Submitted:
01 December 2025
Posted:
05 December 2025
You are already at the latest version
Abstract
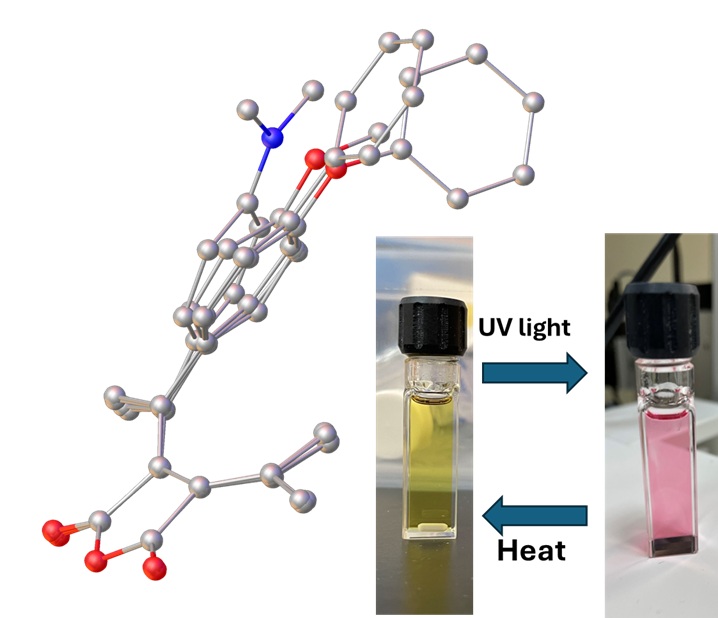
Fulgides are a group of organic compounds that exhibit photochromic properties both in solid state and in solutions. The compounds attracted research attention due to their wide potential applications including photochromic eyewear, smart windows, optical switch, data storage, chemical and biological sensors. We are reporting here the synthesis and crystal structures of fulgides of four different substituents at the para position of a phenyl moiety in the molecules. It was found among the 4 structures that 1) all the 4 compounds packed in space groups of an inversion center; 2) the distance between the two carbon atoms C8 and C11, which form a single C-C bond in the cyclized products, falls in the range of 3.5-3.7 Å; 3) the torsion angle, defined by C6-C3-C4-C11, falls in the range of 23.4o to 32.5o. The fulgides exhibited photochromism. The fulgides should have no ferroelectric property due to their crystallization into centrosymmetric spaces groups.
Keywords:
photochromic
; fulgide
; crystal
; synthesis
; ferroelectric
1. Introduction
Fulgides are a group of organic compounds of succinic anhydride with conjugated bis-methylene moieties as structure A shown in Figure 1.A. When one or more of the substituents contain a double bond (most time in an aromatic moiety like that in E-B) that can undergo a hexatriene eletrocyclic reaction leading to the cyclic product C-B under ultraviolet (UV) light radiation, the molecule will exhibit photochromic property, that is, the molecule E-B will cyclized to form C-B accompanied by the color change of E-B to C-B; Stopping UV radiation, the color of C-B will change back to that of E-B due to the thermal backward reaction of C-B to E-B. The Z-B isomer can isomerize to E-B, while the color change is usually not as significant as E-B to C-B under UV radiation [1].
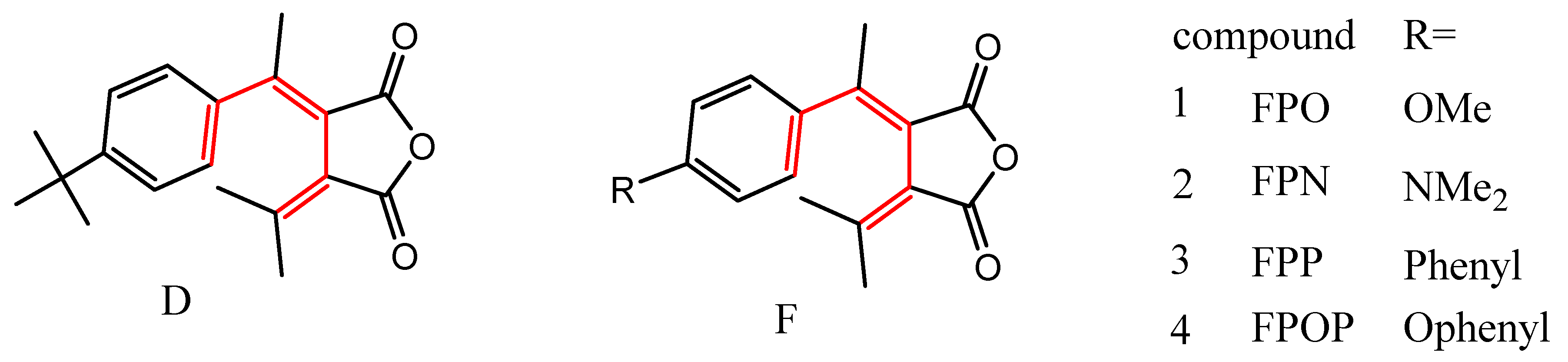
The photochromic fulgides have been attracting much research effort due to their wide potential applications including photochromic eyewear, smart windows, optical switch, data storage, chemical and biological sensors. [2,3]. In addition to the photochromic property, a phenyl fulgide D (Figure 2) was discovered for the first time to exhibit ferroelectricity by Dr. Wang and coworkers the first time in 2023 [4]. Ferroelectric materials can retain polarization orientation and switch by an external electric field. This property is very important for ferroelectric RAM that can store data in its two stable polarization states and for sensors and actuators in industrial automation. Among ferroelectric materials, organic ones are attracting growing research attention due to their flexibility, easiness to synthesis, and process. Although fulgides have been studied for a long time, phenyl fulgides like D have not attracted much attention, especially about their crystal structures [4,5,6,7,8], which are essential for their photochromism and ferroelectricity. Therefore, we designed and synthesized phenyl fulgides F with different substituents at the para position of the phenyl group to explore the electronic and size effect on the molecular packing in crystals and the effect on the photochromic and ferroelectric properties. Here we report the synthesis and crystal structures of the fulgides as our initial research results.
2. Materials and Methods
2.1. Materials and Instrumentation
Reagents and solvent: Compounds FPO, FPN, FPP and FPOP are synthesized in our lab (see synthesis in 3. Results and Discussion). Reagents and solvents are purchased from chemical suppliers and used without further purification: The following reagents and solvents are purchased from Thermo Sceitific Chemicals: diethyl succinate; acetone (Certified ACS), potassium tert-butoxide, reagent grade; acetyl chloride; tert-butanol; 4-methoxyacetophenone, 99%; 4-dimethylaminoacetophenone, 99%; dichloromethane (Certified ACS); deuterated chloroform-d (99.8% atom); petroleum ether, (Certified ACS); hexanes (98.5% Certified ACS). The following chemicals are purchased from Oakwood Chemical: toluene, 99.5%; 4′-phenoxyacetophenone, 97%; chloroform, (Certified ACS reagent). The following chemicals are purchased from TCI: sodium hydride (60% dispersion in paraffin liquid); 4-acetylbiphenyl, 98%.
2.2. Instrumentation
1H and 13C NMR spectra are measured with Bruker, Ascend Evo 400 MHz. Chemical shift in part per million (ppm) is recorded with residual deuterated solvent (CDCl3: 7.260 ppm for 1H, and 77.16 for 13C) signal as reference [9]. Electrospray ionization mass spectrometry (ESI-MS) experiments were performed using a Thermo Scientific Q Exactive Focus. Sample was injected into a 10 µL loop and was transferred to the mass spec using a mobile phase containing 70% methanol and 30% water with 0.1% formic acid at a flow rate of 600 µL/min. The Q Exactive Focus HESI source was operated in full MS in positive mode. A XtaLAB Synergy, Single source at home/near, Eiger2 1M or a XtaLAB Synergy, Dualflex, HyPix diffractometer was used for crystal data collection. The structure was solved with the ShelXT 2018/2 solution program using dual methods and Olex2 1.5 as the graphical interface.
3. Results and Discussion
3.1. Synthesis and Crystallizations
The synthesis of the fulgides has been carried out following a slightly modified pathway illustrated in Scheme 1, which was reported previously.
Diethyl succinate G was reacted with one equivalent acetone H and one equivalent potassium tert-butoxide in tert-butanol under reflux for 8 hours. After removal of solvent tert-butanol with a rotavapor under vacuum, the residue was poured into ice water. The water solution was washed with diethyl ether and was then acidified to pH around 2 and transferred into a separatory funnel, the top layer was collected, and the aqueous phase was washed with ethyl acetate three times. The resulting ethyl acetate solution is combined with the top layer and dried with granule anhydrous sodium sulfate. 3-(ethoxycarbonyl)-4-methylpent-3-enoic acid I yielded when the ethyl acetate solution is condensed to dryness by a rotavapor under vacuum. The carboxylic acid I without further purification was esterified in refluxing ethanol using sulfuric acid as catalyst for 10 hours to yield crude diethyl 2-(propan-2-ylidene) succinate J after removal of solvent from the reaction mixture. The crude product was transferred to a flask and applied to vacuum distillation to remove components of boiling point lower than 60 oC under vacuum of 80 millitorr. The material left in the flask was distillated to almost dryness to yield product J with a purity of around 85% as estimated from 1H NMR spectrum.
Each of the four para-substituted K acetophenone reacted with compound J to produce the corresponding F as detailed in the following. Sodium hydride (1.2 equivalent) was added into the solution of K in toluene, and the suspension was stirred for 24 hours. The reaction mixture was poured into ice water to form a suspension which was transferred into a separatory funnel and the top layer was removed. The aqueous solution was washed with toluene 3 times and then acidified to pH around 2 to yield compound L as precipitate. The precipitate was collected by filtration and dried by a rotavapor and then transferred into a flask. To the flask containing compound L was added ethanol and sodium hydroxide (10 equivalent to L). The reaction mixture was refluxed for 12 hours, cooled down to room temperature, and then the sodium salt of M yielded as precipitate. The precipitate was collected by filtration, washed with chilled ethanol, dried by a rotavapor and transferred into a beaker. Water was added to the beaker until the precipitate was dissolved. The resulting solution was acidified with concentrated hydrochloric acid to pH around 2 to yield compound M as precipitate. The precipitate M was collected by filtration, washed with chilled water, transferred to a round bottom flask and dried with a rotavapor. Acetyl chloride was added to the flask containing M (50 ml acetyl chloride per gram of M), the resulting solution was stirred for 24 hours at room temperature, then the acetyl chloride was removed by a rotavapor. The resulting solid in the flask was applied to silica gel chromatography, fractions that exhibited red color on the wet TLC plate under UV detector were collected and condensed to yield the corresponding F, which was proved to be of E configuration for all the produce fulgides FPO, FPN, FPOP and FPP. FPO was synthesized previously [10], while its crystal structure has not been reported. The products of Z configuration were not the major target of our focus; therefore, they were not isolated from the reaction mixture in this study.
3.1.1. (E)-3-(1-(4-methoxyphenyl) ethylidene)-4-(propan-2-ylidene) dihydrofuran-2,5-dione (FPO)
FPO yielded as yellow plate-shaped crystal;
1H NMR in CDCl3 (chemical shift in ppm): 1.07, singlet, 3 H; 2.11, singlet, 3H, 2.57, singlet, 3 H; 3.70, singlet 3 H; 6.78-6.80, doublet, 2 H; 7.16-7.18, doublet, 2H.
13C NMR in CDCl3 (chemical shift in ppm): 22.64, 22.17, 55.48, 114.55, 119.66, 120.10, 129.37, 134.80, 154.12, 154.57, 160.27, 154.57, 160.27, 163.59, 164.13.
ESI-MS, C16H16O4, [M+H+]: 273.1115, expected: 273.1121
3.1.2. (E)-3-(1-(4-(dimethylamino) phenyl) ethylidene)-4-(propan-2-ylidene) dihydrofuran-2,5-dione (FPN)
FPN yielded as orange plate-shaped crystal.
1H NMR in CDCl3 (chemical shift in ppm): 1.25, singlet, 3 H; 2.23, singlet, 3H, 2.67, singlet, 3 H; 3.01, singlet 3 H; 6.62-6.65, doublet, 2 H; 7.24-7.26, doublet, 2H.
13C NMR in CDCl3 (chemical shift in ppm): 22.42, 22.63, 26.27, 40.21, 111.87, 117.64, 120.83, 129.43, 150.86, 153.23, 155.35, 164.11, 164.45.
ESI-MS, C17H19NO3, [M+H+]: 286.1430, expected: 286.1438
3.1.3. (E)-3-(1-(4-phenoxyphenyl) ethylidene)-4-(propan-2-ylidene) dihydrofuran-2,5-dione (FPOP)
FPOP yielded as yellow block-shaped crystal;
1H NMR in CDCl3 (chemical shift in ppm): 1.14, singlet, 3 H; 2.13, singlet, 3H, 2.57, singlet, 3 H; 2.61, singlet 3 H; 6.89-7.30, multiple peaks, 9H.
13C NMR in CDCl3 (chemical shift in ppm):22.57, 22.60, 26.18, 118.59, 119.61, 119.88, 120.18, 124.27, 129.41, 130.02, 136.93, 153.41, 154.82, 156.00, 158.31, 16.31, 163.93.
ESI-MS, C21H18O4, [M+H+]: 335.1269, expected: 335.1278
3.1.4. (E)-3-(1-([1,1′-biphenyl]-4-yl) ethylidene)-4-(propan-2-ylidene) dihydrofuran-2,5-dione (FPP)
FPP yielded as yellow plate-shaped crystal;
1H NMR in CDCl3 (chemical shift in ppm): 1.08, singlet, 3 H; 2.12, singlet, 3H, 2.64, singlet, 3 H; 7.26-7.30 multiple peak 3 H; 7.34-7.38, multiple peaks, 2 H; 7.51-7.54. multiple peaks, 4 H.
13C NMR in CDCl3 (chemical shift in ppm): 22.64, 22.17, 55.48, 114.55, 119.66, 120.10, 129.37, 134.80, 154.12, 154.57, 160.27, 154.57, 160.27, 163.59, 164.13.
ESI-MS, C21H18O3, [M+H+]: 329.1322, expected: 329.1329
3.2. Crystal Structure and Discussion
A single crystal was picked from the crystals of each fulgide: FPO, FPN, FPOP and FPP and applied to X-ray crystallography analysis. The major parameter and results are summarized in Table 1.
To determine the crystal structure of FPO, a suitable single yellow plate-shaped crystal with dimensions 0.20 × 0.20 × 0.10 mm3 was selected and mounted on a XtaLAB Synergy, Single source at home/near, Eiger2 1M diffractometer. Data was measured using ω scans with Ag Kα radiation. The crystal was kept at a steady T = 110.00 K during data collection. The structure was solved with the ShelXT 2018/2 (Sheldrick, 2018) solution program using dual methods and by using Olex2 1.5 [11] as the graphical interface. The model was refined with ShelXL 2019/2 [12,13] using full matrix least squares minimization on F2. The final wR2 was 0.1426 (all data) and R1 was 0.0473 (I≥2 s(I)). Major parameters and results are listed in Table 1. The structure was determined to be monoclinic within space group of P21/n (No. 14). The molecular packing of FPO in crystal was illustrated in Figure 3. A. The structure of FPO was illustrated in Figure 3. B.
The bond distance of nonhydrogen atoms, selected bond angles and torsions angles were measured and presented in Table 2, Table 3 and Table 4.
The structure of fulgide can be considered of three moieties: the succinic anhydride, the phenyl group, and the conjugated system of C7=C10-C11=C14. The bond lengths among carbons C1-6 in the phenyl group fall into the range 1.385-1.402 Å which is consistent with those in a substituted benzene. The two C=C double bonds C7=C10 and C13=C14 are 1.3662 Å and 1.3578 Å respectively, which are slightly longer than the average C=C length of 1.34 Å due to distorted conjugation. Single C-C bonds as exemplified by C14-C15 and C14-C16 with lengths of 1.5022 Å and 1.4932 Å are slightly shorter than average 1.54 Å due to their adjacent to double bond. The torsion of C7-C10-C11-C14 is -35.8°, which shows the great distortion of the C7=C10-C11=C14 conjugated system. The torsion of C2-C1-C7-C10 is -50.76° shows the C11=C14 double bond is far from perfect conjugation of 0° torsion. The torsions C1-C7-C10-C11 and C13-C10-C11-C12 indicate that the double bond of C7=C10 and C11=C14 are distorted by similar extent of -11.19° and 10.42°. The significant distortion of the conjugated system and the double bonds are caused by the stereo repulsion between the methyl group of C14 and the phenyl group. The stereo strain energy drives the molecule to undergo intramolecular cyclization. The distance between the C14 and C2 was measured to be 3.475 Å.
To see the effect of electron-donating group on the structure of the fulgide, we prepared the FPO’s analogue FPN which has a stronger electron-donating dimethyl amino group than methoxy group at the para position of phenyl group. To determine the crystal structure of FPN, a suitable single orange plate-shaped crystals of dimensions 0.20 × 0.10 × 0.02 mm3 was selected from the crystalized product and mounted on a XtaLAB Synergy, Dualflex, HyPix diffractometer. Data was measured using w scans with Cu Ka radiation. The crystal was kept at a steady T = 100.00(10) K during data collection. The structure was solved and refined as that of FPO. The parameters of determination and crystal structures are listed in Table 1. The molecular packing of FPN in crystal is illustrated in Figure 4.A. The structure of FPN is illustrated in Figure 4.B.
The results showed that the selected bond length in FPN exhibited no change in comparison to those in FPO. Regarding the bond angles, the angle of C12-C11-C10 changed by 16.9°from 105.6°in FPO to 122.5° in FPN. The torsion angles of C2-C1-C7-C10, C1-C7-C10-C11, and C8-C7-C10-C13 changed significantly from -50.8°, -11.2° and -17.4° in FPO to -38.2°, -17.6° and -25.0° in FPN. Overlay of the structure of FPO and FPN by matching C12, C11, C10 and C13 showed clearly that the differences of torsion angles are caused by a rotation around the C1-C7 single bond as shown in Figure 4. C. Such a rotation slightly decreased the distance between the C2 and C14 atoms, the ones forming C-C single bond in cyclization reaction, from 3.475 Å to 3.384 Å.
To study the size effect of the para substitute group, a phenyl group with a larger size that methyl group in FPO was introduced into fulgide FPOP. A single yellow block-shaped crystal of FPOP was subjected to X-ray crystallography. A suitable crystal with dimensions 0.34 × 0.23 × 0.18 mm3 was selected and analyzed by using the same diffractometer, parameters and conditions. The structure was solved and refined as FPO. The parameters of determination and crystal structures are listed in Table 1. The molecular packing of FPOP in crystal is illustrated in Figure 5. A. The structure of FPOP is illustrated in Figure 5. B.
To show the difference in crystal structure caused by the size increasement, the selected bond lengths, bond angles and torsions in FPOP are included in Table 5, Table 6, Table 7 and Table 8 for comparison. The length of bonds related to the conjugated system showed no difference between FPO and FPOP. Among the selected bond angles related to the conjugated system, the angle of C12-C11-C10 increased to 121.8° in FPOP from 105.6° in FPO, like the change observed in FPN. Among the torsion angles, the torsion of C1-C7-C10-C11 changed most to -4.3° from -11.2° in FPO, while C8-C7-C10-C13 changed, with the least extent, to -14.3°from 17.4°in FPO. Such changes are caused by distortion around double bonds C7=C10 and C11-C14 and by slight rotation around bond C1-C7. These changes have led to the decrease of in the distance from 3.475 Å FPO to 3.302 Å FPOP.

Table 5.
The selected bond angles of FPO, FPN, FPOP and FPP.
| Number | Bond Angle | Angle in Degree | |||
|---|---|---|---|---|---|
| FPO | FPN | FPOP | FPP | ||
| 1 | C1-C7-C10 | 122 | 122.1 | 120.7 | 122.1 |
| 2 | C8-C7-C10 | 122.8 | 120.9 | 122.3 | 122.6 |
| 3 | C13-C10-C7 | 120.9 | 120.8 | 121.6 | 122.3 |
| 4 | C11-C10-C7 | 132.5 | 132.2 | 132.1 | 131.4 |
| 5 | C16-C14-C11 | 122.2 | 121.9 | 122.8 | 122.6 |
| 6 | C15-C14-C11 | 123.9 | 123.7 | 121.8 | 123.3 |
| 7 | C12-C11-C10 | 105.6 | 122.5 | 121.8 | 122.9 |
| 8 | C14-C11-C10 | 131.2 | 130.1 | 131.1 | 131.4 |
Table 6.
Selected torsion of FPO, FPN, FPOP and FPP.
| Number | Torsion | Torsion Angles | |||
|---|---|---|---|---|---|
| FPO | FPN | FPOP | FPP | ||
| 1 | C2-C1-C7-C10 | -50.8 | -38.2 | -46.5 | -46 |
| 2 | C1-C7-C10-C11 | -11.2 | -17.6 | -4.3 | -4.9 |
| 3 | C7-C10-C11-C14 | -35.8 | -37 | -39.3 | -41.8 |
| 4 | C10-C11-C14-C16 | 1.5 | 2.2 | -3.6 | -2.4 |
| 5 | C8-C7-C10-C13 | -17.4 | -25 | -14.3 | -15 |
| 6 | C15-C14-C11-C12 | -12.3 | -12.5 | -18.1 | -18.3 |
| 7 | C13-C10-C11-C12 | -10.4 | -10.4 | -14.1 | -16 |
The fulgide FPP with a phenyl substituent of no significant electron-donating properties an analogue of FPO to evaluate both electronic and size effect of the substitution on the crystal structures. A yellow block-shaped single crystal with dimensions 0.62 × 0.50 × 0.32 mm3 of FPP was selected and subjected to single crystal using the same diffractometer. The structure was solved and refined as FPOP. The parameters of determination and crystal structures are listed in Table 1. The molecular packing of FPP in crystal is illustrated in Figure 6. A. The structure of FPP is illustrated in Figure 6. B.
For convenient comparison of the structures, the selected bond lengths, bond angles and torsions are summarized in Table 5-8. The length of bonds related to the conjugated system showed no difference among all analyzed structures. C12-C11-C10 angle in FPP changed to 122.9°, like those in FPN and FPOP, from 105.6°in FPO. The selected torsion angles in FPP are close to those in FPOP, but significantly different from those in FPO and
FPN. The distance between C15 and C5 was measured to be 3.302 Å in comparison to those of 3.475 Å and 3.384 Å in FPO and FPOP respectively. The changes can be easily observed in the overlay of structures of FPO to FPP and FPOP to FPP as shown in Figure 7 A and B. The phenyl group in FBB tilted a little closer to the carbon involved in cyclization as shown in Figure 7A. The phenyl and phenoxy of similar size as substituents led to almost the exact overlap of the phenyl group position, although they oriented quite different in the crystal.
All the four crystal structures have been overlayed by matching the position of the four carbons in the succinic moiety and displayed in Figure 7. C. The succinic moiety showed well overlapping among all the structures. The bond lengths showed no changes. The bond angles related to the conjugate system showed no significant change either. The distortion around the double bond of C7=C10 caused the significant displacement of the carbon C8 and C7. The rotation around C7-C1 led to the tilt of the phenyl moiety. The phenyl moiety in FPOP and FPP overlaps well with each other, while this moieties in FPO and FPN tilted sightly and significantly from those in FPOP and FPP away from the carbon C14. Such distortion contributes most to the distance between C2 and C14, which is most important in the photo cyclization involved in photochromism. All the four fulgides exhibited photochromic property as observed in the purification process of each compound. Although the fulgide D with a tert-butyl group crystalizes into a non-centrosymmetric group of Pc and exhibit ferroelectric property, each of the fulgide: FPO, FPN, FPOP and FPP packed into centrosymmetric group of P21/n, P-1, P21/c and P21/c respectively. The results suggest the new crystalized compounds are not ferroelectric. Such a crystal packing difference among fulgide of similar molecular structures are to be understood.
4. Conclusion
A small group of phenyl fulgides: FPO, FPN, FPOP and FPP have been synthesized and characterized. Single crystal of each fulgide was selected and applied to x-ray crystallography analysis, and the crystal structure was solved. The result of structural analysis showed that all the fulgides crystalized into centrosymmetric space groups. Although the structures of fulgides are quite similar, the distortion of the C=C double bond and the rotation of C-C bond that connect to the phenyl group change the structure notably to a different extent when the electronic property and size of the para substituent changes. The fulgides studied her are not expected to be ferroelectric due to their centrosymmetric space group. The photochromic property has been observed, and further study is underway.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. 1H and 13C NMR and Mass Spectra of the compounds.
Author Contributions
Conceptualization, synthesis, crystallization, writing original draft Y.L, synthesis S.A, X-ray crystallography, data collection, structure solving, refining, N.B and J.R. Photochromic study Y.L and Y.Z. Draft reviewing and correction, all the authors. All the authors have reviewed the draft and agreed to publish it.
Funding
This research is supported by Faculty RISE Program of Prairie View A&M University.
Data Availability Statement
The crystal data has been deposited to the Cambridge Crystallographic Data Centre (CCDC). CCDC 2505338-2505340 and 2505410 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk). For any other information, please contact the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| UV | Ultraviolet |
| NMR | Nuclear Magnetic Resonance |
| ESI-MS | Electrospray Ionization-Mass Spectroscopy |
| TLC | Thin Layer Chromatography |
| FPO | (E)-3-(1-(4-methoxyphenyl) ethylidene)-4-(propan-2-ylidene) dihydrofuran-2,5-dione (FPO) |
| FPN | (E)-3-(1-(4-(dimethylamino) phenyl) ethylidene)-4-(propan-2-ylidene) dihydrofuran-2,5-dione |
| FPOP | (E)-3-(1-(4-phenoxyphenyl) ethylidene)-4-(propan-2-ylidene) dihydrofuran-2,5-dione |
| FPP | (E)-3-(1-([1,1′-biphenyl]-4-yl) ethylidene)-4-(propan-2-ylidene) dihydrofuran-2,5-dione |
References
- Fan, M.-G.; Yu, L.; Zhao, W. Fulgide Family Compounds: Synthesis, Photochromism, and Applications, in Organic Photochromic and Thermochromic Compounds: Volume 1: Main Photochromic Families; Crano, J.C., Guglielmetti, R.J., Eds.; Springer US: Boston, MA, USA, 2002; pp. 141–206. [Google Scholar]
- Irie, M. Photochromism: Memories and SwitchesIntroduction. Chem. Rev. 2000, 100, 1683–1684. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y. Fulgides for Memories and Switches. Chem. Rev. 2000, 100, 1717–1740. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Huang, C.-R.; Xu, Z.-K.; Hu, W.; Li, P.-F.; Xiong, R.-G.; Wang, Z.-X. Photochromic Single-Component Organic Fulgide Ferroelectric with Photo-Triggered Polarization Response. JACS Au 2023, 3, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Kaftory, M. Photochromic and thermochromic compounds. I. Structures of (E) and (Z) isomers of 2-isopropylidene-3- [1-(2-methyl-5-phenyl-3-thienyl)ethylidene]succinic anhydride, C20H18O3S, and the photoproduct 7,7a-dihydro-4,7,7,7a-tetramethyl-2-phenylbenzo [b]thiophene-5,6-dicarboxylic anhydride (P), C20H18O3S. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1984, 40, 1015–1019. [Google Scholar]
- Cohen, M.D.; Kaufman, H.W.; Sinnreich, D.; Schmidt, G.M.J. Photoreactions of di-p-anisylidenefulgide (di-p-anisylidenesuccinic anhydride). J. Chem. Soc. B: Phys. Org. 1970, 1035–1039. [Google Scholar] [CrossRef]
- Boeyens, J.C.A.; Denner, L.; Perold, G.W. Rotamers and isomers in the fulgide series. Part 1. Stereochemistry and conformational analysis of bis-(3,4-dimethoxybenzylidene)succinic anhydrides by X-ray crystallography and molecular mechanics. J. Chem. Soc. Perkin Trans. 2 1988, 1749–1758. [Google Scholar] [CrossRef]
- Boeyens, J.C.A.; Allen, C.C.; Perold, G.W. Rotamers and isomers in the fulgide series. Part 3. Structures of the bis(4-methoxy-3-methylbenzylidene)succinic anhydrides. J. Chem. Soc. Perkin Trans. 2 1993, 1161–1165. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- He, Y.; Gong, F.; De, Y.; Yun, L. Synthesis and Photocroism of Aryl Substituted Fulgide. Acta Chim. Sin. 1996, 54, 716–721. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr C Struct Chem 2015, 71 Pt 1, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT – Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A. the general structure of fulgide. Z-B, E-B and C-B are isomers of a phenyl fulgide involved in photochromism.
Figure 1.
A. the general structure of fulgide. Z-B, E-B and C-B are isomers of a phenyl fulgide involved in photochromism.
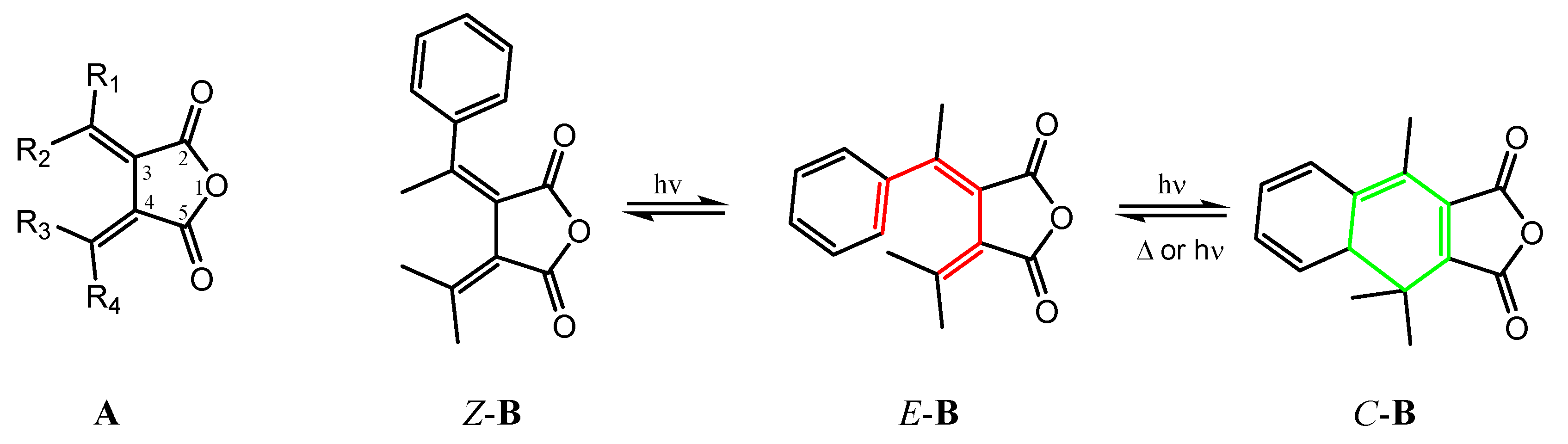
Figure 2.
D: Fulgide reported by Dr. Wand and coworkers [4], F: Fulgides made and studied in this article.
Figure 2.
D: Fulgide reported by Dr. Wand and coworkers [4], F: Fulgides made and studied in this article.
Scheme 1.
The synthetic pathway to phenyl fulgide F.
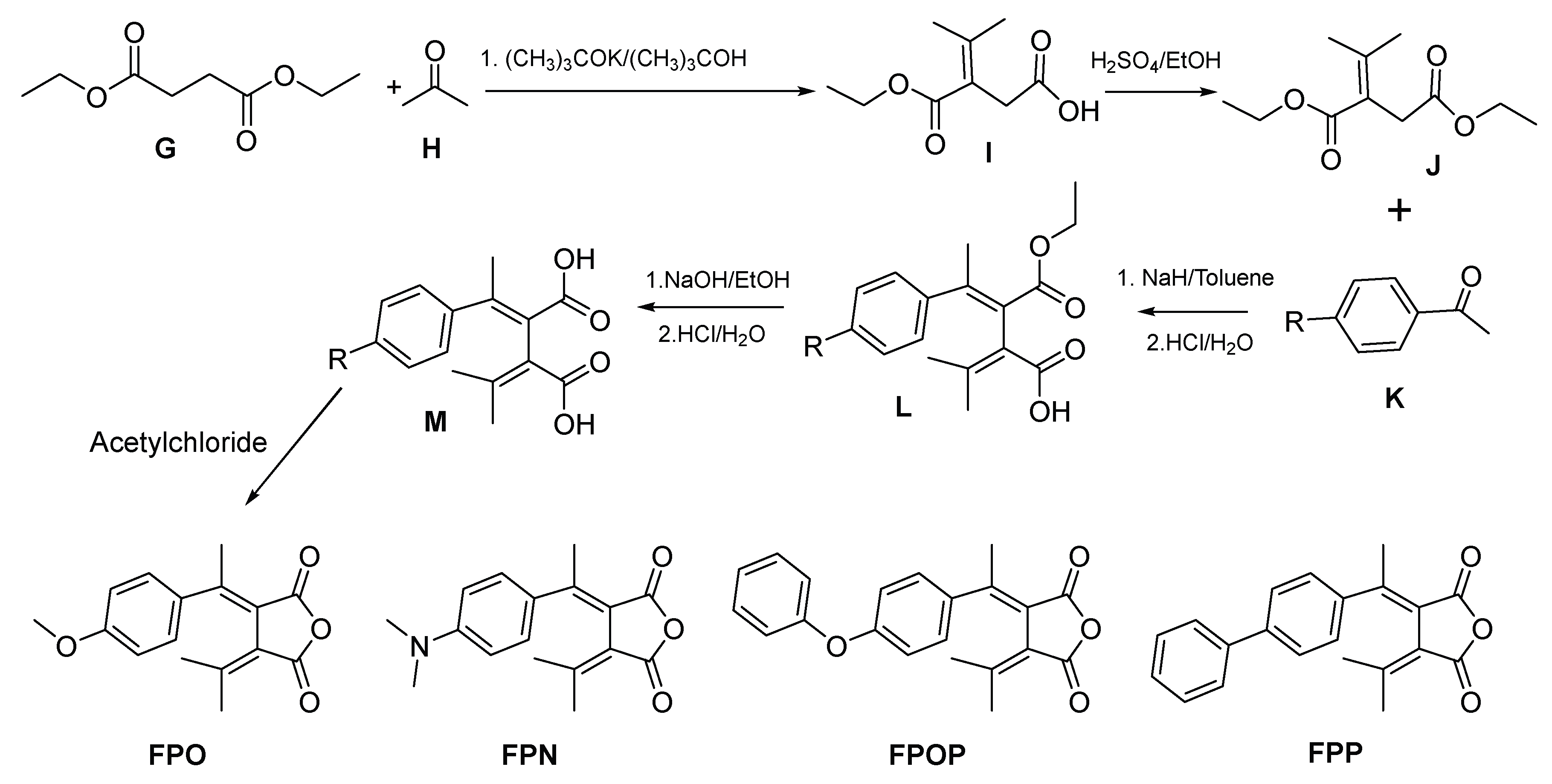
Figure 3.
A. Molecular packing of FPO in the crystal; B. ORTEP diagram of molecule FPO with hydrogen atoms omitted. *Atoms are labeled differently in the other molecules. Therefore, the list of bond length, bond angle, torsion, and distance between atoms and in the context of discussions are based on the labelling of atoms in structure of FPO.
Figure 3.
A. Molecular packing of FPO in the crystal; B. ORTEP diagram of molecule FPO with hydrogen atoms omitted. *Atoms are labeled differently in the other molecules. Therefore, the list of bond length, bond angle, torsion, and distance between atoms and in the context of discussions are based on the labelling of atoms in structure of FPO.
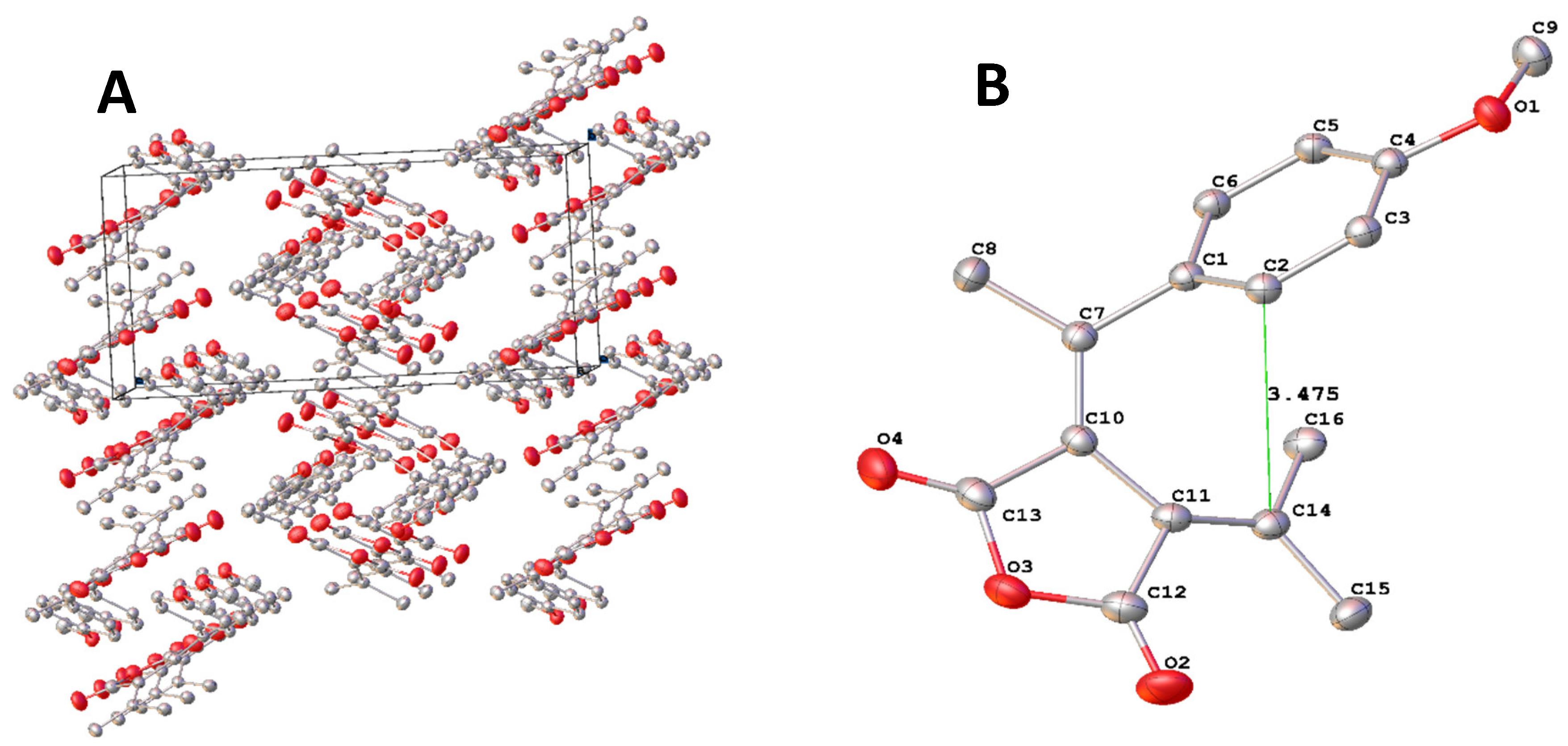
Figure 4.
A. Molecular packing of FPN in the crystal; B. ORTEP diagram of molecule FPN with hydrogen atoms omitted. C. Overlay of FPO and FPN.
Figure 4.
A. Molecular packing of FPN in the crystal; B. ORTEP diagram of molecule FPN with hydrogen atoms omitted. C. Overlay of FPO and FPN.
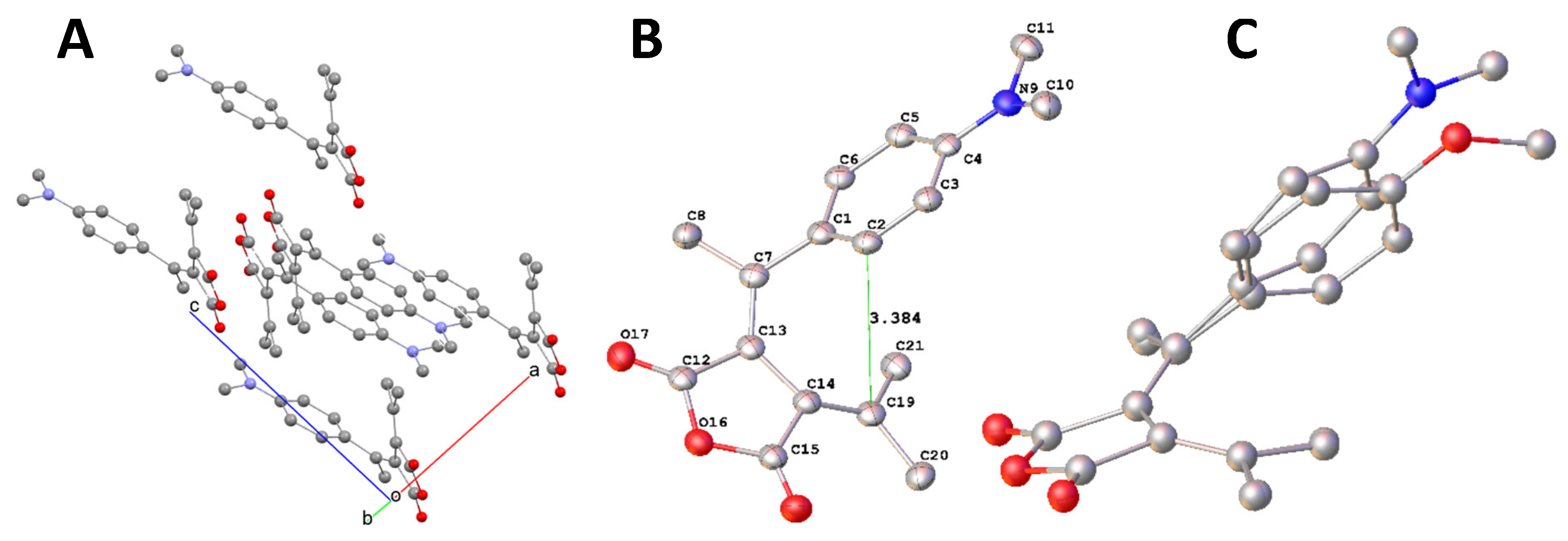
Figure 5.
A. Molecular packing of FPOP in the crystal; B. ORTEP diagram of molecule FPOP with hydrogen atoms omitted. C. Overlay of FPO and FPOP.
Figure 5.
A. Molecular packing of FPOP in the crystal; B. ORTEP diagram of molecule FPOP with hydrogen atoms omitted. C. Overlay of FPO and FPOP.
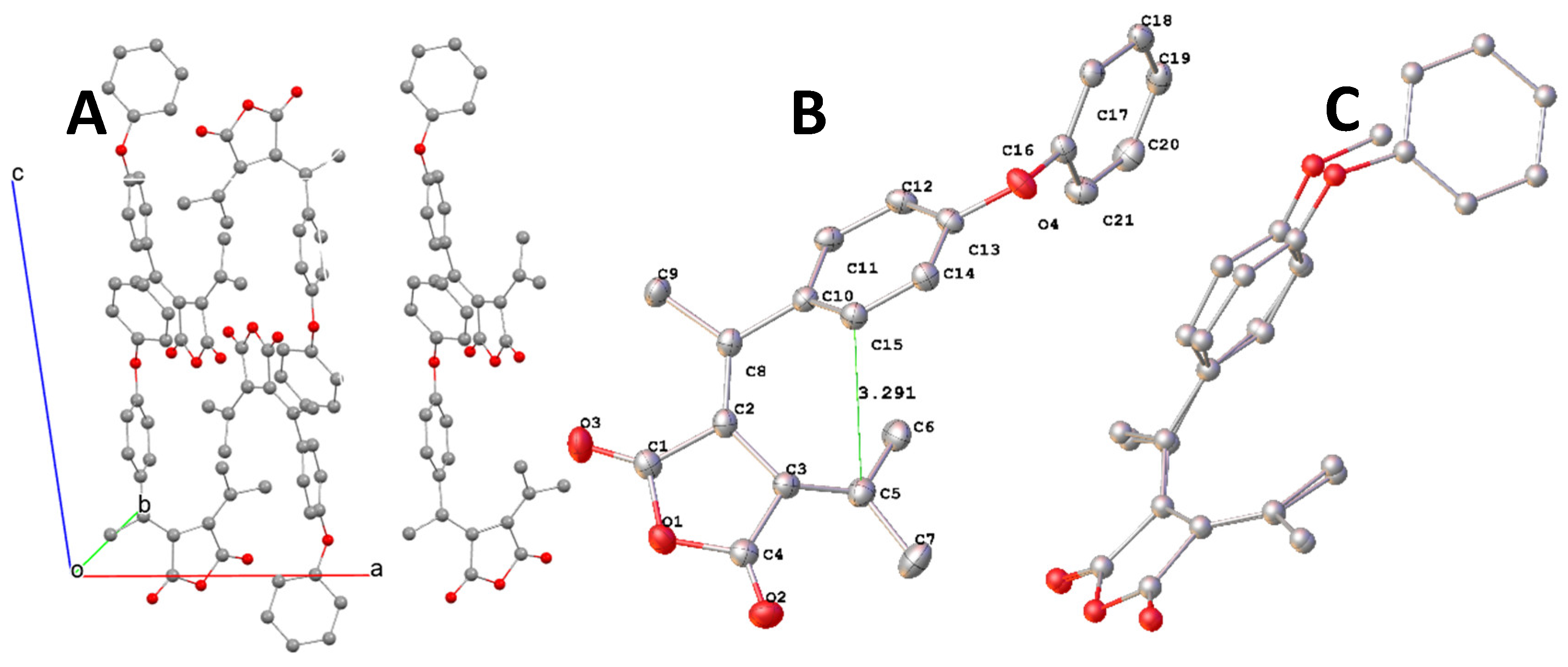
Figure 6.
A. Molecular packing of FPP in the crystal; B. ORTEP diagram of molecule FPP with hydrogen atoms omitted.
Figure 6.
A. Molecular packing of FPP in the crystal; B. ORTEP diagram of molecule FPP with hydrogen atoms omitted.

Figure 7.
A. Overlay of FPO and FPP. B. Overlay of FPOP and FPP. C. Overlay of FPO, FPOP and FPP with hydrogen atoms omitted.
Figure 7.
A. Overlay of FPO and FPP. B. Overlay of FPOP and FPP. C. Overlay of FPO, FPOP and FPP with hydrogen atoms omitted.
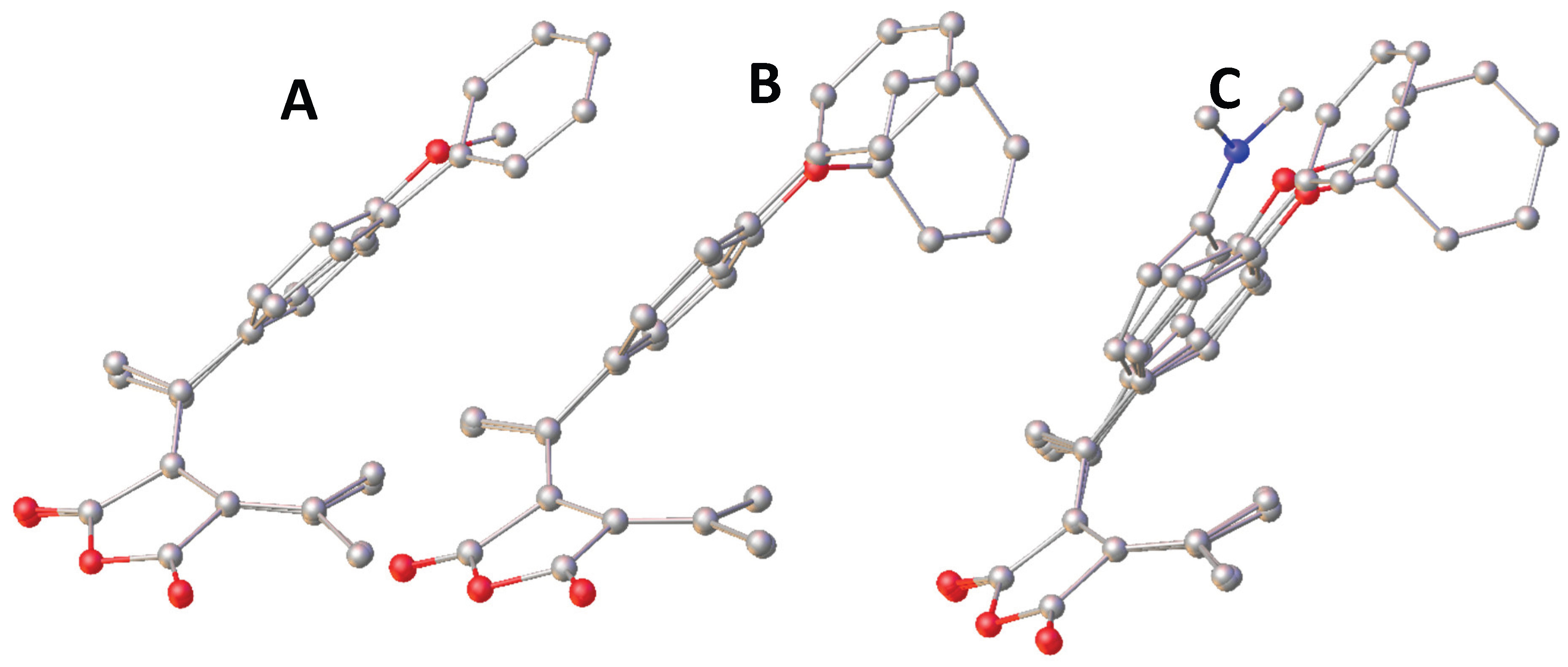
Table 1.
Crystal data and structure refinement of FPO, FPN, FPOP, and FPP*.
| Molecules | FPO | FPN | FPOP | FPP |
|---|---|---|---|---|
| Formula | C16H16O4 | C17H19NO3 | C21H18O4 | C21H18O3 |
| Dcalc./g cm-3 | 1.324 | 1.306 | 1.336 | 1.307 |
| μ/mm-1 | 0.059 | 0.724 | 0.750 | 0.695 |
| Formula Weight | 272.29 | 285.33 | 334.35 | 318.35 |
| Color | yellow | orange | yellow | yellow |
| Shape | plate-shaped | plate-shaped | block-shaped | block-shaped |
| Size/mm3 | 0.20×0.20×0.10 | 0.20×0.10×0.02 | 0.34×0.23×0.18 | 0.62×0.50×0.32 |
| T/K | 110.00 | 100.00(10) | 100.00(10) | 100.00(10) |
| Crystal System | monoclinic | triclinic | monoclinic | monoclinic |
| Space Group | P21/n | P-1 | P21/c | P21/c |
| a/Å | 12.0767(4) | 7.3147(2) | 11.30300(10) | 9.29970(10) |
| b/Å | 7.3269(2) | 9.2104(3) | 9.84180(10) | 23.8381(2) |
| c/Å | 15.5795(4) | 11.0846(4) | 14.99510(10) | 7.81600(10) |
| α/° | 90 | 101.624(3) | 90 | 90 |
| β/° | 97.715(3) | 95.085(3) | 94.7690(10) | 110.9990(10) |
| γ/° | 90 | 94.127(3) | 90 | 90 |
| V/Å3 | 1366.07(7) | 725.49(4) | 1662.31(3) | 1617.63(3) |
| Z | 4 | 2 | 4 | 4 |
| Z’ | 1 | 1 | 1 | 1 |
| Wavelength/Å | 0.56087 | 1.54184 | 1.54184 | 1.54184 |
| Radiation type | Ag Kα | Cu Kα | Cu Kα | Cu Kα |
| Θmin/° | 2.428 | 4.095 | 3.924 | 3.708 |
| Θmax/° | 30.688 | 79.759 | 80.153 | 80.153 |
| Measured Refl’s. | 31177 | 14141 | 29729 | 33706 |
| Indep’t Refl’s | 6750 | 3078 | 3566 | 3488 |
| Refl’s I≥2 σ(I) | 4817 | 2765 | 3437 | 3462 |
| Rint | 0.0414 | 0.0668 | 0.0277 | 0.0234 |
| Parameters | 185 | 195 | 230 | 221 |
| Restraints | 0 | 0 | 0 | 0 |
| Largest Peak | 0.492 | 0.603 | 0.296 | 0.339 |
| Deepest Hole | -0.245 | -0.364 | -0.208 | -0.277 |
| GooF | 1.037 | 1.054 | 1.031 | 1.069 |
| wR2 (all data) | 0.1426 | 0.1985 | 0.0980 | 0.0924 |
| wR2 | 0.1264 | 0.1916 | 0.0971 | 0.0922 |
| R1 (all data) | 0.0732 | 0.0744 | 0.0390 | 0.0369 |
| R1 | 0.0473 | 0.0709 | 0.0380 | 0.0367 |
* CCDC 2505338-2505340 and 2505410 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
Table 2.
Bond length (Å) in molecule of FPO.
| Number | Bond | Length | Number | Bond | Length |
|---|---|---|---|---|---|
| 1 | O1-C4 | 1.3618 | 13 | C4-C5 | 1.3956 |
| 2 | O1-C9 | 1.4314 | 14 | C5-C6 | 1.3915 |
| 3 | O2-C12 | 1.1977 | 15 | C7-C8 | 1.5052 |
| 4 | O3-C12 | 1.3928 | 16 | C7-C10 | 1.3662 |
| 5 | O3-C13 | 1.3934 | 17 | C10-C11 | 1.478 |
| 6 | O4-C13 | 1.1986 | 18 | C10-C13 | 1.4769 |
| 7 | C1-C2 | 1.4023 | 19 | C11-C12 | 1.4752 |
| 8 | C1-C6 | 1.3981 | 20 | C11-C14 | 1.3578 |
| 9 | C1-C7 | 1.4762 | 21 | C14-C15 | 1.5022 |
| 11 | C2-C3 | 1.3852 | 22 | C14-C16 | 1.4932 |
| 12 | C3-C4 | 1.3987 |
Table 3.
selected bond angles (degree) in molecule FPO.
| Number | bond angles | Angle |
|---|---|---|
| 1 | C2-C1-C6 | 118.36 |
| 2 | C2-C1-C7 | 119.75 |
| 3 | C1-C7-C10 | 121.95 |
| 4 | C7-C10-C11 | 132.53 |
| 5 | C10-C11-C14 | 131.21 |
| 6 | C11-C14-C16 | 122.2 |
| 7 | C8-C7-C1 | 115.12 |
| 8 | C15-C14-C16 | 113.79 |
Table 4.
Selected torsion angle (degree) in molecule FPO.
| Number | Torsion | Angle |
|---|---|---|
| 1 | C7-C10-C11-C14 | -35.8 |
| 2 | C13-C10-C11-C12 | -10.42 |
| 3 | C2-C1-C7-C10 | -50.76 |
| 4 | C16-C14-C11-C10 | 1.45 |
| 5 | C1-C7-C10-C11 | -11.19 |
Table 4.
The selected bone lengths of FPO, FPN, FPOP and FPP.
| Number | Bond | Bond Length | |||
|---|---|---|---|---|---|
| FPO | FPN | FPOP | FPP | ||
| 1 | C8-C7 | 1.51 | 1.50 | 1.51 | 1.51 |
| 2 | C1-C7 | 1.48 | 1.47 | 1.48 | 1.48 |
| 3 | C7=C10 | 1.37 | 1.37 | 1.36 | 1.36 |
| 4 | C10-C11 | 1.48 | 1.47 | 1.48 | 1.48 |
| 5 | C11=C14 | 1.36 | 1.36 | 1.36 | 1.35 |
| 6 | C14-C16 | 1.49 | 1.50 | 1.50 | 1.50 |
| 7 | C14-C15 | 1.50 | 1.50 | 1.51 | 1.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



