
Submitted:
02 July 2020
Posted:
03 July 2020
You are already at the latest version
Abstract
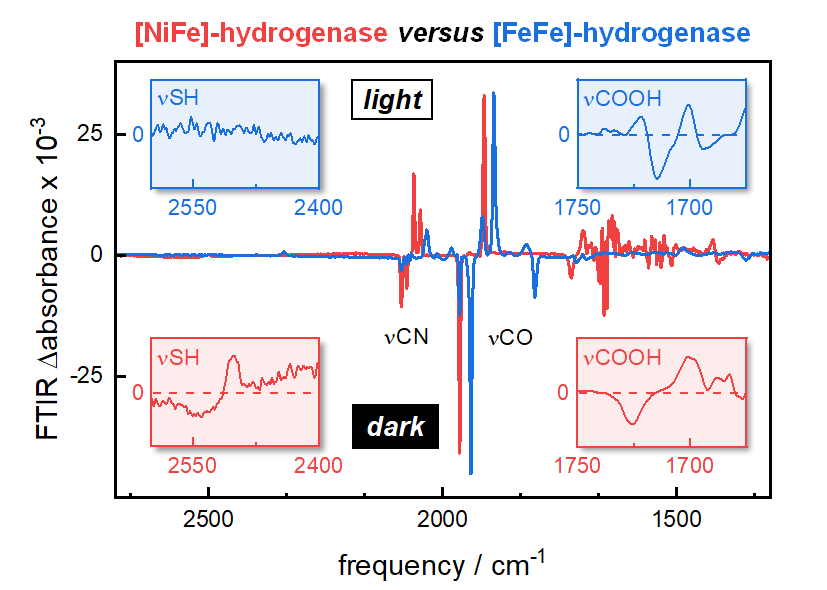
Hydrogenases are iron-sulfur enzymes that catalyze proton reduction and H2 oxidation with outstanding efficiency. They are considered blueprints for the design of transition metal complexes, e.g. as heterogenous catalysts in the context of H2 production from water. Moreover, hydrogenases are biological model systems for metal hydride chemistry and proton-coupled electron transfer. Depending on the composition of the active site cofactor, [NiFe]-hydrogenases are distinguished from [FeFe]-hydrogenases. The former binds a hetero bimetallic nickel/iron site, embedded in the protein by four cysteine ligands. The later, by contrast, carries a homo bimetallic iron/iron site attached to the protein by only a single cysteine. Carbon monoxide and cyanide ligands (CO/CN) at the active site facilitated detailed investigations of hydrogenase catalysis by infrared spectroscopy, owing to strong signals and redox-dependent frequency shifts. However, the details of proton transfer have not been addressed experimentally.We found that specific redox state transitions in [NiFe]- and [FeFe]-hydrogenase can be triggered by visible light to record extremely sensitive ‘light-minus-dark’ infrared difference spectra monitoring key amino acid residues as shown in the ToC figure. As these transitions are coupled to protonation changes, our data allowed investigating dynamic hydrogen-bonding changes that go well beyond the resolution of protein crystallography. In [NiFe]-hydrogenase, photolysis of the bridging hydride ligand in the ‘Ni-C’ state was followed by rapid accumulation of the ‘Ni-SIa’ state and/or ‘Ni-L’ state. Infrared difference spectra in various isotopic media clearly indicated the formation of a protonated cysteine residue as well as hydrogen-bonding changes involving the COOH group of a glutamic acid residue and a ‘dangling water’ molecule. These findings are in excellent agreement with crystallographic analyses of [NiFe]-hydrogenase in the Ni-R state and allowed devising a molecular precise model of catalytic proton transfer. In [FeFe]-hydrogenase, an external redox dye was used to accumulate the ‘Hred’ state over the oxidized resting state ‘Hox’. Infrared difference spectra of wild-type enzyme and numerous amino acid variants indicated hydrogen-bonding changes involving the COOH groups of two glutamic acid residues. Moreover, we noted the deprotonation of an arginine residue. Crystallographic analyses of [FeFe]-hydrogenase in the Hox state failed to explain the rapid proton transfer due to a ‘breach’ in the succession of residues. To this end, our findings facilitated a molecular precise model of ‘discontinued’ proton transfer.The comparison of catalytic proton transfer in bimetallic hydrogenases emphasizes the role of the outer coordination sphere. We suggest that the stable protonation of a nickel-ligating cysteine in [NiFe]-hydrogenase has a crucial influence on the preferred direction of proton flow and catalysis (i.e., H2 oxidation). On the contrary, proton transfer in [FeFe]-hydrogenase involves an adjacent cysteine as a relay group that promotes both proton release and proton uptake. We presume that this causes the notable bidirectionality of [FeFe]-hydrogenase. These observations must guide the design of biomimetic compounds for the production or consumption of H2.
Keywords:
hydrogen
; biocatalysis
; hydride chemistry
; photochemistry
; infrared spectroscopy
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



