Submitted:
20 August 2019
Posted:
21 August 2019
You are already at the latest version
Abstract
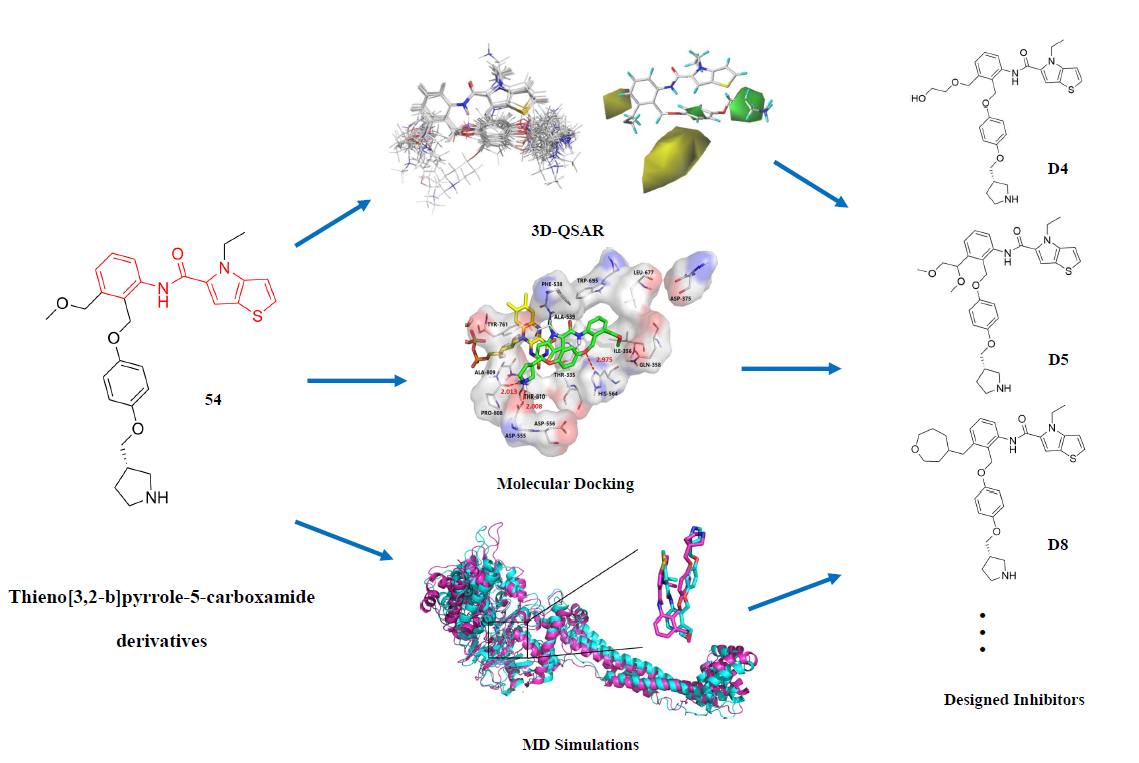
Histone Lysine Specific Demethylase 1 (LSD1) is overexpressed in many cancers and become a new target for anticancer drugs. In recent years, the small molecule inhibitors with various structures targeting LSD1 have been reported. Here we report the binding interaction modes of a series of thieno[3,2-b]pyrrole-5-carboxamides LSD1 inhibitors using molecular docking, three dimensional quantitative structure-activity relationship (3D-QSAR). Comparative molecular field analysis (CoMFA q2=0.783, r2=0.944, r2pred=0.851) and Comparative molecular similarity indices analysis (CoMSIA q2=0.728, r2=0.982, r2pred=0.814) were used to establish 3D-QSAR models, which had good verification and prediction capabilities. Based on the contour maps and the information of molecular docking, 8 novel small molecules were designed in silico, among which compounds D4, D5 and D8 with high predictive activity were subjected to further molecular dynamics simulations (MD), and their possible binding modes were explored. It was found that Asn535 plays a crucial role in stabilizing the inhibitors. Furthermore, the ADME and bioavailability prediction for D4, D5 and D8 were carried out. The results would provide valuable guidance for designing new reversible LSD1 inhibitors in the future.
Keywords:
LSD1
; molecular inhibitors
; thieno[3
; 2-b]pyrrole-5-carboxamide derivatives
; Molecular docking
; 3D-QSAR
; Molecular dynamics simulations
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



