
Submitted:
04 August 2021
Posted:
06 August 2021
You are already at the latest version
Abstract
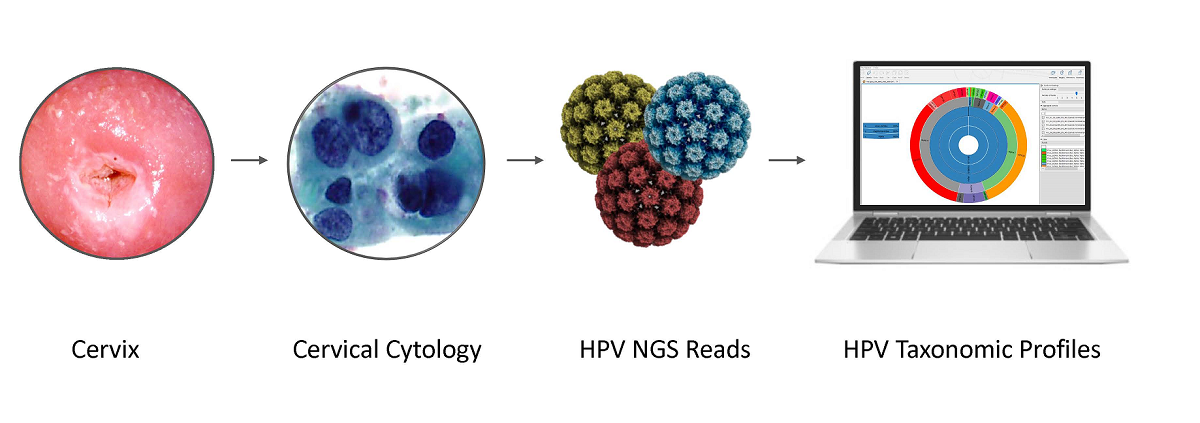
Next-generation sequencing (NGS) has actualized human papillomavirus (HPV) virome profiling for in-depth investigation of viral evolution and pathogenesis. However, viral computational analysis remains a bottleneck due to semantic discrepancies between computational tools and curated reference genomes. To address this, we developed and tested automated workflows for HPV taxonomic profiling and visualization using a customized Papillomavirus database in CLC Microbial Genomics Module. HPV genomes from Papilloma Virus Episteme were customized and incorporated into CLC “ready-to-use” workflows for stepwise data processing to include: 1) Taxonomic Analysis, 2) Estimate Alpha/Beta Diversities, and 3) Map Reads to Reference. Low-grade (n = 95) and high-grade (n = 60) Pap smears were tested with ensuing collective runtimes: Taxonomic Analysis (36 min); Alpha/Beta Diversities (5 sec); Map Reads (45 min). Tabular output conversion to visualizations entailed 1-2 keystrokes. Biodiversity analysis between low- (LSIL) and high-grade squamous intraepithelial lesions (HSIL) revealed loss of species richness and gain of dominance by HPV-16 in HSIL. Integrating clinically relevant, taxonomized HPV reference genomes within automated workflows proved to be an ultra-fast method of virome profiling. The entire process named “HPV DeepSeq” provides a simple, accurate and practical means of NGS data analysis for a broad range of applications in viral research.
Keywords:
Bioinformatics
; Cervical cancer
; Deep Sequencing
; Human papillomavirus
; HPV genotyping
; Metagenome
; Next generation sequencing
; Taxonomic classification
; Virome
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



