Submitted:
10 April 2023
Posted:
11 April 2023
You are already at the latest version
Abstract
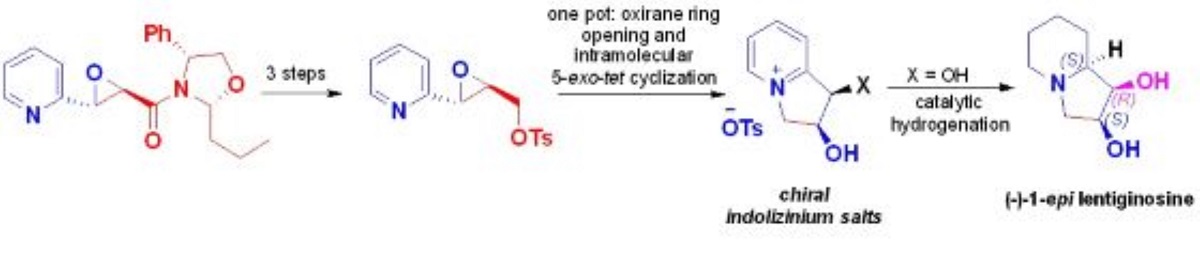
The first diastereoselective synthesis of (–)-1-epi-lentiginosine stars from a common chiral trans-epoxyamide derived from 2-pyridincarbaldehyde is reported. This methodology involves a sequential oxirane ring opening and intramolecular 5-exo-tet cyclization of tosylate tras-epoxyalcohol to afford a diastereomeric mixture of indolizinium salts in a one-pot fashion, followed by regio- and diastereospecific pyridinium ring reduction.
Keywords:
diastereoselective synthesis
; trans-epoxyamides
; indolizinium salts
; lentiginosine
1. Introduction
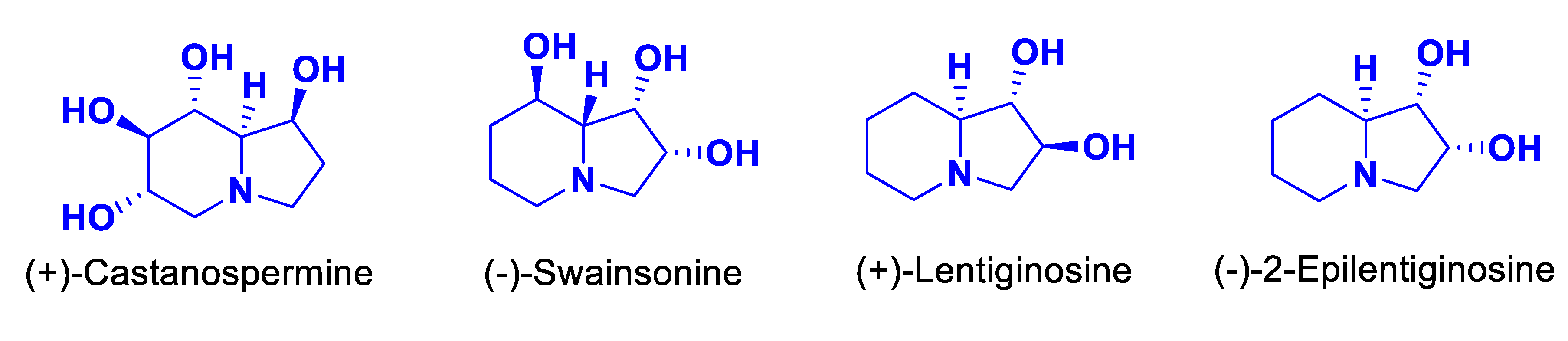
Hydroxylated indolizidines such as (+)-castanospermine, (‒)-swainsonine, (+)-lentiginosine and (‒)-epi-lentiginosine are widely found in plants and microorganisms, and also constitute a class of azasugars that exhibit potent and selective glycosidase inhibitory activities.[1,2] Specifically, Lentiginosine is known not only to be a significant inhibitor of amyloglycosidases but also to have excellent anti-HIV, anti-tumor, and immunomodulatory activities (Figure 1). [1,3,4,5,6] Therefore, several syntheses using a chiral pool [7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27] or enantio- or diastereoselective [28,29,30,31,32,33,34] approaches to lentiginosine have been reported.
Although there are few reports of the use of pyridine derivatives in the synthesis of lentiginosine, this heterocycle has proved to be a valuable starting material for the synthesis of this indolizidine. For example, Zhou reported the use of ethyl 3-(pyridine-2-yl)acrylate N-oxide obtained from picolinaldehyde by the Wittig reaction. Subsequently, the asymmetric dihydroxylation of heteroaromatic acrylate affords the key intermediate for the synthesis of (+)-lentiginosine in an overall yield of 16%.[31] Fruit et al reported a synthesis of (‒)-lentiginosine and its epimers starting from 2-bromopyridine condensed with an enantiomerically pure (R)-2,3-O-isopropylidene glyceraldehyde prepared from D-mannitol. The key step involved quaternization of a completely unprotected pyridinium-polyol unit using the Mitsunobu methodology, followed by PtO2-catalyzed diastereoselective hydrogenation of the pyridinium ring to give the desired dihydroxyindolizidine.[35] Finally, Brandi et al reported the synthesis of (±)-lentiginosine starting from 1-(2-Pyridyl)-2-propen-1-ol. In this methodology, the main steps were a domino process involving the electrophilic addition of bromine to the propen-1-ol derivative and the cyclization of the bromonium ion to give the corresponding indolizinium salt followed by a diastereoselective reduction getting a diastereomeric mixture of tetrahydro derivatives. Nucleophilic substitution (via elimination and addition) finally yields (±)-lentiginosine.[36]
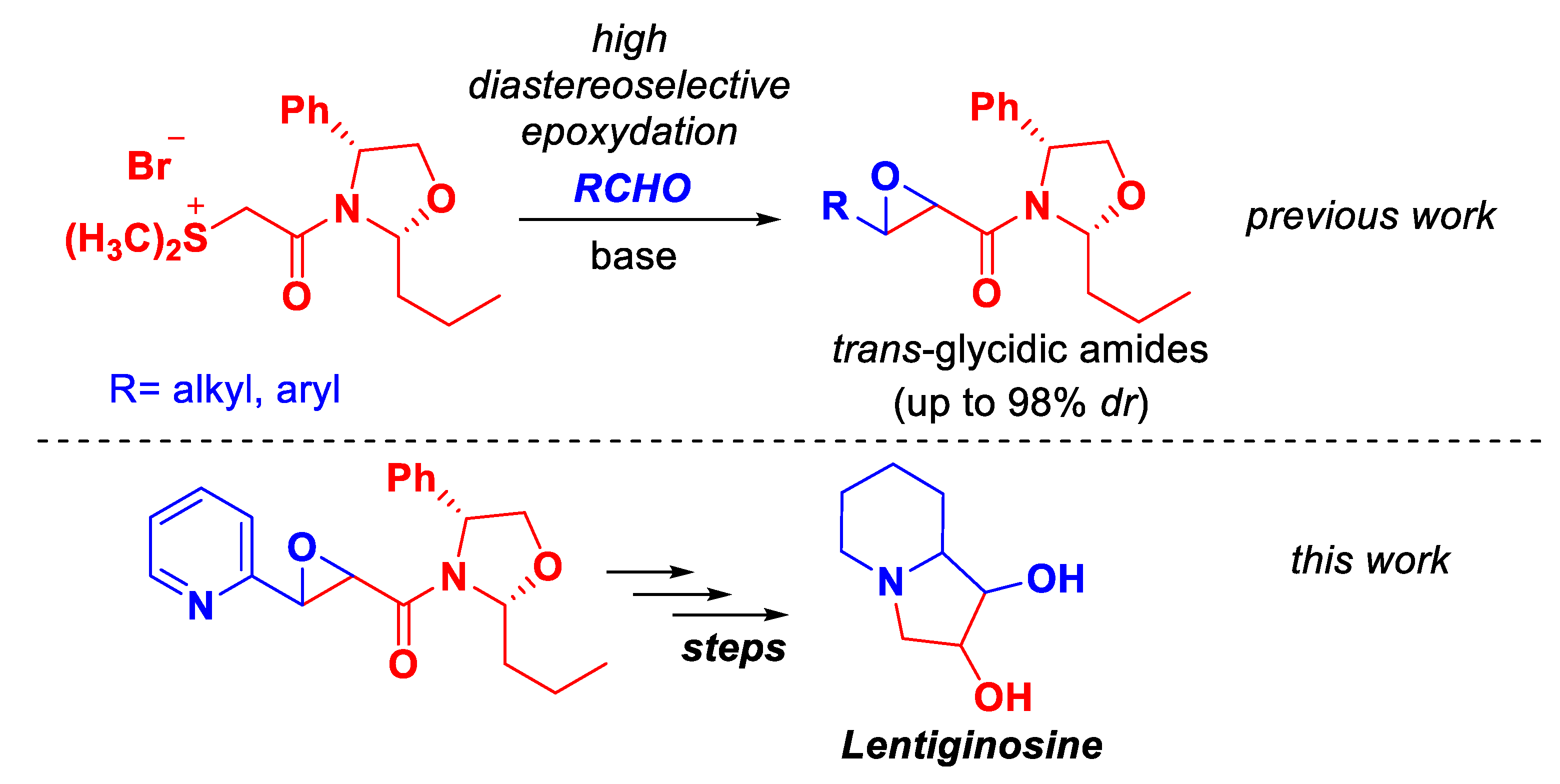
On the other hand, we have shown in a previous report, that chiral N-phenylglycinol-derived 2,4-disubstituted oxazolidines are excellent chiral auxiliaries for the asymmetric condensation of amide-stabilized sulfonium ylides with aldehydes to access at trans-glycidic amides in high diastereoselectivity.[37] Following this work, and in order to highlight our diastereoselective epoxidation methodology, we develop the total synthesis of lentiginosine using trans-glycidic amide as starting material derived from chiral oxazolidine sulfur ylide and 2-pyridincarbaldehyde (Scheme 1).
2. Results
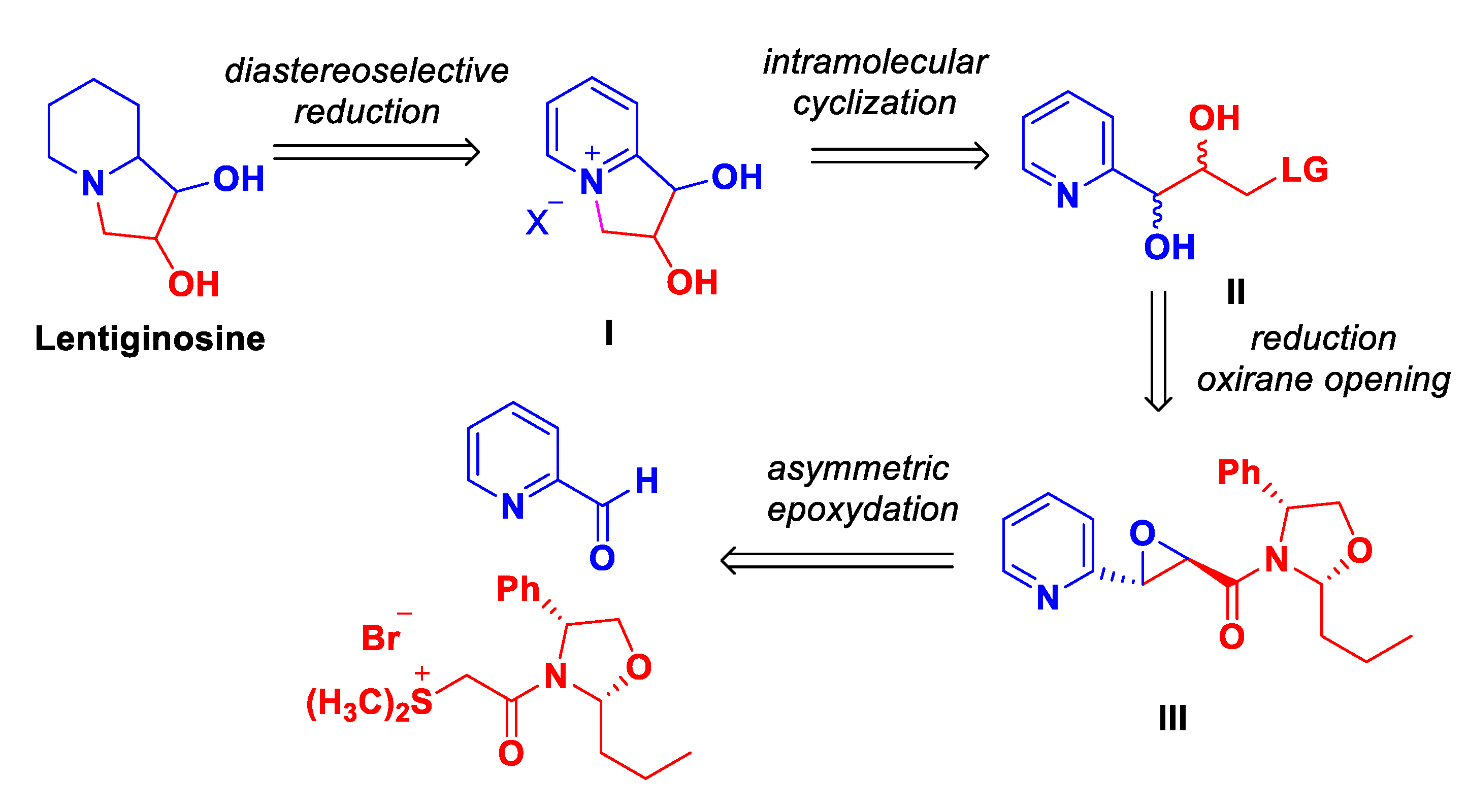
Our retrosynthetic analysis is outlined in Scheme 2. We envisioned that lentiginosine could be accessed via a diastereoselective reduction of indolizinium salt I which can be generated by intramolecular cyclization reaction of the diol II. The intermediate II could be obtained via a reductive remotion of the chiral auxiliary of the glycidic amide III and the opening of the oxirane function. III can be prepared from commercially available 2-pyridine carbaldehyde and the chiral oxazolidine sulfur ylide derived from (R)-(-)-2-phenylglycinol (Scheme 2)
First, we prepared the chiral oxazolidine sulfur ylide 1 and its subsequent condensation with 2-picolinaldehyde to give the desired glycidic amide 2 following our previously reported reaction strategy.[37] Compound 2(a+b) was obtained in a high chemical and stereochemical yield (85% yield, 95:5 d.r.). The major diastereoisomer 2a was separated and we assumed the configuration of the new stereogenic centers as (2R, 3S) according to our previous report (Scheme 3).
With the major diastereoisomer, 2a in hand and continuing with our retrosynthetic plan, the next step consisted of the reductive remotion of the chiral auxiliary. Unfortunately, all attempts to obtain the desired epoxyalcohol were unsuccessful, and the oxirane intermediate 2a was completely degraded.
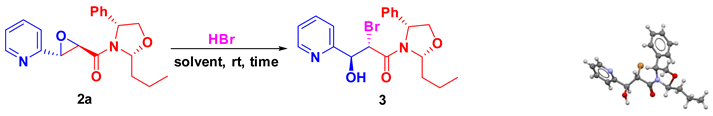
In this way and as mentioned earlier, Brandi et al[36] have reported a racemic synthesis of lentiginosine by cyclization of the bromohydrin intermediate derived from 1-(pyridin-2-yl)prop-2-en-1-ol, for which epoxyamide 2a was treated with HBr to access at the corresponding halohydrin. Pleasingly, the corresponding bromohydrin 3 was obtained when the intermediate 2a was dissolved in DCM and HBr was added at room temperature. This product was detected by NMR spectroscopy as a mixture of rotamers thesince signals coalesced at a higher temperature (see supporting information).
Compound 3 crystallized enabling the unambiguous determination of the absolute stereochemistry of the new stereogenic centers as (2S,3S) and confirmed that a regio- and diastereospecific oxirane opening reaction occurs (Scheme 2).[38] To improve the reaction yield, screening experiments were performed. The best result was obtained when epoxyamide 2a was treated with HBr in CHCl3 at room temperature (entry 2, Table 1). Compound 3 was obtained in 98% yield after chromatographic column purification.
Then, reductive remotion of the chiral auxiliary was performed. Bromohydrin 3 was reacted with LiBH4 under ultrasonic activation conditions. Not only the chiral auxiliary was removed, but 73% of the corresponding epoxyalcohol 4 was obtained as a specific diastereoisomer (Scheme 4).
Tosylation of epoxy alcohol 4, afforded the desired compound 5 in 94 % yield after purification by column chromatography. Immediately, compound 5 was treated with hydrogen halide to promote the formation of the halohydrin. All attempts resulted in the formation of the corresponding diastereomeric mixture of indolizinium salts 6(a+b), obtained by a domino process involving a regiospecific oxirane opening and an intramolecular nucleophilic cyclization reaction favoring the formation of the cis isomer (JH1,H2 = 4.7 Hz), as a result of the more favorable nucleophilic attack on the oxirane ring on the backside. The use of HBr gave the corresponding indolizinium salts 6(a+b) in 80:20 dr. The best diastereoselectivity was obtained with the use of HCl which gave the desired diastereomeric mixture of indolizinium salts 7(a+b) in dr = 90:10. Finally, the use of HI resulted in decreased diastereoselectivity (Scheme 5).
In order to extend the scope of this domino reaction, and to take into account that this reaction occurs in an acidic medium, we decided to investigate this reaction with other nucleophiles catalyzed by a Lewis acid. We were pleased to observe that this reaction occurred in the presence of Ytterbium(III)triflate (10 mol%) with a nucleophile (benzylic alcohol or H2O), in high chemical and stereochemical yields (Scheme 6).
Unfortunately, all attempts to separate the diastereomeric indolizinium salts were unsuccessful. Therefore, we turned our attention to the pyridinium ring reduction without purification, by first using the corresponding inseparable diastereomeric chlorohydrin salt 7(a+b).
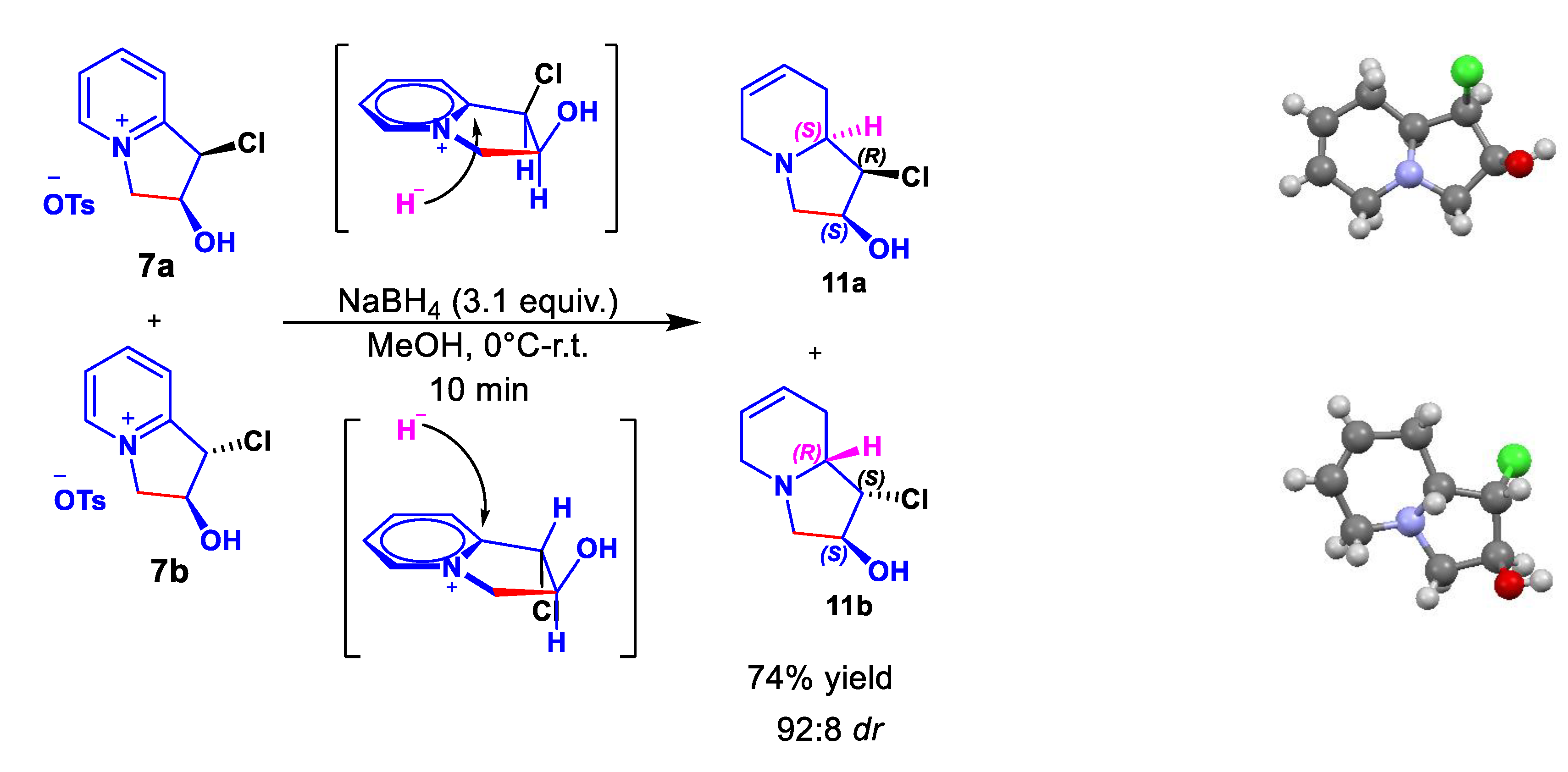
Catalytic hydrogenation of 7(a+b) in the presence of Pd/C afforded a complex mixture and recovery of the starting material despite longer reaction times and different catalysts were tested. Pleasingly, the employ of NaBH4 leads to the tetrahydro derivatives 11(a+b). This mixture was separated by column chromatography and each diastereoisomer was crystallized, which allowed the determination of its absolute stereochemistry by X-ray crystallography.[38] Based on the results obtained we propose that the hydride addition occurs in a diastereospecific manner from the less hindered face of the bicyclic indolizinium salt (Scheme 7).
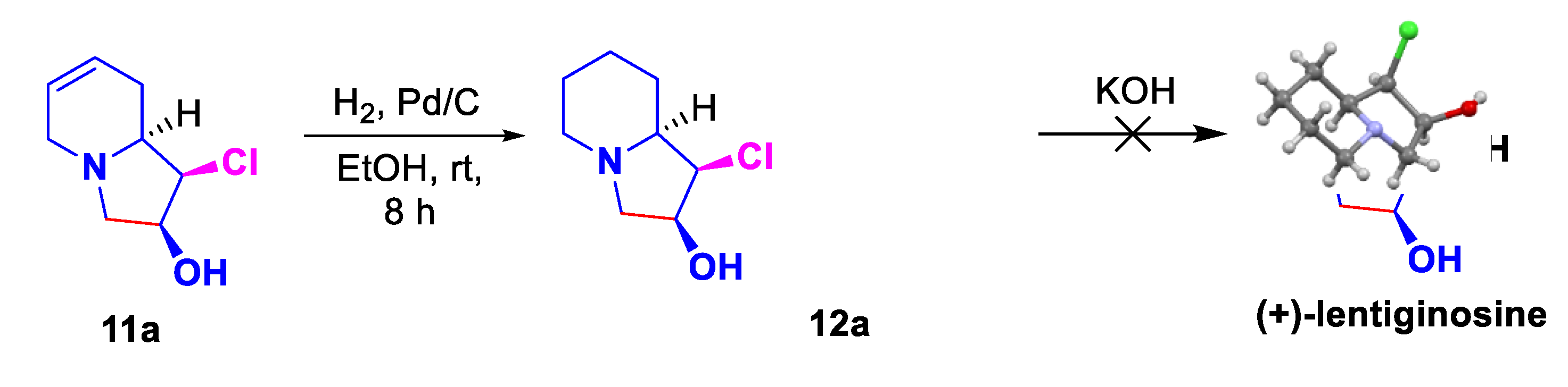
Using the major diastereoisomer 11a, we attempted to complete the total synthesis of (+)-Lentiginosine. Despite the catalytic hydrogenation cleanly affords indolizidine 12a,[38] whose its absolute configuration was determined by x-ray diffraction analysis, the substitution of chlorine atom by OH was unsuccessful (Scheme 8).
Therefore, the total synthesis of (–)-epi-1-lentiginosine was carried out starting from a mixture of indolizinium salts 10(a+b) which was subjected to acidic hydrogenation conditions to afford the desired (–)-1-epi-lentiginosine in 87 % yield after chromatographic purification (Scheme 9).
3. Materials and Methods
3.1. General Information
All reagents and solvents were purchased from commercial sources. The 1H and 13C spectra were determined with a Bruker Avance III Spectrometer (CDCl3 or CD3OD solvents) operating at 500 and 125 MHz, respectively. The chemical shifts were reported in parts per million (ppm), downfield from SiMe4 (δ 0.0) and relative to the signal of chloroform-d (δ 7.26, singlet). Multiplicities were afforded as: s (singlet); d (doublet); t (triplet); q (quartet); dd (doublets of doublet); ddd (doublet of doublets of doublets); or m (multiplets). The number of protons for a given resonance is indicated by nH. Coupling constants were reported as a J value in Hz. Thin layer chromatography (TLC) was used to monitor the reaction on Merck 60 F254 precoated silica gel plate (0.2 mm thickness). Optical rotations were determined at room temperature with a Perkin-Elmer 341 polarimeter, using a 1 dm cell with a total volume of 1mL, and are referenced to the D-line of sodium. Mass spectra were recorded with a JEOL Station JMS-700 instrument at a voltage of 70 eV. X-Ray Diffraction analysis was performed on a diffractometer STOE Stadivari using Ag-Kα radiation (λ = 0.56083 Å, AXO micro-source) and equipped with a Dectris Pilatus-100 K detector. Intensities were collected at 295 K, and structures were refined using the current release of SHELXL (2018/3). The products were purified by column chromatography on silica gel 60 (63-200nm).
3.2. General procedures
Regiospecific oxirane opening.
trans-epoxyamide 2a (0.1g, 1.0 equiv, 0.29 mmol), was dissolved in 1 mL of chloroform at 25 °C, then 3 drops of HBr (48%) were added and the reaction mixture was stirred for 40 minutes. Then, NaHCO3 was added, and the resulting reaction mixture was filtered. Finally, the solvents were evaporated. Product 3 was crystallized in a mixture of petroleum ether: DCM (70:30). Bromohydrin 3 was obtained in 98% yield. (All spectroscopic details are described in ESI).
Reductive remotion of chiral auxiliary.
To a solution of bromohydrin 3 (110 mg, 0.26 mmol) in anhydrous THF (0.08 M) under ultrasonic activation at 5 °C, was added in portions a solution of LiBH4 (2M, THF, 5 equiv.). After 2 h, 1 mL of H2O2 (30%) was added slowly to the reaction mixture followed by addition of NaOH (3 N) solution. The mixture was stirred overnight at room temperature. The crude reaction was then filtered and then the organic layer was dried over anhydrous Na2SO4. After evaporation of the solvent from the filtrate, the residue was subjected to purification by flash column chromatography (SiO2, CH2Cl2/MeOH). Epoxyalcohol 4 was obtained in 73% yield. (All spectroscopic details are described in ESI).
Tosylation of epoxyalcohol 4.
Compound 4 was dissolved in DCM and the resulting solution was cooled to 0°C, then Et3N (0.405 mmol, 0.058 mL) and DMAP (0.0026 g, 0.021 mmol) were added. The resulting mixture was stirred for 10 minutes, then p-TsCl (0.062g, 0.325 mmol) was added in portions and the mixture was stirred for 1 h. Finally, the reaction was quenched by adding a brine solution, and the organic phase was separated and dried over anhydrous Na2SO4, filtered and the solvent was evaporated yielding the desired tosylated epoxyalcohol 5 which was obtained in 94% yield after purification by chromatography (silica gel, AcOEt/petroleum ether). (Spectroscopic details are described in ESI).
General procedure for a one pot regiospecific oxirane opening and intramolecular nucleophilic cyclization reaction
With hydrohalic acids
Compound 5 (0.053g, 0.173 mmol) was dissolved in CHCl3 (1 mL) at room temperature, then the corresponding hydrohalic acid was injected through a needle into the solution and stirring for 3 hours. Finally, the reaction was quenched by adding NaHCO3 until pH = 7. Then, the mixture was filtered, and the solvent was evaporated to get the corresponding inseparable diastereomeric mixture of indolizinium salt which was used without purification for the next reaction. (Spectroscopic details are described in ESI).
With H2O or BnOH as nucleophile
To a solution of compound 5 (0.079g, 0.26 mmol) and the corresponding nucleophile (0.029mL, 0.228mmol) in 1,4-dioxane (1mL) was added ytterbium trifluoromethanesulfonate (0.016g, 0.1 equiv.) under inert atmosphere. The suspension was stirring for 24 hours. Finally, the solvent was evaporated and the desired indolizinium salt was precipitated in AcOEt/petroleum ether given the corresponding inseparable diastereomeric mixture of indolizinium salt.
Diastereospecific reduction of indolizinium salts
To a stirred solution of the corresponding indolizinium salt (0.084 mmol, 29 mg) in methanol (3 mL) at 0 °C was added slowly NaBH4 (10 mg, 0.26 mmol). After, the mixture was stirred for 10 minutes and then a saturated aqueous solution of NaSO4 was added. Next, the resulting mixture was filtered through a celite pad. The solution was evaporated, and the residue was purified by flash chromatography to afford the desired hexahydroindolizin-2-ol. (Spectroscopic details are described in ESI).
Catalytic hydrogenation
To a methanolic solution of the diastereomeric indolizidinium salt 10(a+b) was added concentrated HCl (1 drop) and the resulting mixture was hydrogenated at room temperature in the presence of 10% PtO2. The reaction was stirred overnight. Then, NaOH (3M) solution was added, and the resulting crude reaction was extracted with dichloromethane, dried over anhydrous Na2SO4, filtered, concentrated, and finally subjected to purification by flash column chromatography (SiO2, CH2Cl2/MeOH). The desired (–)-1-epi-lentiginosine was obtained with 87 % yield. (Spectroscopic details are described in ESI).
4. Conclusions
In conclusion, a novel protocol for the diastereoselective synthesis of substituted indolizidinium salts from a common chiral trans-epoxiamide is reported. In addition, the first diastereoselective synthesis of (–)-1-epi-lentiginosine in only 5 steps and 49.7% overall yield starting from trans-epoxyamide 2a is reported. This new, versatile, and diastereoselective access to chiral indolizidine compounds opens the route to the pharmacological investigation of these promising bicyclic cores as well as the design of analogs.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, characterization data, and 1H-NMR and 13C-NMR of compounds 3-12 and (–)-1-epi-lentiginosine.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CONACyT, projects; A1-S-13280, 268178 and VIEP-BUAP.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No applicable.
Acknowledgments
H.R.M. thanks CONACyT for the scholarship (658640). We thank Dr. Sylvain Bernès for the measurement of crystal structures. We thank Hatsumi A. R. for their technical assistance.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Macchi, B.; Minutolo, A.; Grelli, S.; Cardona, F.; Cordero, F.M.; Mastino, A.; Brandi, A. The novel proapoptotic activity of nonnatural enantiomer of Lentiginosine. Grycobiology 2010, 20, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Asano, N. Modern Alkaloids: Structure, Isolation, Synthesis and Biology; Eds.: Fattorusso, E.; Taglialatela-Scafati; Wiley-VCH: Weinheim, Germany, 2008; pp. 111–138. [Google Scholar]
- Pastuszak, I.; Molyneux Russell, J.; James, L.F.; Elbein, A.D. Lentiginosine, a dihydroxyindolizidine alkaloid that inhibits amyloglucosidase. Biochemistry 1990, 29, 1886–1891. [Google Scholar] [CrossRef] [PubMed]
- Brandi, A.; Cicchi, S.; Cordero, F.M.; Fringnoli, R.; Goti, A.; Picasso, S.; Vogel, P. Assignment of the Absolute Configuration of Natural Lentiginosine by Synthesis and Enzymatic Assays of Optically Pure (+) and (-)-Enantiomers. J. Org. Chem. 1995, 60, 6806–6812. [Google Scholar] [CrossRef]
- Dal Piaz, F.; Vassallo, A.; Chini, M.G.; Cordero, F.M.; Cardona, F.; Pisano, C.; Bifulco, G.; Detommasi, N.; Brandi, A. Natural Iminosugar (+)-Lentiginosine Inhibits ATPase and Chaperone Activity of Hsp90. PLoS One 2012, 7, e43316. [Google Scholar] [CrossRef] [PubMed]
- Minutolo, A.; Grelli, S.; Marino-Merlo, F.; Cordero, F.M.; Brandi, A.; Macchi, B.; Mastino, A. D(-)lentiginosine-induced apoptosis involves the intrinsic pathway and is p53-independent. Cell Death Dis. 2012, 3, e358. [Google Scholar] [CrossRef] [PubMed]
- Yoda, H.; Kitayama, H.; Katagiri, T.; Takabe, K. Synthesis of natural lentiginosine employing a cyclic imide with C2-symmetry derived from L-tartaric acid. Tetrahedron: Asymmetry 1993, 4, 1455–1456. [Google Scholar] [CrossRef]
- Ha, D.C.; Yun, C.S.; Lee, Y. Samarium Diiodide-Promoted Cyclization of N-(ω-Iodoalkyl)imides to Polyhydroxylated Indolizidinones and Pyrrolizidinones: Synthesis of (+)-Lentiginosine. J. Org. Chem. 2000, 65, 621–623. [Google Scholar] [CrossRef]
- Yoda, H.; Katoh, H.; Ujihara, Y.; Takabe, K. SmI2-mediated hetero-coupling reaction of lactams with aldehydes; synthesis of indolizidine alkaloids, (-)-δ-coniceine, (+)-5-epiindolizidine 167B and (+)-lentiginosine. Tetrahedron Lett. 2001, 42, 2509–2512. [Google Scholar] [CrossRef]
- Klitzke, C.F.; Pilli, R.A. Enhanced trans diastereoselection in the allylation of cyclic chiral N-acyliminium ions. Synthesis of hydroxylated indolizidines. Tetrahedron Lett. 2001, 42, 5605. [Google Scholar] [CrossRef]
- El-Nezhawy, A.O.H.; El-Diwani, H.I.; Schmidt, R.R. O-(2-Oxopyrrolidin-5-yl)trichloroacetimidates as Amidoalkylating Agents – Synthesis of (+)-Lentiginosine. Eur. J. Org. Chem. 2002, 4137–4142. [Google Scholar] [CrossRef]
- Ichikawa, Y.; Ito, Y.; Nishiyama, T.; Isobe, M. Stereoselective Allyl Amine Synthesis through Enantioselective Addition of Diethylzinc and [1,3]-Chirality Transfer: Synthesis of Lentiginosine and Polyoxamic Acid Derivative. Chem. -Eur. J. 2005, 11, 1949–1957. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, Q.; Zhang, H.-K.; Chen, A. Practical synthesis of trans-dihydroxybutyrolactols as chiral C4 building blocks and their application to the synthesis of polyhydroxylated alkaloids. RSC Adv. 2013, 3, 20298–20307. [Google Scholar] [CrossRef]
- Cordero, L.F.; Cicchi, S.; Goti, A.; Brandi, A. Synthesis of lentiginosine by stereoselective chiral nitrone cycloaddition and thermal rearrangement of strained spiroisoxazolidine. Tetrahedron Lett. 1994, 35, 949–952. [Google Scholar] [CrossRef]
- Socha, D.; Jurczak, M.; Chmielewski, M. Synthesis of polyhydroxyindolizidines from 5,6-dihydro-2H-pyran-2-one. Carbohydr. Res. 2001, 336, 315–318. [Google Scholar] [CrossRef]
- McCaig, A.E.; Meldrum, K.P.; Wightman, R.H. Synthesis of trihydroxylated pyrrolizidines and indolizidines using cycloaddition reactions of functionalized cyclic nitrones, and the synthesis of (+)- and (‒)-lentiginosine. Tetrahedron 1998, 54, 9429–9446. [Google Scholar] [CrossRef]
- Cardona, F.; Moreno, G.; Guarna, F.; Vogel, P.; Schuetz, C.; Merino, P.; Goti, A. New Concise Total Synthesis of (+)-Lentiginosine and Some Structural Analogues. J. Org. Chem. 2005, 70, 6552–6555. [Google Scholar] [CrossRef]
- Yoda, H.; Kawauchi, M.; Takabe, K. Novel Asymmetric Synthesis of an Indolizidine Alkaloid, (+)-Lentiginosine Employing Highly Stereoselective Hydrogenation of α-Hydroxypyrrolidine. Synlett 1998, 137–138. [Google Scholar] [CrossRef]
- Chandra, K.L.; Chandrasekhar, M.; Singh, V.K. Total Synthesis of (‒)- and (+)-Lentiginosine. J. Org. Chem. 2002, 67, 4630–4633. [Google Scholar] [CrossRef]
- Casiraghi, G.; Spanu, P.; Rassu, G.; Pinna, L.; Ulgheri, F. Total Synthesis of All Four Isomers of cis-1,2-Dihydroxypyrrolizidine. J. Org. Chem. 1994, 59, 2906–2909. [Google Scholar] [CrossRef]
- Chaudhari, V.D.; Ajish kumar, K.S.; Dhavale, D.D. Synthesis of (‒)-lentiginosine, its 8a-epimer and dehydroxylated pyrrolizidine alkaloid from d-glucose. Tetrahedron 2006, 62, 4349–4354. [Google Scholar] [CrossRef]
- Kim, I.S.; Zee, O.P.; Jung, Y.H. Regioselective and Diastereoselective Amination of Polybenzyl Ethers Using Chlorosulfonyl Isocyanate: Total Syntheses of 1,4-Dideoxy-1,4-imino-D-arabinitol and (‒)-Lentiginosine. Org. Lett. 2006, 8, 4101–4104. [Google Scholar] [CrossRef] [PubMed]
- Angle, S.R.; Bensa, D.; Belanger, D.S. New Access to Indolizidine and Pyrrolizidine Alkaloids from and Enantiopure Proline: Total Syntheses of (–)-Lentiginosine and (1R,2R,7aR)-Dihydroxypyrrolizidine. J. Org. Chem. 2007, 72, 5592–5597. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, R.; Kokatla, H.P.; Vankar, Y.D. An improved method of ring closing metathesis in the presence of basic amines: application to the formal synthesis of (+)-lentiginosine and other piperidines and carbamino sugar analogs. Tetrahedron Lett. 2011, 52, 781–786. [Google Scholar] [CrossRef]
- Kamal, A.; Vangala, S.R. An expedient total synthesis of optically active piperidine and indolizidine alkaloids (–)-β-conhydrine and (–)-lentiginosine. Tetrahedron 2011, 67, 1341–1347. [Google Scholar] [CrossRef]
- Kim, I.S.; Li, Q.R.; Dong, G.R.; Kim, Y.C.; Hong, Y.J.; Lee, M.; Chi, K.-W.; Oh, J.S.; Jung, Y.H. A Facile Synthesis of Lentiginosine Analogues Based on a Highly Regio- and Diastereoselective Allylic Amination Using Chlorosulfonyl Isocyanate. Eur. J. Org. Chem. 2010, 1569–1573. [Google Scholar] [CrossRef]
- Gurjar, M.K.; Ghosh, L.; Syamala, M.; Jayasree, V. Stereoselective synthesis of (+)- and (–)-lentiginosine. Tetrahedron Lett. 1994, 35, 8871–8872. [Google Scholar] [CrossRef]
- Nukui, S.; Sodeoka, M.; Sasai, H.; Shibasaki, M. Regio- and Stereoselective Functionalization of Optically Active Tetrahydroindolizidine Derivative. Catalytic Asymmetric Syntheses of Lentiginosine, 1,2-Diepilentiginosine, and Gephyrotoxin 209D. J. Org. Chem. 1995, 60, 398–404. [Google Scholar] [CrossRef]
- Rasmussen, M.O.; Delair, P.; Greene, A.E. Enantiocontrolled Preparation of Indolizidines: Synthesis of (–)-2-Epilentiginosine and (+)-Lentiginosine. J. Org. Chem. 2001, 66, 5438–5443. [Google Scholar] [CrossRef]
- Lim, S.H.; Ma, S.; Beak, P. Asymmetric Syntheses of Fused Bicyclic Lactams. J. Org. Chem. 2001, 66, 9056–9062. [Google Scholar] [CrossRef]
- Feng, Z.-X.; Zhou, W.-S. A novel concise total synthesis of (+)-lentiginosine. Tetrahedron Lett. 2003, 44, 497–498. [Google Scholar] [CrossRef]
- Ayad, T.; Genisson, Y.; Baltas, M. Asymmetric syntheses of (-)-lentiginosine and an original pyrrolizidinic analogue thereof from a versatile epoxyamine intermediate. Org. Biomol. Chem. 2005, 3, 2626–2631. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-W.; Hsu, H.-C.; Chang, C.-H.; Tsai, H.-H. T.; Hou, D.-R. Asymmetric Synthesis of (–)-Lentiginosine by Double Aza-Michael Reaction. Eur. J. Org. Chem. 2010, 4771–4773. [Google Scholar] [CrossRef]
- Shao, J.; Yang, J.-S. A Diastereoselective Cyclic Imine Cycloaddition Strategy To Access Polyhydroxylated Indolizidine Skeleton: Concise Syntheses of (+)-/(–)-Lentiginosine and (–)-2-epi-Steviamine. J. Org. Chem. 2012, 77, 7891–7900. [Google Scholar] [CrossRef] [PubMed]
- Azzouz, R.; Fruit, C.; Bischoff, L.; Marsais, F. A Concise Synthesis of Lentiginosine Derivatives Using a Pyridinium Formation via the Mitsunobu Reaction. J. Org. Chem. 2008, 73, 1154–1157. [Google Scholar] [CrossRef]
- Giomi, D.; Alfini, R.; Micoli, A.; Calamai, E.; Faggi, C.; Brandi, A. Synthesis of 1,2-Dihydroxyindolizidines from 1-(2-Pyridyl)-2-propen-1-ol. J. Org. Chem. 2011, 76, 9536–9541. [Google Scholar] [CrossRef]
- Gordillo, P.G.; Aparicio, D.M.; Flores, M.; Mendoza, A.; Orea, L.; Juárez, J.R.; Huelgas, G.; Gnecco, D.; Terán, J.L. Oxazolidine Sulfur Ylides Derived from Phenylglycinol for the Specific and Highly Diastereoselective Synthesis of Aryl and Alkyl trans-Epoxyamides. Eur. J. Org. Chem. 2013, 5561–5565. [Google Scholar] [CrossRef]
- CCDC: 2181510 (3), 2181511 (10a), 2181512 (10b) and 2181513 (11a) see the ESI for details.
Figure 1.
Some hydroxylated indolizidine alkaloids showed potent and selective glycosidase inhibitory activities.
Figure 1.
Some hydroxylated indolizidine alkaloids showed potent and selective glycosidase inhibitory activities.
Scheme 1.
Previous work and this work.
Scheme 2.
Retrosynthetic analysis for the synthesis of lentiginosine.
Scheme 3.
Diastereoselective synthesis of trans-2,3-epoxyamide 2(a+b) derived from 2-picolinaldehyde. Reagents and conditions: i) butyraldehyde, DCM, 0 to rt, 30 min then bromoacetyl bromide, K2CO3; ii) DCM, dimethyl sulfide, rt, 1h; iii) 2-pyridinecarbaldehyde, KOH, THF:H2O, 0°C to rt, 13h.
Scheme 3.
Diastereoselective synthesis of trans-2,3-epoxyamide 2(a+b) derived from 2-picolinaldehyde. Reagents and conditions: i) butyraldehyde, DCM, 0 to rt, 30 min then bromoacetyl bromide, K2CO3; ii) DCM, dimethyl sulfide, rt, 1h; iii) 2-pyridinecarbaldehyde, KOH, THF:H2O, 0°C to rt, 13h.
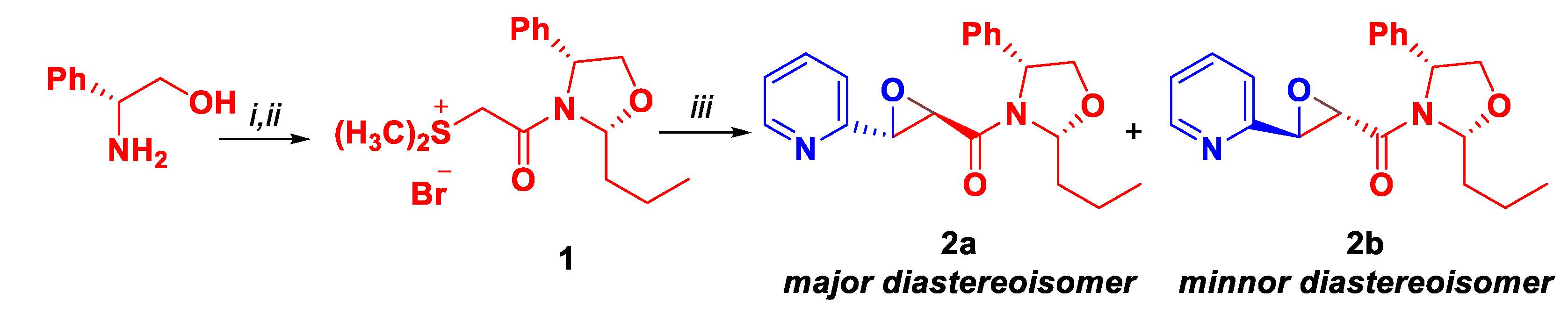
Scheme 4.
Reductive remotion of the chiral auxiliary.
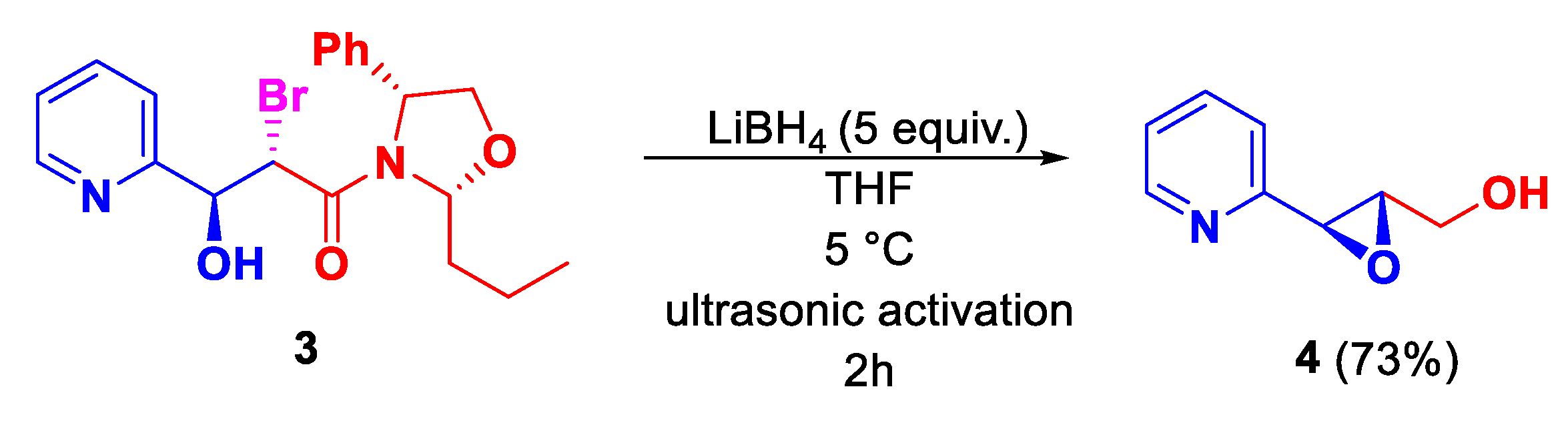
Scheme 5.
Sequential obtention of the inseparable diastereomeric mixture of indolizinium salts.
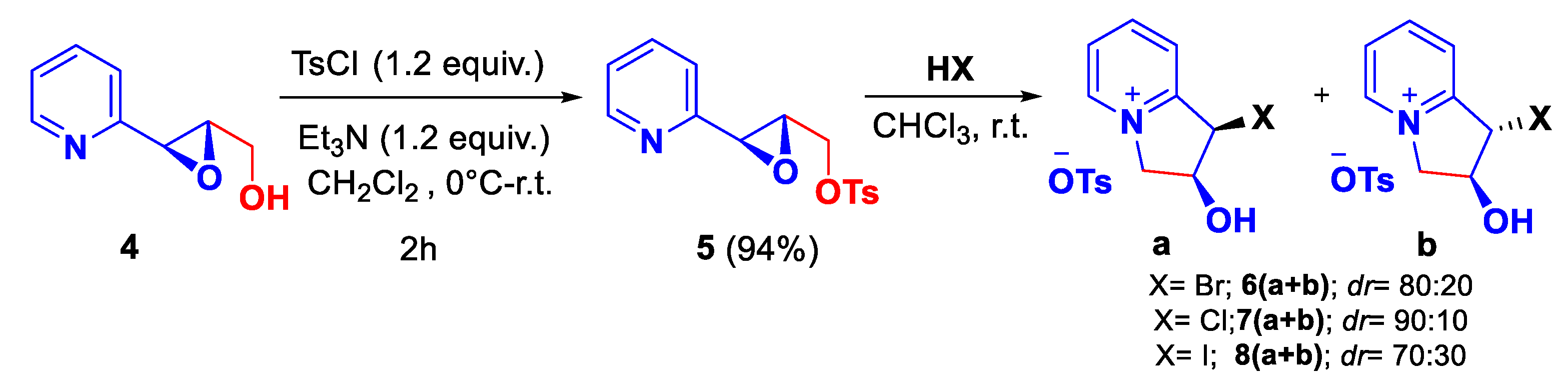
Scheme 6.
One-pot oxirane opening and intramolecular 5-exo-tet cyclization.
Scheme 7.
Diastereospecific reduction of indolizinium salt with NaBH4.
Scheme 8.
Synthesis of (+)-lentiginosine from 11a.
Scheme 9.
Synthesis of (–)-1-epi-lentiginosine from diastereomeric mixture of indolizinium salts 10(a+b).
Scheme 9.
Synthesis of (–)-1-epi-lentiginosine from diastereomeric mixture of indolizinium salts 10(a+b).
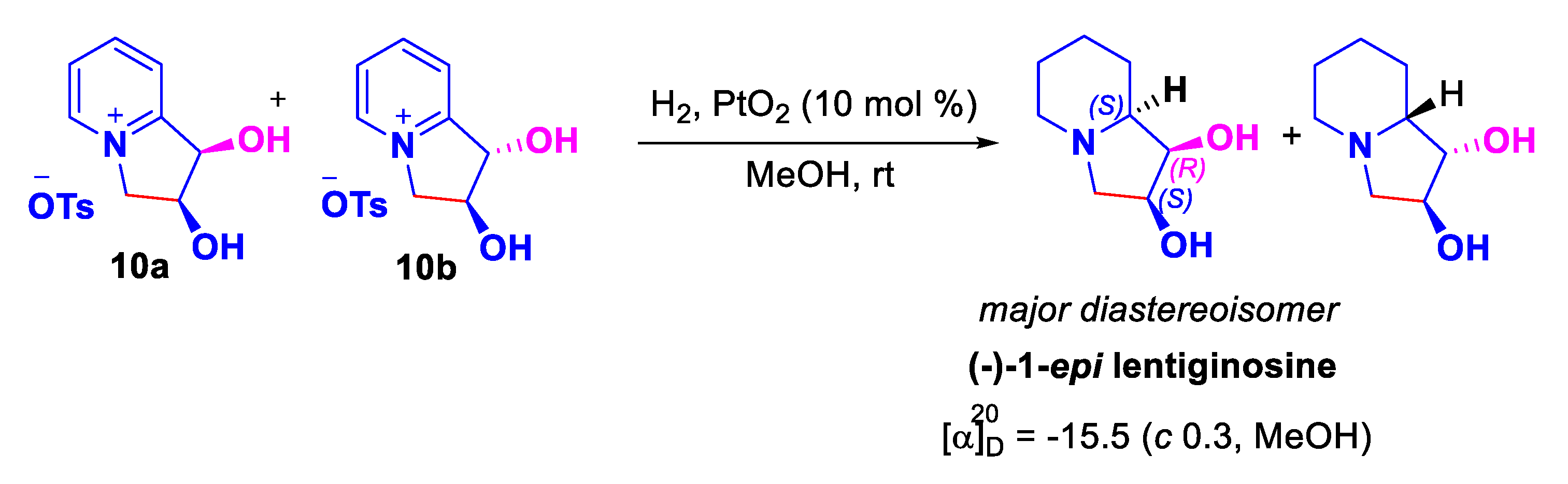

Table 1.
Optimization of the reaction conditions.
| Entry | Solvent | Time | Yield [%] |
|---|---|---|---|
| 1 | THF | 3.5 h | 95 |
| 2 | CHCl3 | 1 h | 98 |
| 3 | Toluene | 3 h | 95 |
| 4 | CCl4 | 40 min | 80 |
| 5 | MeOH | 72 h | NR |
| 6 | CH3CN | 20 h | 80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



