
Submitted:
15 November 2025
Posted:
18 November 2025
You are already at the latest version
Abstract
Cancer immunotherapy has recently become an essential approach for treating cancer, showing considerable promise as a substitute for surgery, radiation therapy, and conventional chemotherapy. It primarily aims to boost the host’s natural defense system to com-bat cancer malignancies by utilizing components of immune checkpoint blockades (ICBs), mainly programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), and along with elements of adoptive cellular therapies (ACTs) like Chimeric Antigen Receptor (CAR) therapy, T Cell Receptor (TCR) therapy and Tu-mor-Infiltrating Lymphocyte (TIL) therapy. However, cancer cells tend to undermine the effectiveness of cancer immunotherapeutic strategies by employing one or more immune evasion mechanisms. The present review briefly discusses the key mechanisms of cancer immune evasion and highlights how the CRISPR/Cas9 systems, as gene editing tools, are set to enhance cancer immunotherapy for treating various challenging cancers. We emphasize that CRISPR/Cas9 systems can be used to explore and positively alter the genes of the immune system, boosting the effectiveness of cancer immunotherapy by editing immune checkpoints, TILs, and CAR-T cells, and disrupting genes facilitating tumors to evade the immune system.
Keywords:
cancer immunotherapy
; immune invasion
; gene editing
; tumor infiltration
; tumor microenvironment
1. Introduction
1.1. Background on Cancer Immunotherapy
Cancer immunotherapy has gained significant attention as a safer and more effective alternative to conventional cancer treatment methods like surgery, radiation, and chemotherapy [1]. Traditional cancer therapies often fail to eliminate metastatic tumors, increasing the risks of recurrence. Additionally, they usually cause damage to surrounding tissues and systemic toxicities from chemotherapy [2,3].
Cancer immunotherapy seeks to stimulate or strengthen the host’s immune system to target and kill malignant tumors. Two principal strategies drive this approach: immune checkpoint blockade (ICB) and adoptive cellular therapies (ACTs). ICB employs immune checkpoint inhibitors (ICIs) to restore T cell activity, primarily by targeting CTLA-4/B7-1/B7-2 and PD-1/PD-L1 on T cells and cancer cells, key regulators of immune response [4,5]. Conversely, ACTs depend on T cells, either autologous or genetically engineered, to enhance their tumor-targeting capabilities. Major ACT strategies include CAR therapy, TCR therapy, and TIL therapy. Although these strategies show promising results in treating specific cancer types, many patients remain inadequately treated due to both intrinsic and acquired tumor resistance. Immunotherapeutic approaches face several limitations, including off-target effects, toxicities, immunosuppressive barriers, and challenges in standardizing manufacturing [6].
A significant obstacle to effective immunotherapy is the ability of tumors to evade the immune system, which diminishes the effectiveness of the treatment [7]. Immune evasion involves a range of strategies through which tumors escape targeting and subsequent destruction by the immune system, even when reactivated by the immunotherapy [8]. One common approach involves minimizing the expression of antigens on cancer cells. Tumor cells may also suppress the immune system’s response to tumors by enhancing the expression of immune checkpoint proteins, like PD-L1, within the tumor microenvironment (TME) [9]. Moreover, tumor cells may further hinder immune cell function by creating a microenvironment rich in specific metabolites and signaling factors, or by depriving immune cells of essential nutrients [8]. Consequently, a thorough understanding of tumor immune evasion mechanisms and strategies to enhance cancer immunotherapy components is necessary.
Emerging gene editing techniques, particularly the CRISPR/Cas9 systems, are well-positioned to address these challenges. The CRISPR/Cas system is already recognized as a versatile instrument against immune evasion, primarily due to its diverse applications. This system has been extensively utilized to study the mechanisms of various cancers [10,11]. CRISPR/Cas9 has also gained traction for in-depth studies of cancer progression mechanisms and has become an essential tool for enhancing cell-based immunotherapies. One remarkable aspect of this endonuclease system is its capability to manipulate and regulate several gene functions by simultaneously targeting multiple loci [12]. Consequently, CRISPR/Cas9-based genetic engineering is considered among the most promising approaches to treating cancer, viral infections, cardiovascular problems, and immunological and genetic diseases [13]. Furthermore, the efficient genomic manipulation ability of CRISPR/Cas9 has enabled the creation of various animal cancer models, a more profound investigation of epigenetic regulation, and, significantly, the genetic manipulation of immune and cancer cells in cancer immunotherapy [1].
This paper discusses how CRISPR/Cas9 systems are utilized to explore and modify genes within the immune system, thereby enhancing the effectiveness of cancer immunotherapy. This is achieved by editing immune checkpoints, engineering TILs and CAR-T cells, and disrupting genes that facilitate tumor evasion. We examine key immune evasion mechanisms, CRISPR-mediated screening of immunomodulatory genes, and immune manipulation strategies. Furthermore, we highlight the innovative CRISPR-based approaches to overcome immune evasion, the challenges and considerations in developing CRISPR-based immunotherapy, and future directions and innovations.
1.2. Gene Editing in Modern Medicine
Genetic engineering emerged in the 1970s, resulting in significant advancements in gene editing technologies. This development enabled unprecedented insights into how single-gene products contribute to disease mechanisms. Since then, gene editing has been extensively utilized in research, medical science, and biotechnology [14,15]. The process typically involves producing double-stranded breaks (DSBs) in DNA using engineered or bacterial nucleases. These breaks can be repaired via homology-directed repair (HDR), which promotes precise integration when a template is available, or through non-homologous end joining (NHEJ), an imprecise process that often causes gene disruptions [16,17,18].
In gene replacement, addition, or inactivation, homologous recombination (HR) is preferred since it utilizes homologous DNA as a template for repair. However, its low efficiency in mammalian cells and many model organisms limits its application [19]. Since DSBs are primarily corrected through HDR, a targeted nuclease strategy was developed to enhance specificity [15]. This method introduces exogenous DNA aligned with the break site, enabling site-specific repair [15,20].
Conversely, the NHEJ mechanism is often deemed error-prone, typically resulting in deletions (indels) or insertions at DSB sites, which can potentially cause frameshift mutations and lead to truncated proteins. NHEJ is easier to implement than homologous recombination (HR), as it does not require a template and relies less on the efficiency of the repair process [23]. As a result , NHEJ is widely used to inactivate one or more genes in cell lines [24].
Genome editing has utilized tools, including Zinc Finger Nucleases (ZFNs) and Transcription Activator-like Effector Nucleases (TALEN). However, the clustered regularly interspaced short palindromic repeat (CRISPR) and associated Cas9 (Cas9) nuclease system has outperformed them in several aspects. Some notable superior properties of the CRISPR/Cas9 system compared to other gene editing tools include operability, scalability, and flexibility [25,26,27]. As such, the CRISPR/Cas9 system is arguably the most advanced gene editing tool in the history of genetic modification. The CRISPR-Cas system originates from the bacterial adaptive immune system. It was brought to limelight in 2013 when it was initially utilized as an instrument for mammalian genome editing [12,28]. The CRISPR/Cas9 technology is a robust, programmable instrument for editing eukaryotic genomes. Its programmable nature enables precise editing of eukaryotic genomes, including pre-transcriptional genomic sequence modification and transcriptional and epigenetic alterations [15,29].
While bacteria evolved CRISPR as a defense system over maybe millions of years, researchers now use it to modulate the immune systems of mice against cancer cells. They are exploring genes linked to tumor immune evasion as potential targets for cancer immunotherapy and disrupting specific genes to enhance the effectiveness of this therapy.
2. Immune Evasion Mechanisms in Cancer
Immune cells are powerful, and cancer immunotherapy holds great potential to destroy malignant tumors. However, aberrant cancer cells continue to develop strategies to escape detection, disrupt immune interaction, and resist destruction. They employ immune evasion strategies to facilitate uncontrolled growth and spread. This section highlights several primary mechanisms.
2.1. The Heterogeneity of Cancer Cells
One key phenomenon that enables immune evasion by cancer cells is their ability to exhibit diverse morphological and phenotypic properties, including multiplication rates, gene expression, morphology, motility, and the potential to metastasize. Within a single tumor, subpopulations may differ considerably in molecular traits and therapeutic susceptibility. This provides them with a selective opportunity to develop resistance to cancer immunotherapy [30]. Resistance to immunotherapy may occur due to tumor cells lacking antigens within a heterogeneous population before therapeutic interventions [31]. For example, when the CAR-T cell treatment method is used against acute lymphoblastic leukemia (ALL), some malignant B cells may not show uniform CD19 expression. A specific study by Rabilloud et al. (2021) discovered that individuals with B-cell acute lymphoblastic leukemia (B-ALL) already possess rare negative subclones of CDThese CD19-negative cells within the heterogeneous cancer population may relapse, proliferate, and pose significant challenges even after CAR-T cell treatment. Thus, cellular heterogeneity enables immune escape and therapeutic resistance [32].
2.2. The Immunosuppressive Nature of the Tumor Microenvironment
Another immune evasion phenomenon of cancer cells involves the cancer cell manipulation of immunosuppressive cells to foster an immunosuppressive environment. Tumor cells establish this environment by attracting immunosuppressive cells that actively diminish the function of immune effector cells. Some notable immunosuppressive cells include myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs) [33,34,35]. Additionally, altering the stromal constituents and extracellular matrix caused by tumor cell stimulation can physically block immune cell infiltration and negatively affect their functions [36].
A critical mutual relationship exists between cancer stem cells (CSCs) and the tumor microenvironment (TME). The TME benefits the CSCs by endowing them with traits that allow them to remain in a stem-like state. This state confers upon them a survival advantage, self-renewal ability, and therapeutic resistance [31]. On the other hand, the CSCs reinforce the immunosuppressive nature of the TME. The CSCs contribute to the presence of essential factors that keep the heterogeneity and immunosuppressed condition of the TME [37,38,39,40]. As such, this complex relationship between TME and the cancer cells is one of the main reasons cancer cells can develop intractable resistance to various modes of cancer therapy.
2.3. Cancer Cells’ Antigens Loss from Mutation and Alternative Splicing
Many individuals diagnosed with B-ALL malignancies and recruited for CAR-T cell therapy, in which CD19 serves as the primary target surface antigen, have been observed to lose the CD19 surface antigen. This phenomenon of antigen loss has also been observed in some malignancies associated with B-cells [41]. Similarly, some clinical studies have reported antigen loss, particularly among patients receiving treatment for multiple myeloma (MM). Some patients in the treatment cohorts showed a reduction in the expression of B-cell maturation antigen (BCMA), the targeted antigen of the administered CAR-T cell treatment strategy [42,43]. These findings highlight the importance of antigen loss mechanisms in undermining the effectiveness of immune-based cancer treatment, particularly in scenarios of tumor relapse after treatment.
Two molecular processes may cause antigen loss in B cell malignancies: alternative splicing and point mutations. For instance, in CD19-positive ALL, a specific mutation in the CD19 gene resulted in the production of truncated proteins. These truncated proteins typically result in either a CD19 antigen protein without the transmembrane domain or a completely non-functional CD19 antigen protein [41,44]. Similarly, the mechanism of alternative splicing can produce either a CD19 lacking the transmembrane domain or with the transmembrane domain, in which the epitope necessary for CAR T cell recognition is missing [45,46,47]. The presence of malignant B cells lacking or expressing altered antigens allows them to escape CAR-T cell detection and destruction, making them especially resistant to immunotherapeutic strategies [31].
2.4. Cancer Cells’ Antigens Masking
Another strategic defense mechanism developed by cancer cells to escape targeting and eventual elimination by the immune system is their ability to use specific molecules to obscure their surface. A significant case that illustrates this mechanism is the CD19-targeting CAR-T cell treatment strategy directed against malignancies related to B-ALL. In this context, tumor relapse following CAR-T cell treatment resulted from the masking of the antigen targeted by the CAR-T cells. Reports indicate that in B-ALL, leukemic cells unexpectedly expressed the CAR construct, mediated by the viral transduction during the cytapheresis process [48]. Consequently, the CAR on the leukemic cell surface bound to its target antigen (CD19), effectively concealing its epitopes. This antigen masking prevented CAR-T cells from recognizing the leukemic cells as foreign, allowing them to evade immune detection and survive despite treatment [48,49].
2.5. Immune Checkpoint and Ligand Activation
Immune cells possess specific receptors that interact with certain molecules to regulate immune responses and prevent autoimmunity through checkpoint signaling. However, tumor cells maneuver these pathways to limit T-cell activity and evade immune destruction [50]. Key components of the immune checkpoints, such as PD-1, PD-L1, and CTLA-4, which have become the central targets in cancer immunotherapy [31].
One immune checkpoint pathway that tumor cells exploit includes the PD-1 and PD-L1 pathways. (PD-1,CD279), is expressed at an elevated level, particularly on activated T-cells, and is crucial for facilitating immune tolerance [51]. PD-L1 and PD-L2 both bind PD-1, but PD-L1 is more extensively studied [52]. In normal tissues, PD-L1 expression is diminished, but tumor tissues can adeptly escape immune system attacks by increasing the expression of PD-L1 ligands [53]. Cancer types reported for elevated expression of PD-L1 include breast and squamous cell head and neck cancer [53].
The relationship between PD-1 and PD-L1 is a well-established mechanism that reduces the immune activity of activated T cells. Therefore, a logical and increasingly recognized technique in immune-based cancer treatment is to block this interaction through anti-PD-L1 and anti-PD-1 antibodies [54,55]. Several studies have highlighted how these antibodies target various tumors by interfering with the PD-1/PD-L1 interactions. For instance, neoadjuvant anti-PD-1 antibodies have shown considerable success stage III/IV melanoma [56]. Meanwhile, in a phase 1 study of pembrolizumab as a PD-1 inhibitor, the subjects with advanced-stage non-small-cell lung cancer patients exhibited a noticeable therapeutic response with minimal side effects [57]. A combined anti-PD1 and platinum therapy This was demonstrated in two Phase 3 clinical trials, improved the overall survival in individuals with recurrent or metastatic head and neck squamous cell carcinoma (HNSCC) in two Phase 3 trials [58,59]. However, some patients demonstrated limited response, indicating possible involvement of other inhibitory mechanisms.
Another highly studied immune inhibitory pathway tumor cells can exploit is (CTLA-4, CD152). (CTLA-4, CD152) is primarily found on regulatory T cells and is principally recognized for negatively regulating the activation of T-cells [60]. CTLA-4 has two ligands, B7-1 and B7-2 (CD80 and CD86), which can also bind to CD28, another receptor found on T cells. While CTL-4 and CD28 can be expressed by CD8+ and CD4+ T cells, their regulatory roles in T-cell activation are opposite [61]. Additionally, regarding interaction strength and efficiency, CTLA-4 exhibits better affinity and avidity for B7-1 and B7-2 compared to CDThis demonstrates that CTLA-4 can compete with CD28 and emphasizes how its regulatory role can negatively impact T-cell activation [62,63]. However, the specific mechanism through which CTLA-4 interacts with its ligands to decrease T-cell activation is poorly understood and requires further extensive studies [60].
The CTLA-4/CD28 pathway attracts significant attention in therapeutic strategies, where fusion proteins and antibodies are among the most viable options [4]. Since the CTLA-4 pathway is crucial for preventing the immune system from damaging self-tissue, enhancing its regulatory role might be a promising approach for managing autoimmune diseases. On the other hand, an approach that blocks or represses the CTLA-4 pathway could be an essential method of encouraging the immune system to exert its lethal action on tumors [4]. Utilizing anti-CTLA-4 antibodies to trigger the immune system to interact and efficiently destroy cancer tissues has accelerated recent innovative cancer therapeutic approaches [31].
Other strategies for immune evasion employed by cancer cells to avoid the attacks and destruction of the immune components of cancer immunotherapy include inducing T cell exhaustion, resisting apoptosis, downregulating antigens, switching lineages, trogocytosis-mediated antigen loss, and apoptosis resistance. These mechanisms are thoroughly reviewed in Figure 1 [31].
3. The CRISPR/Cas 9 Structure and Mechanisms
3.1. Overview of the CRISPR/Cas 9 Mechanisms: Guide RNA, Cas9 Enzyme, and DNA Targeting
The sequences known as clustered regularly interspaced short palindromic repeats (CRISPRs) were first identified in 1987 in Escherichia coli and subsequently discovered in various other bacterial species [64]. Their function was later discovered to be part of the adaptive immune mechanisms of archaea and bacteria, where they defense against invading genetic elements such as bacteriophages. It works through RNA-guided cutting of specific DNA [65,66,67]. The CRISPR Cas system operates through three sequential stages: acquisition, transcription, and interference [67,68]. Owing to its simplicity, precision, and adaptability, the CRISPR/Cas9 has been thoroughly studied for its structure, functionality, and application as a gene editing tool in practical human clinical settings since it was first discovered [69].
Interestingly, scientists are more attracted to the type-II CRISPR/ Cas9 system, particularly for human clinical applications [70]. The CRISPR/ Cas9 relies on Streptococcus pyogenes single Cas protein (SpCas9), which has a key distinguishing feature of targeting specific DNA sequences, making it widely used [70]. However, while SpCas9 remains the most utilized variant, other Cas proteins, such as Cas12, Cas13, and Cas14, have emerged with unique properties, including altered PAM requirements, RNA-targeting capabilities, and smaller sizes, thereby broadening the genome editing and engineering platforms [71].
In the type-II system, a specific DNA segment from the invading genetic elements is integrated into the host (archaea /bacterial) CRISPR locus. This integrated DNA segment is termed a spacer [72]. In the type-II CRISPR/Cas9 system, the locus where the spacer is integrated is transcribed into a long precursor CRISPR RNA (pre-crRNA), which is subsequently processed into short CRISPR-derived RNAs (crRNAs). These crRNAs pair with a trans-activating CRISPR RNA (tracrRNA) to form a dual complex RNA structure that guides the Cas9 protein to a specific target DNA. For simplicity, tracrRNA and crRNS are often combined into a single guide RNA (sgRNA) for simplicity. Within the sgRNA is also a spacer region complementary to the target DNA sequence and a scaffold region that forms a hairpin structure recognized by Cas Once loaded onto Cas9, the sgRNA directs the protein to the complementary sequence on the target DNA, where the Cas9 protein induces a site-specific double-strand break (DSB) [73]. The CRISPR/ Cas9 system is mechanistically made up of two essential elements: a single guide RNA and a Cas9 endonuclease [69].
One striking characteristic of the CRISPR/ Cas9 system is that whenever the need arises to modify the sequence of a target DNA, a sgRNA that matches the desired DNA sequence to be cleaved is designed [69]. The sequences of the sgRNA usually follow this arrangement: a twenty-base sequence at 5’ that matches a target DNA sequence via Watson-Cricks base pairing while at 3’, another approximately twenty-base pair or above, that binds to the Cas 9 protein, which aligns near the protospacer adjacent motifs (PAM) [74,75]. With these sequence arrangements of the sgRNA, two other requirements are necessary for forming a functional sgRNA- Cas-9 complex that can effectively bind and cleave a certain location on the target DNA. First, the exact matching of the first-twenty-base pair of the sgRNA and that of the specific site of the target DNA. Second, the protospacer adjacent motifs must be available precisely adjacent to the target DNA sequence [75,76]. Once all these elements are in place, the binding of the sgRNA to the target DNA with the PAM component occurs, forming the RNA-DNA-hybrid with the cleavage of the target sequence by the RuvC-like and HNH components of the Cas 9 endonuclease [77,78,79]. As highlighted earlier, the DSBs created by endonucleases are repaired by HDR or the NHEJ (Figure 2), with the former being the most sought-after as it allows precise insertion or correction of DNA sequences. NHEJ is an error-prone repair mechanism that frequently causes indels and genome rearrangement [80].
On the other hand, HDR, also known as high-fidelity repair, repairs the CRISPR-induced DSBs by utilizing a homologous DNA template, leading to fewer errors compared to NHEJ [81]. However, a crucial limitation of HDR is that it is a cell-cycle-dependent pathway, primarily active during the S and G2/M phases of the cell cycle. Consequently, scientists are putting significant effort into modulating the cell cycle to increase its efficiency [82]. The CRISPR/Cas9 system has become commonly used for gene editing and genome modification. For instance, it can be utilized to repress or activate the expression of particular genes in CRISPRa (for gene activation) and CRISPRi (for gene repression) systems [83]. Several RNA-guided systems have progressed from basic research stages to various phases of clinical trials [69].
3.2. The CRISPR/Cas 9 System in Immune System Modulation and Cancer Immunotherapy
The adaptability of the CRISPR/Cas9 system is evident in the increasing studies demonstrating its application in cancer immunotherapy. This is mostly achieved by immunomodulating the immune system to improve the success of immune-based cancer treatment methods. We will closely examine how the CRISPR/Cas9 system has been explored to identify and screen immunomodulatory genes, target immune checkpoint molecules, target specific immunomodulators in tumor progression, and manipulate immune cell function, all aimed toward successful cancer immunotherapy.
3.2.1. The CRISPR/Cas 9 Systems: A Tool for the Identification and Screening of Immunomodulatory Genes
Identifying and screening genes directly or indirectly relevant for improving the effectiveness of immune cells in dealing with tumors or changing the TME is essential for the success of cancer immunotherapy. Several vital studies employ the CRISPR/Cas9 system to screen and identify immunomodulatory genes (Figure 3) [84].
Researchers developed a CRISPR/Cas9 immunomodulatory gene-screening tool based on sgRNA library to identify novel immunotherapy targets. The study’s significant findings corroborate the functions of well-known escape molecules such as CD47 and PD-LAdditionally, it reveals that interfering with the IFN-γ signaling pathway may help surpass resistance to cancer immunotherapy, and elements of the IFN-γ signaling pathway, particularly protein tyrosine phosphatase 2 (Ptpn2), might represent a novel, promising target [85]. A subsequent study by different researchers utilized an in vivo CRISPR/Cas9 system to screen CD8+ T cells and discovered that enhancing the capability of the CD8+ T cells to fight breast cancer cells could be significantly improved by CRISPR-mediated repression of the DEAH-Box Helicase 37 (DHX37) gene [86]. Another study aimed to identify potential gain-of-function targets for CD8+ T cells in CAR-T cell engineering. This study utilizes the CRISPR activation screening technique using dead guide RNA (dgRNA). It reveals that CAR-T cells’ cytotoxicity and in vivo efficiency could be significantly improved by overexpression or specific knock-in of the PRODH2 in various cancer types. Furthermore, in addition to potentially influencing metabolic pathways, the increased expression of PRODH2 can significantly enhance CAR-T cells’ mitochondrial and immune functions, as revealed by metabolomic and transcriptomic analysis [87].
Conversely, another study focused on screening for genomic loss-of-function utilizing the CRISPR/Cas9 system. It investigated the function of the death receptor signaling of FADD and TRAILR2 in CAR-T cell therapy against CD19-positive tumor cells. The study supported the significant influences of these signals on CAR T cells’ capacity to manage CD19-positive tumor cells. It provided the foundation for scientists to optimize the therapeutic potential of CAR-Ts [88]. A striking outcome of a specific CRISPR/Cas9 screening study led to concurrent discoveries. The MS4A1 gene unequivocally encodes the target of rituximab, CDThe interferon regulatory factor 8 (IRF8) may impact the antibody-mediated cytotoxicity and phagocytosis of CD20 [89]. These findings emphasized that CRISPR/Cas9 screening techniques may serve as essential instruments in identifying significant genes that could be pivotal targets in cancer immunotherapy.
3.2.2. The CRISPR/Cas 9 System and Immune System Manipulation in Cancer Immunotherapy
The promising applications of the CRISPR/Cas9 system have been extended to manipulating the genes of immune cells for the precise targeting of cancer cells. Based on the CRISPR knockout, selective properties have been endowed on human immune cells to maneuver through the complex tumor microenvironment (TME). This was clearly illustrated by [84] how the CRISPR/Cas9 system can enhance the overall therapeutic effectiveness of engineered T-cell receptors in CAR-T cell therapy. The CRISPR/Cas9 platforms facilitate the massive and efficient manufacture of therapeutic modified T cells and enable the manufacture of tailored modified T cells suitable for application in complex clinical treatments. Additionally, the CRISPR/Cas9 systems can facilitate the genetic manipulation of T cells in laboratory settings and endow the cells with unique properties to identify and fight cancer cells without the necessary interventions of HLA [84].
Meanwhile, the CRISPR/Cas9 technology has been successfully applied to disrupt the activity of PD-1 to enhance the ability of a particular CAR-T cell directed against EGFRvIII of glial cell tumors [90]. Interestingly, the genetic modification of CAR-T cells greatly improved their ability to destroy cancer cells in vitro and the efficient production of pro-inflammatory cytokines. In CAR-T cell therapy, the positive outcomes of the genetically modified CAR-T cells were evident in their ability to prolong survival and enhance cancer control in an animal model. These results highlighted the potential of the CRISPR/Cas9 system to endow the manipulated CAR-T cells in CAR-T cell therapy to surmount the PD-L-mediated immunosuppression, which in turn enhances CAR-T’s effectiveness in the treatment of glial cell tumors [91].
4. CRISPR-Based Approaches to Overcome Immune Evasion
Cancer immunotherapy has indicated a high level of efficacy across various cancer types [92]. However, challenges such as off-target toxicity, an immunosuppressive TME, T-cell exhaustion, and poor T-cell quality hinder cancer immunotherapy from reaching its full potential [93]. CRISPR genome editing has enabled high-throughput identification of immune evasion mechanisms and has enabled the development of strategies to counter them (Table 1). In addition to uncovering immunomodulatory genes, it has also aided in enhancing T-cell activity, suppressing inhibitory receptor expression, and redirecting T-cell antigen specificity by editing chimeric antigen receptors (CARs) or T-cell receptors (TCRs) at the endogenous TCR-⍺ chain (TRAC) locus [94]. This section summarizes key CRISPR strategies for overcoming immune evasion, with insights from preclinical and clinical studies.
4.1. Editing Immune Checkpoints
A major barrier to effective cancer immunotherapy is immune evasion, often driven by T-cell exhaustion. In both human and mouse models, exhausted T cells express high levels of inhibitory receptors such as PD-1 (CD279), CTLA-4 (CD152), LAG3, TIM-3 (HAVCR2), 2B4 (CD244), CD160, and TIGIT, which impair their activation and antitumor function [95]. CRISPR/Cas9-based genome editing offers a powerful strategy to counteract this suppression by disrupting immune checkpoint pathways and enhancing T-cell responses [96]. For instance, CRISPR-mediated deletion of the PD-1 gene in primary T cells has been shown to boost their activation and cytotoxic potential. In cytotoxic T lymphocytes (CTLs), PD-1 knockdown not only improves effector function but also suppresses regulatory T cells (Tregs), further amplifying the immune response.
This strategy has shown particular promise in infection-related cancers, such as Epstein-Barr virus-associated gastric carcinoma (EBVaGC). In these tumors, LMP2A-expressing T cells exhibit elevated levels of PD-L1 (CD274) and PD-L2 (CD273). LMP2A-derived peptides can sensitize peripheral blood lymphocytes, triggering a CTL response against EBVaGC cells [97]. A recent study demonstrated that CRISPR/Cas9-mediated PD-1 knockout in LMP2A-specific T cells significantly enhanced their cytotoxicity. When administered in vivo, these edited T cells improved survival in tumor-bearing mice and exhibited potent antitumor effects, especially when combined with low-dose radiotherapy, highlighting their therapeutic potential in adoptive T-cell therapy for EBVaGC [98]. In addition to surface checkpoints, several intracellular regulators within effector immune cells serve as potential therapeutic targets. One example is the orphan nuclear receptor NR2F6, which functions as an intracellular immune checkpoint by repressing transcription of key cytokines such as TNFα, IL-2, and IFNγ, molecules critical for tumor rejection [99]. CRISPR-based knockout of NR2F6 has been proposed as a novel strategy to enhance T-cell-mediated immunity [100].
CRISPR screening has also identified protein tyrosine phosphatase 1B (PTP1B) as a negative regulator of T-cell function. PTP1B interferes with JAK/STAT signaling by dephosphorylating JAK2 and TYK2, thereby limiting T-cell activation, proliferation, and cytotoxicity. Deleting PTP1B boosts STAT5 signaling, promotes antigen-driven CD8+ T-cell expansion, and inhibits solid tumor growth. Notably, PTP1B inhibition has been shown to enhance the efficacy of both natural and adoptively transferred T cells including CAR T cells and improve responses to PD-1 blockade [101]. Another relevant immune checkpoint is TIGIT, commonly expressed on antitumor T cells such as CD103+ tissue-resident memory (Trm) cells. TIGIT suppresses immune responses by inhibiting the costimulatory receptor CDIt acts synergistically with PD-1, recruiting the phosphatase Shp2, which deactivates CD28 and CD226 signaling pathways, further dampening T-cell function. Dual blockade of PD-L1 and TIGIT disrupts this suppressive network, restoring costimulatory signaling and enhancing antitumor T-cell activity [102]. Together, these studies demonstrate that CRISPR/Cas9 can be leveraged not only to target classical surface checkpoints but also to uncover and modulate intracellular regulators. This dual targeting strategy has the potential to significantly enhance the effectiveness of cancer immunotherapy by overcoming multiple layers of immune evasion.
4.2. Engineering CAR-T Cells and Tumor-Infiltrating Lymphocytes (TILs)
Adoptive cell therapy (ACT) is a promising immunotherapeutic strategy that involves isolating a patient’s immune cells, genetically modifying, and expanding them ex vivo to enhance their antitumor activity and reinfusing them into the patient. ACT aims to overcome immune suppression, including poor T-cell function, inadequate T-cell production, and weak memory T-cell development. The main types of ACT include tumor-infiltrating lymphocyte (TIL) therapy, T-cell receptor (TCR)-based therapy, and chimeric antigen receptor (CAR)-T cell therapy. Other evolving forms of ACT involve engineered macrophages, B cells, dendritic cells, and natural killer (NK) cells [124].
While it is not yet endorsed as a first-line treatment for cancers, the CAR-T cancer therapy method has revolutionized the management of certain aggressive lymphomas and multiple myelomas that have relapsed or are challenging to treat [125]. CAR-T cells combat tumors by releasing cytotoxic molecules (perforins and granzymes) and pro-inflammatory cytokines (e.g., IFN-γ, IL-2), which modify the TME and encourage apoptosis and necrosis in tumor cells [124].
CAR-T therapy has progressed through multiple generations, each built upon the previous one to improve its safety and efficacy, and several CAR-T therapies have gained FDA approval, and many are currently undergoing clinical trials [124,126,127,128,129]. Despite these advances, CAR-T therapy faces limitations. High manufacturing costs, lengthy production timelines, and difficulty in generating high-quality T cells from critically ill patients hinder broader application. Furthermore, disease relapse and variability in autologous CAR-T products contribute to inconsistent outcomes [95,130].
To address these challenges, CRISPR/Cas9 gene editing has emerged as a transformative tool to improve CAR-T safety, efficacy, and scalability. Disruption of co-inhibitory genes like PD-1 and CTLA-4 using CRISPR has been shown to enhance CAR-T cell persistence and antitumor function [131]. Researchers are also integrating CAR-T with chemotherapy, radiotherapy, and ICIs to overcome the TME’s suppressive effects, although these combinations have yet to fully resolve current limitations [132].
CRISPR screens have identified TGF-β and CD7 as critical regulators of CAR-T efficacy in solid and hematologic cancers. TGF-β, secreted by stromal and tumor-associated cells, initiates signaling via TGFBR2, which induces T-cell exhaustion by upregulating immune checkpoints (e.g., PD-1, TIM-3, CTLA-4, LAG3) and promoting Treg-like phenotypes through FOXP3 expression [133]. In a study by Tang et al. (2020), CRISPR-mediated TGFBR2 knockout in CAR-T cells restored their function. Dual knockout of PDCD1 (PD-1) and TGFBR2 further improved T-cell proliferation and resistance to TME suppression in xenograft models, highlighting the value of multiplexed gene editing to enhance CAR-T efficacy [133] In T-cell malignancies, targeting CD7 poses a unique challenge since both malignant and healthy T cells express this marker, risking fratricide. To address this, researchers developed UCART7, an “off-the-shelf” CAR-T product engineered using CRISPR/Cas9 to knock out both CD7 and TRAC (T-cell receptor alpha chain). UCART7 showed strong antitumor activity against T-ALL while avoiding graft-versus-host disease in preclinical models [134].
While CAR-T has had remarkable success in hematologic cancers, its effectiveness against solid tumors remains limited. As a result, attention has shifted toward tumor-infiltrating lymphocyte (TIL) therapy, particularly for solid tumors. TILs are T cells naturally residing within tumor tissue, including both CD4+ and CD8+ αβ T cells [135,136]. Unlike CAR-T therapy, which sources T cells from peripheral blood, TIL therapy harnesses immune cells already primed within the tumor microenvironment. TIL therapy typically involves expanding TILs ex vivo, administering non-myeloablative lymphodepleting chemotherapy (NMA-LD) to enhance their engraftment, and supporting their survival with recombinant human IL-2 [137,138].
The FDA recently approved Amtagvi, a TIL-based therapy, for adult patients with metastatic melanoma previously treated with PD-1 blockers and/or BRAF-targeted therapy [139,140]. While effective in melanoma, TIL therapy still faces challenges in other solid tumors due to a more suppressive TME and potential adverse effects like immunosuppression, infection, and organ toxicity [141].
CRISPR offers powerful strategies to enhance TIL therapy. A CRISPR screen identified SOCS1, a suppressor of cytokine signaling, as a key intracellular brake in T cells. Knockout of SOCS1 increased IFN-γ production, improved cytokine responsiveness, and enhanced the antitumor efficacy of TILs [142]. Additionally, non-viral CRISPR/Cas9 methods have been used to generate PD-1-deficient TILs, enhancing their cytotoxicity across multiple tumor types [143]. These findings demonstrate the significant promise of genetic editing in advancing TIL-based ACT and underscore the need for further exploration to optimize these therapies for clinical use (Figure 4).
4.3. Disrupting Tumor-Intrinsic Immune Evasion Genes
The immunophenotype of the TME significantly influences the response to immune checkpoint inhibitors (ICIs), underscoring the need for detailed profiling of immune signatures and the discovery of new therapeutic targets to enhance immunotherapy outcomes. CRISPR-based functional screens targeting immune-related genes have become powerful tools for identifying such targets. For instance, in triple-negative breast cancer (TNBC), CRISPR screens revealed Lgals2, a key immune evasion regulator [144], SLC7A5, a neutral amino acid transporter essential for citrulline uptake and arginine biosynthesis [145], and DYRK1B, a dual-specificity kinase that supports cell survival and proliferation [146]. Another gene, Cop1, an E3 ubiquitin ligase, was shown to regulate M2 tumor-associated macrophages (TAMs) and response to anti-PD-1 therapy by targeting the oncogene C/ebpδ for degradation [147]. These findings open new avenues for modulating both cancer cell survival and the immunosuppressive microenvironment.
Targeting immunosuppressive components of the TME, particularly TAMs, also holds promise. TAMs, commonly associated with poor prognosis, can be modulated using CRISPR. For example, CD47, a “don’t eat me” signal frequently overexpressed in tumors, interacts with SIRPα on macrophages to prevent phagocytosis. Lin et al. (2022) disrupted CD47 in tumor cells via CRISPR and combined this with IL-12 expression, an immunostimulatory cytokine produced by macrophages and dendritic cells. Engineered tumor cells secreted IL-12, reprogramming TAMs in situ and shifting them toward a pro-inflammatory (M1) phenotype. This dual approach significantly enhanced anti-tumor immune responses and reduced tumor growth [148].
Further supporting this strategy, PGAM5, a mitochondrial phosphatase linked to M2-TAM infiltration, was recently identified as another tumor-intrinsic regulator. CRISPR/Cas9-mediated disruption of PGAM5 in tumor cells reduced M2-TAM recruitment, slowed tumor progression, and strengthened immune responses [149]. These studies highlight the therapeutic potential of targeting tumor-intrinsic genes to reshape the TME and improve immunotherapy efficacy.
5. Challenges and Considerations in CRISPR-Based Immunotherapy Development
5.1. Safety and Off-Target Effects
While CRISPR/Cas9 offers transformative potential in cancer immunotherapy, several challenges must be addressed to ensure its safe and effective application [125]. A major concern is the risk of off-target effects, which can introduce unintended mutations and adverse consequences [150]. Researchers are working to enhance precision through refined delivery systems, most notably, by using CRISPR as a ribonucleoprotein (RNP) complex, which reduces integration risks and improves editing efficiency [151]. Another concern is the uncertainty surrounding long-term effects. For instance, in animal models of tyrosinemia type 1, CRISPR-mediated deletion of the Hpd gene corrected the metabolic disorder but unexpectedly increased hepatocellular carcinoma incidence, emphasizing the need for long-term safety assessments in cancer-prone contexts [152].
To improve gene-editing efficiency and minimize vector size, compact CRISPR systems like Cas14 (Cas12f) have been explored [153]. Despite their smaller size, these nucleases initially suffered from low efficiency [154]. However, an engineered variant, eCas12f1, demonstrated potent anticancer activity by effectively targeting PLK1, a gene critical for cell cycle progression, significantly reducing colony formation in breast cancer cells [155].
5.2. Delivery Challenges
Delivery remains a key challenge in CRISPR therapeutics. Viral and plasmid vectors can trigger immune responses, increase off-target effects, and pose safety concerns due to persistent Cas9 expression [156,157]. However, they sometimes require high doses of RNA therapeutics, leading to inflammation and toxicity. Ongoing research is focused on improving LNP formulations to enhance safety [125].
Another promising delivery method involves extracellular vesicles (EVs), which are naturally compatible with the body and can cross biological barriers, such as the blood–brain barrier. EVs efficiently deliver CRISPR/Cas9 components to tumor cells and immune cells, including in hard-to-reach cancers like glioblastoma [151].
Combining CRISPR with immunotherapy and chemotherapy offers exciting therapeutic potential. For example, a pH-sensitive nanoparticle system using PEI–PLGA was developed to co-deliver CRISPR plasmids and paclitaxel. This system not only knocked down PD-L1 expression via Cas9 targeting of Cdk5 but also enhanced immune activation and tumor suppression by inducing immunogenic cell death and shifting macrophage populations [158].
The use of a rigorous guide RNA (gRNA) design process coupled with electroporation to deliver the ribonucleoprotein (RNP) complex is an emerging strategy to minimize the downstream effects of CRISPR-Cas-based gene editing. Electroporation, a method that involves applying an electrical field to cells to facilitate the delivery of genetic material, is often used in CRISPR-based systems to introduce RNP complexes directly into cells, avoiding the potential risks associated with using viral or plasmid vectors. However, one challenge of electroporation is that it can induce cell damage and death. Despite this, a study by Chamberlain et al. (2022) demonstrated that electroporation could still produce clinically relevant numbers of engineered tumor-infiltrating lymphocytes (TILs) that were ready for infusion [143].
5.3. Ethical Considerations and Regulatory Landscape
CRISPR/Cas technology is advancing at a pace that outstrips existing regulations, raising concerns about its vast yet controversial potential. In light of this, it is crucial to establish appropriate laws and develop ethical guidelines to effectively evaluate both the benefits and risks of this technology [159]. A key ethical concern surrounding CRISPR gene editing is the use of germline cells and embryo editing, which could have unforeseen consequences for future generations. These changes may lead to long-lasting effects, given the uncertainties regarding unintended outcomes beyond the intended edits [160].
Given the uncertainties surrounding CRISPR, several regulatory bodies, including the WHO, UNESCO, and the Declaration on the Human Genome and Human Rights, are working to establish guidelines. While policies vary by country, legally binding agreements such as the “Oviedo Convention” (Convention on Human Rights and Biomedicine) and regulatory agencies like the UK’s HFEA and the U.S. FDA play significant roles in shaping these regulations. The U.S. National Academy of Sciences (NAS) and National Academy of Medicine (NAM) have also emphasized the significance of engaging with the public in discussions regarding the possible advantages and negative consequences that may result from the editing of the human genome [161].
During a meeting held in the United Kingdom in 2015, it was argued that clinical research should be restricted due to fears of misuse. Despite bioethical constraints, the UK’s Authority for Human Embryology and Fertilization approved CRISPR experiments on embryos, sparking significant controversy. This approval raised concerns, particularly regarding the potential for creating genetically modified humans resistant to HIV [162].
One major risk associated with CRISPR is the possibility of unintended or off-target effects, as the single guide RNA used by CRISPR does not require an exact match for Cas9 to induce a double-stranded break. Given the complexity of the human genome, this increases the likelihood of unintended genetic modifications, which could potentially lead to new genetic disorders, including cancer. Moreover, our incomplete understanding of human genes means that modifying one gene could unintentionally disrupt essential biological functions. Some of these unintended effects might not become apparent until future generations, adding complexity to ethical considerations. These risks have sparked global debates and led to regulatory actions, as the scientific community strives to balance CRISPR’s promise for medical advancements with ethical responsibility [163].
6. Future Directions and Innovations
CRISPR/Cas9 system has revolutionized genetic engineering and various treatment approaches, especially in immunotherapy [164]. Although CRISPR-Cas9 holds immense therapeutic potential, concerns regarding off-target effects and immunogenicity persist demanding safe and precise CRISPR-based therapeutics [165,166,167,168]. Prospective developments in CRISPR/Cas9 system involve exploring alternative delivery strategies to mitigate immunogenic responses the engineering of Cas9 variants with improved specificity in genome editing, and optimized guide RNA configurations [169,170,171,172,173]. Additionally, the risk of unintended genomic alterations can be minimized through DNA base editing and prime editing—precision gene editing techniques capable of making precise nucleotide changes without inducing DSBs. These techniques hold exponential promise as an instrument of therapy for controlling disease-causing mutations in the human genome in a programmable manner [165,166,171,174]. CRISPRi/CRISPRa can implement transcriptional regulation, engineer cellular metabolism, and elucidate genotype-phenotype mapping across smaller targeted libraries to genome-wide libraries [175]. Among the numerous advancements in CRISPR technology, CRISPRi, and CRISPRa are particularly significant for their ability to fine-tune gene expression by selectively repressing or activating immune-related genes; these tools hold significant potential for immune modulation, offering enhanced therapeutic strategies against different diseases, including cancer [175,176,177]. CRISPR/Cas9 system has the potential to design single treatment options to disrupt multiple mechanisms utilized by tumors to avoid targeting by the immune system. This approach of finding intervention within a single therapeutic framework promises to improve outcomes of immunotherapy, specifically where tumors resist conventional single-target therapies [69,178]. Moreover, the CRISPR/Cas9 system can help unlock immune responses that are typically suppressed in certain patients by targeting specific genetic alterations within tumor cells [179,180], which will be helpful in designing resilient cancer therapies that counteract specific immune evasion strategies [181]. The ability to modify genes in specific patient populations enables personalized therapeutic strategies, and tailored treatments according to tumor characteristics and an individual’s unique genetic makeup [84,181]. The CRISPR-Cas9 system offers unique precision in manipulating genes involved in drug metabolism, drug resistance, and individualized therapeutic effects, opening new avenues for patient-specific therapies [165,174,179] and advancing pharmacogenomics and personalized drug therapy [165,174]. CRISPR/Cas9 system can provide a more stronger immune cells action against tumors through the modification or disruption of immune checkpoints, notably, PD-1, in T cells [182,183,184]. Similarly, the concurrent use of CRISPR/Cas9 system with cytokine therapies has been reported to provide synergistic effects i.e., a substantial reduction in adverse side effects and enhanced anti-tumor immunity [183] CRISPR/Cas technology has been recently exploited for the development of recombinant viral vector vaccines and live attenuated vaccines for human diseases such as certain viral infections. A novel vaccine candidate has been developed by researchers through the insertion of various viral parts within the appropriate compartments of a phage nanoparticle. This innovative nano vaccine design platform holds promise for the efficient deployment of phage-based vaccinations against a wide range of viral diseases [185,186]. Aside from its significant synergistic role in cancer treatment, immunotherapy, and personalized treatment options based on individualized genetic makeup, the CRISPR/Cas9 technology also holds promise for the treatment of addiction and familial or hereditary disorders [167,168]. Genetic influences involved in drug absorption, distribution, and metabolism have been identified as promising targets for CRISPR-based treatments in the context of addiction. The simultaneous harnessing of CRISPR/Cas9 systems with epigenetic mechanisms has been reported as a recent advancement that may allow the creation of novel therapeutics and addiction treatments. For example, anxiety and alcoholism can be alleviated and disruptive epigenetic modifications can be reversed through the use CRISPR gene modification [167]. Anxiety disorders (AD), major depressive disorders (MDD), autistic spectrum disorders (ASD), attention deficit hyperactivity disorders (ADHD), and schizophrenia (SP) are among the most significant hereditary disorders [26,27,168,187]. Several genes have been identified as faulty that have been linked to these conditions, including ZNRD1, TRIM26, SYNE1, ITIH3, TCF4, DPR1, NT5C2, CACNB2, PPP1R11, CACNA1C, TENM4, CNNM2, ANK3, CSMD1, and AS3MT. These genes are mainly expressed in synaptic transmission, cellular mechanism, neuronal activity, and immune regulation, and play a pivotal role in the development of the above-mentioned disorders [188]. These genetic alterations in the affected subjects present potential targets for CRISPR/Cas9-based treatments for these disorders [26,168].
7. Conclusion
CRISPR/Cas9 genome editing has led to various groundbreaking biomedical research discoveries. As demonstrated by the studies highlighted in this review, its application in cancer immunotherapy has proven to be an effective strategy in enhancing treatment efficacy and mitigating the challenges of conventional ICB and ACT. However, before CRISPR/cas9 modified immunotherapy can be established as a novel therapeutic strategy in oncology, extensive preclinical and clinical trials are needed to give a deeper insight into its safety, efficacy, and long-term effects
Author Contributions
Conceptualization, S.A., M.N., and I.A.; literature review and data curation, S.A., L.A; writing—original draft, S.A., M.N., and R.K.; writing—review and editing, S.A., L.A., L.F., R.K., M.N., and I.A.; illustrations, S.A., L.A; supervision, and funding acquisition, I.A.
Funding
We are grateful to King Fahd University of Petroleum and Minerals (KFUPM), Dhahran, 31261, Saudi Arabia, for financial support towards the APC of this work.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors acknowledge King Fahd University of Petroleum and Minerals (KFUPM), Dhahran, 31261, Saudi Arabia, for financial support for the APC of this work.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Feng, X.; Li, Z.; Liu, Y.; Chen, D.; Zhou, Z. CRISPR/Cas9 technology for advancements in cancer immunotherapy: from uncovering regulatory mechanisms to therapeutic applications. Exp. Hematol. Oncol. 2024, 13, 1–19. [Google Scholar] [CrossRef]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef]
- O’connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2019, 17, 147–167. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Dotti, G.; Savoldo, B. Utilizing cell-based therapeutics to overcome immune evasion in hematologic malignancies. Blood 2016, 127, 3350–3359. [Google Scholar] [CrossRef]
- Allemailem, K.S.; A Alsahli, M.; Almatroudi, A.; Alrumaihi, F.; Alkhaleefah, F.K.; Rahmani, A.H.; Khan, A.A. Current updates of CRISPR/Cas9-mediated genome editing and targeting within tumor cells: an innovative strategy of cancer management. Cancer Commun. 2022, 42, 1257–1287. [Google Scholar] [CrossRef] [PubMed]
- Allemailem, K.S.; Almatroodi, S.A.; Almatroudi, A.; Alrumaihi, F.; Al Abdulmonem, W.; Al-Megrin, W.A.I.; Aljamaan, A.N.; Rahmani, A.H.; Khan, A.A. Recent Advances in Genome-Editing Technology with CRISPR/Cas9 Variants and Stimuli-Responsive Targeting Approaches within Tumor Cells: A Future Perspective of Cancer Management. Int. J. Mol. Sci. 2023, 24, 7052. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Strong, A.; Musunuru, K. Genome editing in cardiovascular diseases. Nat. Rev. Cardiol. 2016, 14, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, D.; Venkataramani, P.; Nandi, S.; Bhattacharjee, S. CRISPR–Cas9 a boon or bane: the bumpy road ahead to cancer therapeutics. Cancer Cell Int. 2019, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- O’DRiscoll, M.; Jeggo, P.A. The role of double-strand break repair — insights from human genetics. Nat. Rev. Genet. 2006, 7, 45–54. [Google Scholar] [CrossRef]
- Rouet, P.; Smih, F.; Jasin, M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc. Natl. Acad. Sci. 1994, 91, 6064–6068. [Google Scholar] [CrossRef]
- Kaniecki, K.; De Tullio, L.; Greene, E.C. A change of view: homologous recombination at single-molecule resolution. Nat. Rev. Genet. 2017, 19, 191–207. [Google Scholar] [CrossRef]
- Verma, P.; Greenberg, R.A. Noncanonical views of homology-directed DNA repair. Genes Dev. 2016, 30, 1138–1154. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Lieber, M.R.; Gu, J.; Lu, H.; Shimazaki, N.; Tsai, A.G. Nonhomologous DNA End Joining (NHEJ) and Chromosomal Translocations in Humans. In Genome Stability and Human Diseases, vol. 50, H.-P. Nasheuer, Ed., in Subcellular Biochemistry, vol. 50., Dordrecht: Springer Netherlands, 2010, pp. 279–296. [CrossRef]
- Delacôte, F.; Lopez, B.S. Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability mintenance: the trans-S double-strand break repair model. Cell Cycle 2008, 7, 33–38. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.-S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, H.; Zhang, H.; Huang, Z.; Tian, R.; Li, L.; Fan, W.; Chen, Y.; Chen, L.; Zhang, S.; et al. The comparison of ZFNs, TALENs, and SpCas9 by GUIDE-seq in HPV-targeted gene therapy. Mol. Ther. - Nucleic Acids 2021, 26, 1466–1478. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., III. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2014, 2014 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: a review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2017, 15, 81–94. [Google Scholar] [CrossRef]
- Li, Y.-R.; Halladay, T.; Yang, L. Immune evasion in cell-based immunotherapy: unraveling challenges and novel strategies. J. Biomed. Sci. 2024, 31, 1–16. [Google Scholar] [CrossRef]
- Rabilloud, T.; Potier, D.; Pankaew, S.; Nozais, M.; Loosveld, M.; Payet-Bornet, D. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat. Commun. 2021, 12, 1–7. [Google Scholar] [CrossRef]
- Li, Y.-R.; Wilson, M.; Yang, L. Target tumor microenvironment by innate T cells. Front. Immunol. 2022, 13, 999549. [Google Scholar] [CrossRef]
- Li, Y.-R.; Yu, Y.; Kramer, A.; Hon, R.; Wilson, M.; Brown, J.; Yang, L. An Ex Vivo 3D Tumor Microenvironment-Mimicry Culture to Study TAM Modulation of Cancer Immunotherapy. Cells 2022, 11, 1583. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. Embo Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Digifico, E.; Belgiovine, C. Macrophages and cancer stem cells: a malevolent alliance. Mol. Med. 2021, 27, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Aramini, B.; Masciale, V.; Grisendi, G.; Banchelli, F.; D’amico, R.; Maiorana, A.; Morandi, U.; Dominici, M.; Haider, K.H. Cancer stem cells and macrophages: molecular connections and future perspectives against cancer. Oncotarget 2021, 12, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Ding, S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Yang, G.; Ye, P.; Cao, N.; Chi, X.; Yang, W.-H.; Yan, X. Macrophages Are a Double-Edged Sword: Molecular Crosstalk between Tumor-Associated Macrophages and Cancer Stem Cells. Biomolecules 2022, 12, 850. [Google Scholar] [CrossRef]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen–specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.-T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 868. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, X.; Tian, Y.; Li, F.; Zhao, X.; Liu, J.; Yao, C.; Zhang, Y. Point mutation in CD19 facilitates immune escape of B cell lymphoma from CAR-T cell therapy. J. Immunother. Cancer 2020, 8, e001150. [Google Scholar] [CrossRef] [PubMed]
- Asnani, M.; Hayer, K.E.; Naqvi, A.S.; Zheng, S.; Yang, S.Y.; Oldridge, D.; Ibrahim, F.; Maragkakis, M.; Gazzara, M.R.; Black, K.L.; et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia 2019, 34, 1202–1207. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; LaNauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef]
- Ruella, M.; Xu, J.; Barrett, D.M.; Fraietta, J.A.; Reich, T.J.; Ambrose, D.E.; Klichinsky, M.; Shestova, O.; Patel, P.R.; Kulikovskaya, I.; et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat. Med. 2018, 24, 1499–1503. [Google Scholar] [CrossRef]
- Warda, W.; Da Rocha, M.N.; Trad, R.; Haderbache, R.; Salma, Y.; Bouquet, L.; Roussel, X.; Nicod, C.; Deschamps, M.; Ferrand, C. Overcoming target epitope masking resistance that can occur on low-antigen-expresser AML blasts after IL-1RAP chimeric antigen receptor T cell therapy using the inducible caspase 9 suicide gene safety switch. Cancer Gene Ther. 2021, 28, 1365–1375. [Google Scholar] [CrossRef]
- Marin-Acevedo, J.A.; Dholaria, B.; Soyano, A.E.; Knutson, K.L.; Chumsri, S.; Lou, Y. Next generation of immune checkpoint therapy in cancer: new developments and challenges. J. Hematol. Oncol. 2018, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and Its Ligands in Tolerance and Immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, J.; Chen, S.; Wang, X.; Xi, Q.; Shen, H.; Zhang, R. Immune checkpoint inhibitors: breakthroughs in cancer treatment. Cancer Biol. Med. 2024, 21, 1–11. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The Next Immune-Checkpoint Inhibitors: PD-1/PD-L1 Blockade in Melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Simon, S.; Labarriere, N. PD-1 expression on tumor-specific T cells: Friend or foe for immunotherapy? OncoImmunology 2017, 7, e1364828–e1364828. [Google Scholar] [CrossRef]
- Huang, A.C.; Orlowski, R.J.; Xu, X.; Mick, R.; George, S.M.; Yan, P.K.; Manne, S.; Kraya, A.A.; Wubbenhorst, B.; Dorfman, L.; et al. A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med. 2019, 25, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Cohen, E.E.W.; Le Tourneau, C.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; Burtness, B.; Zhang, P.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, R.; Yang, A.-G.; Zheng, G. Diversity of immune checkpoints in cancer immunotherapy. Front. Immunol. 2023, 14, 1121285. [Google Scholar] [CrossRef]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: a moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Greene, J.L.; Leytze, G.M.; Emswiler, J.; Peach, R.; Bajorath, J.; Cosand, W.; Linsley, P.S. Covalent Dimerization of CD28/CTLA-4 and Oligomerization of CD80/CD86 Regulate T Cell Costimulatory Interactions. J. Biol. Chem. 1996, 271, 26762–26771. [Google Scholar] [CrossRef]
- Walker, L.S.K.; Sansom, D.M. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef]
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology 2005, 151, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Van Der Oost, J.; Westra, E.R.; Jackson, R.N.; Wiedenheft, B. Unravelling the structural and mechanistic basis of CRISPR–Cas systems. Nat. Rev. Microbiol. 2014, 12, 479–492. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Z.; Qin, Y. CRISPR/Cas9 system: recent applications in immuno-oncology and cancer immunotherapy. Exp. Hematol. Oncol. 2023, 12, 1–18. [Google Scholar] [CrossRef]
- Jiang, W.; Bikard, D.; Cox, D.; Zhang, F.; Marraffini, L.A. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat. Biotechnol. 2013, 31, 233–239. [Google Scholar] [CrossRef]
- Hillary, V.E.; Ceasar, S.A. A Review on the Mechanism and Applications of CRISPR/Cas9/Cas12/Cas13/Cas14 Proteins Utilized for Genome Engineering. Mol. Biotechnol. 2022, 65, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Acquired Resistance Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Deveau, H.; Barrangou, R.; Garneau, J.E.; Labonté, J.; Fremaux, C.; Boyaval, P.; Romero, D.A.; Horvath, P.; Moineau, S. Phage Response to CRISPR-Encoded Resistance in Streptococcus thermophilus. J. Bacteriol. 2008, 190, 1390–1400. [Google Scholar] [CrossRef]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 2006, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Rouet, P.; Smih, F.; Jasin, M. Introduction of Double-Strand Breaks into the Genome of Mouse Cells by Expression of a Rare-Cutting Endonuclease. Mol. Cell. Biol. 1994, 14. [Google Scholar] [CrossRef]
- Li, G.; Yang, X.; Luo, X.; Wu, Z.; Yang, H. Modulation of cell cycle increases CRISPR-mediated homology-directed DNA repair. Cell Biosci. 2023, 13, 1–13. [Google Scholar] [CrossRef]
- He, C.; Han, S.; Chang, Y.; Wu, M.; Zhao, Y.; Chen, C.; Chu, X. CRISPR screen in cancer: status quo and future perspectives. Am J Cancer Res 2021, 11, 1031–1050. [Google Scholar]
- Feng, X.; Li, Z.; Liu, Y.; Chen, D.; Zhou, Z. CRISPR/Cas9 technology for advancements in cancer immunotherapy: from uncovering regulatory mechanisms to therapeutic applications. Exp. Hematol. Oncol. 2024, 13, 1–19. [Google Scholar] [CrossRef]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef]
- Dong, M.B.; Wang, G.; Chow, R.D.; Ye, L.; Zhu, L.; Dai, X.; Park, J.J.; Kim, H.R.; Errami, Y.; Guzman, C.D.; et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 2019, 178, 1189–1204.e23. [Google Scholar] [CrossRef]
- Ye, L.; Park, J.J.; Peng, L.; Yang, Q.; Chow, R.D.; Dong, M.B.; Lam, S.Z.; Guo, J.; Tang, E.; Zhang, Y.; et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022, 34, 595–614.e14. [Google Scholar] [CrossRef] [PubMed]
- Dufva, O.; Koski, J.; Maliniemi, P.; Ianevski, A.; Klievink, J.; Leitner, J.; Pölönen, P.; Hohtari, H.; Saeed, K.; Hannunen, T.; et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020, 135, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Grzelak, L.; Roesch, F.; Vaysse, A.; Biton, A.; Legendre, R.; Porrot, F.; Commère, P.; Planchais, C.; Mouquet, H.; Vignuzzi, M.; et al. IRF8 regulates efficacy of therapeutic anti-CD20 monoclonal antibodies. Eur. J. Immunol. 2022, 52, 1648–1661. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Binder, Z.A.; Lamrani, L.; Marinari, E.; Desai, A.S.; Nasrallah, M.P.; Maloney, E.; Brem, S.; Lustig, R.A.; Kurtz, G.; et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat. Cancer 2024, 5, 517–531. [Google Scholar] [CrossRef]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Smyth, M.J.; Ngiow, S.F.; Ribas, A.; Teng, M.W.L. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. [Google Scholar] [CrossRef]
- Nishino, M.; Giobbie-Hurder, A.; Hatabu, H.; Ramaiya, N.H.; Hodi, F.S. Incidence of Programmed Cell Death 1 Inhibitor–Related Pneumonitis in Patients With Advanced Cancer. JAMA Oncol. 2016, 2, 1607–1616. [Google Scholar] [CrossRef]
- Feng, X.; Li, Z.; Liu, Y.; Chen, D.; Zhou, Z. CRISPR/Cas9 technology for advancements in cancer immunotherapy: from uncovering regulatory mechanisms to therapeutic applications. Exp. Hematol. Oncol. 2024, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Ma, Q.; Yin, W.; Ma, X.; He, Z. CRISPR/Cas9 Gene-Editing in Cancer Immunotherapy: Promoting the Present Revolution in Cancer Therapy and Exploring More. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Allemailem, K.S.; A Alsahli, M.; Almatroudi, A.; Alrumaihi, F.; Al Abdulmonem, W.; A Moawad, A.; Alwanian, W.M.; Almansour, N.M.; Rahmani, A.H.; Khan, A.A. Innovative Strategies of Reprogramming Immune System Cells by Targeting CRISPR/Cas9-Based Genome-Editing Tools: A New Era of Cancer Management. Int. J. Nanomed. 2023, ume 18, 5531–5559. [Google Scholar] [CrossRef]
- Okugawa, K.; Itoh, T.; Kawashima, I.; Takesako, K.; Mazda, O.; Nukaya, I.; Yano, Y.; Yamamoto, Y.; Yamagishi, H.; Ueda, Y. Recognition of Epstein-Barr virus-associated gastric carcinoma cells by cytotoxic T lymphocytes induced in vitro with autologous lymphoblastoid cell line and LMP2-derived, HLA-A24-restricted 9-mer peptide. Oncol. Rep. 2004, 12, 725–731. [Google Scholar] [CrossRef]
- Su, S.; Zou, Z.; Chen, F.; Ding, N.; Du, J.; Shao, J.; Li, L.; Fu, Y.; Hu, B.; Yang, Y.; et al. CRISPR-Cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. OncoImmunology 2016, 6, e1249558. [Google Scholar] [CrossRef] [PubMed]
- Klepsch, V.; Hermann-Kleiter, N.; Baier, G. Beyond CTLA-4 and PD-1: Orphan nuclear receptor NR2F6 as T cell signaling switch and emerging target in cancer immunotherapy. Immunol. Lett. 2016, 178, 31–36. [Google Scholar] [CrossRef]
- Miftah, H.; Benthami, H.; Badou, A. Insights into the emerging immune checkpoint NR2F6 in cancer immunity. J. Leukoc. Biol. 2024, 117. [Google Scholar] [CrossRef] [PubMed]
- Wiede, F.; Lu, K.-H.; Du, X.; Zeissig, M.N.; Xu, R.; Goh, P.K.; Xirouchaki, C.E.; Hogarth, S.J.; Greatorex, S.; Sek, K.; et al. PTP1B Is an Intracellular Checkpoint that Limits T-cell and CAR T-cell Antitumor Immunity. Cancer Discov. 2022, 12, 752–773. [Google Scholar] [CrossRef]
- Banta, K.L.; Xu, X.; Chitre, A.S.; Au-Yeung, A.; Takahashi, C.; O’gOrman, W.E.; Wu, T.D.; Mittman, S.; Cubas, R.; Comps-Agrar, L.; et al. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity 2022, 55, 512–526.e9. [Google Scholar] [CrossRef]
- Wu, M.; Ma, W.; Lv, G.; Wang, X.; Li, C.; Chen, X.; Peng, X.; Tang, C.; Pan, Z.; Liu, R.; et al. DDR1 is identified as an immunotherapy target for microsatellite stable colon cancer by CRISPR screening. npj Precis. Oncol. 2024, 8, 1–11. [Google Scholar] [CrossRef]
- Deng, L.; Yang, L.; Zhu, S.; Li, M.; Wang, Y.; Cao, X.; Wang, Q.; Guo, L. Identifying CDC7 as a synergistic target of chemotherapy in resistant small-cell lung cancer via CRISPR/Cas9 screening. Cell Death Discov. 2023, 9, 1–10. [Google Scholar] [CrossRef]
- Yao, F.; Zhou, S.; Zhang, R.; Chen, Y.; Huang, W.; Yu, K.; Yang, N.; Qian, X.; Tie, X.; Xu, J.; et al. CRISPR/Cas9 screen reveals that targeting TRIM34 enhances ferroptosis sensitivity and augments immunotherapy efficacy in hepatocellular carcinoma. Cancer Lett. 2024, 593, 216935. [Google Scholar] [CrossRef]
- Zhao, D.; Deshpande, R.; Wu, K.; Tyagi, A.; Sharma, S.; Wu, S.-Y.; Xing, F.; O’NEill, S.; Ruiz, J.; Lyu, F.; et al. Identification of TUBB3 as an immunotherapy target in lung cancer by genome wide in vivo CRISPR screening. Neoplasia 2024, 60, 101100. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Yang, K.; Chen, C.; Zhou, Z.; Shen, P.; Jia, Y.; Xue, Y.; Zhang, Z.; Shen, X.; Han, X. Synergistic photothermal cancer immunotherapy by Cas9 ribonucleoprotein-based copper sulfide nanotherapeutic platform targeting PTPN2. Biomaterials 2021, 279, 121233. [Google Scholar] [CrossRef]
- Zou, C.; Liu, X.; Wang, W.; He, L.; Yin, A.; Cao, Z.; Zhu, M.; Wu, Y.; Liu, X.; Ma, J.; et al. Targeting GDF15 to enhance immunotherapy efficacy in glioblastoma through tumor microenvironment-responsive CRISPR-Cas9 nanoparticles. J. Nanobiotechnology 2025, 23, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Dervovic, D.; Malik, A.A.; Chen, E.L.Y.; Narimatsu, M.; Adler, N.; Afiuni-Zadeh, S.; Krenbek, D.; Martinez, S.; Tsai, R.; Boucher, J.; et al. In vivo CRISPR screens reveal Serpinb9 and Adam2 as regulators of immune therapy response in lung cancer. Nat. Commun. 2023, 14, 1–21. [Google Scholar] [CrossRef]
- Ramkumar, P.; Abarientos, A.B.; Tian, R.; Seyler, M.; Leong, J.T.; Chen, M.; Choudhry, P.; Hechler, T.; Shah, N.; Wong, S.W.; et al. CRISPR-based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv. 2020, 4, 2899–2911. [Google Scholar] [CrossRef]
- Boumelha, J.; de Castro, A.; Bah, N.; Cha, H.; Trécesson, S.d.C.; Rana, S.; Tomaschko, M.; Anastasiou, P.; Mugarza, E.; Moore, C.; et al. CRISPR–Cas9 Screening Identifies KRAS-Induced COX2 as a Driver of Immunotherapy Resistance in Lung Cancer. Cancer Res. 2024, 84, 2231–2246. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, Y.; Yang, N.; Mu, M.; Wu, Z.; Li, H.; Tang, X.; Zhong, K.; Zhang, Z.; Huang, C.; et al. Genome-scale CRISPR–Cas9 screen reveals novel regulators of B7-H3 in tumor cells. J. Immunother. Cancer 2022, 10, e004875. [Google Scholar] [CrossRef]
- Jiang, C.; Ward, N.P.; Prieto-Farigua, N.; Kang, Y.P.; Thalakola, A.; Teng, M.; DeNicola, G.M. A CRISPR screen identifies redox vulnerabilities for KEAP1/NRF2 mutant non-small cell lung cancer. Redox Biol. 2022, 54, 102358. [Google Scholar] [CrossRef]
- Ji, P.; Gong, Y.; Jin, M.-L.; Wu, H.-L.; Guo, L.-W.; Pei, Y.-C.; Chai, W.-J.; Jiang, Y.-Z.; Liu, Y.; Ma, X.-Y.; et al. In vivo multidimensional CRISPR screens identify Lgals2 as an immunotherapy target in triple-negative breast cancer. Sci. Adv. 2022, 8, eabl8247. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhao, L.; Lau, Y.S.; Zhang, C.; Han, R. Genome-wide CRISPR screen identifies LGALS2 as an oxidative stress-responsive gene with an inhibitory function on colon tumor growth. Oncogene 2020, 40, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Kim, S.; Kim, Y.; Kim, H.; Jang, D.; Shin, S.; Lee, S.-J.; Kim, J.; Lee, S.E.; Oh, J.; et al. Genome-wide CRISPR screening identifies tyrosylprotein sulfotransferase-2 as a target for augmenting anti-PD1 efficacy. Mol. Cancer 2024, 23, 1–19. [Google Scholar] [CrossRef]
- Wang, S.; Iyer, R.; Han, X.; Wei, J.; Li, N.; Cheng, Y.; Zhou, Y.; Gao, Q.; Zhang, L.; Yan, M.; et al. CRISPR screening identifies the deubiquitylase ATXN3 as a PD-L1–positive regulator for tumor immune evasion. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Yang, J.; Li, Z.; Shen, M.; Wang, Y.; Wang, L.; Li, J.; Yang, W.; Li, J.; Li, H.; Wang, X.; et al. Programmable Unlocking Nano-Matryoshka-CRISPR Precisely Reverses Immunosuppression to Unleash Cascade Amplified Adaptive Immune Response. Adv. Sci. 2021, 8, 2100292. [Google Scholar] [CrossRef]
- Ye, L.; Park, J.J.; Peng, L.; Yang, Q.; Chow, R.D.; Dong, M.B.; Lam, S.Z.; Guo, J.; Tang, E.; Zhang, Y.; et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022, 34, 595–614.e14. [Google Scholar] [CrossRef]
- Dong, M.B.; Wang, G.; Chow, R.D.; Ye, L.; Zhu, L.; Dai, X.; Park, J.J.; Kim, H.R.; Errami, Y.; Guzman, C.D.; et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 2019, 178, 1189–1204.e23. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tokheim, C.; Gu, S.S.; Wang, B.; Tang, Q.; Li, Y.; Traugh, N.; Zeng, Z.; Zhang, Y.; Li, Z.; et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell 2021, 184, 5357–5374.e22. [Google Scholar] [CrossRef]
- Li, F.; Huang, Q.; Luster, T.A.; Hu, H.; Zhang, H.; Ng, W.-L.; Khodadadi-Jamayran, A.; Wang, W.; Chen, T.; Deng, J.; et al. In Vivo Epigenetic CRISPR Screen Identifies Asf1a as an Immunotherapeutic Target in Kras-Mutant Lung Adenocarcinoma. Cancer Discov. 2020, 10, 270–287. [Google Scholar] [CrossRef]
- Wang, G.; Chow, R.D.; Zhu, L.; Bai, Z.; Ye, L.; Zhang, F.; Renauer, P.A.; Dong, M.B.; Dai, X.; Zhang, X.; et al. CRISPR-GEMM Pooled Mutagenic Screening Identifies KMT2D as a Major Modulator of Immune Checkpoint Blockade. Cancer Discov. 2020, 10, 1912–1933. [Google Scholar] [CrossRef]
- Hu, Q.; Xuan, J.; Wang, L.; Shen, K.; Gao, Z.; Zhou, Y.; Wei, C.; Gu, J. Application of adoptive cell therapy in malignant melanoma. J. Transl. Med. 2025, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Youssef, E.; Fletcher, B.; Palmer, D. Enhancing precision in cancer treatment: the role of gene therapy and immune modulation in oncology. Front. Med. 2025, 11, 1527600. [Google Scholar] [CrossRef] [PubMed]
- Vera, D.G.; Waghela, H.; Nuh, M.; Pan, J.; Lulla, P. Approved CAR-T therapies have reproducible efficacy and safety in clinical practice. Hum. Vaccines Immunother. 2024, 20, 2378543. [Google Scholar] [CrossRef] [PubMed]
- O. of the Commissioner, “FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma,” FDA. Accessed: Feb. 13, 2025. [Online]. Available: https://www.fda.gov/news-events/press-announcements/fda-approves-car-t-cell-therapy-treat-adults-certain-types-large-b-cell-lymphoma.
- Leick, M.B.; Maus, M.V.; Frigault, M.J. Clinical Perspective: Treatment of Aggressive B Cell Lymphomas with FDA-Approved CAR-T Cell Therapies. Mol. Ther. 2021, 29, 433–441. [Google Scholar] [CrossRef]
- Sengsayadeth, S.; Savani, B.N.; Oluwole, O.; Dholaria, B. Overview of approved CAR-T therapies, ongoing clinical trials, and its impact on clinical practice. eJHaem 2021, 3, 6–10. [Google Scholar] [CrossRef]
- Tao, R.; Han, X.; Bai, X.; Yu, J.; Ma, Y.; Chen, W.; Zhang, D.; Li, Z. Revolutionizing cancer treatment: enhancing CAR-T cell therapy with CRISPR/Cas9 gene editing technology. Front. Immunol. 2024, 15, 1354825. [Google Scholar] [CrossRef]
- Dötsch, S.; Svec, M.; Schober, K.; Hammel, M.; Wanisch, A.; Gökmen, F.; Jarosch, S.; Warmuth, L.; Barton, J.; Cicin-Sain, L.; et al. Long-term persistence and functionality of adoptively transferred antigen-specific T cells with genetically ablated PD-1 expression. Proc. Natl. Acad. Sci. 2023, 120. [Google Scholar] [CrossRef]
- Agarwal, S.; Aznar, M.A.; Rech, A.J.; Good, C.R.; Kuramitsu, S.; Da, T.; Gohil, M.; Chen, L.; Hong, S.-J.A.; Ravikumar, P.; et al. Deletion of the inhibitory co-receptor CTLA-4 enhances and invigorates chimeric antigen receptor T cells. Immunity 2023, 56, 2388–2407.e9. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. J. Clin. Investig. 2020, 5. [Google Scholar] [CrossRef]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef] [PubMed]
- Presti, D.; Dall’oLio, F.G.; Besse, B.; Ribeiro, J.M.; Di Meglio, A.; Soldato, D. Tumor infiltrating lymphocytes (TILs) as a predictive biomarker of response to checkpoint blockers in solid tumors: A systematic review. Crit. Rev. Oncol. 2022, 177, 103773. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Deng, J.; Rao, S.; Guo, S.; Shen, J.; Du, F.; Wu, X.; Chen, Y.; Li, M.; Chen, M.; et al. Tumor Infiltrating Lymphocyte (TIL) Therapy for Solid Tumor Treatment: Progressions and Challenges. Cancers 2022, 14, 4160. [Google Scholar] [CrossRef]
- Turcotte, S.; Donia, M.; Gastman, B.; Besser, M.; Brown, R.; Coukos, G.; Creelan, B.; Mullinax, J.; Sondak, V.K.; Yang, J.C.; et al. Art of TIL immunotherapy: SITC’s perspective on demystifying a complex treatment. J. Immunother. Cancer 2025, 13, e010207. [Google Scholar] [CrossRef]
- Schoenfeld, A.J.; Lee, S.M.; de Spéville, B.D.; Gettinger, S.N.; Häfliger, S.; Sukari, A.; Papa, S.; Rodriguez-Moreno, J.F.; Finckenstein, F.G.; Fiaz, R.; et al. Lifileucel, an Autologous Tumor-Infiltrating Lymphocyte Monotherapy, in Patients with Advanced Non–Small Cell Lung Cancer Resistant to Immune Checkpoint Inhibitors. Cancer Discov. 2024, 14, 1389–1402. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Lewis, K.D.; Kluger, H.; Hamid, O.; Whitman, E.; Thomas, S.; Wermke, M.; Cusnir, M.; Domingo-Musibay, E.; Phan, G.Q.; et al. Efficacy and safety of lifileucel, a one-time autologous tumor-infiltrating lymphocyte (TIL) cell therapy, in patients with advanced melanoma after progression on immune checkpoint inhibitors and targeted therapies: pooled analysis of consecutive cohorts of the C-144-01 study. J. Immunother. Cancer 2022, 10, e005755. [Google Scholar] [CrossRef]
- Kwong, M.L.M.; Yang, J.C. Lifileucel: FDA-approved T-cell therapy for melanoma. Oncologist 2024, 29, 648–650. [Google Scholar] [CrossRef]
- O. of the Commissioner, “FDA Approves First Cellular Therapy to Treat Patients with Unresectable or Metastatic Melanoma,” FDA. Accessed: Feb. 14, 2025. [Online]. Available: https://www.fda.
- Schlabach, M.R.; Lin, S.; Collester, Z.R.; Wrocklage, C.; Shenker, S.; Calnan, C.; Xu, T.; Gannon, H.S.; Williams, L.J.; Thompson, F.; et al. Rational design of a SOCS1-edited tumor-infiltrating lymphocyte therapy using CRISPR/Cas9 screens. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Chamberlain, C.A.; Bennett, E.P.; Kverneland, A.H.; Svane, I.M.; Donia, M.; Met, Ö. Highly efficient PD-1-targeted CRISPR-Cas9 for tumor-infiltrating lymphocyte-based adoptive T cell therapy. Mol. Ther. - Oncolytics 2022, 24, 417–428. [Google Scholar] [CrossRef]
- Ji, P.; Gong, Y.; Jin, M.-L.; Wu, H.-L.; Guo, L.-W.; Pei, Y.-C.; Chai, W.-J.; Jiang, Y.-Z.; Liu, Y.; Ma, X.-Y.; et al. In vivo multidimensional CRISPR screens identify Lgals2 as an immunotherapy target in triple-negative breast cancer. Sci. Adv. 2022, 8, eabl8247. [Google Scholar] [CrossRef]
- Dunlap, K.N.; Bender, A.; Bowles, A.; Bott, A.J.; Tay, J.; Grossmann, A.H.; Rutter, J.; Ducker, G.S. SLC7A5 is required for cancer cell growth under arginine-limited conditions. Cell Rep. 2025, 44, 115130. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, A.; Füchtbauer, E.-M.; Vahabzadeh, Z.; Soleimani, F.; Rahimi, K.; Nikkhoo, B.; Fakhari, S.; Erfan, M.B.K.; Azarnezhad, A.; Pooladi, A.; et al. CRISPR/Cas9-mediated knockout of DYRK1B in triple-negative breast cancer cells: implications for cell proliferation, apoptosis, and therapeutic sensitivity. Biochem. Eng. J. 2024, 213. [Google Scholar] [CrossRef]
- Wang, X.; Tokheim, C.; Gu, S.S.; Wang, B.; Tang, Q.; Li, Y.; Traugh, N.; Zeng, Z.; Zhang, Y.; Li, Z.; et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell 2021, 184, 5357–5374.e22. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Yang, Z.; Yang, Y.; Peng, Y.; Li, J.; Du, Y.; Sun, Q.; Gao, D.; Yuan, Q.; Zhou, Y.; et al. CRISPR-based in situ engineering tumor cells to reprogram macrophages for effective cancer immunotherapy. Nano Today 2022, 42. [Google Scholar] [CrossRef]
- Wei, X.; Wang, H.; Liu, H.; Wang, J.; Zhou, P.; Li, X.; He, Y.; Li, Y.; Han, D.; Mei, T.; et al. Disruption of tumor-intrinsic PGAM5 increases anti-PD-1 efficacy through the CCL2 signaling pathway. J. Immunother. Cancer 2025, 13, e009993. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Khorramian-Ghahfarokhi, M.; Shafieizadeh, M.; Mahmoudi, E.; Eskandari, F.; Rashidi, M.; Arshi, A.; Mokhtari-Farsani, A. Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy. Mol. Cancer 2024, 23, 1–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, X.; Luo, X.; Zhu, M.; Liu, N.; Li, J.; Zhang, Q.; Zou, C.; Wu, Y.; Cao, Z.; et al. Synergistic strategies for glioblastoma treatment: CRISPR-based multigene editing combined with immune checkpoint blockade. J. Nanobiotechnology 2025, 23, 1–18. [Google Scholar] [CrossRef]
- Chen, T.; Barzi, M.; Furey, N.; Kim, H.R.; Pankowicz, F.P.; Legras, X.; Elsea, S.H.; Hurley, A.E.; Yang, D.; Wheeler, D.A.; et al. CRISPR/Cas9 gene therapy increases the risk of tumorigenesis in the mouse model of hereditary tyrosinemia type I. JHEP Rep. 2025, 7, 101327. [Google Scholar] [CrossRef]
- Takeda, S.N.; Nakagawa, R.; Okazaki, S.; Hirano, H.; Kobayashi, K.; Kusakizako, T.; Nishizawa, T.; Yamashita, K.; Nishimasu, H.; Nureki, O. Structure of the miniature type V-F CRISPR-Cas effector enzyme. Mol. Cell 2021, 81, 558–570.e3. [Google Scholar] [CrossRef]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-J.; Ju, S.; Jung, W.J.; Jeong, T.Y.; Yoon, D.E.; Lee, J.H.; Yang, J.; Lee, H.; Choi, J.; Kim, H.S.; et al. Robust genome editing activity and the applications of enhanced miniature CRISPR-Cas12f1. Nat. Commun. 2025, 16, 1–12. [Google Scholar] [CrossRef]
- Yi, Q.; Ouyang, X.; Zhu, G.; Zhong, J. Letter: The risk-benefit balance of CRISPR-Cas screening systems in gene editing and targeted cancer therapy. J. Transl. Med. 2024, 22, 1–3. [Google Scholar] [CrossRef]
- Mao, K.; Tan, H.; Cong, X.; Liu, J.; Xin, Y.; Wang, J.; Guan, M.; Li, J.; Zhu, G.; Meng, X.; et al. Optimized lipid nanoparticles enable effective CRISPR/Cas9-mediated gene editing in dendritic cells for enhanced immunotherapy. Acta Pharm. Sin. B 2024, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Tu, K.; Deng, H.; Kong, L.; Wang, Y.; Yang, T.; Hu, Q.; Hu, M.; Yang, C.; Zhang, Z. Reshaping Tumor Immune Microenvironment through Acidity-Responsive Nanoparticles Featured with CRISPR/Cas9-Mediated Programmed Death-Ligand 1 Attenuation and Chemotherapeutics-Induced Immunogenic Cell Death. ACS Appl. Mater. Interfaces 2020, 12, 16018–16030. [Google Scholar] [CrossRef]
- Piergentili, R.; Del Rio, A.; Signore, F.; Ronchi, F.U.; Marinelli, E.; Zaami, S. CRISPR-Cas and Its Wide-Ranging Applications: From Human Genome Editing to Environmental Implications, Technical Limitations, Hazards and Bioethical Issues. Cells 2021, 10, 969. [Google Scholar] [CrossRef]
- Stefanoudakis, D. Integrating CRISPR Technology with Key Genetic Markers in Pancreatic Cancer: A New Frontier in Targeted Therapies. SynBio 2025, 3, 1. [Google Scholar] [CrossRef]
- Cribbs, A.P.; Perera, S.M.W. Science and Bioethics of CRISPR-Cas9 Gene Editing: An Analysis Towards Separating Facts and Fiction. Yale J Biol Med 2017, 90, 625–634. [Google Scholar]
- Gonzalez-Avila, L.U.; Vega-López, J.M.; Pelcastre-Rodríguez, L.I.; Cabrero-Martínez, O.A.; Hernández-Cortez, C.; Castro-Escarpulli, G. The Challenge of CRISPR-Cas Toward Bioethics. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- D. Morris, “CRISPR Therapeutics for Pancreatic Cancer,” Mar. 16, 2024, Research Archive of Rising Scholars. [CrossRef]
- Vimal, S.; Madar, I.H.; Thirumani, L.; Thangavelu, L.; Sivalingam, A.M. CRISPR/Cas9: Role of genome editing in cancer immunotherapy. Oral Oncol. Rep. 2024, 10. [Google Scholar] [CrossRef]
- Chehelgerdi, M.; Chehelgerdi, M.; Khorramian-Ghahfarokhi, M.; Shafieizadeh, M.; Mahmoudi, E.; Eskandari, F.; Rashidi, M.; Arshi, A.; Mokhtari-Farsani, A. Comprehensive review of CRISPR-based gene editing: mechanisms, challenges, and applications in cancer therapy. Mol. Cancer 2024, 23, 1–45. [Google Scholar] [CrossRef]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef] [PubMed]
- Akyürek, E.; Uysal, B.; Gülden, G.; Taştan, C. The Effect Of Molecular Genetic Mechanisms On Drug Addiction And Related New Generation CRISPR Gene Engineering Applications. Gene Ed. 2021, 2, 50–60. [Google Scholar] [CrossRef]
- Singh, M.; Agarwal, V.; Jindal, D.; Pancham, P.; Agarwal, S.; Mani, S.; Tiwari, R.K.; Das, K.; Alghamdi, B.S.; Abujamel, T.S.; et al. Recent Updates on Corticosteroid-Induced Neuropsychiatric Disorders and Theranostic Advancements through Gene Editing Tools. Diagnostics 2023, 13, 337. [Google Scholar] [CrossRef]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef]
- Lopes, R.; Prasad, M.K. Beyond the promise: evaluating and mitigating off-target effects in CRISPR gene editing for safer therapeutics. Front. Bioeng. Biotechnol. 2024, 11, 1339189. [Google Scholar] [CrossRef]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef]
- Chen, P.; Zhou, J.; Liu, H.; Zhou, E.; He, B.; Wu, Y.; Wang, H.; Sun, Z.; Paek, C.; Lei, J.; et al. Engineering of Cas12a nuclease variants with enhanced genome-editing specificity. PLOS Biol. 2024, 22, e3002514. [Google Scholar] [CrossRef]
- Kovalev, M.A.; Davletshin, A.I.; Karpov, D.S. Engineering Cas9: next generation of genomic editors. Appl. Microbiol. Biotechnol. 2024, 108, 1–19. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Biswas, R.; Bugde, P.; Li, J.; Liu, D.-X.; Li, Y. Application of CRISPR-Cas9 System to Study Biological Barriers to Drug Delivery. Pharmaceutics 2022, 14, 894. [Google Scholar] [CrossRef]
- Call, S.N.; Andrews, L.B. CRISPR-Based Approaches for Gene Regulation in Non-Model Bacteria. Front. Genome Ed. 2022, 4, 892304. [Google Scholar] [CrossRef] [PubMed]
- Bendixen, L.; Jensen, T.I.; Bak, R.O. CRISPR-Cas-mediated transcriptional modulation: The therapeutic promises of CRISPRa and CRISPRi. Mol. Ther. 2023, 31, 1920–1937. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Singh, S.M. Strategies for precision modulation of gene expression by epigenome editing: an overview. Epigenetics Chromatin 2015, 8, 1–12. [Google Scholar] [CrossRef]
- Djajawi, T.M.; Wichmann, J.; Vervoort, S.J.; Kearney, C.J. Tumor immune evasion: insights from CRISPR screens and future directions. FEBS J. 2023, 291, 1386–1399. [Google Scholar] [CrossRef]
- Mathur, S.; Sutton, J. Personalized medicine could transform healthcare. Biomed. Rep. 2017, 7, 3–5. [Google Scholar] [CrossRef]
- Molla, G.; Bitew, M. Revolutionizing Personalized Medicine: Synergy with Multi-Omics Data Generation, Main Hurdles, and Future Perspectives. Biomedicines 2024, 12, 2750. [Google Scholar] [CrossRef]
- Youssef, E.; Fletcher, B.; Palmer, D. Enhancing precision in cancer treatment: the role of gene therapy and immune modulation in oncology. Front. Med. 2025, 11, 1527600. [Google Scholar] [CrossRef]
- Park, H.; Kang, Y.K.; Shim, G. CRISPR/Cas9-Mediated Customizing Strategies for Adoptive T-Cell Therapy. Pharmaceutics 2024, 16, 346. [Google Scholar] [CrossRef]
- Caforio, M.; Iacovelli, S.; Quintarelli, C.; Locatelli, F.; Folgiero, V. GMP-manufactured CRISPR/Cas9 technology as an advantageous tool to support cancer immunotherapy. J. Exp. Clin. Cancer Res. 2024, 43, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.T.; Greene, L.A.; Mnatsakanyan, H.; Badr, C.E. Revolutionizing Brain Tumor Care: Emerging Technologies and Strategies. Biomedicines 2024, 12, 1376. [Google Scholar] [CrossRef]
- Zahedipour, F.; Zahedipour, F.; Zamani, P.; Jaafari, M.R.; Sahebkar, A. Harnessing CRISPR technology for viral therapeutics and vaccines: from preclinical studies to clinical applications. Virus Res. 2024, 341, 199314. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ananthaswamy, N.; Jain, S.; Batra, H.; Tang, W.-C.; Lewry, D.A.; Richards, M.L.; David, S.A.; Kilgore, P.B.; Sha, J.; et al. A universal bacteriophage T4 nanoparticle platform to design multiplex SARS-CoV-2 vaccine candidates by CRISPR engineering. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.J.; O’dOnovan, M.C. The genetics of neuropsychiatric disorders. Brain Neurosci. Adv. 2018, 2. [Google Scholar] [CrossRef]
- Barešić, A.; Nash, A.J.; Dahoun, T.; Howes, O.; Lenhard, B. Understanding the genetics of neuropsychiatric disorders: the potential role of genomic regulatory blocks. Mol. Psychiatry 2019, 25, 6–18. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Concise illustration of the tumor immune evasion mechanisms. It has been noted that tumor cells may employ one or a combination of different immune evasion strategies to undermine the success of various cancer immunotherapeutic methods, such as TIL and CAR-T cell cancer treatment methods. Some of the key immune evasion mechanisms discussed in the present paper include (A) the heterogeneity of cancer cells, (B) the immunosuppressive nature of the TME, (C) cancer cell antigen loss by mutation and alternative splicing, (D) cancer cell antigen masking, (E) immune checkpoint and ligand activation. Other strategies are only illustrated in this paper but are concisely discussed in the paper referenced here, [31] including (F) resistance to apoptosis, (G) induction of T cell exhaustion, (H) downregulation of antigen presentation, (I) lineage switch and trogocytosis-mediated antigen loss, and (J) tumor-induced immunosuppression [31]. Created with Biorender.com.
Figure 1.
Concise illustration of the tumor immune evasion mechanisms. It has been noted that tumor cells may employ one or a combination of different immune evasion strategies to undermine the success of various cancer immunotherapeutic methods, such as TIL and CAR-T cell cancer treatment methods. Some of the key immune evasion mechanisms discussed in the present paper include (A) the heterogeneity of cancer cells, (B) the immunosuppressive nature of the TME, (C) cancer cell antigen loss by mutation and alternative splicing, (D) cancer cell antigen masking, (E) immune checkpoint and ligand activation. Other strategies are only illustrated in this paper but are concisely discussed in the paper referenced here, [31] including (F) resistance to apoptosis, (G) induction of T cell exhaustion, (H) downregulation of antigen presentation, (I) lineage switch and trogocytosis-mediated antigen loss, and (J) tumor-induced immunosuppression [31]. Created with Biorender.com.
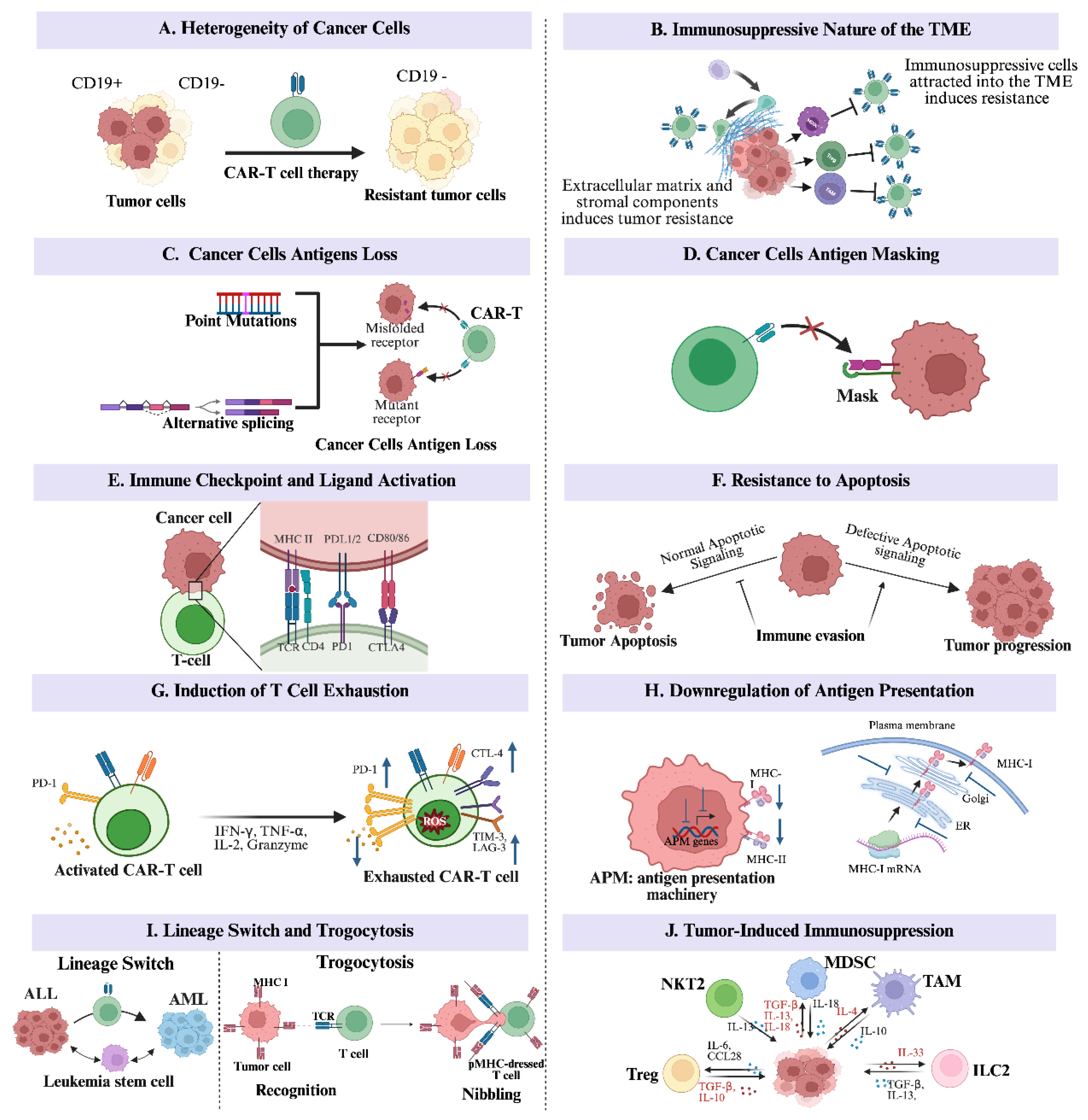
Figure 2.
Illustrates CRISPR/Cas9-driven DSBs and the two DSB repair mechanisms. The guide RNA (sgRNA) first pairs with the target DNA sequence, resulting in a site-specific DSB created by the Cas9 endonucleases. The repair of the DSBs occurs through either NHEJ or HDR mechanisms. Created with Biorender.com.
Figure 2.
Illustrates CRISPR/Cas9-driven DSBs and the two DSB repair mechanisms. The guide RNA (sgRNA) first pairs with the target DNA sequence, resulting in a site-specific DSB created by the Cas9 endonucleases. The repair of the DSBs occurs through either NHEJ or HDR mechanisms. Created with Biorender.com.
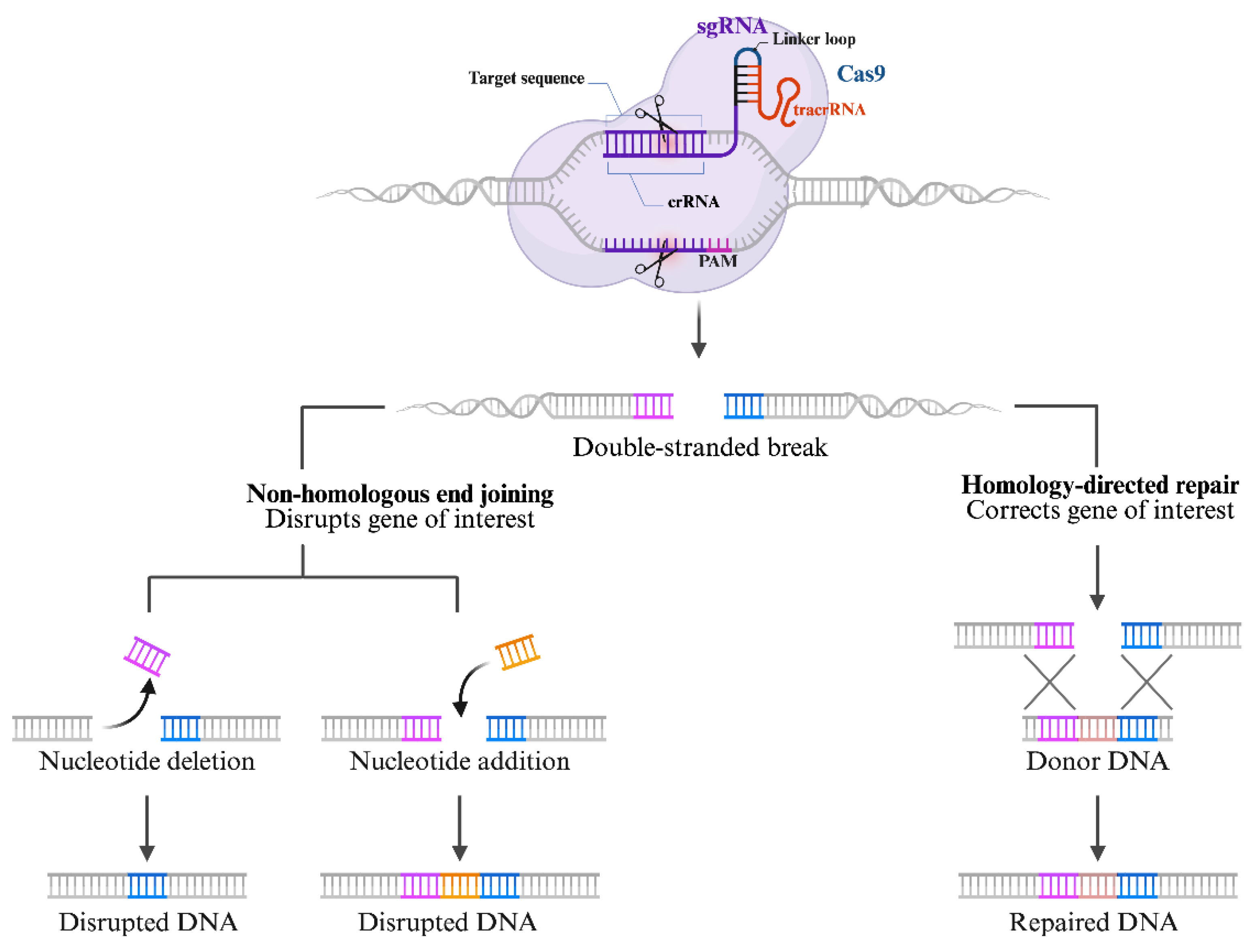
Figure 3.
CRISPR screening approaches used to identify genes involved in cancer and immunotherapy: A. Single-cell Type CRISPR Screening is used to identify genes that regulate immune-related pathways in cancer cells. The sgRNA library is introduced into cancer cells through lentiviral infection, resulting in a mutagenized cell population. Cells are selected based on phenotypic changes using flow cytometry. The sgRNAs present in selected cells are identified using next-generation sequencing (NGS). B. Two-cell type CRISPR Screening investigates essential genes involved in immune cell-mediated cancer killing. Mutagenized cells are co-cultured with tumor-targeting immune cells. Resistant cells are analyzed using high-throughput sequencing. C. Transplantation-based indirect in vivo Screening is used to study tumor progression and immune interactions in a living organism (in vivo) by transplanting genetically modified cancer cells into mouse models. The mice are exposed to different immune pressures. Resulting tumors are analyzed using high-throughput sequencing. D. Autochthonous Direct In Vivo CRISPR Screening is used to study natural tumor development and immune interactions. CRISPR screening method directly induces genetic mutations within a living organism (in vivo) at the target organ site. Mutated cells become cancerous. Target organs are exposed to different immune pressures. Orthotopic tumors are analyzed using high-throughput sequencing. Created with Biorender.com.
Figure 3.
CRISPR screening approaches used to identify genes involved in cancer and immunotherapy: A. Single-cell Type CRISPR Screening is used to identify genes that regulate immune-related pathways in cancer cells. The sgRNA library is introduced into cancer cells through lentiviral infection, resulting in a mutagenized cell population. Cells are selected based on phenotypic changes using flow cytometry. The sgRNAs present in selected cells are identified using next-generation sequencing (NGS). B. Two-cell type CRISPR Screening investigates essential genes involved in immune cell-mediated cancer killing. Mutagenized cells are co-cultured with tumor-targeting immune cells. Resistant cells are analyzed using high-throughput sequencing. C. Transplantation-based indirect in vivo Screening is used to study tumor progression and immune interactions in a living organism (in vivo) by transplanting genetically modified cancer cells into mouse models. The mice are exposed to different immune pressures. Resulting tumors are analyzed using high-throughput sequencing. D. Autochthonous Direct In Vivo CRISPR Screening is used to study natural tumor development and immune interactions. CRISPR screening method directly induces genetic mutations within a living organism (in vivo) at the target organ site. Mutated cells become cancerous. Target organs are exposed to different immune pressures. Orthotopic tumors are analyzed using high-throughput sequencing. Created with Biorender.com.
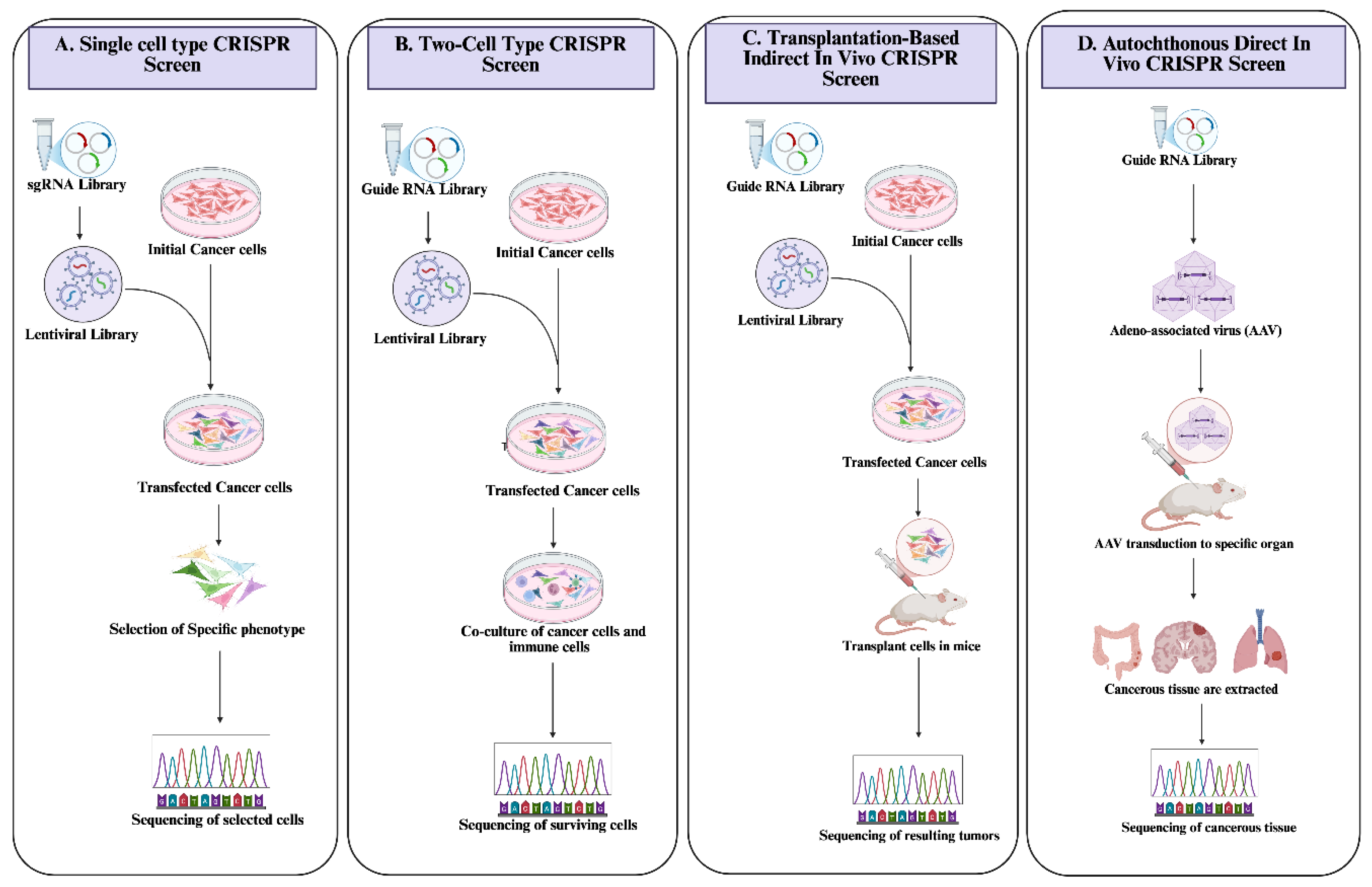
Figure 4.
A schematic of CRISPR/Cas9 technology applications in CAR-T cells and TILs to improve long-term persistence and antitumor activity. Created with Biorender.com.
Figure 4.
A schematic of CRISPR/Cas9 technology applications in CAR-T cells and TILs to improve long-term persistence and antitumor activity. Created with Biorender.com.
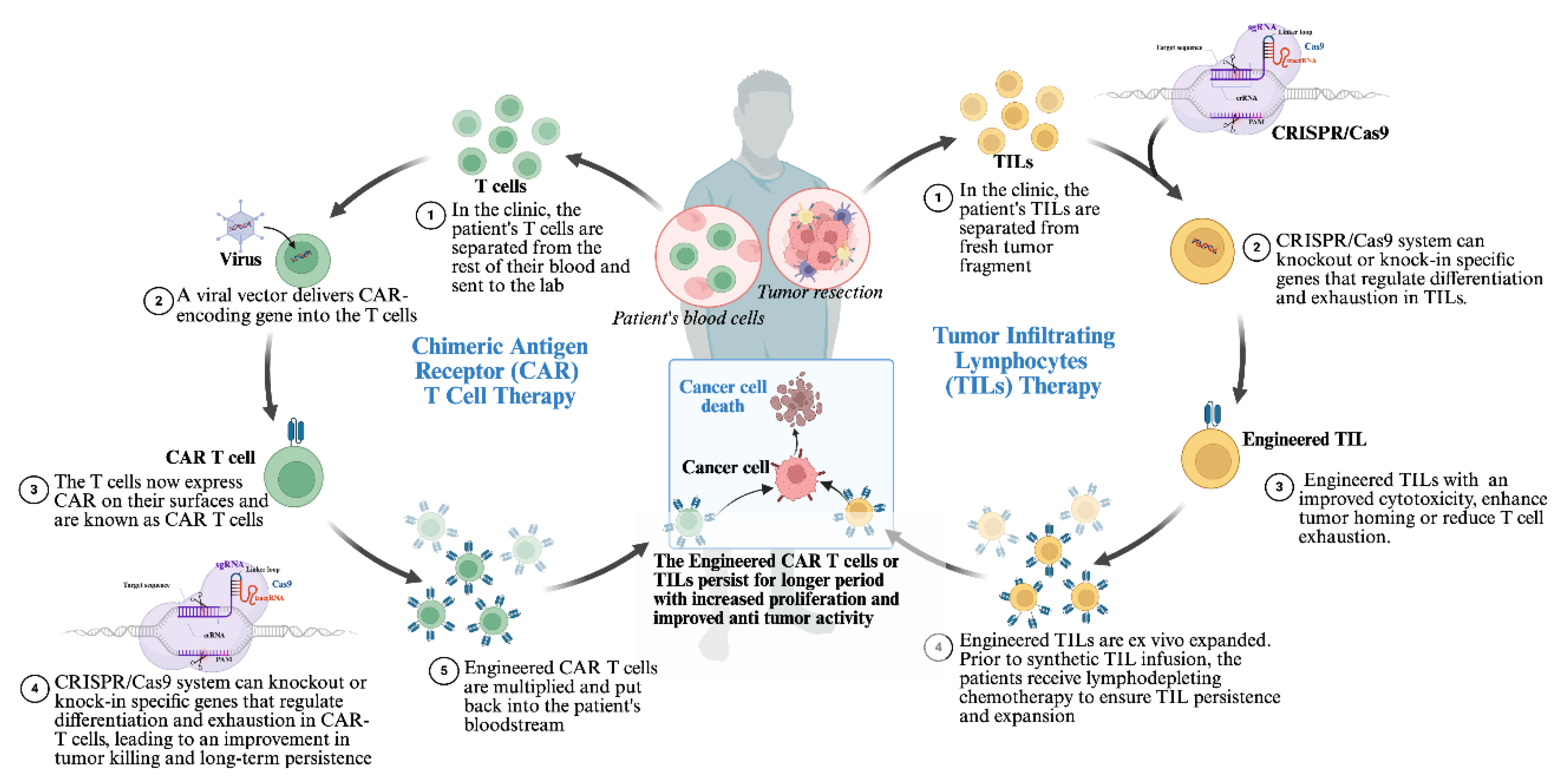

Table 1.
Immunotherapy Sensitizing Target Identified by CRISPR/Cas9 Screening.
| Significant Target | Target Function | Target | CRISPR/Cas 9 Delivery Vector | Therapeutic Implication | Reference |
|---|---|---|---|---|---|
| DDR1 | DDR1 is identified as a collagen receptor involved in colorectal cancer (CRC) liver metastasis and the related stromal response | CRC cells | Lentiviral vector | Blocking DDR1 promoted CD8+ T cell infiltration and increased the sensitivity of microsatellite stable (MSS) CRC mouse models to PD-1 inhibition | [103] |
| CDC7 | A serine/threonine kinase essential for DNA replication, also implicated in small-cell lung cancer (SCLC) resistance | SCLC cell line | Lentiviral vector | Knocking down CDC7 expression in chemotherapy-resistant SCLC cells resulted in a lower IC50 and enhanced the effectiveness of chemotherapy | [104] |
| TRIM34 | It promotes resistance to ferroptosis by inhibiting the degradation of GPX4 mRNA, contributing to the progression of Hepatocellular carcinoma (HCC) | HCC cell line Huh7 | Lentiviral vector | Targeted knockdown of TRIM34 in HCC cells enhanced their response to anti-PD-1 therapy | [105] |
| TUBB3 | TUBB3, a member of the tubulin family that encodes βIII-tubulin, was first recognized as a marker for neurons. Increased expression of TUBB3 is associated with tumor progression | Lung cancer cell lines (CMT167cells and C57BL/6) | Lentiviral vector | Inhibiting TUBB3 makeS cells resistant to anti-PD-1 immunotherapy more vulnerable to T cell-mediated destruction by reducing the expression of PD-L1 | [106] |
| PTPN2 | Ptpn2 encodes a protein tyrosine phosphatase involved in regulating various intracellular processes, including IFNγ signaling, which it inhibits by dephosphorylating STAT1 and JAK1 | Malignant melanoma cell line (A375 cells) | CuS-RNP@PEI nanoparticles | The knockdown of PTPN2 significantly boosted CD8+ T cell infiltration and cytokine production within the tumor microenvironment, leading to a stronger immune response and increased tumor sensitivity to immunotherapy | [107] |
| GDF15 | Recognized as a crucial element in immune evasion | Glioblastoma cells | angiopep-2-decorated, glutathione (GSH)-responsive nanoparticles [ANPSS(Cas9/sgGDF15)] | Deletion of GDF15 in glioblastoma cells alleviated immunosuppression in the TME, thereby enhancing the antitumor efficacy of ICB therapy. | [108] |
| ADAM2 | A cancer testis antigen whose expression suppresses T cell immune response by reducing IFNγ and TNFα signaling. | Lewis Lung Carcinoma (LLC) cell lines | Lentiviral vector | Blocking ADAM2 may improve cancer immunotherapy in tumors that rely on it for immune evasion | [109] |
| HDAC7 and genes involved in the Sec61 pathway | HDAC7 is a crucial transcriptional corepressor, and Sec61 is a translocon. Both have been demonstrated to reduce the expression of BCMA, a key immunotherapy target in multiple myeloma | Multiple Myeloma cell lines | lentiviral vector | Silencing HDAC7 increases BCMA expression, while inhibiting Sec61 improves the antimyeloma effectiveness of a BCMA-targeted antibody-drug conjugate | [110] |
| COX2 | Produces an immunosuppressive molecule prostaglandin E2 (PGE2) | Model of KRAS-mutant lung adenocarcinoma | Lentiviral vector | Targeting the COX2/PGE2 signaling pathway enhances the response of KRAS-mutant lung tumors to anti-PD1 therapy and delays tumor relapse following KRAS inhibition | [111] |
| SP20H | Upregulates the expression of B7-H3 (a receptor that suppresses T cells activation) in tumor cells | Ovarian cancer cells | Lentiviral vector | Deleting the SP20H gene in tumors inhibits tumor growth, increases the ratio of CD8+/CD4+ T cells, and decreases M2 macrophage infiltration in SP20H-deficient tumors. | [112] |
| KEAP1/NRF2 | KEAP1 regulates the degradation of NRF2. | Non-Small Cell Lung Carcinoma Cell | Lentiviral vector | [113] | |
| Lgals2 | Galectin-2 (Lgals2), a β-galactoside-binding lectin, modulates the immune response by regulating inflammation and immune cell interactions. | 4 T1 cell lines | pCDH/Lentiviral vector | Using an inhibitory antibody to block Lgals2 effectively suppresses tumor growth and reactivates the immune system. The study findings suggest that Lgals2 may serve as a potential target for immunotherapy. | [114] |
| Lgals2 | Lgals2, a β-galactoside-binding lectin, modulates the immune response by regulating inflammation and immune cell interactions. | HEK293 cell lines | Lentiviral vector | The overexpression of Gal-2 reduced the proliferation of human colon epithelial cells and diminished H2O2-induced STAT3 phosphorylation. The study highlighted the suppressive function of Gal-2 in colon tumor proliferation and its potential as a therapeutic target in cancer therapy. | [115] |
| TPST2 | TPST2 is an enzyme primarily known for catalyzing the sulfation of tyrosine residues in proteins. This post-transcriptional modification is essential for many cellular processes, including cell signaling and cell adhesion. | Human breast cell lines (MDA-MB-231 and MDA- MB-468) and mouse colon cancer cell line (MC38), | Lentiviral vector | Reducing TPST2 in cancer cells increased the effectiveness of anti-PD1 antibodies in syngeneic tumor models by enhancing tumor-infiltrating lymphocytes. The novel role of TPST2 is proposed as an inhibitor of cancer immunotherapy and one of the targets of immune checkpoint cancer immunotherapy. | [116] |
| ATXN3 | ATXN3 functions as a deubiquitinase for various PD-L1 transcription factors in tumor cells. | LLC1/B16 cells | Lentiviral vector | The specific disruption of ATXN3 resulted in a significant decrease in PD-L1 transcription, ultimately enhancing the immune system’s antitumor effect by working synergistically with checkpoint blockade therapy. The study emphasizes the importance of identifying ATXN3 for its positive regulation of CD274 gene transcription and its potential as a target to enhance the effectiveness of cancer immunotherapy. | [117] |
| Ptpn 2/PD-L1 | Ptpn2 regulates intracellular process including IFNγ signaling while PD-L1 is a ligand that interacts with program cell receptors in immune checkpoint mechanism | B1-F10 cells | PX333 | The disruption of PTPN2 and PD-L1 reinvigorates the immune system by halting the immune checkpoint effect. Additionally, the JAK/STAT pathway is reactivated following the deletion of PTPN2, which promotes the vulnerability of tumor cells to CD+ T cells. | [118] |
| PRODH2 | An enzyme that plays an important role in proline metabolism. | HEK293FT, E0771, and MCF7 cells. | Lentiviral vector | The specific overexpression or knock-in of PRODH2 improved the CAR-T based killing and the in vivo effectiveness in various cancer models. | [119] |
| DHX37 | Plays a key role in RNA metabolism, particularly in the regulation of gene expression | Murine breast cancer cells | Lentiviral vector | Infiltration and degranulation assays highlighted RNA helicase Dhx37 as a critical factor. Knockout of Dhx37 enhanced the efficacy of antigen-specific CD8 T cells in fighting triple-negative breast cancer in vivo. | [120] |
| Cop1 | Known for its role in recruiting M2-type macrophages. | 4 T1 cells | Lentiviral vector | Disruption of Cop 1 inhibits cancer cells’ immune escape and enhances the effectiveness of ICB. | [121] |
| Asf1a | ASF1 is a histone H3-H4 chaperone conserved from yeast to human cells | Lung cancer cell lines | pXPR-GFP-Blast vector | Inhibition of Asf1a sensitizes tumor cells to anti-PD-1 therapy. | [122] |
| KM2D | KM2D encodes a histone H3K4 methyltransferase, one of the genes mutated in cancer patients. | E0771, B16F10 MA1L, LLC, and MB49 cells | AAV vector | The loss of KM2D increases tumor cell sensitivity to ICB by boosting tumor immunogenicity. | [123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



