Submitted:
08 October 2025
Posted:
08 October 2025
You are already at the latest version
Abstract
Triple-negative breast cancer (TNBC), particularly the androgen receptor–low (AR-low) subtype, is one of the most aggressive and hard-to-treat forms of breast cancer, characterized by high index of proliferation, chromosomal instability (CIN), and high prevalence of TP53 mutations. These features fuel intra-tumoral heterogeneity, therapy resistance, and poor clinical outcomes. An integrated framework that encompasses the dysregulated molecular networks and pathways that support the pathobiology of and shape AR-low TNBC has been lacking. In this data-supported review, we synthesize transcriptomic, epigenetic, and mechanistic evidence to show that AR-low and TP53-mutant breast cancers consistently upregulate mitotic kinesin motors, centromeric proteins, and regulators of proteolysis. These modules normally facilitate mitosis and safeguard genomic integrity, but when persistently and excessively activated, they promote unbridled proliferation while undermining fidelity of mitosis. The result is a paradoxical tumor state: rapid proliferation coupled with persistent mitotic errors, CIN, and aneuploidy, which in turn accelerate tumor evolution and adaptation. We propose a unifying model in which a FoxM1–WDR5–ASPM regulatory axis acts as a central driver of this dysregulation. In the absence of AR signaling, this axis locks tumor cells into a hyperproliferative state with cells traversing highly error-prone mitoses. This state involves a steep upregulation of mitotic kinesins, centromeric proteins, and regulators of protein degradation, fueling CIN and intra-tumoral heterogeneity. Loss of TP53 function fuels this dysregulation, and also allows the aneuploid cells to survive and continue dividing, perpetuating the aneuploid state. We also show that breast tumor overexpressing mitotic kinesins, centromeric proteins, and regulators of protein degradation, show characteristic infiltration patterns of immune and stromal cells in their tumor microenvironment, which could impact their prognosis and response to treatments. This perspective highlights how AR-low TNBC and TP53-mutant breast cancers bend core cell cycle machinery to sustain aggressive growth and evolve dynamically. By defining this interconnected regulatory network, we identify a set of actionable vulnerabilities with therapeutic potential and provide a framework for rethinking how to manage high proliferation, genomically unstable breast cancers.
Keywords:
Kinesins
; mitotic
; centromeric proteins
; ubiquitin-proteasome pathway
; aggrephagy
; FOXM1
; WDR5
; ASPM
; proliferation
; genomic instability
; aneuploidy
1. Introduction
Breast cancer remains the most frequently diagnosed malignancy among women in the United States [1]. Its clinical management relies heavily on molecular classification, which is defined by the presence or absence of estrogen and progesterone receptors (ER and PR, respectively), and Human Epidermal growth factor Receptor 2 (HER2) amplification. Four principal breast cancer subtypes are recognized: Luminal A (ER+/PR+/HER2− with Ki67<14%), Luminal B (ER+/PR+/−/HER2+ or ER+/PR+/HER2− with Ki67 ≥14%), HER2-enriched (ER−/PR−/HER2+), and triple-negative breast cancer (TNBC; ER−/PR−/HER2−) [2]. Targeted therapies have significantly improved outcomes for Luminal and HER2-driven cancers, whereas TNBC lacks such options and continues to be managed largely with chemotherapy, surgery, and radiation [3,4]. Among TNBC patients treated with neoadjuvant chemotherapy, those who fail to achieve a pathological complete response frequently relapse within five years, underscoring the aggressive clinical course of this subtype [5].
The burden of breast cancer is not evenly distributed across populations in the US. Although overall incidence of breast cancer is comparable between Black and White women in the US, mortality among Black women is 40% higher, a disparity linked in part to the two-fold higher incidence of TNBC in the Black population (38 vs. 19 per 100,000) [6]. TNBC itself is molecularly heterogeneous, and efforts to stratify patients have included gene expression–based classification systems [7,8]. One proposed strategy has been to separate tumors by androgen receptor (AR) expression, given the clinical precedent of AR-targeted therapy in prostate cancer. Yet, depending on the threshold used, 65–88% of TNBCs are AR-negative, placing them into the so-called quadruple-negative breast cancer (QNBC) category [9].
QNBC tumors display distinct biological and clinical features compared to AR-positive TNBC. They are enriched for basal-like phenotypes, harbor elevated rates of TP53 mutations, present at younger ages, and are associated with poor disease-free survival [10,11]. At the molecular level, QNBCs show enhanced chromosomal instability, centrosome amplification, copy number alterations, and dysregulated miRNA networks [12,13]. Moreover, racial disparities extend into the molecular biology of QNBC: tumors from African-American women exhibit unique expression profiles—including differential expression of E2F1, NFKBIL2, CCL2, TGFB3, CEBPB, PDK1, IL12RB2, IL2RA, and SOS1, and have been reported to overexpress immune checkpoint molecules such as PD-1, PD-L1, and CTLA-4 relative to tumors from White women [10]. These features both compound the aggressiveness of QNBC and limit therapeutic options, further fueling outcome disparities. Despite its clinical relevance, AR-negative TNBC has received far less mechanistic investigation than other breast cancer subtypes. Accumulating data indicate that AR-deficient tumors harbor unique pathobiology, including distinct regulatory networks, high proliferative indices [14,15], and poor prognosis [16], yet they lack well-validated therapeutic targets [17]. This gap highlights the need to define the molecular mechanisms that sustain their aggressive behavior.
Here, we synthesize existing transcriptomic, epigenetic, and functional evidence to map pathways that drive proliferation and genomic instability in AR-low TNBC and TP53-mutant breast cancers. We focus on three interlinked modules—mitotic kinesin motors, centromeric and kinetochore proteins, and proteolysis regulators—that converge to sustain high proliferation while compromising fidelity of chromosomal segregation. We propose a unifying FoxM1–WDR5–ASPM regulatory axis that orchestrates this dysregulation, and discuss how the aberrant activity of this axis fuels tumor evolution, intra-tumoral heterogeneity, and therapy resistance. By framing AR-low TNBC as a disease entity defined by (a) transcriptional chaos, (b) high proliferation, and (c) genomic instability, this review highlights novel vulnerabilities that could guide the development of future therapeutic strategies and help reduce racial disparities in breast cancer outcomes.
2. Different Transcriptional Complexes Ensure Timely Expression of G1/S and G2/M Genes During the Cell Cycle
Cell cycle progression depends on two complex waves of gene expression driven by distinct transcriptional mechanisms: early (G1/S) gene expression for DNA replication, and late (G2/M) cell cycle gene expression for mitotic entry, progression, and exit. G1/S genes typically contain an E2F binding motif in their promoters, and are regulated primarily by the retinoblastoma protein (RB)-E2F complex. By contrast, G2/M genes contain a cell cycle gene homology region (CHR) in their promoters and are primarily regulated by the DREAM complex, comprised of dimerization partner, RB-like, E2F, and multi-vulval class B (MuvB). In the quiescent G0 phase, RB silences target genes by binding to and restraining the activator E2Fs (E2F1, E2F2, and E2F3a), which are positioned at the promoters of the downstream targets. In early G1, CDK4/6-Cyclin D mono-phosphorylates RB, but the latter remains bound to the activator E2F complex. In the late G1 to early S phase, Cyclin E-CDK2-mediated RB hyperphosphorylation causes RB inactivation, allowing for the peak expression of G1/S genes [18].
The MuvB complex—comprised of LIN9, LIN37, LIN52, LIN54, and RBBP4—is a central unifying scaffold for three different, dynamic, and essential transcription complexes that form at distinct phases of the cell cycle to temporally regulate both early and late waves of cell cycle gene expression [19]. During G0 or quiescence, MuvB, binds to p130 (Rb-related protein), E2F4, and DP1, to form the DREAM complex, that acts as a global repressor of both early and late cell cycle genes. In G0, transcription factors B-Myb and FoxM1 undergo proteasomal degradation, and their renewed transcription is blocked by the DREAM complex [20,21]. In G1/S, the DREAM complex is phosphorylated by Cyclin E/CDK2 and dissociates from the promoters of G2/M genes [22], setting MuvB free. This dissociation of the DREAM complex is concomitant with fresh transcription of B-Myb and the formation of B-Myb-MuvB (MMB) complex at the promoters of G2/M genes [23]. Yes-associated protein 1 (YAP1), another transcription cofactor, also promotes re-expression of (i) B-Myb, and (ii) the master regulator transcription factor Forkhead Box M1 (FoxM1) in G1/S [24,25,26] as well as the assembly of MMB at G2/M CHR sites [27]. Epigenetic mechanisms also contribute to transcription of FoxM1 in G1/S; in TNBC for example, the expression of FOXM1 is upregulated by the WD Repeat Domain 5 (WDR5) protein [28]. WDR5 is a core component of histone methyltransferase complexes that catalyze the trimethylation of histone H3 at lysine 4 (H3K4me3) [29]. Promoters marked by H3K4me3 are typically transcriptionally active, with higher H3K4me3 enrichment correlating with a more open chromatin state [30,31,32]. Additionally, methylation at H3K4 may influence how effector proteins bind, thereby shaping downstream biological processes [33]. In TNBC, ChIP analysis has confirmed marked WDR5 and H3K4me3 occupancy at the FOXM1 promoter, and knockdown of WDR5 inhibited FoxM1 expression and H3K4me3 formation in the FoxM1 promoter region, supporting the idea that WDR5 promotes FOXM1 expression epigenetically [28].
The B-Myb–MuvB (MMB) complex is essential for recruiting newly expressed FoxM1 to G2/M gene promoters in late S phase [34]. B-Myb and MuvB exhibit mutual dependence for promoter targeting, and their cooperation is critical for FoxM1 recruitment [35]. FoxM1 interacts with MuvB from late S phase through mitosis. As B-Myb undergoes phosphorylation-dependent degradation during late S and G2/M, its interaction with FoxM1 diminishes. G2/M gene expression is further restrained by the ATR–CHK1 pathway during S phase, which inhibits CDK1 activity, allowing the MMB–FoxM1 complex to persist in a primed but inactive state at CHR sites [25,36]. In G2, CDK1 activation lifts ATR–CHK1 suppression, leading to FoxM1 phosphorylation by CDK1. FoxM1 is also phosphorylated by Polo-like kinase-1 (Plk1). These phosphorylation events activate the MMB-FoxM1 complex and trigger the expeditious degradation of B-Myb. Acetylation then stabilizes FoxM1 [37]. Thus, phosphorylated, and acetylated FoxM1 and MuvB collaborate to drive strong expression of genes essential for G2/M progression and cytokinesis. Importantly, B-Myb proteolysis, FoxM1 phosphorylation and acetylation, and the dynamic reshaping of the MuvB complex coordinate a transcriptional switch, enabling MuvB to adopt a transcriptional activating role at G2/M. Notably, the lack of direct regulatory linkage between E2F1 and FoxM1 reveals that cell cycle progression is not driven by a linear transcription factor cascade. Rather, it is governed by a more broadly integrated regulatory logic involving transcriptional control, post-translational modifications, and targeted proteolysis, that bridges early and late cell cycle transcriptional programs.
3. From Cell Cycle to Tumor Cycle: How Dysregulated Expression of FoxM1 and its Target Genes Underlies Oncogenesis and Disease Progression in Breast Cancer
FoxM1 is an oncogene that regulates apoptosis, drug resistance, DNA damage repair, stem cell renewal, angiogenesis, metastasis, and mitotic spindle maintenance. Aberrantly high activation of FoxM1-driven signaling is essential for development and progression of many types of solid tumors, and FoxM1 overexpression is associated with higher tumor stage, aneuploidy, higher growth fraction, radiotherapy and chemotherapy resistance, metabolic reprogramming, angiogenesis, and poorer disease outcomes in diverse cancer types [38,39,40]. FoxM1 is expressed only in proliferating normal cells and in tumor cells [41,42], which makes it a good therapeutic target in cancer types wherein the tumor biology is strongly influenced by FoxM1 upregulation.
In breast cancer, FoxM1 upregulation drives various facets of tumorigenesis and disease progression [43]. In AR-low TNBC and breast tumors harboring TP53 mutations, FoxM1 overexpression plays a pivotal role in driving the co-upregulation of centrosome amplification and clustering genes, which results in an aggressive disease course, and poor patient outcomes [13]. Loss of TP53 function diminishes expression of p21/CIP1, a key cyclin-dependent kinase (CDK) inhibitor. Under normal conditions, p21 restrains the activities of CDK4/6–Cyclin D and CDK2–Cyclin E complexes, preventing premature phosphorylation of the RB. Recent meta-analysis studies have also confirmed that hundreds of cell cycle genes are repressed by the p53-p21-DREAM pathway and that a subset of these genes are activated by MuvB, B-MYB and FoxM1 in G2/M [44,45]. In TP53-deficient cells, insufficient p21 activity leads to premature hyperphosphorylation and inactivation of RB, and inappropriate activation of G1/S gene transcription. TP53 loss of function also promotes the upregulation of oncogenes such as E2F1 and ATAD2, which drive excessive Cyclin E–CDK2 activity. This CDK2 hyperactivity disrupts the DREAM complex from the promoters of G2/M genes—genes that are particularly sensitive to DREAM-mediated transcriptional repression [44,46].
Concurrently, heightened CDK2 activity induces YAP/TEAD-mediated transcription of B-MYB and FOXM1, while ATAD2 further promotes B-MYB accumulation. B-MYB associates with the MuvB core to form the MMB complex, which binds G2/M gene promoters. Interactions between MMB-bound promoters and YAP/TEAD-bound enhancers culminate in an abnormally high build-up of FOXM1 at G2/M gene promoters during the S/G2 transition. As cells complete DNA replication, CDK1 activity—relieved from ATR-CHK1 inhibition—is able to phosphorylate FOXM1, with PLK1 providing additional activating phosphorylation events. This dual phosphorylation leads to maximal FoxM1 activation at the G2/M boundary. B-MYB is subsequently degraded, and FOXM1 drives the transcription of genes critical for centrosome amplification (e.g., AURKA, CCNA2, CDK1, CEP152, PLK1, PLK4, SASS6, STIL) and clustering (e.g., KIFC1, AURKB, BIRC5/Survivin, CDCA8), alongside genes promoting proliferation, drug resistance, and survival, culminating in poorer patient prognosis [13]. In TNBC, additional mechanisms lead to aberrantly high levels of FoxM1: the androgen receptor (AR) normally upregulates SPDEF, a transcription factor that suppresses FOXM1 expression by disrupting FoxM1’s autoregulatory positive feedback loop. This SPDEF-mediated repression is further reinforced by the p53–p21–DREAM axis. However, in AR-deficient or AR-low TNBC—particularly when combined with TP53 mutation—this regulatory network also collapses, causing massive overexpression of FoxM1 and its target genes, and engendering high levels of CIN, proliferation, chemoresistance, and resistance to apoptosis in AR-low TNBC [13]. Our work also uncovered that because of the above-stated selective advantages conferred by FoxM1 dysregulation, FOXM1 is indispensable for the survival of p53-deficient and AR-low TNBC cells harboring amplified centrosomes [13]. In sum, understanding the nexus of dysregulation around FoxM1 is critical to understanding the tumor biology of AR-low TNBC and TP53-deficient breast tumors.
4. FoxM1 and ASPM Partner Intranuclearly to Drive Expression of G2/M Genes Via Liquid-Liquid Phase Separation
Weighted gene co-expression network analysis [47], identified three “hub” genes—Abnormal Spindle-like Microcephaly-associated gene or ASPM/MCPH5, CDC20, and TTK—which are all known target genes of MMB-FoxM1, whose elevated expression was associated with advanced tumor grades, decreased relapse-free survival (RFS), and lower overall survival (OS). The afore-mentioned study demonstrated that the identified hub genes were strongly correlated with cellular processes such as the cell cycle, DNA replication, homologous recombination, and P53 signaling pathways. Among these “hub” genes, ASPM is of particular interest in the context of breast cancer because it plays conserved and multi-faceted roles in crucial aspects of cell division. In interphase cells, ASPM localizes to centrosomes and the nucleus, and has been reported to play an important role in homologous recombination-mediated DNA repair by increasing the half-life of BRCA1; Inhibition of ASPM destabilizes BRCA1, impairing the efficiency of DNA double-strand break repair [48] and leading to genomic instability. ASPM also copurifies with the Cyclin E/Cdk2 complex and protects Cyclin E from ubiquitination and proteasome-mediated degradation [49].
Interestingly, a mass spectrometry-based screen for phase-separated, chromatin-associated proteins in breast tumor cells, led to the identification of FoxM1 as a preeminent candidate [50]. Liquid-liquid phase separation (LLPS) is a fundamental biological process wherein proteins and nucleic acids de-mix from the cellular environment, and self-assemble into dense, liquid-like subcellular compartments/condensates or hubs through multi-valent interactions. LLPS thus allows compartmentalization of biological processes without requiring membranes. Xie et al., 2025 [50] showed that in breast cancer cells, the sub-nuclear LLPS of FoxM1 with forkhead box (FKH) consensus DNA elements, is critical for FoxM1’s oncogenic function, as it allows FoxM1 to achieve a high local concentration and to effectively compartmentalize the transcriptional machinery, preserve chromatin accessibility and super-enhancer landscapes at CHR promoter sites, thus sustaining high G2/M target gene expression for tumor progression and metastasis. The authors also showed that disrupting this LLPS led to the dissolution of the FoxM1-containing biomolecular condensates, which in turn impaired oncogenic transcription, reduced breast tumor growth, and inhibited metastasis in animal models [50].
Importantly, a recent genome-wide screen to identify intranuclear regulators that boost FoxM1’s transactivation of downstream target genes through LLPS [51] found that an isoform of ASPM physically interacts with FOXM1 within the nucleus, leading to the formation of condensates containing both ASPM and FoxM1 in hepatocellular carcinoma (HCC) cells. Synergistic interactions between intrinsically disordered regions (IDRs) of both ASPM and FoxM1 were essential for this condensate formation. This interaction in turn, enhanced FoxM1 protein’s stability by preventing its ubiquitination, and proteasome-mediated degradation. Furthermore, FOXM1 was shown to transcriptionally activate ASPM expression, creating a "double positive feedback loop" where each protein reciprocally promotes the other's activity and presence. ChIP-sequencing revealed that the great majority of ASPM-annotated genes overlapped with FOXM1-bound genes, indicating that ASPM collaborates with FoxM1 to increase expression of FoxM1 target genes [51]. This coordinated overexpression of ASPM and FoxM1 is strongly linked to poor patient prognosis in HCC [51]. While this relationship has been expounded in HCC, it is plausible that the two proteins interact similarly in other cancer cell types too. ASPM is upregulated in various cancers (including breast, ovarian, prostate, glioblastoma, and hepatocellular carcinoma). A multicohort study [52] demonstrated a significant association between high ASPM expression and aggressive breast cancer features (such as higher tumor grade, higher mitotic score, increased Ki67 labeling index, poor Nottingham Prognostic Index, and lympho-vascular invasion). In multivariate analyses, high ASPM expression was an independent predictor of poorer breast cancer-specific survival and distant metastasis-free survival, and ASPM overexpression was potentially involved in radiotherapy and chemotherapy resistance. The above-mentioned study thus identified ASPM as a promising prognostic marker and a potential therapeutic target for breast cancer [52].
In prostate cancer, higher expression of ASPM was correlated with advanced stages, metastasis, and worse prognosis [53]. A recent study demonstrated that ASPM enhances a stem cell phenotype in prostate cancer cells by augmenting Wnt/β-catenin signaling, thereby promoting tumor aggressiveness. ASPM has also been reported to stimulate Wnt signaling in the developing brain [54]. Similar effects of ASPM upregulation were noted in pancreatic adenocarcinoma [55] and other malignancies, indicating ASPM’s broader role in inducing cancer cell stemness and stimulating disease progression [56]. In malignant gliomas, ASPM expression positively associated with tumor grades and increased in recurrent tumors; knockdown inhibited tumor growth [57]. In hepatocellular carcinoma, ASPM upregulation enhanced metastatic capability and was a marker for vascular invasion, early recurrence, and poor prognosis [58]. In sum, ASPM and FoxM1 contribute to tumor cell proliferation by activating the expression of mitotic genes, and high levels of ASPM, FoxM1, and target genes of the MMB-FoxM1 complex are associated with a poor prognosis in different cancer types.
5. ASPM Preserves Genomic Integrity by Controlling Spindle Orientation and Regulating Microtubule Dynamics at Spindle Poles
The proper orientation of the mitotic spindle hinges on critical molecular connections between dynamic astral microtubules and the actin-rich cell cortex, and mutations in proteins critical for this process are leading causes of the reduced head circumference and intellectual disabilities observed in microcephaly (MCPH). The Abnormal Spindle-like Microcephaly-associated gene (ASPM/MCPH5) is the most frequently mutated gene in MCPH, and the protein it encodes regulates correct orientation of mitotic spindles. The correct orientation of the cleavage plane during cell division is tightly controlled in embryonic and adult tissues, including in the brain, where it contributes to cell fate decisions and proper brain development. ASPM recruits Citron rho-interacting kinase, or CITK—a protein known to play a pivotal role during cytokinesis, especially with midbody formation and abscission, as well as early mitotic events such as regulation of the nucleation and stability of astral microtubules—to the spindle poles, wherein their combined action affects the dynamics of astral microtubules, which is essential for correct spindle positioning [59]. Perturbation of the evolutionarily conserved interaction between ASPM and CITK results in misorientation of the spindle and the division plane of the neuronal progenitor cells. As a result, neuronal progenitor cells prematurely exit mitosis and commit to differentiation, which leads to a reduction in the number of symmetric cell divisions required for adequate neurogenesis, and hence, the microcephaly phenotype [59]. The core mechanism underlying this phenomenon is ASPM's ability to tune Cyclin E ubiquitination by the E3 ligase Skp2, thereby regulating Cyclin E abundance and cell cycle kinetics, particularly the length of the G1 phase and the passage through the Restriction point. ASPM binds to and modulates Cyclin E ubiquitination, which dictates how long cells spend in the G1 phase before dividing [49]. Consistent with reduced Cyclin E/Cdk2 activity, phosphorylated RB-T821 was significantly decreased in murine neural progenitor cells harboring a mutated ASPM locus; this altered RB phosphorylation led to aberrant RB-E2F activity, which affected the balance of gene expression involved in cell cycle progression, self-renewal, and differentiation. These findings suggest that disruptions in the ASPM-mediated Ubiquitin-Cyclin E-Retinoblastoma-E2F signaling pathway impair stem cell maintenance and contribute to microcephaly, independent of previously mentioned mitotic orientation defects.
ASPM also forms a physiological complex with another protein linked to microcephaly— the microtubule severing ATPase katanin (composed of subunits p60 and p80) [60]. The ASPM/katanin complex regulates microtubule dynamics at spindle poles. ASPM performs an evolutionarily conserved function wherein it autonomously tracks growing microtubule minus ends, accumulates at these ends, and inhibits their growth. ASPM and katanin also enhance each other’s minus-end accumulation, and katanin then enhances the minus-end blocking activity of ASPM. ASPM also binds along the length of microtubules, recruits katanin to the microtubule lattice, and promotes katanin-mediated severing of dynamic microtubules [60]. ASPM and katanin depend on each other to co-localize at spindle poles during mitosis, with their strongest overlap from prophase to metaphase. The ASPM/katanin complex primarily controls microtubule disassembly at spindle poles, through a combination of microtubule-severing activity by katanin (enhanced by ASPM) and minus-end blocking activity by ASPM (potentiated by katanin). Blocking minus-end growth might make microtubules more susceptible to depolymerases, which normally promote spindle flux. Disruption of the ASPM-katanin interaction leads to defects in spindle orientation and dysregulation of microtubule dynamics at spindle poles, and reduces poleward microtubule flux [60]. ASPM proteins are highly conserved and show very similar patterns of subcellular localization—to spindle poles, outer regions of the central spindle, microtubule minus-ends, and are critical for multiple aspects of mitotic progression [61]. Mutations results in abnormal chromosome segregation and aneuploidy, defects in spindle pole organization and astral MT stability [61]. Therefore, ASPM not only collaborates with FoxM1 to drive transcriptional aspects of cell proliferation, but also appears to promote genomic integrity (independently of FoxM1) via its microtubule minus-end- and spindle-related activities.
6. Identification of Cell Cycle Regulators that Drive Transcriptional Chaos Downstream of FOXM1, and Support a Highly Proliferative State in AR-Low TNBC
TNBCs in general exhibit high levels of inter- and intra-tumoral heterogeneity, and this heterogeneity is also manifest in the aspect of proliferation. The most widely accepted TNBC classification schema defines four main TNBC molecular subtypes—Basal-like 1 and 2 (BL1 and BL2), Mesenchymal (M), and Luminal Androgen Receptor (LAR)—each endowed with unique gene expression profiles and differential therapeutic responses [8]. Among these subtypes, AR-low TNBC subtypes such as BL1 are more proliferative than AR-high LAR TNBCs [62,63,64]. LAR TNBCs have a lower pathologic complete response (pCR) rate (10%) to neoadjuvant chemotherapy, compared to BL1 TNBCs (52%) [5]. To understand more about the patterns of dysregulation underlying the heterogeneity observed in TNBCs, a 2014 study by Radovich et al. compared RNA sequencing transcriptomic data of 94 TNBCs, 20 micro-dissected normal breast tissues (from reduction mammoplasty), and 10 histologically normal tumor-adjacent tissues, and found that TNBC heterogeneity is attributable to chaos in transcription [65]. Transcriptional chaos refers to the wide range in the numbers of dysregulated genes observed when comparing each individual TNBC to the set of normal tissues. Studies have shown that transcriptional chaos leads to the generation of a wider range of cell states and proteomic profiles [66]. The consequent emergence of greater populational heterogeneity presents clear selective advantages by increasing tumor cell survival rates in environments where multiple stressors exist. Interestingly, studies have shown that the chaotic dynamics of oscillations and a broadening of the distribution in levels of otherwise tightly and periodically expressed transcription factors (such as the Forkhead box M1 protein, FoxM1) can have differential effects on downstream gene networks, enhance the assembly of functional protein complexes, and may end up, paradoxically, upregulating very specific networks and protein modules [66], including those implicated in proliferation. Radovich et al. [65] found that the transcriptional chaos in TNBCs was positively correlated with non-silent DNA mutational load, and chaos analysis identified a network of 146 core cell cycle-regulated genes dysregulated in more than 90% of the TNBCs examined [65]. FoxM1, a “master regulator” transcription factor, was found to directly regulate 61 of the 146 (42%) core genes, and FoxM1 itself was overexpressed 17.2-fold in TNBCs compared to micro-dissected normal tissues [44,65]. Since FoxM1 binds to the promoters of and strongly activates expression of genes critical for G2, proper progression of mitosis, and the M/G1 transition, the authors concluded that FoxM1 overexpression causally drives the profound transcriptional dysregulation that typifies TNBCs.
We reasoned that genes that are associated with, and drive heterogeneity in proliferation, must be discoverable within this dysregulated network of 146 core genes in TNBCs. To specifically uncover genes whose overexpression drives and mechanistically supports high proliferation in AR-low TNBCs, we filtered the set of 146 “FoxM1-regulated TNBC core genes”, for a statistically significant negative correlation between the expression of the core genes and the expression of AR, among TNBCs, using the “Targeted Correlation Analysis” tool of bc-Genexminer [67]. We found that the expression of 82 genes of this core set showed a statistically significant negative correlation with the expression of AR among TNBCs (Suppl. Fig. 1A—1E), meaning that they are overexpressed in AR-low TNBCs. Notably, there was a high degree of positive correlation between the expression of most of these 82 core genes (Suppl. Fig. 1A—1E), which was a reflection of them being co-regulated, directly or indirectly, by FoxM1. We then focused our study on the subset of genes among the 82 (a) that are among the top overexpressed genes in TNBCs, (b) have established roles in the regulation of mitotic progression and cell proliferation, and (c) whose overexpression is associated with poor prognosis. The University of Alabama at Birmingham Cancer data analysis Portal (UALCAN) compares gene expression patterns (TCGA level 3 RNA-sequencing data) between TNBC and other breast cancer types, identifying and ranking the genes most significantly overexpressed in the TNBC subtype [68]. Genes with statistically significant higher expression in TNBC samples are ranked based on the extent of their upregulation (fold change), and we used this platform to determine the overexpression rankings for the 82 genes of interest. Based on the rationale that overexpression of drivers of proliferation is likely to drive a more aggressive disease course and poorer outcomes, we also performed Cox proportional hazards regression analysis for each gene separately, using the Kaplan−Meier Plotter (KM Plotter) online tool [69] to identify the subset of genes whose overexpression results in a poorer prognosis. Based on these preliminary analyses, we identified a group of 15 genes including 5 kinesin motor proteins, 5 centromeric proteins, and 5 proteins that play important roles in the proteolysis of cell cycle regulators. We found that these 15 genes were expressed at a statistically significant higher level among breast tumors that are categorized as “AR-low” compared to “AR-high” breast tumors (Suppl. Fig. 2), indicating that the overexpression of these 15 genes is correlated with low AR expression across all breast tumors. The remainder of this study examined how the overexpression of these 15 cell-cycle regulatory proteins likely supports and provokes high proliferation and chromosomal instability, specifically in AR-low TNBCs, precipitating poor outcomes in this subtype.
Figure 1.
Expression of a set of cell cycle-related proteins that are highly overexpressed in AR-low TNBC is correlated with expression of established markers of proliferation “Targeted” gene expression correlation analysis of 15 cell cycle-regulated genes that are overexpressed in AR-low TNBC, and markers of proliferation (MKI67, PCNA, and MCM2; all RNA sequencing data, TNBC status determined by immunohistochemistry) performed using the bc-GenExMiner online platform. Scatter plots depict Pearson's pairwise correlations and the numbers inside the squares indicate the strength of the observed Pearson’s pairwise correlations. Total n=4421 for each pairwise comparison. Strong negative correlations are depicted in blue, and strong positive correlations are depicted in warm colors. P-Values for all pairwise correlations were statistically significant (p<0.0001).
Figure 1.
Expression of a set of cell cycle-related proteins that are highly overexpressed in AR-low TNBC is correlated with expression of established markers of proliferation “Targeted” gene expression correlation analysis of 15 cell cycle-regulated genes that are overexpressed in AR-low TNBC, and markers of proliferation (MKI67, PCNA, and MCM2; all RNA sequencing data, TNBC status determined by immunohistochemistry) performed using the bc-GenExMiner online platform. Scatter plots depict Pearson's pairwise correlations and the numbers inside the squares indicate the strength of the observed Pearson’s pairwise correlations. Total n=4421 for each pairwise comparison. Strong negative correlations are depicted in blue, and strong positive correlations are depicted in warm colors. P-Values for all pairwise correlations were statistically significant (p<0.0001).
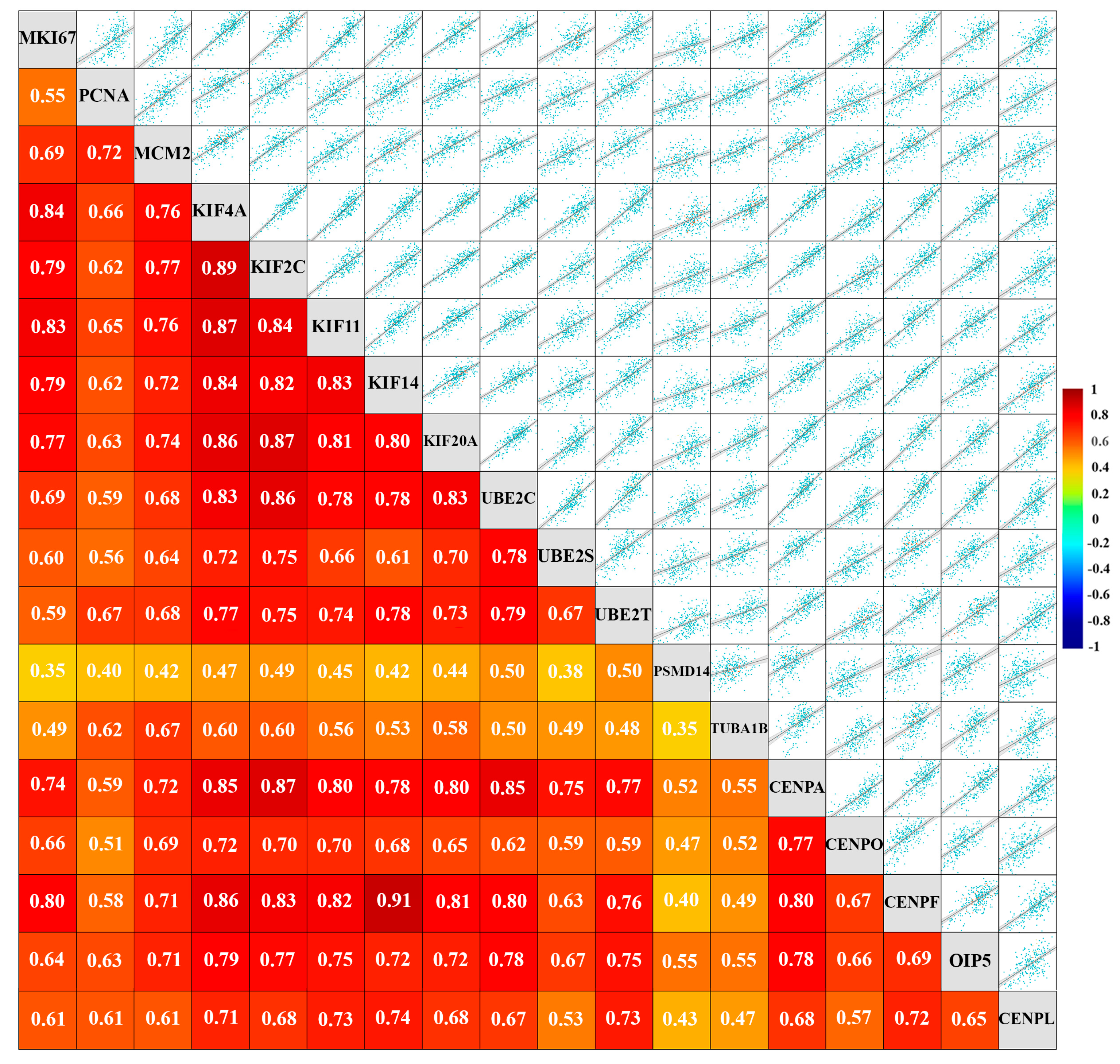
Figure 2.
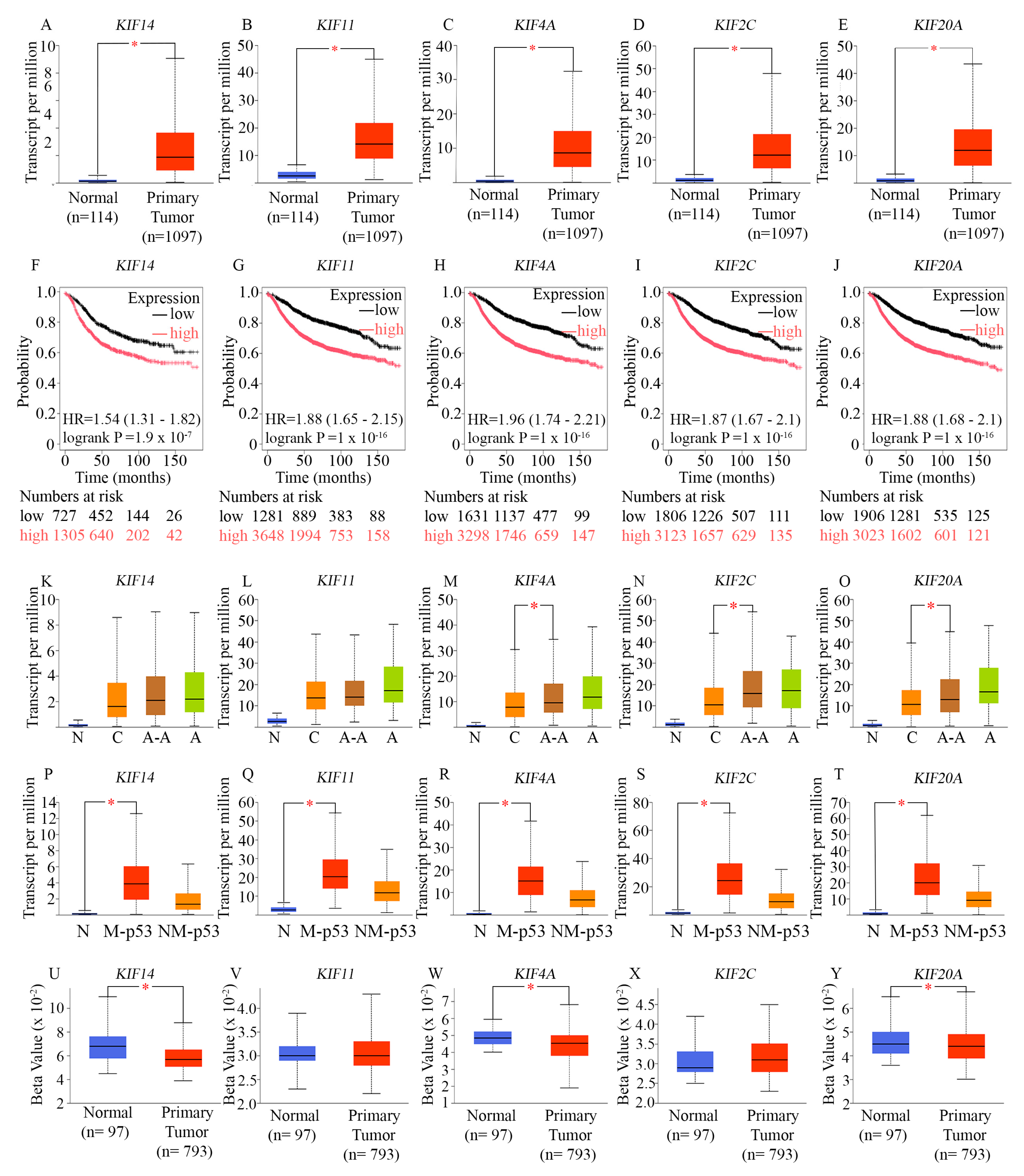
Analysis of the expression levels of genes encoding 5 mitotic kinesins in diverse breast tumors and evaluation of the prognostic significance of their overexpression (A-E) Box-whisker plots comparing the expression levels of KIF14 (A), KIF11 (B), KIF4A (C), KIF2C (D), and KIF20A (E), in primary breast tumor tissues (red boxes) contrasted to normal tissues (blue boxes). The UALCAN platform was used to analyze TCGA level 3 RNA sequencing data. The red asterisk (*) indicates a statistically significant difference (p < 0.05).(F-J) Kaplan–Meier survival analysis evaluating the prognostic significance of the expression of the indicated mitotic kinesins, performed using microarray data from TCGA breast tumors and displayed using the KM Plotter tool. The red line depicts the survival of patients with expression levels of the genes above the cut-point, while the black line represents the recurrence-free survival of patients with expression levels below the cut-point. The analysis was conducted using the KM Plotter’s JetSet optimal microarray probe set and an optimal cutoff for recurrence-free survival over 180 months, without restricting to specific breast cancer subtypes.(K-O) Analysis of the expression levels of the indicated mitotic kinesin genes in breast tumors from patients of different races (self-identified). “N” represents normal breast tissues with a sample size n=114 for all, “C” represents Caucasians with a sample size n=748, “A-A” represents African-Americans with a sample size n=179, and “A” represents Asians with a sample size n=61. Analysis of TCGA RNA sequencing data was performed on the UALCAN platform and visualized using a box-whisker plot. A red asterisk (*) denotes a statistically significant difference in expression between breast tumors from Caucasian and African-American patients (p < 0.05). (P-T) Analysis of the expression levels of the indicated mitotic kinesin genes in breast tumors of differing TP53 mutation status. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the expression of the indicated kinesins in tumors with mutated TP53 (M-p53) contrasted to non-mutated TP53 (NM-p53). A red asterisk (*) denotes a statistically significant difference in expression between the two indicated groups (p < 0.05).(U-Y) Analysis of the promoter methylation profiles of the indicated mitotic kinesin genes in breast tumors. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the methylation level of the promoters of the indicated genes in normal breast tissues (blue boxes) vs breast invasive carcinoma (red boxes). A red asterisk (*) denotes a statistically significant difference in promoter methylation between the two (p < 0.05).
Figure 2.
Analysis of the expression levels of genes encoding 5 mitotic kinesins in diverse breast tumors and evaluation of the prognostic significance of their overexpression (A-E) Box-whisker plots comparing the expression levels of KIF14 (A), KIF11 (B), KIF4A (C), KIF2C (D), and KIF20A (E), in primary breast tumor tissues (red boxes) contrasted to normal tissues (blue boxes). The UALCAN platform was used to analyze TCGA level 3 RNA sequencing data. The red asterisk (*) indicates a statistically significant difference (p < 0.05).(F-J) Kaplan–Meier survival analysis evaluating the prognostic significance of the expression of the indicated mitotic kinesins, performed using microarray data from TCGA breast tumors and displayed using the KM Plotter tool. The red line depicts the survival of patients with expression levels of the genes above the cut-point, while the black line represents the recurrence-free survival of patients with expression levels below the cut-point. The analysis was conducted using the KM Plotter’s JetSet optimal microarray probe set and an optimal cutoff for recurrence-free survival over 180 months, without restricting to specific breast cancer subtypes.(K-O) Analysis of the expression levels of the indicated mitotic kinesin genes in breast tumors from patients of different races (self-identified). “N” represents normal breast tissues with a sample size n=114 for all, “C” represents Caucasians with a sample size n=748, “A-A” represents African-Americans with a sample size n=179, and “A” represents Asians with a sample size n=61. Analysis of TCGA RNA sequencing data was performed on the UALCAN platform and visualized using a box-whisker plot. A red asterisk (*) denotes a statistically significant difference in expression between breast tumors from Caucasian and African-American patients (p < 0.05). (P-T) Analysis of the expression levels of the indicated mitotic kinesin genes in breast tumors of differing TP53 mutation status. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the expression of the indicated kinesins in tumors with mutated TP53 (M-p53) contrasted to non-mutated TP53 (NM-p53). A red asterisk (*) denotes a statistically significant difference in expression between the two indicated groups (p < 0.05).(U-Y) Analysis of the promoter methylation profiles of the indicated mitotic kinesin genes in breast tumors. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the methylation level of the promoters of the indicated genes in normal breast tissues (blue boxes) vs breast invasive carcinoma (red boxes). A red asterisk (*) denotes a statistically significant difference in promoter methylation between the two (p < 0.05).
7. A Set of 15 Cell Cycle-Related Proteins that are Overexpressed in AR-Low TNBC are Associated with High Proliferation
Since deregulated proliferation is a well-known hallmark of cancer [70,71], the measurement of the proliferative fraction of cells in a tumor has prognostic value. To evaluate our hypothesis that the 15 MMB-FoxM1-regulated cell cycle genes identified above are important for driving or supporting the high-proliferation phenotype of AR-low TNBC, we evaluated associations between the expression of the 15 genes of interest and three well-established markers of proliferation in tumors—Ki67, Proliferating Cell Nuclear Antigen (PCNA), and Mini-chromosome Maintenance 2 (MCM2). The Ki67 antigen is expressed universally in the nuclei of all proliferating cells (normal and tumor) in all phases of the cell cycle, but is not expressed in quiescent cells in the G0 phase [72], making it an excellent marker for the proliferative population within a group of cells [73]. In breast cancer, a strong correlation exists between percentage of cells positive for Ki67 expression and histologic grade because both parameters are associated with proliferation [74,75]. Furthermore, higher tumor stages and lymph node positivity are associated with a higher percentage of cells expressing Ki67 [20,75,76], which is associated with poor disease-free survival (DFS) and overall survival (OS) [75,77]. Ki67 serves as a dynamic biomarker for choosing the systemic treatment of early-stage breast cancer [78] and changes in the percentage of Ki67-positive cells in a tumor may be used as an early predictor of treatment efficacy [79]. Despite Ki67 being so widely used as a robust marker for proliferation, its cellular function remains mysteriously unclear. By contrast, the other two markers we chose (PCNA and MCM2) are important participants in the process of DNA replication. In addition to being involved in DNA replication and nucleic acid metabolism, PCNA is also involved in DNA excision repair, cell cycle control, chromatin assembly, and RNA transcription. Since PCNA levels spike during the S and G2/M phases of the cell cycle but is very low in quiescent cells [80], the immunohistochemical staining of PCNA has been extensively used in breast cancer diagnosis and predicting prognosis [80,81]. Increased PCNA expression also correlates with a shorter DFS and OS time in patients with breast cancer [81]. A notable caveat with the use of PCNA is that its expression is not limited to proliferating cells as PCNA is also involved in DNA damage repair [82,83]. MCM proteins are the key factor for initiation of DNA replication. In addition, they are required for replication elongation and are implicated in cohesion, condensation, transcription, and recombination of DNA [84]. The family of MCM proteins mainly include six major proteins, MCM2 through MCM7. Because MCM activity is essential for DNA replication in dividing cells and is lost in quiescence [85], these proteins are also excellent markers for proliferation. Many molecular studies have suggested that increased levels of MCMs may not only be a marker of proliferative malignant cells [86,87] but may also indicate precancerous cells and the potential for recurrence [88,89]. MCM2 is a strong prognostic marker in breast cancer because its high expression is associated with survival, regional recurrence, and distant metastases [90]. Using the “Targeted Correlation Analysis” tool of bc-Genexminer (all RNA sequencing data, TNBC status determined by immunohistochemistry) [104], we examined correlations in expression between the 15 MMB-FoxM1-regulated cell cycle genes and the afore-mentioned established markers of proliferation among TNBC tumors. Our analysis showed a strong statistically significant positive correlation between the expression of all 15 genes and the expression of Ki67, PCNA, and MCM2 among TNBCs (Fig. 1). Among breast cancers, each of these 15 genes were expressed at a statistically significant higher level in Ki67-high breast tumors (Ki67% >25%) compared to Ki67-low breast tumors (Suppl. Fig. 2, A—O), which supported the notion that upregulation of these genes was strongly associated with proliferation among all breast tumors.
A study by Cohen et al. [71] studied cell proliferation in vivo without introducing unwanted biochemical perturbations or relying on transgenic models. The authors wanted to identify a transcriptome signature associated with proliferation status alone (i.e., independent of cellular identity). The authors identified global in vivo circuitries that regulate the mammalian cell cycle; the genes they identified were highly expressed in proliferating cells but minimally expressed in resting cells, resulting in a strong enrichment for cell cycle activators. Six out of the 15 genes of our interest— KIF11, KIF4A, KIF2C, KIF20A, CENPF, UBE2C—as well as FOXM1 and ASPM, were identified by Cohen et al., as part of their 83-gene signature of proliferation, which corroborates the idea that the genes we focused on in the context of AR-low TNBC, are involved more generally in supporting high proliferation in a wider variety of tumor types that are particularly proliferative.
A study by Grant et al. [91] found 96 genes that were cell cycle-regulated in four distinct cell types (U2OS, HeLa, HaCaT, and foreskin fibroblasts), suggesting that these genes played critical role in essential biological processes required for successful cell division. These common genes are often bound by E2F1 (for S phase) or FoxM1 (for G2/M phase) at their promoters. Among the genes whose promoters were bound by FoxM1, and wherein FoxM1 binding stimulated gene transcription, were 9 out of the 15 genes we focused on in our study; the nine genes were KIF11, KIF4A, KIF2C, KIF20A, CENPA, CENPF, OIP5, UBE2C, and UBE2S. ASPM was also found to be part of the 96-gene proliferation signature identified by Grant et al. [91], which suggested that these genes may be components of a transcriptomic signature of cell proliferation and may contribute mechanistically to the highly proliferation fraction observed in AR-low TNBC. Thirteen out of the 15 genes we centered our study on—KIF11, KIF14, KIF2C, KIF20A, KIF4A, CENPA, CENPO, CENPL, CENPF, OIP5, UBE2C, UBE2S, UBE2T—were also identified as MMB-FoxM1 G2/M target genes by Sadasivam et al. [35]. Our study thus identified a group of FoxM1-regulated genes, upregulated in AR-low TNBCs, that are strongly associated with the high proliferation phenotype of AR-low TNBCs. These findings raised the possibility that the overexpression of these genes may be essential to mechanistically support the proliferation needs of AR-low TNBC tumors. The next few sections examine the roles of these proteins in enabling high proliferation in AR-low TNBC and delve into the collateral effect of promoting genomic instability in these tumors.
8. Mitotic Kinesins Overexpressed in AR-low TNBC Normally Ensure Proper Spindle Assembly, Accurate Chromosome Segregation, and Cytokinesis
Mitotic kinesins are ATP-dependent motor proteins crucial for intracellular transport, mitotic spindle formation and function, chromosome segregation, supernumerary centrosome clustering, and cytokinesis [92,93,94,95]. There are at least 16 mitotic kinesins among the 45 kinesins in the human genome; overexpression of mitotic kinesins is commonly observed in tumor cells and is implicated in oncogenesis, chemoresistance, and is associated with more advanced disease stages [94,96,97,98,99,100,101].
Our analysis identified 5 mitotic kinesins (KIF14, KIF11, KIF4A, KIF2C, and KIF20A) among a set of core genes responsible for transcriptional chaos in TNBC, that were (a) overexpressed in AR-low TNBC and AR-low breast cancer, and (b) showed a pattern of expression highly associated with proliferation in AR-low TNBC and AR-low breast tumors. Our examination of the top overexpressed genes among TNBCs (performed using the level 3 RNA sequencing data from UALCAN portal) revealed that 4 out of the 5 mitotic kinesins we identified were ranked among the top 250 genes overexpressed among TNBCs: KIF4A was ranked 10th, KIF2C was ranked 24th, KIF20A was ranked 45th, and KIF11 was ranked 117th [68]. These extremely high rankings suggest the high importance of these kinesins in driving the tumor biology of breast tumors with TN status. The following paragraphs provide a concise description of the main mitotic functions helmed by these kinesins, with a perspective to better understanding their roles in spurring aggressive tumor biology in AR-low TNBC.
KIF14
Kinesin family member 14 (KIF14) is a minus-end-directed motor protein that is localized to the cytoplasm during interphase, to the spindle poles and spindle microtubules during mitosis, and to the midbody during cytokinesis; it regulates key steps in vesicle transport, spindle assembly, chromosome segregation, and cytokinesis [102]. In normal cells, KIF14 localizes to the central spindle and midbody in late mitosis, where it regulates microtubule bundling and interacts with proteins such as PRC1 and CITK to ensure proper execution of cytokinesis [102,103]. KIF14 also contributes to the spindle checkpoint and maintenance of genomic integrity. When KIF14 is suppressed, midbody cleavage fails to occur, inducing cytokinesis failure, and causes a delay in the metaphase-to-anaphase transition, characterized by misaligned chromosomes that oscillate abnormally between the spindle pole body and the metaphase plate, which suggests that KIF14 is essential for these processes [104]. Aberrant KIF14 overexpression, often via gene amplification or transcriptional upregulation, has been reported in several malignancies, including breast, ovarian, and lung cancers, and is associated with poor clinical outcomes [105,106]. In cancer cells, elevated KIF14 enhances proliferation, inhibits apoptosis, and promotes chromosomal instability, underscoring its role as an oncogenic driver and a potential therapeutic target [107]. In breast cancer, KIF14 overexpression was strongly associated with ER-negativity, and predicted worse outcomes [105,108]. Specifically, in TNBC, high levels of KIF14 expression are associated with resistance to chemotherapy [96,109]. KIF14 knockdown significantly reduced AKT phosphorylation and activity, and live-cell imaging of TNBC cells confirmed the transient colocalization of KIF14 with AKT at the plasma membrane, suggesting a role for the tethering of KIF14 to signaling components of the PI3K/AKT1 axis, in AKT activation. Pharmacological inhibition of KIF14 increased chemosensitivity and lowered AKT activity in the TNBC cell lines tested [96]. Thus, KIF14 is a key determinant of TNBC treatment response and disease progression and is a promising therapeutic target. A recent study of morphological heterogeneity (differences in the architectural arrangements of tumor cells) within breast tumors identified hitherto unrecognized “torpedo-like structures” that are characterized by an irregularly elongated, triangular shape with a wide base and pointed tips [110]. The authors found that the localization of KIF14 to the tips of these torpedo-like structures, was significantly associated with increased distant metastases and decreased metastasis-free survival in breast cancer patients; importantly, the mere presence of torpedo-like structures was not associated with metastasis. The transcriptomic profiles of the KIF14-positive cells in these structures suggested a specialization for invasiveness. The authors thus proposed that these KIF14-positive cells were likely metastasis initiating cells [110], suggesting a novel role for KIF14 in promoting aggressive breast tumor phenotypes.
KIF11
Kinesin family member 11 (KIF11/Eg5/Ksp) is a plus end-directed kinesin [111], involved in construction and maintenance of the bipolar spindle because of its role in crosslinking and sliding antiparallel microtubules [93,112]. KIF11 is also the main force generator in centrosome separation, which is essential for bipolar spindle formation [111]. KIF11 overexpression has been implicated in various cancers (gastric, bladder, renal cell, astrocytic, laryngeal squamous cell, oral cancer) and is often associated with advanced stage or poor prognosis [113]. KIF11 has been identified as a potential oncogene that drives the development and progression of breast cancer, strongly associated with poor patient outcomes [113,114]. CDK1 phosphorylates Thr927 in KIF11 [115], to control the motor protein’s association with the spindle and to facilitate centrosome separation and chromosome dynamics. Similarly, Plk1, through direct phosphorylation of intermediary regulators like NEK9 and NEK6/7, activates KIF11 and maintains spindle stability and cytokinesis [116]. Experimental inhibition of KIF11 significantly suppressed breast cancer cell proliferation, migration, and invasion, while promoting apoptosis, both in vitro and in vivo [113]. KIF11's oncogenic role appears to be mediated, at least in part, through the epithelial-to-mesenchymal transition (EMT) process being stimulated, and through activation of key signaling pathways like MAPK/ERK and PI3K/AKT [113]. In breast cancer, KIF11 is one of the targets regulated by the Id proteins (specifically Id1 and Id3), that play key roles in sustaining cancer stem cell phenotypes such as proliferation and self-renewal [117]. KIF11 enhances the self-renewal of breast cancer cells by activating the Wnt/β-catenin signaling pathway [118]. Clinically, high KIF11 expression in TNBC is associated with shorter DFS [60]. KIF11 levels were markedly elevated in the CD44+/CD24− subpopulation of docetaxel-resistant TNBC cells. KIF11 knockdown reduced this cancer stem cell–like fraction, impaired mammosphere formation, and suppressed proliferation by inducing G2/M arrest followed by apoptosis. In docetaxel-resistant TNBC xenografts, a KIF11 inhibitor significantly limited tumor growth. Therefore, KIF11 is a driver of TNBC cell proliferation and self-renewal and is an established therapeutic target in chemo-resistant TNBC [60].
KIF4A
Kinesin family member 4A (KIF4A), a plus-end-directed chromokinesin motor protein, has the ability to bind to both chromatin/DNA and microtubules, and plays an important role in chromosome condensation and segregation, central spindle formation, spindle midzone organization, and cytokinesis [104,119,120,121]. Cdk1-dependent phosphorylation of KIF4A at S1186, and KIF4A’s interaction with condensin 1, are both essential for the formation of the chromosome scaffold structure that serves to assemble the long chromatin fiber into a compact chromatid [122]. KIF4A is also phosphorylated by Aurora kinase B and Plk1, and this reversible modification is required for correct chromosome condensation and decondensation during mitosis [122]. KIF4A overexpression has been implicated in driving a poor prognosis in several cancer types including lung cancer, cervical cancer, hepatocellular carcinoma, and oral cancer [98]. In breast cancer specifically, KIF4A overexpression has been extensively documented to predict a poor prognosis [123,124,125,126,127,128]; notably, KIF4A expression level had a strong prognostic value in both ER-positive and ER-negative breast cancers comparable to or even superior to the prognostic value of tumor size, lymph node invasion, and Elston grade [98]. KIF4A was also identified as a core stem cell marker gene in breast cancer [51]. Another study [129] found that circKIF4A interacts with EIF4A3 to stabilize SDC1 mRNA, which activates the c-src/FAK signaling pathways and promotes TNBC progression; these results further bolster the notion that KIF4A upregulation plays a critical role in TNBC progression. A study aiming to better understand why KIF4A localizes to the nucleus during interphase found that KIF4A rapidly accumulates at sites of DNA damage where it binds to BRCA2 and modulates the BRCA2/Rad51 DNA damage response pathway while inhibiting the enzymatic activity of PARP-1 (i.e., the enzyme that detects and signals DNA damage so that repair can be initiated); the result is high levels of genomic instability in KIF4A-overexpressing tumors, and disease progression [130]. A study that aimed to identify a circRNA-miRNA-mRNA competing endogenous RNA (ceRNA) regulatory network involved in EMT in breast cancer cells, identified two circRNAs (hsa_circRNA_002082 and hsa_circRNA_400031), which appear to act as ceRNAs, that sponge up 10 specific microRNAs (miRNAs) and, consequently, prevent these miRNAs from regulating their 6 target mRNAs, including KIF4A, CENPF, and OIP5 mRNAs (all 3 of which are among the 15 genes we focused on in our study) [131]. The study thus identified KIF4A, CENPF, and OIP5 as among the “hub gene” targets that are significantly overexpressed in breast cancer and are implicated in disease progression and worse patient prognosis [131], and uncovered a key mechanism underlying their upregulation. In TNBC specifically, the circKIF4A-miR-375-KIF4A axis was found to regulate TNBC progression also via the ceRNA mechanism; the authors therefore suggested that circKIF4A may therefore serve as a prognostic biomarker and therapeutic target for TNBC [132].
KIF2C
The kinesin-13 family member 2C (KIF2C) is a plus end-kinesin motor and is also known as the mitotic centromere-associated kinesin (MCAK). KIF2C regulates microtubule dynamics, especially during mitosis, and uniquely, depolymerizes microtubules by disassembling tubulin subunits at the polymer ends; as a result, KIF2C can mediate ciliary disassembly by depolymerizing microtubules within cilia [133]. Studies have found that both the centromeric localization of MCAK as well as its microtubule depolymerization activity are regulated via phosphorylation by multiple kinases—Aurora B [134,135], Aurora A [136], and polo-like kinase-1 (Plk1) [137]. KIF2C depletion or down-regulation led to prominent defects in chromosome congression and segregation due to improper kinetochore attachments in potoroo kidney cells [137] and in normal diploid RPE-1 cells [138]. Conversely, KIF2C overexpression promoted microtubule depolymerization, resulting in increased microtubule detachment from centromeres [139]. Moon et al. [140] reported that fine-tuned regulation of KIF2C is important for cell motility and migration due to its effects on the actin-microtubule interactions and cytoskeletal dynamics, and turnover of focal adhesions. Deregulation of KIF2C impairs cell motility and leads to severe mitotic defects and chromosomal instability [141]. KIF2C is localized on the centromere, and KIF2C activity maintains genomic stability by ensuring proper kinetochore-microtubule attachments [100]. Inhibitors of KIF2C’s microtubule depolymerization activity were sufficient to induce aneuploidy in TNBC models [142]. As KIF2C is the only kinesin protein that localizes to the centromere, it is proposed that KIF2C plays more critical roles in maintaining genomic stability than perhaps other kinesin proteins [143]. Based on their observations that Kif2C knockdown or knockout led to accumulation of endogenous DNA damage, DNA damage hypersensitivity, and reduced double stranded break repair via both non-homologous end-joining and homologous recombination, Zhu et al. [144] concluded that KIF2C also plays an important role in DNA damage repair and maintenance of genomic integrity. Liu et al. [100] found that KIF2C was upregulated in 18 different cancer types. Among breast cancers, KIF2C was upregulated across all molecular subtypes. High KIF2C expression was associated with poor OS in breast cancer, across multiple datasets [100]. Moreover, high-KIF2C breast tumors showed high tumor mutational burden and an immune cell infiltration profile that predicted a more favorable response to immunotherapy. Thus, KIF2C is a potential prognostic biomarker and predictor of immunotherapy response in breast cancer. A study by Jiang et al. [145] found that Doxorubicin resistance in breast cancer was marked by coordinated downregulation of TBX15 and miR-152, and concomitant upregulation of KIF2C, as miR-152 suppression of KIF2C expression was relieved. High levels of KIF2C reduced doxorubicin sensitivity partly via enhanced autophagy. Overexpressed KIF2C also associated with pyruvate kinase M2 (PKM2) and curtailed the latter’s ubiquitination, enhancing PKM2 stability and reinforcing glycolysis/the Warburg effect that undergirds the chemo-resistant phenotype. Thus, KIF2C upregulation drives doxorubicin resistance, proliferation, migration, and invasion in breast cancer by stabilizing PKM2 and promoting glycolysis and autophagy [145].
KIF20A
KIF20A (also known as MKlp2 and RAB6-KIFL), a member of the kinesin superfamily-6, is a plus-end-directed motor protein that localizes to the cleavage furrow, intercellular bridge, and midbody. Additionally, KIF20A helps with microtubule bundle formation, normal spindle formation, chromosome segregation, and cytokinesis regulation. KIF20A protein accumulates mainly in mitotic cells [146]; in metaphase cells, KIF20A is mostly cytosolic and is only recruited to the spindle after the metaphase plate forms, but in anaphase, the chromosomal passenger complex (CPC: complex of Aurora B, INCENP, survivin and borealin) and KIF20A form a complex, which localizes to and promotes the formation of cleavage furrow, via a mechanism enabled by decreased cyclin-dependent kinase 1 (Cdk1) activity [147]. Plk1 also phosphorylates KIF20A [148], which increases KIF20A’s affinity for microtubules, facilitating its attachment and function during late mitosis [99]. During telophase/cytokinesis, KIF20A shows strong localization to the cell cortex at the equator and the midbody, and this localization is essential for the proper formation of the cleavage furrow and execution of cytokinesis [146]. KIF20A relies on myosin-II for its localization to the equatorial cortex, which is in turn required to (i) recruit Aurora B to the equatorial cortex, (ii) promote the highly focused accumulation of active RhoA at the equatorial cortex and stable ingression of the cleavage furrow during cytokinesis [149]. KIF20A-mediated targeting of Aurora B to the cell cortex at the equator, and the formation of a complex between KIF20A and actomyosin filaments, is essential for the maintenance and progression of the ingressing furrow, and successful completion of cytokinesis [149]. In fact, KIF14, KIF4A, and KIF20A all collaborate in the formation and function of the central spindle (also called spindle midzone), the region of overlapping microtubules in the center of the spindle after the chromosomes have separated, and PRC1 has been proposed to act as central spindle matrix protein and receptor for these three kinesins [103,150]. Without this tight spatial and temporal coordination between so many players, and an optimal and balanced level of expression of these proteins, defects in chromosome segregation and cell division could ensue, contributing to aneuploidy.
A study by Miserey-Lenkei et al. [151] showed that functional coupling between actin and microtubule cytoskeletons driven by Myosin II and KIF20A ensures the spatial coordination between RAB6-positive vesicles’ fission from the trans Golgi network membranes and their exit along microtubules, and KIF20A is involved in this fission process. Additionally, siRNA-mediated inhibition of KIF20A and KIF11 each induced lysosomal membrane permeabilization followed by cathepsin-dependent cell death, showing that these kinesin motor proteins also play a role in maintaining lysosomal stability [152]. In the context of breast cancer, high KIF20A expression showed a strong correlation with more aggressive features, such as positive lymph nodes, larger tumor size, high histological grade and Ki67 labeling index; high KIF20A expression was in fact, an independent predictor of poor OS [99,153]. Inhibition of KIF20A led to marked reduction in proliferation and invasion of breast cancer cells [99]. It has also been extensively documented that KIF20A overexpression is associated with therapy resistance in breast cancer. Increased post-radiotherapy levels of KIF20A were linked to higher recurrence rates in breast cancer [153]. Patients with high KIF20A expression also have a higher recurrence rate than those with low KIF20A after chemotherapy. Among breast cancer patients who received anthracycline and/or taxane-containing neoadjuvant chemotherapy, a decreased pCR rate was observed in patients with high KIF20A expression [153]. Single-cell analyses showed that KIF20A was enriched in endothelial cells and fibroblast cells, suggesting that KIF20A may induce treatment resistance by regulating the angiogenic and fibrotic processes in the stroma. Yang et al. [153] also found that genes associated with multi-drug resistance in cancer treatment (ABCB1, ABCC1, ABCG2) were co-overexpressed with KIF20A. Upregulation of KIF20A also plays a role in doxorubicin resistance in breast cancer cells [154]. KIF20A was found to be a direct target of downregulation by miR-153-3, and the synergistic actions of doxorubicin treatment and miR-153-3p overexpression caused downregulation of Rab26 expression and reduced vesicular trafficking, and a decrease in migration/invasion of breast cancer cells, while concomitantly enhancing cell death in these cells [154], supporting the idea that downregulation of KIF20A may be a valuable treatment strategy in breast cancer. Among ER-positive breast cancer patients treated with adjuvant tamoxifen, high expression of KIF20A was identified as an independent factor for predicting worse DFS and OS [155]. Breast cancer cell growth and metastasis are often aided by M2 macrophage polarization. Interestingly, KIF20A and FOXM1 expression levels were markedly elevated in both tumor-associated macrophages and breast cancer cells, and a study by Wang et al. [156] found that direct transcriptional upregulation of KIF20A expression by FOXM1 promotes M2 polarization of macrophages, and proliferation, migration, invasion, and metastasis of breast cancer cells. In TNBC cells in particular, KIF20A knockdown significantly reduced cell viability, proliferation, migration, and invasion [156]. In vivo, KIF20A knockdown suppressed the growth and proliferation of TNBC tumors in nude mice xenograft models. Mechanistically, KIF20A knockdown suppressed expression of proteins in the IL-17 signaling pathway [156]. In sum, KIF20A, KIF14, KIF11, KIF4A and KIF2C all have critical functions necessary for precise spindle organization, regulation of microtubule dynamics, chromosome segregation, cleavage furrow formation and maintenance, and cytokinesis. All the five mitotic kinesins we identified were categorized as components of a “12-gene Mitotic kinesin signature (MKS)” and high expression of MKS genes was correlated with worse RFS, OS and DMFS in breast cancer patients [157], suggesting a strong association between overexpression of these kinesins and aggressive breast cancer phenotypes. Their overexpression drives proliferation and genomic instability which promotes tumor aggressiveness, therapy resistance, and poor survival, making them compelling prognostic biomarkers and therapeutic targets in AR-low breast cancers.
9. Overexpression of Mitotic Kinesins in AR-low and TP53-Mutant Breast Tumors Promotes Proliferation, Chromosomal Instability, and Poor Outcomes
Since (i) the expression levels of the 5 mitotic kinesins (KIF14, KIF11, KIF4A, KIF2C, and KIF20A) that we identified are highly correlated with expression of markers of proliferation not just in TNBC (Fig. 1) but more broadly across breast tumors (Suppl. Fig. 2), and (ii) these 5 kinesin motors are overexpressed not only in AR-low TNBC (Suppl. Fig. 1) but also among AR-low breast tumors more broadly (Suppl. Fig. 3), we decided to examine the patterns of expression of these kinesins more closely.
Among breast cancers, we found significant upregulation of KIF14 (Figure 2A), KIF11 (Figure 2B), KIF4A (Figure 2C), KIF2C (Figure 2D), and KIF20A (Figure 2E) in tumor samples of the UALCAN dataset (Figure 2A–E), compared to normal samples. To examine associations between the expressions of KIF14, KIF11, KIF4A, KIF2C, and KIF20A, and breast cancer patients’ relapse-free survival, we performed Cox proportional hazards regression analysis for each gene separately, using the Kaplan−Meier Plotter (KM Plotter) online tool [69]. All possible cutoff values within the interquartile range were assessed for each gene, and the cut-point that resulted in the lowest log-rank p-value was designated as the optimal choice. Kaplan−Meier plots were then used to visualize associations between gene expression and survival. Our analyses of publicly available microarray data using the (KM Plotter) tool for breast cancer showed that high levels of expression of KIF14 (Figure 2F), KIF11 (Figure 2G), KIF4A (Figure 2H), KIF2C (Figure 2I), and KIF20A (Figure 2J) predicted significantly poorer recurrence-free survival of breast cancer patients, suggesting that upregulation of these mitotic kinesins could potentially contribute to disease progression. Since AR-negative TNBC is more commonly diagnosed among African-American women and is believed to underlie the stark racial disparity in breast cancer outcomes in the US [158], we examined the race-wise expression of the mitotic kinesins KIF14 (Figure 2J), KIF11 (Figure 2K), KIF4A (Figure 2L), KIF2C (Figure. 2M), and KIF20A (Figure 2N), using data from 1102 race-annotated breast tumors in the UALCAN dataset. We found that KIF4A, KIF2C, and KIF20A showed a significantly higher expression level among African-American breast cancer patients compared to Caucasian/White breast cancer patients. Taken together, our analyses suggest that among breast tumors in general, and specifically among AR-low TNBCs, a group of key mitotic kinesins is upregulated, and their upregulation portends poorer outcomes. Furthermore, KIF14 (Figure 2P), KIF11 (Figure 2Q), KIF4A (Figure 2R), KIF2C (Figure 2S), and KIF20A (Figure 2T) were all significantly overexpressed in breast tumors harboring mutant TP53 compared to breast tumors harboring non-mutant TP53. This finding was similar to our earlier data showing that FOXM1 itself was also upregulated in TP53-mutant breast tumors [13]. We then confirmed our findings using the muTarget tool [69] to investigate the effect of mutations in the TP53 coding region (i.e., our input genotype) that have a prevalence of at least 2%, on downstream gene expression in a sample set comprising 305 TP53-mutant and 674 TP53-wild-type breast cancers found in TCGA. In this dataset, KIF14 showed a 1.96-fold upregulation (p = 1.77 × 10−38), KIF11 showed a 1.62-fold upregulation (p = 8.57 × 10−33), KIF4A showed a 1.94-fold upregulation (p = 3.89 × 10−47), KIF2C showed a 2.48-fold upregulation (p = 4.59 × 10−62), KIF20A showed a 1.94-fold upregulation (p = 2.02 × 10−44), in TP53-mutant versus TP53-wild-type breast cancers. These data compellingly indicate that (a) AR-low breast tumors and AR-low TNBCs in particular, (b) Ki67-high breast cancers, and (c) TP53-mutant breast tumors all show a significant upregulation of KIF14, KIF11, KIF4A, KIF2C, and KIF20A.
CHR elements have previously been identified in the promoters of mitotic kinesins [159]. Chromatin Immunoprecipitation (ChIP) assays performed in breast cancer cell lines confirmed strong binding of B-MYB, and FOXM1 to the promoters of KIF14, KIF20A, KIF4A, and moderate binding to the KIF2C promoter [157]. RNA interference (RNAi) experiments showed that depletion of MMB or FOXM1 subunits significantly inhibited the expression of KIF14, KIF4A, KIF20A, and KIF2C, suggesting that these four mitotic kinesins are direct transcriptional targets of MMB-FoxM1 in breast cancer [157]. KIF11 expression was found to be independent of MMB-FoxM1 [157]. To better understand if under-methylation of promoter DNA contributes to the overexpression of these mitotic kinesins, we utilized the UALCAN promoter methylation analysis tool (Figure 2U—Y). The beta value presented indicates the level of DNA methylation ranging from 0 (unmethylated) to 1 (fully methylated). Promoter methylation analysis of KIF4A, KIF14, and KIF20A through the UALCAN database revealed statistically significant hypomethylation at their promoter regions, indicating potential epigenetic regulation. These findings suggest that in addition to transcriptional upregulation mediated by factors such as MMB-FoxM1, epigenetic mechanisms may also contribute to the overexpression of KIF14, KIF4A, and KIF20A.
Overexpression of mitotic kinesins in cancer cells likely alters spindle dynamics, chromosome segregation, and cytokinesis, promoting decreased fidelity of chromosome segregation or chromosomal instability (CIN) and aneuploidy, which are linked to tumorigenesis, chemoresistance, and cancer progression. It turns out that KIF20A and KIF4A are part of the gene expression-based CIN75 chromosomal instability signature that is associated with adverse clinical outcomes in multiple cancers [160]. CIN can spur tumor evolution by resulting in loss of heterozygosity (LOH) of tumor suppressors or by creating imbalances or structural changes that culminate in the overexpression of oncogenes. Aneuploidy (gains/losses of whole chromosomes or large fragments of chromosomes) is prevalent in ~90% of solid tumors [161,162]. Pfister et al. [163] developed a computational method to quantify the degree of aneuploidy or structural rearrangements of large chromosome regions. Shifts in alternate allele frequencies (AAFs) occur as a result of aneuploidy, and these shifts can be assessed using TCGA exome sequencing datasets. In tumors, AAF values reflect a combination of chromosomal copy number in individual cells and the fraction of cells carrying aneuploid genomes. The authors identified heterozygous single nucleotide polymorphisms (SNPs) and calculated their AAFs for 522 human breast tumors from TCGA. The method measures changes in copy number of both whole chromosomes and large fragments of chromosomes, to yield a metric called functional aneuploidy (FA). The standard deviation of these AAF distributions was used as a robust, assumption-free measure of FA, with broader distributions indicating higher prevalence of aneuploidy. The method successfully segregated breast tumors based on their FA levels—the 100 highest FA tumors had an average of 15.6 chromosomes with LOH events, compared to 0.97 in the 100 lowest FA tumors. This study by Pfister et al. [163] identified two major factors associated with high FA in breast tumors: (i) TP53 mutations and (ii) overexpression of specific mitotic transcriptional regulators. Importantly, overexpression of FOXM1, MYBL2, and E2F1 mRNA correlated with high FA scores across all breast tumor subtypes. Furthermore, the DREAM complex, MMB, and FoxM1/MuvB transcriptional complexes regulate the transcription of 92 of the 100 most overexpressed genes among high-FA breast tumors in TCGA, indicating that the overexpression of these three transcription factors and their downstream targets, directly and potently drives high functional aneuploidy. Among the top 100 most overexpressed genes among high-functional aneuploidy breast tumors in TCGA were: KIF14 (ranked 64), KIF4A (ranked 41), KIF2C (ranked 50), KIF20A (ranked 78), FOXM1 (ranked 9), and ASPM (ranked 49). Thus 4 out of the 5 mitotic kinesins we identified as being responsible for transcriptional chaos and high proliferation in AR-low TNBC, are also major drivers of functional aneuploidy in this high-risk, high-need breast cancer subtype, presumably because overexpression of KIFs leads to an increased frequency of lagging chromosomes, and thus, the generation and propagation of functional aneuploidy. These data corroborate the CIN75 study [160] and greatly extend the idea that aneuploidy (and, likely, CIN) in breast cancer more broadly, and AR-low TNBC is particular, is associated with, and likely, causally related to the overexpression of mitotic kinesins.
Our data showed that overexpression of KIF14, KIF11, KIF4A, KIF2C, and KIF20A was associated with mutations in TP53, and Pfister et al. [163] found that TP53 mutations were highly enriched in the high FA tumors. TP53 mutations have previously been connected to aneuploidy in tumors [164,165]. Experimental evidence in Xenopus embryos suggests that TP53 mutations do not directly lower the fidelity of mitosis; instead, TP53's role as the guardian of ploidy prevents the survival and proliferation of cells that (i) have undergone chromosomal missegregation, or (ii) harbor chromosome fragments, by inducing cell cycle arrest, apoptosis, or entosis [163,166]. TP53 loss of function thus leads to the survival and persistence of aneuploid cells, leading to an increased level of functional aneuploidy. To demonstrate causation, Pfister et al. (2018) injected mRNA encoding human MYBL2, E2F1, and FOXM1 into Xenopus laevis embryos. This overexpression was sufficient to increase the rate of lagging anaphase chromosomes in this non-transformed vertebrate tissue and provided evidence for a direct causal link between the overexpression of these factors and reduced mitotic fidelity, and generation of a significantly higher percentage of micronuclei, which often form from lagging chromosomes. In fact, the study found a strong co-association of every combination of TP53 mutations and the overexpression of MYBL2 and FOXM1. Cellular senescence is one of the intrinsic safeguards against cancer progression. Although different triggers induce cellular senescence, p53 is well documented to play a critical role in its induction. Kif2C has been reported to play an important role in the regulation of cellular senescence in human cells through a p53-dependent pathway [167]; TP53 loss of function would presumably compromise this induction of senescence, leading to the persistence of cells with mitotic errors. Taken together, these lines of evidence converge onto the idea that overexpression of mitotic kinesins strongly promotes proliferation in AR-low TNBCs and TP53-mutant breast tumors, although the proliferation involves erroneous chromosome segregation and generation of extensive CIN and aneuploidy.
10. Centromeric Proteins Play Prominent Roles in Establishing Centromere Identity and Function, Enabling Kinetochore Assembly, and Ensuring Accurate Chromosome Segregation During Mitosis
In addition to the 5 mitotic kinesins described above, our analysis identified 5 centromeric proteins (CENPA, CENPO, CENPL, CENPF, and OIP5) among the set of core genes responsible for transcriptional chaos in TNBC. These centromeric proteins were also (a) overexpressed in AR-low TNBC and AR-low breast cancer, and (b) showed a pattern of expression positively associated with high proliferation in AR-low TNBC and AR-low breast tumors. All 5 centromeric proteins are established transcriptional targets of the MMB-DREAM complex [44]. Four out of these five centromeric genes are also ranked among the highest in the UALCAN TCGA database ranking of genes most overexpressed in TNBC, with CENPA ranked 29th most overexpressed in all TNBCs, CENPF 56th, CENPL 220th, and CENPO 239th. Again, these high rankings suggested that upregulation of these genes likely plays a critical role in the tumor biology of TNBCs. To better understand how the overexpression of these genes may undergird and power the aggressive clinical behavior of AR-low TNBCs, we performed an in-depth study of their known cellular functions within human centromeres, described in the sections below.
CENPA
Eukaryotic centromeres are major chromosomal landmarks first defined by Walther Flemming in 1882 as conspicuous constrictions on chromosomes [168]; generally, centromeres in mammalian cells are large ‘regional centromeres’ comprised of AT-rich tandem “satellite” DNA repeats that vary in their sequences and lengths [169]. The DNA sequence in centromeres is neither necessary nor sufficient for centromere formation as centromeres are epigenetically defined by the presence of a centromere-specific histone H3 variant, CENP-A, that plays an indispensable role in centromeric chromatin assembly and centromere specification, in addition to being essential for kinetochore assembly and proper chromosome segregation; in fact, CENPA is considered an epigenetic marker for centromere identity [170,171]. CENPA is deposited in a replication-independent manner by a dedicated histone chaperone HJURP (Holliday junction recognition protein) to replace its canonical counterpart, and forms specific CENPA nucleosomes with histone H4, H2A, and H2B; CENPA-containing nucleosomes are interspersed with canonical histone H3-containing nucleosomes in centromeres [172,173,174,175,176]. Since centromeres are the sites where large megadalton-scale protein assemblies called kinetochores assemble to connect chromosomes to spindle microtubules, mis-targeting of CENP-A to non-centromeric sites leads to abnormal kinetochore formation, which disrupts faithful chromosome segregation. CENPA is loaded onto centromeres by HJURP exclusively in G1 of the cell cycle [177]. Centromeres are typically flanked by pericentromeric constitutive heterochromatin—condensed and transcriptionally inert chromatin domains lacking CENPA—that also serve to define centromeres epigenetically [178]. Historically, the term kinetochore was first introduced in 1934 by Lester Whyland Sharp [179] and it is now clear that a human kinetochore contains more than 100 proteins. Kinetochores regulate chromosome movements during mitosis, act as a central platform for signaling factors that govern the fidelity of chromosome segregation, and are composed of two large submodules: the inner and outer kinetochore [180]. The inner kinetochore, which consists of 16 conserved proteins in vertebrates, assembles on centromeric chromatin, serves as a structural platform for outer kinetochore assembly, and persists with centromeres throughout the cell cycle [177]. The outer kinetochore assembles only during mitosis and plays an essential role in generating and sensing microtubule attachments [181]. The outer kinetochore is quickly dismantled once mitosis concludes, reflecting the highly dynamic and cell-cycle–regulated nature of this structure. Kinetochore dysfunctions commonly lead to CIN and aneuploidy [138,182]. CENPA recruits inner kinetochore proteins, such as CENPB and CENPC [183]. In prometaphase, centromeric chromatin, makes direct contact with inner kinetochore proteins such as CENPC and CENPB, which help establish the foundation of the inner kinetochore complex. CENPC directly binds to CENPA, linking centromeric chromatin to the inner kinetochore, while CENPB recognizes specific DNA sequences within centromeric repeats to stabilize centromere structure. These interactions facilitate the recruitment of additional inner kinetochore components, ultimately connecting to outer kinetochore proteins that interact with spindle microtubules to drive accurate chromosome segregation [183].
Although the centromeric environment generally suppresses transcriptional initiation, active RNA polymerase II complexes, centromere-derived RNAs (cenRNAs), and nascent transcripts have been reported to co-localize with centromeric chromatin [181]. cenRNAs are necessary for the proper formation of CENP-A-containing centromeres [184,185,186,187] and for the formation of pericentromeric heterochromatin [188,189]. The transcripts also play a role in the structure and function of the centromere–kinetochore interface [179]. Importantly, it has recently emerged that CENPA is an m6A reader of cenRNA [190]. The RNA m6A modification—the methylation of adenosine at the nitrogen-6 position in an RNA molecule—is an epitranscriptomic modification, that is the most abundant internal modification of mRNAs and noncoding RNAs, and is associated with almost all aspects of RNA-related processes, including transcription, pre-mRNA splicing and processing, pre-microRNA (pre-miRNA) processing, nuclear export, translation, RNA stability and decay, as well as other biological processes, such as transcriptional regulation, signal transduction, and the DNA damage response [190]. Kang et al. [190] found that the m6A-modification of cenRNA is essential for the stable centromeric localization of CENPA during S phase. Mutations of CENPA at the Leu61 and the Arg63 or removal of cenRNA m6A modification led to loss of centromere-bound CENPA during S phase, compromised centromere integrity, and led to defects in chromosome segregation. Thus, CENPA’s m6A reading ability appears to epigenetically govern centromere integrity [190].
A number of upstream factors influence the expression and stability of CENPA mRNA: A study found that the long non-coding RNA (lncRNA) MBNL1-AS1 was downregulated in breast cancer tissues and cell lines. lncRNAs regulate the expression of target genes by binding to RNA-binding proteins. In vitro and in vivo studies showed that overexpression of MBNL1-AS1 markedly inhibits breast cancer cells’ proliferation and stemness. MBNL1-AS1 normally downregulates CENPA mRNA by directly interacting with the RNA-binding protein Zinc Finger Protein 36 and subsequently decreases the stability of CENPA mRNA [191], ensuring optimal levels of CENPA expression. Upstream, CENPA’s activity is also influenced by DNA damage response pathways, like ATM and ATR, which help coordinate its incorporation during the cell cycle. When DNA damage occurs, these kinases activate checkpoint pathways that can delay CENPA deposition to ensure genome stability before centromere assembly proceeds. This delay helps prevent improper kinetochore formation on damaged DNA, safeguarding chromosome segregation fidelity [192]. Additionally, CENPA incorporation is tightly regulated by factors such as HJURP, which facilitates its deposition specifically at centromeres, ensuring that its function is not disrupted by random chromatin integration [193].
When the centromere is unable to form stable connections to the mitotic spindle, aneuploidy results. A recent study showed that CENPA maintains the integrity of centromere-associated repetitive sequences by ensuring their effective replication in human cells [194]. In the absence of CENPA, generation of DNA–RNA hybrids due to transcription–replication conflicts cause delayed DNA replication, centromere breakage, recombination, and chromosome translocations at centromeres. Thus CENPA-containing centromeric chromatin is specialized to facilitate replication and maintain the integrity of transcribed, noncoding, repetitive centromeric DNA during S phase, and suppress chromosome translocations and their deleterious sequelae [194]. Balachandra et al. [195] provided conclusive evidence that overexpressed CENPA localizes also to non-centromeric regions of chromosomes, and this mis-localization of CENPA causes CIN. The proper localization and function of CENPA thus ensures genome stability.
CENPO
Centromere protein O (CENPO) is a structural centromere protein that plays vital roles in cell proliferation and is essential for several critical aspects of mitosis including centrosome separation and bipolar spindle assembly, kinetochore assembly, accurate chromosome segregation, and checkpoint signaling during mitosis [177,196,197,198]. CENPO is a component of the CENPA-CAD (nucleosome distal) complex (comprised of CENPI, CENPK, CENPL, CENPO, CENPP, CENPQ, CENPR and CENPS, which are all purified in association with CENPA nucleosomes). The CENPA-CAD complex interacts with the CENPA-NAC complex (that includes CENPA, CENPC, CENPH, CENPM, CENPN, CENPT and MLF1IP/CENPU). CENPA-CAD/NAC cooperatively modulate the kinetochore-bound levels of the NDC80 complex (that is essential for microtubule–kinetochore attachment and spindle checkpoint signaling), and they collaborate to drive efficient chromosome segregation during mitosis [197]. CENPO is also involved in incorporation of newly synthesized CENPA into centromeric nucleosomes [199]. Abnormally high expression of CENPO was detected in gastric cancer where CENPO overexpression was associated with high clinical stage, tumor volume, lymph node metastasis, and shorter survival times [200]. CENPO overexpression in ovarian cancer cells drives abnormal proliferation and resistance to apoptosis [201]. Overexpression of CENPO in colorectal cancer was associated with high proliferation [100] while that in bladder cancer was associated with disease progression [126]. In lung adenocarcinoma, CENPO expression correlates with age and advanced TNM stage, and patients with high CENPO expression have poorer OS and DFS [202]. CENPO plays a unique role in spindle damage recovery and is involved in chromatin remodeling and heterochromatin formation [203,204]. Dysregulation of CENPO leads to CIN, a hallmark of cancer, driving aggressive tumor behavior [205].
CENPL
As a component of the CENPA-CAD complex, CENPL is involved in the recruitment of CENPA and in the assembly of centromeric chromatin by interacting with the Mis18 complex, which facilitates the loading of CENPA onto chromatin [177,199]. CENPL, along with other members of the CENPA-CAD complex, also helps assemble kinetochores on top of CENPA-containing centromeric chromatin, to ensure proper chromosome segregation during mitosis. The CENPA-CENPL-CENPO complex interacts with the microtubule motor proteins during the transition from metaphase to anaphase, ensuring accurate alignment of chromosomes on the spindle and their proper segregation [175]. Its dysfunction leads to defective assembly of the kinetochore, misalignment of chromosomes, and eventually to CIN. CENPL also forms a complex with CENPN, and this CENPL-CENPN complex recognizes CENPA nucleosomes; temporally, the cell cycle-dependent loading of CENPN onto centromeres is highly coordinated with structural transitions of the higher-order structure of centromeric chromatin [206]. Mitotic phosphorylation of CENPN by CDK1 regulates the CENPL–CENPN interaction for accurate chromosome segregation; perturbation of CENPN phosphorylation prevents proper chromosome alignment and activates the spindle assembly checkpoint [100]. CENPL is proposed to be an oncogene and breast cancer was among several cancer types exhibiting elevated CENPL expression [207]. High-CENPL breast tumors showed hyperactivation of several oncogenic pathways, especially those related to proliferation [207]. CENPL overexpression has been shown to drive chemoresistance in breast cancer cells [101].
CENPF
CENPF is a very large (380kDa) kinetochore protein that plays a critical role in the dynamic attachment of kinetochores to spindle microtubules, the proper segregation of chromosomes during mitosis, vesicular transport, and even ciliopathies [208,209,210,211,212,213]. CENPF gradually accumulates during the cell cycle until it reaches peak levels in G2 and M phase cells and is rapidly degraded upon completion of mitosis [214]. Upstream, CENPF expression is regulated by cell cycle checkpoints, including CDK1 and other cell division regulators [213]. These ensure that CENPF is appropriately activated during mitosis. CENPF functions downstream by interacting with the microtubule motors such as dynein, stabilizing the kinetochore-microtubule attachment necessary for chromosome segregation. It also coordinates in a complex manner with other kinetochore-associated proteins to ensure integrity of the spindle. A recent study found that in a cancer-specific context, co-occupation of the promoters of centromeric proteins by BMYB-FOXM1 was insufficient to initiate the full FOXM1-associated transcriptional program [215]. The study identified CENPF as a crucial co-regulator with FOXM1 in orchestrating G2/M gene expression and ensuring accurate chromosome segregation. CENPF depletion alters chromatin accessibility at G2/M regulatory loci and disrupts the formation of the FOXM1-MMB transcriptional complex. Notably, this FOXM1–CENPF collaboration appears to selectively co-regulate a subset of G2/M genes to promote cell proliferation [215].
Disruption of CENPF can lead to defects in chromosome segregation, leading to misalignment of chromosomes or aneuploidy [113]. Because of its crucial role in mitosis, CENPF is indispensable in maintaining genome stability. High-resolution electron tomography of the kinetochore–microtubule interface has revealed that the plus-ends of kinetochore-attached microtubules often exhibit flared protofilaments, formed by outwardly curved tubulin strands. These curled protofilaments appear to connect to centromeric chromatin via fine fibrillar linkages approximately 3 nm in diameter and 45–65 nm in length. Such structural features suggest the involvement of filamentous kinetochore proteins that preferentially bind to curved tubulin assemblies. These interactions likely play a critical role in mitotic force generation, as most chromosomes move toward the spindle poles during anaphase by maintaining strong attachments at the plus-ends of kinetochore microtubules. CENP-F has been characterized as a mitotic protein that binds more effectively to curved tubulin oligomers than to intact microtubule lattices [216]. Notably, its N-terminal microtubule-binding domain shows a higher affinity for tubulin curls, and both of its MT-binding domains can engage with dynamic microtubules to harness the mechanical energy generated during tubulin depolymerization [216].
CENPF mis-regulation has been linked to progression of several types of cancer including papillary thyroid cancer [217]. CENPF is overexpressed in breast cancer, where it promotes proliferation, migration, and invasion [218]. Multiple studies have led to the finding that the expression of CENPF is controlled by multiple miRNAs, lncRNAs, and even circular RNAs, whose dysregulation can result in inappropriate overexpression of CENPF in breast cancer. CENPF expression is normally downregulated by miR-28-5p, which is in turn downregulated by a competitively binding cytoplasmic RNA MCM3AP-AS1. MCM3AP-AS1 is highly overexpressed in breast cancer; this overexpression then results in the “sponging up” of miR-28-5p, and upregulation of CENPF expression, driving breast cancer progression [218]. Another study found that the circular RNA Hsa_circ_0002082, which is highly upregulated in breast cancer tissues and cell lines, targets miR-508-3p, which was downregulated in breast cancer. In normal cells, miR-508-3p targets CENPF for downregulation. Thus, upregulation of Hsa_circ_0002082 could also contribute to the overexpression of CENPF in breast cancer [126]. Another study [219] found that the lncRNA LINC00536 and CENPF were overexpressed in breast cancer cells, while miR-4282 was downregulated. LINC00536 binds to and negatively regulates the function of miR-4282. Importantly, CENPF is a target gene that is normally downregulated by miR-4282. Overexpressed LINC00536 thus functions both as an oncogene as well as a ceRNA, that leads to CENPF upregulation in breast cancer [219]. A recent study found that CENPF overexpression promotes breast cancer bone metastasis by activating PI3K-AKT-mTORC1 signaling [220]. High CENPF expression portended worse recurrence-free survival in breast cancer patients receiving neoadjuvant chemotherapy [202]. CENPF was also identified as one of the five embryonic stem cell-specific genes whose expression predicts a high risk of breast cancer recurrence [221]. CENPF is also part of a 14-gene genomic signature of metastatic relapse after adjuvant FEC100 regimen (5-fluorouracil 500 mg/m(2), epirubicin 100 mg/m(2) and cyclophosphamide 500 mg/m(2)) [222]. In breast cancer, high CENP-F expression, correlated with higher standardized uptake value (SUV) detected by 18F-fluorodeoxyglucose positron emission tomography/computed tomography (FDG PET/CT); SUV correlates with proliferation of primary breast cancer, and tumoral SUV levels may serve as a pretherapeutic indicator of aggressiveness of breast cancer [223].
In TNBC, CENPF is overexpressed and high CENPF expression was associated with chemotherapy resistance; conversely, silencing CENPF increased chemosensitivity [202]. It turned out that CENPF regulates TNBC chemoresistance through the RB-E2F1-Chk1 axis, indicating that CENPF is a potential treatment target to overcome chemo-resistance in TNBC. CENPF promotes adriamycin chemoresistance by operating upstream of Chk1 and provoking Chk1-mediated G2/M phase arrest in TNBC, which provides sufficient time for DNA repair to prevent DNA damage from causing apoptosis. CENPF knockdown promotes adriamycin-induced apoptosis as G2/M phase arrest is counteracted. High levels of CENPF bind to RB, triggering E2F1 release. E2F1 transcription factor then binds to the Chk1 promoter and upregulates Chk1 expression in TNBC. In this manner, the RB-E2F1 axis mediates the regulation of Chk1 expression by CENPF, which contributes to chemotherapy resistance in TNBC [202].
Another study found that in TNBC, CENPF level was aberrantly elevated, and knockdown of CENPF suppressed TNBC cell migration and invasion, and lung metastases in mice, suggesting that CENPF promotes TNBC metastasis [224]. CENPF silencing exacerbates arachidonic acid metabolism-induced ferroptosis in TNBC cells, suggesting that CENPF normally inhibits ferroptosis [224]. Furthermore, E2F1 binds to CENPF promoter and activates CENPF expression, which then leads to suppression of ferroptosis. Importantly, arachidonic acid metabolism-induced ferroptosis suppresses TNBC metastasis. The binding of PSMD14 (a deubiquitinase) to E2F1 stabilizes E2F1, which then results in upregulation of CENPF expression, suppression of ferroptosis, and increased metastasis in TNBC [224]. This finding indicating how overexpression of CENPF and PSMD14 can synergize to promote metastasis in TNBC is extremely important given that PSMD14 is also one of the 15 genes we focused on in this paper, based upon its upregulation in AR-low TNBC, and the positive correlation between the expression of PSMD14 and markers of proliferation among breast tumors.
OIP5
OIP5 gene encodes a 25-kDa protein that was originally identified in a yeast two-hybrid screen for proteins that interact with Opa (Neisseria gonorrhoeae opacity-associated) proteins [225]. OIP5's main function deals with the loading of CENP-A onto centromeric chromatin by collaborating with HJURP [226]; this function is important for the structure, stability, and function of the centromere throughout the cell cycle. This is especially critical for the fidelity of chromosome segregation in mitosis, because defects in the function of OIP5 may lead to aberrant alignment and attachment of kinetochores and precipitate chromosome misegegation and aneuploidy [227,228].
OIP5 is historically well established as a Cancer/Testis (C/T) antigen that belongs to a unique class of tumor-associated antigens with expression normally restricted to immune-privileged sites like the testis, and minimal to no expression in normal somatic tissues [229,230]. The blood-testis barrier normally prevents immune recognition of germ cell antigens. For effective immunotherapy, the ideal target antigen in the tumor would demonstrate stable, tumor-specific expression, an absence of expression in normal cells, and be indispensable for cancer cell survival. Evidence indicates that exposure to germ cell antigens has the potential to direct the immune system’s cytolytic capacities against cancer cells expressing that antigen [230]. Based on molecular similarities between gametogenesis and tumorigenesis, it has been hypothesized that cancer cells may inappropriately turn on germline expression programs to promote malignancy [230]. OIP5 is normally expressed in the testis and at low levels in bone marrow, thymus, and colon [231], but is markedly overexpressed in breast cancer [227], glioblastoma [232], colorectal [233], bladder [234], gastric cancers [235], oral cancer [236], and lung and esophageal cancers [237]. The transient expression of OIP5 in NIH3T3 cells resulted in a 2-fold increase in proliferation rate, highlighting its oncogenic properties [233]. OIP5 plays a significant role in promoting glioblastoma progression and metastasis by stabilizing E2F1 signaling through a feedback loop where E2F1 activates OIP5 expression; OIP5 then interacts with and stabilizes E2F1, leading to increased E2F1 signaling that promotes proliferation and metastasis in glioblastoma [228]. In breast cancer, OIP5 expression showed a significant positive correlation with advanced clinical stage [238]. Moreover, OIP5 knockdown inhibited the proliferation of breast cancer cells, and promoted apoptosis. Mechanistically, OIP5 is a direct target of miR-139-5p, and upregulation of OIP5 mRNA turns it into an endogenous molecular sponge that mops up miR-139-5p. Notch1 is a target of regulation by miR-139-5p; as a result, OIP5 overexpression leads to overexpression of Notch1 [238]. Given the very low expression of OIP5 in normal breast tissue and its role in chromatin organization and cell cycle control, dysregulation could have severe oncogenic consequences [239]. These features make C/T antigens, including OIP5, promising targets for immunotherapeutic strategies such as monoclonal antibodies and dendritic cell–based vaccines.
Therefore, the above-described centromeric proteins play pivotal roles in centromere identity establishment and maintenance, kinetochore assembly, formation of high-tension kinetochore-spindle microtubule attachments, proper chromosome alignment at the metaphase plate, and accurate chromosome segregation during anaphase, so that mitosis is error-free. Centromere protein deregulation leads to CIN, a cancer hallmark. Among the five centromeric proteins discussed above, CENPA and CENPO were ranked 34th and 27th, respectively, among the top 100 most overexpressed genes among high-functional aneuploidy breast tumors in TCGA [163]. The CIN75 signature includes OIP5 [160]. CENPF overexpression was significantly associated with markers of CIN including cyclin E, increased telomerase activity, c-Myc amplification and aneuploidy [240]. Taken together, dysregulation of the above-mentioned 5 centromeric proteins favors rapid and uncontrolled proliferation, increased CIN and aneuploidy, and increased chemoresistance, and is correlated with advanced disease stage and poor outcomes in multiple cancer types, including breast cancer; these findings highlight their dual potential as prognostic biomarkers and therapeutic targets.
11. Overexpression of Key Centromeric Proteins in AR-Low and TP53-Mutant Breast Tumors Promotes Proliferation and Poor Outcomes
Since (i) the expression levels of the 5 centromeric proteins (CENPA, CENPO, CENPL, CENPF, and OIP5) that we identified are highly correlated with expression of markers of proliferation not just in TNBC (Fig. 1) but more broadly across breast tumors (Suppl. Fig. 2), and (ii) these 5 centromeric proteins are overexpressed not only in AR-low TNBC (Suppl. Fig. 1) but also among AR-low breast tumors more broadly (Suppl. Fig. 3), we evaluated their patterns of expression more closely.
In breast cancer samples from the UALCAN dataset, we observed marked overexpression of the five centromeric proteins—CENPA, CENPO, CENPL, CENPF, and OIP5—compared to normal breast tissue (Figure 3A–E). To assess the prognostic relevance of these genes, we performed separate Cox proportional hazards regression analyses using the Kaplan–Meier Plotter [69], selecting the optimal expression cut-off based on the most statistically significant log-rank p-value. High expression levels of each centromeric gene were consistently associated with significantly reduced relapse-free survival in breast cancer patients, as visualized by Kaplan–Meier curves (Figure 3F–J).
Given the association of AR-negative TNBC with poorer prognosis in African-American women [158], we next assessed racial disparities in expression using race-annotated data from 1,102 tumors in the UALCAN dataset. CENPA and OIP5 were expressed at significantly higher levels in African-American patients compared to their Caucasian/White counterparts (Figure 3K–O). Collectively, these findings indicate that these centromeric proteins are frequently upregulated in breast cancers—particularly in AR-low TNBCs—and that their elevated expression correlates with worse clinical outcomes.
We further investigated whether expression of these centromeric proteins was influenced by TP53 mutational status of breast tumors. Our analysis of TCGA RNA-seq data through the UALCAN portal revealed that CENPA (Figure 3P), CENPF (Figure 3Q), CENPL (Figure 3R), CENPO (Figure 3S), and OIP5 (Figure 3T) were all significantly upregulated in TP53-mutant tumors compared to those with wild-type TP53, consistent with previous findings on their upstream regulator, FOXM1 overexpression in TP53-mutant breast cancers [13]. To corroborate this, we utilized the muTarget platform [69] to analyze the impact of TP53 mutations (≥2% prevalence) on gene expression in a TCGA cohort of 305 mutant and 674 wild-type samples. The results confirmed substantial upregulation of the centromeric proteins in TP53-mutant tumors: CENPA (2.88-fold; p = 6.91 × 10⁻67), CENPO (1.62-fold; p = 1.11 × 10⁻42), CENPL (1.52-fold; p = 3.75 × 10⁻34), CENPF (1.86-fold; p = 2.29 × 10⁻38), and OIP5 (1.81-fold; p = 2.13 × 10⁻⁴⁴). In cells with functional p53, defects in chromosome segregation normally activate p53-mediated responses, including cellular senescence induction. When a cell harbors p53 loss-of-function mutations, the unchecked entry into mitosis of cells whose centromeres cannot support proper chromosome segregation causes the generation and persistence of aneuploid cells [241]. New studies have found that a reduction in CENPA expression can prompt a p53-dependent cellular senescence response [242]. This response presumably prevents centromere-defective cells from proceeding through mitosis—a process that could potentially lead to the generation of aneuploid cells [243]. Another study also identified p53 as a key determinant of how CENPA impacts cell state, cell identity and therapeutic response [244]. If p53 is functional, CENPA overexpression promotes senescence and radiosensitivity. By contrast, when p53 is inactivated, CENPA overexpression promotes EMT, which is a precursor for tumor cell invasion and metastasis. Thus, CENPA overexpression has an unanticipated function in promoting cell fate reprogramming, with important implications for development and tumor evolution [244]. Evidence now shows that TP53 binds to the CENPA promoter and directly represses CENPA expression [242]. TP53 also binds to HJURP promoter and represses its transcription. In a TP53-deficient background, once HJURP or CENPA is upregulated at the protein level, it can in turn stabilize the other by protecting the other from proteasomal degradation [242]. As a result, CENPA overexpression is accompanied by HJURP overexpression in TP53-deficient contexts, leading to CENPA misincorporation and propagation of CIN [242].
To explore whether reduced promoter DNA methylation may be linked to the elevated expression of these centromeric proteins, we conducted promoter methylation analysis using the UALCAN platform (Figure 3U–Y). This tool reports DNA methylation levels as beta values, which range from 0 (completely unmethylated) to 1 (fully methylated). Analysis of CENPA, CENPL, and OIP5 revealed significantly lower methylation levels at their promoter regions in breast tumor samples, consistent with promoter hypomethylation. These observations suggest that, in addition to transcriptional activation by regulatory complexes such as MMB–FOXM1, epigenetic deregulation may also contribute to the aberrant overexpression of these centromeric proteins. Taken together, these analyses suggest that AR-low breast cancers, particularly AR-low TNBCs, as well as Ki67-high and TP53-mutant breast tumors, are characterized by co-upregulation of CENPA, CENPO, CENPL, CENPF, and OIP5—highlighting their potential contributions to disease progression and poor prognosis.
12. Key Players in Ubiquitin-Dependent Proteolysis and Aggrephagy Modulate the Stability and Activities of Cell Cycle Regulators, Promote Fidelity of Chromosome Segregation, and Help Manage Proteotoxic Stress
In addition to the 5 mitotic kinesins and 5 centromeric proteins described in previous sections, our research also identified 5 genes that play vital roles in the process of protein degradation (UBE2S, UBE2C, UBE2T, PSMD14, and TUBA1B), among the set of core genes responsible for transcriptional chaos in TNBC. These proteolysis-related proteins were also (a) overexpressed in AR-low TNBC (Suppl Fig. 1) and AR-low breast cancer (Suppl Fig. 3), and (b) showed a pattern of expression positively associated with high proliferation in TNBC (Fig. 1) and breast cancer more generally (Suppl. Fig. 2). All 5 proteolysis-related proteins are established transcriptional targets of the MMB-DREAM complex [44]. Three out of these five ubiquitin-proteasome pathway genes are also ranked among the highest in the UALCAN TCGA database ranking of genes most overexpressed in TNBC, with UBE2C ranked 7th most overexpressed, UBE2T ranked 65th most overexpressed, and UBE2S ranked 106th most overexpressed in all TNBCs. Again, these elevated rankings suggest that their overexpression may contribute significantly to the tumor biology of TNBCs.
Ubiquitination is a post-translational modification of proteins that involves the addition of an evolutionarily conserved small protein, ubiquitin (Ub) or ubiquitin-like proteins (UBLs), to lysine side-chains of target (abnormal or inherently short-lived) proteins destined for degradation by the proteasome [245,246]. In addition to marking proteins for degradation, ubiquitination can have different biological outcomes such as altering target proteins’ localization and/or their activity, and promoting or interfering with protein interactions [247,248,249]. As a result, dysregulation of this process can profoundly impact protein stability, activity, and localization. Ubiquitin-dependent protein degradation involves the E1, E2, and E3 enzymes, which carry out ubiquitin activation, conjugation, and ligation, respectively, and sequentially: The C- terminus of Ub is first activated by an E1 (ubiquitin-activating) enzyme and is then transferred onto the active site of an E2 Ub-conjugating enzyme. Subsequently, E3 Ub ligases mediate the transfer of Ub from the active site of the E2 onto a specific lysine residue in the target protein [250]. E2s control crucial aspects of this cascade. The human genome contains 50 E2-encoding genes, and their precise regulation is critical for maintaining homeostasis. By regulating the persistence and stability of oncogenic or tumor suppressor proteins, the ubiquitination pathway can have tumor suppressing or tumor-promoting effects. Ubiquitin-conjugating enzymes E2S (UBE2S), E2C (UBE2C), and E2T (UBE2T) are important members of the E2 family and have been implicated in the tumorigenesis and progression of many cancers. PSMD14 is a deubiquitinase enzyme while TUBA1B is a core player in aggrephagy—a process that removes harmful protein aggregates that arise due to mutations, incomplete mRNA translation, post-translational misfolding, improper protein modifications, and oxidative stress—which impacts tumorigenesis and cancer progression. To elucidate how the overexpression of these genes may contribute to the aggressive clinical phenotype of AR-low TNBCs, we conducted a detailed analysis of their established roles in proteolysis, as outlined in the following sections.
UBE2C
UBE2C is an E2 (Ub-conjugating) enzyme required for the degradation of mitotic regulators in cooperation with the anaphase-promoting complex/cyclosome (APC/ C) [251,252]. UBE2C initiates the Ub chain on the target proteins via the K11 linkage. UBE2C exerts its oncogenic activities by promoting the polyubiquitylation and degradation of several cell cycle–related proteins [253,254]. Two cell culture–based studies also revealed that UBE2C inhibits autophagy [255,256].
High expression of UBE2C is found in many human cancers of the brain, lung, cervix, colon, liver, thyroid, breast, and nasopharynx, and depletion of UBE2C from cancer cells significantly reduces proliferation and induces apoptosis [257,258]. Transgenic mice overexpressing UBE2C were prone to developing carcinogen-induced lung tumors and a broad spectrum of spontaneous tumors [254]. A recent study found that UBE2C likely regulates DEPTOR/mTORC signaling in non-small cell lung cancer, where UBE2C is massively overexpressed and functions as an oncoprotein [259]. The DEP domain–containing mechanistic target of rapamycin (mTOR) interacting protein (DEPTOR), serves as a negative regulator of both mTOR complex 1 (mTORC1) and complex 2 (mTORC2) [260]. DEPTOR acts as an oncoprotein in some contexts and a tumor suppressor in others [260,261,262,263,264,265]. The study by Zhang et al. [259] found that although dispensable for normal lung growth, UBE2C is a growth-essential gene in lung cancer cells harboring a Kras mutation. UBE2C is required for lung tumorigenesis induced by KrasG12D, wherein mTORC signals are activated to cooperate with KrasG12D during the process of lung tumorigenesis. DEPTOR is normally a substrate of the UBE2C-APC/C—CDH1 E2-E3 complex. Overexpressed UBE2C collaborates with CDH1 to promote ubiquitylation and degradation of DEPTOR, thus promoting mTORC1/2 signaling [259]. Our analysis carried out using the bc-GenexMiner platform showed that among TNBCs, there is a statistically significant negative correlation between the expression of UBE2C and DEPTOR (r = −0.27, p<0.0001, n = 293), and a statistically significant positive correlation between the expression of AR and DEPTOR (r = 0.34, p<0.0001, n = 293), suggesting that a similar mechanism could operate in AR-low TNBC leading to downregulation of the tumor suppressor DEPTOR and an increase in mTORC signaling.
The overexpression of UBE2C and the association between high expression of UBE2C and poor prognosis in breast cancer is well documented [266,267,268,269,270,271]. FOXM1, BMYB, and UBE2C are significantly overexpressed in high-grade breast tumors [272]. In breast cancer overall, UBE2S and UBE2C were overexpressed while Numb, the cell fate determinant and tumor suppressor, was found to be downregulated [270]. Numb regulates other tumor suppressors, such as p53 and PTEN, and promotes GLI1 oncogene degradation via ubiquitination [273,274,275,276]. In breast cancer, Numb acts as a tumor suppressor and is a negative regulator of EMT in both human mammary epithelial cells and breast cancer cells [277]. Reduced NUMB expression was significantly associated with elevated EMT in TNBC [278]. In normal mammary epithelial cells and breast cancer cells expressing wild-type p53, NUMB suppresses EMT by stabilizing p53. However, in TNBC cells, loss of NUMB promotes EMT through the activation of Notch signaling pathways. Supporting this mechanism, clinical data reveal a strong association between low NUMB levels, elevated Notch signaling, and the TNBC subtype [278]. Our analysis using the bc-GenexMiner platform also showed that among TNBCs, expression of Numb shows a statistically significant negative correlation with the expression of UBE2S (r = −0.34, p<0.0001, n = 293) and UBE2C (r = −0.31, p<0.0001, n = 293), and a statistically significant positive correlation with the expression of AR (r = 0.26, p<0.0001, n = 293), raising the possibility that AR-low TNBC exploits an analogous mechanism to induce EMT and facilitate metastasis.
UBE2C facilitates cell malignant behavior in lung adenocarcinoma by ubiquitin-dependent degradation of p53 to suppress the p53/p21 signaling pathway [279]. Inactivation of TP53 in mouse stratified epithelia induces spontaneous epidermal tumors with complete penetrance within one year [280]. UBE2C is transcriptionally repressed by wild-type p53 [281]. Importantly, wild-type p53-mediated inhibition of UBE2C is p21-E2F4-dependent. DNA damage-induced wild-type p53 leads to spindle assembly checkpoint arrest by repressing UBE2C [281]. Gene expression profiling of Trp53ΔEC mouse carcinomas and comparison with normal, wild type skin samples, revealed enrichment of human embryonic stem cell genes, Nanog/Sox2/Myc targets, and evidence of EMT, alongside repression of Polycomb targets [282]. Differential expression analysis showed that most upregulated genes were linked to DNA replication, repair, genomic instability, and cell cycle checkpoints. Notably, 20 genes including KIF2C and UBE2C, overlapped between a poor-prognosis 26-gene signature and a TP53 mutation-associated 51-gene signature [282]. This 20-gene set may serve as a biomarker for TP53-mutant or aggressive tumors and holds potential prognostic value for human cancers. These findings also implicated TP53 loss–driven CIN as a key driver of metastatic progression, and shed light on how UBE2C upregulation exacerbates the phenotypes induced by TP53 loss.
FoxM1 overexpression results in an increase of mitotic cell population. UBE2C acts in G2/M checkpoint control, and plays a fundamental role in the maintenance of genetic stability by regulating the degradation of securin, a protein that impairs the premature segregation of chromosomes by binding to and inhibiting the enzyme separase [257]. However, it is noteworthy that there are several other mechanisms that lead to overexpression of UBE2C in addition to upregulation of FoxM1. A recent study identified CLDN19 as an upstream negative regulator of UBE2C in breast cancer cells [283]. CLDN19 expression was significantly reduced in breast tumor tissues, and was associated with UBE2C overexpression and with poor patient survival. UBE2C upregulation has previously been shown to drive EMT and promote gastric cancer metastasis via activation of the Wnt/β-catenin and PI3K/Akt signaling pathways [250]. Notably, Xu et al. [48] demonstrated that in breast cancer cells with low CLDN19 expression, the extracellular matrix (ECM) is a 3D environment that can activate Wnt signaling. Under normal conditions, CLDN19 suppresses UBE2C, thereby preventing ECM-induced activation of the Wnt/β-catenin oncogenic pathway. These findings suggest that CLDN19 inhibits UBE2C-mediated Wnt signaling activation in response to ECM cues in three-dimensional or in vivo contexts. Interestingly, among TNBCs, we found using the bc-GenexMiner online tool that there is a statistically significant negative correlation between the expression of UBE2C and CLDN19 (r = −0.45, p<0.0001, n = 293), and a positive correlation between the expression of AR and CLDN19 (r = 0.28, p<0.0001, n = 293), suggesting that a similar regulatory circuit may also function in AR-low TNBCs.
Epigenetic mechanisms can also play a role in regulating UBE2C levels. In breast cancer cells, miR-196a promotes cell proliferation by upregulating UBE2C [284]. Although the upregulation of mRNA translation via the binding of a miRNA is somewhat unusual, there is precedence for such epigenetic mechanisms of regulation [285,286,287]. Another recent study found that Circ_0059457 had elevated expression in breast cancer tissues and cells, and circ_0059457 knockdown inhibited breast cancer cell proliferation, metastasis, sphere formation ability, and glycolysis [288]. The authors found that UBE2C was a target of downregulation by miR-140-3p, and overexpressed circ_0059457 sponged up miR-140-3p, resulting in UBE2C overexpression [288]. In TNBC, researchers found that LINC01194 promotes the malignant progression by activating the Wnt/β-catenin signaling pathway [289]. Mechanistically, LINC01194 recruits NUMA1 to bind the 3'UTR of UBE2C mRNA, enhancing the mRNA’s stability. In turn, UBE2C stimulates the ubiquitination and degradation of RYR2, a negative regulator of Wnt/β-catenin signaling. This regulatory axis—LINC01194/NUMA1–UBE2C–RYR2—thus drives excessive Wnt pathway activation and TNBC progression [289]. Inhibition of UBE2C sensitizes breast cancer cells to radiation, doxorubicin, and even hormone blocking agents, highlighting the importance of UBE2C as a potential therapeutic target [290]. The potential of UBE2C expression as a readout of proteasome activity, and as a predictive biomarker for therapeutics that target the proteasome, also merits further study.
UBE2S
The human Ub-binding enzyme E2S (UBE2S), is associated with the APC/C and is essential for the Ub conjugation and elongation of Ub chains on target substrate proteins destined for the 26S proteasome [291]. Once UBE2C attaches ubiquitin onto K11 in the target proteins, UBE2S promotes the elongation of ubiquitin chains thereby enabling substrate degradation to proceed. UBE2S executes pivotal functions during mitosis as it is responsible for the timely ubiquitination and degradation of several cell cycle regulators including mitotic cyclins, thereby enabling exit from mitosis [291,292]. UBE2S becomes particularly important for efficient substrate degradation when APC/C activity has been compromised by prolonged spindle-assembly checkpoint (SAC) arrest. The APC/C ubiquitin ligase is the target of the SAC, as it ubiquitinates protein substrates whose degradation regulates progression through and exit from mitosis. Indeed, following release from SAC arrest, UBE2S-depleted cells neither degrade crucial APC/C substrates, nor silence this checkpoint, whereas SAC bypass via BUBR1 depletion or Aurora-B inhibition negates the requirement for UBE2S [291]. Studies demonstrate that cells with high UBE2S expression degrade APC/C substrates more efficiently, facilitating mitotic exit even when it is not appropriate [291]. UBE2S also modulates the stability of the tumor suppressor VHL (Von Hippel-Lindau) under normoxic conditions by promoting VHL’s ubiquitination and degradation [293,294]. VHL destruction then promotes metastasis and proliferation through the VHL/HIF-1α/STAT3 pathway [295]. UBE2S is itself an unstable protein degraded through the proteasome pathway [296]. Together, these findings position UBE2S as a critical orchestrator of mitotic progression and tumor-promoting pathways, whose dysregulation may drive unchecked cell division.
Recent studies have shown that UBE2S is also overexpressed in several cancers and this overexpression is associated with cancer progression, resistance to chemotherapy, and poor prognosis. Aberrantly high expression of UBE2S was observed in cervical cancer, ovarian cancer (where high UBE2S upregulates Wnt/β-catenin signaling, leading to resistance to olaparib in vitro and in vivo, and enhanced proliferation triggered by upregulation of PI3K/AKT/mTOR signaling), breast cancer, oral squamous cell carcinoma (where it promotes the degradation of p53 target, p21), endometrial cancer (where it downregulates Sox6 expression and promotes β-catenin signaling, cell migration, and proliferation), hepatocellular carcinoma (where it enhances the proteolysis of the tumor suppressor, p53), colorectal cancer (where it extends the half-life and promotes the accumulation of β-catenin), and kidney cancer [297,298,299,300,301,302,303,304,305]. In non-small cell lung cancer, UBE2S overexpression was associated with greater metastasis and poorer outcomes, and upregulated the expression of β-catenin, cyclin D1, and MMP7; pharmacological inhibition of Wnt/β-catenin signaling effectively abolished these effects [305]. In lung adenocarcinoma, overexpression of UBE2S leads to enhanced degradation of I-κB proteins that sequester NF-κB dimers in the cytoplasm; the result is increased nuclear translocation of p65/RelA and excessive activation of the NF-κB signaling [306]. In glioblastoma multiforme (GBM), Akt1 physically interacts with and phosphorylates UBE2S at Thr 152, to enhance the latter’s stability. UBE2S also participates in NHEJ-mediated DNA repair process, and thus modulates sensitivity to chemotherapy in GBM [263]. In hepatocellular carcinoma cells, overexpression of UBE2S promoted proliferation, invasion, metastasis, and G1/S phase transition in vitro, and promoted tumor growth significantly in vivo. Mechanistically, UBE2S can enter the nucleus through its nuclear localization signal, where it interacts with TRIM28 (an E3 Ub-ligase); these two proteins work together to enhance the ubiquitination and degradation of the CDK inhibitor p27, thereby promoting cell cycle entry and progression [307]. Thus, UBE2S overexpression drives aggressive tumor biology through a variety of downstream pathways.
UBE2S overexpression is driven by MMB-FoxM1 and E2F2, which bind to its promoter region to enhance transcriptional activity. FOXM1, in particular, has been linked to regulating UBE2S mRNA and protein levels, and FOXM1 knockdown in HCC cells suppresses UBE2S mRNA level, FOXM1 pathway, AKT phosphorylation pathway, and protein ubiquitination regulation. Genes involved in processing of cytotoxic agents and other xenobiotics were enriched in high-UBE2S expression group, which was similar with that in high-FoxM1 expression group based on TCGA-LIHC database, which further supported the idea that UBE2S is upregulated by FOXM1 and increases chemoresistance in HCC cells. Overall, UBE2S is considered to be a critical component of the pathway by which FoxM1 modulates resistance to cytotoxic agents in HCC cells, and potentially in other cancer types [308]. Overexpression of UBE2S is also associated with radiotherapy resistance because of UBE2S’s involvement in DNA damage repair [296]. Inhibition of UBE2S enhances the susceptibility of cervical cancer HeLa cells to etoposide and adriamycin, and heightens the chemosensitivity to topotecan [296].
In breast cancer cells, UBE2S depletion induces changes in cellular morphology and significantly disrupts the formation of actin stress fibers and focal adhesions. UBE2S knockdown suppressed cell spreading, migration, and invasion [301]. In breast cancer, UBE2S overexpression was strongly associated with resistance to Topoisomerase II inhibitors [309]. UBE2S is a component of the 4-gene Ub-related genes signature that was found to be an independent risk factor for poor overall survival among BC patients [310]. UBE2S was also one of the genes associated with improved response to neoadjuvant chemotherapy in breast cancer [311]. In the context of TNBC, UBE2S promotes proliferation and survival, particularly under conditions of prolonged SAC activation [312].
UBE2T
Another ubiquitin-conjugating enzyme E2T (UBE2T), plays a pivotal role in facilitating the repair of DNA damage, including within the Fanconi anemia pathway, with evidence suggesting that regulation of this pathway may occur through a mechanism involving UBE2T self-inactivation [313]. UBE2T monoubiquitinates several proteins of this pathway, including ANCD2 and FANCI [314]. UBE2T also interacts with and colocalizes with the BRCA1/BRCA1-associated RING domain protein (BARD1) complex, and is involved in BRCA1 downregulation [315]. Beyond DNA repair, UBE2T-driven ubiquitination influences multiple cancer-associated signaling pathways: ubiquitination of AKT by UBE2T activates the AKT/β-catenin signaling cascade [236], while ubiquitination of β-catenin promotes β-catenin’s nuclear localization [316]. Furthermore, in lung adenocarcinoma, UBE2T enhances autophagy through modulation of the p53/AMP-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR) axis [317].
UBE2T is overexpressed in breast cancer, and its overexpression predicts poor prognosis [318]. In breast cancer cells, suppression of UBE2T activity leads to downregulation of interferon alpha–inducible protein 6 (IFI6), which in turn triggers DNA replication stress, halts cell cycle progression, and promotes apoptosis. Conversely, forced expression of IFI6 in UBE2T-deficient cells mitigates replication stress and apoptotic signaling, partially rescuing cell proliferation. Moreover, UBE2T mediates resistance to DNA replication stress, apoptosis, and chemotherapeutic agents, and inhibition of UBE2T augments the growth-inhibitory effects of agents that induce DNA replication stress [318]. Our analysis using the bc-GenexMiner platform showed that among TNBCs, the expression of IFI6 showed a positive correlation with the expression of UBE2T (r = 0.22, p<0.0001, n = 293), and a negative correlation with the expression of AR (r = −0.22, p<0.0001, n = 293), suggesting that this pathway may function among AR-low TNBCs.
A recent study that examined breast tumors with an extreme dependence on DNA repair machinery described a DNA damage transcriptomic signature that discriminates patients with poor prognosis. Using transcriptomic mapping combined with gene set enrichment analysis, the authors identified DNA repair networks that are in overdrive in breast cancer. UBE2T was among five genes (a) that were found to be amplified across all breast cancer subtypes, and (b) whose overexpression was consistently associated with unfavorable clinical outcomes regardless of molecular subtype [319]. Another study utilized a novel strategy for predicting risk of breast cancer metastasis and identifying key genomic biomarkers through the integration of machine learning (ML) and explainable artificial intelligence (XAI), especially with the SHapley Additive exPlanations (SHAP) framework, to enhance interpretability and address the “black box” limitation of complex ML systems. Using genomic profiles from primary BC samples, the authors developed a high-accuracy predictive model and pinpointed genes linked to either elevated or reduced metastatic risk. Elastic net feature selection reduced the initial dataset of 24,481 genes to a focused panel of 18 strong biomarker candidates for BC metastasis. Among these, elevated UBE2T expression correlated with an increased likelihood of metastatic progression [320].
UBE2T was also identified as a key regulator of breast cancer stem cell (BCSC) stemness [101]. Genetic ablation of UBE2T markedly impaired BCSC stemness. The authors uncovered that UBE2T partners with the E3 ubiquitin ligase TRIM25 to catalyze polyubiquitination and degradation of CBX6. Reduced CBX6 levels facilitate transcriptional activation of SOX2 and NANOG (which are transcription factors included among the “core pluripotency network" that regulates stem cell self-renewal and differentiation), thereby enhancing BCSC stemness. These findings define the UBE2T–TRIM25–CBX6 signaling axis as a critical modulator of BCSC stemness [101]. Aberrantly high levels of UBE2T in breast cancer also enhance tumor growth, proliferation, migration, invasion, and glycolysis through the PI3K/AKT signaling pathway [321].
In TNBC, UBE2T overexpression has been reported to promote brain metastases; UBE2T upregulation is therefore associated with poor prognosis [322]. Normally, a CDC42-mediated autophagy-dependent pathway promotes the trafficking of the immune checkpoint protein CD276 to lysosomes for degradation; by mediating the ubiquitination and proteasomal degradation of the Rho GTPase CDC42, UBE2T overexpression increases CD276 levels, impairs the antitumor activity of CD8+ T cells, and enables tumor immune escape. Inhibition of UBE2T raises the TNBC sensitivity to immune checkpoint blockade and suppresses breast cancer brain metastases [322]. Higher UBE2T expression levels were predictive of a lower rate of pathological complete response in TNBC patients following neoadjuvant chemotherapy [323].
In addition to transcriptional upregulation via the MMB-FoxM1 pathway, UBE2T levels are also modulated by epigenetic mechanisms. One such mechanism involves micro RNAs: a recent study found that miR-543 exhibited reduced expression in breast cancer tissues and cell lines, whereas UBE2T exhibited abnormally high levels. miR-543 normally downregulates UBE2T expression through the ERK/MAPK pathway, and miR-543 overexpression leads to inhibition of viability, proliferation, migration, and invasion of breast cancer cells [324]. Thus, UBE2T is a promising therapeutic target for breast cancers in general.
PSMD14
The 26S proteasome non–ATP regulatory subunit 14 PSMD14 (also known as RPN11 and POH1) is a deubiquitinating enzyme that plays a significant role in homeostasis, and is implicated in differentiation [325], pluripotency [326], the DNA damage response [327], cellular proliferation and senescence [328], and the pathology of various cancers, including liver, esophageal, and breast cancer [295,329,330,331,332], where PSMD14 overexpression is associated with adverse clinical outcomes. Amplification of the PSMD14 gene can lead to increased expression levels. This amplification is often associated with CIN in cancer cells [100,333]. PSMD14 is known to be a component of the 19S complex of the proteasome and is responsible for substrate deubiquitination. Currently, about a 100 deubiquitinases are known to be encoded by the human genome [334,335]. PSMD14 knockdown caused cell arrest in the G0-G1 phase, and ultimately led to senescence in a broad range of cancer cell lines [328]. A recent study identified PSMD14 as a key regulator of the stability of IRF3 (a critical transcription factor in antiviral innate immune signaling) and type I interferon (IFN) signaling [336]. PSMD14 counteracts NDP52-mediated, virus load–dependent autophagic degradation of IRF3 by deubiquitinating IRF3, thereby preserving basal IRF3 levels and sustaining IFN activation [336].
In ER+ breast cancer, PSMD14 is an ERα deubiquitinase; interestingly, ERα binds to the promoter of PSMD14 and stimulates the latter’s transcription. This positive feedback loop implies that PSMD14 behaves both as an upstream modulator as well as downstream target for ERα signaling in breast cancer [337]. PSMD14 is one of seven genes that comprise a multivariable prognostic model in breast cancer; these 7 genes were identified as being DNA damage response-related genes that were differentially expressed between high- and low-Tumor mutational burden (TMB) breast tumors in the TCGA dataset, and whose overexpression was associated with poor outcomes [338].
In triple-negative breast cancer (TNBC), PSMD14 is markedly upregulated and drives tumor cell proliferation, migration, and invasion [339]. Silencing PSMD14 significantly reduced TNBC growth and metastasis. Mechanistically, PSMD14 deubiquitinates SF3B4 (an RNA-binding protein, or RBP, involved in pre-mRNA splicing), stabilizing the protein and enabling it to form a complex with heterogeneous nuclear ribonucleoprotein complex (HNRNPC), which acts both as an RBP, as well as an m6A reader. The SF3B4-HNRNPC complex then binds m6A-modified FADS1 mRNA, promoting exon 10 inclusion and upregulating FADS1 expression. Elevated FADS1, in turn, activates the Akt/mTOR pathway [339]. Another study found that PSMD14 overexpression also promotes aggressive TNBC phenotypes by modulating ferroptosis sensitivity [224]. Mechanistically, PSMD14 suppresses arachidonic acid metabolism–induced ferroptosis through activation of the E2F1/CENPF signaling axis. Silencing E2F1 downregulates CENPF expression, and CENPF knockdown markedly impairs TNBC cell migration and invasion and enhances ferroptosis cell death. By sustaining E2F1-dependent CENPF expression, PSMD14 acts as a central driver of TNBC progression through ferroptosis inhibition and metastatic pathway activation [224].
TUBA1B
Tubulin α-1b chain (TUBA1B) is an important α-tubulin isoform; this protein is therefore a major component of the microtubule cytoskeleton, and plays critical roles during chromosome segregation and mitotic progression. TUBA1B has also been shown to play multifaceted oncogenic roles, influencing cytoskeletal remodeling, protein degradation, immune cell infiltration, tumor growth, chemoresistance, apoptosis, and patient survival across various malignancies. TUBA1B’s expression levels were significantly elevated in several cancer types, including breast cancer, compared to the corresponding healthy tissue levels [101]. TUBA1B has been observed to impact immune cell infiltration within the tumor microenvironment of liver hepatocellular carcinoma and influence patients’ responsiveness to immunotherapy, or resistance to paclitaxel, thereby significantly affecting their prognosis [340,341]. In glioma, TUBA1B is a key driver of disease progression, with high expression correlating with poor prognosis and aggressive tumor behavior. TUBA1B has been identified as a central gene involved in aggrephagy—a specialized type of lysosome-dependent autophagy that targets and removes misfolded or aggregated proteins [342]. These aberrant proteins, often arising from mutations or cellular stress, are marked as defective and must be cleared to avoid toxic buildup. Normally, the Ub–proteasome system handles the breakdown of such proteins; however, when protein aggregation occurs, the Ub-proteasome system may be unable to process them efficiently. In these situations, aggrephagy serves as an alternative degradation mechanism, serves to clear misfolded protein condensates, and contributes to cancer cell homeostasis [342]. High TUBA1B expression in glioma is linked to increased cell proliferation, migration, autophagy, and apoptosis. Immune profiling indicates an association with cancer-associated fibroblasts and diverse immune cell infiltrates, suggesting a role in shaping the tumor microenvironment. Analysis of TCGA data, stratified by TUBA1B expression, revealed that TUBA1B-high tumors exhibit significant upregulation of cell cycle–related pathways and genes, as shown by KEGG and GSEA analyses [342]. Functionally, TUBA1B enhances glioma growth in xenograft models, promotes tumor stemness, and reduces responsiveness to immunotherapy. These findings indicate that TUBA1B influences glioma biology through (i) intrinsic cell cycle regulation, (ii) enabling cancer cells to better cope with proteotoxic stress, and (iii) by modulating the immune milieu [342]. In sum, TUBA1B overexpression fuels tumor aggressiveness by simultaneously driving intrinsic tumor growth programs, sustaining therapy-resistant populations, and shaping an immune-suppressive, tumor-promoting microenvironment—while bolstering the cell’s ability to cope with stress through enhanced proteostasis mechanisms.
Taken together, overexpression (due to mutations or copy number alterations) of these Ub-proteasome- and aggrephagy-related genes may result in dysregulation of substrate degradation kinetics and proper mitotic progression, undermine normal mitotic checkpoints and safeguards, compromise DNA repair fidelity, and disrupt the balance between protein degradation and stabilization. This convergence contributes to the etiology and progression of cancer, and promotes ongoing CIN, which fuels tumor evolution, intra-tumoral heterogeneity, and therapy resistance. Three out of the five protein degradation-related genes we focused on were found in the list of top 100 most overexpressed genes among high-functional aneuploidy breast tumors in TCGA [163]: UBE2C ranked 6th, UBE2T was ranked 46th, and UBE2S was ranked 47th in this list. The CIN75 gene signature includes UBE2C [160]. Thus, AR-low TNBCs that overexpress these genes are likely to exhibit high levels of aneuploidy and CIN, which is a fuel for aggressive tumor phenotypes.
13. Regulators of Protein Degradation Play Prominent Roles in Driving Poor Outcomes in AR-Low TNBC and TP53-Mutant Breast Tumors
We identified five proteins that play key roles in Ub-related protein degradation and aggrephagy—UBE2S, UBE2C, UBE2T, PSMD14, and TUBA1B—that are significantly overexpressed in AR-low TNBC. The expression levels of these genes correlate strongly with proliferation markers not only in TNBC (Fig. 1) but across breast tumors more broadly (Suppl. Fig. 2). Moreover, these proteins are elevated in both AR-low TNBC (Suppl. Fig. 1) and in AR-low breast cancers overall (Suppl. Fig. 3). These indications prompted a more detailed examination of their expression profiles.
Data from the UALCAN dataset showed that UBE2C (Fig. 4A), UBE2S (Fig. 4B), UBE2T (Fig. 4C), PSMD14 (Fig. 4D), and TUBA1B (Fig. 4E) were all significantly upregulated in breast tumor samples relative to normal tissue. To determine the prognostic relevance of this pattern of overexpression, we assessed relapse-free survival for each gene individually using Cox proportional hazards models in the Kaplan–Meier Plotter tool (Győrffy, 2021). Cutoff points were optimized to achieve the lowest logrank p-values, and survival curves demonstrated that elevated expression of each of these five proteins of interest (Figs. 4F–4J) was associated with a significantly shorter recurrence-free interval, suggesting roles in disease progression. We also compared the expression levels of these genes in breast cancer patients categorized by self-declared race in 1,102 annotated breast tumor samples from UALCAN. The expression levels of UBE2C, UBE2S, UBE2T, and TUBA1B were notably higher in African-American patients than in Caucasian/White patients. Collectively, these findings indicate that in breast cancer—particularly in AR-low TNBC—a subset of proteins that play key roles in protein degradation and cell cycle progression is consistently overexpressed, with higher expression correlating with worse outcomes. Further analysis revealed that all five proteins—UBE2C (Fig. 4P), UBE2S (Fig. 4Q), UBE2T (Fig. 4R), PSMD14 (Fig. 4S), and TUBA1b (Fig. 4T)—were significantly elevated in TP53-mutant tumors compared with TP53–wild-type tumors, consistent with prior results showing FOXM1 overexpression in the TP53-mutant setting [13]. These observations were validated using the muTarget platform, which compared gene expression patterns in in 305 TP53-mutant and 674 TP53-wild-type breast cancers from TCGA. The TP53-mutant group exhibited substantial increases in UBE2C (2.30-fold, p = 1.76 × 10⁻4⁸), UBE2S (1.69-fold, p = 5.60 × 10⁻27), UBE2T (1.61-fold, p = 4.72 × 10⁻30). No data was available for analysis of PSMD14 and TUBA1B levels in this platform. Together, these data strongly support that AR-low breast cancers (including AR-low TNBC), Ki67-high tumors, and TP53-mutated breast tumors share a common phenotype of elevated expression of these proteolysis-regulatory proteins. To assess whether reduced promoter methylation contributes to the elevated expression of these proteolysis-related genes, we employed the UALCAN promoter methylation analysis platform (Figure 4U–Y). In this analysis, the beta value reflects the degree of methylation, ranging from 0 (completely unmethylated) to 1 (fully methylated). Results showed significant promoter hypomethylation for UBE2T and TUBA1B in breast tumors, suggesting that epigenetic mechanisms may contribute the overexpression of these two genes in addition to transcriptional activation driven by regulators such as the MMB–FOXM1 complex. Collectively, these findings indicate that AR-low breast cancers—especially AR-low TNBC—as well as Ki67-high and TP53-mutant breast tumors, exhibit a coordinated overexpression of UBE2C, UBE2S, UBE2T, PSMD14, and TUBA1B, underscoring their likely roles in driving tumor progression and unfavorable clinical outcomes, via a dysregulation of proteolysis pathways.
14. Overexpression of the 15-Gene Set Associated with Proliferation and Genomic Instability is Associated with a characteristic Tumor Microenvironment in Breast Cancer
The tumor microenvironment (TME) is a dynamic and heterogeneous ecosystem composed of immune cells, fibroblasts, adipocytes, and vascular networks. These non-malignant components either promote or restrain anti-tumor immunity through soluble factors, direct cellular interactions, and adaptive phenotypic shifts. Characterizing the immune landscape of the TME is essential for understanding patterns of immune infiltration, their association with gene expression profiles, and their influence on tumor progression. Moreover, the presence and activity of stromal and immune cells within the TME hold prognostic value and may inform the likelihood of response to specific therapeutic strategies.
To gain insights into the composition of the TME in breast tumors that overexpress the 15 genes of our interest, three complementary deconvolution algorithms—TIDE, XCell, and CIBERSORT—were utilized via the TIMER platform (http://timer.cistrome.org/). We examined the Spearman correlation between the expression of the 15 cell cycle regulators at the core of our study, and the infiltration landscape of the TME in breast cancer [125,343] (Fig. 5A–C). Despite methodological differences among the platforms, several convergent patterns emerged, underscoring a consistent pattern of association between gene expression and TME composition.
The Tumor Immune Dysfunction and Exclusion (TIDE) algorithm [344] models two key mechanisms of tumor immune evasion: (i) T cell dysfunction, by detecting prognostic gene expression-based signatures indicative of loss of T cell effector functions within a high cytotoxic T lymphocyte (CTL) tumor environment, and (ii) T cell exclusion, by identifying expression profiles of cells that suppress T cell infiltration, such as cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), and M2 (tumor-associated) macrophages. TIDE analysis (Fig. 5A) revealed that expression of the 15-gene set was positively correlated with CTLs, MDSCs, interferon-γ (IFNG), and CD274 (PD-L1). Expression of the 15 genes was negatively correlated with M2 macrophages, microsatellite instability (MSI) score, and immune dysfunction score. We found no statistically significant association between the expression of the 15 genes, and the Immune Evasion Score. CTLs play a crucial role in directly attacking and eliminating cancer cells [345]; however, exhaustion and dysfunction can occur during cancer progression due to immunosuppression within the TME. CAFs, alternatively activated M2 macrophages, and regulatory T cells can constitute serious immunologic barriers against CTL antitumor immune responses, eventually resulting in CTLs’ exhaustion. A higher number CD8+ CTLs is generally indicative of a favorable response to neoadjuvant therapy in various breast cancer subtypes [346]. Furthermore, studies have revealed that a higher percentage of CD8+ T cells are recruited in the tumor of African-American breast cancer patients compared to their White counterparts, which is suggestive of the mounting of strong T-cell-mediated immune response in African-American patients [347]. MDSCs represent a diverse population of immune cells that mediate tumor-associated immunosuppression. Within the TME, their principal function is to inhibit T-cell activity, either through antigen-dependent mechanisms or via non-specific pathways. Therefore, MDSCs serve as key drivers of T-cell dysfunction and exhaustion [348]. CD274, also known as PD-L1 (programmed cell death-ligand 1), is a transmembrane protein that interacts with the PD-1 (programmed cell death protein 1) receptor on T cells. This interaction inhibits T cell activation and cytokine production. CD274 expression on tumor cells can help them evade the immune system by suppressing T cell activity, making the PD-1—PD-L1 interaction a target for cancer immunotherapy. Interferon-gamma (IFNG or IFN-γ) is a pivotal cytokine in the TME with dual roles in anti-tumor immunity and immune regulation. Produced mainly by activated CD8⁺ CTLs, NK cells, and Th1 CD4⁺ T cells, IFN-γ enhances tumor antigen recognition by upregulating MHC class I and II expression on tumor and antigen-presenting cells, and it activates macrophages to exert cytotoxic effects against tumor cells [349,350]. However, chronic or excessive IFN-γ signaling can paradoxically promote immune escape by inducing immune checkpoint molecules such as PD-L1 on tumor and stromal cells [351,352,353]. Thus, IFN-γ functions as a double-edged sword—bolstering tumor clearance while simultaneously fostering adaptive resistance mechanisms. IFNG expression and CD274 (PD-L1), are both markers of an inflamed TME. Expression of our 15-gene set was further associated with increased levels of MDSCs, suggesting that the inflammatory context may be counterbalanced by the recruitment of immunosuppressive myeloid populations. Conversely, expression of our 15-gene set demonstrated a negative correlation with M2 macrophages, as well as with both the MSI score and the immune dysfunction score. A low TIDE dysfunction score suggests that the tumor microenvironment may be more amenable to immune activity, possibly with less T-cell exhaustion. However, this alone does not guarantee a good prognosis. Cancer prognosis is influenced by a complex interplay of tumor biology, host factors, and treatment effectiveness. Tumor intrinsic mutations can drive rapid growth, metastasis, or resistance to treatments, regardless of the immune response. A negative correlation between the expression of our 15 genes and the TIDE dysfunction score indicates a potentially more favorable immune environment, suggesting better chances of response to immunotherapy. These results also suggest that the TME of AR-low/TP53-mutant breast tumors expressing this gene program differs from the type of TME typically associated with immune-excluded cancers. Overall, expression of the proliferative 15-gene set correlates with features of both immune activation (CD8⁺ T cells, IFNG, PD-L1) and immune suppression (MDSCs), while showing inverse associations with M2 macrophages and immune dysfunction. The strength and direction of these correlations reinforce the notion that breast tumors expressing high levels of mitotic kinesins, centromeric proteins, and proteolysis regulators may attract effector T cells, but they also engage immunosuppressive myeloid populations and likely exploit immune checkpoint pathways.
We next interrogated the relationship between the 15-gene set of interest and immune cell composition using the XCELL algorithm (Fig. 5B). XCELL is an R-based program that provides a more comprehensive view of the complex cellular landscape, using pre-defined gene signatures to assess the enrichment of each cell type [354]. Macrophages in the TME play a complex and multifaceted role, influencing tumor growth, progression, and response to therapy [355]. Our analysis revealed a positive correlation with innate and antigen-presenting immune subsets, including total macrophages, M1 macrophages, and myeloid dendritic cells (both total and activated). By contrast, expression of the 15-gene set demonstrated negative correlations with M2 macrophages. M1 macrophages are generally associated with anti-tumor functions. They secrete pro-inflammatory cytokines like IL-12 and TNF-α, and chemokines, such as CXCL9, CXCL10, and CXCL11, which can stimulate other immune cells like CD8+ T cells and NK cells to kill tumor cells. They also directly mediate tumor cell cytotoxicity and antibody-dependent cell-mediated cytotoxicity. M1 macrophages can inhibit cell proliferation and cause tissue damage through the secretion of pro-inflammatory cytokines and nitric oxide (NO). Some studies, like one focusing on transcriptomically defined M1 macrophages in breast cancer [356], did not find an association between high M1 levels and favorable survival or response to chemotherapy. It appears that the balance of M1 to M2 macrophages, as well as the overall context of the TME, contribute to the ultimate impact on the tumor's progression. In addition, we observed significant positive correlations with common lymphoid progenitors, CD4⁺ memory T cells, and CD4⁺ Th2 cells, suggesting that tumors expressing these 15 genes are enriched for cell types associated with pro-inflammatory activity and adaptive immune priming. It was also inversely associated with several other immune and stromal compartments, including B cells, CAFs, endothelial cells, hematopoietic stem cells, common myeloid progenitors, CD4⁺ central memory T cells, and CD4⁺ effector memory activated T cells. Negative correlations with endothelial cell abundance further implicate these tumors in fostering a TME that is not conducive to angiogenesis. These negative associations indicate that expression of the proliferative gene program coincides with reduced B cell infiltration, lower stromal support, and depletion of certain memory T cell subsets, potentially altering the quality and persistence of immune responses in the TME. Together, these XCELL-derived correlations suggest that tumors with high expression of the 15-gene signature foster a distinct immune milieu characterized by M1 macrophage and dendritic cell predominance, enrichment of CD4⁺ subsets, but reduced stromal and endothelial support and an absence of immunosuppressive M2 macrophage infiltration.
CIBERSORT analysis (Fig. 5C) yielded trends consistent with those observed with TIDE and XCELL: expression of the 15 genes was positively correlated with M0 and M1 macrophages and CD4⁺ activated memory T cells, but negatively correlated with M2 macrophages, monocytes, and activated mast cells. These findings emphasize a skewing of the myeloid compartment toward pro-inflammatory macrophages, coupled with reduced representation of regulatory and tolerogenic subsets. Taken together, the results across all three algorithms converge on a model in which high expression of mitotic kinesins, centromeric proteins, and proteolysis regulators in AR-low TNBC and p53-deficient breast tumors is associated with a TME enriched for effector and pro-inflammatory immune subsets, but with simultaneous evidence of immune evasion pathways (MDSC infiltration, PD-L1 upregulation, loss of immune-stimulatory diversity). This duality may help explain how AR-low TNBC and TP53-mutant tumors maintain aggressive proliferation despite the presence of immune activity, underscoring both the therapeutic vulnerabilities and the adaptive complexity of their TME.
Figure 5.
Tumor Microenvironmental features associated with the 15-Gene set in breast cancer (A) Spearman correlations between expression of the 15-gene cell cycle regulatory gene set and immune cell infiltration, estimated using the TIDE algorithm via the TIMER 2.0 platform. Only statistically significant pairwise correlations are depicted (p<0.05). Statistically significant negative correlations are depicted in shades of blue, and statistically significant positive correlations are depicted in shades of red. White boxes represent the absence of a statistically significant correlation. (B) Spearman correlations between expression of the 15-gene cell cycle regulatory gene set and immune cell infiltration, estimated using the XCELL algorithm via the TIMER 2.0 platform. Only statistically significant pairwise correlations are depicted (p<0.05). Statistically significant negative correlations are depicted in shades of blue, and statistically significant positive correlations are depicted in shades of red. White boxes represent the absence of a statistically significant correlation. (C) Spearman correlations between expression of the 15-gene cell cycle regulatory gene set and immune cell infiltration, estimated using the CIBERSORT algorithm via the TIMER 2.0 platform. Only statistically significant pairwise correlations are depicted (p<0.05). Statistically significant negative correlations are depicted in shades of blue, and statistically significant positive correlations are depicted in shades of red. White boxes represent the absence of a statistically significant correlation.
Figure 5.
Tumor Microenvironmental features associated with the 15-Gene set in breast cancer (A) Spearman correlations between expression of the 15-gene cell cycle regulatory gene set and immune cell infiltration, estimated using the TIDE algorithm via the TIMER 2.0 platform. Only statistically significant pairwise correlations are depicted (p<0.05). Statistically significant negative correlations are depicted in shades of blue, and statistically significant positive correlations are depicted in shades of red. White boxes represent the absence of a statistically significant correlation. (B) Spearman correlations between expression of the 15-gene cell cycle regulatory gene set and immune cell infiltration, estimated using the XCELL algorithm via the TIMER 2.0 platform. Only statistically significant pairwise correlations are depicted (p<0.05). Statistically significant negative correlations are depicted in shades of blue, and statistically significant positive correlations are depicted in shades of red. White boxes represent the absence of a statistically significant correlation. (C) Spearman correlations between expression of the 15-gene cell cycle regulatory gene set and immune cell infiltration, estimated using the CIBERSORT algorithm via the TIMER 2.0 platform. Only statistically significant pairwise correlations are depicted (p<0.05). Statistically significant negative correlations are depicted in shades of blue, and statistically significant positive correlations are depicted in shades of red. White boxes represent the absence of a statistically significant correlation.
15. Model: A FoxM1–WDR5–ASPM Regulatory Axis Drives Proliferation and Chromosomal Instability in AR-Low TNBC and TP53-Mutant Breast Cancer
Our analyses and data in the literature support the following model of the dysregulation that undergirds the high-proliferation phenotype of AR-low TNBC: Normally, the expression of G1/S and G2/M cell cycle genes is tightly controlled by dynamic transcriptional complexes, ensuring orderly DNA replication and mitotic progression. However, in AR-low TNBC and TP53-mutant breast tumors, this tight regulatory balance is disrupted, and induces a transcriptional program that normally regulates proliferation, to be pushed into overdrive. Central to this dysregulation is upregulation of a collaborative axis formed by (a) the transcription factor FoxM1, (b) WDR5—a core subunit of histone methyltransferase complexes that catalyzes H3K4me3, and (c) the mitotic protein ASPM. When overexpressed, these 3 proteins drive the persistent upregulation of a set of genes including 15 “core cell cycle-related genes” essential for proliferation. WDR5 epigenetically modifies the chromatin landscape of the FoxM1 promoter to license persistent FoxM1 transcription, enabling tumor cells to accumulate high levels of this oncogenic transcription factor. Our analyses showed that the expression of WDR5 shows a statistically significant negative correlation with the expression of AR (r = −0.40, p<0.001), and a statistically significant positive correlation with the expression of FoxM1 (r =0.60, p<0.001), among TNBCs. While high levels of WDR5 transcriptionally ensure FoxM1 abundance, ASPM stabilizes FoxM1 protein and amplifies FoxM1 oncogenic activity through (i) phase separation into nuclear condensates that sustain persistent G2/M transcriptional activity, and (ii) feedback regulation. Furthermore, FoxM1 transcriptionally activates ASPM, creating a positive feedback loop in which each protein sustains the other. Consistent with this, we found that the expression of ASPM shows a statistically significant negative correlation with the expression of AR (r = −0.50, p<0.001), and a statistically significant positive correlation with the expression of FoxM1 (r =0.81, p<0.001), among TNBCs. Once upregulated, FoxM1 binds to the MMB-MuvB complex and is activated through phosphorylation by CDK1 and PLK1. This active MMB–FoxM1 complex drives robust transcription of G2/M genes, including kinesins (KIF11, KIF14, KIF4A, KIF2C, KIF20A), centromere-associated proteins (CENPA, CENPO, CENPL, CENPF, OIP5), and cell cycle proteolysis regulators (UBE2C, UBE2S, UBE2T, PSMD14). In healthy cells, the precise regulation of these highly consequential target genes ensures accurate chromosome partitioning and genomic stability. In tumors however, the overexpression of these effectors can promote unchecked proliferation accompanied by erroneous chromosome segregation, and fuel CIN and aneuploidy—processes strongly linked to tumor evolution, therapy resistance, and poor patient survival. ASPM itself regulates spindle orientation and microtubule minus-end dynamics, and its aberrantly elevated expression further compounds the chromosome mis-segregation driven by FoxM1 target gene overexpression. In this context, (i) loss of AR-mediated FoxM1 repression (via SPDEF), and (ii) disruption of the p53–p21–DREAM axis that normally restrains FoxM1 expression, further spike FoxM1 transcription, and increase production of its target effectors. Normally, p53 serves as the “guardian of ploidy”, eliminating aneuploid cells through apoptosis or senescence. In TP53-deficient tumors, however, cells with segregation defects survive and propagate. Collective upregulation of the 15 MMB-FoxM1 target genes we studied (and synergy with p53 deficiency, if it exists) locks cells into a high-proliferation state, with extensive CIN and aneuploidy, and engenders heightened transcriptional chaos and intra-tumoral heterogeneity, that underlie the aggressive tumor biology of AR-low TNBC and TP53-mutant breast cancers.
16. Perspectives
Breast cancers with high proliferation and low AR-related signaling have poor prognosis and unique molecular features with implications for therapy. This study aimed to build a granular portrait of AR-low TNBCs by diving deep into and contextualizing the patterns of dysregulation that drive and support their state of high proliferation. By enhancing our understanding of the processes and drivers undergirding the biology of these tumors, we hoped to unlock insights that will help better manage them. We uncovered that functional modules involving mitotic kinesin motors, centromere and kinetochore components, and proteolysis regulators may have an outsized impact on tumor biology and disease progression in AR-low TNBC and p53-deficient breast cancer, because these emergent modules support and drive a highly proliferative state that is accompanied by mitotic errors and CIN.
CIN—the increased rate of whole or fractional chromosome gains or losses—may at first glance appear to be a feature that should adversely impact tumor cells’ fitness and impair their proliferation. However, CIN facilitates accelerated genomic evolution by generating diverse DNA copy number variants (CNVs) that can be selected during disease progression. The intercellular chromosomal heterogeneity that CIN produces proffers cancer cells the ability to generate and sample a variety of karyotypes in the cells’ quest to adapt to multiple stressors in their environment, while continually selecting for CIN-tolerance and a range of genotypes that may confer other advantages, including survival of aneuploid cells and their rapid proliferation. Several well-established prognostic signatures (e.g., Oncotype Dx, MammaPrint, PAM50, Meta-PCNA, GGI, CIN70, and the 12-gene genomic instability index) include genes that function in mitosis and are involved in cell cycle progression, and are strongly associated with proliferation. Interestingly, they are also strongly associated with CIN and resistance to therapies. In a study that explored the complex relationship between CIN and proliferation, the authors developed a SNP array-based surrogate score used to assess the CIN status of a tumor, called the weighted genomic integrity index (wGII) [357]. This metric measures the percentage of gained and lost genomic material relative to the sample's ploidy, avoiding bias from differing chromosome sizes. The authors found a significant positive correlation between increased chromosomal complexity (assessed by wGII) and the expression of these prognostic signatures. They also found that high CIN status is strongly correlated with increased expression of proliferation markers like MKI67 and the meta-PCNA gene set. The study identified "30 core regulators" whose expression and copy number are associated with increasing wGII scores; importantly, “core regulators” like UBE2C, UBE2T, and KIF14—also highlighted in this review—were amplified in high-CIN tumors and drove co-expression of proliferation modules. CIN genomes thus appear to select for amplifications in regulators such as UBE2C, establishing a “permissive landscape” for ongoing proliferation [357]. Understanding AR-low TNBCs as both high-CIN and high-proliferation (a) demonstrates how ITH is generated, and impacts key cancer phenotypes, and (b) highlights the importance of pursuing CIN attenuation by targeting its core regulators as a therapeutic strategy.
Our findings revealed how this tumor type bends the machinations of the cell cycle at multiple levels and scales to drive, support, and propagate a state typified by more tumor cells entering and traversing the cell cycle albeit in error-prone ways. Indeed, it appears that the benefits of the resulting intra-tumoral karyotypic and phenotypic heterogeneity outweigh the risks that accompany their aggressive proliferation and their faulty mitotic processes. The impacts of this dysregulation permeate so many aspects of the disease’s course, and this study represents a step forward in tallying them. Aside from the myriad epistemic benefits of such a study, understanding what gene expression changes are required to adequately support high rates of proliferation, and what causes underlie the collateral errors and imperfections in the execution of cell division in these tumors, will pave the way for more stringent risk management of diverse cancers predisposed to greater intra-tumoral heterogeneity. While our finding that mitotic kinesin motors, centromeric proteins, and regulators of proteolysis are upregulated in AR-low TNBC was somewhat unsurprising, it also helped us unpack what a tumor needs in order to become highly proliferative. Our findings show how this tumor type manipulates cell cycle machinery, creating a state in which aggressive proliferation and genomic instability are not liabilities but assets for tumor evolution. Our study additionally uncovered that several epigenetic mechanisms, involving promoter hypomethylation, microRNAs and ceRNAs, affect the levels of 15 cell cycle genes of interest, and how dysregulation of these upstream networks also potentially contributes to their overexpression in breast tumors. These insights pave the way to identifying new therapeutic targets for AR-low TNBCs.
By highlighting “Cell cycle patterns” of gene expression enrichment and how dysregulation of each one touches and evokes dysregulation in others, this work strengthened our understanding of how phenotypes such as higher proliferation, CIN, chemoresistance and disease progression then co-arise. This study has thus identified a network of vulnerability with therapeutic potential. For example, disrupting FOXM1's phase-separated state could be a novel therapeutic approach, Also, unlike the MMB-FOXM1 complex itself, mitotic kinesins possess enzymatic activity, making them "druggable targets." This could lead to the development of more specific anti-mitotic drugs with potentially fewer side effects than traditional tubulin-targeting chemotherapies. While we acknowledge that transcriptional chaos and variations in levels of overexpression of these gene groups has a non-linear relationship with phenotypic outcomes, we also emphasize that covariance of these specific cell cycle proteins tips the cell towards a certain state of increased proliferation. By (i) identifying coordinated dysregulation of mitotic motors, centromeric proteins, and proteolysis regulators as a defining signature of AR-low TNBC, (ii) connecting these modules mechanistically to CIN and aneuploidy, (iii) revealing a FoxM1–WDR5–ASPM axis that offers new conceptual and therapeutic entry points, insights from this study open avenues for developing targeted interventions aimed at dismantling the very circuits that sustain their malignant proliferation.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org.
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar]
- Feeley, L.P.; Mulligan, A.M.; Pinnaduwage, D.; Bull, S.B.; Andrulis, I.L. Distinguishing Luminal Breast Cancer Subtypes by Ki67, Progesterone Receptor or TP53 Status Provides Prognostic Information. Mod. Pathol. 2014, 27, 554–561. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M. Response to Neoadjuvant Therapy and Long-Term Survival in Patients with Triple-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J. Triple-Negative Breast Cancer: Role of Specific Chemotherapy Agents. Cancer J. 2010, 16, 53–61. [Google Scholar] [CrossRef]
- Masuda, H.; Baggerly, K.A.; Wang, Y.; Zhang, Y.; Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Valero, V.; Lehmann, B.D.; Pietenpol, J.A.; Hortobagyi, G.N.; et al. Differential Response to Neoadjuvant Chemotherapy among 7 Triple-Negative Breast Cancer Molecular Subtypes. Clin. Cancer Res. 2013, 19, 5533–5540. [Google Scholar] [CrossRef]
- DeSantis, C.E.; Ma, J.; Goding Sauer, A.; Newman, L.A.; Jemal, A. Breast Cancer Statistics, 2017, Racial Disparity in Mortality by State. CA Cancer J. Clin 2017, 67, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M. Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 2, 1688–1698. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Jinna, N.; Jovanovic-Talisman, T.; LaBarge, M.; Natarajan, R.; Kittles, R.; Sistrunk, C.; Rida, P.; Seewaldt, V.L. Racial Disparity in Quadruple Negative Breast Cancer: Aggressive Biology and Potential Therapeutic Targeting and Prevention. Cancers 2022, 14, 4484. [Google Scholar] [CrossRef]
- Davis, M.; Tripathi, S.; Hughley, R.; He, Q.; Bae, S.; Karanam, B.; Martini, R.; Newman, L.; Colomb, W.; Grizzle, W. AR Negative Triple Negative or “Quadruple Negative” Breast Cancers in African American Women Have an Enriched Basal and Immune Signature. PLoS ONE 2018, 13, 0196909. [Google Scholar] [CrossRef]
- Huang, M.; Wu, J.; Ling, R.; Li, N. Quadruple negative breast cancer. Breast Cancer 2020, 27, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Sugita, B.M.; Bortoletto, S.M.; Fonseca, A.S.; Cavalli, L.R.; Aneja, R. QNBC Is Associated with High Genomic Instability Characterized by Copy Number Alterations and miRNA Deregulation. Int. J. Mol. Sci. 2021, 22, 11548. [Google Scholar] [CrossRef] [PubMed]
- Rida, P.; Baker, S.; Saidykhan, A.; Bown, I.; Jinna, N. FOXM1 Transcriptionally Co-Upregulates Centrosome Amplification and Clustering Genes and Is a Biomarker for Poor Prognosis in Androgen Receptor-Low Triple-Negative Breast Cancer. Cancers 2024, 16, 3191. [Google Scholar] [CrossRef]
- Hackbart, H.; Cui, X.; Lee, J.S. Androgen Receptor in Breast Cancer and Its Clinical Implication. Transl. Breast Cancer Res. J. Focus. Transl. Res. Breast Cancer 2023, 4, 30. [Google Scholar] [CrossRef]
- Jinna, N.D.; Alsten, S.; Rida, P.; Seewaldt, V.L.; Troester, M.A. Molecular Features of Androgen-Receptor Low, Estrogen Receptor-Negative Breast Cancers in the Carolina Breast Cancer Study. Breast Cancer Res. Treat. 2023, 201, 171–181. [Google Scholar] [CrossRef]
- Riaz, N.; Idress, R.; Habib, S.; Lalani, E.N. Lack of Androgen Receptor Expression Selects for Basal-Like Phenotype and Is a Predictor of Poor Clinical Outcome in Non-Metastatic Triple Negative Breast Cancer. Front. Oncol. 2020, 10, 1083. [Google Scholar] [CrossRef]
- Barton, V.N.; D’Amato, N.C.; Gordon, M.A.; Christenson, J.L.; Elias, A.; Richer, J.K. Androgen Receptor Biology in Triple Negative Breast Cancer: A Case for Classification as AR+ or Quadruple Negative Disease. Horm. Cancer 2015, 6, 206–213. [Google Scholar] [CrossRef]
- Tedesco, D.; Lukas, J.; Reed, S.I. The pRb-Related Protein P130 Is Regulated by Phosphorylation-Dependent Proteolysis via the Protein-Ubiquitin Ligase SCF(Skp2). Genes. Dev. 2002, 16, 2946–2957. [Google Scholar] [CrossRef]
- Sadasivam, S.; DeCaprio, J.A. The DREAM Complex: Master Coordinator of Cell Cycle-Dependent Gene Expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef]
- Park, H.J.; Costa, R.H.; Lau, L.F.; Tyner, A.L.; Raychaudhuri, P. Anaphase-Promoting Complex/Cyclosome-CDH1-Mediated Proteolysis of the Forkhead Box M1 Transcription Factor Is Critical for Regulated Entry into S Phase. Mol. Cell. Biol. 2008, 28, 5162–5171. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Joo-Okumura, A.; Nakatsukasa, K.; Kamura, T. Hypoxia-Inducible Factor-2α Stabilizes the von Hippel-Lindau (VHL) Disease Suppressor, Myb-Related Protein 2. PLoS ONE 2017, 12, 0175593. [Google Scholar] [CrossRef]
- Schade, A.E.; Oser, M.G.; Nicholson, H.E.; DeCaprio, J.A. Cyclin D-CDK4 Relieves Cooperative Repression of Proliferation and Cell Cycle Gene Expression by DREAM and RB. Oncogene 2019, 38, 4962–4976. [Google Scholar] [CrossRef]
- Osterloh, L.; von Eyss, B.; Schmit, F.; Rein, L.; Hübner, D.; Samans, B.; Hauser, S.; Gaubatz, S. The Human synMuv-like Protein LIN-9 Is Required for Transcription of G2/M Genes and for Entry into Mitosis. EMBO J. 2007, 26, 144–157. [Google Scholar] [CrossRef]
- Pattschull, G.; Walz, S.; Gründl, M.; Schwab, M.; Rühl, E.; Baluapuri, A.; Cindric-Vranesic, A.; Kneitz, S.; Wolf, E.; Ade, C.P. The Myb-MuvB Complex Is Required for YAP-Dependent Transcription of Mitotic Genes. Cell Rep. 2019, 27, 3533–3546.e7. [Google Scholar] [CrossRef] [PubMed]
- Eisinger-Mathason, T.S.; Mucaj, V.; Biju, K.M.; Nakazawa, M.S.; Gohil, M.; Cash, T.P.; Yoon, S.S.; Skuli, N.; Park, K.M.; Gerecht, S. Deregulation of the Hippo Pathway in Soft-Tissue Sarcoma Promotes FOXM1 Expression and Tumorigenesis. Proc. Natl. Acad. Sci. USA 2015, 112, 3402–3411. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.B.; Sun, H.; Robichaux, J.; Pfeifer, M.; McDermott, U.; Travers, J.; Diao, L.; Xi, Y.; Tong, P.; Shen, L. A YAP/FOXM1 Axis Mediates EMT-Associated EGFR Inhibitor Resistance and Increased Expression of Spindle Assembly Checkpoint Components. Sci. Transl. Med. 2020, 12, 4589. [Google Scholar] [CrossRef]
- Kohler, R.; Engeland, K. A-MYB Substitutes for B-MYB in Activating Cell Cycle Genes and in Stimulating Proliferation. Nucleic Acids Res. 2024, 52, 6830–6849. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yang, H.Y.; Zhang, X.L.; Zhang, X.; Huang, Y.Z.; Dai, X.Y.; Shi, L.; Zhou, G.R.; Wei, J.F.; Ding, Q. Kinesin Family Member 23, Regulated by FOXM1, Promotes Triple Negative Breast Cancer Progression via Activating Wnt/β-Catenin Pathway. J. Exp. Clin. Cancer Res. 2022, 41, 168. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L. Regulation of MLL1 H3K4 Methyltransferase Activity by Its Core Components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.T. Active Genes Are Tri-Methylated at K4 of Histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Lauberth, S.M.; Nakayama, T.; Wu, X.; Ferris, A.L.; Tang, Z.; Hughes, S.H. H3K4me3 Interactions with TAF3 Regulate Preinitiation Complex Assembly and Selective Gene Activation. Cell 2013, 152, 1021–1036. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Down, C.F.; Millour, J.; Lam, E.W.-F.; Watson, R.J. Binding of FoxM1 to G2/M Gene Promoters Is Dependent upon B-Myb. Biochim. Biophys. Acta 2012, 1819, 855–862. [Google Scholar] [CrossRef]
- Sadasivam, S.; Duan, S.; DeCaprio, J.A. The MuvB Complex Sequentially Recruits B-Myb and FoxM1 to Promote Mitotic Gene Expression. Genes. Dev. 2012, 26, 474–489. [Google Scholar] [CrossRef]
- Saldivar, J.C.; Hamperl, S.; Bocek, M.J.; Chung, M.; Bass, T.E.; Cisneros-Soberanis, F.; Samejima, K.; Xie, L.; Paulson, J.R.; Earnshaw, W.C. An Intrinsic S/G2 Checkpoint Enforced by ATR. Science 2018, 361, 806–810. [Google Scholar] [CrossRef]
- Kalathil, D.; John, S.; Nair, A.S. FOXM1 and Cancer: Faulty Cellular Signaling Derails Homeostasis. Front. Oncol. 2021, 10, 626836. [Google Scholar] [CrossRef]
- Myatt, S.S.; Lam, E.W. The Emerging Roles of Forkhead Box (Fox) Proteins in Cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Wierstra, I. FOXM1 (Forkhead Box M1) in Tumorigenesis: Overexpression in Human Cancer, Implication in Tumorigenesis, Oncogenic Functions, Tumor-Suppressive Properties, and Target of Anticancer Therapy. Adv. Cancer Res. 2013, 119, 191–419. [Google Scholar]
- Hong, H.; Zhu, H.; Zhao, S.; Wang, K.; Zhang, N.; Tian, Y.; Li, Y.; Wang, Y.; Lv, X.; Wei, T. The novel circCLK3/miR-320a/FoxM1 axis promotes cervical cancer progression. Cell Death Dis. 2019, 10, 950. [Google Scholar] [CrossRef]
- Korver, W.; Roose, J.; Clevers, H. The Winged-Helix Transcription Factor Trident Is Expressed in Cycling Cells. Nucleic Acids Res. 1997, 25, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Kelly, T.F.; Samadani, U.; Lim, L.; Rubio, S.; Overdier, D.G.; Roebuck, K.A.; Costa, R.H. Hepatocyte Nuclear Factor 3/Fork Head Homolog 11 Is Expressed in Proliferating Epithelial and Mesenchymal Cells of Embryonic and Adult Tissues. Mol. Cell. Biol. 1997, 17, 1626–1641. [Google Scholar] [CrossRef]
- Saba, R.; Alsayed, A.; Zacny, J.P.; Dudek, A.Z. The Role of Forkhead Box Protein M1 in Breast Cancer Progression and Re-Sistance to Therapy. Int. J. Breast Cancer 2016, 2016, 9768183. [Google Scholar] [CrossRef]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F Target Gene Analyses Identifies Cell Cycle Gene Regulatory Networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F Target Gene Analyses Identifies Cell Cycle Gene Regulatory Networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Quaas, M.; Müller, G.A.; Engeland, K. P53 Can Repress Transcription of Cell Cycle Genes through a P21(WAF1/CIP1)-Dependent Switch from MMB to DREAM Protein Complex Binding at CHR Promoter Elements. Cell Cycle 2012, 11, 4661–4672. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Huang, X.; Wang, J.; Yang, L.; Kong, Y.; Gao, G.; Zhang, L.; Chen, Z.S.; Xie, X. circKIF4A Acts as a Prognostic Factor and Mediator to Regulate the Progression of Triple-Negative Breast Cancer. Mol. Cancer 2019, 18, 23. [Google Scholar] [CrossRef]
- Xu, S.; Wu, X.; Wang, P.; Cao, S.L.; Peng, B.; Xu, X. ASPM Promotes Homologous Recombination-Mediated DNA Repair by Safeguarding BRCA1 Stability. Iscience 2021, 24, 102534. [Google Scholar] [CrossRef]
- Capecchi, M.R.; Pozner, A. ASPM Regulates Symmetric Stem Cell Division by Tuning Cyclin E Ubiquitination. Nat. Commun. 2015, 6, 8763. [Google Scholar] [CrossRef]
- Xie, F.; Zhou, X.; Ran, Y.; Li, R.; Zou, J.; Wan, S.; Su, P.; Meng, X.; Yan, H.; Lu, H.; et al. Targeting FOXM1 Condensates Reduces Breast Tumour Growth and Metastasis. Nature 2025, 638, 1112–1121. [Google Scholar] [CrossRef]
- Jiang, B.; Zeng, L.; Tan, Y.; Feng, W.; Jiang, Y.; Tang, Y.; Zhu, H.; Xie, L.; Fan, S.; Li, Y. Kinesin Family Member 4 a Drives Cancer Stem Cell Characteristics in Breast Cancer: Insights from scRNA-Seq and Experimental Validation. Int. J. Biol. Macromol. 2025, 1, 144308. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Atallah, N.M.; Makhlouf, S.; Toss, M.S.; Green, A.; Rakha, E. Deciphering the Role of ASPM in Breast Cancer: A Comprehensive Multicohort Study. Cancers 2024, 16, 3814. [Google Scholar] [CrossRef]
- Xie, Y.; Gou, Q.; Wang, Q.; Zhong, X.; Zheng, H. The Role of BRCA Status on Prognosis in Patients with Triple-Negative Breast Cancer. Oncotarget 2017, 8, 87151–87162. [Google Scholar] [CrossRef]
- Buchman, J.J.; Durak, O.; Tsai, L.-H. ASPM Regulates Wnt Signaling Pathway Activity in the Developing Brain. Genes. Dev. 2011, 25, 1909–1914. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.C.; Liao, W.Y.; Chan, T.S.; Chen, W.Y.; Lee, C.T.; Shan, Y.S.; Huang, P.J.; Hou, Y.C.; Li, C.R.; Tsai, K.K. The Differential Distributions of ASPM Isoforms and Their Roles in Wnt Signaling, Cell Cycle Progression, and Pancreatic Cancer Prognosis. J. Pathol. 2019, 249, 498–508. [Google Scholar] [CrossRef]
- Wang, W.Y.; Hsu, C.C.; Wang, T.Y.; Li, C.R.; Hou, Y.C.; Chu, J.M.; Lee, C.T.; Liu, M.S.; Su, J.J.; Jian, K.Y. A Gene Expression Signature of Epithelial Tubulogenesis and a Role for ASPM in Pancreatic Tumor Progression. Gastroenterology 2013, 145, 1110–1120. [Google Scholar] [CrossRef]
- Bikeye, S.-N.N.; Colin, C.; Marie, Y.; Vampouille, R.; Ravassard, P.; Rousseau, A.; Boisselier, B.; Idbaih, A.; Calvo, C.F.; Leuraud, P. ASPM-Associated Stem Cell Proliferation Is Involved in Malignant Progression of Gliomas and Constitutes an Attractive therapeutic target. Cancer Cell Int. 2010, 10, 1. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Pan, H.-W.; Liu, S.-H.; Jeng, Y.-M.; Hu, F.-C.; Peng, S.-Y.; Lai, P.-L.; Hsu, H.-C. ASPM Is a Novel Marker for Vascular Invasion, Early Recurrence, and Poor Prognosis of Hepatocellular Carcinoma. Clin. Cancer Res. 2008, 14, 4814–4820. [Google Scholar] [CrossRef]
- Gai, M.; Bianchi, F.T.; Vagnoni, C.; Vernì, F.; Bonaccorsi, S.; Pasquero, S.; Berto, G.E.; Sgrò, F.; Chiotto, A.M.; Annaratone, L.; et al. ASPM and CITK Regulate Spindle Orientation by Affecting the Dynamics of Astral Microtubules. EMBO Rep. 2016, 17, 1396–1409. [Google Scholar] [CrossRef]
- Jiang, K.; Rezabkova, L.; Hua, S.; Liu, Q.; Capitani, G.; Altelaar, A.F.M.; Heck, A.J.R.; Kammerer, R.A.; Steinmetz, M.O.; Akhmanova, A. Microtubule Minus-End Regulation at Spindle Poles by an ASPM-Katanin Complex. Nat. Cell Biol. 2017, 19, 480–492. [Google Scholar] [CrossRef]
- Razuvaeva, A.V.; Graziadio, L.; Palumbo, V.; Pavlova, G.A.; Popova, J.V.; Pindyurin, A.V.; Bonaccorsi, S.; Somma, M.P.; Gatti, M. The Multiple Mitotic Roles of the ASPM Orthologous Proteins: Insight into the Etiology of ASPM-Dependent Microcephaly. Cells 2023, 12, 922. [Google Scholar] [CrossRef]
- Astvatsaturyan, K.; Yue, Y.; Walts, A.E.; Bose, S. Androgen Receptor Positive Triple Negative Breast Cancer: Clinicopathologic, Prognostic, and Predictive Features. PLoS ONE 2018, 13, e0197827. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Basile, D.; Buono, G.; Placido, S.; Giuliano, M.; Minichillo, S.; Coinu, A.; Martorana, F.; Santo, I.; Del Mastro, L.; et al. Androgen Receptor in Triple Negative Breast Cancer: A Potential Target for the Targetless Subtype. Cancer Treat. Rev. 2018, 68, 102–110. [Google Scholar] [CrossRef]
- Sharifi, M.N.; O’Regan, R.M.; Wisinski, K.B. Is the Androgen Receptor a Viable Target in Triple Negative Breast Cancer in 5 Years? Clin Breast Cancer 2023. [CrossRef]
- Radovich, M.; Clare, S.E.; Atale, R.; Pardo, I.; Hancock, B.A.; Solzak, J.P.; Schneider, B.P. Characterizing the Heterogeneity of Triple-Negative Breast Cancers Using Microdissected Normal Ductal Epithelium and RNA-Sequencing. Breast Cancer Res. Treat. 2013, 143, 57–68. [Google Scholar] [CrossRef]
- Heltberg, M.L.; Krishna, S.; Jensen, M.H. On Chaotic Dynamics in Transcription Factors and the Associated Effects in Differential Gene Regulation. Nat. Commun. 2019, 10, 71. [Google Scholar] [CrossRef]
- Jézéquel, P.; Frénel, J.-S.; Campion, L.; Guérin-Charbonnel, C.; Gouraud, W.; Ricolleau, G.; Campone, M. Bc-GenExMiner 3.0: New Mining Module Computes Breast Cancer Gene Expression Correlation Analyses. Database 2013, 2013, bas060. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Rodriguez, I.P.; BVSK, C.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Győrffy, B. Survival Analysis across the Entire Transcriptome Identifies Biomarkers with the Highest Prognostic Power in Breast Cancer. Comput. Struct. Biotechnol. J. 2021, 19, 4101–4109. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Cohen, M.; Vecsler, M.; Liberzon, A.; Noach, M.; Zlotorynski, E.; Tzur, A. Unbiased Transcriptome Signature of in Vivo Cell Proliferation Reveals Pro- and Antiproliferative Gene Networks. Cell Cycle 2013, 12, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J.; Lemke, H.; Baisch, H.; Wacker, H.H.; Schwab, U.; Stein, H. Cell Cycle Analysis of a Cell Proliferation-Associated Human Nuclear Antigen Defined by the Monoclonal Antibody Ki-67. J. Immunol. 1984, 133, 1710–1715. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, J. Ki-67 and Other Proliferation Markers Useful for Immunohistological Diagnostic and Prognostic Evaluations in Human Malignancies. Semin. Cancer Biol. 1990, 1, 199–206. [Google Scholar]
- Trihia, H.; Murray, S.; Price, K.; Gelber, R.D.; Golouh, R.; Goldhirsch, A.; Coates, A.S.; Collins, J.; Castiglione-Gertsch, M.; Gusterson, B.A. Ki-67 Expression in Breast Carcinoma: Its Association with Grading Systems, Clinical Parameters, and Other Prognostic Factors—A Surrogate Marker? Cancer 2003, 97, 1321–1331. [Google Scholar] [CrossRef]
- Inwald, E.C.; Klinkhammer-Schalke, M.; Hofstädter, F.; Zeman, F.; Koller, M.; Gerstenhauer, M.; Ortmann, O. Ki-67 Is a Prognostic Parameter in Breast Cancer Patients: Results of a Large Population-Based Cohort of a Cancer Registry. Breast Cancer Res. Treat. 2013, 139, 539–552. [Google Scholar] [CrossRef]
- Buxant, F.; Anaf, V.; Simon, P.; Fayt, I.; Noël, J.C. Ki-67 Immunostaining Activity Is Higher in Positive Axillary Lymph Nodes than in the Primary Breast Tumor. Breast Cancer Res. Treat. 2002, 75, 1–3. [Google Scholar] [CrossRef]
- Nishimura, R.; Osako, T.; Okumura, Y.; Hayashi, M.; Toyozumi, Y.; Arima, N. Ki-67 as a Prognostic Marker According to Breast Cancer Subtype and a Predictor of Recurrence Time in Primary Breast Cancer. Exp. Ther. Med. 2010, 1, 747–754. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thürlimann, B.; Senn, H.-J. Strategies for Subtypes—Dealing with the Diversity of Breast Cancer: Highlights of the St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Dowsett, M.; Nielsen, T.O.; A’Hern, R.; Bartlett, J.; Coombes, R.C.; Cuzick, J.; Ellis, M.; Henry, N.L.; Hugh, J.C.; Lively, T.; et al. Assessment of Ki67 in Breast Cancer: Recommendations from the International Ki67 in Breast Cancer Working Group. JNCI J. Natl. Cancer Inst. 2011, 103, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Aaltomaa, S.; Lipponen, P.; Eskelinen, M.; Kosma, V.-M.; Marin, S.; Alhava, E.; Syrjänen, K. Lymphocyte Infiltrates as a Prognostic Variable in Female Breast Cancer. Eur. J. Cancer 1992, 28, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Tahan, S.R.; Neuberg, D.S.; Dieffenbach, A.; Yacoub, L. Prediction of Early Relapse and Shortened Survival in Patients with Breast Cancer by Proliferating Cell Nuclear Antigen Score. Cancer 1993, 71, 3552–3559. [Google Scholar] [CrossRef]
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.-C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef]
- Haneda, H.; Katabami, M.; Miyamoto, H.; Isobe, H.; Shimizu, T.; Ishiguro, A.; Moriuti, T.; Takasaki, Y.; Kawakami, Y. The Relationship of the Proliferating Cell Nuclear Antigen Protein to Cis-Diamminedichloroplatinum(II) Resistance of a Murine Leukemia Cell Line P388/CDDP. Oncology 1991, 48, 234–238. [Google Scholar] [CrossRef]
- Forsburg, S.L. Eukaryotic MCM Proteins: Beyond Replication Initiation. Microbiol. Mol. Biol. Rev. 2004, 68, 109–131. [Google Scholar] [CrossRef] [PubMed]
- Madine, M.A.; Swietlik, M.; Pelizon, C.; Romanowski, P.; Mills, A.D.; Laskey, R.A. The Roles of the MCM, ORC, and Cdc6 Proteins in Determining the Replication Competence of Chromatin in Quiescent Cells. J. Struct. Biol. 2000, 129, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.; Morris, L.S.; Mills, A.D.; Stoeber, K.; Laskey, R.A.; Williams, G.H.; Coleman, N. Minichromosome Maintenance Proteins as Biological Markers of Dysplasia and Malignancy. Clin. Cancer Res. 1999, 5, 2121–2132. [Google Scholar]
- Ishimi, Y.; Okayasu, I.; Kato, C.; Kwon, H.; Kimura, H.; Yamada, K.; Song, S. Enhanced Expression of Mcm Proteins in Cancer Cells Derived from Uterine Cervix. Eur. J. Biochem. 2003, 270, 1089–1101. [Google Scholar] [CrossRef] [PubMed]
- Alison, M.R.; Poulsom, R.; Wright, N.A. Preface to Stem Cells. J. Pathol. 2002, 197, 417–418. [Google Scholar] [CrossRef]
- Hunt, D.P.J.; Freeman, A.; Morris, L.S.; Burnet, N.G.; Bird, K.; Davies, T.W.; Laskey, R.A.; Coleman, N. Early Recurrence of Benign Meningioma Correlates with Expression of Mini-Chromosome Maintenance-2 Protein. Br. J. Neurosurg. 2002, 16, 10–15. [Google Scholar] [CrossRef]
- Gonzalez, M.A.; Pinder, S.E.; Callagy, G.; Vowler, S.L.; Morris, L.S.; Bird, K.; Bell, J.A.; Laskey, R.A.; Coleman, N. Minichromosome Maintenance Protein 2 Is a Strong Independent Prognostic Marker in Breast Cancer. JCO 2003, 21, 4306–4313. [Google Scholar] [CrossRef]
- Grant, G.D.; Brooks, L.; Zhang, X.; Mahoney, J.M.; Martyanov, V.; Wood, T.A.; Sherlock, G.; Cheng, C.; Whitfield, M.L. Identification of Cell Cycle–Regulated Genes Periodically Expressed in U2OS Cells and Their Regulation by FOXM1 and E2F Transcription Factors. MBoC 2013, 24, 3634–3650. [Google Scholar] [CrossRef]
- Vale, R.D.; Schnapp, B.J.; Reese, T.S.; Sheetz, M.P. Organelle, Bead, and Microtubule Translocations Promoted by Soluble Factors from the Squid Giant Axon. Cell 1985, 40, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin Superfamily Motor Proteins and Intracellular Transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Rath, O.; Kozielski, F. Kinesins and cancer. Nat. Rev. Cancer 2012, 12, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Cross, R.A.; McAinsh, A. Prime Movers: The Mechanochemistry of Mitotic Kinesins. Nat. Rev. Mol. Cell Biol. 2014, 15, 257–271. [Google Scholar] [CrossRef]
- Singel, S.M.; Cornelius, C.; Zaganjor, E.; Batten, K.; Sarode, V.R.; Buckley, D.L.; Peng, Y.; John, G.B.; Li, H.C.; Sadeghi, N.; et al. KIF14 Promotes AKT Phosphorylation and Contributes to Chemoresistance in Triple-Negative Breast Cancer. Neoplasia 2014, 16, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Brynychova, V.; Ehrlichova, M.; Hlavac, V.; Nemcova-Furstova, V.; Pecha, V.; Leva, J.; Soucek, P. Genetic and Functional Analyses Do Not Explain the Association of High PRC1 Expression with Poor Survival of Breast Carcinoma Patients. Biomed. Pharmacother. 2016, 83, 857–864. [Google Scholar] [CrossRef]
- Xue, D.; Cheng, P.; Han, M.; Liu, X.; Xue, L.; Ye, C.; Huang, J. An Integrated Bioinformatical Analysis to Evaluate the Role of KIF4A as a Prognostic Biomarker for Breast Cancer. OncoTargets Ther. 2018, 11, 4755–4768. [Google Scholar] [CrossRef]
- Nakamura, M. Characterization of KIF20A as a Prognostic Biomarker and Therapeutic Target for Different Subtypes of Breast Cancer. Int. J. Oncol. 2020, 57, 277–288. [Google Scholar] [CrossRef]
- Liu, S.; Ye, Z.; Xue, V.W.; Sun, Q.; Li, H.; Lu, D. KIF2C Is a Prognostic Biomarker Associated with Immune Cell Infiltration in Breast Cancer. BMC Cancer 2023, 23, 307. [Google Scholar] [CrossRef]
- Wang, F.; Fu, X.; Chang, M.; Wei, T.; Lin, R.; Tong, H.; Zhang, X.; Yuan, R.; Zhou, Z.; Huang, X.; et al. The Interaction of Calcium-Sensing Receptor with KIF11 Enhances Cisplatin Resistance in Lung Adenocarcinoma via BRCA1/Cyclin B1 Pathway. Int. J. Biol. Sci. 2024, 20, 3892. [Google Scholar] [CrossRef]
- Carleton, R.N.; Norton, M.A.P.J.; Asmundson, G.J.G. Fearing the Unknown: A Short Version of the Intolerance of Uncertainty Scale. J. Anxiety Disord. 2007, 21, 105–117. [Google Scholar] [CrossRef]
- Gruneberg, U.; Neef, R.; Li, X.; Chan, E.H.Y.; Chalamalasetty, R.B.; Nigg, E.A.; Barr, F.A. KIF14 and Citron Kinase Act Together to Promote Efficient Cytokinesis. J. Cell Biol. 2006, 172, 363–372. [Google Scholar] [CrossRef]
- Zhu, C.; Jiang, W. Cell Cycle-Dependent Translocation of PRC1 on the Spindle by Kif4 Is Essential for Midzone Formation and Cytokinesis. Proc. Natl. Acad. Sci. USA 2005, 102, 343–348. [Google Scholar] [CrossRef]
- Corson, T.W.; Gallie, B.L. KIF14 mRNA Expression Is a Predictor of Grade and Outcome in Breast Cancer. Int. J. Cancer 2006, 119, 1088–1094. [Google Scholar] [CrossRef]
- Thériault, B.L.; Pajovic, S.; Bernardini, M.Q.; Shaw, P.A.; Gallie, B.L. Kinesin Family Member 14: An Independent Prognostic Marker and Potential Therapeutic Target for Ovarian Cancer. Int. J. Cancer 2012, 130, 1844–1854. [Google Scholar] [CrossRef]
- Singh, A.; Denu, R.A.; Wolfe, S.K.; Sperger, J.M.; Schehr, J.; Witkowsky, T.; Esbona, K.; Chappell, R.J.; Weaver, B.A.; Burkard, M.E.; et al. Centrosome Amplification Is a Frequent Event in Circulating Tumor Cells from Subjects with Metastatic Breast Cancer. Mol. Oncol. 2020, 14, 1898–1909. [Google Scholar] [CrossRef] [PubMed]
- Van ’T Veer, L.J.; Dai, H.; Van De Vijver, M.J.; He, Y.D.; Hart, A.A.M.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene Expression Profiling Predicts Clinical Outcome of Breast Cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef]
- Singel, S.M.; Cornelius, C.; Batten, K.; Fasciani, G.; Wright, W.E.; Lum, L.; Shay, J.W. A Targeted RNAi Screen of the Breast Cancer Genome Identifies KIF14 and TLN1 as Genes That Modulate Docetaxel Chemosensitivity in Triple-Negative Breast Cancer. Clin. Cancer Res. 2013, 19, 2061–2070. [Google Scholar] [CrossRef]
- Gerashchenko, T.S.; Zolotaryova, S.Y.; Kiselev, A.M.; Tashireva, L.A.; Novikov, N.M.; Krakhmal, N.V.; Cherdyntseva, N.V.; Zavyalova, M.V.; Perelmuter, V.M.; Denisov, E.V. The Activity of KIF14, Mieap, and EZR in a New Type of the Invasive Component, Torpedo-Like Structures, Predetermines the Metastatic Potential of Breast Cancer. Cancers 2020, 12, 1909. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Lu, J.; Yang, Z.; Li, E.; Cao, Y.; Xie, L. Mitotic Functions and Characters of KIF11 in Cancers. Biomolecules 2024, 14, 386. [Google Scholar] [CrossRef]
- Kapitein, L.C.; Peterman, E.J.G.; Kwok, B.H.; Kim, J.H.; Kapoor, T.M.; Schmidt, C.F. The Bipolar Mitotic Kinesin Eg5 Moves on Both Microtubules That It Crosslinks. Nature 2005, 435, 114–118. [Google Scholar] [CrossRef]
- Zhou, C.J.; Wang, X.Y.; Han, Z.; Wang, D.H.; Ma, Y.Z.; Liang, C.G. Loss of CENPF Leads to Developmental Failure in Mouse Embryos. Cell Cycle 2019, 18, 2784–2799. [Google Scholar] [CrossRef]
- Pei, Y.Y.; Li, G.C.; Ran, J.; Wei, F.X. Kinesin Family Member 11 Contributes to the Progression and Prognosis of Human Breast Cancer. Oncol. Lett. 2017, 14, 6618–6626. [Google Scholar] [CrossRef]
- Blangy, A.; Lane, H.A.; d’Hérin, P.; Harper, M.; Kress, M.; Nigg, E.A. Phosphorylation by P34cdc2 Regulates Spindle Association of Human Eg5, a Kinesin-Related Motor Essential for Bipolar Spindle Formation in Vivo. Cell 1995, 83, 1159–1169. [Google Scholar] [CrossRef] [PubMed]
- Bertran, M.T.; Sdelci, S.; Regué, L.; Avruch, J.; Caelles, C.; Roig, J. Nek9 Is a Plk1-Activated Kinase That Controls Early Centrosome Separation through Nek6/7 and Eg5. EMBO J. 2011, 30, 2634–2647. [Google Scholar] [CrossRef]
- Thankamony, A.P.; Murali, R.; Karthikeyan, N.; Varghese, B.A.; Teo, W.S.; McFarland, A.; Roden, D.L.; Holliday, H.; Konrad, C.V.; Cazet, A.; et al. Targeting the Id1-Kif11 Axis in Triple-Negative Breast Cancer Using Combination Therapy. Biomolecules 2020, 10, 1295. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.Y.; Li, G.C.; Ran, J.; Wan, X.H.; Wei, F.X.; Wang, L. Kinesin Family Member 11 Enhances the Self-Renewal Ability of Breast Cancer Cells by Participating in the Wnt/β-Catenin Pathway. J. Breast Cancer 2019, 22, 522–532. [Google Scholar] [CrossRef]
- Mazumdar, M.; Sundareshan, S.; Misteli, T. Human Chromokinesin KIF4A Functions in Chromosome Condensation and Segregation. J. Cell Biol. 2004, 166, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Kurasawa, Y.; Earnshaw, W.C.; Mochizuki, Y.; Dohmae, N.; Todokoro, K. Essential Roles of KIF4 and Its Binding Partner PRC1 in Organized Central Spindle Midzone Formation. EMBO J. 2004, 23, 3237–3248. [Google Scholar] [CrossRef]
- Hu, C.K.; Coughlin, M.; Field, C.M.; Mitchison, T.J. KIF4 regulates midzone length during cytokinesis. Curr. Biol. 2011, 21, 815–824. [Google Scholar] [CrossRef]
- Takata, H.; Madung, M.; Katoh, K.; Fukui, K. Cdk1-Dependent Phosphorylation of KIF4A at S1186 Triggers Lateral Chromosome Compaction during Early Mitosis. PLoS ONE 2018, 13, e0209614. [Google Scholar] [CrossRef]
- Colak, D.; Nofal, A.; Albakheet, A.; Nirmal, M.; Jeprel, H.; Eldali, A.; Al-Tweigeri, T.; Tulbah, A.; Ajarim, D.; Malik, O.A.; et al. Age-Specific Gene Expression Signatures for Breast Tumors and Cross-Species Conserved Potential Cancer Progression Markers in Young Women. PLoS ONE 2013, 8, e63204. [Google Scholar] [CrossRef]
- Song, X.; Zhang, T.; Wang, X.; Liao, X.; Han, C.; Yang, C.; Su, K.; Cao, W.; Gong, Y.; Chen, Z.; et al. Distinct Diagnostic and Prognostic Values of Kinesin Family Member Genes Expression in Patients with Breast Cancer. Med. Sci. Monit. 2018, 24, 9442. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for Analysis of Tumor-Infiltrating Immune Cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Y.; Luo, J.; Zhao, W.; Hu, C.; Chen, G. Hsa_circ_0002082 Up-Regulates Centromere Protein F via Abolishing miR-508-3p to Promote Breast Cancer Progression. J. Clin. Lab. Anal. 2022, 36, e24697. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, H.L.; Liu, Y.J. A Novel Five-Gene Signature Related to Clinical Outcome and Immune Microenvironment in Breast Cancer. Front. Genet. 2022, 13, 912125. [Google Scholar] [CrossRef]
- Tang, W.; Zhou, M.; Dorsey, T.H.; Prieto, D.A.; Wang, X.W.; Ruppin, E.; Veenstra, T.D.; Ambs, S. Integrated Proteotranscriptomics of Breast Cancer Reveals Globally Increased Protein-mRNA Concordance Associated with Subtypes and Survival. Genome Med. 2018, 10, 94. [Google Scholar] [CrossRef]
- Luo, P.; Gong, Y.; Weng, J.; Wen, F.; Luo, J.; Hu, C.; Xiao, J.; Shu, J. CircKIF4A Combines EIF4A3 to Stabilize SDC1 Expression to Activate C-Src/FAK and Promotes TNBC Progression. Cell Signal 2023, 108, 110690. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhou, L.; Khidr, L.; Guo, X.E.; Kim, W.; Lee, Y.M.; Krasieva, T.; Chen, P.L. A Novel Role of the Chromokinesin Kif4A in DNA Damage Response. Cell Cycle 2008, 7, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
- Sang, M.; Wu, M.; Meng, L.; Zheng, Y.; Gu, L.; Liu, F.; Sang, M. Identification of Epithelial-Mesenchymal Transition-Related circRNA-miRNA-mRNA ceRNA Regulatory Network in Breast Cancer. Pathol. Res. Pr. 2020, 216, 153088. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The Molecular Machinery of Regulated Cell Death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Miyamoto, T.; Hosoba, K.; Ochiai, H.; Royba, E.; Izumi, H.; Sakuma, T. The Microtubule-Depolymerizing Activity of a Mitotic Kinesin Protein KIF2A Drives Primary Cilia Disassembly Coupled with Cell Proliferation. Cell Rep. 2015, 10, 664–673. [Google Scholar] [CrossRef]
- Andrews, P.D.; Ovechkina, Y.; Morrice, N.; Wagenbach, M.; Duncan, K.; Wordeman, L.; Swedlow, J.R. Aurora B Regulates MCAK at the Mitotic Centromere. Dev. Cell 2004, 6, 253–268. [Google Scholar] [CrossRef]
- Gorbsky, G.J. Mitosis: MCAK under the aura of Aurora B. Curr. Biol. 2004, 14, 346–348. [Google Scholar] [CrossRef]
- Zhang, X.; Ems-McClung, S.C.; Walczak, C.E. Aurora A Phosphorylates MCAK to Control Ran-Dependent Spindle Bipolarity. Mol. Biol. Cell 2008, 19, 2752–2765. [Google Scholar] [CrossRef]
- Stout, J.R.; Rizk, R.S.; Kline, S.L.; Walczak, C.E. Deciphering Protein Function during Mitosis in PtK Cells Using RNAi. BMC Cell Biol. 2006, 7, 26. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Genovese, G.; Compton, D.A. Deviant kinetochore microtubule dynamics underlie chromosomal instability. Curr. Biol. 2009, 19, 1937–1942. [Google Scholar] [CrossRef]
- Ganguly, A.; Yang, H.; Cabral, F. Overexpression of Mitotic Centromere-Associated Kinesin Stimulates Microtubule Detachment and Confers Resistance to Paclitaxel. Mol. Cancer Ther. 2011, 10, 929–937. [Google Scholar] [CrossRef]
- Moon, H.H.; Kreis, N.-N.; Friemel, A.; Roth, S.; Schulte, D.; Solbach, C. Mitotic Centromere-Associated Kinesin (MCAK/KIF2C) Regulates Cell Migration and Invasion by Modulating Microtubule Dynamics and Focal Adhesion Turnover. Cancers 2021, 13, 5673. [Google Scholar] [CrossRef]
- Ritter, A.T.; Asano, Y.; Stinchcombe, J.C.; Dieckmann, N.M.G.; Chen, B.-C.; Gawden-Bone, C.; van Engelenburg, S.; Legant, W.; Gao, L.; Davidson, M.W.; et al. Actin Depletion Initiates Events Leading to Granule Secretion at the Immunological Synapse. Immunity 2015, 42, 864–876. [Google Scholar] [CrossRef]
- Smith, J.C.; Husted, S.; Pilrose, J.; Ems-McClung, S.C.; Stout, J.R.; Carpenter, R.L.; Walczak, C.E. MCAK Inhibitors Induce Aneuploidy in Triple-Negative Breast Cancer Models. Cancers 2023, 15, 3309. [Google Scholar] [CrossRef]
- Manning, A.L.; Ganem, N.J.; Bakhoum, S.F.; Wagenbach, M.; Wordeman, L.; Compton, D.A. The Kinesin-13 Proteins Kif2a, Kif2b, and Kif2c/MCAK Have Distinct Roles during Mitosis in Human Cells. Mol. Biol. Cell 2007, 18, 2970–2979. [Google Scholar] [CrossRef]
- Zhu, S.; Paydar, M.; Wang, F.; Li, Y.; Wang, L.; Barrette, B. Kinesin Kif2C in regulation of DNA double strand break dynamics and repair. Elife 2020, 9, e53402. [Google Scholar] [CrossRef]
- Jiang, C.F.; Xie, Y.X.; Qian, Y.C.; Wang, M.; Liu, L.Z.; Shu, Y.Q.; Bai, X.M.; Jiang, B.H. TBX15/miR-152/KIF2C Pathway Regulates Breast Cancer Doxorubicin Resistance via Promoting PKM2 Ubiquitination. Cancer Cell Int. 2021, 21, 542. [Google Scholar] [CrossRef]
- Hill, E.; Clarke, M.; Barr, F.A. The Rab6-binding kinesin, Rab6-KIFL, is required for cytokinesis. EMBO J. 2000, 19, 5711–5719. [Google Scholar] [CrossRef]
- Verhey, K.J.; Hammond, J.W. Traffic Control: Regulation of Kinesin Motors. Nat. Rev. Mol. Cell Biol. 2009, 10, 765–777. [Google Scholar] [CrossRef]
- Neef, R. Phosphorylation of Mitotic Kinesin-like Protein 2 by Polo-like Kinase 1 Is Required for Cytokinesis. J. Cell Biol. 2003, 162, 863–875. [Google Scholar] [CrossRef]
- Kitagawa, M.; Fung, S.Y.S.; Onishi, N.; Saya, H.; Lee, S.H. Targeting Aurora B to the Equatorial Cortex by MKlp2 Is Required for Cytokinesis. PLoS ONE 2013, 8, e64826. [Google Scholar] [CrossRef]
- Lee, K.Y.; Davies, T.; Mishima, M. Cytokinesis microtubule organisers at a glance. J. Cell Sci. 2012, 125, 3495–3500. [Google Scholar] [CrossRef]
- Miserey-Lenkei, S.; Bousquet, H.; Pylypenko, O. Coupling Fission and Exit of RAB6 Vesicles at Golgi Hotspots through Kinesin-Myosin Interactions. Nat. Commun. 2017, 8, 1254. [Google Scholar] [CrossRef]
- Groth-Pedersen, L.; Aits, S.; Corcelle-Termeau, E.; Petersen, N.H.; Nylandsted, J.; Jäättelä, M. Identification of Cytoskeleton-Associated Proteins Essential for Lysosomal Stability and Survival of Human Cancer Cells. PLoS ONE 2012, 7, e45381. [Google Scholar] [CrossRef]
- Yang, P.; Yang, X.; Wang, D. PSMD14 stabilizes estrogen signaling and facilitates breast cancer progression via deubiquitinating ERα. Oncogene 2024, 43, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.U.; Yang, S.; Azam, N.; Yuan, Z.; Yu, J.; Zhao, C.; Feng, B. Mir-153-3p Modulates the Breast Cancer Cells’ Chemosensitivity to Doxorubicin by Targeting KIF20A. Cancers 2023, 15, 1724. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Li, S.; Gao, W.; Shi, J.; Cheng, M.; Mi, Y.; Liu, Y.; Sang, M.; Li, Z.; Geng, C. KIF20A Is a Prognostic Marker for Female Patients with Estrogen Receptor-Positive Breast Cancer and Receiving Tamoxifen as Adjuvant Endocrine Therapy. Int. J. Gen. Med. 2023, 3623–3635. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ma, R.; Yang, Q.; Yang, L.; Li, X.; Wu, Z. Macrophage Polarization-Based Analysis of the Role of the FOXM1/KIF20A Axis in Breast Cancer Metastasis. Cell Biochem. Biophys. 2025, 1–11. [Google Scholar] [CrossRef]
- Wolter, P.; Hanselmann, S.; Pattschull, G.; Schruf, E.; Gaubatz, S. Central Spindle Proteins and Mitotic Kinesins Are Direct Transcriptional Targets of MuvB, B-MYB and FOXM1 in Breast Cancer Cell Lines and Are Potential Targets for Therapy. Oncotarget 2017, 8, 11160–11172. [Google Scholar] [CrossRef]
- Muhammad, A.; Forcados, G.E.; Katsayal, B.S.; Bako, R.S.; Aminu, S.; Sadiq, I.Z.; Abubakar, M.B.; Yusuf, A.P.; Malami, I.; Fa-ruk, M. Potential Epigenetic Modifications Implicated in Triple- to Quadruple-Negative Breast Cancer Transition: A Re-View. Epigenomics 2022, 14, 711–726. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.A.; Wintsche, A.; Stangner, K.; Prohaska, S.J.; Stadler, P.F.; Engeland, K. The CHR Site: Definition and Genome-Wide Identification of a Cell Cycle Transcriptional Element. Nucleic Acids Res. 2014, 42, 10331–10350. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A Signature of Chromosomal Instability Inferred from Gene Expression Profiles Predicts Clinical Outcome in Multiple Human Cancers. Nat. Genet. 2006, 38, 1043–1048. [Google Scholar] [CrossRef]
- Danielsen, H.E.; Pradhan, M.; Novelli, M. Revisiting Tumour Aneuploidy—the Place of Ploidy Assessment in the Molecular Era. Nat. Rev. Clin. Oncol. 2016, 13, 291–304. [Google Scholar] [CrossRef]
- Roylance, R.; Endesfelder, D.; Gorman, P.; Burrell, R.A.; Sander, J.; Tomlinson, I.; Hanby, A.M.; Speirs, V.; Richardson, A.L.; Birkbak, N.J.; et al. Relationship of Extreme Chromosomal Instability with Long-Term Survival in a Retrospective Analysis of Primary Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2011, 20, 2183–2194. [Google Scholar] [CrossRef] [PubMed]
- Pfister, K.; Pipka, J.L.; Chiang, C.; Liu, Y.; Clark, R.A.; Keller, R.; Skoglund, P.; Guertin, M.J.; Hall, I.M.; Stukenberg, P.T. Identification of Drivers of Aneuploidy in Breast Tumors. Cell Rep. 2018, 23, 2758–2769. [Google Scholar] [CrossRef]
- Li, M.; Fang, X.; Baker, D.J.; Guo, L.; Gao, X.; Wei, Z.; Han, S.; Van Deursen, J.M.; Zhang, P. The ATM–P53 Pathway Suppresses Aneuploidy-Induced Tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 14188–14193. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Compton, D.A. Proliferation of Aneuploid Human Cells Is Limited by a P53-Dependent Mechanism. J. Cell Biol. 2010, 188, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Rizzotto, D.; Villunger, A. P53 Clears Aneuploid Cells by Entosis. Cell Death Differ. 2021, 28, 818–820. [Google Scholar] [CrossRef]
- Gwon, M.-R.; Cho, J.H.; Kim, J.-R. Mitotic Centromere-associated Kinase (MCAK/Kif2C) Regulates Cellular Senescence in Human Primary Cells through a P53-dependent Pathway. FEBS Lett. 2012, 586, 4148–4156. [Google Scholar] [CrossRef]
- Westhorpe, F.G.; Straight, A.F. The Centromere: Epigenetic Control of Chromosome Segregation during Mitosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a015818. [Google Scholar] [CrossRef]
- McKinley, K.L.; Cheeseman, I.M. The Molecular Basis Forcentromere Identity and Function. Nat. Rev. Mol. Cell Biol. 2016, 17, 16–29. [Google Scholar] [CrossRef]
- Van Hooser, A.A.; Ouspenski, I.I.; Gregson, H.C.; Starr, D.A.; Yen, T.J.; Goldberg, M.L.; Yokomori, K.; Earnshaw, W.C.; Sullivan, K.F.; Brinkley, B.R. Specification of Kinetochore-Forming Chromatin by the Histone H3 Variant CENP-A. J. Cell Sci. 2001, 114, 3529–3542. [Google Scholar] [CrossRef]
- De Rop, V.; Padeganeh, A.; Maddox, P.S. CENP-A: The Key Player behind Centromere Identity, Propagation, and Kinetochore Assembly. Chromosoma 2012, 121, 527–538. [Google Scholar] [CrossRef]
- Sullivan, B.A.; Karpen, G.H. Centromeric Chromatin Exhibits a Histone Modification Pattern That Is Distinct from Both Euchromatin and Heterochromatin. Nat. Struct. Mol. Biol. 2004, 11, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Tachiwana, H.; Kagawa, W.; Shiga, T. Crystal Structure of the Human Centromeric Nucleosome Containing CENP-A. Nature 2011, 476, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Nechemia-Arbely, Y.; Fachinetti, D.; Miga, K.H.; Sekulic, N.; Soni, G.V.; Kim, D.H. Human Centromeric CENP-A Chromatin Is a Homotypic, Octameric Nucleosome at All Cell Cycle Points. J. Cell Biol. 2017, 216, 607–621. [Google Scholar] [CrossRef]
- Kixmoeller, K.; Allu, P.K.; Black, B.E. The Centromere Comes into Focus: From CENP-A Nucleosomes to Kine- Tochore Connections with the Spindle. Open Biol. 2020, 10, 200051. [Google Scholar] [CrossRef]
- Stirpe, A.; Heun, P. The Ins and Outs of CENP-A: Chro- Matin Dynamics of the Centromere-Specific Histone. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2023; Volume 135, pp. 24–34. [Google Scholar]
- Foltz, D.R.; Jansen, L.E.; Bailey, A.O. Centromere- Specific Assembly of CENP-a Nucleosomes Is Mediated by HJURP. Cell 2009, 137, 472–484. [Google Scholar] [CrossRef]
- Yang, J.; Li, F. Are All Repeats Created Equal? Understanding DNA Repeats at an Individual Level. Curr. Genet. 2017, 63, 57–63. [Google Scholar] [CrossRef]
- Sharp, L.W. Introduction to Cytology; McGraw-Hill Book Company, inc: New York, 1934. [Google Scholar]
- Tan, A.L.; Rida, P.C.; Surana, U. Essential Tension and Constructive Destruction: The Spindle Checkpoint and Its Regulatory Links with Mitotic Exit. Biochem. J. 2005, 386 Pt 1, 1–13. [Google Scholar] [CrossRef]
- Dong, Q.; Li, F. Cell Cycle Control of Kinetochore Assembly. Nucleus 2022, 13, 210–222. [Google Scholar] [CrossRef]
- Yuen, K.W.; Montpetit, B.; Hieter, P. The Kinetochore and Cancer: What’s the Connection? Curr. Opin. Cell Biol. 2005, 17, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA Sequence-Specific Binding of CENP-B Enhances the Fidelity of Human Centromere Function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef]
- Rošić, S.; Köhler, F.; Erhardt, S. Repetitive Centromeric Satellite RNA Is Essential for Kinetochore Formation and Cell Division. J. Cell Biol. 2014, 207, 335–349. [Google Scholar] [CrossRef]
- Molina, O.; Vargiu, G.; Abad, M.A.; Zhiteneva, A.; Jeyaprakash, A.A.; Masumoto, H.; Kouprina, N.; Larionov, V. Epigenetic Engineering Reveals a Balance between Histone Modifications and Transcription in Kinetochore Maintenance. Nat. Commun. 2016, 7, 13334. [Google Scholar] [CrossRef]
- Bobkov, G.O.M.; Gilbert, N.; Heun, P. Centromere Transcription Allows CENP-A to Transit from Chromatin Association to Stable Incorporation. J. Cell Biol. 2018, 217, 1957–1972. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Yuen, K.W.Y. Point Centromere Activity Requires an Optimal Level of Centromeric Noncoding RNA, Proc. Natl. Acad. Sci. USA 2019, 116, 6270–6279. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.L.; Straight, A.F. RNA-mediated regulation of heterochromatin, Curr. Opin. Cell Biol. 2017, 46, 102–109. [Google Scholar] [CrossRef]
- Janssen, S.U.C.; Karpen, G.H. Heterochromatin: Guardian of the genome, Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef]
- Kang, Z.; Li, R.; Liu, C.; Dong, X.; Hu, Y.; Xu, L.; Liu, X.; Xiang, Y.; Gao, L.; Si, W.; et al. m6A-Modified cenRNA Stabilizes CENPA to Ensure Centromere Integrity in Cancer Cells. Cell 2024, 187, 6035–6054. [Google Scholar] [CrossRef]
- Ding, Y.; Li, Y.; Duan, Y.; Wang, W.; Zheng, W.; Cheng, W.; Qi, Y.; Feng, J.; Chen, Z.; Yu, T.; et al. LncRNA MBNL1-AS1 Represses Proliferation and Cancer Stem-Like Properties of Breast Cancer through MBNL1-AS1/ZFP36/CENPA Axis. J. Oncol. 2022, 2022, 9999343. [Google Scholar] [CrossRef] [PubMed]
- Hédouin, S.; Grillo, G.; Ivkovic, I.; Velasco, G.; Francastel, C. CENP-A Chromatin Disassembly in Stressed and Senescent Murine Cells. Sci. Rep. 2017, 7, 42520. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, M.C.; Kuich, P.H.; Stellfox, M.E.; Ward, J.A.; Bassett, E.A.; Black, B.E.; Foltz, D.R. HJURP Is a CENP-A Chromatin Assembly Factor Sufficient to Form a Functional de Novo Kinetochore. J. Cell Biol. 2011, 194, 229–243. [Google Scholar] [CrossRef]
- Giunta, S.; Hervé, S.; White, R.R.; Wilhelm, T.; Dumont, M.; Scelfo, A.; Gamba, R.; Wong, C.K.; Rancati, G.; Smogorzewska, A.; et al. CENP-A Chromatin Prevents Replication Stress at Centromeres to Avoid Structural Aneuploidy. Proc. Natl. Acad. Sci. USA 2021, 118, e2015634118. [Google Scholar] [CrossRef] [PubMed]
- Balachandra, V.; Shrestha, R.L.; Hammond, C.M.; Lin, S.; Hendriks, I.A.; Sethi, S.C.; Chen, L.; Sevilla, S.; Caplen, N.J.; Chari, R.; et al. DNAJC9 Prevents CENP-A Mislocalization and Chromosomal Instability by Maintaining the Fidelity of Histone Supply Chains. EMBO J. 2024, 43, 2166–2197. [Google Scholar] [CrossRef] [PubMed]
- McAinsh, A.D.; Meraldi, P.; Draviam, V.M.; Toso, A.; Sorger, P.K. The Human Kinetochore Proteins Nnf1R and Mcm21R Are Required for Accurate Chromosome Segregation. EMBO J. 2006, 25, 4033–4049. [Google Scholar] [CrossRef] [PubMed]
- McClelland, S.E.; Borusu, S.; Amaro, A.C.; Winter, J.R.; Belwal, M.; McAinsh, A.D.; Meraldi, P. The CENP-A NAC/CAD Kinetochore Complex Controls Chromosome Congression and Spindle Bipolarity. EMBO J. 2007, 26, 5033–5047. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Winter, J.R.; Garrod, A.J.; Amaro, A.C.; Meraldi, P.; McAinsh, A.D. Kinetochore-Generated Pushing Forces Separate Centrosomes during Bipolar Spindle Assembly. J. Cell Biol. 2009, 184, 365–372. [Google Scholar] [CrossRef]
- Okada, M.; Cheeseman, I.M.; Hori, T.; Okawa, K.; McLeod, I.X.; Yates, J.R.; Desai, A.; Fukagawa, T. The CENP-H–I Complex Is Required for the Efficient Incorporation of Newly Synthesized CENP-A into Centromeres. Nat. Cell Biol. 2006, 8, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Xiong, J.; Li, Z.; Zhang, G.; Tu, Y.; Wang, L. CENPO Expression Regulates Gastric Cancer Cell Proliferation and Is Associated with Poor Patient Prognosis. Mol. Med. Rep. 2019, 20, 3661–3670. [Google Scholar] [CrossRef]
- Si, L.H.; Sun, G.C.; Liu, Z.W.; Gu, S.Y.; Yan, C.H.; Xu, J.Y.; Jia, Y. High Expression Levels of Centromere Protein O Participates in Cell Proliferation of Human Ovarian Cancer. J. Ovarian Res. 2024, 17, 126. [Google Scholar] [CrossRef]
- Wang, D.; Xu, W.; Huang, M.; Ma, W.; Liu, Y.; Zhou, X.; Yang, Q.; Mu, K. CENPF Knockdown Inhibits Adriamycin Chemoresistance in Triple-Negative Breast Cancer via the Rb-E2F1 Axis. Sci. Rep. 2023, 13, 1803. [Google Scholar] [CrossRef]
- Hori, T.; Okada, M.; Maenaka, K.; Fukagawa, T. CENP-O-Class Proteins Form a Stable Complex and Are Required for Proper Kinetochore Function. Mol. Biol. Cell 2008, 19, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Muro, Y.; Sugiura, K.; Ikeno, M.; Yoda, K.; Tomita, Y. CENP-O, a Protein Localized at the Centromere throughout the Cell Cycle, Is a Novel Target Antigen in Systemic Sclerosis. J. Rheumatol. 2009, 36, 781–786. [Google Scholar] [CrossRef]
- Izuta, H.; Ikeno, M.; Suzuki, N.; Tomonaga, T.; Nozaki, N.; Obuse, C.; Kisu, Y.; Goshima, N.; Nomura, F.; Nomura, N.; et al. Comprehensive Analysis of the ICEN (Interphase Centromere Complex) Components Enriched in the CENP-A Chromatin of Human Cells. Genes. Cells 2006, 11, 673–684. [Google Scholar] [CrossRef]
- Fang, J.; Liu, Y.; Wei, Y.; Deng, W.; Yu, Z.; Huang, L.; Teng, Y.; Yao, T.; You, Q.; Ruan, H.; et al. Structural Transitions of Centromeric Chromatin Regulate the Cell Cycle-Dependent Recruitment of CENP-N. Genes. Dev. 2015, 29, 1058–1073. [Google Scholar] [CrossRef]
- Gui, Z.; Tian, Y.; Liu, S.; Yu, T.; Liu, C.; Zhang, L. Highly Expressed CENPL Is Correlated with Breast Cancer Cell Proliferation and Immune Infiltration. Front. Oncol. 2023, 13, 1046774. [Google Scholar] [CrossRef]
- Yang, Z.Y. Mitosin/CENP-F Is a Conserved Kinetochore Protein Subjected to Cytoplasmic Dynein-Mediated Poleward Transport. Cell Res. 2003, 13, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Holt, S.V. Silencing Cenp-F Weakens Centromeric Cohesion, Prevents Chromosome Alignment and Activates the Spindle Checkpoint. J. Cell Sci. 2005, 118, 4889–4900. [Google Scholar] [CrossRef]
- Yang, Z. Silencing Mitosin Induces Misaligned Chromosomes, Premature Chromosome Decondensation before Anaphase Onset, and Mitotic Cell Death. Mol. Cell Biol. 2005, 25, 4062–4074. [Google Scholar] [CrossRef] [PubMed]
- Pooley, R.D. Murine CENPF Interacts with Syntaxin 4 in the Regulation of Vesicular Transport. J. Cell Sci. 2008, 121, 3413–3421. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M. The Kinetochore Protein, CENPF, Is Mutated in Human Ciliopathy and Microcephaly Phenotypes. J. Med. Genet. 2015, 52, 147–156. [Google Scholar] [CrossRef]
- Huang, Y.; Li, D.; Wang, L.; Su, X.; Tang, X. CENPF/CDK1 Signaling Pathway Enhances the Progression of Adrenocortical Carcinoma by Regulating the G2/M-Phase Cell Cycle. J. Transl. Med. 2022, 20, 78. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Winkfein, R.J.; Mack, G.; Rattner, J.B.; Yen, T.J. CENP-F Is a Protein of the Nuclear Matrix That Assembles onto Kinetochores at Late G2 and Is Rapidly Degraded after Mitosis. J. Cell Biol. 1995, 130, 507–518. [Google Scholar] [CrossRef]
- Khurana, S.; Varma, D.; Foltz, D.R. Contribution of CENP-F to FOXM1-Mediated Discordant Centromere and Kinetochore Transcriptional Regulation. Mol. Cell Biol. 2024, 44, 209–225. [Google Scholar] [CrossRef]
- Volkov, V.A.; Grissom, P.M.; Arzhanik, V.K.; Zaytsev, A.V.; Renganathan, K.; McClure-Begley, T.; Old, W.M.; Ahn, N.; McIntosh, J. Centromere Protein F Includes Two Sites That Couple Efficiently to Depolymerizing Microtubules. J. Cell Biol. 2015, 209, 813–828. [Google Scholar] [CrossRef]
- Han, Y.; Xu, S.; Cheng, K.; Diao, C.; Liu, S.; Zou, W.; Bi, Y. CENPF Promotes Papillary Thyroid Cancer Progression by Mediating Cell Proliferation and Apoptosis. Exp. Ther. Med. 2021, 21, 401. [Google Scholar] [CrossRef]
- Chen, Q.; Xu, H.; Zhu, J.; Feng, K.; Hu, C. LncRNA MCM3AP-AS1 Promotes Breast Cancer Progression via Modulating miR-28-5p/CENPF Axis. Biomed. Pharmacother. 2020, 128, 110289. [Google Scholar] [CrossRef]
- Zhou, M.F.; Wang, W.; Wang, L.; Tan, J.D. LINC00536 Knockdown Inhibits Breast Cancer Cells Proliferation, Invasion, and Migration through Regulation of the miR-4282/Centromere Protein F Axis. Kaohsiung J. Med. Sci. 2022, 38, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Huang, J.; Lan, J.; Zhou, K.; Gao, Y.; Song, Z.; Deng, Y.; Liu, L.; Dong, Y.; Liu, X. Overexpression of CENPF Correlates with Poor Prognosis and Tumor Bone Metastasis in Breast Cancer. Cancer Cell Int. 2019, 19, 264. [Google Scholar] [CrossRef]
- Jacquet, E.; Chuffart, F.; Vitte, A.L.; Nika, E.; Mousseau, M.; Khochbin, S.; Rousseaux, S.; Bourova-Flin, E. Aberrant Activation of Five Embryonic Stem Cell-Specific Genes Robustly Predicts a High Risk of Relapse in Breast Cancers. BMC Genom. 2023, 24, 463. [Google Scholar] [CrossRef]
- Campone, M.; Campion, L.; Roché, H.; Gouraud, W.; Charbonnel, C.; Magrangeas, F.; Minvielle, S.; Genève, J.; Martin, A.L.; Bataille, R.; et al. Prediction of metastatic relapse in node-positive breast cancer: Establishment of a clinicogenomic model after FEC100 adjuvant regimen. Breast Cancer Res. Treat. 2008, 109, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Kondoh, N.; Tsuda, H.; Yamamoto, S.; Asakawa, H.; Fukatsu, K.; Kobayashi, T.; Yamamoto, J.; Tamura, K.; Ishida, J.; et al. Expression of Centromere Protein F (CENP-F) Associated with Higher FDG Uptake on PET/CT, Detected by cDNA Microarray, Predicts High-Risk Patients with Primary Breast Cancer. BMC Cancer 2008, 8, 384. [Google Scholar] [CrossRef]
- Zhou, M.; Li, X.; Wang, W.; Wu, J.; Tan, J. PSMD14/E2F1 Axis-Mediated CENPF Promotes the Metastasis of Triple-Negative Breast Cancer Through Inhibiting Ferroptosis. Cancer Sci. 2025. [Google Scholar] [CrossRef]
- Williams, J.M.; Chen, G.C.; Zhu, L. Using the Yeast Two-Hybrid System to Identify Human Epithelial Cell Proteins That Bind Gonococcal Opa Proteins: Intracellular Gonococci Bind Pyruvate Kinase via Their Opa Proteins and Require Host Pyruvate for Growth. Mol. Microbiol. 1998, 27, 171–186. [Google Scholar] [CrossRef]
- Fu, H.; Liu, N.; Dong, Q. SENP6-mediated M18BP1 deSUMOylation regulates CENP-A centromeric localization. Cell Res. 2019, 29, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, M.B.; Shirkoohi, R.; Modarressi, M.H. Cancer/Testis OIP5 and TAF7L Genes Are Up-Regulated in Breast Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 4623–4628. [Google Scholar] [CrossRef]
- He, B.; Gnawali, N.; Hinman, A.W.; Mattingly, A.J.; Osimani, A.; Cimini, D. Chromosomes Missegregated into Micronuclei Contribute to Chromosomal Instability by Missegregating at the next Division. Oncotarget 2019, 10, 2660–2674. [Google Scholar] [CrossRef] [PubMed]
- Ghafouri-Fard, S.; Modarressi, M.H. Cancer-Testis Antigens: Potential Targets for Cancer Immunotherapy. Arch. Iran. Med. 2009, 12, 395–404. [Google Scholar] [PubMed]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Yazarloo, F.; Shirkoohi, R.; Mobasheri, M.B.; Emami, A.; Modarressi, M.H. Expression Analysis of Four Testis-Specific Genes AURKC, OIP5, PIWIL2 and TAF7L in Acute Myeloid Leukemia: A Gender-Dependent Expression Pattern. Med. Oncol. 2013, 30, 368. [Google Scholar] [CrossRef]
- Freitas, M.; Malheiros, S.; Stávale, J.N.; Biassi, T.P.; Zamunér, F.T.; Souza Begnami, M.; Soares, F.A.; Vettore, A.L. Expression of Cancer/Testis Antigens Is Correlated with Improved Survival in Glioblastoma. Oncotarget 2013, 4, 636–646. [Google Scholar] [CrossRef]
- Chun, H.K.; Chung, K.S.; Kim, H.C.; Kang, J.E.; Kang, M.A.; Kim, J.T.; Choi, E.H.; Jung, K.E.; Kim, M.H.; Song, E.Y.; et al. OIP5 Is a Highly Expressed Potential Therapeutic Target for Colorectal and Gastric Cancers. BMB Rep. 2010, 43, 349–354. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Hou, J.; Ping, J. Opa Interacting Protein 5 Acts as an Oncogene in Bladder Cancer. J. Cancer Res. Clin. Oncol. 2017, 143, 2221–2233. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Tanaka, F.; Nagahara, H.; Ieta, K.; Haraguchi, N.; Mimori, K.; Sasaki, A.; Inoue, H.; Yanaga, K.; Mori, M. Opa Interacting Protein 5 (OIP5) Is a Novel Cancer-Testis Specific Gene in Gastric Cancer. Ann. Surg. Oncol. 2007, 14, 885–892. [Google Scholar] [CrossRef]
- Zhu, M.; Takano, A.; Tsevegjav, B.; Yoshitake, Y.; Shinohara, M.; Daigo, Y. Characterization of Opa Interacting Protein 5 as a New Biomarker and Therapeutic Target for Oral Cancer. Int. J. Oncol. 2022, 60, 27. [Google Scholar] [CrossRef]
- Koinuma, J.; Akiyama, H.; Fujita, M. Characterization of an Opa Interacting Protein 5 Involved in Lung and Esophageal Carcinogenesis. Cancer Sci. 2012, 103, 577–586. [Google Scholar] [CrossRef]
- Li, H.C.; Chen, Y.F.; Feng, W.; Cai, H.; Mei, Y.; Jiang, Y.M.; Chen, T.; Xu, K.; Feng, D.X. Loss of the Opa Interacting Protein 5 Inhibits Breast Cancer Proliferation through miR-139-5p/NOTCH1 Pathway. Gene 2017, 603, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kimmins, S.; Kotaja, N.; Davidson, I.; Sassone-Corsi, P. Testis-Specific Transcription Mechanisms Promoting Male Germ-Cell Differentiation. Reproduction 2004, 128, 5–12. [Google Scholar] [CrossRef]
- O’Brien, S.L.; Fagan, A.; Fox, E.J.; Millikan, R.C.; Culhane, A.C.; Brennan, D.J.; McCann, A.H.; Hegarty, S.; Moyna, S.; Duffy, M.J.; et al. CENP-F Expression Is Associated with Poor Prognosis and Chromosomal Instability in Patients with Primary Breast Cancer. Int. J. Cancer 2007, 120, 1434–1443. [Google Scholar] [CrossRef]
- Santaguida, S.; Amon, A. Short- and Long-Term Effects of Chromosome Mis-Segregation and Aneuploidy. Nat. Rev. Mol. Cell Biol. 2015, 16, 473–485. [Google Scholar] [CrossRef]
- Filipescu, D.; Naughtin, M.; Podsypanina, K.; Lejour, V.; Wilson, L.; Gurard-Levin, Z.A.; Orsi, G.A.; Simeonova, I.; Toufektchan, E.; Attardi, L.D.; et al. Essential Role for Centromeric Factors Following P53 Loss and Oncogenic Transformation. Genes. Dev. 2017, 31, 463–480. [Google Scholar] [CrossRef]
- Maehara, K.; Takahashi, K.; Saitoh, S. CENP-A Reduction Induces a P53-Dependent Cellular Senescence Response to Protect Cells from Executing Defective Mitoses. Mol. Cell Biol. 2010, 30, 2090–2104. [Google Scholar] [CrossRef]
- Jeffery, D.; Gatto, A.; Podsypanina, K.; Renaud-Pageot, C.; Ponce Landete, R.; Bonneville, L.; Dumont, M.; Fachinetti, D.; Almouzni, G. CENP-A Overexpression Promotes Distinct Fates in Human Cells, Depending on P53 Status. Commun. Biol. 2021, 4, 417. [Google Scholar] [CrossRef]
- Hershko, A. Ubiquitin: Roles in Protein Modification and Breakdown. Cell 1983, 34, 11–12. [Google Scholar] [CrossRef]
- Wilkinson, K.D. Protein ubiquitination: A regulatory post-translational modification. Anticancer Drug Des 1987.
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.J.; Harper, J.W. DNA damage: Ubiquitin marks the spot. Nat. Struct. Mol. Biol. 2008, 15, 20–22. [Google Scholar] [CrossRef]
- Chen, Z.J.; Sun, L.J. Nonproteolytic Functions of Ubiquitin in Cell Signaling. Mol. Cell 2009, 33, 275–286. [Google Scholar] [CrossRef]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 Ubiquitin Ligases in Cancer and Implications for Therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Ozaki, T.; Miyazaki, K.; Aoyama, M.; Miyazaki, M.; Nakagawara, A. UbcH10 Is the Cancer-Related E2 Ubiquitin- Conjugating Enzyme. Cancer Res. 2003, 63, 4167–4173. [Google Scholar]
- Meyer, H.J.; Rape, M. Processive Ubiquitin Chain Formation by the Anaphase-Promoting Complex. Semin. Cell Dev. Biol. 2011, 22, 544–550. [Google Scholar] [CrossRef]
- Bastians, H. Cell Cycle-Regulated Proteolysis of Mitotic Target Proteins. Mol. Biol. Cell 1999, 10, 3927–3941. [Google Scholar] [CrossRef]
- Van Ree, J.H.; Jeganathan, K.B.; Malureanu, L.; Van Deursen, J.M. Overexpression of the E2 Ubiquitin–Conjugating Enzyme UbcH10 Causes Chromosome Missegregation and Tumor Formation. J. Cell Biol. 2010, 188, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Guo, L. Forkhead Box M1 Positively Regulates UBE2C and Protects Glioma Cells from Autophagic Death. Cell Cycle 2017, 16, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Guo, J. Deregulation of UBE2C-Mediated Autophagy Repression Aggravates NSCLC Progression. Oncogenesis 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Zhang, H.; Cowell, J. Ubiquitin-Conjugating Enzyme UBE2C: Molecular Biology, Role in Tumorigenesis, and Potential as a Bio- Marker. Tumour Biol. 2012, 33, 723–730. [Google Scholar] [CrossRef]
- Shen, Z.; Jiang, X.; Zeng, C.; Zheng, S.; Luo, B.; Zeng, Y. High expression of ubiquitin-conjugating enzyme 2C (UBE2C) corre- lates with nasopharyngeal carcinoma progression. BMC Cancer 2013, 13. [Google Scholar] [CrossRef]
- Zhang, S.; You, X.; Zheng, Y.; Shen, Y.; Xiong, X.; Sun, Y. The UBE2C/CDH1/DEPTOR Axis Is an Oncogene and Tumor Suppressor Cascade in Lung Cancer Cells. J. Clin. Investig. 2023, 133, 162434. [Google Scholar] [CrossRef]
- Peterson, T.R. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef]
- Bi, X.; Pohl, N.M.; Qian, Z.; Yang, G.R.; Gou, Y.; Guzman, G.; Kajdacsy-Balla, A.; Iozzo, R.V.; Yang, W. Decorin-Mediated Inhibition of Colorectal Cancer Growth and Migration Is Associated with E-Cadherin in Vitro and in Mice. Carcinogenesis 2012, 33, 326–330. [Google Scholar] [CrossRef]
- Pei, L. Overexpression of DEP Domain Containing mTOR-Interacting Protein Correlates with Poor Prognosis in Differentiated Thyroid Carcinoma. Mol. Med. Rep. 2011, 4, 817–823. [Google Scholar] [CrossRef]
- Hu, Y. DEPTOR Is a Direct NOTCH1 Target That Promotes Cell Proliferation and Survival in T-Cell Leukemia. Oncogene 2017, 36, 1038–1047. [Google Scholar] [CrossRef]
- Wang, Q. Deptor Is a Novel Target of Wnt/β-Catenin/c-Myc and Contributes to Colorectal Cancer Cell Growth. Cancer Res. 2018, 78, 3163–3175. [Google Scholar] [CrossRef]
- Chen, J. DEPTOR Induces a Partial Epithelial-to-Mesenchymal Transition and Metastasis via Autocrine TGFβ1 Signaling and Is Associated with Poor Prognosis in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2019, 38. [Google Scholar] [CrossRef]
- Loussouarn, D.; Campion, L.; Leclair, F.; Campone, M.; Charbonnel, C.; Ricolleau, G.; Gouraud, W.; Bataille, R.; Jézéquel, P. Validation of UBE2C Protein as a Prognostic Marker in Node-Positive Breast Cancer. Br. J. Cancer 2009. [Google Scholar] [CrossRef]
- Psyrri, A.; Kalogeras, K.T.; Kronenwett, R.; Wirtz, R.M.; Batistatou, A.; Bournakis, E.; Timotheadou, E.; Gogas, H.; Aravantinos, G.; Christodoulou, C.; et al. Prognostic Significance of UBE2C mRNA Expression in High-Risk Early Breast Cancer. A Hellenic Cooperative Oncology Group (HeCOG) Study. Ann. Oncol. 2012. [Google Scholar] [CrossRef]
- Zeng, H.; Irwin, M.L.; Lu, L.; Risch, H.; Mayne, S.; Mu, L.; Deng, Q.; Scarampi, L.; Mitidieri, M.; Katsaros, D.; et al. Physical Activity and Breast Cancer Survival: An Epigenetic Link through Reduced Methylation of a Tumor Suppressor Gene L3MBTL1. Breast Cancer Res. Treat. 2012, 133, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kariri, Y.; Toss, M.S.; Alsaleem, M.; Elsharawy, K.A.; Joseph, C.; Mongan, N.P.; Green, A.R.; Rakha, E.A. Ubiquitin-Conjugating Enzyme 2C (UBE2C) Is a Poor Prognostic Biomarker in Invasive Breast Cancer. Breast Cancer Res. Treat. 2022, 192, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, X.; Zhang, X.; Hu, X. UBE2S and UBE2C Confer a Poor Prognosis to Breast Cancer via Downregulation of Numb. Front. Oncol. 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Chen, Y.; Carpio, A.; Chang, C.; Iyengar, N.M. Incidence, Risk Factors, and Management of Alpelisib-associated Hyperglycemia in Metastatic Breast Cancer. Cancer 2023, 129, 3854–3861. [Google Scholar] [CrossRef] [PubMed]
- Freitas, A.J.A.D.; Causin, R.L.; Calfa, S.; Santana, I.V.V.; Balie, M.; Hirai, W.; Laus, A.C.; Araujo, H.S.; Filho, C.M.H.; de Oliveira Bombarda, F.; et al. Exploring the Nottingham Classification: Assessing Gene Expression Profiles in Breast Cancer Patients and Their Association with Outcomes. Breast Cancer Res. Treat. 2025. [Google Scholar] [CrossRef] [PubMed]
- Colaluca, I.N.; Tosoni, D.; Nuciforo, P.; Senic-Matuglia, F.; Galimberti, V.; Viale, G.; Pece, S.; Fiore, P.P. NUMB controls p53 tumour suppressor activity. Nature 2008. [Google Scholar] [CrossRef]
- Di Marcotullio, L.; Greco, A.; Mazzà, D.; Canettieri, G.; Pietrosanti, L.; Infante, P.; Coni, S.; Moretti, M.; De Smaele, E.; Ferretti, E.; et al. Numb Activates the E3 Ligase Itch to Control Gli1 Function through a Novel Degradation Signal. Oncogene 2011, 30, 65–76. [Google Scholar] [CrossRef]
- Tosoni, D.; Zecchini, S.; Coazzoli, M.; Colaluca, I.; Mazzarol, G.; Rubio, A.; Caccia, M.; Villa, E.; Zilian, O.; Fiore, P.P.; et al. The Numb/P53 Circuitry Couples Replicative Self-Renewal and Tumor Suppression in Mammary Epithelial Cells. J. Cell Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Lou, F.; Yu, F.; Zhang, D.; Wang, C.; Wu, F.; Li, X.; Ping, Y.; Yang, X.; Yang, J.; et al. NUMB Maintains Bone Mass by Promoting Degradation of PTEN and GLI1 via Ubiquitination in Osteoblasts. Bone Res. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Pece, S.; Serresi, M.; Santolini, E.; Capra, M.; Hulleman, E.; Galimberti, V. Loss of Negative Regulation by Numb over Notch Is Relevant to Human Breast Carcinogenesis. J. Cell Biol. 2004, 167, 215–221. [Google Scholar] [CrossRef]
- Zhang, J.; Shao, X.; Sun, H.; Liu, K.; Ding, Z.; Chen, J.; Fang, L.; Su, W.; Hong, Y.; Li, H.; et al. NUMB Negatively Regulates the Epithelial-Mesenchymal Transition of Triple-Negative Breast Cancer by Antagonizing Notch Signaling. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, X. UBE2C Promotes LUAD Progression by Ubiquitin-Dependent Degradation of P53 to Inactivate the P53/P21 Signaling Pathway. Discov. Oncol. 2024, 15, 589. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cruz, A.B.; Santos, M.; Lara, M.F.; Segrelles, C.; Ruiz, S.; Moral, M.; Lorz, C.; Garcia-Escudero, R.; Paramio, J.M. Spontaneous Squamous Cell Carcinoma Induced by the Somatic Inactivation of Retinoblastoma and Trp53 Tumor Suppressors. Cancer Res. 2008, 68, 683–692. [Google Scholar] [CrossRef]
- Bajaj, S.; Alam, S.K.; Roy, K.S.; Datta, A.; Nath, S.; Roychoudhury, S. E2 Ubiquitin-Conjugating Enzyme, UBE2C Gene, Is Reciprocally Regulated by Wild-Type and Gain-of-Function Mutant P53. J. Biol. Chem. 2016. [Google Scholar] [CrossRef]
- García-Escudero, R.; Martínez-Cruz, A.B.; Santos, M.; Lorz, C.; Segrelles, C.; Garaulet, G.; Saiz-Ladera, C.; Costa, C.; Buitrago-Pérez, Á.; Dueñas, M.; et al. Gene Expression Profiling of Mouse P53-Deficient Epidermal Carcinoma Defines Molecular Determinants of Human Cancer Malignancy. Mol. Cancer 2010, 9, 193. [Google Scholar] [CrossRef]
- Xu, J.; Chen, M.; Hu, M.; Wang, H.; Zuo, Z.; Wang, J.; Xie, Z. Claudin 19 Inhibits the Malignant Potential of Breast Cancer Cells by Modulating Extracellular Matrix-Associated UBE2C/Wnt Signaling. Am. J. Cancer Res. 2022, 12, 5552–5563. [Google Scholar] [PubMed]
- Han, Q.; Zhou, C.; Liu, F.; Xu, G.; Zheng, R.; Zhang, X. MicroRNA-196a Post-Transcriptionally Upregulates the UBE2C Proto-Oncogene and Promotes Cell Proliferation in Breast Cancer. Oncol. Rep. 2015. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-Activated microRNA, Regulates E-Cadherin and Cancer Metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef]
- Hussain, M.; Torres, S.; Schnettler, E.; Funk, A.; Grundhoff, A.; Pijlman, G.P.; AA, K.; Asgari, S. West Nile Virus Encodes a microRNA-like Small RNA in the 3′ Untranslated Region Which up-Regulates GATA4 mRNA and Facilitates Virus Replication in Mosquito Cells. Nucleic Acids Res. 2012, 40, 2210–2223. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, G.; Han, L.; Bai, X.; Xi, Z.; Wang, F.; Han, G. Circ_0059457 Promotes Proliferation, Metastasis, Sphere Formation and Glycolysis in Breast Cancer Cells by Sponging miR-140-3p to Regulate UBE2C. Biochem. Genet. 2024, 62, 125–143. [Google Scholar] [CrossRef]
- Zhai, D.; Zhang, M.; Li, Y.; Bi, J.; Kuang, X.; Shan, Z.; Shao, N.; Lin, Y. LINC01194 Recruits NUMA1 to Promote Ubiquitination of RYR2 to Enhance Malignant Progression in Triple-Negative Breast Cancer. Cancer Lett. 2022. [Google Scholar] [CrossRef]
- Rawat, A.; Gopal, G.; Selvaluxmy, G.; Rajkumar, T. Inhibition of Ubiquitin Conjugating Enzyme UBE2C Reduces Proliferation and Sensitizes Breast Cancer Cells to Radiation, Doxorubicin, Tamoxifen and Letrozole. Cell Oncol. 2013, 36, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Mansfeld, J.; Godwin, C.; Matsusaka, T.; Wu, J.; Russell, P. UBE2S Elongates Ubiquitin Chains on APC/C Substrates to Promote Mitotic Exit. Nat. Cell Biol. 2009, 11, 1363–1369. [Google Scholar] [CrossRef] [PubMed]
- Yeong, F.M.; Lim, H.H.; Padmashree, C.G.; Surana, U. Exit from Mitosis in Budding Yeast: Biphasic Inactivation of the Cdc28-Clb2 Mitotic Kinase and the Role of Cdc20. Mol. Cell 2000. [Google Scholar] [CrossRef]
- Jung, C.R.; Hwang, K.S.; Yoo, J.S. E2-EPF UCP Targets pVHL for Degradation and Associates with Tumor Growth and Metastasis. Nat. Med. 2006, 12, 809–816. [Google Scholar] [CrossRef]
- LaGory, E.L.; Giaccia, A.J. The Ever-Expanding Role of HIF in Tumour and Stromal Biology. Nat. Cell Biol. 2016, 18, 356–365. [Google Scholar] [CrossRef]
- Wang, B.; Xu, X.; Yang, Z.; Zhang, L.; Liu, Y.; Ma, A. POH1 contributes to hyperactivation of TGF-beta signaling and facilitates hepatocellular carcinoma metastasis through deubiquitinating TGF-beta receptors and caveolin-1. EBioMedicine 2019, 41, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Hu, L. Molecular and Clinical Characterization of UBE2S in Glioma as a Biomarker for Poor Prognosis and Resistance to Chemo-Radiotherapy. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Tedesco, D.; Zhang, J.; Trinh, L.; Lalehzadeh, G.; Meisner, R.; Yamaguchi, K.D. The Ubiquitin-Conjugating Enzyme E2-EPF Is Overexpressed in Primary Breast Cancer and Modulates Sensitivity to Topoisomerase II Inhibition. Neoplasia 2007, 9, 601–613. [Google Scholar] [CrossRef]
- Roos, F.C.; Evans, A.J.; Brenner, W.; Wondergem, B.; Klomp, J.; Heir, P. Deregulation of E2-EPF Ubiquitin Carrier Protein in Papillary Renal Cell Carcinoma. Am. J. Pathol. 2011, 178, 853–860. [Google Scholar] [CrossRef]
- Liang, J.; Nishi, H.; Bian, M.L.; Higuma, C.; Sasaki, T.; Ito, H. The Ubiquitin-Conjugating Enzyme E2-EPF Is Overexpressed in Cervical Cancer and Associates with Tumor Growth. Oncol. Rep. 2012, 28, 1519–1525. [Google Scholar] [CrossRef]
- Clarke, C.; Madden, S.F.; Doolan, P.; Aherne, S.T.; Joyce, H.; O’Driscoll, L. Correlating Transcriptional Networks to Breast Cancer Survival: A Large-Scale Coexpression Analysis. Carcinogenesis 2013, 34, 2300–2308. [Google Scholar] [CrossRef] [PubMed]
- Ayesha, A.K.; Hyodo, T.; Asano, E.; Sato, N.; Mansour, M.A.; Ito, S.; Hamaguchi, M.; Senga, T. UBE2S Is Associated with Malignant Characteristics of Breast Cancer Cells. Tumour Biol. 2016, 37, 763–772. [Google Scholar] [CrossRef]
- Yoshimura, S.; Kasamatsu, A.; Nakashima, D.; Iyoda, M.; Kasama, H.; Saito, T.; Takahara, T.; Endo-Sakamoto, Y.; Shiiba, M.; Tanzawa, H.; et al. UBE2S Associated with OSCC Proliferation by Promotion of P21 Degradation via the Ubiquitin-Proteasome System. Biochem. Biophys. Res. Commun. 2017, 485, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.H.; Yang, M.; Liu, L.P.; Wu, D.C.; Li, M.Y.; Su, S.G. UBE2S Enhances the Ubiquitination of P53 and Exerts Oncogenic Activities in Hepatocellular Carcinoma. Biochem. Biophys. Res. Commun. 2018, 503, 895–902. [Google Scholar] [CrossRef]
- Lin, M.; Lei, T.; Zheng, J.; Chen, S.; Du, L.; Xie, H. UBE2S mediates tumor progression via SOX6/β-Catenin signaling in endometrial cancer. Int. J. Biochem. Cell Biol. 2019, 109, 17–22. [Google Scholar] [CrossRef]
- Qin, Y.; Du, J.; Fan, C. Ube2S Regulates Wnt/β-Catenin Signaling and Promotes the Progression of Non-Small Cell Lung Cancer. Int. J. Med. Sci. 2020. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, J.; Zhang, Z.; Guo, Y.; Lou, X.; Zhang, L. Diverse Roles of UBE2S in Cancer and Therapy Resistance: Biological Functions and Mechanisms. Heliyon 2024, 10, e24465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.Y.; Liu, Z.K.; Wei, D.; Yong, Y.L.; Lin, P.; Li, H.; Liu, M.; Zheng, N.S.; Liu, K.; Hu, C.X.; et al. UBE2S Interacting with TRIM28 in the Nucleus Accelerates Cell Cycle by Ubiquitination of P27 to Promote Hepatocellular Carcinoma Development. Signal Transduct. Target. Ther. 2021, 6, 64. [Google Scholar] [CrossRef]
- Gui, L.; Zhang, S.; Xu, Y. UBE2S Promotes Cell Chemoresistance through PTEN-AKT Signaling in Hepatocellular Carcinoma. Cell Death Discov. 2021, 7, 357. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, D.; Zhang, J.; Trinh, L.; Lalehzadeh, G.; Meisner, R.; Yamaguchi, K.D. The Ubiquitin-Conjugating Enzyme E2-EPF Is Overexpressed in Primary Breast Cancer and Modulates Sensitivity to Topoisomerase II Inhibition. Neoplasia 2007, 9, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Lu, W.; Chen, B.; Zhao, K. Identification of a Novel Ubiquitination Related Gene Signature for Patients with Breast Cancer. Medicine 2022, 101, e30598. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jin, G.; Qian, J.; Yang, H.; Tang, H.; Meng, X.; Li, Y. Digital Gene Expression Profiling Analysis and Its Application in the Identification of Genes Associated with Improved Response to Neoadjuvant Chemotherapy in Breast Cancer. World J. Surg. Oncol. 2018, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Thu, K.L.; Silvester, J.; Elliott, M.J.; Ba-Alawi, W.; Duncan, M.H.; Elia, A.C.; Mer, A.S.; Smirnov, P.; Safikhani, Z.; Haibe-Kains, B.; et al. Disruption of the Anaphase-Promoting Complex Confers Resistance to TTK Inhibitors in Triple-Negative Breast Cancer. In Proc. Natl. Acad. Sci. USA 2018, 115, E1570–E1577. [Google Scholar] [CrossRef]
- Machida, Y.J.; Machida, Y.; Chen, Y.; Gurtan, A.M.; Kupfer, G.M.; D’Andrea, A.D. UBE2T Is the E2 in the Fanconi Anemia Pathway and Undergoes Negative Autoregulation. Mol. Cell 2006, 23, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Alpi, A.F.; Pace, P.E.; Babu, M.M.; Patel, K.J. Mechanistic Insight into Site-Restricted Monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell 2008, 32, 767–777. [Google Scholar] [CrossRef]
- Ueki, T.; Park, J.H.; Nishidate, T.; Kijima, K.; Hirata, K.; Nakamura, Y. Ubiquitination and Downregulation of BRCA1 by Ubiquitin-Conjugating Enzyme E2T Overexpression in Human Breast Cancer Cells. Cancer Res. 2009, 69, 8752–8760. [Google Scholar] [CrossRef] [PubMed]
- E. , L.; P., M.; E., P.; D, D. UBE2T Promotes Beta-Catenin Nuclear Translocation in Hepatocellular Carcinoma through MAPK/ERK-Dependent Activation. Mol. Oncol. 2022, 16, 1694–1713. [Google Scholar] [CrossRef]
- J. , Z.; H., A.; M., L.; K., C.; J, M. UBE2T Promotes Autophagy via the P53/AMPK/mTOR Signaling Pathway in Lung Adenocarcinoma. J. Transl. Med. 2021, 19. [Google Scholar]
- Dutta, R.; Guruvaiah, P.; Reddi, K.K.; Bugide, S.; Reddy Bandi, D.S.; Edwards, Y.J.K.; Singh, K.; Gupta, R. UBE2T Promotes Breast Cancer Tumor Growth by Suppressing DNA Replication Stress. NAR Cancer 2022, 4, zcac035. [Google Scholar] [CrossRef] [PubMed]
- Cabañas Morafraile, E.; Pérez-Peña, J.; Fuentes-Antrás, J.; Manzano, A.; Pérez-Segura, P.; Pandiella, A.; Galán-Moya, E.M.; Ocaña, A. Genomic Correlates of DNA Damage in Breast Cancer Subtypes. Cancers 2021, 13, 2117. [Google Scholar] [CrossRef]
- Yagin, B.; Yagin, F.H.; Colak, C.; Inceoglu, F.; Kadry, S.; Kim, J. Cancer Metastasis Prediction and Genomic Biomarker Identification through Machine Learning and eXplainable Artificial Intelligence in Breast Cancer Research. Diagnostics 2023, 13, 3314. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Dong, C.; Ma, B. UBE2T Promotes Proliferation, Invasion and Glycolysis of Breast Cancer Cells by Regulating the PI3K/AKT Signaling Pathway. J. Recept. Signal Transduct. Res. 2022, 42, 151–159. [Google Scholar] [CrossRef]
- Shi, L.L.; Chen, Y.; Xie, M.X.; Chen, Q.Z.; Qiao, X.W.; Cheng, Q.H.; Li, L.; Fu, R.; Liang, T.; Jiang, X.; et al. UBE2T/CDC42/CD276 Signaling Axis Mediates Brain Metastasis of Triple-Negative Breast Cancer via Lysosomal Autophagy. J. Immunother. Cancer 2025, 13, 010782. [Google Scholar] [CrossRef]
- Fuentes-Antrás, J.; Alcaraz-Sanabria, A.L.; Morafraile, E.C.; Noblejas-López, M.D.M.; Galán-Moya, E.M.; Baliu-Pique, M.; López-Cade, I.; García-Barberán, V.; Pérez-Segura, P.; Manzano, A.; et al. Mapping of Genomic Vulnerabilities in the Post-Translational Ubiquitination, SUMOylation and Neddylation Machinery in Breast Cancer. Cancers 2021, 13, 833. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.; Zhi, X.; Li, Q.; Yao, L.; Chen, M. The Expression and Role Analysis of Methylation-Regulated Differentially Expressed Gene UBE2C in Pan-Cancer, Especially for HGSOC. Cancers 2022, 14, 3121. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhao, J.; Pan, B.; Ma, G.; Liu, L. UBE2C Overexpression in Melanoma and Its Essential Role in G2/M Transition. J. Cancer 2019, 10, 2176–2184. [Google Scholar] [CrossRef] [PubMed]
- M. , B.S.; B., A.-O.; A., S.; E., A.; E., L.; K., M.-C.; L., F.C.; A., K.A.; R., D.; C., S.J.; et al. Regulation of Pluripotency and Cellular Reprogramming by the Ubiquitin-Proteasome System. Cell Stem Cell 2012, 11, 783–798. [Google Scholar] [CrossRef]
- Butler, L.R.; Densham, R.M.; Jia, J.; Garvin, A.J.; Stone, H.R.; Shah, V. The Proteasomal De-Ubiquitinating Enzyme POH1 Promotes the Double-Strand DNA Break Response. EMBO J. 2012, 31, 3918–3934. [Google Scholar] [CrossRef]
- Byrne, A.; McLaren, R.P.; Mason, P.; Chai, L.; Dufault, M.R.; Huang, Y. Knockdown of Human Deubiquitinase PSMD14 Induces Cell Cycle Arrest and Senescence. Exp. Cell Res. 2010, 316, 258–271. [Google Scholar] [CrossRef]
- B. , W.; A., M.; L., Z.; L., J.W.; Y., Q.; G., X.; B., Q.; Z., Y.; Y., L.; Q., X.; et al. POH1 Deubiquitylates and Stabilizes E2F1 to Promote Tumour Formation. Nat. Commun. 2015, 6, 8704. [Google Scholar] [CrossRef]
- Luo, G.; Hu, N.; Xia, X.; Zhou, J.; Ye, C. RPN11 Deubiquitinase Promotes Proliferation and Migration of Breast Cancer Cells. Mol. Med. Rep. 2017. [Google Scholar] [CrossRef]
- Lv, J.; Zhang, S.; Wu, H.; Lu, J.; Lu, Y.; Wang, F. Deubiquitinase PSMD14 Enhances Hepatocellular Carcinoma Growth and Metastasis by Stabilizing GRB2. Cancer Lett. 2020, 469, 22–34. [Google Scholar] [CrossRef]
- Jing, C.; Li, X.; Zhou, M.; Zhang, S.; Lai, Q.; Liu, D. The PSMD14 Inhibitor Thiolutin as a Novel Therapeutic Approach for Esophageal Squamous Cell Carcinoma through Facilitating SNAIL Degradation. Theranostics 2021, 11, 5847–5862. [Google Scholar] [CrossRef]
- Gong, Y.; Wei, Z.R. Identification of PSMD14 as a Potential Novel Prognosis Biomarker and Therapeutic Target for Osteosarcoma. Cancer Rep. 2022, 5, e1522. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating Enzymes and Drug Discovery: Emerging Opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef] [PubMed]
- De Cesare, V.; Carbajo Lopez, D.; Mabbitt, P.D.; Fletcher, A.J.; Soetens, M.; Antico, O.; Wood, N.T.; Virdee, S. Deubiquitinating enzyme amino acid profiling reveals a class of ubiquitin esterases. Proc. Natl. Acad. Sci. USA 2021, 118, e2006947118. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Jin, S.; Liu, Q.; Zhang, Y.; Ma, L.; Zhao, Z.; Yang, S.; Li, Y.P.; Cui, J. Selective Autophagy Controls the Stability of Transcription Factor IRF3 to Balance Type I Interferon Production and Immune Suppression. Autophagy 2021. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Huang, H.; Zhang, Y.; Wang, Y.; Zhao, J.; Lee, P.; Ma, Y.; Qu, S. Identification and Validation of KIF20A for Predicting Prognosis and Treatment Outcomes in Patients with Breast Cancer. Sci. Rep. 2024. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Yang, T.; Yuan, B.; Yang, Y.; Liu, J.; Wang, X.; Wang, Y.; Ma, T.; Yin, X.; Yu, H.; et al. Prognostic Analysis of Patients with Breast Cancer Based on Tumor Mutational Burden and DNA Damage Repair Genes. Front. Oncol. 2023. [Google Scholar] [CrossRef]
- Yu, Y.; Hu, J.; Wang, W.; Lei, H.; Xi, Z.; Zhang, P.; Zhao, E.; Lu, C.; Chen, H.; Liu, C.; et al. Targeting PSMD14 Combined with Arachidonic Acid Induces Synthetic Lethality via FADS1 m6A Modification in Triple-Negative Breast Cancer. Sci. Adv. 2025, 11, 3173. [Google Scholar] [CrossRef]
- Hu, X.; Zhu, H.; Chen, B.; He, X.; Shen, Y.; Zhang, X.; Chen, W.; Liu, X.; Xu, Y.; Xu, X. Tubulin Alpha 1b Is Associated with the Immune Cell Infiltration and the Response of HCC Patients to Immunotherapy. Diagnostics 2022, 12, 858. [Google Scholar] [CrossRef] [PubMed]
- Lu, C. Increased Alpha-Tubulin1b Expression Indicates Poor Prognosis and Resistance to Chemotherapy in Hepatocellular Carcinoma. Dig. Dis. Sci. 2013, 58, 2713–2720. [Google Scholar] [CrossRef]
- Sun, Z.; Huang, P.; Lin, J.; Jiang, G.; Chen, J.; Liu, Q. The Aggrephagy-Related Gene TUBA1B Influences Clinical Outcomes in Glioma Patients by Regulating the Cell Cycle. Front. Oncol. 2025, 15, 1531465. [Google Scholar] [CrossRef]
- Cui, H.; Zhao, G.; Lu, Y.; Zuo, S.; Duan, D.; Luo, X.; Zhao, H.; Li, J.; Zeng, Z.; Chen, Q.; et al. TIMER3: An Enhanced Resource for Tumor Immune Analysis. Nucleic Acids Res. 2025. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T Cell Dysfunction and Exclusion Predict Cancer Immunotherapy Response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Giles, J.R.; Globig, A.M.; Kaech, S.M.; Wherry, E.J. CD8+ T Cells in the Cancer-Immunity Cycle. Immunity 2023, 56, 2231–2253. [Google Scholar] [CrossRef]
- Seo, A.N.; Lee, H.J.; Kim, E.J.; Kim, H.J.; Jang, M.H.; Lee, H.E.; Kim, Y.J.; Kim, J.H.; Park, S.Y. Tumour-Infiltrating CD8+ Lymphocytes as an Independent Predictive Factor for Pathological Complete Response to Primary Systemic Therapy in Breast Cancer. Br. J. Cancer 2013, 109, 2705–2713. [Google Scholar] [CrossRef] [PubMed]
- Abdou, Y.; Attwood, K.; Cheng, T.D.; Yao, S.; Bandera, E.V.; Zirpoli, G.R.; Ondracek, R.P.; Stein, L.; Bshara, W.; Khoury, T.; et al. Racial Differences in CD8+ T Cell Infiltration in Breast Tumors from Black and White Women. Breast Cancer Res. : BCR 2020, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Tesi, R.J. MDSC; the Most Important Cell You Have Never Heard Of. Trends Pharmacol. Sci. 2019, 40, 4–7. [Google Scholar] [CrossRef]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The Roles of IFN Gamma in Protection against Tumor Development and Cancer Immunoediting. Cytokine Growth Factor. Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-Gamma: An Overview of Signals, Mechanisms and Functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-Regulation of PD-L1, IDO, and T(Regs) in the Melanoma Tumor Microenvironment Is Driven by CD8(+) T Cells. Sci. Transl. Med. 2013. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 1554. [Google Scholar] [CrossRef]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally Portraying the Tissue Cellular Heterogeneity Landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef]
- Schmieder, A.; Michel, J.; Schönhaar, K.; Goerdt, S.; Schledzewski, K. Differentiation and Gene Expression Profile of Tumor-Associated Macrophages. Semin. Cancer Biol. 2012, 22, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Oshi, M.; Tokumaru, Y.; Asaoka, M.; Yan, L.; Satyananda, V.; Matsuyama, R.; Matsuhashi, N.; Futamura, M.; Ishikawa, T.; Yoshida, K.; et al. M1 Macrophage and M1/M2 Ratio Defined by Transcriptomic Signatures Resemble Only Part of Their Conventional Clinical Characteristics in Breast Cancer. Sci. Rep. 2020, 10, 16554. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, D.; Burrell, R.; Kanu, N.; McGranahan, N.; Howell, M.; Parker, P.J.; Downward, J.; Swanton, C.; Kschischo, M. Chromosomal Instability Selects Gene Copy-Number Variants Encoding Core Regulators of Proliferation in ER+ Breast Cancer. Cancer Res. 2014, 74, 4853–4863. [Google Scholar] [CrossRef] [PubMed]
Figure 3.
Analysis of the expression levels of genes encoding 5 centromeric proteins in diverse breast tumors and evaluation of the prognostic significance of their overexpression (A-E) Box-whisker plots comparing the expression levels of CENPA (A), CENPF (B), CENPL (C), CENPO (D), and OIP5 (E), in primary breast tumor tissues (red boxes) contrasted to normal tissues (blue boxes). The UALCAN platform was used to analyze TCGA level 3 RNA sequencing data. The red asterisk (*) indicates a statistically significant difference (p < 0.05). (F-J) Kaplan–Meier survival analysis evaluating the prognostic significance of the expression of the indicated centromeric proteins, performed using microarray data from TCGA breast tumors and displayed using the KM Plotter tool. The red line depicts the recurrence-free survival of patients with expression levels of the genes above the cut-point, while the black line represents the recurrence-free survival of patients with expression levels below the cut-point. The analysis was conducted using the KM Plotter’s JetSet optimal microarray probe set and an optimal cutoff for recurrence-free survival over 180 months, without restricting to specific breast cancer subtypes. (K-O) Analysis of the expression levels of the indicated centromeric genes in breast tumors from patients of different races (self-identified). “N” represents normal breast tissues with a sample size n=114 for all, “C” represents Caucasians with a sample size n=748, “A-A” represents African-Americans with a sample size n=179, and “A” represents Asians with a sample size n=61. Analysis of TCGA RNA sequencing data was performed on the UALCAN platform and visualized using a box-whisker plot. A red asterisk (*) denotes a statistically significant difference in expression between breast tumors from Caucasian and African-American patients (p < 0.05). (P-T) Analysis of the expression levels of the indicated centromeric genes in breast tumors of differing TP53 mutation status. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the expression of the indicated kinesins in tumors with mutated TP53 (M-p53) contrasted to non-mutated TP53 (NM-p53). A red asterisk (*) denotes a statistically significant difference in expression between the two indicated groups (p < 0.05) (U-Y) Analysis of the promoter methylation profiles of the indicated centromeric genes in breast tumors. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the methylation level of the promoters of the indicated genes in normal breast tissues (blue boxes) vs breast invasive carcinoma (red boxes). A red asterisk (*) denotes a statistically significant difference in promoter methylation between the two (p < 0.05).
Figure 3.
Analysis of the expression levels of genes encoding 5 centromeric proteins in diverse breast tumors and evaluation of the prognostic significance of their overexpression (A-E) Box-whisker plots comparing the expression levels of CENPA (A), CENPF (B), CENPL (C), CENPO (D), and OIP5 (E), in primary breast tumor tissues (red boxes) contrasted to normal tissues (blue boxes). The UALCAN platform was used to analyze TCGA level 3 RNA sequencing data. The red asterisk (*) indicates a statistically significant difference (p < 0.05). (F-J) Kaplan–Meier survival analysis evaluating the prognostic significance of the expression of the indicated centromeric proteins, performed using microarray data from TCGA breast tumors and displayed using the KM Plotter tool. The red line depicts the recurrence-free survival of patients with expression levels of the genes above the cut-point, while the black line represents the recurrence-free survival of patients with expression levels below the cut-point. The analysis was conducted using the KM Plotter’s JetSet optimal microarray probe set and an optimal cutoff for recurrence-free survival over 180 months, without restricting to specific breast cancer subtypes. (K-O) Analysis of the expression levels of the indicated centromeric genes in breast tumors from patients of different races (self-identified). “N” represents normal breast tissues with a sample size n=114 for all, “C” represents Caucasians with a sample size n=748, “A-A” represents African-Americans with a sample size n=179, and “A” represents Asians with a sample size n=61. Analysis of TCGA RNA sequencing data was performed on the UALCAN platform and visualized using a box-whisker plot. A red asterisk (*) denotes a statistically significant difference in expression between breast tumors from Caucasian and African-American patients (p < 0.05). (P-T) Analysis of the expression levels of the indicated centromeric genes in breast tumors of differing TP53 mutation status. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the expression of the indicated kinesins in tumors with mutated TP53 (M-p53) contrasted to non-mutated TP53 (NM-p53). A red asterisk (*) denotes a statistically significant difference in expression between the two indicated groups (p < 0.05) (U-Y) Analysis of the promoter methylation profiles of the indicated centromeric genes in breast tumors. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the methylation level of the promoters of the indicated genes in normal breast tissues (blue boxes) vs breast invasive carcinoma (red boxes). A red asterisk (*) denotes a statistically significant difference in promoter methylation between the two (p < 0.05).
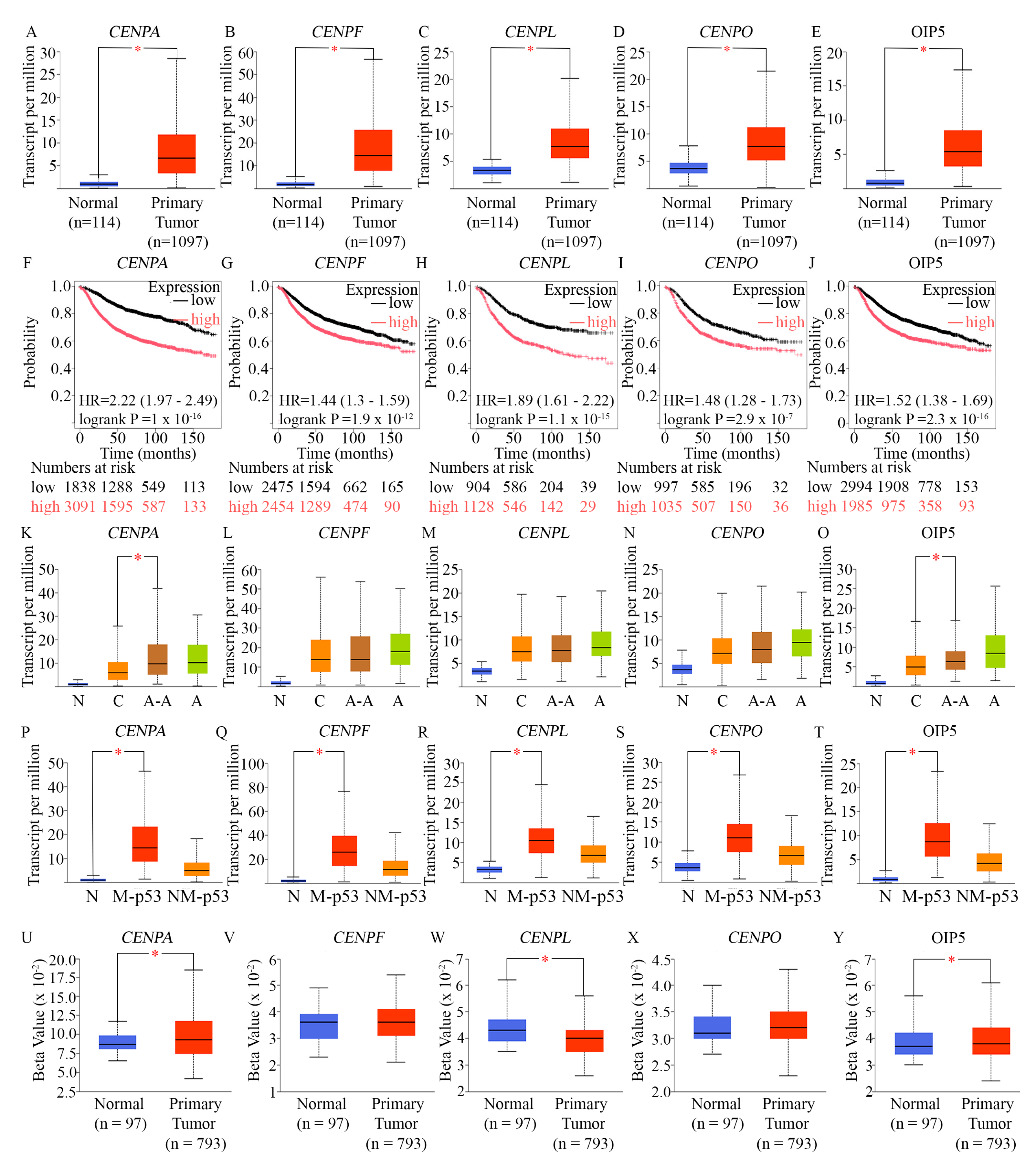
Figure 4.
Analysis of the expression levels of genes encoding 5 proteolysis-regulatory proteins in diverse breast tumors and evaluation of the prognostic significance of their overexpression (A-E) Box-whisker plots comparing the expression levels of UBE2C (A), UBE2S (B), UBE2T (C), PSMD14 (D), and TUBA1B (E), in primary breast tumor tissues (red boxes) contrasted to normal tissues (blue boxes). The UALCAN platform was used to analyze TCGA level 3 RNA sequencing data. The red asterisk (*) indicates a statistically significant difference (p < 0.05). (F-J) Kaplan–Meier survival analysis evaluating the prognostic significance of the expression of the indicated proteolysis-regulatory proteins, performed using microarray data from TCGA breast tumors and displayed using the KM Plotter tool. The red line depicts the recurrence-free survival of patients with expression levels of the genes above the cut-point, while the black line represents the recurrence-free survival of patients with expression levels below the cut-point. The analysis was conducted using the KM Plotter’s JetSet optimal microarray probe set and an optimal cutoff for recurrence-free survival over 180 months, without restricting to specific breast cancer subtypes. (K-O) Analysis of the expression levels of the indicated proteolysis-regulatory genes in breast tumors from patients of different races (self-identified). “N” represents normal breast tissues with a sample size n=114 for all, “C” represents Caucasians with a sample size n=748, “A-A” represents African-Americans with a sample size n=179, and “A” represents Asians with a sample size n=61. Analysis of TCGA RNA sequencing data was performed on the UALCAN platform and visualized using a box-whisker plot. A red asterisk (*) denotes a statistically significant difference in expression between breast tumors from Caucasian and African-American patients (p < 0.05). (P-T) Analysis of the expression levels of the indicated proteolysis-regulatory genes in breast tumors of differing TP53 mutation status. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the expression of the indicated kinesins in tumors with mutated TP53 (M-p53) contrasted to non-mutated TP53 (NM-p53). A red asterisk (*) denotes a statistically significant difference in expression between the two indicated groups (p < 0.05). (U-Y) Analysis of the promoter methylation profiles of the indicated proteolysis-regulatory genes in breast tumors. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the methylation level of the promoters of the indicated genes in normal breast tissues (blue boxes) vs breast invasive carcinoma (red boxes). A red asterisk (*) denotes a statistically significant difference in promoter methylation between the two (p < 0.05).
Figure 4.
Analysis of the expression levels of genes encoding 5 proteolysis-regulatory proteins in diverse breast tumors and evaluation of the prognostic significance of their overexpression (A-E) Box-whisker plots comparing the expression levels of UBE2C (A), UBE2S (B), UBE2T (C), PSMD14 (D), and TUBA1B (E), in primary breast tumor tissues (red boxes) contrasted to normal tissues (blue boxes). The UALCAN platform was used to analyze TCGA level 3 RNA sequencing data. The red asterisk (*) indicates a statistically significant difference (p < 0.05). (F-J) Kaplan–Meier survival analysis evaluating the prognostic significance of the expression of the indicated proteolysis-regulatory proteins, performed using microarray data from TCGA breast tumors and displayed using the KM Plotter tool. The red line depicts the recurrence-free survival of patients with expression levels of the genes above the cut-point, while the black line represents the recurrence-free survival of patients with expression levels below the cut-point. The analysis was conducted using the KM Plotter’s JetSet optimal microarray probe set and an optimal cutoff for recurrence-free survival over 180 months, without restricting to specific breast cancer subtypes. (K-O) Analysis of the expression levels of the indicated proteolysis-regulatory genes in breast tumors from patients of different races (self-identified). “N” represents normal breast tissues with a sample size n=114 for all, “C” represents Caucasians with a sample size n=748, “A-A” represents African-Americans with a sample size n=179, and “A” represents Asians with a sample size n=61. Analysis of TCGA RNA sequencing data was performed on the UALCAN platform and visualized using a box-whisker plot. A red asterisk (*) denotes a statistically significant difference in expression between breast tumors from Caucasian and African-American patients (p < 0.05). (P-T) Analysis of the expression levels of the indicated proteolysis-regulatory genes in breast tumors of differing TP53 mutation status. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the expression of the indicated kinesins in tumors with mutated TP53 (M-p53) contrasted to non-mutated TP53 (NM-p53). A red asterisk (*) denotes a statistically significant difference in expression between the two indicated groups (p < 0.05). (U-Y) Analysis of the promoter methylation profiles of the indicated proteolysis-regulatory genes in breast tumors. This analysis used TCGA RNA sequencing data and was performed via the UALCAN platform. The box-whisker plots display the methylation level of the promoters of the indicated genes in normal breast tissues (blue boxes) vs breast invasive carcinoma (red boxes). A red asterisk (*) denotes a statistically significant difference in promoter methylation between the two (p < 0.05).
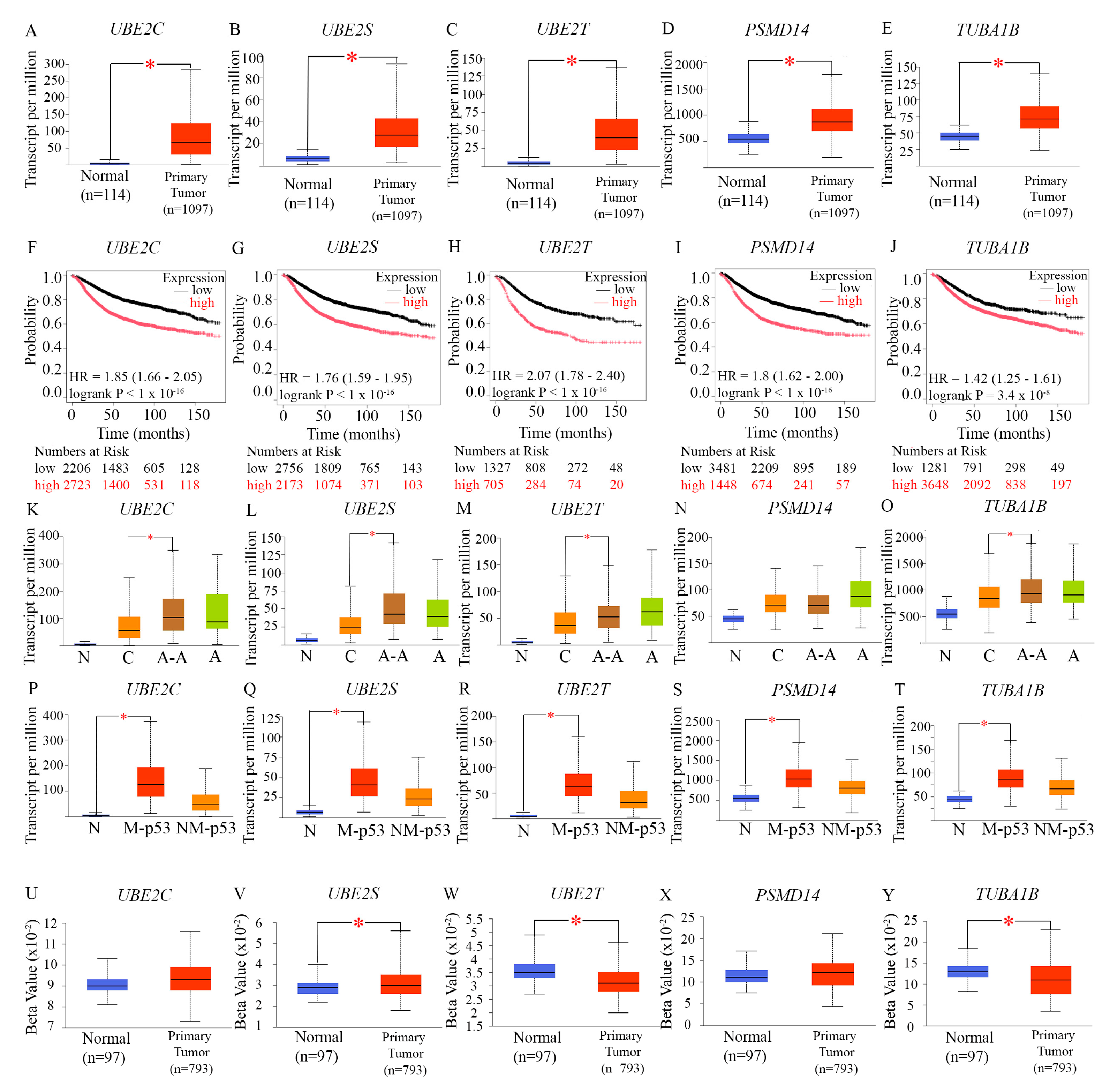
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



