
Submitted:
25 June 2018
Posted:
26 June 2018
You are already at the latest version
Abstract
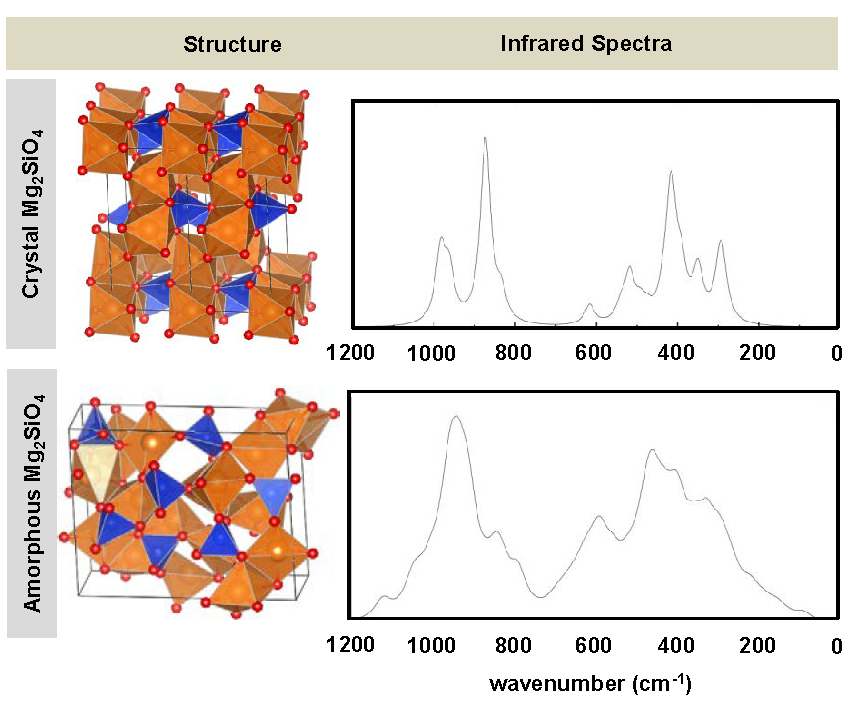
Silicates are among the most abundant and important inorganic materials, not only in the Earth’s crust but also in the interstellar medium in the form of micro-/nano-particles or embedded in the matrices of comets, meteorites and other asteroidal bodies. Although the crystalline phases of silicates are indeed present in Nature, amorphous forms are also highly abundant. Here we report a theoretical investigation of the structural, dielectric and vibrational properties of the amorphous bulk for forsterite (Mg2SiO4) as a silicate test case by a combined approach of classical molecular dynamics (MD) simulations for structure evolution, and periodic quantum mechanical DFT calculations for electronic structure analysis. Using classical MD based on an empirical partial charge rigid ionic model within a melt-quenching scheme at different temperatures performed with the GULP 4.0 code amorphous bulk structures for Mg2SiO4 were generated using as initial guess the crystalline phase. This has been done for bulks with three different unit cell sizes adopting a super-cell approach; i.e., 1×1×2, 2×1×2 and 2×2×2. The radial distribution functions indicated a good degree of amorphisation of the structures. Periodic B3LYP-geometry optimizations performed with the CRYSTAL14 code on the generated amorphous systems were used to analyze their structure, to calculate their high frequency dielectric constants (ε∞), and to simulate the IR, Raman and reflectance spectra, which were compared with the experimental and theoretical crystalline Mg2SiO4. The most significant changes of the physico-chemical properties of the amorphous systems compared to the crystalline ones are presented and discussed (e.g., larger deviations in the bond distances and angles, broadening of the IR bands, etc.), which are consistent with their disordered nature. It is also shown that by increasing the unit cell size the bulk structures present larger degree of amorphisation.
Keywords:
DFT
; periodic simulations
; amorphous minerals
; physico-chemical properties
; super-cell
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



