xia Y (2021) Principal component analysis of alpha-helix deformations in transmembrane proteins. PLOS ONE 16(9): e0257318. [CrossRef]
- Zagrovic B., Jayachandran G.,, et al., How Large is an Helix? Studies of the Radii of Gyration of Helical Peptides by Small-angle X-ray Scattering and Molecular Dynamics, J Mol Biol, [2005] Vol 353, Issue 2, Pags 232-241, ISSN 0022-2836. [CrossRef]
- Deller MC, Kong L, Rupp B. Protein stability: a crystallographer's perspective. Acta Crystallogr F Struct Biol Commun. [2016] Feb;72[Pt 2]:72-95. Epub 2016 Jan 26. PMID: 26841758; PMCID: PMC4741188. [CrossRef]
- S Costantini, G Colonna, AM Facchiano. Amino acid propensities for secondary structures are influenced by the protein structural class. Bioch Biophys Res Comm [2006] 342 [2], 441-451. [CrossRef]
- Yan Y, Tao H, He J, Huang S-Y.* The HDOCK server for integrated protein-protein docking. Nature Protocols, 2020; [CrossRef]
- Lomize AL, Todd SC, Pogozheva ID. (2022) Spatial arrangement of proteins in planar and curved membranes by PPM 3.0. Protein Sci. 31:209-220.
- Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995 May 26;268(5214):1144-9.
- Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res, 35, W522-5, 2007.
- Weller PF, Spencer LA. Functions of tissue-resident eosinophils. Nat Rev Immunol. 2017 Dec;17(12):746-760. Epub 2017 Sep 11. PMID: 28891557; PMCID: PMC5783317. [CrossRef]
- Sharma A, Colonna G., System-Wide Pollution of Biomedical Data: Consequence of the Search for Hub Genes of Hepatocellular Carcinoma Without Spatiotemporal Consideration. Mol Diagn Ther. (2021); 25(1): 9-27. Epub 2021 Jan 21.PMID: 33475988 Review. [CrossRef]
Figure 1.
– The State Diagram shows ORF7b-2 (black) and ORF7b-1 (yellow). ORF7b-2 is a weak polyelectrolyte, negatively charged, with values of FCR < 0.25 and NCPR < 0.25, while ORF7b-1 is a weak polyampholyte, with values of FCR < 0.25 and NCPR < 0.25. Both are in region 1 demonstrate a propensity for ensembles of globules and tadpoles [20,21,81].
Figure 1.
– The State Diagram shows ORF7b-2 (black) and ORF7b-1 (yellow). ORF7b-2 is a weak polyelectrolyte, negatively charged, with values of FCR < 0.25 and NCPR < 0.25, while ORF7b-1 is a weak polyampholyte, with values of FCR < 0.25 and NCPR < 0.25. Both are in region 1 demonstrate a propensity for ensembles of globules and tadpoles [20,21,81].
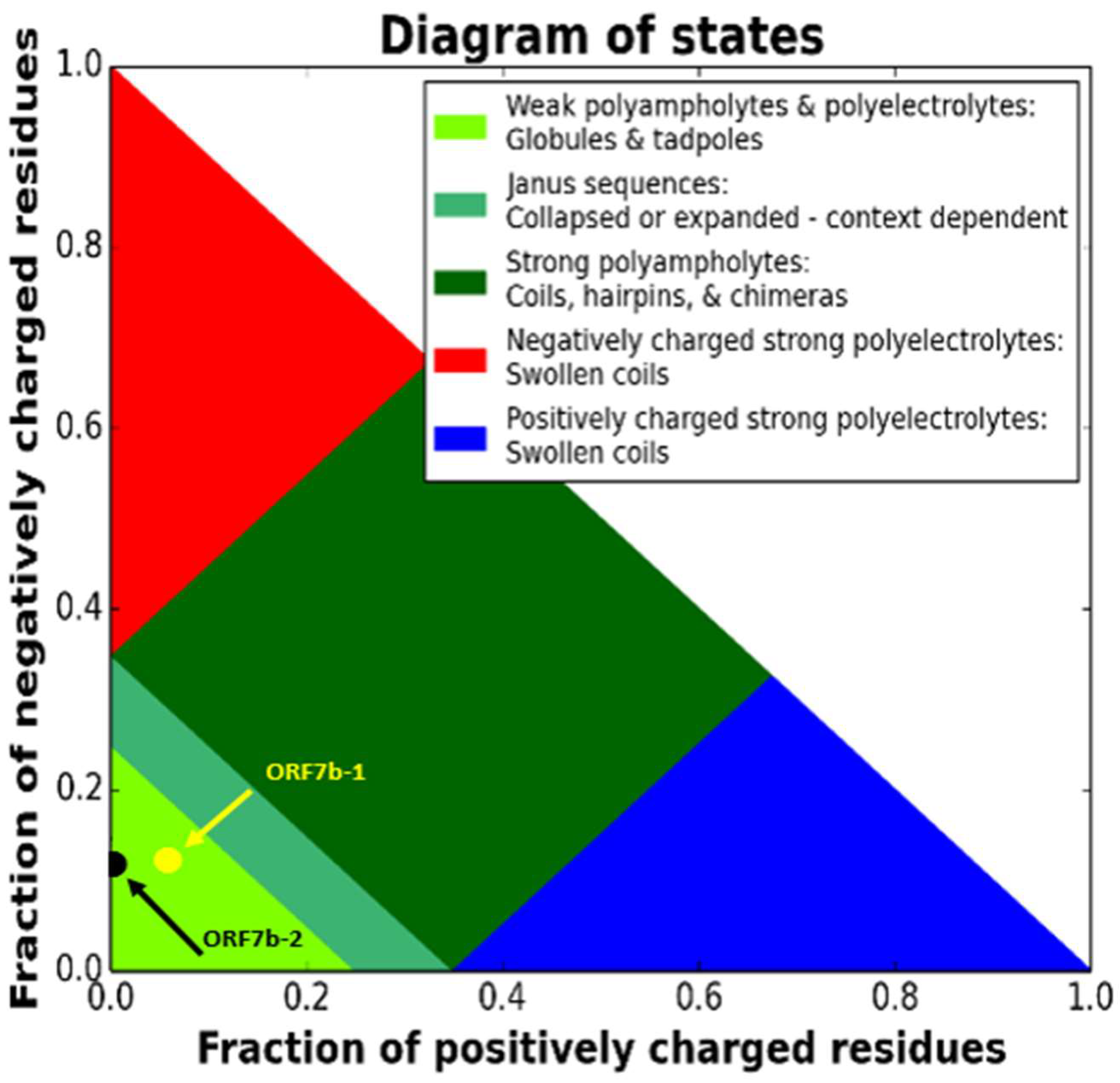
Figure 2.
– Distribution of electrical charges of ORF7b-2 (Top) and ORF7b-1 (Bottom). NCPR, net charge distribution per residue [positive in blue and negative in red], and FCR, the fraction of charged residues. The proteins have a widespread negative surface charge, with fractions of charged residues [FCR] in both terminal segments. Both proteins show a remarkable asymmetry in their charge distribution, with both terminal segments negatively charged. The intensity of the charge distributes a widespread negative charge over the entire structure.
Figure 2.
– Distribution of electrical charges of ORF7b-2 (Top) and ORF7b-1 (Bottom). NCPR, net charge distribution per residue [positive in blue and negative in red], and FCR, the fraction of charged residues. The proteins have a widespread negative surface charge, with fractions of charged residues [FCR] in both terminal segments. Both proteins show a remarkable asymmetry in their charge distribution, with both terminal segments negatively charged. The intensity of the charge distributes a widespread negative charge over the entire structure.
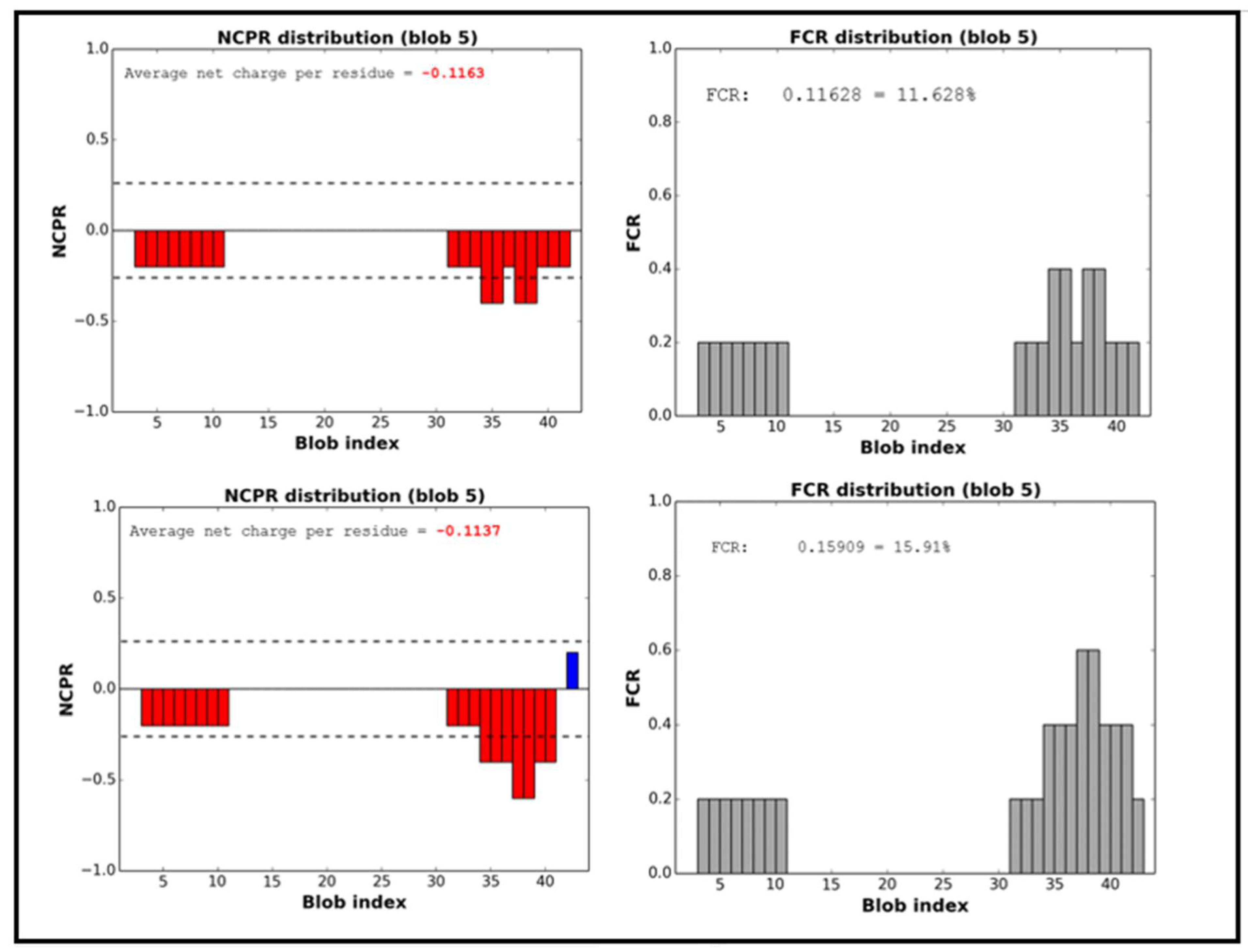
Figure 3.
- The dependence of the net charge [Z] on pH. The figure shows that ORF7b-1 and ORF7b-2 are negatively charged at neutral pH (Z = - 4.08 and – 3.90, respectively). Both remain negatively charged between about pH 4 and 10.
Figure 3.
- The dependence of the net charge [Z] on pH. The figure shows that ORF7b-1 and ORF7b-2 are negatively charged at neutral pH (Z = - 4.08 and – 3.90, respectively). Both remain negatively charged between about pH 4 and 10.
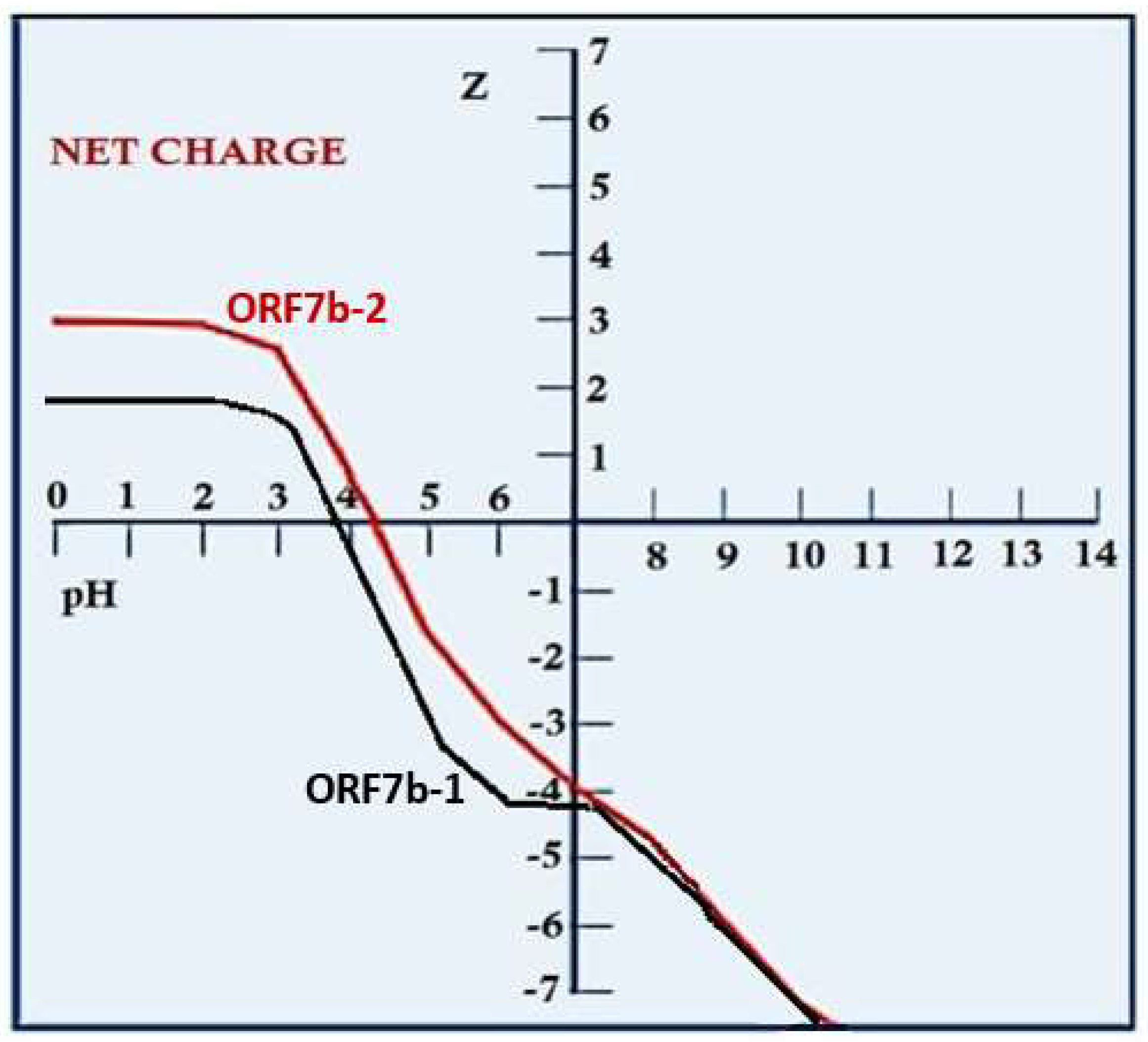
Figure 4.
– The figure shows the best results got from two different platforms for predicting structures, PHYRE2 and PEP-FOLD3. Both use templates for the prediction of the central helical segments [in red] and ab initio methods for the terminal segments [in green]. Experimental details in the Supplements. Folding is presently assumed for neutral pH. [see supplements for details]. The structures have been treated by PyMol (https://pymol.org/2/).
Figure 4.
– The figure shows the best results got from two different platforms for predicting structures, PHYRE2 and PEP-FOLD3. Both use templates for the prediction of the central helical segments [in red] and ab initio methods for the terminal segments [in green]. Experimental details in the Supplements. Folding is presently assumed for neutral pH. [see supplements for details]. The structures have been treated by PyMol (https://pymol.org/2/).
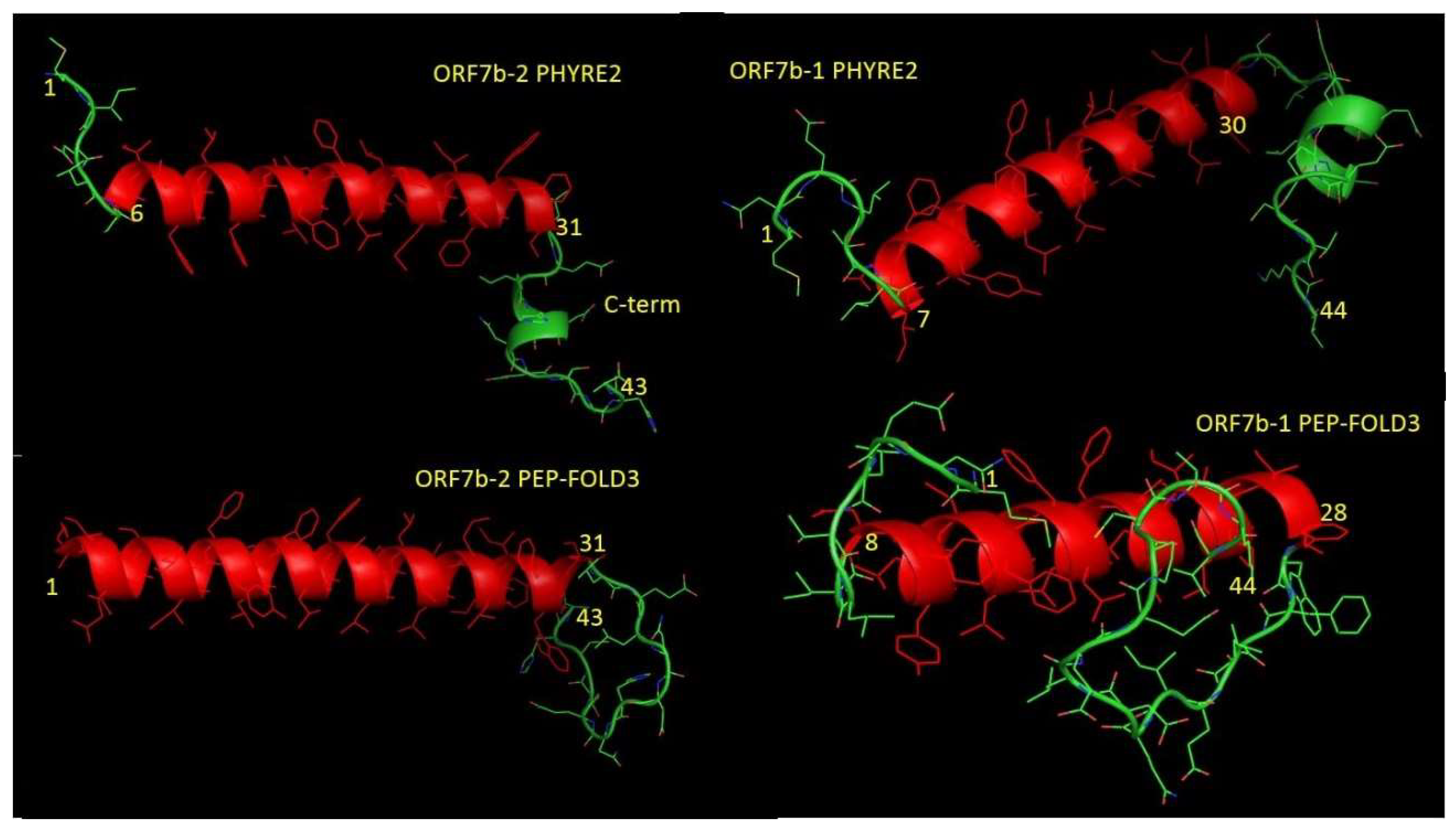
Figure 5.
A – Ramachandran plots of the two 3D models of ORF7b-2. The many residues with anomalous angles in the "extended" zone are all in the terminal sequences. The two modeling systems report similar results. Correct alpha-helical residues are concentrated in the alpha zone [Φ -60° and Ψ -50°]. 3 Glu (top) and 20 Leu (low) are outlier residues. B – Ramachandran plots of the two 3D models of ORF7b-1. We can see residues with anomalous angles are quite spread out, and many are in the terminal sequences. Residues in red are outliers.
Figure 5.
A – Ramachandran plots of the two 3D models of ORF7b-2. The many residues with anomalous angles in the "extended" zone are all in the terminal sequences. The two modeling systems report similar results. Correct alpha-helical residues are concentrated in the alpha zone [Φ -60° and Ψ -50°]. 3 Glu (top) and 20 Leu (low) are outlier residues. B – Ramachandran plots of the two 3D models of ORF7b-1. We can see residues with anomalous angles are quite spread out, and many are in the terminal sequences. Residues in red are outliers.
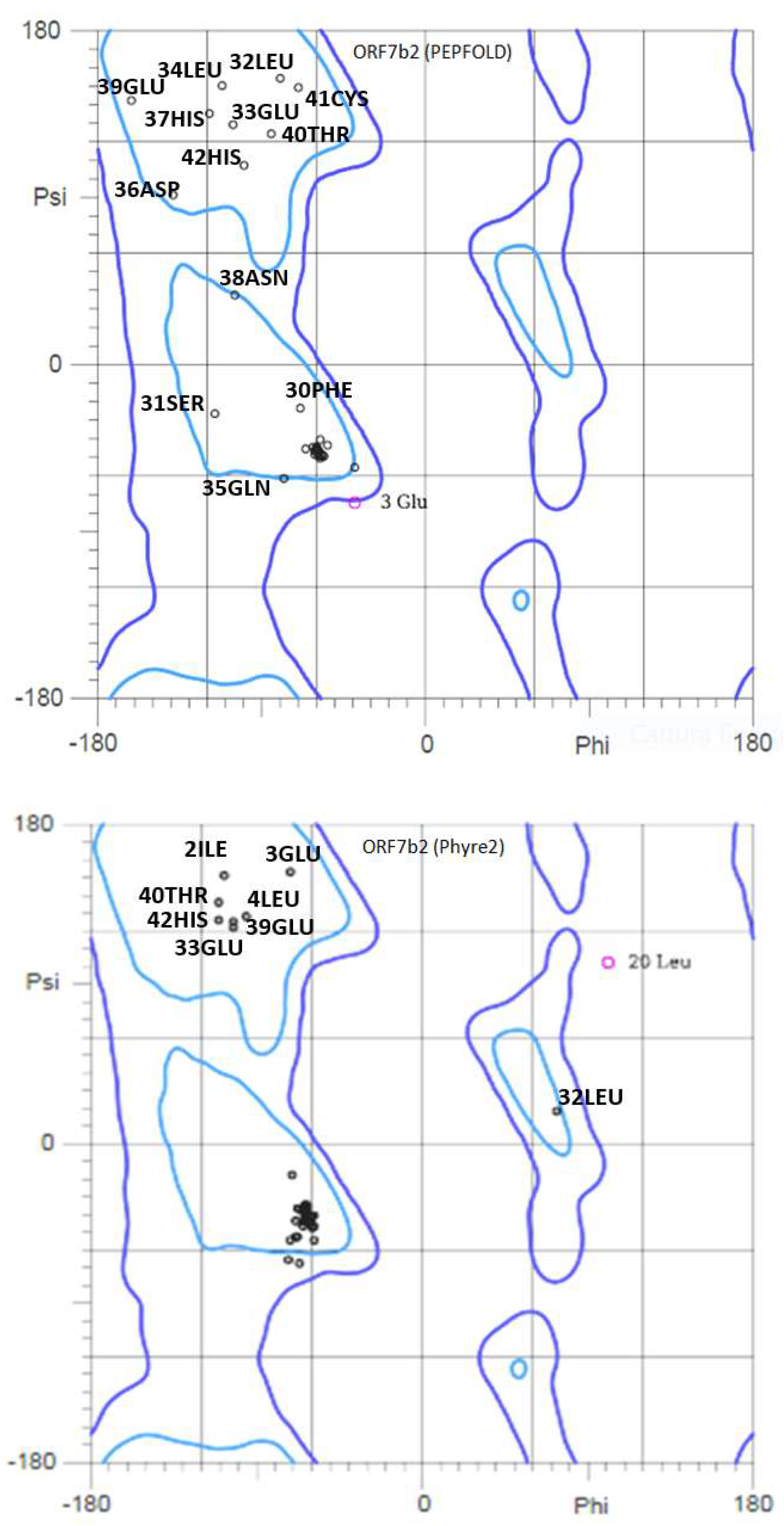
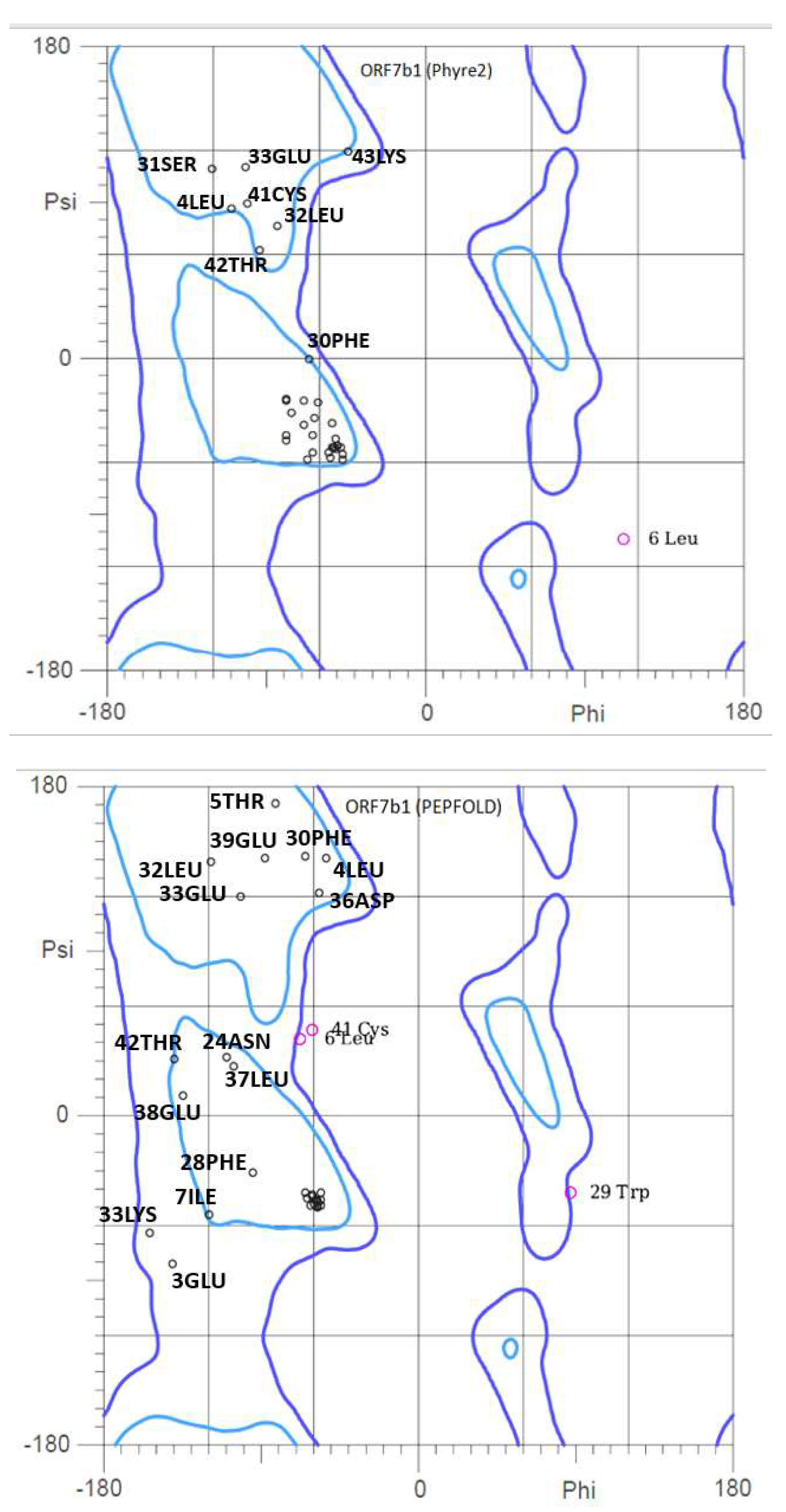
Figure 6.
- The graph shows a graphical representation of the conformational probabilities (0 – 1) for each residue of the two proteins. PEP-FOLD3 is based on the concept of structural alphabet [30], i.e. an ensemble of elementary prototype conformations able to describe the whole diversity of protein structures. The graphical representation shows the probabilities [vertical axis] at each position of the sequence [horizontal axis]. Note that residues correspond to the average of 4 residue. The profile uses the following color code: red: helical, green: extended, blue: coil. The graphs show in conformational terms the effect of the charges on the terminal residues of the two proteins, where at C- level is abundant the extended structure while at N-term level the coil formation.
Figure 6.
- The graph shows a graphical representation of the conformational probabilities (0 – 1) for each residue of the two proteins. PEP-FOLD3 is based on the concept of structural alphabet [30], i.e. an ensemble of elementary prototype conformations able to describe the whole diversity of protein structures. The graphical representation shows the probabilities [vertical axis] at each position of the sequence [horizontal axis]. Note that residues correspond to the average of 4 residue. The profile uses the following color code: red: helical, green: extended, blue: coil. The graphs show in conformational terms the effect of the charges on the terminal residues of the two proteins, where at C- level is abundant the extended structure while at N-term level the coil formation.
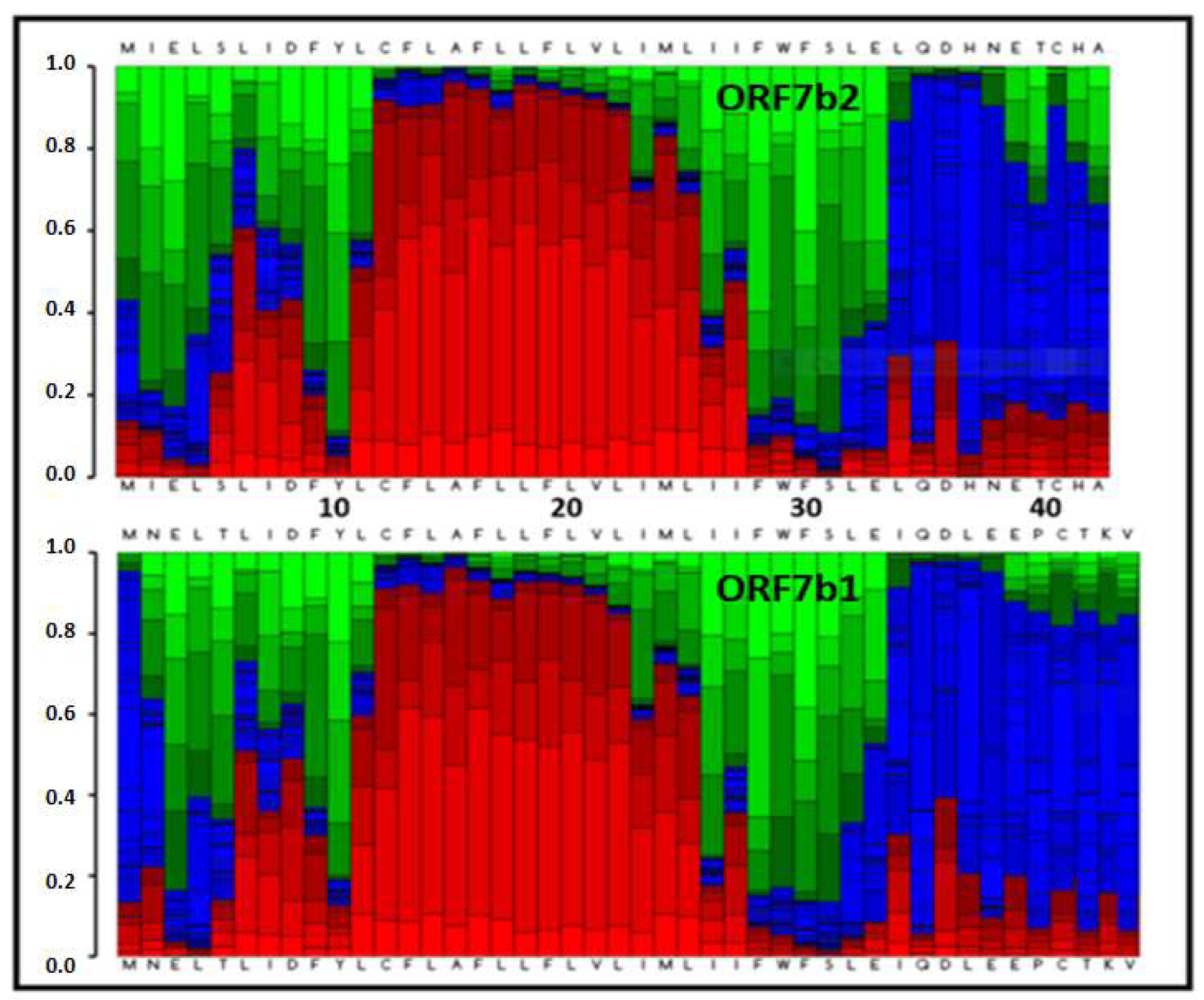
Figure 7.
– Dynamics around the hinge residues of ORF7b-2 [see Table 4]. The hinge position on the model is shown with the residue number. The figures show snapshots of motions from three different views [A, B, and C] and the arrows show the series. Top: Twist movements around residues 9 and 32. Bottom: The backbone shows clear bending movements around residue 21.
Figure 7.
– Dynamics around the hinge residues of ORF7b-2 [see Table 4]. The hinge position on the model is shown with the residue number. The figures show snapshots of motions from three different views [A, B, and C] and the arrows show the series. Top: Twist movements around residues 9 and 32. Bottom: The backbone shows clear bending movements around residue 21.
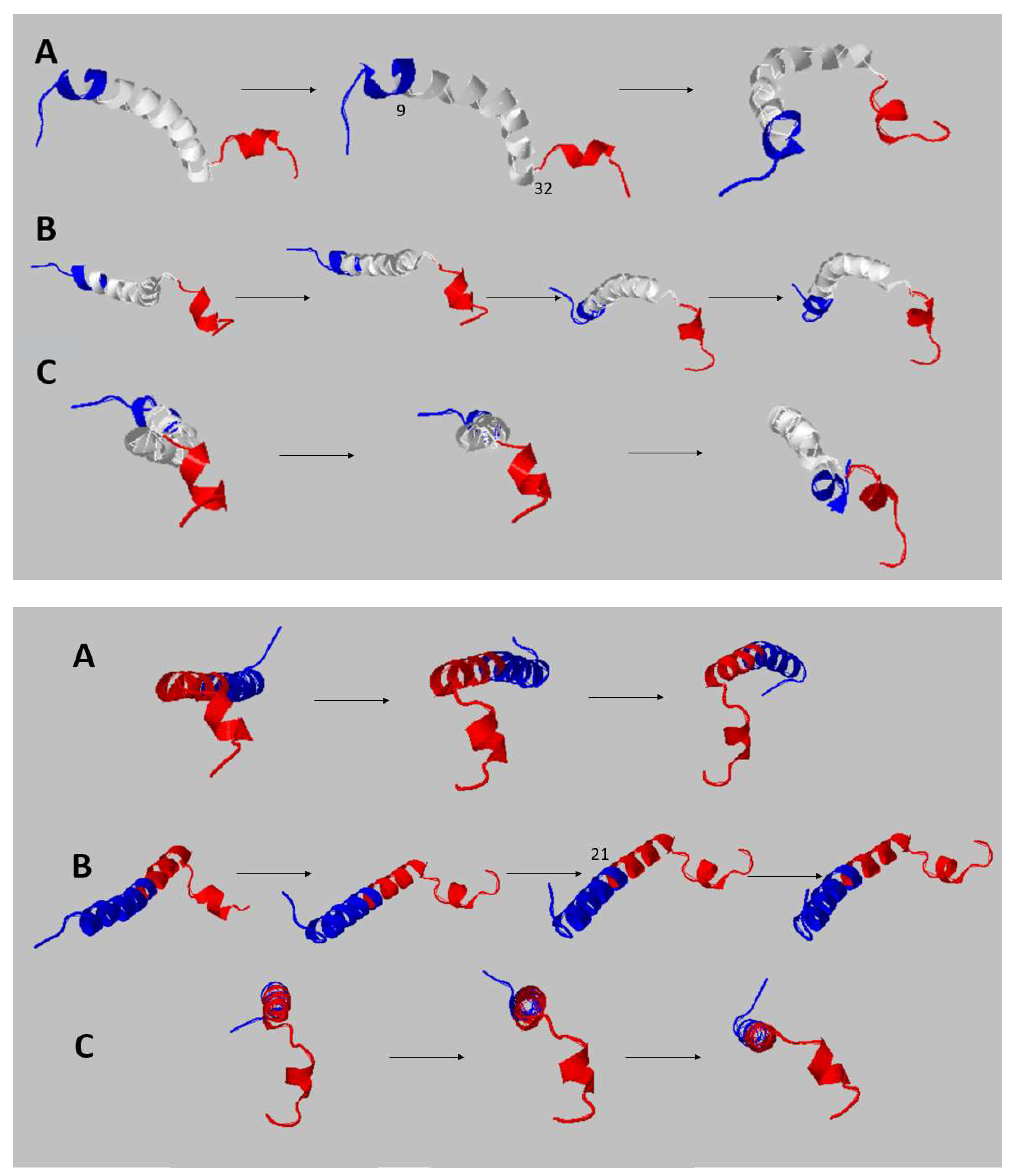
Figure 8.
– Local dynamics of ORF7b-2 - The superimposition of the normal modes shows us the set of local low frequency molecular movements of ORF7b-2. In the upper figure, we have a side view, while in the lower figure we have a view along the major axis of the molecule. The central body of the molecule vibrates [displacement of about 12Å] but remains quite organized, although it deforms, and also shows clear bending at the tails. In the bottom figure, both terminal segments show large fluctuations and displacements of the residues of a few tens of angstroms. We underline that the structural stability of transmembrane proteins relies on a Gly-driven tight packing of the transmembrane helices. A sequence property of ORF7b2 is that it does not exhibit Gly, thus its helix has a low tendency to compact, as confirmed by the dynamic properties of its helix in this normal-mode analysis.
Figure 8.
– Local dynamics of ORF7b-2 - The superimposition of the normal modes shows us the set of local low frequency molecular movements of ORF7b-2. In the upper figure, we have a side view, while in the lower figure we have a view along the major axis of the molecule. The central body of the molecule vibrates [displacement of about 12Å] but remains quite organized, although it deforms, and also shows clear bending at the tails. In the bottom figure, both terminal segments show large fluctuations and displacements of the residues of a few tens of angstroms. We underline that the structural stability of transmembrane proteins relies on a Gly-driven tight packing of the transmembrane helices. A sequence property of ORF7b2 is that it does not exhibit Gly, thus its helix has a low tendency to compact, as confirmed by the dynamic properties of its helix in this normal-mode analysis.
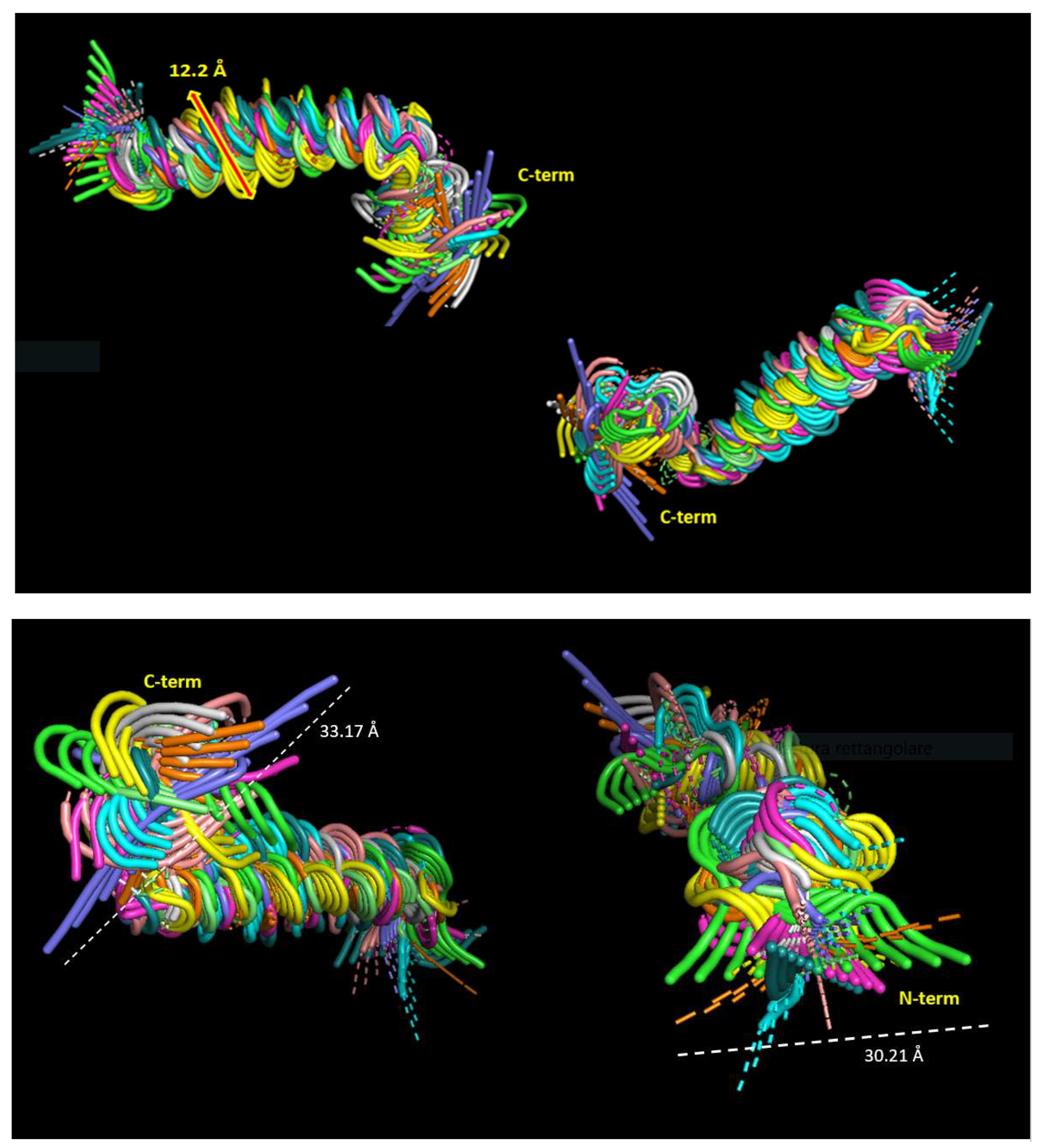
Figure 9.
– The ribbon diagram of ORF7b-2 shows two views from which we can appreciate the strong distortion of the dipole (red) and mass moment (greenish) vectors. The dipole vector is not parallel to the main axis of the protein and points outwards with a tilt of 24°. Both vectors begin at the center-of-mass origin of the protein. The origin of the red dipole line corresponds with the net negative charge and the far end with the net positive charge of the dipole moment. Since the dipole is equivalent to a charge of +0.5 at the N term and -0.5 at the C term, missing positively charged residues at or near the C-cap end with the helix dipole suggests a destabilizing effect because of the lack of favorable interactions with negatively charged residues. This should make stable insertion into a membrane quite difficult. The distances stated in the figure approximate a central helix of 39.07 A and the C-terminal movable element of 17.04 A. Both segments will generate solids of rotation which will converge into the global prolate ellipsoid of the molecule.
Figure 9.
– The ribbon diagram of ORF7b-2 shows two views from which we can appreciate the strong distortion of the dipole (red) and mass moment (greenish) vectors. The dipole vector is not parallel to the main axis of the protein and points outwards with a tilt of 24°. Both vectors begin at the center-of-mass origin of the protein. The origin of the red dipole line corresponds with the net negative charge and the far end with the net positive charge of the dipole moment. Since the dipole is equivalent to a charge of +0.5 at the N term and -0.5 at the C term, missing positively charged residues at or near the C-cap end with the helix dipole suggests a destabilizing effect because of the lack of favorable interactions with negatively charged residues. This should make stable insertion into a membrane quite difficult. The distances stated in the figure approximate a central helix of 39.07 A and the C-terminal movable element of 17.04 A. Both segments will generate solids of rotation which will converge into the global prolate ellipsoid of the molecule.
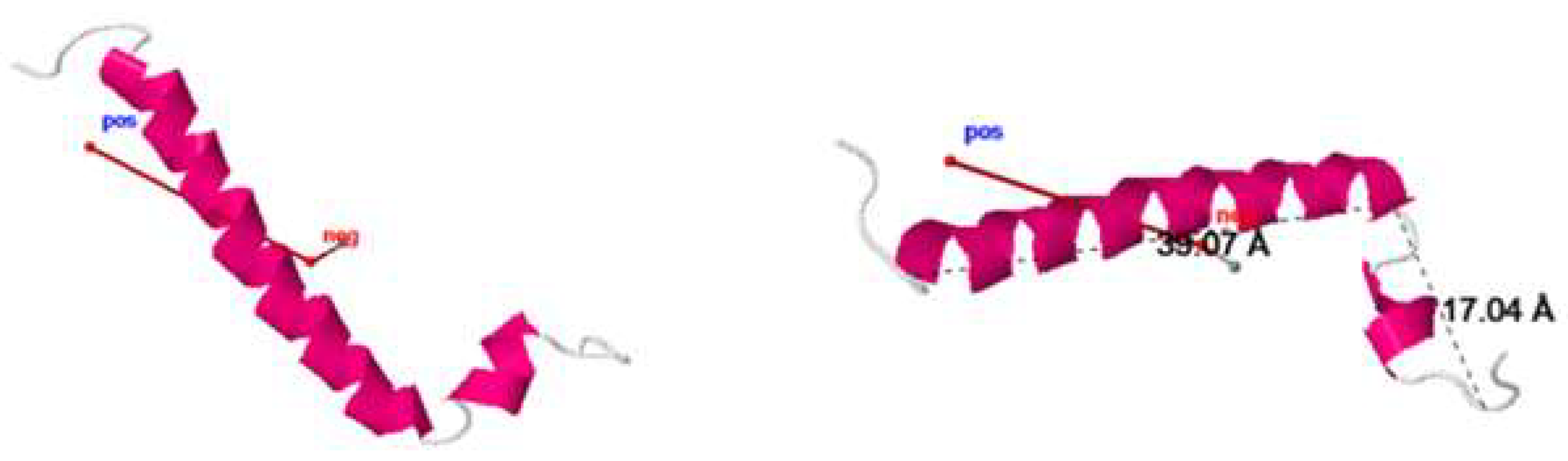
Figure 10.
- The figure shows the trend of the ORF7b-2 molecular dynamics simulation in water. Around 25 ns is when the Protein comes to equilibrium. The simulation shows that the protein is stable in an aqueous environment and the conformational adaptation towards the structural organization at equilibrium shows that the gradual conformational changes of settlement generate electrostatic surfaces very different from each other in terms of charge and extension. The electrostatic surfaces were calculated with DelPhi (see Methods). The small dimensions of the molecule show how even minimal conformational changes are easily reflected in electrostatic variations of its surface.
Figure 10.
- The figure shows the trend of the ORF7b-2 molecular dynamics simulation in water. Around 25 ns is when the Protein comes to equilibrium. The simulation shows that the protein is stable in an aqueous environment and the conformational adaptation towards the structural organization at equilibrium shows that the gradual conformational changes of settlement generate electrostatic surfaces very different from each other in terms of charge and extension. The electrostatic surfaces were calculated with DelPhi (see Methods). The small dimensions of the molecule show how even minimal conformational changes are easily reflected in electrostatic variations of its surface.
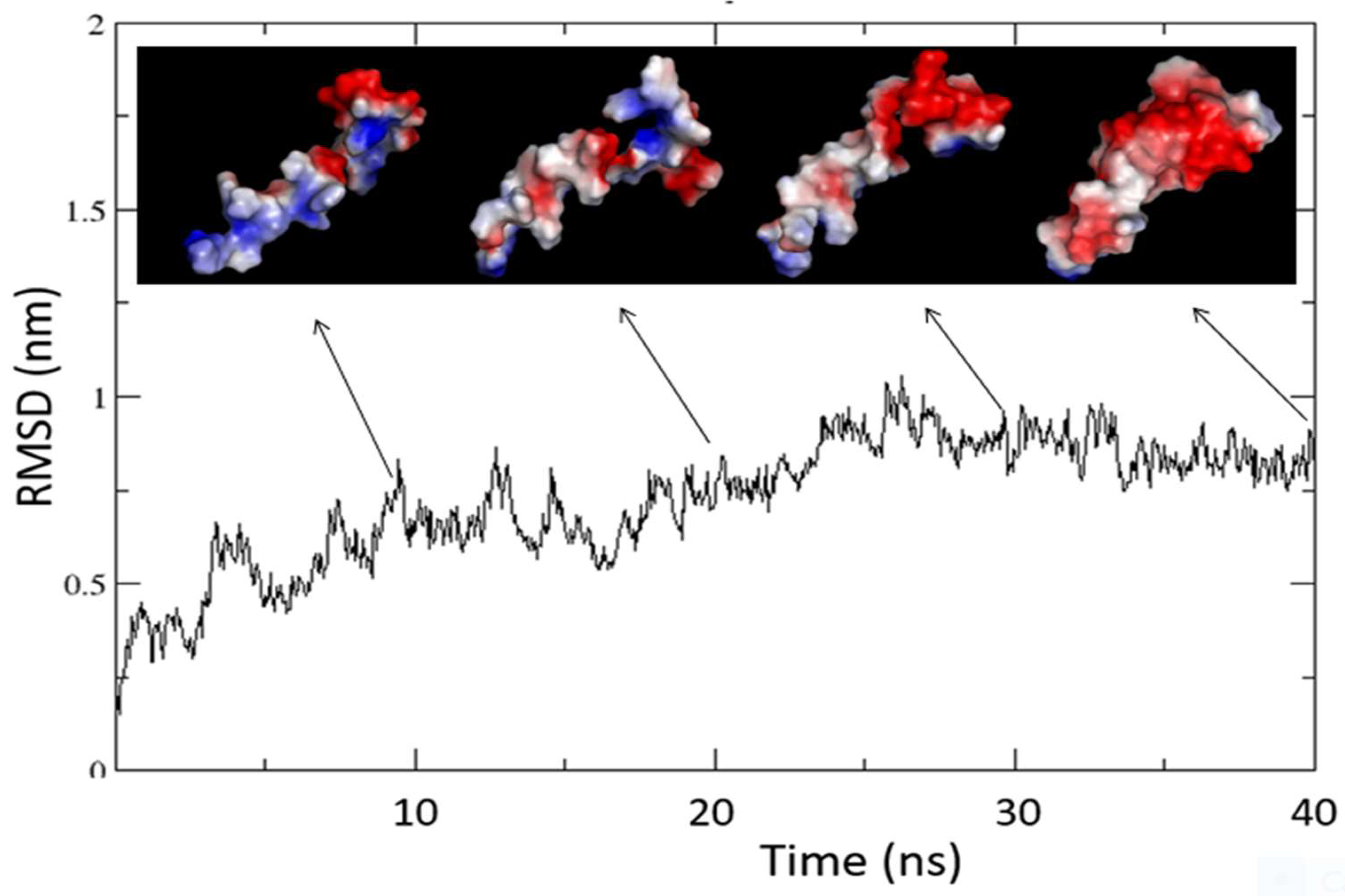
Figure 11.
- The figure shows the main structural features of the ORF7b-2 model got from molecular dynamics in water at neutral pH. The helix extends from L6 to W29 demonstrates bending centered on residues L17 and W21. The representation of its surface shows that the two opposite sides of the protein possess different electrostatic characteristics. One side is covered by a diffuse negative charge (in red) while the other side shows both charged ends (the positive charge in blue is that of the NH3+ terminal) with the central surface predominantly hydrophobic. Electrostatic surfaces were calculated by DelPhi and shown through PyMol.
Figure 11.
- The figure shows the main structural features of the ORF7b-2 model got from molecular dynamics in water at neutral pH. The helix extends from L6 to W29 demonstrates bending centered on residues L17 and W21. The representation of its surface shows that the two opposite sides of the protein possess different electrostatic characteristics. One side is covered by a diffuse negative charge (in red) while the other side shows both charged ends (the positive charge in blue is that of the NH3+ terminal) with the central surface predominantly hydrophobic. Electrostatic surfaces were calculated by DelPhi and shown through PyMol.
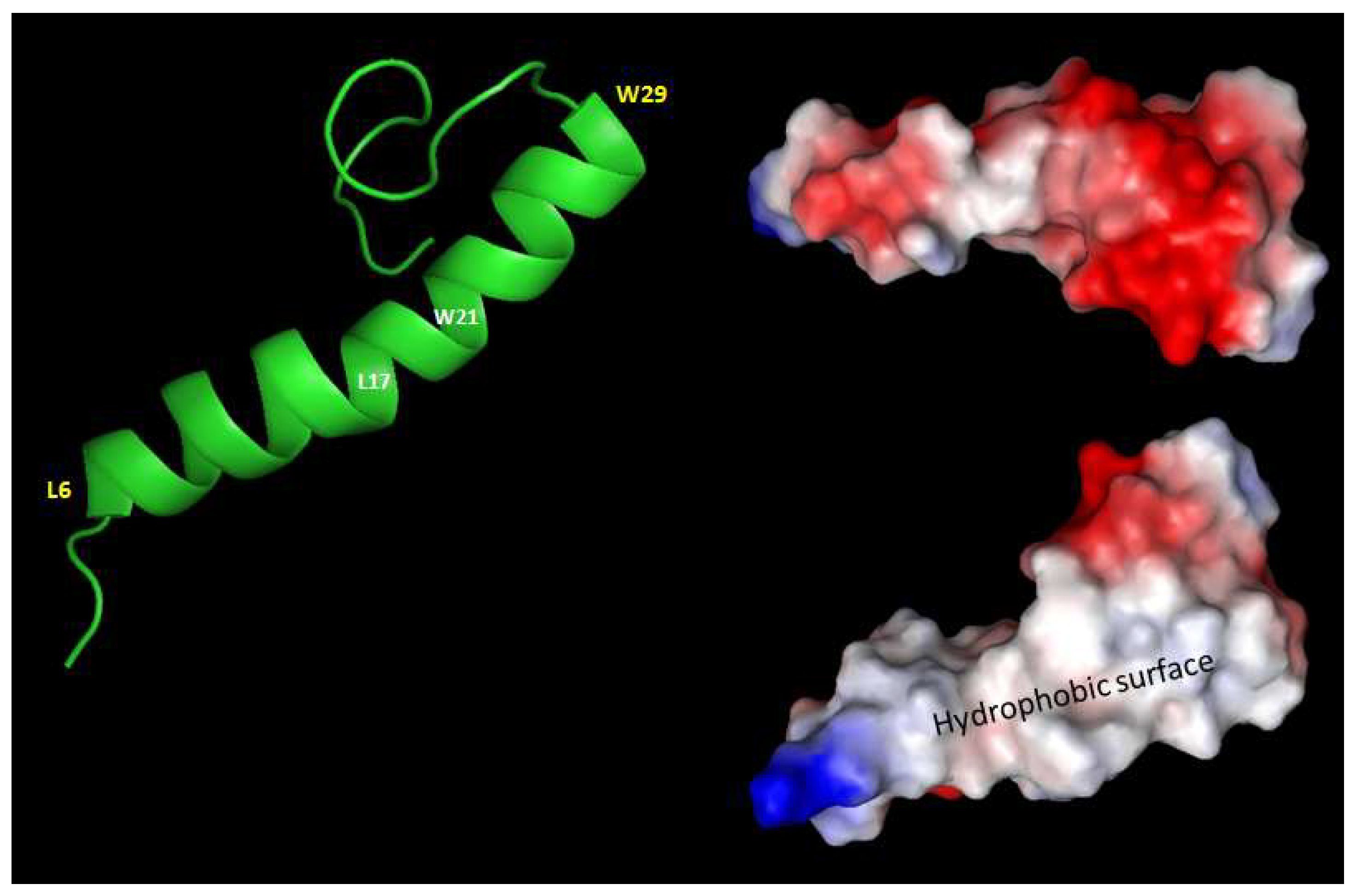
Figure 12.
- The figure shows the trend of the molecular dynamics of the dimer in the membrane. For greater clarity, the structures at the various times are shown without the membrane (we showed structures inside the membrane in the Supplements). The graph contains as an inset the evolution of the total helicity during the 100 ns of simulation. The two graphs show in the same time interval (35 – 55 ns) a transition, quite super-imposable, which suggests a sudden change of structural organization with a concomitant loss of total helicity and an increase in the average distance between the atoms of the global system. In a single experiment, the dynamics were forced up to 200 ns with no variation.
Figure 12.
- The figure shows the trend of the molecular dynamics of the dimer in the membrane. For greater clarity, the structures at the various times are shown without the membrane (we showed structures inside the membrane in the Supplements). The graph contains as an inset the evolution of the total helicity during the 100 ns of simulation. The two graphs show in the same time interval (35 – 55 ns) a transition, quite super-imposable, which suggests a sudden change of structural organization with a concomitant loss of total helicity and an increase in the average distance between the atoms of the global system. In a single experiment, the dynamics were forced up to 200 ns with no variation.
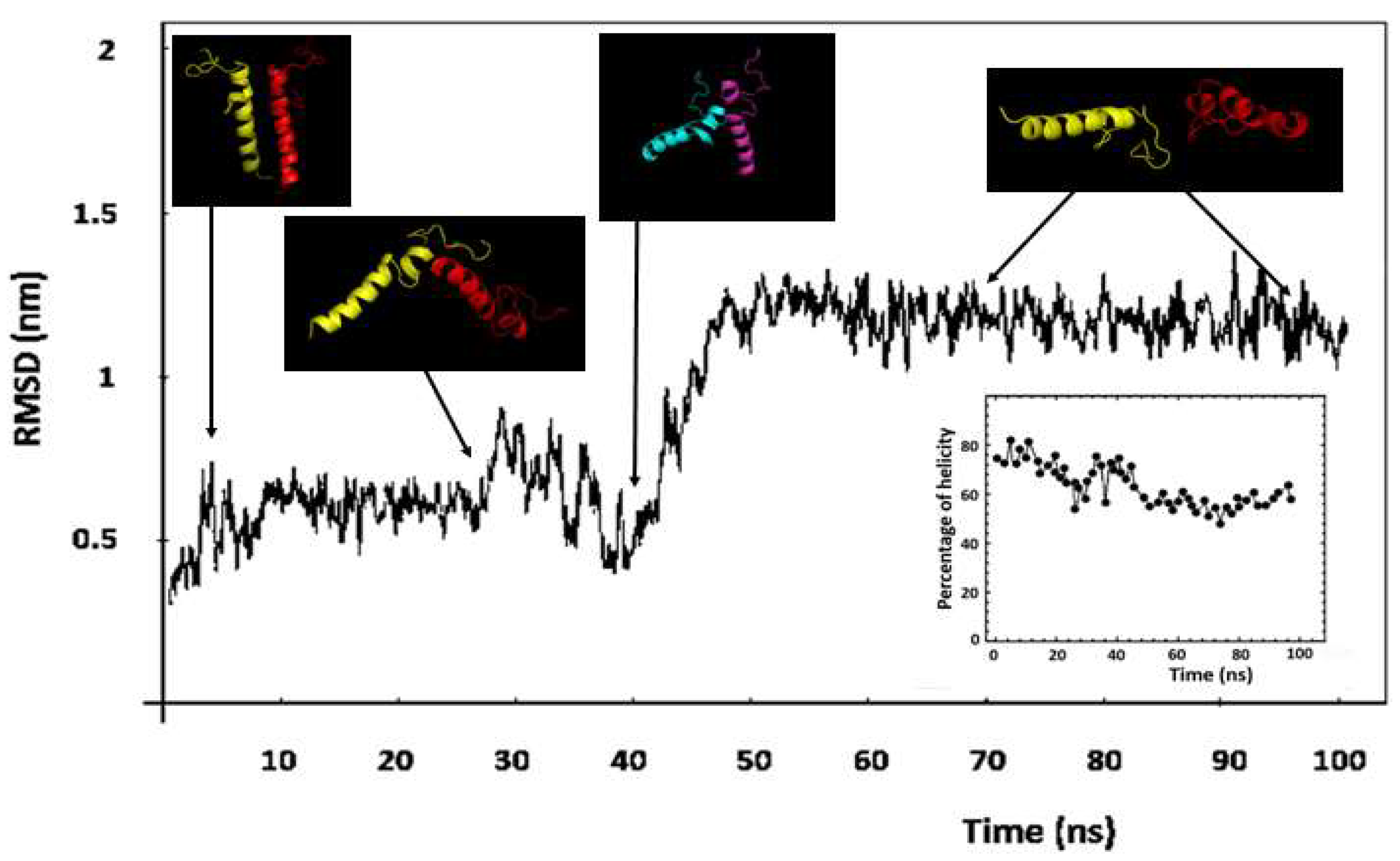

Table 1.
- Amino acid composition.
| ORF7b-2* | ORF7b-1** | ||||
|---|---|---|---|---|---|
| Amino acid | Number of residues | Percentage % |
Number of residues |
Percentage % |
|
| Ala [A] | 2 | 4.7 | 1 | 2.3 | |
| Asn [N] | 1 | 2.3 | 1 | 2.3 | |
| Asp [D] | 2 | 4.7 | 2 | 4.5 | |
| Cys [C] | 2 | 4.7 | 2 | 4.5 | |
| Gln [Q] | 1 | 2.3 | 1 | 2.3 | |
| Glu [E] | 3 | 7.0 | 4 | 9.1 | |
| His [H] | 2 | 4.7 | - | - | |
| Ile [I] | 5 | 11.6 | 5 | 11.4 | |
| Leu [L] | 11 | 25.6 | 11 | 25.0 | |
| Lys [K] | - | - | 1 | 2.3 | |
| Met [M] | 2 | 4.7 | 2 | 4.5 | |
| Phe [F] | 6 | 14.0 | 6 | 13.6 | |
| Pro [P] | - | - | 1 | 2.3 | |
| Ser [S] | 2 | 4.7 | 1 | 2.3 | |
| Thr [T] | 1 | 2.3 | 2 | 4.5 | |
| Trp [W] | 1 | 2.3 | 1 | 2.3 | |
| Tyr [Y] | 1 | 2.3 | 1 | 2.3 | |
| Val [V] | 1 | 2.3 | 2 | 4.5 | |
In red the negative residues, in blue the positive ones. *Total number of negatively charged residues [Asp + Glu]: 5, and of positively charged residues [Arg + Lys]: 0. Instability index [99,104] is computed to be 50.96, this classifies the protein as unstable. ** Total number of negatively charged residues [Asp + Glu]: 6, and of positively charged residues [Arg + Lys]: 1. Instability index is computed to be 39.77, this classifies the protein as stable. Data computed by ProtParam – Expasy - https://web.expasy.org/protparam/. Chemical parameters from Swiss-Prot or TrEMBL. https://www.uniprot.org/.
Table 2.
– Protein Sequence.
| Protein | Sequence 5 10 15 20 25 30 35 40 |
|---|---|
| ORF7b-2 | MIELSLIDFYLCFLAFLLFLVLIMLIIFWFSLELQDHNETCHA |
| ORF7b-1 | MNELTLIDFYLCFLAFLLFLVLIMLIIFWFSLEIQDLEEPCTKV |
Table 3.
- Charge distribution analysis of ORF7b-1 and ORF7b-2.
| Physical-chemical parameters |
ORF7b-1 |
ORF7b-2 |
Notes |
|
|---|---|---|---|---|
| N [MW] | 44 [5301.51] | 43 [5179.31] | Number of residues and M.W. |
|
| f- | 0.13636 | 0.11628 | Fraction of negative residues | |
| f+ | 0.02273 | 0.00000 | Fraction of positive residues | |
| FCR | 0.15909 | 0.11628 | Fraction of charged residues | |
| NCPR | -0.11364 | -0.11628 | Net charge per residue | |
| Sigma | 0.08117 | 0.11628 | Charge asymmetry | |
| K | 0.35577 | 0.25372 | Charge patterning parameter | |
| Delta | 0.03182 | 0.01706 | ||
| Max Delta | 0.08945 | 0.06725 | ||
| pI | 3.72 | 4.32 | Isoelectric point | |
| AH | -0.83 | -0.98 | Average hydrophilicity | |
| Phase Plot Region | 1 | 1 | [see State diagram] | |
| Phase Plot Annotation | Globule/Tadpole | Globule/Tadpole | Prolate elongated structures | |
| Polymeric State | Weak polyampholyte | Polyanion (Weak negative polyelectrolyte) |
Table 4.
– ORF7b-2 HINGE RESIDUES.
| Slowest mode 1 | |||
|---|---|---|---|
| Rigid Part No | Residues | Score | Hinge residues |
| 1 | 1-20 | 0.88 | 20 |
| 2 | 21-43 | 0.9 | 20 |
| Slowest mode 2 | |||
| Rigid Part No | |||
| 1 | 1-9 | 0.68 | 9 |
| 2 | 10-32 | 0.82 | 32 |
| 3 | 33-43 | 0.85 | 32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



