Submitted:
23 August 2016
Posted:
23 August 2016
You are already at the latest version
Abstract
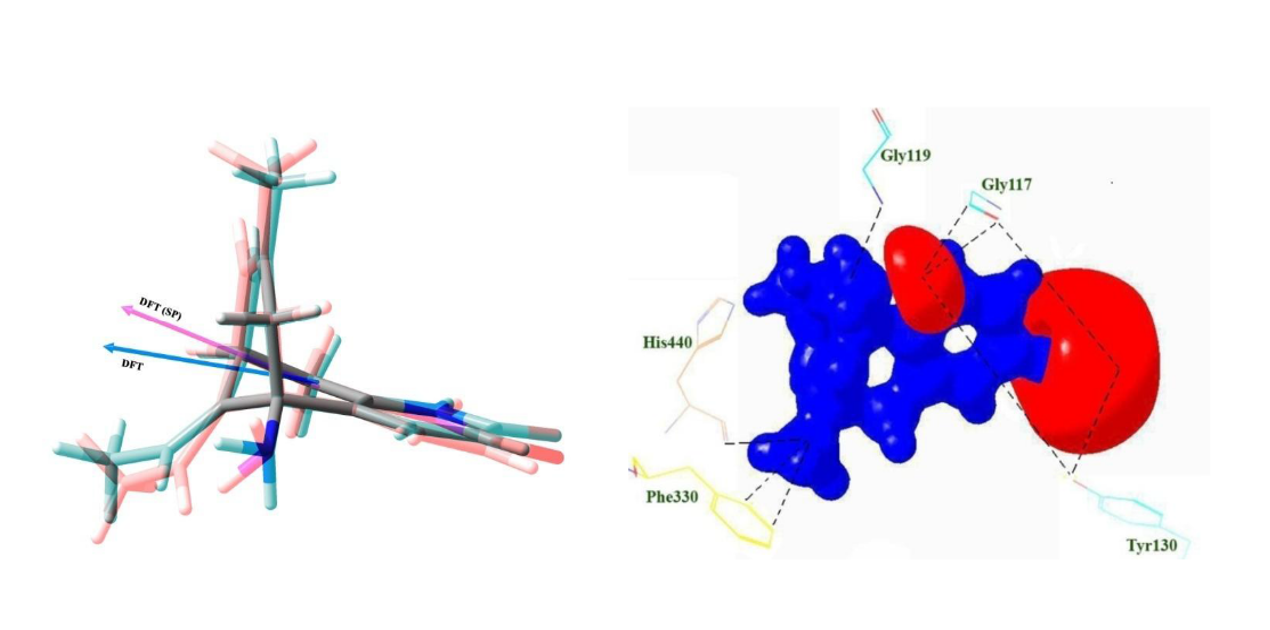
Huperzine A is an herbal reversible inhibitor of Acetylcholinesterase (AChE). A molecular docking analysis on Huperzine A molecule has been carried out to understand its structure, conformational flexibility, intermolecular interaction and the binding affinity in the active site of AChE enzyme. Further, the charge density distribution of huperzine A molecule (lifted from the active site of AChE) was determined from the high level quantum chemical calculations coupled with charge density analysis. The binding affinity of Huperzine A towards AChE was calculated from the molecular docking; the lowest docked energy is -8.46 kcal/mol. In the active site, huperzine A molecule interacts with acyl binding pocket-Phe330 of AChE, that is, the bicyclo ring group of huperzine A forms an intermolecular interaction with the oxygen atom of main chain of the amino acid residue Phe330 at the distances 3.02 and 3.25 Å respectively. On the other hand, a gas phase study on huperzine A molecule also performed using HF and DFT (B3LYP) methods with the basis set 6-311G**. The molecular structure, conformation, and the charge density distribution of huperzine A molecule in the gas phase have determined using quantum chemical calculations and the charge density analysis. The comparative studies between the gas phase and the active site forms of huperzine A molecule, explicitly reveals the degree of conformational modification and the charge density redistribution of huperzine A when present in the active site. The dipole moment of the molecule in the active site is 6.85 D, which is slightly higher than its gas phase value (5.91 D). The electrostatic potential (ESP) surface of active site molecule clearly shows the strong electronegative and positive ESP regions of the molecule, which are the expected strong reactive locations of the molecule.
Keywords:
huperzine A-AChE
; molecular docking
; intermolecular interaction
; quantum chemical calculation
; charge density distribution
; atomic charges
; dipole moment
; electrostatic potential
; toxicity analysis
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



