Submitted:
20 December 2025
Posted:
22 December 2025
You are already at the latest version
Abstract
Rapid phylogenomic analysis is essential for outbreak surveillance and large-scale viral comparative genomics, yet conventional alignment-based workflows remain computationally intensive and difficult to deploy at scale. Covary is a computational framework designed for large-scale biological sequence analysis. It is a translation-aware, alignment-free machine learning framework that encodes genomic information into biologically informed vector representations, enabling efficient genome-scale comparison without multiple sequence alignment (MSA). Here, Covary was applied to thousands-scale analysis of outbreak-causing viral genomes to assess its scalability and biological resolution. A total of 4,000 complete genomes of SARS-CoV-2, dengue virus, measles virus, and alphainfluenza virus were retrieved from the NCBI Viral Genomes Resource, of which 3,831 passed quality filtering and were analyzed using Covary. Results showed that Covary rapidly processed all genomes and consistently grouped sequences according to expected taxonomic assignments and known ingroup structure, including SARS-CoV-2 Pango lineages, dengue virus subtypes, measles virus geographic origin, and alphainfluenza virus clades. Covary completed the analysis in 45 minutes on free-tier Google Colab, inferring genome-wide relationships using modest computational resources. These results demonstrate that Covary enables rapid, alignment-free phylogenomic analysis of thousands of outbreak-causing viral genomes without requiring advanced computational infrastructure. In conclusion, Covary represents a scalable, deploy-ready machine learning pipeline for genome-informed outbreak surveillance and monitoring systems.
Keywords:
1. Introduction
2. Materials and Methods
3. Results
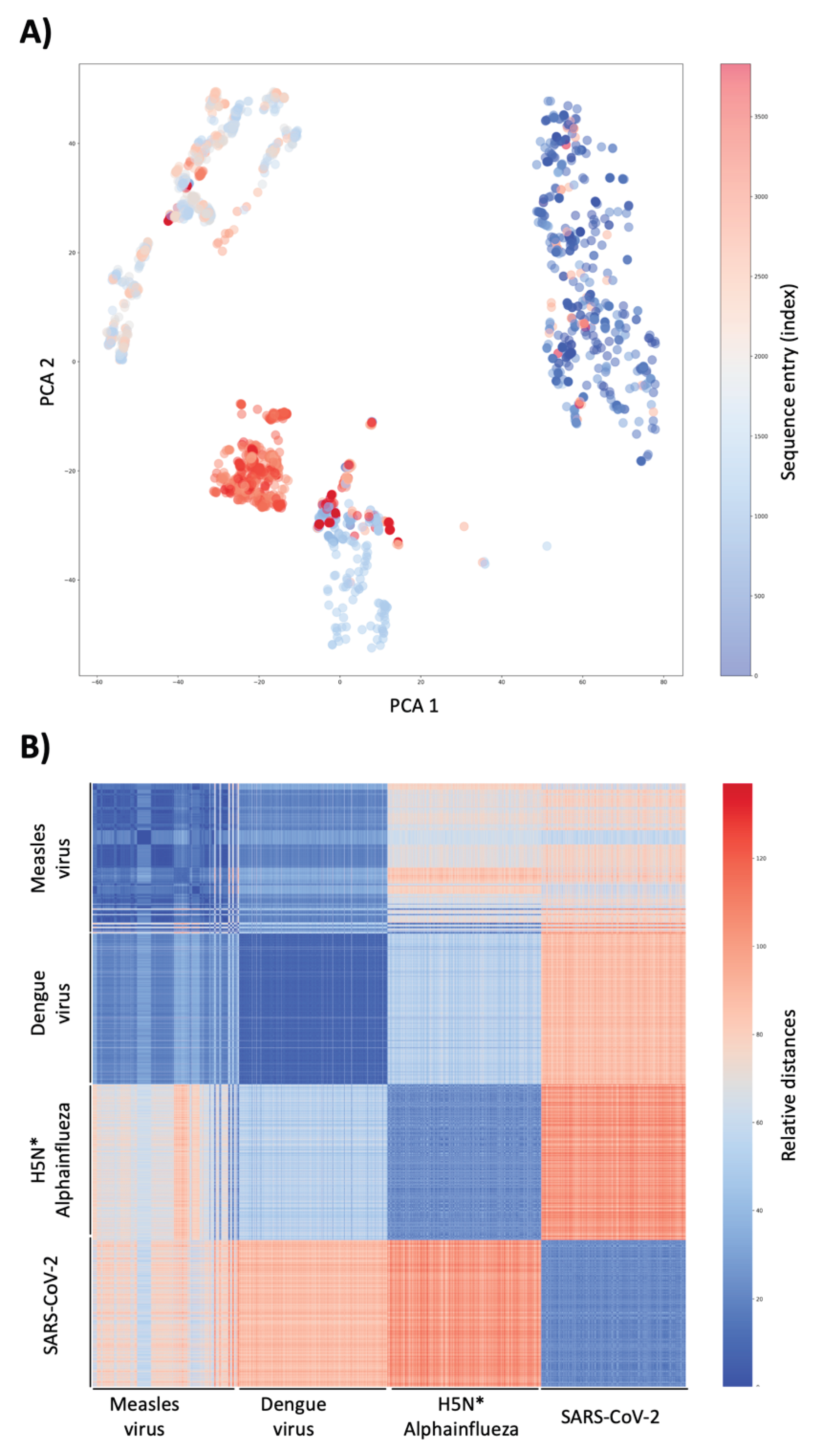
3.1. Genome-Scale Embedding Reveals Clear Inter-Viral Separation
3.2. Heatmap Visualization Confirms Structured Clustering of Viral Embeddings
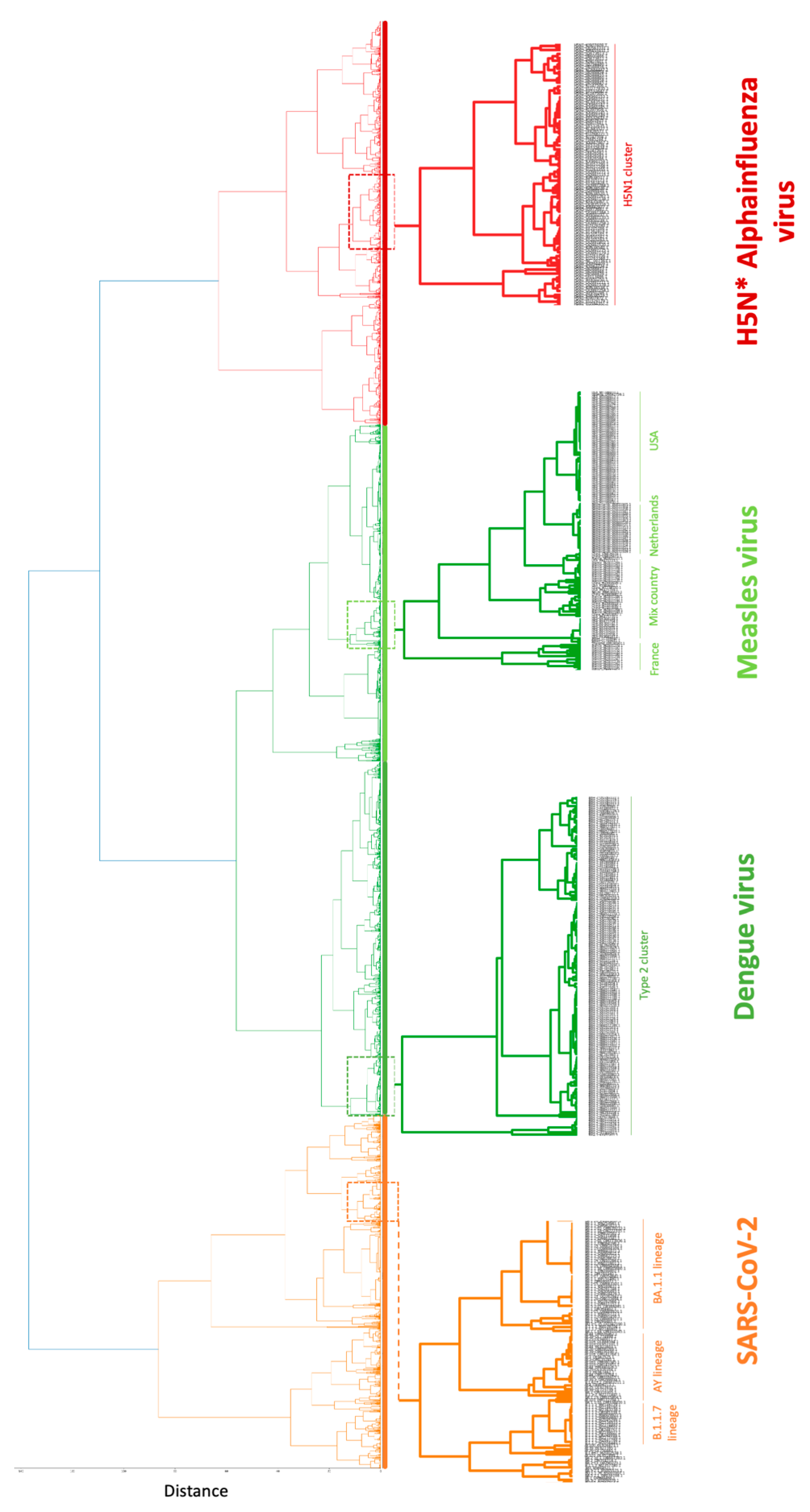
3.3. Hierarchical Clustering Resolves Viral Classification and Ingroup Structure
3.4. Ingroup Diversification Reflects Known Epidemiological and Lineage Structure
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brister, J. R.; Ako-adjei, D.; Bao, Y.; Blinkova, O. NCBI Viral Genomes Resource. Nucleic Acids Research 2014, 43, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- De los Santos, M. Covary: A translation-aware framework for alignment-free phylogenetics using machine learning; Cold Spring Harbor Laboratory., 2025a. [Google Scholar] [CrossRef]
- De los Santos, M. I. TIPs-VF: An augmented vector-based representation for variable-length DNA fragments with sequence, length, and positional awareness; Cold Spring Harbor Laboratory., 2025. [Google Scholar] [CrossRef]
- Gangavarapu, K.; Latif, A. A.; Mullen, J. L.; Alkuzweny, M.; Hufbauer, E.; Tsueng, G.; Haag, E.; Zeller, M.; Aceves, C. M.; Zaiets, K.; Cano, M.; Zhou, X.; Qian, Z.; Sattler, R.; Matteson, N. L.; Levy, J. I.; Lee, R. T. C.; Freitas, L.; Maurer-Stroh, S.; Hughes, L. D. Outbreak.info genomic reports: scalable and dynamic surveillance of SARS-CoV-2 variants and mutations. Nature Methods 2023, 20, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Gardy, J. L.; Loman, N. J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nature Reviews Genetics 2017, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Shen, Y.; Xu, X.; Li, J.; Wang, X. Spatial–temporal pattern and drivers associated with measles resurgence from 2018 to 2023: a global perspective from 192 countries. BMJ Public Health 2025, 3, e001912. [Google Scholar] [CrossRef] [PubMed]
- Ma, K. C.; Castro, J.; Lambrou, A. S.; Rose, E. B.; Cook, P. W.; Batra, D.; Cubenas, C.; Hughes, L. J.; MacCannell, D. R.; Mandal, P.; Mittal, N.; Sheth, M.; Smith, C.; Winn, A.; Hall, A. J.; Wentworth, D. E.; Silk, B. J.; Thornburg, N. J.; Paden, C. R. Genomic Surveillance for SARS-CoV-2 Variants: Circulation of Omicron XBB and JN.1 Lineages — United States, May 2023–September 2024. MMWR. Morbidity and Mortality Weekly Report 2024, 73, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Plowright, R. K.; Parrish, C. R.; McCallum, H.; Hudson, P. J.; Ko, A. I.; Graham, A. L.; Lloyd-Smith, J. O. Pathways to zoonotic spillover. Nature Reviews Microbiology 2017, 15, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Saada, B.; Zhang, T.; Siga, E.; Zhang, J.; Magalhães Muniz, M. M. Whole-Genome Alignment: Methods, Challenges, and Future Directions. Applied Sciences 2024, 14, 4837. [Google Scholar] [CrossRef]
- Shen, Y.; Liu, Y.; Krafft, T.; Wang, Q. Progress and challenges in infectious disease surveillance and early warning. Medicine Plus 2025, 2, 100071. [Google Scholar] [CrossRef]
- Tariq, F.; Irfan, M.; Farooq, S.; Iqbal, H.; Atia-tul-Wahab; Khan, I. A.; Iftner, T.; Choudhary, M. I. Dynamics and genetic variation of dengue virus serotypes circulating during the 2022 outbreak in Karachi. Scientific Reports 2025, 15. [Google Scholar] [CrossRef] [PubMed]
- Voznica, J.; Zhukova, A.; Boskova, V.; Saulnier, E.; Lemoine, F.; Moslonka-Lefebvre, M.; Gascuel, O. Deep learning from phylogenies to uncover the epidemiological dynamics of outbreaks. Nature Communications 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, Z.; Li, H.; Wang, X.; Li, B.; Ren, X.; Zeng, Z.; Zhang, X.; Liu, S.; Hu, P.; Qi, W.; Liao, M. Biological Characterizations of H5Nx Avian Influenza Viruses Embodying Different Neuraminidases. Frontiers in Microbiology 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Jun, S.-R.; Leuze, M.; Ussery, D.; Nookaew, I. Viral Phylogenomics Using an Alignment-Free Method: A Three-Step Approach to Determine Optimal Length of k-mer. Scientific Reports 2017, 7. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



