Submitted:
25 March 2026
Posted:
28 March 2026
You are already at the latest version
Abstract
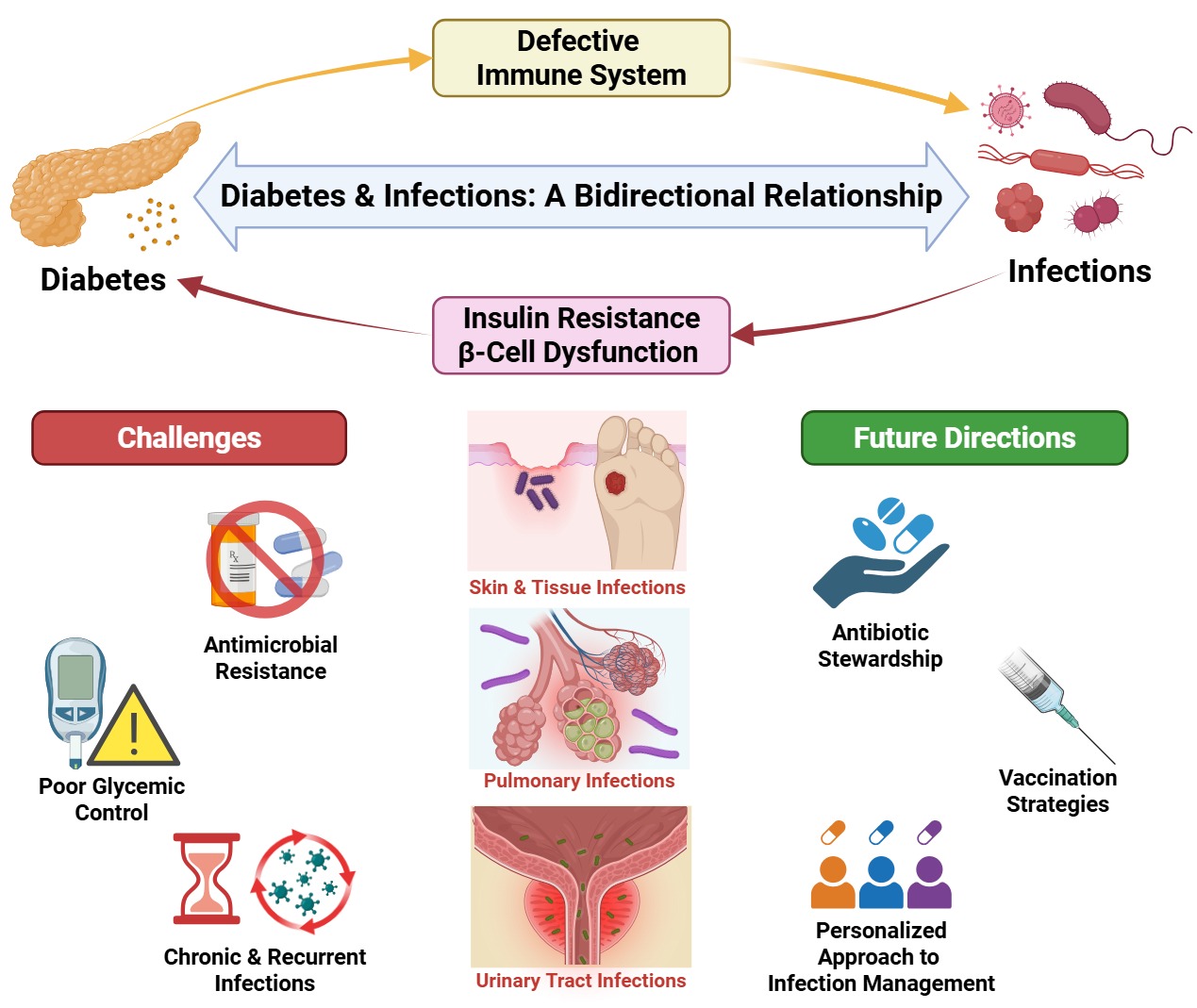
Diabetes mellitus (DM) is a rapidly growing global health burden and a major driver of infection-related morbidity and mortality. Chronic hyperglycemia disrupts both innate and adaptive immunity through impaired complement activity, dysfunctional dendritic cells and natural killer cells, altered macrophage polarization and efferocytosis, and neutrophil defects including reduced chemotaxis, impaired phagocytosis, and dysregulated NETosis. These immune abnormalities, compounded by endothelial dysfunction and microvascular disease, increase susceptibility to severe and recurrent infections such as urinary tract infections, tuberculosis, pneumonia, skin and soft tissue infections, and invasive fungal diseases. Emerging evidence also supports a bidirectional relationship in which infections may precipitate or aggravate DM via mechanisms including molecular mimicry, β-cell injury, chronic inflammation, and gut microbiota dysbiosis, contributing to insulin resistance and β-cell dysfunction. Recurrent infections and frequent exposure to broad-spectrum antibiotics, together with altered pharmacokinetics, chronic wounds with biofilm formation, and prolonged healthcare exposure, create strong selective pressure for antimicrobial resistance (AMR) in diabetic populations. Using clinical and scientific based evidence, this review explores mechanisms linking DM, infection risk, and AMR, highlighting implications for the diagnosis, therapy, stewardship, and vaccination, as well as outlines key research gaps including improved AMR surveillances stratified by diabetes status and integrated predictive models incorporating glycemic control, host factors, and microbial genomics.
Keywords:
diabetes mellitus (DM)
; hyperglycemia
; infection/infectious diseases bacterial and viral infections
1. Literature Search Strategy
A comprehensive literature search was conducted to identify studies examining the relationship between diabetes mellitus, infection susceptibility, and antimicrobial resistance. Electronic databases including PubMed, Scopus, and Web of Science were systematically searched from database inception until March 2026.
The search strategy combined Medical Subject Headings (MeSH) and free-text terms related to diabetes, infections, immune dysfunction, and antimicrobial resistance. Key search terms included:
- -
- “diabetes mellitus” OR “hyperglycemia”
- -
- “infection susceptibility” OR “infectious diseases”
- -
- “immune dysfunction” OR “immune dysregulation”
- -
- “antimicrobial resistance” OR “antibiotic resistance” OR “multidrug-resistant organisms”
- -
- “microbiome” OR “viral infections” OR “bacterial infections”
These terms were combined using Boolean operators (AND, OR) to identify relevant studies. Reference lists of included articles and relevant review papers were also manually screened to identify additional eligible studies.
Studies were considered eligible if they investigated the relationship between diabetes mellitus and infection susceptibility, examined immunological or metabolic mechanisms linking diabetes and infections, or explored associations between diabetes and antimicrobial resistance.
Priority was given to systematic reviews, meta-analyses, randomized controlled trials, and large observational studies, while mechanistic and experimental studies were also included to provide insights into underlying Immunometabolic pathways.
Only English-language articles involving human subjects or relevant experimental models were included. Studies with insufficient methodological details, duplicate publications, or articles unrelated to the interaction between diabetes and infectious diseases were excluded.
2. Introduction
Diabetes mellitus (DM) is a group of metabolic disorders characterized by impaired cellular glucose utilization and increased endogenous production, resulting in hyperglycemia [1]. It is broadly classified into two main types: type 1 and type 2 DM. Type 1 DM is an autoimmune condition characterized by the progressive destruction of insulin-producing β-cells in the pancreatic islets, resulting in little to no insulin production. In contrast, type 2 DM arises from insulin resistance, which impairs cellular responsiveness to insulin and leads to elevated blood glucose levels [2].
Given the rising risk factors currently, diabetes mellitus has become one of the most prevalent chronic diseases globally. According to the WHO, the number of people with diabetes rose from 200 million in 1990 to 830 million in 2022 with a rapid increase observed in low- and middle-income countries (LMICs) [3]. Regarding pathophysiology, hyperglycemia has been cited as one of the foremost components of DM in promoting chronic diabetes complications. It induces long-term epigenetic alterations that affect multiple physiological systems. These modifications contribute to dysregulated inflammatory responses, impaired immune functions, and increased oxidative stresses and ultimately leading to micro and macrovascular chronic complications [4].
Importantly, hyperglycemia-induced immune dysfunction is a key contributor to the increased susceptibility of diabetic patients to various infections. Chronic high blood glucose levels impair both innate and adaptive immunity through multiple interconnected mechanisms [5]. Within the innate immune system, hyperglycemia compromises host defense by impairing the function of natural killer cells, disrupting the complement cascade, increasing the generation of reactive oxygen species, and promoting excessive release of pro-inflammatory cytokines. It also increases the risk of skin lesions and ulcers, which serve as entry points for pathogens. In parallel, the adaptive immune system is weakened through the impaired function of antigen-presenting cells, a reduction in T helper cell populations, and further cytokine dysregulation—ultimately blunting the adaptive immune response. Together, these disturbances create an immunocompromised environment that significantly heightens the risk of infection in individuals with diabetes [6].
The increased susceptibility to infections amongst individuals with DM often necessitates more frequent and prolonged courses of antimicrobial therapy. As a result, bacteria begin to develop the ability to survive in the presence of these antimicrobial agents—a phenomenon widely known as antimicrobial resistance (AMR). This occurs through various mechanisms, including modifying the drug’s target, inactivating the antimicrobial agent, actively pumping the drug out of the cell, and enhancing bacterial survival by reducing outer membrane permeability, altering porin activity, and increasing the function of multidrug efflux pumps [7].
Accordingly, while diabetes mellitus may be associated with an increased risk AMR, the relationship remains not fully established. For instance, a systemic review by Dai et al. reported a higher prevalence of multidrug-resistant organism (MDRO) infections in patients with complications of DM [8]. In contrast, findings by Jagadeesan et al. indicate no significant difference in AMR patterns between patients with and without diabetes [9]. These inconsistencies may reflect differences in study populations (hospital vs. community settings), variations in prior antibiotic exposure, and heterogeneity in laboratory definitions and surveillance methods for resistant organisms [10]. However, the relationship between diabetes and antimicrobial resistance remains controversial, with some studies demonstrating increased risk of multidrug-resistant infections in diabetic populations while others report no significant differences compared with non-diabetic individuals. Therefore, understanding the relationship between diabetes mellitus and the development of AMR in various infections is crucial for identifying common pathogens and guiding clinicians in the appropriate use of antibiotics in diabetic patients.
3. Diabetes Mellitus as a Risk Factor for Infections
Among the established complications of DM, is the increased susceptibility to infections, driven mainly by the interplay of several mechanisms, including the chronic hyperglycemic state, impaired immunity, and vascular insufficiency [11]. Chronic hyperglycemia, as the hallmark of DM, initiates immune dysfunction and vascular damage, thereby heightening susceptibility to infections [12]. It exerts its effects on multiple components of the innate immune system, starting with the first line of defense; the complement system [13]. Hyperglycemia has been shown to reduce the binding of mannose-binding lectin (MBL) and other lectin pathway proteins to their targets in a dose-dependent manner, impairing lectin complement pathway activity. Additionally, it decreases the activity of the ficolin-3-mediated lectin and alternative pathways, while reducing levels of complement activation markers such as C4d and soluble C5b-9 [14].
Beyond the complement system, hyperglycemia impairs the function of dendritic cell which is the key antigen-presenting cells that bridge innate and adaptive immunity (Figure 1). Additionally, in patients with DM, both the myeloid and plasmacytoid dendritic cell populations are reduced, compromising antigen presentation and increasing susceptibility to opportunistic infections [14]. In contrast, natural killer (NK) cells which detect and eliminate virus-infected and tumor cells displays reduced functional activity in hyperglycemic states, including impaired degranulation and decreased expression of activating receptors such as NKG2D and NKp46. While reports on NK cell counts in diabetes vary, the most consistent finding is a marked reduction in cytotoxic function rather than changes in cell number [15]. This leads to reduced degranulation and cytotoxic activity [16]. It also triggers the unfolded protein response (UPR) in NK cells, inducing apoptosis and weakening their immune function [17].
Another critical component of innate immunity affected by hyperglycemia is macrophages. In hyperglycemic states, macrophages exhibit impaired activation, characterized by decreased expression of key surface markers such as ICAM-1 and CD86, along with diminished production of pro-inflammatory cytokines including TNF-α, IL-6, IL-1β, IFN-γ, and IL-12 upon stimulation. These cytokines are critical for pathogen clearance and adaptive immune activation. Meanwhile, the production of nitric oxide is increased, which overall weakens pathogen clearance [18].
In addition, polarization of macrophages is altered referring to the shift from the M1 pro-inflammatory phenotype to the M2 phenotype, which is tissue-repair oriented and marked by elevated levels of IL-10 and Arginase 1[19]. This imbalance in the M1/M2 ratio decreases the number of macrophages actively involved in clearing pathogens and favors a less needed repair response at this stage, leading to ineffective immune regulation and chronic inflammation [20]. This is exacerbated by impaired efferocytosis, a specialized function of macrophages that involves the phagocytosis of dead and damaged cells [21]. When efferocytosis is compromised, apoptotic and secondary necrotic cells accumulate and release pro-inflammatory cytokines, triggering a cycle of prolonged inflammation. This complex interaction between impaired efferocytosis and imbalanced cytokine secretion creates a state of sustained inflammation and delayed tissue repair [22] (Figure 1).
Notably, inflammation normally triggers the rapid recruitment of neutrophils as the first cells to be deplored at infection sites subsequently contributing to pathogen clearance through chemotaxis, phagocytosis, and the generation of reactive oxygen radicals [23]. However, in patients with DM, these functions are impaired at multiple levels. Starting with recruitment, DM impairs neutrophil migration to infection sites due to endothelial cell damage caused by oxidative stress, dyslipidemia, and impaired angiogenesis and vasodilation. Even when some neutrophils succeed in migrating through the endothelial layer into infected tissue, their chemotactic ability is reduced [24]. Neutrophils circulating in hyperglycemic environments experience increased oxidative stress and undergo spontaneous apoptosis and NETosis, a form of neutrophil cell death characterized by the release of neutrophil extracellular traps (NET)—web-like structures composed of DNA and antimicrobial proteins. While NETosis is normally protective, its overactivation disrupts immune balance and contributes to vascular dysfunctions [25] (Figure 1).
Increased reactive oxygen species (ROS) in diabetic-related hyperglycemia arise through several mechanisms, including increased polyol (sorbitol) pathway flux, advanced glycation end-product (AGE) formation, activation of protein kinase C (PKC) isoforms, and increased hexosamine pathway flux [26]. These elevated ROS levels, particularly hydrogen peroxide, trigger downstream processes. In DM, AGEs bind to their receptor (RAGE), further amplifying oxidative stress and inducing autophagy in neutrophils. This heightened autophagic activity in addition to the activation of other pathways like PKC and PAD4 contributes to excessive NETosis [27]. These immune disruptions—especially the release of ROS and persistent dysregulated NETosis—directly cause vascular injury by promoting inflammation, endothelial damage, and impaired tissue repair, laying the foundation for diabetic micro- and macrovascular complications[28]. In DM wounds, excessive NETs impair re-epithelialization and delay healing. Similarly, in diabetic retinopathy, NETs promote leukocyte adhesion to the endothelium, vascular occlusion, and retinal damage. Furthermore, in atherosclerosis, NETs worsen endothelial dysfunction, activate macrophages, and drive pro-inflammatory Th17 responses, accelerating plaque formation and contributing to vascular insufficiency [29].
In addition to disrupting innate immunity and promoting vascular injury, DM also impairs the adaptive immune response, affecting both its humoral and cellular branches summarized in Figure 2. Humoral immunity is compromised primarily through non-enzymatic glycation, where elevated glucose levels lead to the formation of covalent sugar adducts with various proteins, including immunoglobulins. This modification alters the structure and function of IgG antibodies, increasing their molecular mass and rendering them glycated and dysfunctional. Consequently, vaccine-induced antibodies in patient with diabetes are less effective at neutralizing pathogens and fail to adequately support phagocytosis, leading to reduced vaccine efficacy and increased susceptibility to infections. Additionally, fewer IgG molecules has been observed, linked to impaired germinal center formation and diminished T and B cell activity within these centers—ultimately hindering antibody production and class-switch recombination [14].
In addition to humoral immunity, the other integral cellular immunity represented by T cell functions are similarly affected. It has been established that DM alters T cells development and reduces T cell receptors (TCR) diversity, impairing the ability to recognize new antigens and mount effective immune responses. Upon stimulation, T cells from diabetic individuals produce lower levels of key cytokines such as IL-2, IL-6, IL-17, and TNF-α, and show reduced expansion of CD4+ Th17 cells. This weakens neutrophil recruitment and phagocytic responses. Cytotoxic CD8+ T cell function is diminished as well, as evidenced by reduced expression of perforin, granzyme B, and CD107a, along with lower production of IFN-γ and other effector cytokines [6]. Diabetes may also induce intrinsic lymphocyte dysfunction through mitochondrial DNA damage, further decreasing IFN-producing cells against viruses like CMV, EBV, and influenza thereby compounding susceptibility to infection [30].
The combined effects of chronic hyperglycemia, immune dysfunction, and vascular compromise in DM substantially increase patients' susceptibility to infections. These infections are not only more frequent but also tend to be more severe, prolonged, and often complicated by poor or delayed healing. Among the leading observed infections in individuals with DM include tuberculosis (TB), urinary tract infections (UTIs), sinopulmonary infections such as pneumonia, genital infections, hepatobiliary infections, skin and soft tissue infections, and even possibly sepsis [31] (Figure 3). From multiple studies it has been established that DM significantly increases the risk of both upper and lower UTIs. A meta-analysis by Dai et al. reported that the prevalence of asymptomatic bacteriuria in patients with type 2 diabetes is approximately 23.7% [32]. Similarly, another recent meta-analysis by Tegegne et al. found a high prevalence of UTIs among diabetic patients, with significant associations with female sex, illiteracy, and a prior history of UTIs [33]. Intriguingly, despite the plausible notion that hyperglycemia is the main driver of UTIs in patients with DM, some studies highlighted the diminished protective roles of urinary antimicrobial peptides which are directly downregulated by DM [34,35]. In contrast, despite the perceived association that sodium glucose cotransporter -2 inhibitors (SGLT2 inhibitors) accentuate hyperglycosuria predisposing patients with DM to UTIs, multiple studies refuted that association citing other related genital infections as opposed to UTIs while others upheld their benefits over highlighted risks [36,37]. While, examining that important pathophysiology, it is worth noting at one point that the FDA previously issued a warning in 2018 of the use of SGLT-2 inhibitors following being associated with infections in the genital area such as the notorious Fournier gangrene which is associated with significant morbidity and mortality.
Examining pathogen factors, patients with DM are more likely to be infected with highly virulent pathogens, such as Klebsiella species and β-lactamase-producing Escherichia coli, which contribute to increased severity and complexity of these infections. Several pathophysiological mechanisms have been proposed to explain this increased susceptibility, including glucosuria, which facilitates bacterial growth in the urinary tract; impaired immune function, which compromises bacterial clearance; structural and functional changes in the urothelium that enhance bacterial adhesion; and autonomic neuropathy leading to bladder dysfunction and urinary stasis [38]. Furthermore, multiple studies highlighted that DM enhances both virulence and pathogenicity of Klebsiella pneumoniae frequently shifting the pathogen from colonization to invasive state including leading to invasive forms of infections such as gas-forming abscesses [39]. The highlighted association and susceptibility are multifactorial with host factors underpinned by immune dysfunctions, pathogen factors of specific clones such as K1/K2 serotypes with certain capsular polysaccharides as well as environmental factors mainly associated metabolic hyperglycemia that facilitates virulence [40].
Distinctively, DM has been strongly linked to the increased risk of TB infection spectrums. A meta-analysis by Zhou et al. demonstrated a positive association between DM and latent TB infection (LTBI), indicating that individuals with diabetes are at higher risk not only for developing active TB but also for harboring LTBIs [41]. To support that observation, epidemiological data from South Asia reported by Gautam et al. reveal that up to a quarter of TB patients have comorbid DM, with prevalence rates ranging from 11% in Bangladesh to 24% in Sri Lanka. Notably, diabetic individuals with TB consistently experience poorer treatment outcomes [42]. Diabetes significantly increases susceptibility to TB through multiple mechanisms, including impaired innate and adaptive immune responses, chronic inflammation, and elevated bacterial loads, all of which contribute to more severe disease manifestations and treatment challenges [43].
Another common respiratory infection to which patients with diabetes are particularly susceptible is RTIs such as pneumonia. A meta-analysis by Brunetti et al., involving over 14 million patients, demonstrated that type 2 diabetes is associated with a significantly increased risk of developing community-acquired pneumonia (CAP), with a pooled relative risk of 1.64 compared to non-diabetic individuals [44]. Similarly, a meta-analysis by Silverii et al. reported that diabetic populations have markedly higher odds of invasive pneumococcal disease (OR 2.42), pneumococcal pneumonia (OR 2.98), intensive care unit (ICU) admission (OR 2.09), and case-fatality (OR 1.61) [45]. The increased susceptibility to pneumonia in diabetes arises from multiple mechanisms, including impaired lung dendritic cell function driven by metabolic and epigenetic alterations, as well as a diminished antiviral immune response characterized by elevated viral loads and dysregulated immunity [46].
4. Infectious Pathogens as Triggers or Aggravators of DM
Diabetes mellitus is known to increase susceptibility to various infections via inducing an immunosuppressed state. However, recent evidence shows that the interaction between DM and infections is rather more complex and bidirectional, as infectious agents seem to have correlations to the induction and aggravation of both T1DM and T2DM. Numerous viral and bacterial infections have been linked to triggering a hyperglycemic state. Notably, molecular mimicry appears to be one of the key mechanisms by which this phenomenon occurs. For example, Dolton et al. [2024] reported that bacterial proteins from Klebsiella oxytoca induce T cells to target preproinsulin (PPI)-expressing β-cells in HLA-A*24:02 individuals [47]. Those lymphocytes cross-react with endogenous PPI on the insulin producing cells, breaching the self-tolerance mechanisms due to microbial antigen mimicry. Such example demonstrates the plausibility of infection-induced autoimmunity against the insulin producing cells in T1DM. Additionally, other mechanisms by which infections induce a diabetic phenotype are well reported in the literature. Vu et al. [2015] conducted a study on rabbit models and isolated in vitro adipocytes probing the mechanisms by which exposure to Staphylococcus aureus infection can precipitate features of T2DM [48]. Prolonged exposure to S. aureus superantigens in rabbit models, primarily the toxic shock syndrome toxin-1 (TSST-1), prompted systemic inflammation along with impaired glucose tolerance and hyperinsulinemia, which are hallmark features of insulin resistance and metabolic dysfunction in T2DM. Similar findings where corroborated in vitro where TSST-1 impaired insulin signaling in isolated adipocytes. This reveals that persistent infection can trigger diabetic status via mechanisms of toxin exposure and systemic inflammation acting as environmental stressors. Collectively, these examples and various others from the body of literature pinpoint the involvement of infections in both autoimmune and metabolic forms of diabetes. This highlights the potential disease prevention approaches grounded in modulating microbial exposure and minimizing infection risk in high-risk populations.
Additionally, microbial involvement in disease state goes far beyond infections, imbalances in the gut microbiota composition, otherwise called dysbiosis, has also been linked to T2DM (Figure 4). According to Wen et al. [2025], patients with T2DM have altered gut microbiota with high abundance of proteobacteria, firmicutes and ruminococcus, and a notable insufficiency of beneficial bacteria like bifidobacteria and Bacteroides [49]. This microbial dysbiosis can trigger systemic inflammation by disrupting the production of short-chain fatty acids (SCFAs) such as butyrate, impairing the integrity of the intestinal barrier, and promoting toxin spread which can eventually manifest as β-cell dysfunction and insulin resistance (49,50). Bacterial pathogens like Fusobacterium nucleatum and Ruminococcus gnavus have been implicated in reduction in SCFAs, increased inflammation and sugar absorption from the intestines, which culminate in metabolic disturbance and exacerbated insulin resistance [49]. This prompted exploring therapeutic strategies to improve insulin sensitivity by targeting gut microbiota. Fecal microbiota transplantation (FMT) has shown merit in improving insulin sensitivity and increasing SCFA-producing bacteria in both metabolic syndrome and T2DM patients (51,52). A 6-week long clinical trial conducted in 2012 showed that healthy donor intestinal microbiota FMT improved insulin sensitivity (median rate of glucose disappearance changed from 26.2 to 45.3 mol/kg/min; P <.05) [51]. Nevertheless, probiotics were also explored as an alternative intervention to FMT. In a clinical trial by Khalili et al. [2019], daily supplementation with Lactobacillus casei for eight weeks led to improved glycemic control in T2DM participants[53]. The probiotic showed marked reductions in fasting blood glucose, insulin levels, and insulin resistance compared to the placebo group. Additionally, the probiotic group exhibited increased serum levels of sirtuin 1 (SIRT1) and decreased levels of fetuin-A—both of which are biomarkers linked to glucose metabolism and insulin sensitivity. Also, palacios et al. [2020] showed that administering multi-strain probiotics with metformin together for 12 weeks can reduce fasting plasma glucose, improve insulin resistance, and reverse gut microbiota imbalances in prediabetics and T2DM patients[54].
5. Potential Infectious Agents Triggering DM
A variety of viral pathogens have been implicated in triggering or exacerbating DM through mechanisms including direct β-cell cytotoxicity, molecular mimicry, chronic inflammation, and metabolic dysregulation. Table 1 summarizes commonly studied viruses and their proposed mechanisms in relation to T1DM and T2DM. (Rajsfus et al., 2023; Yoneda et al., 2017).
These viral infections can predispose hyperglycemic states via their tropism to the insulin-producing pancreatic β-cells. Vuorinen et al. [1992], conducted a study using Islet-like cell clusters (ICCs) prepared from human fetal pancreases[55]. The ICCs were infected with mumps or coxsackie B3 virus and became dysfunctional and unable to produce insulin 7 days post-infection, which demonstrates how viral infection can alter β-cell function and precipitate diabetes. In addition, viral proteins can share structural similarities with key β-cell autoantigens like GAD65 and IA-2, thereby triggering autoreactive T-cell responses in genetically susceptible individuals [56]. In T2DM, the role of viral infections is less well established but is gaining attention. Chronic viral infections such as hepatitis C virus (HCV), hepatitis B virus (HBV) and HIV have been linked to the development of insulin resistance in T2DM [56]. Hepatitis C virus (HCV) has been shown to contribute to T2DM development by disrupting glucose and lipid metabolism. Importantly, the metabolic and immunologic effects of these viral infections can persist even after the acute infection has been resolved, indicating that dysglycemia and diabetes risk can persist beyond viral clearance. This occurs through altered secretion of hepatokines such as fetuin-A and selenoprotein P, which may help explain the significantly higher prevalence of T2DM among HCV-infected individuals compared to the general population (57,58). Notably, HIV infection also contributes to metabolic dysregulation through the actions of viral proteins such as Vpr and Nef that can block the action of insulin and impair glycemic metabolism [56]. Similarly, SARS-CoV-2 has been associated with the onset of insulin resistance and hyperglycemia in both diabetic and non-diabetic individuals via triggering adipose tissue disease [56]. The virus can infect adipocytes and alter the secretion of adipokines like adiponectin, further disrupting lipid and glycemic metabolic process (59,60). Unsworth et al. [2020] have also reported that following COVID-19 exposure or infection, the prevalence of new-onset T1DM was notably increased [60]. Overall, the evidence underscores a significant viral contribution to the pathogenesis of both type 1 and type 2 DM.
6. Role of Antimicrobial Resistance in DM Population
Diabetes mellitus (DM), and in particular type 2 diabetes mellitus (T2DM), constitutes a substantial global health challenge, currently afflicting over 530 million individuals worldwide, with this number anticipated to rise further by 2045 [61]. In addition to its well-documented metabolic sequelae, DM markedly increases susceptibility to severe and recurrent infections. This heightened vulnerability arises from a multifaceted constellation of immunological impairments, vascular dysfunction, neuropathic changes, and various biochemical disturbances. The frequent necessity for repeated courses of broad-spectrum antibiotics to manage these infections substantially contributes to the emergence and propagation of antimicrobial resistance (AMR) [5]. The intersection of DM and AMR thus engenders a perilous synergism, posing significant threats to individual clinical outcomes and exerting considerable strain on global public health infrastructures (Figure 5).
6.1. Increased Antibiotic Exposure Among Patients with DM
Individuals living with T2DM exhibit a disproportionately high burden of infections affecting multiple organ systems, including skin and soft tissues, urinary tract, lower respiratory tract, and gastrointestinal systems. This heightened susceptibility is principally attributable to the deleterious effects of chronic hyperglycemia on both innate and adaptive immune functions. Hyperglycemia induces non-enzymatic glycation of immunoglobulins, thereby impairing opsonization and complement activation, while neutrophil dysfunction manifested as reduced chemotaxis, phagocytosis, and intracellular microbial killing, constitutes a hallmark immunopathological feature in this population. Additionally, diabetic vasculopathy and peripheral neuropathy contribute to compromised tissue perfusion and impaired wound healing, further exacerbating infection risk, particularly in the lower extremities [62].
From an epidemiological perspective, individuals with diabetes are approximately 1.5 to 3 times more likely than counterparts with no diabetes to require hospitalization for infectious diseases[63]. Among the most prevalent infections in this group are diabetic foot infections (DFIs), which affect roughly one-quarter of patients with diabetes during their lifetime, as well as recurrent urinary tract infections (UTIs) frequently linked to autonomic neuropathy-related bladder dysfunction and glycosuria [64]. The chronicity and recurrence of these infections often necessitate prolonged or repeated courses of antibiotic therapy, driven in part by biofilm formation and impaired vascular supply [65]. This clinical reality results in markedly increased antibiotic exposure among patients with diabetes, who rank among the highest consumers of antibiotics globally. The empirical and often extended use of broad-spectrum antibiotics, motivated by concerns over rapid clinical deterioration and complications, exerts a potent selective pressure favoring the emergence and propagation of antimicrobial resistance (AMR). This is evident by a comprehensive meta-analysis by Carrillo-Larco and colleagues confirming that individuals with diabetes have significantly higher odds of infections caused by antibiotic-resistant pathogens, including methicillin-resistant Staphylococcus aureus (MRSA) and extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae [63].
This trend is particularly pronounced in LMICs where microbiological diagnostic capacity and antibiotic stewardship programs may be limited, fostering widespread empirical antibiotic administration and acceleration of resistance [66]. Furthermore, regional studies from Peru and India have documented alarming resistance profiles in isolates from diabetic wounds and UTIs, with resistance rates up to 80% against major antibiotic classes and a concerning rise in carbapenem resistance, necessitating more aggressive and complex therapeutic regimens [67,68]. Moreover, the recurrent nature of infections in patients with diabetes establishes a vicious cycle whereby repeated antibiotic use disrupts the commensal microbiota and promotes colonization with multidrug-resistant organisms (MDROs). Consequently, patients with diabetes may serve as reservoirs facilitating the transmission of resistant pathogens within healthcare settings and the community, thereby amplifying the broader public health challenge posed by AMR [69]. Therefore, patients with diabetes should be prioritized as a key target group in antimicrobial stewardship programs, with strategies aimed at minimizing unnecessary antibiotic exposure and monitoring for resistant infections.
6.2. Altered Pharmacokinetics and Dosing Challenges
Diabetes engenders profound pharmacokinetic and pharmacodynamic challenges that substantially undermine the therapeutic effectiveness of antimicrobials and facilitate the emergence of resistant microbial strains. From an anatomical perspective, microvascular and macrovascular complications commonly observed in DM, particularly peripheral arterial disease and endothelial dysfunction, markedly diminish tissue perfusion, especially in distal extremities. This compromised vascular supply restricts antibiotic delivery to infected tissues, often resulting in subtherapeutic drug concentrations, incomplete bacterial eradication, and the consequent propagation of persistent infections and antimicrobial resistance [70]. For example, β-lactam antibiotics—which require sustained time-dependent exposure above the minimum inhibitory concentration (MIC) to be effective—may fail to reach therapeutic levels in poorly perfused diabetic tissues, increasing the risk of treatment failure and resistance development. At the biochemical level, chronic hyperglycemia promotes the formation of advanced glycation end products (AGEs), which alter plasma protein binding dynamics and disrupt tissue structural integrity, thereby affecting the distribution and bioavailability of antibiotics. Furthermore, hyperglycemia-induced perturbations in cellular membrane permeability and the function of various efflux transporters, such as P-glycoprotein and organic anion transporters, further modify drug absorption and elimination processes[5]. These metabolic derangements underscore the necessity for individualized dosing strategies to adequately address altered drug pharmacokinetics.
Renal impairment, a prevalent sequela in approximately 40% of patients with longstanding DM due to diabetic nephropathy, critically affects the clearance of renally excreted antimicrobials, including β-lactams, vancomycin, and aminoglycosides. Inadequate dose adjustments in the setting of renal dysfunction can lead either to supratherapeutic drug levels, heightening the risk of nephrotoxicity, or to subtherapeutic concentrations, thereby promoting the selection of resistant pathogens [71]. Moreover, concomitant hepatic dysfunction, frequently arising from comorbid non-alcoholic fatty liver disease (NAFLD) in individuals with diabetes, further complicates antimicrobial metabolism, particularly for agents primarily processed via hepatic pathways, such as macrolides and linezolid [72]. Additionally, obesity, a common comorbidity in type 2 DM, augments the volume of distribution for lipophilic antibiotics (e.g., fluoroquinolones and macrolides), necessitating weight-adjusted dosing regimens to attain appropriate therapeutic plasma concentrations [73,74] Failure to adequately consider these complex physiological and metabolic alterations through precise, individualized dosing strategies risks suboptimal drug exposure at infection sites. Such inadequacies not only predispose patients to therapeutic failure but also contribute to the selective enrichment of resistant bacterial populations, thereby exacerbating the global challenge of AMR among individuals with DM [74].
6.3. Hospital-Acquired Infections and AMR in DM
Individuals with DM are predisposed to prolonged hospitalizations, often necessitated by the management of severe infections, chronic wounds, and the requirement for invasive surgical interventions, including debridement procedures and limb amputations [5,62]. Such extended durations of inpatient care substantially increase their vulnerability to hospital-acquired infections (HAIs), particularly those attributable to MDROs (74,75). For example, in a study of diabetic foot infection patients with multidrug-resistant organisms, mean hospital stay was 19.15 ± 12.95 days for MDRO cases compared to 17.65 ± 8.05 days for non-MDRO cases (Chander et al., 2017). A comprehensive systematic review and meta-analysis conducted by Carrillo-Larco et al. [2021] revealed that individuals with DM possess markedly elevated odds of infection with methicillin-resistant Staphylococcus aureus (MRSA), extended-spectrum β-lactamase (ESBL)-producing Enterobacteriaceae, and carbapenem-resistant pathogens, relative to counterparts with no diabetes [63]. This observation is further substantiated by region-specific studies; for instance, a cross-sectional study in Peru documented carbapenem resistance rates as high as 78% among isolates from diabetic foot infections (DFIs), thereby highlighting the global intensification of antimicrobial resistance (AMR) [68]. In parallel, Hamdan et al. [2024] reported a high prevalence of resistant uropathogens, notably Escherichia coli and Klebsiella pneumoniae, among women with diabetes in Qatar, demonstrating significant resistance to third-generation cephalosporins and fluoroquinolones [66]. In the Indian context, Kande et al. [2021] identified a significantly higher incidence of ESBL-producing and carbapenem-resistant uropathogens in individuals with diabetes compared to controls with no diabetes [67]. Similarly, Coşkun et al. [2024] observed that 90% of DFIs in Turkey were polymicrobial in nature, predominantly comprising MDROs resistant to first-line antimicrobial agents [65]. Polymicrobial communities in diabetic foot infections frequently form structured biofilms, which limit antibiotic penetration and allow organisms to share resistance determinants, making empiric therapy more difficult and increasing the likelihood of treatment failure.
The elevated susceptibility to HAIs among individuals with DM is further compounded by frequent utilization of invasive medical devices, such as urinary catheters and central venous catheters, as well as repeated administration of broad-spectrum antibiotics [41,76]. These factors collectively not only complicate clinical management but also promote nosocomial transmission and environmental persistence of resistant pathogens, thereby positioning patients with diabetes as potential reservoirs for the dissemination of antimicrobial resistance [5,74].
6.4. Chronic Wounds, Biofilm Formation, and Resistance
Diabetic foot ulcers (DFUs), alongside other chronic wounds, exemplify the intricate interplay between DM and the escalating global challenge of AMR. The pathophysiological milieu characteristic of diabetic patients, comprising neuropathy-induced sensory deficits, autonomic dysfunction resulting in xerosis and cutaneous fissuring, and both macrovascular and microvascular ischemic changes, collectively gives rise to a highly conducive environment for the development and persistence of chronic wounds [62,70].
From a microbiological perspective, these chronic wounds are frequently colonized by complex polymicrobial consortia, which include Staphylococcus aureus, Pseudomonas aeruginosa, Enterococcus species, and various multidrug-resistant Gram-negative bacilli [65,74,75]. Notably, these microbial communities often exist within biofilms, highly organized aggregates of microorganisms embedded within a self-produced extracellular polymeric matrix, which afford them up to a 1,000-fold increase in antibiotic tolerance relative to their planktonic counterparts and enable evasion of host immune mechanisms [5,77,78]. The inherent heterogeneity of biofilms further exacerbates treatment complexity, as metabolically quiescent subpopulations known as "persister" cells can withstand antimicrobial assaults and subsequently re-establish infection following the cessation of therapy [74,79].
Moreover, the distinct microenvironment of diabetic wounds compounds therapeutic difficulties. Factors such as localized hypoxia and tissue acidosis compromise phagocyte function and diminish the efficacy of antimicrobial agents [62,70]. Additionally, the frequent necessity for extensive and often combinatorial antibiotic regimens to manage these infections fosters the emergence and selection of resistant strains [41,63].
6.5. Poor Glycemic Control Complicating Infection Management
Chronic hyperglycemia profoundly disrupts host immune function, thereby heightening vulnerability to infections and complicating clinical management strategies. At the molecular level, persistent hyperglycemia impairs neutrophil oxidative burst activity, diminishes complement system efficacy, and induces non-enzymatic glycation of immunoglobulins, collectively undermining opsonization and phagocytic mechanisms [62,80]. Simultaneously, sustained hyperglycemia fosters a chronic pro-inflammatory milieu, characterized by elevated circulating concentrations of tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and various other cytokines. This persistent inflammatory state paradoxically attenuates acute immune responses, consistent with the concept of immune exhaustion [62,81].
Clinically, the literature robustly links inadequate glycemic control with increased infection incidence, impaired wound healing, and elevated infection-related mortality rates. For instance, Critchley et al. [2018] reported a dose-response association between elevated glycated hemoglobin (HbA1c) levels and higher risks of pneumonia, skin and soft tissue infections, sepsis, and infection-related deaths [12]. Moreover, a bidirectional interplay between antibiotic use and dysglycemia further exacerbates this clinical scenario. Extended antibiotic exposure has been shown to worsen glycemic dysregulation, thereby accelerating diabetes progression and further increasing infection susceptibility. Zhou et al. [2021], in a comprehensive meta-analysis, demonstrated that cumulative antibiotic exposure is associated with a significantly elevated risk of developing type 2 diabetes, implicating a vicious cycle involving gut dysbiosis, metabolic dysfunction, and selective pressure favoring antimicrobial resistance [82]. This creates a self-reinforcing loop in which infections lead to increased antibiotic use, antibiotics impair metabolic control, and worsening glycemia further heightens vulnerability to infection. Recognizing and interrupting this cycle is essential for improving outcomes in diabetic patients. Collectively, these interconnected mechanisms highlight the paramount importance of rigorous glycemic control alongside prudent antibiotic stewardship in diabetic populations, to mitigate the dual threat posed by infectious complications and the propagation of antimicrobial resistance.
7. Discussion: Clinical and Public Health Implications
7.1. Diagnostic and Therapeutic Challenges
Patients with diabetes mellitus (DM) often present atypically when infected, leading to delayed diagnosis and treatment. Hyperglycemia can impair leukocyte function, blunt inflammatory responses, and alter clinical presentations — for instance, masking the classic signs of infection like fever or leukocytosis [83]. Infections such as mucormycosis, emphysematous pyelonephritis, and diabetic foot infections are particularly insidious in diabetic patients and may be advanced by the time they are recognized.
Therapeutic challenges also arise due to altered pharmacokinetics and pharmacodynamics in DM patients. For example, diabetic nephropathy may necessitate dose adjustments for renally-excreted antimicrobials, while peripheral vascular disease may impair antibiotic delivery to infected tissues. Furthermore, glucose dysregulation itself can reduce treatment efficacy and prolong recovery.
Since diabetes mellitus significantly alters the clinical presentation and management of infectious diseases, introducing both diagnostic and therapeutic challenges that have broad implications for patient outcomes and public health systems. One major issue is the atypical presentation of infections in diabetic individuals, often characterized by a blunted inflammatory response, absence of fever, and reduced perception of pain due to peripheral neuropathy [69]. These atypical features may lead to delayed recognition of infections such as diabetic foot ulcers, urinary tract infections, and even severe conditions like osteomyelitis or sepsis. In addition, overlapping symptoms between infection and diabetes complications—such as fatigue, neuropathy, or confusion—can obscure timely diagnosis. Laboratory parameters like white blood cell count and C-reactive protein may also be less reliable in the context of immune dysfunction and chronic inflammation [84].
Managing infections in individuals with diabetes mellitus presents numerous therapeutic difficulties due to the interplay between metabolic dysregulation, immune compromise, and the pharmacologic complexities of antimicrobial therapy. As previously discussed in Section 8.2, altered pharmacokinetics and pharmacodynamics in diabetes further complicate antimicrobial therapy by limiting drug penetration into infected tissues and disrupting optimal therapeutic exposure. Infections in diabetic patients, particularly diabetic foot ulcers, urinary tract infections, and respiratory infections, tend to be more persistent, polymicrobial, and recurrent, necessitating longer treatment courses and broader-spectrum antimicrobial agents [85]. Recent systematic reviews (2021–2023) confirm that diabetic foot infections are increasingly associated with multidrug-resistant organisms, including MRSA and ESBL-producing Gram-negative bacteria, which complicates empirical therapy and increases the risk of treatment failure and hospital readmissions [86].
Another major therapeutic concern is the alteration of antibiotic pharmacokinetics in diabetic individuals. For example, studies have shown a delayed time to maximum plasma concentration (Tmax) for rifampicin in diabetic patients with tuberculosis, which may necessitate therapeutic drug monitoring or adjusted dosing strategies to achieve therapeutic levels [87]. Additionally, diabetic nephropathy—common in long-standing diabetes—can impair renal clearance of antibiotics, increasing the risk of toxicity if standard dosing is used [88,89]. This underscores the importance of individualized dosing regimens based on renal and hepatic function, especially in critically ill diabetic patients who often require modified or continuous infusion dosing for time-dependent antibiotics.
Drug–drug interactions between antidiabetic and antimicrobial agents further complicate treatment. Sulfonylureas, such as glipizide and glyburide, are frequently implicated in severe hypoglycemia when combined with certain antibiotics, including fluoroquinolones, sulfonamides, tetracyclines, and macrolides. A Korean nationwide study found that these combinations were associated with a significantly higher risk of hypoglycemia, with adjusted odds ratios exceeding 2.5 for fluoroquinolones and sulfonamides [90]. In the U.S., nearly 38% of sulfonylurea-antibiotic co-prescriptions lacked clear indications, highlighting poor antimicrobial stewardship and the need for better prescriber awareness [91]. Some antibiotics also directly affect glucose metabolism; for instance, fluoroquinolones have been shown to induce both hypo- and hyperglycemia by stimulating insulin release via ATP-sensitive potassium channels in pancreatic β-cells, a risk so serious that the FDA issued safety warnings [92].
Moreover, antibiotics such as rifampicin act as enzyme inducers (e.g., CYP3A4 and CYP2C9), leading to reduced plasma concentrations of sulfonylureas and meglitinides, which can result in loss of glycemic control and new or worsening hyperglycemia [93,94]. Conversely, CYP inhibitors such as clarithromycin and fluconazole can reduce the metabolism of antidiabetic drugs, increasing the risk of hypoglycemia. These bidirectional metabolic interactions demand vigilant monitoring of blood glucose and often necessitate temporary adjustments of antidiabetic therapy when starting or stopping antibiotic treatment.
Taken together, these therapeutic challenges necessitate a personalized and multidisciplinary approach to infection management in diabetic patients. Clinicians must not only select appropriate empiric antibiotics based on local resistance patterns and comorbidities but also account for metabolic status, organ function, and potential interactions with existing antidiabetic regimens. Routine glycemic monitoring, renal dose adjustments, and coordinated care between infectious disease specialists, endocrinologists, and pharmacists are essential to improving outcomes and minimizing iatrogenic harm.
7.2. Need for Tailored Antibiotic Stewardship in Patients with DM
Patients with diabetes mellitus are disproportionately affected by recurrent and complicated infections, such as diabetic foot infections (DFIs), which frequently lead to the overuse or misuse of antibiotics. This population is particularly prone to infections caused by multidrug-resistant organisms including MRSA, ESBL-producing Gram-negative bacilli, and carbapenem-resistant strains [95]. Inappropriate empiric therapy without culture guidance is still common in many healthcare settings [96], resulting in higher morbidity, longer hospital stays, and increased healthcare costs.
To address these issues, antibiotic stewardship (ASP) efforts tailored to patients with DM are promising. Interventions using syndrome-specific ASP protocols and electronic medical record–guided prescribing have successfully reduced the use of broad-spectrum antibiotics, particularly anti-pseudomonal agents, without compromising patient outcomes [97]. Studies also highlight the importance of multidisciplinary care models, where infectious disease specialists collaborate with endocrinologists and wound care teams to guide optimal antimicrobial strategies [98].
Of note, international guidelines, such as those from the IWGDF/IDSA, emphasize culture-driven therapy, early de-escalation strategies, and integration of local antibiograms in treating DFIs [99]. Moreover, diagnostic tools like nasal PCR for MRSA colonization have enabled clinicians to avoid unnecessary vancomycin use when results are negative [100]. Distinctively, the WHO’s AWaRe framework supports these principles by classifying antibiotics into Access, Watch, and Reserve groups to encourage the use of narrower-spectrum agents whenever possible [101].
In summary, effective ASP in patients with DM requires a multifaceted approach—combining diagnostics, multidisciplinary coordination, guideline adherence, and individualized glycemic control. These measures collectively reduce the selection pressure for resistance and improve long-term clinical outcomes [102].
7.3. Screening and Vaccination Strategies as Preventive Measures
People with diabetes are at notably higher risk of a range of severe, vaccine-preventable infections—most notably influenza, pneumococcal disease, hepatitis B, COVID-19, shingles, and RSV—due to chronic immune impairment and hyperglycemia-related inflammation. Seasonal influenza vaccination has been shown to reduce hospitalizations for pneumonia and influenza by approximately 43% and all-cause hospitalizations by 28% in working-age adults with diabetes (RR reductions: PI 43%, ALL 28%) [103,104] CDC/ACIP guidelines recommend annual inactivated influenza vaccination for everyone ≥ 6 months, with particular emphasis on those with DM.
For Streptococcus pneumoniae, patients with DM are more susceptible to invasive disease and pneumococcal pneumonia with increased morbidity, length of hospital stay as well as mortality [105]. Comprehensive immunization program (CIP ) advocates either one dose of PCV20 or a sequence of PCV15 followed by PPSV23 vaccines for adults with DM ≥ 19 years of age. This has been reflected on the updated 2023 ACIP guidelines which provides clear schedules tailored to risk profiles [106].Similarly, vaccination against hepatitis B virus (HBV) is warranted for adults with DM under 60 years of age and may be considered for older individuals with additional risk factors (AACE guidelines). Conversely, COVID-19 vaccination, including updated booster doses, remains critical, as individuals with DM face increased risk of severe outcomes and mortality. Additionally, adults ≥ 50 years should receive the recombinant zoster vaccine, since diabetics are 1.6 times more likely to develop shingles compared to the general population (AACE) [107].Emerging approvals of RSV vaccines for adults ≥ 60 years also apply to those with diabetes, given their elevated risk of RSV hospitalization.
Beyond vaccination, latent tuberculosis infection screening is advised for diabetic individuals in high-endemic areas to prevent progression to active TB. Regular HBV/HCV serology is recommended in those with shared medical-device use or originating from endemic regions. Routine foot, skin, oral examinations, as well as screening for asymptomatic bacteriuria in women with frequent UTIs, can support early detection and prevent serious infections. To enhance implementation, integrating vaccine reminders and standing orders into electronic medical records, combined with pharmacist- and nurse-led vaccine delivery programs, has demonstrable success in increasing adult vaccination rates. Moreover, public health campaigns aimed at healthcare providers and patients effectively address misinformation and access barriers, thereby improving uptake. Collectively, these strategies—comprehensive immunization, targeted infectious disease screening, and proactive monitoring—are essential to reducing both infection burden and adverse outcomes in people with diabetes and bolstering prevention in the general population.
8. Knowledge Gaps and Future Research Directions
8.1. Understanding the Mechanism and the Causative Pathogens:
Despite growing research on the intersection between diabetes, infection susceptibility, and antimicrobial resistance, several key knowledge gaps remain. These gaps limit the development of targeted prevention strategies and precision antimicrobial stewardship approaches for diabetic populations. Recent studies have pointed out that hypoglycemia drives resistance mechanisms in the AMR independently from the immune dysfunction [108]. DM patients may experience systemic hyperglycemia with localized hypoglycemia in areas of infection due to vascular complications, as microvascular impairment reduces perfusion and consequently restricts the delivery of glucose and oxygen to infected tissues, creating a nutrient-limited environment accelerated by the nutritional competition with the pathogen [109]. Such an environment will force the pathogen to shift from glycolysis to purine metabolism, reducing the levels of guanosine and impairing the normal homeostatic processes of the microorganisms targeted by traditional antibiotics[110]. The mechanisms are based on rescue experiments where Edwardsiella tarda selected for ceftazidime resistance, cultured in glucose-deficient environments, were re-sensitized after adding either glucose or guanosine [111].
Deeper research should be conducted on how different pathogens develop resistance in patients with or without DM. For example, groundbreaking research demonstrated that Staphylococcus aureus was more likely to develop resistance in mice with DM [74], and a meta-analysis of 10,994 diabetic foot infection patients showed MRSA prevalence of 16.78% [112]. However, recent studies revealed that MRSA reinfection was 20.1 times more likely in non-diabetic patients than not [113], suggesting a complex dynamic beyond just immune suppression. Other studies demonstrated that 54.3% of Enterobacteriaceae isolates from urinary tract for diabetic patients were ESBL-producing [114]. Further genotypic characterization of E. coli from patients with DM demonstrated that 64.3% of ESBL-producing isolates harbored blaCTX-M genes, proving that isolates with such genes had beta-lactam antibiotics. This provides evidence for the correlation between genotypic and phenotypic trends and being and potential target for diagnosis and therapy[115].
8.2. Vaccine Research in the Interface of DM, Infection and AMR:
The associated suppression in antibody production and hyperinflammatory neutrophil recruitment in diabetic hosts could explain the consistent decrease in vaccine effectiveness [116]. For example, vaccine effectiveness of COVID-19 disease ranged between 33-91% in those with DM compared to 42-94% in the total population [117]. Therefore, recent studies have promised that tailored therapeutic strategies based on glycemic status, age, BMI, and kidney function could improve vaccine efficacy[118]. In addition, bacterial vaccines can reduce the impact of AMR on diabetic patients, by reducing the need and quantity of antibiotics [119]. Recent development in mRNA vaccine granted the ability to rapidly develop vaccines and target heterogeneous pathogen populations with multiple mutations [120].
8.3. Advanced Diagnostic Methods:
Current antimicrobial resistance surveillance methods rarely stratify by the status of DM or lack the ability to track the development of AMR in such populations [120]. Furthermore, there is a lack of integrating simultaneous glucose levels, infection, and resistance markers measurement in the point of care to guide targeted therapy. Incorporating HbA1c—a key indicator of chronic glycemic control—into AMR prediction models may be particularly valuable, as poor glycemic control is strongly linked to both infection susceptibility and antibiotic treatment failure. Further research could employ machine learning frameworks to integrate clinical variables (e.g., HbA1c and comorbidities), metabolic indices, and microbial genomic profiles to better predict AMR development in diabetic patients, by integrating clinical, metabolic and microbial variables [121].
8.4. Methodological Approaches:
Many research papers around the topic of AMR highlighted multiple methodological limitations, including heterogeneity in the study methodology, sequencing, duration and timing of the sampling [84]. The scarcity of high-quality assessments of research outcomes and lack of guideline recommendations further hinders the value of such papers.
In addition, most of the current papers are retrospective, obstructing forming any conclusion. Also, many temporal factors should be taken into consideration, such as extended follow-up times, multiple time point sampling, and accounting for seasonal variations. Such factors call for more comprehensive methods for monitoring diabetes and infection status, such as continuous glucose monitoring[82], optimizing insulin treatment with resistance profiling of serial bacterial culture and using biomarker kinetics to assess the host’s response[122].
Such a sophisticated issue requires multivariable modeling, however, the sheer number of variables available to factor in have increased the methodological heterogeneity across papers. Therefore, standardization of variable selection, outcome definition, and validation methods is necessary to transform such a theory into clinical practice.
Furthermore, a recent systematic review revealed that most AMR studies are observational with multiple confounders, making it difficult to substantiate causations[123]. These unaccounted confounders often include key clinical factors such as duration of DM, level of glycemic control (e.g., HbA1c), presence of diabetic complications, and prior antibiotic exposure, all of which strongly influence both infection severity and AMR risk. Thus, to establish causality, mediation analysis and randomized controlled trials could be implemented. The fast advancement in omics technologies, such as single cell and spatial transcriptome, tied with machine learning and advance time series analysis, could further enhance the ability to deduce the relationship [124].
9. Conclusions
Diabetes mellitus and infection are linked through a complex, bidirectional interface leading to major clinical and public health consequences. From numerous published research, there is accumulating evidence that support, exposure to infectious pathogens and microbiome perturbations contribute to the onset or worsening of both T1DM and T2DM through inflammatory and immune-metabolic mechanisms. Following manifesting DM, immune dysfunction and vascular compromise substantially increase the frequency, severity, and persistence of infections, while recurrent infection and repeated antibiotic exposure amplify selective pressure for AMR. The driving force for that are chronic wounds, biofilms formation, altered drug pharmacokinetics, and repeated and prolonged contact with healthcare. These interactions translate into diagnostic delays, higher complication rates, and more difficult antimicrobial decision-making, underscoring the need for diabetes-focused ASP, culture-guided de-escalation, individualized dosing strategies, as well as optimized preventive measures such as vaccination and targeted screening in high-risk settings. Future research should adopt latest development in genomics, and artificial intelligence using all available host clinical, metabolic data as well as pathogens microbiological variables and the context of the surrounding environment to decipher the interface between DM, infection and AMR.
Author Contributions
F.A.Z. conceptualized and led the review, coordinated the project, conducted the literature search, synthesized the findings, and drafted the manuscript. M.H. contributed to the literature review, drafting of assigned sections, and critical revision of the manuscript. A.A. contributed to the literature review, drafting of assigned sections, and critical revision of the manuscript. R.D. contributed to the literature review, drafting of assigned sections, and critical revision of the manuscript. K.N.A.S. contributed to the literature review, drafting of assigned sections, and critical revision of the manuscript. A.A.A. contributed to the literature review, drafting of assigned sections, and critical revision of the manuscript. A.A. contributed to critical revision of the manuscript and was responsible for figure design and preparation. H.A.H. supervised the work, provided critical feedback on manuscript drafts, guided the intellectual direction of the review, and approved the final version. M.S.A. supervised the work, provided critical feedback on manuscript drafts, guided the intellectual direction of the review, and approved the final version. All authors read and approved the final manuscript.
Funding
The authors declare that no financial support was received for this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
AACE – American Association of Clinical Endocrinology
ACE2 – Angiotensin-Converting Enzyme 2
ACIP – Advisory Committee on Immunization Practices
ADA – American Diabetes Association
AGE – Advanced Glycation End-Product
AMR – Antimicrobial Resistance
APCs – Antigen-Presenting Cells
ATP – Adenosine Triphosphate
AWaRe – Access, Watch, Reserve (WHO Antibiotic Classification)
BMI – Body Mass Index
β-cells – Pancreatic Beta Cells
CAP – Community-Acquired Pneumonia
CD – Cluster of Differentiation
CDC – Centers for Disease Control and Prevention
CI – Confidence Interval
CKD – Chronic Kidney Disease
CMV – Cytomegalovirus
Cmax – Maximum Plasma Concentration
COVID-19 – Coronavirus Disease 2019
CRP – C-Reactive Protein
CYP – Cytochrome P450
DC – Dendritic Cell
DDD – Defined Daily Dose
DFI – Diabetic Foot Infection
DFU – Diabetic Foot Ulcer
DM – Diabetes Mellitus
DNA – Deoxyribonucleic Acid
E. coli – Escherichia coli
EBV – Epstein–Barr Virus
EMR – Electronic Medical Record
ESBL – Extended-Spectrum β-Lactamase
FBG – Fasting Blood Glucose
FDA – Food and Drug Administration
FMT – Fecal Microbiota Transplantation
GAD65 – Glutamic Acid Decarboxylase 65
GFR – Glomerular Filtration Rate
HAI – Hospital-Acquired Infection
Hb – Hemoglobin
HbA1c – Glycated Hemoglobin A1c
HBV – Hepatitis B Virus
HCV – Hepatitis C Virus
HIV – Human Immunodeficiency Virus
HLA – Human Leukocyte Antigen
IA-2 – Islet Antigen-2
ICAM-1 – Intercellular Adhesion Molecule-1
ICC (cells) – Islet-Like Cell Clusters
ICU – Intensive Care Unit
IDSA – Infectious Diseases Society of America
IFN – Interferon
IFN-γ – Interferon Gamma
Ig – Immunoglobulin
IgG – Immunoglobulin G
IL – Interleukin
IL-1 – Interleukin 1
IL-1β – Interleukin-1 Beta
IL-2 – Interleukin 2
IL-6 – Interleukin 6
IL-10 – Interleukin 10
IL-12 – Interleukin-12
IL-17 – Interleukin 17
IWGDF – International Working Group on the Diabetic Foot
K. oxytoca – Klebsiella oxytoca
K. pneumoniae – Klebsiella pneumoniae
LMICs – Low- and Middle-Income Countries
LTBI – Latent Tuberculosis Infection
M1 macrophage – Classically Activated Pro-Inflammatory Macrophage Phenotype
M2 macrophage – Alternatively Activated Tissue Repair Macrophage Phenotype
MBL – Mannose-Binding Lectin
MHC – Major Histocompatibility Complex
MDRO – Multidrug-Resistant Organism
MIC – Minimum Inhibitory Concentration
MRSA – Methicillin-Resistant Staphylococcus aureus
mRNA – Messenger Ribonucleic Acid
NAFLD – Non-Alcoholic Fatty Liver Disease
NET – Neutrophil Extracellular Trap
NETosis – Neutrophil Extracellular Trap Formation
NK – Natural Killer Cells
NKp46 – Natural Killer Cell p46-Related Antigen
NKG2D – Natural Killer Group 2 Member D Receptor
OR – Odds Ratio
PAD4 – Peptidyl Arginine Deiminase 4
P. aeruginosa – Pseudomonas aeruginosa
PCV15/PCV20 – Pneumococcal Conjugate Vaccine (15/20-Valent)
PCR – Polymerase Chain Reaction
PD – Pharmacodynamics
PK – Pharmacokinetics
PKC – Protein Kinase C
PPI – Preproinsulin
PPSV23 – Pneumococcal Polysaccharide Vaccine (23-Valent)
RAGE – Receptor for Advanced Glycation End Products
RNA – Ribonucleic Acid
ROS – Reactive Oxygen Species
RR – Relative Risk
RSV – Respiratory Syncytial Virus
S. aureus – Staphylococcus aureus
SARS-CoV-2 – Severe Acute Respiratory Syndrome Coronavirus 2
SCFA – Short-Chain Fatty Acid
SGLT-2 – Sodium-Glucose Cotransporter-2
SIRT1 – Sirtuin 1
SSTI – Skin and Soft Tissue Infection
T1DM – Type 1 Diabetes Mellitus
T2DM – Type 2 Diabetes Mellitus
TB – Tuberculosis
TCR – T-Cell Receptor
Th cell – T Helper Cell
Th17 – T Helper 17 Cells
Tmax – Time to Maximum Plasma Concentration
TNF-α – Tumor Necrosis Factor Alpha
TSST-1 – Toxic Shock Syndrome Toxin-1
UPR – Unfolded Protein Response
UTI – Urinary Tract Infection
VZV – Varicella-Zoster Virus
WHO – World Health Organization
References
- American Diabetes Association Professional Practice Committee, ElSayed NA, McCoy RG, Aleppo G, Balapattabi K, Beverly EA, et al. 2. Diagnosis and Classification of Diabetes: Standards of Care in Diabetes—2025. Diabetes Care. 2025 Jan;48(Supplement_1):S27–S49. [CrossRef]
- Antar SA, Ashour NA, Sharaky M, Khattab M, Ashour NA, Zaid RT, et al. Diabetes mellitus: Classification, mediators, and complications; A gate to identify potential targets for the development of new effective treatments. Biomedicine & Pharmacotherapy. 2023 Dec;168:115734. [CrossRef]
- Diabetes. World health organization [Internet]. 2023. Available from: https://doi-org.qulib.idm.oclc.org/10.1016/j.biopha.2023.115734.
- Yang T, Qi F, Guo F, Shao M, Song Y, Ren G, et al. An update on chronic complications of diabetes mellitus: from molecular mechanisms to therapeutic strategies with a focus on metabolic memory. Molecular Medicine. 2024;30(1):71.
- Akash MSH, Rehman K, Fiayyaz F, Sabir S, Khurshid M. Diabetes-associated infections: development of antimicrobial resistance and possible treatment strategies. Arch Microbiol. 2020 Jul;202(5):953–65. [CrossRef]
- Holt RIG, Cockram CS, Ma RCW, Luk AOY. Diabetes and infection: review of the epidemiology, mechanisms and principles of treatment. Diabetologia. 2024 Jul;67(7):1168–80. [CrossRef]
- Da Silva Dantas A. Antimicrobial resistance. Mol Microbiol. 2022 May;117(5):959–60. [CrossRef]
- Dai J, Jiang C, Chen H, Chai Y. Assessment of the Risk Factors of Multidrug-Resistant Organism Infection in Adults With Type 1 or Type 2 Diabetes and Diabetic Foot Ulcer. Can J Diabetes. 2020 Jun;44(4):342–9. [CrossRef]
- Jagadeesan S, Tripathi BK, Patel P, Muthathal S. Urinary tract infection and Diabetes Mellitus—Etio-clinical profile and antibiogram: A North Indian perspective. J Family Med Prim Care. 2022;11(5):1902–6.
- Carrillo-Larco RM, Anza-Ramírez C, Saal-Zapata G, Villarreal-Zegarra D, Zafra-Tanaka JH, Ugarte-Gil C, et al. Type 2 diabetes mellitus and antibiotic-resistant infections: a systematic review and meta-analysis. J Epidemiol Community Health. 2022;76(1):75–84.
- Uthaya Kumar A, Ahmad Zan M, Ng CL, Chieng S, Nathan S. Diabetes and Infectious Diseases with a Focus on Melioidosis. Curr Microbiol. 2024 Jul;81(7):208. [CrossRef]
- Critchley JA, Carey IM, Harris T, DeWilde S, Hosking FJ, Cook DG. Glycemic Control and Risk of Infections Among People With Type 1 or Type 2 Diabetes in a Large Primary Care Cohort Study. Diabetes Care. 2018 Oct;41(10):2127–35. [CrossRef]
- Ajjan RA, Schroeder V. Role of complement in diabetes. Mol Immunol. 2019 Oct;114:270–7. [CrossRef]
- Daryabor G, Atashzar MR, Kabelitz D, Meri S, Kalantar K. The Effects of Type 2 Diabetes Mellitus on Organ Metabolism and the Immune System. Front Immunol. 2020 Jul;11:1582. [CrossRef]
- Mxinwa V, Dludla P V, Nyambuya TM, Mokgalaboni K, Mazibuko-Mbeje SE, Nkambule BB. Natural killer cell levels in adults living with type 2 diabetes: a systematic review and meta-analysis of clinical studies. BMC Immunol. 2020;21(1):51.
- Berbudi A, Rahmadika N, Tjahjadi AI, Ruslami R. Type 2 Diabetes and its Impact on the Immune System. Curr Diabetes Rev. 2020 May;16(5):442–9. [CrossRef]
- Berrou J, Fougeray S, Venot M, Chardiny V, Gautier JF, Dulphy N, et al. Natural Killer Cell Function, an Important Target for Infection and Tumor Protection, Is Impaired in Type 2 Diabetes. Ahlenstiel G, editor. PLoS One. 2013 Apr;8(4):e62418. [CrossRef]
- Sharma S, Kishen A. Dysfunctional crosstalk between macrophages and fibroblasts under LPS-infected and hyperglycemic environment in diabetic wounds. Sci Rep. 2025 May;15(1):17233. [CrossRef]
- Zhang B, Yang Y, Yi J, Zhao Z, Ye R. Hyperglycemia modulates M1/M2 macrophage polarization via reactive oxygen species overproduction in ligature-induced periodontitis. J Periodontal Res. 2021 Oct;56(5):991–1005. [CrossRef]
- Zhao Y, Jiang Y, Wang F, Sun L, Ding M, Zhang L, et al. High glucose promotes macrophage switching to the M1 phenotype via the downregulation of STAT-3 mediated autophagy. Jia Z, editor. PLoS One. 2024 Dec;19(12):e0314974. [CrossRef]
- Razi S, Yaghmoorian Khojini J, Kargarijam F, Panahi S, Tahershamsi Z, Tajbakhsh A, et al. Macrophage efferocytosis in health and disease. Cell Biochem Funct. 2023 Mar;41(2):152–65. [CrossRef]
- Rong B, Jiang H, Zhu W, Yang G, Zhou X, Lyu Z, et al. Unraveling the role of macrophages in diabetes: Impaired phagocytic function and therapeutic prospects. Medicine. 2025 Feb;104(8):e41613. [CrossRef]
- Tsalamandris S, Antonopoulos AS, Oikonomou E, Papamikroulis GA, Vogiatzi G, Papaioannou S, et al. The Role of Inflammation in Diabetes: Current Concepts and Future Perspectives. European Cardiology Review. 2019 Apr;14(1):50–9. [CrossRef]
- Darwitz BP, Genito CJ, Thurlow LR. Triple threat: how diabetes results in worsened bacterial infections. Richardson AR, editor. Infect Immun. 2024 Sep;92(9):e00509–23. [CrossRef]
- Yang S, Wang S, Chen L, Wang Z, Chen J, Ni Q, et al. Neutrophil Extracellular Traps Delay Diabetic Wound Healing by Inducing Endothelial-to-Mesenchymal Transition via the Hippo pathway. Int J Biol Sci. 2023;19(1):347–61. [CrossRef]
- Papachristoforou E, Lambadiari V, Maratou E, Makrilakis K. Association of Glycemic Indices (Hyperglycemia, Glucose Variability, and Hypoglycemia) with Oxidative Stress and Diabetic Complications. Sardu C, editor. J Diabetes Res. 2020 Oct;2020:1–17. [CrossRef]
- Zhu Y, Xia X, He Q, Xiao QA, Wang D, Huang M, et al. Diabetes-associated neutrophil NETosis: pathogenesis and interventional target of diabetic complications. Front Endocrinol (Lausanne). 2023 Aug;14:1202463. [CrossRef]
- An Y, Xu B tuo, Wan S rong, Ma X mei, Long Y, Xu Y, et al. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc Diabetol. 2023 Sep;22(1):237. [CrossRef]
- Njeim R, Azar WS, Fares AH, Azar ST, Kfoury Kassouf H, Eid AA. NETosis contributes to the pathogenesis of diabetes and its complications. J Mol Endocrinol. 2020 Nov;65(4):R65–R76. [CrossRef]
- Van Niekerk G, Davis T, Patterton H, Engelbrecht A. How Does Inflammation-Induced Hyperglycemia Cause Mitochondrial Dysfunction in Immune Cells? BioEssays. 2019 May;41(5):1800260. [CrossRef]
- Barkai LJ, Sipter E, Csuka D, Baló T, Nébenführer Z, Máthé A, et al. 2-es típusú diabetesesek és nem cukorbetegek területen szerzett, belgyógyászati osztályos felvételt igénylő bakteriális infekcióinak klinikai összehasonlítása. Orv Hetil. 2019 Oct;160(41):1623–32. [CrossRef]
- Dai M, Hua S, Yang J, Geng D, Li W, Hu S, et al. Incidence and risk factors of asymptomatic bacteriuria in patients with type 2 diabetes mellitus: a meta-analysis. Endocrine. 2023;82(2):263–81.
- Tegegne KD, Wagaw GB, Gebeyehu NA, Yirdaw LT, Shewangashaw NE, Kassaw MW. Prevalence of urinary tract infections and risk factors among diabetic patients in Ethiopia, a systematic review and meta-analysis. Amaravadi SK, editor. PLoS One. 2023 Jan;18(1):e0278028. [CrossRef]
- Depta J, Małkowska P, Wysokińska M, Todorska K, Sierawska O, Hrynkiewicz R, et al. Therapeutic role of antimicrobial peptides in diabetes mellitus. Biologics. 2022;2(1):92–106.
- Mohanty S, Kamolvit W, Scheffschick A, Björklund A, Tovi J, Espinosa A, et al. Diabetes downregulates the antimicrobial peptide psoriasin and increases E. coli burden in the urinary bladder. Nat Commun. 2022;13(1):4983.
- Wu MZ, Guo R, Chandramouli C, Liu L, Tung AMO, Tsang CTW, et al. Urinary tract infection and continuation of sodium-glucose cotransporter-2 inhibitors in diabetic patients. Eur Heart J. 2025;ehaf788.
- Puckrin R, Saltiel MP, Reynier P, Azoulay L, Yu OHY, Filion KB. SGLT-2 inhibitors and the risk of infections: a systematic review and meta-analysis of randomized controlled trials. Acta Diabetol. 2018;55(5):503–14.
- Pishdad R, Auwaerter PG, Kalyani RR. Diabetes, SGLT-2 inhibitors, and urinary tract infection: a review. Curr Diab Rep. 2024;24(5):108–17.
- Todd K, Gunter K, Bowen JM, Holmes CL, Tilston-Lunel NL, Vornhagen J. Type-2 diabetes mellitus enhances Klebsiella pneumoniae pathogenesis. BioRxiv. 2024.
- Lee CH, Chen IL, Chuah SK, Tai WC, Chang CC, Chen FJ, et al. Impact of glycemic control on capsular polysaccharide biosynthesis and opsonophagocytosis of Klebsiella pneumoniae: implications for invasive syndrome in patients with diabetes mellitus. Virulence. 2016;7(7):770–8.
- Zhou G, Guo X, Cai S, Zhang Y, Zhou Y, Long R, et al. Diabetes mellitus and latent tuberculosis infection: an updated meta-analysis and systematic review. BMC Infect Dis. 2023;23(1):770.
- Gautam S, Shrestha N, Mahato S, Nguyen TPA, Mishra SR, Berg-Beckhoff G. Diabetes among tuberculosis patients and its impact on tuberculosis treatment in South Asia: a systematic review and meta-analysis. Sci Rep. 2021;11(1):2113.
- Bisht MK, Dahiya P, Ghosh S, Mukhopadhyay S. The cause–effect relation of tuberculosis on incidence of diabetes mellitus. Front Cell Infect Microbiol. 2023 Jun;13:1134036. [CrossRef]
- Brunetti VC, Ayele HT, Yu OHY, Ernst P, Filion KB. Type 2 diabetes mellitus and risk of community-acquired pneumonia: a systematic review and meta-analysis of observational studies. Canadian Medical Association Open Access Journal. 2021;9(1):E62–E70.
- Silverii GA, Gabutti G, Tafuri S, Sarti F, Pratesi A, Clerico A, et al. Diabetes as a risk factor for pneumococcal disease and severe related outcomes and efficacy/effectiveness of vaccination in diabetic population. Results from meta-analysis of observational studies. Acta Diabetol. 2024 Apr;61(8):1029–39. [CrossRef]
- Herder C, Roden M, Venteclef N. Diabetes and pulmonary infection: how hyperglycaemia shapes the immune system. Signal Transduct Target Ther. 2024 Mar;9(1):67. [CrossRef]
- Dolton G, Bulek A, Wall A, Thomas H, Hopkins JR, Rius C, et al. HLA A*24:02–restricted T cell receptors cross-recognize bacterial and preproinsulin peptides in type 1 diabetes. Journal of Clinical Investigation. 2024 Jul;134(18). [CrossRef]
- Vu BG, Stach CS, Kulhankova K, Salgado-Pabón W, Klingelhutz AJ, Schlievert PM. Chronic superantigen exposure induces systemic inflammation, elevated bloodstream endotoxin, and abnormal glucose tolerance in rabbits: Possible role in diabetes. mBio. 2015;6(2). [CrossRef]
- Wen X, Qi LM, Zhao K. Influence of gut bacteria on type 2 diabetes: Mechanisms and therapeutic strategy. World J Diabetes. 2025 Jul;16(1). [CrossRef]
- Sadagopan A, Mahmoud A, Begg M, Tarhuni M, Fotso M, Gonzalez NA, et al. Understanding the Role of the Gut Microbiome in Diabetes and Therapeutics Targeting Leaky Gut: A Systematic Review. Cureus. 2023 Jul. [CrossRef]
- Vrieze A, Nood E Van, Holleman F, Salojärvi J, Kootte RS, Bartelsman JFWM, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012;143(4). [CrossRef]
- Ng SC, Xu Z, Mak JWY, Yang K, Liu Q, Zuo T, et al. Microbiota engraftment after faecal microbiota transplantation in obese subjects with type 2 diabetes: a 24-week, double-blind, randomised controlled trial. Gut. 2022 Jul;71(4):716–23. [CrossRef]
- Khalili L, Alipour B, Jafar-Abadi MA, Faraji I, Hassanalilou T, Abbasi MM, et al. The effects of Lactobacillus casei on glycemic response, serum sirtuin1 and fetuin-a levels in patients with type 2 diabetes mellitus: a randomized controlled trial. Iran Biomed J. 2019;23(1):68.
- Palacios T, Vitetta L, Coulson S, Madigan CD, Lam YY, Manuel R, et al. Targeting the intestinal microbiota to prevent type 2 diabetes and enhance the effect of metformin on glycaemia: a randomised controlled pilot study. Nutrients. 2020;12(7):2041.
- Vuorinen T, Nikolakaros G, Smell O, Hyypia T, Vainionpaa R. Mumps and Coxsackie B3 Virus Infection of Human Fetal Pancreatic Islet-like Cell Clusters. Pancreas. 1992 Jul;7(4):460–4. [CrossRef]
- Rajsfus BF, Mohana-Borges R, Allonso D. Diabetogenic viruses: linking viruses to diabetes mellitus. Heliyon. 2023 Jul;9(4). [CrossRef]
- Ali SA, Nassif WMH, Abdelaziz DHA. Alterations in serum levels of fetuin A and selenoprotein P in chronic hepatitis C patients with concomitant type 2 diabetes: A case-control study. Clin Res Hepatol Gastroenterol. 2016 Jul;40(4):465–70. [CrossRef]
- Ambachew S, Eshetie S, Geremew D, Endalamaw A, Melku M. Prevalence of Type 2 Diabetes Mellitus among Hepatitis C Virus-Infected Patients: A Systematic Review and Meta-Analysis. Dubai Diabetes and Endocrinology Journal. 2018;24(1–4):29–37. [CrossRef]
- Nhau PT, Gamede M, Sibiya N. COVID-19-Induced Diabetes Mellitus: Comprehensive Cellular and Molecular Mechanistic Insights. Pathophysiology. Multidisciplinary Digital Publishing Institute (MDPI); 2024. p. 197–209. [CrossRef]
- Unsworth R, Wallace S, Oliver NS, Yeung S, Kshirsagar A, Naidu H, et al. New-onset type 1 diabetes in children during COVID-19: Multicenter regional findings in the U.K. Diabetes Care. American Diabetes Association Inc.; 2020. p. e170–1. [CrossRef]
- Arabi A, Nasrallah D, Mohsen S, Abugharbieh L, Al-Hashimi D, AlMass S, et al. The interplay between vitamin D status, subclinical inflammation, and prediabetes. Heliyon. 2024 Aug;10(15):e35764. [CrossRef]
- Tae-Bong K, Yasmin H, Youngmin L, Hyunjhung J, Joohee K, Soohyun K. Diabetes and bacterial infection. Int J Clin Endocrinol Metab. 2022 Sep 24;8(1):001–8. [CrossRef]
- Carrillo-Larco RM, Anza-Ramírez C, Saal-Zapata G, Villarreal-Zegarra D, Zafra-Tanaka JH, Ugarte-Gil C, et al. Type 2 diabetes mellitus and antibiotic-resistant infections: a systematic review and meta-analysis. J Epidemiol Community Health (1978). 2022 Jan;76(1):75–84. [CrossRef]
- Lipsky BA, Senneville É, Abbas ZG, Aragón-Sánchez J, Diggle M, Embil JM, et al. Guidelines on the diagnosis and treatment of foot infection in persons with diabetes (IWGDF 2019 update). Diabetes Metab Res Rev. 2020 Mar 16;36(S1). [CrossRef]
- Coşkun B, Ayhan M, Ulusoy S, Guner R. Bacterial Profile and Antimicrobial Resistance Patterns of Diabetic Foot Infections in a Major Research Hospital of Turkey. Antibiotics. 2024 Jun 27;13(7):599. [CrossRef]
- Hamdan A, AbuHaweeleh MN, Al-Qassem L, Kashkoul A, Alremawi I, Hussain U, et al. Prevalence of Antimicrobial Resistance Among the WHO’s AWaRe Classified Antibiotics Used to Treat Urinary Tract Infections in Diabetic Women. Antibiotics. 2024 Dec 14;13(12):1218. [CrossRef]
- Kande S, Patro S, Panigrahi A, Khora PK, Pattnaik D. Prevalence of uropathogens and their antimicrobial resistance pattern among adult diabetic patients. Indian J Public Health. 2021;65(3):280–6. [CrossRef]
- Moya-Salazar J, Chamana JM, Porras-Rivera D, Goicochea-Palomino EA, Salazar CR, Contreras-Pulache H. Increase in antibiotic resistance in diabetic foot infections among peruvian patients: a single-center cross-sectional study. Front Endocrinol (Lausanne). 2023;14:1267699. [CrossRef]
- Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: A review of pathogenesis. Indian J Endocrinol Metab. 2012;16(Suppl1):S27–S36. [CrossRef]
- Jeffcoate WJ, Harding KG. Diabetic foot ulcers. Lancet. 2003 May 3;361(9368):1545–51. [CrossRef]
- Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KAM, Zoungas S, et al. Diabetic kidney disease. Nat Rev Dis Primers. 2015 Jul 30;1:15018. [CrossRef]
- Byrne CD, Targher G. NAFLD: A multisystem disease. J Hepatol. 2015 Apr;62(1):S47–64. [CrossRef]
- Pai MP, Paloucek FP. The origin of the “ideal” body weight equations. Ann Pharmacother. 2000 Sep;34(9):1066–9. [CrossRef]
- Shook JC, Genito CJ, Darwitz BP, Tyson KJ, Velez AZ, Bridwell SK, et al. Diabetes potentiates the emergence and expansion of antibiotic resistance. Sci Adv. 2025 Feb 14;11(7). [CrossRef]
- Trivedi U, Parameswaran S, Armstrong A, Burgueno-Vega D, Griswold J, Dissanaike S, et al. Prevalence of Multiple Antibiotic Resistant Infections in Diabetic versus Nondiabetic Wounds. J Pathog. 2014;2014:1–6. [CrossRef]
- CP SR, KM R, Ali SM, P DrLM. Antimicrobial Resistance Pattern of Infections in Patients with Type II Diabetes Mellitus. Int J Pharm Sci Rev Res. 2023 Jun;80(1). [CrossRef]
- Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999 May 21;284(5418):1318–22. [CrossRef]
- Lebeaux D, Ghigo JM, Beloin C. Biofilm-Related Infections: Bridging the Gap between Clinical Management and Fundamental Aspects of Recalcitrance toward Antibiotics. Microbiology and Molecular Biology Reviews. 2014 Sep;78(3):510–43. [CrossRef]
- James GA, Swogger E, Wolcott R, Pulcini E deLancey, Secor P, Sestrich J, et al. Biofilms in chronic wounds. Wound Repair and Regeneration. 2008 Jan 13;16(1):37–44. [CrossRef]
- Delamaire M, Maugendre D, Moreno M, Le Goff MC, Allannic H, Genetet B. Impaired leucocyte functions in diabetic patients. Diabet Med. 1997 Jan;14(1):29–34. [CrossRef]
- Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011 Feb;11(2):98–107. [CrossRef]
- Shen Y, Zhang L, Fan X, Zhou J. Effectiveness of remote continuous glucose monitoring on adverse outcomes among patients with diabetes complicated with COVID-19. J Diabetes Investig. 2021 Oct 20;12(10):1923–4. [CrossRef]
- Casqueiro J, Casqueiro J, Alves C. Infections in patients with diabetes mellitus: A review of pathogenesis. Indian J Endocrinol Metab. 2012;16(Suppl1):S27–36.
- Maity S, Leton N, Nayak N, Jha A, Anand N, Thompson K, et al. A systematic review of diabetic foot infections: pathogenesis, diagnosis, and management strategies. Frontiers in Clinical Diabetes and Healthcare. 2024 Aug 6;5. [CrossRef]
- Peleg AY, Weerarathna T, McCarthy JS, Davis TME. Common infections in diabetes: Pathogenesis, management and relationship to glycaemic control. Diabetes Metab Res Rev. 2007;23(1):3–13. [CrossRef]
- Mavrogeni E, Papanas N. Microbial patterns and resistance in diabetic foot infections: A systematic update. J Clin Med. 2023;12(3):632. [CrossRef]
- Sharma SK, Dhooria S, Agarwal R. Pharmacokinetics of antitubercular drugs in patients with diabetes mellitus: A meta-analysis. International Journal of Tuberculosis and Lung Disease. 2022;26(8):740–7. [CrossRef]
- Falagas ME, Kompoti M. Obesity and infection. Lancet Infect Dis. 2006;6(7):438–46. [CrossRef]
- Han HJ, Lee S, Kim JH, Kim Y. Optimizing antibiotic dosing in patients with diabetic nephropathy: A clinical perspective. Journal of Infection and Chemotherapy. 2023;29(4):283–91. [CrossRef]
- Kim KO, Lee JY, Jung HY, Lee H. Risk of hypoglycemia associated with the concurrent use of sulfonylureas and antibiotics: A nationwide case-crossover study. J Clin Pharm Ther. 2020;45(1):47–55. [CrossRef]
- Parekh N, Husain A, Wilbur K. Co-prescribing patterns of sulfonylureas and interacting antibiotics in the US outpatient setting. Diabetes Therapy. 2020;11(6):1271–80. [CrossRef]
- Thangaraju P, Venkatesan S, Subramaniyan A. Fluoroquinolone-induced dysglycemia: A review of mechanisms and cases. J Basic Clin Pharm. 2021;12(2):90–6. [CrossRef]
- Niemi M, Backman JT, Neuvonen M, Neuvonen PJ, Kivistö KT. Rifampin decreases the plasma concentrations and effects of repaglinide. Clin Pharmacol Ther. 2000;68(5):495–500.
- Park J, Kim K, Park P, Park C, Shin J. Effect of rifampin on the pharmacokinetics and pharmacodynamics of gliclazide. Clin Pharmacol Ther. 2003;74(4):334–40.
- Lin H, Zhao X, Wu Y, others. Distribution of microbes and antimicrobial susceptibility in patients with diabetic foot infections in South China. Front Endocrinol (Lausanne). 2023;14:1113622. [CrossRef]
- Surya E, Wijaya R, Hadi U. Evaluation of the use of antimicrobial therapy for treating diabetic foot infections in an Indonesian referral hospital: A retrospective cohort study. Infectious Diseases in Clinical Practice. 2023;31(5):e368. [CrossRef]
- Taylor M, Rajiv K, Stoner B. Impact of a syndrome-specific antibiotic stewardship intervention on antipseudomonal antibiotic use in inpatient diabetic foot infection management. Journal of Antimicrobial Chemotherapy. 2023;78(1):88–96. [CrossRef]
- Choi T, Osuagwu UL, Tran C, others. Impact of multidisciplinary care of diabetic foot infections for inpatients at Campbelltown Hospital. BMC Health Serv Res. 2023;23:1126. [CrossRef]
- Fonseca L, Lipsky BA, others. IWGDF/IDSA Guidelines on the Diagnosis and Treatment of Diabetic Foot Infections. Diabetes Metab Res Rev. 2024. [CrossRef]
- Raiss Y, Bachour H, Leleiko W. Clinical utility of methicillin-resistant Staphylococcus aureus nasal PCR to streamline antimicrobial use in treatment of diabetic foot infection with or without osteomyelitis. BMC Infect Dis. 2023;23:297. [CrossRef]
- World Health Organization. AWaRe classification of antibiotics for evaluation and monitoring of use [Internet]. World Health Organization; 2021. Available from: https://www.who.int/publications/i/item/2021-aware-classification.
- Monnet DL, Tängdén T. Systemic Antimicrobial Therapy for Diabetic Foot Infections: An Overview of Systematic Reviews. J Glob Antimicrob Resist. 2024. [CrossRef]
- Vamos EP, Pape UJ, Bottle A, Majeed A, Millett C. Effectiveness of influenza vaccination in working-age adults with diabetes: A population-based cohort study. BMC Med. 2015;13:60. [CrossRef]
- Liu G, Pang Y, Lv M, Lu M, Huang Y, Ge F, et al. Effectiveness of influenza vaccination on hospitalization outcomes among older patients with diabetes. Vaccine. 2024;42(25):126142.
- Ghit A. Pneumococcal vaccination in diabetic patients: review from clinical practice. Egypt J Intern Med. 2023;35(1):16.
- Kobayashi M. Pneumococcal vaccine for adults aged≥ 19 years: recommendations of the advisory committee on immunization practices, United States, 2023. MMWR Recommendations and reports. 2023;72.
- Poirrier JE, Meyers JL, Nagar SP, Patterson BJ, Glasser LI, Jabbour SA. Herpes zoster incidence and burden in adults with type 2 diabetes in the US: a retrospective database analysis. Diabetes Care. 2022;45(11):2585–93.
- Tadesse BT, Keddy KH, Rickett NY, Zhusupbekova A, Poudyal N, Lawley T, et al. Vaccination to Reduce Antimicrobial Resistance Burden—Data Gaps and Future Research. Clinical Infectious Diseases. 2023 Dec 20;77(Supplement_7):S597–607. [CrossRef]
- Orasanu G, Plutzky J. The Pathologic Continuum of Diabetic Vascular Disease. J Am Coll Cardiol. 2009 Feb;53(5):S35–42. [CrossRef]
- Hobbs JK, Boraston AB. (p) ppGpp and the stringent response: an emerging threat to antibiotic therapy. ACS Infect Dis. 2019;5(9):1505–17.
- Xiang J, Wang S wen, Tao Y, Ye J zhou, Liang Y, Peng X xian, et al. A glucose-mediated antibiotic resistance metabolic flux from glycolysis, the pyruvate cycle, and glutamate metabolism to purine metabolism. Front Microbiol. 2023 Oct 17;14. [CrossRef]
- Stacey HJ, Clements CS, Welburn SC, Jones JD. The prevalence of methicillin-resistant Staphylococcus aureus among diabetic patients: a meta-analysis. Acta Diabetol. 2019 Aug 6;56(8):907–21. [CrossRef]
- Lavery LA, Reyes MC, Suludere M, Najafi B, Sideman M, Siah MC, et al. Methicillin-resistant Staphylococcus aureus in diabetic and non-diabetic foot infections. Int Wound J. 2024 Sep 13;21(9). [CrossRef]
- Alzahrani OM, Uddin F, Mahmoud SF, Alswat AS, Sohail M, Youssef M. Resistance to Some New Drugs and Prevalence of ESBL- and MBL-Producing Enterobacteriaceae Uropathogens Isolated from Diabetic Patients. 2022 Dec 16;12(12):2125. [CrossRef]
- Adjei DN, Mughogho TS, Michael OT, Saidu S, Amegatcher G, Forson AO. Characterization of the Phenotypic and Genotypic Antibiotic Resistance Markers in Escherichia coli (E. coli) Associated With Diabetes and Nondiabetic Patients. Int J Microbiol. 2025;2025(1):3694023.
- Johnson RM, Ardanuy J, Hammond H, Logue J, Jackson L, Baracco L, et al. Diet-induced obesity and diabetes enhance mortality and reduce vaccine efficacy for SARS-CoV-2. J Virol. 2023 Nov 30;97(11). [CrossRef]
- van den Berg JM, Remmelzwaal S, Blom MT, van Hoek BACE, Swart KMA, Overbeek JA, et al. Effectiveness of COVID-19 Vaccines in Adults with Diabetes Mellitus: A Systematic Review. Vaccines (Basel). 2022 Dec 22;11(1):24. [CrossRef]
- Mojaddidi MA, Aboonq M, Alqahtani SA. Glycemic control and vaccine response: the role of mucosal immunity after vaccination in diabetic patients. Front Immunol. 2025 May 8;16. [CrossRef]
- Lim CG, Shin SH, Han Y, Heo YH, Kim I, Lee JH, et al. Abstract LB352: Unraveling therapeutic potentials of mRNA-based cancer vaccine concurrently targeting heterogeneous KRAS mutant cancer. Cancer Res. 2024 Apr 5;84(7_Supplement):LB352–LB352. [CrossRef]
- Hamers RL, Dobreva Z, Cassini A, Tamara A, Lazarus G, Asadinia KS, et al. Global knowledge gaps on antimicrobial resistance in the human health sector: A scoping review. International Journal of Infectious Diseases. 2023 Sep;134:142–9. [CrossRef]
- Chen C, Li SL, Xu YY, Liu J, Graham DW, Zhu YG. Characterising global antimicrobial resistance research explains why One Health solutions are slow in development: An application of AI-based gap analysis. Environ Int. 2024 May;187:108680. [CrossRef]
- Bahloul M, Bradii S, Turki M, Bouchaala K, Ben Hamida C, Chelly H, et al. The value of sepsis biomarkers and their kinetics in the prognosis of septic shock due to bacterial infections. Anaesthesiol Intensive Ther. 2021 Aug 23;53(4):312–8. [CrossRef]
- van Kessel SAM, Wielders CCH, Schoffelen AF, Verbon A. Enhancing antimicrobial resistance surveillance and research: a systematic scoping review on the possibilities, yield and methods of data linkage studies. Antimicrob Resist Infect Control. 2025 Mar 29;14(1):25. [CrossRef]
- Patharkar A, Cai F, Al-Hindawi F, Wu T. Predictive modeling of biomedical temporal data in healthcare applications: review and future directions. Front Physiol. 2024 Oct 15;15. [CrossRef]
Figure 1.
Chronic hyperglycemia disrupts key components of innate immunity, including dendritic cells, natural killer cells, macrophages, and neutrophils. These alterations impair antigen presentation, reduce cytotoxic and phagocytic activity, promote dysfunctional inflammatory responses, and ultimately weaken host defense against infections.
Figure 1.
Chronic hyperglycemia disrupts key components of innate immunity, including dendritic cells, natural killer cells, macrophages, and neutrophils. These alterations impair antigen presentation, reduce cytotoxic and phagocytic activity, promote dysfunctional inflammatory responses, and ultimately weaken host defense against infections.

Figure 2.
Hyperglycemia impairs adaptive immunity by reducing T-cell activation, cytokine production, and cytotoxic function, while also disrupting B-cell responses. Non-enzymatic glycation of immunoglobulins compromises antibody-mediated neutralization, opsonization, and complement activation, leading to increased susceptibility to infections.
Figure 2.
Hyperglycemia impairs adaptive immunity by reducing T-cell activation, cytokine production, and cytotoxic function, while also disrupting B-cell responses. Non-enzymatic glycation of immunoglobulins compromises antibody-mediated neutralization, opsonization, and complement activation, leading to increased susceptibility to infections.

Figure 3.
Diabetes mellitus predisposes individuals to a wide range of infections involving multiple organ systems, including respiratory, urinary, hepatobiliary, skin and soft tissue, and bloodstream infections. Impaired immunity, vascular insufficiency, and hyperglycemia-related tissue changes contribute to increased susceptibility, severity, and recurrence of these infections.
Figure 3.
Diabetes mellitus predisposes individuals to a wide range of infections involving multiple organ systems, including respiratory, urinary, hepatobiliary, skin and soft tissue, and bloodstream infections. Impaired immunity, vascular insufficiency, and hyperglycemia-related tissue changes contribute to increased susceptibility, severity, and recurrence of these infections.

Figure 4.
Alterations in gut microbiota composition (dysbiosis) reduce short-chain fatty acid production and impair intestinal barrier integrity, leading to systemic inflammation and metabolic dysfunction. These changes promote insulin resistance and β-cell impairment, contributing to the development and progression of diabetes mellitus.
Figure 4.
Alterations in gut microbiota composition (dysbiosis) reduce short-chain fatty acid production and impair intestinal barrier integrity, leading to systemic inflammation and metabolic dysfunction. These changes promote insulin resistance and β-cell impairment, contributing to the development and progression of diabetes mellitus.

Figure 5.
Multiple diabetes-related factors—including recurrent infections, frequent broad-spectrum antibiotic use, impaired immunity, altered pharmacokinetics, and poor tissue perfusion—contribute to the development of antimicrobial resistance. Chronic wounds and prolonged healthcare exposure further promote colonization with multidrug-resistant organisms.
Figure 5.
Multiple diabetes-related factors—including recurrent infections, frequent broad-spectrum antibiotic use, impaired immunity, altered pharmacokinetics, and poor tissue perfusion—contribute to the development of antimicrobial resistance. Chronic wounds and prolonged healthcare exposure further promote colonization with multidrug-resistant organisms.


Table 1.
Viral Pathogens Implicated in the Onset or Exacerbation of Diabetes Mellitus and Proposed Mechanisms.
Table 1.
Viral Pathogens Implicated in the Onset or Exacerbation of Diabetes Mellitus and Proposed Mechanisms.
| Virus / Virus Group | Associated Diabetes Type | Proposed Mechanism |
|---|---|---|
| Coxsackievirus B | T1DM | Direct β-cell infection & cytolysis; induces autoimmune T-cell activation. |
| Enteroviruses (e.g., Echovirus) | T1DM | Trigger molecular mimicry leading to autoreactive T-cells against β-cell antigens. |
| Cytomegalovirus (CMV) | T1DM & T2DM (less established) | Chronic infection induces persistent inflammation impairing β-cell function. |
| Epstein–Barr Virus (EBV) | T1DM (associative) | Alters immune tolerance; may promote autoimmunity via B-cell dysregulation. |
| Rubella (Congenital infection) | T1DM | Damages pancreatic islet development in utero. |
| Mumps Virus | T1DM | Infects pancreatic tissue and reduces insulin secretion. |
| Rotavirus | T1DM | Shares peptide similarity with β-cell autoantigens (molecular mimicry). |
| Varicella-Zoster Virus (VZV) | T1DM (rare/associative) | Viral inflammation may contribute to autoimmune activation. |
| Influenza A/B | T2DM (acute dysglycemia) | Triggers systemic cytokine surge → transient insulin resistance. |
| HBV / HCV | T2DM | Alter hepatic insulin signaling through hepatokine dysregulation. |
| HIV | T2DM | Viral proteins impair glucose uptake; antiretrovirals further worsen metabolic profiles. |
| SARS-CoV-2 | T1DM & T2DM (new onset cases reported) | Infects ACE2-expressing β-cells & adipose tissue → impaired insulin secretion + increased insulin resistance. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



