Submitted:
31 January 2023
Posted:
03 February 2023
You are already at the latest version
Abstract
The synthesis of nitrogen-based heterocycles has always been considered essential in the development of pharmaceuticals in medicine and agriculture. This explains why various synthetic approaches have been proposed in recent decades. However, performing as methods often imply harsh conditions or the employment of toxic solvents and dangerous reagents. Mechanochemistry is undoubtedly one of the most promising technologies currently used for reducing any possible environmental impact due to the worldwide interest in developing solvent-free synthetic pathways. Following this line, we propose a renewed mechanochemical protocol for synthesizing various heterocyclic classes by exploiting the reducing proprieties and the electrophilic nature of thiourea dioxide (TDO). Simultaneously using the ready availability and low cost of a component of the textile industry such as TDO and all the advantages brought by a green technique such as mechanochemistry, we plotted the route towards a more sustainable and eco-friendly methodology for obtaining heterocyclic moieties.
Keywords:
heterocycles
; thiourea dioxide
; TDO
; mechanochemistry
1. Introduction
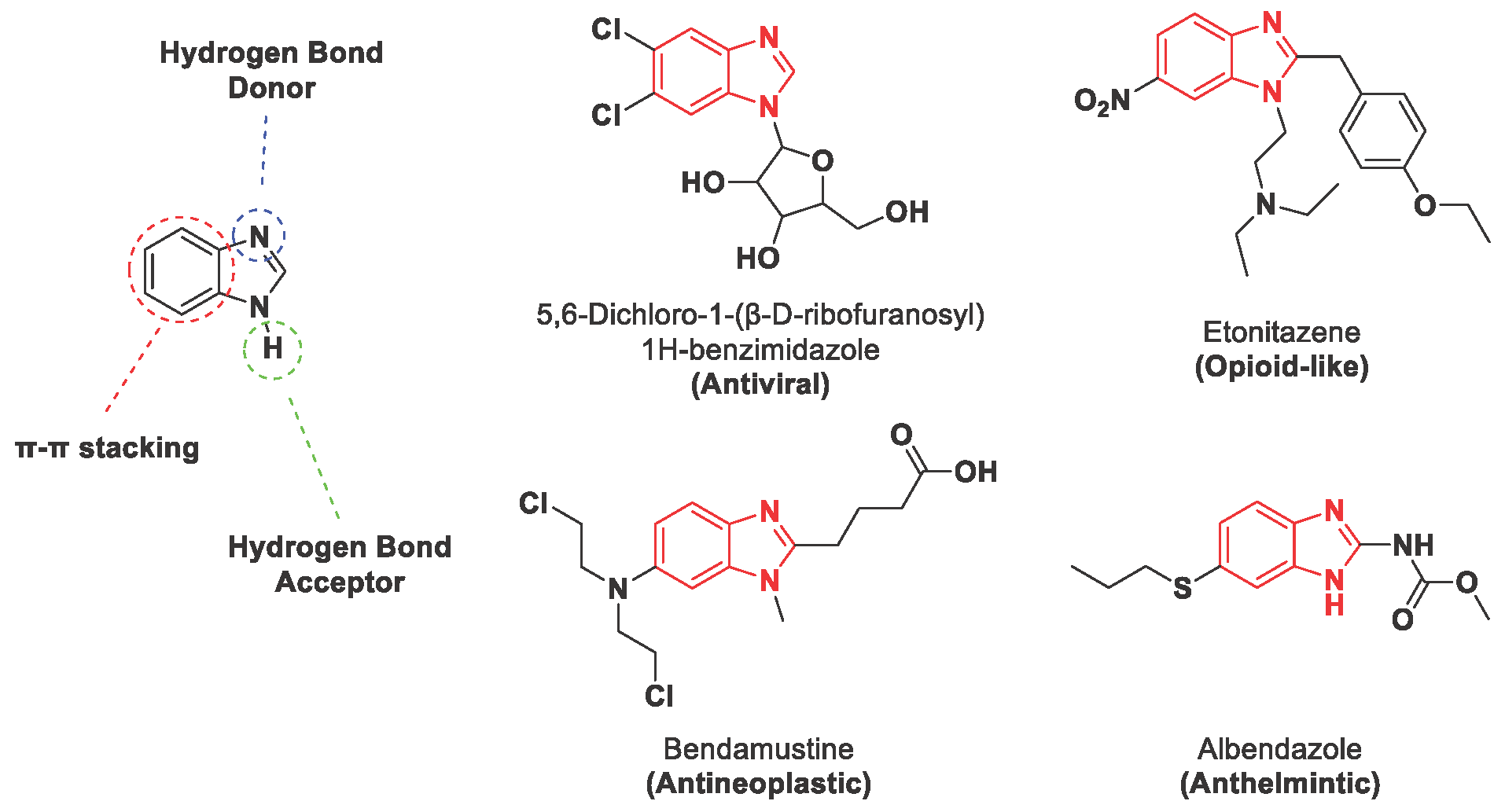
Heterocycles are ubiquitous in biologically active compounds, natural products, and common pharmaceuticals [1,2,3,4,5,6,7,8], representing a highly privileged structural motif. For example, common biocides [9], fungicides [10], antitumoral [11,12], and analgesics [13,14,15], to mention a few, contain a benzimidazole or benzothiazole moiety in their structure. Furthermore, the benzimidazole scaffold represents a benchmark for synthesizing new potential agents against various cancer or infectious diseases, even those that still cannot be effectively treated [16,17]. This is due to the benzimidazole ring’s astonishing proprieties that simultaneously possess a hydrophobic unit and two hydrogen bonding domains proper citation of hydrogen bond donor and acceptor should be given (Figure 1) [18].
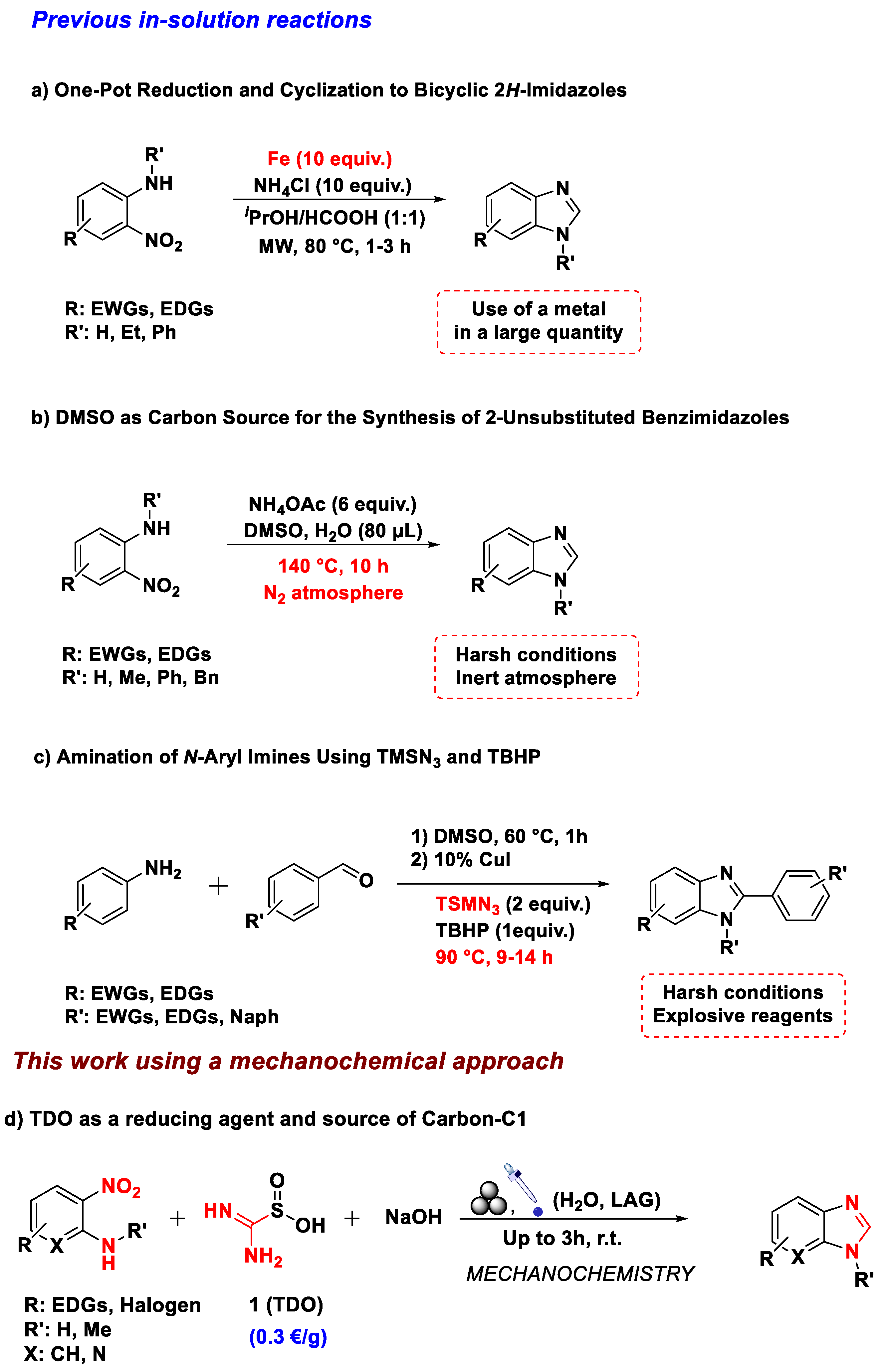
Accordingly, several endeavors have been dedicated to the synthesis of such compounds. Among all the reported methods, using either formaldehyde or a relative surrogate has proved an efficient and valuable choice in preparing different heterocycles containing the benzimidazole core [19,20,21,22,23,24,25]. However, they usually suffer from some limitations, such as the employment of additives like metal catalysts, harsh reaction conditions, or the use of toxic solvents (Scheme 1).
In this context, thiourea dioxide (TDO), a solid surrogate for formaldehyde, could allow us to overcome many of the shortcomings mentioned above. TDO is a cheap and commercial compound commonly employed in textile industries for bleaching processes [26]. Moreover, it has also been exploited in the past for analytical measurements [27,28,29]. Finally, it can also be synthesized, as documented by Barnett (Scheme 2) [30], Lai [31], and Kluttz [32], and different studies, albeit out of date, have already been made concerning its both electrophilic and reducing nature [33,34,35,36,37,38].
As a matter of fact, TDO is insoluble in most organic solvents as well as in water, and its reactivity can be triggered by raising the temperature. Because of this, it is typically used in mixtures of methanol and basic or heated water [39,40].
In 2021, Wu’s team documented benzimidazoles’ synthesis through thiourea dioxide’s (TDO, 1) electrophilic character in a solvent-based process [41]. The procedure was carried out in the water at ≃ 60 °C, providing around ten benzimidazoles in satisfactory yields and a solvent screening on the control reaction proving the inefficacy of TDO in organic solvents due to a solubility matter. Moreover, the use and removal of solvents during a chemical synthesis represent a significant portion of organic pollution and process energy consumption.
Benzimidazoles are generally synthesized from aniline derivatives, which in turn, are often prepared from the corresponding nitrobenzenes. Furthermore, the synthetic methods to produce anilines still involve classical methodologies, and there is currently a worldwide interest in proposing new synthetic routes to improve the sustainability and applicability of such a process [42,43,44,45]. Reducing an aromatic nitro moiety usually requires an acid environment in the presence of a metal [46,47,48,49] or the employment of gaseous hydrogen [50,51,52,53]. Despite entirely performing as methods, all the concerns regarding these methodologies must be remarked. Starting from metals, this procedure is usually run under an acidic environment [54,55,56,57,58], and consequently, it may not apply to specific substrates sensitive to such conditions.
Moreover, the use of metals is often associated with various risks concerning human health and the environment due to their well-established toxicity. For example, with molecular gaseous hydrogen, its use is related to an explosion hazard because of its high reactivity [54,55]. Furthermore, since this procedure is exergonic (ΔrG° < 0), a high amount of heat is typically released during an industrial process [56]. Such heat generation can be challenging to remove and may also induce expensive cooling costs or lower the reaction yields due to reactor hot spots [57,58]. Other methods rely on electrochemistry [58,59], enzymatic processes [55], rare-earth elements [60], and hydrogen transfer reagents [60,61,62,63] which all need specific reaction conditions and advanced equipment. Few reductions in a basic environment have been described in the literature [64,65,66]; the most recurrent one is undoubtedly the employment of Zn powder in the presence of NaOH for synthesizing azobenzene [67]. Unfortunately, a basic environment generally implies other sub-products formed during the redox process [56,66,68]. Their presence complicates the obtainment of the corresponding anilines making the entire procedure complex and cumbersome.
Contrarily, the reducing processes of TDO are associated with releasing nontoxic side products, mainly urea and sodium sulfite [36].
The above-mentioned drawbacks prompted us to assess the feasibility of grinding the reaction under solvent-free conditions. Indeed, ball-milling remains an impressive technology in this regard [69,70,71,72]. Furthermore, many mechanochemical strategies described astonishing advantages like high reaction efficiency, prevention of harsh reaction conditions, and minimization of organic solvents [73,74,75].
2. Results and Discussion
2.1. TDO as a reducing agent
Firstly, the reducing properties of TDO (Compound 1) on nitrobenzenes were explored to outline and screen all the mechanochemical parameters for this step. Considering the few existing techniques for reducing nitrobenzene with TDO [76,77], we had to lay the groundwork for a methodology with broader applicability. We set the mechanochemical procedure at a 1.0 mmol scale using nitrobenzene as a reference substrate. We milled TDO 1 (1.0 mmol), nitrobenzene 2a (1.0 mmol), and NaOH (1.0 mmol) inside a 10 mL stainless steel (SS) vessel equipped with two balls (Ø = 7 mm, 2.67 g) of the same material, running the reaction for 1.0 hour (Scheme 3).
Unfortunately, we detected the only presence of the starting material through a GC-MS analysis (Table 1, entry 1). Several process parameters have been investigated to overcome these failures, and the whole optimization process is summarized in Table 1. To begin with, we raised the ratio of 1 and NaOH (Table 1, entries 2 and 3), which allowed us to convert 2a into aniline 3a with a 21% yield (Table 1, entry 3). Then, prompted by these results, we tried to increase both the reaction time and the reducing mixture equivalents (TDO and NaOH). In the first case, the yield was even lower in a 2-hour milling with a 5% yield (GC-analysis), probably due to further reactivity of the formed aniline with the redox intermediates (Table 1, entry 4). In the latter one, 2a was consumed entirely, but 3a was obtained with only a 29% yield together with other process intermediates (Table 1, entry 5). Lastly, we ran the reducing process at 70 °C to accelerate the kinetics of the reaction, but we only obtained the corresponding symmetric diazobenzene (PhN=NPh, Table 1, entry 6).
So, after all these failed attempts, we considered using drops of different solvents to run a Liquid Assisted Grinding (LAG) [78,79,80,81,82] in a 90-minute procedure. Consistently, less polar, like decane or toluene, and polar solvents, like acetone or isopropanol, did not permit a reasonable conversion rate (Table 1, entries 7-10). Lastly, methanol and water were used, as in analogous solvent-based procedures. However, in this case, the ratio of solvent/reagents was drastically cut down compared to the already reported methodologies (LAG, η = 0.44 µL/mg). Contrasting what is known, methanol used under LAG conditions produced a complex mixture of aniline and nitrobenzene reduction process intermediates (Table 1, entry 11). Water, instead, led to excellent yields of 3a (Table 1, entry 12). Its amount, however, was found to be a critical parameter since the conversion rate dramatically dropped when η = 0.22 µL/mg (Table 1, entry 13). Contrarily, a little increase in the reaction time of up to 2 hours allowed a quantitative conversion of 2a to 3a (Table 1, entry 14). Concerning the bases, weaker ones like sodium carbonate and sodium bicarbonate did not allow a comparable result (Table 1, entries 15 and 16), proving that NaOH plays a crucial role in the mechanochemical process - as already insight by Hawkes for similar reactions in solution [83]. Lowering the NaOH amount negatively affected the reaction performance as well (Table 1, entry 17).
With the optimized conditions in hand, we extended the entire procedure to other nitrobenzenes to validate this mechanochemical process. In the case of activated substrates 2b and 2c, the process smoothly proceeded to a complete conversion within 2 hours, while for more complex substrates like 2s, the process was completed only in 3 hours (Scheme 4).
Reducing instead 2-nitroaniline 2d, many unpredicted outcomes showed up, as summarized in Table 2. In this case, we synthesized the o-phenylenediamine 3d with a 37% yield in 90 minutes without a LAG (Table 2, entry 1). Such different behavior can be ascribed to the positive effects of an EDG. However, unexpectedly, prolonging the reaction time to 2 hours under neat grinding conditions resulted in the formation of the corresponding benzimidazole, albeit in low yields (Table 2, entry 2). For the sake of completeness, we also tried to reduce in neat conditions other substrates having a comparable charge distribution (Table 2, entries 3 and 4). With 2- nitro anisole, we obtained the corresponding aniline in a 54% isolated yield. At the same time, the employment of 2-nitro phenol resulted in a mixture of various unidentified products, likely generated by the high phenoxide reactivity.
To better understand several critical details of the process, we have to better focus on several points of the process. First, under LAG conditions, the presence of methanol resulted in the concurrent formation of 3d and various reaction intermediates. At the same time, water use was associated with obtaining the desired product with a nearly quantitative yield in 2 hours (Table 2, entries 5 and 6). These different outcomes can be reconducted to the role covered by water as a better proton source. On the other hand, water, to some extent, also inhibits the final cyclization pathway that leads to benzimidazole formation. The reduction of the nitro group and the construction of the benzimidazole ring both consume TDO, with the latter being kinetically faster. As a result, any attempt to decrease the NaOH equivalents failed because of the high reactivity of the formed o-phenylenediamine towards the TDO that was still present in the reaction medium (Table 2, entry 7). Once these issues are focused on, we can draw conclusions based on the above and after a long, meticulous exploratory study. Six equivalents of NaOH promote the sluggish kinetics of the reduction reaction to the detriment of the cyclization reaction, resulting in a complete reduction of the nitro group (Table 2, entry 6).

Table 2.
Optimization process for the reduction of 2d to 3d.
| Entry | TDO eq. | Base eq. | Reaction Time (h) | Additivesb | Yieldsa |
|---|---|---|---|---|---|
| 1 | 3 | 6 | 1.5 | / | 37% |
| 2c | 3 | 6 | 2 | / | 45% |
| 3d | 3 | 6 | 2 | / | 54% |
| 4e | 3 | 6 | 2 | / | Complex mixture |
| 5f | 3 | 6 | 2 | MeOH | 0% |
| 6 | 3 | 6 | 2 | H2O | 98% |
| 7g | 3 | 3 | 2 | H2O | 1% |
All the reactions were carried out with the same experimental parameters unless otherwise specified: 2-nitroaniline 2d (1.0 mmol), compound 1 (3.0 mmol), and NaOH (3.0 – 6.0 mmol) in a SS jar (10.0 mL) equipped with two balls (SS, Ø = 7.0 mm, 2.67 g) at a frequency of 30 Hz. a The yields were calculated by GC-MS analysis. b The additive quantity was 250 μL. c The o-phenylenediamine reacted with 1 and formed the benzimidazole 4d yielding 20% and other unidentified subproducts. d The starting material was 2-nitro anisole. e The starting material was 2-nitro phenol. f The signals in the spectra were attributed to 2-((2-nitrophenyl)diazenyl)aniline and 2,2′-(hydrazine-1,2-diyl)dianiline. g 2-nitroaniline 2d and benzimidazole 4d were detected with a yield of 75% and 24%, respectively.
Once we understood the reactivity of 2-nitroanilines, we also extended this process to other 2-nitroaniline derivatives (Scheme 5). As a result, o-phenylenediamines 3e and 3f were successfully synthesized in a 2-hour ongoing process, whereas the substrates 3g-j and 3o-p needed a longer reaction time of 3 hours. These outcomes were utterly in line with Hammett’s parameters and steric hindrance on the aromatic ring of the starting materials.
2.2. TDO as an electrophile
Clarified the role of TDO 1 as a reducing agent, we thoroughly analyzed its application as a green solid surrogate of formaldehyde for synthesizing aza-heterocycles. Its electrophilicity is connected to the presence of the two nitrogen atoms depleting the central carbon atom in terms of electron density. Furthermore, an excellent leaving group like the sulfur moiety makes the whole molecule more prone to a nucleophilic attack. We accomplished the heterocycle synthesis by establishing two separate procedures: a single-step process based on the employment of phenylenediamines (procedure A) and a double-step methodology starting from 2-nitroanilines (procedure B).
2.2.1. Procedure A
The reactivity of phenylenediamines toward compound 1 has already been reported in the literature for solvent-based processes restricted to a few very reactive substrates. [41]. We investigated the mechanosynthesis of heterocycles from less reactive substrates, in this case, the ones that possess a lower electron density in their aromatic ring (Scheme 6). All the reactions were conducted using a LAG, where water was used as the additive (η =0.44). Starting from the o-phenylenediamines 3l-n, we obtained the corresponding heterocycles in low yields due to the low reactivity of such compounds. Deactivating groups either on the ring or on the nitrogen atom drastically affected the ring-closure process, as evidenced by the poor yields obtained for compound 3l-n. Remarkably, diamines 3q and 3r enabled to access molecular framework of biological interest in modest to good yields [84,85,86].
2.2.2. Procedure B
With the optimal conditions for reducing 2-nitroanilines in hands, we wondered whether a double-step methodology could be feasible. Bearing in mind that a basic environment consumes compound 1 for forming the reducing specie, we realized that we needed a refill of it (fresh adding) for running the second step. In addition, we added a small quantity of water because the process proved to perform poorly through neat grinding, as formerly stated. After these considerations, we shaped the procedure that allowed us to convert 2-nitroanilines into aza-heterocycles (Scheme 7). The first step was run under fine-tuned conditions so that the newly formed o-phenylenediamine was ready for the forthcoming ring-closure step. This last stage was successfully accomplished with a refill of 1 (3.0 mmol) and water (250 μL, η = 0.44), and it was run for further 2 hours for the substrates 2d-f. The 2-nitroanilines 2g-k and 2o-p required a longer reaction time of 3 hours instead.
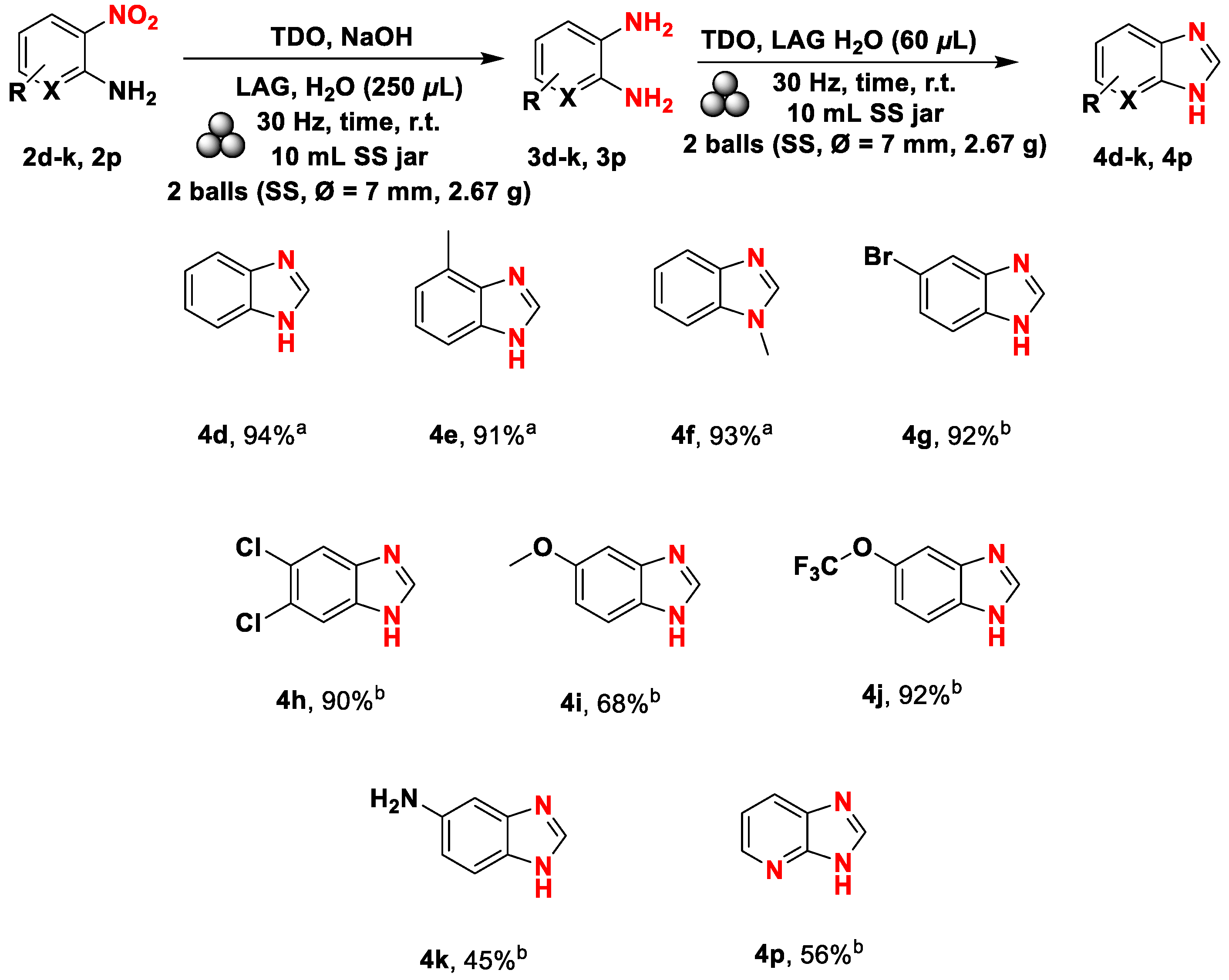
The reaction proceeded well in all the considered cases and provided benzimidazoles 4d-4j in near-quantitative yields except for compounds 4k and 4p (Scheme 7).
To assess the green footprint of this mechanochemical procedure, we calculated the green metrics for our methodology and compared them with a previously reported solvent-based process. The results highlight a substantial improvement, in a green chemistry framework, of the proposed mechanochemical technique concerning the solvent-based approach (see the paragraph “Green Metrics” in the SI for further details) [87].
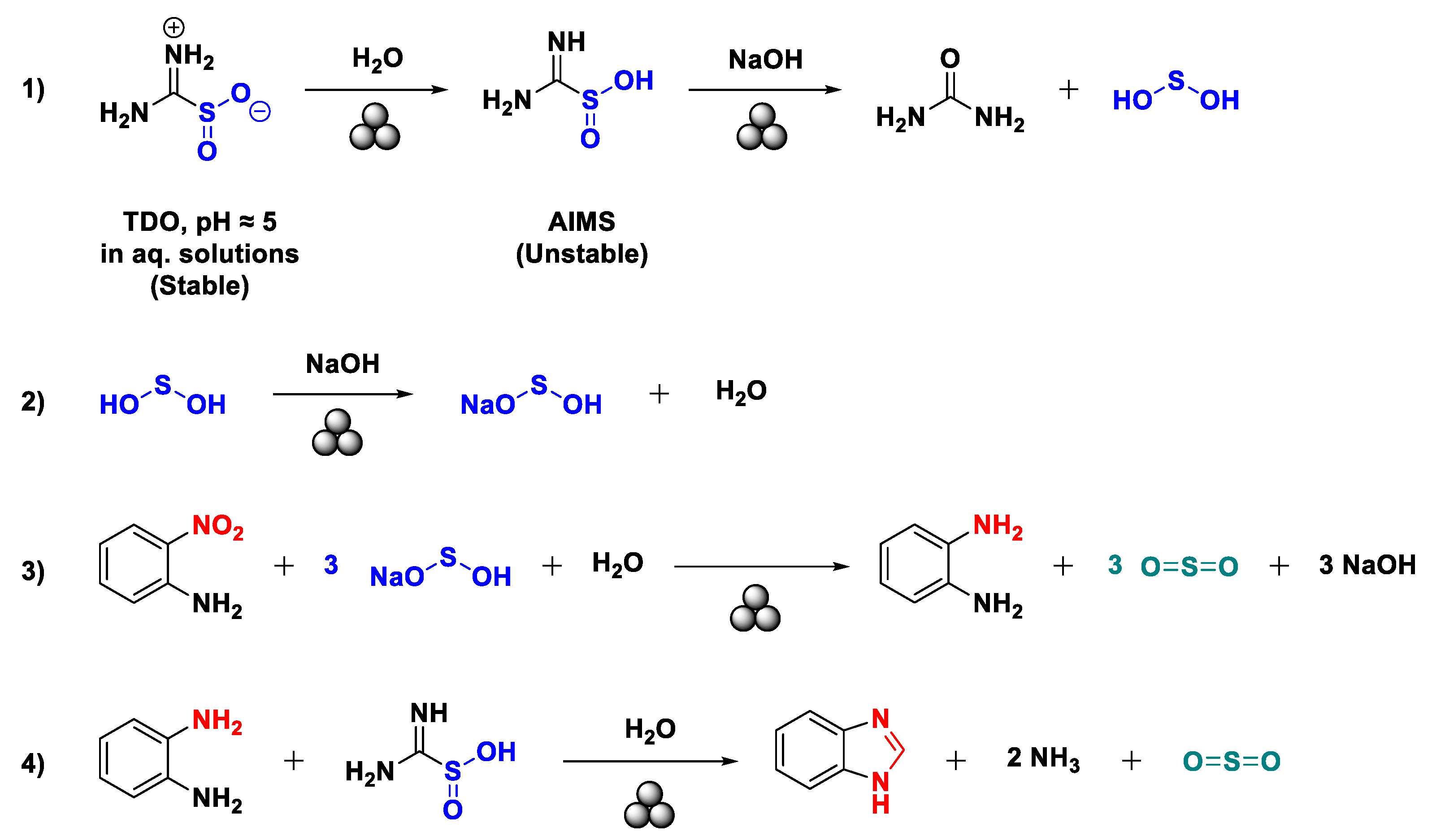
All the syntheses presented are easy to accomplish and proceed with a first redox process followed by a final ring closure and aromatization step (Scheme 8). The reducing ability of compound 1 has been widely described, and it exploits the formation of sulfoxylic acid [35,36,37,88,89]. Such a chemical specie can be formed only from the tautomer of compound 1, aminoiminomethanesulfinic acid (AIMS), usually only observed in an aqueous medium (Scheme 8, pathway 1) [90,91,92,93]. After being formed, it is converted to its more stable form, sodium hydrogen sulfoxylate, by the remaining equivalents of NaOH (Scheme 8, pathway 2). It is hard to imagine that a specie like sodium sulfoxylate could be formed due to the low acidity of sodium hydrogen sulfoxylate [88]. This last compound is then ready to take part in the redox process and will be reduced to gaseous sulfur dioxide (Scheme 8, pathway 3).
Nonetheless, we cannot completely rule out the presence of sodium dithionite and sodium bisulfite as reducing agents. The former can be formed by TDO degradation [38,94], and the sulfur dioxide may generate the latter in the presence of water [95]. Ultimately, adding fresh TDO to the reaction medium let the ring be closed to the corresponding aromatic heterocycle (Scheme 8, pathway 4).
Concerning water (250 μL) used to perform the LAG process (η = 0.44), it plays three fundamental roles in the mechanochemical redox reaction. Firstly, it enables the formation of the active tautomer AIMS, as described by Dittmer [92] and Krug [93]. Secondly, the complete consumption of 1 to sodium sulphoxylate in a strong basic environment avoids other collateral processes like the auto condensation of TDO to cyclic derivatives, as reported in the literature [96]. Thirdly, the presence of water might prevent the formation of other undesired redox intermediates because it acts as a proton donor [97]. Lastly, it probably permits sodium hydroxide to participate extensively in concert with TDO due to its high solubility in water. This may also explain the poorer performance of the mechanochemical process when methanol is employed for a LAG approach.
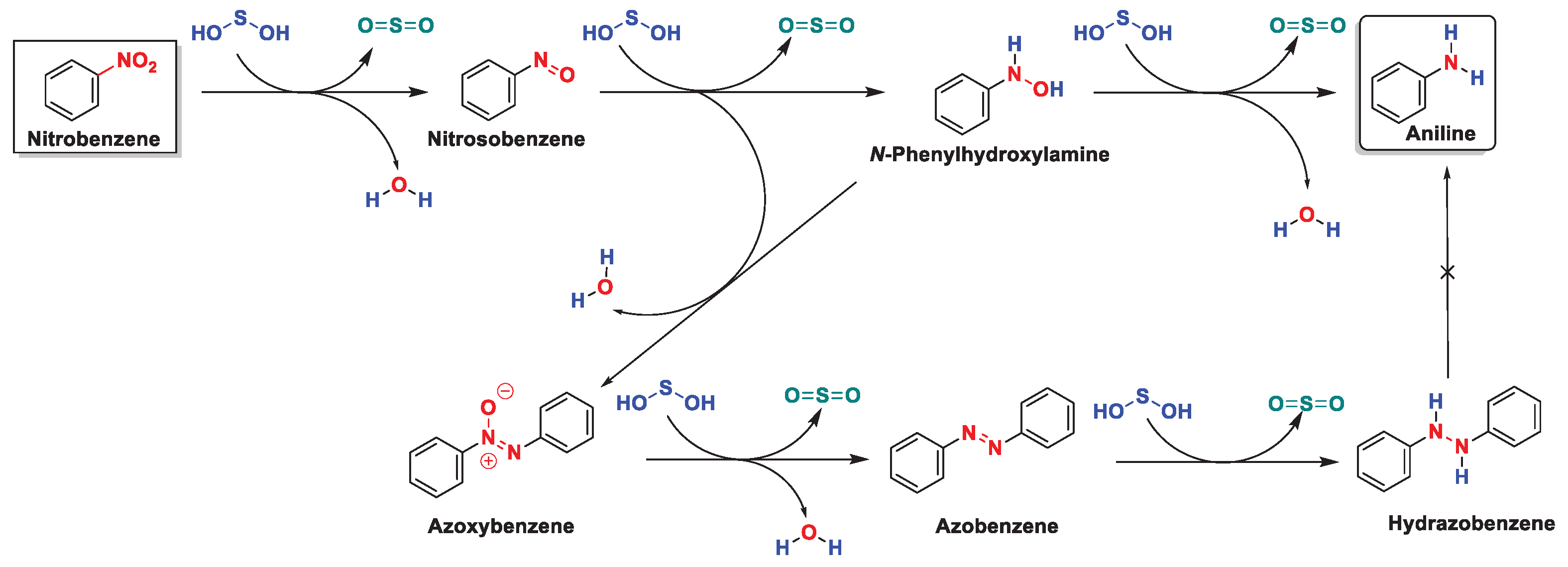
To conclude, we also studied the reactivity of hydrazobenzene under our optimized conditions. Unfortunately, conducting the reaction with 1 mmol of hydrazobenzene with TDO (3 mmol), NaOH (6 mmol), and water (250 μL) only yielded the starting material as described by Huang [77]. Therefore, the entire redox process is wholly described in Scheme 9.
Considering the ring-closure step, the electrophilic nature intrinsic to compound 1 makes possible the formation of a heterocycle (Scheme 10a,b). We firmly support the idea that, in the presence of NaOH, cyanimide cannot be formed by the degradation of compound 1 [98]. Therefore, the only possible pathway for heterocycle synthesis is the release of formamidine through a dismutative process (Scheme 10a pathway 1) [99,100]. After undergoing a nucleophilic attack from the o-phenylenediamine, formamidine allows the generation of an N-arylformamidine intermediate that will go through a second nucleophilic attack from the other nitrogen atom (Scheme 10a, pathway 2). The resulting 2-amino dehydrobenzimidazole then extrudes the NH2 moiety as ammonia for forming the benzimidazole structure (Scheme 10a, pathway 3).
Nevertheless, we cannot rule out a reaction mechanism based on the supposed carbenoid structure of TDO as well [90,101,102,103] (Scheme 10b). In this case, the reaction mechanism initially follows a diverse pathway based on the minor charge separation between the two moieties of TDO (Scheme 10b, pathway 1) [91,104]. Then, after forming an N-arylformamidine intermediate, the process continues as mentioned above (Scheme 10b, pathway 1).
We ruled out a possible deamination approach from the corresponding 2-aminobenzimidazole for the following reasons. Firstly, when we milled commercial 2-aminobenzimidazole in the presence of NaOH, we did not see any change in the starting material’s nature. Secondly, the thermodynamic stability of such a substrate prevents any structure alteration under our mild conditions [105]. Thirdly, its synthesis should be associated with a dehydrogenative process that is unlikely in our reaction medium. Finally, once the entire process is finished, the desired product needs to be extracted from the reaction mixture.
3. Materials and Methods
3.1. Materials
Commercially available reagents were purchased from Acros (Geel, Belgium), Aldrich (Darmstadt, Germania), Strem Chemicals (Newburyport, USA), Alfa-Aesar (Haverhill, USA), and TCI Europe (Zwijndrecht, Belgium) and used as received. All of the reactions were monitored by thin-layer chromatography (TLC) performed on glass-backed silica gel 60 F254, 0.2 mm plates (Merck, Darmstadt, Germania), and compounds were visualized under UV light (254 nm) or using cerium ammonium molybdate solution with subsequent heating. The eluents were technical grade. The mechanochemical reactions were performed using a Retsch Mixer Mill MM 500 VARIO apparatus (horizontal vibratory mill). The reagents were milled using a stainless-steel grinding jar (10 mL) equipped with two balls (Ø = 7.00 mm, 2.67 g) of the same material. The 1H- and 13C-NMR spectra were recorded on a Bruker (Billerica, USA) Avance III HD 600 MHz NMR spectrometer at 298 K. Proton chemical shifts are expressed in parts per million (ppm, δ scale) and are referred to as the residual hydrogen in the solvent (CDCl3, 7.27 ppm or DMSO-d6 2.54 ppm). Carbon chemical shifts are expressed in parts per million (ppm, δ scale) and are referenced to the carbon resonances of the NMR solvent (CDCl3, 77.0 ppm or DMSO-d6 39.5 ppm). GC-MS analyses were performed on an Agilent 5977B MS interfaced to the GC 7890B equipped with a DB-5ms column (J & W, New Brighton, UK). Yields refer to pure, isolated materials.
3.2. General procedure A for anilines and o-phenylenediamines 3a-j, 3o-p, 3s synthesis from 2- nitroanilines 2a-j, 2o-p, 2s
A 10 mL stainless steel jar equipped with two stainless steel milling balls (7 mm diameter, 2.67 g) was filled with nitrobenzenes 2a-j, 2o-p, 2s (1.0 mmol), NaOH (6.0 mmol), 1 (3.0 mmol), and 250 μL of distilled water. The vessel was then closed, and the mechanochemical reaction was conducted, ranging from 60 to 180 min at a frequency of 30 Hz. Whenever necessary, further purification through flash column chromatography has been made. Lastly, the solvent was removed under reduced pressure to afford the pure anilines 3a-j, 3o-p, 3s.
3.3. General procedure B for heterocycles 4l-n, 4q-r synthesis from o-phenylenediamines 3l-n, 3q-r
A 10 mL stainless steel jar equipped with two stainless steel milling balls (7 mm diameter, 2.67 g) was filled with o-phenylenediamines 3l-n, 3q-r (1.00 mmol), 1 (2.00 mmol), and 200 μL of distilled water. The vessel was then closed, and the mechanochemical reaction was conducted, ranging from 60 min to 180 min at a frequency of 30 Hz. At the end of the reaction, an additional silica pad (SiO2, heptane/ethyl acetate/methanol = 1:1:0→6:3:1) was made to purify the reaction mixture. Lastly, the solvent was removed under reduced pressure to afford the pure heterocycle 4l-n, 4q-r.
3.4. General procedure C for heterocycles 4d-k, 4p synthesis from 2-nitroanilines 2d-k, 2p
A 10 mL stainless steel jar equipped with two stainless steel milling balls (7 mm diameter, 2.67 g) was filled with 2-nitroanilines 2d-k, 2p (1.0 mmol), NaOH (6.0 mmol), 1 (3.0 mmol), and 250 μL of distilled water. The vessel was then closed, and the mechanochemical reaction was conducted, ranging from 60 to 180 min at a frequency of 30 Hz. After that, an additional refill of 1 (3.0 mmol) extra refill of 1 (3.0 mmol) was made, and 60 μL of distilled water and the mechanochemical reaction was made run ranging from 60 to 180 min at a frequency of 30 Hz. At the end of the reaction, an additional silica pad (SiO2, heptane/ethyl acetate/methanol = 1:1:0→6:3:1) was made to purify the reaction mixture. Lastly, the solvent was removed under reduced pressure to afford the pure heterocycle 4d-k, 4p.
4. Conclusions
This work has thoroughly explained a mechanochemical protocol for synthesizing heterocycles rediscovering an old forgotten solid reagent as thiourea dioxide (TDO). Not only did the dual nature of such a compound allow us to propose both reducing and ring-closure procedures, but it also allowed us to merge these two processes in a one-pot technique starting from 2-nitroanilines. By avoiding a mixture of methanol and basic water like in the in-solution methods, we were also able to analyze the reducing properties of TDO, as nobody has ever reported. The reaction is easy to perform and allows to obtain of the desired products with yields ranging from low to excellent. In addition, this methodology provided an alternative pathway for synthesizing scaffolds of biological and pharmaceutical interest, like benzimidazole derivatives, and valuable building blocks with potential application in drug design, like perimidines and imidazopyridines.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, writing—review and editing, A.P.; validation, formal analysis, investigation, data curation, F.C., and F.B.; LD supervision; review, and editing, L. D.; funding acquisition, A.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by MIUR Italy, PRIN 2017 project (grant number: 2017B7MMJ5_001) “MultIFunctional poLymer cOmposites based on groWn matERials (MIFLOWER) and Fondazione di Sardegna (FdS, F72F20000230007).
Data Availability Statement
The data presented in this study are available in Supplementary Materials.
Acknowledgments
We acknowledge the CeSAR (Centro Servizi Ricerca d’Ateneo) core facility of the University of Cagliari and Dr. Sandrina Lampis for assistance with the generation of the 1H- and 13C-NMR spectroscopic data. We also thank Gianluigi Corrias for the technical support in managing the ball mills and jars. Finally, we would like to dedicate our entire work to the memory of Professor Adolfo Lai. What he documented 50 years ago began a journey that has finally reached its end, and we are proud to have taken part in it.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Elattar, K.M.; Mert, B.D.; Monier, M.; El-Mekabaty, A. Advances in the chemical and biological diversity of heterocyclic systems incorporating pyrimido[1,6-a]pyrimidine and pyrimido[1,6-c]pyrimidine scaffolds. RSC Adv. 2020, 10, 15461–15492. [Google Scholar] [CrossRef] [PubMed]
- Hammouda, M.M.; Gaffer, H.E.; Elattar, K.M. Insights into the medicinal chemistry of heterocycles integrated with a pyrazolo[1,5-a]pyrimidine scaffold. RSC Med Chem 2022, 13, 1150–1196. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, N.; Naim, M.J.; Alam, M.J.; Nawaz, F.; Ahmed, S.; Alam, O. Benzimidazole Scaffold as Anticancer Agent: Synthetic Approaches and Structure-Activity Relationship. Arch Pharm (Weinheim) 2017, 350. [Google Scholar] [CrossRef]
- Tahlan, S.; Kumar, S.; Kakkar, S.; Narasimhan, B. Benzimidazole scaffolds as promising antiproliferative agents: a review. BMC Chem 2019, 13, 66. [Google Scholar] [CrossRef] [PubMed]
- Singla, P.; Luxami, V.; Paul, K. Benzimidazole-biologically attractive scaffold for protein kinase inhibitors. RSC Adv. 2014, 4, 12422–12440. [Google Scholar] [CrossRef]
- Mudi, P.K.; Mahanty, A.K.; Kotakonda, M.; Prasad, S.; Bhattacharyya, S.; Biswas, B. A benzimidazole scaffold as a promising inhibitor against SARS-CoV-2. J. Biomol. Struct. Dyn. 2022, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Gutiérrez, A.; Curiel-Quesada, E.; Correa-Basurto, J.; Martínez-Muñoz, A.; Reyes-Arellano, A. N-Heterocycles Scaffolds as Quorum Sensing Inhibitors. Design, Synthesis, Biological and Docking Studies. Int. J. Mol. Sci. 2020, 21, 9512. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, S.K.; Ibrahim, N.A.; El-Kaed, S.A.; El-Helw, E.A.E. New potential fungicides pyrazole-based heterocycles derived from 2-cyano-3-(1,3-diphenyl-1H-pyrazol-4-yl) acryloyl isothiocyanate. J. Sulfur Chem. 2021, 42, 529–546. [Google Scholar] [CrossRef]
- Merel, S.; Benzing, S.; Gleiser, C.; Di Napoli-Davis, G.; Zwiener, C. Occurrence and overlooked sources of the biocide carbendazim in wastewater and surface water. Environ. Pollut. 2018, 239, 512–521. [Google Scholar] [CrossRef]
- Han, P.; Rios-Miguel, A.B.; Tang, X.; Yu, Y.; Zhou, L.-J.; Hou, L.; Liu, M.; Sun, D.; Jetten, M.S.M.; Welte, C.U.; et al. Benzimidazole fungicide biotransformation by comammox Nitrospira bacteria: Transformation pathways and associated proteomic responses. J. Hazard. Mater. 2023, 445, 130558. [Google Scholar] [CrossRef]
- Chu, B.; Liu, F.; Li, L.; Ding, C.; Chen, K.; Sun, Q.; Shen, Z.; Tan, Y.; Tan, C.; Jiang, Y. A benzimidazole derivative exhibiting antitumor activity blocks EGFR and HER2 activity and upregulates DR5 in breast cancer cells. Cell. Death Dis. 2015, 6, e1686. [Google Scholar] [CrossRef]
- Son, D.S.; Lee, E.S.; Adunyah, S.E. The Antitumor Potentials of Benzimidazole Anthelmintics as Repurposing Drugs. Immune Netw 2020, 20, e29. [Google Scholar] [CrossRef]
- Gaba, M.; Singh, S.; Mohan, C. Benzimidazole: an emerging scaffold for analgesic and anti-inflammatory agents. Eur. J. Med. Chem. 2014, 76, 494–505. [Google Scholar] [CrossRef]
- Ersan, S.; Nacak, S.; Noyanalpan, N.; Yeşilada, E. Studies on analgesic and anti-inflammatory activities of 1-dialkylaminomethyl-2-(p-substituted phenyl)-5-substituted benzimidazole derivatives. Arzneimittelforschung 1997, 47, 834–836. [Google Scholar]
- Ujváry, I.; Christie, R.; Evans-Brown, M.; Gallegos, A.; Jorge, R.; de Morais, J.; Sedefov, R. DARK Classics in Chemical Neuroscience: Etonitazene and Related Benzimidazoles. ACS Chem. Neurosci. 2021, 12, 1072–1092. [Google Scholar] [CrossRef] [PubMed]
- Satija, G.; Sharma, B.; Madan, A.; Iqubal, A.; Shaquiquzzaman, M.; Akhter, M.; Parvez, S.; Khan, M.A.; Alam, M.M. Benzimidazole based derivatives as anticancer agents: Structure activity relationship analysis for various targets. J. Heterocycl. Chem. 2022, 59, 22–66. [Google Scholar] [CrossRef]
- Almalki, A.S.A.; Nazreen, S.; Elbehairi, S.E.I.; Asad, M.; Shati, A.A.; Alfaifi, M.Y.; Alhadhrami, A.; Elhenawy, A.A.; Alorabi, A.Q.; Asiri, A.M.; et al. Design, synthesis, anticancer activity and molecular docking studies of new benzimidazole derivatives bearing 1,3,4-oxadiazole moieties as potential thymidylate synthase inhibitors. New J. Chem. 2022, 46, 14967–14978. [Google Scholar] [CrossRef]
- Hosamani, K.M.; Hiremath, V.B.; Keri, R.S.; Harisha, R.S.; Halligudi, S.B. Synthesis of novel 2-alkyl substituted oleobenzimidazole derivatives using ethylene glycol as solvent. Can. J. Chem. 2008, 86, 1030–1033. [Google Scholar] [CrossRef]
- Hanan, E.J.; Chan, B.K.; Estrada, A.A.; Shore, D.G.; Lyssikatos, J.P. Mild and General One-Pot Reduction and Cyclization of Aromatic and Heteroaromatic 2-Nitroamines to Bicyclic 2H-Imidazoles. Synlett 2010, 2010, 2759–2764. [Google Scholar] [CrossRef]
- Nale, D.B.; Bhanage, B.M. N-Substituted Formamides as C1-Sources for the Synthesis of Benzimidazole and Benzothiazole Derivatives by Using Zinc Catalysts. Synlett 2015, 26, 2835–2842. [Google Scholar] [CrossRef]
- Zhu, X.; Zhang, F.; Kuang, D.; Deng, G.; Yang, Y.; Yu, J.; Liang, Y. K2S as Sulfur Source and DMSO as Carbon Source for the Synthesis of 2-Unsubstituted Benzothiazoles. Org. Lett. 2020, 22, 3789–3793. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, D.; Sadhu, P.; Punniyamurthy, T. Copper(I)-Catalyzed Regioselective Amination of N-Aryl Imines Using TMSN3 and TBHP: A Route to Substituted Benzimidazoles. J. Org. Chem. 2015, 80, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Dadwal, S.; Kumar, M.; Bhalla, V. “Metal-Free” Nanoassemblies of AIEE-ICT-Active Pyrazine Derivative: Efficient Photoredox System for the Synthesis of Benzimidazoles. J. Org. Chem. 2020, 85, 13906–13919. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Mondal, A.; Srimani, D. Selective Synthesis of 2-Substituted and 1,2-Disubstituted Benzimidazoles Directly from Aromatic Diamines and Alcohols Catalyzed by Molecularly Defined Nonphosphine Manganese(I) Complex. J. Org. Chem. 2018, 83, 9553–9560. [Google Scholar] [CrossRef] [PubMed]
- Caron, S.; Jones, B.P.; Wei, L. Preparation of Substituted Benzimidazoles and Imidazopyridines Using 2,2,2-Trichloroethyl Imidates. Synthesis 2012, 44, 3049–3054. [Google Scholar] [CrossRef]
- Fischer, K.; Marquardt, K.; Schlüter, K.; Gebert, K.; Borschel, E.-M.; Heimann, S.; Kromm, E.; Giesen, V.; Schneider, R.; Lee Wayland Jr., R. Textile Auxiliaries. In Ullmann’s Encycl. Ind. Chem.
- Obtemper, S.I.; Zlobin, V.K. Application of Formamidinesulfinic acid for Separate Spectrophotometric Determination of para Nitrophenol, ortho Nitrophenol and meta Nitrophenol in their Mutual Presence. 1974, 29, 609-611.
- Obtemper, S.I.; Zlobin, V.K. Use of Thiourea Dioxide in Organic-Analysis-Determination of Nitric-Acid Esters, Nitroso and Azo-Compounds. 1974, 247-249.
- Koniecki, W.B.; Linch, A.L. Determination of Aromatic Nitro Compounds. Anal. Chem. 1958, 30, 1134–1137. [Google Scholar] [CrossRef]
- de Barry Barnett, E. VII.—The action of hydrogen dioxide on thiocarbamides. J. Chem. Soc., Trans. 1910, 97, 63–65. [Google Scholar] [CrossRef]
- De Filippo, D.; Ponticelli, G.; Trogu, E.F.; Lai, A. Spectrochemical study of aminoiminomethanesulphinic acid and related NN′-substituted derivatives. J. Chem. Soc., Perkin Trans. II 1972, 1500–1502. [Google Scholar] [CrossRef]
- Havel, J.J.; Kluttz, R.Q. A Synthesis of Formamidinesulfinic Acids and Formamidines. Synth. Comm. 1974, 4, 389–393. [Google Scholar] [CrossRef]
- Dictionary. In Gardner’s Commercially Important Chemicals; 2005; pp. 2-682.
- Pu, S.; Liang, Q.; Luo, X.; Luo, J. Convenient Two-step One-pot Synthesis of Benzimidazoles Using 2-nitroanilines and Thiourea Dioxide. J. Chem. Res. 2014, 38, 118–120. [Google Scholar] [CrossRef]
- Hamad, M.O.; Kiptoo, P.K.; Stinchcomb, A.L.; Crooks, P.A. Synthesis and hydrolytic behavior of two novel tripartate codrugs of naltrexone and 6β-naltrexol with hydroxybupropion as potential alcohol abuse and smoking cessation agents. Bioorg. Med. Chem. 2006, 14, 7051–7061. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.; Gupta, S.; Sureshbabu, P.; Sabiah, S.; Kandasamy, J. A metal free reduction of aryl-N-nitrosamines to the corresponding hydrazines using a sustainable reductant thiourea dioxide. Green Chem. 2016, 18, 6215–6221. [Google Scholar] [CrossRef]
- Chatterjie, N.; Umans, J.G.; Inturrisi, C.E. Reduction of 6-ketones of the morphine series with formamidinesulfinic acid. Stereoselectivity opposite to that of hydride reductions. J. Org. Chem. 1976, 41, 3624–3625. [Google Scholar] [CrossRef] [PubMed]
- Svarovsky, S.A.; Simoyi, R.H.; Makarov, S.V. Reactive oxygen species in aerobic decomposition of thiourea dioxides . J. Chem. Soc., Dalton Trans. 2000, 511–514. [Google Scholar] [CrossRef]
- He, F.-S.; Yang, M.; Ye, S.; Wu, J. Sulfonylation from sodium dithionite or thiourea dioxide. Chin. Chem. Lett. 2021, 32, 461–464. [Google Scholar] [CrossRef]
- Verma, S.; Singh, R.; Tripathi, D.; Gupta, P.; Bahuguna, G.M.; Jain, S.L. Thiourea dioxide with TBHP: a fruitful and greener recipe for the catalytic oxidation of alcohols. RSC Advances 2013, 3, 4184–4188. [Google Scholar] [CrossRef]
- Zhou, L.H.; Jin, Y.J.; Ma, L.F.; Huang, W.H.; Wu, Y. Highly Efficient and Catalyst-Free Synthesis of Benzimidazoles in Aqueous Media. Russ. J. Org. Chem. 2021, 57, 825–830. [Google Scholar] [CrossRef]
- Kahl, T.; Schröder, K.-W.; Lawrence, F.R.; Marshall, W.J.; Höke, H.; Jäckh, R. Aniline. In Ullmann’s Encycl. Ind. Chem.
- Available online: https://archive.vn/20020219104231/http://www.the-innovation-group.com/ChemProfiles/Aniline.
- Formenti, D.; Ferretti, F.; Scharnagl, F.K.; Beller, M. Reduction of Nitro Compounds Using 3d-Non-Noble Metal Catalysts. Chem. Rev. 2019, 119, 2611–2680. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bu, J.; Wang, J.; Sun, C.; Zhao, D.; Sheng, G.; Xie, X.; Sun, M.; Yu, L. Highly Efficient Hydrogenation of Nitrobenzene to Aniline over Pt/CeO2 Catalysts: The Shape Effect of the Support and Key Role of Additional Ce3+ Sites. ACS Catal. 2020, 10, 10350–10363. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, Y.; Prashad, M.; Repič, O.; Blacklock, T.J. A Practical and Chemoselective Reduction of Nitroarenes to Anilines Using Activated Iron. Adv. Synth. Catal. 2005, 347, 217–219. [Google Scholar] [CrossRef]
- Anjali, K.; Ahmed, M.; Christopher, J.; Sakthivel, A. Rhodium-calix[4]pyrrole and rhodium-tetraphenyl porphyrin: preparation, surface grafting and their catalytic application in nitro-benzene reduction. Dalton Trans. 2018, 47, 12353–12361. [Google Scholar] [CrossRef]
- Srilakshmi, C.; Saraf, R.; Prashanth, V.; Rao, G.M.; Shivakumara, C. Structure and Catalytic Activity of Cr-Doped BaTiO3 Nanocatalysts Synthesized by Conventional Oxalate and Microwave Assisted Hydrothermal Methods. Inorg. Chem. 2016, 55, 4795–4805. [Google Scholar] [CrossRef] [PubMed]
- Mondal, P.; Purkait, M.K. Green synthesized iron nanoparticle-embedded pH-responsive PVDF-co-HFP membranes: Optimization study for NPs preparation and nitrobenzene reduction. Sep. Sci. Technol. 2017, 52, 2338–2355. [Google Scholar] [CrossRef]
- Leng, F.; Gerber, I.C.; Lecante, P.; Moldovan, S.; Girleanu, M.; Axet, M.R.; Serp, P. Controlled and Chemoselective Hydrogenation of Nitrobenzene over Ru@C60 Catalysts. ACS Catal. 2016, 6, 6018–6024. [Google Scholar] [CrossRef]
- Xiong, W.; Zhou, S.; Zhao, Z.; Hao, F.; Cai, Z.; Liu, P.; Zhang, H.; Luo, H. Highly uniform Ni particles with phosphorus and adjacent defects catalyze 1,5-dinitronaphthalene hydrogenation with excellent catalytic performance. Front. Chem. Sci. Eng. 2021, 15, 998–1007. [Google Scholar] [CrossRef]
- Gong, W.; Lin, Y.; Chen, C.; Al-Mamun, M.; Lu, H.-S.; Wang, G.; Zhang, H.; Zhao, H. Nitrogen-Doped Carbon Nanotube Confined Co–Nx Sites for Selective Hydrogenation of Biomass-Derived Compounds. Adv. Mater. 2019, 31, 1808341. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; chen, Y.; Zhao, Z.; Deng, H.; Zhou, D.; Wei, C.; Nie, R.; Xia, Q. Highly selective one-step hydrogenation of nitrobenzene to cyclohexylamine over the supported 10% Ni/carbon catalysts doped with 3‰ Rh. RSC Advances 2016, 6, 15354–15361. [Google Scholar] [CrossRef]
- Diao, S.; Qian, W.; Luo, G.; Wei, F.; Wang, Y. Gaseous catalytic hydrogenation of nitrobenzene to aniline in a two-stage fluidized bed reactor. Appl. Catal. A: Gen. 2005, 286, 30–35. [Google Scholar] [CrossRef]
- Krishnan, S.; Patel, P.N.; Balasubramanian, K.K.; Chadha, A. Yeast supported gold nanoparticles: an efficient catalyst for the synthesis of commercially important aryl amines. New J. Chem. 2021, 45, 1915–1923. [Google Scholar] [CrossRef]
- Daems, N.; Wouters, J.; Van Goethem, C.; Baert, K.; Poleunis, C.; Delcorte, A.; Hubin, A.; Vankelecom, I.F.J.; Pescarmona, P.P. Selective reduction of nitrobenzene to aniline over electrocatalysts based on nitrogen-doped carbons containing non-noble metals. Appl. Catal. B: Environ. 2018, 226, 509–522. [Google Scholar] [CrossRef]
- Niknam, T.; Bornapour, M.; Gheisari, A.; Bahmani-Firouzi, B. Impact of heat, power and hydrogen generation on optimal placement and operation of fuel cell power plants. Int. J. Hydrog. Energy 2013, 38, 1111–1127. [Google Scholar] [CrossRef]
- Sheng, X.; Wouters, B.; Breugelmans, T.; Hubin, A.; Vankelecom, I.F.J.; Pescarmona, P.P. Cu/CuxO and Pt nanoparticles supported on multi-walled carbon nanotubes as electrocatalysts for the reduction of nitrobenzene. Appl. Catal. B: Environ. 2014, 147, 330–339. [Google Scholar] [CrossRef]
- Sheng, X.; Wouters, B.; Breugelmans, T.; Hubin, A.; Vankelecom, I.F.J.; Pescarmona, P.P. Pure and Alloyed Copper-Based Nanoparticles Supported on Activated Carbon: Synthesis and Electrocatalytic Application in the Reduction of Nitrobenzene. ChemElectroChem 2014, 1, 1198–1210. [Google Scholar] [CrossRef]
- Zhang, T.; Xie, Z.; Jiang, L.; Zhao, W.; Cao, S.; Wang, B.; Si, R.; Zhang, R.; Liu, Y.; Zhao, Z. Selective transfer hydrogenation coupling of nitroaromatics to azoxy/azo compounds by electron-enriched single Ni-N4 sites on mesoporous N-doped carbon. Chem. Eng. J. 2022, 443, 136416. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Yang, B.; Ma, L.; Wang, N.; Wei, X. Facile Synthesis of a Novel Heterogeneous Rh/COF Catalyst and Its Application in Tandem Selective Transfer Hydrogenation and Monomethylation of Nitro Compounds with Methanol. Ind. Eng. Chem. Res. 2022, 61, 1066–1077. [Google Scholar] [CrossRef]
- Moran, M.J.; Martina, K.; Baricco, F.; Tagliapietra, S.; Manzoli, M.; Cravotto, G. Tuneable Copper Catalysed Transfer Hydrogenation of Nitrobenzenes to Aniline or Azo Derivatives. Adv. Synth. Catal. 2020, 362, 2689–2700. [Google Scholar] [CrossRef]
- Xu, D.; Liu, R.; Li, J.; Zhao, H.; Ma, J.; Dong, Z. Atomically dispersed Co-N4 sites anchored on N-doped carbon for aqueous phase transfer hydrogenation between nitroarenes and saturated N-heterocycles. Appl. Catal. B: Environ. 2021, 299, 120681. [Google Scholar] [CrossRef]
- Dai, X.; Cui, X.; Yuan, H.; Deng, Y.; Shi, F. Cooperative transformation of nitroarenes and biomass-based alcohols catalyzed by CuNiAlOx. RSC Adv. 2015, 5, 7970–7975. [Google Scholar] [CrossRef]
- Liu, H.; Khuan Chuah, G.; Jaenicke, S. Alumina-entrapped Ag catalyzed nitro compounds coupled with alcohols using borrowing hydrogen methodology. Phys. Chem. Chem. Phys. 2015, 17, 15012–15018. [Google Scholar] [CrossRef]
- Wei, R.P.; Shi, F. Controllable synthesis of azoxybenzenes and anilines with alcohol as the reducing agent promoted by KOH. Synth. Commun. 2019, 49, 688–696. [Google Scholar] [CrossRef]
- Bigelow, H.E.; Robinson, D.B. Org. Synth. 1942, 22, 28. [CrossRef]
- Srilakshmi, C.; Vijay Kumar, H.; Praveena, K.; Shivakumara, C.; Muralidhar Nayak, M. A highly efficient iron doped BaTiO3 nanocatalyst for the catalytic reduction of nitrobenzene to azoxybenzene. RSC Adv. 2014, 4, 18881–18884. [Google Scholar] [CrossRef]
- Mateti, S.; Mathesh, M.; Liu, Z.; Tao, T.; Ramireddy, T.; Glushenkov, A.M.; Yang, W.; Chen, Y.I. Mechanochemistry: A force in disguise and conditional effects towards chemical reactions. Chem. Comm. 2021, 57, 1080–1092. [Google Scholar] [CrossRef] [PubMed]
- Cuccu, F.; De Luca, L.; Delogu, F.; Colacino, E.; Solin, N.; Mocci, R.; Porcheddu, A. Mechanochemistry: New Tools to Navigate the Uncharted Territory of “Impossible” Reactions. ChemSusChem 2022, 15, e202200362. [Google Scholar] [CrossRef] [PubMed]
- Achar, T.K.; Bose, A.; Mal, P. Mechanochemical synthesis of small organic molecules. Beilstein J. Org. Chem. 2017, 13, 1907–1931. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Mal, P. Mechanochemistry of supramolecules. Beilstein J. Org. Chem. 2019, 15, 881–900. [Google Scholar] [CrossRef] [PubMed]
- Shearouse, W.C.; Korte, C.M.; Mack, J. A two-step ball milling method synthesizes and purifies α,β-unsaturated esters. Green Chem. 2011, 13, 598–601. [Google Scholar] [CrossRef]
- Do, J.-L.; Mottillo, C.; Tan, D.; Štrukil, V.; Friščić, T. Mechanochemical Ruthenium-Catalyzed Olefin Metathesis. J. Am. Chem. Soc. 2015, 137, 2476–2479. [Google Scholar] [CrossRef] [PubMed]
- Hermann, G.N.; Bolm, C. Mechanochemical Rhodium(III)-Catalyzed C–H Bond Amidation of Arenes with Dioxazolones under Solventless Conditions in a Ball Mill. ACS Catal. 2017, 7, 4592–4596. [Google Scholar] [CrossRef]
- Nakagawa, K.M.; Kawamura, S.; Minami, K. Reduction of Organic Compounds with Thiourea Dioxide. II Reduction of Aromatic Nitro Compounds and Synthesis of Hydrazo Compounds. Yakugaku Zasshi 1977, 97, 1253–1256. [Google Scholar] [CrossRef]
- Huang, S.-L.; Chen, T.-Y. Reduction of Organic Compounds with Thiourea Dioxide II. The Reduction of Organic Nitrogen Compounds. J. Chin. Chem. Soc. 1975, 22, 91–94. [Google Scholar] [CrossRef]
- Do, J.-L.; Friščić, T. Mechanochemistry: A Force of Synthesis. ACS Central Science 2017, 3, 13–19. [Google Scholar] [CrossRef]
- Friščić, T.; Childs, S.L.; Rizvi, S.A.A.; Jones, W. The role of solvent in mechanochemical and sonochemical cocrystal formation: a solubility-based approach for predicting cocrystallisation outcome. CrystEngComm 2009, 11, 418–426. [Google Scholar] [CrossRef]
- Tan, D.; García, F. Main group mechanochemistry: from curiosity to established protocols. Chemical Society Reviews 2019, 48, 2274–2292. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.L.; Cao, Q.; Browne, D.L. Mechanochemistry as an emerging tool for molecular synthesis: what can it offer? Chem. sci. 2018, 9, 3080–3094. [Google Scholar] [CrossRef]
- Howard, J.L.; Brand, M.C.; Browne, D.L. Switching chemoselectivity: using mechanochemistry to alter reaction kinetics. Ang. Chem. 2018, 130, 16336–16340. [Google Scholar] [CrossRef]
- Lewis, D.; Mama, J.; Hawkes, J. An Investigation into the Structure and Chemical Properties of Formamidine Sulfinic Acid. Appl. Spectrosc. 2014, 68, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.-C.; Lu, Y.-T.; Mai, Y.-W.; Zhang, C.; Xia, J.; Yao, P.-F.; Wang, H.-G.; Huang, S.-L.; Huang, Z.-S. Design, synthesis and biological evaluation of novel perimidine o-quinone derivatives as non-intercalative topoisomerase II catalytic inhibitors. Bioorg. Chem. 2019, 91, 103131. [Google Scholar] [CrossRef] [PubMed]
- Dymińska, L. Imidazopyridines as a source of biological activity and their pharmacological potentials—Infrared and Raman spectroscopic evidence of their content in pharmaceuticals and plant materials. Bioorg. Med. Chem. 2015, 23, 6087–6099. [Google Scholar] [CrossRef]
- Scribner, A.; Dennis, R.; Hong, J.; Lee, S.; McIntyre, D.; Perrey, D.; Feng, D.; Fisher, M.; Wyvratt, M.; Leavitt, P.; et al. Synthesis and biological activity of imidazopyridine anticoccidial agents: part I. Eur. J. Med. Chem. 2007, 42, 1334–1357. [Google Scholar] [CrossRef]
- Blazquez-Barbadillo, C.; González, J.F.; Porcheddu, A.; Virieux, D.; Menéndez, J.C.; Colacino, E. Benign synthesis of therapeutic agents: domino synthesis of unsymmetrical 1,4-diaryl-1,4-dihydropyridines in the ball-mill. Green Chem. Lett. Rev. 2022, 15, 881–892. [Google Scholar] [CrossRef]
- Makarov, S.V.; Sal’nikov, D.S.; Pogorelova, A.S. Acid-base properties and stability of sulfoxylic acid in aqueous solutions. Russ. J. Inorg. Chem. 2010, 55, 301–304. [Google Scholar] [CrossRef]
- Büeseken, J. Étude sur les Oxydes de Thiourée, I. Sur le dioxyde de thiourée. Recl. Trav. Chim. Pays-Bas 1936, 55, 1040–1043. [Google Scholar] [CrossRef]
- Sullivan, R.A.L.; Hargreaves, A. The crystal and molecular structure of thiourea dioxide. Acta Crystallogr. 1962, 15, 675–682. [Google Scholar] [CrossRef]
- Kis, Z.; Makarov, S.V.; Silaghi-Dumitrescu, R. Computational investigations on the electronic structure and reactivity of thiourea dioxide: sulfoxylate formation, tautomerism and dioxygen liberation. J. Sulfur Chem. 2010, 31, 27–39. [Google Scholar] [CrossRef]
- Grady, B.J.; Dittmer, D.C. Reaction of perfluoroaryl halides with reduced species of sulfur dioxide (HSO2−, SO22−, S2O42−). J. Fluor. Chem. 1990, 50, 151–172. [Google Scholar] [CrossRef]
- Krug, P. Thiourea Dioxide (Formamidinesulphinic Acid) A New Reducing Agent for Textile Printing. J. Soc. Dye. 1953, 69, 606–611. [Google Scholar] [CrossRef]
- Makarov, S.V.; Horváth, A.K.; Silaghi-Dumitrescu, R.; Gao, Q. Recent Developments in the Chemistry of Thiourea Oxides. Chem. Eur. J. 2014, 20, 14164–14176. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, H.F.; Mattern, J.A.; Fernelius, W.C. Sulfites and Pyrosulfites of the Alkali Metals. In Inorg. Synth.; Inorganic Syntheses; 1946; pp. 162-167.
- Miller, A.E.; Bischoff, J.J.; Pae, K. Chemistry of aminoiminomethanesulfinic and-sulfonic acids related to the toxicity of thioureas. Chem. Res. Toxicol. 1988, 1, 169–174. [Google Scholar] [CrossRef]
- Surasani, S.R.; Maity, S. Deciphering Intermediates and Additives Effect on the Reduction of Nitrobenzene by SmI2. ChemistrySelect 2017, 2, 598–603. [Google Scholar] [CrossRef]
- Böeseken, J. Etude sur les Oxydes de Thiouree. IV. Recl. Trav. Chim. Pays-Bas 1948, 67, 603–621. [Google Scholar] [CrossRef]
- Knopp, C. Zur verwendung von aminoiminomethanesulfinsaure ais antioxidans. Sci. Pharm 1983, 51, 283–290. [Google Scholar]
- Brown, D. A new synthesis of formamidine. J. Appl. Chem. 1952, 2, 202–203. [Google Scholar] [CrossRef]
- Dunitz, J.D. The structure of sodium dithionite and the nature of the dithionite ion. Acta Crystallogr. 1956, 9, 579–586. [Google Scholar] [CrossRef]
- Hartwig, U.; Pritzkow, H.; Rall, K.; Sundermeyer, W. Bis (trifluoromethyl) sulfene (CF3)2C SO2, Isolated as Adduct. Angew. Chem., Int. Ed. Engl. 1989, 28, 221–223. [Google Scholar] [CrossRef]
- Weber, H.P.; Craven, B.M. Structure and charge density of the 1: 1 complex of thiourea with parabanic acid at 298 K. Acta Crystallogr. B: Struct. Sci. 1987, 43, 202–209. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, N.L.; Pai, C.T. Charge density study of thiourea S, S-dioxide. Inorg. Chem. 1990, 29, 3256–3259. [Google Scholar] [CrossRef]
- Singh, P.K.; Silakari, O. Benzimidazole: Journey from Single Targeting to Multitargeting Molecule. In Key Heterocycle Cores for Designing Multitargeting Molecules; Elsevier: 2018; pp. 31-52.
- Wang, S.; Gao, Q.; Wang, J. Thermodynamic analysis of decomposition of thiourea and thiourea oxides. J. Phys. Chem. B 2005, 109, 17281–17289. [Google Scholar] [CrossRef]
Figure 1.
Benzimidazole features with common benzimidazoles pharmaceutical scaffolds.
Scheme 1.
A comparison between this work and previous methodologies.
Scheme 2.
TDO synthesis and structure, as reported by Barnett.
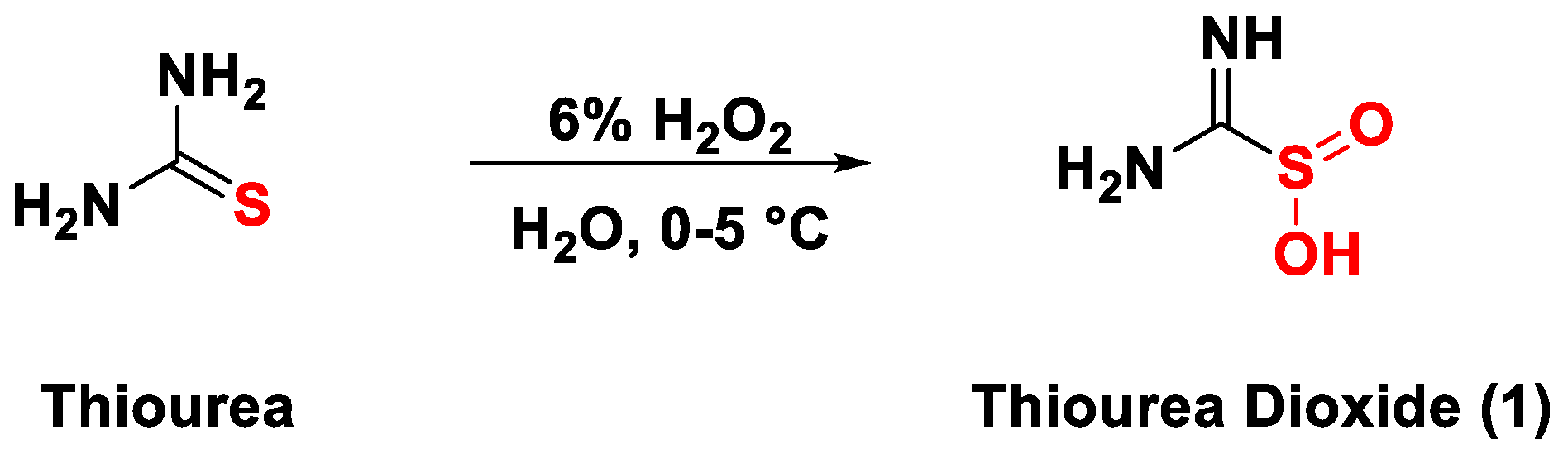
Scheme 3.
General Scheme of the reaction.
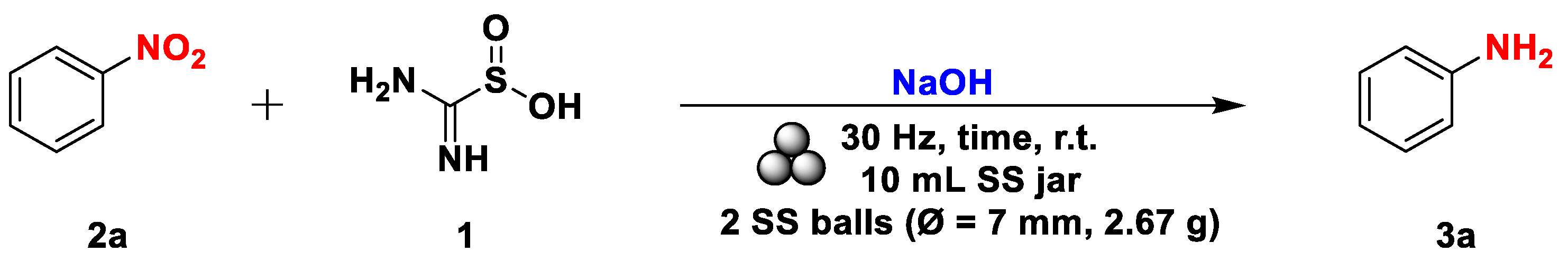
Scheme 4.
Mechanochemical synthesis of anilines from the corresponding nitrobenzenes. The yields were determined on isolated products. a 120 min reaction time. b 180 min of reaction time.
Scheme 4.
Mechanochemical synthesis of anilines from the corresponding nitrobenzenes. The yields were determined on isolated products. a 120 min reaction time. b 180 min of reaction time.
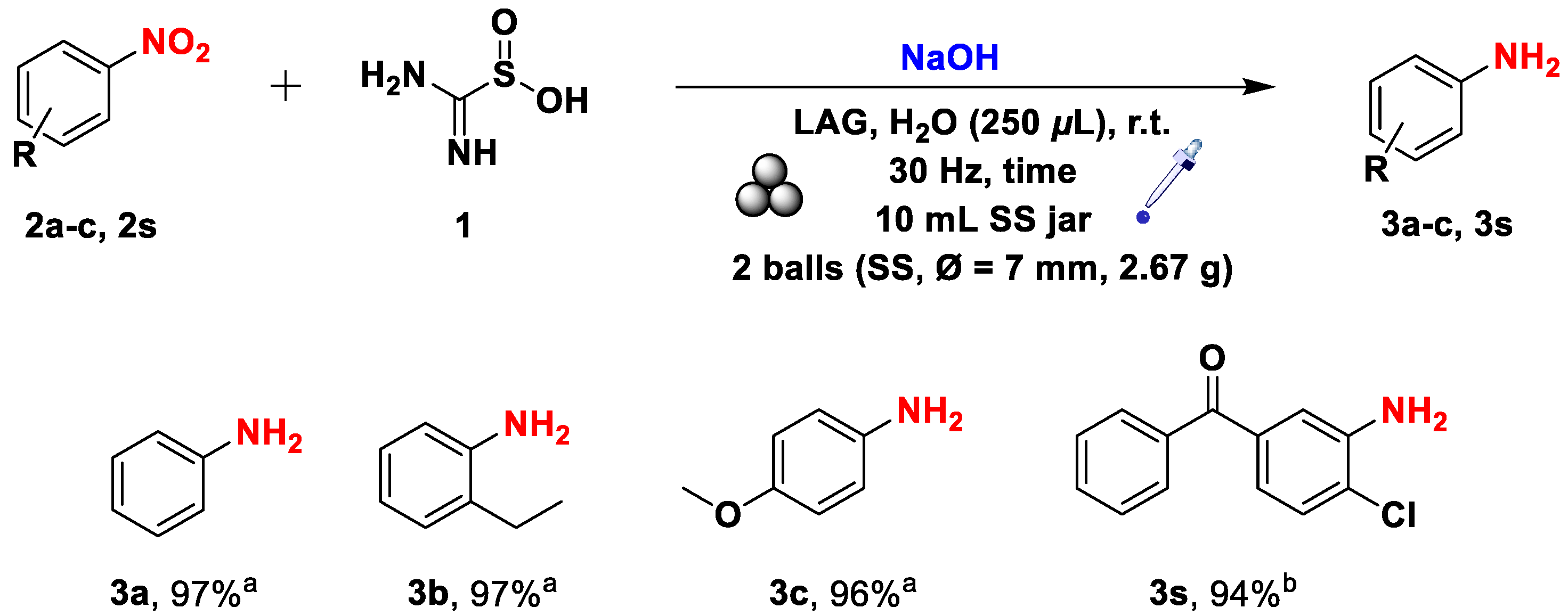
Scheme 5.
Mechanochemical synthesis of o-phenylenediamines from the corresponding 2-nitroanilines. The reported yields refer to pure isolated compounds. a 120 min reaction time. b 180 min of reaction time. c The product was purified through a silica pad.
Scheme 5.
Mechanochemical synthesis of o-phenylenediamines from the corresponding 2-nitroanilines. The reported yields refer to pure isolated compounds. a 120 min reaction time. b 180 min of reaction time. c The product was purified through a silica pad.
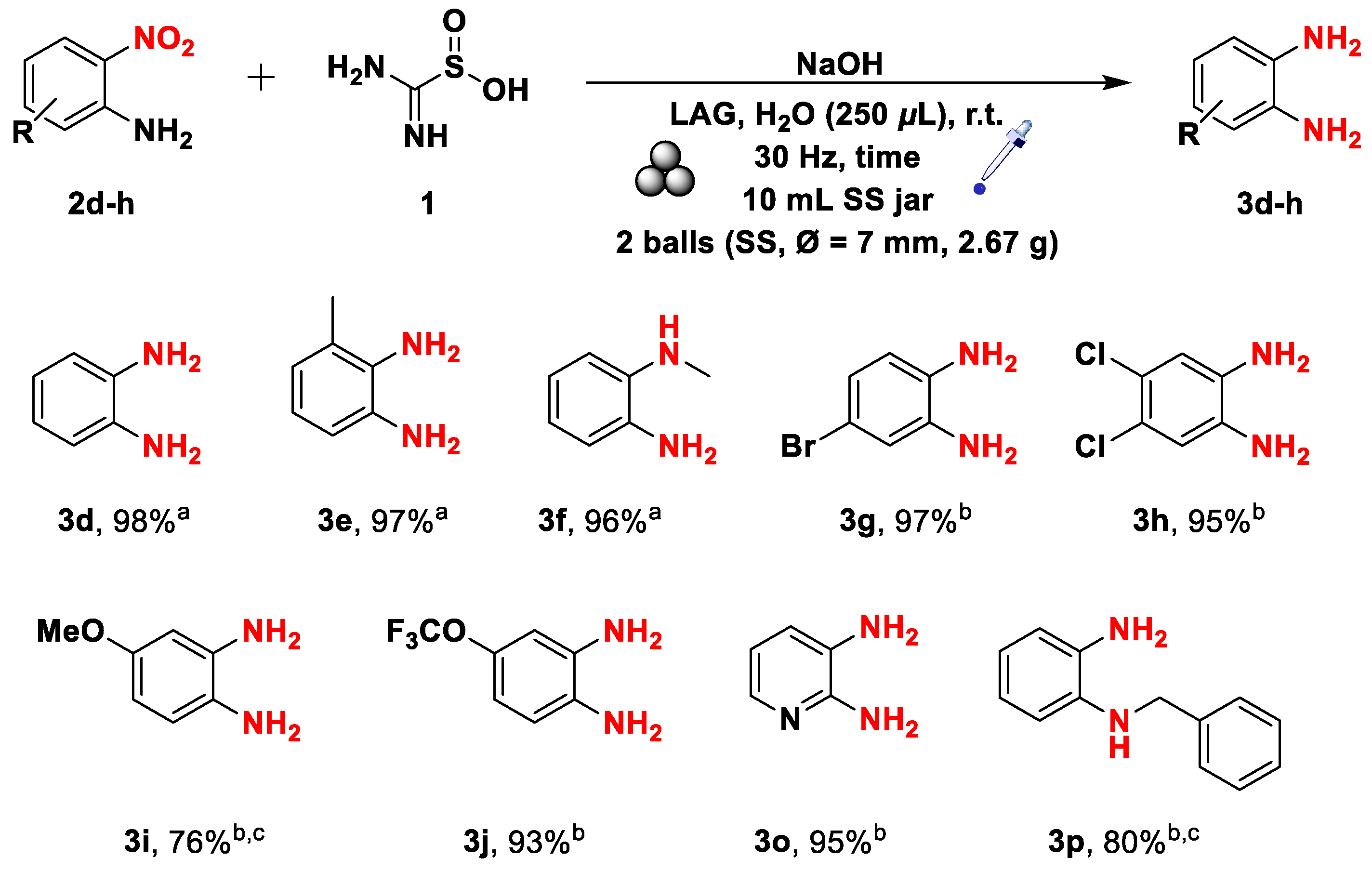
Scheme 6.
Mechanochemical synthesis of aza-heterocycles from the corresponding phenylenediamines. The yields were determined on isolated products.
Scheme 6.
Mechanochemical synthesis of aza-heterocycles from the corresponding phenylenediamines. The yields were determined on isolated products.
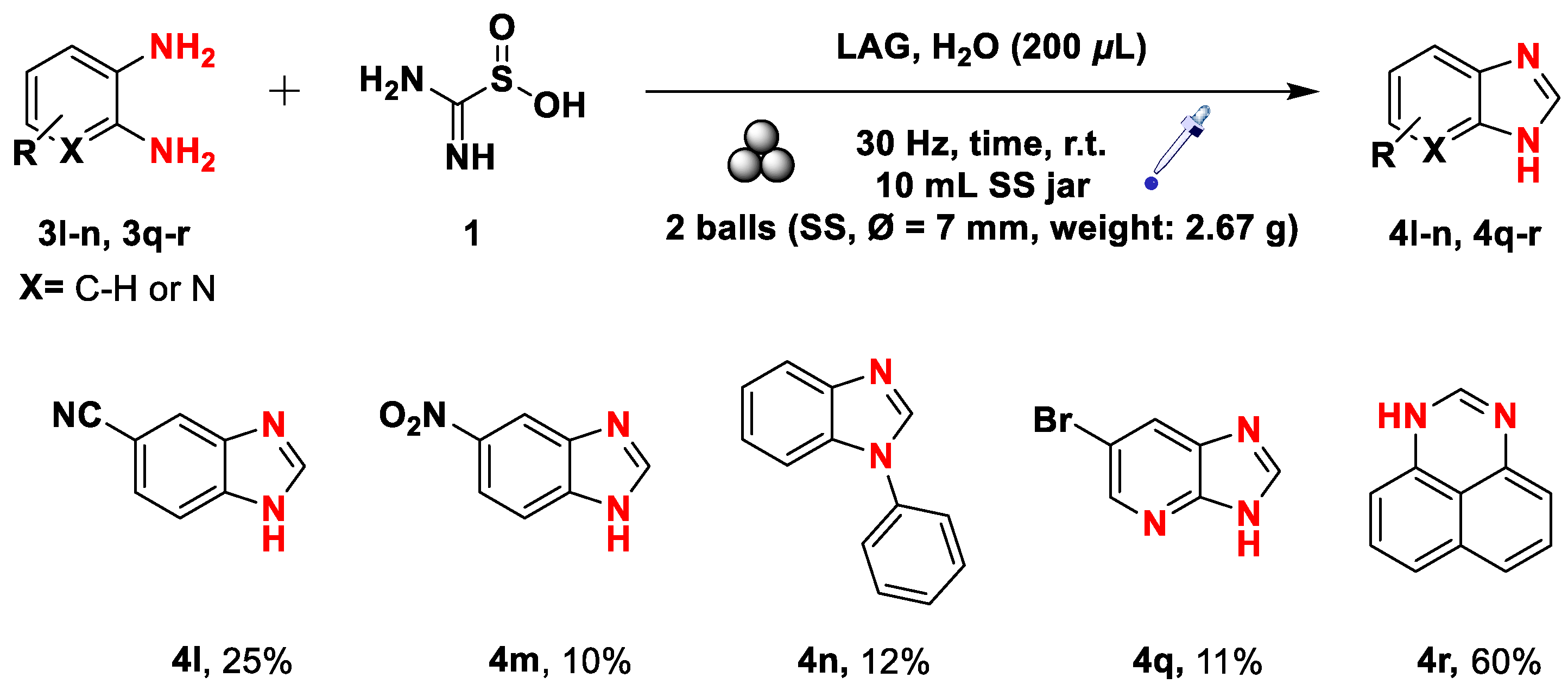
Scheme 7.
Mechanochemical synthesis of aza-heterocycles from the corresponding 2-nitroanilines through a double-step procedure. Yields refer to pure isolated compounds. a 2 hours of reaction time were required. b 3 hours of reaction time were needed.
Scheme 7.
Mechanochemical synthesis of aza-heterocycles from the corresponding 2-nitroanilines through a double-step procedure. Yields refer to pure isolated compounds. a 2 hours of reaction time were required. b 3 hours of reaction time were needed.
Scheme 8.
Mechanochemical reaction steps under LAG condition. Water (250 μL, η = 0.44) was added in one single shot at the start of the reaction.
Scheme 8.
Mechanochemical reaction steps under LAG condition. Water (250 μL, η = 0.44) was added in one single shot at the start of the reaction.
Scheme 9.
The assumed redox mechanism for the mechanochemical reduction of nitrobenzene 2a into aniline 3a.
Scheme 9.
The assumed redox mechanism for the mechanochemical reduction of nitrobenzene 2a into aniline 3a.
Scheme 10a.
The assumed reaction mechanism for the mechanochemical synthesis of 4a from 2a through the release of formamidine.
Scheme 10a.
The assumed reaction mechanism for the mechanochemical synthesis of 4a from 2a through the release of formamidine.
Scheme 10b.
The assumed reaction mechanism for the mechanochemical synthesis of 4a from 2a through the carbenoid pathway.
Scheme 10b.
The assumed reaction mechanism for the mechanochemical synthesis of 4a from 2a through the carbenoid pathway.
Table 1.
Optimization process for the reduction of 2a to 3a.
| Entry | TDO eq. | Base eq. | Reaction Time (h) | Additivesb | Yieldsa |
|---|---|---|---|---|---|
| 1 | 1 | 1 | 1 | / | 0% |
| 2 | 3 | 6 | 1 | / | 2% |
| 3 | 6 | 6 | 1 | / | 21% |
| 4 | 6 | 6 | 2 | / | 5% |
| 5 | 10 | 10 | 2 | / | 29% |
| 6c | 6 | 6 | 2 | / | 0% |
| 7 | 3 | 6 | 1.5 | Decane | 2% |
| 8 | 3 | 6 | 1.5 | Toluene | 4% |
| 9 | 3 | 6 | 1.5 | iPrOH | 3% |
| 10 | 3 | 6 | 1.5 | Acetone | 5% |
| 11d | 3 | 6 | 1.5 | MeOH | Complex Mixture |
| 12 | 3 | 6 | 1.5 | H2O | 89% |
| 13e | 3 | 6 | 1.5 | H2O | 0% |
| 14 | 3 | 6 | 2 | H2O | 97% |
| 15f | 3 | 6 | 1.5 | H2O | 20% |
| 16g | 3 | 6 | 1.5 | H2O | 1% |
| 17h | 3 | 3 | 2 | H2O | <5% |
All the reactions were carried out with the same experimental parameters unless otherwise specified: nitrobenzene (1.0 mmol), compound 1 (1.0-10.0 mmol), and NaOH (1.0-10.0 mmol) in a SS jar (10.0 mL) equipped with two balls (SS, Ø = 7.0 mm, 2.67 g) at a frequency of 30 Hz. a The yields were calculated by GC-MS analysis. b The additive quantity was 250 μL. c The reaction was run at 70 °C; the only product obtained was the corresponding azobenzene. d The desired product was obtained only in traces. e The amount of water was reduced to 125 μL. The main product found was azoxybenzene; the remaining peaks were attributed to nitrobenzene f Na2CO3 was used instead of NaOH. g NaHCO3 was used instead of NaOH. h The SM presence was not detected, and the 1H NMR spectra presented signals which cannot be attributed to specific compounds.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



