
Submitted:
29 March 2023
Posted:
31 March 2023
You are already at the latest version
Abstract
Despite extensive research, the links between the accumulation of glycosaminoglycans (GAGs) and the clinical features seen in patients suffering from various forms of Mucopolysaccharidoses (MPSs) have yet to be further elucidated. This is particularly true for the neuropathology of these disorders, even though the neurological symptoms are currently incurable, even in the cases where a dis-ease-specific therapeutic approach does exist. One of the best ways to get insights on the molecular mechanisms driving that pathogenesis is the analysis of patient-derived cells. Yet, not every pa-tient-derived cell holds potential to recapitulate relevant disease features. For the neurodegenerative forms of these diseases in particular, it is challenging to grow neuronal cultures that accurately represent them because of the obvious inability to access live neurons. This scenario changed sig-nificantly since Yamanaka et al. published their protocol for induction of pluripotent stem cells (SC) from adult human fibroblasts. From then on, a series of differentiation protocols to generate neurons from induced pluripotent stem cells (iPSC) was developed and extensively used for disease mod-eling. Currently, human iPSC and iPSC-derived cell models have been generated for several MPS and numerous lessons were learnt from their analysis. Here we review most of those studies, not only listing the currently available MPS iPSC lines and their derived models, but also summarizing how they were generated and the major information different groups have gathered from their analysis. Finally, and taking into account that iPSC generation is a laborious/expensive protocol that holds significant limitations, we also comment on a tempting alternative to establish MPS pa-tient-derived neuronal cells in a much more expedite way by taking advantage of the existence of a population of multipotent SC in human dental pulp, to establish mixed neuronal and glial cultures.
Keywords:
Mucopolysaccharidoses
; Disease Modeling
; in vitro models
; induced Pluripotent Stem Cells (iPSCs)
; Dental Pulp Stem Cells (DPSC)
1. Introduction
Lysosomal storage disorders (LSD) are a group of rare diseases caused by mutations in genes that encode lysosomal enzymes, lysosomal membrane proteins or transporters and in a few cases by other cell proteins that are important for lysosomal function. This leads to an accumulation of undegraded substrates, which ultimately causes a broad range of highly debilitating clinical symptoms affecting multiple organs/systems, including the central nervous system (CNS) [1]. Among the LSDs that may present with severe neurological phenotypes, are Mucopolysaccharidoses (MPSs), which are caused by impaired degradation of glycosaminoglycans (GAGs), with consequent intralysosomal accumulation of undegraded products [2]. Quite remarkably, none of the available therapies for this sub-group of disorders works over the neurological symptoms. Instead, they are limited to treating non-neurological signs [3]. Thus, there is an urgent need for the development of new ones that can tackle the neuronal pathogenesis. A crucial step towards the development of those approaches is the existence of suitable disease models, which can be used to both further understand the pathophysiological mechanisms that underlie the phenotype and adequately test those therapeutic strategies in vitro. Here we will review some of those models and the major results that other groups have published on the pathophysiological mechanisms underlying this particular subset of LSDs. We will highlight the different patient samples they used to start with, and the protocols they relied on. Particular attention will be given to the induced Pluripotent Stem Cells (iPSCs) potential to mimic disease-relevant phenotypes and to the methods others have used to assess them. Finally, we will also mention a few studies, which have provided in vitro proof of principle on the potential of ex vivo genetically-corrected iPSC -derived cells for therapeutic purposes.
Overall, the results here reviewed strongly support the utility of iPSCs for the study of MPSs. Still, iPSCs generation is a laborious and expensive protocol. Furthermore, the use of iPSCs has a number of limitations, which should not be ignored. That is why in our lab we are addressing the question of whether alternative sources of stem cells (SC) may exist, holding a similar potential for disease modeling in these rare yet life-threatening genetic disorders. In fact, recent studies have shown that dental pulp provides a niche for diverse arrays of dental mesenchymal stem cells (MSCs), and they are now being established in our laboratory for the study of LSDs, particularly MPSs. This approach is non-invasive, cost-effective, and can be established in any laboratory with standard cell culture conditions. And as we will briefly highlight in this manuscript, it may provide another potentially effective approach for investigating cellular and gene expression changes that occur in monogenic diseases.
2. Lysosomal Storage Diseases
Lysosomes have in their composition around 60 acidic hydrolases responsible for the degradation of a variety of substrates including proteins, lipids, carbohydrates and nucleic acids [4,5]. When one or more lysosomal enzymes fails to fulfill its function, the substrate(s) it would degrade starts to accumulate in a process which, eventually will result in cellular toxicity and even cell death [6,7,8]. In general, those enzymatic dysfunctions have a genetic origin, as they are caused by mutations in any of the genes that encodes for the defective protein. This sort of monogenic disorders characterized by intralysosomal substrate accumulation constitutes a large group collectively known as LSDs [9]. This group comprehends around 70 disorders being almost all characterized by a recessive autosomal pattern of inheritance. Currently, only three exceptions are known, all of them X-linked.
Classically, LSDs are classified into different subgroups depending on the substrate that is accumulated [10]. According to that classification, we can distinguish five major groups of LSDs: Sphingolipidoses (those which accumulate sphingolipids), Mucopolysaccharidoses (those which accumulate GAGs), Oligosaccharidoses (those which accumulate oligosaccharides), Sialic Acid disorders (those which accumulate sialic acid) and Mucolipidoses (which accumulate a number of different substrates, namely of mucopolysaccharides, sphingolipids and glycolipids). But not all LSDs fit into this traditional classification. That is why we can usually find (at least) two extra categories in most of the tables where these disorders are listed: the so-called Neuronal Ceroid Lipofuscinoses (NCLs) and a general category coined Miscellaneous (whose disorders may accumulate substrates as diverse as polysaccharides and amino acids) [11]. There is, however, an obvious link between the majority of the referred disorders: the neuronal storage of undegraded or partially degraded substances, with subsequent cell death in the brain. Accumulation within this system result into a panoply of symptoms including neurocognitive decline, blindness, seizures and, ultimately, premature death. Still, not every LSD shows a obvious CNS involvement. Some LSDs present in a much more multisystemic way and, for some the milder forms may actually lack neurological symptoms. Symptoms like hepatosplenomegaly, cardiomyopathy, fibroelastosis, dysostosis multiplex and cervical spinal cord strangulation are often part of the LSD phenotype, and may be the only clinical manifestations in a number of patients [12]. In general, the clinical manifestations depend on the substrate accumulated and on the site where that accumulation occurs. Furthermore, depending on the specific function of the enzyme, which is either missing or dysfunctional, and on its level of deficiency, storage may accumulate at different rates, causing the disease progression to be significantly different [12].
Generically, LSDs are rare diseases. Nevertheless, when considered as a whole, their prevalence may be as high as 1 in 5,000 [10]. Depending on the group and/or subgroup of diseases, there are differences in the severity of symptoms, rate of progression, and organs/systems affected. Still, regardless of their overall severity, LSDs are characterized by a relentless progression of symptoms and no cure is yet known for any of these disorders. There are, however, four different approaches, which have been explored for a number of them and some of them have actually reached the clinic [13]: Enzyme Replacement Therapy (ERT) [13]; Hematopoietic Stem Cell Transplantation (HSCT) [13]; Substrate Reduction Therapy (SRT) [10,13] and Chaperone Therapy [13,14]. It should be noticed, however, that these therapies are only available for a restrict number of LSDs and, even in the cases where a therapeutic option is available, it may fail to address all of the disease’s symptoms, as we will extensively discuss throughout this review.
The most widely used therapeutic approach in the field is also the first one to have been developed: ERT. Briefly, ERT relies on a very simple principle: if LSDs are caused by an enzyme deficiency, one may overcome them by simply giving the enzyme that is missing to the patients who suffer from its dysfunction. Easier said than done, but still, a number of recombinant enzymes are now available in the market and being used by different LSD patients worldwide [15]. Those ERT formulations are administrated intravenously in a periodic manner. Briefly, the recombinant enzyme gets internalized into the cells by the so-called mannose-6 phosphate receptors (M6PR), and reaches the lysosomes through the mannose-6-phosphate pathway, where it may fulfill its function. The existence of M6PRs within the plasma membrane also allows for subcellular cross correction. Meaning: the recombinant enzyme may move from one cell to the next one, thus maximizing its therapeutic effect [15]. However, ERT does hold a series of drawbacks, for instances it may lead to the production of antibodies against the synthetic enzyme. Furthermore, recombinant enzymes do not reach all organs/systems. For example, traditional ERT does not reach the CNS, thus being a real therapeutic option only for non-neurologic diseases or for their non-neurological forms. Despite their limitations, ERTs for Gaucher Disease [16], Fabry Disease [17], Acid Lipase Deficiency [18], Ceroid lipofuscinosis type 2 [19], Niemann-Pick diseases type A/B [20] , α- Mannosidosis [21], and MPS I, II, IV, VI, and VII [22] are, nowadays, a reality and numerous patients have benefited from them over the last decades. Additional clinical trials with novel enzymes and alternative delivery routes are also ongoing [23]. Overall, ERT is not a cure, but it does significantly increase enzyme activity in many disorders, thus improving their associated clinical symptoms [24].
Another therapeutic approach for LSDs, which has been around for a few decades now, with very good results for a few diseases is HSCT [25]. Briefly, we can distinguish 3 types of HSCT: allogenic (when the transplanted cells are derived from a healthy and fully-matched donor); syngeneic (when the transplanted cells are derived from an identical twin); and autologous (when the transplanted cells are derived from the patient before the procedure). While allogeneic HSCT is the standard of care these days for a few LSDs, either syngeneic or autologous transplants are virtually better options, as they work around some of the acute complications associated with HSCT such as veno-occlusive disease of the liver, acute and chronic graft versus host disease, and opportunistic infectious conditions. In those two cases, however, the cells which are collected need to be genetically modified ex vivo to a normal function. Currently, those approaches are under clinical trial for a few LSDs [26,27,28,29,30]. Regardless of the HSCT type, in terms of procedure, its principle is simple: first, the patient needs to receive some type of therapy that will inhibit the immune system (to prevent rejection); then the modified cells are injected in the patient. Due to their stemness potential, the graft cells, which are capable of synthesizing functional target enzymes, will rapidly proliferate and differentiate providing a natural, endogenous source of the enzyme, which was previously missing [31].
Still, this approach does not seem to be effective for a number of LSDs where, in theory, it should work [32]. There are, however, a few diseases for which this procedure is highly recommended and does show exceptional results if performed soon enough. That is the case of one particular form of MPS: the Hurler syndrome (the severe forms of MPS I). Allogeneic HSCT is still considered the "standard of care" for patients suffering from that syndrome. Nevertheless, this procedure in only effective when performed at the very initial stages of the disorder. In fact, it has only been shown to enhance the cognitive function in patients with less than 9 months [9,10,25]. Even though Hurler seems to be the perfect example on the success of HSCT, there are some general considerations we can draw for other LSDs to which may apply. Usually, visceral symptoms can be improved, whereas skeletal lesions remain relatively unaffected. The effect on neurologic symptoms varies. Still, HSCT remains a viable treatment option in those LSDs where data supportive of disease stabilization or amelioration is known (reviewed in [33]).
But there are two other, more recent approaches, which may be used to overcome the LSD-associated pathology. The first one is SRT, with licensed products available for Gaucher Disease and Niemann-Pick Type C. Again, its rationale is quite straightforward: it promotes an overall reduction of the accumulated substrate(s) by inhibiting its biosynthesis, thus ameliorating the associated phenotype(s). Unlike ERT, the presently available substrate reduction drugs are orally administrated, and some of them have the ability to cross Blood-Brain Barrier (BBB) achieving an effect on CNS [34]. Still, this option has a slower onset efficacy, and so far, it is restricted to sphingolipidoses. The conjugation of SRT with other therapies may significantly improve the treatment of LSDs [10,34].
Finally, there is also the so-called chaperone therapy. Pharmacological chaperones are small molecules defined by their ability to help a protein to fold correctly [35]. By doing so, those molecules will help their target protein escape proteasomal degradation and reach an adequate subcellular destination, where it can exert its function. Basically, this molecule binds to the misfolded protein in the endoplasmic reticulum (ER) forming a stable complex that prevents the misfolding. When the complex arrives to the lysosome, dissociation occurs. As a result, a functional or partially functional protein gets internalized into that organelle, where it can exert its activity [14]. It is worth mentioning that this sort of therapeutic approach may only work for disease-causing missense mutations. So far, Fabry disease (one of the most common LSDs worldwide) is the only LSD with an approved chaperone therapy: migalastat (Galafold®, Amicus Therapeutics). This drug is currently being used in the clinic for a significant number of Fabry disease patients, all harboring missense mutations that cause misfolding of α-galactosidase, and has been shown to improve the associated cardiac and renal symptoms [36,37]. And, while no other chaperone molecule has reached the clinic so far, several studies are being performed in other LSDs [38,39,40,41,42].
3. Mucopolysaccharidoses
Among the LSDs in need for better and more effective therapeutic options are the Mucopolysaccharidoses (MPSs). The MPSs subgroup includes seven different disease types, all of them accumulating glycosaminoglycans (or GAGs) as the primary substrate. An overview of each individual disorder is described below.
MPS I is one of the most common forms of MPS and the first MPS type treated with ERT (available since 2003) [43]. At a clinical level, MPS I may be divided into three subtypes: Hurler (OMIM #607014), Hurler-Scheie (OMIM #607015), and Scheie (OMIM #607016) depending on the disease severity [44]. Hurler syndrome is the most severe form of them all and Scheie is the mildest, with Hurler/Scheie being somehow intermediate phenotype but in general, type I has an incidence of 0.11 [45] to 3.62 [46] per 100,000 live births (reviewed in [47]). As the majority of LSDs, MPS I is characterized by a progressive pattern that includes several stages of clinical manifestations. In this multisystemic disease during the first 6 months of life, the children present symptoms such as coarse facies hepatosplenomegaly, and upper airway obstructions that usually evolve to more specific and severe symptoms associated to a constant increase in the accumulation of GAGs in the soft tissues, bones, spleen and liver. Overall, dysostosis multiplex is considered the most common clinical symptom of MPS I [48]. Regardless of the clinical presentation, IDUA is the affected gene in this disorder. Mutations in this gene, which encodes for α-L-iduronidase (IDUA; EC 3.2.1.76), lead to an enzyme deficiency that ultimately results in heparan and dermatan sulfate (HS and DS, respectively) accumulation [49]. To date, 359 mutations are identified for this gene [50,51], and currently, there are two possible therapeutic options: ERT and HSCT, which is only used in the most severe form of the disease and, preferably in the first years of life [52]. Regarding ERT, there is only one recombinant enzyme approved for MPS I: laronidase (Aldurazyme®, Genzyme). As every other ERT, this recombinant enzyme is injected into the blood circulation, which leads to the correction of the enzyme deficiency in various organs and tissues, except the brain, once it does not cross the BBB [53,54].
MPS II (OMIM #309900), or Hunter syndrome, is the only X-linked MPS disease; all the other MPSs are autosomal. Thus, in the Hunter syndrome, males are the most affected, with a prevalence of 0.1 [55] to 2.16 [56] in 100,000 live births (reviewed in [47]). Two forms of the disease may be distinguished: neuronopathic and non-neuronopathic, being the most severe the CNS-associated [57]. Regarding clinical manifestations, the skeletal, cardiac and respiratory systems are the ones mostly affected. In the most severe cases, adding up to the symptoms affecting the previously referred systems, there is also an involvement of the CNS. Usually, for the neuronopathic form, the average life expectancy is around 10-15 years of age, while the individuals who suffer from the attenuated one may live beyond 50 years [58]. Regardless of the subtype, MPS II is caused by mutations in the IDS gene, which encodes the enzyme iduronate 2-sulfatase (IDS; EC 3.1.6.13). The IDS gene is split into 9 exons, spanning approximately 24 kb [59]. There are 817 mutations identified to date, which may cause this syndrome ([50]). The iduronate 2-sulfatase deficiency leads to the accumulation of two substrates: HS and DS. Regarding MPS II therapeutics, ERT with idursulfase (Elaprase®, Shire) is the first choice for patients with this condition [60].
MPS type III, also known as Sanfilippo syndrome, may be subdivided into 4 subtypes: IIIA (OMIM #252900), IIIB (OMIM #252920), IIIC (OMIM #252930), and IIID (OMIM #252940). Each particular subtype is associated to a unique enzymatic defect: MPS IIIA is caused by the deficiency of the enzyme Heparan-N-sulfatase (SGSH, EC 3.10.1.1); MPS IIIB, by its turns is caused by defects in the enzyme N-acetylglucosaminidase (NAGLU, EC 3.2.1.50); in MPS IIIC the protein involved is the transmembrane enzyme, acetyl-CoA:Glucosamine N-acetyltransferase (HGSNAT, EC 2.3.1.78) and, finally, the MPS IIID is caused by defects in N-acetyl-glucosamine-6-sulfatase (GNS, EC 3.1.6.14). Regardless of the enzymatic defect itself, all of them are associated with a severe deterioration of neurological function [61], which results in a number of clinical symptoms either directly or indirectly related to a CNS dysfunction, such as behavior problems, sleep disturbances, hearing impairment, development regression, recurrent infections in the respiratory tract, and facial dysmorphology [62,63]. The general prevalence is 0.06 [64] to 1.89 [65] in 100,000 live births (reviewed in [47]), with subtypes A and B being more common for most populations than C and D [66]. Regardless of the affected genes, the stored substrate is always HS.
Various mutations were already identified for the different forms of MPS III [67]: in the case of SGSH gene (with a total of 8 exons and associated with subtype IIIA), 163 mutations have already been identified; in subtype IIIB, 215 mutations have already been identified in any of the 6 exons that constitute the NAGLU gene, or their surrounding intronic sequences; in the HGSNAT gene, 93 mutations along the 18 exons and their respective introns are known to cause the deficiency observed in subtype IIIC. Finally, in subtype IIID, where the GNS gene (which spans 14 exons) is mutated, only 25 mutations were identified [50]. Unfortunately, there is no approved treatment for these neurologic diseases. On the one hand, while it has already been attempted by several different teams, HSCT has proven virtually no benefit over the neurocognitive symptoms [68,69,70,71,72]. On the other hand, ERT is hard to apply, once classically formulated enzymes do not penetrate the CNS. Moreover, in the case of MPS IIIC, for example, ERT is not an option, once the deficient enzyme is a transmembrane protein.
There are, however teams attempting brain-specific delivery of both ERT and chemical compounds for MPS type III. In general, there are three strategies to increase the delivery (reviewed in [73]): enzymatic modulation, route(s) of administration [74,75,76], and increase of enzyme dosage. In addition, cellular and genetic therapies represent approaches that have gained importance when it comes to BBB delivery (reviewed in [77]). Targeting brain cells through enzymatic modulation consists of the combination of the enzyme with protein/peptides than can facilitate BBB crossing (reviewed in [78,79]). In the cellular and genetic therapies field, among other possibilities, gene therapy with the use of adeno-associated virus has been stealing a lot of attention with extensive works to reach the BBB and have the intended effect [75,80,81,82]. Besides the modifications above referred, substrate reduction therapy (SRT) constitutes also an alternative to overcome the BBB [83,84,85]. The development of a valuable treatment has reached very high levels of need so that regulatory initiatives to support the development of a possible treatment are commonly found [61,67,86,87].
There are two different forms of MPS IV, each one caused by a single enzymatic defect: N-acetyl-galactosamine-6-sulfatase (GALNS; EC 3.1.6.4) deficiency underlies MPS IVA (OMIM #253000) while β-galactosidase (EC 3.2.1.23) defects cause MPS IVB (OMIM #253010). The involved genes are GALNS and GLB1, respectively. MPS IV, or Morquio Syndrome, has an incidence of 0.07 [64,88] to 3.62 [46] in 100,000 live births (reviewed in [47]). Unlike MPS III, which is almost exclusively a neurological syndrome, the skeleton is the main affected system in MPS IV, with the substrate accumulating predominantly in the cartilage and bones. Consequently, the major clinical manifestations observed are bone deformations, short stature, and mobility alterations [89]. In both cases, keratan sulfate (KS) and chondroitin-6-sulfate (C6S) are the accumulated substrates. So far, approximately 467 mutation have been described in the GALNS gene [50], composed of 14 exons, all associated with MPS IVA [90,91]. Concerning type IVB, 263 ([50]) mutations are known to cause this disorder. The only FDA-approved treatment for MPS IV is elosulfase alfa (Vimizim®; BioMarin Pharmaceutical Inc.) that is used in MPS IVA patients. All other options are symptomatic and mostly consist in surgical approaches to prevent spinal cord damage or other skeleton issues, for example, spinal decompression surgery [92].
Yet another form of MPS, usually coined as Maroteaux-Lamy Syndrome, is MPS type VI (OMIM #253220). 242 mutations in the ARSB gene (which spans 8 exons) are known to cause this disorder ([50]). The frequency for this disorder is estimated between 0.0132 [93] and 7.85 [46] in 100,000 live births (reviewed in [47]). Even though being a multisystemic condition, MPS VI does not affect intelligence, and, like Morquio syndrome, the skeleton is the most affected system [94]. Thus, the clinical manifestations are very similar to those described above including short stature, low body weight and impaired pulmonary and motor functions [95]. To counteract the DS storage promoted by the deficiency of Arylsulfatase B (EC 3.1.6.12) activity, galsulfase (Naglazyme®, BioMarin Pharmaceutical Inc) is the drug approved and currently employed in patients. HSCT may also be possible; however, additional safety studies are needed [95,96,97].
MPS type VII (OMIM #253220) or Sly syndrome occurs with an estimated frequency of 0.02 [64,98,99,100] to 0.29 [55] per 100,000 live births (reviewed in [47]). Several systems/organs are involved in this disease with clinical features affecting organs as diverse as the eyes, lungs, heart, musculoskeletal, spleen, etc. Thus, the most common symptoms are described as coarse facial features, increased of cranial circumference, reduced of pulmonary function, obstructive airway disease, dystosis multiplex, decrease of mobility, joint contractures, abdominal abnormalities, short stature and hepatomegaly/splenomegaly. There may also be a neurological involvement as testified by recurrent observations of limited vocabulary and mental retardation in several MPS VII patients [101]. Overall, these symptoms are caused by an ubiquous accumulation of several different GAGs, namely DS, HS, and CS, as a consequence of the deficient activity deficiency of β-glucuronidase (GUS: β-D-glucuronoside glucuronosohydrolase, EC 3.2.1.31). The GUSB gene (12 exons) [102] with 81 mutations identified so far ([50]), is the one affected in this disorder [103]. The approved drug for this pathology is vestronidase alfa (Mepsevii™, Ultragenyx), which is indicated in both pediatric and adult cases [104].
Finally, MPS IX or Natowicz disease (OMIM #601492) is an ultra-rare disorder. The first report was published in 1996, with the described patient presenting a number of clinical manifestations associated to joint and skeletal systems [105]. This disorder is caused by a deficiency in the enzyme hyaluronidase 1 (HYAL1; EC 3.2.1.35) due to mutations in the HYAL1 gene, which leads to the accumulation of yet another substrate: hyaluronan. Due to the rareness of the disorder, very few mutations have been reported to date (only 7 [50]), and a possible treatment is very challenging [106].
In general, even though the molecular bases and biochemical defects underlying MPS diseases are well defined, knowledge is still lacking on the pathophysiological mechanisms that actually trigger the appearance of different symptoms in the different organs and systems. And, even though much has been learnt over the last decades, from the study of individual patients and, particularly, from the generation and extensive characterization of bona fide in vivo models, truth is we haven’t still fully understood the whole physiological cascade, which underlies some of MPSs’ most challenging phenotypes, namely those which affect the CNS. And this is particularly relevant since no therapeutic exists to ameliorate them. Still, finding an in vitro model that could recapitulate the disease-relevant features is also challenging once live neurons are inaccessible cells. Indeed, for almost a century, patient-derived fibroblasts were gold standard for in vitro studies in MPSs, as in all other LSDs. These cells were relatively easy to access, since a simple skin biopsy would be enough to obtain them and remarkably, they did display the cellular phenotype that actually coined these diseases as “storage” disorders: the presence of undegraded or partially degraded substrates. Nevertheless, fibroblasts may also fail to recapitulate disease-relevant features, which are only evident in other particular cell types, of higher pathological significance such as neurons. A viable option is to generate the neurons from a patient-derived cell line, which involves extracting the cell from the patient and differentiating it into neuronal cells. Indeed, there are two possible ways to do this process: iPSCs and mesenchymal stem cells (MSCs) from the patient.
4. Modeling Mucopolysaccharidoses with induced pluripotent stem cells (iPSCs)
Human iPSC generation in particular started its journey in 2007, when Yamanaka et al. [107] first generated those cells from human somatic fibroblasts using a remarkable method, which relies in the retroviral transduction of 4 independent transcription factors into patients’ fibroblasts: Oct-3/4, Sox2, Klf4, and c-Myc. Remarkably, the cells that resulted from this experiment showed numerous similarities with human embryonic stem cells (hESCs) including morphology, proliferation capacity, gene expression pattern and in vitro differentiation potential. Ever since this hallmark report was published, the search for novel and improved protocols for cells reprogramming advanced at an outstanding pace, with various optimizations being published in order to generate virtually every cell of interest from iPSC of different origins [108].
Over the past few years, in vitro models derived from iPSCs have been unraveling some enigmatic aspects of MPSs. In particular, the subtypes that present neurological involvement appear as the ones with the greatest need for additional knowledge and new therapeutic solutions.
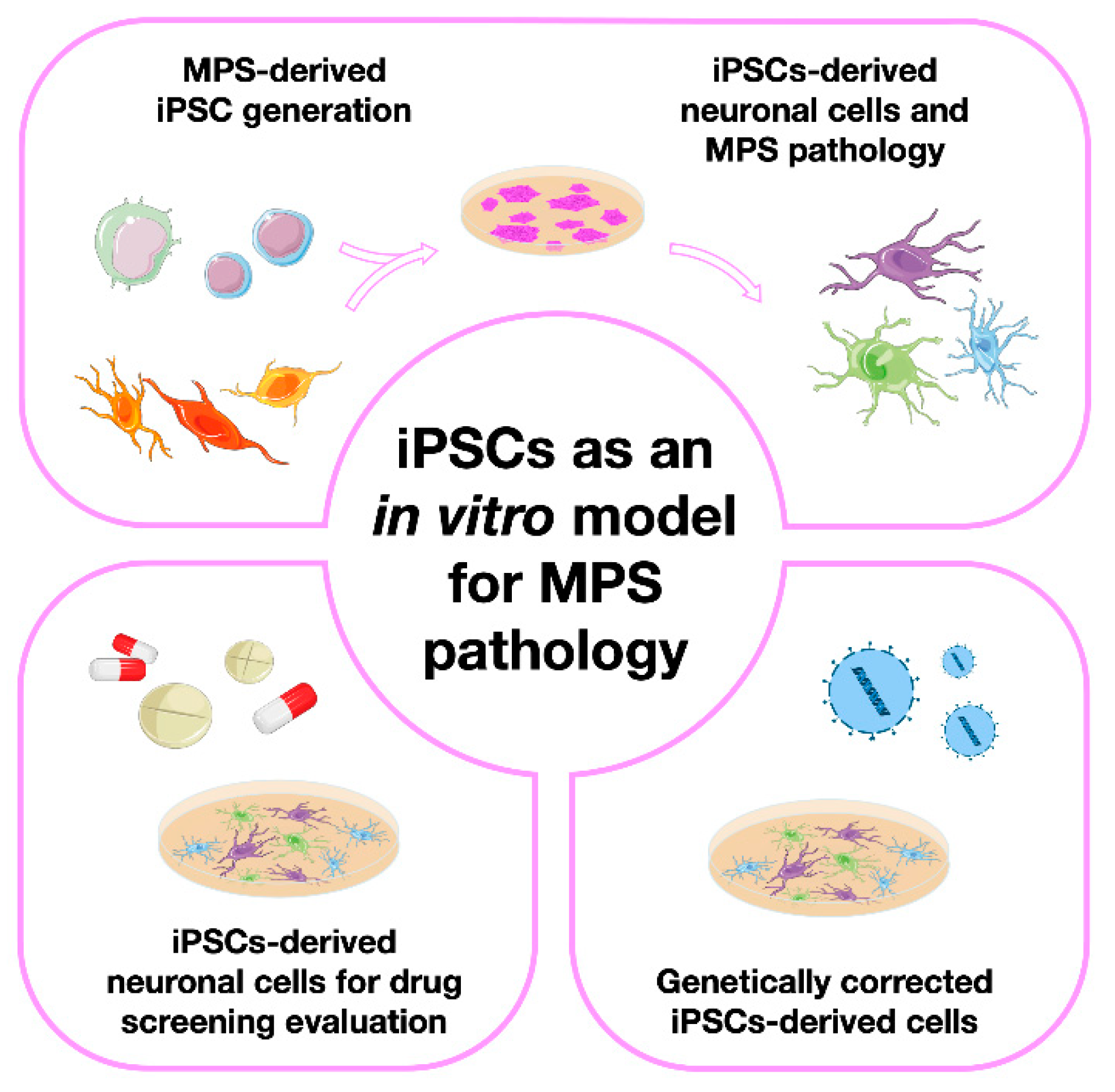
Here we will review numerous studies attempting not only MPS-derived iPSC generation, but also their subsequent differentiation into relevant cell types. We have divided those studies into four major groups, each one of them having a dedicated section in this review (Error! Reference source not found.). First, we will address the papers in which only iPSCs were generated, briefly discussing the methods used to characterize them. Then, we will focus on those papers where iPSCs were further differentiated into either neural precursor cells or totally differentiated neurons, highlighting the disease modeling potential of those lines by showing the numerous pathophysiological insights one can get with a few simple cellular assays. Then, we will go through the papers where those cells were used for in vitro drug screening, commenting not only on the results obtained but also on the advantages or disadvantages of the use of those particular cells for therapy development. Finally on the last iPSC-devoted section, we will refer to a few studies were the therapeutic potential of these particular SC was addressed. Meaning: we will summarize the papers where instead of generating iPSCs to further understand one particular disorder or genotype or to serve as a drug-screening platform, the authors have actually created them for gene therapy.
Figure 1.
The four major types of studies involving the generation and characterization of induced pluripotent stem cells (iPSCs) as in vitro models for Mucopolysaccharidoses (MPS), grouped together according to their ultimate aims: 1) papers on iPSC generation and characterization; 2) papers describing their subsequent differentiation into different types of neuronal cells; 3) papers on the use of iPSCs and their derived cell models for drug screening and, 4) papers on the use of genetically corrected iPSCs for therapeutic purposes.
Figure 1.
The four major types of studies involving the generation and characterization of induced pluripotent stem cells (iPSCs) as in vitro models for Mucopolysaccharidoses (MPS), grouped together according to their ultimate aims: 1) papers on iPSC generation and characterization; 2) papers describing their subsequent differentiation into different types of neuronal cells; 3) papers on the use of iPSCs and their derived cell models for drug screening and, 4) papers on the use of genetically corrected iPSCs for therapeutic purposes.
4.1. The basic studies: iPSC generation from different MPS patient-derived cell sources
The first MPS-derived iPSCs were generated in 2011 when Thomas Lemonnier and colleagues [109] reprogrammed fibroblasts from two patients suffering from MPS IIIB into pluripotent stem cells (PSCs). As in any other iPSC generation report, the resulting SC were extensively analysed and characterized. In this particular study the authors confirmed a positive expression of three particular markers (SSEA4, Nanog and TRA-1-60) and the differentiation ability of those cells, thus proving their pluripotency nature. Additionally, the authors have also provided information on the karyotype presented by those cells. This is a relevant assessment whenever a novel iPSC line is generated but it should also be considered later on, when using the same iPSC line after several passages, or after having one particular iPSC cell line in culture for a long period. In fact, long-term iPSCs culture is known to result in chromosomal abnormalities, changes in gene expression and cellular functions, and even increases the risk of the iPSCs being tumorigenic. As genomic alterations present potential risks in the overall applications of iPSCs, it is crucial to monitor the genomic integrity of iPSCs lines. That is why iPSC karyotype analysis is such an important step on the validation of this type of cell models, and nowadays considered as a routine procedure by all the groups working with iPSC technology [109].
But these weren’t the only published MPS IIIB-derived iPSCs reported in the literature so far. Two other MPS IIIB patient-derived iPSCs lines were generated from skin fibroblasts by Vallejo-Diez et al. in 2018 [110]. In that particular study additional pluripotency markers were also assessed besides the previously referred Nanog and TRA-1-60: Oct-3/4; Sox2; TRA-1-81 were also analyzed. As in the previous study, karyotype was also assessed, and the associated mutation confirmed. But the authors have actually went one step further in terms of SC characterization analysing the differentiation ability of the generated iPSCs by evaluating the formation of embryoid bodies after 10 days of differentiation, using specific markers for the 3 germ layers. The same characterization was carried out for MPS IIIA-derived cells, where the same team was the first to create patient-derived iPSCs for that particular disease [111].
Regarding MPS IIIC, Noelia Benetò et al. have also generated iPSC lines, but this time using a slightly different protocol from the previously described ones. Instead of using patient-derived cell lines, these authors have created isogenic HGSNAT-mutated lines from healthy iPSCs using CRISPR/Cas9. This technology allows to create such lines in human cells which have the genetic background of the wild-type cells but differ by the genetic modification of interest. These isogenic pairs are powerful tools for understanding gene function. While circumventing confounding effects of genetic background, they allow for genotype-phenotype correlation studies [112]. To prove the reliability of this model, they measured HGSNAT enzyme activity and assessed the differentiation capacity of the generated SC. This last parameter, was studied by inducing the formation of embryoid bodies and their subsequent differentiation into three germ layers. The formation of these structures is a characteristic of pluripotent SC and serves as a platform for the intended differentiations [113].
Still, and even though the neurological involvement is a major hallmark of the Sanfilippo syndrome, almost every other MPS may present with severe neurological symptoms. Types I and II in particular have even specifically recognized clinical forms where the CNS is strongly affected, presenting with major clinical symptoms. Thus, these disorders would also strongly benefit from the development of appropriate neuronal cell models to study them. Furthermore, they would also allow for tissue- or cell-specific drug screening assays. Generating iPSC lines from those disorders is a rational step towards that first goal and that is probably one of the reasons why iPSCs lines from both disorders have also been created and subsequently published in the past few years.
Regarding MPS type II, in 2016, Eszter Varga et al. collected peripheral blood mononuclear cells (PBMCs) from phenotypically affected patients with 1-, 3-, and 7-year old [114,115,116] and an unaffected carrier mutation woman with 39-year old [117]. Then, all PBMCs patients’ cells were subjected to induction of the pluripotent stage, originating disease-specific iPSCs, which were extensively characterized as required by the technology itself.
Last but not least, MPS type I has also been modeled with the help of this revolutionary technology. In 2019, Lito S. et al. [118] and Suga M et al. [119] have reprogrammed and characterized dermal fibroblasts and PBMCs, respectively, into iPSC lines. The fibroblast-derived pluripotent cells were obtained from a patient with the Hurler form of the disease, whereas PBMCs were collected from a patient suffering from Scheie.
4.2. Moving one step further: generation of neuronal models from MPS-derived iPSCs
As we have already referred, the neurological involvement, which places such a tremendous burden over patients suffering from several forms of MPS, may be further explored by differentiating iPSCs into different types of neuronal or pre-neuronal populations. And in fact, most works published up until now are not only focused on reprogramming different types of patient-derived cells (namely fibroblasts and PBMCs) but also on the differentiation step, searching for disease-relevant features in those cells. Ultimately, these models may also allow for the discovery of novel hallmarks related or non-related with neuropathology. Considering the intrinsic nature of all MPSs, lysosomal pathology is probably the more crucial parameter to study, once the enzymatic defect will primarily affect this organelle.
Thus, when it comes to disease phenotype assessments, some markers have been particularly relevant in the LSD field, namely the lysosome-associated membrane proteins 1 and 2 (LAMP-1 and LAMP-2). These two proteins are heavily investigated once they represent the major components of the lysosome membrane. For example, in the study involving the first MPS-derived iPSCs [109], which were generated from MPS IIIB patients’ samples, the accumulation of storage lesions was intensively analyzed through LAMP-1 and Golgi matrix protein 130 (GM130) detection. A prominent fluorescence of both markers was detected in patient-derived iPSCs, and the vesicles observed by microscopy were revealed to have a heterogenous content. This was actually the first study to describe Golgi Complex impairment in the MPS pathology. Most importantly, beyond iPSCs generation, this group has also investigated the differentiation into Neural Stem Cells (NSCs) by adding specific growth factors to the original iPSC culture, namely fibroblast growth factor 2 (FGF2), and endothelial growth factor (EGF). When this protocol was initiated, the development of neurospheres became evident and after 2 weeks of non-adherent growth, the authors measured both the expression of Nestin (a neural progenitor marker) and total GAGs storage, having observed increased storage levels. Interestingly, the higher LAMP-1 and GM130 expressions previously seen in iPSCs did not translate to the floating neurospheres. However, the gene expression profile showed significant alterations in several pathways including the Wnt signal transduction pathway and transforming growth factor β (TGFβ) signaling, as well as genes encoding proteins associated with cell adhesion, Golgi apparatus and lysosomes. Curiously, that higher LAMP-1 and GM130 fluorescence was seen again as soon as neurosphere adhesion was performed, and during the final process of neuronal differentiation. This observation was also accompanied by vesicle storage positive to LAMP-1 and Ganglioside GM3. These results reflect the existence of a modest cellular pathology during the neurodifferentiation of this iPSC model. This study was the first comprehensive characterization of MPS-affected neuronal cells in vitro [109].
To the best of our knowledge, the second report on the differentiation of MPS-derived iPSCs into neuronal cells, was the work of Bruyerè and collaborators, in 2015 [120], where these authors correlated two independent models of the disease: one in vitro and another in vivo. For the in vitro studies they used patient- and control-derived iPSCs, further differentiated into neural precursor cells (NPCs), while for the in vivo differences they used a mouse model. Their goal was to investigate how HS saccharides accumulation impact the focal adhesions (FAs). They saw that activation of FAs occurred when neural cells from healthy individuals were submitted to exogenous soluble HS fragments. Consequently, this activation becomes constitutive in MPS IIIB, once those fragments are accumulated. Constitutive activation of FA, by its in turn, affects the polarization as well as the oriented migration of those cells [120].
Later, in 2015 Canals et al. [121] performed the differentiation of MPS IIIC-derived iPSCs into neuronal cells. Their goal was to verify if early functional alterations could be visible before the appearance of disease-related phenotypes. Briefly, iPSCs lines generated spherical neural masses (SNMs), whose expression patterns included PAX6, Nestin and Sox2. The existence of active neurons was also proven by the presence of microtubule-associated protein 2 (MAP2) and Synapsin (SYP), which are dendritic and synaptic markers, respectively. Besides the formation of mature neurons, an astrocytic-related marker Glial Fibrillary Acidic Protein (GFAP) was noticed. That observation further reinforced the neurogenic capacity of these cells. Regarding the neuronal cultures generated, as expected GAG accumulation was shown to have a progressive pattern, becoming significant only after 9 weeks. These observations document a marked difference between the patient's fibroblasts and iPSCs-derived neurons: the patients’ fibroblasts presented a double amount of accumulated GAGs, right from the first cell culture, when compared to iPSCs-derived neurons. Networks activities were also evaluated to verify whether there were differences between Sanfilippo’s- and the control- iPSCs-derived neurons. Through calcium imaging, the spontaneous activity of Sanfilippo-derived neurons was shown to gradually decrease between the 6 and 9 weeks. Concerning degradation of effective connectivity, which was determined by identifying causal influences among neurons through generalized transfer entropy, an information theory method that allows drawing a functional map of neuronal interactions in the network, the authors reported that, quite differently from the controls analyzed, in the Sanfilippo neurons, strong connections were only established within a subset of neurons remaining the rest of them disconnected or poorly connected [121].
At a technical level, the authors used two different protocols, one relying on neuronal induction medium without any extra supplementation, and another where that medium was supplemented with N2 and B27, two chemically-defined supplements recommended for growth and survival of neuronal cells, and observed significant differences in the time it took for them to generate neurons. In fact, while it took several weeks in neuronal induction medium to arise mature neurons, when supplementing that same medium with N2 and B27, it took only 3-5 weeks to distinguish synapses between neurons. Moreover, the neuronal activity and effective connectivity analyses they performed were nicely designed and described, and could be applicable to virtually any other neurodegenerative disease in which iPSC-based models are available [121].
Five years later, Benetó et al. [122], took advantage of the existence of a few previously reported iPSC cell lines to generate neuronal and astrocytic models of Sanfilippo syndrome type C for disease modeling and drug development: two isogenic MPS IIIC mutant lines [95], one wild-type control (from a healthy donor), and one MPS IIIC-derived line [103]. Again, all four lines were differentiated into neurons and astrocytes through lentiviral transduction and promoting into the cells the overexpression of neurogenin 2 (Ngn2) in the case of neurons (named iNs) and Sox2/Nuclear Factor one B (NfIb) in the case of astrocytes (named iAs). To confirm cell identity, the authors performed a characterization of the specific markers: in the generated neurons, they detected an increase in neural stem cell markers, namely tubulin β-3 (TUBB3), SYP, MAP2, and Neuron-specific Class III β-Tubulin (Tuj1). In the astrocytes, they observed that the expression of astrocytic-specific genes namely GFAP, Aldehyde Dehydrogenase 1 Family Member L1 (ALDH1L1), calcium-binding protein B (S100B) and vimentin (VIM) increased during the astrocytic differentiation. In addition, disease-relevant features were assessed through LAMP-2 staining and HS quantification. On the LAMP-2 immunocytochemistry assays, the authors have clearly seen an intensity increase in all disease lines compared to the wild-type one. In the case of HS accumulation, they only present results for neurons, where, as expected, increased substrate storage could be observed [122] .
Still on Sanfilippo syndrome, for the most frequent type, MPS IIIA, a comprehensive study was carried out by R. J. Lehmann et al. in 2021 [123], to investigate the ability of fibroblasts-derived iPSCs to differentiate into a neuronal cell line and discover intrinsic mechanisms of the disease. After properly characterizing the pluripotency phase, the authors performed a neurodifferentiation protocol. Two main parameters were assessed: the FGF2 signaling pathway and the neurogenesis process. Interestingly, at the beginning of this study, a curious fact was noticed: when the FGF2 supplement was added to the medium, the proliferation rate of the MPS IIIA iPSC-derived NPC culture increased significantly. Remarkably, however, even with the supplementary-FGF2, the signaling pathway of this factor is still reduced, when compared to controls. So, understanding this event became a priority for this team. In fact, the FGF2 signaling pathway only occurs when this factor binds to a possible receptor. Since FGF2 also binds to HS, this may suggest that this GAG has a key role in neurogenesis and in the homeostasis of the CNS. Taking this into account, the subsequent step was to investigate the relationship between the accumulated HS in the MPS IIIA and that lower proliferation rate. They verified that the affinity of HS MPS IIIA to FGF2 was similar to the HS present in the control, meaning that the accumulation does not alter the affinity. So, a possible explanation for decreased FGF2 signaling is that once the FGF2 binds to the accumulated HS, it does not interact with the proper receptors (cell-surface HS and Fibroblast growth factors receptors), thus affecting not only cell proliferation without supplementary FGF2 but also the signaling pathway. To investigate the disorder's impact on the neurogenesis process, control and disease cells were analyzed regarding both morphological parameters and expression patterns. At a structural level, the formation of cell bodies aggregation and cell extensions was seen in both cell lines. However, as already seen by other authors in SC models for other neurological disorders, in MPS IIIA cells, those characteristics were less frequent. Regarding the expression profiles, the genes evaluated were Nestin, TUBB3, Hyperphosphorilated neurofilament (NF-H), and neuron-specific enolase (NSE). In general, the increase/decrease pattern during the four weeks of neuronal induction was consistent between the controls and the disease cell lines; nonetheless, the disease cells showed consistently lower levels of all markers. This pattern was seen both in the absolute values themselves and in differences during the period of the procedure. Attention was also paid to the model’s capacity to recapitulate disease-relevant features. So, the same parameters, which were initially assessed in fibroblasts, were also analysed after the neurodifferentiation protocol. Not surprisingly, the MPS IIIA cells exhibited higher levels of HS, a consequence of lower enzyme activity compared to the controls, further validating the disease modeling value of this kind of cells [123].
As previously stated, though, other MPS apart from the Sanfilippo syndrome may benefit from the development of disease-specific neuronal cell models, and from the pathophysiological insights one may gain from them. Thus, some of the most stricking reports on iPSC-derived neuronal and astrocytic models for MPSs, actually came from MPS II. In 2019, Kobolák et al. [124] have even proposed a novel neuropathology model using this approach and observed similar results regarding the impact of HS as the ones described above for MPS IIIA. They used the iPSCs originally published in a number of publications already reviewed in the previous section [114,115,116,117] that were differentiated into NPCs and terminal differentiated (TD) neuronal cells. Briefly, those iPSCs were derived from two affected siblings. As expected, both individuals shared the same nonsense mutation. Also included in this study was their mother, a carrier for the same causal mutation, and an unrelated patient with a different mutation (missense). Finally, the authors have also included cells from an unrelated non-carrier, which were used as a control. At the neurodifferentiation stage, neither the patients-derived nor the healthy cells had differences in the expression of specific neuronal markers. Briefly, for NPCs the authors assessed Nestin, Sox1, and PAX6; for terminal differentiated neuronal cells, on the other hand, they checked TUBB3, MAP2, and Neurofilament 200 KDa (NF200). An exhaustive characterization of those cells was done showing that mature neurons exhibited postsynaptic density protein 95 (PSD95) expression, an indicator of activated synapses. Astrocytes, on the other hand, were shown positive to GFAP and Aquaporin 4 (AQP4) markers [124]. Interestingly, according to these authors’ results, the proliferation capacity of NPCs seems to be a distinctive factor between the controls and the patients’ cells once, after 8 passages the proliferation capacity of the MPS II-derived cells slowed down or even stopped and the PAX6 and Sox1 expression decreased, independently of bFGF and EGF presence in the cell culture. Meanwhile, the control-derived NPCs maintained the proliferation rate up until passage 12. Actually, the authors considered this event to be related to the overall MPS II brain pathology: in normal conditions HS binds at a proper rate to transcription factors, not harming the proper function of these ones. However, in the case of storage, the accumulated HS usually binds at a higher rate to transcription factors, including the one with a key role in NPCs proliferation, FGF2. This overlink prevents the accomplishment of the transcription factor function. As a response, the cells start to differentiate into neurons, occurring the appearance of anticipated neurites when compared with control cells [124].
One of the essential aims of this work was to verify if some of the disease hallmarks were already present in the NPC stage. Thus, the authors have performed several analyses and, remarkably, they realized that GAG accumulation was not evident. Interestingly however, it was even reduced compared with both controls (carrier and non-carrier). They hypothesized that this phenomenon could be related to the lower levels of the early endosomal marker RAB5, (which is translated in a lower endocytosis level) and to the normal levels of the late endosomal marker RAB7, and of the lysosomal marker Cathepsin D, in addition to the higher LAMP-2 expression. The existence of those factors is reflected into functional exocytosis by patients’ cells: GAGs and GAG fragments are expelled to the extracellular space, which could explain the appearance of GAG accumulation in cerebrospinal fluid. This whole pattern changed however, when mature neurons and astrocytes were analyzed. In fact, for those mature neurons and astrocytes differentiated from cells harboring the frameshift/PTC mutation, GAGs accumulation was (quite) evident. Importantly, however, the levels of Rab7, Rab5, and LAMP-2, were still similar to those observed in controls, indicating a non-influence of endosomal-lysosomal system over substrate accumulation. It should also be stressed that for the cells harboring the missense mutation, all assessed parameters were comparable to those seen in the controls. While somehow unexpected, these results highlight the intrinsic potential of these sort of cell-based patient-derived models as they allow for more accurate comparisons between the effect of different disease-causing mutations over several subcellular parameters, ultimately allowing for more precise genotype-phenotype correlation. Also noteworthy, regardless of the analyzed genotype, all terminal differentiated neuronal cells (neurons and astrocytes) showed a significantly increased of the autophagy marker LC3-I, revealing the involvement of this pathway in disease cytopathology. Additionally, an accumulation of autophagosomes, as well as a lower ratio of LC3-II/LC3-I, was also detected [124].
Regardless of the cell differentiation status, a common point in the cytopathology of MPS II from NPCs and TDs was the presence of ER stress with the occurrence of dilated ER cisterns. In NPCs, the authors have observed a significantly higher level of XBP1, a well-known ER stress marker. For TDs, even more events related to this stress were observed, namely: depletion of ER luminal Ca2+ storage, higher ion concentration in the cytoplasm, and a higher sensitivity to apoptosis. Concerning cell death, they noticed a higher rate of apoptosis in astrocytes rather than other TDs. It is known that this cell type plays an important role in supporting the differentiation and survival of cortical neurons. Therefore, if they are not functional, cell death and neurodegeneration may occur [124].
Also for MPS I, a few studies exist where iPSCs were differentiated into NSCs and from where curious insights were gathered. An interesting study was performed in 2018, by Swaroop et al. [126], where after generating iPSCs and NSCs from all MPS I subtypes, the authors addressed the question of whether those three subtypes could be distinguished from each other, while extensively characterizing each one of them. In the characterization step, they observed a normal iPSCs and NSCs morphology, karyotype, and growth rate in all three. Still, differences among the MPS I subtypes were quite evident, when it came to the disease’s hallmarks. Regarding enzyme activity, for example, all NSCs-MPS I types exhibited a lower rate when compared with controls. However, the levels observed in the Hurler-derived cells were remarkably lower than the others. The same happened when DS and HS accumulation and lysosomal enlargement were evaluated: the values for the Hurler subtype were much higher. Also noteworthy, when the authors compared those cell lines by differential expression (DE), about 3036 genes were found to be significantly changed between patients and controls. Remarkably, however, out of those, 42% were Hurler Syndrome exclusive. Not surprisingly, those genes were involved in GAG homeostasis, dysregulation of the lysosomal pathway and autophagy [108]. Overall, these results strongly supported the idea that one can nicely characterize and distinguish different forms of the same disorder, by evaluating iPSC-derived models, as they recapitulate at the subcellular level the severity seen in patients [126].
Four years later, another interesting study was conducted by Lito S. et al. [127] focusing on the most severe form of the disease alone (Hurler). In that paper, besides reprogramming dermal fibroblasts into iPSCs and generating NSCs, the authors went one step beyond and created an isogenic control from these iPSCs by reestablishing IDUA expression to avoid any type of variability that could emerge from the comparison with iPSC control cell lines derived from other individuals. Then those isogenic cells were also differentiated into NSCs. As a matter of fact, these cells showed a total functional enzyme both in iPSCs phase as well as when differentiated into NSCs. Through comparison with isogenic ones, they could see the most evident hallmark of these disorders: GAG accumulation. Furthermore, at the end of a three weeks-neuronal differentiation protocol (where FGF2 and EGF were removed from the media), they saw a higher migration in vitro of rescued-enzyme NSCs as well as neurite outgrowth when compared to deficient iPSCs-derived NSCs. In turn, proliferation capacity during three weeks of neurodifferentiation, did not change significantly between the two cell conditions. They hypothesize that due to the strong binding properties of CS and HS when accumulation occurs, these storage products bind to molecules responsible for neurite outgrowth and cell migration, preventing their binding with the proper receptors, and accomplishing the right function. Also, these aspects were accompanied by an evaluation of gene expression patterns. Biological processes associated with pathways of TGFβ, focal adhesions, PI3K-AKT signaling, Hippo signaling, RAP1 signaling pathway, extracellular matrix interaction, and calcium signaling were altered with around 173 downregulated and 167 upregulated genes. In general, these migration defects and gene expression changes seen in patients affected by monogenic diseases are associated with a cause-effect relationship, where the genotype presents as a cause and the phenotype as an effect. However, based on these results, the authors purpose that the reverse may also occur, presenting a bidirectional pattern [127].
4.3. iPSCs-derived neuronal cells for drug screening/ therapies evaluation
As we have already referred, MPS iPSC-derived neuronal cells have been generated not only to model MPS and study their pathology. Indeed, one of their crucial goals is to work as a platform to test future therapeutics. Thus, several research and development groups, some of them already mentioned in the previous sections, have been using those cells to test a number of compounds that may allegedly hold promise for the treatment of this LSD class.
Starting, again, with the Sanfilippo syndrome, one of the studies referred before [122], besides intending at the development of neuronal and astrocytic models derived from MPS IIIC iPSCs, also aimed at testing an SRT approach that had already given positive results in MPS IIIC fibroblasts [128]. That was the work of Benetó and co-workers, back in 2015, and the approach they wanted to test consisted on the use of a siRNA against one of the genes responsible for GAGs biosynthesis (the EXTL2 gene) as a genetically triggered SRT. Still, while its application in the generated neuronal and astrocyte cells revealed a great success in the reduction of mRNA levels of this gene (about 75%), when the HS levels were analyzed in neurons, no difference in substrate accumulation could be detected. Curiously, this parameter was not measured in astrocytes, and it is actually a future perspective of this the group to test it as well. A few years ago, this team has also reported an siRNA-driven SRT approach against EXTL3 (another gene involved in GAGs biosynthesis) with positive results in fibroblast disease cells [128], and they proposed to assess its effect in the same neuronal and astrocytic models but, to the best of our knowledge, no follow-up studies have been published so far. Altogether, however, the results they published so far, further highlight the need to develop suitable cell models for drug testing, by clearly demonstrating there may be significant differences between the results obtained in vitro in fibroblasts vs neurons using the exact same therapeutic molecule. In fact, fibroblasts are the classical human cellular model in LSDs, but there are significant metabolic differences between fibroblasts and neural cell types. Furthermore, fibroblasts are dividing cells, while neurons are not. This means that even though fibroblasts accumulate undegraded materials, storage can be underestimated due to dilution by cell division, when compared with that of non-dividing cells [122].
One year later, Huang W. et al. [129] published a comprehensive work, which went all the way from the iPSCs generation and characterization up until the generation of (iPSC-derived) MPS IIIB neuronal cells. While it goes far beyond the scope of this review to go through the extensive characterization analysis and pathophysiological assessments the authors performed on both types of cells, we would like to briefly highlight the therapeutic assessment they made in vitro using these models. Briefly, they examined the effects of three possible therapeutic agents: ERT with recombinant NAGLU (rhNAGLU), δ-tocopherol (DT), and hydroxypropyl-β-cyclodextrin (HPBCD). When rhNAGLU was applied to NSCs, a dose-dependent decrease in enlarged lysosomes was readily observed; the same happened when testing DT and HPBCD in addition to a dose-dependent reduction in the lipidic accumulation. In fact, those two compounds have already positive results in Niemann-Pick disease type C and, more recently, also in other LSDs [130,131,132]. Due to this observation, both compounds were also evaluated in MPS II iPSCs-derived NSCs by Hong et al. [133]. In the case of DT, the results showed a reduction of lipid accumulation after three days, but in a dose-dependent manner; in turn, when evaluating the lysosomal accumulation it was revealed only a 7% reduction. The HPBCD results were not so encouraging, once it had virtually no effect on primary and secondary accumulation. As previously anticipated, however, when NSCs were treated with recombinant enzyme for MPS II (rhIDS), a marked reduction of lipid accumulation was also observed.
Curiously, the effect of rhIDS enzyme was also the target of a study developed in 2018 by Rybová et al. [134]. This study also contemplated reprogramming MPS II PBMCs into iPSCs and their subsequent differentiation into NPCs, neurons, astrocytes, and oligodendrocytes. Having all those cells properly characterized, the authors moved on to evaluate the effect of rhIDS over GAG levels. Remarkably, however, their results showed that despite achieving 10-fold higher enzyme activity levels, the treatment could not reverse the exponential growth of GAGs levels, even though some decrease could be seen [134].
4.4. Genetically corrected MPSs-derived iPSCs
Finally, we will also mention a few studies, which have provided in vitro proof of principle on the potential of ex vivo genetically-corrected iPSCs for therapeutic purposes.
The proof of concept study on the therapeutic use of iPSC for autologous HSCT was published in 2015, by Griffin and co-workers, who attempted ex vivo gene therapy using patient iPSC-derived NSCs to reverse brain pathology in MPS VII [135]. Those authors assessed the engraftment potential of MPS VII NSCs genetically corrected with a transposon vector, by transplanting those cells in a previously reported mouse model for the disease, the so-called NOD/SCID/MPS VII model. Briefly, they injected intraventricularly genetically corrected GFP-labelled NSCs into different neonatal mice populations, either suffering or not from MPS VII. Remarkably, the authors observed similar levels of cell distribution in both pathological and non-pathological contexts, demonstrating that engraftment properties are not influenced by disease. Importantly, transplanted cells survived and remained in the immature stage (Nestin-positive) for over 4 months. However, the proliferation rate reduced dramatically with the total disappearance of proliferation markers after 4 weeks of transplantation. It is worth mentioning that the authors chose to work with neonatal mice for these initial assessments because they provide a more hospitable environment for engraftment relative to the adult brain. Then, to test whether similar results could be obtained in older animals, they injected ex vivo corrected MPS VII iPSC-NSCs in diseased mice adult brains. Again, the immature stage remained with a Nestin-positive pattern. In the adult mice, however, the authors also addressed a number of pathology aspects, in order to address the therapeutic potential of this approach. And, in fact, they did detect GUSB activity but only near to the injection site of the hemisphere receiving corrected cells. Additionally, they also verified a high reduction in neuroinflammation after only 1 month of transplantation in that same region. Basically, they showed that xenotransplantation of ex vivo corrected MPS VII-derived NSCs into a mouse homolog of the human disease, can reverse pathologic lesions surrounding the engrafted cells. But, more relevant than the particular results they saw in this disease and their accurate analysis, is the innovation potential they hold and the new avenues they open, by showing that genetically corrected iPSC-derived NSCs may indeed may have potential to treat MPSs [135].
Then, in 2018 Clarke et al. described a somehow similar approach, attempting to use genetically corrected NSCs derived from iPSCs as a transplantation approach to the treatment of MPS IIIB [136]. Briefly, Naglu–/– mouse embryonic fibroblasts were reprogrammed into iPSCs and later differentiated in NSCs. Those cells were then corrected ex vivo, through lentiviral transduction of the full-length human NAGLU cDNA. This lead to an obvious overexpression of the gene in the corrected NSCs, which resulted in a 4-fold increase in enzyme activity and in a 14-fold higher level of secreted NAGLU when compared with wild-type. Importantly, before they attempted HSCT of those genetically corrected cells, the authors confirmed in vitro whether secreted NAGLU could enter in Naglu-/- cells in an M6P-dependent way, and verified that corrected cells were indeed able to “cross-correct” enzyme-deficient ones. Additionally, they also addressed whether there was a difference in lysosomal enlargement between genetically corrected NSCs and unmodified Naglu-/--derived NSCs. Curiously, they could not see any differences. However, when both cell lines were allowed to differentiate into mature neural cells, the ones derived from genetically corrected NSCs did show a significant decrease. Only then did the authors move to in vivo studies. Basically, they did virtually the same previous teams had done before: ex vivo genetically modified cells were injected into newborn Naglu–/– mice to understand whether they would promote an amelioration of the animals’ phenotype. But there is one remarkable aspect about this study that shoud be highlighted: this team has evaluated two independent protocols: intracerebroventricular (ICV) and intraparenchymal (directly in the striatum), and the pathological aspects they analysed were microglial activation, astrocytosis, and lysosomal dysfunction/storage material. All these aspects were analyzed through immunostaining of CD68, GFAP, and LAMP-1, respectively [136].
Again, we will not review in detail all their observations, but we would like to stress that, from this team’s observations regarding the two administration routes attempted, intraparenchymal was the one shown to have better engraftment. Still, it should be stressed that, at 2 months of age, there was high variability in the pathophysiology results in both ICV and intraparenchymal approaches. Importantly, however, the follow-up results after long-term transplantation of the corrected NSCs into Naglu-/- mice were much more evident. The evaluation of the long-term effect was performed after 9 months of transplantation with the intraparenchymal administration route. In general, NAGLU activity was detected in the majority of engrafted animals. Furthermore, all pathological hallmarks evaluated were more pronounced in non-transplanted Naglu–/– mice. In grafted Naglu–/– mice, however, CD68, and GFAP levels were significantly lower in some regions of the brain. A similar pattern was observed after LAMP-1 staining, meaning that transplanted mice showed a significant decrease in storage material, a reduction in astrocyte activation, and complete prevention of microglial activation within the area of engrafted cells and neighboring regions, with beneficial effects extending partway along the rostrocaudal axis of the brain. Altogether, this study provided evidence that the transplantation of genetically corrected iPSCs-derived NSCs, may indeed represent a potential treatment for MPS IIIB and this is particularly relevant since no approved therapeutic approach exists for this neurological MPS [136,137].
The latest ex vivo gene therapy experience to be performed in MPS models is extremely recent. It was pubished in 2022 [137] and took advantage of results we have just reviewed, for MPS IIIB [136]. In fact, the same team, which originally published the proof of principle on the potential of ex vivo corrected NSCs to positively impact the brain neuropathology in Naglu-/- mice, later extended that study by using a modified Naglu enzyme with the fusion protein IFGII (named NAGLU-IGFII) for the ex vivo correction of the NSCs. This modified/chimeric enzyme, had already been described to allow a greater cellular uptake via IGFII binding sites on the M6PR. Again, the overall process of NSCs generation was performed as well as their lentivirus transduction of the NAGLU-IGFII sequence. Having confirmed that the modified NAGLU-IGFII enzyme could also be secreted and taken up, just like the unmodified enzyme they had previously reported [136], the authors moved on to in vivo studies. Briefly, they engrafted modified cells into the brain of newborn mice and evaluated the long-term therapeutic effect of that approach, 9 months post-transplantation. First, they confirmed the remaining capability of engrafted NSCs to generate different subtypes of CNS-associated cells through positive staining of several markers: NeuN and MAP2 for neurons; GFAP for astrocytes; and O4 for oligodendrocytes. Once more, the success of the engraftment could be better since there was a high variability in the enzyme activity between sections of the brain in different animals. However, the range of enzyme activity was increased by 10%, compared to Naglu–/– mice, which could be promising once it is reported that sometimes only an increase of 1-5% is sufficient for a proper enzyme activity correction. In the case of pathophysiological events, glial activation and storage accumulation, measured, respectively through the staining of CD68/GFAP and LAMP-1, revealed a pattern similar to that of wild type animals. Both effects were more pronounced in closer injection sites [137]. Furthermore, the authors also assessed a parameter, which had not yet been looked at in previous studies: the downregulation of MAP2. MAP2 is now known to have a relevant/significant role in the microtubule stabilization of dendritic processes. Its downregulation is heavily associated with dementia in Alzheimer's disease. Dementia is also a primary symptom in MPS IIIB and, remarkably, when Naglu–/– mice were stained for MAP2, the results have shown that MAP2 was reduced when compared with wild type. 9 months post-transplantation, this downregulation was actually reversed, with treated animals presenting MAP2 levels similar to those observed in Naglu+/– mice. Moreover, the accumulation of aggregates of synaptophysin, which is a known indicator of axonal damage in inflammatory conditions, was higher in Naglu–/– mice than in wild type and engrafted animals [137].
Overall, even though the efficacy of this therapeutic approach must be improved to reach all brain sections and counteract the Sanfilippo-associated neuroimmune response throughout the whole brain, truth is that, once more, this team has gathered evidence on the possibility of ex vivo gene therapy, with remarkable ameliorated MPS IIIB phenotypic aspects. Moreover, this was the first report documenting a significant reduction of the neuronal marker MAP2 and accumulation of synaptophysin-positive aggregates, both well-known to be related with neuropathophysiology [137].
Then again, even MPSs, which already benefit from the existent ERTs, may ultimately benefit from this sort of approaches. Therefore, ex vivo gene therapy experiments have also been performed in MPS I. In fact, in 2019, Miki et al. [138] have generated iPSCs from Idua–/– mouse embryonic fibroblasts. Then, the authors performed the ex vivo correction of those cells by CRISPR/Cas9 technology and verified that the resulting levels of enzyme activity were significantly restored with values comparable to the wild-type iPSCs. While exploratory and not yet attempted in vivo these results further validate the overall potential of iPSCs and iPSC-derived cells for gene therapy in MPSs.
Table 1 summarizes the works performed until the moment with iPSCs technology.
5. An alternative approach to model Mucopolysaccharidoses
Regardless of its ultimate purpose, in general, the rationale followed in all the studies reviewed so far is the same: first, differentiated cells from patients with the target disease are reprogrammed into iPSCs and, then, differentiated again but into disease-relevant cell lines, thus creating a viable cell model for neuronopathic MPS. This technology, as described above, is undoubtedly contributing to increase the knowledge on the pathophysiology of MPSs with neurological involvement and, consequently, with no treatment available. Nevertheless, while iPSC technology proves to be quite valuable and promising, it also involves some disadvantages. Those positive and negative considerations are recapitulated in the Figure 2.
That is why, alternative protocols and additional sources of SC should also be considered, especially those, which are naturally-occurring (Figure 3).
An excellent option would be to take advantage of patients' MSCs, reducing the possibility of errors and avoiding the long, laborious and expensive pluripotency induction phase. In fact, those cells represent a suitable alternative once they can be differentiated into any of the three germ layers: endodermal, mesodermal, and ectodermal, as long as they are cultured in proper media. To be considered a MSC, the cell needs to fulfill a number of criteria (Figure 4).
Bone Marrow Mesenchymal Stem Cells (BMMSCs) are the more often used ones. However, the patient's wellness remains an essential issue, due to invasive procedure [140,142].
An interesting study [143] in 2000 introduced to the world a possible new source of SC: the dental pulp. The dental pulp is an oral non-mineralized tissue with various cell types, localized in the central pulp cavity and mostly comprises soft tissue with nervous/vascular lymphatic elements [144]. Inside it, we may find the so-called Dental Pulp Stem Cells (DPSC). Those cells have an ectodermal origin derived from neural crest cells [145], more specifically from peripheral nerve-associated glia [146].In that original study [143], those recently discovered SC were compared to BMMSCs, and the evidence they gathered showed that those DPSCs exhibit a higher proliferation rate when compared to BMMSCs, while expressing the same pluripotency markers. Thus, this pivotal study became a launching pad for the subsequent exploration of these cells. The impossibility of generating adipocyte cells in the original study was the only lack in classifying DPSCs as MSCs. However, over the following years, more evidence was gathered proving their stem nature. Ultimately, in 2002, the same group that originally assessed their MSCs features, was actually able to promote the adipogenic differentiation of those cells using a more specific induction medium. They also confirmed that human DPSC are capable of self- renewal after an in vivo transplant [147].
After a few years of constant research, a terminology was established that is still used today, which allows us to distinguish between the different SC populations that reside inside the dental tissue (Figure 5). Indeed, depending on the source of the oral cavity from which they are extracted, five different types of stem cells may be distinguished: DPSCs, Stem Cells From Deciduous Teeth (SHEDs) [148], Stem Cells From Apical Papilla (SCAPs) [149], Periodontal Ligament Stem Cells (PDLSCs) [149], and Dental Follicle Stem Cells (DFSCs- precursor cells of PDLSCs) [149,150,151].
Besides the different oral cavity source, we can distinguish those SCs by their proliferation rate and potential to differentiation into the several cells. Regarding the proliferation rate, the Follicle-derived ones seem to have the highest, closely followed by SHEDs, SCAPs, PSLSCs and DPSCs [152,153,154,155,156,157,158,159]. In Table 2 , we review some experiences done so far, to identify the better cell type for each kind of differentiation.
We are now in 2023, and the existence of these different SCs populations has now been known for over 20 years. So, DPSC and SHEDs in particular, have been generated from pulp for some time. Nevertheless, the majority of the studies involving those cells have focused on their differentiation into chondrocytes for dental repair, with the eventual goal of re-growing teeth from multipotent dental mesenchymal stem cells (DMSC) [147,165]. Also addressed by a few teams is the potential they hold for stroke therapy. The first study to investigate DPSC in an animal model of stroke dates back to 2009 and used a mechanical extraction method to obtain cells from human third molars. The cells extracted from those teeth were shown to efficiently express the nuclear receptor related 1 protein, which is essential for the dopaminergic system of the brain, and promote, when transplanted, motor functional recovery [166]. After this pivotal study was performed, a few others followed, always relying on the use of SC from different dental pulp sources, and being tested in vivo in rat models of focal cerebral ischaemia via the transient occlusion of the middle cerebral artery. While it falls completely out of the scope of this review to summarize all those studies, it is worth mentioning that most of them showed really promising results (reviewed in [167]). Curiously, those cells have been shown to enhance poststroke functional recovery through a non-neural replacement mechanism, i.e., via DPSC-dependent paracrine effects ([168]; reviewed in [167]). And that is probably one of the reasons why this sort of cells have been addressed for their therapeutic potential on many other disorders, affecting various different organs such as kidney (acute renal injury [169,170] and nefritis [171]); lungs (acute lung injury [172]); brain (Parkinson’s disease [173,174],Alzheimer’s disease [175], cerebral ischemia [176,177]); spinal cord (spinal cord injury [178,179,180,181]); liver (liver fibrosis [182,183,184]); heart (acute myocardial infarction [185,186]); muscle (muscular dystrophy [187,188,189]); bone (calvarial defect [171,190,191,192], and osteoporosis [193]); skin (wound injury [194,195]); pancreas (diabetes [196,197]) eye (glaucoma [198], cornea trauma [184]) and immune system (rheumatoid arthritis [199], autoimmune encephalomyelitis [200] and systemic lupus erythematosus (reviewed in [201,202]).
And if it is true that, for most of these injuries, the evidence gathered so far comes from in vivo studies alone, when it comes to the use of DMSC in oral diseases, the scenario is significantly different, with two clinical studies on pulp regeneration having been launched within the past several years that have achieved breakthroughs in humans (reviewed in [201]). Overall, the results are so good and the possibilities so vast that soon a commercial interest was found in this type of cells. In fact, due to their easy accessibility and favorable therapeutic applications, cell/tissue banking in the dental field are now a reality in several countries, with some of the most well-known ones being BioEDEN (Austin, Texas), Store-a-Tooth (Lexington, Kentucky), Cell Technology (Japan) or the Tooth Bank (Brownsburg, Indiana) (reviewed in [158,203]). And as exciting as these results and perspectives may sound per se, we believe that the overall potential of these SCs goes far beyond their properties for tissue repair and regeneration. We think, as other authors have also highlighted before, these cells also hold an exceptional potential for neurogenetic disease cell modeling and basic research. In general, DMSC have a neural crest origin, which makes them a useful source of primary cells for modeling virtually any neurological disorders at the molecular level [204]. Given our interest in LSDs, their monogenic nature and the extremely high prevalence of severe neurological phenotypes in this group of disorders, we considered DMSCs as a perfect model to study these disorders.
Interestingly, while their modelling potential has never been addressed for LSDs, as advantageous as it may sound, truth is DPSC are not totally unknown in the field. In fact, back in 2015, Jackson et al. [205] suggested that human MSCs derived from Bone Marrow and Dental Pulp could work as an alternative to the use of Hematopoietic Stem Cells, in standard transplantation approaches for the treatment of MPSs. Similarly to what has been discussed in the last session in which we summarized the studies published so far in MPS using iPSCs, in this particular publication, it was the therapeutic potential of the MSC per se, which was analysed. Actually, none of the MSCs analyzed derived from MPS patients. Instead, all studies were performed in MSCs obtained from healthy donors. This meant that neither the BMMSCs nor the DPSCs they established had any MPS-related enzymatic defect. Instead, all analysed cell lines (MSCs and HSCs) were able to produce the different MPS-associated enzymes in the cell layer and secrete low levels of each and every one of them into the surrounding media, the same being true for the used HSCs. However, MSCs were found to produce significantly higher levels of the majority of MPS enzymes assayed when compared to HSCs, a result that can be considered particularly relevant for therapeutic purposes.
But these authors have done more than just characterizing the normal levels of MPS-related enzymes secreted by different types of wild type SCs, namely BMMSCs, DPSCs and HSCs. They also attempted to overexpress, through lentivirus transduction, four different lysosomal enzymes in those same cell lines, to check whether their secretion levels were somewhat similar. Importantly, the evidence they gathered further supported the idea that MSCs had higher secretion and production levels of MPS enzymes when compared to HSCs. Also noteworthy, the lentivirus transduction was more efficient in MSCs compared with HSCs.
Then, the authors moved on to investigate in vitro the cross correction potential of MPS enzymes secreted from those two different sorts of MSCs in MPS patients’ derived fibroblasts, and after confirming the reduction of GAGs accumulation, they also verified that this cross-correction was reached in an M6P-dependent way.
Finally, they also addressed the differentiation ability of the MSCs tested, verifying that both transduced and non-transduced cells maintained that capacity, with only slight differences in the neurogenic process, which appeared to have a slower differentiation pattern in transduced MSCs. As expected, however, MSCs derived from dental pulp had a premature upregulation on mature neuron markers, when compared with those derived from bone marrow.
Altogether, these results provided the in vitro proof of principle on the therapeutic potential of DPSCs and BMSCs as an isolated therapy or even combined therapy with the standard HSCTs. To the best of our knowledge, no follow-up studies or in vivo assessments have yet been published on this subject, even though its overall results seem extremely promising.
To the best of our knowledge, MPS patient-derived DPSC had never been used for differentiation into specific cell types even though they represent a natural source of SC that may be used to investigate human disease especially for the infantile forms of these disorders. In fact, taking into account that the most severe forms of MPSs are pediatric, there is one particular population of SC in the dental pulp that seems particularly suitable to study them: SHEDs. Among their numerous advantages, which include a high proliferation rate and the greater tendency to generate both skeletal and brain cells, SHEDs collection does not require the active removal of teeth, only their natural fall, and this is certainly an advantage for children who may already be dealing with undue stress and pain.
It is also worth mentioning that, while this review focuses on the insights one can get over the neuropathology of MPS by studying iPSCs, and we have only commented on the neuronal differentiation potential of SC from different sources, such as the DPSC, their differentiation capacity to osteogenic fates is also known and successful protocols are published. This is quite relevant for MPS disease modelling because some of these diseases present with a marked skeletal phenotype, which fails to be corrected by the currently available therapies. Thus, by implementing the method here envisaged, one may also pave the way for additional applications of DPSC. For example, we may easily foresee their differentiation into chondrocytes, one of the major components of cartilage and primary site of accumulation in several LSD.
In general, the higher the number of genotypes we collect the larger the spectrum of future applications our DPSC-derived LSD neuronal cultures may have not only in our lab but also for other researchers in the field. In addition, with the advances of new gene editing technologies, such as CRISPR/Cas, base editing, prime editing and the "older" transcription activator-like effector nucleases (TALEN) and zinc finger nucleases (ZFN), arised the possibility to generate pairs of isogenic lines that facilitate the study of the function of a given gene and the role that different mutations play in the pathophysiological mechanisms of the respective diseases. This approach has been increasingly applied to iPSC lines and could also be very useful in the case of our DPSC-derived cell lines.
Still another naturally-occurring source of SCs are human urine-derived stem cells (USCs), a type of MSCs with proliferation and multi-potent differentiation potential that can be readily obtained from voided urine using an non-invasive protocol and with minimum ethical restriction. These cells express surface markers of MSCs, but not of hematopoietic stem cells, express the stemness-related genes Nanog and Oct3/4 and show telomerase activity, not forming teratomas in vivo after being subcutaneously implanted in nude mice [206,207,208,209]. When cultured in appropriate media, USCs may differentiate into endothelial, osteogenic, chondrogenic, adipogenic, skeletal myogenic, and neurogenic lineages. Interestingly, USCs may be established from individuals of any age, despite Gao et al. have shown that those isolated from children (5 to 14 year-old) have higher proliferation, lower tendency to senescence, and stronger osteogenic capacity than those from middle-aged (30 to 40 years-old) and elder (65 to 75 year-old) individuals [208]. This property allows to significantly expand the cohort of patients accessible to be studied. Overall, USCs are yet another alternative source of SCs that can be used as a valuable in vitro model to study genetic diseases, with potential applications in regenerative medicine, cell therapy, diagnostic testing and drug screening [210].
6. Conclusions
Disease models are essential tools to both identify and study the pathological mechanisms that underlie the development of a disease. They are also a pre-requisite for proper drug development. Indeed, it is essential to have a relevant study model, which reproduces the pathological features of the disease to design and evaluate new therapeutic strategies. And this need goes all the way, from the early in vitro assessments to the investigative in vivo pre-clinical studies.
Over the last decades, amazing advances have been made on the attempt to model the neuropathology of MPSs in vitro, mostly relying on the establishment and subsequent differentiation of disease-specific human iPSCs. And this is certainly true for the larger LSD field, where multiple studies have identified neural progenitor cell migration and differentiation defects, substrate accumulation, axon growth and myelination defects, impaired calcium homeostasis, and altered electrophysiological properties, all using patient-derived iPSCs (reviewed in [211]). So, not even 20 years after iPSCs generation was first described and attempted, their potential to provide mechanistic insights to unravel the pathophysiology associated with neurodevelopment in these rare pathologies is well-established. However, several challenges do remain. That is why we consider it may be useful to contemplate additional sources of patient-derived pluripotent or multipotent cell lines, namely those which are naturally occurring, such as the dental pulp SC derived from human permanent and deciduous teeth. Those cells will also allow for subsequent differentiation into mixed neuronal and glial cultures, which may be analyzed with virtually the exact same methods many authors have been performing to address neuropathology in MPS-derived iPSCs. Finally, regardless of the original source of the SC we are considering, in an era where personalized medicine and mutation-specific therapeutic approaches are gaining momentum, those SC-derived models will also constitute optimal platforms for in vitro drug testing.
Author Contributions
Conceptualization, S.A. and M.F.C; NCBI Pubmed search, S.C., J.I.S., M.F.C. and S.A.; manuscript structure, S.C., M.F.C. and S.A.; original draft preparation, S.C. and M.F.C.; additional writing: J.I.S., M.G., H.D., L.V.M., L.M., M.E. and S.A.; digital figures: H.D. and M.G.; analogue figure: M.F.C; critical review, S.C., J.I.S., M.G., H.D., L.V.M., L.M., M.E., M.F.C. and S.A.; editing, S.C., J.I.S. and M.F.C.; funding acquisition, S.A. and M.F.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work is partially supported by the Portuguese Society for Metabolic Disorders (SPDM - Bolsa SPDM de apoio à investigação Dr. Aguinaldo Cabral 2018; 2019DGH1629/ SPDM2018I&D), Sanfilippo Children's Foundation (2019DGH1656/ SCF2019I&D) and FCT (EXPL/BTM-SAL/0659/2021).
Acknowledgments
UIDB/00211/2020 - Centro de Estudos de Ciência Animal/ Center for the Study of Animal Science. LA/P/0059/2020 - Laboratório Associado para Ciência Animal e Veterinária/Associate Laboratory for Animal and Veterinary Sciences.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Scerra, G.; De Pasquale, V.; Scarcella, M.; Caporaso, M.G.; Pavone, L.M.; D’Agostino, M. Lysosomal Positioning Diseases: Beyond Substrate Storage. Open Biol. 2022, 12. [Google Scholar] [CrossRef]
- Leal, A.F.; Benincore-Flórez, E.; Rintz, E.; Herreño-Pachón, A.M.; Celik, B.; Ago, Y.; Alméciga-Díaz, C.J.; Tomatsu, S. Mucopolysaccharidoses: Cellular Consequences of Glycosaminoglycans Accumulation and Potential Targets. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Sahin, O.; Thompson, H.P.; Goodman, G.W.; Li, J.; Urayama, A. Mucopolysaccharidoses and the Blood–Brain Barrier. Fluids Barriers CNS 2022, 19. [Google Scholar] [CrossRef]
- Schröder, B.A.; Wrocklage, C.; Hasilik, A.; Saftig, P. The Proteome of Lysosomes. Proteomics 2010, 10, 4053–4076. [Google Scholar] [CrossRef]
- Manuscript, A. Lübke et Al., 2008 Lysosome.Pdf. 2010, 1793, 625–635.
- Platt, F.M. Emptying the Stores: Lysosomal Diseases and Therapeutic Strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef]
- La Cognata, V.; Guarnaccia, M.; Polizzi, A.; Ruggieri, M.; Cavallaro, S. Highlights on Genomics Applications for Lysosomal Storage Diseases. Cells 2020, 9, 1–15. [Google Scholar] [CrossRef]
- Parenti, G.; Medina, D.L.; Ballabio, A. The Rapidly Evolving View of Lysosomal Storage Diseases. EMBO Mol. Med. 2021, 13, 1–21. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal Storage Diseases: From Pathophysiology to Therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Platt, F.M.; d’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal Storage Diseases. Nat. Rev. Dis. Prim. 2018, 4. [Google Scholar] [CrossRef]
- Rajkumar, V.; Dumpa, V. Lysosomal Storage Disease. Available online: https://www.ncbi.nlm.nih.gov/books/NBK563270/ (accessed on 29 November 2021).
- Boustany, R.M.N. Lysosomal Storage Diseases - The Horizon Expands. Nat. Rev. Neurol. 2013, 9, 583–598. [Google Scholar] [CrossRef]
- Kleppin, S. Enzyme Replacement Therapy for Lysosomal Storage Diseases. J. Infus. Nurs. 2020, 43, 243–245. [Google Scholar] [CrossRef]
- Suzuki, Y. Chaperone Therapy for Molecular Pathology in Lysosomal Diseases. Brain Dev. 2021, 43, 45–54. [Google Scholar] [CrossRef]
- Desnick, R.J.; Schuchman, E.H. Enzyme Replacement Therapy for Lysosomal Diseases: Lessons from 20 Years of Experience and Remaining Challenges; 2012; Vol. 13. [Google Scholar]
- Beutler, E. Enzyme Replacement in Gaucher Disease. PLoS Med. 2004, 1, 118–121. [Google Scholar] [CrossRef]
- Li, X.; Ren, X.; Zhang, Y.; Ding, L.; Huo, M.; Li, Q. Fabry Disease: Mechanism and Therapeutics Strategies. Front. Pharmacol. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Pastores, G.M.; Hughes, D.A. Lysosomal Acid Lipase Deficiency: Therapeutic Options. Drug Des. Devel. Ther. 2020, 14, 591–601. [Google Scholar] [CrossRef]
- Lewis, G.; Morrill, A.M.; Conway-Allen, S.L.; Kim, B. Review of Cerliponase Alfa: Recombinant Human Enzyme Replacement Therapy for Late-Infantile Neuronal Ceroid Lipofuscinosis Type 2. J. Child Neurol. 2020, 35, 348–353. [Google Scholar] [CrossRef]
- Diaz, G.A.; Jones, S.A.; Scarpa, M.; Mengel, K.E.; Giugliani, R.; Guffon, N.; Batsu, I.; Fraser, P.A.; Li, J.; Zhang, Q.; et al. One-Year Results of a Clinical Trial of Olipudase Alfa Enzyme Replacement Therapy in Pediatric Patients with Acid Sphingomyelinase Deficiency. Genet. Med. 2022, 24, 2209. [Google Scholar] [CrossRef]
- Ceccarini, M.R.; Codini, M.; Conte, C.; Patria, F.; Cataldi, S.; Bertelli, M.; Albi, E.; Beccari, T. Alpha-Mannosidosis: Therapeutic Strategies. Int. J. Mol. Sci. 2018, 19, 1–11. [Google Scholar] [CrossRef]
- Monica, P.-P.; David R, B.; Paul, H. Current and New Therapies for Mucopolysaccharidoses. Pediatr. Neonatol. 2022, 1–8. [Google Scholar]
- Kido, J.; Sugawara, K.; Nakamura, K.; Tomanin, R.; Azzo, A.D. Gene Therapy for Lysosomal Storage Diseases : Current Clinical Trial Prospects. 2023, 1–16.
- Li, M. Enzyme Replacement Therapy: A Review and Its Role in Treating Lysosomal Storage Diseases. Pediatr. Ann. 2018, 47, e191–e197. [Google Scholar] [CrossRef]
- Barriga, F.; Ramírez, P.; Wietstruck, A.; Rojas, N. Hematopoietic Stem Cell Transplantation: Clinical Use and Perspectives. Biol. Res. 2012, 45, 307–316. [Google Scholar] [CrossRef]
- Biffi, A. Hematopoietic Stem Cell Gene Therapy for Storage Disease: Current and New Indications. Mol. Ther. 2017, 25, 1155–1162. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science (80-. ). 2013, 341. [Google Scholar] [CrossRef]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral Haemopoietic Stem-Cell Gene Therapy in Early-Onset Metachromatic Leukodystrophy: An Ad-Hoc Analysis of a Non-Randomised, Open-Label, Phase 1/2 Trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy. Science (80-. ). 2009, 326, 818–823. [Google Scholar] [CrossRef]
- Nagree, M.S.; Felizardo, T.C.; Faber, M.L.; Rybova, J.; Rupar, C.A.; Foley, S.R.; Fuller, M.; Fowler, D.H.; Medin, J.A. Autologous, Lentivirus-modified, T-rapa Cell “Micropharmacies” for Lysosomal Storage Disorders. EMBO Mol. Med. 2022, 14, 1–13. [Google Scholar] [CrossRef]
- Saccardi, R.; Gualandi, F. Hematopoietic Stem Cell Transplantation Procedures. Autoimmunity 2008, 41, 570–576. [Google Scholar] [CrossRef]
- Boelens, J.J.; Prasad, V.K.; Tolar, J.; Wynn, R.F.; Peters, C. Current International Perspectives on Hematopoietic Stem Cell Transplantation for Inherited Metabolic Disorders. Pediatr. Clin. North Am. 2010, 57, 123–145. [Google Scholar] [CrossRef]
- Malatack, J.J.; Consolini, D.M.; Bayever, E. The Status of Hematopoietic Stem Cell Transplantation in Lysosomal Storage Disease. Pediatr. Neurol. 2003, 29, 391–403. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Ulrich Hartl, F. Molecular Chaperone Functions in Protein Folding and Proteostasis; 2013; Vol. 82. [Google Scholar]
- Fan, J.Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated Transport and Maturation of Lysosomal α-Galactosidase A in Fabry Lymphoblasts by an Enzyme Inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef]
- Hughes, D.A.; Nicholls, K.; Shankar, S.P.; Sunder-Plassmann, G.; Koeller, D.; Nedd, K.; Vockley, G.; Hamazaki, T.; Lachmann, R.; Ohashi, T.; et al. Oral Pharmacological Chaperone Migalastat Compared with Enzyme Replacement Therapy in Fabry Disease: 18-Month Results from the Randomised Phase III ATTRACT Study. J. Med. Genet. 2017, 54, 288–296. [Google Scholar] [CrossRef]
- Clemente, F.; Martínez-Bailén, M.; Matassini, C.; Morrone, A.; Falliano, S.; Caciotti, A.; Paoli, P.; Goti, A.; Cardona, F. Synthesis of a New β-Galactosidase Inhibitor Displaying Pharmacological Chaperone Properties for GM1 Gangliosidosis. Molecules 2022, 27, 1–21. [Google Scholar] [CrossRef]
- Stütz, A.E.; Thonhofer, M.; Weber, P.; Wolfsgruber, A.; Wrodnigg, T.M. Pharmacological Chaperones for β-Galactosidase Related to GM1-Gangliosidosis and Morquio B: Recent Advances. Chem. Rec. 2021, 21, 2980–2989. [Google Scholar] [CrossRef]
- Matsuhisa, K.; Imaizumi, K. Loss of Function of Mutant Ids Due to Endoplasmic Reticulum-Associated Degradation: New Therapeutic Opportunities for Mucopolysaccharidosis Type Ii. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Pan, X.; Taherzadeh, M.; Bose, P.; Heon-Roberts, R.; Nguyen, A.L.A.; Xu, T.; Pará, C.; Yamanaka, Y.; Priestman, D.A.; Platt, F.M.; et al. Glucosamine Amends CNS Pathology in Mucopolysaccharidosis IIIC Mouse Expressing Misfolded HGSNAT. J. Exp. Med. 2022, 219. [Google Scholar] [CrossRef]
- González-Cuesta, M.; Herrera-González, I.; García-Moreno, M.I.; Ashmus, R.A.; Vocadlo, D.J.; García Fernández, J.M.; Nanba, E.; Higaki, K.; Ortiz Mellet, C. Sp2-Iminosugars Targeting Human Lysosomal β-Hexosaminidase as Pharmacological Chaperone Candidates for Late-Onset Tay-Sachs Disease. J. Enzyme Inhib. Med. Chem. 2022, 37, 1364–1374. [Google Scholar] [CrossRef]
- De Ponti, G.; Donsante, S.; Frigeni, M.; Pievani, A.; Corsi, A.; Bernardo, M.E.; Riminucci, M.; Serafini, M. MPSI Manifestations and Treatment Outcome: Skeletal Focus. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef]
- Lorne A Clarke, M. Mucopolysaccharidosis Type I. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1162/.
- Lin, H.Y.; Lin, S.P.; Chuang, C.K.; Niu, D.M.; Chen, M.R.; Tsai, F.J.; Chao, M.C.; Chiu, P.C.; Lin, S.J.; Tsai, L.P.; et al. Incidence of the Mucopolysaccharidoses in Taiwan, 1984-2004. Am. J. Med. Genet. Part A 2009, 149, 960–964. [Google Scholar] [CrossRef]
- Moammar, H.; Cheriyan, G.; Mathew, R.; Al-Sannaa, N. Incidence and Patterns of Inborn Errors of Metabolism in the Eastern Province of Saudi Arabia, 1983-2008. Ann. Saudi Med. 2010, 30, 271–277. [Google Scholar] [CrossRef]
- Celik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.S.; Eisengart, J.B.; Lund, T.C.; Orchard, P.J.; Swietlicka, M.; Wesley, J.; McIvor, R.S. Mucopolysaccharidosis Type I: A Review of the Natural History and Molecular Pathology. Cells 2020, 9, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Medline Plus Mucopolysacchaidosis Type I. Available online: https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-i/.
- HGMD. Available online: https://www.hgmd.cf.ac.uk/ac/index.php.
- Carbajal-Rodríguez, L.M.; Pérez-García, M.; Rodríguez-Herrera, R.; Rosales, H.S.; Olaya-Vargas, A. Long-Term Evolution of Mucopolysaccharidosis Type I in Twins Treated with Enzyme Replacement Therapy plus Hematopoietic Stem Cells Transplantation. Heliyon 2021, 7, e07740. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Muschol, N.; Keenan, H.A.; Dant, M.; Muenzer, J. Improvement in Time to Treatment, but Not Time to Diagnosis, in Patients with Mucopolysaccharidosis Type I. Arch. Dis. Child. 2021, 106, 674–679. [Google Scholar] [CrossRef] [PubMed]
- NIH; GARD Mucopolysaccharidosis Type I. Available online: https://rarediseases.info.nih.gov/diseases/10335/mucopolysaccharidosis-type-i.
- Williams, I.M.; Pineda, R.; Neerukonda, V.K.; Stagner, A.M. Mucopolysaccharidosis Type I–Associated Corneal Disease: A Clinicopathologic Study. Am. J. Ophthalmol. 2021, 231, 39–47. [Google Scholar] [CrossRef]
- Applegarth, D.A.; Toone, J.R.; Brian Lowry, R. Incidence of Inborn Errors of Metabolism in British Columbia, 1969-1996. Pediatrics 2000, 105, 109. [Google Scholar] [CrossRef]
- Krabbi, K.; Joost, K.; Zordania, R.; Talvik, I.; Rein, R.; Huijmans, J.G.M.; Verheijen, F. V.; Õunap, K. The Live-Birth Prevalence of Mucopolysaccharidoses in Estonia. Genet. Test. Mol. Biomarkers 2012, 16, 846–849. [Google Scholar] [CrossRef]
- NORD Mucopolysaccharidosis Type II. Available online: https://rarediseases.org/rare-diseases/mucopolysaccharidosis-type-ii-2/.
- Mohamed, S.; He, Q.Q.; Singh, A.A.; Ferro, V. Mucopolysaccharidosis Type II (Hunter Syndrome): Clinical and Biochemical Aspects of the Disease and Approaches to Its Diagnosis and Treatment. Adv. Carbohydr. Chem. Biochem. 2020, 77, 71–117. [Google Scholar]
- Wilson, P.J.; Meaney, C.A.; Hopwood, J.J.; Morris, C.P. Sequence of the Human Iduronate 2-Sulfatase (IDS) Gene. Genomics 1993, 17, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Pantoom, S.; Petters, J.; Pandey, A.K.; Hermann, A.; Lukas, J. A Molecular Genetics View on Mucopolysaccharidosis Type II. Mutat. Res. - Rev. Mutat. Res. 2021, 788, 108392. [Google Scholar] [CrossRef]
- Kong, W.; Yao, Y.; Zhang, J.; Lu, C.; Ding, Y.; Meng, Y. Update of Treatment for Mucopolysaccharidosis Type III (Sanfilippo Syndrome). Eur. J. Pharmacol. 2020, 888. [Google Scholar] [CrossRef]
- Valstar, M.J.; Ruijter, G.J.G.; van Diggelen, O.P.; Poorthuis, B.J.; Wijburg, F.A. Sanfilippo Syndrome: A Mini-Review. J. Inherit. Metab. Dis. 2008, 31, 240–252. [Google Scholar] [CrossRef]
- Bodamer, O.A.; Giugliani, R.; Wood, T. The Laboratory Diagnosis of Mucopolysaccharidosis III (Sanfilippo Syndrome): A Changing Landscape. Mol. Genet. Metab. 2014, 113, 34–41. [Google Scholar] [CrossRef]
- Josahkian, J.A.; Trapp, F.B.; Burin, M.G.; Michelin-Tirelli, K.; de Magalhães, A.P.P.S.; Sebastião, F.M.; Bender, F.; De Mari, J.F.; Brusius-Facchin, A.C.; Leistner-Segal, S.; et al. Updated Birth Prevalence and Relative Frequency of Mucopolysaccharidoses across Brazilian Regions. Genet. Mol. Biol. 2021, 44, 1–6. [Google Scholar] [CrossRef]
- Poorthuis, B.J.H.M.; Wevers, R.A.; Kleijer, W.J.; Groener, J.E.M.; de Jong, J.G.N.; van Weely, S.; Niezen-Koning, K.E.; van Diggelen, O.P. The Frequency of Lysosomal Storage Diseases in The Netherlands. Hum. Genet. 1999, 105, 151. [Google Scholar] [CrossRef]
- Medline Plus Mucopolysaccharidosis Type III. Available online: https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-iii/.
- Benetó, N.; Vilageliu, L.; Grinberg, D.; Canals, I. Sanfilippo Syndrome: Molecular Basis, Disease Models and Therapeutic Approaches. Int. J. Mol. Sci. 2020, 21, 1–20. [Google Scholar] [CrossRef] [PubMed]
- NORD Mucopolysaccharidosis Type III. Available online: https://rarediseases.org/rare-diseases/mucopolysaccharidosis-type-iii/.
- Köhn, A.F.; Grigull, L.; du Moulin, M.; Kabisch, S.; Ammer, L.; Rudolph, C.; Muschol, N.M. Hematopoietic Stem Cell Transplantation in Mucopolysaccharidosis Type IIIA: A Case Description and Comparison with a Genotype-Matched Control Group. Mol. Genet. Metab. Reports 2020, 23, 100578. [Google Scholar] [CrossRef] [PubMed]
- Hoogerbrugge, P.M.; Brouwer, O.F.; Bordigoni, P.; Cornu, G.; Kapaun, P.; Ortega, J.J.; O’Meara, A.; Souillet, G.; Frappaz, D.; Blanche, S.; et al. Allogeneic Bone Marrow Transplantation for Lysosomal Storage Diseases. Lancet 1995, 345, 1398–1402. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, E.G.; Lockman, L.A.; Balthazor, M.; Krivit, W. Neuropsychological Outcomes of Several Storage Diseases with and without Bone Marrow Transplantation. J. Inherit. Metab. Dis. 1995, 18, 413–429. [Google Scholar] [CrossRef] [PubMed]
- Sivakumur, P.; Wraith, J.E. Bone Marrow Transplantation in Mucopolysaccharidosis Type IIIA: A Comparison of an Early Treated Patient with His Untreated Sibling. J. Inherit. Metab. Dis. 1999, 22, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Okuyama, T. Novel Enzyme Replacement Therapies for Neuropathic Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Beard, H.; Chidlow, G.; Neumann, D.; Nazri, N.; Douglass, M.; Trim, P.J.; Snel, M.F.; Casson, R.J.; Hemsley, K.M. Is the Eye a Window to the Brain in Sanfilippo Syndrome? Acta Neuropathol. Commun. 2020, 8, 1–16. [Google Scholar] [CrossRef]
- Belur, L.R.; Romero, M.; Lee, J.; Podetz-Pedersen, K.M.; Nan, Z.; Riedl, M.S.; Vulchanova, L.; Kitto, K.F.; Fairbanks, C.A.; Kozarsky, K.F.; et al. Comparative Effectiveness of Intracerebroventricular, Intrathecal, and Intranasal Routes of AAV9 Vector Administration for Genetic Therapy of Neurologic Disease in Murine Mucopolysaccharidosis Type I. Front. Mol. Neurosci. 2021, 14, 1–12. [Google Scholar] [CrossRef]
- Grover, A.; Crippen-Harmon, D.; Nave, L.; Vincelette, J.; Wait, J.C.M.; Melton, A.C.; Lawrence, R.; Brown, J.R.; Webster, K.A.; Yip, B.K.; et al. Translational Studies of Intravenous and Intracerebroventricular Routes of Administration for CNS Cellular Biodistribution for BMN 250, an Enzyme Replacement Therapy for the Treatment of Sanfilippo Type B. Drug Deliv. Transl. Res. 2020, 10, 425–439. [Google Scholar] [CrossRef]
- Christensen, C.L.; Ashmead, R.E.; Choy, F.Y.M. Cell and Gene Therapies for Mucopolysaccharidoses: Base Editing and Therapeutic Delivery to the CNS. Diseases 2019, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Safary, A.; Akbarzadeh Khiavi, M.; Omidi, Y.; Rafi, M.A. Targeted Enzyme Delivery Systems in Lysosomal Disorders: An Innovative Form of Therapy for Mucopolysaccharidosis. Cell. Mol. Life Sci. 2019, 76, 3363–3381. [Google Scholar] [CrossRef] [PubMed]
- Sahin, O.; Thompson, H.P.; Goodman, G.W.; Li, J.; Urayama, A. Mucopolysaccharidoses and the Blood-Brain Barrier. Fluids Barriers CNS 2022, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Tordo, J.; O’Leary, C.; Antunes, A.S.L.M.; Palomar, N.; Aldrin-Kirk, P.; Basche, M.; Bennett, A.; D’Souza, Z.; Gleitz, H.; Godwin, A.; et al. A Novel Adeno-Associated Virus Capsid with Enhanced Neurotropism Corrects a Lysosomal Transmembrane Enzyme Deficiency. Brain 2018, 141, 2014–2031. [Google Scholar] [CrossRef] [PubMed]
- Marcó, S.; Haurigot, V.; Bosch, F. In Vivo Gene Therapy for Mucopolysaccharidosis Type III (Sanfilippo Syndrome): A New Treatment Horizon. Hum. Gene Ther. 2019, 30, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Gray, A.L.; O’Leary, C.; Liao, A.; Agúndez, L.; Youshani, A.S.; Gleitz, H.F.; Parker, H.; Taylor, J.T.; Danos, O.; Hocquemiller, M.; et al. An Improved Adeno-Associated Virus Vector for Neurological Correction of the Mouse Model of Mucopolysaccharidosis IIIA. Hum. Gene Ther. 2019, 30, 1052–1066. [Google Scholar] [CrossRef]
- De Pasquale, V.; Sarogni, P.; Pistorio, V.; Cerulo, G.; Paladino, S.; Pavone, L.M. Targeting Heparan Sulfate Proteoglycans as a Novel Therapeutic Strategy for Mucopolysaccharidoses. Mol. Ther. - Methods Clin. Dev. 2018, 10, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Park, C.; Maeng, S. Mucopolysaccharidoses I and II: Brief Review of Therapeutic Options and Supportive/Palliative Therapies. Biomed Res. Int. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.; Santos, J.; Matos, L.; Alves, S. Genetic Substrate Reduction Therapy: A Promising Approach for Lysosomal Storage Disorders. Diseases 2016, 4, 33. [Google Scholar] [CrossRef]
- Andrade, F.; Aldámiz-Echevarría, L.; Llarena, M.; Couce, M.L. Sanfilippo Syndrome: Overall Review. Pediatr. Int. 2015, 57, 331–338. [Google Scholar] [CrossRef]
- Jakobkiewicz-banecka, J.; Gabig-ciminska, M.; Kloska, A.; Malinowska, M.; Piotrowska, E.; Banecka-majkutewicz, Z.; Banecki, B.; Wegrzyn, A. Glycosaminoglycans and Mucopolysaccharidosis Type III Table 1. Birth Prevalence of MPS III. 2016, 1393–1409. [Google Scholar]
- Malm, G.; Lund, A.M.; Månsson, J.E.; Heiberg, A. Mucopolysaccharidoses in the Scandinavian Countries: Incidence and Prevalence. Acta Paediatr. Int. J. Paediatr. 2008, 97, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Politei, J.; Schenone, A.B.; Guelbert, N.; Fainboim, A.; Szlago, M. Morquio Disease (Mucopolysaccharidosis Type IV-A): Clinical Aspects, Diagnosis and New Treatment with Enzyme Replacement Therapy. Arch. Argent. Pediatr. 2015, 113, 359–364. [Google Scholar]
- Hendriksz, C.J.; Berger, K.I.; Giugliani, R.; Harmatz, P.; Kampmann, C.; Mackenzie, W.G.; Raiman, J.; Villarreal, M.S.; Savarirayan, R. International Guidelines for the Management and Treatment of Morquio a Syndrome. Am. J. Med. Genet. Part A 2015, 167, 11–25. [Google Scholar] [CrossRef]
- Khan, S.; Alméciga-Díaz, C.J.; Sawamoto, K.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Mason, R.W.; Orii, T.; Tomatsu, S. Mucopolysaccharidosis IVA and Glycosaminoglycans. Mol. Genet. Metab. 2017, 120, 78–95. [Google Scholar] [CrossRef]
- NORD Mucopolysaccharidosis Type IV. Available online: https://rarediseases.org/rare-diseases/morquio-syndrome/.
- Jurecka, A.; Ługowska, A.; Golda, A.; Czartoryska, B.; Tylki-Szymańska, A. Prevalence Rates of Mucopolysaccharidoses in Poland. J. Appl. Genet. 2015, 56, 205–210. [Google Scholar] [CrossRef]
- NORD Maroteaux Lamy Syndrome. Available online: https://rarediseases.org/rare-diseases/maroteaux-lamy-syndrome/.
- Vi, M. Disease Name with Synonyms. Orphanet J. Rare Dis. 2010. [Google Scholar]
- Tomanin, R.; Karageorgos, L.; Zanetti, A.; Al-Sayed, M.; Bailey, M.; Miller, N.; Sakuraba, H.; Hopwood, J.J. Mucopolysaccharidosis Type VI (MPS VI) and Molecular Analysis: Review and Classification of Published Variants in the ARSB Gene. Hum. Mutat. 2018, 39, 1788–1802. [Google Scholar] [CrossRef] [PubMed]
- Harmatz, P.R.; Shediac, R. Mucopolysaccharidosis VI: Pathophysiology, Diagnosis and Treatment. Front. Biosci. - Landmark 2017, 22, 385–406. [Google Scholar] [CrossRef] [PubMed]
- Poup, H.; Ledvinová, J.; Berná, L.; Dvo, L.; Ko, V.; Elleder, M. The Birth Prevalence of Lysosomal Storage Disorders in the Czech Republic : Comparison with Data in Different Populations. 2010, 387–396. [Google Scholar]
- Federhen, A.; Pasqualim, G.; de Freitas, T.F.; Gonzalez, E.A.; Trapp, F.; Matte, U.; Giugliani, R. Estimated Birth Prevalence of Mucopolysaccharidoses in Brazil. Am. J. Med. Genet. Part A 2020, 182, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Rohrbach, M.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; et al. Epidemiology of Mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Montaño, A.M.; Lock-Hock, N.; Steiner, R.D.; Graham, B.H.; Szlago, M.; Greenstein, R.; Pineda, M.; Gonzalez-Meneses, A.; çoker, M.; Bartholomew, D.; et al. Clinical Course of Sly Syndrome (Mucopolysaccharidosis Type VII). J. Med. Genet. 2016, 53, 403–418. [Google Scholar] [CrossRef]
- Medline Plus Mucopolysaccharidosis Type VII. Available online: https://medlineplus.gov/genetics/condition/mucopolysaccharidosis-type-vii/.
- Tomatsu, S.; Montano, A.M.; Dung, V.C.; Grubb, J.H.; Sly, W.S. Mutations and Polymorphisms in GUSB Gene in Mucopolysaccharidosis VII (Sly Syndrome). Hum. Mutat. 2009, 30, 511–519. [Google Scholar] [CrossRef] [PubMed]
- NORD Mucopolysaccharidosis Type VII. Available online: https://rarediseases.org/rare-diseases/sly-syndrome/.
- Of, A.; Report, C. Clinical and Biochemical Manifestations of Hyaluronidase Deficiency. New Engl. J. Med. Fig. 1996, 335, 1029–1033. [Google Scholar]
- Society, M. MPS IX. Available online: https://www.mpssociety.org.uk/mps-ix.
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Malik, N.; Rao, M.S. A Review of the Methods for Human IPSC Derivation. Methods Mol. Biol. 2013, 997, 23–33. [Google Scholar]
- Lemonnier, T.; Blanchard, S.; Toli, D.; Roy, E.; Bigou, S.; Froissart, R.; Rouvet, I.; Vitry, S.; Heard, J.M.; Bohl, D. Modeling Neuronal Defects Associated with a Lysosomal Disorder Using Patient-Derived Induced Pluripotent Stem Cells. Hum. Mol. Genet. 2011, 20, 3653–3666. [Google Scholar] [CrossRef]
- Vallejo-Diez, S.; Fleischer, A.; Martín-Fernández, J.M.; Sánchez-Gilabert, A.; Bachiller, D. Generation of Two Induced Pluripotent Stem Cells Lines from a Mucopolysaccharydosis IIIB (MPSIIIB) Patient. Stem Cell Res. 2018, 33, 180–184. [Google Scholar] [CrossRef]
- Vallejo, S.; Fleischer, A.; Martín, J.M.; Sánchez, A.; Palomino, E.; Bachiller, D. Generation of Two Induced Pluripotent Stem Cells Lines from Mucopolysaccharydosis IIIA Patient: IMEDEAi004-A and IMEDEAi004-B. Stem Cell Res. 2018, 32, 110–114. [Google Scholar] [CrossRef]
- Ben Jehuda, R.; Shemer, Y.; Binah, O. Genome Editing in Induced Pluripotent Stem Cells Using CRISPR/Cas9. Stem Cell Rev. Reports 2018, 14, 323–336. [Google Scholar] [CrossRef]
- Benetó, N.; Cozar, M.; García-Morant, M.; Creus-Bachiller, E.; Vilageliu, L.; Grinberg, D.; Canals, I. Generation of Two Compound Heterozygous HGSNAT-Mutated Lines from Healthy Induced Pluripotent Stem Cells Using CRISPR/Cas9 to Model Sanfilippo C Syndrome. Stem Cell Res. 2019, 41, 101616. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Nemes, C.; Bock, I.; Varga, N.; Fehér, A.; Dinnyés, A.; Kobolák, J. Generation of Mucopolysaccharidosis Type II (MPS II) Human Induced Pluripotent Stem Cell (IPSC) Line from a 1-Year-Old Male with Pathogenic IDS Mutation. Stem Cell Res. 2016, 17, 482–484. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Nemes, C.; Bock, I.; Varga, N.; Fehér, A.; Kobolák, J.; Dinnyés, A. Generation of Mucopolysaccharidosis Type II (MPS II) Human Induced Pluripotent Stem Cell (IPSC) Line from a 3-Year-Old Male with Pathogenic IDS Mutation. Stem Cell Res. 2016, 17, 479–481. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Nemes, C.; Bock, I.; Varga, N.; Fehér, A.; Kobolák, J.; Dinnyés, A. Generation of Mucopolysaccharidosis Type II (MPS II) Human Induced Pluripotent Stem Cell (IPSC) Line from a 7-Year-Old Male with Pathogenic IDS Mutation. Stem Cell Res. 2016, 17, 463–465. [Google Scholar] [CrossRef]
- Varga, E.; Nemes, C.; Kovács, E.; Bock, I.; Varga, N.; Fehér, A.; Dinnyés, A.; Kobolák, J. Generation of Human Induced Pluripotent Stem Cell (IPSC) Line from an Unaffected Female Carrier of Mucopolysaccharidosis Type II (MPS II) Disorder. Stem Cell Res. 2016, 17, 514–516. [Google Scholar] [CrossRef]
- Lito, S.; Burda, P.; Baumgartner, M.; Sloan-Béna, F.; Táncos, Z.; Kobolák, J.; Dinnyés, A.; Krause, K.H.; Marteyn, A. Generation of Human Induced Pluripotent Stem Cell Line UNIGEi001-A from a 2-Years Old Patient with Mucopolysaccharidosis Type IH Disease. Stem Cell Res. 2019, 41, 101604. [Google Scholar] [CrossRef]
- Suga, M.; Kondo, T.; Imamura, K.; Shibukawa, R.; Okanishi, Y.; Sagara, Y.; Tsukita, K.; Enami, T.; Furujo, M.; Saijo, K.; et al. Generation of a Human Induced Pluripotent Stem Cell Line, BRCi001-A, Derived from a Patient with Mucopolysaccharidosis Type I. Stem Cell Res. 2019, 36, 101406. [Google Scholar] [CrossRef]
- Bruyère, J.; Roy, E.; Ausseil, J.; Lemonnier, T.; Teyre, G.; Bohl, D.; Etienne-Manneville, S.; Lortat-Jacob, H.; Heard, J.M.; Vitry, S. Heparan Sulfate Saccharides Modify Focal Adhesions: Implication in Mucopolysaccharidosis Neuropathophysiology. J. Mol. Biol. 2015, 427, 775–791. [Google Scholar] [CrossRef]
- Canals, I.; Soriano, J.; Orlandi, J.G.; Torrent, R.; Richaud-Patin, Y.; Jiménez-Delgado, S.; Merlin, S.; Follenzi, A.; Consiglio, A.; Vilageliu, L.; et al. Activity and High-Order Effective Connectivity Alterations in Sanfilippo C Patient-Specific Neuronal Networks. Stem Cell Reports 2015, 5, 546–557. [Google Scholar] [CrossRef]
- Benetó, N.; Cozar, M.; Castilla-Vallmanya, L.; Zetterdahl, O.G.; Sacultanu, M.; Segur-Bailach, E.; García-Morant, M.; Ribes, A.; Ahlenius, H.; Grinberg, D.; et al. Neuronal and Astrocytic Differentiation from Sanfilippo C Syndrome IPSCs for Disease Modeling and Drug Development. J. Clin. Med. 2020, 9. [Google Scholar] [CrossRef]
- Lehmann, R.J.; Jolly, L.A.; Johnson, B. V.; Lord, M.S.; Kim, H.N.; Saville, J.T.; Fuller, M.; Byers, S.; Derrick-Roberts, A.L.K. Impaired Neural Differentiation of MPS IIIA Patient Induced Pluripotent Stem Cell-Derived Neural Progenitor Cells. Mol. Genet. Metab. Reports 2021, 29. [Google Scholar] [CrossRef]
- Kobolák, J.; Molnár, K.; Varga, E.; Bock, I.; Jezsó, B.; Téglási, A.; Zhou, S.; Lo Giudice, M.; Hoogeveen-Westerveld, M.; Pijnappel, W.P.; et al. Modelling the Neuropathology of Lysosomal Storage Disorders through Disease-Specific Human Induced Pluripotent Stem Cells. Exp. Cell Res. 2019, 380, 216–233. [Google Scholar] [CrossRef]
- Yick, L.W.; Ma, O.K.F.; Ng, R.C.L.; Kwan, J.S.C.; Chan, K.H. Aquaporin-4 Autoantibodies from Neuromyelitis Optica Spectrum Disorder Patients Induce Complement-Independent Immunopathologies in Mice. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, M.; Brooks, M.J.; Gieser, L.; Swaroop, A.; Zheng, W. Patient IPSC-Derived Neural Stem Cells Exhibit Phenotypes in Concordance with the Clinical Severity of Mucopolysaccharidosis I. Hum. Mol. Genet. 2018, 27, 3612–3626. [Google Scholar] [CrossRef] [PubMed]
- Lito, S.; Sidibe, A.; Ilmjarv, S.; Burda, P.; Baumgartner, M.; Wehrle-Haller, B.; Krause, K.H.; Marteyn, A. Induced Pluripotent Stem Cells to Understand Mucopolysaccharidosis. I: Demonstration of a Migration Defect in Neural Precursors. Cells 2020, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Canals, I.; Benetó, N.; Cozar, M.; Vilageliu, L.; Grinberg, D. EXTL2 and EXTL3 Inhibition with SiRNAs as a Promising Substrate Reduction Therapy for Sanfilippo C Syndrome. Sci. Rep. 2015, 5, 3–7. [Google Scholar] [CrossRef]
- Huang, W.; Cheng, Y.S.; Yang, S.; Swaroop, M.; Xu, M.; Huang, W.; Zheng, W. Disease Modeling for Mucopolysaccharidosis Type IIIB Using Patient Derived Induced Pluripotent Stem Cells. Exp. Cell Res. 2021, 407, 112785. [Google Scholar] [CrossRef]
- Marín, T.; Contreras, P.; Castro, J.F.; Chamorro, D.; Balboa, E.; Bosch-Morató, M.; Muñoz, F.J.; Alvarez, A.R.; Zanlungo, S. Vitamin E Dietary Supplementation Improves Neurological Symptoms and Decreases C-Abl/P73 Activation in Niemann-Pick C Mice. Nutrients 2014, 6, 3000–3017. [Google Scholar] [CrossRef]
- Val, E. C ELL -B ASED D RUG D EVELOPMENT, S CREENING, AND T OXICOLOGY Induced Pluripotent Stem Cells Reveal Functional Differences Between Drugs Currently Investigated in Patients With Hutchinson-Gilford Progeria Syndrome. 2014, 1–10. [CrossRef]
- Cheng, Y.S.; Yang, S.; Hong, J.; Li, R.; Beers, J.; Zou, J.; Huang, W.; Zheng, W. Modeling Cns Involvement in Pompe Disease Using Neural Stem Cells Generated from Patient-Derived Induced Pluripotent Stem Cells. Cells 2021, 10, 1–14. [Google Scholar] [CrossRef]
- Hong, J.; Cheng, Y.S.; Yang, S.; Swaroop, M.; Xu, M.; Beers, J.; Zou, J.; Huang, W.; Marugan, J.J.; Cai, X.; et al. IPS-Derived Neural Stem Cells for Disease Modeling and Evaluation of Therapeutics for Mucopolysaccharidosis Type II. Exp. Cell Res. 2022, 412, 113007. [Google Scholar] [CrossRef] [PubMed]
- Rybová, J.; Ledvinová, J.; Sikora, J.; Kuchař, L.; Dobrovolný, R. Neural Cells Generated from Human Induced Pluripotent Stem Cells as a Model of CNS Involvement in Mucopolysaccharidosis Type II. J. Inherit. Metab. Dis. 2018, 41, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.A.; Anderson, H.C.; Wolfe, J.H. Ex Vivo Gene Therapy Using Patient IPSC-Derived NSCs Reverses Pathology in the Brain of a Homologous Mouse Model. Stem Cell Reports 2015, 4, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.; Pearse, Y.; Kan, S. hsin; Le, S.Q.; Sanghez, V.; Cooper, J.D.; Dickson, P.I.; Iacovino, M. Genetically Corrected IPSC-Derived Neural Stem Cell Grafts Deliver Enzyme Replacement to Affect CNS Disease in Sanfilippo B Mice. Mol. Ther. - Methods Clin. Dev. 2018, 10, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Pearse, Y.; Clarke, D.; Kan, S.; Le, S.Q.; Sanghez, V.; Luzzi, A.; Pham, I.; Nih, L.R.; Cooper, J.D.; Dickson, P.I.; et al. Brain Transplantation of Genetically Corrected Sanfilippo Type B Neural Stem Cells Induces Partial Cross-Correction of the Disease. Mol. Ther. - Methods Clin. Dev. 2022, 27, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Vazquez, L.; Yanuaria, L.; Lopez, O.; Garcia, I.M.; Ohashi, K.; Rodriguez, N.S. Induced Pluripotent Stem Cell Derivation and Ex Vivo Gene Correction Using a Mucopolysaccharidosis Type 1 Disease Mouse Model. Stem Cells Int. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, J.; Liang, Z.; Gao, C.; Niu, Q.; Wu, F.; Zhang, L. Mesenchymal Stem Cells and Their Microenvironment. Stem Cell Res. Ther. 2022, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Berebichez-Fridman, R.; Montero-Olvera, P.R. Sources and Clinical Applications of Mesenchymal Stem Cells State-of-the-Art Review. Sultan Qaboos Univ. Med. J. 2018, 18, e264–e277. [Google Scholar] [CrossRef] [PubMed]
- Macrin, D.; Joseph, J.P.; Pillai, A.A.; Devi, A. Eminent Sources of Adult Mesenchymal Stem Cells and Their Therapeutic Imminence. Stem Cell Rev. Reports 2017, 13, 741–756. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.T.J.; Gronthos, S.; Shi, S. Critical Reviews in Oral Biology & Medicine: Mesenchymal Stem Cells Derived from Dental Tissues vs. Those from Other Sources: Their Biology and Role in Regenerative Medicine. J. Dent. Res. 2009, 88, 792–806. [Google Scholar] [PubMed]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal Human Dental Pulp Stem Cells (DPSCs) in Vitro and in Vivo. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13625–13630. [Google Scholar] [CrossRef] [PubMed]
- Nuti, N.; Corallo, C.; Chan, B.M.F.; Ferrari, M.; Gerami-Naini, B. Multipotent Differentiation of Human Dental Pulp Stem Cells: A Literature Review. Stem Cell Rev. Reports 2016, 12, 511–523. [Google Scholar] [CrossRef]
- Koussoulakou, D.S.; Margaritis, L.H.; Koussoulakos, S.L. A Curriculum Vitae of Teeth: Evolution, Generation, Regeneration. Int. J. Biol. Sci. 2009, 12, 329–334. [Google Scholar] [CrossRef]
- Kaukua, N.; Shahidi, M.K.; Konstantinidou, C.; Dyachuk, V.; Kaucka, M.; Furlan, A.; An, Z.; Wang, L.; Hultman, I.; Ährlund-Richter, L.; et al. Glial Origin of Mesenchymal Stem Cells in a Tooth Model System. Nature 2014, 513, 551–554. [Google Scholar] [CrossRef]
- Gronthos, S.; Brahim, J.; Li, W.; Fisher, L.W.; Cherman, N.; Boyde, A.; Denbesten, P.; Robey, P.G.; Shi, S. Stem Cell Properties Of. J. Dent. Res. 2002, 81, 531–535. [Google Scholar] [CrossRef]
- Miura, M.; Gronthos, S.; Zhao, M.; Lu, B.; Fisher, L.W.; Robey, P.G.; Shi, S. SHED: Stem Cells from Human Exfoliated Deciduous Teeth. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 5807–5812. [Google Scholar] [CrossRef]
- Al Madhoun, A.; Sindhu, S.; Haddad, D.; Atari, M.; Ahmad, R.; Al-Mulla, F. Dental Pulp Stem Cells Derived From Adult Human Third Molar Tooth: A Brief Review. Front. Cell Dev. Biol. 2021, 9, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Handa, K.; Saito, M.; Yamauchi, M.; Kiyono, T.; Sato, S.; Teranaka, T.; Narayanan, A.S. Cementum Matrix Formation in Vivo by Cultured Dental Follicle Cells. Bone 2002, 31, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Morsczeck, C.; Götz, W.; Schierholz, J.; Zeilhofer, F.; Kühn, U.; Möhl, C.; Sippel, C.; Hoffmann, K.H. Isolation of Precursor Cells (PCs) from Human Dental Follicle of Wisdom Teeth. Matrix Biol. 2005, 24, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Yildirim, S.; Zibandeh, N.; Genc, D.; Ozcan, E.M.; Goker, K.; Akkoc, T. The Comparison of the Immunologic Properties of Stem Cells Isolated from Human Exfoliated Deciduous Teeth, Dental Pulp, and Dental Follicles. Stem Cells Int. 2016, 2016, 11–13. [Google Scholar] [CrossRef]
- Qu, G.; Li, Y.; Chen, L.; Chen, Q.; Zou, D.; Yang, C.; Zhou, Q. Comparison of Osteogenic Differentiation Potential of Human Dental-Derived Stem Cells Isolated from Dental Pulp, Periodontal Ligament, Dental Follicle, and Alveolar Bone. Stem Cells Int. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Kunimatsu, R.; Nakajima, K.; Awada, T.; Tsuka, Y.; Abe, T.; Ando, K.; Hiraki, T.; Kimura, A.; Tanimoto, K. Comparative Characterization of Stem Cells from Human Exfoliated Deciduous Teeth, Dental Pulp, and Bone Marrow–Derived Mesenchymal Stem Cells. Biochem. Biophys. Res. Commun. 2018, 501, 193–198. [Google Scholar] [CrossRef]
- Park, J.Y.; Jeon, H.J.; Kim, T.Y.; Lee, K.Y.; Park, K.; Lee, E.S.; Choi, J.M.; Park, C.G.; Jeon, S.H. Comparative Analysis of Mesenchymal Stem Cell Surface Marker Expression for Human Dental Mesenchymal Stem Cells. Regen. Med. 2013, 8, 453–466. [Google Scholar] [CrossRef]
- Guo, L.; Li, J.; Qiao, X.; Yu, M.; Tang, W.; Wang, H.; Guo, W.; Tian, W. Comparison of Odontogenic Differentiation of Human Dental Follicle Cells and Human Dental Papilla Cells. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Bakopoulou, A.; Leyhausen, G.; Volk, J.; Tsiftsoglou, A.; Garefis, P.; Koidis, P.; Geurtsen, W. Comparative Analysis of in Vitro Osteo/Odontogenic Differentiation Potential of Human Dental Pulp Stem Cells (DPSCs) and Stem Cells from the Apical Papilla (SCAP). Arch. Oral Biol. 2011, 56, 709–721. [Google Scholar] [CrossRef]
- Shi, S.; Miura, M.; Seo, B.M.; Robey, P.G.; Bartold, P.M.; Gronthos, S. The Efficacy of Mesenchymal Stem Cells to Regenerate and Repair Dental Structures. Orthod. Craniofacial Res. 2005, 8, 191–199. [Google Scholar] [CrossRef]
- Eleuterio, E.; Trubiani, O.; Sulpizio, M.; Di Giuseppe, F.; Pierdomenico, L.; Marchisio, M.; Giancola, R.; Giammaria, G.; Miscia, S.; Caputi, S.; et al. Proteome of Human Stem Cells from Periodontal Ligament and Dental Pulp. PLoS One 2013, 8. [Google Scholar] [CrossRef]
- Winning, L.; El Karim, I.A.; Lundy, F.T. A Comparative Analysis of the Osteogenic Potential of Dental Mesenchymal Stem Cells. Stem Cells Dev. 2019, 28, 1050–1058. [Google Scholar] [CrossRef]
- Ullah, I.; Subbarao, R.B.; Kim, E.J.; Bharti, D.; Jang, S.J.; Park, J.S.; Shivakumar, S.B.; Lee, S.L.; Kang, D.; Byun, J.H.; et al. In Vitro Comparative Analysis of Human Dental Stem Cells from a Single Donor and Its Neuronal Differentiation Potential Evaluated by Electrophysiology. Life Sci. 2016, 154, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Patil, R.; Kumar, B.M.; Lee, W.J.; Jeon, R.H.; Jang, S.J.; Lee, Y.M.; Park, B.W.; Byun, J.H.; Ahn, C.S.; Kim, J.W.; et al. Multilineage Potential and Proteomic Profiling of Human Dental Stem Cells Derived from a Single Donor. Exp. Cell Res. 2014, 320, 92–107. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Xu, W.; Chen, H.; Li, S.; Dou, R.; Shen, H.; Liu, X.; Liu, X.; Hong, Y.; He, J. Comparison of the Differentiation of Dental Pulp Stem Cells and Periodontal Ligament Stem Cells into Neuron-like Cells and Their Effects on Focal Cerebral Ischemia. Acta Biochim. Biophys. Sin. (Shanghai). 2020, 52, 1016–1029. [Google Scholar] [CrossRef] [PubMed]
- Morsczeck, C.; Völlner, F.; Saugspier, M.; Brandl, C.; Reichert, T.E.; Driemel, O.; Schmalz, G. Comparison of Human Dental Follicle Cells (DFCs) and Stem Cells from Human Exfoliated Deciduous Teeth (SHED) after Neural Differentiation in Vitro. Clin. Oral Investig. 2010, 14, 433–440. [Google Scholar] [CrossRef]
- Bueno, D.F.; Sunaga, D.Y.; Kobayashi, G.S.; Aguena, M.; Raposo-Amaral, C.E.; Masotti, C.; Cruz, L.A.; Pearson, P.L.; Passos-Bueno, M.R. Human Stem Cell Cultures from Cleft Lip/Palate Patients Show Enrichment of Transcripts Involved in Extracellular Matrix Modeling By Comparison to Controls. Stem Cell Rev. Reports 2011, 7, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.L.; Chen, M.F.; Liao, C.H.; Pang, C.Y.; Lin, P.Y. A Simple and Efficient Method for Generating Nurr1-Positive Neuronal Stem Cells from Human Wisdom Teeth (TNSC) and the Potential of TNSC for Stroke Therapy. Cytotherapy 2009, 11, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Gancheva, M.R.; Kremer, K.L.; Gronthos, S.; Koblar, S.A. Using Dental Pulp Stem Cells for Stroke Therapy. Front. Neurol. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Leong, W.K.; Henshall, T.L.; Arthur, A.; Kremer, K.L.; Lewis, M.D.; Helps, S.C.; Field, J.; Hamilton-Bruce, M.A.; Warming, S.; Manavis, J.; et al. Human Adult Dental Pulp Stem Cells Enhance Poststroke Functional Recovery Through Non-Neural Replacement Mechanisms. Stem Cells Transl. Med. 2012, 1, 177–187. [Google Scholar] [CrossRef]
- Hattori, Y.; Kim, H.; Tsuboi, N.; Yamamoto, A.; Akiyama, S.; Shi, Y.; Katsuno, T.; Kosugi, T.; Ueda, M.; Matsuo, S.; et al. Therapeutic Potential of Stem Cells from Human Exfoliated Deciduous Teeth in Models of Acute Kidney Injury. PLoS One 2015, 10, 1–18. [Google Scholar]
- Barros, M.A.; Martins, J.F.P.; Maria, D.A.; Wenceslau, C.V.; De Souza, D.M.; Kerkis, A.; Câmara, N.O.S.; Balieiro, J.C.C.; Kerkis, I. Immature Dental Pulp Stem Cells Showed Renotropic and Pericyte-Like Properties in Acute Renal Failure in Rats. Cell Med. 2015, 7, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Makino, Y.; Yamaza, H.; Akiyama, K.; Hoshino, Y.; Song, G.; Kukita, T.; Nonaka, K.; Shi, S.; Yamaza, T. Cryopreserved Dental Pulp Tissues of Exfoliated Deciduous Teeth Is a Feasible Stem Cell Resource for Regenerative Medicine. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, H.; Hashimoto, N.; Matsushita, Y.; Matsubara, K.; Yamamoto, N.; Hasegawa, Y.; Ueda, M.; Yamamoto, A. Factors Secreted from Dental Pulp Stem Cells Show Multifaceted Benefits for Treating Acute Lung Injury in Mice. Cytotherapy 2015, 17, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Matsubara, K.; Sakai, K.; Ito, M.; Ohno, K.; Ueda, M.; Yamamoto, A. Dopaminergic Differentiation of Stem Cells from Human Deciduous Teeth and Their Therapeutic Benefits for Parkinsonian Rats. Brain Res. 2015, 1613, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Simon, C.; Gan, Q.F.; Kathivaloo, P.; Mohamad, N.A.; Dhamodharan, J.; Krishnan, A.; Sengodan, B.; Palanimuthu, V.R.; Marimuthu, K.; Rajandas, H.; et al. Deciduous DPSCs Ameliorate MPTP-Mediated Neurotoxicity, Sensorimotor Coordination and Olfactory Function in Parkinsonian Mice. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef]
- Mita, T.; Furukawa-Hibi, Y.; Takeuchi, H.; Hattori, H.; Yamada, K.; Hibi, H.; Ueda, M.; Yamamoto, A. Conditioned Medium from the Stem Cells of Human Dental Pulp Improves Cognitive Function in a Mouse Model of Alzheimer’s Disease. Behav. Brain Res. 2015, 293, 189–197. [Google Scholar] [CrossRef]
- Inoue, T.; Sugiyama, M.; Hattori, H.; Wakita, H.; Wakabayashi, T.; Ueda, M. Stem Cells from Human Exfoliated Deciduous Tooth-Derived Conditioned Medium Enhance Recovery of Focal Cerebral Ischemia in Rats. Tissue Eng. - Part A 2013, 19, 24–29. [Google Scholar] [CrossRef]
- Yamagata, M.; Yamamoto, A.; Kako, E.; Kaneko, N.; Matsubara, K.; Sakai, K.; Sawamoto, K.; Ueda, M. Human Dental Pulp-Derived Stem Cells Protect against Hypoxic-Ischemic Brain Injury in Neonatal Mice. Stroke 2013, 44, 551–554. [Google Scholar] [CrossRef]
- Matsubara, K.; Matsushita, Y.; Sakai, K.; Kano, F.; Kondo, M.; Noda, M.; Hashimoto, N.; Imagama, S.; Ishiguro, N.; Suzumura, A.; et al. Secreted Ectodomain of Sialic Acid-Binding Ig-like Lectin-9 and Monocyte Chemoattractant Protein-1 Promote Recovery after Rat Spinal Cord Injury by Altering Macrophage Polarity. J. Neurosci. 2015, 35, 2452–2464. [Google Scholar] [CrossRef]
- Nicola, F. do C.; Marques, M.R.; Odorcyk, F.; Arcego, D.M.; Petenuzzo, L.; Aristimunha, D.; Vizuete, A.; Sanches, E.F.; Pereira, D.P.; Maurmann, N.; et al. Neuroprotector Effect of Stem Cells from Human Exfoliated Deciduous Teeth Transplanted after Traumatic Spinal Cord Injury Involves Inhibition of Early Neuronal Apoptosis. Brain Res. 2017, 1663, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Taghipour, Z.; Karbalaie, K.; Kiani, A.; Niapour, A.; Bahramian, H.; Nasr-Esfahani, M.H.; Baharvand, H. Transplantation of Undifferentiated and Induced Human Exfoliated Deciduous Teeth-Derived Stem Cells Promote Functional Recovery of Rat Spinal Cord Contusion Injury Model. Stem Cells Dev. 2012, 21, 1794–1802. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, X.; Sun, L.; Guo, W.; Tian, W. Potential of Human Dental Stem Cells in Repairing the Complete Transection of Rat Spinal Cord. J. Neural Eng. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Cho, Y.A.; Lee, Y.M.; Lee, S.Y.; Bae, W.J.; Kim, E.C. PIN1 Suppresses the Hepatic Differentiation of Pulp Stem Cells via Wnt3a. J. Dent. Res. 2016, 95, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Yamaza, T.; Alatas, F.S.; Yuniartha, R.; Yamaza, H.; Fujiyoshi, J.K.; Yanagi, Y.; Yoshimaru, K.; Hayashida, M.; Matsuura, T.; Aijima, R.; et al. In Vivo Hepatogenic Capacity and Therapeutic Potential of Stem Cells from Human Exfoliated Deciduous Teeth in Liver Fibrosis in Mice. Stem Cell Res. Ther. 2015, 6, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Prins, Henk-Jan, Schulten, Engelbert Bruggenkate, C. Tissue Engineering and Regenerative Medicine T ISSUE E NGINEERING AND R EGENERATIVE M EDICINE Concise Review : New Frontiers in MicroRNA-Based Tissue Regeneration. Stem Cells Transl. Med. 2014; 969–976. [CrossRef]
- Yamaguchi, S.; Shibata, R.; Yamamoto, N.; Nishikawa, M. Dental Pulp-Derived Stem Cell Conditioned Medium Reduces Cardiac Injury Following Ischemia- Reperfusion. Nat. Publ. Gr. 2015, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Erdugo, A.; O, E.L.L.L.E.D.; Orrijos, J.O.S.A.; A, R.A.P.A.Y. T ISSUE -S PECIFIC S TEM C ELLS Human Dental Pulp Stem Cells Improve Left Ventricular Function, Induce Angiogenesis, and Reduce Infarct Size in Rats with Acute Myocardial Infarction ´, a A MPARO R UIZ, b AND. 2008, 638–645. [CrossRef]
- Martínez-Sarrà, E.; Montori, S.; Gil-Recio, C.; Núñez-Toldrà, R.; Costamagna, D.; Rotini, A.; Atari, M.; Luttun, A.; Sampaolesi, M. Human Dental Pulp Pluripotent-like Stem Cells Promote Wound Healing and Muscle Regeneration. Stem Cell Res. Ther. 2017, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kerkis, I.; Ambrosio, C.E.; Kerkis, A.; Martins, D.S.; Zucconi, E.; Fonseca, S.A.S.; Cabral, R.M.; Maranduba, C.M.C.; Gaiad, T.P.; Morini, A.C.; et al. Early Transplantation of Human Immature Dental Pulp Stem Cells from Baby Teeth to Golden Retriever Muscular Dystrophy (GRMD) Dogs: Local or Systemic? J. Transl. Med. 2008, 6, 1–13. [Google Scholar] [CrossRef]
- Pisciotta, A.; Riccio, M.; Carnevale, G.; Lu, A.; De Biasi, S.; Gibellini, L.; La Sala, G.B.; Bruzzesi, G.; Ferrari, A.; Huard, J.; et al. Stem Cells Isolated from Human Dental Pulp and Amniotic Fluid Improve Skeletal Muscle Histopathology in Mdx/SCID Mice. Stem Cell Res. Ther. 2015, 6, 1–15. [Google Scholar] [CrossRef]
- Levi, M.; Feitosa, T.; Ii, L.F.; Cristina, P.; Iii, B.B.; Valverde, C.; Iv, W.; X, M.A.M.; Eduardo, C.; Ix, A. Successful Transplant of Mesenchymal Stem Cells in Induced Osteonecrosis of the Ovine Sucesso No Transplante de Células Tronco Mesenquimais Em Ovinos Com Osteonecrose Induzida Da Cabeça Do Fêmur. Resultados Preliminares. Acta Cir. Bras. 2010, 25, 416–422. [Google Scholar]
- Novais, A.; Lesieur, J.; Sadoine, J.; Slimani, L.; Baroukh, B.; Saubaméa, B.; Schmitt, A.; Vital, S.; Poliard, A.; Hélary, C.; et al. Priming Dental Pulp Stem Cells from Human Exfoliated Deciduous Teeth with Fibroblast Growth Factor-2 Enhances Mineralization Within Tissue-Engineered Constructs Implanted in Craniofacial Bone Defects. Stem Cells Transl. Med. 2019, 8, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Asutay, F.; Polat, S.; Gül, M.; Subaşi, C.; Kahraman, S.A.; Karaöz, E. The Effects of Dental Pulp Stem Cells on Bone Regeneration in Rat Calvarial Defect Model: Micro-Computed Tomography and Histomorphometric Analysis. Arch. Oral Biol. 2015, 60, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Shi, X.; Xiao, F.; Yang, Y.; Zhang, X.; Wang, L.S.; Wu, C.T.; Wang, H. Transplantation of Hepatocyte Growth Factor-Modified Dental Pulp Stem Cells Prevents Bone Loss in the Early Phase of Ovariectomy-Induced Osteoporosis. Hum. Gene Ther. 2018, 29, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Yamada, Y.; Ebisawa, K.; Nakamura, S.; Okabe, K.; Umemura, E.; Hara, K.; Ueda, M. Stem Cells from Human Exfoliated Deciduous Teeth (SHED) Enhance Wound Healing and the Possibility of Novel Cell Therapy. Cytotherapy 2011, 13, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Nishino, Y.; Ebisawa, K.; Yamada, Y.; Okabe, K.; Kamei, Y.; Ueda, M. Human Deciduous Teeth Dental Pulp Cells with Basic Fibroblast Growth Factor Enhance Wound Healing of Skin Defect. J. Craniofac. Surg. 2011, 22, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Izumoto-Akita, T.; Tsunekawa, S.; Yamamoto, A.; Uenishi, E.; Ishikawa, K.; Ogata, H.; Iida, A.; Ikeniwa, M.; Hosokawa, K.; Niwa, Y.; et al. Secreted Factors from Dental Pulp Stem Cells Improve Glucose Intolerance in Streptozotocin-Induced Diabetic Mice by Increasing Pancreatic β-Cell Function. BMJ Open Diabetes Res. Care 2015, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kanafi, M.M.; Rajeshwari, Y.B.; Gupta, S.; Dadheech, N.; Nair, P.D.; Gupta, P.K.; Bhonde, R.R. Transplantation of Islet-like Cell Clusters Derived from Human Dental Pulp Stem Cells Restores Normoglycemia in Diabetic Mice. Cytotherapy 2013, 15, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.; Hill, L.J.; Blanch, R.J.; Ward, K.; Logan, A.; Berry, M.; Leadbeater, W.; Scheven, B.A. Mesenchymal Stromal Cell-Mediated Neuroprotection and Functional Preservation of Retinal Ganglion Cells in a Rodent Model of Glaucoma. Cytotherapy 2016, 18, 487–496. [Google Scholar] [CrossRef]
- Ishikawa, J.; Takahashi, N.; Matsumoto, T.; Yoshioka, Y.; Yamamoto, N.; Nishikawa, M.; Hibi, H.; Ishigro, N.; Ueda, M.; Furukawa, K.; et al. Factors Secreted from Dental Pulp Stem Cells Show Multifaceted Benefits for Treating Experimental Rheumatoid Arthritis. Bone 2016, 83, 210–219. [Google Scholar] [CrossRef]
- Shimojima, C.; Takeuchi, H.; Jin, S.; Parajuli, B.; Hattori, H.; Suzumura, A.; Hibi, H.; Ueda, M.; Yamamoto, A. Conditioned Medium from the Stem Cells of Human Exfoliated Deciduous Teeth Ameliorates Experimental Autoimmune Encephalomyelitis. J. Immunol. 2016, 196, 4164–4171. [Google Scholar] [CrossRef]
- Sui, B.; Wu, D.; Xiang, L.; Fu, Y.; Kou, X.; Shi, S. Dental Pulp Stem Cells: From Discovery to Clinical Application. J. Endod. 2020, 46, S46–S55. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Mao, J.; Liu, Y. Pulp Stem Cells Derived from Human Permanent and Deciduous Teeth: Biological Characteristics and Therapeutic Applications. Stem Cells Transl. Med. 2020, 9, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Tomokiyo, A.; Hasegawa, D.; Hamano, S.; Sugii, H.; Maeda, H. Insight into the Role of Dental Pulp Stem Cells in Regenerative Therapy. Biology (Basel). 2020, 9, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Victor, A.K.; Reiter, L.T. Dental Pulp Stem Cells for the Study of Neurogenetic Disorders. Hum. Mol. Genet. 2017, 26, R166–R171. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.; Derrick Roberts, A.; Martin, E.; Rout-Pitt, N.; Gronthos, S.; Byers, S. Mucopolysaccharidosis Enzyme Production by Bone Marrow and Dental Pulp Derived Human Mesenchymal Stem Cells. Mol. Genet. Metab. 2015, 114, 584–593. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Liu, G.; Shi, Y.; Wu, R.; Yang, B.; He, T.; Fan, Y.; Lu, X.; Zhou, X.; Liu, H.; et al. Multipotential Differentiation of Human Urine-Derived Stem Cells: Potential for Therapeutic Applications in Urology. Stem Cells 2013, 31, 1840–1856. [Google Scholar] [CrossRef]
- Falzarano, M.S.; D’Amario, D.; Siracusano, A.; Massetti, M.; Amodeo, A.; La Neve, F.; Maroni, C.R.; Mercuri, E.; Osman, H.; Scotton, C.; et al. Duchenne Muscular Dystrophy Myogenic Cells from Urine-Derived Stem Cells Recapitulate the Dystrophin Genotype and Phenotype. Hum. Gene Ther. 2016, 27, 772–783. [Google Scholar] [CrossRef]
- Gao, P.; Han, P.; Jiang, D.; Yang, S.; Cui, Q.; Li, Z. Effects of the Donor Age on Proliferation, Senescence and Osteogenic Capacity of Human Urine-Derived Stem Cells. Cytotechnology 2017, 69. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Rossi, R.; Grilli, A.; Fang, M.; Osman, H.; Sabatelli, P.; Antoniel, M.; Lu, Z.; Li, W.; Selvatici, R.; et al. Urine-Derived Stem Cells Express 571 Neuromuscular Disorders Causing Genes, Making Them a Potential in Vitro Model for Rare Genetic Diseases. Front. Physiol. 2021, 12, 716471. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Ferlini, A. Urinary Stem Cells as Tools to Study Genetic Disease: Overview of the Literature. J. Clin. Med. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Sabitha, K.R.; Chandran, D.; Shetty, A.K.; Upadhya, D. Delineating the Neuropathology of Lysosomal Storage Diseases Using Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Dev. 2022, 31, 221–238. [Google Scholar] [CrossRef] [PubMed]
Figure 2.
Advantages and limitations of iPSCs.
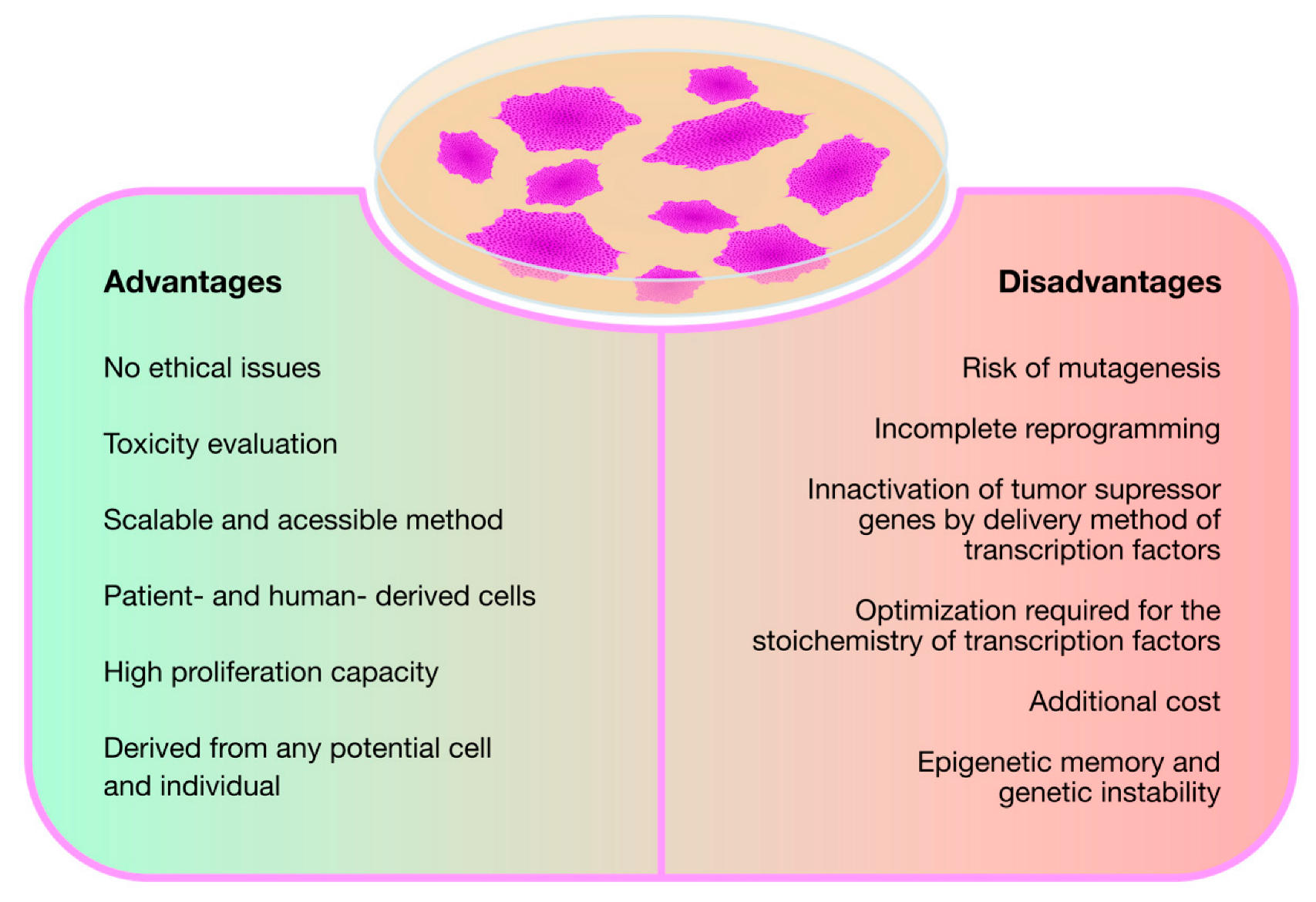
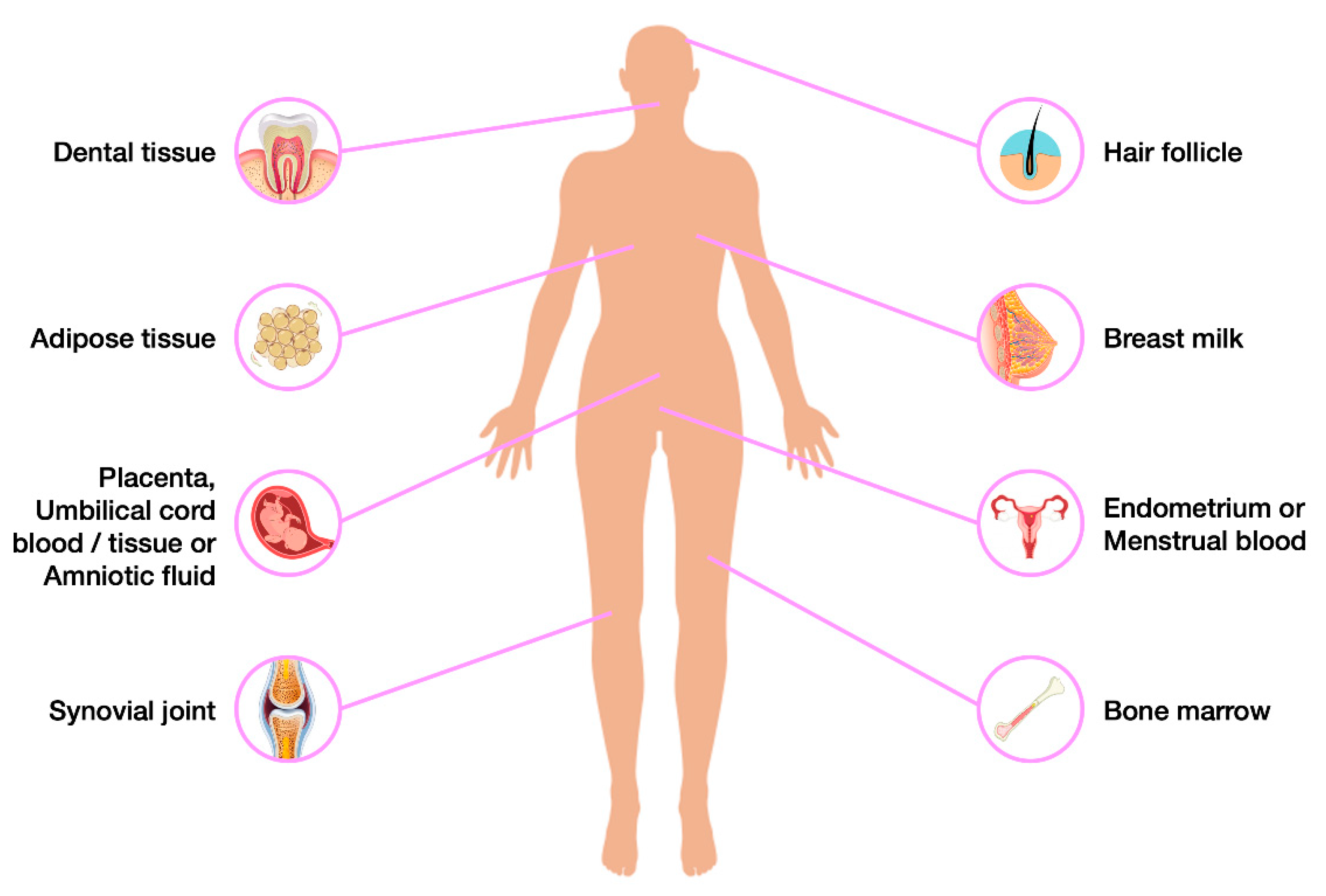
Figure 4.
Minimal Requirements for identification of Mesenchymal Stem Cells (MSCs).
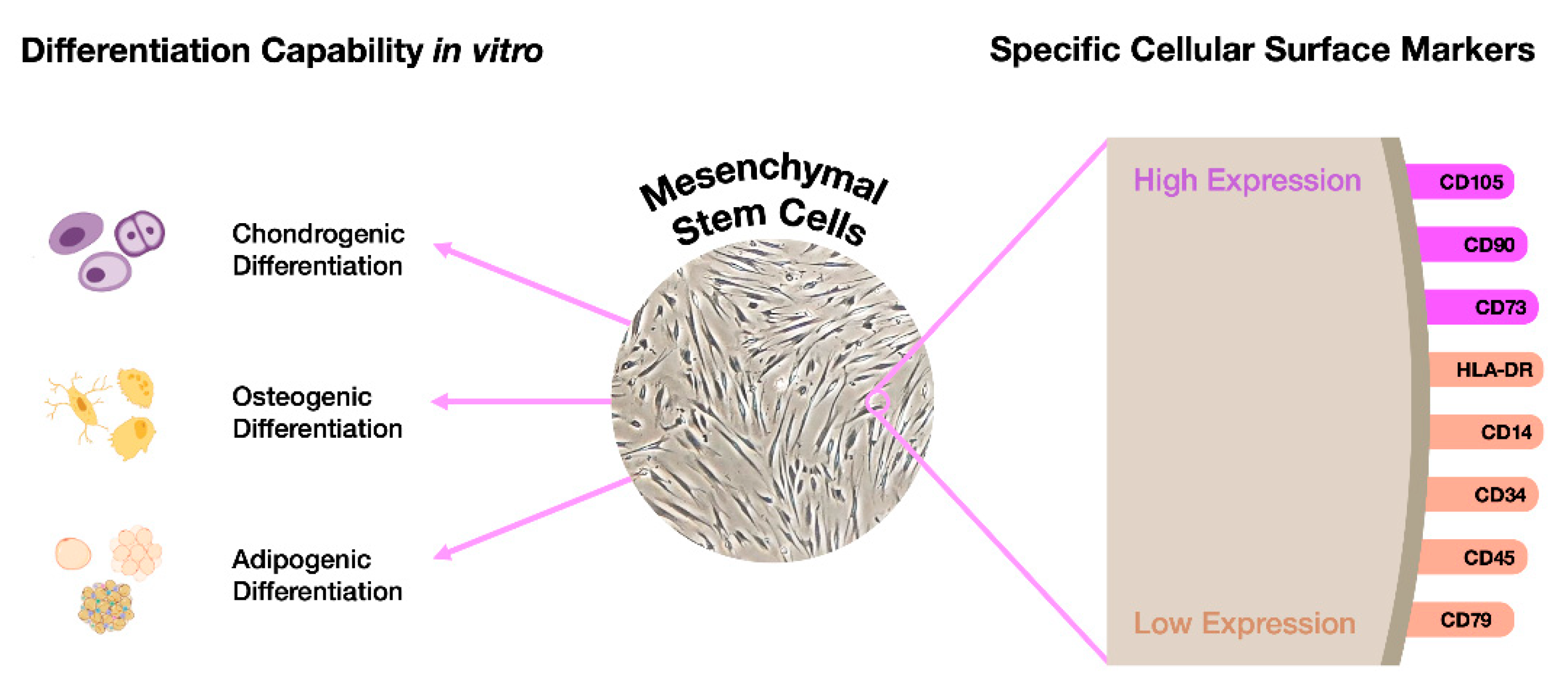
Figure 5.
Schematic drawing illustrating the sources of Dental Mesenchymal Stem Cells in oral cavity. Abbreviations: GMSCs, gingiva-derived MSCs; DFPCs, dental follicle progenitor cells; SHED, stem cells from exfoliated deciduous teeth; SCAPs, stem cells from the apical papilla; DPSCs, dental pulp stem cells; PDLSCs, periodontal ligament stem cells.
Figure 5.
Schematic drawing illustrating the sources of Dental Mesenchymal Stem Cells in oral cavity. Abbreviations: GMSCs, gingiva-derived MSCs; DFPCs, dental follicle progenitor cells; SHED, stem cells from exfoliated deciduous teeth; SCAPs, stem cells from the apical papilla; DPSCs, dental pulp stem cells; PDLSCs, periodontal ligament stem cells.
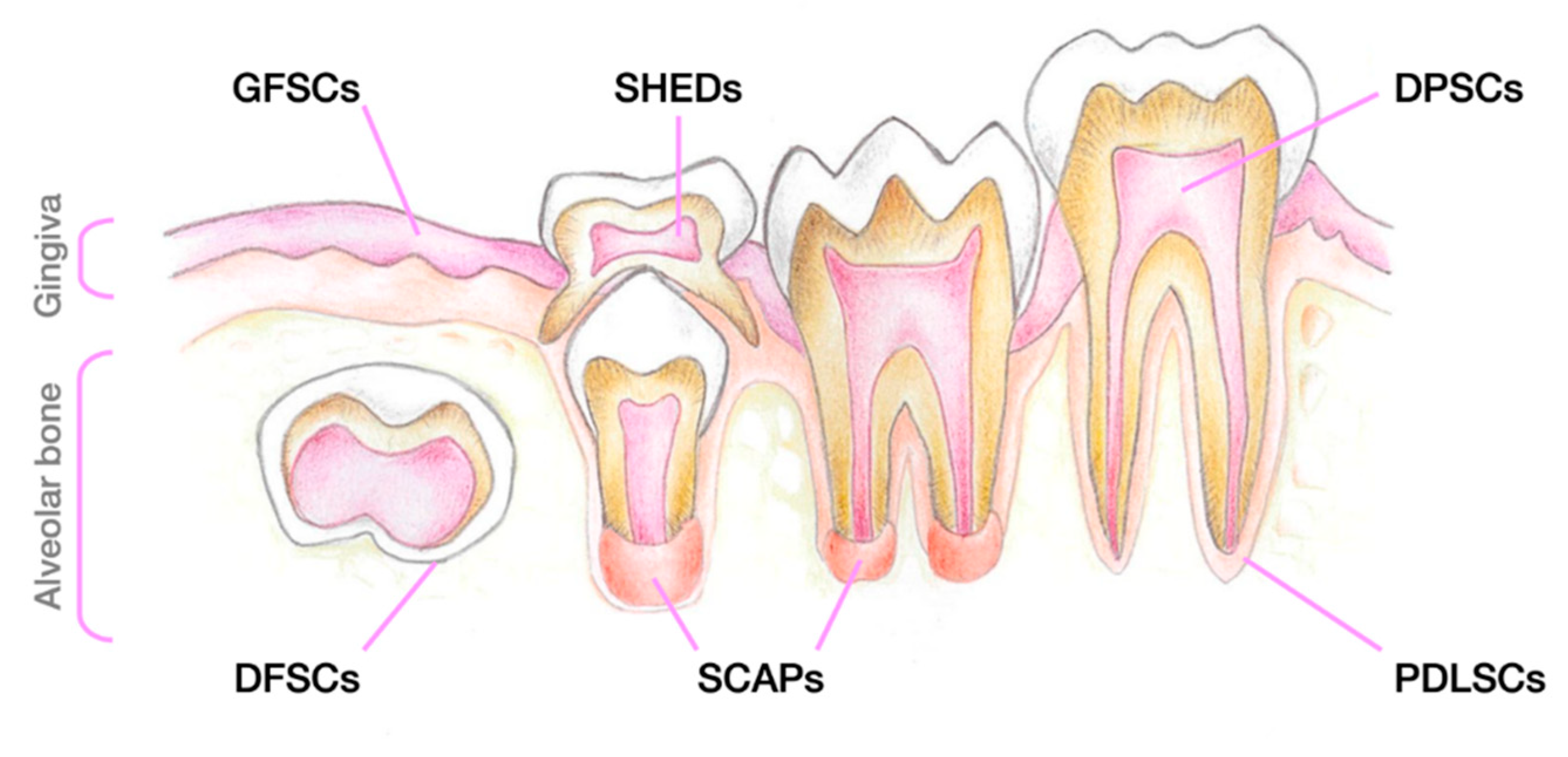

Table 1.
Works performed in Mucopolysaccharidoses (MPSs) using Induced Pluripotent Stem Cells (iPSCs) technology.
Table 1.
Works performed in Mucopolysaccharidoses (MPSs) using Induced Pluripotent Stem Cells (iPSCs) technology.
| Disorder | Affected gene | Defective Enzyme | Stored substrate | Subtype | Generation of MPS-derived iPSCs | Drug Screening | Ex vivo gene therapy | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Source | iPSC | NPC | Mature Neurons | |||||||
| MPS I | IDUA | α-L-iduronidase | DS and HS | Hurler | Fibroblasts | [118,126,127] | [126,127] | |||
| Mouse Embryonic Fibroblasts | [138] | [138] | ||||||||
| Hurler/Scheie | Fibroblasts | [126] | [126] | |||||||
| Scheie | Fibroblasts | [119] | ||||||||
| PBMCs | [126] | [126] | ||||||||
| MPS II | IDS | Iduronate-2-sulfatase | DS and HS | Fibroblasts | [123,133] | [123,133] | [133] | |||
| PBMCs | [114,115,116,134] | [124,134] | [124,134] | [134] | ||||||
| MPS III | SGSH | Sulfamidase | HS | A | Fibroblasts | [111] | ||||
| NAGLU | α-N-acetylglucosaminidase | B | Fibroblasts | [109,110,120,129] | [109,120,129] | [129] | [129] | |||
| Mouse Embryonic Fibroblasts | [136,137] | [136,137] | [136,137] | |||||||
| HGSNAT | N-acetyltransferase | C | Fibroblasts | [113,121] | [121,122] | [121,122] | [121,122] | |||
| MPS VII | GUSB | β-Glucuronidase | DS, HS, and CS | Mouse Embryonic Fibroblasts | [135] | [135] | [135] | |||
Table 2.
Potential for differentiation of the different Stem Cells derived from the dental tissue.
| Type of Differentiation | Potential Differentiation | References |
|---|---|---|
| Osteogenic | PDLSCs>DFSCs/SHEDs>DPSCs>SCAPs | [148,153,159,160,161,162] |
| Chondrogenic | DPSCs>SCAPs/DFSCs/PDLSCs | [153,161,162] |
| Adipogenic | DFSCs>DPSCs/SCAPs>PDLSCs | [153,162] |
| Neurogenic | SHEDs>PDLSCs>DPSCs>DFSCs>SCAPs | [161,163,164] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



