Submitted:
28 January 2026
Posted:
28 January 2026
You are already at the latest version
Abstract
Development of new polymers, that cannot be achieved by using conventional catalysts has been the central research objective, and copolymerization is an effective strategy to modify the materials’ (thermal, physical, mechanical and electronic) properties. Modified half-titanocenes, Cp’TiX2(Y) (Cp’ = cyclopentadienyl; X = Cl, Me etc.; Y = anionic donor such as phenoxide, ketimide, amidinate etc.), are known to be the effective catalysts. This review introduces several selected efforts for efficient synthesis of ethylene copolymers containing cyclic olefins, biobased conjugated dienes, disubstituted α-olefins including effect of cocatalysts. Moreover, here introduces analysis using XAS (X-ray absorption spectroscopy), which has been recognized as powerful method providing direct information of the catalytically active species, such as coordination numbers and the distances of the coordinated atoms as well as oxidation state and the geometry of the metal centre in catalyst solution.
Keywords:
titanium catalysts
; polymerization
; polyolefin
; copolymerization
; half-titanocene
; homogeneous catalysts
; XANES
; EXAFS
; ethylene
1. Introduction
Polyolefins [linear high density polyethylene (HDPE), linear low density polyethylene (LLDPE), low density polyethylene (LDPE), and isotactic polypropylene (PP)] are the dominant commodity thermoplastics, accounting for over 50% of global plastic production in the world; the growth of the world production still has been observed especially in Asia [1]. Transition metal catalyzed olefin coordination insertion polymerization is the key technology for the industrial production [2,3,4,5,6,7,8]. The development of new polymers, that cannot be achieved by using conventional catalysts (such as Ziegler-Natta, metallocene catalysts), has been considered as the central research objective [2,3,4,5,6,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23]. Copolymerization has been taken into consideration as an effective strategy, because their thermal, physical, mechanical and electronic properties of the resulting copolymers can be modified by the individual components, incorporation of two (or three) monomer units [4,6,14,15,16,17]. Design, development of the molecular catalysts, that display highly active with better comonomer incorporations, has thus been the pivotal challenge [2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23].
Discovery of metallocene catalysts (called single-site catalysts), bearing two cyclopentadienyl (Cp’) ligands of type Cp’2MX2 (M = Ti, Zr, Hf; X = halogen etc.), marked an importance of the catalyst design, since the resultant copolymer possesses uniform composition with controlled molecular weight due to the formation of single active species in the catalytic polymerization [5,9,10,11,12]. The fact displayed a different feature in the copolymerization using the Ziegler-Natta catalysts (called multi-site catalysts), which generate several catalytically active species in situ; as a results, Ziegler-Natta catalysts afford the copolymer with multiple compositions [5,9,10,11,12]. The high oxidation state cationic alkyl species, Cp’2M+R (R = alkyl), formed by treating with borate or methylaluminoxane (MAO) through alkyl abstraction, have been proposed as the active species in this catalysis (Scheme 1) [5,9,10,11,12,13,14,15,16,17,24,25,26,27]. In this catalysis, nature of the active species, such as basic geometry, electronic and steric bulk of the ligand sets, directly affects the catalytic activity, stereo specificity, and the comonomer incorporation in the copolymerization.
It is widely recognized in ethylene copolymerization with α-olefin that bridged (ansa, linked) metallocene catalysts generally showed better α-olefin incorporation than the unbridged analogues; the fact was explained as due to different coordination environment imposed by the bridging framework (Scheme 1) [5,9,10,11,12,13,14,15,16,17,28,29]. Accordingly, linked (ansa, bridged) half-titanocenes exemplified as constrained geometry catalyst (CGC), [Me2Si(C5Me4)(NtBu)]TiCl2 [12,13,28,29,30,31,32,33], exhibit enhanced ability in α-olefin incorporation in the copolymerization. Interestingly, CGC exhibited styrene incorporation in the ethylene copolymerization, although the incorporation level typically remains below 50 mol% [33,34,35]. This limitation was later overcome by the bimetallic system (bimetallic CGC, Scheme 1), which enabled synthesis of the copolymer with high styrene content (76 mol%) [21,22,36,37].
In contrast, it has been widely known that ordinary half-titanocenes containing one Cp’ of type, Cp’TiX3, are effective catalysts for synthesis of syndiotactic polystyrene (SPS, Scheme 1), that is inaccessible by conventional radical, ionic (anionic, cationic) polymerization [38,39,40]. However, these Cp’TiX3 catalysts generally display low catalytic activity for olefin polymerization, and the (attempted) ethylene/styrene copolymerization yielded a mixture of polyethylene, SPS, and the copolymer in small amount [34,41]. The behaviour displays the sharp contrast from the facts that CGC and ordinary metallocene catalysts showed the negligible catalytic activities [34,36,37]. Moreover, as described above, CGC predominantly produce the ethylene/styrene copolymers [33,34,35,36,37]; these facts led to an assumption that the active species in olefin polymerization differ from those in the syndiospecific styrene polymerization, as described in detail below.
It is well known that modified half-titanocenes featuring anionic donor ligands, Cp’TiX2(Y) (Scheme 2, Y = phenoxide, ketimide, amidinate, phosphinimide, iminoimidazolide, iminoimidazolidide etc.) [6,14,15,16,17], first introduced by our team [42,43], display remarkably distinctive catalytic properties, particularly in the copolymerization of ethylene (or propylene, α-olefin) with sterically encumbered olefins, cyclic olefins, and with aromatic vinyl monomers (Scheme 2) [14,15,16,17]. The phenoxide (1) [42,43,44,45] and the ketimide (2) [46,47,48,49,50,51,52] catalysts have proven distinctively effective for synthesis of new (co)polymers [6,14,15,16,17]; it has become clear that effective catalyst for the targeted (co)polymerization can be finely tuned through modification of cyclopentadienyl (Cp’) and the anionic donor (Y) ligands. Later, the η1-amidinate analogue (3) [4,53,54,55] enabled industrial-scale production of ethylene propylene diene terpolymer (EPDM) [4], as chlorine-free synthetic rubber without deep cooling, characteristic of conventional (Ziegler type) vanadium-based catalyst systems, therefore highlighting the practical significance of this catalyst platform [37]. Moreover, interestingly, the phenoxide catalysts exhibit remarkable activities for SSP [56,57], and exclusively afforded ethylene/styrene copolymers in the copolymerization [56,58]. Moreover, these catalysts enabled synthesis of a series of ethylene copolymers with the other aromatic vinyl monomers (Scheme 2) [59]. The observed characteristics represent quite different from those observed by metallocene and linked half-titanocene catalysts.
This review thus introduces several selected recent efforts for synthesis of ethylene copolymers containing cyclic olefins [60,61,62,63,64,65,66,67,68], biobased conjugated dienes [69,70], disubstituted α-olefins [44,71,72,73,74] including effect of cocatalysts [75]. Moreover, here also introduces analysis results by synchrotron XAS (X-ray absorption spectroscopy) [27,76,77] which has been recognized as useful to obtain information not only of valence (oxidation state) and the basic structure (geometry) through XANES (X-ray Absorption Near Edge Structure), but also number of coordination atoms and the distance to the cantered metal through EXAFS (Extended X-ray Absorption Fine Structure) analysis.
2. Efficient Synthesis of Cyclic Olefin Copolymers (COCs)
Certain amorphous cyclic olefin copolymers (COCs) are polymeric materials with high transparency in the UV-vis region, thermal resistance (possessing high glass transition temperature, Tg), humidity resistance (negligible water absorption), low dielectric constants, and then dimensional stability [78,79,80,81,82,83,84,85,86], as commercialized as TOPAS® [87] and APEL® [88], ethylene-based copolymers with norbornene (NBE) and tetracyclododecene (TCD), respectively, for optical lens and films for medical applications. There have been extensive number of reports for ethylene/NBE copolymerization using group 4 transition-metal complexes, including metallocenes [89,90,91,92,93,94,95], bridged half-metallocenes (so-called constrained geometry type) [96,97,98,99], modified non-bridged half-metallocenes [60,61,62,63], and the others called post metallocene catalysts [100,101,102,103,104,105]. However, reports for the efficient synthesis of high molar mass the ethylene/NBE copolymers possessing high Tg values (high NBE contents) with efficient and random NBE incorporation remain scarce [61,63], Moreover, vanadium-based catalyst systems, such as VOCl3 or VO(OEt)Cl2 together with EtAlCl2·Et2AlCl cocatalyst, which have been employed in the commercial production of ethylene/TCD copolymers, require deep-cooling conditions because of limited thermal stability of these catalyst systems [106]. The efficient synthesis of high molar mass ethylene/TCD copolymers still have been limited by the ketimide analogue [63,64], whereas copolymerization employing conventional metallocene catalysts [107,108,109,110] and the known linked half-titanocene catalyst (CGC) [64] generally suffers from low catalytic activity and/or inefficient TCD incorporation. The catalyst development for synthesis of high molecular weights COCs possessing high glass transition temperature maintaining promising optical property (birefringence, with low water absorption, dielectric constants, and dimensional stability) has been an attractive subject [78,79,80,81,82,83,84,85,86].
As described above, ethylene/NBE copolymers are known to be commercialized COC, as TOPAS® [87], using metallocene–MAO catalysts (Scheme 3). In general, the copolymerization by ordinary metallocenes (SBI-Zr) and linked half-titanocene catalysts (CGC) afforded amorphous poly(ethylene-co-NBE)s with uniform compositions possessing a sole glass transition temperature (Tg) by DSC thermograms [79,80,81,82,83,84,85]. However, both the activity and the Mn values in the resultant copolymers decreased upon increase in the NBE/ethylene feed molar ratios (increasing the NBE concentration charged and/or lowering ethylene pressure) [89,90,91,92,93,94,95,96,97,98,99], as exemplified in Table 1 [61,82]. The fluorenyl-substituted CGC (Flu-CGC) enables living NBE polymerization in the presence of dried MAO or MMAO (obtained as white solid by removal of AlMe3 and AliBu3 from the commercially available samples) with much improved NBE incorporation [99]; the resultant copolymers with α-olefins (1-octene, 1-decene, and 1-dodecene) possessed gradient NBE incorporation (monomer sequences in the copolymers) due to different reactivity between two monomers [6,84].
The ketimide-modified half-titanocene, CpTiCl2(N=CtBu2) (2a, Scheme 3) first demonstrated efficient synthesis of ethylene random copolymers with high NBE contents possessing high Mn values (Table 1) [61]. The activities (productivities) did not change significantly even over 30 min and increased at elevated temperature (60 ºC); the copolymerization conducted at 80 ºC did not show the significant decrease in the activity [61]. In contrast to the observation in the copolymerization using the metallocene, CGC catalysts, the activity by 2a–MAO catalyst rather increased upon increase in the initial NBE feed concentration, the catalyst afforded the high molar mass copolymers [61]; Al cocatalyst (MAO, MMAOs) does not significantly affect the activity and the NBE incorporation. Therefore, this catalysis method using 2a gave the high molar mass copolymers with high NBE contents (58.8-73.5 mol%, Table 1) with uniform compositions [61]. A linear correlation between the Tg values and the NBE content in the copolymers was demonstrated even at high NBE contents, strongly suggesting the NBE random incorporation. The random NBE incorporation was also demonstrated by their microstructural analyses of the copolymers, consisting of NBE isolated, alternating, repeat incorporation without stereoregularity [6,61,63,80,82].
The imidazolin-2-iminato modified half-titanocene, CpTiCl2[1,3-tBu2(CHN)2C=N] (4), also exhibited promising capabilities in the copolymerization, probably due to the strong σ-donating nature. The catalyst (4) showed high catalytic activity affording high molar mass copolymers with rather efficient NBE incorporation as well as uniform composition [65]. The activities by 4 were rather low compared to the ketimide analogue (2a) and further ligand modification might be required, suggesting that the anionic donor ligand play an important role toward both the catalytic activity and the NBE incorporation.
More recently, as summarized in Table 1, improved both catalytic activities and NBE incorporation from the above ketimide catalyst (2a) could be achieved by introduction of trialkylsilyl group on the cyclopentadienyl fragment, (RC5H4)TiCl2(N=CtBu2) [R = SiMe3 (2c) SiEt3 (2d)]. These catalysts displayed remarkable activities (25700-91400 kg-polymer/mol-Ti·h) in the copolymerization at 50 ºC to afford high molar mass copolymers with high NBE contents (NBE 36.2-72.7 mol%) possessing high Tg value (238 ºC) [63]. The observed results seem interesting contrast to those in copolymerization using the tert-BuC5H4 analogue (2b, Table 1).
As described above, ethylene/TCD copolymers are known to be commercialized COC, as APEL® [88], which have been produced using vanadium-based catalyst systems (VOCl3–EtAlCl2·Et2AlCl etc.) under deep-cooling conditions [106]. It seemed that the conventional (metallocene, linked-half-titanocene) catalysts face difficulty for synthesis of the high molar mass ethylene/TCD copolymers at moderate temperature (>50 ºC, Scheme 4) [107,108,109,110].
The ketimide-modified half-titanocene, (tBuC5H4)TiCl2(N=CtBu2) (2b), demonstrated synthesis of the copolymers with remarkable activities (43700 kg-polymer/mol-Ti·h) to afford the copolymers possessing high Tg values (108-203 ºC) with uniform compositions (Scheme 4, Table 2) [64]. The Cp analogue (2a) showed lower catalytic activity than 2b, and the Mn value in the resultant copolymer was low compared to that prepared by 2b under the same conditions; modification of cyclopentadienyl fragment thus plays a key role [64]. The imidazolin-2-iminato analogue (4) also displayed rather low capability for the copolymerization [64]. The ketimide analogue seems to be thus promising for synthesis for COCs.
More recently, as seen in the ethylene/NBE copolymerization, the catalyst capability could be improved by introduction of trialkylsilyl group on the cyclopentadienyl fragment, (RC5H4)TiCl2(N=CtBu2) [R = SiMe3 (2c) SiEt3 (2d), Table 2] [63]. In particular, the SiEt3 analogue (2d) showed higher catalytic activities at 50 °C, affording the high molecular weight copolymers with uniform composition possessing sole Tg values. The TCD contents in the copolymer increased with increase in the TCD concentration charged (as well as lowering the ethylene pressure), and these catalysts (2c,d) demonstrated synthesis of the copolymers with TCD content higher than 50 mol% (Tg = 255 °C, TCD 52.3 mol%) [63]. The results clearly indicate that further improvement in the catalyst capability can be achieved by the ligand modification.
Basic structure (size, strain etc.) in cyclic olefin directly affects not only the efficiency of cyclic olefin incorporation in the (ethylene) copolymerization, but also the properties in the resultant COCs (thermal and tensile properties, dielectric constant etc.) [6,84]. As described in the introduction, limited number of reports were known for successful synthesis of (rather high molar mass) amorphous ethylene copolymers with so called low strained cyclic olefins such as cyclohexene (CHE) [66,68], cycloheptene (CHP) [66,67], and cis-cyclooctene (COE) [66,67]. Recently, as shown in Scheme 5, synthesis of a series of amorphous ethylene and propylene copolymers with CPE, CHE, CHP, COE, tricyclo[6.2.1.0(2,7)]undeca-4-ene (TCUE), and with TCD were demonstrated [66,67]. Linear relationships between the Tg values and the cyclic olefin contents were demonstrated in all cases. These results thus strongly suggest that Tg values were affected by the cyclic structure in COCs (except the copolymers with CPE, COE). The Tg values in the propylene copolymers in the region of low cyclic olefin content (up to 25 mol%) seemed rather high compared to those in the ethylene copolymers [66]. This is due to rather high Tg value in the atactic polypropylene.
The cyclic olefin copolymers by copolymerizing norbornene (NBE) and tetracyclododecene (TCD) with various linear α-olefins, such as 1-hexane (HX), 1-octane (OC), and 1-dodecene (DD), using ketimide half-titanocene catalysts Cp’TiCl2(N=CtBu2) [Cp’ = or Cp (2a), tBuC5H4 (2b)] (Scheme 6) [65]. The Cp analogue (2a) exhibited superior to the tBuC5H4 derivative (2b), exhibited over 10 times higher activity and more efficient comonomer incorporation under same condition. While these half-titanocene catalysts successfully produced high molecular weight TCD/α-olefins copolymers [65]. The conventional metallocene and Flu-CGC failed to synthesize the high molecular weight copolymers, yielding only low molecular weight oligomers [111,112,113,114].
A critical finding in this copolymer system is the linear relationship observed between the cyclic olefin content and glass transition temperature across all synthesized polymers. In copolymers with TCD exhibited higher Tg values than their NBE counterparts at equivalent cyclic olefin concentrations (exceeding 200 °C). The Tg values are further influenced by the α-olefins chain length, decreasing in the order of 1-hexene > 1-octene > 1-dodecene. Additionally, the reactive functionality was demonstrated by introducing via the incorporation of 1,7-octadiene (OD), which adds terminal olefinic double bonds to the side chains. In this term, the tBuC5H4 analogue (2b) proved more suitable than Cp analogue (2a) because the latter caused unwanted isomerization of the terminal double bonds into internal olefins [65].
3. Ethylene Copolymerization with Sterically Encumbered Olefins
Ethylene copolymerization with 1,1-disubstituted α-olefins such as isobutene (IB), 2-methyl-1-pentene (2M1P) has rarely been reported in metal-catalyzed olefin coordination insertion polymerization [115]; these monomers have long been regarded as “traditionally unreactive” in this field. However, these ethylene copolymers are expected to show enhanced weather resistance compared with conventional ethylene/α-olefin copolymers, because of absence of reactive tertiary C–H bonds in the polymer backbone, which are removed by oxidative or UV-induced degradation. For instance, the ethylene/IB copolymerization using [Et(indenyl)2]ZrCl2 gave the copolymer with low (<2.8 mol%) IB content even under large excess IB concentration conditions (charge molar ratio of ethylene/IB = 1/4000) [116].
Ordinary CGC showed the negligible IB incorporation in the ethylene copolymerization, the IB incorporation was much improved by using cyclododecylamide analogue (CGC-CDD, Scheme 7) [117]; the fluorenyl analogue (Flu-CGC-CDD, Scheme 7) later showed the IB incorporation [118]. However, the copolymerization with 2M1P by CGC-CDD afforded the polymer possessing large PDI (polydispersity index, Mw/Mn = 5.90) [117]. The bimetallic CGC, consisting of dinuclear catalyst and dianionic borate catalyst system (Scheme 7), showed improved IB incorporation from the mononuclear CGC (IB 3.1 mol% vs 15.2 mol%: ethylene 1 atm, IB 1.3 M, 24 ºC), but showed low activity decreased and the obtained polymer possessed rather large PDI (Mw/Mn = 3.67) [119,120]. The catalyst system incorporated methylenecyclopentene and methylenecyclohexene in the ethylene copolymerization. The attempted copolymerization with 2-methyl-2-butene gave the copolymer by incorporation as 2-methyl-1-butene formed by olefin isomerization [120]. Reports for synthesis of high molar mass ethylene copolymers with 1,1-disubstituted α-olefin (except IB) have still remained scarce.
Synthesis of ethylene/2M1P copolymers was achieved by the phenoxide modified half-titanocene catalyst with rather efficient 2M1P incorporation (1a, Mn = 3.30-13.0×104, Mw/Mn = 1.70-1.90) [71,73]. As summarized the selected data in Table 3, 2M1P content in the resultant copolymer increased upon increasing upon increase in the 2M1P concentration charged or lowering ethylene pressure (6 → 4 atm) with decrease in the activity [71,73]. The resultant copolymers possessed uniform compositions confirmed by DSC thermograms and their microstructural analysis by 13C NMR spectra revealed that 2M1P was incorporated in an isolated and/or alternating manner (without repeat insertions). Use of the tert-BuC5H4 analogue (1c) led to decrease in both the activity and the 2M1P incorporation explained as due to the steric bulk [71,73]. Later, the activity by the SiEt3 analogue (1b) increased at elevated temperature (up to 80 ºC) [75]. In contrast, the Cp–ketimide analogue (2a) showed poor 2M1P incorporation, suggesting that nature of anionic donor ligand (as well as steric bulk on Cp’) affect the comonomer incorporation. Moreover, as described above, CGC also showed poor 2M1P incorporation under the same conditions.
The Cp*–phenoxide analogue (1a) also showed efficient VCH incorporation in the ethylene copolymerization [72], whereas incorporation of branched (γ-disubstituted) α-olefins in the copolymerization using the conventional catalysts seemed difficult [72]. However, as summarized in Table 3, 1a showed negligible tert-butyl ethylene incorporation, and showed rather poor VTMS incorporation compared to the Cp–ketimide analogue (2a), CGC; the VTMS incorporation was improved by using the tert-BuC5H4 analogue (1c) but the resultant copolymer possessed rather low Mn values compared with those prepared by 2a [121]. Synthesis of ethylene copolymers with tert-butyl ethylene (TBE) was achieved by the tert-BuC5H4 analogue (1d) and the 1,2,4-Me3C5H2 analogue (1e); the observed effect on Cp’ was the same as that in the ethylene copolymerization with cyclohexane [68].
– Effect of Borate Cocatalysts toward Activity, Comonomer Incorporation in Alkane Solvent –
Recently, it has been recognized that solvent coordination (toluene vs alkane) in addition to catalyst–cocatalyst interaction (nuclearity effect) affect both the catalytic activity, comonomer incorporation in olefin polymerization [75,122,123]. For instance, [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC), Cp*TiMe2(O-2,6-iPr2-4-RC6H2) [R = H (1a’), SiEt3 (1b’)]–borate, [A(H)]+[BAr4]- (Ar = C6F5 or C10F7, B1–B6 in Scheme 8), catalyst systems conducted in methylcyclohexane (MCH) exhibited better comonomer incorporation than those conducted in toluene (in the presence of MAO, borate cocatalysts) in the ethylene copolymerization with 2M1P, 1-dodecene, VCH [75].
The activity by CGC in the E/2M1P (co)polymerization (ethylene 4 atm, 25 ºC) in MCH was affected by the borate cocatalyst employed and increased in the order: activity = 149 kg-polymer/mol-Ti·h (B1) < 768 (B6) < 2660 (B5) < 3770 (B2) < 6810 (B3). Interestingly, CGC–B3 catalyst system in MCH afforded the copolymer (2M1P 0.4 mol%, by DSC thermogram and 13C NMR spectrum), whereas CGC‒MAO catalyst system in toluene showed negligible 2M1P incorporation under the same conditions [75]. It seems that the 2M1P incorporations (estimated by the Tm values) were also affected by the borate cocatalyst employed. Similarly, the activities by 1a’ and 1b’ were affected by the borate cocatalyst employed, and 1b’‒B5 catalyst system showed the highest activity (5060 kg-polymer/mol-Ti·h) and the resultant copolymer possessed higher 2M1P content than that conducted in toluene by 1b’‒MAO catalyst system (6.0 mol% vs 3.1 mol%) [75]. In contrast, 1b’–B1 catalyst system showed low activity affording the polymer with two compositions estimated by DSC thermograms. It should be noted that the 2M1P incorporation was affected by the borate cocatalyst employed: B5 showed better 2M1P incorporation than B3. It was shown that conducting these copolymerizations in MCH in the presence of borate cocatalysts (B2‒B6) showed better 2M1P incorporation than that conducting in toluene in the presence of MAO [75].
Moreover, the activity by CGC conducted in MCH was affected by the borate cocatalyst employed. CGC‒B5 catalyst system in MCH exhibited higher catalytic activity and better VCH incorporation than CGC‒MAO catalyst system in toluene [75]. Note that no significant differences in the VCH contents in the copolymers were observed when these polymerizations using CGC‒borate catalyst systems were conducted in toluene; the VCH incorporation was thus affected by the solvent (toluene vs MCH). The results thus suggest that the observed difference in MCH would be due to a weak cation‒anion interaction without coordination of toluene (and amine or ether, exhibited A formed after treatment of CGC with borate) to the assumed cationic alkyl species [122,123,124]. Non-coordinating oxonium ion, especially HO+(n-C14H29)2·O(n-C14H29)2 (B5) containing long alkyl chains, was preferred than the ammonium salts (probably due to poor coordination ability of O(n-C14H29)2 to the assumed cationic species) [75]. As expected for better anion delocalization (BAr4-), perfluorinated naphthyl borate, B(C10F7)4-, showed higher activity than B(C6F5)4-.
Scheme 8.
Effect of borate cocatalysts (B1‒B6) in Ethylene copolymerization with 2-methyl-1-pentene (2M1P), vinylcyclohexane (VCH) by [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC), Cp*TiMe2(O-2,6-iPr2-4-R’C6H2) [R’ = H (1a’), SiEt3 (1b’)] in methylcyclohexane (MCH) [75].
Scheme 8.
Effect of borate cocatalysts (B1‒B6) in Ethylene copolymerization with 2-methyl-1-pentene (2M1P), vinylcyclohexane (VCH) by [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC), Cp*TiMe2(O-2,6-iPr2-4-R’C6H2) [R’ = H (1a’), SiEt3 (1b’)] in methylcyclohexane (MCH) [75].
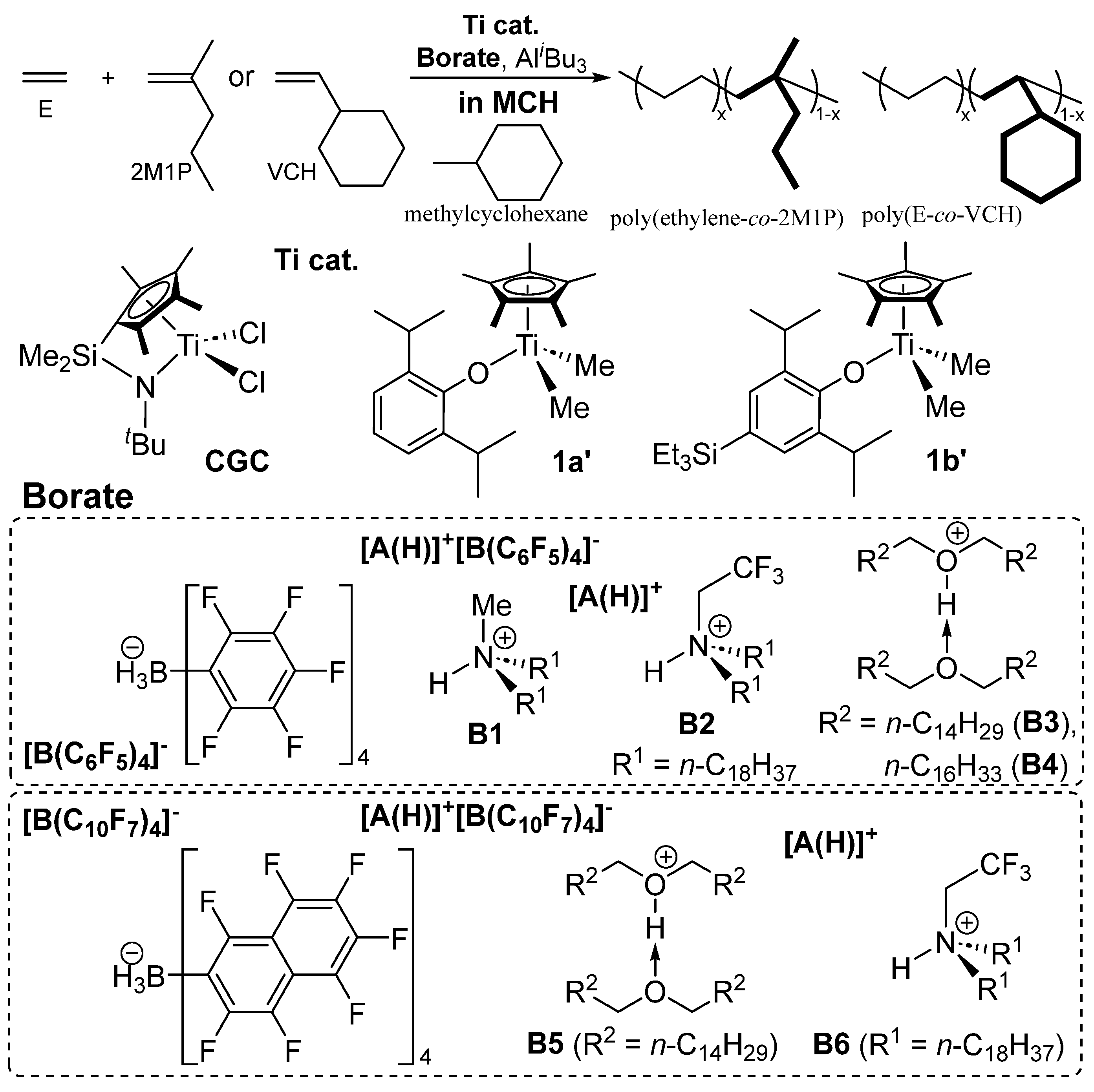

Table 4.
Ethylene copolymerization with 2-methyl-1-pentene (2M1P) or vinylcyclohexane (VCH) by [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC), Cp*TiMe2(O-2,6-iPr2-4-R’-C6H2) [R = H (1a’), SiEt3 (1b’)]‒borate cocatalyst systems [at 25 °C, ethylene 4 atm, 10 min (2M1P); ethylene 6 atm 6 min (VCH)].a.
Table 4.
Ethylene copolymerization with 2-methyl-1-pentene (2M1P) or vinylcyclohexane (VCH) by [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC), Cp*TiMe2(O-2,6-iPr2-4-R’-C6H2) [R = H (1a’), SiEt3 (1b’)]‒borate cocatalyst systems [at 25 °C, ethylene 4 atm, 10 min (2M1P); ethylene 6 atm 6 min (VCH)].a.
| cat. (µmol) |
solvent | Al cocat. | borate | comonomer | activityb |
Mnc ×10-4 |
Mw/ Mnc |
Tmd / °C |
cont.e / mol% |
| CGC (1.0) | MCH | AliBu3f | B1 | 2M1P | 149 | 6.88 | 3.09 | 131 | |
| CGC (1.0) | MCH | AliBu3f | B2 | 2M1P | 3770 | 38.9 | 5.88 | 122 | |
| CGC (1.0) | MCH | AliBu3f | B3 | 2M1P | 6810 | 44.9 | 6.42 | 120 | 0.4 |
| CGC (1.0) | MCH | AliBu3f | B5 | 2M1P | 2660 | 32.0 | 5.66 | 122 | |
| CGC (1.0) | MCH | AliBu3f | B6 | 2M1P | 768 | 26.1 | 5.23 | 126 | |
| CGC (0.1) | toluene | MAO | --- | 2M1P | 5090 | 32.4 | 3.66 | 129 | trace |
| 1a’ (1.0) | MCH | AliBu3 | B3 | 2M1P | 4210 | 6.28 | 1.93 | 99.0 | 5.5 |
| 1a’ (1.0) | MCH | AliBu3 | B4 | 2M1P | 1280 | 7.87 | 1.84 | 96.9, 122 | |
| 1a’ (1.0) | MCH | AliBu3 | B5 | 2M1P | 4260 | 5.76 | 1.76 | 93.6 | 6.8 |
| 1a’ (1.0) | MCH | AliBu3 | B6 | 2M1P | 1870 | 4.93 | 1.89 | 94.5 | |
| 1a’ (0.05) | toluene | MAO | --- | 2M1P | 11200 | 8.41 | 2.25 | 111 | 2.6 |
| 1b’ (1.0) | MCH | AliBu3 | B1 | 2M1P | 547 | 7.04 | 2.47 | 99.5, 121 | |
| 1b’ (1.0) | MCH | AliBu3 | B2 | 2M1P | 2680 | 3.82 | 1.96 | 98.7 | |
| 1b’ (1.0) | MCH | AliBu3 | B3 | 2M1P | 4040 | 4.11 | 1.90 | 101 | 5.0 |
| 1b’ (1.0) | MCH | AliBu3 | B4 | 2M1P | 3980 | 3.21 | 2.41 | 98.4 | |
| 1b’ (1.0) | MCH | AliBu3 | B5 | 2M1P | 5060 | 5.41 | 1.84 | 97.9 | 6.0 |
| 1b’ (0.05) | toluene | MAO | --- | 2M1P | 18600 | 8.34 | 2.05 | 109 | 3.1 |
| CGC (0.05) | MCH | AliBu3 | B2 | VCH | 16800 | 7.70 | 1.70 | 87.5 | 8.8 |
| CGC (0.05) | MCH | AliBu3 | B3 | VCH | 44000 | 6.31 | 1.65 | 86.0 | 9.0 |
| CGC (0.05) | MCH | AliBu3 | B4 | VCH | 10600 | 5.93 | 1.75 | 87.0 | 8.7 |
| CGC (0.05) | MCH | AliBu3 | B5 | VCH | 69000 | 8.65 | 1.98 | 81.4 | 9.7 |
| CGC (0.05) | toluene | MAO | --- | VCH | 39400 | 21.4 | 3.38 | 92.5 | 6.0 |
| CGC (0.05) | toluene | AliBu3 | B1 | VCH | 1090 | 10.8 | 2.28 | 98.6 | |
| CGC (0.05) | toluene | AliBu3 | B2 | VCH | 16500 | 13.0 | 2.30 | 96.3 | 5.6 |
| CGC (0.05) | toluene | AliBu3 | B3 | VCH | 14100 | 13.1 | 2.59 | 96.9 | 5.6 |
| CGC (0.05) | toluene | AliBu3 | B4 | VCH | 21100 | 8.55 | 2.21 | 97.0 | 5.4 |
| CGC (0.05) | toluene | AliBu3 | B5 | VCH | 31000 | 13.8 | 3.07 | 94.2 | 5.8 |
| 1b’ (0.01) | toluene | MAO | --- | VCH | 224000 | 17.5 | 2.43 | (-15.1) | 24.1 |
aConditions: 2-Methyl-1-pentene (2M1P) 5.0 mL (1.35 M) or vinylcyclohexane (VCH) 5.0 mL (1.22 M), 2M1P (or VCH) + methylcyclohexane (MCH) or toluene total 30.0 mL, AliBu3 [0.55 mmol/L hexane, Al/Ti = 1000 (molar ratio)] or MAO 3.0 mmol, borate (borate/Ti molar ratio = 1.0). bActivity = kg-polymer/mol-Ti·h. cGPC data in o-dichlorobenzene vs polystyrene standards (Mn in g/mol). dBy DSC thermograms. e2M1P or VCH content (mol%) estimated by 13C NMR spectra. fAl/Ti = 500, molar ratio.
4. Synthesis of Biobased Polyolefins: Copolymerization of Biobased Conjugated Dienes
Development of functional polymers from renewable feedstocks has been an important subject in circular economy [125,126,127,128,129,130,131,132]. Cyclic monoterpenes consisting of two isoprene units as formula of C10H16 shown in Scheme 9, can be considered as promising monomers obtained from the abundant plant oil [133,134]. However, the successful synthesis by coordination insertion polymerization has been scarce until recently [69,135], whereas there are the reports by ionic (cationic, radical) polymerization.
(1,2,4-Me3C5H2)TiCl2(O-2,6-iPr2C6H3) (1e) enabled synthesis of ethylene/limonene copolymers possessing rather high molecular weights (Mn = 4.00-12.6×104) with unimodal molecular weight distributions [135]. As observed in the ethylene/2M1P copolymerization (Table 3), increase in the limonene concentration led to decrease in the activity and the Mn value in the copolymer, while simultaneously increasing the limonene content in the copolymer. In contrast, the Cp* analogues (1a,b) showed less efficient limonene incorporation and CGC showed the negligible incorporation. Microstructural analysis revealed resonances consistent with 1,2-limonene insertion followed by cyclization, favoring a 2’,1’-insertion (than 1’,2’-insertion) pathway (Scheme 9) [135].
The Cp* analogues (1a,b)–MAO catalyst gave high molar mass poly(ethylene-co-β-pinene)s (Mn = 2.10-22.4×104) possessing unimodal molecular weight distributions (Mw/Mn = 1.53-2.32). The activity was influenced by the β-pinene concentration charged, ethylene pressure, Al/Ti molar ratio and the temperature. The 1,2,4-Me3C5H2 analogue (1e) showed the low activities compared with 1a,b, affording the copolymers possessing rather low Mn values [135]. The attempted copolymerization by CGC gave polymers with negligible β-pinene incorporation.
β-Myrcene (myrcene, My), a promising biobased linear terpene, has been polymerized by radical or anionic polymerization and reports concerning the metal catalyzed ethylene copolymerization have been limited by scandium catalysts [136] or half-titanocene catalysts [69], whereas there are reports for the styrene/My copolymerization [137,138,139,140,141,142,143]. The scandium catalysts, however, afforded copolymers possessing multi-block microstructures, expressed as poly(E-bl-My), possessing melting temperature of polyethylene segment (133 ºC, My 9 mol%) [136]. Phenoxide-modified Cp* analogues (1a,b) demonstrated synthesis of high molar mass (semicrystalline or amorphous) random ethylene/My copolymers with efficient My incorporation [69]. The microstructural analysis using 13C NMR spectra of the resultant (unsaturated) copolymers and the saturated copolymers prepared by olefin hydrogenation. The analysis data revealed that the copolymers possessed cyclopentane units containing My pendant arm (-CH2CH=CMe2); which are presumably formed by 2,1- or 1,4-My insertion and subsequent cyclization after ethylene insertion (Scheme 9) [69]. In general, formation of ƞ3-allyl intermediate, considered as dormant, disturbs proceeding further olefin insertion. In this catalysis, the subsequent cyclization enabled to proceed the copolymerization with random My incorporation. The elongation (tensile strain) at break increased with increasing the My contents accompanied with decrease in the tensile stress (strength); the copolymer showed promising elastic behaviors as biobased elastomers [69]. The catalysts also enabled synthesis of ethylene/isoprene copolymers possessing cyclopentane and cyclohexane units [70].
5. Analysis of Catalytically Active Species Through XAS (X-Ray Absorption Spectra)
5.1. Introduction: XAS for Analysis of Catalytically Active Species
Identification of catalytically active species and the reaction chemistry are prerequisite not only for clear understanding the catalysis mechanism, but also for catalyst design through the structural and electronic insight. Single crystal X-ray diffraction analysis provides a structural information of the proposed intermediate(s) in solid state, although such species are often (sometimes) required by certain stabilization for isolation and the information is limited to be in solid state. Mechanistic studies are also supported by reaction chemistry, stoichiometric and/or catalytic reactions, using model complexes (and the isolated species in the catalytic reaction) and computational studies. However, we often observed that the isolated species exhibit apparently low catalytic activities or are inactive, due to required stabilization for the isolation and/or isolated species are indeed dormant in the catalysis cycle.
Nuclear magnetic resonance (NMR) spectroscopy is the principal technique for characterization of diamagnetic inorganic and organometallic compounds in (partially) deuterated solvent. Electron spin resonance (ESR) spectroscopy has been powerful method for analysis of the paramagnetic species that exhibit negligible or broadened NMR signals [144,145]. However, these methods still provide limited access for obtainment of real/clear structural information (image) in catalysis solution. Moreover, ESR often lacks quantitative analysis/reliability [144,145,146], and a possibility of co-presence of “ESR silent” species cannot be excluded. For example, vanadium(III) species with 3d2 electron configuration (S = 1, triplet, S = spin quantum number) are ESR silent due to an interaction between the two unpaired electrons, while vanadium(IV) dimers coupled antiferromagnetically also become ESR-silent through spin-orbit coupling (SOC) [146,147].
Analysis by XAS (X-ray absorption spectroscopy), XANES (XANES = X-ray Absorption Near Edge Structure) and EXAFS (EXAFS = Extended X-ray Absorption Fine Structure), performed at synchrotron facilities provides information of not only oxidation state and the basic geometry around the metal centre (by XANES), but also kind of atoms and the distances connected (coordinated) to the metal centre (by EXAFS). More recently, we see growth of number of reports identifying homogeneous, molecular catalysis through XAS analysis [27,76,148,149,150,151,152,153,154,155,156,157,158,159], whereas the methods are common in heterogeneous catalysis [160,161,162,163,164]. The methods are recognized as a useful analysis method especially of catalysis research with early transition metals, as exemplified in analysis of paramagnetic vanadium(III) species and titanium(III) species that cannot be observed especially by NMR spectroscopy [27,76,148,149,153,154,158].
Although we need to use the appropriate synchrotron facility (e.g. SPring-8, BL01B1 beamline), we do not need any specified apparatus, and the sample preparations are possible on site in the drybox. Moreover, we demonstrated good reproducibility (in the independent runs even conducted during the different beam time) and no severe x-ray radiation damages during data acquisition were observed [27,76]. Herein, we first describe basics in XAS analysis for uninitiated researchers and introduce selected results in analysis of catalytically active species in olefin polymerization and syndiospecific styrene polymerization using half-titanocene catalysts.
5.2. Basics in XANES Spectra
It has been known that pre-edge peak intensity and edge absorption (exemplified in Figure 1) in XANES spectra are influenced by the oxidation state and the basic geometry around the centred metal [27,148,149,160,161,162,163,164,165,166,167,168,169,170,171,172,173]. For basic introductory, V-K edge XANES spectra of vanadium oxides with various oxidation states are shown in Figure 1a [174]. The basic geometries [octahedral (Oh), square pyramidal (C4v)], tetrahedral (Td)] and their selected irreducible and relating functions and point groups are also placed in the figure. Pre-edge absorption is known to provide information about basic (local) geometry around the metal center reflected by s → d transitions (quadrupole transition); dipole transition (s → p) is also considered depending on degree of p–d orbital hybridization [166,169,175,176]. For instance, complexes with Td and C4v symmetries display higher pre-edge peak intensity than those in Oh symmetry. The observed difference can be explained as due to a difference in the possibility of a p–d orbital hybridization, realized from a table in irreducible representations and the relating functions in the complexes with these symmetries (Figure 1b).
The complex with tetrahedral geometry (Td) leads to increase degree of electric dipole transition of a 1s electron to the p–d hybridized orbital [166,169,175,176]. This is because that px, py, pz orbitals and dxy, dyz, dzx orbitals in Td symmetry (or px, py orbitals and dyz, dzx orbitals in C4v symmetry) belong to the irreducible representation, leading to formation of a p–d hybridized orbital with increased electric dipole transition of a 1s electron occurs to the hybridized orbital [169,176]. However, no irreducible representations containing d and p orbitals are present for the complexes with octahedral geometry (Oh point group). Therefore, the intensity of the pre-edge peak is generally weak, because only the relatively weak quadrupole transition (s → d transitions) is allowed, while the dipole transition (s → p) is forbidden; distortion of the symmetry (symmetry breaking), however, enables p–d hybridization that allows the dipole transition. Therefore, simple prediction of the pre-edge peak intensity could be possible without theoretical calculations by using the character tables of group theory. V2O3 and V2O4 also fold a distorted octahedral geometry fold a distorted octahedral geometry around vanadium, whereas V5+ ions in V2O5 fold distorted tetragonal pyramid [27,148,149,174]. Observed pre-edge peaks [at 5468.4 and 5469.8 eV (V2O3), 5470.0 eV (VO2), 5470.8 eV in V2O5] are, as described above, generally assigned as due to a transition from 1s to 3d + 4p [166,169,175,176].
Figure 2a shows Ti K-edge XANES spectra in toluene for CpTiCl3, Cp*TiX3 (X = OCH3, Cl) measured at 25 ºC (Ti K-edge 4.97 keV, through use of synchrotron radiation at SPring-8, BL01B1 beamline). These spectra show rather strong pre-edge absorption at 4965-4967 eV due to tetrahedral geometry around titanium, as described above. Rather strong absorption bands called shoulder-edge at 4977.0 eV in Cp*TiCl3 and CpTiCl3, whereas no such absorption was seen in Cp*Ti(OCH3)3; the absorption was due to presence of Ti–Cl bond, known as a shakedown peak assigned to the metal 1s to 4p coupled with the ligand to metal charge transfer (LMCT) [170,171,172,173]. No significant differences in the spectra were seen between CpTiCl3 and Cp*TiCl3, because of the similar geometry and number of chloride ligands around titanium.
Figure 2b shows Ti K-edge XANES spectra in toluene solution (at 25 °C, conc. 50 μmol-Ti/mL) for Cp’TiCl2(O-2,6-iPr2C6H3) [Cp’ = Cp, tBuC5H4 (1d), Cp* (1a)], CpTiCl2(N=CtBu2) (2a), and [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC) [27]. The XANES spectrum of 1a shows strong pre-edge absorptions at 4966.2 and 4967.6 eV and these are similar to those in the dimethyl complex, Cp’TiMe2(O-2,6-iPr2C6H3) (1a’), which showed at 4966.1 and 4967.7 eV. As described above, these are generally considered as due to a transition from 1s to 3d + 4p [166,169,175,176]. Moreover, as described above, 1a shows an absorption maxima at 4978.0 eV (called as shoulder-edge) ascribed to a presence of Ti–Cl bond [76,170,171,172,173]. Interestingly, as described above, positions of the edge peak (absorption) and the intensities in the half-titanocenes chosen in Figure 2b are close, whereas CGC shows rather high intensity at the pre-edge absorption at 4967.6 eV. Moreover, the spectra in toluene solution were highly analogous to those measured in solid (tablets with boron nitride) [76], strongly suggesting that these are Ti(IV) complexes with (a distorted) tetrahedral geometry in solid and solution [76].
5.3. XAS Analysis for Exploring Active Species in Olefin Polymerization and Syndiospecific Styrene Polymerization by Half-Titanocene Catalysts
The high oxidation state group 4 cationic metal-alkyl species, Cp’2M+R (R = alkyl) or Cp’M+(Y)R (Y = anionic ancillary donor), formed from the dialkyl analogues by reaction with borate or MAO through alkyl abstraction, play an essential role as the catalytically active species in this catalysis cycle (Scheme 10) [5,9,10,11,12,13,14,15,16,17,24,25,26,27]. However, effective catalysts for olefin polymerization (especially metallocene, linked-half-titanocene) display poor (negligible) capability for synthesis of syndiotactic polystyrene (SPS) [34,36,37,38,39]. In contrast, half-titanocene catalysts, Cp’TiX3 (X = F, Cl, OMe etc.), enables high catalytic activities for syndiospecific styrene polymerization (SSP) [38,39,40], whereas these catalysts exhibited low catalytic activities for ethylene polymerization and afforded a mixture of polyethylene, SPS and the copolymer, poly(ethylene-co-styrene) [34,41]. It has thus been recognized that the active species between olefin polymerization and SSP are different. The oxidation state of the active species in the SSP had thus been proposed as cationic Ti(III) species [177,178,179] or neutral Ti(III) species [56,57,180,181]. However, these mechanistic studies lacked “direct evidence” until recently, especially from the catalyst solution in the presence of styrene and MAO.
As described above, XANES (XANES = X-ray Absorption Near Edge Structure) and EXAFS (EXAFS = Extended X-ray Absorption Fine Structure) analyses provide direct information of not only oxidation state and the basic geometry around the metal centre (by XANES), but also kind of atoms and the distances connected (coordinated) to the metal centre (by EXAFS). Therefore, here introduces summary of the mechanistic studies reported recently [27,76].
Figure 3a shows the XANES spectra for the phenoxide-modified half-titanocene dichloride (1a) and dimethyl complexes (1a’), and the catalyst solution in the presence of MAO and 1-hexene, exhibiting catalytic activities for 1-hexene polymerisation in situ [27,76]. These complexes (1a,1a’) exhibit high catalytic activities for ethylene polymerization [14,15,16,17,42,43,44], α-olefin polymerization [182], and ethylene copolymerization with various monomers (as described above) [14,15,16,17,42,43,44].
As shown in Figure 3a, the pre-edge peak positions and edge absorption of 1a [4966.2 and 4967.6 eV (pre-edge), 4978.0 eV (shoulder-edge)] did not change upon addition of MAO, whereas intensity in the shoulder edge absorption in 1a, ascribed to Ti–Cl bond, decreased upon addition of MAO (especially 50 equiv) [76]. No apparent changes in the spectra were observed even upon addition of excess 1-hexene (200 equiv). Moreover, the spectra did not change when the dimethyl complex (1a’) was added MAO (10 equiv). These results strongly suggest that the oxidation state as well as basic geometry around titanium preserved under these conditions even addition of MAO and 1-hexene (excess amounts). Similarly, as shown in Figure 3b, no significant differences in the XANES spectra were seen when the 1d was treated with d-MAO and 1-hexene [76]. These results also suggest the same possibility that major species formed as Ti(IV) species upon addition of MAO.
Although no significant changes in the XANES spectra were observed, the corresponding EXAFS spectra, shown in Figure 4 [oscillations (left) and the FT-EXAFS spectra (right)], provide the clearer image, information in the reactions. The XANES spectra for Cp*TiCl2(OAr) (1a) and the catalyst solution in the presence of MAO (10 and 50 equiv) showed no distinct spectral changes in toluene solution and in solid state (in solid, disk with boron nitride) measured at 25 °C (Figure 4a,b). The summary of coordination number (C.N.) and the distance (Å) in the atoms connected to titanium analyzed through the curve-fitting are summarized in Table 5 [76].
The Ti–C (on Cp’ coordinated) and Ti–O (phenoxide) bonds were preserved even after treating 1a with MAO (10 and 50 equiv). In contrast, C.N. of Ti–Cl decreased by treating with MAO (Figure 4d, Table 5), clearly indicating that the Ti–Cl bonds were reacted (dissociated) with MAO (alkylation). Since separation of Ti–C and Ti–O bond seemed difficult (distances are close) in the analysis, observed increase in the C.N. of Ti–O bond upon addition of MAO could be explained as due to a subsequent formation of Ti–C bond [76]. These data also suggest that the Ti–Cl bonds in 1a was reacted with MAO without dissociation of the Ti–O bond to form dimethyl complexes; the data support that the Ti(IV)-alkyl species play a role in this catalysis [14,15,16,17,76].
As described above, the oxidation state of the active species in the SSP has been proposed as cationic or neutral Ti(III) species, Cp’Ti+(R)(styrene) [177,178,179] or Cp’Ti(R)(Y)(styrene) [56,57,180,181] (Scheme 10). The cationic Ti(III) species were proposed based on the results in ESR measurement [177], reaction chemistry (formation of SPS) [179], and theoretical support [178]. The neutral Ti(III) species were proposed based on the results of (i) polymerization data (ligand effect, anionic donor) [56,57], (ii) step (co)polymerization [181], and (iii) theoretical support [180]. However, until recently, there were no “direct evidence” from the catalyst solution containing MAO and styrene [27,76,183].
Figure 5 shows Ti K-edge XANES spectra (in toluene at 25 ºC) for Cp’TiCl2(OAr) [Cp’ = tBuC5H4 (1d, left), Cp (1e, right)], and the spectra of solutions containing 1d or 1e and MAO (50 equiv), styrene (200 equv), which produce SPS in situ. The XANES spectrum of 1d shows pre-edge absorptions (4966.5, 4967.5 eV) and a shoulder-edge absorption (4977.9 eV, presence of Ti–Cl bond) [27,170,171,172,173]. Addition of MAO (50 equiv) did not lead the significant spectral changes except decrease in the intensity of the shoulder-edge absorption in 1d was observed, suggesting dissociation of Ti–Cl bond by alkylation.76
In contrast, noteworthy, styrene addition (200 equiv) into the solution (1d and MAO 50 equiv) led to the clear low energy shift (2.2 eV on the basis of the inflection point) in the edge absorption accompanied with decrease in intensities of two pre-edge peaks. The results thus strongly suggest that complex 1d was reduced by addition of styrene (not by MAO). Moreover, the low energy shift in the edge absorption was observed upon increase amount of styrene charged (100→200 equiv) [76]. Moreover, as shown in Figure 5b, the solution containing 1e showed the similar change when 200 equiv of styrene and 50 equiv of MAO were added into a toluene solution, which also produce SPS in situ [57]. These analysis data strongly suggest a formation of Ti(III) species from Ti(IV), derived from 1d, accompanied with the structural changes upon addition of styrene.
Figure 6 shows EXAFS oscillations and the FT-EXAFS spectra (in toluene at 25 ºC) for (tBuC5H4)TiCl2(OAr) (1d), and the catalyst solution in the presence of MAO (50 equiv) and styrene (200 equiv), which showed the significant changes in the oxidation state upon addition of styrene in the XANES spectra (Figure 5). The analysis data by curve fitting are summarized in Table 6 [76].
Apparent change in the EXAFS oscillations from a toluene solution of 1d to the solution with addition of MAO and styrene strongly suggest certain structural change (accompanied with change in the oxidation state). As observed by 1a, C.N. of Ti–Cl diminished along with observation of new Ti–C bond which is formed by methylation with MAO, whereas C.N. and bond lengths in Ti–C bonds corresponding to Cp’ were unchanged before/after the reaction. Importantly, preservation of the Ti–O bond (corresponding to phenoxide ligand) was confirmed even after 1d was treated with MAO upon addition of styrene (200 equiv). The analysis data clearly demonstrate formation of the catalytically active species for SSP without dissociation of the Ti–O bond accompanied with reduction; the results thus strongly suggest an existence of the phenoxide species, (tBuC5H4)Ti(R)(OAr) [or CpTi(R)(OAr)], in the solution [27,76]. The result is a good agreement with our previous assumption on the basis of two step ethylene/styrene (co)polymerization (Scheme 11) [181] as well as effect of anionic donor ligand in SSP [57] that the neutral Ti(III) species containing phenoxide ligand thus play a role for the SSP [181].
Moreover, the proposed catalytically active species based on XAS analysis as well as step ethylene/styrene (co)polymerization (Scheme 11a) was further supported by theoretical calculation [27,76,183]. DFT calculation was conducted for syndiospecific styrene insertion step of the three possible models of the active species containing the phenoxide ligand, neutral Ti(III), cationic Ti(IV), and Ti(IV)–B(C6F5)4 catalysts (shown in Scheme 11b). The calculation was conducted from the Ti catalysts containing the phenylpropyl group, as a model of polystyrene in chain-propagation step (R; styrene + Ti, zero), and the styrene coordinated models (IM), and inserted model (P) in which styrene was inserted to the phenylpropyl moiety [76]. As shown in Scheme 11b, the results clearly indicate that the neutral Ti(III) catalyst exhibits the lower activation energy than the others, thus strongly support the above mechanism. The computational analysis also led to a conclusion that the neutral Ti(III) species containing the phenoxide ligand is more efficient and plausible catalyst model; formation of the Ti(III) species, proposed in Scheme 11, is a key to promote the SSP in an efficient manner [76]. Further DFT calculation results revealed that the neutral Ti(III) active species has high selectivity in SSP; the orientation of styrene (si-/h-/re-)insertions related to the stereoselectivity of polystyrene. The spin density analysis indicated the neutral Ti(III) center promote an electron transfer from the polymeric chain to the incoming styrene [183].
6. Concluding Remarks and Outlook
Olefin polymerization by group 4 transition metal catalysts has been the key technology for production of polyolefins, commodity plastics in our daily life. Development of new polymers, that cannot be achieved by using conventional catalysts has been the central research objective and copolymerization is an effective strategy. In this reviewing article, modified half-titanocenes, Cp’TiX2(Y) (Y = anionic donor such as phenoxide, ketimide, amidinate etc.) [6,14,15,16,17], contributed a significant effort for efficient, exclusive synthesis of ethylene copolymers containing cyclic olefins (COCs) [60,61,62,63,64,65,66,67,68], biobased conjugated dienes [69,70], disubstituted α-olefins [44,71,72,73,74], and aromatic vinyl monomers [34,41,56,58,59], ethylene/α-olefin copolymers containing hydroxy group [44] etc. More recently, we realized that the catalysts containing unsymmetric iminoimidazolide ligands exhibited promising capabilities for synthesis of COCs [184]. Moreover, modification of phenoxide para-position enhanced the activity in ethylene polymerization and the copolymerization with α-olefin [45]. These results strongly suggest that the catalysts of this type will continue to contribute the development of new polyolefins. Moreover, this manuscript introduces analysis of catalytically active species through XAS (X-ray absorption spectroscopy). We highly hope that the analysis method can be used for more researchers in the near future for better understanding in catalysis cycle.
Author Contributions
Conceptualization, supervision, project administration, funding acquisition, data curation, KN; writing—original draft preparation, visualization, KN and KJ.; writing—review and editing, KN. All authors have read and agreed to the published version of the manuscript.
Funding
The projects by KN were partly supported by Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS, Nos. 13555253, 18350055, 21350054, 24350049, 15H03812, 15K14225, 18H01982, 18K18981, 19KK0139, 21H01942, 25K01583), Grant-in-Aid for Scientific Research on Innovative Areas (No. 26105003, "3D Active-Site Science") from The Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan. The synchrotron XAFS analysis were performed at the SPring-8 beam lines of BL01B1 with the approval of Japan Synchrotron Radiation Research Institute (JASRI, 2017A1512, 2018A1245, 2019A1233, 2020A0618, 2021A1435, 2021B1594, 2022A1276, 2022B1669, 2023A1765).
Data Availability Statement
We encourage all authors of articles published in MDPI journals to share their research data. In this section, please provide details regarding where data supporting reported results can be found, including links to publicly archived datasets analyzed or generated during the study. Where no new data were created, or where data is unavailable due to privacy or ethical restrictions, a statement is still required. Suggested Data Availability Statements are available in section “MDPI Research Data Policies” at https://www.mdpi.com/ethics.
Acknowledgments
KN thanks to Dr. S. Kikkawa and Prof. S. Yamazoe (Tokyo Metropolitan University, TMU) for big support in XAS analysis, and Prof. N. Nakatani (TMU) for support in computational analysis. KJ expresses Tokyo Metropolitan government (Tokyo Global Partner Scholarship Program) for pre-doctoral fellowship.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Plastics Europe. Plastics the Fast Facts 2025 . Available online: https://plasticseurope.org/knowledge-hub/plastics-the-fast-facts-2025/ (accessed on January 2026).
- Selected recent reviews and book chapter, see refs 2–8: Baier, M. C.; Zuideveld, M. A.; Mecking S. Post-metallocenes in the industrial production of polyolefins. Angew. Chem. Int. Ed. 2014, 53, 9722–9744. [CrossRef]
- Stürzel, M.; Mihan, S.; Mülhaupt, R. From multisite polymerization catalysis to sustainable materials and all-polyolefin composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef] [PubMed]
- van Doremaele, G.; van Duin, M.; Valla, M.; Berthoud, A. On the development of titanium κ1-amidinate complexes, commercialized as keltan ACETM technology, enabling the production of an unprecedented large variety of EPDM polymer structures. J. Polym. Sci., Part A: Polym. Chem. 2017, 55, 2877–2891. [Google Scholar] [CrossRef]
- Handbook of Transition Metal Polymerization Catalysts, 2nd Ed.; Hoff, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Nomura, K.; Kitphaitun, S. Catalysis for a Sustainable Environment: Reactions, Processes and Applied Technologies; Pombeiro, A. J. L., Sutradhar, M., Alegria, E. C. B. A., Eds.; John Wiley & Sons, Ltd.: Chichester, West Sussex, UK, 2024; pp. 323–338. [Google Scholar] [CrossRef]
- Tan, C.; Zhou, C.; Chen, C. Material properties of functional polyethylenes from transition-metal-catalyzed ethylene–polar monomer copolymerization. Macromolecules 2022, 55, 1910–1922. [Google Scholar] [CrossRef]
- Chen, C. Designing catalysts for olefin polymerization and copolymerization: Beyond electronic and steric tuning. Nat. Rev. Chem. 2018, 2, 6–14. [Google Scholar] [CrossRef]
- Selected pioneering reviews for metallocene catalysts, see refs 9–12: Brintzinger, H. H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth R. M. Stereospecific olefin polymerization with chiral metallocene catalysts. Angew. Chem., Int. Ed. Engl. 1995, 34, 1143–1170. [CrossRef]
- Kaminsky, W. New polymers by metallocene catalysis. Macromol. Chem. Phys. 1996, 197, 3907–3945. [Google Scholar] [CrossRef]
- Kaminsky, W.; Arndt, M. Metallocenes for polymer catalysis. Adv. Polym. Sci. 1997, 127, 143–187. [Google Scholar] [CrossRef]
- Suhm, J.; Heinemann, J.; Wörner, C.; Müller, P.; Stricker, F.; Kressler, J.; Okuda, J.; Mülhaupt, R. Novel polyolefin materials via catalysis and reactive processing. Macromol. Symp. 1998, 129, 1–28. [Google Scholar] [CrossRef]
- Pioneering reviews for linked half-titanocene catalysts, see refs 12,13: McKnight, A. L.; Waymouth, R. M. Group 4 ansa-cyclopentadienyl-amido catalysts for olefin polymerization. Chem. Rev. 1998, 98, 2587–2598. [CrossRef]
- Selected reviews for modified half-titanocene catalysts, refs. 4,6,14–17: Nomura, K.; Liu, J.; Padmanabhan, S.; Kitiyanan, B. Nonbridged half-metallocenes containing anionic ancillary donor ligands: New promising candidates as catalysts for precise olefin polymerization. J. Mol. Catal. A: Chem. 2007, 267, 1–29. [CrossRef]
- Nomura, K. Half-titanocenes containing anionic ancillary donor ligands as promising new catalysts for precise olefin polymerization. Dalton Trans. 2009, 8811–8823. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Liu, J. Half-titanocenes for precise olefinpolymerisation: Effects of ligand substituents and some mechanistic aspects. Dalton Trans. 2011, 40, 7666–7682. [Google Scholar] [CrossRef] [PubMed]
- Nomura, K.; Liu, J. In Organometallic Reactions and Polymerisation; Osakada, K., Ed.; The Lecture Notes in Chemistry, vol. 85; Springer: Berlin, 2014; pp 51–88.
- Selected reviews for called post metallocene catalysts, refs. 2,7,8,18–20: Britovsek, G. J. P.; Gibson, V. C.; Wass, D. F. The search for new-generation olefin polymerization catalysts: Life beyond metallocenes. Angew. Chem., Int. Ed. Engl. 1999, 38, 428–447. [CrossRef]
- Gibson, V. C.; Spitzmesser, S. K. Advances in non-metallocene olefin polymerization catalysis. Chem. Rev. 2003, 103, 283–316. [Google Scholar] [CrossRef]
- Coates, G. W.; Hustad, P. D.; Reinartz, S. Catalysts for the living insertion polymerization of alkenes: Access to new polyolefin architectures using Ziegler-Natta chemistry. Angew. Chem. Int. Ed. 2002, 41, 2236–2257. [Google Scholar] [CrossRef]
- Delferro, M.; Marks, T. J. Multinuclear olefin polymerization catalysts. Chem. Rev. 2011, 111, 2450–2485. [Google Scholar] [CrossRef]
- McInnis, J. P.; Delferro, M.; Marks, T. J. Multinuclear group 4 catalysis: Olefin polymerization pathways modified by strong metal–metal cooperative effects. Acc. Chem. Res. 2014, 47, 2545–2557. [Google Scholar] [CrossRef]
- Valente, A.; Mortreux, A.; Visseaux, M.; Zinck, P. Coordinative chain transfer polymerization. Chem. Rev. 2013, 113, 3836–3857. [Google Scholar] [CrossRef]
- Macchioni, A. Ion pairing in transition-metal organometallic chemistry. Chem. Rev. 2005, 105, 2039–2074. [Google Scholar] [CrossRef]
- Bochmann, M. The chemistry of catalyst activation: The case of group 4 polymerization catalysts. Organometallics 2010, 29, 4711–4740. [Google Scholar] [CrossRef]
- Kaminsky, W. Discovery of methylaluminoxane as cocatalyst for olefin polymerization. Macromolecules 2012, 45, 3289–3297. [Google Scholar] [CrossRef]
- Yi, J.; Nakatani, N.; Nomura, K. Solution XANES and EXAFS analysis of active species of titanium, vanadium complex catalysts in ethylene polymerisation/dimerisation and syndiospecific styrene polymerization. Dalton Trans. 2020, 49, 8008–8028. [Google Scholar] [CrossRef]
- Suhm, J.; Schneider, M. J.; Mülhaupt, R. Influence of metallocene structures on ethene copolymerization with 1-butene and 1-octene. J. Mol. Catal. A: Chem. 1998, 128, 215–227. [Google Scholar] [CrossRef]
- Suhm, J.; Schneider, M. J.; Mülhaupt, R. Temperature dependence of copolymerization parameters in ethene/1-octene copolymerization using homogeneous rac-Me2Si(2-MeBenz[e]Ind)2ZrCl2/MAO catalyst. J. Polym. Sci. A 1997, 35, 735–740. [Google Scholar] [CrossRef]
- For example, see refs 30-33: Canich, J. A. M.; Hlatky, G. G.; Turner, H. W. U.S. Patent 5,422,236, 1990.
- Canich, J. A. M. U.S. Patent 5,026,798, 1991.
- Stevens, J. C.; Timmers, F. J.; Wilson, D. R.; Schmidt, G. F.; Nickias, P. N.; Rosen, R. K.; Knight, G. W.; Lai, S.-Y. Eur. Pat. Appl. 416,815, 1991.
- Stevens, J. C.; Neithamer, D. R. Eur. Pat. Appl. 418,022, 1991.
- Nomura, K. Syndiotactic Polystyrene: Synthesis, Characterization, Processing, and Applications; Schellenberg, J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, 2010; pp. 60–91. [Google Scholar]
- Arriola, D. J.; Bokota, M.; Campbell, R. E., Jr.; Klosin, J.; LaPointe, R. E.; Redwine, O. D.; Shankar, R. B.; Timmers, F. J.; Abboud, K. A. Penultimate effect in ethylene−styrene copolymerization and the discovery of highly active ethylene−styrene catalysts with increased styrene reactivity. J. Am. Chem. Soc. 2007, 129, 7065–7076. [Google Scholar] [CrossRef]
- Guo, N.; Li, L.; Marks, T. J. Bimetallic catalysis for styrene homopolymerization and ethylene−styrene copolymerization: Exceptional comonomer selectivity and insertion regiochemistry. J. Am. Chem. Soc. 2004, 126, 6542–6543. [Google Scholar] [CrossRef]
- Guo, N.; Stern, C. L.; Marks, T. J. Bimetallic effects in homopolymerization of styrene and copolymerization of ethylene and styrenic comonomers: Scope, kinetics, and mechanism. J. Am. Chem. Soc. 2008, 130, 2246–2261. [Google Scholar] [CrossRef]
- Tomotsu, N.; Ishihara, N. Novel catalysts for syndiospecific polymerization of styrene. Catal. Surv. Jpn. 1997, 1, 89–110. [Google Scholar] [CrossRef]
- Tomotsu, N.; Ishihara, N.; Newman, T. H.; Malanga, M. T. Syndiospecific polymerization of styrene. J. Mol. Catal. A 1998, 128, 167–190. [Google Scholar] [CrossRef]
- Schellenberg, J. Recent transition metal catalysts for syndiotactic polystyrene. Prog. Polym. Sci. 2009, 34, 688–718. [Google Scholar] [CrossRef]
- Zhang, H.; Nomura, K. Living copolymerization of ethylene with styrene catalyzed by (cyclopentadienyl)(ketimide)titanium(IV) complex−MAO catalyst system: Effect of anionic ancillary donor ligand. Macromolecules 2006, 39, 5266–5274. [Google Scholar] [CrossRef]
- Nomura, K.; Naga, N.; Miki, M.; Yanagi, K.; Imai, A. Synthesis of various non-bridged Cp-aryloxy titanium(IV) complexes of the type CpTi(OAr)X2, and the catalytic alkene polymerization: Important role of substituents on both aryloxy and cyclopentadienyl groups. Organometallics 1998, 17, 2152–2154. [Google Scholar] [CrossRef]
- Nomura, K.; Naga, N.; Miki, M.; Yanagi, K. Olefin polymerization by (cyclopentadienyl)(aryloxy)titanium(IV) complexes–cocatalyst system. Macromolecules 1998, 31, 7588–7597. [Google Scholar] [CrossRef]
- Kitphaitun, S.; Yan, Q.; Nomura, K. Effect of SiMe3, SiEt3 para-substituents for exhibiting high activity, introduction of hydroxy group in ethylene copolymerization catalyzed by phenoxide-modified half-titanocenes. Angew. Chem. Int. Ed. 2020, 59, 23072–23076. [Google Scholar] [CrossRef]
- Gao, J.; Sun, W.-H.; Nomura, K. Synthesis of new phenoxide modified half-titanocene catalysts for ethylene polymerization. Catalysts 2025, 15, 840–851. [Google Scholar] [CrossRef]
- Zhang, S.; Piers, W. E.; Gao, X.; Parvez, M. The mechanism of methane elimination in B(C6F5)3-initiated monocyclopentadienyl-ketimide titanium and related olefin polymerization catalysts. J. Am. Chem. Soc. 2000, 122, 5499–5509. [Google Scholar] [CrossRef]
- McMeeking, J.; Gao, X.; Spence, R. E. v. H.; Brown, S. J.; Jerermic, D. U.S. Patent 6,114,481, Sept 5, 2000.
- Nomura, K.; Fujita, K.; Fujiki, M. Effects of cyclopentadienyl fragment in ethylene, 1-hexene, and styrene polymerizations catalyzed by half-titanocenes containing ketimide ligand of the type, Cp′TiCl2(NCtBu2). Catal. Commun. 2004, 5, 413–417. [Google Scholar] [CrossRef]
- Nomura, K.; Fujita, K.; Fujiki, M. Olefin polymerization by (cyclopentadienyl)(ketimide)titanium(IV) complexes of the type, Cp′TiCl2(NCtBu2)-methylaluminoxane (MAO) catalyst systems. J. Mol. Catal. A 2004, 220, 133–144. [Google Scholar] [CrossRef]
- Dias, A. R.; Duarte, M. T.; Fernandes, A. C.; Fernandes, S.; Marques, M. M.; Martins, A. M.; da Silva, J. F.; Rodrigues, S. S. Titanium ketimide complexes as α-olefin homo- and copolymerisation catalysts: X-ray diffraction structures of [TiCp′(NCtBu2)Cl2] (Cp′=Ind, Cp*). J. Organomet. Chem. 2004, 689, 203–213. [Google Scholar] [CrossRef]
- Martins, A. M.; Marques, M. M.; Ascenso, J. R.; Dias, A. R.; Duarte, M. T.; Fernandes, A. C.; Fernandes, S.; Ferreira, M. J.; Matos, I.; Oliveira, M. C.; Rodrigues, S. S.; Wilson, C. J. Titanium and zirconium ketimide complexes: synthesis and ethylene polymerisation catalysis. Organomet. Chem. 2005, 690, 874–884. [Google Scholar] [CrossRef]
- Ferreira, M. J.; Martins, A. M. Group 4 ketimide complexes: Synthesis, reactivity and catalytic applications. Coord. Chem. Rev. 2006, 250, 118–132. [Google Scholar] [CrossRef]
- Ijpeij, E. G.; Zuideveld, M. A.; Arts, H. J.; van der Burgt, F.; van Doremaele, G. H. J. WO Patent 2007, 031, 295, 2007.
- Ijpeij, E. G.; Windmuller, P. J. H.; Arts, H. J.; van der Burgt, F.; van Doremaele, G. H. J.; Zuideveld, M. A. WO Patent 2005, 090, 418, 2005.
- Ijpeij, E. G.; Coussens, B.; Zuideveld, M. A.; van Doremaele, G. H. J.; Mountford, P.; Lutzc, M.; Spek, A. L. Synthesis, solid state and DFT structure and olefin polymerization capability of a unique base-free dimeric methyl titanium dication. Chem. Commun. 2010, 46, 3339–3341. [Google Scholar] [CrossRef]
- Nomura, K.; Komatsu, T.; Imanishi, Y. Syndiospecific styrene polymerization and efficient ethylene/styrene copolymerization catalyzed by (cyclopentadienyl)(aryloxy)titanium(IV) complexes−MAO system. Macromolecules 2000, 33, 8122–8124. [Google Scholar] [CrossRef]
- Byun, D. J.; Fudo, A.; Tanaka, A.; Fujiki, M.; Nomura, K. Effect of cyclopentadienyl and anionic ancillary ligand in syndiospecific styrene polymerization catalyzed by nonbridged half-titanocenes containing aryloxo, amide, and anilide ligands: Cocatalyst systems. Macromolecules 2004, 37, 5520−5530. [Google Scholar] [CrossRef]
- Nomura, K.; Okumura, H.; Komatsu, T.; Naga, N. Ethylene/styrene copolymerization by various (cyclopentadienyl)(aryloxy)titanium(IV) complexes−MAO catalyst systems. Macromolecules 2002, 35, 5388−5395. [Google Scholar] [CrossRef]
- Aoki, H.; Nomura, K. Synthesis of amorphous ethylene copolymers with 2-vinylnaphthalene, 4-vinylbiphenyl and 1-(4-vinylphenyl)naphthalene. Macromolecules 2021, 54, 83−93. [Google Scholar] [CrossRef]
- Nomura, K.; Tsubota, M.; Fujiki, M. Efficient ethylene/norbornene copolymerization by (Aryloxo)(indenyl)titanium(IV) complexes−MAO catalyst system. Macromolecules 2003, 36, 3797–3799. [Google Scholar] [CrossRef]
- Nomura, K.; Wang, W.; Fujiki, M.; Liu, J. Notable norbornene (NBE) incorporation in ethylene–NBE copolymerization catalysed by nonbridged half-titanocenes: Better correlation between NBE incorporation and coordination energy. Chem. Commun. 2006, 2659–2661. [Google Scholar] [CrossRef] [PubMed]
- Apisuk, W.; Trambitas, A. G.; Kitiyanan, B.; Tamm, M.; Nomura, K. Efficient ethylene/norbornene copolymerization by half-titanocenes containing imidazolin-2-iminato ligands and MAO catalyst systems. J. Polym. Sci. Part A: Polym. Chem. 2013, 51, 2575–2580. [Google Scholar] [CrossRef]
- Kawatsu, M.; Fujioka, T.; Losio, S.; Tritto, I.; Nomura, K. Trialkylsilyl-cyclopentadienyl)titanium(IV) dichloride complexes containing ketimide ligands, Cp′TiCl2(N=CtBu2) (Cp′ = Me3SiC5H4, Et3SiC5H4), as efficient catalysts for ethylene copolymerisation with norbornene and tetracyclododecene. Catal. Sci. Technol. 2024, 15, 2757–2765. [Google Scholar] [CrossRef]
- Apisuk, W.; Ito, H.; Nomura, K. Efficient synthesis of cyclic olefin copolymers with high glass transition temperatures by ethylene copolymerization with tetracyclododecene using (tert-BuC5H4)TiCl2(N=CtBu2)–MAO Catalyst. J. Polym. Sci., Part A: Polym. Chem. 2016, 54, 2662–2667. [Google Scholar] [CrossRef]
- Zhao, W.; Nomura, K. Copolymerizations of norbornene and tetracyclododecene with α-olefins by half-titanocene catalysts: Efficient synthesis of highly transparent, thermal resistance polymers. Macromolecules 2016, 49, 59–70. [Google Scholar] [CrossRef]
- Okabe, M.; Nomura, K. Propylene/cyclic olefin copolymers with cyclopentene, cyclohexene, cyclooctene, tricyclo[6.2.1.0(2,7)]undeca-4-ene, and tetracyclododecene: The synthesis and effect of cyclic structure on thermal properties. Macromolecules 2023, 56, 81–91. [Google Scholar] [CrossRef]
- Harakawa, H.; Okabe, M.; Nomura, K. The synthesis of cyclic olefin copolymers (COCs) by ethylene copolymerisations with cyclooctene, cycloheptene, and with tricyclo[6.2.1.0(2,7)]undeca-4-ene: The effect of cyclic monomer structures on thermal properties. Polym. Chem. 2020, 11, 5590–5600. [Google Scholar] [CrossRef]
- Wang, W.; Fujiki, M.; Nomura, K. Copolymerization of ethylene with cyclohexene (CHE) catalyzed by nonbridged half-titanocenes containing aryloxo ligand: Notable effect of both cyclopentadienyl and anionic donor ligand for efficient CHE incorporation. J. Am. Chem. Soc. 2005, 127, 4582–4583. [Google Scholar] [CrossRef]
- Kitphaitun, S.; Chaimongkolkunasin, S.; Manit, J.; Makino, R.; Kadota, J.; Hirano, H.; Nomura, K. Ethylene/myrcene copolymers as new bio-based elastomers prepared by coordination polymerization using titanium catalysts. Macromolecules 2021, 54, 10049–10058. [Google Scholar] [CrossRef]
- Guo, L.; Makino, R.; Shimoyama, D.; Kadota, J.; Hirano, H.; Nomura, K. Synthesis of ethylene/isoprene copolymers containing cyclopentane/cyclohexane units as unique elastomers by half-titanocene catalysts. Macromolecules 2023, 56, 899–914. [Google Scholar] [CrossRef]
- Nomura, K.; Itagaki, K.; Fujiki, M. Efficient incorporation of 2-methyl-1-pentene in copolymerization of ethylene with 2-methyl-1-pentene catalyzed by nonbridged half-titanocenes. Macromolecules 2005, 38, 2053–2055. [Google Scholar] [CrossRef]
- Nomura, K.; Itagaki, K. Efficient incorporation of vinylcylohexane in ethylene/vinylcyclohexane copolymerization catalyzed by nonbridged half-titanocenes. Macromolecules 2005, 38, 8121–8123. [Google Scholar] [CrossRef]
- Itagaki, K.; Fujiki, M.; Nomura, K. Effect of cyclopentadienyl and anionic donor ligands on monomer reactivities in copolymerization of ethylene with 2-methyl-1-pentene by nonbridged half-titanocenes−cocatalyst systems. Macromolecules 2007, 40, 6489–6499. [Google Scholar] [CrossRef]
- Khan, F. Z.; Kakinuki, K.; Nomura, K. Copolymerization of ethylene with tert-butylethylene using nonbridged half-titanocene-cocatalyst systems. Macromolecules 2009, 42, 3767–3773. [Google Scholar] [CrossRef]
- Kitphaitun, S.; Fujimoto, T.; Ochi, Y.; Nomura, K. Effect of borate cocatalysts toward activity and comonomer incorporation in ethylene copolymerization by half-titanocene catalysts in methylcyclohexane. ACS Org. Inorg. Au. 2022, 2, 386–391. [Google Scholar] [CrossRef]
- Nomura, K.; Izawa, I.; Yi, J.; Nakatani, N.; Aoki, H.; Ina, T.; Mitsudome, T.; Tomotsu, N.; Yamazoe, S. Solution XAS analysis for exploring active species in syndiospecific styrene polymerization and 1-hexene polymerization using half-titanocene–MAO catalysts: Significant changes in the oxidation state in the presence of styrene. Organometallics 2019, 38, 4497–4507. [Google Scholar] [CrossRef]
- Sun, Z.; Unruean, P.; Aoki, H.; Kitiyanan, B.; Nomura, K. Phenoxide-modified half-titanocenes supported on star-shaped ROMP polymers as efficient catalyst precursors for ethylene copolymerization. Organometallics 2020, 39, 2998–3009. [Google Scholar] [CrossRef]
- Cherdron, H.; Brekner, M. -J.; Osan, F. Cycloolefin-copolymere: Eine neue klasse transparenter thermoplaste. Angew. Makromol. Chem. 1994, 223, 121–133. [Google Scholar] [CrossRef]
- Tritto, I.; Boggioni, L.; Ferro, D. R. Metallocene catalyzed ethene- and propene-co-norbornene polymerization: Mechanisms from a detailed microstructural analysis. Coord. Chem. Rev. 2006, 250, 212–241. [Google Scholar] [CrossRef]
- Nomura, K. Nonbridged half-titanocenes containing anionic ancillary donor ligands: Promising new catalysts for precise synthesis of cyclic olefin copolymers (COCs). Chin. J. Polym. Sci. 2008, 26, 513–523. [Google Scholar] [CrossRef]
- Li, X.; Hou, Z. Organometallic catalysts for copolymerization of cyclic olefins. Coord. Chem. Rev. 2008, 252, 1842–1869. [Google Scholar] [CrossRef]
- Zhao, W.; Nomura, K. Design of efficient molecular catalysts for synthesis of cyclic olefin copolymers (COC) by copolymerization of ethylene and α-olefins with norbornene or tetracyclododecene. Catalysts 2016, 6, 175–190. [Google Scholar] [CrossRef]
- Boggioni, L.; Tritto, I. State of the art of cyclic olefin polymers. MRS Bull. 2013, 38, 245–251. [Google Scholar] [CrossRef]
- Nomura, K. Development of half-titanocene catalysts for synthesis of cyclic olefin copolymers. Polyolefin J. 2023, 10, 59–70. [Google Scholar] [CrossRef]
- Wang, W.; Qu, S.; Li, X.; Chen, J.; Guo, Z.; Sun, W.-H. Transition metal complex catalysts promoting copolymers of cycloolefin with propylene/higher olefins. Coord. Chem. Rev. 2023, 494, 215351. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, J.; Zhang, Y.; Cui, L.; Chi, Y.; Jian, Z. Advances in high refractive index cycloolefin-containing polymeric materials. Macromolecules 2025, 58, 10949–10962. [Google Scholar] [CrossRef]
- Polyplastics, *!!! REPLACE !!!*. Topas: High-Performance Cyclic Olefin Copolymer. Available online: https://www.polyplastics.com/en/product/topas.vm (accessed on January 2026).
- Mitsui Chemicals. ApelTM Cyclic Olefin Copolymer. Available online: https://jp.mitsuichemicals.com/en/special/apel/ (accessed on January 2026).
- Selected initial reports, refs. 89-95: Ruchatz, D.; Fink, G. Ethene-norbornene copolymerization using homogeneous metallocene and half-sandwich catalysts: Kinetics and relationships between catalyst structure and polymer structure: 1. Kinetics of the ethene-norbornene copolymerization using the [(isopropylidene)(η5-inden-1-ylidene-η5-cyclopentadienyl)]zirconium dichloride/methylaluminoxane catalyst. Macromolecules 1998, 31, 4669–4673. [CrossRef]
- Ruchatz, D.; Fink, G. Ethene−norbornene copolymerization using homogenous metallocene and half-sandwich catalysts: Kinetics and relationships between catalyst structure and polymer structure: 2. Comparative study of different metallocene- and half-sandwich/methylaluminoxane catalysts and analysis of the copolymers by 13C nuclear magnetic resonance spectroscopy. Macromolecules 1998, 31, 4674–4680. [Google Scholar] [CrossRef]
- Arndt, M.; Beulich, I. C1-Symmetric metallocenes for olefin polymerisation, 1. Catalytic performance of [Me2C(3-tertBuCp)(Flu)]ZrCl2 in ethene/norbornene copolymerisation. Macromol. Chem. Phys. 1998, 199, 1221–1232. [Google Scholar] [CrossRef]
- Provasoli, A.; Ferro, D. R.; Tritto, I.; Boggioni, L. The conformational characteristics of ethylene-norbornene copolymers and their influence on the 13C NMR spectra. Macromolecules 1999, 32, 6697–6706. [Google Scholar] [CrossRef]
- Tritto, I.; Marestin, C.; Boggioni, L.; Zetta, L.; Provasoli, A.; Ferro, D. R. Ethylene-norbornene copolymer microstructure: Assessment and advances based on assignments of 13C NMR spectra. Macromolecules 2000, 33, 8931–8944. [Google Scholar] [CrossRef]
- Tritto, I.; Marestin, C.; Boggioni, L.; Brintzinger, H. H.; Ferro, D. R. Stereoregular and stereoirregular alternating ethylene-norbornene copolymers. Macromolecules 2001, 34, 5770–5777. [Google Scholar] [CrossRef]
- Tritto, I.; Boggioni, L.; Ferro, D. R. Alternating isotactic ethylene−norbornene copolymers by C1-symmetric metallocenes: Determination of the copolymerization parameters and mechanistic considerations on the basis of pentad analysis. Macromolecules 2004, 37, 9681–9693. [Google Scholar] [CrossRef]
- Selected initial reports, refs. 96-99: Harrington, B. A.; Crowther, D. J. Stereoregular, alternating ethylene–norbornene copolymers from monocyclopentadienyl catalysts activated with non-coordinating discrete anions. J. Mol. Catal. A: Chem. 1998, 128, 79–84. [CrossRef]
- McKnight, A. L.; Waymouth, R. M. Ethylene/norbornene copolymerizations with titanium CpA catalysts. Macromolecules 1999, 32, 2816–2825. [Google Scholar] [CrossRef]
- Thorshaug, K.; Mendichi, R.; Tritto, I.; Trinkle, S.; Friedrich, C.; Mülhaupt, R. Poly(ethene-co-norbornene) obtained with a constrained geometry catalyst. A study of reaction kinetics and copolymer properties. Macromolecules 2002, 35, 2903–2911. [Google Scholar] [CrossRef]
- Hasan, T.; Ikeda, T.; Shiono, T. Ethene−norbornene copolymer with high norbornene content produced by ansa-fluorenylamidodimethyltitanium complex using a suitable activator. Macromolecules 2004, 37, 8503–8509. [Google Scholar] [CrossRef]
- Selected initial reports, refs. 100-105: Altamura, P.; Grassi, A. Crystalline alternating sequences identified in ethylene-co-norbornene polymers produced by the (η5-C2B9H11)Zr(NEt2)2(NHEt2)-AliBu3 catalyst. Macromolecules 2001, 34, 9197–9200. [CrossRef]
- Yoshida, Y.; Mohri, J.; Ishii, S.; Mitani, M.; Saito, J.; Matsui, S.; Makio, H.; Nakano, T.; Tanaka, H.; Onda, M.; Yamamoto, Y.; Mizuno, A.; Fujita, T. Living copolymerization of ethylene with norbornene catalyzed by bis(pyrrolide−imine) titanium complexes with MAO. J. Am. Chem. Soc. 2004, 126, 12023–12032. [Google Scholar] [CrossRef]
- Li, X.-F.; Dai, K.; Ye, W.-P.; Pan, L.; Li, Y.-S. New titanium complexes with two β-enaminoketonato chelate ligands: Syntheses, structures, and olefin polymerization activities. Organometallics 2004, 23, 1223–1230. [Google Scholar] [CrossRef]
- Marconi, R.; Ravasio, A.; Boggioni, L.; Tritto, I. Silyl-terminated ethylene-co-norbornene copolymers by organotitanium-based catalysts. Macromol. Rapid Commun. 2009, 30, 39–44. [Google Scholar] [CrossRef]
- He, L. P.; Liu, J. L.; Li, Y. G.; Liu, S. R.; Li, Y. S. High-temperature living copolymerization of ethylene with norbornene by titanium complexes bearing bidentate [O, P] ligands. Macromolecules 2009, 42, 8566–8570. [Google Scholar] [CrossRef]
- Yang, X. H.; Wang, Z.; Sun, X. L.; Tang, Y. Synthesis, characterization, and catalytic behaviours of β-carbonylenamine-derived [O−NS]TiCl3 complexes in ethylene homo- and copolymerization. Dalton Trans. 2009, 8945–8954. [Google Scholar] [CrossRef] [PubMed]
- For example, Mitsui Chemicals Co. Production of Cyclic Olefin Copolymers with Tetracyclododecene (TCD) Using Vanadium Catalysts. Jpn. Pat. JP 2001-106730, 2001; Jpn. Pat. JP 2006-022266, 2006; Jpn. Pat. JP 2008-248171, 2008.
- Kaminsky, W.; Bark, A. Copolymerization of ethene and dimethanooctahydronaphthalene with aluminoxane containing catalysts. Polym. Int. 1992, 28, 251–253. [Google Scholar] [CrossRef]
- Kaminsky, W.; Engehausen, R.; Kopf, J. A tailor-made metallocene for the copolymerization of ethene with bulky cycloalkenes. Angew. Chem. Int. Ed. Engl. 1995, 34, 2273–2275. [Google Scholar] [CrossRef]
- Goodall, B. L.; McIntosh, L. H.; Rhodes, L. F. New catalysts for the polymerization of cyclic olefins. Macromol. Symp. 1995, 89, 421–432. [Google Scholar] [CrossRef]
- Donner, M.; Fernandes, M.; Kaminsky, W. Synthesis of copolymers with sterically hindered and polar monomers. Macromol. Symp. 2006, 236, 193–202. [Google Scholar] [CrossRef]
- Selected initial reports, see refs 111-114: Hasan, T.; Ikeda, T.; Shiono, T. Random Copolymerization of propene and norbornene with ansa-fluorenylamidodimethyltitanium-based catalysts. Macromolecules 2005, 38, 1071–1074. [CrossRef]
- Cai, Z.; Nakayama, Y.; Shiono, T. Living random copolymerization of propylene and norbornene with ansa-fluorenylamidodimethyltitanium complex: Synthesis of novel syndiotactic polypropylene-b-poly(propylene-ran-norbornene). Macromolecules 2006, 39, 2031–2033. [Google Scholar] [CrossRef]
- Shiono, T.; Sugimoto, M.; Hasan, T.; Cai, Z.; Ikeda, T. Random copolymerization of norbornene with higher 1-alkene with ansa-fluorenylamidodimethyltitanium catalyst. Macromolecules 2008, 41, 8292–8294. [Google Scholar] [CrossRef]
- Cai, Z.; Harada, R.; Nakayama, Y.; Shiono, T. Highly active living random copolymerization of norbornene and 1-alkene with ansa-fluorenylamidodimethyltitanium derivative: Substituent effects on fluorenyl ligand. Macromolecules 2010, 43, 4527–4531. [Google Scholar] [CrossRef]
- Pino, P.; Giannini, U.; Porri, L. Encyclopedia of Polymer Science and Engineering, 2nd ed.; Mark, H. F., Bikales, N. M., Overberger, C. C., Menges, G., Eds.; Wiley-Interscience: New York, 1987; Vol. 8, pp. 155–179. [Google Scholar]
- Kaminsky, W.; Bark, A.; Spiehl, R.; Möller-Linderhof, N.; Niedoba, S. Transition Metals and Organometallics as Catalysts for Olefin Polymerization; Kaminsky, W., Sinn, H., Eds.; Springer-Verlag: Berlin, 1988; pp. 291–301. [Google Scholar]
- Shaffer, T. D.; Canich, J. A. M.; Squire, K. R. metallocene-catalyzed copolymerization of ethylene and isobutylene to substantially alternating copolymers. Macromolecules 1998, 31, 5145–5147. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Sogo, Y.; Cai, Z.; Shiono, T. Copolymerization of ethylene with 1,1-disubstituted olefins catalyzed by ansa-(fluorenyl)(cyclododecylamido)dimethyltitanium complexes. J. Polym. Sci., Part A: Polym. Chem. 2013, 51, 1223–1229. [Google Scholar] [CrossRef]
- Li, H.; Li, L.; Marks, T. J.; Liable-Sands, L.; Rheingold, A. L. Catalyst/cocatalyst nuclearity effects in single-site olefin polymerization. significantly enhanced 1-octene and isobutene comonomer enchainment in ethylene polymerizations mediated by binuclear catalysts and cocatalysts. J. Am. Chem. Soc. 2003, 125, 10788–10789. [Google Scholar] [CrossRef]
- Li, H.; Li, L.; Schwartz, D. J.; Metz, M. V.; Marks, T. J.; Liable-Sands, L.; Rheingold, A. L. Coordination copolymerization of severely encumbered isoalkenes with ethylene: enhanced enchainment mediated by binuclear catalysts and cocatalysts. J. Am. Chem. Soc. 2005, 127, 14756–14768. [Google Scholar] [CrossRef]
- Nomura, K.; Kakinuki, K.; Fujiki, M.; Itagaki, K. Direct precise functional group introduction into polyolefins: Efficient incorporation of vinyltrialkylsilanes in ethylene copolymerizations by nonbridged half-titanocenes. Macromolecules 2008, 41, 8974–8976. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, J.; Wang, Y.; Pickens, D. B.; Motta, A.; Wang, Q. J.; Chung, Y.-W.; Lohr, T. L.; Marks, T. J. Highly branched polyethylene oligomers via group IV-catalysed polymerization in very nonpolar media. Nat. Catal. 2019, 2, 236–242. [Google Scholar] [CrossRef]
- Sian, L.; Dall’Anese, A.; Macchioni, A.; Tensi, L.; Busico, V.; Cipullo, R.; Goryunov, G. P.; Uborsky, D.; Voskoboynikov, A. Z.; Ehm, C.; Rocchigiani, L.; Zuccaccia, C. Role of solvent coordination on the structure and dynamics of ansa-zirconocenium ion pairs in aromatic hydrocarbons. Organometallics 2022, 41, 547–560. [Google Scholar] [CrossRef]
- Baumann, R.; Davis, W. M.; Schrock, R. R. Synthesis of titanium and zirconium complexes that contain the tridentate diamido ligand, [((t-Bu-d6)N-o-C6H4)2O]2- ([NON]2-) and the living polymerization of 1-hexene by activated [NON]ZrMe2. J. Am. Chem. Soc. 1997, 119, 3830–3831. [Google Scholar] [CrossRef]
- Gandini, A. Polymers from renewable resources: A challenge for the future of macromolecular materials. Macromolecules 2008, 41, 9491–9504. [Google Scholar] [CrossRef]
- Coates, G. W.; Hillmyer, M. A. A virtual issue of Macromolecules: Polymers from renewable resources. Macromolecules 2009, 42, 7987–7989. [Google Scholar] [CrossRef]
- Yao, K.; Tang, C. Controlled polymerization of next-generation renewable monomers and beyond. Macromolecules 2013, 46, 1689–1712. [Google Scholar] [CrossRef]
- Satoh, K. Controlled/living polymerization of renewable vinyl monomers into bio-based polymers. Polym. J. 2015, 47, 527–536. [Google Scholar] [CrossRef]
- Wang, Z.; Yuan, L.; Tang, C. Sustainable elastomers from renewable biomass. Acc. Chem. Res. 2017, 50, 1762–1773. [Google Scholar] [CrossRef]
- Polymers from Plant Oils, 2nd ed.; Gandini, A., Lacerda, T. M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ; Scrivener Publishing LLC: Beverly, MA, 2019. [Google Scholar]
- Fagnani, D. E.; Tami, J. L.; Copley, G.; Clemons, M. N.; Getzler, Y. D. Y. L.; McNeil, A. J. 100th Anniversary of macromolecular science viewpoint: Redefining sustainable polymers. ACS Macro Lett. 2021, 10, 41–53. [Google Scholar] [CrossRef]
- Haque, F. M.; Ishibashi, J. S. A. A.; Lidston, C. A. L. L.; Shao, H.; Bates, F. S.; Chang, A. B.; Coates, G. W.; Cramer, C. J.; Dauenhauer, P. J.; Dichtel, W. R.; Ellison, C. J.; Gormong, E. A.; Hamachi, L. S.; Hoye, T. R.; Jin, M; Kalow, J. A. Kim, H. J.; Kumar, G.; LaSalle, C. J.; Liffland, S.; Lipinski, B. M.; Pang, Y.; Parveen, R.; Peng, X.; Popowski, V.; Prebihalo, E. A.; Reddi, Y.; Reineke, T. M.; Sheppard, D. T.; Swartz, J. L.; Tolman, W. B.; Vlaisavljevich, B.; Wissinger, J.; Xu, S.; Hillmyer, M. A. Defining the macromolecules of tomorrow through synergistic sustainable polymer research. Chem. Rev. 2022, 122, 6322–6373. [CrossRef]
- Wilbon, P. A.; Chu, F.; Tang, C. Progress in renewable polymers from natural terpenes, terpenoids, and rosin. Macromol. Rapid Comm. 2013, 34, 8–37. [Google Scholar] [CrossRef]
- Thomsett, M. R.; Storr, T. E.; Monaphan, O. R.; Stockman, R. A.; Howdle, S. M. Progress in the synthesis of sustainable polymers from terpenes and terpenoids. Green Materials 2016, 4, 115–134. [Google Scholar] [CrossRef]
- Kawamura, K.; Nomura, K. Ethylene copolymerization with limonene and β-pinene: new bio-based polyolefins prepared by coordination polymerization. Macromolecules 2021, 54, 4693–4703. [Google Scholar] [CrossRef]
- Ren, X.; Guo, F.; Fu, H.; Song, Y.; Li, Y.; Hou, Z. Scandium-catalyzed copolymerization of myrcene with ethylene and propylene: Convenient syntheses of versatile functionalized polyolefins. Polym. Chem. 2018, 9, 1223–1233. [Google Scholar] [CrossRef]
- For example, refs. 137-143: Georges, S.; Touré, A. O.; Visseaux, M.; Zinck, P. Coordinative chain transfer copolymerization and terpolymerization of conjugated dienes. Macromolecules 2014, 47, 4538–4547. [CrossRef]
- Liu, B.; Li, L.; Sun, G.; Liu, D.; Li, S.; Cui, D. Isoselective 3,4-(co)polymerization of bio-renewable myrcene using NSN-ligated rare-earth metal precursor: an approach to a new elastomer. Chem. Commun. 2015, 51, 1039–1041. [Google Scholar] [CrossRef] [PubMed]
- Naddeo, M.; Buonerba, A.; Luciano, E.; Grassi, A.; Proto, A.; Capacchione, C. Stereoselective polymerization of biosourced terpenes β-myrcene and β-ocimene and their copolymerization with styrene promoted by titanium catalysts. Polymer 2017, 131, 151–159. [Google Scholar] [CrossRef]
- Laur, E.; Welle, A.; Vantomme, A.; Brusson, J.-M.; Carpentier, J.-F.; Kirillov, E. Stereoselective copolymerization of styrene with terpenes catalyzed by an ansa-lanthanidocene catalyst: Access to new syndiotactic polystyrene-based materials. Catalysts 2017, 7, 361–372. [Google Scholar] [CrossRef]
- Li, W.; Zhao, J.; Zhang, X.; Gong, D. Capability of PN3-type cobalt complexes toward selective (co-)polymerization of myrcene, butadiene, and isoprene: Access to biosourced polymers. Ind. Eng. Chem. Res. 2019, 58, 2792–2800. [Google Scholar] [CrossRef]
- González-Zapata, J. L.; Enríquez-Medrano, F. J.; López González, H. R.; Revilla-Vázquez, J.; Carrizales, R. M.; Georgouvelas, D.; Valencia, L.; Díaz de León Gómez, R. E. Introducing random bio-terpene segments to high cis-polybutadiene: making elastomeric materials more sustainable. RSC Adv. 2020, 10, 44096–44102. [Google Scholar] [CrossRef]
- Lamparelli, D. H.; Paradiso, V.; Monica, F. D.; Proto, A.; Guerra, S.; Giannini, L.; Capacchione, C. Toward more sustainable elastomers: Stereoselective copolymerization of linear terpenes with butadiene. Macromolecules 2020, 53, 1665–1673. [Google Scholar] [CrossRef]
- Talsi, E.; Bryliakov, K. Application of EPR and NMR Spectroscopy in Homogeneous Catalysis; CRC Press, Taylor & Francis: Boca Raton, FL, USA, 2017. [Google Scholar]
- Goswami, M.; Chirila, A.; Rebreyend, C.; de Bruin, B. EPR spectroscopy as a tool in homogeneous catalysis research. Top Catal. 2015, 58, 719–750. [Google Scholar] [CrossRef]
- Boča, R. Zero-field splitting in metal complexes. Coord. Chem. Rev. 2004, 248, 757–815. [Google Scholar] [CrossRef]
- Krzystek, J.; Ozarowski, A.; Telser, J.; Crans, D. C. High-frequency and -field electron paramagnetic resonance of vanadium(IV, III, and II) complexes. Coord. Chem. Rev. 2015, 301–302, 123–133. [Google Scholar] [CrossRef]
- Selected initial reports, see refs 148-159: Nomura, K.; Mitsudome, T.; Yamazoe, S. Direct observation of catalytically active species in reaction solution by x-ray absorption spectroscopy (XAS). Jpn. J. Appl. Phys. 2019, 58, 100502. [CrossRef]
- Nomura, K. Solution x-ray absorption spectroscopy (XAS) for analysis of catalytically active species in reactions with ethylene by homogeneous (imido)vanadium(V) complexes–Al cocatalyst systems. Catalysts 2019, 9, 1016. [Google Scholar] [CrossRef]
- Bartlett, S. A.; Moulin, J.; Tromp, M.; Reid, G.; Dent, A. J.; Cibin, G.; McGuinness, D. S.; Evans, J. Activation of [CrCl3{R-SN(H)S-R}] catalysts for selective trimerization of ethene: A freeze-quench Cr K-edge XAFS study. ACS Catal. 2014, 4, 4201–4204. [Google Scholar] [CrossRef]
- Takaya, H.; Nakajima, S.; Nakagawa, N.; Isozaki, K.; Iwamoto, T.; Imayoshi, R.; Gower, N. J.; Adak, L.; Hatakeyama, T.; Honma, T.; Takagaki, M.; Sunada, Y.; Nagashima, H.; Hashizume, D.; Takahashi, O.; Nakamura, M. Bull. Chem. Soc. Jpn. 2015, 88, 410–418. [CrossRef]
- Nomura, K.; Mitsudome, T.; Igarashi, A.; Nagai, G.; Tsutsumi, K.; Ina, T.; Omiya, T.; Takaya, H.; Yamazoe, S. Synthesis of (adamantylimido)vanadium(V) dimethyl complex containing (2-anilidomethyl)pyridine ligand and selected reactions: Exploring the oxidation state of the catalytically active species in ethylene dimerization. Organometallics 2017, 36, 530–542. [Google Scholar] [CrossRef]
- Nagai, G.; Mitsudome, T.; Tsutsumi, K.; Sueki, S.; Ina, T.; Tamm, M.; Nomura, K. Effect of Al cocatalyst in ethylene and ethylene/norbornene (co)polymerization by (imido)vanadium dichloride complexes containing anionic N-heterocyclic carbenes having weakly coordinating borate moiety. J. Jpn. Petrol. Inst. 2017, 60, 256–262. [Google Scholar] [CrossRef]
- Nomura, K.; Oshima, M.; Mitsudome, T.; Harakawa, H.; Hao, P.; Tsutsumi, K.; Nagai, G.; Ina, T.; Takaya, H.; Sun, W. -H.; Yamazoe, S. Synthesis and structural analysis of (imido)vanadium dichloride complexes containing 2-(2’-benz-imidazolyl)pyridine ligands: effect of al cocatalyst for efficient ethylene (co)polymerization. ACS Omega 2017, 2, 8660−8673. [Google Scholar] [CrossRef]
- Nomura, K.; Tsutsumi, K.; Nagai, G.; Omiya, T.; Ina, T.; Yamazoe, S.; Mitsudome, T. S. Solution XAS analysis of various (imido)vanadium(V) dichloride complexes containing monodentate anionic ancillary donor ligands: Effect of aluminium cocatalyst in ethylene/norbornene (co)polymerization. J. Jpn. Petrol. Inst. 2018, 61, 282−287. [Google Scholar] [CrossRef]
- Kuboki, M.; Nomura, K. (Arylimido)niobium(V) complexes containing 2-pyridylmethylanilido ligand as catalyst precursors for ethylene dimerization that proceeds via cationic Nb(V) species. Organometallics 2019, 38, 1544−1559. [Google Scholar] [CrossRef]
- Hirano, M.; Sano, K.; Kanazawa, Y.; Komine, N.; Maeno, Z.; Mitsudome, T.; Takaya, H. Mechanistic insights on pd/cu-catalyzed dehydrogenative coupling of dimethyl phthalate. ACS Catal. 2018, 8, 5827−5841. [Google Scholar] [CrossRef]
- Nomura, K.; Nagai, G.; Izawa, I.; Mitsudome, T.; Tamm, M.; Yamazoe, S. XAS analysis of reactions of (arylimido)vanadium(v) dichloride complexes containing anionic NHC that contains a weakly coordinating B(C6F5)3 moiety (WCA-NHC) or phenoxide ligands with Al alkyls: A Potential ethylene polymerization catalyst with WCA-NHC ligands. ACS Omega 2019, 4, 18833−18845. [Google Scholar] [CrossRef]
- Agata, R.; Takaya, H.; Matsuda, H.; Nakatani, N.; Takeuchi, K.; Iwamoto, T.; Hatakeyama, T.; Nakamura, N. Iron-catalyzed cross coupling of aryl chlorides with alkyl grignard reagents: Synthetic scope and FeII/FeIV Mechanism supported by x-ray absorption spectroscopy and density functional theory calculations. Bull. Chem. Soc. Jpn. 2019, 92, 381−390. [Google Scholar] [CrossRef]
- For examples, refs. 160-164: Thomas, J. M.; Sankar, G. J. The role of XAFS in the in situ and ex situ elucidation of active sites in designed solid catalysts. Synch. Rad. 2001, 8, 55−60. [CrossRef]
- Dent, A. J. Development of time-resolved XAFS instrumentation for quick EXAFS and energy-dispersive EXAFS measurements on catalyst systems. Top. Catal. 2002, 18, 27−35. [Google Scholar] [CrossRef]
- Thomas, J. M.; Catlow, C. R. A.; Sankar, G. Determining the structure of active sites, transition states and intermediates in heterogeneously catalysed reactions. Chem. Commun. 2002, 24, 2921−2925. [Google Scholar] [CrossRef]
- Bare, S. R.; Ressler, T. Chapter 6 characterization of catalysts in reactive atmospheres by X-ray absorption spectroscopy. Adv. Catal. 2009, 52, 339−465. [Google Scholar] [CrossRef]
- XAFS Techniques for Catalysts, Nanomaterials, and Surfaces; Iwasawa, Y., Asakura, K., Tada, M., Eds.; Springer: Cham, Switzerland, 2017. [Google Scholar]
- Srivastava, U. C.; Nigam, H. L. X-ray absorption edge-structure of compounds of some transition elements. Coord. Chem. Rev. 1973, 9, 275–310. [Google Scholar] [CrossRef]
- Wong, J.; Lytle, F. W.; Messmer, R. P.; Maylotte, D. H. K-edge absorption spectra of selected vanadium compounds. Phys. Rev. B 1984, 30, 5596−5610. [Google Scholar] [CrossRef]
- Wu, Z.; Xian, D. C.; Natoli, C. R.; Marceli, A.; Paris, E.; Mottana, A. Symmetry dependence of x-ray absorption near-edge structure at the metal edge of transition metal compounds. Appl. Phys. Lett. 2001, 79, 1918−1920. [Google Scholar] [CrossRef]
- Rehr, J. J.; Ankudinov, A. L. Progress in the theory and interpretation of XANES. Coord. Chem. Rev. 2005, 249(1−2), 131−140. [Google Scholar] [CrossRef]
- Yamamoto, T. Assignment of pre-edge peaks in K-edge x-ray absorption spectra of 3d transition metal compounds: electric dipole or quadrupole? X-Ray Spectrom. 2008, 37, 572−584. [Google Scholar] [CrossRef]
- Glatzel, P.; Smolentsev, G.; Bunker, G. The electronic structure in 3d transition metal complexes: Can we measure oxidation states? J. Phys.: Conf. Ser. 2009, 190, 012046. [Google Scholar] [CrossRef]
- Yi, J.; Nakatani, N.; Nomura, K.; Hada, M. Time-dependent DFT study of the K-edge spectra of vanadium and titanium complexes: effects of chloride ligands on pre-edge features. Phys. Chem. Chem. Phys. 2020, 22, 674−682. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Kosugi, N.; Kuroda, H. Polarized xanes spectra of CuCl2 · 2H2O: Further evidence for shake-down phenomena. Chem. Phys. 1986, 103, 101−109. [Google Scholar] [CrossRef]
- Bair, R. A.; Goddard, W. A., III. Ab initio studies of the x-ray absorption edge in copper complexes: I. Atomic Cu2+ and Cu(II)Cl2. Phys. Rev. B 1980, 22, 2767−2776. [Google Scholar] [CrossRef]
- Hu, P.; Hu, P.; Vu, T. D.; Li, M.; Wang, S.; Ke, Y.; Zeng, X.; Mai, L.; Long, Y. Vanadium oxide: Phase diagrams, structures, synthesis, and applications. Chem.Rev 2023, 123, 4353−4415. [Google Scholar] [CrossRef]
- Asakura, H.; Shishido, T.; Yamazoe, S.; Teramura, K.; Tanaka, T. Structural analysis of group V, VI, and VII metal compounds by XAFS. J. Phys. Chem. C 2011, 115, 23653−23663. [Google Scholar] [CrossRef]
- Yamamoto, T. What is the origin of pre-edge peaks in K-edge XANES spectra of 3d transition metals: electric dipole or quadrupole? Adv. X-Ray. Chem. Anal. Japan 2007, 38, 45−65. [Google Scholar]
- Grassi, A.; Zambelli, A.; Laschi, F. Reductive decomposition of cationic half-titanocene(IV) complexes, precursors of the active species in syndiospecific styrene polymerization. Organometallics 1996, 15, 480−482. [Google Scholar] [CrossRef]
- Minieri, G.; Corradini, P.; Guerra, G.; Zambelli, A.; Cavallo, L. A theoretical study of syndiospecific styrene polymerization with Cp-based and Cp-free titanium catalysts: 2. Mechanism of chain-end stereocontrol. Macromolecules 2001, 34, 5379−5385. [Google Scholar] [CrossRef]
- Mahanthappa, M. K.; Waymouth, R. M. Titanium-mediated syndiospecific styrene polymerizations: Role of oxidation state. J. Am. Chem. Soc. 2001, 123, 12093−12094. [Google Scholar] [CrossRef]
- Tomotsu, N.; Shozaki, H.; Aida, M.; Takeuchi, M.; Yokota, K.; Aoyama, Y.; Ikeuchi, S.; Inoue, T. Future Technology for Polyolefin and Olefin Polymerization Catalysis; Terano, M., Shiono, T., Eds.; Technology and Education Publishers: Tokyo, 2002; pp. 49–54. [Google Scholar]
- Zhang, H.; Byun, D.-J.; Nomura, K. Tuning the active species from syndiospecific styrene polymerisation to ethylene/styrene copolymerisation by (aryloxo)(cyclopentadienyl)titanium complexes–MAO catalysts. Dalton Trans 2007, 1802–1806. [CrossRef]
- Nomura, K.; Komatsu, T.; Imanishi, Y. Polymerization of 1-hexene, 1-octene catalyzed by Cp′TiCl2(O-2,6-iPr2C6H3)–MAO system: Unexpected increase of the catalytic activity for ethylene/1-hexene copolymerization by (1,3-tBu2C5H3)TiCl2(O-2,6-iPr2C6H3)–MAO catalyst system. J. Mol. Catal. A: Chem. 2000, 152, 249‒252. [Google Scholar] [CrossRef]
- Yi, J.; Nakatani, N.; Tomotsu, N.; Nomura, K.; Hada, M. Theoretical study of reaction mechanism for half-titanocene-catalyzed styrene polymerization, ethylene polymerization, and styrene-ethylene copolymerization: Roles of the neutral Ti(III) and the cationic Ti(IV) species. Organometallics 2021, 40, 643‒653. [Google Scholar] [CrossRef]
- Jantawan, K.; Groth, L.; Frank, R.; Chatchaipaiboon, K.; Tamm, M.; Nomura, K. Half-titanocenes bearing unsymmetric imidazolin-2-iminato ligand that exhibit efficient cyclic olefin incorporation in ethylene copolymerization. ACS Polym. Au submitted revision.
Scheme 1.
Group 4 transition metal complex catalysts for olefin polymerization and syndiospecific styrene polymerization (SSP).
Scheme 1.
Group 4 transition metal complex catalysts for olefin polymerization and syndiospecific styrene polymerization (SSP).
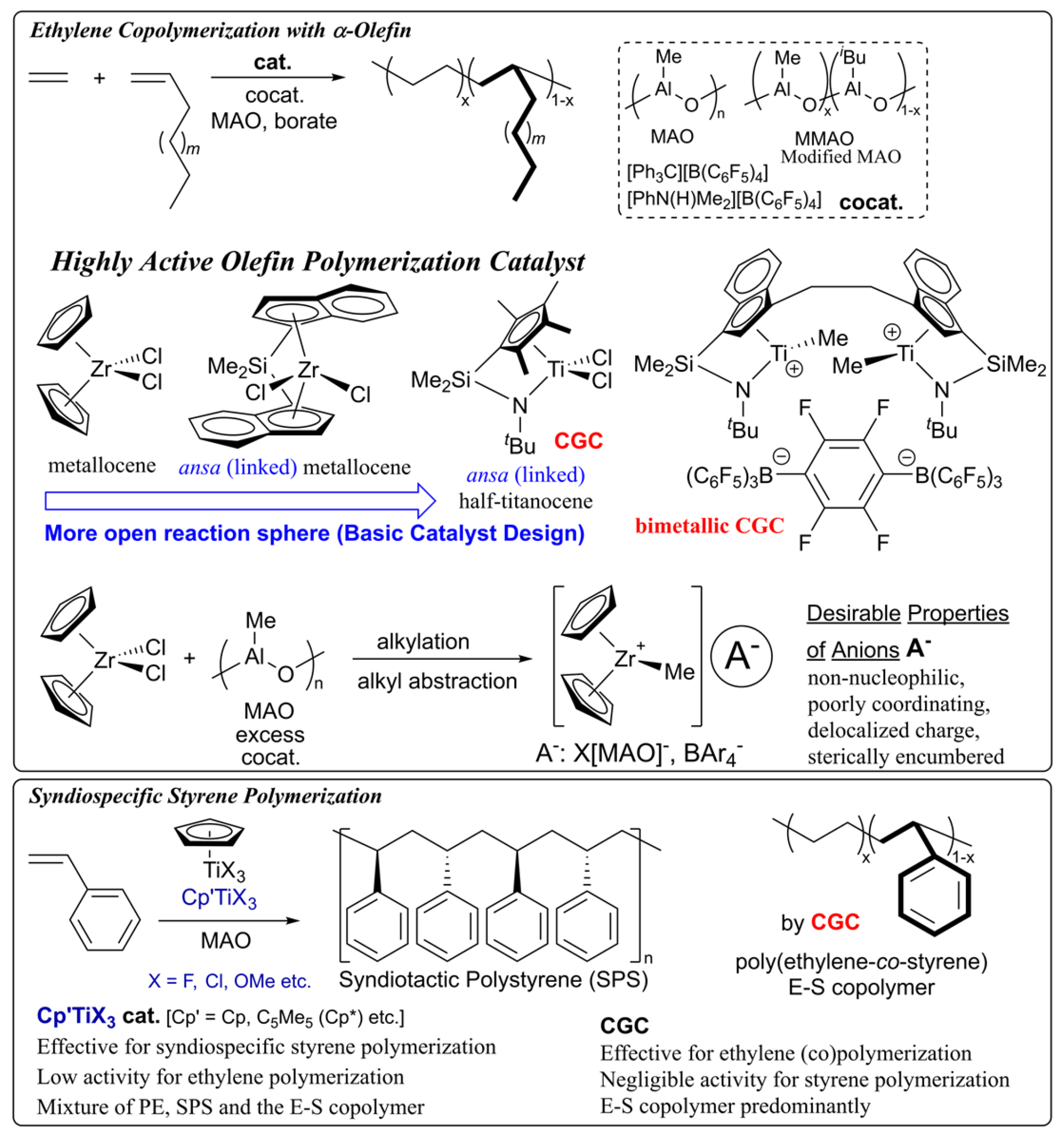
Scheme 2.
Nonbridged half-titanocenes: Basic catalyst design and selected examples of catalysts, copolymers.
Scheme 2.
Nonbridged half-titanocenes: Basic catalyst design and selected examples of catalysts, copolymers.
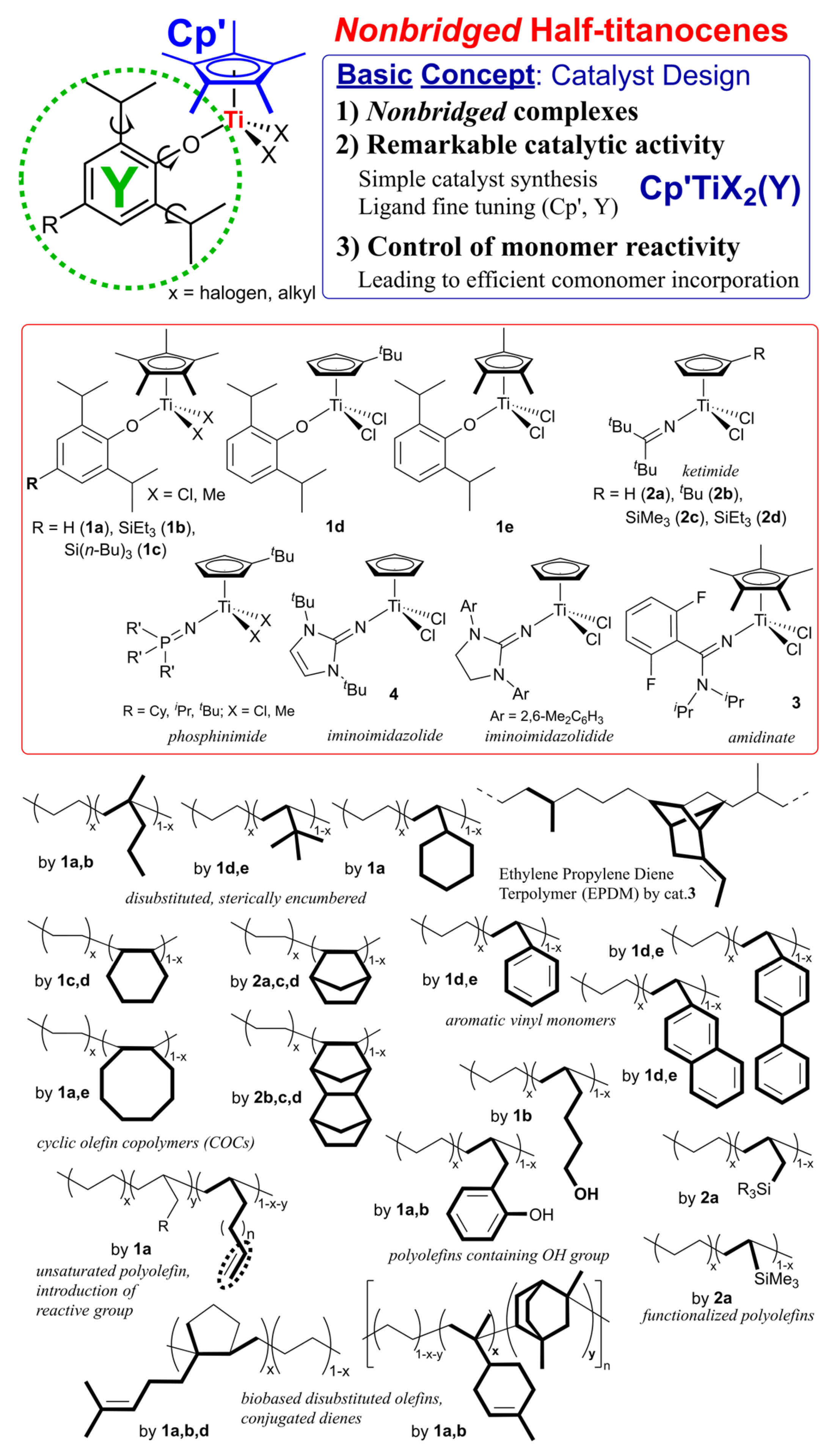
Scheme 3.
Ethylene copolymerization with norbornene (NBE).
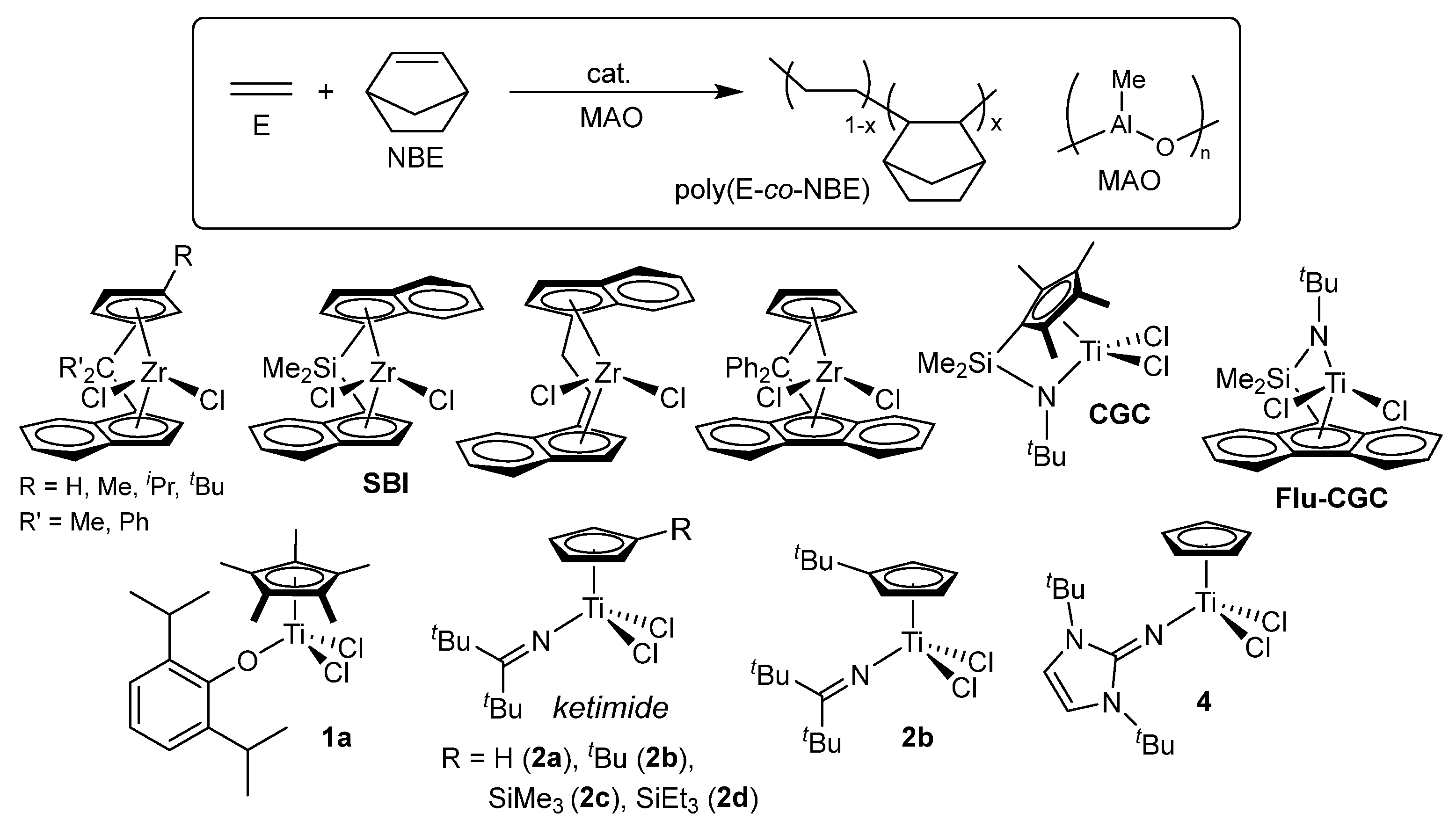
Scheme 4.
Catalysts reported for ethylene copolymerization with tetracyclododecene (TCD).
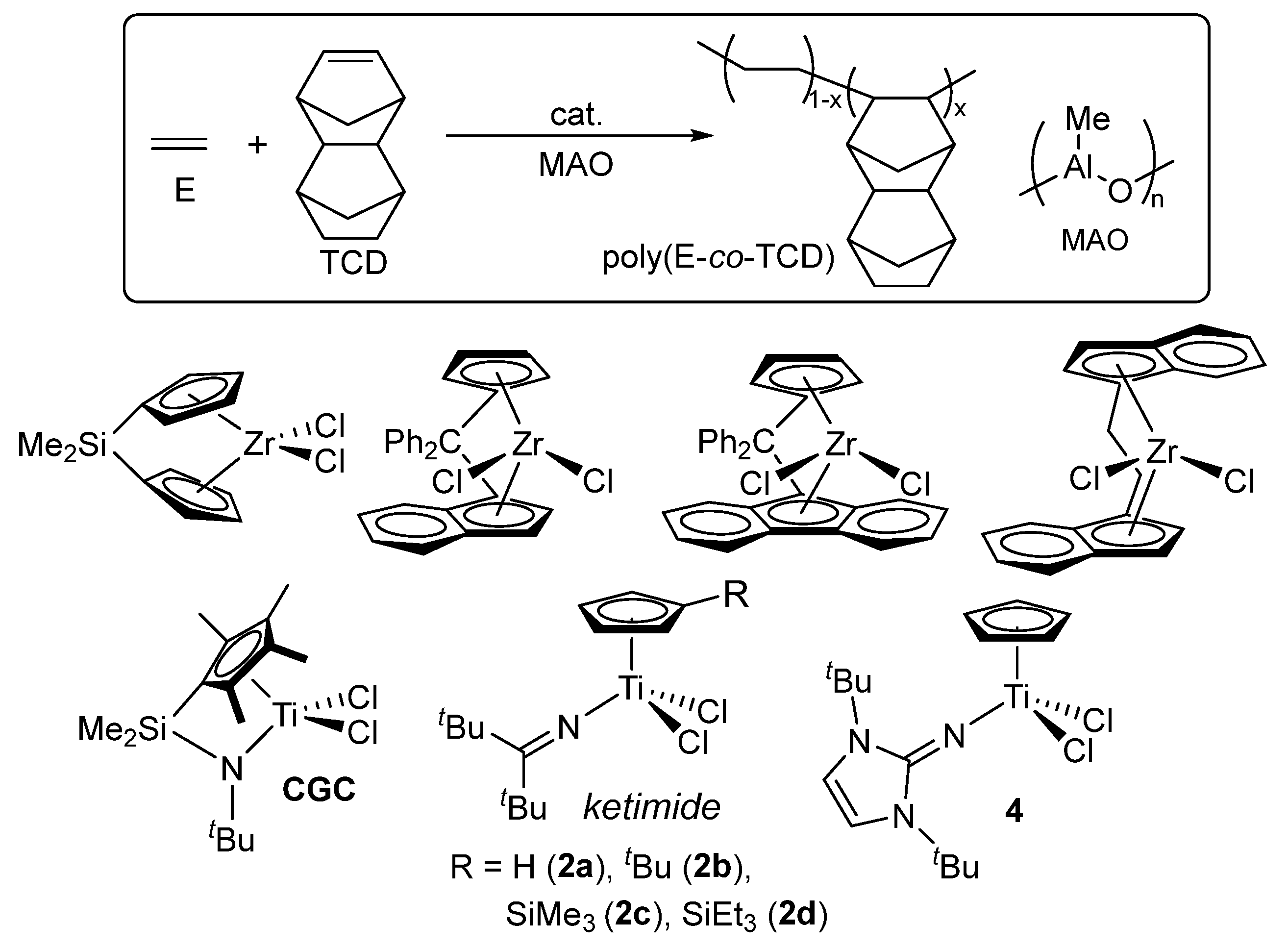
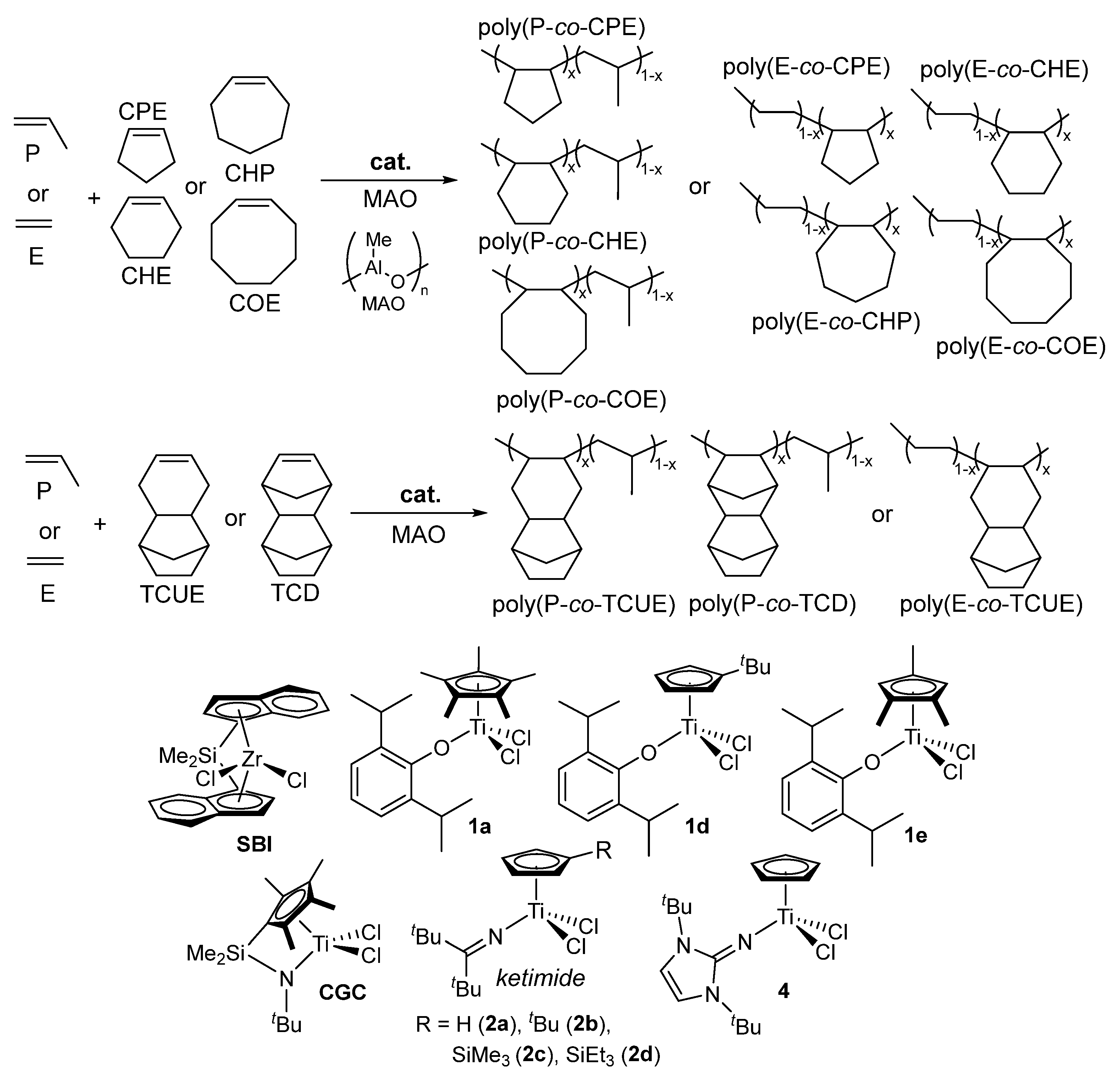
Scheme 6.
Copolymerization of norbornene (NBE), tetracyclododecene (TCD) with α-olefin (1-hexene, 1-octene, 1-dodecene, 1,7-octadiene) [65].
Scheme 6.
Copolymerization of norbornene (NBE), tetracyclododecene (TCD) with α-olefin (1-hexene, 1-octene, 1-dodecene, 1,7-octadiene) [65].
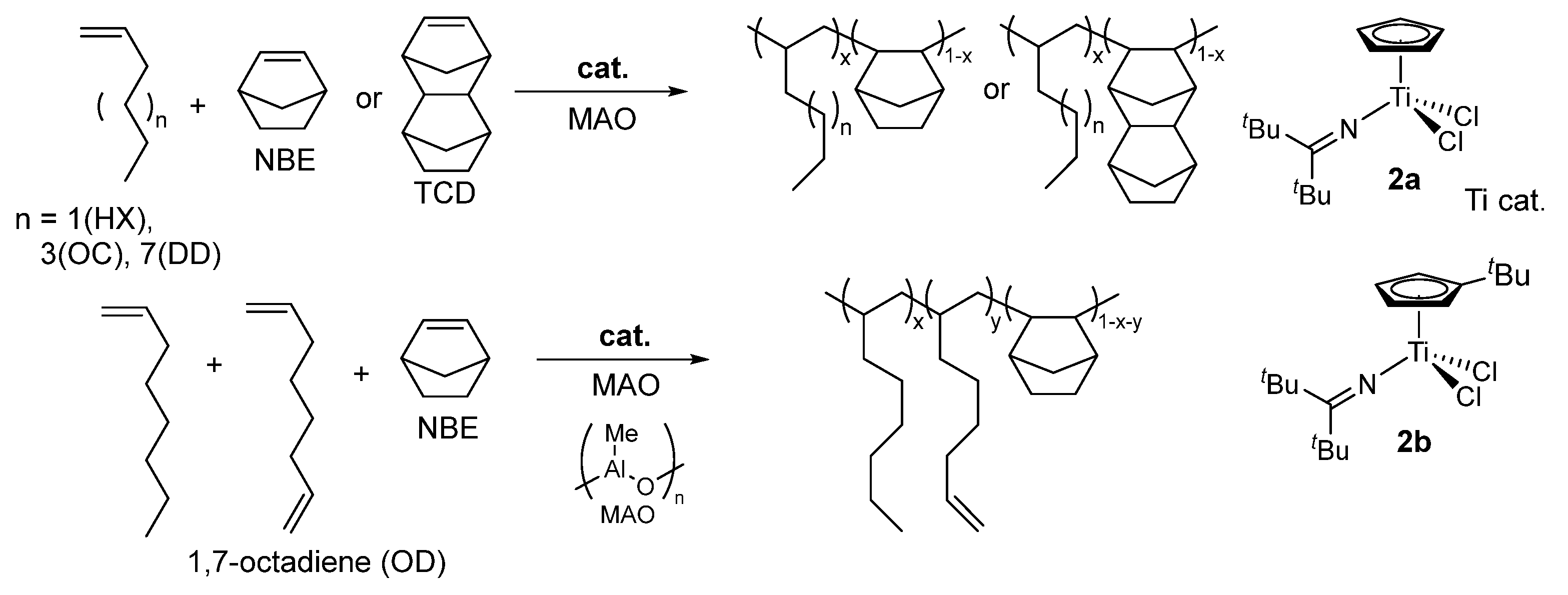
Scheme 7.
Ethylene copolymerization with α-olefins containing steric bulk [71,72,73,74,75,116,117,118,119,120,121].
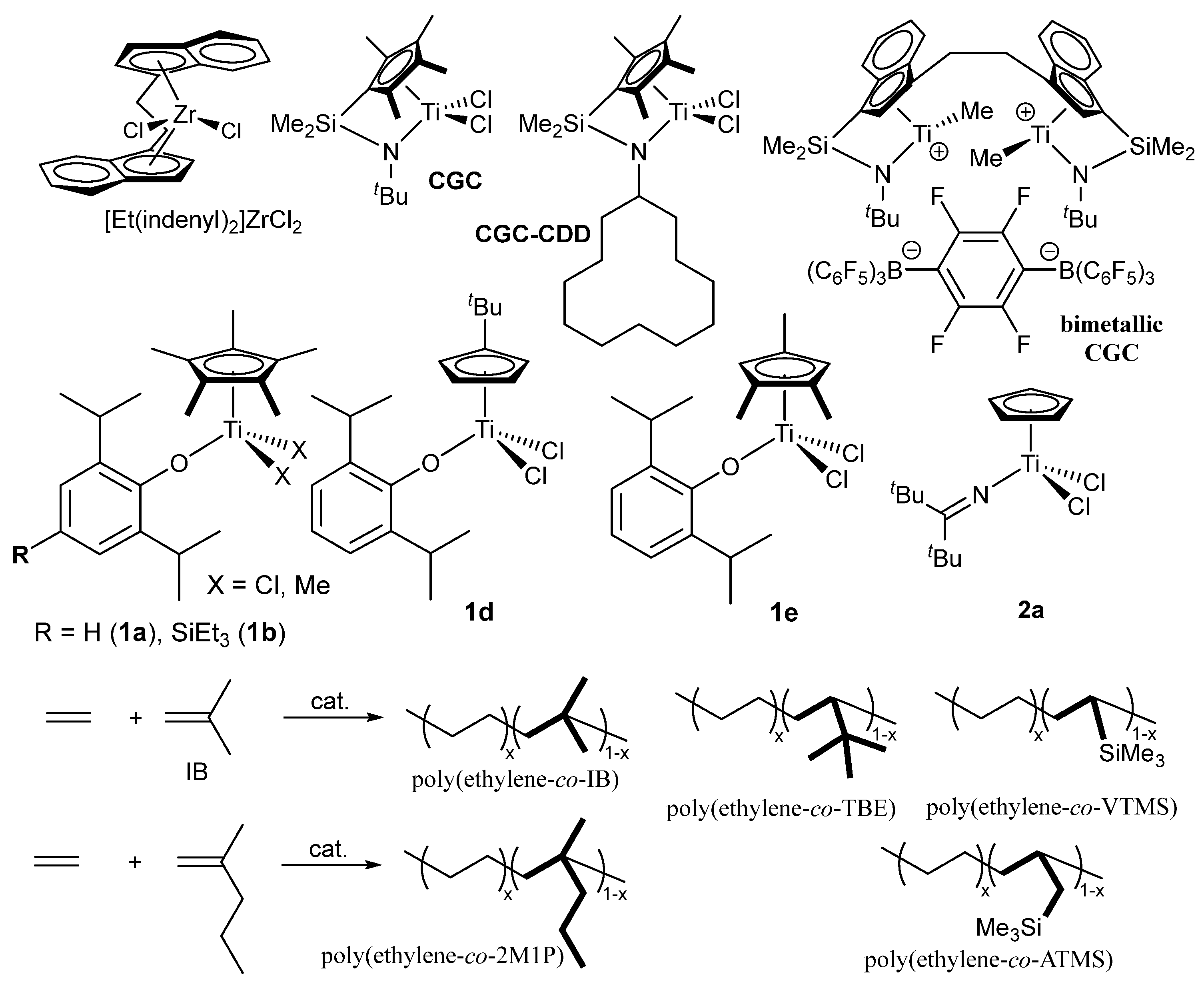
Scheme 9.
Synthesis of biobased ethylene copolymers with limonene, β-pinene, and myrcene [69].
Scheme 9.
Synthesis of biobased ethylene copolymers with limonene, β-pinene, and myrcene [69].
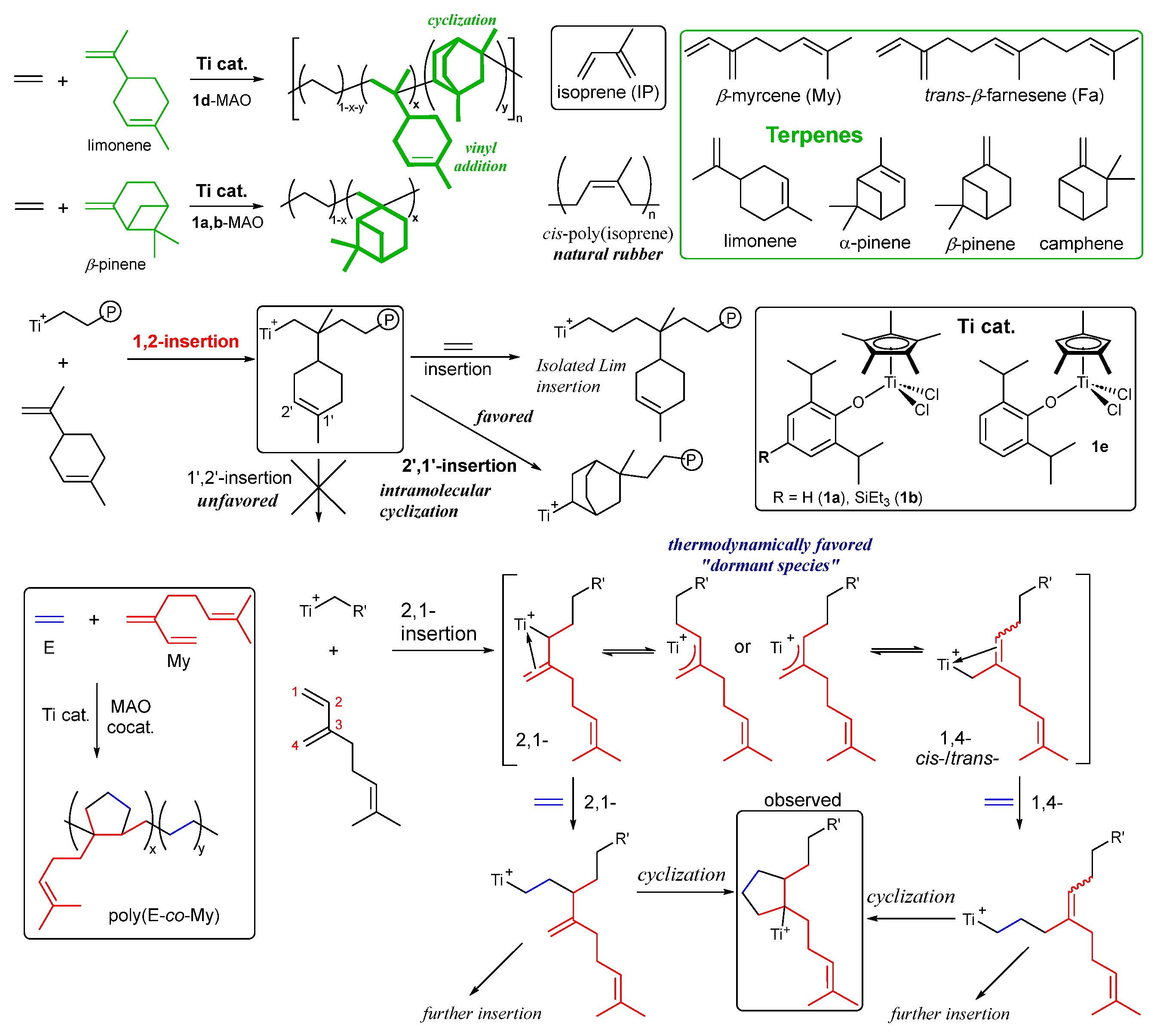
Figure 1.
(a) V K-edge spectra (5.46 keV, by using synchrotron radiation at SPring-8, BL01B1 beamline) for vanadium oxides with various oxidation state, (b) selected irreducible and relating functions and point groups in octahedral, square pyramidal and tetrahedral complexes [27,148].
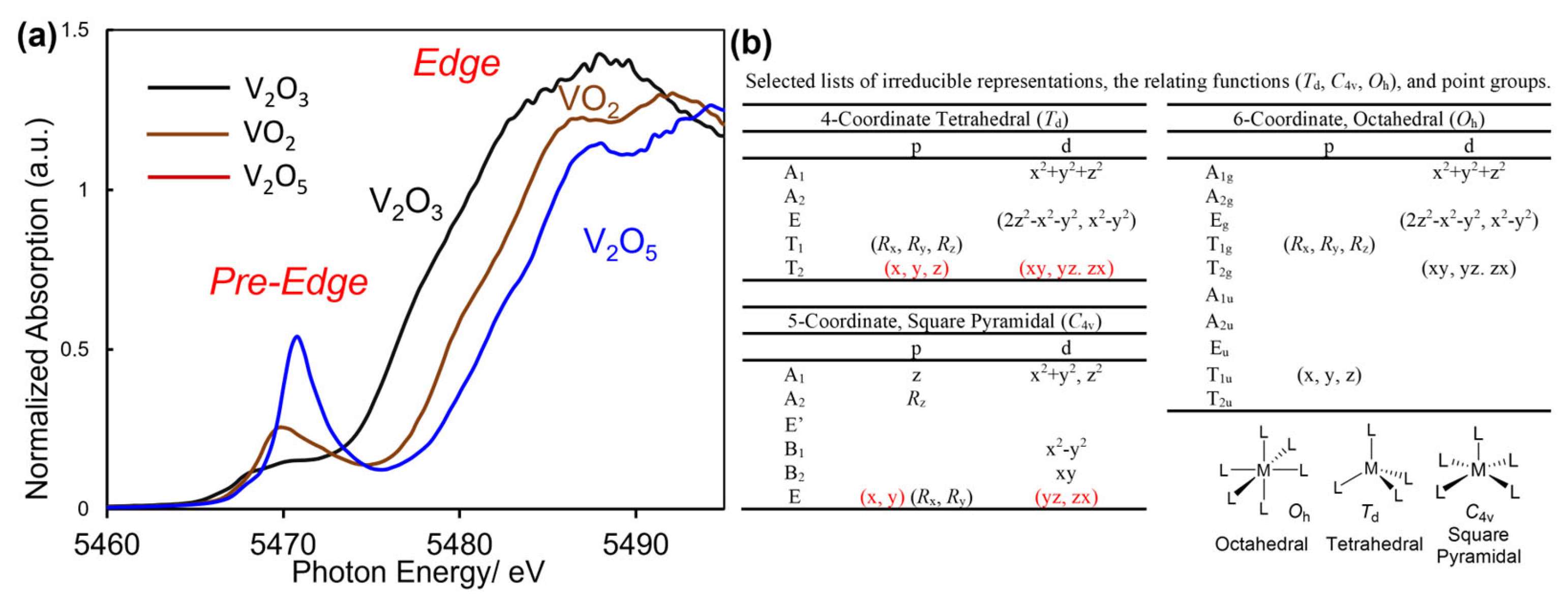
Figure 2.
The solution-phase Ti K-edge XANES spectra (in toluene at 25 ºC, Ti K-edge 4.97 keV, through the use of synchrotron radiation at SPring-8, BL01B1 beamline) for (a) CpTiCl3, Cp*TiX3 (X = OCH3, Cl), and (b) Cp’TiCl2(O-2,6-iPr2C6H3) [Cp’ = Cp, tBuC5H4 (1d), Cp* (1a)], CpTiCl2(N=CtBu2) (2a), and [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC) [27,76].
Figure 2.
The solution-phase Ti K-edge XANES spectra (in toluene at 25 ºC, Ti K-edge 4.97 keV, through the use of synchrotron radiation at SPring-8, BL01B1 beamline) for (a) CpTiCl3, Cp*TiX3 (X = OCH3, Cl), and (b) Cp’TiCl2(O-2,6-iPr2C6H3) [Cp’ = Cp, tBuC5H4 (1d), Cp* (1a)], CpTiCl2(N=CtBu2) (2a), and [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC) [27,76].
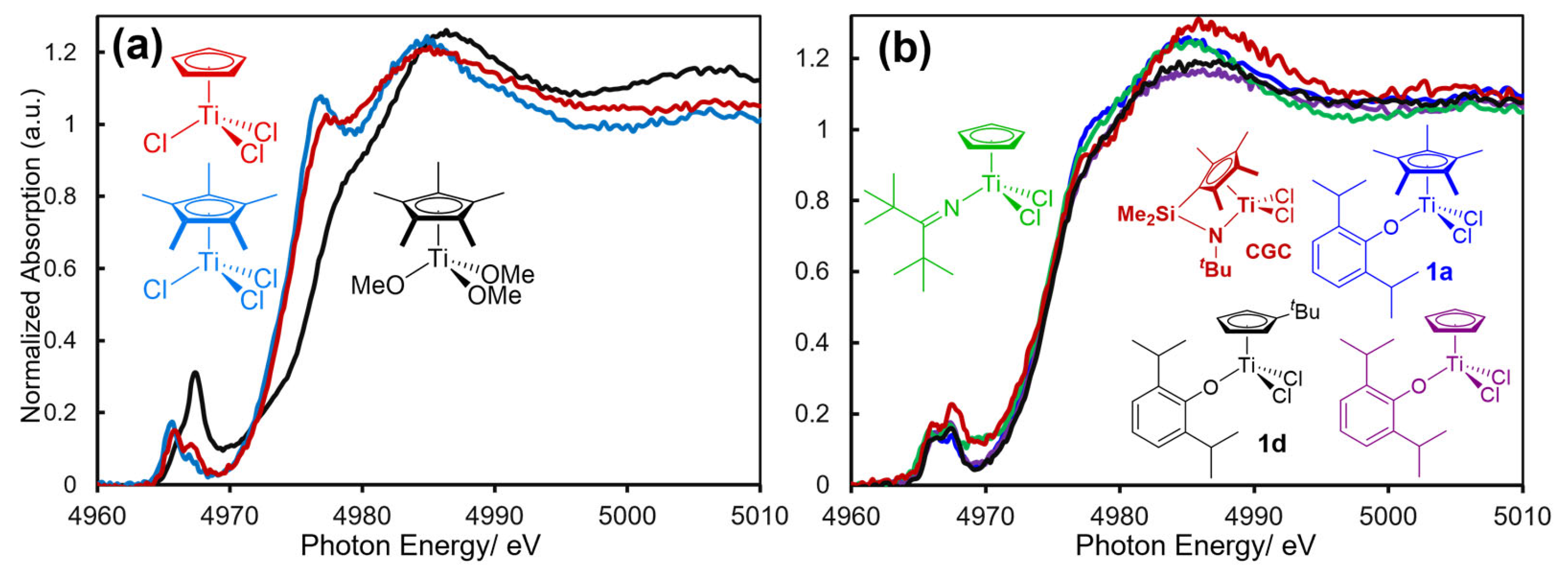
Scheme 10.
Proposed catalytically active species in olefin polymerization and syndiospecific styrene polymerisation (SSP).
Scheme 10.
Proposed catalytically active species in olefin polymerization and syndiospecific styrene polymerisation (SSP).
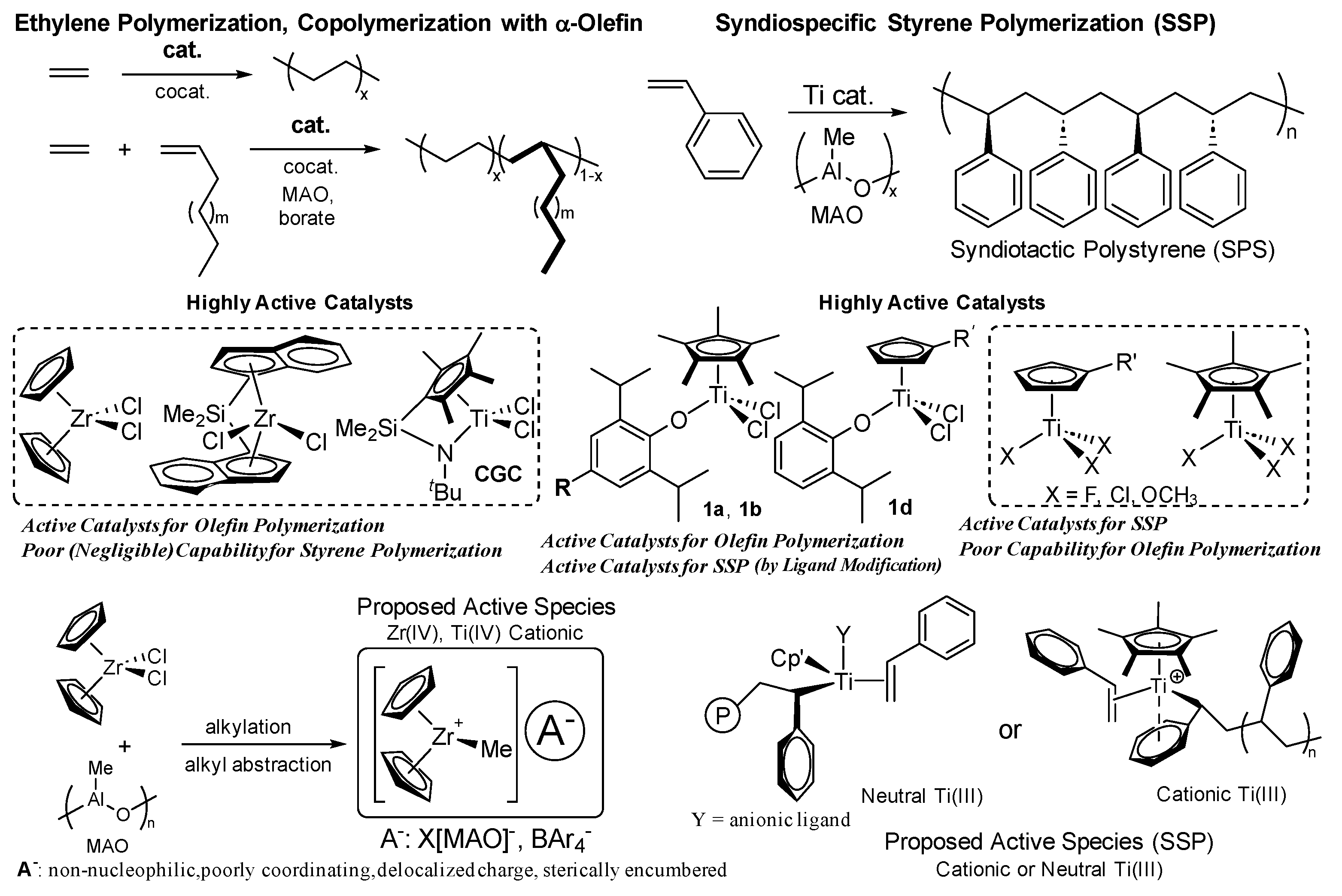
Figure 3.
Ti K-edge XANES spectra (in toluene at 25 ºC) for (a) Cp*TiX2(O-2,6-iPr2C6H3) [X = Cl (1a), Me (1a’)] or (b) (tBuC5H4)TiCl2(O-2,6-iPr2C6H3) (1d), and the spectra upon addition of MAO, and 1-hexene [76].
Figure 3.
Ti K-edge XANES spectra (in toluene at 25 ºC) for (a) Cp*TiX2(O-2,6-iPr2C6H3) [X = Cl (1a), Me (1a’)] or (b) (tBuC5H4)TiCl2(O-2,6-iPr2C6H3) (1d), and the spectra upon addition of MAO, and 1-hexene [76].
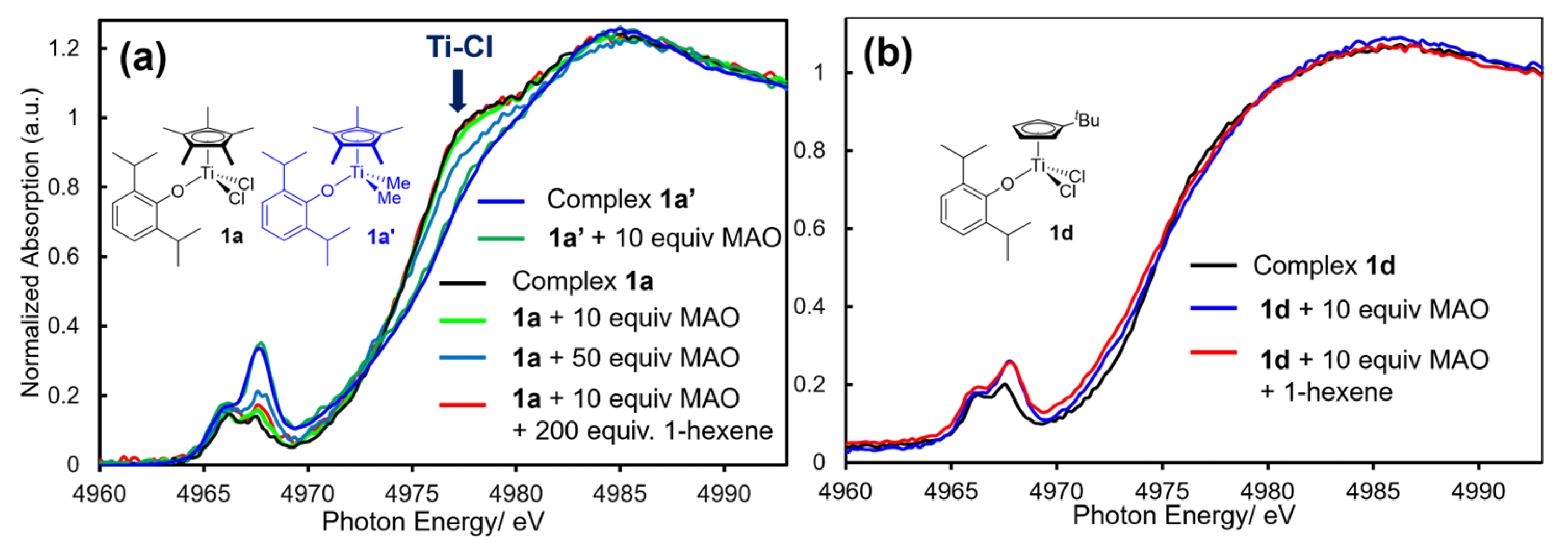
Figure 4.
Ti K-edge (a) EXAFS oscillations and (b) FT-EXAFS spectra (in toluene at 25 ºC) for Cp*TiCl2(OAr) (1a) by treating with MAO (10 or 50 equiv) [76].
Figure 4.
Ti K-edge (a) EXAFS oscillations and (b) FT-EXAFS spectra (in toluene at 25 ºC) for Cp*TiCl2(OAr) (1a) by treating with MAO (10 or 50 equiv) [76].
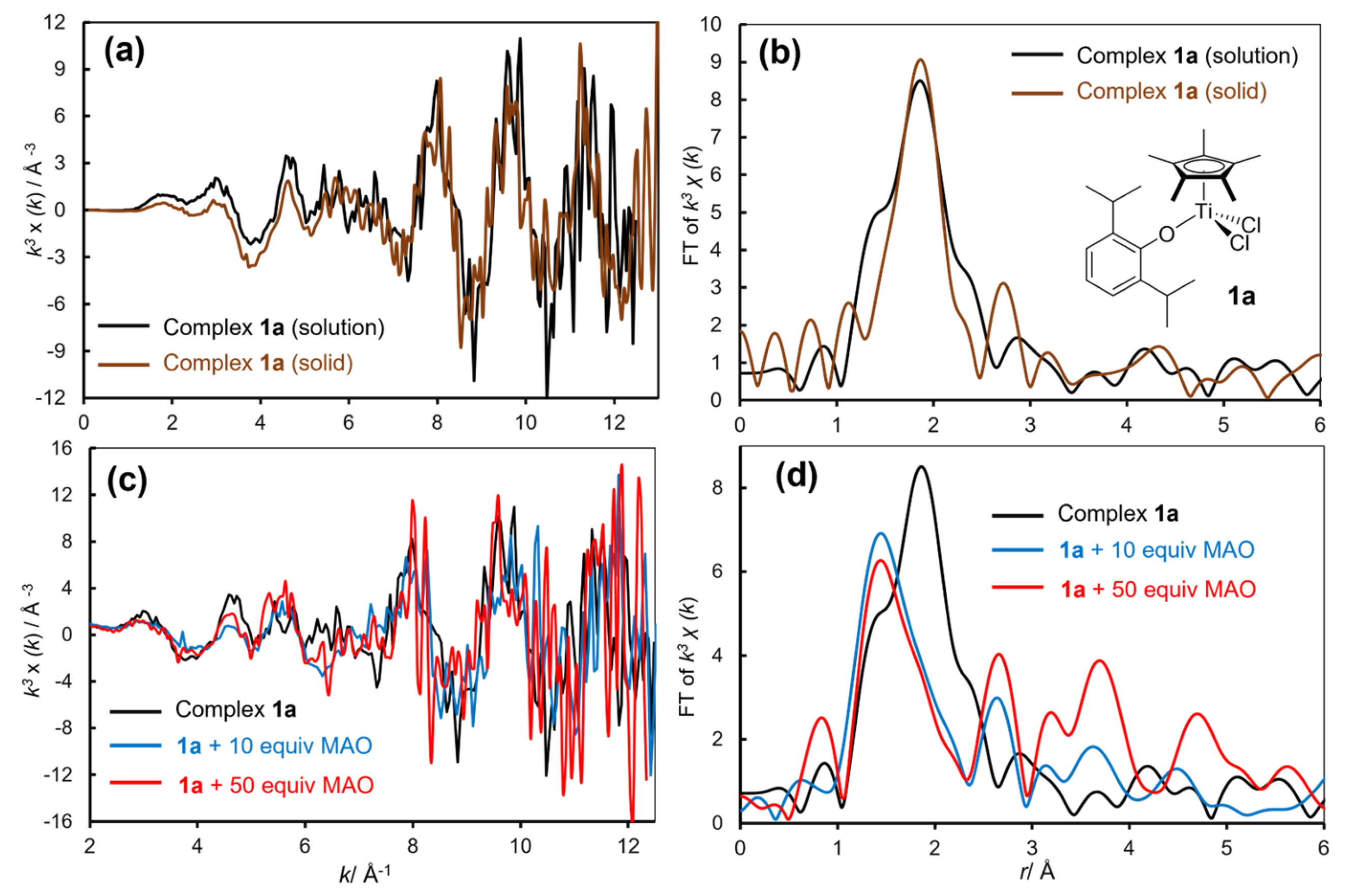
Figure 5.
Ti K-edge XANES spectra (in toluene at 25 ºC) for Cp’TiCl2(O-2,6-iPr2C6H3) [Cp’ = tBuC5H4 (1d), Cp (1e)], and the spectra for the catalyst solutions with addition of MAO, styrene [76].
Figure 5.
Ti K-edge XANES spectra (in toluene at 25 ºC) for Cp’TiCl2(O-2,6-iPr2C6H3) [Cp’ = tBuC5H4 (1d), Cp (1e)], and the spectra for the catalyst solutions with addition of MAO, styrene [76].
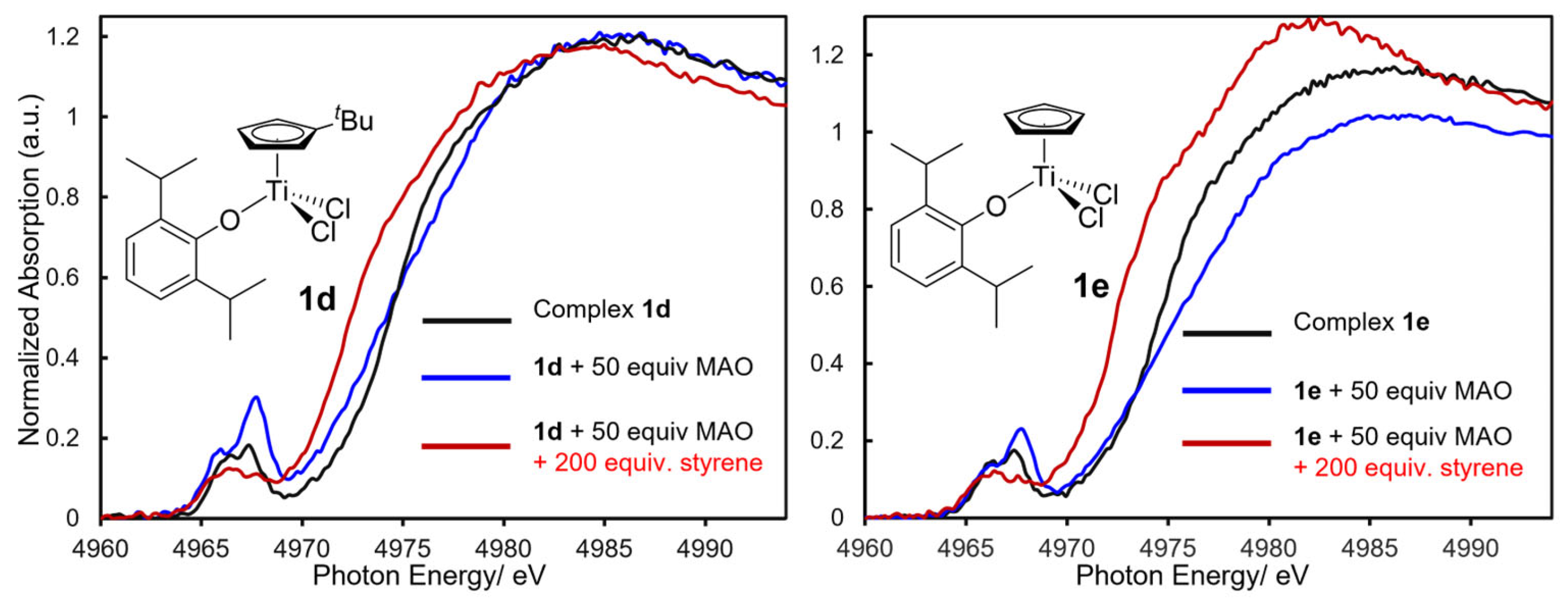
Figure 6.
Ti K-edge (in toluene at 25 ºC) (a) EXAFS oscillations and (b) FT-EXAFS spectra for (tBuC5H4)TiCl2(OAr) (1d) upon addition of MAO (50 equiv) and styrene (200 equiv) [76].
Figure 6.
Ti K-edge (in toluene at 25 ºC) (a) EXAFS oscillations and (b) FT-EXAFS spectra for (tBuC5H4)TiCl2(OAr) (1d) upon addition of MAO (50 equiv) and styrene (200 equiv) [76].
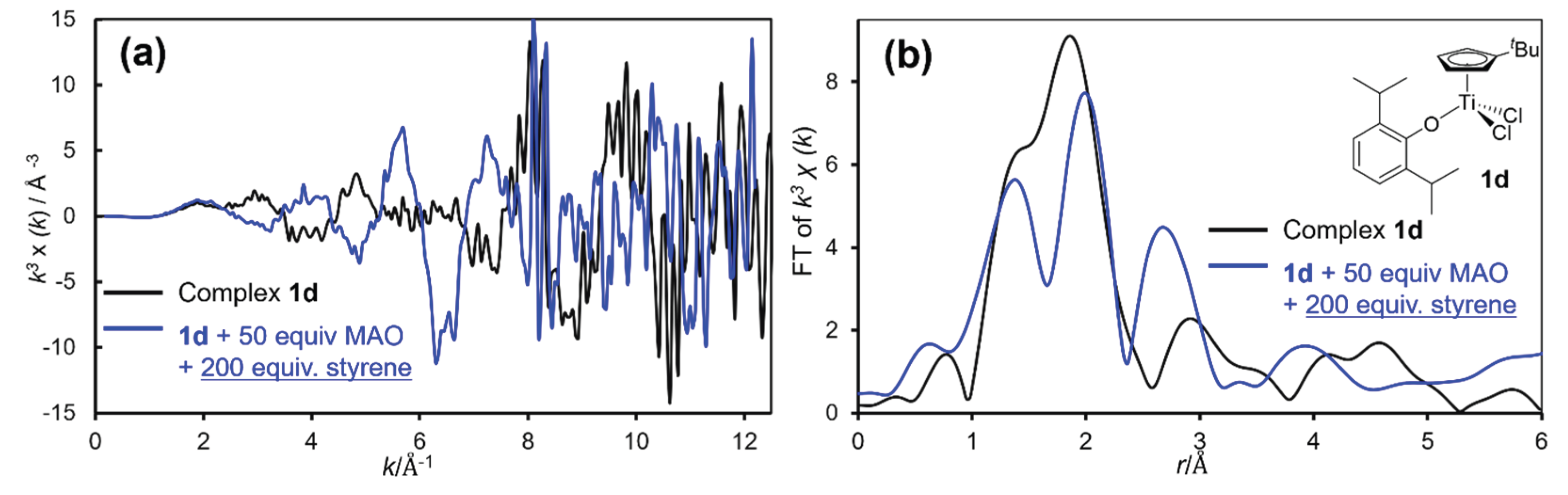
Scheme 11.
(a) Proposed active species for syndiospecific styrene polymerization (SSP) confirmed by two step copolymerization, XAS analysis [76,181]. (b) The energy profiles for styrene insertion to assumed three species containing phenoxide ligand [76].
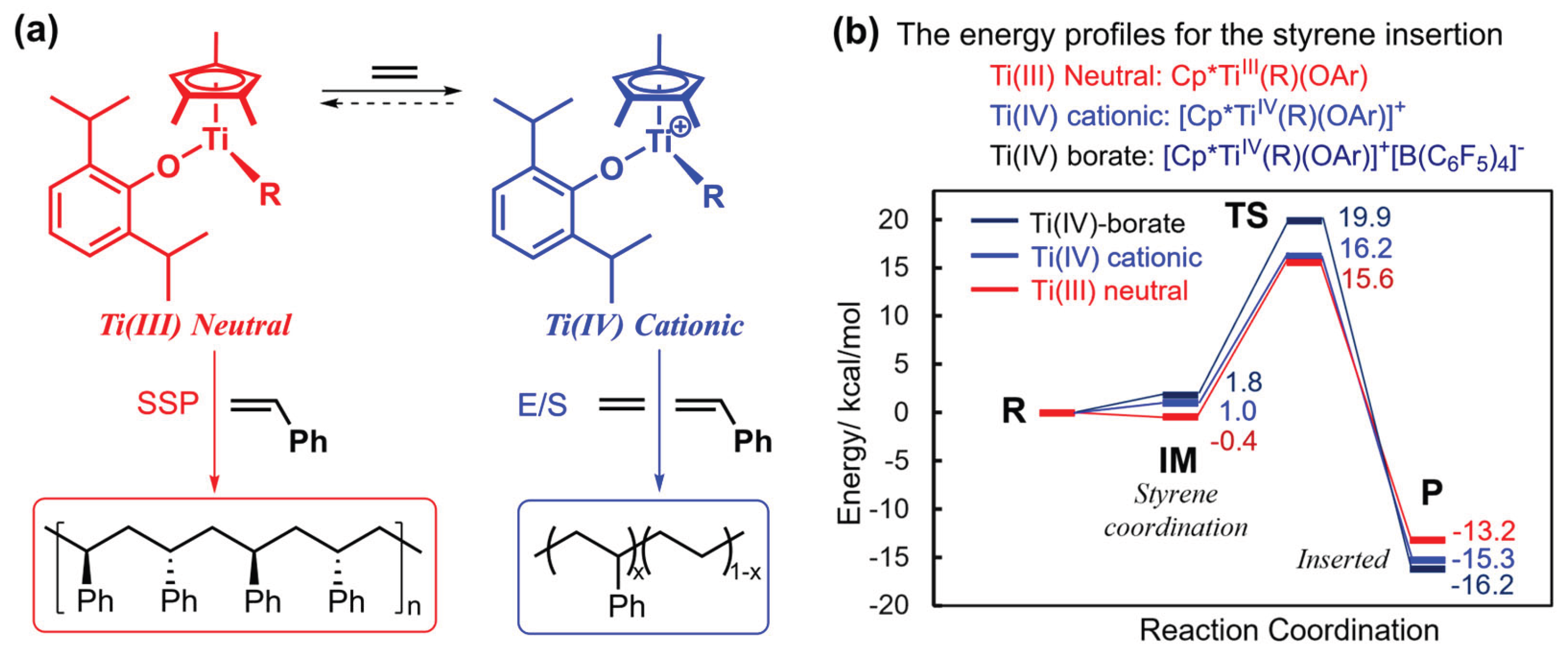
Table 1.
Ethylene (E) copolymerization with norbornene (NBE) by [Me2Si(indenyl)2]ZrCl2 (SBI), [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC), Cp’TiCl2(N=CtBu2) [Cp’ = Cp (2a), tBuC5H4 (2b), Me3SiC5H4 (2c), Et3SiC5H4 (2d)], and CpTiCl2[1,3-tBu2(CHN)2C=N] (4)–MAO catalysts (in toluene, ethylene 2 or 4 atm, 10 min) [61,62,63].a.
Table 1.
Ethylene (E) copolymerization with norbornene (NBE) by [Me2Si(indenyl)2]ZrCl2 (SBI), [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC), Cp’TiCl2(N=CtBu2) [Cp’ = Cp (2a), tBuC5H4 (2b), Me3SiC5H4 (2c), Et3SiC5H4 (2d)], and CpTiCl2[1,3-tBu2(CHN)2C=N] (4)–MAO catalysts (in toluene, ethylene 2 or 4 atm, 10 min) [61,62,63].a.
| cat. (μmol) | temp. / ºC |
E / atm |
NBEb / M |
activityc |
Mnd ×10-4 |
Mw/ Mnd |
Tge / ºC |
NBEf / mol% |
| SBI (0.10) | 25 | 4 | 0.2 | 28900 | 23.1 | 2.02 | 10.8 | |
| SBI (0.10) | 25 | 4 | 1.0 | 4860 | 22.9 | 2.37 | 29.5 | |
| CGC (0.50) | 25 | 4 | 0.2 | 2460 | 21.1 | 1.88 | 9.6 | |
| CGC (0.50) | 25 | 4 | 1.0 | 2000 | 12.8 | 2.15 | 26.5 | |
| 2a (0.02) | 80 | 4 | 1.0 | 133000 | 33.8 | 2.34 | 61.7 | |
| 2a (0.02) | 60 | 4 | 1.0 | 194000 | 47.5 | 2.20 | 51.2 | |
| 2a (0.02) | 40 | 4 | 1.0 | 48900 | 62.0 | 2.37 | 45.9 | |
| 2a (0.02) | 25 | 4 | 1.0 | 40200 | 71.9 | 2.92 | 40.7 | |
| 2a (0.02)g | 25 | 4 | 1.0 | 59700 | 61.3 | 2.18 | 41.0 | |
| 2a (0.01)h | 25 | 2 | 2.5 | 90000 | 32.3 | 2.09 | 58.8 | |
| 2a (0.01)h | 25 | 2 | 5.0 | 85800 | 34.0 | 2.00 | 65.8 | |
| 2a (0.01)h | 25 | 2 | 10.0 | 31500 | 44.4 | 2.01 | 73.5 | |
| 2b (0.01)i | 25 | 4 | 1.0 | 68400 | 62.4 | 2.78 | 38.2 | |
| 2b (0.10)i | 25 | 4 | 5.0 | 15000 | 17.5 | 2.05 | 52.7 | |
| 2c (0.02)i | 25 | 4 | 1.0 | 28300 | 79.5 | 1.82 | 99.6 | 36.2 |
| 2c (0.05)i | 50 | 2 | 6.0 | 32700 | 55.2 | 2.04 | 238 | 72.7j |
| 2d (0.01)i | 25 | 4 | 1.0 | 49800 | 82.9 | 2.06 | 94.1 | 37.5j |
| 2d (0.01)i | 50 | 4 | 1.0 | 91400 | 110 | 1.94 | 113 | 42.1j |
| 4 (0.20) | 25 | 4 | 1.0 | 6180 | 108 | 2.53 | 31.4 | |
| 4 (0.20) | 80 | 4 | 1.0 | 5780 | 80.0 | 2.35 | 36.9 |
aConditions: Toluene+NBE total 50 mL, d-MAO 0.5-3.0 mmol. bInitial NBE feed conc. (mmol/mL). cActivity in kg-polymer/mol-M·h (M = Ti, Zr). dGPC data in o-dichlorobenzene vs PS stds. eBy DSC thermograms. fNBE content (mol%) estimated by 13C NMR spectra. gTime 30 min. hToluene+NBE total 10 mL. iToluene+NBE total 30 mL. jEstimated on the basis of the plots of Tg and NBE content.
Table 2.
Ethylene copolymerization with tetracyclododecene (TCD) by Me2Si(C5Me4)(NtBu)]TiCl2 (CGC), Cp’TiCl2(N=CtBu2) [Cp’ = Cp (2a), tBuC5H4 (2b), Me3SiC5H4 (2c), Et3SiC5H4 (2d)]–MAO catalysts (in toluene, ethylene 4 or 6 atm, 10 min) [63,64].a.
| cat. (μmol) | E / atm |
TCDb / M |
temp. / ºC |
activityc |
Mnd ×10-5 |
Mw /Mnd |
Tge / ºC |
TCDf / mol% |
| CGC (0.05) | 6 | 1.0 | 25 | 13900 | 14.3 | 1.58 | 56 | |
| 2a (0.80) | 6 | 2.0 | 25 | 1650 | 1.92 | 1.41 | 150 | |
| 2b (0.02) | 6 | 1.0 | 25 | 43700 | 5.88 | 1.60 | 108 | 25.6 |
| 2b (0.02) | 6 | 2.0 | 25 | 23900 | 6.38 | 1.50 | 153 | 32.8 |
| 2b (0.02) | 6 | 2.0 | 40 | 27800 | 6.43 | 1.67 | 170 | 33.5g |
| 2b (0.02) | 6 | 2.0 | 60 | 33300 | 6.53 | 1.72 | 177 | 35.3 |
| 2b (0.02) | 6 | 3.0 | 25 | 16800 | 6.43 | 1.61 | 171 | 33.6g |
| 2b (0.02) | 6 | 4.0 | 60 | 22400 | 6.08 | 1.61 | 203 | 36.7 |
| 2c (0.02) | 6 | 1.0 | 25 | 33400 | 17.3 | 2.03 | 102 | 23.7g |
| 2c (0.05) | 6 | 4.0 | 25 | 7710 | 6.41 | 2.03 | 202 | 42.5g |
| 2c (0.10) | 4 | 4.0 | 50 | 2560 | 3.90 | 1.95 | 255 | 52.3g |
| 2d (0.02) | 6 | 1.0 | 25 | 55900 | 7.29 | 1.88 | 119 | 26.9g |
| 2d (0.04) | 6 | 4.0 | 25 | 15700 | 5.40 | 2.41 | 203 | 41.9 |
| 2d (0.10) | 4 | 2.0 | 50 | 9550 | 5.76 | 1.53 | 186 | 38.3 |
| 2d (0.20) | 4 | 3.0 | 50 | 7790 | 3.98 | 1.72 | 234 | 48.3g |
| 2d (0.10) | 4 | 4.0 | 50 | 3790 | 2.79 | 1.79 | 244 | 50.3g |
aPolymerization conditions: Toluene and TCD total 30 mL, d-MAO 3.0 mmol. bInitial TCD concentration in mmol/mL. cActivity = kg-polymer/mol-Ti·h. dGPC data in o-dichlorobenzene vs PS standards. eBy DSC thermograms. fTCD content (mol%) estimated by 13C NMR spectra. gEstimated based on the plots of Tg and TCD content.
Table 3.
Ethylene (E) copolymerization with 2-methyl-1-pentene (2M1P), tert-butylethylene (TBE), and with vinyltrimethylsilane (VTMS) by Cp’TiCl2(OAr) [OAr = O-2,6-iPr2C6H3, Cp’ = Cp* (1a), tBuC5H4 (1d)], CpTiCl2(N=CtBu2) (2a), [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC)–MAO catalysts (in toluene at 25 °C, 10 min).a.
Table 3.
Ethylene (E) copolymerization with 2-methyl-1-pentene (2M1P), tert-butylethylene (TBE), and with vinyltrimethylsilane (VTMS) by Cp’TiCl2(OAr) [OAr = O-2,6-iPr2C6H3, Cp’ = Cp* (1a), tBuC5H4 (1d)], CpTiCl2(N=CtBu2) (2a), [Me2Si(C5Me4)(NtBu)]TiCl2 (CGC)–MAO catalysts (in toluene at 25 °C, 10 min).a.
| cat. (μmol) | E/ atm | comonomer/ Mb | activityc | Mnd×10-4 | Mw/Mnd | cont.e/ mol% | |
| 1a (0.10) | 4 | --- | - | 12100 | 40.0 | 3.70 | |
| 1a (0.50) | 6 | 2M1P | 1.35 | 6980 | 13.0 | 1.70 | |
| 1a (0.50)f | 6 | 2M1P | 1.35 | 8460 | 12.0 | 2.10 | 3.3 |
| 1a (0.50)f | 6 | 2M1P | 2.70 | 5760 | 10.0 | 1.80 | 5.7 |
| 1a (0.50)f | 4 | 2M1P | 1.35 | 4240 | 6.50 | 2.00 | 5.0 |
| 1a (0.50)f | 4 | 2M1P | 2.70 | 2680 | 4.90 | 1.60 | 9.4 |
| 1d (2.0) | 6 | 2M1P | 1.35 | 573 | 5.80 | 2.00 | |
| 2a (0.20) | 6 | 2M1P | - | 19100 | 53.0 | 2.10 | |
| 2a (0.20) | 6 | 2M1P | 1.35 | 10100 | 43.0 | 2.00 | |
| 2a (0.20) | 6 | 2M1P | 2.70 | 6960 | 34.0 | 1.80 | 0.3 |
| CGC (1.0)g | 6 | 2M1P | 2.70 | 1840 | 12.0 | 2.40 | 0.3 |
| CGC (1.0)g | 4 | 2M1P | 1.35 | 1420 | 9.70 | 2.50 | |
| CGC (1.0)g | 4 | 2M1P | 2.70 | 1320 | 7.40 | 2.40 | 0.4 |
| 1a (0.20) | 6 | TBE | 1.29 | 14100 | 15.5 | 2.00 | trace |
| 1a (0.50) | 6 | VTMS | 1.15 | 1870 | 30.5 | 1.90 | 5.1 |
| 1d (1.0) | 6 | TBE | 2.58 | 3310 | 11.7 | 2.50 | 1.3 |
| 1d (1.0)h | 4 | TBE | 1.60 | 2020 | 8.17 | 2.40 | 3.3 |
| 1d (2.0)h | 2 | TBE | 1.60 | 918 | 7.27 | 2.10 | 4.4 |
| 1d (2.0)h | 2 | TBE | 3.90 | 756 | 4.04 | 2.00 | 6.8 |
| 1d (0.20) | 6 | VTMS | 1.15 | 92 | 1.41 | 2.80 | 13.6 |
| 2a (0.20) | 6 | TBE | 1.29 | 7830 | 71.1 | 1.90 | trace |
| 2a (0.20) | 6 | TBE | 2.58 | 6000 | 68.7 | 1.90 | trace |
| 2a (1.0) | 6 | VTMS | 1.15 | 3730 | 57.3 | 2.30 | 11.9 |
| 2a (1.0) | 4 | VTMS | 1.15 | 1560 | 42.2 | 2.30 | 18.7 |
| CGC (0.25) | 6 | TBE | 1.29 | 3020 | 32.0 | 2.00 | none |
| CGC (0.25) | 6 | VTMS | 1.15 | 2280 | 36.7 | 2.50 | 10.4 |
aConditions: Comonomer+toluene total 30 mL, d-MAO 3.0 mmol. bInitial comonomer feed conc. (mmol/mL). cActivity in kg-polymer/mol-Ti·h. dGPC data in o-dichlorobenzene vs PS stds. eComonomer content (mol%) estimated by 13C NMR spectra. fMAO 4.5 mmol. gPolymerization 6 min. hTBE+toluene total 10 mL.
Table 5.
Summary of analysis data (by curve fitting) for toluene solution of Cp*TiCl2(O-2,6-iPr2C6H3) (1a), and 1a treated with MAO (10 and 50 equiv) [76].
Table 5.
Summary of analysis data (by curve fitting) for toluene solution of Cp*TiCl2(O-2,6-iPr2C6H3) (1a), and 1a treated with MAO (10 and 50 equiv) [76].
| atoma | Cp*TiCl2(OAr) (1a) | 1a + 10 equiv MAOe | 1a + 50 equiv MAOe | ||||||
| C.N.b | r (Å)c | D.W.d | C.N.b | r (Å)c | D.W.d | C.N.b | r (Å)c | D.W.d | |
| O | 1.3(1) | 1.803(6) | 0.0013(10) | 2.3(2) | 1.81(1) | 0.0035 (12) | 3.4(6) | 1.87(1) | 0.0059(25) |
| C | 4.7(9) | 2.42(1) | 0.0030(19) | 5.5(9) | 2.13(2) | 0.0047(22) | 4.5(9) | 2.14(1) | 0.0040(30) |
| Cl | 2.4(3) | 2.269(7) | 0.0047(13) | 0.9(3) | 2.18(3) | 0.0062(49) | |||
aAtom: Neighbor atom. bC.N.: coordination number. cr: bond length. dD.W.: Debye-Waller factor. eFor analysis, fixed ΔE values were used. R factors = 11.4%, 14.8%, and 12.9%, respectively, for complex 1a, 1a treated with MAO (10 equiv, 50 equiv).
Table 6.
Summary of analysis data (by curve fitting) for toluene solution of (tBuC5H4)TiCl2(O-2,6-iPr2C6H3) (1d), and 1d with addition of MAO (50 equiv) and styrene (200 equiv) at 25 °C [76].
Table 6.
Summary of analysis data (by curve fitting) for toluene solution of (tBuC5H4)TiCl2(O-2,6-iPr2C6H3) (1d), and 1d with addition of MAO (50 equiv) and styrene (200 equiv) at 25 °C [76].
| atoma | (tBuC5H4)TiCl2(OAr)e (1d) | 1d + 50 equiv MAO + 200 equiv styrenee | ||||
| C.N.b | r (Å)c | D.W.d | C.N.b | r (Å)c | D.W.d | |
| O | 1.3(1) | 1.76(1) | 0.0010(5) | 0.7(4) | 1.80(2) | 0.0064(54) |
| Cl | 1.7(1) | 2.25(1) | 0.0012(4) | --- | --- | --- |
| C1 | 5.2(9) | 2.41(2) | 0.0055(45) | 5.2(3) | 2.40(1) | 0.0040(15) |
| C2 | --- | --- | --- | 1.3(6) | 1.95(3) | 0.0034(28) |
aAtom: neighbor atom. bC.N.: coordination number. cr: bond length. dD.W.: Debye-Waller factor. eFor analysis, fixed ΔE values were used. R factors = 10.7%, 7.5%, respectively, for complex 1d, 1d treated with MAO and styrene.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



