Submitted:
01 June 2026
Posted:
02 June 2026
You are already at the latest version
Abstract
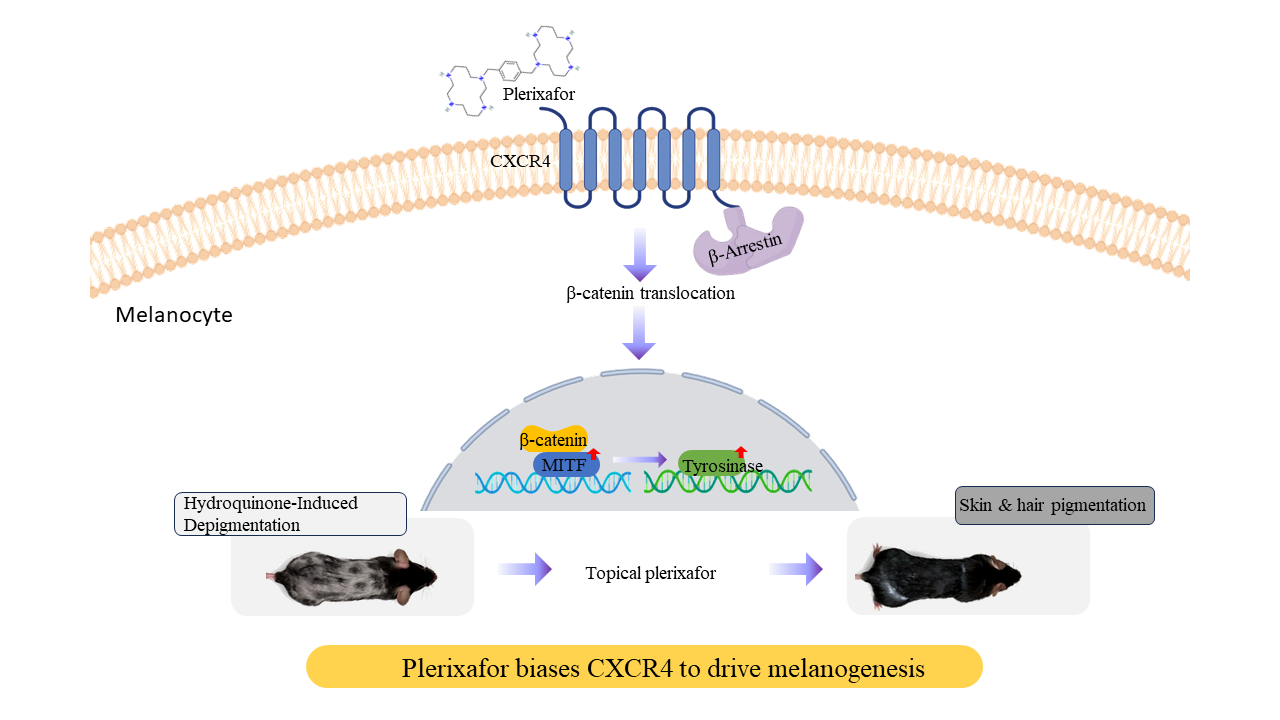
Plerixafor is a clinically approved CXCR4 antagonist that mobilizes hematopoietic stem cells by disrupting CXCL12/CXCR4 retention signaling. However, its biochemical effects on melanocytes and pigmentation remain unexplored. We investigated how plerixafor modulates CXCR4 sig-naling in melanocytes and evaluated its potential as a pro-melanogenic agent using in vitro and in vivo approaches. Human PIG1 melanocytes were treated with 10 nM plerixafor with or without hydro-quinone (HQ), followed by qPCR for MITF and tyrosinase expression, flow cytometry for CXCR4/CXCR7 and integrin profiling, transwell migration assays, β-arrestin siRNA knockdown, Western blotting, subcellular fractionation, and ChIP-qPCR for β-catenin binding to MITF regu-latory regions. A murine HQ-induced depigmentation model was used to test topical plerixafor on pigmentation, hair follicles, melanogenic gene expression, and systemic safety markers. Plerixafor significantly increased MITF and tyrosinase mRNA and enhanced melanocyte migration, while counteracting HQ-induced suppression of melanogenic genes. Plerixafor reduced cell-surface CXCR4 (consistent with β-arrestin–mediated receptor internalization) without altering CXCR7, c-KIT, or N-cadherin. β-arrestin knockdown abolished plerixafor-induced ERK phosphorylation and melanogenic responses, confirming β-arrestin dependence. Plerixafor promoted β-catenin nuclear translocation and direct β-catenin occupancy at MITF promoter/enhancer TCF/LEF motifs. In vivo, topical plerixafor restored HQ-induced depigmentation, increased hair follicle number and melanin content, and upregulated cutaneous MITF and tyrosinase without hepatic, renal, or inflammatory toxicity. Plerixafor functions as a biased CXCR4 ligand in melanocytes, engaging a β-arrestin–β-catenin–MITF signaling axis to drive melanogenesis and repigmentation. These findings identify β-arrestin–dependent CXCR4 signaling as a tractable pharmacologic mechanism for therapeutic repigmentation in pigmentary disorders.
Keywords:
CXCR4
; β-arrestin
; β-catenin
; MITF
; plerixafor
; repigmentation
1. Introduction
The fully differentiated human melanocyte, mainly found in the basal layer of the skin’s epidermis, is essential in this layer by producing melanin within its organelles and transferring these pigment-filled organelles to neighboring keratinocytes. Melanocytes respond to various signals, including UV radiation, hormones, and paracrine signaling from surrounding tissues, to control the amount and type of melanin produced [1]. Dysregulation of these factors can lead to pigmentation disorders which melanocytes are lost or dysfunctional. Research indicates that melanocytes are primarily located in reservoirs, particularly within hair follicles. However, the number of functional melanocytes and the ability to produce melanin tend to decrease with age. This decline contributes to the lightening of skin and hair color observed in older individuals [2]. Current treatments for pigmentation disorders often involve skin grafting or cell therapy using autologous melanocytes. However, these methods are limited by the poor proliferative ability of melanocytes obtained from skin biopsies. Recent advances in generating melanocytes from human pluripotent stem cells have demonstrated potential, providing a more sustainable source for cell therapy [3,4]. Promoting the generation of healthy melanocytes would likely involve enhancing or mimicking the signals that attract and support melanoblast/melanocyte survival and differentiation, which could potentially involve controlled manipulation of chemokine signaling [5]. Understanding the biochemical mechanisms by which CXCR4 antagonists modulate cellular phenotypes beyond stem cell mobilization may reveal novel therapeutic applications. Biased GPCR ligands that selectively activate β-arrestin pathways while blocking G-protein signaling represent a pharmacologically distinct mode of action with tissue-specific consequences that remain incompletely characterized in melanocytes.
Plerixafor (Mozobil®, AMD3100) is a medication primarily used in medicine to mobilize hematopoietic stem cells (HSCs) [6]. It blocks the binding of stromal cell-derived factor-1α (SDF-1α, also known as CXCL12) to CXCR4 [7,8]. This disruption leads to the mobilization or release of these cells from their niche into the peripheral circulation, resulting in their release and subsequent vulnerability to external factors. The CXCL12/CXCR4 signaling axis acts as a primary homing and retention signal. CXCL12 signals primarily through two receptors: CXCR4 and CXCR7. Both are G protein-coupled transmembrane receptors, but they differ in their signaling mechanisms and physiological roles [9]. Pleiotropic effects of the CXCR4/CXCR7/CXCR12 pathway have been reported in various physiopathological processes, as well as in malignant diseases [10,11]. Plerixafor is considered a biased antagonist of the CXCR4 receptor. It effectively blocks the binding of CXCL12 and prevents the activation of the G-protein signaling pathway, which is the canonical pathway responsible for the retention and survival signals within the stem cell niche [12]. Plerixafor also acts as a β-arrestin-biased agonist. Binding to CXCR4 promotes the recruitment of the β-arrestin protein that typically triggers CXCR4 internalization. CXCR7 acts as a decoy receptor, influencing CXCR4 binding to CXCL12 and thereby limiting excessive signaling, which helps maintain the balance between cell movement, survival, and differentiation [13,14].
Our laboratory previously screened a library of 1,500 FDA-approved drugs and unexpectedly discovered that plerixafor promotes melanin production in B16F10 cells. Studies have revealed its intriguing effects on skin biology and hair follicle dynamics. Treatment with plerixafor protects against the development of UV-induced skin cancer in murine models, highlighting the therapeutic potential of antagonizing this chemokine axis. Plerixafor promotes wound healing in diabetic patients and inhibits tumor proliferation [15,16,17]. CXCL12 enhances, while plerixafor relieves itch and pain sensations, in the allergic contact dermatitis mouse model [18]. Recently, Zheng et al. identified that silencing the CXCL12/CXCR4 axis stimulates hair growth in androgenetic alopecia [19]. CXCR4 expression in primary melanocytes was variable but consistent [20]. Therefore, we hypothesized that plerixafor acts as a β-arrestin–biased CXCR4 ligand in melanocytes, engaging a β-catenin–MITF transcriptional program to promote melanogenesis and repigmentation. To test this, we combined in vitro pathway dissection in human PIG1 melanocytes with an in vivo hydroquinone-induced depigmentation model to define the CXCR4-dependent mechanisms and therapeutic potential of plerixafor in pigmentation disorders. Hydroquinone (HQ) is a phenolic depigmenting agent that functions primarily as an alternate substrate and competitive inhibitor of tyrosinase, diverting tyrosine oxidation toward non-melanogenic quinones and thereby suppressing melanin synthesis in active melanocytes [21,22].
2. Materials and Methods
2.1. Melanocyte Culture and Treatment
Human PIG1 cells (purchased from ATCC) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., NY, USA), 100 U/mL penicillin, and 100 μg/mL streptomycin. Maintain the cells in a humidified incubator at 37°C with 5% CO2. Passage cells are regularly maintained using TrypLE Express (Thermo Scientific, MA, USA) to achieve approximately 80% confluency. PIG1 cells (1×106 /mL) were allowed to grow for 16 hours before treatment. The cells were treated with hydroquinone (1 μM) (AlfaAesar, Thermo Fisher Scientific, MA, USA) for 8 hours, followed by the replacement with plerixafor (10 nM) (MedChemExpress, NJ, USA) for 16 hours. The non-cytotoxic and pharmacokinetic dose of 1-10 nM of plerixafor was chosen to treat cells [23]. This concentration range approximates the nanomolar plasma levels achieved clinically during stem cell mobilization and has been established as non-cytotoxic in primary cells, allowing mechanistic interrogation of CXCR4 biased signaling without off-target toxicity.
2.2. Cell Migration Assay
PIG1 cells were seeded at a density of 1×104 cells per well in the upper chamber of a transwell plate with a pore size of 5 μm. The lower chamber contains a medium sup-plemented with plerixafor for 24 hours, allowing for migration. The number of migrated cells was fixed with methanol, stained with a 0.2% crystal violet solution, and counted under a microscope at a magnification of ×200 in five random fields per well. The migra-tion ratio was calculated as the % of trans-migrated cells compared with the DMSO ve-hicle control. This assay was repeated in triplicate for each experimental condition.
2.3. siRNA Transfection
β-arrestin1/2 siRNA oligonucleotides were purchased from Tri-I Biotech Inc., Taiwan. Their nucleotide sequences were as follows: β1-arrestin, 5’-AAAGCCUUCUGCGCGGAGAAU-3’ and β2-arrestin, 5’-AAGGACCGCAAAGUGUUUGUG-3’. PIG1 cells were seeded in six-well plates, and transfection was performed when the cell confluence reached approximately 60%. siRNAs (20 nM) were mixed with Lipofectamine RNAiMAX Reagent (Invitrogen, Thermo Fisher Scientific Inc.) according to the manufacturer’s protocol and used for transfection. Transfection efficiency was assessed using Western blot analysis.
2.4. Flow Cytometry
PIG1 cells (1 x 106/ mL) were harvested and incubated with 100 ng of fluorescently con-jugated monoclonal antibodies N-Cadherin (Genetex, CA, USA), c-kit (YR145; abcam), CXCR4 (HL2612; Genetex, CA, USA), CXCR7 (C1C2; Genetex, CA, USA), Integrin alpha 2 (EPR5788; abcam), Integrin alpha v (272-17E6; abcam), Integrin beta 1 (12G10; abcam) for 2 h. Cells were washed to remove unbound antibodies and then resuspended in PBS for analysis. A total of 30,000 events were recorded by the Attune NxT flow cytometer (Thermo Fisher Scientific Inc.). For each surface marker, a positive gate was established based on the control sample by setting a threshold at the mean fluorescence intensity (MFI). This gate was then applied uniformly to all treatment conditions within the same experiment. The % of positive cells was defined as the proportion of events exceeding this threshold. Data are also presented as fold change of MFI relative to vehicle controls. A minimum of three independent experiments was performed for statistical analysis.
2.5. RNA Isolation, Quantitative PCR (QPCR)
Total RNA was extracted using the TRIzol method, as per the manufacturer’s instructions. The RNA was then reverse-transcribed into cDNA using the PrimeScript RT kit. The resulting cDNA was subjected to quantitative fluorescence PCR (qPCR) according to the guidelines of the SYBR Premix Ex Taq II kit (TaKaRa, Tokyo, Japan). The RT-qPCR was conducted on a 7500 instrument (Applied Biosystems, Foster City, CA, USA). The ex-pression level was normalized against that of 18S rRNA, and the relative transcription levels were calculated using the relative quantification method (2-ΔΔCT). The primers are listed in Table S1.
2.6. Chromatin Immunoprecipitation-Quantitative PCR (ChIP-qPCR) Assay
The ChIP-qPCR assay was performed using the SimpleChIP® Plus Enzymatic Chromatin IP Kit (Cell Signaling Technology Inc.), according to the manufacturer’s instructions. The cells were harvested and fixed in 1% formaldehyde with vacuum infiltration. Crosslinking was quenched by adding glycine (0.125 M). The chromatin was collected and sonicated, and then immunoprecipitated using an anti-β-catenin antibody. The immunoprecipitated DNA fragments were analyzed by quantitative PCR using specific primers (Table S2). Enriched values were normalized with the level of the IgG group.
2.7. Western Blot Analysis
Proteins of PIG1 were immunoblotted with specific primary antibodies against ERK (1:3000; EPR17526; abcam), pERK (1:1000; EPR19401; abcam), β-catenin (1:1000; D10A8; Cell Signaling Technology, Danvers, MA, USA), β-arrestin1/2 (1:1000; D24H9; Cell Sig-naling Technology), β-actin (1:10000; Genetex, CA, USA), GAPDH (1:10000; Genetex, CA, USA), Lamin B1 (1:5000; Genetex, CA, USA), and then incubated with the corresponding secondary antibodies. The luminescence intensity of bands was quantified by using ImageJ.
2.8. Animals Treatments
All animal experiments were approved by the Institutional Animal Care and Use Com-mittee of China Medical University (Approval No. CMUIACUC-2024-115) and conducted in accordance with the International Standards on Animal Welfare. Female C57BL/6 mice were purchased from the National Center for Biomodels (NCB, Taiwan). Mice were sensitized to 2% HQ by applying a DMSO solution of HQ onto shaved flanks (100 μl/mouse) for three weeks. The mice were divided into 4 groups (5 mice each) using stratified randomization. For the experimental groups, 10 mM or 0.1 mM of plerixafor was applied to the mice’s backs daily for 14 days. Topical concentrations of 0.1–10 mM were selected based on preliminary dose-finding experiments and to achieve local tissue exposure sufficient for CXCR4 engagement without systemic effects. The control group received DMSO at the same interval. All mice were sacrificed and sampled on the day 37.
2.9. Specimens and Immunohistochemistry
The skin tissue (1 cm²) of mice was harvested by administering pentobarbital (200 mg/kg, IP). The skin samples were fixed in a 4% buffered neutral formalin solution for 24 hours at room temperature, and then embedded in paraffin. Serial sections were floated in warm water containing 2% gelatin to prevent them from peeling off the sample. The 5-mm par-affin cross-sections were deparaffinized, rehydrated, and stained with hematoxylin & eosin (H&E) for histochemical analysis or with the Fontana-Masson Stain Kit for staining melanin. For each mouse, three non-overlapping fields per section were acquired from the interscapular area at 200× magnification. The image quantification was performed using ImageJ software [24]. Values from individual fields were averaged to obtain a single value per animal, and one-way ANOVA compared group means with post-hoc testing.
2.10. Inflammatory Cytokine Activity
To assess the effect of plerixfor on proinflammatory markers, the Mouse Inflammation Antibody Array Membrane (40 targets) (Abcam 133999, Cambridge, UK) was used, following the manufacturer’s protocol. Briefly, 250 µg of serum from each sample was diluted in 1 mL of blocking buffer and applied to each array membrane. After washing, biotin-conjugated antibodies were added, followed by incubation with streptavidin con-jugated to horseradish peroxidase. The membranes were then visualized with a Chemi-Doc imaging system (Bio-Rad, Hercules, USA). Densitometric analysis of the signals was performed using the Protein Array Analyzer tool in ImageJ software (Research Services Branch, National Institute of Mental Health, Bethesda, USA). Array data were normalized according to the manufacturer’s instructions. Heatmaps of the results were generated using Microsoft Excel.
2.11. Statistical Analysis
All quantitative data were obtained from at least three independent experiments and are presented as the mean ± standard deviation (SD). Results were statistically evaluated using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test or Stu-dent’s t-test, using the Statistical Package for the Social Sciences (SPSS) version 21 (IBM, Armonk, NY, USA). Significance thresholds were set at p < 0.05 *, p < 0.01 **, and p < 0.005 *** for comparisons with the control. Comparisons within the HQ group are denoted as p < 0.05 #, p < 0.01 ##, and p < 0.005 ### at equivalent thresholds.
3. Results
3.1. Plerixafor Enhanced Melanogenesis
We examined the pro-melanogenic activity of plerixafor by measuring the expression of the key melanogenic regulator MITF at 24 h, tyrosinase at 48 h, and the melanosome structural protein PMEL at 48 h using qPCR in PIG1 cells. Plerixafor markedly upregulated MITF and tyrosinase mRNA levels compared with the control group, while maintaining PMEL expression close to basal levels. In contrast, HQ treatment substantially suppressed MITF, tyrosinase, and PMEL transcripts, consistent with its inhibitory effect on tyrosinase activity and melanogenic gene expression; however, co-treatment with plerixafor largely reversed the HQ-induced downregulation of MITF and tyrosinase, with only a modest restoration of PMEL expression. These findings suggest that plerixafor stimulates melanogenesis predominantly by enhancing the MITF/tyrosinase transcriptional program, thereby promoting melanin production, and can counteract HQ-mediated transcriptional suppression even though it may not directly relieve enzymatic inhibition of tyrosinase.
Figure 1.
Plerixafor reverses HQ-induced suppression of melanogenic genes in PIG1 cells. PIG1 cells were treated with HQ, plerixafor, or their combination, and MITF (24 h), tyrosinase (TYR, 48 h), and PMEL (48 h) mRNA levels were quantified by qPCR using 18S rRNA as an internal control. Relative expression levels were normalized to the control (set as 1.0) and are shown as the mean ± SD from at least three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.005 versus control; # p < 0.05, ## p < 0.01, ### p < 0.005 versus HQ.
Figure 1.
Plerixafor reverses HQ-induced suppression of melanogenic genes in PIG1 cells. PIG1 cells were treated with HQ, plerixafor, or their combination, and MITF (24 h), tyrosinase (TYR, 48 h), and PMEL (48 h) mRNA levels were quantified by qPCR using 18S rRNA as an internal control. Relative expression levels were normalized to the control (set as 1.0) and are shown as the mean ± SD from at least three independent experiments. * p < 0.05, ** p < 0.01, *** p < 0.005 versus control; # p < 0.05, ## p < 0.01, ### p < 0.005 versus HQ.

3.2. Effects of Plerixafor and HQ on PIG1 Cell Migration and Surface Marker Expression
The migration ability in PIG1 cells was enhanced by plerixafor (Figure 2). Surface expression of CXCR4, CXCR7, c-KIT, N-cadherin, and integrin subunits was assessed by flow cytometry. In control PIG1 cells, CXCR4-positive cells constituted approximately 23% of the population. Following plerixafor treatment, the percentage of CXCR4-positive cells decreased significantly to 10% (p< 0.05), consistent with receptor internalization [7]. In contrast, the rate of CXCR7-positive cells remained unchanged across treatment groups, indicating that plerixafor selectively modulates CXCR4. (Figure 3A). For melanocyte lineage markers, c-KIT and N-cadherin expression remained stable, with no significant change following HQ or plerixafor treatment.
Integrin profiling revealed that HQ treatment reduced the percentage of integrin β1-positive cells from 48% to 8% (p<0.05). Conversely, integrin αv-positive cells increased from 49% to 99% following HQ treatment (p<0.05), suggesting a shift in integrin heterodimer composition. Plerixafor did not significantly alter integrin β1 or αv expression compared with HQ-treated cells, indicating that plerixafor-induced migration occurs independently of integrin modulation. (Figure 3B). These results suggest that HQ shifts the balance of integrins β1/αv, which can influence cell motility; however, the actual outcomes depend on how the integrin is reconfigured and the specific composition of the extracellular matrix [25].
3.3. Plerixafor Enhances β-Catenin Activation
We further investigated the mechanism in PIG1 cells responding to plerixafor, which blunts CXCR4-driven retention in combination with integrin modulation. Plerixafor exhibits biased antagonism at CXCR4 by selectively blocking G protein–mediated signaling while still promoting β-arrestin recruitment to the receptor. [26]. We use siRNA to knock down the level of β-arrestin to dissect pathway specificity in melanocytes in plerixafor signaling. Transfection with si-β-arrestin markedly reduced β-arrestin protein levels, as confirmed by western blotting (Figure 4A). β-arrestin knockdown markedly re-duces basal and plerixafor-mediated ERK phosphorylation, indicating that when G-protein signaling is blocked by plerixafor, any residual ERK activity is β-arrestin–dependent Nevertheless, the loss of plerixafor responsiveness in β-arrestin–deficient cells supports a critical role for β-arrestin in this pathway.
Among key transcription factors binding the MITF Promoter, a notable β-catenin trans-location was detected in subcellular fractionation, compared with vehicle controls (Figure 4B). To determine the direct association with MITF regulatory sequences, we performed ChIP using anti-β-catenin antibodies, followed by qPCR with primers targeting the MITF promoter/enhancer regions that contain putative TCF/LEF binding motifs. ChIP-qPCR revealed a significant enrichment of β-catenin at these MITF regulatory loci in stimulated cells versus HQ (5- to 8-fold, p<0.05), indicating that nuclear β-catenin directly binds to MITF-associated chromatin regions. These data showed that β-catenin translocation to the nucleus enables direct transcriptional regulation of MITF by β-catenin–TCF/LEF complexes, linking signaling to MITF-mediated transcriptional programs (Figure 4C).
3.4. Plerixafor Improved Re-Pigmentation in a Mouse Model Treated with HQ
Using HQ caused visible depigmentation on the backs of C57BL/6 mice. Topically applied plerixafor improved repigmentation and increased the expression of MITF and tyrosinase in skin tissue. Significant difference in the number of hair follicles was observed between the experimental groups. Fontana-Masson staining was used to visualize and measure melanin, revealing that HQ eliminated hair follicles and their melanin content by ap-proximately 50% compared with controls. In contrast, topical plerixafor treatment re-stored the eliminated hair follicles, resulting in a 1.5-fold increase in their number and melanin content (p<0.05), consistent with restoration of pigment production. Figure 5 provides the principal functional readout of pigment restoration in vivo, integrating follicular melanocyte activation and tissue-level melanin deposition in the HQ-treated mouse model. These structural changes did not differ following concentration variation; it ap-pears that the 0.1 mM topical dose is sufficient to affect melanogenesis and enhance hair follicle regeneration. Skin tissue analysis and HE staining revealed no local skin irritation or histologic inflammation. Biochemical tests revealed that serum aspartate aminotransferase (AST) and creatinine levels remained within normal ranges, confirming that the animal study did not cause liver or kidney damage and did not induce toxicity. The spleen-to-body weight ratio showed no significant difference compared to the control group. This in vivo mouse model therefore provides the primary tissue-level evidence that plerixafor restores melanin deposition and repigmentation.
Cytokine antibody array analysis demonstrated that neither HQ alone nor plerixafor, given individually or in HQ+plerixafor combination, altered the inflammatory cytokine profile compared with control. Across all 40 pro-inflammatory cytokines measured, signal intensities in the plerixafor group remained within the same range as those in the control groups, without consistent upward or downward trends, indicating that plerixafor treatment, alone or combined with HQ, did not measurably modulate this panel of inflammation-related mediators under the tested conditions (Figure 6).
4. Discussion
CXCL12/CXCR4 represents a prototypical chemokine axis in which biased ligands can differentially engage G protein– versus β-arrestin–dependent signaling to modify cellular phenotypes. Plerixafor is clinically used as a CXCR4 antagonist for hematopoietic stem cell mobilization, but its biased actions and downstream signaling consequences in melanocytes have not been defined. Elucidating how plerixafor reprograms CXCR4 signaling at the biochemical and molecular levels, and how this translates into changes in melanocyte behavior and skin pigmentation, is therefore of interest for both GPCR bi-ased-signaling pharmacology and drug repurposing. Our results show that plerixafor, a CXCR4 antagonist with biased signaling properties, has a strong pro-melanogenic effect by increasing MITF and tyrosinase expression in human melanocytes. The increase in MITF and tyrosinase mRNA levels after exposure to plerixafor directly supports its role in promoting melanogenic gene programs. Importantly, plerixafor countered HQ-induced suppression of melanogenesis, restoring MITF and tyrosinase expression and providing a biological rescue against depigmentation agents. It would be helpful to test the efficacy of plerixafor in combination with standard therapies (NB-UVB, topical steroids/JAK inhibitors) to determine if plerixafor enhances repigmentation in future work.
Melanocyte stem cells serve as the source of melanocyte replenishment in adult skin. They are found in specific skin niches, such as the hair follicle bulge region, the epidermal basement membrane, and sweat glands [27]. Homeostasis of melanocytes depends on the closely coordinated signaling of melanocyte stem cells, paracrine factor communication, transcriptional networks, and immune/stress responses to regulate melanocyte survival, quantity, and melanin production, thereby maintaining healthy skin pigmentation [28]. Further research is necessary to understand how recruiting these progenitor cells in specific situations can provide pigment cell sources for repigmenting depigmented areas. Yamada, T. et al. reported that CXCL12 attracts melanocyte stem cells to the proper position, facilitating the maintenance of their undifferentiated state [29]. The same team re-ported that differentiation of melanocytes occurs through the activation of β-catenin and migration to the epidermis to complete repigmentation in mice [30]. Our study showed that the CXCL12-CXCR4 axis antagonist plerixafor activates a β-arrestin–dependent signaling pathway associated with β-catenin nuclear translocation, interaction with T-cell-specific factor/lymphoid enhancer-binding factor 1 (TCF/LEF-1), and binding to the TCF/LEF-1 responsive region of the MITF promoter, suggesting that plerixafor blunts the CXCL12-mediated retention signal and promotes melanogenic differentiation-associated signaling. Moreover, topically applied plerixafor enhances the re-pigmentation of depigmented skin and promotes hair follicle growth in mice, also re-vealing the maturation and migration of pigment cells.
β-arrestin is essential for plerixafor signaling in melanocytes as evidenced by the com-plete loss of plerixafor effects when β-arrestin was silenced. In this study, a dual siRNA approach targeting β-arrestin1 and β-arrestin2 was used to obtain a more robust knock-down than either single siRNA alone. Future work will use complementary approaches, such as alternative siRNA sequences or rescue experiments, to further validate knock-down specificity. Plerixafor antagonizes the CXCL12-CXCR4–ERK pathway by fully blocking G-protein activation at CXCR4, while simultaneously engaging a β-arrestin-biased route that supports alternative downstream β-catenin/MITF without restoring canonical ERK signaling. This β-arrestin dependence, coupled with CXCR4 internalization, indicates that plerixafor operates via biased signaling rather than canonical G protein pathways. Future studies dissecting alternative β-arrestin–scaffolded effectors (e.g., PI3K, Src, Rho GTPases) [31] may clarify which downstream nodes drive melano-genesis versus migration. The nuclear translocation of β-catenin, observed through sub-cellular fractionation and ChIP-qPCR analysis, reveals a key downstream mechanism by which plerixafor boosts MITF transcriptional activity. β-catenin directly binds to the TCF/LEF motif [CTTTGAT] located near the transcriptional start site on the MITF promoter/enhancer, thereby linking CXCR4 antagonism to canonical Wnt signaling pathways that promote melanogenic gene expression. This molecular insight improves un-der-standing of how CXCR4 signaling influences melanocyte biology beyond chemotaxis and retention.
A previously reported study revealed that CXCL12 binding to CXCR4 receptors in-duces the activation of integrins [32]. In essence, plerixafor’s effect on cell migration was evidenced by increased motility in PIG1 cells, independent of changes in the expression of melanocyte surface markers c-KIT and N-cadherin. Notably, plerixafor reduced CXCR4 surface expression, consistent with prior studies showing receptor internalization driven by β-arrestin recruitment under biased antagonism. CXCR7 levels remained stable, underscoring a selective modulation of CXCR4 by plerixafor. Additionally, HQ treatment reduced the surface levels of integrin β1. It concomitantly increased integrin αv expression, consistent with a shift in available heterodimer pairs toward αv-containing receptors when β1 is limiting, as described for epithelial and carcinoma cells [33]. This pattern is compatible with altered adhesion to fibronectin-rich or vitronectin-rich matrices and, in principle, could influence melanocyte anchorage and migration [34]. Still, integrin regulation appears to play a permissive rather than primary role in the pro-migratory effects of plerixafor. Although both HQ and plerixafor alone induced a comparable reduction of CXCR4 at the cell surface, their downstream consequences for melanocyte behavior ap-pear to be mechanistically distinct. In our system, HQ primarily disrupts the αv/β1 integrin balance, which may alter cell–matrix interactions and create a ‘primed’ state that is more responsive to chemokine cues, but it does not by itself strongly activate CXCR4–β-arrestin signaling or ERK/β-catenin–MITF pathways. By contrast, plerixafor acts as a β-arrestin–biased CXCR4 ligand, driving robust β-arrestin–dependent ERK activation and β-catenin–MITF signaling and thereby enhancing melanocyte motility, even in the context of HQ-induced integrin imbalance. Thus, rather than further modifying the integrin repertoire, plerixafor appears to exploit an HQ-sensitized background to promote chemokine receptor–driven migration predominantly through CXCR4–β-arrestin signaling. Future work using integrin-blocking antibodies or defined ECM coatings will be required to dissect this crosstalk more precisely.
In our system, HQ decreased the expression of melanogenic genes, reduced CXCR4 sur-face levels, and altered cytoskeletal organization, thereby changing melanocyte motility, which is consistent with previous reports [35,36]. While plerixafor-induced CXCR4 downregulation can be attributed to β-arrestin-mediated receptor internalization, the mechanism underlying HQ-induced CXCR4 reduction requires further investigation, although it may involve altered receptor stability under oxidative stress. Because PIG1 cells contain little to no detectable melanin, they were used in this study primarily as a mechanistic human melanocyte model for transcriptional and signaling analyses, and a 2% hydroquinone solution was applied to mice to reliably induce stable hypopigmenta-tion of the hair and skin for tissue-level evaluation of pigment restoration. Given that plerixafor treatment generated round, melanin-containing hair follicles, it is inferred that plerixafor promoted hair follicle regeneration, maturation of melanogenic progenitor cells, and melanin production in both hair follicles and the epidermis. In addition, the in vivo study demonstrated consistent pigmentary efficacy at two distinct topical doses of plerixafor (0.1 mM and 10 mM), both of which improved repigmentation and increased melanogenic readouts under the present experimental conditions. Although these data support reproducible biological activity across a 100-fold dose range, a full dose–response analysis will still be required in future studies to more precisely define the minimum effective dose and therapeutic window. Additionally, the WNT/β-catenin signaling pathway is one of the earliest and most critical pathways for hair follicle induction [37]. In this study, topical plerixafor at the tested doses did not alter serum AST or creatinine levels, spleen-to-body-weight ratio, or the profile of 40 pro-inflammatory cytokines in mouse serum, suggesting a lack of overt systemic toxicity and measurable systemic inflammation under the experimental conditions. Histological examination of treated skin also did not reveal local inflammatory infiltrates or tissue damage, indicating that plerixafor’s pigmentary and hair follicle effects occur without clear histopathological evidence of dermal toxicity in this model. The functional role of plerixafor related to hair follicles remains an area in need of in-depth exploration and explanation, and more comprehensive toxicology studies will be required to rigorously define the safety profile of topical plerixafor for the future.
5. Conclusions
In summary, this study demonstrates that plerixafor enhances melanogenesis-related re-sponses by engaging a β-arrestin–dependent CXCR4 signaling pathway associated with β-catenin–dependent MITF transcriptional regulation. This signaling mechanism pro-motes melanogenesis-associated gene expression and melanocyte migration while counteracting the effects of depigmenting agents, offering a promising strategy for therapeutic repigmentation and treatment of pigmentary disorders. Further research should investigate the interplay between integrin dynamics and CXCR4 signaling in melanocyte motility, as well as the long-term effects of plerixafor in clinical pigmentation disorders.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, Huey-Chun Huang, Tsong-Min Chang; Methodology, Tsong-Min Chang, Ting-Ya Yang; Writing, Review & Editing, Huey-Chun Huang, Tsong-Min Chang; Funding Acquisition, Huey-Chun Huang.
Funding
The Ministry of Science and Technology funded this study under grant numbers NSTC 112-2635-B-039-004 and a grant from China Medical University, Taiwan (CMU112-MF-104). We appreciate the experiments and data analysis performed through the use of the Medical Research Core Facilities Office of Research & Development at China Medical University, Taichung, Taiwan, ROC.
Conflicts of Interest
The authors declare no competing financial interests.
Abbreviations
The following abbreviations are used in this manuscript:
| HQ | Hydroquinone |
| Tyr | Tyrosinase |
| MITF | Microphthalmia-associated transcription factor |
| PMEL | Premelanosome protein |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
References
- Snyman, M.; Walsdorf, R.E.; Wix, Sophia N.; Gill, J.G. The metabolism of melanin synthesis—From melanocytes to melanoma. 2024, 37, 438–452. [Google Scholar] [CrossRef]
- Sun, Q.; Lee, W.; Hu, H.; Ogawa, T.; De Leon, S.; Katehis, I.; Lim, C.H.; Takeo, M.; Cammer, M.; Taketo, M.M.; et al. Dedifferentiation maintains melanocyte stem cells in a dynamic niche. Nature 2023, 616, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Kobori, C.; Takagi, R.; Yokomizo, R.; Yoshihara, S.; Mori, M.; Takahashi, H.; Javaregowda, P.K.; Akiyama, T.; Ko, M.S.H.; Kishi, K.; et al. Functional and long-lived melanocytes from human pluripotent stem cells with transient ectopic expression of JMJD3. Stem Cell Res. Ther. 2023, 14, 242. [Google Scholar] [CrossRef]
- Coutant, K.; Magne, B.; Ferland, K.; Fuentes-Rodriguez, A.; Chancy, O.; Mitchell, A.; Germain, L.; Landreville, S. Melanocytes in regenerative medicine applications and disease modeling. J. Transl. Med. 2024, 22, 336. [Google Scholar] [CrossRef]
- Jia, X.; He, L. Unveiling the dual-edged roles of melanocytes: Key insights into skin photoaging and therapeutic avenues. Pharmacol. Res. 2025, 218, 107848. [Google Scholar] [CrossRef]
- De Clercq, E. Mozobil® (Plerixafor, AMD3100), 10 years after its approval by the US Food and Drug Administration. Antivir. Chem. Chemother. 2019, 27, 2040206619829382. [Google Scholar] [CrossRef]
- Jørgensen, A.S.; Daugvilaite, V.; De Filippo, K.; Berg, C.; Mavri, M.; Benned-Jensen, T.; Juzenaite, G.; Hjortø, G.; Rankin, S.; Våbenø, J.; et al. Biased action of the CXCR4-targeting drug plerixafor is essential for its superior hematopoietic stem cell mobilization. Commun. Biol. 2021, 4, 569. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Van Noorden, C.J.F.; Molenaar, R.J. CXCR4 Antagonists as Stem Cell Mobilizers and Therapy Sensitizers for Acute Myeloid Leukemia and Glioblastoma? Biology 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Xia, Y.; Zuo, K.; Wang, Y.; Zhang, S.; Kuang, D.; Duan, Y.; Zhao, X.; Wang, G. Crosstalk between SDF-1/CXCR4 and SDF-1/CXCR7 in cardiac stem cell migration. Sci. Rep. 2015, 5, 16813. [Google Scholar] [CrossRef]
- Shi, Y.; Riese, D.J., 2nd; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front Pharmacol. 2020, 11, 574667. [Google Scholar] [CrossRef] [PubMed]
- Britton, C.; Poznansky, M.C.; Reeves, P. Polyfunctionality of the CXCR4/CXCL12 axis in health and disease: Implications for therapeutic interventions in cancer and immune-mediated diseases. Faseb J. 2021, 35, e21260. [Google Scholar] [CrossRef]
- Sison, E.A.; Magoon, D.; Li, L.; Annesley, C.E.; Rau, R.E.; Small, D.; Brown, P. Plerixafor as a chemosensitizing agent in pediatric acute lymphoblastic leukemia: efficacy and potential mechanisms of resistance to CXCR4 inhibition. Oncotarget 2014, 5, 8947–8958. [Google Scholar] [CrossRef] [PubMed]
- Scala, S. Molecular Pathways: Targeting the CXCR4–CXCL12 Axis—Untapped Potential in the Tumor Microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef]
- Heinrich, E.L.; Lee, W.; Lu, J.; Lowy, A.M.; Kim, J. Chemokine CXCL12 activates dual CXCR4 and CXCR7-mediated signaling pathways in pancreatic cancer cells. J. Transl. Med. 2012, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Ii, M.; Qin, G.J.; Hamada, H.; Asai, J.; Takenaka, H.; Sekiguchi, H.; Renault, M.A.; Jujo, K.; Katoh, N.; et al. CXCR4 Antagonist AMD3100 Accelerates Impaired Wound Healing in Diabetic Mice. J. Investig. Dermatol. 2012, 132, 711–720. [Google Scholar] [CrossRef]
- Jacquelot, N.; Duong, C.P.M.; Belz, G.T.; Zitvogel, L. Targeting Chemokines and Chemokine Receptors in Melanoma and Other Cancers. Front Immunol. 2018, 9, 2480. [Google Scholar] [CrossRef]
- Liu, H.; Liu, H.; Deng, X.; Chen, M.; Han, X.; Yan, W.; Wang, N. CXCR4 antagonist delivery on decellularized skin scaffold facilitates impaired wound healing in diabetic mice by increasing expression of SDF-1 and enhancing migration of CXCR4-positive cells. Wound Repair Regen. 2017, 25, 652–664. [Google Scholar] [CrossRef]
- Su, W.; Yu, J.; Liu, Q.; Ma, L.; Huang, Y. CXCL12/CXCR4 signaling induced itch and pain sensation in a murine model of allergic contact dermatitis. Mol. Pain 2020, 16, 1744806920926426. [Google Scholar] [CrossRef]
- Zheng, M.; Oh, S.H.; Choi, N.; Choi, Y.J.; Kim, J.; Sung, J.-H. CXCL12 inhibits hair growth through CXCR4. Biomed. Pharmacother. 2022, 150, 112996. [Google Scholar] [CrossRef]
- McConnell, A.T.; Ellis, R.; Pathy, B.; Plummer, R.; Lovat, P.E.; O’Boyle, G. The prognostic significance and impact of the CXCR4–CXCR7–CXCL12 axis in primary cutaneous melanoma. British Journal of Dermatology 2016, 175, 1210–1220. [Google Scholar] [CrossRef]
- Searle, T.; Al-Niaimi, F.; Ali, F.R. Hydroquinone: myths and reality. Clin. Exp. Dermatol. 2021, 46, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yang, Y.F.; Yanhua, L.; Zhang, Y.; Haixia, S. Therapeutic effects of topical Mycophenolate mofetil on hydroquinone-induced depigmentation in Guinea pigs and mice. Ann. Med. 2025, 57, 2544880. [Google Scholar] [CrossRef]
- Lemery, S.J.; Hsieh, M.M.; Smith, A.; Rao, S.; Khuu, H.M.; Theresa, D.; Viano, J.M.; Cook, L.; Goodwin, R.; Boss, C.; et al. A pilot study evaluating the safety and CD34+ cell mobilizing activity of escalating doses of plerixafor in healthy volunteers. Br. J. Haematol. 2011, 153, 66–75. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Hayashida, T.; Jones, J.C.R.; Lee, C.K.; Schnaper, H.W. Loss of beta1-integrin enhances TGF-beta1-induced collagen expression in epithelial cells via increased alphavbeta3-integrin and Rac1 activity. J. Biol. Chem. 2010, 285, 30741–30751. [Google Scholar] [CrossRef] [PubMed]
- Clift, I.C.; Bamidele, A.O.; Rodriguez-Ramirez, C.; Kremer, K.N.; Hedin, K.E. β-Arrestin1 and Distinct CXCR4 Structures Are Required for Stromal Derived Factor-1 to Downregulate CXCR4 Cell-Surface Levels in Neuroblastoma. Mol. Pharmacol. 2014, 85, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zuo, Y.; Li, S.; Li, C. Melanocyte stem cells in the skin: Origin, biological characteristics, homeostatic maintenance and therapeutic potential. Clin. Transl. Med. 2024, 14, e1720. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Fisher, D.E. Melanocyte biology and skin pigmentation. Nature 2007, 445, 843–850. [Google Scholar] [CrossRef]
- Yamada, T.; Hasegawa, S.; Hasebe, Y.; Kawagishi-Hotta, M.; Arima, M.; Iwata, Y.; Kobayashi, T.; Numata, S.; Yamamoto, N.; Nakata, S.J.A.o.D.R. CXCL12 regulates differentiation of human immature melanocyte precursors as well as their migration. 2019, 311, 55–62. [Google Scholar] [CrossRef]
- Yamada, T.; Hasegawa, S.; Inoue, Y.; Date, Y.; Yamamoto, N.; Mizutani, H.; Nakata, S.; Matsunaga, K.; Akamatsu, H.J.J.o.I.D. Wnt/β-catenin and kit signaling sequentially regulate melanocyte stem cell differentiation in UVB-induced epidermal pigmentation. 2013, 133, 2753–2762. [Google Scholar] [CrossRef]
- Miller, W.E.; Lefkowitz, R.J. Expanding roles for β-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr. Opin. Cell Biol. 2001, 13, 139–145. [Google Scholar] [CrossRef]
- Hartmann, T.N.; Burger, J.A.; Glodek, A.; Fujii, N.; Burger, M.J.O. CXCR4 chemokine receptor and integrin signaling co-operate in mediating adhesion and chemoresistance in small cell lung cancer (SCLC) cells. 2005, 24, 4462–4471. [Google Scholar] [CrossRef]
- Hayashida, T.; Jones, J.C.; Lee, C.K.; Schnaper, H.W. Loss of beta1-integrin enhances TGF-beta1-induced collagen expression in epithelial cells via increased alphavbeta3-integrin and Rac1 activity. J. Biol. Chem. 2010, 285, 30741–30751. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, L.; Grenman, R.; Heino, J.; Larjava, H. Integrins α5β1, αvβ1, and αvβ6 Collaborate in Squamous Carcinoma Cell Spreading and Migration on Fibronectin. Exp. Cell Res. 2000, 255, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Fabian, I.M.; Sinnathamby, E.S.; Flanagan, C.J.; Lindberg, A.; Tynes, B.; Kelkar, R.A.; Varrassi, G.; Ahmadzadeh, S.; Shekoohi, S.; Kaye, A.D. Topical Hydroquinone for Hyperpigmentation: A Narrative Review. Cureus 2023, 15, e48840. [Google Scholar] [CrossRef]
- Inoue, Y.; Hasegawa, S.; Yamada, T.; Date, Y.; Mizutani, H.; Nakata, S.; Matsunaga, K.; Akamatsu, H. Analysis of the Effects of Hydroquinone and Arbutin on the Differentiation of Melanocytes. Biol. Pharm. Bull. 2013, 36, 1722–1730. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Zhu, Z.; Sun, X.; Fu, X. Functional hair follicle regeneration: an updated review. Signal Transduct. Target. Ther. 2021, 6, 66. [Google Scholar] [CrossRef]
Figure 2.
Plerixafor promotes PIG1 cell migration. Scale bar 100 µm. The results are presented as the mean ± SD from at least three independent experiments. P-values are indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.005 compared to the control. #p < 0.05 compared to the HQ group.
Figure 2.
Plerixafor promotes PIG1 cell migration. Scale bar 100 µm. The results are presented as the mean ± SD from at least three independent experiments. P-values are indicated as follows: * p < 0.05, ** p < 0.01, *** p < 0.005 compared to the control. #p < 0.05 compared to the HQ group.

Figure 3.
Plerixafor modulates integrin expression and CXCR receptor levels in PIG1 cells. (A) Representative histograms showing fluorescence intensity distributions for CXCR4, CXCR7, c-KIT, and N-cadherin; gray-lined histograms represent vehicle controls. (B) Integrin subunit expression (αv, α2, β1). The right panels show quantification as fold change in mean fluorescence intensity (MFI) relative to controls. Data represent mean ± SD from three independent experiments. Statistical significance: *p<0.05, **p<0.01, ***p<0.001 vs control; #p<0.05 vs HQ group.
Figure 3.
Plerixafor modulates integrin expression and CXCR receptor levels in PIG1 cells. (A) Representative histograms showing fluorescence intensity distributions for CXCR4, CXCR7, c-KIT, and N-cadherin; gray-lined histograms represent vehicle controls. (B) Integrin subunit expression (αv, α2, β1). The right panels show quantification as fold change in mean fluorescence intensity (MFI) relative to controls. Data represent mean ± SD from three independent experiments. Statistical significance: *p<0.05, **p<0.01, ***p<0.001 vs control; #p<0.05 vs HQ group.

Figure 4.
Plerixafor activates β-arrestin–dependent ERK and β-catenin–MITF signaling in PIG1 melanocytes. (A) Representative Western blots showing phosphorylated ERK1/2 (pERK) and total ERK1/2 (ERK) in PIG1 cells transfected with control siRNA or β arrestin1/2 siRNA and treated with plerixafor or vehicle. GAPDH is shown as the loading control. The right panel shows densitometric quantification of the p ERK/ERK ratio, normalized to the control group. ##p < 0.01 vs. si-control. (B) Western blot analysis of β catenin in cytoplasmic and nuclear fractions from PIG1 cells treated with plerixafor or vehicle. GAPDH and lamin B1 are shown as cytoplasmic and nuclear loading controls, respectively. (C) ChIP qPCR analysis showing β catenin binding to MITF promoter/enhancer regions containing TCF/LEF motifs following plerixafor treatment. Fold enrichment is shown relative to the IgG control. Data are presented as mean ± SD. **p < 0.01, ***p < 0.001 vs. the control group.
Figure 4.
Plerixafor activates β-arrestin–dependent ERK and β-catenin–MITF signaling in PIG1 melanocytes. (A) Representative Western blots showing phosphorylated ERK1/2 (pERK) and total ERK1/2 (ERK) in PIG1 cells transfected with control siRNA or β arrestin1/2 siRNA and treated with plerixafor or vehicle. GAPDH is shown as the loading control. The right panel shows densitometric quantification of the p ERK/ERK ratio, normalized to the control group. ##p < 0.01 vs. si-control. (B) Western blot analysis of β catenin in cytoplasmic and nuclear fractions from PIG1 cells treated with plerixafor or vehicle. GAPDH and lamin B1 are shown as cytoplasmic and nuclear loading controls, respectively. (C) ChIP qPCR analysis showing β catenin binding to MITF promoter/enhancer regions containing TCF/LEF motifs following plerixafor treatment. Fold enrichment is shown relative to the IgG control. Data are presented as mean ± SD. **p < 0.01, ***p < 0.001 vs. the control group.

Figure 5.
Plerixafor induces re-pigmentation in HQ-treated C57BL/6 mice. (A) Represented H&E stain photographs. (B) Fontana–Masson–stained skin sections and quantified melanin content. Scale bar 100 µm. (C) The spleen weight and body weight ratio are shown in. (D) The expression of the indicated genes in mouse skin tissue was measured by qPCR. These results are presented as the mean ± SD from three independent experiments. P-values are shown as follows: * p < 0.05, ** p < 0.01, *** p < 0.005 compared to the HQ group.
Figure 5.
Plerixafor induces re-pigmentation in HQ-treated C57BL/6 mice. (A) Represented H&E stain photographs. (B) Fontana–Masson–stained skin sections and quantified melanin content. Scale bar 100 µm. (C) The spleen weight and body weight ratio are shown in. (D) The expression of the indicated genes in mouse skin tissue was measured by qPCR. These results are presented as the mean ± SD from three independent experiments. P-values are shown as follows: * p < 0.05, ** p < 0.01, *** p < 0.005 compared to the HQ group.

Figure 6.
Effect of Plerixafor on proinflammatory cytokine protein levels in the serum of mice. The cytokine expression is displayed as heatmaps, with dark green indicating no expression and red showing the highest levels observed.
Figure 6.
Effect of Plerixafor on proinflammatory cytokine protein levels in the serum of mice. The cytokine expression is displayed as heatmaps, with dark green indicating no expression and red showing the highest levels observed.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



