Submitted:
25 January 2026
Posted:
26 January 2026
You are already at the latest version
Abstract
Aging is accompanied by profound alterations in immune function that collectively drive increased susceptibility to infection, reduced vaccine efficacy, impaired tissue repair, and heightened risk of age-related diseases (ARDs). These alterations are characterized by the coexistence of immunosenescence and inflammaging. Rather than reflecting isolated cellular defects, immune aging emerges as a systems-level reprogramming of immune networks that disrupts the initiation, resolution, and regenerative phases of inflammatory responses. In particular, aging is associated with impaired resolution of inflammation, defective efferocytosis, reduced responsiveness to pro-resolving signals, and diminished regenerative capacity, leading to persistent inflammatory milieus and tissue damage. This review summarizes recent advances in the mechanisms underlying immune dysfunction in aging, with a focus on how chronic inflammation, failed resolution, and defective repair reinforce one another. We discuss how alterations in innate and adaptive immunity, immunometabolism, cellular senescence, and immune–tissue interactions drive inflammaging and contribute to major ARDs, including cancer, neurodegenerative, and cardiometabolic diseases. Finally, we highlight emerging therapeutic strategies aimed at restoring immune balance and resolution. By adopting a systems-level and network-based perspective, this review underscores immune aging as a modifiable driver of ARDs and identifies key knowledge gaps and future directions toward interventions that promote healthy aging and extended healthspan.
Keywords:
immunosenescence
; inflammaging
; age-related diseases
; pro-resolving lipid mediators
; impaired resolution of inflammation
; systems-level and network-based perspective
; therapy
1. Introduction
Aging is associated with profound and progressive changes in immune system composition and function that affect both innate and adaptive immunity. This process, commonly referred to as immunosenescence, encompasses reduced immune responsiveness, impaired host defense against infections, diminished vaccine efficacy, and increased cancer incidence. Concomitantly, aging is characterized by a chronic, low-grade inflammatory state termed inflammaging, which persists in the absence of overt infection and contributes to functional decline across multiple tissues [1,2,3,4,5,6]. Together, immunosenescence and inflammaging represent interconnected hallmarks of immune aging that critically influence susceptibility to ARDs.
Accumulating evidence indicates that immune dysfunction is not merely a passive consequence of aging but a central driver of ARD pathogenesis, including cancer, neurodegenerative disorders, cardiovascular disease, and metabolic syndromes [4,7,8,9,10]. Aging-associated immune remodeling leads to altered leukocyte composition, impaired immune regulation, and sustained production of pro-inflammatory mediators [2,11]. Importantly, aging does not simply exaggerate inflammatory responses; rather, it fundamentally alters their kinetics and resolution. In young organisms, inflammation is typically transient and followed by an active resolution phase that restores tissue homeostasis. In contrast, aging disrupts this tightly regulated sequence, favoring persistent inflammation and tissue damage [12,13,14,15].
Resolution of inflammation is now recognized as an active and highly coordinated process involving specialized pro-resolving lipid mediators (SPMs), effective clearance of apoptotic cells through efferocytosis, and reprogramming of immune cells toward anti-inflammatory and reparative phenotypes [16,17]. Multiple components of these resolution pathways are compromised with age. Reduced biosynthesis of SPMs, impaired responsiveness to pro-resolving signals, and defective macrophage efferocytosis have been documented in aged tissues and experimental models [18,19,20]. These defects result in delayed termination of inflammatory responses and promote a sustained inflammatory milieu that predisposes tissues to chronic injury, fibrosis, and functional decline.
In parallel, immune aging interferes with regenerative and repair processes that are essential for maintaining tissue integrity. Immune cells play critical roles in coordinating regeneration by regulating stem- and progenitor-cell activation, angiogenesis, and extracellular matrix remodeling [12,21,22]. However, aging-associated changes in immune cell phenotype and plasticity shift the balance from regenerative to maladaptive repair responses, often favoring fibrosis over functional tissue restoration [23,24]. The convergence of impaired inflammatory resolution and defective regeneration therefore represents a key mechanistic link between immune aging and the progression of ARDs.
Despite increasing recognition of immune dysfunction as a fundamental hallmark of aging, therapeutic strategies to effectively modulate immune aging remain limited. Most current interventions aim to broadly suppress inflammation, with variable efficacy and potential adverse effects [25]. Emerging evidence suggests that approaches targeting the restoration of inflammatory resolution and regenerative immune functions—rather than indiscriminate immunosuppression—may offer more precise and durable benefits for healthy aging [8,26,27].
In this review, we summarize recent advances in the understanding of immune dysfunction during aging, with a particular focus on the mechanisms underlying impaired resolution and regeneration of inflammation. We discuss how these processes contribute to the pathophysiology of major age-related diseases and highlight emerging therapeutic strategies aimed at restoring immune balance. By reframing immune aging as a failure of coordinated inflammatory control rather than excessive inflammation alone, this review aims to provide a mechanistic framework for the development of targeted interventions against immune-driven aging pathologies.
2. Hallmarks of Immune Dysfunction in Aging
Immune aging is characterized by coordinated yet heterogeneous changes affecting both innate and adaptive immune compartments (Figure 1). Rather than a uniform decline, aging induces a profound remodeling of immune cell composition, phenotype, and function, leading to reduced immune resilience and dysregulated inflammatory responses. These alterations collectively underlie the increased susceptibility to infections, impaired vaccine responses, chronic inflammation, and heightened risk of ARD observed in older individuals.
2.1. Alterations in Innate Immunity
The innate immune system undergoes substantial functional and phenotypic changes with age (Figure 1, left). Hematopoietic stem cell aging promotes a myeloid-biased differentiation program, resulting in increased production of monocytes and granulocytes at the expense of lymphoid progenitors [10,28,29]. While myeloid cell numbers may be preserved or even increased, their functional capacity is often compromised.
Aged macrophages display reduced plasticity and an altered activation spectrum, with impaired transitions between pro-inflammatory and pro-resolving phenotypes. Defects in phagocytosis and efferocytosis—the clearance of apoptotic cells—are consistently observed and contribute to prolonged inflammation and secondary necrosis [30,31]. In parallel, aged macrophages show dysregulated cytokine production, characterized by elevated basal levels of pro-inflammatory mediators and blunted responses to acute stimuli.
Neutrophil function is also markedly altered during aging. Although neutrophil counts are often maintained, aged neutrophils exhibit impaired chemotaxis, reduced microbial killing, and altered formation of neutrophil extracellular traps (NETs), which may further propagate tissue damage and inflammation [32]. Dendritic cells display reduced antigen uptake, impaired migration, and diminished capacity to prime naïve T cells, thereby weakening the bridge between innate and adaptive immunity [6,29,33,34,35,36].
Collectively, these innate immune alterations contribute to ineffective pathogen clearance, persistent inflammatory signaling, and impaired orchestration of downstream immune responses.
2.2. Remodeling of Adaptive Immunity
Adaptive immune aging is dominated by a progressive loss of diversity and flexibility. Thymic involution, which begins early in adulthood and accelerates with age, leads to a marked reduction in naïve T-cell output (Figure 1, right). As a consequence, the peripheral T-cell pool becomes increasingly dominated by memory and highly differentiated effector T cells, many of which exhibit features of replicative senescence or functional exhaustion [1,11,27,37,38].
Senescent T cells display reduced proliferative capacity, altered signal transduction, and skewed cytokine profiles, often favoring pro-inflammatory responses [6]. Accumulation of late-differentiated CD8⁺ T cells with limited antigen specificity has been linked to impaired immune surveillance and increased susceptibility to viral infections and malignancies [39,40,41]. Regulatory T-cell function may also be altered with age, contributing to impaired immune tolerance and chronic inflammation [42].
B-cell aging is characterized by reduced generation of naïve B cells, diminished antibody diversity, and impaired class-switch recombination and somatic hypermutation [43,44,45]. These changes result in weaker and less durable humoral responses to infection and vaccination, alongside an increased propensity for autoantibody production. Together, adaptive immune remodeling leads to reduced responsiveness to new antigens while sustaining inflammatory activity driven by antigen-experienced cells.
2.3. Systemic Consequences: Loss of Immune Homeostasis
The cumulative effects of innate and adaptive immune aging result in a systemic loss of immune homeostasis. Basal inflammatory tone is elevated, driven by senescent immune and non-immune cells, persistent innate immune activation, and defective resolution mechanisms [12,46]. This chronic inflammatory environment not only exacerbates tissue damage but also feeds back to further impair immune cell function, establishing self-sustaining inflammatory loops.
Importantly, immune aging is associated with reduced immune adaptability—the capacity to mount effective responses to acute challenges while efficiently terminating them [47]. This loss of flexibility underlies the paradoxical coexistence of immune deficiency and chronic inflammation in older individuals [25]. Rather than representing isolated defects, these hallmarks reflect a global reprogramming of immune networks that predisposes aging tissues to persistent inflammation, impaired repair, and progressive functional decline [48,49].
3. Inflammaging: Sources, Amplifiers, and Feedback Loops
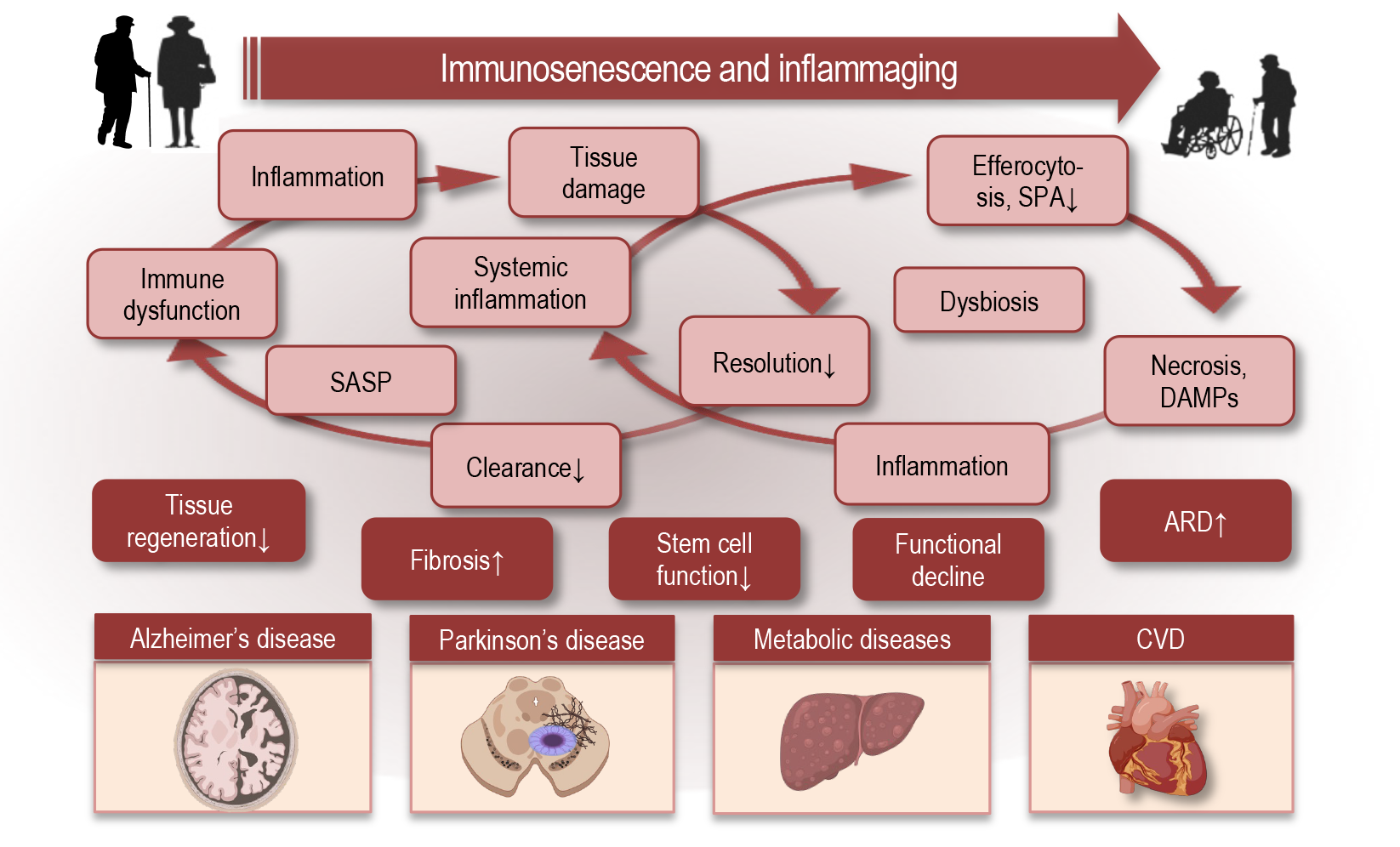
Inflammaging is defined as a chronic, systemic, low-grade inflammatory state that progressively increases with age and contributes to the pathogenesis of multiple ARDs including cardiovascular disease, metabolic syndrome, neurodegeneration, and frailty (Figure 2). While the term initially described an age-associated elevation in circulating inflammatory markers, it now encompasses a complex network of interlinked sources and amplifiers whose dynamic interactions create self-perpetuating feedback loops that sustain chronic inflammation and diminish immune homeostasis [50].
3.1. Cellular Sources of Inflammatory Mediators
A central contributor to inflammaging is the accumulation of senescent cells across tissues (Figure 2A). These cells adopt a senescence-associated secretory phenotype (SASP), releasing pro-inflammatory cytokines (e.g., IL-6, IL-1β, TNF-α), chemokines, growth factors, matrix metalloproteinases, and extracellular matrix fragments that alter local and systemic immune environments. SASP factors not only propagate inflammation locally but also recruit and activate immune cells, linking cellular senescence to chronic immune activation [13,48].
In addition to senescent stromal and epithelial cells, aged immune cells themselves acquire pro-inflammatory phenotypes. For example, age-related polarization shifts in macrophages favor sustained production of pro-inflammatory cytokines, and NK cells exhibit altered cytokine secretion and impaired cytotoxicity, further contributing to inflammatory load [51].
3.2. Amplifiers of Inflammation
Beyond primary inflammatory sources, inflammaging is sustained and intensified by multiple endogenous amplifiers that progressively shift immune signaling toward a chronically activated state (Figure 2B). A central amplifier is mitochondrial dysfunction, which emerges as a hallmark of aging across immune and non-immune cells. Age-associated mitochondrial damage leads to increased production of reactive oxygen species (ROS) and the cytosolic release of mitochondrial DNA (mtDNA), both of which act as potent danger-associated molecular patterns (DAMPs) [52,53]. These signals activate redox-sensitive transcriptional programs and innate immune sensors, including NF-κB, NLRP3 inflammasomes, and the cGAS–STING pathway, thereby reinforcing inflammatory gene expression and cytokine release even in the absence of infection [54,55].
Chronic dysregulation of innate immune sensing further amplifies inflammaging. With age, pattern recognition receptors such as Toll-like receptors and cytosolic nucleic acid sensors exhibit heightened basal activity and impaired negative feedback control. Endogenous ligands derived from damaged cells, extracellular matrix breakdown, and protein aggregates continuously stimulate these receptors, maintaining a pro-inflammatory transcriptional baseline. This altered signaling threshold favors persistent cytokine production while simultaneously impairing responsiveness to acute immune challenges, thereby contributing to immune rigidity [56,57].
The aging gut microbiome represents another powerful amplifier of systemic inflammation. Age-related dysbiosis is characterized by reduced microbial diversity, loss of beneficial commensals, and expansion of pro-inflammatory taxa [58,59]. Concurrent deterioration of epithelial barrier integrity increases translocation of microbial products such as lipopolysaccharide into the circulation, resulting in tonic activation of innate immune pathways [48,60]. Recent human cohort and mechanistic studies demonstrate that gut-derived inflammatory signals strongly correlate with systemic inflammaging markers and predict frailty and multimorbidity in older adults [61,62].
Metabolic reprogramming of immune cells also plays a critical amplifying role. Aging is associated with shifts in immunometabolic pathways that favor sustained glycolysis, altered lipid handling, and impaired mitochondrial oxidative phosphorylation in myeloid cells [63]. These metabolic states support prolonged inflammatory cytokine production and reduce the capacity for phenotype switching toward anti-inflammatory or pro-resolving states. Importantly, metabolic dysfunction and inflammation are mutually reinforcing, creating a feed-forward loop that stabilizes inflammatory immune phenotypes [64,65].
Together, these amplifiers convert transient inflammatory stimuli into persistent inflammatory signaling, progressively locking immune cells and tissues into a pro-inflammatory state that resists resolution.
3.3. Feedback Loops Sustaining Chronic Inflammation
Inflammaging is ultimately maintained by interconnected feedback loops that link immune dysfunction, tissue damage, and impaired clearance mechanisms (Figure 2C). One of the most prominent loops involves cellular senescence and immune surveillance. Senescent cells accumulate with age and secrete a SASP, rich in inflammatory cytokines, chemokines, and matrix-remodeling enzymes. While senescent cells are normally cleared by immune mechanisms, aging compromises this clearance capacity. As a result, senescent cells persist, continue to release SASP factors, and further activate immune cells, reinforcing chronic inflammation and promoting the spread of senescence to neighboring cells [48,52].
A second major feedback loop operates at the level of inflammation resolution. Aging impairs key resolution pathways, including efferocytosis and the biosynthesis and signaling of specialized pro-resolving mediators. Defective clearance of apoptotic immune cells leads to secondary necrosis and release of additional DAMPs, which further stimulate innate immune receptors and prolong inflammation. This failure to properly terminate inflammatory responses transforms otherwise self-limiting immune reactions into chronic inflammatory states that progressively damage tissue architecture [30,31,66].
The microbiome–immune axis also forms a self-sustaining loop in aging. Chronic immune activation alters gut barrier function and microbial composition, while dysbiosis and microbial translocation further stimulate systemic inflammation. This reciprocal interaction perpetuates low-grade immune activation and contributes to sustained elevations of circulating inflammatory mediators, which in turn negatively affect distant organs, including the brain, vasculature, and musculoskeletal system [58,67,68,69,70].
At the systemic level, persistent inflammatory signaling interferes with tissue regeneration and repair, favoring fibrotic remodeling over functional restoration. Pro-inflammatory cytokines impair stem and progenitor cell function, disrupt extracellular matrix dynamics, and promote maladaptive wound-healing responses [71,72]. Over time, these processes lead to progressive functional decline and increased vulnerability to age-related diseases, closing a final feedback loop in which inflammation both drives and is reinforced by tissue dysfunction [73,74].
Collectively, these feedback mechanisms demonstrate that inflammaging is not the result of isolated defects but reflects a network-level reprogramming of immune–tissue interactions. Once established, these loops stabilize chronic inflammation, erode immune adaptability, and accelerate biological aging. In metabolic tissues, for instance, inflammatory cytokines impair insulin signaling and lipid homeostasis, increasing the risk of type 2 diabetes [71,75].
3.4. Systemic and Organ-Level Consequences
Chronic low-grade inflammation, or inflammaging, exerts systemic effects that contribute to multi-organ dysfunction and age-related diseases (Figure 2D). Elevated circulating cytokines, including IL-6, TNF-α, and CRP, correlate with frailty, multimorbidity, and mortality in older adults [14,39,47]. These inflammatory signals accelerate biological aging, as measured by epigenetic clocks and other biomarkers of physiological dysregulation [76].
At the organ level, chronic inflammation drives structural and functional alterations. In the cardiovascular system, it promotes endothelial dysfunction and atherogenesis [77].
In metabolic tissues, inflammatory cytokines impair insulin signaling and lipid homeostasis, increasing the risk of type 2 diabetes [75,78]. In the brain, sustained neuroinflammation accelerates neurodegeneration and contributes to Alzheimer’s pathology [79,80]. Skeletal muscle and connective tissues are similarly affected: chronic inflammation disrupts regeneration, accelerates sarcopenia, and contributes to frailty [81,82].
Thus, across organ systems, persistent inflammatory signaling favors maladaptive repair, including fibrosis, which reduces tissue elasticity and functional reserve. This establishes a self-reinforcing cycle in which inflammation both drives organ declne and is amplified by tissue dysfunction. Inflammaging, therefore, represents a network-level perturbation linking immune aging to systemic and organ-specific pathophysiology, increasing vulnerability to cardiovascular, metabolic, neurodegenerative, and musculoskeletal disorders, and ultimately contributing to reduced lifespan.
4. Impaired Resolution of Inflammation in Aging
4.1. Resolution as an Active, Regulated Process
The termination of inflammation is an active and tightly regulated biological process that is essential for restoring tissue homeostasis following immune activation (Figure 3). Resolution is not achieved through the passive decay of pro-inflammatory signals but instead requires the coordinated engagement of molecular and cellular programs that suppress further leukocyte recruitment, promote the clearance of inflammatory cells, and initiate tissue repair. This concept has fundamentally reshaped our understanding of inflammatory responses, establishing resolution as a distinct immunological phase governed by dedicated signaling pathways rather than as a mere endpoint of inflammation [83].
Central to this process is a lipid mediator class switch that favors the production of SPM, which actively counter-regulate inflammatory signaling, limit neutrophil infiltration, and promote macrophage reprogramming toward reparative phenotypes [84]. Through receptor-mediated mechanisms, SPM suppress pro-inflammatory gene expression while stimulating pathways linked to efferocytosis, tissue remodeling, and restoration of homeostasis [85]. In parallel, resolution signaling creates a permissive microenvironment for tissue repair by supporting stem and progenitor cell survival, activation, and regenerative capacity. Effective resolution therefore represents a distinct immunological state, genetically and metabolically programmed, that is as critical for tissue health as the initial inflammatory response itself (Figure 3, right).
4.2. Age-Related Defects in Resolution Pathways
Aging is associated with a progressive decline in the efficiency of resolution pathways, resulting in prolonged inflammatory responses and incomplete restoration of tissue homeostasis. Experimental models and human studies demonstrate that aged tissues exhibit a reduced capacity to generate SPM following inflammatory challenge, leading to delayed termination of leukocyte recruitment and extended inflammatory duration [31]. In parallel, aging alters receptor expression and downstream signaling responsiveness in immune cells, further blunting pro-resolving signaling.
One of the most consistent defects observed with aging is impaired efferocytosis by macrophages (Figure 3, left). Aged macrophages display reduced phagocytic capacity, defective cytoskeletal remodeling, and altered metabolic programming, all of which compromise efficient clearance of apoptotic inflammatory cells [86]. Because successful efferocytosis feeds back to enhance SPM production and suppress inflammatory cytokine release, its impairment creates a feed-forward loop that perpetuates inflammation.
Delayed clearance of neutrophils and monocytes further exacerbates these defects. Persisting inflammatory cells undergo secondary necrosis, releasing danger-associated molecular patterns that sustain innate immune activation and prevent the timely transition toward resolution. As a result, aging skews inflammatory responses toward a prolonged, low-grade but self-sustaining state characterized by resolution failure rather than exaggerated initiation [87].
4.3. Consequences of Failed Resolution
The inability to effectively resolve inflammation has profound consequences for tissue integrity and organismal health. Persistent inflammatory infiltrates maintain elevated levels of cytokines, proteases, and reactive oxygen species that disrupt normal tissue architecture and compromise organ function. In this context, inflammatory responses increasingly shift from adaptive host defense to maladaptive tissue injury [88].
Resolution failure promotes aberrant wound-healing programs characterized by sustained fibroblast activation, excessive extracellular matrix deposition, and progressive fibrosis (Figure 3, left). These processes have been documented across multiple organs, including the cardiovascular system, lung, liver, and kidney, where unresolved inflammation accelerates structural remodeling and functional decline [83]. Importantly, fibrotic remodeling further impairs tissue elasticity and regenerative capacity, increasing susceptibility to subsequent inflammatory insults.
At the systemic level, chronic resolution defects contribute to persistent immune activation and reinforce inflammaging. Tissue damage resulting from unresolved inflammation generates additional danger signals that perpetuate immune stimulation, establishing a self-reinforcing cycle in which inflammation both drives and is amplified by tissue dysfunction. Impaired resolution therefore represents a central mechanistic link between immune aging, chronic inflammation, and the progression of ARD [26,89,90].
5. Defective Regeneration and Tissue Repair in the Aging Immune System
Effective tissue regeneration following injury relies on a tightly coordinated dialogue between immune cells, stromal compartments, and resident stem and progenitor cells. In young organisms, inflammatory responses are rapidly followed by resolution programs that actively promote repair through immune cell reprogramming, growth factor release, and extracellular matrix remodeling. With aging, this coordination becomes progressively disrupted, resulting in delayed or incomplete regeneration and a shift toward maladaptive repair outcomes [91,92].
Aging alters the functional plasticity of innate immune cells that orchestrate tissue repair. Macrophages, which normally transition from inflammatory to reparative phenotypes during resolution, exhibit impaired phenotypic switching in aged tissues, maintaining pro-inflammatory transcriptional programs while failing to adequately support angiogenesis, matrix remodeling, and progenitor cell activation [83,93]. This persistent inflammatory bias interferes with the regenerative microenvironment required for effective tissue restoration.
In parallel, chronic exposure to inflammatory cytokines directly compromises stem and progenitor cell function. Experimental and human studies demonstrate that prolonged signaling through pathways such as TNF, IL-1, and IFN disrupts stem cell quiescence, reduces self-renewal capacity, and biases differentiation toward dysfunctional or senescent states. These effects are further amplified by aging-associated changes in the extracellular matrix, where inflammatory remodeling alters mechanical and biochemical cues essential for regeneration [94,95].
Defective immune-mediated clearance mechanisms also contribute to impaired repair. Inefficient removal of apoptotic cells and tissue debris sustains local danger signaling, reinforcing inflammatory activation and preventing the establishment of a pro-regenerative niche. As unresolved inflammation persists, fibroblasts and myofibroblasts are preferentially activated, promoting excessive matrix deposition and fibrotic remodeling rather than functional tissue replacement [86,91,92].
At the systemic level, defective regeneration feeds back into immune aging. Tissue dysfunction and fibrosis generate persistent stress and damage signals that perpetuate immune activation, reinforcing inflammaging and further impairing regenerative responses. This establishes a self-sustaining loop in which immune dysfunction limits repair, and failed repair in turn amplifies immune dysregulation. As a result, impaired tissue regeneration represents not only a consequence but also a driver of immune aging and age-related disease progression [96,97,98].
6. Immune Aging and the Pathophysiology of Age-Related Diseases
The chronic immune dysregulation and defective resolution described in the previous sections do not remain confined to isolated tissues. Instead, they propagate systemic alterations that compromise multiple organs, creating a permissive environment for the development and progression of ARDs. Persistent low-grade inflammation, impaired efferocytosis, defective SPM signaling, and maladaptive tissue repair collectively generate a network of immune–tissue interactions that underlie many age-associated pathologies. By integrating these shared mechanisms, immune aging emerges not only as a contributor but also as a central driver of ARDs, linking cellular and molecular defects to organ-level dysfunction and clinical disease. This section summarizes the current understanding of how immune aging contributes to the pathophysiology of key ARDs and highlights common immune mechanisms that underlie these disorders.
6.1. Cancer
Immune surveillance plays a central role in controlling emerging neoplasms, yet immune aging degrades this capacity (Figure 4). With advancing age, thymic involution reduces the output of naïve T cells and narrows T-cell receptor diversity, limiting the repertoire available to recognize diverse tumor antigens and diminishing effective cytotoxic T-cell responses against emerging neoplastic cells [99].
This is compounded by an increase in exhausted and senescent T-cell phenotypes within the peripheral pool, which express inhibitory markers and exhibit reduced proliferative and effector capacities, further impairing anti-tumor immunity [100].
Innate immune cells are also affected by aging in ways that promote a tumor-permissive environment. Natural killer cells, key effectors of early anti-tumor responses, display altered subset distributions and diminished cytotoxicity in older adults, undermining their ability to eliminate transformed cells [1,101]. Macrophages in aged tissues often adopt immunosuppressive or tumor-associated phenotypes, driven in part by chronic inflammatory cues and SASP, which can support angiogenesis, matrix remodeling, and tumor growth rather than tumor clearance. The accumulation of immunosuppressive regulatory T cells with age and age-related defects in dendritic-cell antigen presentation further compromise adaptive anti-tumor responses, creating a microenvironment that facilitates immune evasion and tumor progression [102,103,104].
Inflammaging contributes mechanistically to tumor development and progression by sustaining a pro-tumorigenic milieu. Chronic exposure to inflammatory cytokines such as IL-6, IL-1β, and TNF-α promotes cellular proliferation and survival signaling, stimulates angiogenesis, and can induce DNA damage and epigenetic alterations in non-malignant cells, accelerating oncogenesis [96,101,102,103]. The prevalence of low-grade inflammation in older adults thus converges with immunosenescence to tip the balance from effective surveillance toward immune tolerance of tumor cells [100,105,106].
6.2. Neurodegenerative Disorders
Neurodegenerative disorders represent a major class of ARDs in which immunosenescence plays a critical and increasingly well-defined role (Figure 5). Aging is associated with profound changes in both central and peripheral immune responses that affect brain homeostasis, resilience, and vulnerability to neurodegeneration [12]. Microglia, the resident innate immune cells of the central nervous system, undergo age-associated priming characterized by heightened basal inflammatory tone, exaggerated responses to secondary stimuli, and impaired ability to return to homeostatic states [107]. This primed phenotype promotes sustained production of pro-inflammatory cytokines and reactive oxygen species, contributing to synaptic dysfunction and neuronal stress [12,65,107].
Aging also compromises key resolution and clearance mechanisms within the brain. Microglial phagocytic capacity declines with age, reducing efficient removal of protein aggregates such as amyloid-β and α-synuclein, as well as apoptotic neurons and synaptic debris. This decline is accompanied by altered microglial phenotypes and reduced lysosomal and metabolic efficiency, further limiting effective clearance. Impaired removal of cellular and proteinaceous debris amplifies neuroinflammation and facilitates the accumulation of toxic protein species that drive disease progression in Alzheimer’s and Parkinson’s disease [79,108]. In parallel, reduced responsiveness to pro-resolving signals further delays termination of inflammatory responses, reinforcing a chronic inflammatory milieu within neural tissues [18].
Peripheral immune aging contributes additional layers of dysregulation that intersect with CNS pathology. Increased systemic inflammation, altered monocyte phenotypes, and age-related disruption of blood–brain barrier integrity enhance immune–brain crosstalk and permit greater infiltration of peripheral immune cells into the central nervous system. Once within neural tissues, these cells can adopt pro-inflammatory states, amplify local cytokine and chemokine signaling, and interact with resident microglia, thereby accelerating neurodegenerative cascades [12,65,109,110]. Moreover, senescent cells accumulating in the aging brain and periphery release inflammatory mediators that further amplify microglial activation and neuronal vulnerability [12,111].
Collectively, these processes establish a self-reinforcing loop in which immune aging promotes chronic neuroinflammation, impaired clearance, and neuronal damage, while ongoing neurodegeneration generates additional danger signals that perpetuate immune activation [65,111]. As in other ARDs, neurodegenerative disorders therefore emerge not solely from intrinsic neuronal defects but from progressive dysregulation of immune–tissue interactions that accompany aging, positioning immune aging as a central driver of disease onset and progression.
6.3. Cardiovascular and Metabolic Disorders
Cardiovascular and metabolic disorders are among the most prevalent and debilitating consequences of aging, and immune aging contributes directly to their pathophysiology through shared mechanisms of chronic inflammation, innate and adaptive immune dysregulation, and maladaptive tissue responses (Figure 6). The interplay of inflammaging and immunosenescence creates a pro-inflammatory vascular milieu that drives endothelial dysfunction, vascular remodeling, and the progression of atherosclerosis, hypertension, and related cardiovascular diseases (CVDs) in older adults [112].
Age-associated chronic low-grade inflammation is characterized by elevated cytokines such as IL-6, IL-1β, and TNF-α, as well as high-sensitivity C-reactive protein (hsCRP), which collectively contribute to vascular injury, oxidative stress, and impaired endothelial homeostasis [10,112,113]. Monocyte and macrophage populations in aged individuals exhibit increased pro-inflammatory polarization and reduced capacity for efferocytosis, contributing to plaque progression and instability in atherosclerosis [10].
Dysfunctional T- and B-cell responses further compound vascular inflammation; aging skews T-cell subpopulations toward senescent and pro-inflammatory phenotypes, and diminishes regulatory mechanisms that ordinarily restrain chronic inflammatory signaling in vascular tissues. Such adaptive immune changes contribute both to sustained vascular inflammation and to the breakdown of immune tolerance, which can promote autoantibody production and maladaptive immune responses linked to plaque instability and cardiovascular events [114].
Metabolic disorders, including insulin resistance and type 2 diabetes, are closely tangled with immune aging. Chronic inflammatory signaling emanating from adipose tissue macrophages and senescent immune cells alters insulin signaling pathways and lipid metabolism, creating a state of “metaflammation” that predisposes older individuals to metabolic dysregulation [115]. Inflammaging-driven oxidative stress and mitochondrial dysfunction within metabolic tissues further exacerbate metabolic impairment and promote cardiometabolic risk [116]. Age-related changes in the gut microbiome and increased microbial translocation also contribute to systemic metabolic inflammation, linking gut immune interactions with metabolic and cardiovascular dysregulation [117].
Importantly, these mechanisms do not operate in isolation. The influence of immune aging on cardiovascular and metabolic disorders reflects a self-reinforcing network in which chronic inflammation promotes tissue dysfunction and metabolic derangement, and in turn, tissue stress and altered metabolic signals sustain immune activation [10]. This integrated perspective illustrates how immune aging shapes not only the risk of CVDs and metabolic syndromes but also their progression through shared immunological pathways such as persistent low-grade inflammation, impaired resolution of inflammation, and maladaptive immune–tissue interactions.
7. Therapeutic Strategies to Modulate Immune Aging
As immune aging emerges not only as a hallmark of chronological aging but also as a driver of multiple age-related pathologies, therapeutic strategies aimed at restoring immune competence and rebalancing inflammatory networks have gained traction. These approaches span pharmacological, cellular, lifestyle, nutritional, and microbiome-targeted interventions, reflecting the multifactorial nature of immune aging and its complex mechanistic underpinnings.
One key class of interventions targets chronic inflammation and senescent cells to mitigate both immunosenescence and inflammaging. Pharmacological agents such as inhibitors of mTOR (e.g., rapamycin) have been shown to modulate signaling pathways associated with chronic inflammation, reduce SASP intensity, and enhance aspects of antiviral gene expression in older adults, suggesting that dampening inflammatory circuitry can partially rejuvenate immune responses [118,119,120]. Senolytic drugs such as dasatinib combined with quercetin, fisetin, and BCL-2 inhibitors like navitoclax have demonstrated preclinical efficacy in selectively eliminating senescent cells, thereby reducing pro-inflammatory SASP and improving systemic immune homeostasis; early clinical investigations are underway to assess their potential to delay or prevent age-related dysfunction [121,122,123,124,125].
Complementary pharmacological strategies involve modulation of key inflammatory signaling pathways and immune checkpoints. Inhibitors of p38 MAPK can restore macrophage function and promote pro-resolving phenotypes, enhancing efferocytosis and reducing unresolved inflammatory burden [126,127,128]. The development of pro-resolving and pro-efferocytic nanoparticle-based therapies demonstrates strong potential for the treatment of chronic inflammatory diseases such as atherosclerosis, and more broadly, these nanoparticle platforms may be adaptable to a range of inflammatory and degenerative conditions, underscoring the versatile therapeutic promise of nanoparticle-based approaches [129].
Beyond pharmacotherapy, cellular and regenerative approaches are being explored. Mesenchymal stem cells (MSCs) have potent immunomodulatory properties that can dampen excessive inflammation and foster tissue repair; recent literature highlight their potential to counteract immunosenescence and support regenerative processes across aged tissues [130,131,132,133]. Strategies aimed at rejuvenating hematopoietic stem cells or enhancing thymic function — such as IL-7 administration or bioengineered thymic organoids — hold promise for restoring adaptive immune output and diversity, although translational research is still emerging [134].
Lifestyle and nutritional interventions remain foundational and translationally accessible strategies to modulate immune aging [48,135]. Regular physical activity, adequate sleep, and structured stress management have all been associated with reduced pro-inflammatory cytokines, improved innate and adaptive immune function, and enhanced vaccine responsiveness in older adults [121,136,137,138,139]. Dietary supplementation with micronutrients such as vitamins C, D, E, zinc, and selenium has been shown to improve key immune cell functions, including chemotaxis, phagocytosis, and proliferative capacity in aging populations, and correcting micronutrient deficiencies can blunt chronic inflammation that drives immunosenescence [48,140,141,142,143,144,145]. Omega-3 polyunsaturated fatty acids serve as precursors for pro-resolving lipid mediators and have been linked to reduced systemic inflammation and enhanced resolution capacity in both humans and mice [146,147,148,149,150].
Modulation of the gut microbiome also represents a promising therapeutic avenue [151,152,153]. Diets rich in fiber and prebiotics, targeted probiotic supplementation, and microbiome-directed interventions can enhance gut barrier integrity, promote beneficial microbial taxa, and reduce translocation-induced inflammaging, thereby influencing systemic immune function and inflammatory set points [48,154,155].
Finally, personalized and emerging approaches that integrate immunoprofiling and multi-omics tools are being developed to tailor interventions to individual immune vulnerabilities [156]. Such strategies may combine dietary, pharmacological, and lifestyle interventions with targeted support for metabolic and epigenetic pathways relevant to immune resilience, offering the potential to optimize immune aging trajectories and extend healthspan [48,156,157,158,159,160].
Collectively, these diverse strategies underscore that immune aging is modifiable at multiple levels. While no single intervention will universally reverse immune aging, a multimodal approach that combines mechanistic pharmacology with lifestyle, nutrition, and microbiome targeting holds promise for attenuating immunosenescence, enhancing resolution, and ultimately reducing the burden of age-related diseases.
8. Systems-Level and Network Perspectives on Immune Aging
Immune aging is best understood not as a collection of isolated defects, but as a complex, interconnected reconfiguration of immune and tissue networks that alters how the body responds to internal and external stressors. Aging causes coordinated changes in innate and adaptive immunity, metabolic pathways, and inter-organ communication, creating a web of interactions whose emergent properties differ fundamentally from those of younger systems. Modern multi-omics and deep immunophenotyping studies emphasize this network behavior, showing that age-associated immune changes involve coordinated shifts across cell subsets, cytokine profiles, and systemic signatures rather than single pathway perturbations [161].
Central to this perspective is the concept that immune resilience—the ability to mount effective responses and return to homeostasis—is a network property arising from interaction among cellular populations, signaling pathways, and tissue niches. With aging, network dynamics shift toward a state dominated by inflammaging and reduced responsiveness to perturbations. Elements of this rewiring include persistent pro-inflammatory feedback loops, increased representation of regulatory and suppressive cell phenotypes, and altered communication between immune cells and tissues such as bone marrow, adipose, and the gut mucosa. These integrated changes impact not only host defense but also tissue maintenance, metabolic regulation, and repair processes, contributing to multisystem dysfunctions characteristic of ARD [12,25,65,161,162].
A critical feature emerging from systems analyses is the interdependence of metabolic and immune networks. Age-related metabolic shifts influence immune cell energy states, redox balance, and mediator synthesis, which in turn shape cytokine networks and cell–cell interactions. For example, dysregulated NF-κB and nutrient-sensing pathways such as mTOR and AMPK, detected across multiple immune subsets, can simultaneously affect inflammatory output and cellular metabolism, linking systemic metabolic aging with immune dysfunction [8]. Such network nodes serve as integration points where metabolic, inflammatory, and aging signals converge, highlighting opportunities for interventions that target foundational cross-talk rather than discrete pathways.
Systems approaches also underscore the role of tissue niches and inter-organ communication in immune aging. The gut microbiome, thymus, lymphoid architecture, and stromal environments each function as network hubs that influence systemic immune behavior. Aging-associated remodeling in these niches—such as microbiome dysbiosis and thymic involution—alters antigen exposure, cytokine gradients, and developmental signals, which ripple through the immune network and modulate disease susceptibility across organs [154].
Finally, systems-level frameworks help explain observed heterogeneity in aging trajectories. Integrative immune profiling reveals that individuals diverge in their immune network states, with some maintaining more resilient, youthful signatures while others show pronounced pro-inflammatory and dysregulated profiles even at similar chronological ages. These network signatures predict differences in vaccine responses, frailty, and disease risk, providing a basis for precision approaches to modulate immune aging [12,48,65,161].
Together, these insights emphasize that immune aging is not the failure of a single cell type or pathway but a reprogramming of interacting immune and physiological networks. Understanding and intervening in these network dynamics offers a path toward restoring immune balance, enhancing resilience, and mitigating the multi-organ impacts of aging.
9. Knowledge Gaps and Future Directions
Despite substantial progress in defining the cellular and molecular features of immune aging, major knowledge gaps remain that limit translation into effective interventions. A central unresolved issue is the causal hierarchy among immunosenescence, inflammaging, impaired resolution, and defective regeneration. While these processes clearly reinforce one another, the temporal sequence and dominant drivers likely vary across tissues and individuals, and remain poorly defined. Longitudinal human studies integrating immune, metabolic, and tissue-level data are still scarce, constraining our ability to distinguish primary mechanisms from downstream consequences.
Another major gap concerns the resolution phase of inflammation in aging. Although reduced pro-resolving capacity is increasingly recognized as a key determinant of chronic inflammation, the precise defects in resolution circuits—whether at the level of mediator biosynthesis, receptor signaling, cellular responsiveness, or tissue context—are incompletely characterized in humans. Moreover, resolution biology has largely been studied in acute inflammation models, and its role in chronic, low-grade inflammatory states typical of aging requires further mechanistic and clinical investigation.
The heterogeneity of immune aging represents both a challenge and an opportunity. Individuals of similar chronological age can exhibit markedly different immune network states, inflammatory burdens, and disease susceptibility. The determinants of this heterogeneity—including genetics, early-life exposures, microbiome composition, metabolic status, and environmental stressors—are not yet fully integrated into unified models of immune aging. Future studies must move beyond population averages toward stratified and personalized approaches that identify distinct immune aging trajectories.
At a methodological level, there remains a need for systems-level integration across scales. While multi-omics technologies have expanded rapidly, linking molecular signatures to functional immune behaviors, tissue outcomes, and clinical phenotypes remains difficult. Improved computational models, network-based analyses, and experimentally testable frameworks are required to translate complex datasets into actionable biological insight.
Finally, therapeutic development faces unresolved questions regarding timing, specificity, and safety. It is unclear when during the aging process immune-targeted interventions are most effective, whether treatments should aim to suppress inflammation, enhance resolution, restore immune diversity, or combine these strategies, and how to avoid compromising host defense. Addressing these gaps will be essential for designing interventions that improve healthspan without increasing vulnerability to infection or malignancy.
10. Conclusions
Immune aging is a central, systems-level process that shapes the trajectory of aging and the development of ARDs. Rather than reflecting isolated cellular defects, immunosenescence and inflammaging emerge from a coordinated reprogramming of immune networks, characterized by chronic inflammatory activation, impaired resolution, defective regeneration, and maladaptive immune–tissue feedback loops. These alterations compromise immune adaptability, disrupt tissue homeostasis, and promote progressive functional decline across multiple organs.
A key insight emerging from recent work is that aging-associated inflammation is not merely excessive, but incompletely resolved. Defects in efferocytosis, resolution signaling, and regenerative responses transform otherwise protective immune reactions into persistent drivers of tissue damage and fibrosis. This failure to terminate and repair inflammation provides a unifying mechanism linking immune aging to diverse ARDs, including cancer, neurodegeneration, and cardiometabolic disease.
Viewing immune aging through a systems and network lens offers a powerful framework for understanding its complexity and for identifying intervention points with broad impact. Therapeutic strategies that restore immune balance—by enhancing resolution, modulating metabolic–immune cross-talk, reducing senescence-associated inflammation, and preserving tissue repair capacity—hold promise for altering aging trajectories and extending healthspan.
Ultimately, targeting immune aging represents an opportunity not only to treat individual diseases, but to modify a fundamental biological process underlying multiple age-related pathologies. Continued integration of mechanistic biology, systems-level analysis, and carefully designed human studies will be essential to translate these insights into effective and safe interventions for aging populations.
Author Contributions
Conceptualization, L.M. and S.D.B.; writing—original draft preparation, S.D.B. and L.M.; writing—review and editing, L.M. and S.D.B.; visualization, L.M.; supervision, L.M. All authors have read and agreed to the published version of the manuscript.
Funding
Open Access funding provided by the Max Planck Society.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The Figures were partly created using icons from BioRender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goyani, P.; Christodoulou, R.; Vassiliou, E. Immunosenescence: Aging and Immune System Decline. Vaccines (Basel) 2024, 12. [Google Scholar] [CrossRef]
- Müller, L.; Fulop, T.; Pawelec, G. Immunosenescence in vertebrates and invertebrates. Immun Ageing 2013, 10, 12. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci 2014, 69 Suppl 1, S4–9. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Pratico, D.; Tang, D.; Zhou, S.; Franceschi, C.; Ren, J. Immunosenescence and inflammaging: Mechanisms and role in diseases. Ageing Res Rev 2024, 101, 102540. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol 2017, 8, 1960. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S.; Pawelec, G. The Immune System and Its Dysregulation with Aging. Subcell Biochem 2019, 91, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, S.; Müller, L.; Wenger, E.; Duzel, S.; Pawelec, G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci Biobehav Rev 2017, 75, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, B.; Alu, A.; Hong, W.; Lei, H.; He, X.; Shi, H.; Cheng, P.; Yang, X. Immunosenescence: signaling pathways, diseases and therapeutic targets. Signal Transduction and Targeted Therapy 2025, 10, 250. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S. Aging brain: exploring the interplay between bone marrow aging, immunosenescence, and neuroinflammation. Front Immunol 2024, 15, 1393324. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S. Inflammaging, immunosenescence, and cardiovascular aging: insights into long COVID implications. Front Cardiovasc Med 2024, 11, 1384996. [Google Scholar] [CrossRef]
- Pawelec, G. Hallmarks of human “immunosenescence”: adaptation or dysregulation? Immun Ageing 2012, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Di Benedetto, S.; Müller, V. From Homeostasis to Neuroinflammation: Insights into Cellular and Molecular Interactions and Network Dynamics. Cells 2025, 14. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, E.; Cox, L.S. Interconnections between Inflammageing and Immunosenescence during Ageing. Cells 2022, 11. [Google Scholar] [CrossRef]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and inflammaging in the aging process: age-related diseases or longevity? Ageing Res Rev 2021, 71, 101422. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Savill, J. Resolution of inflammation: the beginning programs the end. Nature Immunology 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Julliard, W.A.; Myo, Y.P.A.; Perelas, A.; Jackson, P.D.; Thatcher, T.H.; Sime, P.J. Specialized pro-resolving mediators as modulators of immune responses. Semin Immunol 2022, 59, 101605. [Google Scholar] [CrossRef]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Chiang, N.; Serhan, C.N. Specialized pro-resolving mediator network: an update on production and actions. Essays Biochem 2020, 64, 443–462. [Google Scholar] [CrossRef]
- Doyle, R.; Sadlier, D.M.; Godson, C. Pro-resolving lipid mediators: Agents of anti-ageing? Seminars in Immunology 2018, 40, 36–48. [Google Scholar] [CrossRef]
- Rymut, N.; Heinz, J.; Sadhu, S.; Hosseini, Z.; Riley, C.O.; Marinello, M.; Maloney, J.; MacNamara, K.C.; Spite, M.; Fredman, G. Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J 2020, 34, 597–609. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef]
- Eming, S.A.; Wynn, T.A.; Martin, P. Inflammation and metabolism in tissue repair and regeneration. Science 2017, 356, 1026–1030. [Google Scholar] [CrossRef]
- Hams, E.; Bermingham, R.; Fallon, P.G. Macrophage and Innate Lymphoid Cell Interplay in the Genesis of Fibrosis. Front Immunol 2015, 6, 597. [Google Scholar] [CrossRef] [PubMed]
- Bektas, A.; Schurman, S.H.; Sen, R.; Ferrucci, L. Aging, inflammation and the environment. Exp Gerontol 2018, 105, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.Q.T.; Cho, K.A. Targeting immunosenescence and inflammaging: advancing longevity research. Exp Mol Med 2025, 57, 1881–1892. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Huo, T.; Lu, M.; Zhao, Y.; Zhang, J.; He, W.; Chen, H. Recent Advances in Aging and Immunosenescence: Mechanisms and Therapeutic Strategies. Cells 2025, 14. [Google Scholar] [CrossRef]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front Immunol 2016, 7, 502. [Google Scholar] [CrossRef]
- Bleve, A.; Motta, F.; Durante, B.; Pandolfo, C.; Selmi, C.; Sica, A. Immunosenescence, Inflammaging, and Frailty: Role of Myeloid Cells in Age-Related Diseases. Clin Rev Allergy Immunol 2023, 64, 123–144. [Google Scholar] [CrossRef]
- Sendama, W. The effect of ageing on the resolution of inflammation. Ageing Research Reviews 2020, 57, 101000. [Google Scholar] [CrossRef]
- Arnardottir, H.H.; Dalli, J.; Colas, R.A.; Shinohara, M.; Serhan, C.N. Aging delays resolution of acute inflammation in mice: reprogramming the host response with novel nano-proresolving medicines. J Immunol 2014, 193, 4235–4244. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nature Reviews Immunology 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Mahbub, S.; Brubaker, A.L.; Kovacs, E.J. Aging of the Innate Immune System: An Update. Curr Immunol Rev 2011, 7, 104–115. [Google Scholar] [CrossRef]
- Müller, L.; Pawelec, G. As we age: Does slippage of quality control in the immune system lead to collateral damage? Ageing Res Rev 2015, 23, 116–123. [Google Scholar] [CrossRef]
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fulop, T. Innate immunosenescence: effect of aging on cells and receptors of the innate immune system in humans. Semin Immunol 2012, 24, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kim, S.J.; Lei, Y.; Wang, S.; Wang, H.; Huang, H.; Zhang, H.; Tsung, A. Neutrophil extracellular traps in homeostasis and disease. Signal Transduction and Targeted Therapy 2024, 9, 235. [Google Scholar] [CrossRef]
- Di Benedetto, S.; Derhovanessian, E.; Steinhagen-Thiessen, E.; Goldeck, D.; Müller, L.; Pawelec, G. Impact of age, sex and CMV-infection on peripheral T cell phenotypes: results from the Berlin BASE-II Study. Biogerontology 2015, 16, 631–643. [Google Scholar] [CrossRef]
- Thomas, R.; Wang, W.; Su, D.M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun Ageing 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. Immune Aging and Its Implication for Age-Related Disease Progression. Physiology 2025, 40, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Czesnikiewicz-Guzik, M.; Lee, W.W.; Cui, D.; Hiruma, Y.; Lamar, D.L.; Yang, Z.Z.; Ouslander, J.G.; Weyand, C.M.; Goronzy, J.J. T cell subset-specific susceptibility to aging. Clin Immunol 2008, 127, 107–118. [Google Scholar] [CrossRef]
- Moller, S.H.; Hsueh, P.C.; Yu, Y.R.; Zhang, L.; Ho, P.C. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging. Cell Metab 2022, 34, 378–395. [Google Scholar] [CrossRef]
- Raynor, J.; Lages, C.S.; Shehata, H.; Hildeman, D.A.; Chougnet, C.A. Homeostasis and function of regulatory T cells in aging. Curr Opin Immunol 2012, 24, 482–487. [Google Scholar] [CrossRef]
- Ratliff, M.; Alter, S.; Frasca, D.; Blomberg, B.B.; Riley, R.L. In senescence, age-associated B cells secrete TNFalpha and inhibit survival of B-cell precursors. Aging Cell 2013, 12, 303–311. [Google Scholar] [CrossRef]
- Weksler, M.E. Changes in the B-cell repertoire with age. Vaccine 2000, 18, 1624–1628. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Garcia, D.; Blomberg, B.B. B Cell Immunosenescence. Annu Rev Cell Dev Biol 2020, 36, 551–574. [Google Scholar] [CrossRef]
- Huntington, N.D.; Gray, D.H. Immune homeostasis in health and disease. Immunol Cell Biol 2018, 96, 451–452. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Mu, W.C.; Markov, N.T.; Fuentealba, M.; Halaweh, H.; Senchyna, F.; Manwaring-Mueller, M.N.; Winer, D.A.; Furman, D. Immunological biomarkers of aging. J Immunol 2025, 214, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Di Benedetto, S. Immunosenescence and inflammaging: Mechanisms and modulation through diet and lifestyle. Front Immunol 2025, 16, 1708280. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Maestre, I.; Harris, A.S.; Amor, C. Aging and immunity: the age-old tango. Genes Dev 2025, 39, 948–974. [Google Scholar] [CrossRef]
- Franceschi, C.; Olivieri, F.; Moskalev, A.; Ivanchenko, M.; Santoro, A. Toward precision interventions and metrics of inflammaging. Nature Aging 2025, 5, 1441–1454. [Google Scholar] [CrossRef]
- Kamroo, A.; Kakroudi, M.H.; Sarmadian, A.J.; Firouzabadi, A.; Mousavi, S.; Yazdanpanah, N.; Saleki, K.; Rezaei, N. Immunosenescence and organoids: pathophysiology and therapeutic opportunities. Immunity & Ageing 2025, 22, 46. [Google Scholar] [CrossRef] [PubMed]
- Karpuzoglu, E.; Holladay, S.D.; Gogal, R.M., Jr. Inflammaging: triggers, molecular mechanisms, immunological consequences, sex differences, and cutaneous manifestations. Front Immunol 2025, 16, 1704203. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Loygorri, J.I.; Villarejo-Zori, B.; Viedma-Poyatos, A.; Zapata-Munoz, J.; Benitez-Fernandez, R.; Frutos-Lison, M.D.; Tomas-Barberan, F.A.; Espin, J.C.; Area-Gomez, E.; Gomez-Duran, A.; et al. Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat Commun 2024, 15, 830. [Google Scholar] [CrossRef]
- Gulen, M.F.; Samson, N.; Keller, A.; Schwabenland, M.; Liu, C.; Glück, S.; Thacker, V.V.; Favre, L.; Mangeat, B.; Kroese, L.J.; et al. cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature 2023, 620, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Hu, Z.; Xu, D.; Bian, H.; Lv, J.; Zhu, X.; Zhang, Q.; Su, L.; Yin, H.; Lu, T.; et al. STING signaling in inflammaging: a new target against musculoskeletal diseases. Front Immunol 2023, 14, 1227364. [Google Scholar] [CrossRef]
- Li, X.; Li, C.; Zhang, W.; Wang, Y.; Qian, P.; Huang, H. Inflammation and aging: signaling pathways and intervention therapies. Signal Transduction and Targeted Therapy 2023, 8, 239. [Google Scholar] [CrossRef]
- Shaw, A.C.; Joshi, S.; Greenwood, H.; Panda, A.; Lord, J.M. Aging of the innate immune system. Curr Opin Immunol 2010, 22, 507–513. [Google Scholar] [CrossRef]
- Caetano-Silva, M.E.; Shrestha, A.; Duff, A.F.; Kontic, D.; Brewster, P.C.; Kasperek, M.C.; Lin, C.H.; Wainwright, D.A.; Hernandez-Saavedra, D.; Woods, J.A.; et al. Aging amplifies a gut microbiota immunogenic signature linked to heightened inflammation. Aging Cell 2024, 23, e14190. [Google Scholar] [CrossRef]
- Strasser, B.; Ticinesi, A. Intestinal microbiome in normal ageing, frailty and cognition decline. Curr Opin Clin Nutr Metab Care 2023, 26, 8–16. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S. Bridging the brain and gut: neuroimmune mechanisms of neuroinflammation and therapeutic insights. Front Cell Neurosci 2025, 19, 1590002. [Google Scholar] [CrossRef]
- Wen, N.N.; Sun, L.W.; Geng, Q.; Zheng, G.H. Gut microbiota changes associated with frailty in older adults: A systematic review of observational studies. World J Clin Cases 2024, 12, 6815–6825. [Google Scholar] [CrossRef]
- Xu, Y.; Mao, T.; Wang, Y.; Qi, X.; Zhao, W.; Chen, H.; Zhang, C.; Li, X. Effect of Gut Microbiota-Mediated Tryptophan Metabolism on Inflammaging in Frailty and Sarcopenia. J Gerontol A Biol Sci Med Sci 2024, 79. [Google Scholar] [CrossRef]
- Maurmann, R.M.; Schmitt, B.L.; Mosalmanzadeh, N.; Pence, B.D. Mitochondrial dysfunction at the cornerstone of inflammatory exacerbation in aged macrophages. Explor Immunol 2023, 3, 442–452. [Google Scholar] [CrossRef]
- Ginefra, P.; Hope, H.C.; Lorusso, G.; D’Amelio, P.; Vannini, N. The immunometabolic roots of aging. Current Opinion in Immunology 2024, 91, 102498. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S.; Müller, V. The dual nature of neuroinflammation in networked brain. Front Immunol 2025, 16, 1659947. [Google Scholar] [CrossRef]
- Whittington, R.A.; Planel, E.; Terrando, N. Impaired Resolution of Inflammation in Alzheimer’s Disease: A Review. Front Immunol 2017, 8, 1464. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.; A.D., N. Ageing of the gut microbiome: Potential influences on immune senescence and inflammageing. Ageing Res Rev 2021, 68, 101323. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Cheng, G.; Hardy, M. Gut microbiome, short-chain fatty acids, alpha-synuclein, neuroinflammation, and ROS/RNS: Relevance to Parkinson’s disease and therapeutic implications. Redox Biol 2024, 71, 103092. [Google Scholar] [CrossRef]
- Kohler, C.A.; Maes, M.; Slyepchenko, A.; Berk, M.; Solmi, M.; Lanctot, K.L.; Carvalho, A.F. The Gut-Brain Axis, Including the Microbiome, Leaky Gut and Bacterial Translocation: Mechanisms and Pathophysiological Role in Alzheimer’s Disease. Curr Pharm Des 2016, 22, 6152–6166. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, K.J.; Moloney, G.M.; Keane, L.; Clarke, G.; Cryan, J.F. The gut microbiota-immune-brain axis: Therapeutic implications. Cell Rep Med 2025, 6, 101982. [Google Scholar] [CrossRef]
- Nirenjen, S.; Narayanan, J.; Tamilanban, T.; Subramaniyan, V.; Chitra, V.; Fuloria, N.K.; Wong, L.S.; Ramachawolran, G.; Sekar, M.; Gupta, G.; et al. Exploring the contribution of pro-inflammatory cytokines to impaired wound healing in diabetes. Front Immunol 2023, 14, 1216321. [Google Scholar] [CrossRef]
- Josephson, A.M.; Bradaschia-Correa, V.; Lee, S.; Leclerc, K.; Patel, K.S.; Muinos Lopez, E.; Litwa, H.P.; Neibart, S.S.; Kadiyala, M.; Wong, M.Z.; et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc Natl Acad Sci U S A 2019, 116, 6995–7004. [Google Scholar] [CrossRef]
- Riparini, G.; Mackenzie, M.; Naz, F.; Brooks, S.; Jiang, K.; Deewan, A.; Dulek, B.; Islam, S.; Ko, K.D.; Tsai, W.L.; et al. Epigenetic dysregulation in aged muscle stem cells drives mesenchymal progenitor expansion via IL-6 and Spp1 signaling. Nature Aging 2025, 5, 2399–2416. [Google Scholar] [CrossRef]
- Mlynarska, E.; Kowalik, A.; Krajewska, A.; Krupinska, N.; Marcinkowska, W.; Motor, J.; Przybylak, A.; Tlustochowicz, K.; Rysz, J.; Franczyk, B. Inflammaging and Senescence-Driven Extracellular Matrix Remodeling in Age-Associated Cardiovascular Disease. Biomolecules 2025, 15. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro-Machado, E.; Gurgul-Convey, E.; Marzec, M.T. Immunometabolism in type 2 diabetes mellitus: tissue-specific interactions. Arch Med Sci 2023, 19, 895–911. [Google Scholar] [CrossRef]
- Meier, H.C.S.; Mitchell, C.; Karadimas, T.; Faul, J.D. Systemic inflammation and biological aging in the Health and Retirement Study. GeroScience 2023, 45, 3257–3265. [Google Scholar] [CrossRef] [PubMed]
- Spray, L.; Richardson, G.; Haendeler, J.; Altschmied, J.; Rumampouw, V.; Wallis, S.B.; Georgiopoulos, G.; White, S.; Unsworth, A.; Stellos, K.; et al. Cardiovascular inflammaging: Mechanisms, consequences, and therapeutic perspectives. Cell Reports Medicine 2025, 6, 102264. [Google Scholar] [CrossRef]
- Rohm, T.V.; Meier, D.T.; Olefsky, J.M.; Donath, M.Y. Inflammation in obesity, diabetes, and related disorders. Immunity 2022, 55, 31–55. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamed, H.G.; Hassan, A.A.; Sakraan, A.A.; Al-Deeb, R.T.; Mousa, D.M.; Aboul Ezz, H.S.; Noor, N.A.; Khadrawy, Y.A.; Radwan, N.M. Brain interleukins and Alzheimer’s disease. Metab Brain Dis 2025, 40, 116. [Google Scholar] [CrossRef]
- Carr, L.; Mustafa, S.; Collins-Praino, L.E. The Hallmarks of Ageing in Microglia. Cell Mol Neurobiol 2025, 45, 45. [Google Scholar] [CrossRef]
- Antuna, E.; Cachan-Vega, C.; Bermejo-Millo, J.C.; Potes, Y.; Caballero, B.; Vega-Naredo, I.; Coto-Montes, A.; Garcia-Gonzalez, C. Inflammaging: Implications in Sarcopenia. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef]
- Damanti, S.; Senini, E.; De Lorenzo, R.; Merolla, A.; Santoro, S.; Festorazzi, C.; Messina, M.; Vitali, G.; Sciorati, C.; Manfredi, A.A.; et al. Molecular constraints of sarcopenia in the ageing muscle. Front Aging 2025, 6, 1588014. [Google Scholar] [CrossRef]
- Panezai, J.; Van Dyke, T.E. Resolution of inflammation: Intervention strategies and future applications. Toxicol Appl Pharmacol 2022, 449, 116089. [Google Scholar] [CrossRef] [PubMed]
- Perez-Hernandez, J.; Chiurchiu, V.; Perruche, S.; You, S. Regulation of T-Cell Immune Responses by Pro-Resolving Lipid Mediators. Front Immunol 2021, 12, 768133. [Google Scholar] [CrossRef] [PubMed]
- Peh, H.Y.; Chen, J. Pro-resolving lipid mediators and therapeutic innovations in resolution of inflammation. Pharmacol Ther 2025, 265, 108753. [Google Scholar] [CrossRef]
- Boucher, D.M.; Robichaud, S.; Lorant, V.; Leon, J.S.; Suliman, I.; Rasheed, A.; Susser, L.I.; Emerton, C.; Geoffrion, M.; De Jong, E.; et al. Age-Related Impairments in Immune Cell Efferocytosis and Autophagy Hinder Atherosclerosis Regression. Arterioscler Thromb Vasc Biol 2025, 45, 481–495. [Google Scholar] [CrossRef]
- Doran, A.C. Inflammation Resolution: Implications for Atherosclerosis. Circ Res 2022, 130, 130–148. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef]
- Dalli, J.; Colas, R.A.; Serhan, C.N. Novel n-3 Immunoresolvents: Structures and Actions. Scientific Reports 2013, 3, 1940. [Google Scholar] [CrossRef]
- Marques, R.M.; Gonzalez-Nunez, M.; Walker, M.E.; Gomez, E.A.; Colas, R.A.; Montero-Melendez, T.; Perretti, M.; Dalli, J. Loss of 15-lipoxygenase disrupts Treg differentiation altering their pro-resolving functions. Cell Death & Differentiation 2021, 28, 3140–3160. [Google Scholar] [CrossRef]
- Yue, Z.; Nie, L.; Zhang, P.; Chen, Q.; Lv, Q.; Wang, Q. Tissue-resident macrophage inflammaging aggravates homeostasis dysregulation in age-related diseases. Cell Immunol 2021, 361, 104278. [Google Scholar] [CrossRef]
- Li, N.; Chen, Y.; Wang, Q.; Liu, X.; Han, C.; Qu, C.; Guan, X.; Zou, W.; Wang, X.; Li, A.; et al. Microenvironment-driven satellite cell regeneration and repair in aging-related sarcopenia: mechanisms and therapeutic frontiers. Stem Cell Res Ther 2025, 16, 545. [Google Scholar] [CrossRef]
- Kushioka, J.; Chow, S.K.; Toya, M.; Tsubosaka, M.; Shen, H.; Gao, Q.; Li, X.; Zhang, N.; Goodman, S.B. Bone regeneration in inflammation with aging and cell-based immunomodulatory therapy. Inflamm Regen 2023, 43, 29. [Google Scholar] [CrossRef]
- Rahman, F.A.; Angus, S.A.; Stokes, K.; Karpowicz, P.; Krause, M.P. Impaired ECM Remodeling and Macrophage Activity Define Necrosis and Regeneration Following Damage in Aged Skeletal Muscle. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Duong, L.; Radley, H.G.; Lee, B.; Dye, D.E.; Pixley, F.J.; Grounds, M.D.; Nelson, D.J.; Jackaman, C. Macrophage function in the elderly and impact on injury repair and cancer. Immunity & Ageing 2021, 18, 4. [Google Scholar] [CrossRef]
- Singh, A.; Schurman, S.H.; Bektas, A.; Kaileh, M.; Roy, R.; Wilson, D.M., 3rd; Sen, R.; Ferrucci, L. Aging and Inflammation. Cold Spring Harb Perspect Med 2024, 14. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Pulido, S.; Yousefzadeh, M.J.; Mittelbrunn, M. Aging reshapes the adaptive immune system from healer to saboteur. Nat Aging 2025, 5, 1393–1403. [Google Scholar] [CrossRef]
- Salminen, A. Increased immunosuppression impairs tissue homeostasis with aging and age-related diseases. Journal of Molecular Medicine 2021, 99, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tang, D. Immunosenescence promotes cancer development: from mechanisms to treatment strategies. Cell Communication and Signaling 2025, 23, 128. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.S.; Burton Sojo, G.; Sun, H.; Friedland, B.N.; McNamara, M.E.; Schmidt, M.O.; Wellstein, A. The Role of Aging and Senescence in Immune Checkpoint Inhibitor Response and Toxicity. Int J Mol Sci 2024, 25. [Google Scholar] [CrossRef]
- Ikeda, H.; Togashi, Y. Aging, cancer, and antitumor immunity. International Journal of Clinical Oncology 2022, 27, 316–322. [Google Scholar] [CrossRef]
- Lian, J.; Yue, Y.; Yu, W.; Zhang, Y. Immunosenescence: a key player in cancer development. Journal of Hematology & Oncology 2020, 13, 151. [Google Scholar] [CrossRef]
- Liu, Q.; Li, J.; Sun, X.; Lin, J.; Yu, Z.; Xiao, Y.; Li, D.; Sun, B.; Bao, H.; Liu, Y. Immunosenescence and cancer: molecular hallmarks, tumor microenvironment remodeling, and age-specific immunotherapy challenges. J Hematol Oncol 2025, 18, 81. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Qin, P.; Li, F.; Zhu, H.; You, H.; Zhang, Y.; Xu, B.; Li, T.; Zhang, F.; Han, L.; et al. The inter-link of ageing, cancer and immunity: findings from real-world retrospective study. Immunity & Ageing 2023, 20, 75. [Google Scholar] [CrossRef]
- Gao, D.; Kan, P.; He, Y.; Sun, S.; Tang, L.; Yang, F. Senescent immune cells in the tumor microenvironment: emerging insights into cancer immunotherapy resistance. Front Immunol 2025, 16, 1656733. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Cheng, J. Cellular Senescence and Immunosenescence in Melanoma: Insights From the Tumor Microenvironment. Cancer Med 2025, 14, e71223. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Muccigrosso, M.M.; Godbout, J.P. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology 2015, 96, 29–41. [Google Scholar] [CrossRef]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: mechanism and potential therapeutic targets. Signal Transduction and Targeted Therapy 2023, 8, 359. [Google Scholar] [CrossRef]
- Alkhalifa, A.E.; Al-Ghraiybah, N.F.; Odum, J.; Shunnarah, J.G.; Austin, N.; Kaddoumi, A. Blood-Brain Barrier Breakdown in Alzheimer’s Disease: Mechanisms and Targeted Strategies. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front Cell Neurosci 2021, 15, 661838. [Google Scholar] [CrossRef]
- Müller, L.; Di Benedetto, S. Neuroimmune crosstalk in chronic neuroinflammation: microglial interactions and immune modulation. Front Cell Neurosci 2025, 19, 1575022. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Pratico, D.; Vinciguerra, M.; Lip, G.Y.H.; Franceschi, C.; Ren, J. Inflammaging: mechanisms and role in the cardiac and vasculature. Trends Endocrinol Metab 2023, 34, 373–387. [Google Scholar] [CrossRef]
- Barcena, M.L.; Aslam, M.; Pozdniakova, S.; Norman, K.; Ladilov, Y. Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Snijckers, R.P.M.; Foks, A.C. Adaptive immunity and atherosclerosis: aging at its crossroads. Front Immunol 2024, 15, 1350471. [Google Scholar] [CrossRef]
- Acierno, C.; Frontuto, M.; De Stefano, G.F.; Erezanu, A.; Limone, A.; Morella, S.; Picaro, F.; Palazzo, D.; Gilio, M. From Steatosis to Immunosenescence: The Impact of Metabolic Dysfunction on Immune Aging in HIV and Non-HIV Populations. Biomedicines 2025, 13. [Google Scholar] [CrossRef] [PubMed]
- Mapuskar, K.A.; London, B.; Zacharias, Z.R.; Houtman, J.C.D.; Allen, B.G. Immunometabolism in the Aging Heart. J Am Heart Assoc 2025, 14, e039216. [Google Scholar] [CrossRef]
- Spray, L.; Richardson, G.; Haendeler, J.; Altschmied, J.; Rumampouw, V.; Wallis, S.B.; Georgiopoulos, G.; White, S.; Unsworth, A.; Stellos, K.; et al. Cardiovascular inflammaging: Mechanisms, consequences, and therapeutic perspectives. Cell Rep Med 2025, 6, 102264. [Google Scholar] [CrossRef]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. Author Correction: MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2021, 23, 564–565. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.S.; Huang, Y.W.; Lee, T.H.; Chang, L.; Huang, Z.D.; Wu, T.Y.; Wang, T.J.; Ho, C.S. Rapamycin Alleviates Protein Aggregates, Reduces Neuroinflammation, and Rescues Demyelination in Globoid Cell Leukodystrophy. Cells 2023, 12. [Google Scholar] [CrossRef]
- Wang, F.; Song, Y.; Liu, P.; Ma, F.; Peng, Z.; Pang, Y.; Hu, H.; Zeng, L.; Luo, H.; Zhang, X. Rapamycin suppresses neuroinflammation and protects retinal ganglion cell loss after optic nerve crush. Int Immunopharmacol 2023, 119, 110171. [Google Scholar] [CrossRef]
- Hieber, C.; Grabbe, S.; Bros, M. Counteracting Immunosenescence-Which Therapeutic Strategies Are Promising? Biomolecules 2023, 13. [Google Scholar] [CrossRef]
- Kiss, T.; Nyul-Toth, A.; DelFavero, J.; Balasubramanian, P.; Tarantini, S.; Faakye, J.; Gulej, R.; Ahire, C.; Ungvari, A.; Yabluchanskiy, A.; et al. Spatial transcriptomic analysis reveals inflammatory foci defined by senescent cells in the white matter, hippocampi and cortical grey matter in the aged mouse brain. Geroscience 2022, 44, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: from discovery to translation. J Intern Med 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Burdusel, D.; Doeppner, T.R.; Surugiu, R.; Hermann, D.M.; Olaru, D.G.; Popa-Wagner, A. The Intersection of Epigenetics and Senolytics in Mechanisms of Aging and Therapeutic Approaches. Biomolecules 2024, 15. [Google Scholar] [CrossRef]
- De Maeyer, R.P.H.; van de Merwe, R.C.; Louie, R.; Bracken, O.V.; Devine, O.P.; Goldstein, D.R.; Uddin, M.; Akbar, A.N.; Gilroy, D.W. Blocking elevated p38 MAPK restores efferocytosis and inflammatory resolution in the elderly. Nat Immunol 2020, 21, 615–625. [Google Scholar] [CrossRef]
- Raza, A.; Crothers, J.W.; McGill, M.M.; Mawe, G.M.; Teuscher, C.; Krementsov, D.N. Anti-inflammatory roles of p38alpha MAPK in macrophages are context dependent and require IL-10. J Leukoc Biol 2017, 102, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Hota, S.S.; Sarma, P.; Bhattacharyya, A.; Medhi, B.; Reddy, D.H. Recent advances in molecular pathways and therapeutic implications targeting neuroinflammation for Alzheimer’s disease. Inflammopharmacology 2021, 29, 1669–1681. [Google Scholar] [CrossRef]
- Patel, Y.; Manturthi, S.; Tiwari, S.; Gahunia, E.; Courtemanche, A.; Gandelman, M.; Cote, M.; Gadde, S. Development of Pro-resolving and Pro-efferocytic Nanoparticles for Atherosclerosis Therapy. ACS Pharmacol Transl Sci 2024, 7, 3086–3095. [Google Scholar] [CrossRef]
- Wang, X.; Guo, D.; He, C.; Wang, X.; Wei, Y.; Zhang, F.; Wang, L.; Yang, Y. Clinical application of mesenchymal stem cells in immunosenescence: a qualitative review of their potential and challenges. Stem Cell Research & Therapy 2025, 16, 265. [Google Scholar] [CrossRef]
- Han, X.; Liao, R.; Li, X.; Zhang, C.; Huo, S.; Qin, L.; Xiong, Y.; He, T.; Xiao, G.; Zhang, T. Mesenchymal stem cells in treating human diseases: molecular mechanisms and clinical studies. Signal Transduct Target Ther 2025, 10, 262. [Google Scholar] [CrossRef]
- Song, N.; Scholtemeijer, M.; Shah, K. Mesenchymal Stem Cell Immunomodulation: Mechanisms and Therapeutic Potential. Trends Pharmacol Sci 2020, 41, 653–664. [Google Scholar] [CrossRef]
- Muller, L.; Tunger, A.; Wobus, M.; von Bonin, M.; Towers, R.; Bornhauser, M.; Dazzi, F.; Wehner, R.; Schmitz, M. Immunomodulatory Properties of Mesenchymal Stromal Cells: An Update. Front Cell Dev Biol 2021, 9, 637725. [Google Scholar] [CrossRef]
- Nguyen, T.Q.T.; Cho, K.A. Targeting immunosenescence and inflammaging: advancing longevity research. Experimental & Molecular Medicine 2025, 57, 1881–1892. [Google Scholar] [CrossRef]
- Strasser, B.; Wolters, M.; Weyh, C.; Kruger, K.; Ticinesi, A. The Effects of Lifestyle and Diet on Gut Microbiota Composition, Inflammation and Muscle Performance in Our Aging Society. Nutrients 2021, 13. [Google Scholar] [CrossRef]
- Martínez-Albert, E.; Lutz, N.D.; Hübener, R.; Dimitrov, S.; Lange, T.; Born, J.; Besedovsky, L. Sleep promotes T-cell migration towards CCL19 via growth hormone and prolactin signaling in humans. Brain, Behavior, and Immunity 2024, 118, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, A.C.; De Nys, L.; Brindle, R.C.; Drayson, M.T. Physical activity and sleep relate to antibody maintenance following naturalistic infection and/or vaccination in older adults. Brain, Behavior, & Immunity - Health 2023, 32, 100661. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Zisapel, N. New perspectives on the role of melatonin in human sleep, circadian rhythms and their regulation. Br J Pharmacol 2018, 175, 3190–3199. [Google Scholar] [CrossRef] [PubMed]
- Bjorklund, G.; Shanaida, M.; Lysiuk, R.; Antonyak, H.; Klishch, I.; Shanaida, V.; Peana, M. Selenium: An Antioxidant with a Critical Role in Anti-Aging. Molecules 2022, 27. [Google Scholar] [CrossRef]
- Ecarnot, F.; Maggi, S. The impact of the Mediterranean diet on immune function in older adults. Aging Clin Exp Res 2024, 36, 117. [Google Scholar] [CrossRef]
- Bischoff-Ferrari, H.A.; Gängler, S.; Wieczorek, M.; Belsky, D.W.; Ryan, J.; Kressig, R.W.; Stähelin, H.B.; Theiler, R.; Dawson-Hughes, B.; Rizzoli, R.; et al. Individual and additive effects of vitamin D, omega-3 and exercise on DNA methylation clocks of biological aging in older adults from the DO-HEALTH trial. Nature Aging 2025, 5, 376–385. [Google Scholar] [CrossRef]
- Chambers, E.S.; Vukmanovic-Stejic, M.; Turner, C.T.; Shih, B.B.; Trahair, H.; Pollara, G.; Tsaliki, E.; Rustin, M.; Freeman, T.C.; Mabbott, N.A.; et al. Vitamin D(3) replacement enhances antigen-specific immunity in older adults. Immunother Adv 2021, 1, ltaa008. [Google Scholar] [CrossRef] [PubMed]
- Ghaseminejad-Raeini, A.; Ghaderi, A.; Sharafi, A.; Nematollahi-Sani, B.; Moossavi, M.; Derakhshani, A.; Sarab, G.A. Immunomodulatory actions of vitamin D in various immune-related disorders: a comprehensive review. Front Immunol 2023, 14, 950465. [Google Scholar] [CrossRef] [PubMed]
- Rastgoo, S.; Pourvali, K.; Raeissadat, S.A.; Eslamian, G.; Zand, H. Co-administration of vitamin D and N-acetylcysteine to modulate immunosenescence in older adults with vitamin D deficiency: a randomized clinical trial. Front Immunol 2025, 16, 1570441. [Google Scholar] [CrossRef]
- Barry, A.R.; Dixon, D.L. Omega-3 fatty acids for the prevention of atherosclerotic cardiovascular disease. Pharmacotherapy 2021, 41, 1056–1065. [Google Scholar] [CrossRef]
- Djuricic, I.; Calder, P.C. Beneficial Outcomes of Omega-6 and Omega-3 Polyunsaturated Fatty Acids on Human Health: An Update for 2021. Nutrients 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Azzolino, D.; Bertoni, C.; De Cosmi, V.; Spolidoro, G.C.I.; Agostoni, C.; Lucchi, T.; Mazzocchi, A. Omega-3 polyunsatured fatty acids and physical performance across the lifespan: a narrative review. Front Nutr 2024, 11, 1414132. [Google Scholar] [CrossRef]
- Bodur, M.; Yilmaz, B.; Agagunduz, D.; Ozogul, Y. Immunomodulatory Effects of Omega-3 Fatty Acids: Mechanistic Insights and Health Implications. Mol Nutr Food Res 2025, 69, e202400752. [Google Scholar] [CrossRef]
- Kerlikowsky, F.; Kruger, K.; Hahn, A.; Schuchardt, J.P. Multimicronutrient and omega-3 fatty acid supplementation reduces low-grade inflammation in older participants: An exploratory study. Nutr Res 2025, 140, 46–58. [Google Scholar] [CrossRef]
- Pluta, R.; Ulamek-Koziol, M.; Januszewski, S.; Czuczwar, S.J. Gut microbiota and pro/prebiotics in Alzheimer’s disease. Aging (Albany NY) 2020, 12, 5539–5550. [Google Scholar] [CrossRef]
- Yu, L.W.; Agirman, G.; Hsiao, E.Y. The Gut Microbiome as a Regulator of the Neuroimmune Landscape. Annu Rev Immunol 2022, 40, 143–167. [Google Scholar] [CrossRef]
- Zhang, T.; Gao, G.; Kwok, L.Y.; Sun, Z. Gut microbiome-targeted therapies for Alzheimer’s disease. Gut Microbes 2023, 15, 2271613. [Google Scholar] [CrossRef]
- Dou, L.; Peng, Y.; Zhang, B.; Yang, H.; Zheng, K. Immune Remodeling during Aging and the Clinical Significance of Immunonutrition in Healthy Aging. Aging Dis 2024, 15, 1588–1601. [Google Scholar] [CrossRef]
- Silva, Y.P.; Bernardi, A.; Frozza, R.L. The Role of Short-Chain Fatty Acids From Gut Microbiota in Gut-Brain Communication. Front Endocrinol (Lausanne) 2020, 11, 25. [Google Scholar] [CrossRef]
- Nourazarain, A.; Vaziri, Y. Nutrigenomics meets multi-omics: integrating genetic, metabolic, and microbiome data for personalized nutrition strategies. Genes Nutr 2025, 20, 30. [Google Scholar] [CrossRef]
- Dolan, M.; Libby, K.A.; Ringel, A.E.; van Galen, P.; McAllister, S.S. Ageing, immune fitness and cancer. Nature Reviews Cancer 2025, 25, 848–872. [Google Scholar] [CrossRef]
- McGee, K.C.; Sullivan, J.; Hazeldine, J.; Schmunk, L.J.; Martin-Herranz, D.E.; Jackson, T.; Lord, J.M. A combination nutritional supplement reduces DNA methylation age only in older adults with a raised epigenetic age. Geroscience 2024, 46, 4333–4347. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.D.; Nagvekar, R.; Pogson, A.N.; Brunet, A. Brain aging and rejuvenation at single-cell resolution. Neuron 2025, 113, 82–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fan, D.; Yang, Y.; Gimple, R.C.; Zhou, S. Integrative multi-omics approaches to explore immune cell functions: Challenges and opportunities. iScience 2023, 26, 106359. [Google Scholar] [CrossRef] [PubMed]
- Riemann, L.; Gutierrez, R.; Odak, I.; Barros-Martins, J.; Roesner, L.M.; Leon Lara, X.; Falk, C.; Schulz, T.F.; Hansen, G.; Werfel, T.; et al. Integrative deep immune profiling of the elderly reveals systems-level signatures of aging, sex, smoking, and clinical traits. EBioMedicine 2025, 112, 105558. [Google Scholar] [CrossRef] [PubMed]
- Cisneros, B.; Garcia-Aguirre, I.; Unzueta, J.; Arrieta-Cruz, I.; Gonzalez-Morales, O.; Dominguez-Larrieta, J.M.; Tamez-Gonzalez, A.; Leyva-Gomez, G.; Magana, J.J. Immune system modulation in aging: Molecular mechanisms and therapeutic targets. Front Immunol 2022, 13, 1059173. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hallmarks of immune dysfunction in aging. Immune aging is characterized by coordinated yet heterogeneous alterations across innate (left) and adaptive (right) immune compartments. Rather than a uniform decline, aging drives extensive remodeling of immune cell composition, phenotype, and function, resulting in reduced immune resilience and dysregulated inflammatory responses. These changes promote impaired resolution and defective repair, establishing self-sustaining inflammatory loops that contribute to chronic inflammation and age-related pathology. Abbreviations: BM: bone marrow; TC: T cell; mTC: memory T cell; eTC: effector T cell; sTC: senescent T cell; BC: B cell; mBC: memory BC; aC: apoptotic cell; Mf: macrophages; Nf: neutrophil; NET: neutrophil extracellular trap; Treg: regulatory T cell; DC: dendritic cell; AG: antigen; aAb: autoreactive antibody; CSR: class-switch recombination; SHM: somatic hypermutation.
Figure 1.
Hallmarks of immune dysfunction in aging. Immune aging is characterized by coordinated yet heterogeneous alterations across innate (left) and adaptive (right) immune compartments. Rather than a uniform decline, aging drives extensive remodeling of immune cell composition, phenotype, and function, resulting in reduced immune resilience and dysregulated inflammatory responses. These changes promote impaired resolution and defective repair, establishing self-sustaining inflammatory loops that contribute to chronic inflammation and age-related pathology. Abbreviations: BM: bone marrow; TC: T cell; mTC: memory T cell; eTC: effector T cell; sTC: senescent T cell; BC: B cell; mBC: memory BC; aC: apoptotic cell; Mf: macrophages; Nf: neutrophil; NET: neutrophil extracellular trap; Treg: regulatory T cell; DC: dendritic cell; AG: antigen; aAb: autoreactive antibody; CSR: class-switch recombination; SHM: somatic hypermutation.
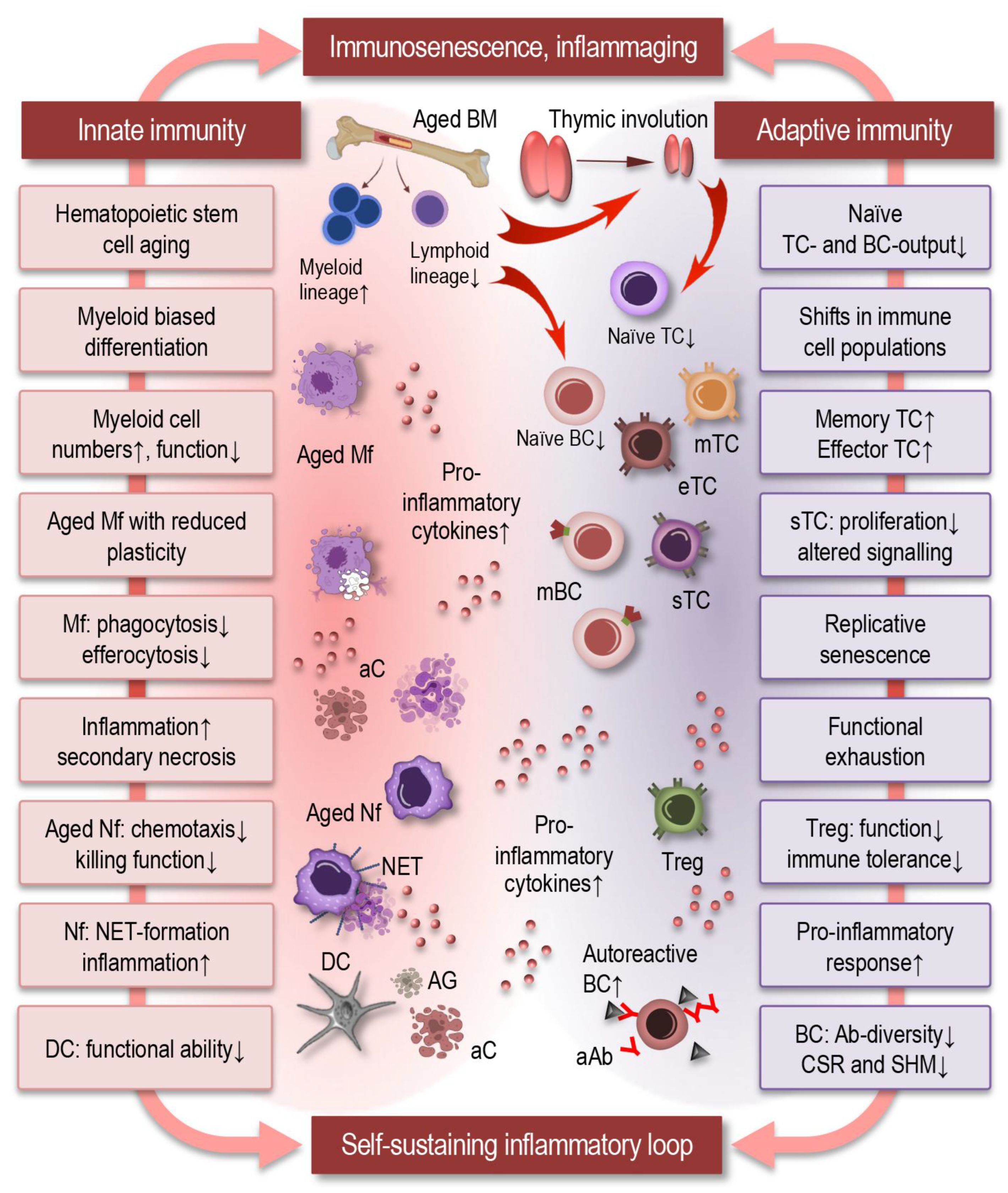
Figure 2.
Inflammaging: sources and feedback loops. Inflammaging is a chronic, low-grade, age-associated inflammatory state driven by interconnected sources (A), amplifiers (B), and self-perpetuating feedback loops (C) that disrupt immune homeostasis. This persistent imbalance promotes the development of multiple ARDs (D), including cardiovascular, metabolic, and neurodegenerative disorders. Abbreviations: SASP: senescence-associated secretory phenotype; IL: interleukin; TNF: tumor necrosis factor; DAMP: danger-associated molecular pattern; SPM: pro-resolving lipid mediators; CVD: cardiovascular disease.
Figure 2.
Inflammaging: sources and feedback loops. Inflammaging is a chronic, low-grade, age-associated inflammatory state driven by interconnected sources (A), amplifiers (B), and self-perpetuating feedback loops (C) that disrupt immune homeostasis. This persistent imbalance promotes the development of multiple ARDs (D), including cardiovascular, metabolic, and neurodegenerative disorders. Abbreviations: SASP: senescence-associated secretory phenotype; IL: interleukin; TNF: tumor necrosis factor; DAMP: danger-associated molecular pattern; SPM: pro-resolving lipid mediators; CVD: cardiovascular disease.
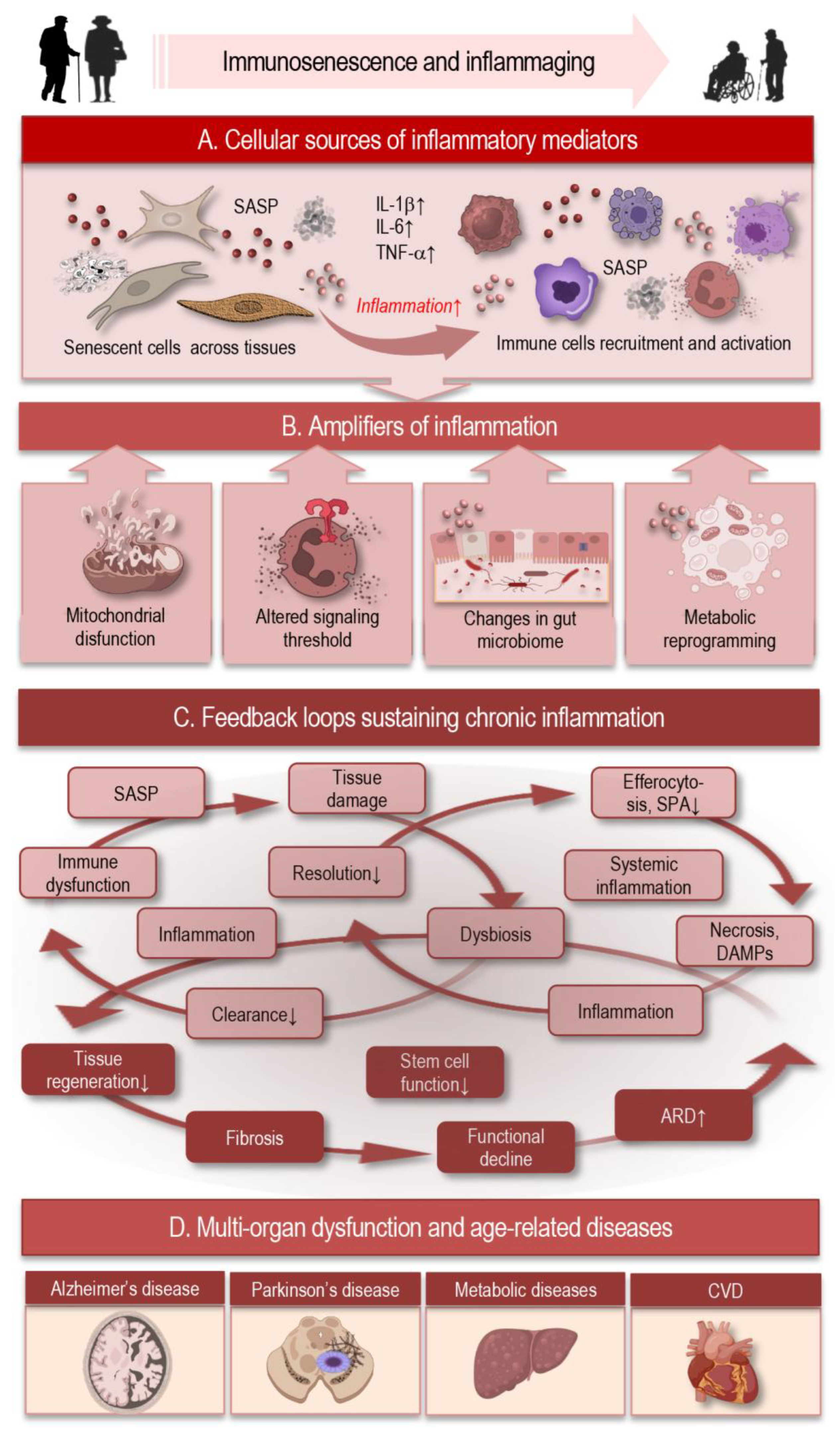
Figure 3.
Inflammation and its impaired resolution during aging. The termination of inflammation is an active and tightly regulated biological process that is essential for restoring tissue homeostasis following immune activation. Inflammation is initiated by neutrophil (Nf) and monocyte (Mo) recruitment and inflammatory cytokine (IC) release, followed by neutrophil apoptosis or necrosis. Effective resolution (right) is driven by specialized pro-resolving mediators (SPM), which limit further neutrophil infiltration, promote monocyte (Mo) recruitment, and induce macrophage (Mf) efferocytosis and reprogramming toward reparative (M2) phenotypes, enabling tissue repair, remodeling, and restoration of homeostasis, including support of stem cell (SC)–mediated regeneration. In aging (left), macrophages display reduced phagocytic capacity, leading to impaired efferocytosis. Because efferocytosis promotes SPM production and suppresses inflammatory cytokines, its impairment creates a feed-forward loop that perpetuates inflammation. This results in defective resolution, persistent immune cell recruitment, fibrosis, accumulation of necrotic debris, and sustained low-grade inflammation, collectively contributing to inflammaging and tissue dysfunction.
Figure 3.
Inflammation and its impaired resolution during aging. The termination of inflammation is an active and tightly regulated biological process that is essential for restoring tissue homeostasis following immune activation. Inflammation is initiated by neutrophil (Nf) and monocyte (Mo) recruitment and inflammatory cytokine (IC) release, followed by neutrophil apoptosis or necrosis. Effective resolution (right) is driven by specialized pro-resolving mediators (SPM), which limit further neutrophil infiltration, promote monocyte (Mo) recruitment, and induce macrophage (Mf) efferocytosis and reprogramming toward reparative (M2) phenotypes, enabling tissue repair, remodeling, and restoration of homeostasis, including support of stem cell (SC)–mediated regeneration. In aging (left), macrophages display reduced phagocytic capacity, leading to impaired efferocytosis. Because efferocytosis promotes SPM production and suppresses inflammatory cytokines, its impairment creates a feed-forward loop that perpetuates inflammation. This results in defective resolution, persistent immune cell recruitment, fibrosis, accumulation of necrotic debris, and sustained low-grade inflammation, collectively contributing to inflammaging and tissue dysfunction.
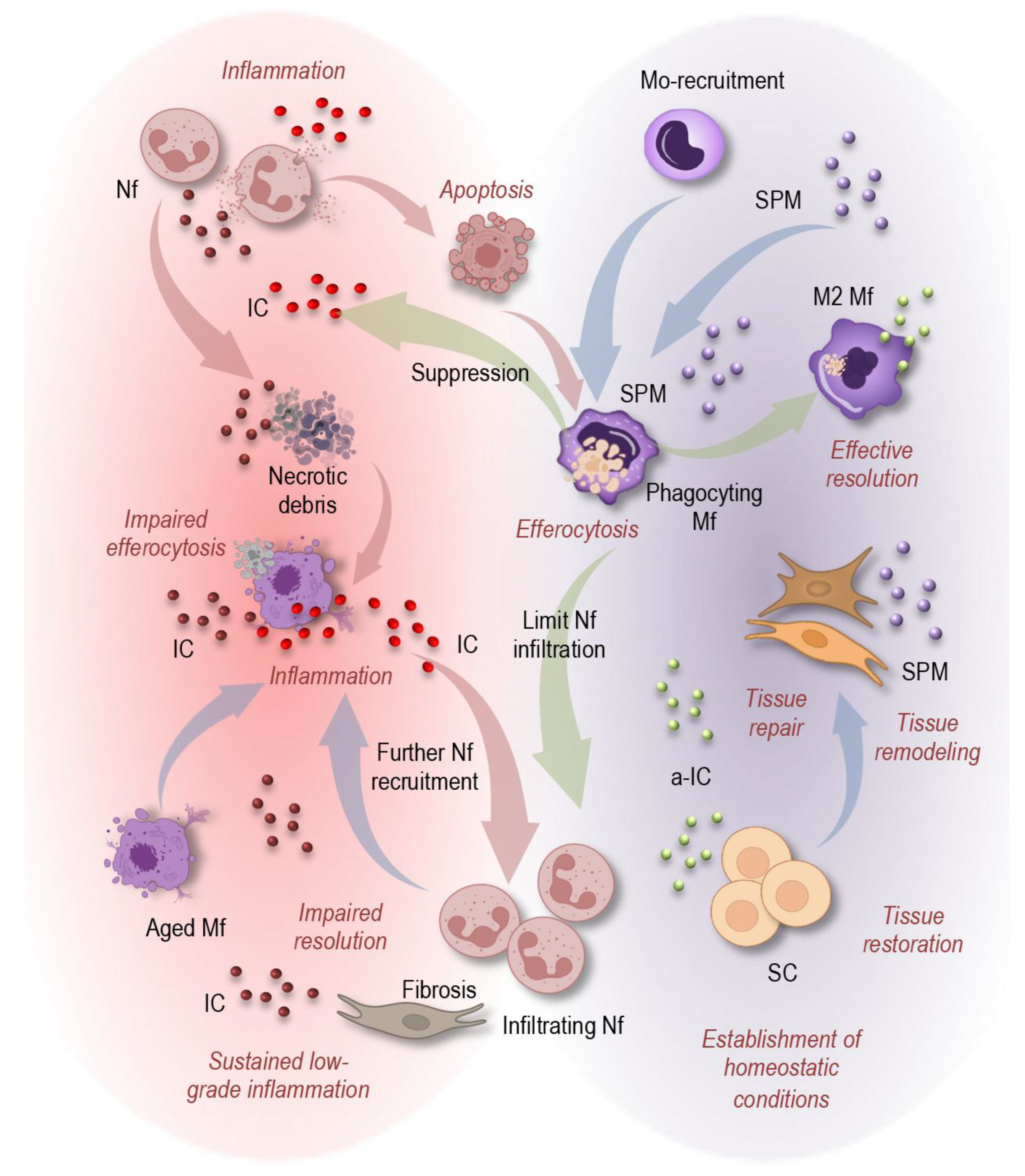
Figure 4.
Immune aging compromises immune surveillance and promotes a tumor-permissive environment, leading to immune tolerance of malignant cells and accelerating oncogenesis. Abbreviations: TC: T cell; mTC: memory T cell; CD: cluster of differentiation; sTC: senescent T cell; Mf: macrophages; NK: natural killer cells; Treg: regulatory T cell; DC: dendritic cell; AG: antigen; DNA: deoxyribonucleic acid.
Figure 4.
Immune aging compromises immune surveillance and promotes a tumor-permissive environment, leading to immune tolerance of malignant cells and accelerating oncogenesis. Abbreviations: TC: T cell; mTC: memory T cell; CD: cluster of differentiation; sTC: senescent T cell; Mf: macrophages; NK: natural killer cells; Treg: regulatory T cell; DC: dendritic cell; AG: antigen; DNA: deoxyribonucleic acid.
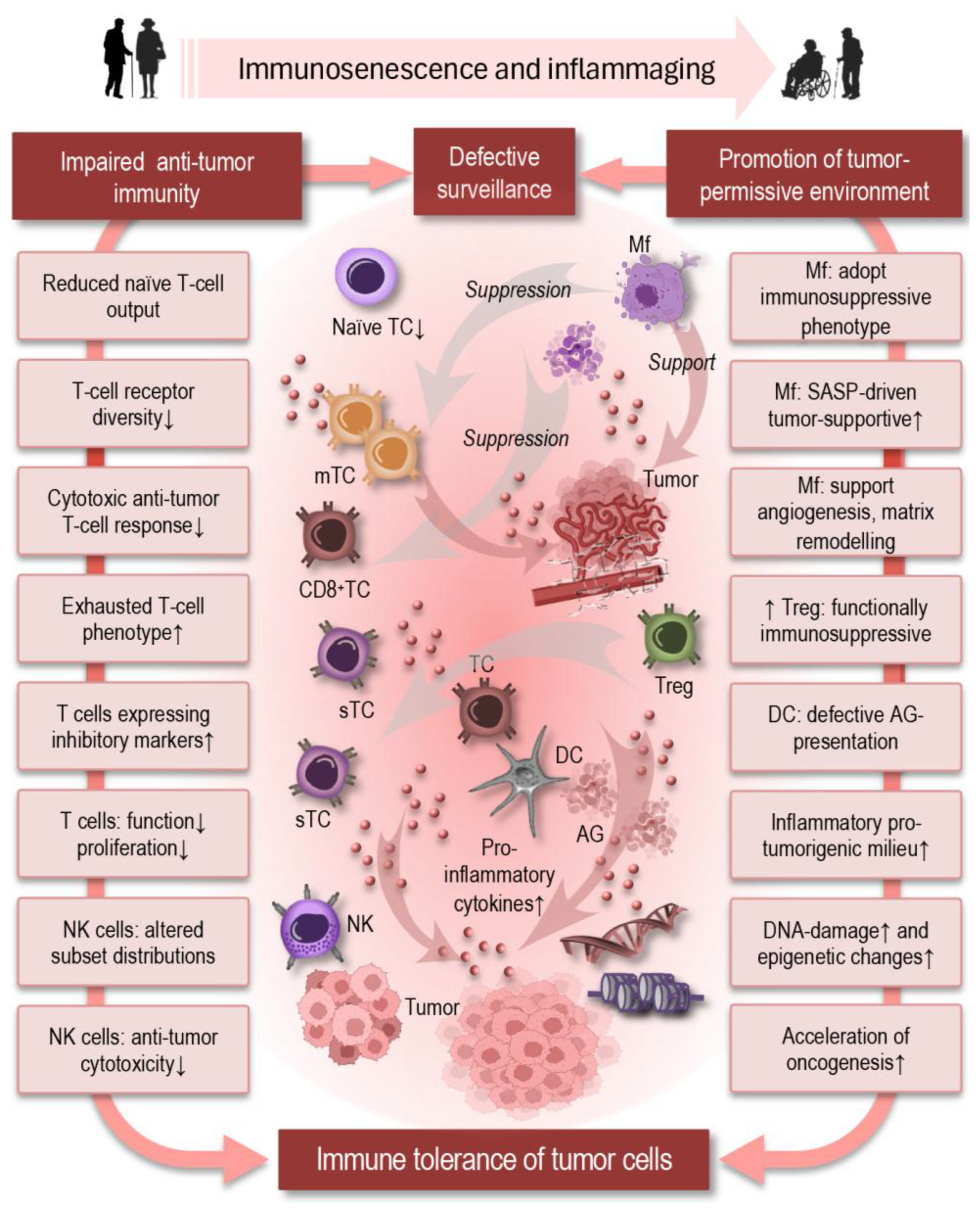
Figure 5.
Aging profoundly alters central and peripheral immune responses, affecting brain homeostasis and resilience. Key clearance mechanisms decline, as microglial phagocytic capacity, lysosomal function, and metabolic efficiency decrease, impairing the removal of protein aggregates (amyloid-β, α-synuclein), apoptotic neurons, and synaptic debris. This amplifies neuroinflammation and promotes accumulation of toxic proteins that drive Alzheimer’s and Parkinson’s disease progression. Together, these changes create a self-reinforcing loop in which immune aging sustains chronic neuroinflammation, impaired clearance, and neuronal damage, while ongoing neurodegeneration generates additional danger signals that perpetuate immune activation. Abbreviations: MG: microglia; aMG: activated microglia; Am-β: amyloid-β; dN: degenerating neuron; IL: interleukin; TNF: tumor necrosis factor; α-Sy: α-synuclein; Mo: monocyte; M1: M1 macrophage; dBBB: disrupted blood-brain barrier; BV: blood vessel; pIC: peripheral immune cell.
Figure 5.
Aging profoundly alters central and peripheral immune responses, affecting brain homeostasis and resilience. Key clearance mechanisms decline, as microglial phagocytic capacity, lysosomal function, and metabolic efficiency decrease, impairing the removal of protein aggregates (amyloid-β, α-synuclein), apoptotic neurons, and synaptic debris. This amplifies neuroinflammation and promotes accumulation of toxic proteins that drive Alzheimer’s and Parkinson’s disease progression. Together, these changes create a self-reinforcing loop in which immune aging sustains chronic neuroinflammation, impaired clearance, and neuronal damage, while ongoing neurodegeneration generates additional danger signals that perpetuate immune activation. Abbreviations: MG: microglia; aMG: activated microglia; Am-β: amyloid-β; dN: degenerating neuron; IL: interleukin; TNF: tumor necrosis factor; α-Sy: α-synuclein; Mo: monocyte; M1: M1 macrophage; dBBB: disrupted blood-brain barrier; BV: blood vessel; pIC: peripheral immune cell.
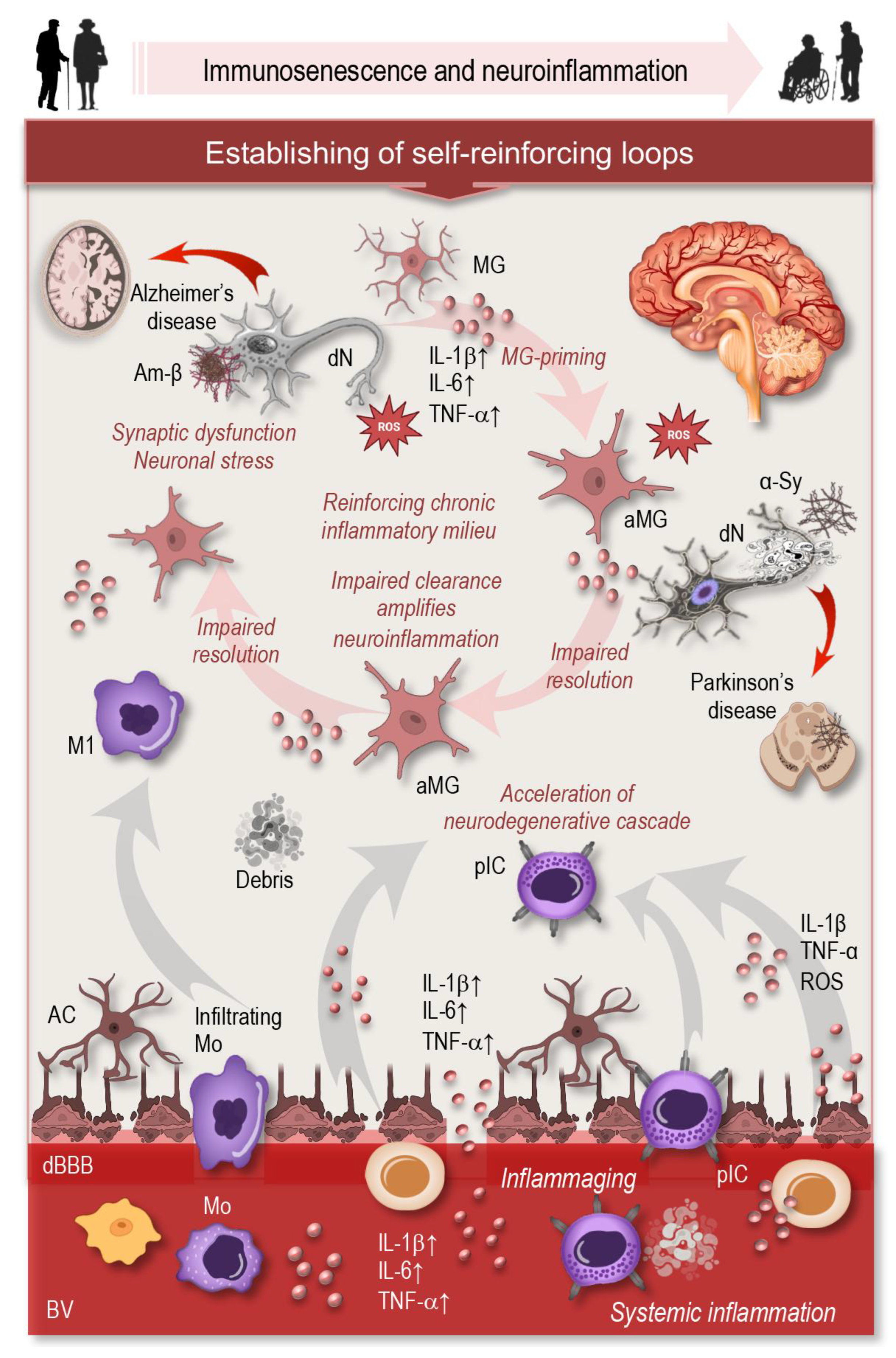
Figure 6.
Immune aging drives cardiovascular and metabolic dysfunction through chronic inflammation, innate and adaptive immune dysregulation, and maladaptive tissue responses. Inflammaging-associated oxidative stress, mitochondrial dysfunction, and age-related gut dysbiosis amplify metabolic impairment and cardiometabolic risk. These processes form a self-reinforcing network, in which chronic inflammation promotes tissue dysfunction and metabolic derangement, while tissue stress and altered metabolic signals sustain immune activation. Abbreviations: IC: immune cell; IL: interleukin; TNF: tumor necrosis factor; CRP: C-reactive protein; mTC: memory T cell; sTC: senescent T cell; Mo: monocyte; aAB: autoreactive antibody; Mf: macrophages; Mth: mithochondria; LPS: lipopolysaccharide.
Figure 6.
Immune aging drives cardiovascular and metabolic dysfunction through chronic inflammation, innate and adaptive immune dysregulation, and maladaptive tissue responses. Inflammaging-associated oxidative stress, mitochondrial dysfunction, and age-related gut dysbiosis amplify metabolic impairment and cardiometabolic risk. These processes form a self-reinforcing network, in which chronic inflammation promotes tissue dysfunction and metabolic derangement, while tissue stress and altered metabolic signals sustain immune activation. Abbreviations: IC: immune cell; IL: interleukin; TNF: tumor necrosis factor; CRP: C-reactive protein; mTC: memory T cell; sTC: senescent T cell; Mo: monocyte; aAB: autoreactive antibody; Mf: macrophages; Mth: mithochondria; LPS: lipopolysaccharide.
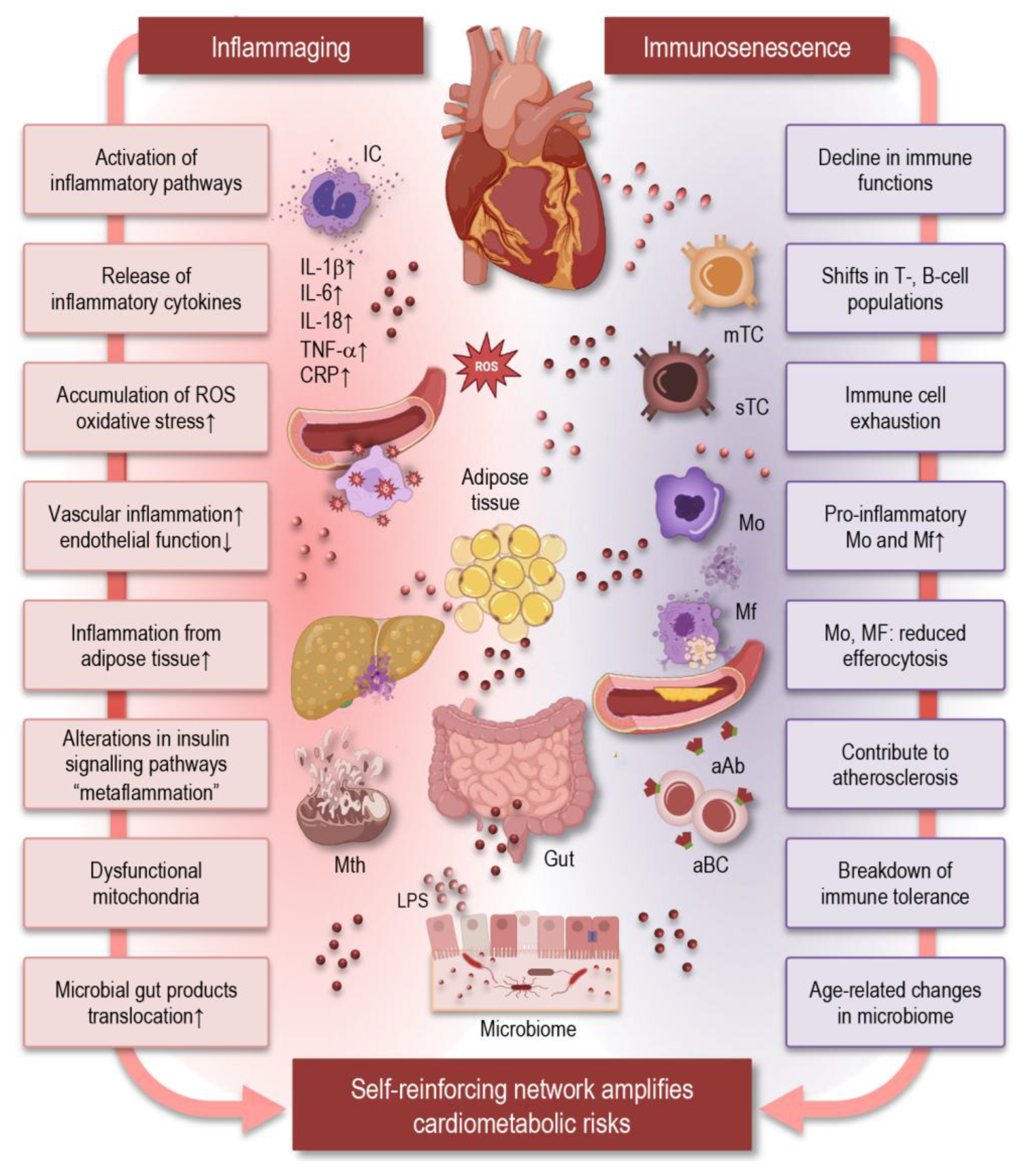
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



