
Submitted:
29 December 2025
Posted:
29 December 2025
You are already at the latest version
Abstract
Chimeric antigen receptor (CAR) immunotherapy has made significant strides, particularly in hematological malignancies, with five CAR T therapies gaining FDA approval. However, its application in solid tumors remains challenging due to issues like poor CAR T cell trafficking, an immunosuppressive tumor microenvironment (TME), and therapy-related toxicity. CAR Natural Killer (NK) cells present an alternative with advantages such as multiple mechanisms for targeting cancer and reduced side effects. Additionally, macrophages, which naturally infiltrate tumors, are under investigation for CAR therapy to overcome the limitations seen in CAR T and CAR NK approaches. In this review, our primary focus will be on solid tumors, as they present unique challenges in CAR T cell therapy, such as poor trafficking and the immunosuppressive tumor microenvironment. However, we will also address some hematological malignancies, particularly those with poor prognoses, where similar issues of CAR T cell dysfunction and lack of persistence are observed. By covering both solid tumors and certain blood cancers, we aim to provide a comprehensive understanding of the barriers and potential strategies for improving CAR T cell therapies across different malignancies.
Keywords:
CAR-T cell therapy
; solid tumors
; γδ CAR-T cells
; immunosuppressive tumor microenvironment
; NK CAR cells
; precision medicine
1. Introduction
CAR-T cell therapy has revolutionized cancer treatment by harnessing the immune system to target and destroy cancer cells. Initially focused on hematologic malignancies like B-cell acute lymphocytic leukemia (B-ALL), non-Hodgkin lymphoma (NHL), and multiple myeloma (MM), this approach has shown remarkable results [1,2,3]. Six CAR-T cell products have received FDA approval for treating relapsed/refractory B-cell malignancies, including Tisagenlecleucel (Kymriah), Axicabtagene ciloleucel (Yescarta), and Ciltacabtagene autoleucel (Carvykti), among others [4,5,6,7,8,9,10,11,12,13,14,15,16].
Future CAR-T therapy developments aim to enhance efficacy through multi-targeting. By designing CAR-T cells that recognize multiple antigens, researchers hope to overcome tumor heterogeneity and reduce relapse risks, leading to more durable responses [17,18]. Additionally, efforts to improve CAR-T cell persistence and longevity are underway, focusing on strategies such as incorporating co-stimulatory domains and cytokine modulation to prolong anti-tumor activity [19].
Expanding CAR-T therapy to solid tumors presents challenges like the immunosuppressive tumor microenvironment and tumor heterogeneity. However, novel CAR constructs with enhanced targeting capabilities, along with combinatorial approaches using checkpoint inhibitors, are being explored to overcome these obstacles [20,21].
Addressing safety concerns, including cytokine release syndrome (CRS) and neurotoxicity, remains a critical focus. Future research will refine protocols, optimize dosing, and develop predictive biomarkers to minimize toxicities while maintaining efficacy [20,21].
CD19 is a vital target in B-cell malignancies, with several FDA-approved CAR-T therapies targeting this antigen [22,23,24]. However, CD19 antigen loss has prompted the exploration of alternative targets like CD20 and CD22, showing promising results in preventing antigen escape and improving outcomes [25,26,27,28,29,30].
2. Preparation of CAR-T Cells
CAR-T cell therapy involves several key steps. T cells are first collected from the patient’s peripheral blood or from healthy donors [1,31]. These T cells are then genetically engineered to express CARs, which include an extracellular domain (scFv) for antigen recognition, a transmembrane domain for anchoring, and an intracellular signaling domain to activate T cells upon antigen detection [32,33]. CAR-T cells can recognize tumor antigens independently of MHC presentation, allowing them to target cancer cells directly [34]. CAR-T cells have evolved through several generations to enhance their effectiveness: first-generation CARs have limited capacity due to the lack of co-stimulatory signals, second-generation CARs incorporate co-stimulatory domains to improve proliferation and cytokine release, and third-generation CARs combine multiple co-stimulatory molecules to further boost T cell activity [35,36]. The modified T cells undergo extensive in vitro expansion before being re-infused into the patient following lymphodepletion chemotherapy [1,37,38]. Once in the patient, CAR-T cells recognize specific tumor antigens and rapidly proliferate to mount an anti-tumor response [4].
3. Generations of CAR-T Cells
CAR-T cells have evolved through multiple generations to enhance their efficacy in cancer treatment. First-generation CARs consist of a single-chain variable fragment (scFv) linked to the CD3ζ intracellular signaling domain, providing the essential signal for T cell activation. However, these CARs lack costimulatory molecules, resulting in reduced activation and antitumor efficacy in vivo [39]. Second-generation CARs improve upon this by incorporating costimulatory domains such as CD28, 4-1BB, OX-40, ICOS, and CD134, which prevent T cell exhaustion, promote sustained proliferation, and enhance cytokine secretion, significantly boosting their effectiveness in targeting and eliminating cancer cells [40,41,42]. Third-generation CARs further enhance T cell survival and cytokine production by incorporating multiple costimulatory domains, though they carry risks of off-target effects and excessive cytokine production, with limited clinical data available [43,44]. Fourth-generation CARs, derived from second-generation designs, add domains that regulate cytokine expression, such as IL-12, IL-18, or IL-21, improving CAR-T cell cytotoxicity, proliferation, and memory T cell differentiation, leading to more effective and sustained antitumor responses in preclinical models [45,46,47].
4. Toxicities in CAR-T Cell Therapy
CAR-T cell therapy can lead to several toxicities. Cytokine release syndrome (CRS) is a common and potentially severe complication, driven by the release of pro-inflammatory cytokines like IL-6 [48,49,50]. Management of CRS includes the use of tocilizumab and corticosteroids, particularly in severe cases [51,52,53]. CAR-T cell therapy may also cause T and B lymphocyte aplasia, increasing the risk of infections, which are particularly prevalent within the first 30 days post-infusion [1,55,56]. Prophylactic measures, including immunoglobulin supplementation and antiviral drugs, are critical to managing these risks [57,62]. CRS-related coagulopathy, characterized by abnormal coagulation and hyperfibrinolysis, is another complication that requires early management to mitigate risks like disseminated intravascular coagulation [63,64,65]. Cytopenias, including anemia and thrombocytopenia, can persist for months after CAR-T therapy and are often managed with transfusions and growth factors [4,66,67]. Antigen mutation and loss pose challenges in treatment efficacy, with innovative strategies like dual-targeting CARs and γδ T cells being explored to address these issues [35,68,69,70,71,72,73,74,75,76]. Neurotoxicity, including encephalopathy and seizures, is another significant concern, often linked to CRS, and requires strategies for prediction and management [80,81,82,83,84]. Graft-versus-host disease (GVHD), particularly in allogeneic CAR-T therapy, remains a critical area of ongoing research to improve the safety profile of CAR-T treatments [81,85].
5. Management of Toxicities in CAR-T Cell Therapy
5.1. Management of Cytokine-Release Syndrome (CRS)
CRS management involves multiple strategies to control symptoms and prevent complications. Tocilizumab, an IL-6 receptor antagonist, is the primary treatment [36]. Corticosteroids are added for severe cases to reduce inflammation [88]. Anakinra, an IL-1 receptor antagonist, shows promise for managing both CRS and CRES [88]. GM-CSF inhibition may alleviate CRS and enhance CAR-T efficacy [89]. Reducing tumor burden with chemotherapy or radiotherapy before CAR-T infusion can decrease CRS severity [36]. Understanding the mechanisms of CRS, including CAR-T activation and cytokine release, aids in developing targeted therapies [87].
5.2. Management of CRS-Related Coagulopathy:
Coagulopathy in CRS requires regular monitoring of coagulation parameters like PT, aPTT, fibrinogen, and D-dimer levels [59]. Risk stratification helps identify patients at higher risk for coagulopathy, tailoring monitoring and treatment [81]. Individualized treatment, including anticoagulants, replacement therapy, and supportive care, is crucial [90]. Managing underlying conditions, such as infections, and multidisciplinary collaboration are essential for optimal care [82,91].
5.3. Management of T and B Lymphocytes Aplasia and Infections
Immunoglobulin supplementation is vital to restore humoral immunity after B cell depletion [92,93,94]. Infection monitoring and prophylaxis, especially for herpesvirus, are necessary [81]. Differentiating CRS from infections is critical, with IL-6 levels helping in the diagnosis [81]. Antiviral prophylaxis prevents HBV reactivation in patients with resolved infections [81]. Dose-escalation regimens may reduce infection risk during high-dose CAR-T therapy [86].
5.4. Management of Cytopenias
Cytopenias post-CAR-T therapy are managed with cytokine support (e.g., IL-6, G-CSF) [88], blood transfusions [95], and growth factors like G-CSF and erythropoietin [96]. Supportive care, including hydration and nutrition, is essential for recovery [97]. Adjusting CAR-T dose and conditioning regimens may help reduce cytopenia severity [98].
5.5. Management of GVHD
GVHD is managed with immunosuppressive medications like corticosteroids and calcineurin inhibitors [99,100,101,102]. Anti-thymocyte globulin (ATG) may be used in conditioning regimens to prevent GVHD [99]. T-cell depletion techniques and photopheresis are also used to manage the condition [100]. Supportive care, including nutrition and infection prevention, is crucial.
6. CAR-T Cells and Targeted Antigens in Cancer Treatments: Importance, Future Insights, Strengths, and Weaknesses
Chimeric antigen receptor (CAR) T-cell therapy has revolutionized cancer treatment, particularly for hematologic malignancies. By engineering T cells to express receptors that specifically target antigens on cancer cells, CAR-T therapy enables the immune system to recognize and eliminate cancerous cells. The success of CAR-T cells largely depends on selecting the appropriate target antigens, which vary across different types of cancer. This study explores the importance of targeted antigens in CAR-T therapy, future insights, and the strengths and weaknesses of this approach.
6.1. Importance of Targeted Antigens
Target antigens are crucial in determining the specificity and efficacy of CAR-T cells. These antigens are typically proteins expressed on the surface of cancer cells but not on normal cells, minimizing the risk of off-target effects. For instance, CD19 is a well-established target in B-cell malignancies like acute lymphoblastic leukemia (ALL) and chronic lymphocytic leukemia (CLL). Targeting CD19 has resulted in high response rates and durable remissions in these diseases [101]. Similarly, B-cell maturation antigen (BCMA) is targeted in multiple myeloma, demonstrating significant efficacy [102]. The choice of antigen directly impacts the success of the therapy, as it determines the ability of CAR-T cells to recognize and destroy cancer cells while sparing healthy tissues.
6.2. Future Insights
The future of CAR-T therapy lies in expanding its application beyond hematologic malignancies to solid tumors, which present unique challenges. Solid tumors often have a heterogeneous expression of antigens and an immunosuppressive microenvironment that hinders CAR-T cell efficacy. Researchers are exploring the development of CAR-T cells targeting multiple antigens simultaneously to overcome tumor heterogeneity and reduce the likelihood of antigen escape. Another promising area is the engineering of CAR-T cells with enhanced persistence and resistance to the immunosuppressive tumor microenvironment. Additionally, there is growing interest in using allogeneic CAR-T cells derived from healthy donors, which could make the therapy more accessible and reduce costs [103].
6.3. Strengths of CAR-T Therapy
One of the primary strengths of CAR-T therapy is its ability to provide personalized and highly specific treatment. By targeting antigens unique to cancer cells, CAR-T cells can effectively eliminate malignant cells with minimal impact on normal tissues. The durability of the responses seen in hematologic malignancies, particularly in cases where patients have failed other therapies, highlights the potential of CAR-T therapy to achieve long-term remissions. Furthermore, advancements in genetic engineering and cell manufacturing processes continue to improve the safety and efficacy of CAR-T products. Table 1 CAR-T cells and the targeted antigens for different types of cancer treatments [104,105].
6.4. Weaknesses of CAR-T Therapy
Despite its success, CAR-T therapy has limitations. One significant challenge is the management of toxicities, such as cytokine release syndrome (CRS) and neurotoxicity, which can be life-threatening. The selection of appropriate antigens is also critical, as targeting antigens that are not entirely specific to cancer cells can lead to off-target effects and damage to healthy tissues. Another weakness is the limited effectiveness of CAR-T therapy in solid tumors due to factors like antigen heterogeneity, physical barriers within the tumor microenvironment, and immune evasion mechanisms. Moreover, the high cost and complex manufacturing process of CAR-T cells limit their accessibility to patients.
This table highlights some of the key CAR-T cell therapies, their targeted antigens, the cancer types they treat, and any relevant notes regarding their approval status or investigational stage.
6.5. bb21217 CAR-T Cell Therapy
Bb21217 CAR-T cell therapy represents an innovative treatment for multiple myeloma, targeting the B-cell maturation antigen (BCMA) on myeloma cells. This therapy involves engineering T cells to express a chimeric antigen receptor (CAR) that binds to BCMA, enabling the targeted destruction of cancerous cells. A key feature of bb21217 is the gene-editing step that enhances the CAR-T cells with a memory-like phenotype, improving their persistence and long-term effectiveness. Early clinical trials have shown promising outcomes, with significant response rates in patients with relapsed or refractory multiple myeloma. However, further studies are needed to fully assess the long-term efficacy, persistence of the memory-like CAR-T cells, and safety profile, particularly concerning side effects like cytokine release syndrome and neurotoxicity. Bb21217 offers hope for patients with limited treatment options, but ongoing research is crucial for its broader clinical application [106].
6.6. CART-PSMA Therapy
CART-PSMA (Chimeric Antigen Receptor T cells targeting Prostate-Specific Membrane Antigen) represents a promising and innovative therapy for advanced or treatment-resistant prostate cancer. This approach leverages the patient’s immune system by engineering T cells to express a chimeric antigen receptor (CAR) that targets PSMA, a protein highly expressed on prostate cancer cells. Early clinical trials have shown significant tumor regression, with some patients achieving partial or complete remission. CART-PSMA offers a favorable side effect profile compared to traditional therapies, though challenges remain, particularly in managing treatment-related toxicities such as cytokine release syndrome (CRS) and neurotoxicity. Additionally, the long-term effectiveness and potential for tumor escape mechanisms are still under investigation. Despite these challenges, CART-PSMA therapy provides hope for patients with advanced prostate cancer and demonstrates the potential of CAR-T cell therapies in treating solid tumors, which have been more difficult to address with this approach [107].
Ongoing and future clinical trials are crucial in several areas to optimize CART-PSMA therapy, improve its safety and efficacy, and explore its potential in combination with other therapies. Here are some key focus areas for these trials:
1. Safety and Toxicity Management: Current and future trials are focused on developing strategies to manage and mitigate the toxicities associated with CART-PSMA therapy, such as cytokine release syndrome (CRS) and neurotoxicity. These studies are investigating different dosing regimens, preconditioning treatments, and the use of adjunct therapies to reduce the incidence and severity of these side effects [108].
2. Enhancing Durability of Response: Trials are also exploring methods to enhance the durability of the response to CART-PSMA therapy. This includes research into the persistence of CAR-T cells in the body, and strategies to prevent tumor escape mechanisms, where cancer cells lose or alter PSMA expression to evade detection by CAR-T cells. Modifying the CAR-T cells to have memory-like properties or combining them with checkpoint inhibitors may improve long-term outcomes [109].
3. Combination Therapies: Combining CART-PSMA with other therapies is a major area of research. Trials are investigating the combination of CART-PSMA with immune checkpoint inhibitors (like PD-1/PD-L1 inhibitors) to enhance the anti-tumor response and overcome the immunosuppressive tumor microenvironment. Other combinations being explored include CART-PSMA with traditional therapies such as chemotherapy, radiation, and androgen deprivation therapy (ADT), as well as novel agents targeting other aspects of tumor biology [110].
4. Targeting Tumor Heterogeneity: Another focus is on addressing tumor heterogeneity, where different populations of cancer cells within the same tumor express varying levels of PSMA or other antigens. Trials are exploring the use of multi-targeted CAR-T cells that can recognize and attack multiple tumor antigens simultaneously, reducing the likelihood of tumor escape and relapse [111].
These ongoing and future clinical trials will be critical in refining CART-PSMA therapy, making it safer, more effective, and potentially applicable to a broader range of cancers. The outcomes of these studies will help determine the optimal use of CART-PSMA in clinical practice and may lead to new standards of care for patients with advanced prostate cancer and other PSMA-expressing tumors.
6.7. CAR-EGFRvIII Therapy
CAR-EGFRvIII is a promising CAR-T therapy targeting the epidermal growth factor receptor variant III (EGFRvIII), a mutation commonly found in glioblastoma, an aggressive brain tumor. EGFRvIII is selectively expressed on tumor cells, reducing the risk of off-target effects. By genetically engineering T cells to recognize and attack EGFRvIII-expressing cells, CAR-EGFRvIII aims to destroy the tumor. Early clinical studies have shown the therapy’s ability to infiltrate brain tumors, generating optimism for treating glioblastoma. However, its efficacy has been limited due to tumor heterogeneity and adaptive resistance, where not all tumor cells express EGFRvIII, allowing some to evade destruction. To overcome these challenges, researchers are exploring combination therapies, such as pairing CAR-EGFRvIII with immune checkpoint inhibitors, and developing CAR-T cells that target multiple antigens. Modifying CAR-T cells to resist the tumor microenvironment or incorporating cytokine engineering are also being investigated. Ongoing research aims to enhance CAR-EGFRvIII’s effectiveness, offering hope for glioblastoma treatment [112,113].
Researchers are exploring several combination therapies and strategies to enhance the effectiveness of CAR-EGFRvIII therapy, particularly for challenging cancers like glioblastoma. Some examples include:
1. Checkpoint Inhibitors: Combining CAR-EGFRvIII with immune checkpoint inhibitors, such as PD-1 or CTLA-4 inhibitors, can help overcome the immunosuppressive tumour microenvironment and prevent the tumour from evading the immune response [113].
2. Radiation Therapy: Radiation can upregulate the expression of EGFRvIII and other tumour antigens, potentially increasing the effectiveness of CAR-T cells. It can also alter the tumour microenvironment, making it more conducive to CAR-T cell activity [113].
3. Targeting Multiple Antigens: Developing CAR-T cells that target multiple tumour-specific antigens simultaneously can reduce the likelihood of tumour escape due to antigen loss or heterogeneity. For instance, dual-targeted CAR-T cells could simultaneously target EGFRvIII and another glioblastoma-associated antigen [113].
4. Gene Editing and Cytokine Engineering: Enhancing CAR-T cells with additional genetic modifications, such as the incorporation of cytokine genes (e.g., IL-12) or using CRISPR technology to enhance T cell persistence and reduce exhaustion, is another promising approach. These strategies aim to improve the durability of responses and overcome the adaptive resistance that can occur with CAR-EGFRvIII monotherapy [113].
6.8. MUC1-CAR-T Cell Therapy
MUC1-CAR-T cell therapy is a promising advancement in cancer treatment, targeting the overexpressed and aberrantly glycosylated MUC1 antigen found in cancers such as breast, ovarian, pancreatic, and non-small cell lung cancers (NSCLC). Preclinical studies have shown significant tumor regression in breast cancer models and improved survival in pancreatic cancer models, attributed to the specific targeting of MUC1 with minimal off-target effects. MUC1-CAR-T therapy has also demonstrated efficacy in challenging malignancies like ovarian cancer and NSCLC. However, translating this therapy to clinical practice faces challenges, including the immunosuppressive tumor microenvironment and the potential for tumor antigen escape. Researchers are exploring combination therapies, including checkpoint inhibitors and cytokine engineering, to enhance the effectiveness and persistence of MUC1-CAR-T cells in overcoming these obstacles and providing a new therapeutic option for patients with advanced cancers [114].
MUC1-CAR-T cell therapy is a cutting-edge treatment for epithelial-derived solid tumors, particularly targeting triple-negative breast cancer (TNBC), where the MUC1 antigen is present in 95% of cases. Despite challenges in distinguishing between tumor-associated MUC1 (tMUC1) and its normal counterpart, significant progress has been made. Notably, second-generation MUC28z CAR-T cells, derived from the TAB004 antibody, have shown success in eliminating TNBC cells in preclinical studies. These cells maintain strong tumor-killing abilities even when expressing inhibitory markers like PD1. In animal models, a single dose led to significant tumor reduction, suggesting potential for repeated dosing. MUC28z CAR-T cells also show promise for treating other tMUC1-expressing cancers, such as pancreatic ductal adenocarcinoma, with ongoing research focusing on enhancing their effectiveness [115].
6.9. Mesothelin-CAR-T Cell Therapy
Mesothelin-CAR-T cell therapy is a promising approach for treating mesothelin-expressing cancers, including mesothelioma, pancreatic, and ovarian cancers. Mesothelin is overexpressed in several malignancies, making it an ideal target for CAR-T therapy. Initial studies have shown significant tumor regression in preclinical models and early-phase clinical trials, with promising results in patients with advanced, treatment-resistant cancers. The therapy’s specificity for mesothelin-expressing tumors has resulted in a favorable safety profile. However, challenges like the immunosuppressive tumor microenvironment and the potential for antigen escape remain. Researchers are exploring combination therapies, multi-targeted CAR-T cells, and gene editing to enhance efficacy. Future developments may include “armored” CAR-T cells and off-the-shelf CAR-T products, potentially improving accessibility and scalability. As clinical trials progress, Mesothelin-CAR-T therapy could offer new hope for patients with hard-to-treat cancers [116,117].
6.10. ALLO-501 Therapy
ALLO-501 is a pioneering allogeneic CAR-T cell therapy targeting CD19-expressing malignancies, initially developed for relapsed or refractory non-Hodgkin lymphoma (NHL). Its application is also being explored in other B-cell malignancies like B-cell acute lymphoblastic leukemia (B-ALL) and chronic lymphocytic leukemia (CLL), where CD19 is overexpressed on malignant B cells. ALLO-501’s off-the-shelf availability offers a significant advantage, providing an immediate treatment option for patients, particularly those with rapidly progressing B-ALL or difficult-to-treat CLL relapses. Early clinical trials have shown promising remission rates, leading to further research in both pediatric and adult populations. Additionally, ALLO-501 is being studied in other CD19-positive lymphomas, such as mantle cell lymphoma and follicular lymphoma. Future research is anticipated to explore combination therapies to enhance its efficacy, potentially overcoming resistance mechanisms and expanding its use across various hematologic malignancies [118,119,120].
6.11. ALLO-501A Therapy
The ALPHA2 study explores ALLO-501A, an allogeneic anti-CD19 CAR T cell therapy, in non-HLA matched patients with relapsed/refractory large B-cell lymphoma (LBCL) who have undergone at least two prior treatments. The therapy incorporates Cellectis technologies, including disrupted TCRα and edited CD52 genes, to mitigate graft-versus-host disease (GvHD) and enable selective depletion of host T cells with ALLO-647. Of the 11 patients enrolled, 10 were treated with ALLO-501A following lymphodepletion. The study observed manageable safety, with common adverse events being hematologic, and no GvHD or immune cell-associated neurotoxicity syndrome (ICANS) reported. Two patients achieved complete remission (CR), demonstrating early signs of efficacy. The study continues to enroll for further evaluation of safety, clinical activity, and biomarker data [121].
6.12. JCARH125 Therapy
JCARH125 is a chimeric antigen receptor (CAR) T-cell therapy designed to target B cell maturation antigen (BCMA), which is highly expressed on multiple myeloma cells. This innovative therapy offers new hope for patients with relapsed or refractory multiple myeloma, particularly those who have exhausted other treatment options. The EVOLVE study, a Phase 1/2 clinical trial, was initiated to evaluate the safety, tolerability, and preliminary efficacy of JCARH125 in such patients. Early results were promising, showing significant anti-tumor activity with many patients achieving partial or complete responses, despite their heavily pretreated status. While some side effects like cytokine release syndrome (CRS) and neurotoxicity were observed, they were manageable and did not overshadow the overall positive outcomes. JCARH125’s specificity for BCMA minimizes off-target effects, enhancing its safety and efficacy. The ongoing study focuses on optimizing dosing, managing side effects, and exploring long-term response durability. Researchers are also investigating potential combination therapies to boost efficacy and overcome resistance mechanisms. JCARH125 represents a significant advancement in treating relapsed or refractory multiple myeloma, with continued research poised to refine and expand its use [122,123,124].
6.13. CT053 Therapy
CT053 is an advanced CAR T-cell therapy targeting BCMA for relapsed/refractory multiple myeloma (RRMM). In a Phase 1 study conducted in China with 24 heavily pretreated RRMM patients, CT053 showed a promising overall response rate (ORR) of 87.5%, with 79.2% achieving complete or stringent complete responses (CR/sCR). The median duration of response was 21.8 months, and median progression-free survival (PFS) was 18.8 months, with six-month and 12-month PFS rates of 87% and 60.9%, respectively. Although 13 patients experienced disease progression, nine maintained persistent CR/sCR, indicating potential long-lasting responses. The safety profile was manageable, with the most common severe adverse events being hematological toxicities. Cytokine release syndrome (CRS) occurred in 62.5% of patients, all low-grade and self-limiting. CAR-BCMA T-cell expansion peaked between days 7 and 21 and persisted up to 341 days. CT053 offers significant efficacy, durable responses, and the potential for long-term disease control in heavily pretreated RRMM patients, positioning it as a valuable treatment option [125].
7. Other Cells That Belong to the Arsenal of CAR-T Cells
7.1. CD22 CAR-T Cell Therapy
CD22 CAR-T cells are an innovative approach in cancer immunotherapy, particularly for B-cell malignancies like acute lymphoblastic leukemia (ALL) and non-Hodgkin lymphoma (NHL). These engineered T cells target CD22, a protein on B cells, including those that have lost CD19 expression, which often leads to relapse after CD19-directed CAR-T therapy. By focusing on CD22, this therapy provides an alternative or complementary strategy to maintain remission. Clinical trials have shown significant response rates in relapsed or refractory B-cell malignancies. However, challenges like antigen escape, where cancer cells reduce CD22 expression, remain a concern. Researchers are exploring combination therapies, dual-targeted CAR-T cells (targeting both CD19 and CD22), and optimizing CAR constructs to improve outcomes. CD22 CAR-T cells offer a valuable addition to CAR-T therapy, especially for patients with limited options after CD19-targeted treatments [126,127,128].
7.2. CD20 CAR-T Cell Therapy
CD20 CAR-T cell therapy is an innovative cancer immunotherapy targeting CD20, a protein on B cells found in malignancies like non-Hodgkin lymphoma (NHL) and chronic lymphocytic leukemia (CLL). Traditionally targeted by monoclonal antibodies like rituximab, CD20 is now a focus of CAR-T cell technology, offering a potent and sustained anti-tumor response. CD20 CAR-T cells are engineered by modifying a patient’s T cells to specifically bind to the CD20 antigen on cancer cells, enabling them to locate and destroy these cells. This therapy is particularly beneficial for patients who have relapsed or are refractory to standard treatments, including those resistant to CD19-targeted CAR-T therapies. Clinical trials have shown high response rates, with many patients achieving complete or partial remissions. However, challenges like antigen escape, where cancer cells downregulate CD20, persist. Research is ongoing to enhance efficacy through combination therapies and multi-targeted CAR-T cells. CD20 CAR-T therapy offers hope for improved outcomes in challenging cases [129,130].
7.3. HER2 CAR-T Cell Therapy
HER2 CAR-T cell therapy is a novel immunotherapy targeting the human epidermal growth factor receptor 2 (HER2), overexpressed in cancers like breast, gastric, ovarian, and lung cancers. HER2 is a key oncogenic driver, making it an ideal target for therapies aiming to eradicate tumor cells. In this approach, a patient’s T cells are genetically engineered to express a CAR that specifically binds to HER2 on cancer cells, allowing effective recognition and destruction of these cells. This therapy shows promise, particularly for patients who have relapsed or are refractory to standard treatments, including those unresponsive to HER2-targeted monoclonal antibodies like trastuzumab. Clinical trials have shown significant anti-tumor responses, especially in solid tumors, with some patients achieving partial or complete remissions. However, challenges like on-target, off-tumor toxicity due to HER2 expression in normal tissues pose risks of severe side effects. Research focuses on improving the therapy’s specificity and safety through strategies like dose optimization, local delivery, and combination therapies to minimize these risks while enhancing efficacy. Additionally, ongoing studies are exploring the potential of integrating HER2 CAR-T cells with other emerging immunotherapies, such as checkpoint inhibitors, to further boost their effectiveness. HER2 CAR-T therapy represents a significant advancement in cancer treatment, offering new hope for patients with limited options and paving the way for future innovations in solid tumor immunotherapy [131,132,133]. Figure 1 [133] shows the molecular docking simulation of anti-HER2 mAbs with homology model of human HER2.
7.4. GD2 CAR-T Cell Therapy
GD2 CAR-T cell therapy is a promising approach for treating cancers expressing the disialoganglioside GD2, particularly neuroblastoma, a common pediatric cancer. GD2 is a surface glycolipid found on malignancies such as neuroblastoma, osteosarcoma, and small cell lung cancer, while being minimally present in normal tissues, making it an ideal target for CAR-T therapy. In this treatment, a patient’s T cells are genetically engineered to express a chimeric antigen receptor (CAR) that targets GD2 on cancer cells. Once infused, these modified CAR-T cells attack GD2-expressing tumor cells. Early clinical trials have shown potential in inducing tumor regression, especially in neuroblastoma, where traditional treatments often fail. However, challenges include on-target, off-tumor effects like neuropathic pain and neurotoxicity due to GD2’s presence on peripheral nerves. Researchers are working on strategies to reduce these side effects, such as dose adjustments and combination therapies. Despite these hurdles, GD2 CAR-T therapy represents a significant advancement, offering new hope for patients with GD2-expressing cancers who have limited treatment options [134,135,136].
7.5. GPC3 CAR-T Cell Therapy
GPC3 CAR-T cell therapy is a promising immunotherapy targeting glypican-3 (GPC3), a protein overexpressed in hepatocellular carcinoma (HCC) and other solid tumors, while being absent in most normal adult tissues. This selective expression makes GPC3 an ideal target for CAR-T therapy. In this approach, a patient’s T cells are genetically engineered to express a CAR that specifically recognizes GPC3 on cancer cells. Once reintroduced, these CAR-T cells target and destroy GPC3-expressing tumors with high precision. Preclinical studies have demonstrated significant tumor regression, and early-phase clinical trials in advanced HCC patients have shown promising results, with some patients responding to the therapy after exhausting other options. However, challenges include potential on-target, off-tumor toxicity due to low GPC3 expression in some normal tissues and the immunosuppressive tumor microenvironment in HCC, which may limit CAR-T effectiveness. Ongoing research aims to improve safety and efficacy, explore combination therapies, and enhance CAR-T cell engineering, offering hope for patients with limited treatment options and potential applications in other GPC3-expressing cancers [137,138,139,140].
7.6. ROR1 (Receptor Tyrosine Kinase-like Orphan Receptor 1) CAR-T Cell Therapy
ROR1 CAR-T cell therapy is an emerging cancer immunotherapy targeting the ROR1 protein, an oncofetal antigen expressed during fetal development and aberrantly in several cancers, including chronic lymphocytic leukemia (CLL), mantle cell lymphoma, and solid tumors like breast cancer and lung adenocarcinoma. This therapy involves harvesting and genetically modifying a patient’s T cells to express a CAR specific to ROR1, allowing them to recognize and destroy ROR1-expressing cancer cells. Preclinical studies have shown potent anti-tumor activity, leading to early-phase clinical trials evaluating its safety and efficacy in humans. ROR1 CAR-T therapy’s potential lies in its ability to target a wide range of malignancies due to ROR1’s broad expression across different cancer types. However, challenges such as the immunosuppressive tumor microenvironment, potential resistance mechanisms, and the risk of on-target, off-tumor effects remain. Researchers are exploring strategies to enhance the durability, specificity, and persistence of ROR1 CAR-T cells, aiming to mitigate risks and improve patient outcomes. This therapy holds significant promise for treating various ROR1-expressing cancers, offering new hope for patients with limited treatment options and possibly expanding into other malignancies [141,142,143].
7.7. NY-ESO-1 CAR-T Cell Therapy
NY-ESO-1 CAR-T cell therapy is a promising cancer immunotherapy targeting the cancer-testis antigen NY-ESO-1, which is highly expressed in cancers like melanoma, synovial sarcoma, multiple myeloma, and certain lung cancers. This therapy involves genetically modifying a patient’s T cells to target NY-ESO-1, allowing them to specifically attack and kill cancer cells while minimizing harm to healthy tissues. Early clinical trials have shown success, particularly in treating synovial sarcoma, with some patients achieving durable responses and significant tumor regression. However, challenges include potential on-target, off-tumor toxicity and the immunosuppressive tumor microenvironment, which can limit the therapy’s effectiveness. Ongoing research aims to enhance the efficacy, persistence, and safety of NY-ESO-1 CAR-T therapy through combination treatments and improved CAR designs, offering hope for patients with limited treatment options and potentially expanding its application to other NY-ESO-1-expressing cancers [144,145].
7.8. NKG2D CAR-T Cell Therapy
NKG2D CAR-T cell therapy is an innovative cancer immunotherapy that leverages the natural killer (NK) cell receptor, NKG2D, to enhance T cells’ ability to recognize and destroy cancer cells. Unlike traditional CAR-T therapies, which target a single tumor-associated antigen, NKG2D CAR-T cells are engineered to target multiple stress-induced ligands (such as MICA, MICB, and ULBPs) overexpressed on a variety of tumor cells. This multi-target approach is particularly effective against cancers with high heterogeneity, where different cells within the same tumor express different antigens, reducing the likelihood of immune evasion. NKG2D CAR-T cells have shown promising results in preclinical studies, demonstrating the ability to induce tumor regression and improve survival in animal models of cancers like leukemia, lymphoma, multiple myeloma, and solid tumors, including ovarian, pancreatic, and colorectal cancers. However, challenges such as potential on-target, off-tumor effects, where normal cells expressing NKG2D ligands may be damaged, and the immunosuppressive tumor microenvironment could limit their effectiveness. Researchers are exploring combination therapies and genetic modifications to enhance the persistence and function of NKG2D CAR-T cells. Ongoing clinical trials aim to validate their safety and efficacy in patients, offering hope for a versatile and potent cancer treatment [146,147,148,149,150].
8. Methods for Studying the Binding of CAR-T Cells to Antigens and Their Importance
8.1. Methods for Studying the Binding of CAR-T Cells to Antigens and Their Importance
CAR-T cell therapy has emerged as a revolutionary approach to cancer treatment, particularly for hematologic malignancies. These therapies rely on the precise interaction between CAR-T cells and their target antigens, which are typically expressed on the surface of cancer cells. Understanding and studying the binding of CAR-T cells to these antigens is crucial for improving the efficacy and safety of CAR-T therapies. This essay explores the current methods used to study CAR-T cell binding to antigens, their significance, and the need for future advancements in this field [1,2,3].
Current Methods for Studying CAR-T Cell Binding
1. Surface Plasmon Resonance (SPR):
Surface plasmon resonance is a widely used technique for studying the binding kinetics of CAR-T cells to their target antigens. SPR measures the interaction in real-time by detecting changes in the refractive index near the surface of a sensor chip. CAR-T cells or their receptors are immobilized on the chip, and the antigen is flowed over the surface. SPR provides valuable data on the binding affinity, association, and dissociation rates, which are critical for understanding the strength and duration of the CAR-T cell’s engagement with its target [151].
2. Flow Cytometry:
Flow cytometry is another essential tool for analyzing CAR-T cell binding. This technique involves labeling CAR-T cells and target cells (or antigens) with fluorescent markers. By passing the cells through a laser, researchers can quantify the binding interactions between CAR-T cells and antigens based on fluorescence intensity. Flow cytometry allows for the assessment of binding efficiency and specificity, providing insights into how well CAR-T cells recognize and bind to their targets [152].
3. Enzyme-Linked Immunosorbent Assay (ELISA):
ELISA is a biochemical technique used to detect and quantify the binding of CAR-T cells to their antigens. In this method, the target antigen is immobilized on a plate, and CAR-T cells or their receptors are introduced. After washing away non-bound components, an enzyme-linked secondary antibody is added, which produces a detectable signal proportional to the amount of binding. ELISA is useful for screening and comparing different CAR constructs based on their binding abilities [153].
4. Microscopy-Based Techniques:
Advanced microscopy techniques, such as confocal and fluorescence microscopy, allow for the visualization of CAR-T cell binding to antigens at the cellular level. These methods enable the study of spatial distribution, clustering, and dynamics of CAR-antigen interactions within the context of the cellular environment. High-resolution microscopy can reveal details about the formation of the immunological synapse between CAR-T cells and target cells, which is crucial for effective cytotoxic activity [154].
8.2. Importance of Studying CAR-T Cell Binding
Understanding the binding interactions between CAR-T cells and their target antigens is fundamental for several reasons:
1. Optimization of CAR Design:
The binding affinity and specificity of CAR-T cells are critical determinants of therapeutic efficacy and safety. High-affinity binding may enhance the effectiveness of CAR-T cells, but it could also increase the risk of off-target effects and toxicity. Conversely, low-affinity binding might reduce the therapeutic impact. By studying these interactions, researchers can optimize CAR constructs to achieve the desired balance between efficacy and safety [155].
2. Predicting Clinical Outcomes:
The strength and stability of CAR-T cell binding can influence clinical outcomes, such as response rates and durability of remission. By correlating binding characteristics with patient outcomes, researchers can identify biomarkers of success or failure, aiding in the selection of the most promising CAR-T candidates for clinical use [156].
3. Understanding Resistance Mechanisms:
Tumor cells can develop resistance to CAR-T cell therapy through various mechanisms, including antigen loss or modification. Studying the binding interactions helps researchers understand how tumors evade CAR-T cell recognition and can guide the development of strategies to overcome resistance, such as dual-targeting CARs or combination therapies [157].
8.3. Future Directions: The Need for New Methods
While current methods provide valuable insights into CAR-T cell binding, there is a growing need for more advanced and precise techniques. Future methods should address several key challenges:
1. Single-Cell Analysis:
Heterogeneity among CAR-T cells and tumor cells is a significant challenge in understanding binding interactions. New methods that allow for single-cell analysis could provide more detailed information about the variability in binding affinities and their impact on therapeutic outcomes [158].
2. In Vivo Imaging:
Most current methods study CAR-T cell binding in vitro, which may not fully capture the complexity of interactions within the tumor microenvironment. Developing in vivo imaging techniques that can monitor CAR-T cell binding and activity in real-time within a living organism would greatly enhance our understanding of therapy dynamics and efficacy [159].
3. Artificial Intelligence (AI) and Machine Learning:
Integrating AI and machine learning with existing methods could help analyze large datasets from binding studies more effectively. These technologies could identify patterns and correlations that are not immediately apparent, leading to better predictions of clinical outcomes and the design of more effective CAR-T therapies [160].
10. Advancements in NK CAR Cells for Cancer Immunotherapy
Natural Killer (NK) cells are a critical component of the innate immune system, recognized for their ability to target and destroy malignant cells without prior sensitization. Recent advancements in immunotherapy have led to the development of Chimeric Antigen Receptor (CAR) technology, originally applied to T cells but now being explored with NK cells. The potential of NK CAR cells in cancer treatment is immense, combining the innate cytotoxic abilities of NK cells with the specificity and adaptability of CAR technology.
10.1. Introduction to NK Cells and CAR Technology
NK cells are unique in their ability to recognize and eliminate cancer cells through both direct cytotoxicity and the production of cytokines that stimulate other immune cells [161,162]. Unlike T cells, NK cells do not require antigen presentation via the major histocompatibility complex (MHC), allowing them to target a broader range of tumor cells, including those that evade T cell detection [163]. The introduction of CAR technology to NK cells aims to enhance their natural tumor-targeting capabilities by equipping them with engineered receptors that recognize specific tumor antigens [164]. Figure 2 shows soluble factors in the bi-directional crosstalk between NK cells and immune cells in the tumor microenvironment (TME).
NK cells’ anti-tumor activities are influenced by a range of cytokines and soluble factors produced by immune cells within the TME, which can either enhance or suppress their functions.
(A) Neutrophils: Neutrophils can inhibit NK cell function by downregulating NKp46 expression through the release of Cathepsin G and reactive oxygen species (ROS). They also hinder NK cell activity by depleting arginine and releasing neutrophil extracellular traps (NETs), which block IFN-γ production and cytotoxicity. On the other hand, neutrophil-derived elastase, lactoferrin, and interleukin (IL)-12 boost NK cell functions. Additionally, IFN-γ produced by NK cells can suppress the angiogenic potential of tumor-associated neutrophils.
(B) Macrophages: M1-polarized macrophages in the tumor microenvironment activate NK cells by secreting cytokines such as IL-12, IL-15, IL-18, and TNF-α. In contrast, M2-polarized macrophages inhibit NK cell activation through the release of TGF-β. NK cells, in turn, can modulate macrophage activity by releasing IFN-γ, TNF-α, GM-CSF, and IL-10.
(C) T Cells: NK cell-derived IFN-γ promotes the polarization of CD4+ T cells towards a Th1 response and enhances the cytotoxic functions of CD8+ T cells. In response, T cells produce IL-2 and IL-15, which are essential for NK cell survival and activation.
(D) Regulatory T Cells (Treg): NK cell-mediated tumor killing and IFN-γ production are negatively impacted by immunosuppressive molecules like TGF-β and adenosine, which are released by Tregs. Tregs also suppress NK cell functions by competing for IL-2 and can directly kill NK cells through granzyme B.
(E) Monocytes: Patrolling monocytes contribute to NK cell-mediated anti-metastatic activity by releasing IL-15.
(F) Dendritic Cells (DCs): NK cell interaction with dendritic cells leads to mutual activation. Mature DCs secrete cytokines such as IL-12, IL-15, IL-18, CXCL9, and CXCL10, which promote NK cell activation and recruitment. Furthermore, NK cells help optimize DC-driven immune responses by eliminating immature DCs. In return, NK cells facilitate the activation and recruitment of DCs within the tumor bed by releasing IFN-γ, TNF-α, CCL5, XCL1, and XCL2. This content was partly created using BioRender.com.
CAR technology involves genetically modifying immune cells to express receptors that bind to antigens on tumor cells, leading to their destruction. Initially developed for T cells, CAR-T cell therapy has shown remarkable success in treating hematological malignancies [165]. However, the use of CAR technology with NK cells offers several advantages, including reduced risk of graft-versus-host disease (GVHD) and the ability to target a broader range of tumor cells [166].
10.2. Development of NK CAR Cells
The first generation of NK CAR cells was developed by transducing NK cells with CAR constructs similar to those used in T cells. These constructs typically consist of an extracellular antigen recognition domain, a hinge region, a transmembrane domain, and an intracellular signaling domain [167]. The most common target for these NK CAR cells has been CD19, a B cell antigen widely expressed in B cell malignancies [168]. Early studies demonstrated that NK CAR cells could effectively target and eliminate CD19-positive tumor cells, leading to tumor regression in preclinical models [169].
Further advancements have focused on optimizing the CAR constructs specifically for NK cells. For example, researchers have explored the use of NK cell-specific signaling domains, such as those derived from the NKG2D receptor, which enhance the cytotoxic activity of NK CAR cells [170]. Additionally, the inclusion of cytokines, such as IL-15, in the CAR constructs has been shown to improve the proliferation and persistence of NK CAR cells in vivo, leading to more sustained anti-tumor responses [171].
10.3. Challenges in the Development of NK CAR Cells
Despite the promising potential of NK CAR cells, several challenges remain in their development and clinical application. One of the primary challenges is the efficient transduction of NK cells with CAR constructs. Unlike T cells, NK cells are more resistant to genetic modification, which can limit the efficiency of CAR expression [172]. To address this, researchers have explored various viral and non-viral transduction methods, as well as the use of transposon-based systems, to improve the transduction efficiency of NK cells [173].
Another significant challenge is the persistence of NK CAR cells in vivo. Unlike T cells, which can persist for extended periods and provide long-term protection against cancer recurrence, NK cells have a shorter lifespan and may require repeated infusions to maintain therapeutic efficacy [174]. Strategies to enhance the persistence of NK CAR cells include the use of cytokines, such as IL-2 and IL-15, and the co-administration of other immune-modulating agents [175].
The tumor microenvironment (TME) also poses a significant challenge to the efficacy of NK CAR cells. Tumors can create an immunosuppressive environment that inhibits the activity of immune cells, including NK cells. This can be mediated by various factors, such as the expression of checkpoint molecules (e.g., PD-L1) and the presence of regulatory cells (e.g., Tregs, MDSCs) that suppress NK cell activity [176]. Researchers are exploring combination therapies that target these immunosuppressive mechanisms, such as the use of checkpoint inhibitors alongside NK CAR cells, to enhance their anti-tumor efficacy [177].
10.4. Clinical Applications of NK CAR Cells
The clinical application of NK CAR cells is still in its early stages, but initial results from clinical trials have been promising. One of the most significant advantages of NK CAR cells is their potential to be used as an “off-the-shelf” therapy. Unlike T cells, which must be derived from the patient (autologous therapy), NK cells can be derived from healthy donors or cell lines and used in multiple patients (allogeneic therapy) [178]. This reduces the time and cost associated with generating CAR-T cells and makes NK CAR cell therapy more accessible to a broader range of patients [179].
Several clinical trials are currently underway to evaluate the safety and efficacy of NK CAR cells in various cancers. For example, a phase I clinical trial is evaluating the use of NK CAR cells targeting CD19 in patients with relapsed or refractory B cell malignancies [180]. Preliminary results have shown that NK CAR cells are well-tolerated and can induce complete remissions in some patients, even those who have failed previous CAR-T cell therapy [181].
Other trials are exploring the use of NK CAR cells targeting different antigens, such as GD2 in neuroblastoma and HER2 in solid tumors [182,183]. These studies are crucial in determining the broader applicability of NK CAR cells across different cancer types and will provide valuable insights into their safety and efficacy in a clinical setting [184].
10.5. Future Directions
The future of NK CAR cell therapy lies in overcoming the current challenges and optimizing their use in clinical practice. One promising area of research is the development of multi-specific CARs that target multiple tumor antigens simultaneously. This approach could reduce the likelihood of tumor escape, where cancer cells lose the targeted antigen and evade immune detection [185]. Additionally, researchers are exploring the combination of NK CAR cells with other immunotherapies, such as checkpoint inhibitors and immune-modulating agents, to enhance their efficacy and overcome resistance mechanisms [186].
11. The Potential of γδ T Cells in Mitigating GVHD
To address these challenges, researchers are increasingly exploring the potential of γδ T cells in combination with CAR-T therapy. γδ T cells are known for their unique ability to recognise stressed or transformed cells without the need for antigen presentation via MHC molecules, reducing the likelihood of triggering adverse immune responses such as GVHD [187,188]. Their intrinsic properties, including their capacity to produce anti-inflammatory cytokines and their ability to home in on tumour sites, make them an attractive candidate for enhancing the safety and efficacy of CAR-T cell therapy [189].
The incorporation of γδ T cells into CAR-T therapy represents a novel approach that could not only mitigate the risks associated with CAR-T therapy but also improve its therapeutic outcomes. By leveraging the strengths of both cell types, this strategy holds the potential to revolutionize cancer immunotherapy, offering more effective and safer treatment options for patients [190].
12. Exploration of Other Important Cell Types That Also Hold Promise as Sources for Universal CAR-T Therapies
In the quest to develop universal CAR-T cell therapies, various cell types are being explored for their potential to overcome the limitations of current patient-specific approaches. Among these, TCR knockout conventional T cells, natural killer T (NKT) cells, and double-negative (CD4- CD8-) T cells have shown promise as potential sources [191].
TCR knockout conventional T cells are engineered to lack the T-cell receptor, preventing them from recognizing and attacking host tissues, thus reducing the risk of Graft-versus-Host Disease (GVHD) [192]. Their strength lies in their ability to be universally applicable, as they do not require extensive donor-recipient matching. However, their main weakness is the complexity of the gene-editing process required to produce them, which can increase production costs and introduce potential off-target effects [193].
Natural Killer T (NKT) cells combine features of both T cells and natural killer (NK) cells, allowing them to recognize glycolipid antigens presented by the CD1d molecule. Their unique ability to produce a rapid and potent immune response against tumors makes them a strong candidate for CAR-T therapy. Additionally, NKT cells have a natural resistance to GVHD, further supporting their use in an allogeneic setting. However, the rarity of NKT cells in peripheral blood and the challenges in expanding them to sufficient numbers for therapeutic use are significant drawbacks [194].
Double-negative (CD4- CD8-) T cells are a subset of T cells that do not express the CD4 or CD8 co-receptors, reducing the risk of GVHD. Their inherent resistance to inducing GVHD and their ability to target a broad range of tumors make them attractive for universal CAR-T therapies. However, their limited understanding and the challenges in isolating and expanding these cells pose hurdles to their widespread use [195].
Each of these cell types, TCR knockout T cells, NKT cells, and double-negative T cells, offers unique strengths and faces specific challenges in the development of universal CAR-T therapies. Their potential to minimize GVHD while maintaining anti-tumor efficacy positions them as promising candidates in the evolution of CAR-T therapy.
12.1. TCR Knockout T Cells
The current generation of CAR-T therapies faces significant challenges, including patient-specific production, high costs, and potential adverse effects such as Graft-versus-Host Disease (GVHD). To address these issues, researchers are exploring alternative cell types that could serve as universal sources for CAR-T therapies. One such promising avenue is the use of T-cell receptor (TCR) knockout conventional T cells.
TCR knockout T cells are engineered to lack the T-cell receptor, which is responsible for recognizing antigens presented by the major histocompatibility complex (MHC) on the surface of cells. By eliminating the TCR, these T cells are rendered incapable of recognizing and attacking host tissues, thereby reducing the risk of GVHD when used in an allogeneic (donor-derived) setting. This makes them an attractive candidate for the development of universal CAR-T therapies, as they could potentially be used in any patient without the need for extensive matching or individualization [196,197].
The creation of TCR knockout T cells involves advanced gene-editing techniques, such as CRISPR/Cas9, to precisely delete the genes encoding the TCR [198,199]. Once the TCR is knocked out, these cells can be engineered to express a chimeric antigen receptor, allowing them to target and destroy cancer cells with high specificity [197]. Studies have shown that these modified T cells retain their functionality and potency in attacking cancer cells, while their universal applicability could significantly reduce the cost and complexity of CAR-T therapy production. TCR knockout conventional T cells represent a promising source for the development of universal CAR-T therapies [196]. Their potential to minimize GVHD, combined with their effectiveness in targeting cancer cells, positions them as a key innovation in the quest to make CAR-T therapy more accessible and broadly applicable [200].
12.2. Natural Killer T (NKT) Cells
Natural Killer T (NKT) cells have emerged as a promising candidate for universal CAR-T cell therapy due to their unique characteristics, which combine the properties of both T cells and natural killer (NK) cells. NKT cells can recognize glycolipid antigens presented by the CD1d molecule, allowing them to respond rapidly and robustly to a broad range of tumor cells. This ability makes them particularly effective in targeting cancers that may be resistant to conventional T cell-based therapies [200].
One of the key advantages of using NKT cells in CAR-T therapy is their natural resistance to inducing Graft-versus-Host Disease (GVHD). This resistance makes NKT cells an attractive option for allogeneic CAR-T therapies, where cells from a donor are used to treat a patient without the need for strict donor-recipient matching. Additionally, NKT cells can be readily engineered to express chimeric antigen receptors, enhancing their ability to target and destroy tumor cells with precision [201].
Randomized controlled trials (RCTs) exploring NKT-based CAR-T therapies have shown encouraging results, demonstrating significant anti-tumor activity with reduced incidences of GVHD compared to conventional CAR-T approaches [188]. These trials also highlight the potential for NKT cells to be expanded ex vivo to sufficient numbers for therapeutic use, addressing one of the primary challenges of this approach [202]. Despite these advances, further research and larger RCTs are needed to fully establish the safety and efficacy of NKT cells as a universal CAR-T therapy, but the early results are promising, offering a new avenue for effective cancer treatment.
12.3. Invariant Natural Killer T (iNKT) Cells
Invariant natural killer T (iNKT) cells are rare yet highly versatile immune cells with significant antitumor capabilities. These cells not only directly kill tumor cells but also enhance antitumor responses through adjuvant effects, superior tumor infiltration, and the inhibition of tumor-associated macrophages. Due to their potent immunoregulatory functions, iNKT cells are increasingly being viewed as a promising platform for CAR-based cancer immunotherapy. Unlike other immune cells, allogeneic iNKT cells do not cause graft-versus-host disease (GvHD), making them an ideal candidate for developing universal CAR-T cell therapies. The potential to create a universally applicable therapeutic product from CAR-iNKT cells opens new avenues for more effective and broadly applicable cancer treatments [203,204].
12.4. Double-Negative (CD4- CD8-) T Cells
Double-negative (CD4- CD8-) T cells, a unique subset of T cells lacking both CD4 and CD8 co-receptors, are gaining attention as a potential source for universal CAR-T cell therapy. These cells exhibit several advantageous characteristics that make them promising candidates for allogeneic applications, where cells from a donor are used to treat a patient [196, 197, 205_ 207]. Double-negative (CD4- CD8-) T cells offer significant potential for CAR-T cell therapy due to their resistance to causing Graft-versus-Host Disease (GVHD), a serious complication in allogeneic therapies [208]. Their lack of CD4 and CD8 markers, which are typically involved in recognizing and attacking host tissues, reduces the risk of harmful immune responses [206]. This makes them ideal candidates for universal CAR-T therapies, allowing broader patient application without strict donor-recipient matching [209]. In addition to their safety profile, double-negative T cells exhibit strong anti-tumor activity. They can be engineered with chimeric antigen receptors (CARs) to effectively target and eliminate cancer cells [205,209]. Despite challenges in isolating and expanding these cells to therapeutic levels, they represent a promising avenue for safer and more accessible CAR-T therapies.
12.5. CAR Macrophages Cells
CAR macrophages (CAR-Ms) represent an emerging frontier in cancer immunotherapy, designed to overcome the limitations associated with CAR T and CAR NK cells, particularly in solid tumors [210]. Macrophages naturally infiltrate tumor sites and play a critical role in modulating the tumor microenvironment (TME). CAR-Ms are engineered to express chimeric antigen receptors (CARs), enabling them to specifically target and eliminate cancer cells while also enhancing their inherent immune functions [211,212]. Unlike CAR T cells, which often struggle with trafficking to and persisting within solid tumors, CAR-Ms are adept at navigating the complex TME and can exert sustained anti-tumor effects [213].
One of the key advantages of CAR-Ms is their dual functionality. They not only phagocytose and kill cancer cells directly but also act as antigen-presenting cells, stimulating a broader immune response against the tumor. Moreover, CAR-Ms can be engineered to shift the TME from an immunosuppressive to an immunostimulatory state, thereby enhancing the efficacy of other immune cells. Despite these promising features, CAR-Ms face challenges such as ensuring sustained activation and avoiding the induction of an overly aggressive immune response that could lead to toxicity [214,215,216,217]. Ongoing research focuses on optimizing CAR-M design, improving manufacturing processes, and evaluating safety and efficacy in preclinical models, with the goal of advancing CAR-Ms into clinical trials for solid tumors and select hematological malignancies [217].
12.6. CAR Neutrophils Cells
CAR-neutrophils, engineered from human pluripotent stem cells using CRISPR/Cas9, are designed to target glioblastoma (GBM) by crossing the blood-brain barrier. These cells are modified to express anti-GBM CAR constructs, enabling them to deliver tumor-responsive nanodrugs without inducing additional inflammation. This innovative approach demonstrates strong anti-tumor activity, reduced off-target effects, and prolonged lifespan in tumor-bearing mice, offering a promising GBM treatment strategy [218].
A chemically-defined protocol enables large-scale production of neutrophils from human pluripotent stem cells (hPSCs). CAR constructs significantly boost the antitumor activity of these hPSC-derived neutrophils without altering their phenotype. Notably, anti-PSMA CAR-neutrophils demonstrate superior effectiveness against prostate cancer, highlighting their potential in targeted cancer therapies [219].
Neutrophils, though naturally cytotoxic against cancer, face limitations due to their short lifespan and resistance to genetic modification. By engineering human pluripotent stem cells with synthetic CARs, researchers created CAR neutrophils with enhanced tumor-targeting abilities, offering a promising new platform for myeloid cell-based cancer therapies [220].
12.7. CD5 Knockout on CAR T Cells
Most patients treated with FDA-approved CAR T cells eventually experience disease progression, and the therapy is not curative for solid cancers or some hematological malignancies like T cell lymphomas. A key barrier is CAR T cell dysfunction and lack of persistence after infusion. This study demonstrates that knocking out CD5 using CRISPR-Cas9 enhances CAR T cell activation, leading to improved antitumor effects, increased cytotoxicity, and better in vivo expansion and persistence, suggesting CD5 as a potential target for improving T cell therapies [221].
T cell malignancies have high recurrence and mortality rates, with CD5 expressed in about 85% of cases. However, CD5 expression on CAR-T cells causes fratricide. To address this, researchers developed a biepitopic CAR (FHVH3/VH1) targeting CD5 using novel human heavy-chain variable domains. By employing CRISPR-Cas9 for CD5 knockout (CD5KO), they optimized CAR-T cell production, resulting in enhanced and longer-lasting efficacy in preclinical models, with reduced cytokine release and high specificity, making it promising for further development [222].
12.8. Mucosal-Associated Invariant T (MAIT) Cells
MAIT cells, a conserved T cell subpopulation, are mostly CD161+CD8+ and possess a semi-constant TCR combining TRAV1 with TRAJ33, TRAJ12, or TRAJ20 [223]. They recognize vitamin B2 derivatives via MR1 on APCs, promoting cytokine secretion and cytotoxic activity. In antitumor immunity, MAIT cells kill tumor cells through TCR or NK receptors and modulate immune responses by interacting with DC, Tumor-Associated Macrophages (TAM), and Myeloid-Derived Suppressor Cells (MDSC) [224].
CAR (Chimeric Antigen Receptor) therapy, which is commonly used to modify T cells to target cancer cells, is theoretically possible for MAIT (Mucosal-Associated Invariant T) cells. MAIT cells have unique properties, such as their ability to recognize microbial metabolites presented by MR1 molecules, and they exhibit both innate and adaptive immune features [225,226].
12.9. CAR MAIT Cells
Dogan et al., 2022 explore using MAIT (Mucosal-Associated Invariant T) cells in CAR-T therapy for targeting B cell lymphoma and breast cancer. Researchers engineered MAIT cells with anti-CD19 and anti-Her2 CARs, showing high cytotoxicity against tumor cells, comparable to CD8+ CAR-T cells, though with lower IFN-γ expression. Additionally, when activated by the vitamin B2 metabolite 5-ARU through the MR1 protein, MAIT cells effectively killed MR1-expressing tumor cells in a dose-dependent manner. This approach highlights the potential for developing novel, off-the-shelf cancer immunotherapy strategies using MAIT cells that could rival conventional methods [227].
12.10. Fibroblast Activation Protein (FAP) CAR-T Cells
Fibroblast Activation Protein (FAP) CAR-T cells represent a cutting-edge approach to cancer therapy by targeting the tumor stroma rather than the cancer cells directly. FAP. CAR-T cells are specifically designed to target the FAP, a surface protein, predominantly expressed on cancer-associated fibroblasts (CAFs) within the tumor microenvironment [228,229]. Research is ongoing to optimize FAP CAR-T cells, with efforts focused on improving their specificity, enhancing their persistence in the tumor microenvironment, and combining them with other therapeutic modalities [228,229]. Approaches such as dual-targeting CAR-T cells, which target both FAP and a tumor-specific antigen, are also being explored to increase efficacy while minimizing off-tumor effects [228,229,230].
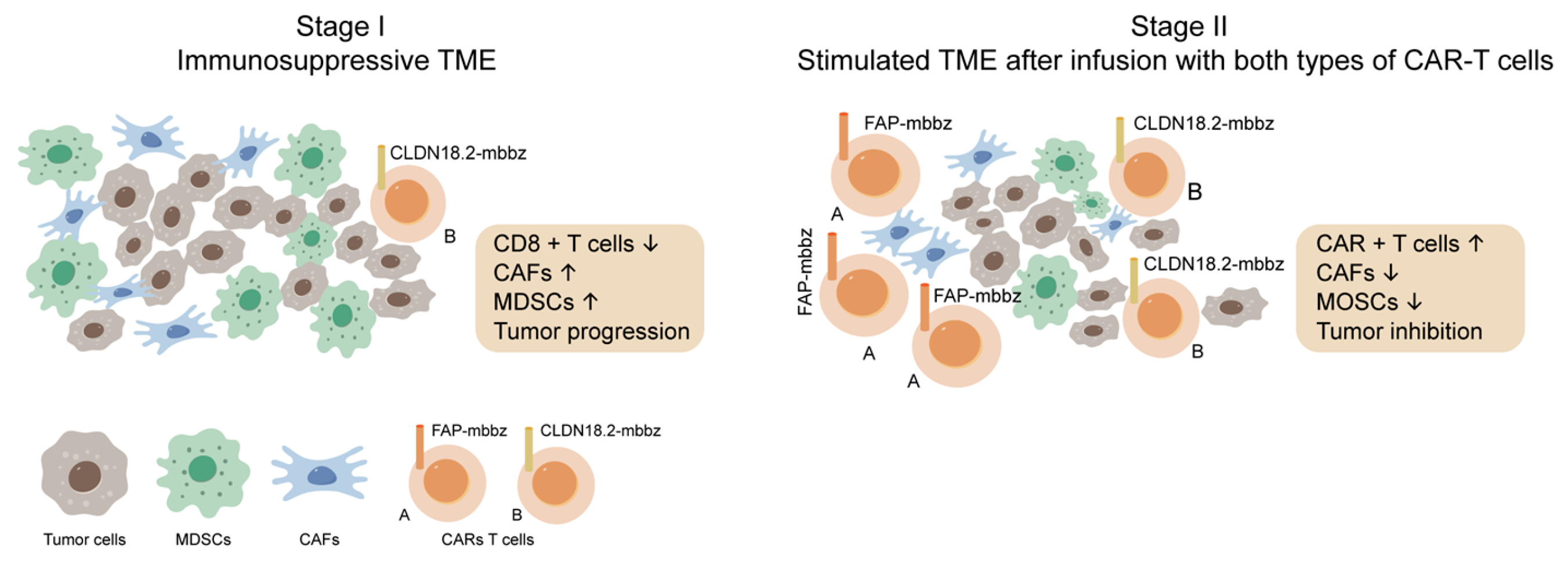
Figure 3.
“Sequential Therapy enhances the antitumor ability of CAR-T by suppressing the infiltration of MDSCs. Left: CAFs are crucial in TME, forming an immune surveillance barrier and perpetuating tumor-promoting. The CAFs could also promote recruit monocytes from the bone marrow, such as MDSCs, to form a tumor-suppressing microenvironment, suppressing CAR-T cell function. Right: FAP-targeted CAR-T cells eliminate CAFs via specific recognition of FAP on CAFs in the tumor microenvironment. Further, CLDN18.2-targeted CAR-T cells could also increase cytotoxic T cells and inhibit the recruiting of MDSCs. With an improved immune suppressive microenvironment, the antitumor effect of the sequential infusion of CLDN18.2-targeted CAR-T cells were enhanced”. Modified originally from [229].
Figure 3.
“Sequential Therapy enhances the antitumor ability of CAR-T by suppressing the infiltration of MDSCs. Left: CAFs are crucial in TME, forming an immune surveillance barrier and perpetuating tumor-promoting. The CAFs could also promote recruit monocytes from the bone marrow, such as MDSCs, to form a tumor-suppressing microenvironment, suppressing CAR-T cell function. Right: FAP-targeted CAR-T cells eliminate CAFs via specific recognition of FAP on CAFs in the tumor microenvironment. Further, CLDN18.2-targeted CAR-T cells could also increase cytotoxic T cells and inhibit the recruiting of MDSCs. With an improved immune suppressive microenvironment, the antitumor effect of the sequential infusion of CLDN18.2-targeted CAR-T cells were enhanced”. Modified originally from [229].
12.10. Is There Any Probability of the Creations of CAR TAM Cells
CAR (Chimeric Antigen Receptor) therapy, primarily used to modify T cells to target cancer cells [1], is theoretically possible for TAMs, but it presents unique challenges and considerations:
1. TAMs:
- CAR-TAMs: It is possible to engineer macrophages to express CARs targeting specific tumor antigens [210,211,212]. This approach could help reprogram TAMs to act against the tumor rather than supporting it. For example, CAR-TAMs could be designed to recognize tumor antigens, enabling them to phagocytose tumor cells or modulate the tumor microenvironment to inhibit tumor growth and promote anti-tumor immunity. However, engineering macrophages is more complex than T cells due to their different biology and the challenges in efficiently transducing macrophages with viral vectors used in CAR therapy [210,231].
Challenges:
- Cell Plasticity: TAMs are highly plastic and can change their phenotype based on environmental signals [233], which might complicate CAR design and functionality.
- Immunosuppresive nature: The mechanisms of immunosuppresion employed by TAMs are targeted towards inhibiting the activity of the adaptive immune system, including NK cells and T cells [233].
Current Research:
While the concept of CAR-modified TAMs is intriguing, most CAR research currently focuses on T cells and, to a lesser extent, NK cells [128,130,131,134,137,141,146,192,193,194]. Research into macrophage is still in early stages, but it holds promise for developing novel immunotherapies that target the tumor microenvironment more effectively. TAMs help tumors grow by using various strategies. These cells naturally suppress the immune system, which allows the tumor to evade immune attacks. They also support the formation of new blood vessels (neoangiogenesis) that supply the tumor with nutrients and oxygen. Additionally, they contribute to the process of epithelial-mesenchymal transition (EMT), where cancer cells become more mobile and invasive. TAMs can also change the way cells metabolize energy, further supporting tumor survival and growth. The interaction between these cell types strengthens their pro-tumor activities, making them attractive targets for developing new cancer therapies [234,235].
13. Clinical Trials of CAR-T Cell Therapy of Solid Tumors
13.1. Clinical Trials
The development of CAR-T cell therapies has marked a significant advancement in the field of cancer treatment, particularly in hematologic malignancies [1,2]. However, the translation of this success to solid tumors has been met with considerable challenges [222]. First-generation CAR-T cells targeting antigens such as carbonic anhydrase IX (CAIX), neuronal cell adhesion molecule L1 (NCAM-L1)/CD171, folate receptor alpha (FR-α), and GD2, despite demonstrating feasibility, have largely failed to produce significant clinical benefits. The limited activity and frequent toxicity observed with these therapies have led to disappointing outcomes. This lack of success can be attributed to several factors, including the inherent quality of the CAR-T cells and the immunosuppressive nature of the tumor microenvironment (TME), which acts as a significant barrier to effective treatment [236].
To overcome these challenges, research has shifted towards the development of more advanced CAR-T cell generations. Fourth-generation CAR-T cells, also known as TRUCKs (T cells Redirected for Universal Cytokine Killing), have been engineered to secrete transgenic cytokines such as IL-7, IL-12, IL-15, and IL-18 [165]. These cytokines enhance T-cell activation and stimulate the recruitment of innate immune cells, which are crucial for eliminating antigen-negative cancer cells within the tumor. For example, 4H11-28z/IL-12 CAR-T cells, which are specific for the MUC-16ecto antigen, have shown promising results in preclinical trials [237]. This antigen is highly expressed on ovarian tumor cells, and the engineered CAR-T cells that target it have demonstrated enhanced antitumor efficacy in Scid-Beige mice with human ovarian cancer xenografts [238]. The success was marked by increased survival rates, prolonged T-cell persistence in the TME, and elevated systemic levels of IFN-γ, which are all indicators of a robust antitumor response.
Furthermore, to mitigate the risk of systemic toxicity, some CAR-T cells have been designed with inducible cytokine expression. These CAR-T cells use an expression cassette driven by the NFAT/IL-2 promoter, which releases IL-12 upon CAR binding to the target antigen. This inducible system ensures that IL-12 production occurs only at the tumor site, providing high local cytokine levels while minimizing systemic exposure. Once the CAR-T cells disengage from the target antigen, IL-12 production ceases, effectively providing a “shutdown” mechanism to prevent off-target effects [239].
In addition to TRUCKs, several other innovative CAR-T cell types are under investigation. These include universal CARs, which lack endogenous TCR or MHC, self-destructive CARs that can be deactivated through external signals, and methotrexate (MTX)-conditional CAR-T cells, which offer the ability to modulate the antitumor effect through the administration of MTX. These advancements represent the next frontier in CAR-T cell therapy, offering hope for more effective and safer treatments for solid tumors. Clinical trials are actively exploring the potential of second-, third-, and fourth-generation autologous CAR-T cells targeting EGFR, mesothelin, GD2, and other markers in the treatment of various cancers [240,241].
These advanced CAR-T therapies are designed to improve efficacy and safety in combating tumors. Additionally, allogeneic CAR-T cells, specifically P-MUC1C-ALLO1, are undergoing Phase I clinical trials (NCT05239143) for patients with advanced or metastatic solid tumors. This allogeneic approach aims to provide off-the-shelf solutions, potentially overcoming limitations associated with autologous CAR-T therapies and broadening the accessibility of these innovative treatments [242]. Figure 4 shows clinical trials of CAR-T cell therapy of solid tumors.
13. Conclusion
CAR-T therapy for solid tumors is in its early clinical development stages, but preclinical studies and early-phase trials have shown promise. CAR-T cells targeting antigens like HER2, EGFRvIII, and mesothelin have demonstrated potential in treating breast cancer, glioblastoma, and mesothelioma, respectively. Ongoing research aims to refine these strategies, with the hope that CAR-T therapy will eventually offer effective treatment options for solid tumors, particularly for cancers that are difficult to treat [242,243]. Despite significant challenges, advancements in target selection, CAR design, and combination therapies are paving the way for CAR-T cell therapy to become a powerful tool in oncology. In studying CAR-T cell binding to antigens, conventional methods such as flow cytometry, ELISA, and immunohistochemistry are widely used due to their established protocols and accessibility. While these techniques provide valuable insights, they may lack the sensitivity and throughput required for more detailed analyses. Advanced methods like surface plasmon resonance (SPR), single-cell RNA sequencing (scRNA-seq), and CRISPR screening offer higher resolution data, enabling deeper understanding of CAR-T cell interactions with target antigens. NK CAR cells, combining the innate cytotoxicity of NK cells with CAR technology, represent a promising advancement in cancer immunotherapy, potentially targeting a broad range of tumor cells with minimal adverse effects. Ongoing research and clinical trials are crucial to overcoming challenges in their development and expanding their clinical application. Universal CAR-T cells offer a transformative approach by overcoming the limitations of patient-specific therapies. Using TCR knockout T cells, NKT cells, and double-negative T cells, these universal CAR-T cells promise broader applicability, reduced risk of GVHD, and lower production costs. As research advances, universal CAR-T cells could revolutionize cancer treatment by making it more effective, accessible, and scalable for a wider patient population.
Author Contributions
The manuscript was conceptualized by AJV and planning and discussion were conducted by all authors. AJV wrote the initial draft of the manuscript. All authors investigated and reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study did not receive any external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data are available within the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhang, X; Zhu, L; Zhang, H; Chen, S; Xiao, Y. CAR-T cell therapy in hematological malignancies: current opportunities and challenges. Front Immunol. 2022, 13, 927153. [Google Scholar] [CrossRef]
- Schuster, SJ; Bishop, MR; Tam, CS; Waller, EK; Borchmann, P; McGuirk, JP; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Sang, W; Shi, M; Yang, J; Cao, J; Xu, L; Yan, D; et al. Phase II trial of co-administration of CD19- and CD20-targeted chimeric antigen receptor T cells for relapsed and refractory diffuse large B cell lymphoma. Cancer Med. 2020, 9, 5827–38. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, SS; Locke, FL; Bartlett, NL; Lekakis, LJ; Miklos, DB; Jacobson, CA; et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017, 377, 2531–44. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, CA; Chavez, JC; Sehgal, AR; William, BM; Munoz, J; Salles, G; et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022, 23, 91–103. [Google Scholar] [CrossRef]
- Locke, FL; Ghobadi, A; Jacobson, CA; Miklos, DB; Lekakis, LJ; Oluwole, OO; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Watanabe, N; Mo, F; McKenna, MK. Impact of manufacturing procedures on CAR T cell functionality. Front Immunol. 2022, 13, 876339. [Google Scholar] [CrossRef] [PubMed]
- Labbé, RP; Vessillier, S; Rafiq, QA. Lentiviral vectors for T cell engineering: clinical applications, bioprocessing and future perspectives. Viruses 2021, 13, 1528. [Google Scholar] [CrossRef]
- Kamdar, M; Solomon, SR; Arnason, J; Johnston, PB; Glass, B; Bachanova, V; et al. Lisocabtagene maraleucel versus standard of care with salvage chemotherapy followed by autologous stem cell transplantation as second-line treatment in patients with relapsed or refractory large B-cell lymphoma (TRANSFORM): results from an interim analysis of an open-label, randomised, phase 3 trial. Lancet 2022, 399, 2294–308. [Google Scholar] [CrossRef]
- Sehgal, A; Hoda, D; Riedell, PA; Ghosh, N; Hamadani, M; Hildebrandt, GC; et al. Lisocabtagene maraleucel as second-line therapy in adults with relapsed or refractory large B-cell lymphoma who were not intended for haematopoietic stem cell transplantation (PILOT): an open-label, phase 2 study. Lancet Oncol. 2022, 23, 1066–77. [Google Scholar] [CrossRef]
- Kharfan-Dabaja, MA; Yassine, F; Moustafa, MA; Iqbal, M; Murthy, H. Lisocabtagene maraleucel in relapsed or refractory diffuse large B cell lymphoma: what is the evidence? Hematol Oncol Stem Cell Ther. 2022, 15, 168–75. [Google Scholar] [CrossRef]
- Munshi, NC; Anderson, LD, Jr.; Shah, N; Madduri, D; Berdeja, J; Lonial, S; et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021, 384, 705–16. [Google Scholar] [CrossRef] [PubMed]
- Hansen, DK; Sidana, S; Peres, LC; Leitzinger, CC; Shune, L; Shrewsbury, A; et al. Idecabtagene vicleucel for relapsed/refractory multiple myeloma: real-world experience from the myeloma CAR T consortium. J Clin Oncol. 2023, 41, 2087–97. [Google Scholar] [CrossRef]
- Jagannath, S; Lin, Y; Goldschmidt, H; Reece, D; Nooka, A; Senin, A; et al. KarMMa-RW: comparison of idecabtagene vicleucel with real-world outcomes in relapsed and refractory multiple myeloma. Blood Cancer J. 2021, 11, 116. [Google Scholar] [CrossRef]
- Berdeja, JG; Madduri, D; Usmani, SZ; Jakubowiak, A; Agha, M; Cohen, AD; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet 2021, 398, 314–24. [Google Scholar] [CrossRef]
- Mi, JQ; Zhao, W; Jing, H; Fu, W; Hu, J; Chen, L; et al. Phase II, open-label study of ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor-T-cell therapy, in Chinese patients with relapsed/refractory multiple myeloma (CARTIFAN-1). J Clin Oncol. 2023, 41, 1275–84. [Google Scholar] [CrossRef]
- Lindo, L; Wilkinson, LH; Hay, KA. Befriending the hostile tumor microenvironment in CAR T-cell therapy. Front Immunol. 2020, 11, 618387. [Google Scholar] [CrossRef] [PubMed]
- Bell, M; Gottschalk, S. Engineered cytokine signaling to improve CAR T cell effector function. Front Immunol. 2021, 12, 684642. [Google Scholar] [CrossRef] [PubMed]
- López-Cantillo, G; Urueña, C; Camacho, BA; Ramírez-Segura, C. CAR-T cell performance: how to improve their persistence? Front Immunol 2022, 13, 878209. [Google Scholar] [CrossRef]
- Corti, C; Venetis, K; Sajjadi, E; Zattoni, L; Curigliano, G; Fusco, N. CAR-T cell therapy for triple-negative breast cancer and other solid tumors: preclinical and clinical progress. Expert Opin Investig Drugs 2022, 31, 593–605. [Google Scholar] [CrossRef]
- Guzman, G; Reed, MR; Bielamowicz, K; Koss, B; Rodriguez, A. CAR-T therapies in solid tumors: opportunities and challenges. Curr Oncol Rep. 2023, 25, 479–89. [Google Scholar] [CrossRef]
- Roex, G; Campillo-Davo, D; Flumens, D; Shaw, PAG; Krekelbergh, L; De Reu, H; et al. Two for one: targeting BCMA and CD19 in B-cell malignancies with off-the-shelf dual-CAR NK-92 cells. J Transl Med. 2022, 20, 124. [Google Scholar] [CrossRef]
- Viardot, A; Locatelli, F; Stieglmaier, J; Zaman, F; Jabbour, E. Concepts in immuno-oncology: tackling B cell malignancies with CD19-directed bispecific T cell engager therapies. Ann Hematol. 2020, 99, 2215–29. [Google Scholar] [CrossRef]
- Hay, KA; Turtle, CJ. Chimeric antigen receptor (CAR) T cells: lessons learned from targeting of CD19 in B-cell malignancies. Drugs 2017, 77, 237–45. [Google Scholar] [CrossRef]
- Strati, P; Neelapu, SS. CAR-T failure: beyond antigen loss and T cells. Blood 2021, 137, 2567–8. [Google Scholar] [CrossRef] [PubMed]
- Mulgaonkar, A; Udayakumar, D; Yang, Y; Harris, S; Öz, OK; Ramakrishnan Geethakumari, P; et al. Current and potential roles of immuno-PET/-SPECT in CAR T-cell therapy. Front Med. 2023, 10, 1199146. [Google Scholar] [CrossRef] [PubMed]
- Duell, J; Leipold, AM; Appenzeller, S; Fuhr, V; Rauert-Wunderlich, H; Da Via, M; et al. Sequential antigen loss and branching evolution in lymphoma after CD19- and CD20-targeted T-cell-redirecting therapy. Blood 2024, 143, 685–96. [Google Scholar] [CrossRef] [PubMed]
- Al-Haideri, M; Tondok, SB; Safa, SH; Maleki, AH; Rostami, S; Jalil, AT; et al. CAR-T cell combination therapy: the next revolution in cancer treatment. Cancer Cell Int. 2022, 22, 365. [Google Scholar] [CrossRef]
- Liu, Z; Lei, W; Wang, H; Liu, X; Fu, R. Challenges and strategies associated with CAR-T cell therapy in blood malignancies. Exp Hematol Oncol. 2024, 13, 22. [Google Scholar] [CrossRef]
- Baird, JH; Frank, MJ; Craig, J; Patel, S; Spiegel, JY; Sahaf, B; et al. CD22-directed CAR T-cell therapy induces complete remissions in CD19-directed CAR-refractory large B-cell lymphoma. Blood 2021, 137, 2321–5. [Google Scholar] [CrossRef]
- Maude, SL; Laetsch, TW; Buechner, J; Rives, S; Boyer, M; Bittencourt, H; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018, 378, 439–48. [Google Scholar] [CrossRef]
- Maus, MV; June, CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res. 2016, 22, 1875–84. [Google Scholar] [CrossRef]
- Sadelain, M; Brentjens, R; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–98. [Google Scholar] [CrossRef]
- June, CH; Sadelain, M. Chimeric antigen receptor therapy. N Engl J Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Lee, DW; Kochenderfer, JN; Stetler-Stevenson, M; Cui, YK; Delbrook, C; Feldman, SA; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 2015, 385, 517–28. [Google Scholar] [CrossRef] [PubMed]
- Maude, SL; Frey, N; Shaw, PA; Aplenc, R; Barrett, DM; Bunin, NJ; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014, 371, 1507–17. [Google Scholar] [CrossRef] [PubMed]
- Levine, BL; Miskin, J; Wonnacott, K; Keir, C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017, 4, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Turtle, CJ; Hanafi, LA; Berger, C; Hudecek, M; Pender, B; Robinson, E; et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016, 8, 355ra116. [Google Scholar] [CrossRef]
- Heuser, C; Hombach, A; Lösch, C; Manista, K; Abken, H. T-cell activation by recombinant immunoreceptors: impact of the intracellular signalling domain on the stability of receptor expression and antigen-specific activation of grafted T cells. Gene Ther. 2003, 10, 1408–19. [Google Scholar] [CrossRef]
- Guedan, S; Posey, AD, Jr.; Shaw, C; Wing, A; Da, T; Patel, PR; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Finney, HM; Akbar, AN; Lawson, ADG. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004, 172, 104–13. [Google Scholar] [CrossRef]
- Song, DG; Ye, Q; Poussin, M; Harms, GM; Figini, M; Powell, DJ, Jr. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 2012, 119, 696–706. [Google Scholar] [CrossRef]
- Guercio, M; Orlando, D; Di Cecca, S; Sinibaldi, M; Boffa, I; Caruso, S; et al. CD28.OX40 co-stimulatory combination is associated with long in vivo persistence and high activity of CAR.CD30 T-cells. Haematologica 2021, 106, 987–99. [Google Scholar] [CrossRef]
- Wei, C; Xia, K; Xie, Y; Ye, S; Ding, Y; Liu, Z; et al. Combination of 4-1BB and DAP10 promotes proliferation and persistence of NKG2D(bbz) CAR-T cells. Front Oncol. 2022, 12, 893124. [Google Scholar] [CrossRef]
- Luo, Y; Chen, Z; Sun, M; Li, B; Pan, F; Ma, A; et al. IL-12 nanochaperone-engineered CAR T cell for robust tumor-immunotherapy. Biomaterials 2022, 281, 121341. [Google Scholar] [CrossRef] [PubMed]
- Avanzi, MP; Yeku, O; Li, X; Wijewarnasuriya, DP; van Leeuwen, DG; Cheung, K; et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018, 23, 2130–41. [Google Scholar] [CrossRef]
- Chmielewski, M; Abken, H. CAR T cells releasing IL-18 convert to T-Bethigh FoxO1low effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 2017, 21, 3205–19. [Google Scholar] [CrossRef] [PubMed]
- Obstfeld, AE; Frey, NV; Mansfield, K; Lacey, SF; June, CH; Porter, DL; et al. Cytokine release syndrome associated with chimeric-antigen receptor T-cell therapy: clinicopathological insights. Blood 2017, 130, 2569–72. [Google Scholar] [CrossRef]
- Dong, R; Jiang, S; Chen, Y; Ma, Y; Sun, L; Xing, C; et al. Prognostic significance of cytokine release syndrome in B cell hematological malignancies patients after chimeric antigen receptor T cell therapy. J Interferon Cytokine Res. 2021, 41, 469–76. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M; Camisa, B; Barbiera, G; Falcone, L; Purevdorj, A; Genua, M; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018, 24, 739–48. [Google Scholar] [CrossRef]
- Qi, C; Tian, S; Wang, J; Ma, H; Qian, K; Zhang, X. Co-expression of CD40/CD40L on XG1 multiple myeloma cells promotes IL-6 autocrine function. Cancer Invest. 2015, 33, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T; van der Stegen, SJC; Eyquem, J; Hamieh, M; Piersigilli, A; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med. 2018, 24, 731–8. [Google Scholar] [CrossRef] [PubMed]
- Mehta, P; Cron, RQ; Hartwell, J; Manson, JJ; Tattersall, RS. Silencing the cytokine storm: the use of intravenous anakinra in haemophagocytic lymphohistiocytosis or macrophage activation syndrome. Lancet Rheumatol. 2020, 2, e358–67. [Google Scholar] [CrossRef]
- Li, P; Zhou, L; Ye, S; Zhang, W; Wang, J; Tang, X; et al. Risk of HBV reactivation in patients with resolved HBV infection receiving anti-CD19 chimeric antigen receptor T cell therapy without antiviral prophylaxis. Front Immunol. 2021, 12, 638678. [Google Scholar] [CrossRef]
- Wei, J; Zhao, J; Han, M; Meng, F; Zhou, J. SARS-CoV-2 infection in immunocompromised patients: humoral versus cell-mediated immunity. J Immunother Cancer 2020, 8, e000862. [Google Scholar] [CrossRef] [PubMed]
- de Haas, PA; Young, RD. Attention styles of hyperactive and normal girls. J Abnorm Child Psychol. 1984, 12, 531–46. [Google Scholar] [CrossRef]
- Bonilla, FA; Bernstein, IL; Khan, DA; Ballas, ZK; Chinen, J; Frank, MM; et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005, 94, S1–63. [Google Scholar] [CrossRef]
- Chapel, H; Cunningham-Rundles, C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009, 145, 709–27. [Google Scholar] [CrossRef]
- Park, JH; Romero, FA; Taur, Y; Sadelain, M; Brentjens, RJ; Hohl, TM; et al. Cytokine release syndrome grade as a predictive marker for infections in patients with relapsed or refractory B-cell acute lymphoblastic leukemia treated with chimeric antigen receptor T cells. Clin Infect Dis. 2018, 67, 533–40. [Google Scholar] [CrossRef]
- Mak, JWY; Law, AWH; Law, KWT; Ho, R; Cheung, CKM; Law, MF. Prevention and management of hepatitis B virus reactivation in patients with hematological malignancies in the targeted therapy era. World J Gastroenterol. 2023, 29, 4942–61. [Google Scholar] [CrossRef] [PubMed]
- Wei, J; Zhu, X; Mao, X; Huang, L; Meng, F; Zhou, J. Severe early hepatitis B reactivation in a patient receiving anti-CD19 and anti-CD22 CAR T cells for the treatment of diffuse large B-cell lymphoma. J Immunother Cancer 2019, 7, 315. [Google Scholar] [CrossRef]
- Murthy, H; Iqbal, M; Chavez, JC; Kharfan-Dabaja, MA. Cytokine release syndrome: current perspectives. Immunotargets Ther. 2019, 8, 43–52. [Google Scholar] [CrossRef]
- Johnsrud, A; Craig, J; Baird, J; Spiegel, J; Muffly, L; Zehnder, J; et al. Incidence and risk factors associated with bleeding and thrombosis following chimeric antigen receptor T-cell therapy. Blood Adv. 2021, 5, 4465–75. [Google Scholar] [CrossRef]
- Wang, Y; Qi, K; Cheng, H; Cao, J; Shi, M; Qiao, J; et al. Coagulation disorders after chimeric antigen receptor T cell therapy: analysis of 100 patients with relapsed and refractory hematologic malignancies. Biol Blood Marrow Transplant. 2020, 26, 865–75. [Google Scholar] [CrossRef]
- Shao, M; Yu, Q; Teng, X; Guo, X; Wei, G; Xu, H; et al. CRS-related coagulopathy in BCMA targeted CAR-T therapy: a retrospective analysis in a phase I/II clinical trial. Bone Marrow Transplant. 2021, 56, 1642–50. [Google Scholar] [CrossRef]
- Mavroudi, I; Papadaki, V; Pyrovolaki, K; Katonis, P; Eliopoulos, AG; Papadaki, HA. The CD40/CD40 ligand interactions exert pleiotropic effects on bone marrow granulopoiesis. J Leukoc Biol. 2011, 89, 771–83. [Google Scholar] [CrossRef]
- Xing, L; Wang, Y; Liu, H; Gao, S; Shao, Q; Yue, L; et al. Case report: sirolimus alleviates persistent cytopenia after CD19 CAR-T-cell therapy. Front Oncol. 2021, 11, 798352. [Google Scholar] [CrossRef]
- Benjamin, R; Graham, C; Yallop, D; Jozwik, AC; Mirci-Danicar, OC; Lucchini, G; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet 2020, 396, 1885–94. [Google Scholar] [CrossRef] [PubMed]
- Pan, J; Tang, K; Luo, Y; Seery, S; Tan, Y; Deng, B; et al. Sequential CD19 and CD22 chimeric antigen receptor T-cell therapy for childhood refractory or relapsed B-cell acute lymphocytic leukaemia: a single-arm, phase 2 study. Lancet Oncol. 2023, 24, 1229–41. [Google Scholar] [CrossRef] [PubMed]
- Shah, NN; Fry, TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019, 16, 372–85. [Google Scholar] [CrossRef] [PubMed]
- Xu, X; Sun, Q; Liang, X; Chen, Z; Zhang, X; Zhou, X; et al. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef]
- Schultz, L. Chimeric antigen receptor T cell therapy for pediatric B-ALL: narrowing the gap between early and long-term outcomes. Front Immunol. 2020, 11, 1985. [Google Scholar] [CrossRef]
- Leahy, AB; Newman, H; Li, Y; Liu, H; Myers, R; DiNofia, A; et al. CD19-targeted chimeric antigen receptor T-cell therapy for CNS relapsed or refractory acute lymphocytic leukaemia: a post-hoc analysis of pooled data from five clinical trials. Lancet Haematol. 2021, 8, e711–22. [Google Scholar] [CrossRef]
- Hayden, PJ; Roddie, C; Bader, P; Basak, GW; Bonig, H; Bonini, C; et al. Management of adults and children receiving CAR T-cell therapy: 2021 best practice recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE) and the European Haematology Association (EHA). Ann Oncol. 2022, 33, 259–75. [Google Scholar] [CrossRef]
- Ganapathy, T; Radhakrishnan, R; Sakshi, S; Martin, S. CAR γδ T cells for cancer immunotherapy. Is the field more yellow than green? Cancer Immunol Immunother. 2023, 72, 277–86. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, HR; Mirzaei, H; Lee, SY; Hadjati, J; Till, BG. Prospects for chimeric antigen receptor (CAR) γδ T cells: a potential game changer for adoptive T cell cancer immunotherapy. Cancer Lett. 2016, 380, 413–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, S; Deng, B; Yin, Z; Lin, Y; An, L; Liu, D; et al. Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell acute lymphoblastic leukemia after allogeneic transplantation. Am J Hematol. 2021, 96, 671–9. [Google Scholar] [CrossRef]
- Dai, H; Wu, Z; Jia, H; Tong, C; Guo, Y; Ti, D; et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol. 2020, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Li, W; Ding, L; Shi, W; Wan, X; Yang, X; Yang, J; et al. Safety and efficacy of co-administration of CD19 and CD22 CAR-T cells in children with B-ALL relapse after CD19 CAR-T therapy. J Transl Med. 2023, 21, 213. [Google Scholar] [CrossRef] [PubMed]
- Gust, J; Hay, KA; Hanafi, LA; Li, D; Myerson, D; Gonzalez-Cuyar, LF; et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017, 7, 1404–19. [Google Scholar] [CrossRef]
- Lee, DW; Santomasso, BD; Locke, FL; Ghobadi, A; Turtle, CJ; Brudno, JN; et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant. 2019, 25, 625–38. [Google Scholar] [CrossRef]
- Neelapu, SS; Tummala, S; Kebriaei, P; Wierda, W; Gutierrez, C; Locke, FL; et al. Chimeric antigen receptor T-cell therapy—assessment and management of toxicities. Nat Rev Clin Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, BD; Park, JH; Salloum, D; Riviere, I; Flynn, J; Mead, E; et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia. Cancer Discov. 2018, 8, 958–71. [Google Scholar] [CrossRef] [PubMed]
- Rubin, DB; Danish, HH; Ali, AB; Li, K; LaRose, S; Monk, AD; et al. Neurological toxicities associated with chimeric antigen receptor T-cell therapy. Brain 2019, 142, 1334–48. [Google Scholar] [CrossRef] [PubMed]
- Brudno, JN; Kochenderfer, JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 2016, 127, 3321–30. [Google Scholar] [CrossRef]
- Grupp, SA; Kalos, M; Barrett, D; Aplenc, R; Porter, DL; Rheingold, SR; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013, 368, 1509–18. [Google Scholar] [CrossRef]
- Hay, KA; Hanafi, LA; Li, D; Gust, J; Liles, WC; Wurfel, MM; et al. Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 2017, 130, 2295–306. [Google Scholar] [CrossRef]
- Lee, DW; Gardner, R; Porter, DL; Louis, CU; Ahmed, N; Jensen, M; et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–95. [Google Scholar] [CrossRef]
- Teachey, DT; Lacey, SF; Shaw, PA; Melenhorst, JJ; Maude, SL; Frey, N; et al. Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov. 2016, 6, 664–79. [Google Scholar] [CrossRef]
- Karschnia, P; Jordan, JT; Forst, DA; Arrillaga-Romany, IC; Batchelor, TT; Baehring, JM; et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood 2019, 133, 2212–22. [Google Scholar] [CrossRef]
- Frey, NV; Shaw, PA; Hexner, EO; Pequignot, E; Gill, S; Luger, SM; et al. Optimizing chimeric antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol. 2020, 38, 415–22. [Google Scholar] [CrossRef]
- Hu, S. Overview of immunosuppressive therapy in solid organ transplantation. Mayo Clin Proc. 2019, 94, 1975–86. [Google Scholar] [CrossRef]
- Kruetzmann, S; Rosado, MM; Weber, H; Germing, U; Tournilhac, O; Peter, HH; et al. Human immunoglobulin M memory B cells controlling Streptococcus pneumoniae infections are generated in the spleen. J Exp Med. 2003, 197, 939–45. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S; Patel, P. Approach to immunodeficiency diseases. Mayo Clin Proc. 2020, 95, 544–50. [Google Scholar] [CrossRef]
- Author Index. Vox Sang. 2011, 101, 135–42. [CrossRef]
- Panitsas, FP; Theodoropoulou, M; Kouraklis, A; Karakantza, M; Theodorou, GL; Zoumbos, NC; et al. Adult chronic idiopathic thrombocytopenic purpura (ITP) is the manifestation of a type-1 polarized immune response. Blood 2004, 103, 2645–7. [Google Scholar] [CrossRef] [PubMed]
- Smith, TJ; Bohlke, K; Lyman, GH; Carson, KR; Crawford, J; Cross, SJ; et al. Recommendations for the use of WBC growth factors: American Society of Clinical Oncology clinical practice guideline update. J Clin Oncol. 2015, 33, 3199–212. [Google Scholar] [CrossRef] [PubMed]
- Turtle, CJ; Hanafi, LA; Berger, C; Gooley, TA; Cherian, S; Hudecek, M; et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016, 126, 2123–38. [Google Scholar] [CrossRef]
- Penack, O; Holler, E; van den Brink, MRM. Graft-versus-host disease: regulation by microbe-associated molecules and innate immune receptors. Blood 2010, 115, 1865–72. [Google Scholar] [CrossRef]
- Malard, F; Mohty, M. Updates in chronic graft-versus-host disease management. Am J Hematol. 2023, 98, 1637–44. [Google Scholar] [CrossRef]
- Maude, SL; Laetsch, TW; Buechner, J; Rives, S; Boyer, M; Bittencourt, H; et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018, 378, 439–48. [Google Scholar] [CrossRef]
- Cohen, AD; Garfall, AL; Stadtmauer, EA; Lacey, SF; Lancaster, E; Vogl, DT; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019, 129, 2210–21. [Google Scholar] [CrossRef]
- Brudno, JN; Maric, I; Hartman, SD; Rose, JJ; Wang, M; Lam, N; et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J Clin Oncol. 2018, 36, 2267–80. [Google Scholar] [CrossRef]
- Schuster, SJ; Bishop, MR; Tam, CS; Waller, EK; Borchmann, P; McGuirk, JP; et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Munshi, NC; Anderson, LD, Jr.; Shah, N; Madduri, D; Berdeja, J; Lonial, S; et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021, 384, 705–16. [Google Scholar] [CrossRef]
- Raje, N; Berdeja, J; Lin, Y; Siegel, D; Jagannath, S; Madduri, D; et al. Anti-BCMA CAR T-cell therapy bb21217 in patients with relapsed or refractory multiple myeloma: updated results of a multicenter phase I CRB-402 study. Blood 2019, 134, 927. [Google Scholar] [CrossRef]
- McKean, M; Carabasi, MH; Stein, MN; Schweizer, MT; Luke, JJ; Narayan, V; et al. Safety and early efficacy results from a phase 1, multicenter trial of PSMA-targeted armored CAR T cells in patients with advanced mCRPC. J Clin Oncol. 2022, 40, 5079. [Google Scholar] [CrossRef]
- Zahid, A; Siegler, EL; Kenderian, SS. CART Cell Toxicities: New Insight into Mechanisms and Management. Clin Hematol Int. 2020, 2(4), 149–55. [Google Scholar] [CrossRef]
- Narayan, V; Barber-Rotenberg, JS; Jung, IY; Lacey, SF; Rech, AJ; Davis, MM; et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. 2022, 28(4), 724–34. [Google Scholar] [CrossRef] [PubMed]
- Serganova, I; Moroz, E; Cohen, I; Moroz, M; Mane, M; Zurita, J; et al. Enhancement of PSMA-Directed CAR Adoptive Immunotherapy by PD-1/PD-L1 Blockade. Mol Ther Oncolytics 2016, 4, 41–54. [Google Scholar] [CrossRef]
- Lauri, C; Chiurchioni, L; Russo, VM; Zannini, L; Signore, A. PSMA Expression in Solid Tumors beyond the Prostate Gland: Ready for Theranostic Applications? J Clin Med. 2022, 11(21), 6590. [Google Scholar] [CrossRef]
- Sampson, JH; Choi, BD; Sanchez-Perez, L; Suryadevara, CM; Snyder, DJ; Flores, CT; et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin Cancer Res. 2014, 20, 972–84. [Google Scholar] [CrossRef]
- O’Rourke, DM; Nasrallah, MP; Desai, A; Melenhorst, JJ; Mansfield, K; Morrissette, JJD; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Posey, AD, Jr.; Schwab, RD; Boesteanu, AC; Steentoft, C; Mandel, U; Engels, B; et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 2016, 44, 1444–54. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R; Yazdanifar, M; Roy, LD; Whilding, LM; Gavrill, A; Maher, J; et al. CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front Immunol. 2019, 10, 1149. [Google Scholar] [CrossRef] [PubMed]
- Beatty, GL; O’Hara, MH; Lacey, SF; Torigian, DA; Nazimuddin, F; Chen, F; et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, PS; Cherkassky, L; Villena-Vargas, J; Colovos, C; Servais, E; Plotkin, J; et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014, 6, 261ra151. [Google Scholar] [CrossRef]
- Roddie, C; O’Reilly, M; Dias Alves Pinto, J; Vispute, K; Lowdell, M. Manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy 2019, 21, 327–40. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, SS; Nath, R; Munoz, J; Tees, M; Miklos, DB; Frank, MJ; et al. ALPHA study: ALLO-501 produced deep and durable responses in patients with relapsed/refractory non-Hodgkin’s lymphoma comparable to autologous CAR T. Blood 2021, 138, 3878. [Google Scholar] [CrossRef]
- Shah, NN; Fry, TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. 2019, 16, 372–85. [Google Scholar] [CrossRef]
- Locke, FL; Malik, S; Tees, MT; Neelapu, SS; Popplewell, L; Abramson, JS; et al. First-in-human data of ALLO-501A, an allogeneic chimeric antigen receptor (CAR) T-cell therapy and ALLO-647 in relapsed/refractory large B-cell lymphoma (R/R LBCL): ALPHA2 study. J Clin Oncol. 2021, 39, 2529. [Google Scholar] [CrossRef]
- Raje, N; Berdeja, J; Lin, Y; Siegel, D; Jagannath, S; Madduri, D; et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019, 380, 1726–37. [Google Scholar] [CrossRef]
- Mailankody, S; Htut, M; Lee, KP; Bensinger, W; Devries, T; Piasecki, J; et al. JCARH125, anti-BCMA CAR T-cell therapy for relapsed/refractory multiple myeloma: initial proof of concept results from a phase 1/2 multicenter study (EVOLVE). Blood 2018, 132, 957. [Google Scholar] [CrossRef]
- Abramson, HN. B-cell maturation antigen (BCMA) as a target for new drug development in relapsed and/or refractory multiple myeloma. Int J Mol Sci. 2020, 21, 5192. [Google Scholar] [CrossRef]
- Hao, S; Jin, J; Jiang, S; Li, Z; Zhang, W; Yang, M; et al. Two-year follow-up of investigator-initiated phase 1 trials of the safety and efficacy of fully human anti-BCMA CAR T cells (CT053) in relapsed/refractory multiple myeloma. Blood 2020, 136, 27–8. [Google Scholar] [CrossRef]
- Frank, MJ; Baird, JH; Kramer, AM; Srinagesh, HK; Patel, S; Brown, AK; et al. CD22-directed CAR T-cell therapy for large B-cell lymphomas progressing after CD19-directed CAR T-cell therapy: a dose-finding phase 1 study. Lancet 2024, 404, 353–63. [Google Scholar] [CrossRef] [PubMed]
- Srinagesh, HK; Jackson, C; Shiraz, P; Jeyakumar, N; Hamilton, MP; Egeler, E; et al. A phase 1 clinical trial of NKTR-255 with CD19-22 CAR-T cell therapy for refractory B-cell acute lymphoblastic leukemia. Blood 2024. [Google Scholar] [CrossRef]
- Wu, X; Cao, Z; Chen, Z; Wang, Y; He, H; Xiao, P; et al. Infectious complications in pediatric patients undergoing CD19+CD22+ chimeric antigen receptor T-cell therapy for relapsed/refractory B-lymphoblastic leukemia. Clin Exp Med. 2024, 24, 87. [Google Scholar] [CrossRef] [PubMed]
- Wu, X; Sun, X; Deng, W; Xu, R; Zhao, Q. Combination therapy of targeting CD20 antibody and immune checkpoint inhibitor may be a breakthrough in the treatment of B-cell lymphoma. Heliyon 2024, 10, e34068. [Google Scholar] [CrossRef]
- Xue, F; Zheng, P; Yang, F; Liu, R; Feng, S; Guo, Y; et al. Salvage CD20-SD-CART therapy in aggressive B-cell lymphoma after CD19 CART treatment failure. Front Oncol. 2024, 14, 1376490. [Google Scholar] [CrossRef]
- Budi, HS; Ahmad, FN; Achmad, H; Ansari, MJ; Mikhailova, MV; Suksatan, W; et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor (CAR) for tumor immunotherapy; recent progress. Stem Cell Res Ther. 2022, 13(1), 40. [Google Scholar] [CrossRef]
- Shabaneh, TB; Stevens, AR; Stull, SM; Shimp, KR; Seaton, BW; Gad, EA; et al. Systemically administered low-affinity HER2 CAR T cells mediate antitumor efficacy without toxicity. J Immunother Cancer 2024, 12(2), e008566. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G; Fu, S; Zhang, Y; Li, S; Guo, Z; Ouyang, D; et al. Antibody recognition of human epidermal growth factor receptor-2 (HER2) juxtamembrane domain enhances anti-tumor response of chimeric antigen receptor (CAR)-T cells. Antibodies 2024, 13, 45. [Google Scholar] [CrossRef]
- Locatelli, F; Quintarelli, C. GD2 target antigen and CAR T cells: does it take more than two to tango? Clin Cancer Res. 2024, 30, 3361–3. [Google Scholar] [CrossRef] [PubMed]
- Gargett, T; Truong, NT; Gardam, B; Yu, W; Ebert, LM; Johnson, A; et al. Safety and biological outcomes following a phase 1 trial of GD2-specific CAR-T cells in patients with GD2-positive metastatic melanoma and other solid cancers. J Immunother Cancer 2024, 12, e008659. [Google Scholar] [CrossRef]
- Kinoshita, S; Ishii, M; Ando, J; Kimura, T; Yamaguchi, T; Harada, S; et al. Rejuvenated iPSC-derived GD2-directed CART cells harbor robust cytotoxicity against small cell lung cancer. Cancer Res Commun. 2024, 4, 723–37. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L; Li, Y; Zheng, D; Zheng, Y; Cui, Y; Qin, L; et al. Bispecific CAR-T cells targeting FAP and GPC3 have the potential to treat hepatocellular carcinoma. Mol Ther Oncolytics 2024, 32, 200817. [Google Scholar] [CrossRef]
- Zheng, X; Liu, X; Lei, Y; Wang, G; Liu, M. Glypican-3: a novel and promising target for the treatment of hepatocellular carcinoma. Front Oncol. 2022, 12, 824208. [Google Scholar] [CrossRef]
- Cao, G; Zhang, G; Liu, M; Liu, J; Wang, Q; Zhu, L; et al. GPC3-targeted CAR-T cells secreting B7H3-targeted BiTE exhibit potent cytotoxicity activity against hepatocellular carcinoma cell in the in vitro assay. Biochem Biophys Res. 2022, 31, 101324. [Google Scholar] [CrossRef]
- Ortiz, MV; Roberts, SS; Glade Bender, J; Shukla, N; Wexler, LH. Immunotherapeutic targeting of GPC3 in pediatric solid embryonal tumors. Front Oncol. 2019, 9, 108. [Google Scholar] [CrossRef]
- Yin, L; Chen, GL; Xiang, Z; Liu, YL; Li, XY; Bi, JW; et al. Current progress in chimeric antigen receptor-modified T cells for the treatment of metastatic breast cancer. Biomed Pharmacother. 2023, 162, 114648. [Google Scholar] [CrossRef] [PubMed]
- Meng, S; Li, M; Qin, L; Lv, J; Wu, D; Zheng, D; et al. The onco-embryonic antigen ROR1 is a target of chimeric antigen T cells for colorectal cancer. Int Immunopharmacol. 2023, 121, 110402. [Google Scholar] [CrossRef]
- Tran, TM; Chand Thakuri, BK; Nurmukhambetova, S; Lee, JJ; Hu, P; Tran, NQ; et al. Armored TGFβRIIDN ROR1-CAR T cells reject solid tumors and resist suppression by constitutively-expressed and treatment-induced TGFβ1. J Immunother Cancer 2024, 12, e008261. [Google Scholar] [CrossRef]
- Liu, X; Xu, Y; Xiong, W; Yin, B; Huang, Y; Chu, J; et al. Development of a TCR-like antibody and chimeric antigen receptor against NY-ESO-1/HLA-A2 for cancer immunotherapy. J Immunother Cancer 2022, 10, e004035. [Google Scholar] [CrossRef]
- Rochigneux, P; Chanez, B; De Rauglaudre, B; Mitry, E; Chabannon, C; Gilabert, M. Adoptive cell therapy in hepatocellular carcinoma: biological rationale and first results in early phase clinical trials. Cancers 2021, 13, 271. [Google Scholar] [CrossRef]
- Mai, Q; He, B; Deng, S; Zeng, Q; Xu, Y; Wang, C; et al. Efficacy of NKG2D CAR-T cells with IL-15/IL-15Rα signaling for treating Epstein-Barr virus-associated lymphoproliferative disorder. Exp Hematol Oncol. 2024, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Wang, D; Dou, L; Sui, L; Xue, Y; Xu, S. Natural killer cells in cancer immunotherapy. MedComm 2024, 5, e626. [Google Scholar] [CrossRef]
- Fuertes, MB; Domaica, CI; Zwirner, NW. Leveraging NKG2D ligands in immuno-oncology. Front Immunol. 2021, 12, 713158. [Google Scholar] [CrossRef]
- Zhang, Y; Zhang, C; He, M; Xing, W; Hou, R; Zhang, H. Co-expression of IL-21-enhanced NKG2D CAR-NK cell therapy for lung cancer. BMC Cancer 2024, 24, 119. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez-Navarro, M; Fernández, A; Escudero, A; Esteso, G; Campos-Silva, C; Navarro-Aguadero, MÁ; et al. NKG2D-CAR memory T cells target pediatric T-cell acute lymphoblastic leukemia in vitro and in vivo but fail to eliminate leukemia initiating cells. Front Immunol. 2023, 14, 1187665. [Google Scholar] [CrossRef]
- Mallis, RJ; Brazin, KN; Duke-Cohan, JS; Hwang, W; Wang, JH; Wagner, G; et al. NMR: an essential structural tool for integrative studies of T cell development, pMHC ligand recognition and TCR mechanobiology. J Biomol NMR 2019, 73, 319–32. [Google Scholar] [CrossRef]
- Shepherd, A; Bennychen, B; Ahmed, Z; Weeratna, RD; McComb, S. A flow cytometry-based method for assessing CAR cell binding kinetics using stable CAR Jurkat cells. Bio-protocol 2024, 14, e5021. [Google Scholar] [CrossRef]
- Potthoff, B; McBlane, F; Spindeldreher, S; Sickert, D. A cell-based immunogenicity assay to detect antibodies against chimeric antigen receptor expressed by tisagenlecleucel. J Immunol Methods 2020, 476, 112692. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z; Wang, J; Zhao, Y; Jin, J; Si, W; Chen, L; et al. 3D live imaging and phenotyping of CAR-T cell mediated-cytotoxicity using high-throughput Bessel oblique plane microscopy. Nat Commun. 2024, 15, 6677. [Google Scholar] [CrossRef] [PubMed]
- Ghorai, SK; Pearson, AN. Current strategies to improve chimeric antigen receptor T (CAR-T) cell persistence. Cureus 2024, 16, e65291. [Google Scholar] [CrossRef] [PubMed]
- Cappell, KM; Kochenderfer, JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. 2023, 20, 359–71. [Google Scholar] [CrossRef]
- Srivastava, S; Riddell, SR. Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J Immunol. 2018, 200, 459–68. [Google Scholar] [CrossRef]
- Huang, S; Wang, X; Wang, Y; Wang, Y; Fang, C; Wang, Y; et al. Deciphering and advancing CAR T-cell therapy with single-cell sequencing technologies. Mol Cancer 2023, 22, 80. [Google Scholar] [CrossRef]
- Si, X; Xiao, L; Brown, CE; Wang, D. Preclinical evaluation of CAR T cell function: in vitro and in vivo models. Int J Mol Sci. 2022, 23, 3154. [Google Scholar] [CrossRef]
- Hort, S; Herbst, L; Bäckel, N; Erkens, F; Niessing, B; Frye, M; et al. Toward rapid, widely available autologous CAR-T cell therapy—artificial intelligence and automation enabling the smart manufacturing hospital. Front Med. 2022, 9, 913287. [Google Scholar] [CrossRef]
- Portale, F; Di Mitri, D. NK Cells in Cancer: Mechanisms of Dysfunction and Therapeutic Potential. Int J Mol Sci. 2023, 24(11), 9521. [Google Scholar] [CrossRef]
- Yu, Y. The Function of NK Cells in Tumor Metastasis and NK Cell-Based Immunotherapy. Cancers 2023, 15(8), 2323. [Google Scholar] [CrossRef]
- Zhou, Y; Cheng, L; Liu, L; Li, X. NK cells are never alone: crosstalk and communication in tumour microenvironments. Mol Cancer 2023, 22(1), 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, W; Liu, Y; He, Z; Li, L; Liu, S; Jiang, M; et al. Breakthrough of solid tumor treatment: CAR-NK immunotherapy. Cell Death Discov. 2024, 10(1), 40. [Google Scholar] [CrossRef] [PubMed]
- Uscanga-Palomeque, AC; Chávez-Escamilla, AK; Alvizo-Báez, CA; Saavedra-Alonso, S; Terrazas-Armendáriz, LD; Tamez-Guerra, RS; et al. CAR-T Cell Therapy: From the Shop to Cancer Therapy. Int J Mol Sci. 2023, 24(21), 15688. [Google Scholar] [CrossRef]
- Li, T; Niu, M; Zhang, W; Qin, S; Zhou, J; Yi, M. CAR-NK cells for cancer immunotherapy: recent advances and future directions. Front Immunol. 2024, 15, 1361194. [Google Scholar] [CrossRef]
- Bashiri Dezfouli, A; Yazdi, M; Pockley, AG; Khosravi, M; Kobold, S; Wagner, E; et al. NK Cells Armed with Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development. Cells 2021, 10(12), 3390. [Google Scholar] [CrossRef] [PubMed]
- Daher, M; Melo Garcia, L; Li, Y; Rezvani, K. CAR-NK cells: the next wave of cellular therapy for cancer. Clin Transl Immunol. 2021, 10(4), e1274. [Google Scholar] [CrossRef]
- Li, H; Song, W; Li, Z; Zhang, M. Preclinical and clinical studies of CAR-NK-cell therapies for malignancies. Front Immunol. 2022, 13, 992232. [Google Scholar] [CrossRef]
- Kong, JC; Sa’ad, MA; Vijayan, HM; Ravichandran, M; Balakrishnan, V; Tham, SK; et al. Chimeric antigen receptor-natural killer cell therapy: current advancements and strategies to overcome challenges. Front Immunol. 2024, 15, 1384039. [Google Scholar] [CrossRef]
- Ma, S; Caligiuri, MA; Yu, J. Harnessing IL-15 signaling to potentiate NK cell-mediated cancer immunotherapy. Trends Immunol. 2022, 43(10), 833–47. [Google Scholar] [CrossRef]
- Schmidt, D; Ebrahimabadi, S; Gomes, KRS; de Moura Aguiar, G; Cariati Tirapelle, M; Nacasaki Silvestre, R; et al. Engineering CAR-NK cells: how to tune innate killer cells for cancer immunotherapy. Immunother Adv. 2022, 2(1), ltac003. [Google Scholar] [CrossRef] [PubMed]
- Matosevic, S. Viral and Nonviral Engineering of Natural Killer Cells as Emerging Adoptive Cancer Immunotherapies. J Immunol Res. 2018, 2018, 4054815. [Google Scholar] [CrossRef]
- Islam, R; Pupovac, A; Evtimov, V; Boyd, N; Shu, R; Boyd, R; et al. Enhancing a Natural Killer: Modification of NK Cells for Cancer Immunotherapy. Cells 2021, 10(5), 1058. [Google Scholar] [CrossRef]
- Tarannum, M; Romee, R; Shapiro, RM. Innovative Strategies to Improve the Clinical Application of NK Cell-Based Immunotherapy. Front Immunol. 2022, 13, 859177. [Google Scholar] [CrossRef]
- Lu, C; Liu, Y; Ali, NM; Zhang, B; Cui, X. The role of innate immune cells in the tumor microenvironment and research progress in anti-tumor therapy. Front Immunol. 2023, 13, 1039260. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y; Liu, J. Emerging roles of CAR-NK cell therapies in tumor immunotherapy: current status and future directions. Cell Death Discov. 2024, 10(1), 318. [Google Scholar] [CrossRef]
- Lu, H; Zhao, X; Li, Z; Hu, Y; Wang, H. From CAR-T Cells to CAR-NK Cells: A Developing Immunotherapy Method for Hematological Malignancies. Front Oncol. 2021, 11, 720501. [Google Scholar] [CrossRef] [PubMed]
- Li, W; Wang, X; Zhang, X; Aziz, AuR; Wang, D. CAR-NK Cell Therapy: A Transformative Approach to Overcoming Oncological Challenges. Biomolecules 2024, 14(8), 1035. [Google Scholar] [CrossRef]
- Keshavarz, A; Salehi, A; Khosravi, S; Shariati, Y; Nasrabadi, N; Kahrizi, MS; et al. Recent findings on chimeric antigen receptor (CAR)-engineered immune cell therapy in solid tumors and hematological malignancies. Stem Cell Res Ther. 2022, 13(1), 482. [Google Scholar] [CrossRef]
- Rossi, F; Fredericks, N; Snowden, A; Allegrezza, MJ; Moreno-Nieves, UY. Next Generation Natural Killer Cells for Cancer Immunotherapy. Front Immunol. 2022, 13, 886429. [Google Scholar] [CrossRef] [PubMed]
- Fares, J; Davis, ZB; Rechberger, JS; Toll, SA; Schwartz, JD; Daniels, DJ; et al. Advances in NK cell therapy for brain tumors. NPJ Precis Oncol. 2023, 7(1), 17. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y; Liu, J. Emerging roles of CAR-NK cell therapies in tumor immunotherapy: current status and future directions. Cell Death Discov. 2024, 10(1), 318. [Google Scholar] [CrossRef]
- Valeri, A; García-Ortiz, A; Castellano, E; Córdoba, L; Maroto-Martín, E; Encinas, J; et al. Overcoming tumor resistance mechanisms in CAR-NK cell therapy. Front Immunol. 2022, 13, 953849. [Google Scholar] [CrossRef]
- Wang, W; Liu, Y; He, Z; Li, L; Liu, S; Jiang, M; et al. Breakthrough of solid tumor treatment: CAR-NK immunotherapy. Cell Death Discov. 2024, 10(1), 40. [Google Scholar] [CrossRef]
- Li, J; Hu, H; Lian, K; Zhang, D; Hu, P; He, Z; et al. CAR-NK cells in combination therapy against cancer: A potential paradigm. Heliyon 2024, 10(5), e27196. [Google Scholar] [CrossRef]
- Allemailem, KS; Alsahli, MA; Almatroudi, A; Alrumaihi, F; Al Abdulmonem, W; Moawad, AA; et al. Innovative Strategies of Reprogramming Immune System Cells by Targeting CRISPR/Cas9-Based Genome-Editing Tools: A New Era of Cancer Management. Int J Nanomedicine 2023, 18, 5531–59. [Google Scholar] [CrossRef]
- Lv, Z; Luo, F; Chu, Y. Strategies for overcoming bottlenecks in allogeneic CAR-T cell therapy. Front Immunol. 2023, 14, 1199145. [Google Scholar] [CrossRef]
- Tang, C; Zhang, Y. Potential alternatives to αβ-T cells to prevent graft-versus-host disease (GvHD) in allogeneic chimeric antigen receptor (CAR)-based cancer immunotherapy: A comprehensive review. Pathol Res Pract. 2024, 262, 155518. [Google Scholar] [CrossRef]
- Yazdanifar, M; Barbarito, G; Bertaina, A; Airoldi, I. γδ T Cells: The Ideal Tool for Cancer Immunotherapy. Cells 2020, 9(5), 1305. [Google Scholar] [CrossRef] [PubMed]
- Lonez, C; Breman, E. Allogeneic CAR-T Therapy Technologies: Has the Promise Been Met? Cells 2024, 13(2), 146. [Google Scholar] [CrossRef]
- Caldwell, KJ; Gottschalk, S; Talleur, AC. Allogeneic CAR Cell Therapy-More Than a Pipe Dream. Front Immunol. 2021, 11, 618427. [Google Scholar] [CrossRef]
- Song, P; Zhang, Q; Xu, Z; Shi, Y; Jing, R; Luo, D. CRISPR/Cas-based CAR-T cells: production and application. Biomark Res. 2024, 12(1), 54. [Google Scholar] [CrossRef] [PubMed]
- Courtney, AN; Tian, G; Metelitsa, LS. Natural killer T cells and other innate-like T lymphocytes as emerging platforms for allogeneic cancer cell therapy. Blood 2023, 141(8), 869–76. [Google Scholar] [CrossRef] [PubMed]
- Hillhouse, EE; Delisle, JS; Lesage, S. Immunoregulatory CD4(-)CD8(-) T cells as a potential therapeutic tool for transplantation, autoimmunity, and cancer. Front Immunol. 2013, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z; Zheng, Y; Sheng, J; Han, Y; Yang, Y; Pan, H; et al. CD3+CD4-CD8- (Double-Negative) T Cells in Inflammation, Immune Disorders and Cancer. Front Immunol. 2022, 13, 816005. [Google Scholar] [CrossRef]
- Liu, XC; Sun, KN; Zhu, HR; Dai, YL; Liu, XF. Diagnostic and prognostic value of double-negative T cells in colorectal cancer. Heliyon 2024, 10(14), e34645. [Google Scholar] [CrossRef]
- Wei, W; Chen, ZN; Wang, K. CRISPR/Cas9: A Powerful Strategy to Improve CAR-T Cell Persistence. Int J Mol Sci. 2023, 24(15), 12317. [Google Scholar] [CrossRef]
- Rezalotfi, A; Fritz, L; Förster, R; Bošnjak, B. Challenges of CRISPR-Based Gene Editing in Primary T Cells. Int J Mol Sci. 2022, 23(3), 1689. [Google Scholar] [CrossRef]
- Zhang, P; Zhang, G; Wan, X. Challenges and new technologies in adoptive cell therapy. J Hematol Oncol. 2023, 16(1), 97. [Google Scholar] [CrossRef]
- Hadiloo, K; Tahmasebi, S; Esmaeilzadeh, A. CAR-NKT cell therapy: a new promising paradigm of cancer immunotherapy. Cancer Cell Int. 2023, 23(1), 86. [Google Scholar] [CrossRef] [PubMed]
- Shissler, SC; Bollino, DR; Tiper, IV; Bates, JP; Derakhshandeh, R; Webb, TJ. Immunotherapeutic strategies targeting natural killer T cell responses in cancer. Immunogenetics 2016, 68(8), 623–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y; Wang, G; Chai, D; Dang, Y; Zheng, J; Li, H. iNKT: A new avenue for CAR-based cancer immunotherapy. Transl Oncol. 2022, 17, 101342. [Google Scholar] [CrossRef]
- Fujii, SI; Shimizu, K. Exploiting Antitumor Immunotherapeutic Novel Strategies by Deciphering the Cross Talk between Invariant NKT Cells and Dendritic Cells. Front Immunol. 2017, 8, 886. [Google Scholar] [CrossRef]
- Wu, Z; Zheng, Y; Sheng, J; Han, Y; Yang, Y; Pan, H; et al. CD3+CD4-CD8- (Double-Negative) T Cells in Inflammation, Immune Disorders and Cancer. Front Immunol. 2022, 13, 816005. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K; Voelkl, S; Heymann, J; Przybylski, GK; Mondal, K; Laumer, M; et al. Isolation and characterization of human antigen-specific TCR alpha beta+ CD4(-)CD8- double-negative regulatory T cells. Blood 2005, 105(7), 2828–35. [Google Scholar] [CrossRef]
- Brandt, D; Hedrich, CM. TCRαβ+CD3+CD4-CD8- (double negative) T cells in autoimmunity. Autoimmun Rev. 2018, 17(4), 422–30. [Google Scholar] [CrossRef]
- Ye, H; Chang, Y; Zhao, X; Huang, X. Characterization of CD3+CD4-CD8- (double negative) T cells reconstitution in patients following hematopoietic stem-cell transplantation. Transpl Immunol. 2011, 25(4), 180–6. [Google Scholar] [CrossRef]
- Vasic, D; Lee, JB; Leung, Y; Khatri, I; Na, Y; Abate-Daga, D; et al. Allogeneic double-negative CAR-T cells inhibit tumor growth without off-tumor toxicities. Sci Immunol. 2022, 7(70), eabl3642. [Google Scholar] [CrossRef]
- Hadiloo, K; Taremi, S; Heidari, M; Esmaeilzadeh, A. The CAR macrophage cells, a novel generation of chimeric antigen-based approach against solid tumors. Biomark Res. 2023, 11(1), 103. [Google Scholar] [CrossRef] [PubMed]
- Basak, U; Sarkar, T; Mukherjee, S; Chakraborty, S; Dutta, A; Dutta, S; et al. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front Immunol. 2023, 14, 1295257. [Google Scholar] [CrossRef]
- Li, N; Geng, S; Dong, ZZ; Jin, Y; Ying, H; Li, HW; et al. A new era of cancer immunotherapy: combining revolutionary technologies for enhanced CAR-M therapy. Mol Cancer 2024, 23(1), 117. [Google Scholar] [CrossRef]
- Sadri, M; Heidari, S; Faridzadeh, A; Roozbehani, M; Toosi, S; Mahmoudian, RA; et al. Potential applications of macrophages in cancer immunotherapy. Biomed Pharmacother. 2024, 178, 117161. [Google Scholar] [CrossRef]
- Pan, K; Farrukh, H; Chittepu, VCSR; Xu, H; Pan, CX; Zhu, Z. CAR race to cancer immunotherapy: from CAR T, CAR NK to CAR macrophage therapy. J Exp Clin Cancer Res. 2022, 41(1), 119. [Google Scholar] [CrossRef] [PubMed]
- Yu, T; Lu, Y; Fang, J; Jiang, X; Lu, Y; Zheng, J; et al. Chimeric antigen receptor-based immunotherapy in breast cancer: Recent progress in China. Cancer 2024, 130(S8), 1378–91. [Google Scholar] [CrossRef]
- Dong, X; Fan, J; Xie, W; Wu, X; Wei, J; He, Z; et al. Efficacy evaluation of chimeric antigen receptor-modified human peritoneal macrophages in the treatment of gastric cancer. Br J Cancer 2023, 129(3), 551–62. [Google Scholar] [CrossRef]
- Chen, K; Liu, ML; Wang, JC; Fang, S. CAR-macrophage versus CAR-T for solid tumors: The race between a rising star and a superstar. Biomol Biomed. 2024, 24(3), 465–76. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y; Cai, X; Syahirah, R; Yao, Y; Xu, Y; Jin, G; et al. CAR-neutrophil mediated delivery of tumor-microenvironment responsive nanodrugs for glioblastoma chemo-immunotherapy. Nat Commun. 2023, 14(1), 2266. [Google Scholar] [CrossRef]
- Harris, JD; Chang, Y; Syahirah, R; Lian, XL; Deng, Q; Bao, X. Engineered anti-prostate cancer CAR-neutrophils from human pluripotent stem cells. J Immunol Regen Med. 2023, 20, 100074. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y; Syahirah, R; Wang, X; Jin, G; Torregrosa-Allen, S; Elzey, BD; et al. Engineering chimeric antigen receptor neutrophils from human pluripotent stem cells for targeted cancer immunotherapy. Cell Rep. 2022, 40(3), 111128. [Google Scholar] [CrossRef]
- Patel, RP; Ghilardi, G; Zhang, Y; Chiang, YH; Xie, W; Guruprasad, P; et al. CD5 deletion enhances the antitumor activity of adoptive T cell therapies. Sci Immunol. 2024, 9(97), eadn6509. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z; Mu, W; Zhao, Y; Jia, X; Liu, J; Wei, Q; et al. The rational development of CD5-targeting biepitopic CARs with fully human heavy-chain-only antigen recognition domains. Mol Ther. 2021, 29(9), 2707–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z; Wang, H; D’Souza, C; Sun, S; Kostenko, L; Eckle, SB; et al. Mucosal-associated invariant T-cell activation and accumulation after in vivo infection depends on microbial riboflavin synthesis and co-stimulatory signals. Mucosal Immunol. 2017, 10(1), 58–68. [Google Scholar] [CrossRef]
- Li, YR; Wilson, M; Yang, L. Target tumor microenvironment by innate T cells. Front Immunol. 2022, 13, 999549. [Google Scholar] [CrossRef]
- Chengalroyen, MD. Current Perspectives and Challenges of MAIT Cell-Directed Therapy for Tuberculosis Infection. Pathogens 2023, 12(11), 1343. [Google Scholar] [CrossRef] [PubMed]
- Legoux, F; Salou, M; Lantz, O. MAIT Cell Development and Functions: the Microbial Connection. Immunity 2020, 53(4), 710–23. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M; Karhan, E; Kozhaya, L; Placek, L; Chen, X; Yigit, M; et al. Engineering Human MAIT Cells with Chimeric Antigen Receptors for Cancer Immunotherapy. J Immunol. 2022, 209(8), 1523–31. [Google Scholar] [CrossRef]
- Ruixin, S.; Yifan, L.; Yansha, S.; Min, Z.; Yiwei, D.; Xiaoli, H.; Bizhi, S.; Hua, J.; Zonghai, L. Dual targeting chimeric antigen receptor cells enhance antitumour activity by overcoming T cell exhaustion in pancreatic cancer. In British journal of pharmacology; Advance online publication, 2024; Volume 10.1111/bph. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Y.; Wang, P.; Li, S.; Dong, Y.; Zhou, M.; Shi, B.; Jiang, H.; Sun, R.; Li, Z. FAP-targeted CAR-T suppresses MDSCs recruitment to improve the antitumor efficacy of claudin18.2-targeted CAR-T against pancreatic cancer. Journal of translational medicine 2023, 21(1), 255. [Google Scholar] [CrossRef]
- Bughda, R.; Dimou, P.; D’Souza, R.R.; Klampatsa, A. Fibroblast Activation Protein (FAP)-Targeted CAR-T Cells: Launching an Attack on Tumor Stroma. ImmunoTargets and therapy 2021, 10, 313–323. [Google Scholar] [CrossRef]
- Wei, F; Liu, H; Wang, Y; Li, Y; Han, S. Engineering macrophages and their derivatives: A new hope for antitumor therapy. Biomed Pharmacother. 2024, 177, 116925. [Google Scholar] [CrossRef]
- Sloas, C; Gill, S; Klichinsky, M. Engineered CAR-Macrophages as Adoptive Immunotherapies for Solid Tumors. Front Immunol. 2021, 12, 783305. [Google Scholar] [CrossRef]
- Davidov, V; Jensen, G; Mai, S; Chen, SH; Pan, PY. Corrigendum: Analyzing One Cell at a TIME: Analysis of Myeloid Cell Contributions in the Tumor Immune Microenvironment. Front Immunol. 2021, 11, 645213. [Google Scholar] [CrossRef]
- Ugel, S; De Sanctis, F; Mandruzzato, S; Bronte, V. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015, 125(9), 3365–76. [Google Scholar] [CrossRef]
- Szebeni, GJ; Vizler, C; Nagy, LI; Kitajka, K; Puskas, LG. Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. Int J Mol Sci. 2016, 17(11), 1958. [Google Scholar] [CrossRef]
- Kakarla, S; Gottschalk, S. CAR T Cells for Solid Tumors. Cancer J. 2014, 20, 151–55. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M; Purdon, TJ; Spriggs, D; Koneru, S; Brentjens, RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology 2015, 4(3), e994446. [Google Scholar] [CrossRef]
- Chen, X; Sandrine, IK; Yang, M; Tu, J; Yuan, X. MUC1 and MUC16: critical for immune modulation in cancer therapeutics. Front Immunol. 2024, 15, 1356913. [Google Scholar] [CrossRef] [PubMed]
- Smole, A; Benton, A; Poussin, MA; Eiva, MA; Mezzanotte, C; Camisa, B; et al. Expression of inducible factors reprograms CAR-T cells for enhanced function and safety. Cancer Cell. 2022, 40(12), 1470–87.e7. [Google Scholar] [CrossRef]
- Esmaeilzadeh, A; Tahmasebi, S; Athari, SS. Chimeric antigen receptor-T cell therapy: Applications and challenges in treatment of allergy and asthma. Biomed Pharmacother. 2020, 123, 109685. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M; O’Cearbhaill, R; Pendharkar, S; Spriggs, DR; Brentjens, RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16 ecto directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. 2015, 13, 1–11. [Google Scholar] [CrossRef]
- Kuznetsova, AV; Glukhova, XA; Popova, OP; Beletsky, IP; Ivanov, AA. Contemporary Approaches to Immunotherapy of Solid Tumors. Cancers 2024, 16, 2270. [Google Scholar] [CrossRef] [PubMed]
- Justiz-Vaillant, A.; Pandit, B.R.; Unakal, C.; Vuma, S.; Akpaka, P.E. A Comprehensive Review About the Use of Monoclonal Antibodies in Cancer Therapy. Antibodies 2025, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Justiz-Vaillant, A.; Gopaul, D.; Soodeen, S.; Unakal, C.; Thompson, R.; Pooransingh, S.; Arozarena-Fundora, R.; Asin-Milan, O.; Akpaka, P.E. Advancements in Immunology and Microbiology Research: A Comprehensive Exploration of Key Areas. Microorganisms 2024, 12, 1672. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
“Molecular docking simulation of anti-HER2 mAbs with homology model of human HER2. (A) The docking model of the antibody and antigen. The four domains of Her2 are presented in different colors. The variable regions of heavy and light chains of trastuzumab and Ab8 are represented in violet and orange, respectively. The different domains of HER2 are labelled. (B) The predominant residues in the interaction between Ab8 and HER2.” Taken and modified from [133].
Figure 1.
“Molecular docking simulation of anti-HER2 mAbs with homology model of human HER2. (A) The docking model of the antibody and antigen. The four domains of Her2 are presented in different colors. The variable regions of heavy and light chains of trastuzumab and Ab8 are represented in violet and orange, respectively. The different domains of HER2 are labelled. (B) The predominant residues in the interaction between Ab8 and HER2.” Taken and modified from [133].
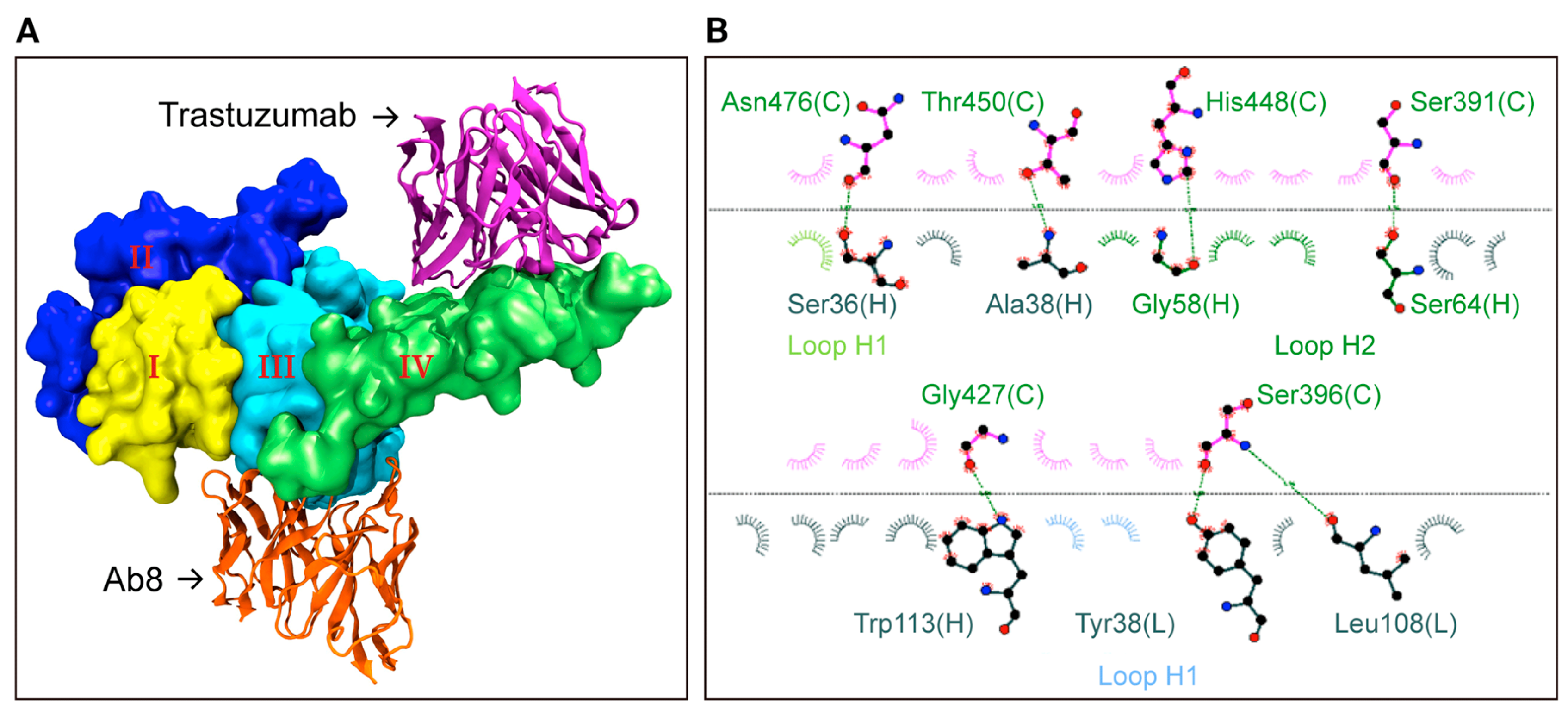
Figure 2.
Soluble Factors in the Bi-Directional Crosstalk between NK Cells and Immune Cells in the Tumor Microenvironment (TME). Taken and modified from [161].
Figure 2.
Soluble Factors in the Bi-Directional Crosstalk between NK Cells and Immune Cells in the Tumor Microenvironment (TME). Taken and modified from [161].
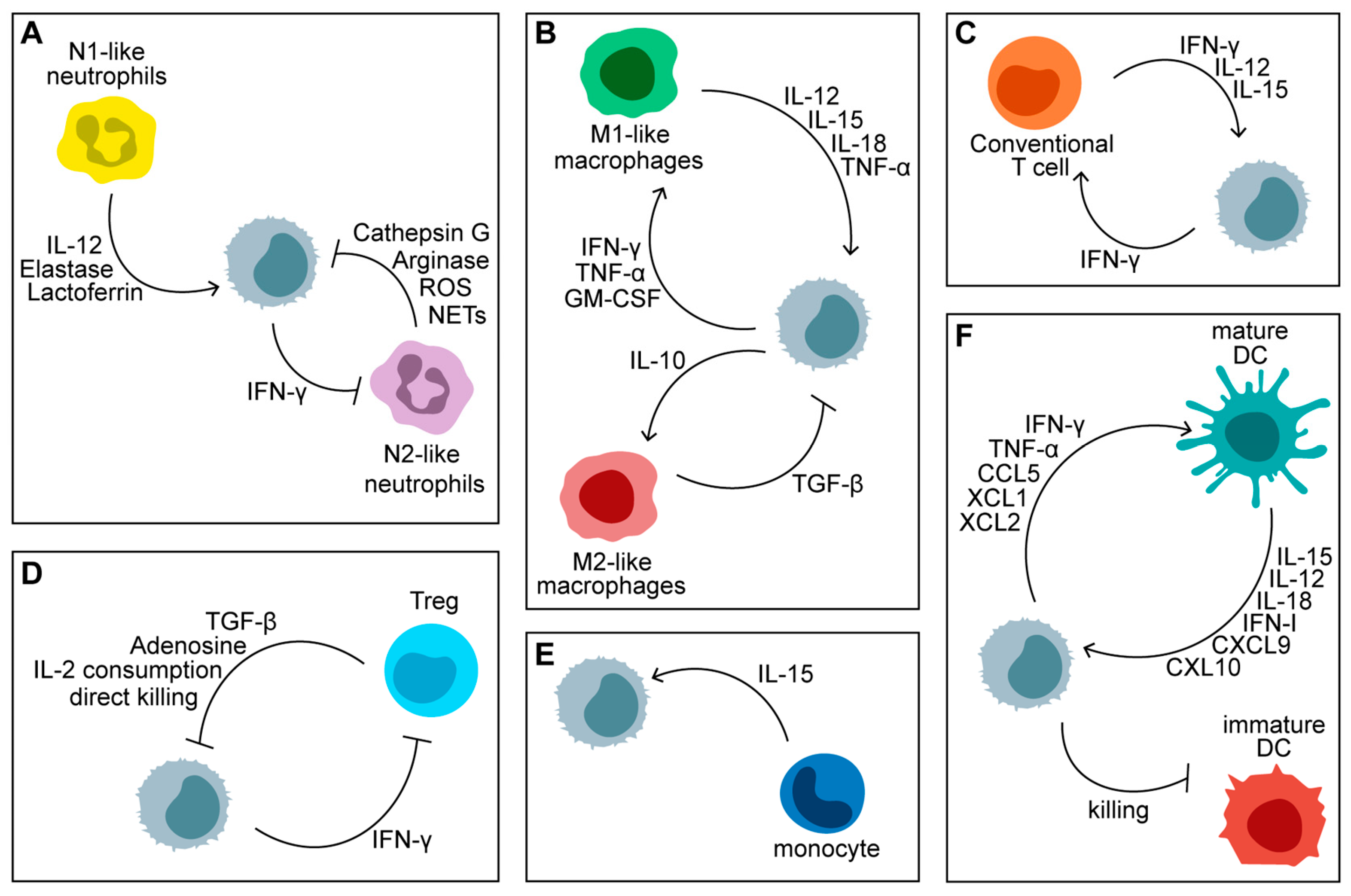
Figure 4.
Clinical trials of CAR-T cell therapy of solid tumors. Modified from [242].
Figure 4.
Clinical trials of CAR-T cell therapy of solid tumors. Modified from [242].
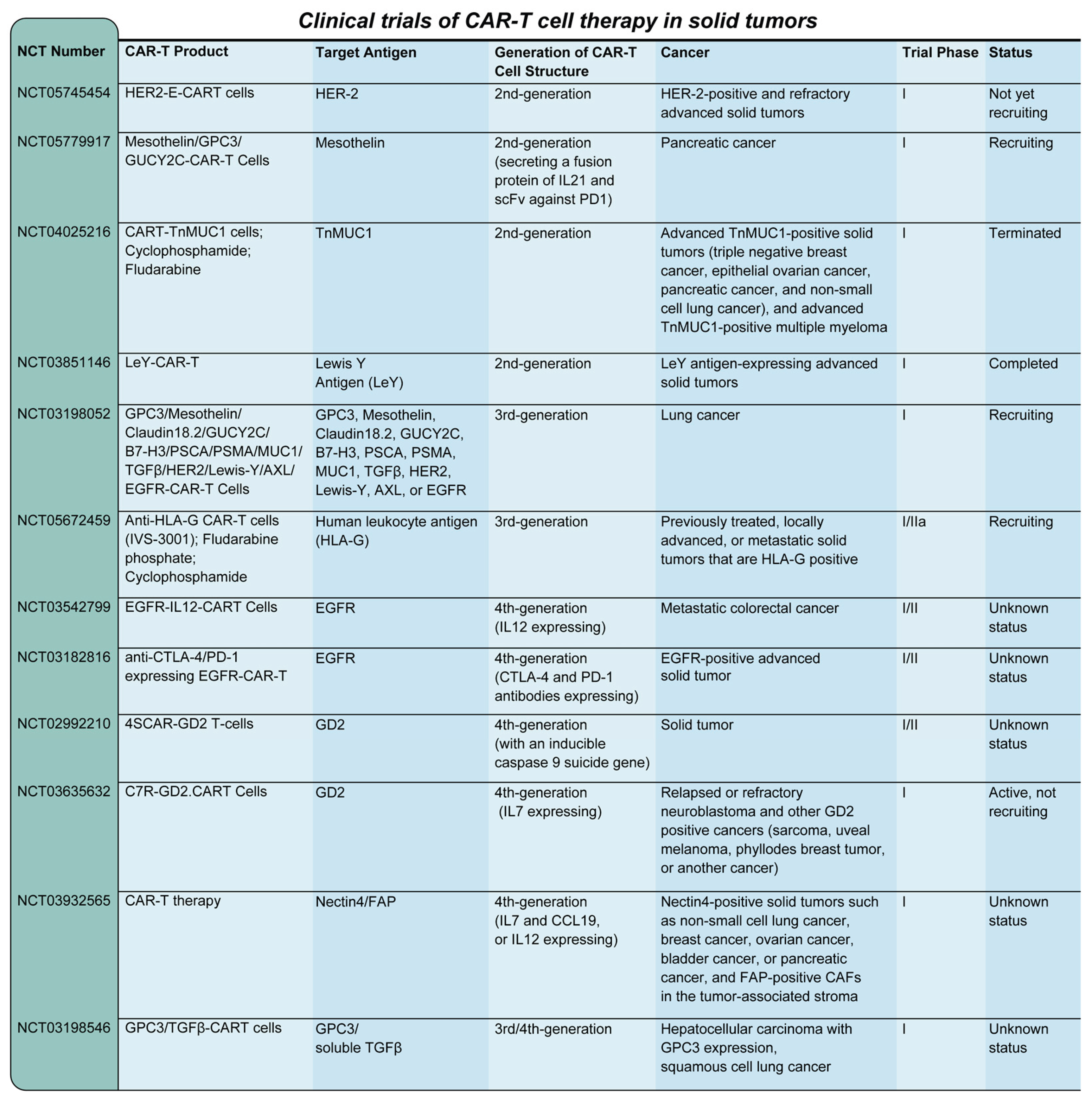

| CAR-T cell Therapy | Targeted antigen |
Cancer Type | Notes |
|---|---|---|---|
| bb21217 | BCMA | Multiple myeloma | Enhanced persistence CAR-T therapy; in clinical trials |
| CTL019 | CD19 | B-cell malignancies | Early version of Kymriah; basis for FDA-approved therapy |
| CART-PSMA | PSMA | Prostate cancer | Investigational CAR-T therapy; in early-phase trials |
| CAR-EGFRvIII | EGFRvIII | Glioblastoma | Investigational CAR-T therapy targeting specific EGFR mutation |
| Mesothelin-CAR-T | Mesothelin | Mesothelioma, ovarian cancer |
Experimental CAR-T therapy targeting mesothelin |
| MUC1-CAR-T | MUC1 | Various solid tumours |
Investigational CAR-T therapy for multiple epithelial cancers |
| ALLO-501 (Allogeneic) | CD19 | Non-Hodgkin lymphoma | Allogeneic CAR-T therapy under investigation; uses donor-derived cells |
| JCARH125 | BCMA | Multiple myeloma | Experimental CAR-T therapy; in clinical trials |
| CT053 | BCMA | Multiple myeloma | Investigational CAR-T therapy; in clinical trials |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



