
Submitted:
15 November 2018
Posted:
19 November 2018
You are already at the latest version
Abstract
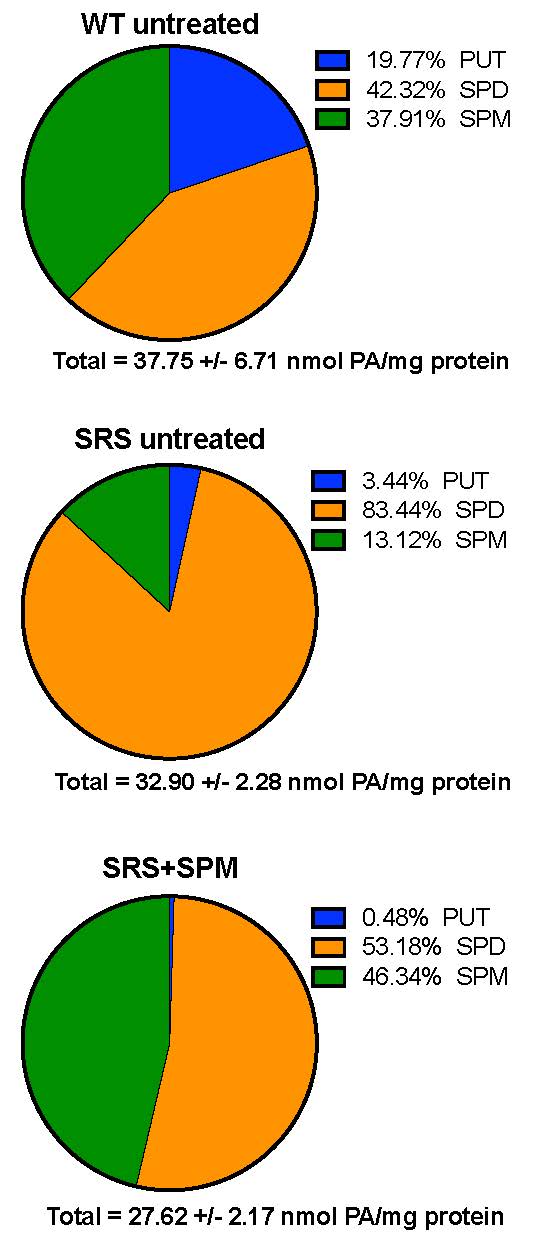
Loss-of-function mutations of the spermine synthase gene (SMS) result in Snyder-Robinson Syndrome (SRS), a recessive X-linked syndrome characterized by intellectual disability, osteoporosis, hypotonia, speech abnormalities, kyphoscoliosis, and seizures. As SMS catalyzes the biosynthesis of the polyamine spermine from its precursor spermidine, SMS deficiency causes a lack of spermine with an accumulation of spermidine. As polyamines, spermine and spermidine play essential cellular roles that require tight homeostatic control to ensure normal cell growth, differentiation, and survival. Using patient-derived lymphoblast cell lines, we sought to comprehensively investigate the effects of SMS deficiency on polyamine homeostatic mechanisms including polyamine biosynthetic and catabolic enzymes, derivatives of the natural polyamines, and polyamine transport activity. In addition to decreased spermine and increased spermidine in SRS cells, ornithine decarboxylase activity and its product putrescine were significantly decreased. Treatment of SRS cells with exogenous spermine revealed that polyamine transport was active, as the cells accumulated spermine, decreased their spermidine level, and established a spermidine-to-spermine ratio within the range of wild type cells. SRS cells also demonstrated elevated levels of tissue transglutaminase, a change associated with certain neurodegenerative diseases. These studies form a basis for further investigations into the leading biochemical changes and properties of SMS-mutant cells that potentially represent therapeutic targets for the treatment of Snyder-Robinson Syndrome.
Keywords:
snyder-robinson syndrome
; spermine synthase
; X-linked intellectual disability
; polyamine transport
; spermidine
; spermine
; transglutaminase
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



