Submitted:
24 June 2026
Posted:
26 June 2026
You are already at the latest version
Abstract
Aortitis—inflammation of the aortic wall—presents at the interface of vasculitis, infection, structural aortic disease, and cardiovascular risk. It may occur in giant cell arteritis (GCA), Takayasu arteritis, IgG4-related disease, drug-induced injury, infection, or as an isolated finding after aortic surgery. Modern imaging detects aortic inflammation more frequently, but the main challenge is classification rather than detection: determining whether disease is infectious or immune-mediated, active or dominated by fixed structural damage, systemic or isolated, and whether the dominant threat is aneurysm, dissection, undertreated infection, or avoidable immunosuppression. This review considers aortitis as a high-risk vascular syndrome requiring aetiology-first classification rather than descriptive labelling. Before escalating immunosuppression, infection must be actively excluded and inflammatory activity distinguished from fixed vascular damage. Treatment should be individualised according to phenotype, age, vascular territory, comorbidity, and toxicity risk, with surveillance continuing even after symptoms and inflammatory markers improve. Optimal care depends on multidisciplinary assessment integrating rheumatology, infectious diseases, vascular surgery, radiology, and cardiology expertise. Progress will require standardised imaging definitions, registries linking inflammatory control with structural vascular outcomes, and validation of artificial intelligence (AI) tools before clinical adoption.
Keywords:
aortitis
; large‐vessel vasculitis
; giant cell arteritis
; Takayasu arteritis
; infectious aortitis
; IgG4‐related disease
; multimodality imaging
; multidisciplinary care
; personalised medicine
; aortic aneurysm
1. Introduction
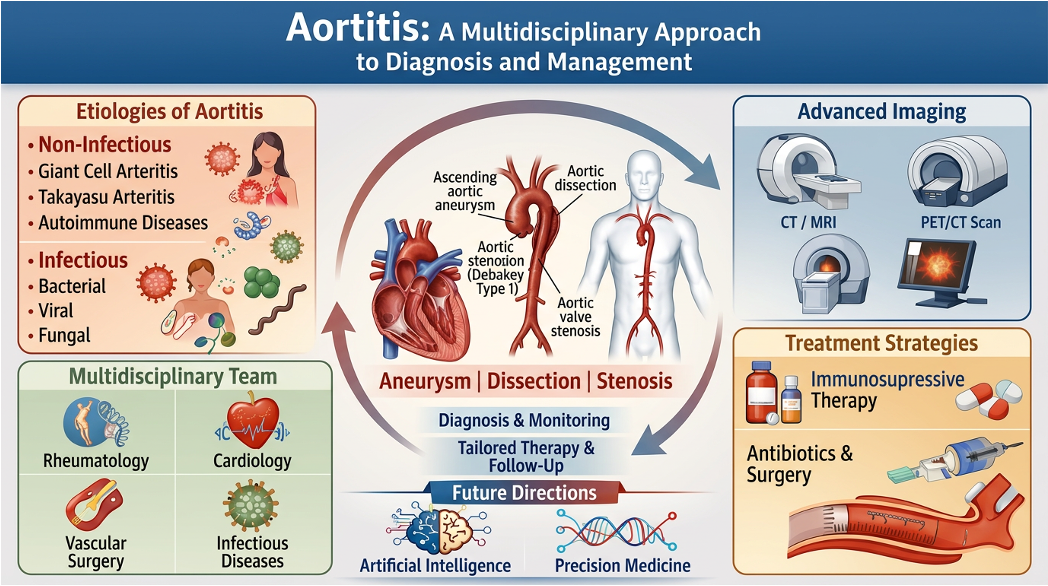
Aortitis is often introduced as inflammation of the aortic wall—a simple anatomical term. In practice, however, the label is rarely simple, as the same radiological or histological finding may reflect giant cell arteritis (GCA), Takayasu arteritis, IgG4-related disease, immune-checkpoint inhibitor (ICI) toxicity, granulocyte-colony stimulating factor (G-CSF) exposure, or a localised inflammatory lesion identified only after thoracic aortic surgery [1,2,3,4,5]. Rather than a final diagnosis, aortitis is better understood as a warning signal requiring clarification of cause, inflammatory activity, anatomical distribution, structural risk, and treatment implications.
The stakes are high because the aorta has little tolerance for diagnostic delay. Ongoing inflammation may cause mural thickening, fibrosis, elastic fibre fragmentation, and matrix degradation. These changes may progress to aneurysm formation, dissection, or rupture, while branch-vessel stenosis and aortic regurgitation develop alongside systemic complications including coronary or cerebral ischaemia, renovascular hypertension, and limb claudication that necessitate repeated vascular procedures [1,2,6,7,8]. However, early presentation is often non-specific, as fever, malaise, weight loss, fatigue, chest or abdominal discomfort, and raised inflammatory markers may be attributed to infection, malignancy, polymyalgia rheumatica, degenerative aortic disease, or postoperative inflammation. By the time pulse deficits, bruits, blood pressure asymmetry, organ ischaemia, aneurysm, or dissection become evident, irreversible vascular damage may already have occurred.
A major difficulty is that aortitis rarely fits within a single specialty. Rheumatologists usually approach it through large-vessel vasculitis, whereas infectious diseases physicians focus on microbial seeding, source control, and antimicrobial treatment. Radiologists characterise wall thickening, metabolic uptake, and structural complications, and surgeons may first encounter the disease as unexpected inflammation during aortic repair. Cardiologists manage blood pressure control, valve complications, and long-term cardiovascular risk. Each perspective brings essential expertise, although none alone provides a complete picture of the disease.
This review frames aortitis as a high-risk vascular syndrome requiring cause-first classification, distinction between active inflammation and established damage, individualised treatment, and long-term surveillance. Management should be guided not only by diagnostic label but by vascular anatomy, infection risk, inflammatory activity, structural vulnerability, treatment toxicity, comorbidity, and patient priorities. A 75-year-old patient, for example, with GCA-related aortitis, diabetes, osteoporosis, and expanding aneurysm requires different decisions than a 30-year-old patient with Takayasu arteritis, critical stenosis, and preserved renal function. Both have “aortitis”, but they do not have the same syndrome.
2. Review Scope and Approach
The aim of this review is to synthesise clinically important concepts across rheumatology, cardiovascular medicine, infectious diseases, imaging, pathology and aortic surgery. The emphasis is on decision points that affect patient care that include distinguishing infection from immune-mediated disease, interpreting imaging in context, deciding when immunosuppression is justified, recognising structural risk and designing follow-up pathways. Reviews, major guidelines, observational cohorts, clinical trials and contemporary reports of emerging phenotypes were prioritised where they informed these practical questions.
The evidence base is uneven, with practical consequences for appropriate care. Randomised trials, for example, exist for glucocorticoid tapering and tocilizumab in GCA but are limited for isolated aortitis, Takayasu arteritis, infectious aortitis, and surgically discovered disease. Several recommendations in this field rely on cohort data, expert consensus, and extrapolation from broader large-vessel vasculitis or aortic disease literature [7,9,10,11]. This limitation is acknowledged throughout the review.
3. Aortitis Is a Syndrome, not a Single Disease
While distinguishing infectious from non-infectious disease is essential, this distinction alone does not guide treatment. Infectious aortitis, though uncommon, is immediately dangerous and may arise through haematogenous spread, septic embolisation, contiguous infection, infection of atherosclerotic plaques or aneurysms, or seeding of a previously injured aortic segment [7,12,13]. The pathogen spectrum is also broad, encompassing Salmonella species, Staphylococcus aureus, Streptococcus species, and other Gram-negative organisms, alongside infections caused by syphilis, tuberculosis, Q fever, and fungal agents [7,12,13]. Regardless of the organism, management depends on prompt recognition, microbiological sampling, targeted antimicrobial therapy, and early vascular surgical evaluation in the presence of aneurysm, pseudoaneurysm, dissection, rupture, or uncontrolled sepsis [7,12,13].
Reports of less common pathogens reinforce the same principle, showing that infectious aortitis requires prompt recognition and source control while its presentation may vary considerably. Escherichia coli infection may manifest with periaortic fluid, pseudoaneurysm formation, perforation, or delayed abscess development. Burkholderia pseudomallei endograft infections underscore how geography, environmental exposure, antimicrobial resistance patterns, and source control intersect in complex ways. Listeria monocytogenes periaortitis can remain culture-negative, only becoming apparent through tissue sampling or molecular diagnostics, while hypervirulent Klebsiella pneumoniae strains can cause metastatic infection with mycotic aneurysm formation [14,15,16,17]. Clinically, these examples show that infectious aortitis behaves differently and should not be treated simply as a vasculitis mimic. It progresses differently, requires different decisions, and carries different consequences if missed.
When infection has been excluded, non-infectious aortitis is usually considered within the framework of large-vessel vasculitis. GCA usually affects patients over 50 years of age and may involve cranial vessels, the aorta and its major branches. Takayasu arteritis typically presents earlier in life, has a strong female predominance and frequently involves the aortic arch and its branches, with stenosis, occlusion, aneurysm or renovascular hypertension [18,19,20,21,22,23,24]. These patterns are helpful, but they should not be applied rigidly. GCA can present predominantly with extracranial large-vessel disease and few cranial symptoms and Takayasu arteritis may be diagnosed only after vascular damage has developed. Histological aortitis may also be found in resected aortic tissue without an obvious systemic syndrome [3,25,26].
Beyond the major large-vessel vasculitides, other immune-mediated and inflammatory aetiologies complicate the differential diagnosis. IgG4-related aortitis and periaortitis may cause adventitial or periaortic fibroinflammatory thickening, aneurysm, retroperitoneal fibrosis or multi-organ disease [27,28]. Behcet disease, systemic lupus erythematosus, sarcoidosis, spondyloarthritis and other systemic inflammatory disorders may involve the aorta. Drug-associated aortitis with G-CSF and ICI is recognised and challenges the older binary distinction between idiopathic autoimmunity and infection [29,30,31].
Among these entities, IgG4-related aortitis requires careful diagnostic assessment. Serum IgG4 alone is neither sensitive nor specific enough to define the diagnosis, and IgG4-positive plasma cells in tissue must be interpreted in clinicopathological context. A defensible diagnosis requires coherent imaging, evidence of organ involvement where present, supportive histology such as IgG4-rich lymphoplasmacytic inflammation, storiform fibrosis or obliterative phlebitis when available, exclusion of infection and malignancy, and longitudinal response to treatment [27,28]. This matters because relapse risk, surveillance and steroid-sparing strategies may differ from those used in GCA or Takayasu arteritis.
The entity of clinically isolated aortitis illustrates the difficulty of rigid disease classification. Some patients have histological aortitis unexpectedly identified during surgery for thoracic aneurysm or dissection [3,25,26]. Some remain without systemic disease, whereas others later develop GCA, polymyalgia rheumatica, another rheumatic diagnosis, new vascular lesions or further need for intervention [3,25,26,32]. These findings are not incidental; they suggest that localised aortic inflammation at presentation may still have prognostic consequences. At the time of surgery, the patient may not fit a conventional vasculitis category and may still need full vascular mapping and long-term follow-up.
The practical question is therefore not only “Is there aortitis?” but clinicians also need to establish the most likely cause, whether infection has been reasonably excluded, whether the process is active or not, which aortic segments and branch vessels are involved, whether aneurysm, stenosis, dissection, valve disease or rupture risk is present, whether systemic disease is evident, what treatment risk is acceptable, and what surveillance is required after symptoms improve. This sequence makes the label clinically useful.
The major clinical categories of aortitis are summarised in Figure 1.
4. Clinical Presentation: Why Diagnosis Is Often Delayed
Aortitis is often diagnosed late because it rarely presents with a single pathognomonic symptom. Early in the disease course, patients may experience non-specific constitutional symptoms including fever, night sweats, malaise, fatigue, anorexia, and weight loss, alongside pain affecting the chest, back, abdomen, or joints. Laboratory evaluation typically reveals raised inflammatory markers, but these findings remain non-specific and may equally suggest infection, malignancy, systemic autoimmune disease, degenerative vascular pathology, or postoperative inflammation [1,2]. In older patients, headache, scalp tenderness, jaw claudication, visual symptoms or polymyalgia rheumatica may point toward GCA, but large-vessel disease may occur with minimal cranial involvement [18,19,23,24]. In younger patients, Takayasu arteritis may begin with constitutional symptoms before limb claudication, reduced pulses, bruits, blood-pressure discrepancies, hypertension, cerebrovascular symptoms or renal artery disease become apparent [20,21,22].
Clinical examination remains underused, although simple bedside findings, including asymmetrical or absent pulses, inter-limb blood pressure discrepancies, vascular bruits over major arteries, signs of aortic regurgitation, limb ischaemia, or unexplained hypertension in the context of systemic inflammation, can substantially narrow the differential diagnosis before any imaging is performed. Aortitis should enter the differential when an aneurysm appears at an atypical age, expands unexpectedly rapidly, displays periaortic inflammatory changes, develops after recurrent vascular procedures, accompanies otherwise unexplained systemic inflammation, or demonstrates wall thickening out of proportion to atherosclerotic burden.
The absence of these signs does not exclude aortitis, as aortic inflammation may remain silent until aneurysm, dissection, rupture, stenosis or embolisation occurs. Wider access to computed tomography angiography (CTA), magnetic resonance angiography (MRA), vascular ultrasound scan (USS) and FDG-PET has improved detection of subclinical large-vessel disease, but it has also created a new problem as clinicians now see more abnormal aortas than they can confidently classify [10,33,34,35].
The consequences of delayed recognition are tangible and serious. A patient with systemic symptoms may receive empirical treatment and later present with an aneurysm. Another patient with infectious aortitis may receive corticosteroids for presumed vasculitis before blood cultures, tissue sampling or source control have been pursued. A third may undergo aortic surgery, receive a pathology report showing aortitis and then leave hospital without a structured follow-up plan. These failures are clinically likely consequences of fragmented pathways.
5. Pathobiology: Active Inflammation, Infection and Structural Legacy
Distinguishing active inflammation from the structural legacy of prior injury is clinically critical. Active inflammation reflects ongoing immune or microbial injury that may respond to immunosuppression, antimicrobial therapy, source control or treatment of an underlying disease. Structural legacy refers to fibrosis, medial degeneration, elastic lamina disruption, calcification, aneurysm, stenosis, occlusion, dissection or postoperative change that can persist after inflammation has become clinically quiet. The distinction is imperfect in practice, but it directly affects whether clinicians escalate medication, operate or observe.
In large-vessel vasculitis, immune activation appears to originate in the adventitia and vasa vasorum, where dendritic cells activate and recruit T lymphocytes and macrophages, triggering a cascade of cytokine amplification, granulomatous inflammation, vascular smooth muscle dysfunction, neoangiogenesis, and progressive matrix degradation [36,37,38,39]. Key pathways implicated in this process include interleukin-6 (IL-6), IL-17, interferon-gamma, granulocyte-macrophage colony-stimulating factor, and matrix metalloproteinases [36,37,38,39,40]. Histological examination typically reveals infiltration by lymphocytes and macrophages, with multinucleated giant cells forming granulomas with scarring and disrupted elastic laminae [25,26,36]. Chronic disease may progress to fibrosis and loss of elasticity even when systemic inflammation seems controlled.
GCA and Takayasu arteritis share several immunopathological themes, but their clinical trajectories differ. Takayasu arteritis often causes wall thickening, stenosis, occlusion, aneurysm and vascular insufficiency in younger patients [20,21,22]. GCA may present with cranial ischaemia, polymyalgia or systemic symptoms, while aortic dilatation and aneurysm may develop later [8,18,19,23,24]. The main danger is assuming that clinical remission means the aorta is structurally safe as aortitis can leave a structurally vulnerable aortic wall long after fever, pain and C-reactive protein (CRP) have improved.
In contrast to immune-mediated disease, infectious aortitis follows a different pathobiological logic. Microbial invasion of a predisposed or damaged aortic wall may rapidly cause suppurative inflammation, necrosis, pseudoaneurysm, rupture and death [7,12,13]. Multiple factors increase susceptibility to microbial invasion, including pre-existing atherosclerotic plaques, aneurysmal segments, prosthetic material, prior vascular interventions, systemic immunosuppression, and ongoing bacteraemia. In this setting, the principal therapeutic error is premature immunosuppression. When patients present with fever, ongoing bacteraemia, endocarditis risk, recent infections, systemic immunosuppression, prosthetic graft material, or high-risk vascular anatomy, wall thickening or FDG uptake should not be reflexively attributed to autoimmune vasculitis without first excluding infection.
Treatment itself may obscure assessment, as glucocorticoids can normalise inflammatory markers before vascular inflammation has fully settled. IL-6 blockade may suppress CRP despite persistent disease activity. Conversely, CTA or MRA may continue to show wall thickening or enhancement after active inflammation has declined [9,10,33,39]. The relevant question is not whether a single marker is abnormal, but whether the total evidence supports active disease, fixed damage, infection or a mixed state. Current practice still lacks a validated method that reliably separates these categories in every patient.
The main pathobiological mechanisms linking inflammation, infection and structural vascular damage are shown in Figure 2.
6. Diagnosis: Assembling Evidence from Imperfect Tools
Diagnosis begins with suspicion, but no single history item, laboratory result, imaging sign or histological feature is sufficient across all patients. Initial clinical evaluation should explore a broad range of features including constitutional symptoms, cranial manifestations suggestive of GCA, polymyalgia, limb claudication, neurological deficits, and evidence of renal or mesenteric ischaemia. The history should document chest or abdominal pain, aortic valve dysfunction, recent infections, or endocarditis risk, alongside details of immunosuppression, vascular grafts, previous aortic surgery, or recent cancer therapy including ICI or G-CSF. Associated autoimmune features, oral or genital ulceration, and relevant family history should complete the assessment [1,2,3,4,5,29,30,31].
Diagnostic evaluation must account for patient-specific variables that alter both disease probability and test interpretation. For example, age influences differential diagnosis as GCA is rare below 50 years while Takayasu arteritis is uncommon beyond 40 years. Sex, ethnicity, and geographic origin may also affect disease prevalence. Immunosuppression, cancer therapy, vascular grafts, and recent infection increase the probability of infectious aortitis, and diabetes, chronic kidney disease, and osteoporosis increase steroid toxicity risk and influence treatment decisions even before final diagnosis. A diagnostic pathway that ignores these variables may misclassify disease or recommend treatment that is unsafe and/or unnecessary for that patient.
In aortitis, laboratory investigations support rather than confirm the diagnosis. Inflammatory markers are often raised in untreated inflammatory aortitis, but they are non-specific and may be normal in treated disease, isolated vascular disease or disease suppressed by IL-6 inhibition [9,33,39]. Additional laboratory abnormalities, such as anaemia, thrombocytosis, leucocytosis, renal impairment, or deranged liver function, may provide supporting context. Blood cultures should also be obtained when infection is suspected. Additional investigations, including serological testing for syphilis, tuberculosis, Q fever, fungal pathogens, hepatitis, and HIV, alongside autoimmune panels, complement levels, urinalysis, and IgG subclass quantification, should be guided by clinical phenotype rather than ordered routinely without clinical direction [1,7,27,28].
CTA often serves as the initial major imaging investigation owing to its speed, widespread availability, and capacity to define luminal anatomy, assess wall thickening, detect aneurysm or dissection, characterise calcification and thrombus, map branch-vessel involvement, and guide surgical planning [7,10]. While its strength lies in structural definition, its weakness is biological ambiguity. CTA, for example, can show that a wall is thickened or an aorta is aneurysmal, but it does not always distinguish active inflammation from scar, atherosclerosis, infection or postoperative change.
MRA is valuable in younger patients and for longitudinal surveillance because it avoids ionising radiation and can provide vessel wall information, including oedema and enhancement [10,41]. This is especially useful in Takayasu arteritis, where repeat imaging may be required over decades. However, MRI findings depend on protocol, timing, disease stage, treatment exposure and reader expertise. Persistent enhancement is not always equivalent to clinically meaningful active inflammation.
USS is central in suspected GCA, particularly for temporal and axillary artery assessment in experienced centres [10]. It is non-invasive, accessible and useful for cranial and extracranial branches. Limitations include operator dependence, restricted visualisation of deep thoracic and abdominal aortic segments, and inability to provide comprehensive whole-aorta assessment. In aortitis, USS should be treated as one part of a multimodality strategy rather than as a complete vascular assessment.
FDG-PET provides metabolic information and may identify active inflammation in the aorta and major branches, especially when systemic symptoms are prominent or the distribution of disease is unclear [10,33,34,35]. It may also detect occult malignancy or infection in selected contexts. However, FDG-PET interpretation remains challenging due to false-positive uptake from atherosclerotic plaque inflammation, concurrent infection, occult malignancy, postoperative changes, and technical factors affecting image acquisition and quantification. Low-grade uptake can be difficult to interpret, and treatment may reduce signal before structural risk has resolved.
Conventional angiography is now less central diagnostically because it visualises luminal change rather than vessel-wall inflammation and is invasive. It remains relevant when endovascular planning or intervention is required. Histopathology can be decisive when tissue is available, but it is usually obtained opportunistically after aortic surgery. Surgical specimens warrant careful examination for patterns of granulomatous, lymphoplasmacytic, suppurative, necrotising, or IgG4-rich inflammation, with microbiological stains and tissue cultures performed whenever infection remains a diagnostic consideration [25,26,27,28,42]. A pathology report showing aortitis should not close the case, but it should trigger clinical, microbiological, rheumatological and vascular imaging assessment.
Because many imaging findings are non-specific, a formal mimics checkpoint should be incorporated into the diagnostic pathway. Aortic wall thickening, periaortic soft tissue abnormalities, FDG uptake, and elevated inflammatory markers are non-specific findings that may arise from large-vessel vasculitis, infection, IgG4-related disease, malignancy, atherosclerotic inflammation, postoperative healing, retroperitoneal fibrosis, drug-induced injury, or concurrent overlapping processes [1,2,3,4,5,27,28,29,30,31]. A stronger diagnosis rests on concordance between phenotype, microbiology, serology, disease distribution, tissue findings where available and longitudinal behaviour. Discordance should prompt reassessment rather than automatic escalation of immunosuppression.
A practical diagnostic approach to suspected aortitis is proposed in Figure 3.
7. Management: Cause Before Escalation
Across all disease subtypes, treatment decisions should be individualised according to patient characteristics. A 70-year-old patient with diabetes, osteoporosis, and previous myocardial infarction should receive different glucocorticoid dosing and earlier steroid-sparing therapy than a younger patient without comorbidity. A patient with chronic kidney disease may not tolerate certain immunosuppressive agents. A patient with active malignancy receiving ICI requires oncology-rheumatology coordination to balance cancer control against vascular risk. Surgical candidacy depends not only on aneurysm size but on frailty, lung function, prior sternotomy, and tissue quality. These variables are part of safe and effective care, not secondary considerations.
Treatment should be guided by cause, activity, structural risk and patient vulnerability. A basic but often missed principle is that infectious aortitis must be actively considered before immunosuppression is escalated. When infection is suspected, management priorities include immediate broad-spectrum intravenous antimicrobials, prompt blood culture collection, pathogen-directed therapy once organisms are identified, and early vascular surgical evaluation [7,12,13]. In patients with aneurysm, pseudoaneurysm, dissection, rupture, persistent sepsis or high-risk anatomy, medical therapy alone is often insufficient. Contemporary aortic disease guidance supports open repair for infectious aortitis associated with aneurysm or dissection, while endovascular repair may be considered in selected high-risk patients, with prolonged antimicrobial therapy and suppressive treatment in selected unrepaired or recurrent cases [7,43].
A critical limitation is that negative microbiological cultures do not exclude infectious aortitis. Multiple factors can yield falsely negative cultures despite active infection, including prior antibiotic exposure, slow-growing organisms, graft-associated biofilms, and loculated periaortic abscesses with poor vascular communication. In patients with systemic sepsis, prosthetic material, saccular aneurysm, periaortic fluid, adjacent infection, immunosuppression or atypical imaging, source control and microbiological diagnosis should remain priorities even when inflammatory markers or blood cultures are initially negative [7,14,15,16].
For non-infectious large-vessel vasculitis, glucocorticoids remain the usual initial treatment for active disease. EULAR recommendations and the ACR/Vasculitis Foundation guideline support high-dose glucocorticoid induction in active GCA and Takayasu arteritis, followed by tapering according to disease control and toxicity risk [9,10,11]. While this traditional approach is effective, it comes at a cost. Prolonged glucocorticoid therapy is associated with substantial morbidity, increasing risks of infection, new-onset diabetes, hypertension, accelerated osteoporosis, cataract formation, metabolic weight gain, neuropsychiatric symptoms, progressive frailty, and compounded cardiovascular risk. In a disease defined by vascular vulnerability, treatment toxicity cannot be treated as a secondary issue.
Steroid-sparing agents have improved care in large-vessel vasculitis but do not eliminate long-term vascular risk. Tocilizumab increases sustained remission and reduces glucocorticoid exposure in GCA [44,45]. Methotrexate has also been evaluated as adjunctive therapy, although the effect is more modest [46]. Adalimumab did not show clear steroid-sparing benefit in a randomised trial [47]. Translating trial outcomes into aortitis-specific protection requires caution because many trials focus on relapse, remission and steroid dose rather than late aneurysm formation, aortic dilatation, dissection, rupture or reintervention. Tocilizumab also suppresses CRP, making clinicians more dependent on symptoms, imaging and structured review [9,33,45,48].
Newer targeted therapies may expand treatment options. Mavrilimumab, which targets granulocyte-macrophage colony-stimulating factor receptor alpha, showed encouraging efficacy signals in a phase 2 trial in GCA [40]. Upadacitinib has shown efficacy in a phase 3 GCA trial when combined with a glucocorticoid taper [49]. These developments signal a move from broad immunosuppression toward pathway-directed therapy. These agents should be interpreted cautiously, however, as it remains uncertain whether they prevent late aortic structural complications, perform consistently in extracranial large-vessel disease, or benefit clinically isolated aortitis.
The evidence base for Takayasu arteritis management remains limited compared to that for GCA. Glucocorticoids may induce remission, but relapse, progression and vascular complications are common [20,21,22]. Methotrexate, azathioprine, mycophenolate, tumour necrosis factor inhibitors and tocilizumab are used in practice, but the evidence base is weaker than in GCA and relies heavily on observational cohorts, small trials and expert consensus [9,11,22,50,51]. Surgery or endovascular treatment may be needed for critical stenosis, organ-threatening ischaemia, uncontrolled renovascular hypertension, aneurysm or dissection. Outcomes are generally better when active inflammation is controlled where feasible, but delay may allow irreversible organ injury. The practical tension is clear: intervene too early and restenosis or complications may follow but wait too long and vascular damage becomes fixed.
From a management perspective, clinically isolated aortitis poses difficult treatment decisions. Routine immunosuppression for every patient with histological aortitis after aortic surgery is not supported by strong evidence, but simple reassurance is unsafe [3,25,26]. A reasonable approach includes complete baseline imaging of the aorta and major branches, exclusion of infection and systemic inflammatory disease, cardiovascular risk optimisation, specialist vasculitis assessment and serial imaging. Immunosuppression should be individualised to evidence of persistent active inflammation, systemic disease, progression, high-risk anatomy or recurrent vascular lesions rather than applied reflexively.
A cause-first management and follow-up pathway is outlined in Figure 4.
8. Surgery, Endovascular Repair and Timing
Aortitis requires clinicians to judge not only inflammatory activity but also the structural consequences of aortic disease. Multiple variables inform these decisions, including absolute diameter, expansion rate, symptomatic presentation, aortic valve involvement, dissection risk, branch-vessel ischaemia, infection status, patient frailty, and local surgical expertise. Inflammatory activity changes how these variables should be interpreted. An expanding aneurysm in active vasculitis differs fundamentally from stable degenerative disease, a pseudoaneurysm in infectious aortitis is not routine elective pathology, and stenotic lesions in Takayasu arteritis may restenose if treated during active inflammation.
Open surgical repair remains essential for many complex aortic complications, especially when tissue quality, infection, arch involvement, valve disease or extensive reconstruction is relevant. Endovascular therapy has changed the management of aortic disease and may reduce immediate physiological stress in selected patients. In inflammatory or infectious aortitis, however, endovascular repair should not be treated as a straightforward less-invasive substitute. It may be a bridge, a definitive option in selected high-risk patients or a hazard if infected tissue remains uncontrolled [7,12,13].
Every major aortic operation in which aortitis is suspected should also be treated as a diagnostic opportunity. Tissue should be examined carefully, and pathology should be linked to postoperative vascular medicine or rheumatology follow-up. Conversely, every unexpected pathology report showing aortitis should trigger reassessment rather than be treated as a minor operative finding. Care is strongest when the operating room, pathology department, imaging team, rheumatology service, infectious diseases team and long-term surveillance pathway are connected.
9. Multidisciplinary Care and Patient Communication
In aortitis, multidisciplinary care is central to risk management, with each speciality contributing distinct expertise to diagnosis, treatment and surveillance. Rheumatologists diagnose systemic vasculitis and guide immunosuppression while infectious diseases specialists protect against inadvertent steroid use in unrecognised infection. Radiologists and nuclear medicine physicians differentiate active inflammation from structural damage while standardising longitudinal assessment. Vascular and cardiothoracic surgeons determine operative candidacy and optimal timing while cardiologists address hypertension, valvular complications, coronary risk, and sustained surveillance. Pathologists characterise tissue phenotypes, and primary care physicians maintain therapeutic continuity across transitions of care.
Such coordination is best understood as a risk-management process rather than administrative formality, because a patient with aortitis and an expanding aneurysm needs more than a diameter measurement. They need assessment of inflammatory activity, infection risk, tissue quality, valve involvement, branch-vessel disease, treatment toxicity, perioperative steroid and imaging follow-up. Fragmented care is especially dangerous because aortitis may become clinically quiet before the aorta becomes structurally safe.
An effective clinical pathway should include several core elements. First, aetiological classification should be documented explicitly and complete baseline imaging of the thoracoabdominal aorta and major branches should be established. Infection exclusion should be addressed where clinically appropriate and therapeutic targets should be clearly defined, whether inflammatory control, antimicrobial treatment, structural intervention, or a combination of these. Cardiovascular risk factors require optimisation, and glucocorticoid toxicity mitigation strategies must be implemented, and serial imaging surveillance should be scheduled systematically. Finally, clear criteria should specify when multidisciplinary reassessment is triggered—when symptoms recur, inflammatory markers rise, imaging findings evolve, or aortic dimensions progress. These elements should be standard rather than exceptional.
A centralised aortic multidisciplinary team with defined outputs is preferable to informal advice. Each review should state the working aetiology, whether infection has been reasonably excluded, whether imaging suggests activity, damage or both, whether the dominant risk is inflammation, sepsis, aneurysm growth, stenosis, rupture, graft infection or treatment toxicity, which specialty owns the next action and when repeat imaging is required. This shifts multidisciplinary care from good intention to accountable clinical practice.
Patient communication is also part of quality as aortitis can be difficult to explain because patients may feel well while carrying a dangerous vascular lesion. Clinicians should be direct as normal blood tests are not the final goal. The goal is to prevent aneurysm growth, dissection, rupture, ischaemia, repeated procedures, treatment toxicity and missed infection. This framing helps patients understand why surveillance continues even after symptoms improve.
10. Evidence Gaps and Research Priorities
The first problem is definitional. Studies inconsistently distinguish between radiographically detected aortitis, histopathologically confirmed inflammation, clinically isolated disease, periaortic involvement, GCA-associated aortitis, Takayasu arteritis, IgG4-related disease, infectious aetiologies, and drug-induced injury [2,3,25,26,52]. Without shared definitions that specify aetiology, anatomical segment, inflammatory activity, structural damage and mode of detection, the literature will continue to produce results that are difficult to compare and hard to apply.
Monitoring remains a major weakness because symptoms, inflammatory markers and imaging each have important limitations when interpreted in isolation. Inflammatory markers are insufficient on their own, symptoms may be absent until complications occur, and imaging, although anatomically and metabolically informative, remains expensive and not always specific [2,3,10,33]. Composite activity scores in large-vessel vasculitis do not fully capture aortic-wall biology or reliably predict late aneurysm. The field needs biomarkers that reflect vascular inflammation more directly, imaging scores reproducible across scanners and readers, and endpoints that distinguish inflammatory relapse from structural progression.
Outcome selection also remains problematic as trials often measure what is feasible rather than outcomes most consequential to patients. While sustained remission, relapse prevention, and glucocorticoid dose reduction remain important, patients ultimately require protection from clinically meaningful complications: irreversible visual loss, progressive aneurysmal dilatation, aortic dissection or rupture, critical vascular stenosis, repeated interventions, accumulating disability, and premature mortality. These outcomes require longer follow-up, larger cohorts and registry-based collaboration. The rarity of aortitis makes research challenging, but it should not justify prolonged dependence on small retrospective series.
A further gap lies between surgical and immunology datasets. Surgical aortic cohorts may under-report inflammatory phenotype, immunosuppression, PET or MRI activity and long-term vasculitis outcomes. Vasculitis cohorts may under-report operative details, graft outcomes, restenosis, reintervention and pathology [25,26,42]. This separation highlights the need for multidisciplinary registries that capture both inflammatory control and structural aortic outcomes. Major aortitis studies should assess not only whether inflammation improves, but whether the aorta remains structurally safe.
Patient-centred outcomes remain under-represented in aortitis research, including persistent fatigue, medication adverse effects, psychological burden from aneurysm surveillance, claudication-related functional impairment, reproductive health concerns in younger patients and the cumulative toll of serial imaging. Similarly, quality of life, treatment satisfaction, and the quality of shared decision-making remain inadequately characterised. Aortitis is not only a vessel-wall disease. It is a long-term condition in which patients live with uncertainty about inflammation, structural progression, and future procedures. Future research should prioritise outcomes that matter to patients alongside traditional endpoints such as remission and aneurysm growth.
The literature also exposes several underdeveloped questions: the relative effectiveness of open versus endovascular techniques in infected aortic tissue, optimal antimicrobial duration stratified by pathogen and clinical response, the capacity of FDG-PET to distinguish residual infection from sterile post-treatment inflammation, consensus diagnostic criteria for IgG4-related aortitis, systematic pharmacovigilance for ICI and G-CSF-associated disease, and evidence-based surveillance intervals following surgically discovered clinically isolated aortitis [7,10,49,50,51,52]. These questions are clinically consequential because they determine whether patients receive timely source control and surveillance or move through care without a coherent plan.
Equity and access require separate attention because geographic and institutional variation affects access to vascular ultrasound, advanced imaging such as PET and MRI, specialised vascular medicine and rheumatology services, infectious diseases input and experienced aortic surgical teams. Patients diagnosed in centres without integrated pathways may receive delayed classification, inconsistent imaging and variable follow-up. Standardisation should not only improve academic comparability but it should reduce geographic and institutional variation in care.
11. Artificial Intelligence and Precision Medicine: Promise and Limits
Aortitis brings together the kinds of data that artificial intelligence tools are often proposed to handle, including clinical phenotype, biochemical inflammatory markers, longitudinal aortic dimensions, vessel-wall imaging characteristics, quantitative FDG uptake, treatment history, histopathological patterns, and long-term clinical outcomes. Computational approaches could assist with automated aortic segmentation, precise growth-rate quantification, standardised PET interpretation, structured reporting, disease flare prediction, and individualised complication-risk stratification.
A cautious interpretation is warranted, as a model trained on one centre’s scanner protocols, referral patterns, ethnic mix, disease definitions and treatment practices may not perform reliably in other settings. Aortitis datasets also have several systematic weaknesses, including selection bias from non-uniform imaging practices, inconsistent definitions of disease flare, remission, inflammatory activity and structural progression, and marked under-representation of rarer disease subtypes. Explainability is essential if an algorithm may influence immunosuppression, surgery or lifelong surveillance, while equity matters because a tool that performs poorly in under-imaged or under-represented populations could widen diagnostic delay rather than reduce it.
The near-term role of artificial intelligence should therefore be supportive, not autonomous. It may help quantify what clinicians already inspect, flag interval change, reduce interobserver variation and generate risk estimates for multidisciplinary review. It should not decide whether aortitis is infectious or immune-mediated, whether immunosuppression is safe or whether a borderline aneurysm should be repaired. Without validated data integration, precision medicine in aortitis risks becoming terminology rather than practice.
12. Conclusions
Modern imaging has made aortitis more visible, but not necessarily easier to classify. CTA, MRI, USS, FDG-PET and surgical pathology reveal disease that would previously have been missed. Visibility, however, has not solved classification, monitoring or treatment. The main task remains to determine whether aortic inflammation is infectious or immune-mediated, systemic or isolated, active or quiescent with residual vascular damage, anatomically stable or structurally dangerous.
High-quality care requires treating aortitis as a high-risk vascular syndrome rather than a diagnostic footnote. Every patient needs disciplined aetiological assessment, complete vascular mapping, infection exclusion when appropriate, tailored anti-inflammatory or antimicrobial therapy, cardiovascular risk optimisation and long-term imaging surveillance. Multidisciplinary care is therefore not an optional refinement, but the most coherent response to a disease that repeatedly crosses specialty boundaries.
Future progress should be judged by clinically meaningful outcomes: earlier diagnosis, fewer missed infections, lower cumulative glucocorticoid exposure, better prediction of aneurysm and dissection, fewer avoidable procedures, safer timing of intervention and more equitable access to specialist imaging and expertise. Until the evidence reaches that standard, clinicians should remain both ambitious and cautious and they should treat active disease decisively, respect infection, monitor structural risk after symptoms settle and avoid pretending that one test, one specialty or one drug can provide a complete solution.
Author Contributions
Conceptualization, G.P.G., K.S. and S.K.; methodology, K.S., S.K., M.N. and G.P.G.; investigation, K.S., S.K., M.N. and I.T.; resources, F.T., K.L., P.G., A.G., I.T., N.I. and G.P.G.; data curation, K.S., S.K. and M.N.; formal analysis, K.S., S.K., M.N. and N.I.; writing—original draft preparation, G.P.G., K.S. and S.K.; writing—review and editing, G.P.G., K.S., S.K., F.T., K.L., P.G., A.G., M.N. and I.T.; visualization, K.S., S.K., M.N. and I.T.; validation, F.T., K.L., P.G., A.G., M.N., I.T., N.I. and G.P.G.; supervision, G.P.G., F.T. and K.L.; project administration, G.P.G., K.S. and S.K. All authors have read and agreed to the published version of the manuscript. Georgios P. Georghiou and Klitia Socratous contributed equally to this paper and share joint first authorship.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analysed in this study. Data sharing is not applicable to this article.
Acknowledgments
During the preparation of this manuscript, the authors used OpenAI ChatGPT for assistance and support in developing graphical elements. No generative artificial intelligence tool was used to generate primary data, perform analyses, or replace author interpretation. The authors reviewed and edited all outputs and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACR | American College of Rheumatology |
| AI | Artificial intelligence |
| CRP | C-reactive protein |
| CTA | Computed tomography angiography |
| EULAR | European Alliance of Associations for Rheumatology |
| FDG-PET | Fluorodeoxyglucose positron emission tomography |
| GCA | Giant cell arteritis |
| G-CSF | Granulocyte-colony stimulating factor |
| HIV | Human immunodeficiency virus |
| ICI | Immune-checkpoint inhibitor |
| IgG4 | Immunoglobulin G4 |
| IL-6 | Interleukin-6 |
| IL-17 | Interleukin-17 |
| MRA | Magnetic resonance angiography |
| MRI | Magnetic resonance imaging |
| PET | Positron emission tomography |
| USS | Ultrasound scan |
References
- Gornik, H.L.; Creager, M.A. Aortitis. Circulation 2008, 117, 3039–3051. [CrossRef]
- Pugh, D.; Grayson, P.; Basu, N.; Dhaun, N. Aortitis: recent advances, current concepts and future possibilities. Heart 2021, 107, 1620–1629. [CrossRef]
- Clifford, A.H. Incidentally detected noninfectious thoracic aortitis: a clinical approach. Cleve. Clin. J. Med. 2024, 91, 621–633. [CrossRef]
- Shchetynska-Marinova, T.; Amendt, K.; Sadick, M.; Keese, M.; Sigl, M. Aortitis—An Interdisciplinary Challenge. In Vivo 2021, 35, 41–52. [CrossRef]
- Sharma, S.; Pandey, N.N.; Sinha, M.; Chandrashekhara, S.H. Etiology, Diagnosis and Management of Aortitis. Cardiovasc. Intervent. Radiol. 2020, 43, 1821–1836. [CrossRef]
- Staniforth, E.; Dubey, S.; Ttofi, I.; Perinparajah, V.; Ttofi, J.; Vijjhalwar, R.; Uberoi, R.; Sideso, E.; Krasopoulos, G. Aortitis Increases the Risk of Surgical Complications and Re-Operations After Major Aortic Surgery. J. Cardiovasc. Dev. Dis. 2024, 11, 405. [CrossRef]
- Isselbacher, E.M.; Preventza, O.; Hamilton Black, J., 3rd; Augoustides, J.G.; Beck, A.W.; Bolen, M.A.; et al. 2022 ACC/AHA guideline for the diagnosis and management of aortic disease. J. Am. Coll. Cardiol. 2022, 80, e223–e393. [CrossRef]
- Nuenninghoff, D.M.; Hunder, G.G.; Christianson, T.J.H.; McClelland, R.L.; Matteson, E.L. Incidence and Predictors of Large-Artery Complication (Aortic Aneurysm, Aortic Dissection, and/or Large-Artery Stenosis) in Patients with Giant Cell Arteritis: A Population-Based Study over 50 Years. Arthritis Rheum. 2003, 48, 3522–3531. [CrossRef]
- Hellmich, B.; Agueda, A.; Monti, S.; Buttgereit, F.; de Boysson, H.; Brouwer, E.; et al. 2018 update of the EULAR recommendations for the management of large vessel vasculitis. Ann. Rheum. Dis. 2020, 79, 19–30. [CrossRef]
- Dejaco, C.; Ramiro, S.; Bond, M.; Bosch, P.; Ponte, C.; Mackie, S.L.; et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice: 2023 update. Ann. Rheum. Dis. 2024, 83, 741–751. [CrossRef]
- Maz, M.; Chung, S.A.; Abril, A.; Langford, C.A.; Gorelik, M.; Guyatt, G.; et al. 2021 American College of Rheumatology/Vasculitis Foundation guideline for the management of GCA and Takayasu arteritis. Arthritis Rheumatol. 2021, 73, 1349–1365. [CrossRef]
- Sorelius, K.; Wanhainen, A.; Furebring, M.; Björck, M.; Gillgren, P.; Mani, K. Nationwide study of the treatment of mycotic abdominal aortic aneurysms comparing open and endovascular repair. Circulation 2016, 134, 1822–1832.
- Bossone, E.; Pluchinotta, F.R.; Andreas, M.; Blanc, P.; Citro, R.; Limongelli, G.; et al. Aortitis. Vasc. Pharmacol. 2016, 80, 1–10.
- Abdelkareem, H.; Jabarin, S.A.; Farhoud, A.; Schwartz, Y.; Goldin, I.; Munter, G. Escherichia coli-induced aortitis and aortic perforation: a case study and literature review. Am. J. Case Rep. 2025, 26, e949202. [CrossRef]
- Cheok, S.; Gan, L.S.C.; Chung, S.J.; Ch’ng, J.K. Aortic endograft infection secondary to Burkholderia pseudomallei: a case report and review of the literature. J. Vasc. Surg. Cases Innov. Tech. 2021, 7, 421–424. [CrossRef]
- Foulex, A.; Coen, M.; Cherkaoui, A.; Lazarevic, V.; Gaia, N.; Leo, S.; et al. Listeria monocytogenes infectious periaortitis: a case report from the infectious disease standpoint. BMC Infect. Dis. 2019, 19, 326. [CrossRef]
- Kim, M.; Yoo, J.R.; Oh, H.; Kim, Y.R.; Lee, K.H.; Heo, S.T. The First Case of Abdominal Mycotic Aneurysm Caused by K1 Hypervirulent Klebsiella pneumoniae in a Healthy Adult. Acute Crit. Care 2021, 36, 390–394. [CrossRef]
- Salvarani, C.; Cantini, F.; Hunder, G.G. Polymyalgia rheumatica and giant-cell arteritis. Lancet 2008, 372, 234–245.
- Warrington, K.J.; Matteson, E.L. Management Guidelines and Outcome Measures in Giant Cell Arteritis (GCA). Clin. Exp. Rheumatol. 2007, 25, S137–S141.
- Seyahi, E. Takayasu arteritis: an update. Curr. Opin. Rheumatol. 2017, 29, 51–56.
- Comarmond, C.; Biard, L.; Lambert, M.; Mekinian, A.; Ferfar, Y.; Kahn, J.E.; et al. Long-term outcomes and prognostic factors of complications in Takayasu arteritis: a multicenter study of 318 patients. Circulation 2017, 136, 1114–1122. [CrossRef]
- Mason, J.C. Takayasu arteritis: advances in diagnosis and management. Nat. Rev. Rheumatol. 2010, 6, 406–415.
- Prieto-Gonzalez, S.; Arguis, P.; Garcia-Martinez, A.; Espígol-Frigolé, G.; Tavera-Bahillo, I.; Butjosa, M.; et al. Large vessel involvement in biopsy-proven GCA: prospective study in 40 newly diagnosed patients using CT angiography. Ann. Rheum. Dis. 2012, 71, 1170–1176.
- Kermani, T.A.; Warrington, K.J. Aortic involvement in GCA. Curr. Opin. Rheumatol. 2013, 25, 24–31.
- Miller, D.V.; Isotalo, P.A.; Weyand, C.M.; Edwards, W.D.; Aubry, M.C.; Tazelaar, H.D. Surgical pathology of noninfectious ascending aortitis: a study of 45 cases with emphasis on an isolated variant. Am. J. Surg. Pathol. 2006, 30, 1150–1158.
- Miller, D.V.; Maleszewski, J.J. The pathology of large-vessel vasculitides. Clin. Exp. Rheumatol. 2011, 29, S92–S98.
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-related disease. N. Engl. J. Med. 2012, 366, 539–551. [CrossRef]
- Wallace, Z.S.; Naden, R.P.; Chari, S.; Choi, H.; Della-Torre, E.; Dicaire, J.F.; et al. The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Arthritis Rheumatol. 2020, 72, 7–19. [CrossRef]
- Mukai, T.; Kubo, S.; Morita, Y.; Yamamoto, M.; Ikeda, M. Aortitis Which Developed after the Administration of Granulocyte-Colony Stimulating Factor. Mod. Rheumatol. Case Rep. 2020, 4, 74–78. [CrossRef]
- Lyon, A.R.; Yousaf, N.; Battisti, N.M.L.; Moslehi, J.; Larkin, J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018, 19, e447–e458.
- Michel, L.; Rassaf, T.; Totzeck, M. Cardiotoxicity from immune checkpoint inhibitors. Int. J. Cardiol. Heart Vasc. 2019, 25, 100420. [CrossRef]
- Grayson, P.C.; Cuthbertson, D.; Carette, S.; Hoffman, G.S.; Khalidi, N.A.; Koening, C.L.; et al. New Features of Disease after Diagnosis in 6 Forms of Systemic Vasculitis. J. Rheumatol. 2013, 40, 1905–1912. [CrossRef]
- Grayson, P.C.; Alehashemi, S.; Bagheri, A.A.; Civelek, A.C.; Cupps, T.R.; Kaplan, M.J.; et al. 18F-fluorodeoxyglucose positron emission tomography as an imaging biomarker in a prospective, longitudinal cohort of patients with large-vessel vasculitis. Arthritis Rheumatol. 2018, 70, 439–449.
- Blockmans, D.; De Ceuninck, L.; Vanderschueren, S.; Knockaert, D.; Mortelmans, L.; Bobbaers, H. Repetitive 18F-fluorodeoxyglucose positron emission tomography in GCA: a prospective study of 35 patients. Arthritis Rheum. 2006, 55, 131–137.
- Slart, R.H.J.A.; Writing Group; Reviewer Group; Members of EANM Cardiovascular; Infection and Inflammation Committees. FDG-PET/CT(A) imaging in large vessel vasculitis and polymyalgia rheumatica: joint procedural recommendation. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1250–1269.
- Weyand, C.M.; Goronzy, J.J. Immune mechanisms in medium and large-vessel vasculitis. Nat. Rev. Rheumatol. 2013, 9, 731–740.
- Weyand, C.M.; Goronzy, J.J. Clinical practice: giant-cell arteritis and polymyalgia rheumatica. N. Engl. J. Med. 2014, 371, 50–57.
- Watanabe, R.; Zhang, H.; Berry, G.; Goronzy, J.J.; Weyand, C.M. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am. J. Physiol. Heart Circ. Physiol. 2017, 312, H1052–H1059.
- Samson, M.; Corbera-Bellalta, M.; Audia, S.; Planas-Rigol, E.; Martin, L.; Bonnotte, B.; et al. Recent advances in our understanding of GCA pathogenesis. Autoimmun. Rev. 2017, 16, 833–844.
- Cid, M.C.; Unizony, S.H.; Blockmans, D.; Brouwer, E.; Dagna, L.; Dasgupta, B.; et al. Efficacy and safety of mavrilimumab in GCA: a phase 2, randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2022, 81, 653–661. [CrossRef]
- Tso, E.; Flamm, S.D.; White, R.D.; Schvartzman, P.R.; Mascha, E.; Hoffman, G.S. Takayasu arteritis: utility and limitations of magnetic resonance imaging in diagnosis and treatment. Arthritis Rheum. 2002, 46, 1634–1642.
- Maleszewski, J.J. Inflammatory ascending aortic disease: perspectives from pathology. J. Thorac. Cardiovasc. Surg. 2015, 149, S176–S183.
- Sörelius, K.; Budtz-Lilly, J.; Mani, K.; Wanhainen, A. Systematic Review of the Management of Mycotic Aortic Aneurysms. Eur. J. Vasc. Endovasc. Surg. 2019, 58, 426–435. [CrossRef]
- Villiger, P.M.; Adler, S.; Kuchen, S.; Wermelinger, F.; Dan, D.; Fiege, V.; et al. Tocilizumab for induction and maintenance of remission in GCA: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016, 387, 1921–1927. [CrossRef]
- Stone, J.H.; Tuckwell, K.; Dimonaco, S.; Klearman, M.; Aringer, M.; Blockmans, D.; et al. Trial of tocilizumab in giant-cell arteritis. N. Engl. J. Med. 2017, 377, 317–328. [CrossRef]
- Mahr, A.D.; Jover, J.A.; Spiera, R.F.; Hernández-García, C.; Fernández-Gutiérrez, B.; Lavalley, M.P.; et al. Adjunctive methotrexate for treatment of GCA: an individual patient data meta-analysis. Arthritis Rheum. 2007, 56, 2789–2797.
- Seror, R.; Baron, G.; Hachulla, E.; Larroche, C.; Puéchal, X.; Maurier, F.; et al. Adalimumab for steroid sparing in giant-cell arteritis: results of a multicentre randomised controlled trial. Ann. Rheum. Dis. 2014, 73, 2074–2081.
- Unizony, S.; Arias-Urdaneta, L.; Miloslavsky, E.; Arvikar, S.; Khosroshahi, A.; Keroack, B.; et al. Tocilizumab for the Treatment of Large-Vessel Vasculitis (Giant Cell Arteritis, Takayasu Arteritis) and Polymyalgia Rheumatica. Arthritis Care Res. (Hoboken) 2012, 64, 1720–1729. [CrossRef]
- Blockmans, D.; Penn, S.K.; Setty, A.R.; Tarzynski-Potempa, R.; Schmidt, W.A.; Hellmich, B.; et al.; SELECT-GCA Study Group. A phase 3 trial of upadacitinib for giant-cell arteritis. N. Engl. J. Med. 2025, 392, 2013–2024. [CrossRef]
- Tombetti, E.; Mason, J.C. Takayasu Arteritis: Advanced Understanding Is Leading to New Horizons. Rheumatology (Oxford) 2019, 58, 206–219. [CrossRef]
- Hoffman, G.S.; Merkel, P.A.; Brasington, R.D.; Lenschow, D.J.; Liang, P. Anti-tumor necrosis factor therapy in patients with difficult-to-treat Takayasu arteritis. Arthritis Rheum. 2004, 50, 2296–2304.
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; et al. 2012 revised International Chapel Hill Consensus Conference nomenclature of vasculitides. Arthritis Rheum. 2013, 65, 1–11. [CrossRef]
Figure 1.
Classification of aortitis according to infectious, non-infectious, drug-associated and clinically isolated phenotypes. The figure highlights that aortitis is a syndrome requiring aetiology-first classification rather than descriptive labelling alone.
Figure 1.
Classification of aortitis according to infectious, non-infectious, drug-associated and clinically isolated phenotypes. The figure highlights that aortitis is a syndrome requiring aetiology-first classification rather than descriptive labelling alone.

Figure 2.
Pathophysiology of aortitis. Immune-mediated and infectious pathways converge on aortic wall inflammation, medial injury, extracellular matrix degradation and progressive structural complications including aneurysm formation, stenosis, dissection and rupture.
Figure 2.
Pathophysiology of aortitis. Immune-mediated and infectious pathways converge on aortic wall inflammation, medial injury, extracellular matrix degradation and progressive structural complications including aneurysm formation, stenosis, dissection and rupture.

Figure 3.
Diagnostic algorithm for suspected aortitis. The pathway emphasises clinical suspicion, infection exclusion, laboratory assessment, multimodality imaging, tissue assessment where available, and reassessment when clinical, microbiological, serological or imaging findings are discordant.
Figure 3.
Diagnostic algorithm for suspected aortitis. The pathway emphasises clinical suspicion, infection exclusion, laboratory assessment, multimodality imaging, tissue assessment where available, and reassessment when clinical, microbiological, serological or imaging findings are discordant.

Figure 4.
Management and follow-up pathway for aortitis. Treatment should be guided by aetiology, inflammatory activity, structural risk and patient vulnerability, with multidisciplinary reassessment and long-term imaging surveillance even after symptoms and inflammatory markers improve.
Figure 4.
Management and follow-up pathway for aortitis. Treatment should be guided by aetiology, inflammatory activity, structural risk and patient vulnerability, with multidisciplinary reassessment and long-term imaging surveillance even after symptoms and inflammatory markers improve.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



