Submitted:
30 September 2025
Posted:
01 October 2025
You are already at the latest version
Abstract
Deaminases are versatile enzymes present across all domains of life, playing pivotal roles in metabolism, cellular and organ development, heme and vitamin biosynthesis, toxin modulation, antibiotic degradation, the modification of nucleobases, and the mutation of RNA and DNA. This review explores the structural and functional diversity of deaminases, highlighting their mechanisms and evolutionary adaptations. Deaminases catalyze the removal of amine groups, typically using metal cations and proton shuttles to facilitate hydroxyl group incorporation, with some reactions employing pyridoxal 5'-phosphate (PLP) as a cofactor. The review categorizes deaminases based on their substrates, including porphobilinogen, amino sugars, amino acids, and nucleobases. Particular emphasis is placed on nucleobase deaminases due to their roles in RNA/DNA editing and mutagenesis which have significant implications in immune response, cancer progression, and viral defense mechanisms. Structural insights reveal the diverse evolutionary pathways of these enzymes, from simple single-domain forms to complex multi-domain configurations that enable processive deamination of polynucleotides. Through a comprehensive analysis of deaminase families—PBGD, sugar deaminases, amino acid deaminases, and nucleobase deaminases—this review underscores their biological significance and potential applications in agriculture, medicine, and biotechnology, providing a foundational understanding for future research.
Keywords:
deaminase structural evolution
; deamination
; nucleic acid deaminases
; structure-function mechanisms
1. Introduction
Deaminases are critical enzymes found across all domains of life, influencing metabolism, plant and organ development, vitamin and heme biosynthesis, toxin creation or reduction, antibiotic degradation, and RNA/DNA mutation [1,2,3,4,5,6,7,8,9,10]. The core reaction involves the replacement of an amine-containing molecule with a hydroxyl group, yielding a deaminated product and ammonia or ammonium as a byproduct (Figure 1; Supplementary Table 1 and 2; Scheme 1) [11,12]. This process is facilitated by various mechanisms, often involving one or more coordinated metal cations to hydrolyze water and introduce a hydroxyl group, along with single or dual-proton shuttles for proton transfer [13,14,15,16,17,18,19,20]. Pyridoxal 5’-phosphate (PLP), commonly used in deamination with lysine, also plays roles in decarboxylation, racemization, and transamination, though alternative deamination mechanisms exist [21,22,23,24,25]. Deaminases, a diverse enzyme group, have evolved through varied structures, enabling them to interact with similar chemical species and diversifying their functional capabilities [26]. This review explores deaminases acting on porphobilinogen, amino sugars, amino acids, nucleobases, and their derivatives (Figure 2).
Nucleobase deaminases exhibit remarkable diversity, targeting guanine, adenine, cytosine, nucleosides, nucleotides, and polynucleotides like RNA and DNA as well as their analogues and derivatives, including those involved in vitamin biosynthesis [13,14,15,16,17,18,19,20]. These deaminases are particularly significant due to their mutagenic effects in immune cells, cancers, and viruses, akin to methylation and acetylation [27,28]. Understanding the structure and function of deaminases is crucial given the prevalence of these reactions in various biological processes.
This review aims to showcase the structural and functional diversity of deaminases, especially those acting on nucleic acids. We present a comprehensive list of deaminases categorized by substrate and protein superfamily, covering porphobilinogen deaminase (PBGD), sugar deaminases, amino acid deaminases (both PLP-dependent and independent), and nucleobase, nucleoside, and nucleotide deaminases from dUTPase, AHS, and CDA superfamilies. By examining these enzymes, we highlight their evolutionary adaptations and structural diversity. Understanding deamination mechanisms and their evolutionary trajectories could enhance our knowledge in fields like agriculture, metabolism, cellular development, cancer, immunology, virology, bacterial antagonism, and antibiotic resistance. This comprehensive overview underscores the importance of deaminases and their diverse roles across different biological systems.
2. Tetrapyrroles Biosynthesis Pathway Deaminases
Porphobilinogen deaminase (PBGD), also known as hydroxymethylbilane synthase (HMBS), is a crucial enzyme present in all non-viral organisms. It catalyzes the conversion of porphobilinogen into the linear tetrapyrrole 1-hydroxymethylbilane, a precursor to uroporphyrinogen III. This compound can further transform into heme, chlorophyll, cobalamin (Vitamin B12), or siroheme, depending on the organism (Scheme 2) [29]. These tetrapyrroles are vital for functions such as oxygen transport, photosynthesis, metabolism, DNA synthesis, and fluorescence, which are essential for the survival of all organisms. The formation of porphobilinogen follows two pathways: the Shemin pathway in fungi, mammals, and α-proteobacteria, which utilizes succinyl-CoA and glycine, and the C5 pathway in plants, archaea, and most bacteria, which uses glutamyl-tRNA. Despite different origins, both pathways converge at the biosynthesis of uroporphyrinogen III, leading to various end products.
PBGD’s central role involves converting four porphobilinogen molecules into the tetrapyrrole 1-hydroxymethylbilane, using the dipyrromethane (DPM) cofactor and its three flexible domains (Figure 3A-B) [6,29,30,31]. This mechanism is conserved across species, including humans [6,32,33,34,35], Arabidopsis thaliana [30], Escherichia coli [31,36], Priestia megaterium [37,38], and Vibrio cholerae [39]. DPM is covalently bound to a cysteine residue (C261 in humans), forming the base of the growing product [6]. Domains 1 and 2 consist of three α-helices and a mixed β-sheet, while domain 3 includes two α-helices, an antiparallel β-sheet, and the cysteine-DPM binding site [30]. A groove between domains 1 and 2 extends to domain 3, enabling flexibility and maintaining structural integrity.
Although the exact mechanism of PBGD remains unclear, it involves successive elongation of the substrate through electrophilic addition to the distal pyrrole subunit and subsequent deprotonation. In humans, the conserved residues R26, R173, R167, S28, Q34, S96, and N169 are critical for catalysis, likely forming salt bridges with porphobilinogen to stabilize the substrate (Figure 3C) [6,35]. R26 and Q34 are proposed catalytic residues, conserved across species despite only a 45% sequence identity between humans and E. coli [33]. After forming the hexapyrrole, the bond between DPM and the first added pyrrole subunit is hydrolyzed, releasing 1-hydroxymethylbilane [6]. This product must cyclize via uroporphyrinogen III synthase to avoid forming uroporphyrinogen I, a condition observed in acute intermittent porphyria [32].
In summary, while PBGD’s exact mechanism of action remains elusive, its structural conservation across species underscores its essential role in tetrapyrrole biosynthesis. PBGD uniquely utilizes its three domains and covalently linked DPM cofactor to synthesize critical molecules like heme and its analogs (Scheme 2) [29,30]. This enzyme’s flexibility and conserved structure enable it to maintain vital biosynthetic pathways despite incomplete understanding of its functional dynamics.
3. Amino Sugar Deaminases
Glucosamine-6-phosphate (GlcN6P) is a pivotal intermediate in various metabolic and biosynthetic pathways, also serving roles in carbon and nitrogen storage [40,41]. This molecule is most extensively studied in Escherichia coli, where 10 structural variants have been identified [42,43,44,45,46]. Homologues of GlcN6P deaminase have been characterized in several other bacteria, including Bacillus subtilis [40], Streptococcus mutans [47], Borreliella burgdorferi [48], Vibrio cholerae [49,50], Klebsiella pneumoniae [51], Pasteurella multocida [52], Haemophilus influenzae [53], and even in humans [12]. GlcN6P plays a role in glycolysis, N-acetylglucosamine synthesis, bacterial peptidoglycan synthesis, gram-positive teichoic acid synthesis, purine synthesis, the pentose phosphate pathway (precursors to nucleic acids), yeast trehalose, glycogen, cell wall synthesis, chitin glycosylation pathways, mannosylation pathways, and kidney function in mammals [12,40,41].
The enzyme GlcN6P synthase converts fructose-6-phosphate (Fru6P) and glutamine into GlcN6P and glutamate, while GlcN6P deaminase reverses this reaction, producing Fru6P and ammonium (Scheme 3). GlcN6P deaminase is a homohexameric protein complex (~266 residues) and functions as an allosteric enzyme, activated by N-acetyl-D-glucosamine-6-phosphate (GlcNAc6P), which regulates amino sugar synthesis [42]. It operates as a K-system enzyme under Michaelis-Menten kinetics, where the allosterically bound relaxed (R) state exhibits higher activity and substrate affinity than the non-allosterically bound tense (T) state (Figure 4A-C) [42,43,44].
Structurally, each subunit contains a core parallel β-sheet of seven strands surrounded by eight α-helices, with an additional antiparallel β-sheet and an α-helix (residues 163-183) forming the active site lid, varying across species. The allosteric site in the T-state, absent of GlcNAc6P, is wider and neutral. Upon GlcNAc6P binding, shifts in residues R158 and K160 narrow the site, tightening the active site through movement of D141, H143, and E148, along with the stationary D72, the key catalytic residues [43,44,45]. The dual-proton shuttle formed by these residues facilitates the opening of the GlcN6P sugar ring, stabilizing the substrate as H143 and D72 reposition the proton from O5 to O1, elongating the sugar and enabling further reactions [40,42,44,47]. The cis-enolate-ammonium intermediate is disrupted by D72, adding a proton to C1 and hydroxyl to C2, ultimately releasing Fru6P and ammonium. This reaction is reversible and important for mitigating toxic ammonium buildup. The conserved catalytic residues D72 and H143 are found across bacterial and human structures, with D141 and E148 varying in some species (asparagine in B. subtilis and S. mutans, leucine in K. pneumoniae) [40,47,51]. Despite structural variations, these enzymes uniformly catalyze the deamination and linearization of sugar rings through conserved active sites.
Interestingly, distinct structures of GlcN6P deaminase exist in hyperthermophilic organisms Thermotoga maritima and Pyrococcus furiosus which thrive in hydrothermal vents and are among the most ancient extant species [54,55,56]. These enzymes form dimers instead of hexamers and feature β-sheets with five parallel strands surrounded by α-helices, aligning with the isomerase domain of GlcN6P synthase (Figure 4D) [56,57,58]. Their catalytic pockets house critical residues from each dimer H240/227, E224/211, and K326/321, enabling sugar ring opening and deamination across T. maritima and P. furiosus, respectively (Figure 4E). The catalytic lysine, though obstructing the pocket in resting state, appears flexible, suggesting an active site opening mechanism. P. furiosus GlcN6P deaminase, capable of catalyzing the reverse reaction, shows a preference for deamination [56,57,58]. This structural adaptation likely evolved from GlcN6P synthase to suit extreme thermal conditions.
In summary, GlcN6P deaminase demonstrates an intriguing case of convergent evolution, where most bacterial species and humans utilize a homohexameric enzyme, while hyperthermophilic species adopt a dimeric structure [12,40,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56]. Both enzyme types exhibit a similar mechanism involving positive residues interacting with oxygen and negative residues acting as a proton shuttle, resulting in substrate deamination and linearization. The homohexameric enzymes rely on allosteric activation via GlcNAc6P, whereas the hyperthermophilic enzymes depend on dimerization for catalytic function. This evolutionary divergence reflects adaptations to varying environmental pressures, highlighting the versatility of GlcN6P deaminase in maintaining critical metabolic functions.
4. Amino Acid Derivative Deaminases
Amino acids are commonly recognized as the fundamental building blocks of proteins; however, deamination has enabled them to serve additional biological functions. This is particularly evident in enzymes utilizing the cofactor pyridoxal 5’-phosphate (PLP), such as threonine deaminase which contributes to branched-chain amino acid synthesis; serine deaminase (also called serine dehydratase, SDH) which regulates neurotransmission co-agonists and gluconeogenesis in chickens; L-arginine deaminase which participates in the biosynthesis of non-proteogenic amino acid antibiotics; and enzymes involved in plant hormone metabolism [22,23,24,25,59,60]. Despite structural variability across species, key residues such as catalytic lysine remain highly conserved.
Threonine deaminase (TD) in bacteria and plants shares homologous structural features with serine deaminase in rodents and primates, suggesting an evolutionary relationship wherein serine deaminases may have originated from TDs [22,23,61,62,63,64,65,66,67,68]. Notably, some animal liver serine deaminases retain threonine deamination capability. These enzymes consist of two domains featuring β-sheets and α-helices with the catalytic pocket situated between them, where PLP is covalently bound to lysine (Figure 5A, B). The deamination reaction involves dehydration of threonine and serine into aminocrotonate and α-aminoacrylate, respectively, which are then hydrolyzed non-enzymatically to form α-ketobutyrate or pyruvate with free ammonia (Scheme 4; Figure 5C) [23,62,66,67]. Interestingly, human serine deaminase residue A65 (conserved across homologs) coordinates a water molecule for deamination of the PLP-bound substrate, suggesting that TDs and SDHs may play a more direct role in deamination beyond simply facilitating hydrolysis [67].
A unique serine deaminase structure has been identified in chickens, diverging from canonical TD/SDH enzymes and sharing similarities with threonine aldolases and Paraburkholderia xenovorans serine deaminase (Figure 5.D) [60,69]. This enzyme features a TIM β-barrel domain linked to a β-sandwich domain surrounded by α-helices, with the catalytic lysine-PLP complex and a Zn2+ cofactor coordinating multiple water molecules. This architecture allows for deamination to aminoacrylate and subsequently pyruvate, possibly through Zn-mediated water activation. Another notable deaminase, 1-aminocyclopropane-1-carboxylic acid (ACC) deaminase, facilitates plant hormone regulation and metabolic pathways, utilizing a catalytic pocket situated between two α/β-sheet folds of four and six strands, where a six-stranded domain covalently binds PLP (Figure 5E) [2,24,70,71,72]. Although deamination occurs similarly to PLP-linked deamination, the precise mechanism of ACC ring opening remains unresolved with proposed pathways involving either direct proton addition or nucleophilic addition, leading to proton abstraction [71,72].
A structurally similar enzyme, MppP from Streptomyces wadayamensis, features a lysine bound PLP catalytic pocket residing between two β-sheets of seven and four strands, each containing multiple α-helices with an N-terminal α-helix potentially shielding the substrate and influencing catalysis (Figure 5F) [9,25]. The N-terminal α-helix appears to shield the substrate from solvent and potentially plays a role in catalytic activity. MppP acts as an L-arginine deaminase, producing either 2-oxo-5-guanidinovaleric acid through direct deamination or 2-oxo-4-hydroxy-5-guanidinovaleric acid via a hydroxylation-dependent pathway, leading to biosynthesis of L-enduracididine, a non-proteinogenic amino acid.
PLP-dependent proteins are known for decarboxylation and racemization; however, PLP-dependent deaminases contribute to neurotransmitter synthesis and plant metabolism [59,68]. For instance, aromatic L-amino acid decarboxylase (AAAD) catalyzes the production of aromatic monoamines in animals, whereas in plants, decarboxylation-dependent oxidative deamination produces aromatic acetaldehydes, such as phenylacetaldehyde from L-phenylalanine in Arabidopsis thaliana and 4-hydroxy-phenylacetaldehyde from L-tyrosine in Rhodiola rosea (Figure 5G). The catalytic pocket of AAAD resides between two of its domains, one predominantly helical and the other composed of β-sheets surrounded by α-helices. Typically, histidine and tyrosine facilitate Cα protonation after decarboxylation, but these plant species replace tyrosine with phenylalanine, allowing dioxygen interaction with the carbanion and enabling alternate deamination routes via a pre-existing PLP enzyme to produce aromatic acetaldehydes, ammonia, and hydrogen peroxide.
Several amino acid deaminases function without PLP, employing alternative mechanisms. One such enzyme is L-amino acid deaminase (LAAD), a membrane-bound deaminase in Proteus bacteria. LAAD consists of three domains: an FAD-binding domain, a substrate-binding domain, and a membrane insertion module (Figure 5H) [73,74]. The catalytic reaction involves oxidation of the α-amino acid by FAD, forming an α-imino acid and FADH2, followed by hydrolysis to generate an α-keto acid and ammonia (Scheme 4). Proteus myxofaciens and Proteus vulgaris prefer hydrophobic amino acids, though P. vulgaris also deaminates positively charged and polar residues. Interestingly, P. myxofaciens lacks peroxide byproducts in FADH2 turnover, unlike P. vulgaris, suggesting an alternative electron acceptor, possibly a cytochrome b-like protein.
Another non-PLP-dependent deaminase, reactive intermediate deaminase A (RidA), functions across all domains of life to mitigate cellular toxicity caused by enamine and imine intermediates produced by TDs and SDHs (Scheme 4) [75,76,77,78]. RidA proteins form homotrimers, each featuring a six-stranded β-sheet and two α-helices, which together assemble into a β-barrel, with catalytic pockets positioned at inter-subunit interfaces (Figure 5I) [76,77,78,79,80,81,82]. While the exact mechanism of RidA remains unclear, conserved arginine and tyrosine residues stabilize the imino acid, while glutamate likely acts as a proton shuttle, although production of a Schiff base intermediate with the arginine has also been proposed. RidA is believed to accelerate spontaneous hydrolysis, preventing interference with other PLP-dependent enzymes. Specificity varies across RidA homologs with RidA-1 preferring non-aromatic hydrophobic residues, while RidA-2 favours glutamate and aromatic imino acids [75,78,82]. Interestingly, Pseudomonas AmnE, a 2-aminomuconate deaminase creating 4-oxalocrotonate is involved in nitrogen metabolism, sharing structural similarities with RidA, including a trimeric β-barrel configuration and conserved catalytic residues [83]. Unlike RidA, AmnE utilises phenylalanine instead of tyrosine and forms hexamers via 18 inter-subunit hydrogen bonds.
Finally, another non-PLP deaminase, HypD, catalyzes the deamination of 1-pyrroline-4-hydroxy-2-carboxylate (HPC) to α-ketoglutarate semialdehyde in proline metabolism [84]. HypD features an α/β-barrel with C-terminal α-helices, where a lysine residue within the β-barrel is thought to form Schiff base intermediates with HPC, as demonstrated in unpublished structural data (PDB: 4O0K) (Figure 5J) [84,85,86].
In summary, amino acid deaminases can be categorized into PLP-dependent and non-PLP-dependent enzymes. Despite structural variability, PLP-dependent deaminases share a conserved mechanism involving transient covalent bonding between PLP and lysine, leading to an enamine intermediate that undergoes spontaneous hydrolysis to yield an imino acid and finally an α-keto acid [23,62,66,67]. In contrast, non-PLP deaminases employ diverse catalytic strategies: LAAD bypasses the enamine intermediate via FAD oxidation, and HypD utilizes Schiff base intermediates [73,74,84]. Spontaneous imino acid hydrolysis, while essential for deamination, poses risks to PLP enzymes, which RidA mitigates through a cation-anion residue pair at trimeric interfaces, akin to dUTPase superfamily deaminases [75,76,77,78]. Despite differences in structure and substrate specificity, amino acid deaminases exhibit a convergent evolution of reaction mechanisms across biological systems.
5. Guanine Derivative Deaminases
The deamination of the nucleic acid guanine is crucial across all species for purine metabolism and, in eukaryotes, for facilitating dendritic arborization [5,87]. Guanine deaminases (GDA) are divided into two distinct superfamilies: the amidohydrolase superfamily (AHS), which includes enzymes found in eukaryotes like fungi and some bacteria, and the cytidine deaminase (CDA) superfamily, found in plants, archaea, and some bacteria. Both superfamilies deaminate guanine to xanthine (Scheme 5A) [17,88].
The AHS GDAs are notable for their structure, consisting of two domains: a distorted TIM β-barrel domain housing the catalytic pocket with a metal cationic cofactor, and a smaller PDZ (β-sandwich)-binding domain important for protein-protein interactions from the enzyme’s N- and C-termini (Figure 6.A-B) [87,88,89,90]. This structure has been solved for several species, including humans, Saccharomyces cerevisiae, Bradyrhizobium japonicum, and Clostridium acetobutylicum. Typically, a Zn2+ cation is at the centre of the TIM β-barrel, coordinated by three histidines and an aspartate, which becomes protonated while the Zn gains a hydroxyl (Figure 6C) [5,91,92,93,94]. The guanine enters the catalytic pocket through an opening between α-helices 3 and 8, which closes once the substrate is inside, allowing the reaction to occur via a dual proton shuttle involving aspartate, histidine, and glutamate while oxygen is transferred from the Zn [5,94]. Though these deaminases primarily target guanine, they can also deaminate ammeline and ammelide at lower efficiency [90,94]. An engineered ammelide deaminase showed a 100-fold increase in ammelide deamination by introducing a GNGV loop in human GDA with an arginine-to-asparagine mutation, anchoring ammelide for deamination [90]. Some organisms have adapted their enzymes for different substrates by altering the arginine and phenylalanine dyad, which stabilizes the substrate and caps its size [94,95]. Mutations in this dyad, such as cysteine-serine or threonine-threonine, enable the enzymes to deaminate 8-oxoguanine and isoxanthopterin, producing 8-oxoxanthine and 7-oxylumazine, respectively [89,94,95].
The CDA superfamily GDAs, found in plants, archaea, and some bacteria, evolved distinct structures [17,88]. Their structure includes an αβα layered fold with a single Zn cation, resembling the adenosine deaminase TadA [88,96]. This αβα-fold comprises five β-strands and three α-helices on one side of the catalytic pocket, with three additional α-helices on the opposite side of the β-sheet (Figure 6D-E) [17,88,97,98]. Differences in the protein structures of Bacillus subtilis and Nitrosomonas europaea include variations in the loop 3 and the presence of an α-helix on the side of the catalytic pocket. Both proteins form homodimers across catalytic pocket α-helices, facing one another with the pockets’ directionality oriented ~120° from each other. N. europaea’s structure features an extended N-terminal αβ subunit binding the dimers together, while B. subtilis’ uses α5 to traverse the inter-domain space, with β5 and α6 contributing to the opposing dimer’s structure (Figure 6D-F). The catalytic pocket stabilizes guanine using asparagine and 2 phenylalanine residues on surrounding loops, with a C-terminal tyrosine aiding guanine stabilization. The catalytic residues include a histidine on α2 and two cysteines on α3 which coordinate the Zn holding the hydroxyl. A dual proton shuttle involves glutamate of α2 and either glutamate of Loop 7 in N. europaea or aspartate of α5 in B. subtilis, (which occupy the same relative state with respect to one another), protonating guanine’s N3 and N2 residues, respectively, allowing the release of ammonia (Figure 6F-G). N. europaea retains a small capacity for ammeline deamination, which is eliminated by mutating glutamate to aspartate [88,98].
Guanosine deaminase (GSDA), unique to plants, deaminates guanosine to xanthosine, essential for catabolizing ribonucleosides to produce amino acids and other critical molecules (Scheme 5B and Figure 6H) [18,99]. GSDA shares structural similarities with N. europaea GDA, minus the N-terminal binding region and featuring a shorter α5, which is perpendicular in directionality. Unlike dual-proton shuttle enzymes, GSDA employs a single proton shuttle, utilizing Zn-OH, Zn coordinating residues, and α2 glutamate, with loops 5 and 7 repurposed for hydrogen bonding with the ribose sugar. This glutamate acts on guanosine’s N2, facilitating ammonia release.
In summary, guanine-derived deaminases can be categorized into AHS and CDAS superfamilies, differing slightly from adenine and cytosine deaminases by acting on the nucleobase ring’s third carbon instead of the first. Both groups require metal cofactors and coordinating residues to stabilize Zn2+ with its hydroxyl and facilitate dual proton-shuttling (except GSDA which uses a single proton-shuttle) [5,88,91,92,93,94,96]. The AHS deaminases use their PDZ domain for multimerization and a TIM β-barrel domain for substrate processing, whereas CDA GDAs and GSDA use an αβα fold and a C-terminal loop to form enclosed pockets [17,18,87,88,89,90,97,98,99]. Notably, some CDA members extend their dimerization surfaces, with B. subtilis using part of its C-terminal on the opposing dimer and N. europaea employing an extended N-terminal αβ subunit. These structural differences highlight the diversity in enzyme adaptation across species.
6. Adenine Deaminases and Derivatives
6.1. Free adenine-Derived Nucleo -Base/-Side Deaminases
Adenine deaminases (ADEase) play a crucial role in purine metabolism, converting adenine to hypoxanthine as part of the nucleotide and ammonia salvaging pathway in non-eukaryotic organisms (Scheme 6A) [1,100,101,102,103,104]. Similarly, adenosine deaminases (ADA) facilitate the conversion of adenosine to inosine and ammonia across many non-plant eukaryotes (Scheme 6B). In mammals, this reaction is significant for regulating cell growth in the gastrointestinal tract, fetal-maternal interface, and immune system. The structural composition of ADEase includes the TIM β-barrel domain and the small PDZ (β-sandwich)-binding domain, similar to the AHS GDA enzymes [100,101,105]. However, there are distinct variations in the loops and α-helices surrounding the core structure and in the coordination of two divalent metal cations (Figure 7A). Unlike guanine or cytidine deaminases of the AHS superfamily, each cation is coordinated by two histidines and either an aspartate or glutamate. Intriguingly, Agrobacterium fabrum contains an additional domain with a twisted β-sheet surrounded by α-helices of unknown function (Figure 7B-C). This domain lies opposite the catalytic pocket, appearing to protect it, and includes a third divalent metal cation coordinated by two histidines, one glutamate, and one aspartate [100,101]. A. fabrum shows a preference for divalent Fe over Mn or Zn; however, this preference reduces ADE deamination activity, and the oxidative PTMs and loss of function seen with Fe are not observed when Mn or Zn are present as cofactors.
Some bacterial deaminases exhibit higher activity on other adenine-derived compounds such as adenosine or methylthioadenosine, leading to their classification as adenosine or methylthioadenosine deaminases [106,107]. These enzymes typically contain a single divalent zinc ion. Certain AHS proteins lacking the PDZ domain discovered in bacteria were initially believed to function as ADAs but were later found to deaminate adenine, 2,6-diaminopurine, and N-6-methyladenine [108]. Notably, bacteria typically possess only one adenine/adenosine deaminase per species. Additionally, some bacteria lacking the ability to produce menaquinone (vitamin K2) through the shikimate or the traditional futalosine pathways have evolved to utilize aminofutalosine deaminase (AFLDA) [109,110]. AFLDA is a PDZ-containing AHS protein that deaminates 6-aminofutalosine to futalosine using a divalent zinc or iron and a dual-proton shuttle. AFLDA can deaminate various adenine/adenosine-based compounds and may represent the original menaquinone production pathway. However, it is rare in the human gut microbiome, with Helicobacter pylori being an exception.
Adenosine deaminases (ADA), part of the AHS, differ from ADEase in that they lack the PDZ domain seen in ADEase and guanine deaminase [1,105]. ADA is known to interact with CD26 (dipeptidyl peptidase-4/adenosine deaminase complexing protein 2), which is inhibited by HIV gp120 and Tat, facilitating T cell proliferation through the regulation of extracellular adenosine (Figure 7D-E) [111,112]. Extracellular adenosine acts as an inhibitor of T-lymphocytes and requires coordinated divalent zinc for ADA activity. ADA’s dual role depends on its location: extracellular adenosine attenuates inflammation, while intracellular adenosine is essential for energy production and purine metabolism [20]. ADA binds CD26 at α4 and α6, leaving the active site unblocked [111,113]. ADA deficiency in mammals leads to severe combined immunodeficiency due to impaired differentiation and maturation of lymphoid lineages [114,115,116,117]. Conversely, ADA inhibition is being researched as a potential cancer treatment through both nucleoside mimics and non-nucleoside inhibitors. In certain Plasmodium species, lacking critical de novo purine biosynthesis pathways necessitates adenosine to inosine conversion to produce GMP and AMP, which can be inhibited using various mechanisms [118]. Humans possess two ADA isoforms: hADA1 and hADA2 [119,120,121]. hADA1 is monomeric, located in the cytosol and nucleus, and attaches in an ecto-form to CD26. hADA2, less active than hADA1, is secreted into plasma and sera as a dimer. This dimerization is facilitated by four additional N-terminal α-helices and multiple ADA core helices interactions, along with a three β-strand insert that may act as a receptor-binding region. hADA2 acts as an extracellular adenosine deaminase growth factor, inducing monocyte-dependent CD4 T cell proliferation and uniquely prompting monocytes to differentiate into macrophages.
Plants lack ADEase or ADA but contain an intracellular dAMPD (deoxyadenosine monophosphate deaminase) [104]. This enzyme includes the AHS TIM β-barrel and functions as a dimer with an N-terminal linker and transmembrane domain, facilitating purine metabolism. Similarly, Arabidopsis thaliana has a related protein, MAPDA (N6-methyl-adenosine monophosphate deaminase), which deaminates N6-methyl-adenosine monophosphate to inosine and methylamine using zinc and an aspartate (Scheme 6C; Figure 7F) [122,123]. MAPDA is crucial for purine metabolism, affecting embryo maturation, germination, and sprouting in plants. MAPDA differs from ADA in its phosphate anchor pocket and hydrophobic pockets. Specific mutations in the pocket, such as phenylalanine to leucine and valine to phenylalanine, enable adenosine activity at the cost of N6-methyl-adenosine monophosphate activity.
Deamination reactions of adenine and adenosine also occur in multifunctional proteins like YlmD and YfiH, found in Bacillales and Enterobacterales genera, respectively [124]. These proteins are chemically resolved in prokaryotes and play a role in the mitochondrial purine nucleotide cycle, where it is known as FAMIN (fatty acid metabolism-immunity nexus), though its structure remains unresolved. These proteins catalyze five reactions: (1) adenosine and phosphate to adenine and α-D-ribose-1-phosphate; (2) adenosine and water to inosine and ammonia; (3) inosine and phosphate to hypoxanthine and α-D-ribose-1-phosphate; (4) guanosine and phosphate to guanine and α-D-ribose-1-phosphate; (5) S-methyl-5’-thioadenosine and phosphate to adenine and S-methyl-5-thio-α-D-ribose-1-phosphate. YfiH aids in peptidoglycan synthesis by removing serine in UDP-MurNAc-Ser, replacing it with alanine [125]. These multifunctional deaminases possess an α/β/β/α fold with a central β-turn allowing for two antiparallel β-sheets, surrounded by a small 3 antiparallel strand β-sheet, one α-helix on one side, and three on the other, with two helices broken and redirecting [124,125,126,127,128,129]. Additionally, transient small helices appear depending on the structure. The mechanism of action for these proteins is unknown; however, inosine interacts with a pocket containing arginine, cysteine, and two histidines, with H47 as a gatekeeper [124,125]. YfiH’s interaction with UDP-MurNAc-Ser occurs at a nearby groove interacting with the β-turn. The I254V mutation in eukaryotic FAMIN, which downregulates adenosine phosphorolysis but upregulates deamination, increases the risk of Crohn’s disease and leprosy.
In summary, free adenine-derived deaminases predominantly belong to the AHS superfamily but exhibit greater diversity than their cytosine or guanine counterparts. This diversity includes most ADEases containing two metal cations which can be Fe, Zn, or Mn, with A. fabrum having three metal cations and an extra domain [100,101,105]. Additionally, deaminases acting on other adenine-derived substrates, including AFLDA, ADA, and others, contain a single cation with a dual proton-shuttle [106,107,108,109,110]. Some, including human ADA, lack the common PDZ domain seen in other AHS deaminases [1,105,108]. While most deaminases function intracellularly, hADA isoforms also act extracellularly, either bound to CD26 or secreted [104,119,120,121,122,123]. Free adenine and adenosine can also be deaminated by non-AHS deaminases such as FAMIN, YlmD, and YfiH, which process various reactions beyond deamination [124,125,126,127,128,129]. These AHS deaminases are notable for their diversity compared to those deaminating guanine and cytosine derivatives, as well as their ability to catalyze multiple reactions with distinct structural differences.
6.2. Adenosine tRNA Deaminases
As nucleobase deamination evolved, proteins capable of deaminating residues within larger macromolecular polynucleotides, such as tRNA, emerged. The deamination of adenosine in tRNA is crucial for cell viability. In prokaryotes and plant chloroplasts, this function is performed by TadA (tRNA adenosine deaminases) which targets one specific tRNA, whereas in eukaryotes, ADATs (adenosine deaminases of tRNA) target 7-8 different tRNAs [19,130,131,132,133,134]. TadA specifically deaminates tRNA-Arg2 (AGC motif) at position A34 to produce inosine (I34) at the wobble position, enabling the tRNA to pair with multiple codons containing A, C, or U [19,131,132]. TadA belongs to the CDA superfamily and shares the αβα-fold characteristic of GDA. It forms a homodimer at a similar angle as GDA but lacks the N-terminal binding region seen in GSDA (Figure 8A) [131,132,135]. The catalytic pocket of TadA contains a divalent Zinc ion coordinated by a histidine and two cysteines, which also bind a hydroxyl group (Figure 8B) [132,136]. Additionally, a catalytic glutamate is protonated. TadA interacts with the tRNA-anticodon through polar and aromatic residues across loops 4, 6, and 8 from the trans-dimer, while the cis-dimer uses an extended α6 at the C-terminus to interact with the larger tRNA fold [19,130]. The unique FFxxxR motif in the C-terminal α6 above the substrate-binding groove is crucial for substrate recognition, aiding in the specificity for tRNA nucleotides U33-G37 through hydrogen bonds and π-stacking interactions.
Due to its conserved structure and function, TadA has been utilized as an adenosine base editor (ABE) by linking it to Cas9 [137,138]. Modifications such as a proline mutation or truncation in the α6 disrupt tRNA binding, allowing single-stranded DNA binding instead. Further modifications could potentially convert TadA into a cytidine base editor (CBE). Mutations such as G122H in loop 8 increase pocket flexibility, while V82T in the β3-strand potentially enhances proton transfer efficiency. Dual mutations like E27G and I49N in loop1 and α2 (which breaks up the traditional loop3), respectively, modify the hydrogen bonding network around the pocket.
The heterodimeric ADAT2/3 in eukaryotes evolved from the homodimeric TadA with an expanded range of deamination sequences, relying more on the global tRNA structure than the local anticodon specificity seen in TadA [19,133,139]. Both TadA and ADAT2/3 bind the ASL (anticodon stem loop) of tRNA and deaminate the wobble position of arginine (ACG anticodon) [133,134,140]. However, ADAT2/3’s shorter amino acids lead to the loss of specific hydrogen bonds, allowing eukaryotes to also deaminate tRNA anticodons for threonine (AGU), alanine (AGC), proline (AGG), serine (AGA), leucine (AAG), isoleucine (AAU), and valine (AAC). Interestingly, ADAT1, which lacks a chemically derived structure, is known to deaminate alanine’s tRNA and resembles ADAR proteins in size [130,141]. ADAT2 is the active component of the ADAT2/3 dimer, while ADAT3, despite its pseudo-catalytic domain, plays a structural support role (Figure 8C-D) [133,134,140]. The N-terminal lobe of ADAT3 features an α+β fold with four central β-strands in a sheet. Upon binding, ADAT2 contacts the ASL, while ADAT3’s pseudo-catalytic domain interacts with the T-arm, and its lobe interacts with the D-arm [133]. In yeast, ADAT2 contains additional N-terminal regions, either extra α-helices or a β-sheet surrounded by α-helices, possibly derived from an inactivated nucleotide kinase, enhancing solubility and catalytic stability [134,139]. Homodimerization of ADAT2 mimics the positioning seen in ADAT2/3 and TadA but remains inactive without ADAT3 [133,134,140,142].
ADAT2 shares several interactions with TadA, including H-bond networks involving loops 1, 3, and α3 [140]. Nucleotides 32 and 38 form salt bridges with loop 8, and ADAT3’s hydrogen bonding with nucleotides 33, 35, 36, and 37 mirrors TadA’s opposing monomer. However, ADAT2 does not contact nucleotides 38-40, a task performed by ADAT3 [133]. Unique to ADAT2 is an RY-gate formed by arginine in loop 8 and tyrosine in α6, stabilizing and facilitating substrate entry and whose pi-cationic interactions must be broken. The unique KR-motif in an extra C-terminal loop stabilizes tRNA’s major groove phosphates, supporting nucleotides 28-31. Mutations disrupting the RY-gate and KR-motif abrogate deamination, except when tyrosine is replaced by phenylalanine. ADAT3’s inactivity is due to the pseudo-catalytic domain’s pocket being blocked by C-terminal cysteines or aspartate and proline [134,140]. In higher eukaryotes, the catalytic glutamate proton shuttle is replaced by valine, further inhibiting activity. ADAT3’s N-terminal lobe acts as an RNA recognition motif (RRM), essential for tRNA binding. This lobe resembles a ferredoxin-like domain (FLD), common in archaea’s CDAT8. The expansion of ADAT’s tRNA motifs compared to TadA results from relaxed specific bonds in ADAT2 with the addition of ADAT3’s lobe. If ADAT3 lacks the N-terminal lobe, C-terminal tail, or contains the inactive catalytic glutamate mutated to valine, deamination is abolished [19,134,140]. The mutation V144M in the lobe causes intellectual disabilities by creating steric clashes that prevent tRNA binding to the lobe without deamination. Steric clashes near the lobe-pseudo-catalytic domain interface permit tRNA binding but prevent the deamination needed for wobble position mutations. ADAT3’s lobe is connected to the core via two linkers, but 10% of ADAT3 remains unstructured and thus unresolved, possibly aiding in multi-tRNA interactions at the cost of higher degradation when unbound to ADAT2 [133,134].
In summary, tRNA adenosine deaminases share similarities with other CDA superfamily members like GDA and GSDA, except they do not require a lid due to their substrate size [19,131,132,133,135,139]. These tRNA deaminases utilize a single proton shuttle with Zn2+ coordinated by a histidine and two cysteines. The main difference lies in their substrate specificity; bacterial TadA deaminates tRNA-Arg2 (AGC motif), while eukaryotic ADATs target multiple tRNA codons [19,131,132,133,134,140]. This difference is attributed to TadA forming a homodimer with the C-terminal α6, containing the critical FFxxxR motif critical for substrate recognition [19,130]. Conversely, ADAT2/3 forms a heterodimer where ADAT2 is active, and ADAT3’s large N-terminal lobe acts as an RRM, providing extensive tRNA interactions necessary for less specific substrate recognition than TadA [133,134,140,142]. These enzymes are notable for their ability to react with highly organized nucleic acid structures, varying in specificity due to these interactions.
6.3. Adenosine dsRNA Deaminases
ADARs (dsRNA-specific adenosine deaminases) are unique to metazoans and have diverse roles in RNA metabolism [143,144,145,146,147]. They are involved in alternative splicing, crucial for proper brain function, miRNA editing and sequestration, UTR editing, RNA virus editing, and retrotransposon editing, including LINE-1 and Alu elements. There are three ADAR proteins, each containing a C-terminal deaminase domain, but with varying other regions [144]. ADAR1 is expressed in both the nucleus and the cytoplasm and includes three dsRNA-binding motifs (dsRBM) in the centre of the protein along with one Z-DNA-binding domain at its N-terminus in its constitutive state, and two such domains in its induced state. Nuclear ADAR2 contains two N-terminal dsRBMs, conserved in ADAR3, which evolved from ADAR2. However, ADAR3 does not dimerize, is catalytically inactive, has an extra arginine-rich region, and inhibits ADAR1 and ADAR2 by sequestering their substrate. Deficiencies in ADAR1 and ADAR2 lead to autoimmune disorders like Aicardi-Goutières Syndrome and Dyschromatosis Symmetrica Hereditaria, or neurological disorders such as epilepsy, seizures, and ALS [148].
Z-DNA, formed transiently following RNA polymerase movement, is stabilized by negative supercoiling and targeted by interferon-induced ADAR1 through its Z-DNA-binding domains, Zα and Zβ [144,149,150,151]. Zα, which only appears in the induced state, binds Z-DNA’s anti-syn nucleobase conformation using an α+β (winged) helix-turn-helix motif (similar to the protein ZBP1), with two key tyrosines at positions 136 and 177 enabling interaction with syn conformation DNA, especially guanosine via CH–π interaction (Figure 9A) [149,150,152,153,154]. In contrast, Zβ lacks these tyrosines and cannot bind Z-DNA, and B-DNA is precluded from both binding domains due to major and minor groove steric clashes. Zα may also interact with RNA, targeting Z-RNA found in some viruses and rRNA to halt certain protein translations [151,155]. The α3 of Zα stabilizes B-Z and Z-Z DNA junctions, with the C-terminal end of α3 being critical for B-Z junctions and proline residues 192-193 in the loop after β3 binding Z-Z junctions with imperfect CpG repeats [156,157,158,159]. The constitutive Zβ, compared to the induced Zα, has a hydrophobic core, shorter α1, and an extra α4 that binds a metal cation, allowing Zβ to dimerize with Zα, thereby stabilizing the Z-DNA-binding Zα [154].
ADAR dsRBMs share a mixed α/β fold with a conserved αβββα topology, where α-helices pack tightly against an anti-parallel β-sheet [160]. In ADAR2, dsRBM interactions with dsRNA and some mRNA stem-loops at R/G editing sites, like GluR-2 (a neurotransmitter), involve α1, β1, and β2 contacting the dsRNA minor groove, while β3 and the N-terminal of α2 interact with major groove phosphates (Figure 9B) [160,161,162]. These two dsRBMs, which do not dimerize, interact with RNA at a 120° rotation, affecting the minor groove creating potential space for ADAR2 dimerization (Figure 9C). DsRBM1 recognizes conserved pentaloop structures, and dsRBM2 recognizes cytosine bases near editing sites. ADAR1’s extra dsRBM3 contains unique N- and C-terminal sequences, brought together by an extra N-terminal α-helix that connects to α1 but turns away from the fold, forming ADAR1’s nuclear localization signal via transportin1 [160]. This extra α-helix, not part of the domain’s hydrophobic core or scaffolding, and doesn’t affect binding, but it significantly alters the positions of the N- and C-terminal regions to bring them together.
The catalytic domains of ADARs are similar in size, having evolved from ADATs, but only ADAR2’s structure has been chemically resolved [141,163]. These cores of the deaminases retain the CDA superfamily fold with multiple insertions and an extended C-terminus [148,163,164,165,166,167]. The common active site elements—histidine, glutamate, two cysteines, and coordinated zinc—remain, though one cysteine that is in the same 3-dimentional location is separated from the next by 64 amino acids due to a long insertion. Modifications include an extended α2 with the catalytic histidine and glutamate, incorporating another α-helix and two β-strands forming a sheet (Figure 9D). The β3-α3 (now α5) loop is extended by a long loop, an α-helix, and three β-strands forming a sheet, plus an extra α-helix between α5 and β4, with the traditional C-terminal α-helix of ADAT replaced by a long loop continuing into two β-strands extending the anti-parallel side of the canonical β-sheet, and four more stable α-helices. These α-helices, contacting others from different insertions as well as α2 and α5, hold inositol hexakisphosphate (IHP), essential for ADARs and ADAT1 stability [163]. Adenosine specificity over cytidine is achieved through steric clash of T375, preventing cytidine from entering the pocket [164]. Sequence specificity likely comes from a disordered loop adjacent to the first catalytic cysteine. This differs in ADAR1, which contacts the dsRNA minor groove near the editing site, supported by a U:A wobble. The +1 position specificity is determined by G489 which interacts with the minor groove in U:A but not G:C pairings [167].
Interestingly, non-Watson-Crick pairings like Gsyn:Ganti and Gsyn:3-deaza-dAanti at position +1, using both Watson-Crick and Hoogsteen sides, allow G489 entry and subsequent deamination. ADARs must employ base-flipping to access nucleobases within the dsRNA [148,163,164,165,167]. This highly conserved mechanism involves E488 penetrating the helix via the minor groove, stabilizing the empty adenosine site with interactions with the orphan base, sugars, and flanking nucleobases (Figure 9E) [164]. Supporting interactions include K376 and R455 with phosphates, T375 with the sugar, and V351 and catalytic residues with the pocket’s adenosine. ADARs are proposed as gene-editing Cas tools with mutations like E448Y, which cannot base-flip but could induce base flipping at sites with an orphan base creating an abasic site preventing off-target mutations [164,165,166]. Furthermore, E488Q interacts with cytidine orphan bases. Dimerization of ADAR2, utilizing the second insertion’s α-helix, allows one member to perform base flipping and catalysis, while the other stabilizes the RNA through dsRBM interactions [148].
In summary, ADARs are large CDA superfamily deaminases. While all contain the deaminase domain, they differ in the number of Z-domains and dsRBMs [144]. ADAR1 is unique with one or two Z-domains, depending on its state, and while all ADARs have dsRBMs, ADAR1 includes three compared to two in ADAR2 and ADAR3. The Z-domains of ADAR1, winged α+β helix-turn-helix motifs, are critical for binding syn guanines and stabilizing B-Z and Z-Z junctions [149,150,152,153,154,156,157]. Meanwhile, dsRBMs have distinct roles: recognizing pentaloop structures in dsRBM1 of ADAR2, cytidines near editing sites in dsRBM2 of ADAR2, and nuclear localization in ADAR1’s dsRBM3 [160,161,162]. Targeting dsRNA involves base flipping, where E488 penetrates the RNA helix’s minor groove, allowing target adenosine entry into the catalytic pocket [148,163,164,165,167]. Overall, ADARs stand out among deaminases for their size and multiple domains necessary for dsDNA and dsRNA interactions, as well as their unique capabilities to modify double-helix structures.
7. Cytosine Deaminases and Derivatives
7.1. Free Cytosine-Derived Nucleobase Deaminases
The deamination of cytosine has been characterized in both bacteria and single-celled fungi, but not in multicellular eukaryotes, as a pathway for pyrimidine salvaging; although, these two CoDA (cytosine deaminase) proteins come from separate deaminase lineages [168,169]. Both enzymes deaminate cytosine to produce uracil and ammonia (Scheme 7A). The bacterial CoDA (bCoDA) forms homohexameric complexes that maintain the same AHS fold as seen in both GDA and some ADEases, containing the distorted TIM β-barrel and PDZ β-sandwich domain [14,168,170]. Although the structural fold is similar to ADEases and ADA, this similarity is undetectable in BLAST searches, with only 13% identity between ADA and bCoDA [168]. Most divergence occurs in the loops and helices. The divalent cation, typically iron, is coordinated by histidines at positions 61, 63, 214, and 246, along with aspartate 313 (Figure 10A). When iron is replaced by zinc, the enzyme’s activity drops to 10% of its iron-based activity. The catalysis of bCoDA utilizes a dual-proton shuttle involving E217-H246-D313 [168,170]. The glutamate acts on the nitrogen at position 3, initiating the reaction by breaking one bond in the double bond with carbon 4 and allowing hydroxyl bonding. The aspartate then protonates the amino group of cytosine. This process is aided by loop 6 and α1, which move by 5Å upon binding cytosine, burying 750Å2 of the protein’s surface area and creating a stable hydrophobic environment [168]. Mutating glutamine 156, which stabilizes cytosine on nitrogen 1 and oxygen on position 2, or catalytic residues H246, E217, and D313, kills activity [171]. However, mutations H246Q and D313N retain low but detectable activity, possibly due to the ability to coordinate with Fe and the inefficient reaction completion by E217, respectively [170]. Bacterial CoDA can also catalyze isocytosine to uridine, 3-oxauracil to malonate semialdehyde, and isoguanine to xanthine efficiently at pH 7.7. Additionally, in Klebsiella pneumoniae and mutated E. coli with D314A, the pocket allows the catalysis of 5-methylcytosine to thymine [170,172,173].
Meanwhile, yeast CoDA (yCoDA) is part of the CDA superfamily with a fold and dimerization pattern similar to TadA and GSDA [169,171,174,175,176]. The catalytic pocket of yCoDA is maintained with divalent zinc, histidine, and two cysteines as coordinating residues, along with a glutamate proton shuttle (Figure 10B). The secondary structure is similar to GSDA, except for the insertion of an α-helix between α3 and β3, and the modification of the C-terminal α-helix. Unlike the C-terminal α-helix of GSDA, which ends in a loop over the pocket, or TadA, which ends in a long straight helix, yCoDA has a proline at position 149 acting as a helix breaker, allowing the α-helix to bend over the pocket [18,99,131,135,169,171,174,175,176]. This α-helix acts as a gate for cytosine entry, stabilizing it in place, fully enclosed in the pocket, and weakening after uracil formation for release, while halting cytidine entry due to lack of space [171]. Inhibition occurs through 2-hydroxypyrimidine (that lacks the amino group on the 4th carbon seen in cytosine), which starts the reaction by adding hydrogen to position 3 and hydroxyl to position 4, creating 4I)-hydroxyl-3,4-dihydropyrimidine. Without the amino group, the reaction’s second half cannot complete, remaining in stasis. Bacterial CoDA has greater thermostability than yCoDA [169,175]. Mutating A23L/I140L/V108I on α1, β4, and α8 increases yCoDA’s stability at temperatures up to 50 °C. Although inefficient, cancer gene therapy with mutated bCoDA or yCoDA has been proposed, acting as a suicide gene by catalyzing 5-fluorocytosine to toxic 5-fluorouracil [14,169,177].
Intriguingly, Mycolicibacterium smegmatis produces a protein known as SDA (selective deaminase), which defends against xenobiotic nucleotide analogues acting as herbicides through improper genome inclusion [178]. SDA cannot deaminate natural nucleobases but can deaminate molecules with s-triazine scaffolds, such as acetoguanide, 5-azacytosine, and ammeline, and shows lower activity on isoguanine (Scheme 7B; Figure 10C). Structurally, SDA is closest to yeast cytosine deaminase, sharing the same fold except for the lack of a C-terminal helix gate, replaced by a loop, and the insertion of an α-helix at the C-terminal end of β4 akin to those seen in GDA and GSDA [17,18,178]. This unique α-helix forms a flap over the catalytic site, elongating the catalytic pocket [178]. Meanwhile, critical residues E27, F29 with catalytic H56, and N46, coordinate the substrate, create π-stacking, and anchor the substrate, respectively.
In summary, CoDA is similar to guanine deaminases, having both CDA and AHS members. Although AHS bCoDA is structurally similar to ADA, it only shares 13% identity, using a single Fe or Zn cation with a dual proton-shuttle to effectuate reactions and a PDZ similar to AHS GDA for multimerization [14,168,170]. Meanwhile, CDAS yCoDA is similar to TadA with a single proton-shuttle and GDA in its use of a lid, though yCoDA uses a C-terminal α-helix instead of a loop [18,99,131,135,169,171,174,175,176]. Some species like M. smegmatis use this CDAS structure without the lid, with an additional α-helix creating space for deaminating various xenobiotic nucleotide analogues [178]. These deaminases showcase complex evolution through divergence from other members.
7.2. Free Cytosine-Derived Nucleoside Deaminases
Cytidine nucleosides are deaminated by the pyrimidine salvaging protein cytidine deaminase (CDA), of which there are two forms [179,180,181,182,183,184]. The most common form is T-CDA, which forms tetramers and exists across gram-positive prokaryotes, archaea, and eukaryotes, including animals. These enzymes perform the reaction converting cytidine to uridine (Scheme 8A). The CDA superfamily fold is named after these proteins’ secondary structure, featuring the α/β/α-fold [15,179,180,181,182,183,185,186,187,188]. This fold includes an α-helix on the non-catalytic side, a core parallel/anti-parallel 5 β-strand sheet, and five α-helices on the other side, with α3 and α4 forming the catalytic pocket, which faces the other members of the tetramers (Figure 11A). The tetrameric conformation is maintained by the inward positioning of α2-α6, facing the other members. Each member’s pocket faces the same direction while remaining inverted to the two direct contacts at either side, with α6 supporting the adjacent catalytic pocket. This conformation is a dimer of dimers with two-fold symmetry, requiring conserved hydrophobic interactions and contacts in Mycobacterium tuberculosis involving residues Y24, S25, Y51, R93, Q94, E98, and L124, of which L124 is species-specific [180,186]. Notably, D117 and D121 interact with R114 and point toward the central cavity between the tetramer’s domains.
These catalytic pockets utilize a single proton shuttle via glutamate, with the divalent zinc coordinated by three cysteine thiolates from α3 and α4 (Figure 11B) [179,183]. R56 (from B. subtilis) neutralizes the cysteines’ negative charges, maintaining their proper position. Typically, this arginine adopts a gauche+ conformation, interacting with cysteines from α3 and α4 [15]. However, a 130° rotation to gauche- creates a negative environment around the zinc, disrupting the cofactor-product bond and facilitating uridine release. Mutations such as R56A and R56Q impair catalytic efficiency due to disrupted catalytic pocket stability, while R56D and C53H inactivate T-CDA, and C53H/R56Q mutations lead to inactive monomers due to disrupted tetramerization [183]. T-CDA shows 45% structural conservation between B. subtilis and H. sapiens and can deaminate both deoxycytidine and cytidine, although it cannot deaminate nucleotides [180]. In M. tuberculosis, three regions relate to the catalytic pocket: region 1 (V22-F27) stabilizes the protein’s quaternary structure; region 2 (T42-C56) aids in quaternary structure and forms the pocket entrance; and region 3 (V110-F123) interacts with the adjacent subunit, with F123 involved in π-π stacking of the substrate (Figure 11C) [187]. Region 3, bordering α6, is active in substrate selectivity, acting as a motile “flap” [181]. In Saccharomyces cerevisiae, this region is permissive to larger substrates, enabling RNA deamination.
Some yeast, including Aspergillus terreus, contain BSD (blasticidin S deaminase), which forms a tetramer similar to T-CDA but deaminates blasticidin S, an antibiotic and antifungal agent inhibiting translocation during protein synthesis by binding guanine in the P-site of the 50S ribosome (Figure 11D) [8,15,183,189]. BSD’s secondary structure resembles T-CDA but lacks α2 and α6. It has a similar numbering system to AID/APOBECs with catalytic residues on α2 and α3. Blasticidin S comprises a cytosine head attached to glucuronic acid, linked to N-methyl β-arginine, binding across loops 1, 3, and 5 (Scheme 8B; Figure 11E). The substrate is deaminated to diaminohydroxyblasticidin S with glutamate and zinc coordinated by three cysteines, similar to T-CDA. The essential arginine is replaced by leucine, and an alanine on α3 adapts to an arginine to maintain the necessary interaction. This arginine, along with threonine and glycine from loop 3 backbones, interacts with a chlorine anion, leaving no internal pocket between subunits as seen in T-CDA. BSD uses Y126 instead of phenylalanine in T-CDA, as the neighbouring subunit’s pocket lid, for π-π stacking with the head and polar interactions with glucuronic acid.
A separate group, D-CDA, exists in gram-negative prokaryotes and some plants like A. thaliana, containing three domains instead of one [179,184,190]. D-CDA emerged from ancestral T-CDA duplication and maintains pyrimidine salvaging function. In plants, D-CDA can mutate cauliflower mosaic virus (CaMV), potentially playing a role in plant immunity. Its domains include an unknown N-terminal domain of three α-helices, a catalytic domain resembling T-CDA subunits, and an inactive pseudo-catalytic C-terminal domain unable to hold a divalent cation due to replaced pseudo-catalytic residues (Figure 11F-G) [179,184,190,191,192,193,194]. The catalytic and pseudo-catalytic domains form a dimer similar to T-CDA’s tetrameric form with two-fold pseudo-symmetry. The dimeric interface involves hydrogen bonds and salt bridging across catalytic domains (α2, α4, and α5-α6 loop) and pseudo-catalytic domains (β2-α2 and β3-α3 loops) [179,184]. The pseudo-catalytic domain of D-CDA is 29 residues shorter, lacking residues after the β4 strand [179,184,190,191,192,193,194]. The key residues F24, V26, and E44 which interact with the substrate, and F125 which interacts with the substrate of the T-CDA’s opposing subunit, are conserved or homologous in D-CDA’s catalytic domain [179]. In D-CDA’s catalytic [191,193CDA superfamily members, while adjacent T127 resembles A3B-CD1 and A3F-CD1 of the APOBEC family, often a serine in other cytosine derivative deaminases and ADAR, but glutamate in adenosine, guanine, and guanosine deaminases [8,88,99,131,134,163,171,179,191,192,195,196,197,198]. D-CDA does not require conformational change for reaction as T127’s backbone carbonyl interacts with the substrate, facilitating reaction, while P128 geometry aids uridine carbonyl release [191,193].
In summary, CDA superfamily proteins feature the classic α/β/α-fold [15,179,180,181,182,183,184,185,186,187,188,190,191,192,193,194]. T-CDA and D-CDA have lids from neighbouring monomers; T-CDA forms a tetramer with arginines and aspartates directed toward a central cavity, while D-CDA forms a dimer with pseudo-catalytic domains mimicking T-CDA. T-CDA uses three cysteines and R56 to stabilize zinc coordination [179,183]. BSD in A. terreus uses similar residues and an arginine on α3 but lacks T-CDA or D-CDA’s lid due to blasticidin S’s larger substrate size [8,15,183,189]. Despite D-CDA’s divergence from T-CDA, structural similarities persist due to their essential role in pyrimidine salvage pathways.
7.3. Free Cytosine-Derived Nucleotide Deaminases
Most deaminases that bind free deoxycytidine nucleotides function in the dTTP synthesis pathway, which generates 70-80% of the dUMP required for DNA synthesis [16,199,200]. In certain gram-negative prokaryotes, this process is facilitated by dCTPD (deoxycytidine triphosphate deaminase), which catalyzes the conversion of dCTP to dUTP. This pathway progresses through the formation of dUMP, dTMP, and ultimately dTTP. Structurally, dCTPD consists of 14 β-strands with nine forming a distorted β-barrel from two β-sheets, three α-helices, and four η-helices (Figure 12A) [16,199,201,202,203]. The catalytic pockets of dCTPD are formed at the interface between two subunits, necessitating the formation of homotrimeric complexes. These trimers can further assemble into a homohexameric complex, stabilizing two rings of active trimers. This structure aligns with the dUTPase-like (deoxyuridine triphosphatase) superfamily [16,199,200]. When the catalytic pocket of dCTPD forms through subunit interaction, dCTP and divalent magnesium ions enter the pocket [16,199,201]. The magnesium ion binds to the triphosphate group, shielding the C-terminus of the protein from negative charge.
The phosphates participate in a hydrogen bonding network: the α and β phosphates interact with arginines and serines, the γ phosphate interacts with lysine and tyrosine, and the magnesium ion, stabilized by an aspartate residue, interacts within this network (Figure 12B) [16,199,201,204]. The deamination reaction occurs without the involvement of a metal cation, instead utilizing R115 as a cation and E138 as a proton shuttle in E. coli [16,199,201]. The substrate is stabilized by a complex hydrogen bond network and π-π stacking by W131, preventing the accommodation of the 2’ hydroxyl group of ribose in the catalytic pocket. In dUTPase, this position is occupied by tyrosine. Inorganic phosphate and dTTP, byproducts of this pathway, inhibit dCTP entry [16,199]. However, dTTP binds with low affinity in most dUTPases. The protein’s C-terminus is essential for catalytic activity, as it closes the pocket during the reaction, although it does not directly interact with the ligand [199]. Mutations in the catalytic pocket, such as R115 and S111C, result in inactivity due to disrupted interactions [201]. Mutations like S111T and E138D reduce activity with S111T specifically blocking dTTP inhibition due to steric hindrance. An intriguing variant of dCTPD is found in Agrobacterium fabrum, featuring a double domain duplication that prevents proper trimer formation [205]. Despite this, it retains a single intact catalytic pocket and forms multimerization interfaces elsewhere on the protein surface.
In some gram-negative prokaryotes and archaea, DCD:DUT (bifunctional deaminase/diphosphatase) has evolved from dUTPases to become bifunctional [204,206]. Unlike dCTPD, some species retain dUTPase activity. This bifunctionality allows dCTP to be converted into dUMP, PPi, and NH2, maintaining low dUTP concentrations to prevent toxic misincorporation into DNA [200,204]. In M. tuberculosis, DCD:DUT is located in the unique waxy layer of its cell wall, while Bacillus halodurans possesses genes for both DCD:DUT and dCMPD, providing multiple pathways for dTTP synthesis via deamination [200,206]. Like dCTPD, DCD:DUT maintains a homotrimeric quaternary structure that can form dimers, creating a distorted β-barrel surrounded by α and η-helices, and uses C-terminal residues to close the active site (Figure 12C) [200,204,206,207]. The deamination process does not require a cation and uses homologous residues for catalysis [204,207]. However, the catalytic arginine and glutamate essential for deamination are absent in ancestral dUTPase.
Both DCD:DUT and dCTPD contain four motifs in their catalytic pockets, where motifs 1, 2, and 4 bind phosphates from one subunit, and motif 3 recognizes the pyrimidine from another subunit. A fifth motif on the C-terminus is crucial for catalysis in DCD:DUT and dUTPases [16,204,207]. In dUTPase, the C-terminus wraps around the adjacent subunit to the next catalytic pocket, involving all three subunits in each substrate’s processing. In contrast, in dCTPD and DCD:DUT, the C-terminus interacts with the nearest pocket. The C-terminus is crucial for pyrophosphatase activity in DCD:DUT and dUTPase, although it remains intrinsically disordered when no substrate is bound. The hydrogen bond network in DCD:DUT stabilizes the α-phosphate of the substrate, with an aspartate coordinating the magnesium ion, which also activates water for phosphate hydrolysis [200,206].
In contrast to the dUTPase superfamily proteins DCD:DUT and dCTPD, which are prevalent in archaea and gram-negative prokaryotes, the CDA superfamily protein dCMPD (deoxycytidylate deaminase) is utilized by gram-positive prokaryotes, eukaryotes, and some viruses like T4 bacteriophage, cyanophage S-TIM5, and PBCV-1 [16,195,208,209,210,211,212]. This pathway offers an alternative for dTTP synthesis, converting dCMP into dUMP for thymidylate synthase, crucial for DNA synthesis [195]. The CDA fold of dCMPD features a five-strand β-sheet flanked by α-helices, with an intriguing subdomain insertion in the loop between β2 and the first catalytic α-helix, varying by species contain 0-2 α-helices [195,208,209,211,212]. The protein forms homohexameric complexes with two interfaces: one across the subdomain and three α-helices, and the other involving α1 and β2 interacting with counterparts on adjacent subunits (Figure 12D). The subdomain provides extra pocket space for dCMP binding through a hydrogen bonding network. A tyrosine (or phenylalanine in T-CDA) in the β4-α4 loop interacts with substrate specificity via planar interactions and phosphate hydrogen bonding, blocking 5-methyldeoxycytidine and deoxythymidine entry due to steric hindrance [195]. The PBCV1 virus features a tryptophan instead of tyrosine on the N-terminus of α4, allowing catalytic activity with dCTP and dCDP [212]. The catalytic pocket contains histidine and two cysteines coordinating a divalent zinc ion with a glutamate proton shuttle (Figure 12E) [195,208,209,210]. T4 bacteriophage and gram-positive prokaryotes have an additional divalent zinc coordinated by a histidine and two cysteines within the subdomain.
Cyanophage S-TIM5 lacks a coordinated zinc in the subdomain, instead relying on a disulfide bond between C22 in loop 1 and C54 of the subdomain for stability (Figure 12F) [211]. This subdomain also contains an allosteric site opposite the substrate-binding region, accommodating dCTP or dTTP with a divalent magnesium coordinating the triphosphate [195,208,209,211,212]. dCTP presence is required for dCMP activity, except in mammals where low catalysis occurs without dCTP, while dTTP presence abolishes activity (Figure 12G-H). The concentration of dCTP, the pathway’s starting point, and dTTP, the end product, influence activity through the allosteric site. Retaining its global structure, binding causes a shift in a network of hydrogen bonds, π-π stacking, and salt-bridging, which only occurs with dCTP’s γ-phosphate. At the hexamer interfaces, a tyrosine (or tryptophan in cyanophage) at the β2 terminus enhances dTTP binding via π-π stacking, while dCTP promotes a more compact hexameric complex [195,208,209,210,211,212].
In summary, free cytidine nucleotide deaminases are categorized into the dUTPase superfamily, acting on triphosphates, and the CDA superfamily, acting on monophosphates. dCTPD and DCD:DUT from the dUTPase superfamily deaminate dCTP using a basic trimeric arrangement, forming catalytic pockets at subunit interfaces [16,199,200,201,202,203,204,206,207]. The deamination reaction employs an arginine coordinating the hydroxyl and glutamate proton shuttle with DCD:DUT featuring an additional C-terminal region for phosphatase activity [16,199,201,204,207]. Meanwhile, hexameric dCMPD, with its diverse pocket subdomain (equivalent to APOBECs loop 3), regulates substrate binding and acts as an allosteric site, modulating activity on dCMP based on dCTP or dTTP presence [195,208,209,211,212]. This subdomain uniqueness distinguishes dCMPD from other CDA family members, showcasing a novel mechanism for securing a free nucleic acid, while dUTPase enzymes are only similar to RidA.
7.4. Free Cytosine-Derived Vitamin B2 Pathway Deaminases
Riboflavin (vitamin B2) synthesis is a crucial precursor process for the formation of coenzymes FMN (flavin mononucleotide) and FAD (flavin adenine dinucleotide) [214,215,216,217,218,219]. This synthesis begins through pathways originating from either the pentose phosphate pathway or the purine pathway, with the latter featuring two distinct routes depending on the organism’s domain of life. The process consistently starts with DAROPP (2,5-diamino-6-ribosylamino-4-(3H)-pyrimidinone 5’-phosphate) and culminates in ARIPP (5-amino-6-(D-ribitylamino-2,4(1H,3H)-pyrimidinedione 5’-phosphate), necessitating both a deamination and a reduction step. In bacteria and plants, the protein RibG (sometimes referred to as RibD) catalyzes the deamination of DAROPP through its N-terminal deaminase domain (RibG-D) to produce AROPP (5-amino-6-ribosylamino-2,4(1H,3H)-pyrimidinedione 5’-phosphate) (Scheme 9). This is followed by the action of its C-terminal reductase domain (RibG-R), which, using NADPH, reduces AROPP to ARIPP. Conversely, fungi and some archaea use two distinct proteins for these transformations: Rib7 reduces DAROPP to DARIPP (2,5-diamino-6-ribitylamino-4(3H)-pyrimidinone 5’-phosphate), which is then deaminated to ARIPP by Rib2. Notably, animals lack these biosynthetic pathways and must obtain riboflavin through their diet [218]. RibG-D adopts the canonical CDA fold with its β-sheet and α2 and α3 helices, (which house the usual histidine, cysteines, glutamate catalytic residues, and a divalent zinc ion), being relatively shorter (Figure 13A) [7,214,215,216,217,220]. Unique to RibG-D is a short η-helix inserted in the α2-β2 loop and an extended β3-α3 loop, positioning one catalytic cysteine midway. Furthermore, similar to the C-terminal α-helix of TadA, ADAT, GDA, and GSDA, it features an α5-helix adjacent to α1, transitioning into α6, which serves as the linker to the reductase domain—this α6-helix being crucial for structural stability even in Rib2, despite it lacking a direct linking function (Figure 13B) [218]. However, Rib2 contains an additional η-helix in the β1-β2 loop, and the β-sheet and α2 are longer.
Substrate specificity in RibG-D is facilitated by key residues like K79, which interacts with H76 and T80 in the β3-α3 loop, stabilizing the phosphate group via D101 and N103 in the β4-α4 loop interacting with the ribosyl (Figure 13A) [214,215,216,217,221]. K79 is required for proper binding of DAROPP but requires the circularization of the ribose precluding the binding of DARIPP bound by Rib2. In Rib2, key residues include R105 (lysine in RibG-D) and ribose-hydroxyl-interacting L106 and E136 (D101 and N103 in RibG-D), offering specificity to DARIPP (Figure 13B) [218]. RibG-R features a central β-sheet of 9 strands flanked by two α-helices per side and four η-helices near RibG-D [7,214,215,216,217,220,221,222]. RibG-R utilises a lysine and glutamate in the β-sheet playing crucial roles alongside NADPH in stabilizing AROPP [214,215,221]. This stabilization facilitates the formation of a Schiff base intermediate and the subsequent protonation of the ribose to ARIPP. Rib7, used by archaea and fungi, shares the core structure with RibG-R but exhibits diversity in loop conformations and lacks the catalytic lysine, making it specific for AROPP [215,217,218]. Structural variations are seen in how RibG proteins form complexes. In E. coli and Acinetobacter baumannii, RibG forms homodimers with interactions across RibG-R domains, whereas in B. subtilis, a ring-like tetrameric structure is observed, employing additional interfaces between RibG-D domains (Figure 13C) [7,214,215,216]. The absence of tetramerization in E. coli may be attributed to steric clashes and the lack of potential salt-bridges in RibG-D.
In summary, RibG-D and Rib2 are both members of the CDA superfamily, performing deamination in the riboflavin biosynthesis pathway [214,215,216,217,218,219]. The substrates DAROPP and DARIPP, similar in structure to guanosine, differ mainly in their sugar conformations—ring or linear. Both enzymes exhibit the typical CDA structure and catalytic residues with secondary structure modifications such as η-helices enhancing substrate accommodation [7,214,215,216,217,218,220]. Substrate binding is mediated by specific residues, with K79 in RibG-D replaced by R105 in Rib2 interacting with catalytic residues, as well as structural adaptation for binding either ring (RibG-D’s D101 and N103) or linear (Rib2’s L106 and E136) sugar forms [214,215,216,217,218,221]. RibG, combining both deaminase and reductase functions, contrasts with the separate Rib2 and Rib7 proteins, showcasing the evolutionary divergence in the riboflavin synthesis pathway [7,214,215,216,217,220,221,222].
7.5. Cytidine RNA Deaminases
Among non-APOBEC cytidine deaminases, two notable RNA deaminases include those acting on tRNA in the hyperthermophilic archaeon Methanopyrus kandleri and mRNA in plant mitochondria and chloroplasts, mediated through the CDAT8 (cytidine deaminase of tRNA 8) and DYW domains, respectively [140,223,224]. These enzymes belong to the modified CDA superfamily and employ divalent zinc along with the histidine, cysteine, and glutamate catalytic pocket characteristic of this enzyme family. Both enzymes are connected to other domains that facilitate their specific activities.
In M. kandleri, 30 out of 34 tRNAs contain a cytidine at the 8th position, which needs deamination to ensure proper folding and functionality for translation [224]. Typically, uridine occupies this position in tRNA to maintain its tertiary structure, as it is located between the acceptor stem and D-arm. The necessity of cytidine at position 8 could be due to genetic drift or thermodynamic advantages, either at the genetic level, during primary transcription, or in the folding and modification of tRNA, especially given the organism’s survival at extreme temperatures of 110 °C. In contrast, a U to C mutation at this position in humans destabilizes tRNA, causing mitochondrial myopathy and impairing the initiation and elongation processes of ribosomal translation. However, some eukaryotic and plant mitochondria contain position 8 uridine, but no known protein is known to cause it.
CDAT8 comprises an N-terminal CDA deaminase domain characterized by a short α1 and α2 and an additional β-sheet with two strands beside α1 (Figure 14A). This domain is followed by an FLD domain, similar to the lobe seen in ADAT3, consisting of a four-stranded β-sheet with two α-helices [140,224]. The FLD domain is linked to a C-terminal THUMP domain, which also contains a β-sheet of four strands and two α-helices. CDAT8 functions as an asymmetric dimer with dimerization interfaces across the CDA and FLD domains (Figure 14B). This asymmetry results in the blockage of one catalytic pocket, allowing the other to remain accessible for deamination. Shortened substrates, lacking the tRNA anticodon or D-arm, can be deaminated, whereas full-length tRNA binding is facilitated by dimerization, placing the acceptor stem’s 3’ CCA motif near the THUMP domain and positioning C8 close to the CDA’s catalytic pocket (Figure 14C) [224]. This interaction is stabilized by a positively charged groove formed between these regions.
In plants, the DYW domain plays a crucial role in maintaining homeostasis within mitochondria and chloroplasts [223]. The DYW protein features two substrate recognition pentatricopeptide repeat (PPR) domains—E1 and E2—at its N-terminus, with the DYW deaminase domain located at the C-terminus. This DYW domain retains the CDA fold with the hallmark zinc-binding motif but notably lacks the canonical α1 observed in other CDA family deaminases (Figure 14D). A unique aspect of DYW is the absence of the conserved proline preceding the first catalytic cysteine in the CDA superfamily. Furthermore, it contains a PG box motif and the DYW motif at the C-terminal loop beyond β5, which stabilizes a secondary zinc. Additionally, a gating subdomain is inserted in the β2-α2 loop, comprising an α-helix and a β-finger, which may sometimes be replaced by two α-helices [223,225,226].
This gating mechanism, common across land plants, enables autoinhibition through β-finger shifts, while DYW2 of the same species contains a different mechanism [223]. These β-finger shifts result in lysine interactions with water and glutamate in the catalytic pocket, along with π-π stacking of histidines, which destabilizes the catalytic geometry (Figure 14E-F). Upon activation, the β-finger shift allows the lysine to interact elsewhere, positioning the catalytic histidine to better coordinate zinc, thereby facilitating water’s proximity to glutamate and zinc for catalysis. The PG-box and DYW motifs stabilize the RNA backbone, while gate state determination relies on interactions between R895 and either D922, forming the inactive gate, or D872, forming the active gate [223,226].
In summary, cytidine RNA deaminases include CDAT8, which targets tRNA, and DYW, which acts on mRNA [140,223,224]. Both enzymes maintain the CDA fold but require additional structural features. CDAT8 consists of three domains and necessitates dimerization for tRNA binding, with its FLD domain resembling ADAT3’s N-terminal lobe that stabilizes tRNA [140,224]. DYW is part of a larger enzyme complex with two PPR domains for substrate recognition [223]. The gating mechanism, involving a supplementary α-helix and β-finger, utilizes polar charged and π-π interactions, while the PG-box and DYW motifs stabilize the RNA backbone. These proteins facilitate the deamination of distinct RNA structures via multiple domains, producing uridine through controlled mechanisms—either asymmetric dimerization or a gate subdomain.
7.6. Cytidine DNA Interbacterial Toxin Deaminases
Prokaryotes employ various strategies to attack one another, one of which involves the release of toxins via bacterial secretion systems [10,226,227]. In Burkholderia cenocepacia, the DddA (dsDNA deaminase toxin A), and in Pseudomonas syringae, the SsdA (ssDNA deaminase toxin A), are notable examples of such toxins. These gram-negative bacteria use the type VI secretion system (T6SS) to transfer these toxins into adjacent cells, leading to mutations that cause chromosomal degradation, DNA replication arrest, or even the acquisition of resistance genes through sublethal mutations.
DddA features a repeating PAAR motif near the N-terminus that facilitates secretion, followed by an RHS (rearrangement hotspot), and culminating in a toxic deaminase domain (Figure 15A) [10]. This domain belongs to the CDA superfamily and includes the canonical catalytic residues and zinc, with a preference for TC motifs in DNA. Structurally, the deaminase domain of DddA deviates from the typical CDA fold as it lacks the N-terminal α-helix supporting the β-sheet [10,227]. Instead, it is stabilized by two alternative β-strands forming a β-sheet in place of the canonical α1. Additionally, DddA features an extended β2-α1 loop and, similar to T-CDA, a direct β4-β5 loop without an intervening α-helix, leading to an extended C-terminal loop. In contrast, SsdA’s deaminase domain resembles that of the DYW domain but lacks the DYW motif and secondary zinc (Figure 15B) [226]. Instead, SsdA includes two additional α-helices at the N-terminus, one replacing the canonical α1 and the other located in the α1-β1 loop. DddA exhibits high activity on dsDNA, particularly in regions with base mismatches, while SsdA shows low activity on dsDNA but a stronger effect on ssDNA [10,226,227].
The precise mechanism of cytidine entry into the catalytic pocket for deamination remains unclear [227]. However, a phenylalanine residue in α3 displaces the dT in the -1 position which, in turn, displaces the dC, allowing entry into the pocket (Figure 15C). A T-G mismatch stabilizes the dsDNA. DddA interacts with dsDNA by widening the minor groove by 9Å, involving interactions across all loops near the catalytic pocket, the C-terminal loop, and α2, which stabilizes the groove and the DNA backbone, enabling base flipping. DddA’s potential for genetic engineering has been explored, particularly in conjunction with Cas9 and genetically modified TadA, for applications in the nucleus, mitochondria, and chloroplasts [10,227,228]. On the other hand, SsdA’s interactions with ssDNA are akin to those observed in DYW domains with RNA [226]. Interestingly, the PG-box motif of the DYW domain is replaced by an SWG motif in SsdA. Both DddA and SsdA require mechanisms to prevent self-damage in their own cells which are accomplished through inhibitory proteins, DddAi and SsdAi, respectively (Figure 15D-E). These inhibitors bind the catalytic pocket and grooves of their respective deaminases, mimicking the geometry of the DNA substrate [10,226,227]. DddAi consists of two α-helices and two β-sheets made up of two or seven β-strands, respectively, while SsdAi is composed entirely of nine α-helices.
In summary, interbacterial toxin deaminases like DddA and SsdA represent a fascinating mechanism of bacterial warfare through genotoxic deamination, facilitated by secretion systems [10,226,227]. Both belong to the CDA superfamily with DddA targeting dsDNA and SsdA primarily affecting ssDNA. DddA features a repeating PAAR motif, while SsdA incorporates an SWG motif instead of the DYW domain’s PG-box. Like all dsDNA/dsRNA reactive deaminases, DddA has evolved mechanisms to circumvent the challenge of double-stranded interactions through base flipping, facilitated by a phenylalanine inserted in into the dsDNA. A critical challenge for these deaminases is preventing self-reactivity, given that bacterial genomes are not segregated from the rest of the cell by a nucleus. This issue is resolved by specific inhibitory proteins that prevent genotoxic activity within their own cells.
7.7. AID/APOBEC Family Deaminases
One of the most well-known and studied groups of cytidine deaminases is the AID/APOBEC (activation-induced cytidine deaminase/apolipoprotein B mRNA editing catalytic polypeptide-like) family of deaminases within the CDA superfamily [198,229,230,231,232,233,234,235]. These enzymes deaminate cytidine to uridine in polynucleotides, causing mutations across metazoans. Across evolution, APOBECs have created divergent proteins found in various species, with APOBEC4 believed to be the oldest (Figure 16A) [230,236]. APOBEC4, not yet structurally resolved or functionally understood, is found across metazoans, dating back to cnidarians, and existing separately in some algal species. AID (also known as AICDA) evolved in jawless vertebrates and plays a critical role in increasing variability in the adaptive immune system [229,237,238]. In B-cell nuclei, AID regulates antibodies via somatic hypermutation (SHM) of the variable region, enhancing diversity, and class switch recombination (CSR), producing antibodies with diverse effector functions. APOBEC2, found in jawed vertebrates, acts on mRNA and ssDNA in cardiac and muscle development [26,198,230]. APOBEC1 evolved as a mammalian nuclear protein with recent discoveries of orthologues in some avians and lizards [230,239,240]. It deaminates DNA and RNA, notably creating a premature stop codon in mammalian APOB-100 mRNA to produce APOB-48, regulating low-density lipoproteins (LDLs) and lipid uptake. APOBEC1 also mutates protein-coding sequences and 3’UTRs. APOBEC3, which developed later in placental mammals, has diverged into three subtypes: A3Z1, A3Z2, and A3Z3 which act as mediators of the innate immune system against endogenous viruses and zoonotic viral incursions [241,242,243,244,245,246]. In humans, all APOBEC3 genes are located on chromosome 22 [239,247].
Species such as bovines, sheep, and goats maintain one of each A3 subtype (Figure 16B) [242,243,244,245,246]. However, rodents, felines, and pigs have lost the A3Z1 subtype, while primates, equines, and bats have further duplications of their A3Zs, some becoming dual-domain due to neighbouring duplications. Primates have seven APOBEC3s (A3A-H), with three being single-domain and four double-domain proteins (Figure 16A) [244,248,249]. The double-domain proteins have an inactive pseudo-catalytic domain (CD1) and an active catalytic domain (CD2). A3A, A3C, and A3H are single-domain proteins across A3Z subtypes 1, 2, and 3, respectively [247,248,249,250,251]. Double-domain proteins A3D (also called A3E) and A3F contain both domains from A3Z2, while A3B and A3G maintain CD1 from A3Z2 and CD2 from A3Z1. Notably, A3H has multiple haplotypes, such as seven in humans and thirteen in pig-tailed macaques [235]. A3A, expressed in myeloid lineages, and A3B, mainly in erythrocytes, B-cells, and plasma cells, are primarily nuclear except during the M phase of the cell cycle [231,239,252,253,254,255,256,257]. A3C, A3F, A3G, and A3H, found across many cell types, notably thymocytes, are located in both the cytoplasm and nucleus in varying amounts. A3A, A3D, A3F, A3G, and A3H expressions are induced by interferon-α, while A3B and A3C are not [253,257,258,259].
The APOBEC3 lineage significantly affects endogenous retrotransposons like LINE-1, Alu elements, LTR retroelements, coronaviruses, HPV, Rous sarcoma virus, parvoviruses, HSV1, CMV, Epstein-Barr virus, and hepatitis B virus [13,231,253,260,261,262,263]. APOBEC3s are best known for restricting lentiviruses like HIV and SIV, acting as zoonotic barriers. A3C, A3D, A3F, A3G, and A3H (haplotypes 2, 5, 7) mutate HIV/SIV by incorporating into budding virions, interacting with RNA, and either processively mutating the genome or stalling reverse transcription [13,231,234,235,248,250,264,265,266,267]. HIV/SIV counteracts this using the protein Vif (Viral infectivity factor) which binds APOBEC3, recruiting the E3 ubiquitin ligase complex for proteasomal degradation. Different interfaces are used during Vif counteraction, with one interface for A3C, A3D-CD2, and A3F-CD2, another for A3G-CD1, and another for A3H (Figure 17A-F) [268,269]. For A3C, A3D, and A3F, Vif binds α2, α3, and loop 4 of APOBEC through polar and planar contacts on three F-boxes with CBF-β’s (core binding factor-β) aid (Figure 17G) [250,251,270,271]. A3H, which targets viruses with G-rich regions during RNA priming, binds near the region seen in A3C, A3D, and A3F on α3 and α4 [235]. A3G interacts with Vif at the DPD motif on α4 in humans (KPD in most primates), acting as a zoonotic barrier (Figure 17H) [234,272,273,274]. Full interaction between Vif and A3G requires branched dsRNA, allowing both proteins to bind it and each other [267,274].
The AID/APOBEC family of deaminases are renowned not only for their functional effects but also for maintaining a modified form of the CDA superfamily fold, known as the APOBEC fold [198,229,230,231,232,233,234,235]. This fold features an α/β/α structure with a central β-sheet composed of five strands (Figure 18A). The catalytic pocket contained on α-helices 2 and 3, containing one histidine, two cysteines coordinating a divalent zinc, and a glutamate proton shuttle, is flanked by α-helix 4 on one side. On the opposite side of the β-sheet, α-helices 1, 5, and 6 are positioned perpendicularly to the helices forming the catalytic pocket. Differences from the CDA fold include the additions of α5 and α6, the shortening and angling of α1, the addition of an N-terminal loop, and the formation of a DNA/RNA-binding groove, created by the loops α1-β1, β2-α2, β3-α3, and β4-α4, referred to as loops 1, 3, 5, and 7, respectively (Figure 18B) [13,198,252,267,273,275,276,277].
Across the AID/APOBEC lineage, structural variations are observed in the fold. Generally, the lengths of loops 1 and 3 show significant variation [197,198,230,249,250,255,267,271,275,278]. AID is unique with an extra α7 at the C-terminus, whose function and positioning remain unresolved (Figure 18C) [275]. APOBEC1 contains a distinct hydrophobic subdomain at its C-terminal end which aids in dimerization and occludes the nuclear export signal on α6 when dimerized (Figure 18D) [230]. APOBEC2 stands out with extended β1 and β2 strands [26,198,230]. Among primates, A3B, A3D, A3F, and A3G are double-domain proteins, each having an inactive pseudo-catalytic domain (CD1) and an active catalytic domain (CD2), linked by a flexible linker (Figure 18F,G) [244,248,249]. Intriguingly, A3Z1s such as A3A, A3B-CD2, and A3G-CD2 possess a distinctive bulge in their β2-strand, forming a β2-bulge-β2’ crucial for catalysis [231,239,248,249,253,279,280,281,282]. This bulge can manifest as either a loop or an η-helix, potentially important for dimerization.
These structural variations allow AID/APOBEC deaminases to target diverse substrates across multiple cellular locations [229,248,249]. Each protein’s cellular localization and substrate specificity centred on 3 base nucleotide motifs play critical roles in their biological functions. This specificity can lead to genomic informational decay and double-strand breaks when acting on viral or chromosomal polynucleotides. AID/APOBEC substrate specificity is linked to loop 7, which interacts with the -1 and -2 positions of the target motif (Figure 18E) [229,278]. These proteins bind their substrates via a specific and non-specific manner around the substrate-binding groove [231,232,247,249,252,274]. This groove passes through the loop 1/3 interface, over the catalytic pocket and loop 5, through the loop 1/7 interface, and over α6, creating a U-shaped curve in the DNA. While ssDNA and ssRNA can enter these grooves, the grooves spatial arrangement determines the deamination capability of a substrate type [275,283]. For instance, A3G can accommodate the C2’-endo sugar pucker common in B-DNA but cannot accommodate the C3’-endo sugar pucker found in A-RNA. A key residue, known as the gatekeeper, binds and stabilizes the 5’-phosphate of the deaminated cytidine [275]. The gatekeeper residue varies among AID/APOBECs and species, with human AID, A3B-CD2, and A3F-CD1 having arginine; APOBEC1 and A3H having lysine; A3A and A3G-CD2 having histidine; A3C, A3D-CD2, and A3F-CD2 having asparagine; A3B-CD1 and A3D-CD1 having serine; and A3G-CD1 having leucine [230,238,252,255,267,270,275,284,285].
Little is known about the substrate specificity of APOBEC2 and APOBEC4 [230,286]. APOBEC2 appears to interact with transcription factors through cooperative molecular mechanisms. APOBEC1, although capable of deaminating ssDNA independently, requires cofactors like A1CF (APOBEC1 complementation factor) or RBM47 (RNA-binding protein 47) to deaminate C6666 in APOB mRNA, creating a stop codon (CAA to UAA) in the 27S “editosome” complex [230]. AID shows a preference for the WRCA motif, particularly favouring 3 and 6 nucleotides after forming guanine quadruplexes (G4) occurring at immunoglobulin switch regions, aiding class switch recombination (CSR) [229,275,287]. Interactions with G4 are facilitated by the removal of α7 and DNA-binding of the “assistant patch” on α6, absent in APOBEC3s lacking G4 preferences [275].
APOBEC3 proteins, particularly of the A3Z2 and A3Z3 subtypes, are known to processively deaminate ssDNA in a 3’ to 5’ direction, a process enhanced in double-domain APOBECs [13,197,266,288,289,290,291,292,293]. These proteins have greater substrate contact and intersegmental transfer capabilities compared to single-domain APOBECs. Double-domain APOBEC3s can rotate independently due to their linker, adopting conformations such as the compact “globular state” and the extended “dumbbell state” for multiple nucleic acid interactions (Figure 18F-G) [234,294]. Most APOBEC3s prefer the TTCA motif but can also deaminate other TC motifs [229,239,247,251,252,253,256,265,295,296,297,298,299]. This activity involves loop 7, containing hydrophobic and planar residues.
A3A and A3H can deaminate both deoxycytidine and 5-methyldeoxycytidine to deoxythymidine, though only dC is observed in A3H from Pan troglodytes [253,264,277]. A3A, the most active deaminase among human APOBEC3s, deaminates ssDNA efficiently, binds ssRNA more effectively than ssDNA without deamination, and is even capable of deaminating dCTP with low efficiency [231,247,248,253]. A3H binds RNA complexes, like those seen in A3F and A3G’s CD1s, with RNA-binding defective mutants becoming DNA “hypermutators,” potentially due to loop 7 interactions being freed up [235,255]. A3H’s cytoplasmic localization is influenced by positive surface patches interacting with RNA, including double-stranded RNA [255,277]. A3D-CD2 shows potential for a stabilizing disulfide bond between loop 3 and α2 known from A3C and A3F-CD2, missing in inactive CD1s of other APOBEC3s, which are also A3Z2s [197,233,250,273,284,285]. Instead, these APOBEC3s contain an α2 extension that shifts the catalytic architecture. Uniquely, A3G targets the CCCA motif, deaminating the cytidine at the 0 position early and the -1 position subsequently, without affecting the -2 position, facilitated by two aspartates in loop 7 during 3’ to 5’ processivity [13,196,229,253,266,279,280,295]. Across all APOBEC3s, preference for deoxyadenosine at the +1 position is attributed to a combination of hydrogen bonding and π-π stacking interactions that stabilize the polynucleotide [283,295].
AID/APOBECs are also capable of non-catalytically interacting with nucleotides, each other, and viral factors [197,198,250]. AID and APOBEC1 contain extra C-terminal residues comprising AID’s α7 and APOBEC1’s HD domain made of two β-strands in a β-hairpin and three α-helices, allowing dimer formation (Figure 18H) [230,275]. For AID, dimerization requires G4, whereas in APOBEC1, the HD domain forms a four-strand β-sheet from the hairpins, creating a large positive surface, even though dimerization is not required for activity. A3A dimerizes through N-terminal loops, involving two histidines interacting with three metal ions, creating a groove with two lysines where both monomers cooperatively bind ssDNA until the product is formed, at which point they are thought to dissociate (Figure 18I) [248]. APOBEC2 dimerization is unique, involving 12 hydrogen bonds between each monomer’s β2 strands (Figure 18J) [198]. This forms an extended β-sheet across the dimer of 10 strands with the C-terminal ends of each α2 contacting each other head-on, creating a pseudo double-length helix. This dimer positions the catalytic pockets of each monomer in opposite directions, although APOBEC2 can also form head-to-head dimers, creating a larger tetrameric complex. A3F-CD2, A3G-CD1, and A3G-CD2 monomers show multiple potential dimerization sites, including some involving A3G-CD2 with an extra zinc in loop 3, coordinated by two histidines [234,273,276,282,300]. However, not all dimerization forms are possible in double-domain forms due to the degree of plasticity between the domains. Double-domain A3G does not require substrate for dimerization and can form either a head-to-head dimer across substrate-binding grooves or through a mirrored CD1-CD1 dimerization across loops 1 and 7 with α6, creating a positive electrostatic potential between two R24 residues critical for RNA and DNA interactions [234,248]. Although double-domain APOBECs have enhanced substrate binding and processivity, they are more susceptible to forming high molecular weight complexes called P-bodies, unlike single-domain APOBECs, except A3H (Figure 18K) [197,255,277]. In P-bodies, A3H interact with each other and RNA using multiple planar residues, requiring RNAse A for dissociation. A3H susceptibility to P-bodies is partly due to its unique dimerization mode involving dsRNA, forming a complex network of π-π stacking with GC-rich palindrome-like sequences [235,255,264]. A similar dimerization mechanism was found with dsRNA and A3G between its CD1s, where overhangs stretch across to the CD2s [267].
In summary, AID/APOBECs are metazoan CDAS members with functions in homeostasis, cellular development, antibody diversification, and antiviral restriction [26,198,229,230,231,232,233,234,235,237,238,239,240,242,243,244,245,246]. Antiviral A3s have diversified across placental mammals, acting as a zoonotic barrier by mutating viruses like HIV/SIV, which counteracts with Vif proteins [13,231,253,260,261,262,263]. These proteins bind different A3 surfaces, using cellular machinery for proteasomal degradation. All AID/APOBECs use a modified α/β/α fold with a DNA-binding groove formed by loops 1, 3, 5, and 7, and the catalytic pocket [198,229,230,231,232,233,234,235]. The gatekeeper residue over the pocket stabilizes or blocks polynucleotides in the groove and pocket [275]. Each AID/APOBEC has substrate specificity for a preferred nucleic acid motif determined by loop 7 [229,248,249,278]. Despite maintaining the CDA fold, some members have duplications creating two domains, as seen in some A3s, while others have extra secondary structures like APOBEC1’s HD-domain and AID’s α7 [230,242,243,244,245,246,275]. Together, AID/APOBECs form a diverse set of single-stranded polynucleotide-mutating enzymes critical for immunological and homeostatic host survival.
8. Conclusion
Deaminases encompass a broad range of enzymes that utilize various reaction mechanisms and cofactors to replace ammonia or ammonium with oxygen [11,12]. These enzymes are found across all domains of life, including archaea, prokaryotes, eukaryotes, and some viruses [2,14,18,70,134,183,190,195,207,208,211,275,303]. Many deaminases target specific substrates, cycling them one at a time, such as GlcN6P deaminase, amino acid deaminases, GDA, GSDAs, ADEase, AFLDA, ADA, TadA/ADATs, CoDAs, CDAs, dCTPD, DCD:DUT, dCMP, and RibG/Rib2. In contrast, enzymes like PBGD, ADARs, and AID/APOBECs can catalyze multiple deamination reactions. For example, PBGD performs four sequential reactions using flexible domains to form tetrapyrroles, while ADARs and AID/APOBECs engage in processive reactions on RNA/DNA via specific substrate-binding grooves. Non-nucleobase deaminases like PBGD possess unique structures, such as a three-domain configuration with DPM as an initiating scaffold for tetrapyrrole formation. GlcN6P deaminase features a distinct lid that, combined with a dual-proton shuttle, drives catalysis.
Amino acid deaminases have evolved through both convergent and divergent pathways, typically using lysine and PLP for deamination, whereas LAAD utilizes FADH2, and RidA’s mechanism remains under investigation. Nucleobase deaminases generally derive from the AHS or CDA superfamilies, with exceptions like dCTPD and DCD:DUT originating from dUTPases. These exceptions often feature proton shuttles and non-metal cations for deamination as opposed to the metal cofactor of AHS and CDAS. Non-plant eukaryotic and some prokaryotic deaminases like GDA, ammelide deaminase, ADEase, AFLDA, dAMPD, and bCoDA commonly exhibit a distorted TIM β-barrel structure, incorporating 1 or more divalent metal cations, single or dual-proton shuttles, and variations in the PDZ β-sandwich domain or additional domains.
The CDA superfamily fold is prevalent among nucleobase derived deaminases, including plant, archaeal, and bacterial GDA, GSDA, TadA/ADATs, ADARs, yCoDA, CDAs, dCMPD, RibG/Rib2, CDAT8, DYW, interbacterial DNA toxin deaminases, and AID/APOBECs. Many of these enzymes function as multimers with CDAs often forming dimers or tetramers for pocket stability through C-terminal interactions. TadA/ADATs, essential for tRNA editing at the wobble site, require multimerization to maintain nucleotide and tRNA structure. Processive deaminases like AID/APOBEC and ADAR, however, use wider substrate-binding grooves or binding domains, respectively, without needing direct monomer interactions for catalysis. ADAR and DddA face the unique challenge of dsRNA/dsDNA binding, requiring base flipping for deamination. Despite different base-flipping mechanisms, their deamination processes align with those of other CDA superfamily enzymes.
In conclusion, deaminases demonstrate evolutionary diversity in mechanisms and structures, facilitating a range of biological reactions and offering valuable insights into enzyme functionality across different contexts.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: RCSB Deaminases Overview; Table S2: RCSB-PDB References of Deaminases across all types and species.
Author Contributions
Conceptualization, D.N.G.H. and M.L.; software, D.N.G.H.; investigation, D.N.G.H.; writing—original draft preparation, D.N.G.H.; writing—review and editing, D.N.G.H and M.L..; visualization, D.N.G.H.; supervision, M.L..; project administration, D.N.G.H.; funding acquisition, M.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Canadian Institute of Health Research (CIHR), grant number MOP111132, and the International Development Research Center (IDRC), grant number 108405001. The Natural Sciences and Engineering Research Council of Canada provides a discovery grant to M.L. The Leukemia & Lymphoma Society of Canada (LLSC) provides an operating grant to M.L., and supported D.H. with a fellowship, 2018–2021.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors are grateful to the aid of Gladdale Huebert as well as the other members of the Larijani Lab for editing this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the review; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| A1CF | APOBEC1 complementation factor |
| AAAD | Aromatic L-amino acid decarboxylase |
| ABE | Adenosine base editor |
| ACC | 1-aminocyclopropane-1-carboxylic acid |
| ACCD | 1-aminocyclopropane-1-carboxylic acid deaminase |
| ADA | Adenosine deaminases |
| ADAR | dsRNA-specific adenosine deaminases |
| ADATs | Adenosine deaminases of tRNA (Eukaryotic) |
| ADEase | Adenine deaminases |
| AFLDA | Aminofutalosine deaminase |
| AID | Activation-induced cytidine deaminase |
| APOBEC | Apolipoprotein B mRNA editing catalytic polypeptide-like |
| ARIPP | 5-amino-6-(D-ribitylamino-2,4(1H,3H)-pyrimidinedione 5’-phosphate |
| AROPP | 5-amino-6-ribosylamino-2,4(1H,3H)-pyrimidinedione 5’-phosphate |
| BSD | Blasticidin S deaminase |
| CaMV | Cauliflower mosaic virus |
| CBE | Cytidine base editor |
| CBF-β | Core binding factor-β |
| CD | Catalytic domain |
| CDA | Cytidine deaminase |
| CDAT8 | Cytidine deaminase of tRNA 8 |
| CoDA | Cytosine deaminase |
| CSR | Class switch recombination |
| dAMPD | Deoxyadenosine monophosphate deaminase |
| DARIPP | 2,5-diamino-6-ribitylamino-4(3H)-pyrimidinone 5’-phosphate |
| DAROPP | 2,5-diamino-6-ribosylamino-4-(3H)-pyrimidinone 5’-phosphate |
| DCD:DUT | Bifunctional deaminase/diphosphatase |
| D-CDA | Dimeric cytidine deaminase |
| dCTPD | deoxycytidine triphosphate deaminase |
| DddA | dsDNA deaminase toxin A |
| DPM | Dipyrromethane |
| dsRBM | dsRNA-binding motifs |
| FAD | Flavin adenine dinucleotide |
| FAMIN | Fatty acid metabolism-immunity nexus |
| FMN | Flavin mononucleotide |
| GDA | Guanine deaminases |
| GlcN6P | Glucosamine-6-phosphate |
| GlcNAc6P | N-acetyl-D-glucosamine-6-phosphate |
| GSDA | Guanosine deaminase |
| HMBS | Hydroxymethylbilane synthase |
| HPC | 1-pyrroline-4-hydroxy-2-carboxylate |
| IHP | Inositol hexakisphosphate |
| LAAD | L-amino acid deaminase |
| LDL | Low-density lipoprotein |
| MAPDA | N6-methyl-adenosine monophosphate deaminase |
| PBGD | Porphobilinogen deaminase |
| PLP | Pyridoxal 5’-phosphate |
| PPR | Pentatricopeptide repeat |
| RBM47 | RNA-binding protein 47 |
| RHS | rearrangement hotspot |
| RidA | Reactive intermediate deaminase A |
| SHM | Somatic hypermutation |
| SsdA | ssDNA deaminase toxin A |
| TadA | tRNA adenosine deaminases (prokaryotic and plant chloroplast) |
| T-CDA | Tetrameric cytidine deaminase |
| TD | Threonine deaminase |
| Vif | Viral infectivity factor |
References
- Wilson, D.K., F.B. Rudolph, and F.A. Quiocho, Atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and immunodeficiency mutations. Science, 1991. 252(5010): p. 1278-84.
- Ose, T., et al., Reaction intermediate structures of 1-aminocyclopropane-1-carboxylate deaminase: insight into PLP-dependent cyclopropane ring-opening reaction. J Biol Chem, 2003. 278(42): p. 41069-76.
- Slater, P.G., et al., Characterization of Xenopus laevis guanine deaminase reveals new insights for its expression and function in the embryonic kidney. Dev Dyn, 2019. 248(4): p. 296-305.
- Tasca, C.I., et al., Neuromodulatory Effects of Guanine-Based Purines in Health and Disease. Front Cell Neurosci, 2018. 12: p. 376.
- Egeblad, L., et al., Pan-pathway based interaction profiling of FDA-approved nucleoside and nucleobase analogs with enzymes of the human nucleotide metabolism. PLoS One, 2012. 7(5): p. e37724.
- Sato, H., et al., Crystal structures of hydroxymethylbilane synthase complexed with a substrate analog: a single substrate-binding site for four consecutive condensation steps. Biochem J, 2021. 478(5): p. 1023-1042.
- Dawson, A., et al., Structure of diaminohydroxyphosphoribosylaminopyrimidine deaminase/5-amino-6-(5-phosphoribosylamino)uracil reductase from Acinetobacter baumannii. Acta Crystallogr Sect F Struct Biol Cryst Commun, 2013. 69(Pt 6): p. 611-.
- Kumasaka, T., et al., Crystal structures of blasticidin S deaminase (BSD): implications for dynamic properties of catalytic zinc. J Biol Chem, 2007. 282(51): p. 37103-11.
- Han, L., et al., Streptomyces wadayamensis MppP is a PLP-Dependent Oxidase, Not an Oxygenase. Biochemistry, 2018. 57(23): p. 3252-3264.
- Mok, B.Y., et al., A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature, 2020. 583(7817): p. 631-637.
- Maiti, A., et al., Structure of the catalytically active APOBEC3G bound to a DNA oligonucleotide inhibitor reveals tetrahedral geometry of the transition state. Nat Commun, 2022. 13(1): p. 7117.
- Arreola, R., et al., Two mammalian glucosamine-6-phosphate deaminases: a structural and genetic study. FEBS Lett, 2003. 551(1-3): p. 63-70.
- Holden, L.G., et al., Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature, 2008. 456(7218): p. 121-4.
- Mahan, S.D., et al., Random mutagenesis and selection of Escherichia coli cytosine deaminase for cancer gene therapy. Protein Eng Des Sel, 2004. 17(8): p. 625-33.
- Teh, A.H., et al., The 1.48 A resolution crystal structure of the homotetrameric cytidine deaminase from mouse. Biochemistry, 2006. 45(25): p. 7825-33.
- Johansson, E., et al., Structures of dCTP deaminase from Escherichia coli with bound substrate and product: reaction mechanism and determinants of mono- and bifunctionality for a family of enzymes. J Biol Chem, 2005. 280(4): p. 3051-9.
- Bitra, A., A. Biswas, and R. Anand, Structural basis of the substrate specificity of cytidine deaminase superfamily Guanine deaminase. Biochemistry, 2013. 52(45): p. 8106-14.
- Jia, Q., et al., Substrate Specificity of GSDA Revealed by Cocrystal Structures and Binding Studies. Int J Mol Sci, 2022. 23(23).
- Losey, H.C., A.J. Ruthenburg, and G.L. Verdine, Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat Struct Mol Biol, 2006. 13(2): p. 153-9.
- Kinoshita, T., et al., Structural basis of compound recognition by adenosine deaminase. Biochemistry, 2005. 44(31): p. 10562-9.
- Liang, J., et al., Current Advances on Structure-Function Relationships of Pyridoxal 5’-Phosphate-Dependent Enzymes. Front Mol Biosci, 2019. 6: p. 4.
- Gallagher, D.T., et al., Structure and control of pyridoxal phosphate dependent allosteric threonine deaminase. Structure, 1998. 6(4): p. 465-75.
- Yamada, T., et al., Crystal structure of serine dehydratase from rat liver. Biochemistry, 2003. 42(44): p. 12854-65.
- Yao, M., et al., Crystal structure of 1-aminocyclopropane-1-carboxylate deaminase from Hansenula saturnus. J Biol Chem, 2000. 275(44): p. 34557-65.
- Han, L., et al., Streptomyces wadayamensis MppP Is a Pyridoxal 5’-Phosphate-Dependent L-Arginine alpha-Deaminase, gamma-Hydroxylase in the Enduracididine Biosynthetic Pathway. Biochemistry, 2015. 54(47): p. 7029-40.
- Conticello, S.G., The AID/APOBEC family of nucleic acid mutators. Genome Biol, 2008. 9(6): p. 229.
- Nabel, C.S., S.A. Manning, and R.M. Kohli, The curious chemical biology of cytosine: deamination, methylation, and oxidation as modulators of genomic potential. ACS Chem Biol, 2012. 7(1): p. 20-30.
- Borzooee, F., et al., Viral subversion of APOBEC3s: Lessons for anti-tumor immunity and tumor immunotherapy. Int Rev Immunol, 2018. 37(3): p. 151-164.
- Layer, G., et al., Structure and function of enzymes in heme biosynthesis. Protein Sci, 2010. 19(6): p. 1137-61.
- Roberts, A., et al., Insights into the mechanism of pyrrole polymerization catalysed by porphobilinogen deaminase: high-resolution X-ray studies of the Arabidopsis thaliana enzyme. Acta Crystallogr D Biol Crystallogr, 2013. 69(Pt 3): p. 471-85.
- Louie, G.V., et al., Structure of porphobilinogen deaminase reveals a flexible multidomain polymerase with a single catalytic site. Nature, 1992. 359(6390): p. 33-9.
- Song, G., et al., Structural insight into acute intermittent porphyria. FASEB J, 2009. 23(2): p. 396-404.
- Gill, R., et al., Structure of human porphobilinogen deaminase at 2.8 A: the molecular basis of acute intermittent porphyria. Biochem J, 2009. 420(1): p. 17-25.
- Pluta, P., et al., Structural basis of pyrrole polymerization in human porphobilinogen deaminase. Biochim Biophys Acta Gen Subj, 2018. 1862(9): p. 1948-1955.
- Bustad, H.J., et al., Characterization of porphobilinogen deaminase mutants reveals that arginine-173 is crucial for polypyrrole elongation mechanism. iScience, 2021. 24(3): p. 102152.
- Helliwell, J.R., et al., Time-resolved and static-ensemble structural chemistry of hydroxymethylbilane synthase. Faraday Discuss, 2003. 122: p. 131-44; discussion 171-90.
- Azim, N., et al., Structural evidence for the partially oxidized dipyrromethene and dipyrromethanone forms of the cofactor of porphobilinogen deaminase: structures of the Bacillus megaterium enzyme at near-atomic resolution. Acta Crystallogr D Biol Crystallogr, 2014. 70(Pt 3): p. 744-51.
- Guo, J., et al., Structural studies of domain movement in active-site mutants of porphobilinogen deaminase from Bacillus megaterium. Acta Crystallogr F Struct Biol Commun, 2017. 73(Pt 11): p. 612-620.
- Funamizu, T., Chen, M., Tanaka, Y., Ishimori, K., Uchida, T., Porphobilinogen deaminase from Vibrio Cholerae. 2016.
- Vincent, F., G.J. Davies, and J.A. Brannigan, Structure and kinetics of a monomeric glucosamine 6-phosphate deaminase: missing link of the NagB superfamily? J Biol Chem, 2005. 280(20): p. 19649-55.
- Flores, C.L. and C. Gancedo, Construction and characterization of a Saccharomyces cerevisiae strain able to grow on glucosamine as sole carbon and nitrogen source. Sci Rep, 2018. 8(1): p. 16949.
- Oliva, G., et al., Structure and catalytic mechanism of glucosamine 6-phosphate deaminase from Escherichia coli at 2.1 A resolution. Structure, 1995. 3(12): p. 1323-32.
- Horjales, E., et al., The allosteric transition of glucosamine-6-phosphate deaminase: the structure of the T state at 2.3 A resolution. Structure, 1999. 7(5): p. 527-37.
- Rudino-Pinera, E., et al., Structural flexibility, an essential component of the allosteric activation in Escherichia coli glucosamine-6-phosphate deaminase. Acta Crystallogr D Biol Crystallogr, 2002. 58(Pt 1): p. 10-20.
- Bustos-Jaimes, I., et al., On the role of the conformational flexibility of the active-site lid on the allosteric kinetics of glucosamine-6-phosphate deaminase. J Mol Biol, 2002. 319(1): p. 183-9.
- Rudino-Pinera, E., Rojas-Trejo, S.P., Horjales, E., Glucosamine-6-Phosphate Deaminase Complexed with the Allosteric Activator N-Acetyl-Glucoamine-6-Phosphate both in the Active and Allosteric sites. 2009.
- Liu, C., et al., Ring-opening mechanism revealed by crystal structures of NagB and its ES intermediate complex. J Mol Biol, 2008. 379(1): p. 73-81.
- (SSGCID), S.S.G.C.f.I.D., Crystal structure of glucosamine-6-phosphate deaminase from Borrelia burgdorferi. 2009.
- Maltseva, N., Kim, Y., Kwon, K., Anderson, W.F., Joachimiak, A., CSGID, Center for Structural Genomics of Infectious Diseases (CSGID), Crystal structure of glucosamine-6-phosphate deaminase from Vibrio cholerae. 2014.
- Chang, C., Maltseva, N., Kim, Y., Kwon, K., Anderson, W.F., Joachimiak, A., Center for Structural Genomics of Infectious Diseases (CSGID), Crystal structure of tertiary complex of glucosamine-6-phosphate deaminase from Vibrio cholerae with BETA-D-GLUCOSE-6-PHOSPHATE and FRUCTOSE-6-PHOSPHATE. 2016.
- Minasov, G., Shuvalova, L., Kiryukhina, O., Dubrovska, I., Satchell, K.J.F., Joachimiak, A., Center for Structural Genomics of Infectious Diseases (CSGID), 2.05 Angstrom Resolution Crystal Structure of Hypothetical Protein KP1_5497 from Klebsiella pneumoniae. 2018.
- Subramanian, R., Srinivasachari, S., Glucosamie-6-phosphate Deaminase from Pasturella multocida. 2021.
- Subramanian, R., Srinivasachari, S., Glucosamine-6-phosphate Deaminase from H. influenzae. 2021.
- Stetter, G.F.a.K.O., Pyrococcus furiosus sp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100 °C. Arch. Microbiol., 1986. 145: p. 56-61.
- Blamey, J.M. and M.W. Adams, Characterization of an ancestral type of pyruvate ferredoxin oxidoreductase from the hyperthermophilic bacterium, Thermotoga maritima. Biochemistry, 1994. 33(4): p. 1000-7.
- Kim, K.J., et al., The crystal structure of a novel glucosamine-6-phosphate deaminase from the hyperthermophilic archaeon Pyrococcus furiosus. Proteins, 2007. 68(1): p. 413-7.
- (JCSG), J.C.f.S.G., Crystal structure of Glucosamine-6-phosphate deaminase (TM0813) from Thermotoga maritima at 1.8 A resolution. 2002.
- Teplyakov, A., et al., Channeling of ammonia in glucosamine-6-phosphate synthase. J Mol Biol, 2001. 313(5): p. 1093-102.
- Torrens-Spence, M.P., et al., Structural basis for divergent and convergent evolution of catalytic machineries in plant aromatic amino acid decarboxylase proteins. Proc Natl Acad Sci U S A, 2020. 117(20): p. 10806-10817.
- Tanaka, H., et al., Crystal structure of a zinc-dependent D-serine dehydratase from chicken kidney. J Biol Chem, 2011. 286(31): p. 27548-58.
- Goto, M., RIKEN Structural Genomics/Proteomics Initiative (RSGI), Crystal Structure of T.th. HB8 Threonine deaminase. 2004.
- Simanshu, D.K., H.S. Savithri, and M.R. Murthy, Crystal structures of Salmonella typhimurium biodegradative threonine deaminase and its complex with CMP provide structural insights into ligand-induced oligomerization and enzyme activation. J Biol Chem, 2006. 281(51): p. 39630-41.
- Gonzales-Vigil, E., et al., Adaptive evolution of threonine deaminase in plant defense against insect herbivores. Proc Natl Acad Sci U S A, 2011. 108(14): p. 5897-902.
- Sun, L., et al., Crystallization and preliminary crystallographic analysis of human serine dehydratase. Acta Crystallogr D Biol Crystallogr, 2003. 59(Pt 12): p. 2297-9.
- Bharath, S.R., et al., Crystal structures of open and closed forms of d-serine deaminase from Salmonella typhimurium - implications on substrate specificity and catalysis. FEBS J, 2011. 278(16): p. 2879-91.
- Urusova, D.V., et al., Crystal structure of D-serine dehydratase from Escherichia coli. Biochim Biophys Acta, 2012. 1824(3): p. 422-32.
- Wang, C.Y., et al., Modulating the function of human serine racemase and human serine dehydratase by protein engineering. Protein Eng Des Sel, 2012. 25(11): p. 741-9.
- Deka, G., et al., Structural studies on the decameric S. typhimurium arginine decarboxylase (ADC): Pyridoxal 5’-phosphate binding induces conformational changes. Biochem Biophys Res Commun, 2017. 490(4): p. 1362-1368.
- (JCSG), J.C.f.S.G., Crystal structure of a putative d-serine deaminase (bxe_a4060) from burkholderia xenovorans lb400 at 2.00 A resolution. 2009.
- Fujino, A., et al., Structural and enzymatic properties of 1-aminocyclopropane-1-carboxylate deaminase homologue from Pyrococcus horikoshii. J Mol Biol, 2004. 341(4): p. 999-1013.
- Karthikeyan, S., et al., Structural analysis of 1-aminocyclopropane-1-carboxylate deaminase: observation of an aminyl intermediate and identification of Tyr 294 as the active-site nucleophile. Angew Chem Int Ed Engl, 2004. 43(26): p. 3425-9.
- Karthikeyan, S., et al., Structural analysis of Pseudomonas 1-aminocyclopropane-1-carboxylate deaminase complexes: insight into the mechanism of a unique pyridoxal-5’-phosphate dependent cyclopropane ring-opening reaction. Biochemistry, 2004. 43(42): p. 13328-39.
- Motta, P., et al., Structure-Function Relationships in l-Amino Acid Deaminase, a Flavoprotein Belonging to a Novel Class of Biotechnologically Relevant Enzymes. J Biol Chem, 2016. 291(20): p. 10457-75.
- Ju, Y., et al., Crystal structure of a membrane-bound l-amino acid deaminase from Proteus vulgaris. J Struct Biol, 2016. 195(3): p. 306-315.
- Niehaus, T.D., et al., Genomic and experimental evidence for multiple metabolic functions in the RidA/YjgF/YER057c/UK114 (Rid) protein family. BMC Genomics, 2015. 16(1): p. 382.
- Liu, X., et al., Crystal structures of RidA, an important enzyme for the prevention of toxic side products. Sci Rep, 2016. 6: p. 30494.
- Burman, J.D., et al., The crystal structure of Escherichia coli TdcF, a member of the highly conserved YjgF/YER057c/UK114 family. BMC Struct Biol, 2007. 7: p. 30.
- Digiovanni, S., et al., Two novel fish paralogs provide insights into the Rid family of imine deaminases active in pre-empting enamine/imine metabolic damage. Sci Rep, 2020. 10(1): p. 10135.
- Deriu, D., et al., Structure and oligomeric state of the mammalian tumour-associated antigen UK114. Acta Crystallogr D Biol Crystallogr, 2003. 59(Pt 9): p. 1676-8.
- Manjasetty, B.A., et al., Crystal structure of Homo sapiens protein hp14.5. Proteins, 2004. 54(4): p. 797-800.
- Kwon, S., et al., Crystal structure of the reactive intermediate/imine deaminase A homolog from the Antarctic bacterium Psychrobacter sp. PAMC 21119. Biochem Biophys Res Commun, 2020. 522(3): p. 585-591.
- Visentin, C., et al., Apis mellifera RidA, a novel member of the canonical YigF/YER057c/UK114 imine deiminase superfamily of enzymes pre-empting metabolic damage. Biochem Biophys Res Commun, 2022. 616: p. 70-75.
- Chen, Y., et al., A Unique Homo-Hexameric Structure of 2-Aminomuconate Deaminase in the Bacterium Pseudomonas species AP-3. Front Microbiol, 2019. 10: p. 2079.
- Chen, S., et al., L-Hydroxyproline and d-Proline Catabolism in Sinorhizobium meliloti. J Bacteriol, 2016. 198(7): p. 1171-81.
- (SSGCID), S.S.G.C.f.I.D., Crystal structure of1-pyrroline-4-hydroxy-2-carboxylate deaminase from Brucella melitensis ATCC 23457. 2013.
- (SSGCID), S.S.G.C.f.I.D., Crystal structure of 1-pyrroline-4-hydroxy-2-carboxylate deaminase from Brucella melitensis with covalently bound substrate. 2013.
- Fernandez, J.R., B. Byrne, and B.L. Firestein, Phylogenetic analysis and molecular evolution of guanine deaminases: from guanine to dendrites. J Mol Evol, 2009. 68(3): p. 227-35.
- Bitra, A., et al., Identification of function and mechanistic insights of guanine deaminase from Nitrosomonas europaea: role of the C-terminal loop in catalysis. Biochemistry, 2013. 52(20): p. 3512-22.
- Hall, R.S., et al., Discovery and structure determination of the orphan enzyme isoxanthopterin deaminase. Biochemistry, 2010. 49(20): p. 4374-82.
- Murphy, P.M., et al., Alteration of enzyme specificity by computational loop remodeling and design. Proc Natl Acad Sci U S A, 2009. 106(23): p. 9215-20.
- Kumaran, D., Burley, S.K., Swaminathan, S., New York SGX Research Center for Structural Genomics (NYSGXRC), Crystal Structure of Guanine Deaminase from C. acetobutylicum with bound guanine in the active site. TBA.
- Agarwal, R., Burley, S.K., Swaminathan, S., New York SGX Research Center for Structural Genomics (NYSGXRC), Crystal structure of guanine deaminase from Bradyrhizobium japonicum. 2007.
- Moche, M., et al., Human Guanine Deaminase (Guad) in Complex with Zinc and its Product Xhantine. 2007.
- Shek, R., et al., Structural Determinants for Substrate Selectivity in Guanine Deaminase Enzymes of the Amidohydrolase Superfamily. Biochemistry, 2019. 58(30): p. 3280-3292.
- Hall, R.S., et al., The hunt for 8-oxoguanine deaminase. J Am Chem Soc, 2010. 132(6): p. 1762-3.
- Mariam, J., et al., Deciphering protein microenvironment by using a cysteine specific switch-ON fluorescent probe. Org Biomol Chem, 2021. 19(23): p. 5161-5168.
- Liaw, S.H., et al., Crystal structure of Bacillus subtilis guanine deaminase: the first domain-swapped structure in the cytidine deaminase superfamily. J Biol Chem, 2004. 279(34): p. 35479-85.
- Singh, J., et al., Structure guided mutagenesis reveals the substrate determinants of guanine deaminase. J Struct Biol, 2021. 213(3): p. 107747.
- Jia, Q., et al., The C-terminal loop of Arabidopsis thaliana guanosine deaminase is essential to catalysis. Chem Commun (Camb), 2021. 57(76): p. 9748-9751.
- Kamat, S.S., et al., Catalytic mechanism and three-dimensional structure of adenine deaminase. Biochemistry, 2011. 50(11): p. 1917-27.
- Kamat, S.S., et al., The catalase activity of diiron adenine deaminase. Protein Sci, 2011. 20(12): p. 2080-94.
- Wilson, D.K. and F.A. Quiocho, A pre-transition-state mimic of an enzyme: X-ray structure of adenosine deaminase with bound 1-deazaadenosine and zinc-activated water. Biochemistry, 1993. 32(7): p. 1689-94.
- Terasaka, T., et al., A highly potent non-nucleoside adenosine deaminase inhibitor: efficient drug discovery by intentional lead hybridization. J Am Chem Soc, 2004. 126(1): p. 34-5.
- Han, B.W., et al., Membrane association, mechanism of action, and structure of Arabidopsis embryonic factor 1 (FAC1). J Biol Chem, 2006. 281(21): p. 14939-47.
- Sugadev, R., Kumaran, D., Burley, S.K., Swaminathan, S., New York SGX Research Center for Structural Genomics (NYSGXRC), Crystal structure of an adenine deaminase. 2006.
- Guan, R., et al., Methylthioadenosine deaminase in an alternative quorum sensing pathway in Pseudomonas aeruginosa. Biochemistry, 2012. 51(45): p. 9094-103.
- Hitchcock, D.S., et al., Structure-guided discovery of new deaminase enzymes. J Am Chem Soc, 2013. 135(37): p. 13927-33.
- Goble, A.M., et al., Pa0148 from Pseudomonas aeruginosa catalyzes the deamination of adenine. Biochemistry, 2011. 50(30): p. 6589-97.
- Goble, A.M., et al., Deamination of 6-aminodeoxyfutalosine in menaquinone biosynthesis by distantly related enzymes. Biochemistry, 2013. 52(37): p. 6525-36.
- Feng, M., et al., Aminofutalosine Deaminase in the Menaquinone Pathway of Helicobacter pylori. Biochemistry, 2021. 60(24): p. 1933-1946.
- Terasaka, T., et al., Structure-based design, synthesis, and structure-activity relationship studies of novel non-nucleoside adenosine deaminase inhibitors. J Med Chem, 2004. 47(15): p. 3730-43.
- Niu, W., et al., The role of Zn2+ on the structure and stability of murine adenosine deaminase. J Phys Chem B, 2010. 114(49): p. 16156-65.
- Weihofen, W.A., et al., Crystal structures of HIV-1 Tat-derived nonapeptides Tat-(1-9) and Trp2-Tat-(1-9) bound to the active site of dipeptidyl-peptidase IV (CD26). J Biol Chem, 2005. 280(15): p. 14911-7.
- Terasaka, T., et al., Structure-based design and synthesis of non-nucleoside, potent, and orally bioavailable adenosine deaminase inhibitors. J Med Chem, 2004. 47(11): p. 2728-31.
- Weihofen, W.A., et al., Crystal structure of CD26/dipeptidyl-peptidase IV in complex with adenosine deaminase reveals a highly amphiphilic interface. J Biol Chem, 2004. 279(41): p. 43330-5.
- Terasaka, T., et al., Rational design of non-nucleoside, potent, and orally bioavailable adenosine deaminase inhibitors: predicting enzyme conformational change and metabolism. J Med Chem, 2005. 48(15): p. 4750-3.
- Kinoshita, T., T. Tada, and I. Nakanishi, Conformational change of adenosine deaminase during ligand-exchange in a crystal. Biochem Biophys Res Commun, 2008. 373(1): p. 53-7.
- Vedadi, M., et al., Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol Biochem Parasitol, 2007. 151(1): p. 100-10.
- Ma, M.T., et al., Catalytically active holo Homo sapiens adenosine deaminase I adopts a closed conformation. Acta Crystallogr D Struct Biol, 2022. 78(Pt 1): p. 91-103.
- Jaruwat, A., et al., Crystal structure of Plasmodium falciparum adenosine deaminase reveals a novel binding pocket for inosine. Arch Biochem Biophys, 2019. 667: p. 6-13.
- Gao, Z.W., et al., Distinct Roles of Adenosine Deaminase Isoenzymes ADA1 and ADA2: A Pan-Cancer Analysis. Front Immunol, 2022. 13: p. 903461.
- Jia, Q. and W. Xie, Alternative conformation induced by substrate binding for Arabidopsis thalianaN6-methyl-AMP deaminase. Nucleic Acids Res, 2019. 47(6): p. 3233-3243.
- Wu, B., et al., Structure of Arabidopsis thaliana N(6)-methyl-AMP deaminase ADAL with bound GMP and IMP and implications for N(6)-methyl-AMP recognition and processing. RNA Biol, 2019. 16(10): p. 1504-1512.
- Cader, M.Z., et al., FAMIN Is a Multifunctional Purine Enzyme Enabling the Purine Nucleotide Cycle. Cell, 2020. 180(2): p. 278-295 e23.
- Lee, M.S., et al., Structural Basis for the Peptidoglycan-Editing Activity of YfiH. mBio, 2022. 13(1): p. e0364621.
- Minasov, G., Shuvalova, L., Mondragon, A., Taneja, B., Moy, S.F., Collart, F.R., Anderson, W.F., Midwest Center for Structural Genomics (MCSG), 1.8 A CRYSTAL STRUCTURE OF AN UNCHARACTERIZED B. STEAROTHERMOPHILUS PROTEIN. 2004.
- Seetharaman, J., Swaminathan, S., Burley, S.K., New York SGX Research Center for Structural Genomics (NYSGXRC), Crystal structure of protein yfiH from Salmonella enterica serovar Typhi, Pfam DUF152. 2003.
- Seetharaman, J., Swaminathan, S., Burley, S.K., New York SGX Research Center for Structural Genomics (NYSGXRC), Crystal structure of protein yfiH from Shigella flexneri, Pfam DUF152. 2004.
- Almeida, L.R., Grejo, M.P., Mulinari, E.J., Santos, J.C., Camargo, S., Bernardes, A., Muniz, J.R.C., Crystal structure of Bacillus licheniformis hypothetical protein YfiH. 2018.
- Elias, Y. and R.H. Huang, Biochemical and structural studies of A-to-I editing by tRNA:A34 deaminases at the wobble position of transfer RNA. Biochemistry, 2005. 44(36): p. 12057-65.
- Kuratani, M., et al., Crystal structure of tRNA adenosine deaminase (TadA) from Aquifex aeolicus. J Biol Chem, 2005. 280(16): p. 16002-8.
- Kim, J., et al., Structural and kinetic characterization of Escherichia coli TadA, the wobble-specific tRNA deaminase. Biochemistry, 2006. 45(20): p. 6407-16.
- Dolce, L.G., et al., Structural basis for sequence-independent substrate selection by eukaryotic wobble base tRNA deaminase ADAT2/3. Nat Commun, 2022. 13(1): p. 6737.
- Liu, X., et al., Crystal structure of the yeast heterodimeric ADAT2/3 deaminase. BMC Biol, 2020. 18(1): p. 189.
- Liu, H., et al., Structure of a tRNA-specific deaminase with compromised deamination activity. Biochem J, 2020. 477(8): p. 1483-1497.
- Lee, W.H., et al., Crystal structure of the tRNA-specific adenosine deaminase from Streptococcus pyogenes. Proteins, 2007. 68(4): p. 1016-9.
- Lapinaite, A., et al., DNA capture by a CRISPR-Cas9-guided adenine base editor. Science, 2020. 369(6503): p. 566-571.
- Lam, D.K., et al., Improved cytosine base editors generated from TadA variants. Nat Biotechnol, 2023.
- Liu, X., et al., Functional and structural investigation of N-terminal domain of the SpTad2/3 heterodimeric tRNA deaminase. Comput Struct Biotechnol J, 2021. 19: p. 3384-3393.
- Ramos-Morales, E., et al., The structure of the mouse ADAT2/ADAT3 complex reveals the molecular basis for mammalian tRNA wobble adenosine-to-inosine deamination. Nucleic Acids Res, 2021. 49(11): p. 6529-6548.
- Keegan, L.P., et al., Adenosine deaminases acting on RNA (ADARs): RNA-editing enzymes. Genome Biol, 2004. 5(2): p. 209.
- Welin, M., Tresaugues, L., Andersson, J., Arrowsmith, C.H., Berglund, H., Collins, R., Dahlgren, L.G., Edwards, A.M., Flodin, S., Flores, A., Graslund, S., Hammarstrom, M., Johansson, A., Johansson, I., Karlberg, T., Kotenyova, T., Lehtio, L., Moche, M., Nilsson, M.E., Nyman, T., Olesen, K., Persson, C., Sagemark, J., Schueler, H., Thorsell, A.G., van der Berg, S., Wisniewska, M., Wikstrom, M., Nordlund, P., Crystal structure of human tRNA-specific adenosine-34 deaminase subunit ADAT2. 2008.
- Hood, J.L. and R.B. Emeson, Editing of neurotransmitter receptor and ion channel RNAs in the nervous system. Curr Top Microbiol Immunol, 2012. 353: p. 61-90.
- Barraud, P. and F.H. Allain, ADAR proteins: double-stranded RNA and Z-DNA binding domains. Curr Top Microbiol Immunol, 2012. 353: p. 35-60.
- Wulff, B.E. and K. Nishikura, Modulation of microRNA expression and function by ADARs. Curr Top Microbiol Immunol, 2012. 353: p. 91-109.
- Piontkivska, H., et al., ADAR Editing in Viruses: An Evolutionary Force to Reckon with. Genome Biol Evol, 2021. 13(11).
- Orecchini, E., et al., ADAR1 restricts LINE-1 retrotransposition. Nucleic Acids Res, 2017. 45(1): p. 155-168.
- Thuy-Boun, A.S., et al., Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res, 2020. 48(14): p. 7958-7972.
- Schwartz, T., et al., Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science, 1999. 284(5421): p. 1841-5.
- Schwartz, T., et al., Structure of the DLM-1-Z-DNA complex reveals a conserved family of Z-DNA-binding proteins. Nat Struct Biol, 2001. 8(9): p. 761-5.
- Placido, D., et al., A left-handed RNA double helix bound by the Z alpha domain of the RNA-editing enzyme ADAR1. Structure, 2007. 15(4): p. 395-404.
- Ha, S.C., et al., Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Nature, 2005. 437(7062): p. 1183-6.
- Schade, M., et al., The solution structure of the Zalpha domain of the human RNA editing enzyme ADAR1 reveals a prepositioned binding surface for Z-DNA. Proc Natl Acad Sci U S A, 1999. 96(22): p. 12465-70.
- Athanasiadis, A., et al., The crystal structure of the Zbeta domain of the RNA-editing enzyme ADAR1 reveals distinct conserved surfaces among Z-domains. J Mol Biol, 2005. 351(3): p. 496-507.
- Feng, S., et al., Alternate rRNA secondary structures as regulators of translation. Nat Struct Mol Biol, 2011. 18(2): p. 169-76.
- Kim, D., et al., Sequence preference and structural heterogeneity of BZ junctions. Nucleic Acids Res, 2018. 46(19): p. 10504-10513.
- Ha, S.C., et al., The structures of non-CG-repeat Z-DNAs co-crystallized with the Z-DNA-binding domain, hZ alpha(ADAR1). Nucleic Acids Res, 2009. 37(2): p. 629-37.
- de Rosa, M., et al., Crystal structure of a junction between two Z-DNA helices. Proc Natl Acad Sci U S A, 2010. 107(20): p. 9088-92.
- Park, C., et al., Dual conformational recognition by Z-DNA binding protein is important for the B-Z transition process. Nucleic Acids Res, 2020. 48(22): p. 12957-12971.
- Barraud, P., et al., A bimodular nuclear localization signal assembled via an extended double-stranded RNA-binding domain acts as an RNA-sensing signal for transportin 1. Proc Natl Acad Sci U S A, 2014. 111(18): p. E1852-61.
- Stefl, R., et al., Structure and specific RNA binding of ADAR2 double-stranded RNA binding motifs. Structure, 2006. 14(2): p. 345-55.
- Stefl, R., et al., The solution structure of the ADAR2 dsRBM-RNA complex reveals a sequence-specific readout of the minor groove. Cell, 2010. 143(2): p. 225-37.
- Macbeth, M.R., et al., Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science, 2005. 309(5740): p. 1534-9.
- Matthews, M.M., et al., Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol, 2016. 23(5): p. 426-33.
- Monteleone, L.R., et al., A Bump-Hole Approach for Directed RNA Editing. Cell Chem Biol, 2019. 26(2): p. 269-277 e5.
- Doherty, E.E., et al., Rational Design of RNA Editing Guide Strands: Cytidine Analogs at the Orphan Position. J Am Chem Soc, 2021. 143(18): p. 6865-6876.
- Doherty, E.E., et al., ADAR activation by inducing a syn conformation at guanosine adjacent to an editing site. Nucleic Acids Res, 2022. 50(19): p. 10857-10868.
- Ireton, G.C., et al., The structure of Escherichia coli cytosine deaminase. J Mol Biol, 2002. 315(4): p. 687-97.
- Ireton, G.C., M.E. Black, and B.L. Stoddard, The 1.14 A crystal structure of yeast cytosine deaminase: evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure, 2003. 11(8): p. 961-72.
- Hall, R.S., et al., Three-dimensional structure and catalytic mechanism of cytosine deaminase. Biochemistry, 2011. 50(22): p. 5077-85.
- Ko, T.P., et al., Crystal structure of yeast cytosine deaminase. Insights into enzyme mechanism and evolution. J Biol Chem, 2003. 278(21): p. 19111-7.
- Hitchcock, D.S., et al., Rescue of the orphan enzyme isoguanine deaminase. Biochemistry, 2011. 50(25): p. 5555-7.
- Hitchcock, D.S., et al., Discovery of a bacterial 5-methylcytosine deaminase. Biochemistry, 2014. 53(47): p. 7426-35.
- Ireton, G.C. and B.L. Stoddard, Microseed matrix screening to improve crystals of yeast cytosine deaminase. Acta Crystallogr D Biol Crystallogr, 2004. 60(Pt 3): p. 601-5.
- Korkegian, A., et al., Computational thermostabilization of an enzyme. Science, 2005. 308(5723): p. 857-60.
- Korkegian, A.M., Stoddard, B.L., Yeast Cytosine Deaminase D92E Triple Mutant bound to transition state analogue HPY. 2006.
- Fuchita, M., et al., Bacterial cytosine deaminase mutants created by molecular engineering show improved 5-fluorocytosine-mediated cell killing in vitro and in vivo. Cancer Res, 2009. 69(11): p. 4791-9.
- Gaded, V. and R. Anand, Selective Deamination of Mutagens by a Mycobacterial Enzyme. J Am Chem Soc, 2017. 139(31): p. 10762-10768.
- Johansson, E., et al., Crystal structure of the tetrameric cytidine deaminase from Bacillus subtilis at 2.0 A resolution. Biochemistry, 2002. 41(8): p. 2563-70.
- Chung, S.J., J.C. Fromme, and G.L. Verdine, Structure of human cytidine deaminase bound to a potent inhibitor. J Med Chem, 2005. 48(3): p. 658-60.
- Xie, K., et al., The structure of a yeast RNA-editing deaminase provides insight into the fold and function of activation-induced deaminase and APOBEC-1. Proc Natl Acad Sci U S A, 2004. 101(21): p. 8114-9.
- Taylor, L., Crystal structure of a plant cytidine deaminase. 2016.
- Johansson, E., et al., Structural, kinetic, and mutational studies of the zinc ion environment in tetrameric cytidine deaminase. Biochemistry, 2004. 43(20): p. 6020-9.
- Liu, W., et al., Biochemical and structural analysis of the Klebsiella pneumoniae cytidine deaminase CDA. Biochem Biophys Res Commun, 2019. 519(2): p. 280-286.
- Levdikov, V.M., Blagova, E.V., Fogg, M.J., Brannigan, J.A., Moroz, O.V., Wilkinson, A.J., Wilson, K.S., Crystal Structure of Cytidine Deaminase Cdd-2 (BA4525) from Bacillus Anthracis at 2.40A Resolution. 2005.
- Sanchez-Quitian, Z.A., et al., Structural and functional analyses of Mycobacterium tuberculosis Rv3315c-encoded metal-dependent homotetrameric cytidine deaminase. J Struct Biol, 2010. 169(3): p. 413-23.
- Sanchez-Quitian, Z.A., Rodrigues-Junior, E., Rehm, J.G., Eichler, P., Trivella, D.B.B., Bizarro, C.V., Basso, L.A., Santos, D.S., Functional and structural evidence for the catalytic role played by glutamate-47 residue in the mode of action of Mycobacterium tuberculosis cytidine deaminase. Rsc Adv, 2015. 5: p. 830-840.
- Baugh, L., et al., Increasing the structural coverage of tuberculosis drug targets. Tuberculosis (Edinb), 2015. 95(2): p. 142-8.
- (SSGCID), S.S.G.C.f.I.D., Crystal structure of Blasticidin S Deaminase from Coccidioides Immitis. 2010.
- Wang, J., et al., Crystal structure of Arabidopsis thaliana cytidine deaminase. Biochem Biophys Res Commun, 2020. 529(3): p. 659-665.
- Xiang, S., et al., Transition-state selectivity for a single hydroxyl group during catalysis by cytidine deaminase. Biochemistry, 1995. 34(14): p. 4516-23.
- Xiang, S., et al., Cytidine deaminase complexed to 3-deazacytidine: a “valence buffer” in zinc enzyme catalysis. Biochemistry, 1996. 35(5): p. 1335-41.
- Xiang, S., et al., The structure of the cytidine deaminase-product complex provides evidence for efficient proton transfer and ground-state destabilization. Biochemistry, 1997. 36(16): p. 4768-74.
- Minasov, G., Wawrzak, Z., Skarina, T., Wang, Y., Grimshaw, S., Papazisi, L., Savchenko, A., Anderson, W.F., Center for Structural Genomics of Infectious Diseases (CSGID), 2.2 Angstrom Crystal Structure of Cytidine deaminase from Vibrio cholerae in Complex with Zinc and Uridine. 2012.
- Almog, R., et al., Three-dimensional structure of the R115E mutant of T4-bacteriophage 2’-deoxycytidylate deaminase. Biochemistry, 2004. 43(43): p. 13715-23.
- Maiti, A., et al., Crystal Structure of a Soluble APOBEC3G Variant Suggests ssDNA to Bind in a Channel that Extends between the Two Domains. J Mol Biol, 2020. 432(23): p. 6042-6060.
- Xiao, X., et al., Structural determinants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res, 2017. 45(12): p. 7494-7506.
- Prochnow, C., et al., The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature, 2007. 445(7126): p. 447-51.
- Johansson, E., et al., Regulation of dCTP deaminase from Escherichia coli by nonallosteric dTTP binding to an inactive form of the enzyme. FEBS J, 2007. 274(16): p. 4188-98.
- Oehlenschlaeger, C.B., et al., Bacillus halodurans Strain C125 Encodes and Synthesizes Enzymes from Both Known Pathways To Form dUMP Directly from Cytosine Deoxyribonucleotides. Appl Environ Microbiol, 2015. 81(10): p. 3395-404.
- Thymark, M., et al., Mutational analysis of the nucleotide binding site of Escherichia coli dCTP deaminase. Arch Biochem Biophys, 2008. 470(1): p. 20-6.
- Abendroth, J., et al., SAD phasing using iodide ions in a high-throughput structural genomics environment. J Struct Funct Genomics, 2011. 12(2): p. 83-95.
- Baugh, L., et al., Combining functional and structural genomics to sample the essential Burkholderia structome. PLoS One, 2013. 8(1): p. e53851.
- Johansson, E., et al., Structure of the bifunctional dCTP deaminase-dUTPase from Methanocaldococcus jannaschii and its relation to other homotrimeric dUTPases. J Biol Chem, 2003. 278(30): p. 27916-22.
- Zhang, R., Dong, A., Xu, X., Savchenko, A., Edwards, A.M., Joachimiak, A., Midwest Center for Structural Genomics (MCSG), Crystal structure of 2’-deoxycytidine 5’-triphosphate deaminase from Agrobacterium tumefaciens. 2007.
- Helt, S.S., et al., Mechanism of dTTP inhibition of the bifunctional dCTP deaminase:dUTPase encoded by Mycobacterium tuberculosis. J Mol Biol, 2008. 376(2): p. 554-69.
- Huffman, J.L., et al., Structural basis for recognition and catalysis by the bifunctional dCTP deaminase and dUTPase from Methanococcus jannaschii. J Mol Biol, 2003. 331(4): p. 885-96.
- Hou, H.F., et al., Crystal structures of Streptococcus mutans 2’-deoxycytidylate deaminase and its complex with substrate analog and allosteric regulator dCTP x Mg2+. J Mol Biol, 2008. 377(1): p. 220-31.
- Li, Y., et al., Mechanism of the allosteric regulation of Streptococcus mutans 2’-deoxycytidylate deaminase. Acta Crystallogr D Struct Biol, 2016. 72(Pt 7): p. 883-91.
- Siponen, M.I., Moche, M., Arrowsmith, C.H., Berglund, H., Bountra, C., Collins, R., Dahlgren, L.G., Edwards, A.M., Flodin, S., Flores, A., Graslund, S., Hammarstrom, M., Johansson, A., Johansson, I., Karlberg, T., Kotenyova, T., Lehtio, L., Nilsson, M.E., Nyman, T., Persson, C., Sagemark, J., Schuler, H., Thorsell, A.G., Tresaugues, L., Van Den Berg, S., Weigelt, J., Welin, M., Wikstrom, M., Wisniewska, M., Nordlund, P., The Crystal Structure of Human Dcmp Deaminase. 2008.
- Marx, A. and A. Alian, The first crystal structure of a dTTP-bound deoxycytidylate deaminase validates and details the allosteric-inhibitor binding site. J Biol Chem, 2015. 290(1): p. 682-90.
- Li, Y.H., et al., Structural basis of a multi-functional deaminase in chlorovirus PBCV-1. Arch Biochem Biophys, 2022. 727: p. 109339.
- Lovgreen, M.N., Harris, P., Ucar, E., Willemoes, M., Dttp Inhibition of the Bifunctional Dctp Deaminase- Dutpase from Mycobacterium Tuberculosis is Ph Dependent: Kinetic Analyses and Crystal Structure of A115V Variant. 2011.
- Stenmark, P., et al., The crystal structure of the bifunctional deaminase/reductase RibD of the riboflavin biosynthetic pathway in Escherichia coli: implications for the reductive mechanism. J Mol Biol, 2007. 373(1): p. 48-64.
- Chen, S.C., et al., Complex structure of Bacillus subtilis RibG: the reduction mechanism during riboflavin biosynthesis. J Biol Chem, 2009. 284(3): p. 1725-31.
- Chen, S.C., et al., Crystal structure of a bifunctional deaminase and reductase from Bacillus subtilis involved in riboflavin biosynthesis. J Biol Chem, 2006. 281(11): p. 7605-13.
- Chen, S.C., et al., Evolution of vitamin B2 biosynthesis: eubacterial RibG and fungal Rib2 deaminases. Acta Crystallogr D Biol Crystallogr, 2013. 69(Pt 2): p. 227-36.
- Chen, S.C., et al., Crystal structures of Aspergillus oryzae Rib2 deaminase: the functional mechanism involved in riboflavin biosynthesis. IUCrJ, 2021. 8(Pt 4): p. 549-558.
- Averianova, L.A., et al., Production of Vitamin B2 (Riboflavin) by Microorganisms: An Overview. Front Bioeng Biotechnol, 2020. 8: p. 570828.
- (JCSG), J.C.f.S.G., Crystal structure of a diaminohydroxyphosphoribosylaminopyrimidine deaminase/ 5-amino-6-(5-phosphoribosylamino)uracil reductase (tm1828) from thermotoga maritima at 1.80 A resolution. 2006.
- Hajian, B., et al., Drugging the Folate Pathway in Mycobacterium tuberculosis: The Role of Multi-targeting Agents. Cell Chem Biol, 2019. 26(6): p. 781-791 e6.
- Bonomi, H.R., Cerutti, M.L., Posadas, D.M., Goldbaum, F.A., Klinke, S., C-terminal domain of RibD from Brucella abortus (5-amino-6-ribosylamino-2,4(1H,3H)-pyrimidinedione 5’-phosphate reductase). 2021.
- Takenaka, M., et al., DYW domain structures imply an unusual regulation principle in plant organellar RNA editing catalysis. Nat Catal, 2021. 4(6): p. 510-522.
- Randau, L., et al., A cytidine deaminase edits C to U in transfer RNAs in Archaea. Science, 2009. 324(5927): p. 657-9.
- Toma-Fukai, S., et al., Structural insight into the activation of an Arabidopsis organellar C-to-U RNA editing enzyme by active site complementation. Plant Cell, 2023. 35(6): p. 1888-1900.
- de Moraes, M.H., et al., An interbacterial DNA deaminase toxin directly mutagenizes surviving target populations. Elife, 2021. 10.
- Yin, L., K. Shi, and H. Aihara, Structural basis of sequence-specific cytosine deamination by double-stranded DNA deaminase toxin DddA. Nat Struct Mol Biol, 2023.
- Cho, S.I., et al., Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell, 2022. 185(10): p. 1764-1776 e12.
- Pham, P., et al., Structural analysis of the activation-induced deoxycytidine deaminase required in immunoglobulin diversification. DNA Repair (Amst), 2016. 43: p. 48-56.
- Wolfe, A.D., et al., The structure of APOBEC1 and insights into its RNA and DNA substrate selectivity. NAR Cancer, 2020. 2(4): p. zcaa027.
- Yaping Liu, W.L., Chunxi Wang, Chunyang Cao, Two different kinds of interaction modes of deaminase APOBEC3A with single-stranded DNA in solution detected by nuclear magnetic resonance. Protein Science, 2021. 31(2).
- Shi, K., et al., Conformational Switch Regulates the DNA Cytosine Deaminase Activity of Human APOBEC3B. Sci Rep, 2017. 7(1): p. 17415.
- Shaban, N.M., et al., 1.92 Angstrom Zinc-Free APOBEC3F Catalytic Domain Crystal Structure. J Mol Biol, 2016. 428(11): p. 2307-2316.
- Yang, H., et al., Understanding the structural basis of HIV-1 restriction by the full length double-domain APOBEC3G. Nat Commun, 2020. 11(1): p. 632.
- Bohn, J.A., et al., APOBEC3H structure reveals an unusual mechanism of interaction with duplex RNA. Nat Commun, 2017. 8(1): p. 1021.
- Krishnan, A., et al., Diversification of AID/APOBEC-like deaminases in metazoa: multiplicity of clades and widespread roles in immunity. Proc Natl Acad Sci U S A, 2018. 115(14): p. E3201-E3210.
- Quinlan, E.M., et al., Biochemical Regulatory Features of Activation-Induced Cytidine Deaminase Remain Conserved from Lampreys to Humans. Mol Cell Biol, 2017. 37(20).
- Ghorbani, A., et al., Ancestral reconstruction reveals catalytic inactivation of activation-induced cytidine deaminase concomitant with cold water adaption in the Gadiformes bony fish. BMC Biol, 2022. 20(1): p. 293.
- Shi, K., et al., Crystal Structure of the DNA Deaminase APOBEC3B Catalytic Domain. J Biol Chem, 2015. 290(47): p. 28120-30.
- Damsteegt, E.L., A. Davie, and P.M. Lokman, The evolution of apolipoprotein B and its mRNA editing complex. Does the lack of editing contribute to hypertriglyceridemia? Gene, 2018. 641: p. 46-54.
- Modenini, G., P. Abondio, and A. Boattini, The coevolution between APOBEC3 and retrotransposons in primates. Mob DNA, 2022. 13(1): p. 27.
- Hayward, J.A., et al., Differential Evolution of Antiretroviral Restriction Factors in Pteropid Bats as Revealed by APOBEC3 Gene Complexity. Mol Biol Evol, 2018. 35(7): p. 1626-1637.
- Salter, J.D., R.P. Bennett, and H.C. Smith, The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem Sci, 2016. 41(7): p. 578-594.
- Sawyer, S.L., M. Emerman, and H.S. Malik, Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol, 2004. 2(9): p. E275.
- Munk, C., A. Willemsen, and I.G. Bravo, An ancient history of gene duplications, fusions and losses in the evolution of APOBEC3 mutators in mammals. BMC Evol Biol, 2012. 12: p. 71.
- Nakano, Y., et al., A conflict of interest: the evolutionary arms race between mammalian APOBEC3 and lentiviral Vif. Retrovirology, 2017. 14(1): p. 31.
- Byeon, I.J., et al., Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry, 2016. 55(21): p. 2944-59.
- Bohn, M.F., et al., The ssDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Structure, 2015. 23(5): p. 903-11.
- Kouno, T., et al., Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat Commun, 2017. 8: p. 15024.
- Kitamura, S., et al., The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat Struct Mol Biol, 2012. 19(10): p. 1005-10.
- Siu, K.K., et al., Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat Commun, 2013. 4: p. 2593.
- Shi, K., et al., Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat Struct Mol Biol, 2017. 24(2): p. 131-139.
- Byeon, I.J., et al., NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat Commun, 2013. 4: p. 1890.
- Hirabayashi, S., et al., APOBEC3B is preferentially expressed at the G2/M phase of cell cycle. Biochem Biophys Res Commun, 2021. 546: p. 178-184.
- Shaban, N.M., et al., The Antiviral and Cancer Genomic DNA Deaminase APOBEC3H Is Regulated by an RNA-Mediated Dimerization Mechanism. Mol Cell, 2018. 69(1): p. 75-86 e9.
- Vasudevan, A.A.J., et al., Loop 1 of APOBEC3C regulates its antiviral activity against HIV-1. bioRxiv preprint, 2020: p. 1-67.
- Koning, F.A., et al., Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol, 2009. 83(18): p. 9474-85.
- Mohanram, V., et al., IFN-alpha induces APOBEC3G, F, and A in immature dendritic cells and limits HIV-1 spread to CD4+ T cells. J Immunol, 2013. 190(7): p. 3346-53.
- Peng, G., et al., Induction of APOBEC3 family proteins, a defensive maneuver underlying interferon-induced anti-HIV-1 activity. J Exp Med, 2006. 203(1): p. 41-6.
- Milewska, A., et al., APOBEC3-mediated restriction of RNA virus replication. Sci Rep, 2018. 8(1): p. 5960.
- Wang, S.M. and C.T. Wang, APOBEC3G cytidine deaminase association with coronavirus nucleocapsid protein. Virology, 2009. 388(1): p. 112-20.
- Cheng, A.Z., et al., Epstein-Barr virus BORF2 inhibits cellular APOBEC3B to preserve viral genome integrity. Nat Microbiol, 2019. 4(1): p. 78-88.
- Shaban, N.M., et al., Cryo-EM structure of the EBV ribonucleotide reductase BORF2 and mechanism of APOBEC3B inhibition. Sci Adv, 2022. 8(17): p. eabm2827.
- Matsuoka, T., et al., Structural basis of chimpanzee APOBEC3H dimerization stabilized by double-stranded RNA. Nucleic Acids Res, 2018.
- Cheng, C., Zhang, T., Wang, C., Lan, W., Ding, J., Cao, C., Crystal Structure of Cytidine Deaminase Human APOBEC3F Chimeric Catalytic Domain in Complex with DNA. Chinese Journal of Chemistry, 2019. 36(12).
- Yan, X., et al., Structural Investigations on the Interactions between Cytidine Deaminase Human APOBEC3G and DNA. Chem Asian J, 2019. 14(13): p. 2235-2241.
- Yang, H., et al., Structural basis of sequence-specific RNA recognition by the antiviral factor APOBEC3G. Nat Commun, 2022. 13(1): p. 7498.
- Wang, J., et al., Simian Immunodeficiency Virus Vif and Human APOBEC3B Interactions Resemble Those between HIV-1 Vif and Human APOBEC3G. J Virol, 2018. 92(12).
- Delviks-Frankenberry, K.A., B.A. Desimmie, and V.K. Pathak, Structural Insights into APOBEC3-Mediated Lentiviral Restriction. Viruses, 2020. 12(6).
- Nakashima, M., et al., Structural Insights into HIV-1 Vif-APOBEC3F Interaction. J Virol, 2016. 90(2): p. 1034-47.
- Hu, Y., et al., Structural basis of antagonism of human APOBEC3F by HIV-1 Vif. Nat Struct Mol Biol, 2019. 26(12): p. 1176-1183.
- Kouno, T., et al., Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat Struct Mol Biol, 2015. 22(6): p. 485-91.
- Xiao, X., et al., Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat Commun, 2016. 7: p. 12193.
- Ito, F., et al., Structural basis for HIV-1 antagonism of host APOBEC3G via Cullin E3 ligase. Sci Adv, 2023. 9(1): p. eade3168.
- Qiao, Q., et al., AID Recognizes Structured DNA for Class Switch Recombination. Mol Cell, 2017. 67(3): p. 361-373 e4.
- Bohn, M.F., et al., Crystal structure of the DNA cytosine deaminase APOBEC3F: the catalytically active and HIV-1 Vif-binding domain. Structure, 2013. 21(6): p. 1042-50.
- Ito, F., et al., Understanding the Structure, Multimerization, Subcellular Localization and mC Selectivity of a Genomic Mutator and Anti-HIV Factor APOBEC3H. Sci Rep, 2018. 8(1): p. 3763.
- Shi, K., et al., Active site plasticity and possible modes of chemical inhibition of the human DNA deaminase APOBEC3B. FASEB Bioadv, 2020. 2(1): p. 49-58.
- Chen, K.M., et al., Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature, 2008. 452(7183): p. 116-9.
- Furukawa, A., et al., Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J, 2009. 28(4): p. 440-51.
- Harjes, E., et al., An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J Mol Biol, 2009. 389(5): p. 819-32.
- Shandilya, S.M., et al., Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure, 2010. 18(1): p. 28-38.
- Maiti, A., et al., Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat Commun, 2018. 9(1): p. 2460.
- Jumper, J., et al., Highly accurate protein structure prediction with AlphaFold. Nature, 2021. 596(7873): p. 583-589.
- Varadi, M., et al., AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res, 2022. 50(D1): p. D439-D444.
- Lorenzo, J.P., et al., APOBEC2 is a Transcriptional Repressor required for proper Myoblast Differentiation. bioRxiv, 2021: p. 2020.07.29.223594.
- Tang, C., A. Krantsevich, and T. MacCarthy, Deep learning model of somatic hypermutation reveals importance of sequence context beyond hotspot targeting. iScience, 2022. 25(1): p. 103668.
- Adolph, M.B., R.P. Love, and L. Chelico, Biochemical Basis of APOBEC3 Deoxycytidine Deaminase Activity on Diverse DNA Substrates. ACS Infect Dis, 2018. 4(3): p. 224-238.
- Senavirathne, G., et al., Single-stranded DNA scanning and deamination by APOBEC3G cytidine deaminase at single molecule resolution. J Biol Chem, 2012. 287(19): p. 15826-35.
- Pham, P., et al., Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature, 2003. 424(6944): p. 103-7.
- Wong, L., et al., APOBEC1 cytosine deaminase activity on single-stranded DNA is suppressed by replication protein A. Nucleic Acids Res, 2021. 49(1): p. 322-339.
- Adolph, M.B., et al., Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization. Nucleic Acids Res, 2017. 45(6): p. 3378-3394.
- Adolph, M.B., et al., Enzyme cycling contributes to efficient induction of genome mutagenesis by the cytidine deaminase APOBEC3B. Nucleic Acids Res, 2017. 45(20): p. 11925-11940.
- Gorle, S., et al., Computational Model and Dynamics of Monomeric Full-Length APOBEC3G. ACS Cent Sci, 2017. 3(11): p. 1180-1188.
- Ziegler, S.J., et al., Insights into DNA substrate selection by APOBEC3G from structural, biochemical, and functional studies. PLoS One, 2018. 13(3): p. e0195048.
- Mitra, M., et al., Sequence and structural determinants of human APOBEC3H deaminase and anti-HIV-1 activities. Retrovirology, 2015. 12: p. 3.
- Wittkopp, C.J., et al., A Single Nucleotide Polymorphism in Human APOBEC3C Enhances Restriction of Lentiviruses. PLoS Pathog, 2016. 12(10): p. e1005865.
- Silvas, T.V. and C.A. Schiffer, APOBEC3s: DNA-editing human cytidine deaminases. Protein Sci, 2019. 28(9): p. 1552-1566.
- Hix, M.A. and G.A. Cisneros, Computational Investigation of APOBEC3H Substrate Orientation and Selectivity. J Phys Chem B, 2020. 124(19): p. 3903-3908.
- Lu, X., et al., Crystal structure of DNA cytidine deaminase ABOBEC3G catalytic deamination domain suggests a binding mode of full-length enzyme to single-stranded DNA. J Biol Chem, 2015. 290(7): p. 4010-21.
- Kitamura, S., Ode, H., Nakashima, M., Imahashi, M., Naganawa, Y., Ibe, S., Yokomaku, Y., Watanabe, N., Suzuki, A., Sugiura, W., Iwatani, Y., Crystal structure of the human APOBEC3C having HIV-1 Vif-binding interface. 2011.
- Li, Y.L., et al., The structural basis for HIV-1 Vif antagonism of human APOBEC3G. Nature, 2023. 615(7953): p. 728-733.
- Larson, E.T., et al., Structures of substrate- and inhibitor-bound adenosine deaminase from a human malaria parasite show a dramatic conformational change and shed light on drug selectivity. J Mol Biol, 2008. 381(4): p. 975-88.
Scheme 1.
Generalized deaminase reactions. Deamination reactions that occur producing (A,C) ammonia or (B,D) ammonium as a byproduct while (A,B) produce a double bonded oxygen alone and (C,D) produce a double bonded oxygen with an added hydrogen.
Scheme 1.
Generalized deaminase reactions. Deamination reactions that occur producing (A,C) ammonia or (B,D) ammonium as a byproduct while (A,B) produce a double bonded oxygen alone and (C,D) produce a double bonded oxygen with an added hydrogen.
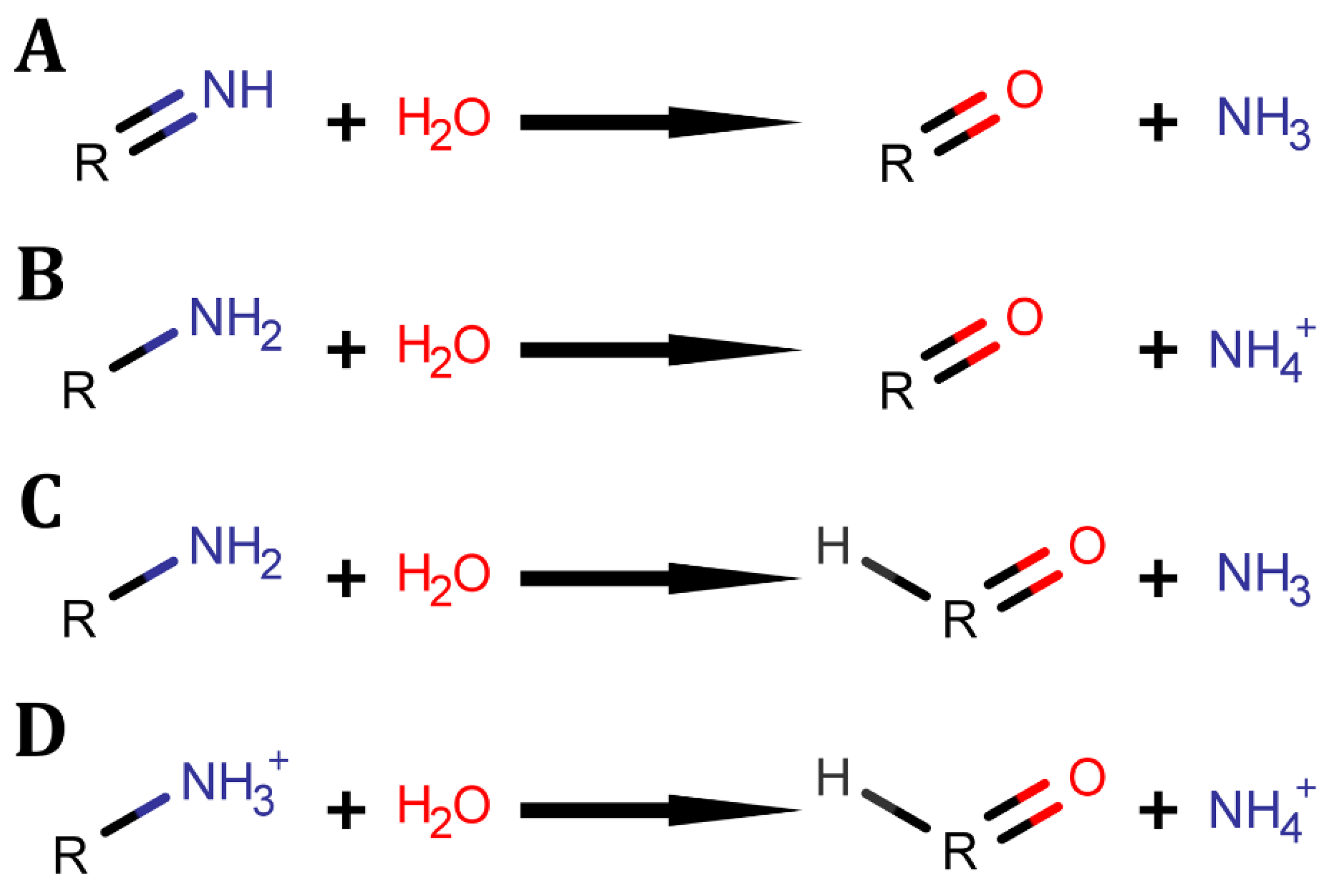
Figure 1.
Deaminase across domains of life. Evolution of deaminases across clades of life shown through labelling of different deaminases where colours denote protein superfamilies and substrate targets as shown in the legend, while white box within section denotes absence of the protein across the domain.
Figure 1.
Deaminase across domains of life. Evolution of deaminases across clades of life shown through labelling of different deaminases where colours denote protein superfamilies and substrate targets as shown in the legend, while white box within section denotes absence of the protein across the domain.

Figure 2.
Deaminase substrates by protein groups. The deaminase substrates across porphobilinogen deaminases (orange), glucosamine-6-phosphate deaminase (grey), amino acid deaminases (green), dUTPase superfamily deaminases (olive), amidohydrolase superfamily (red), cytidine deaminase superfamily (blue), and substrates of both AHS and CDA superfamilies (purple). Porphobilinogen deaminase: (A) porphobilinogen. Glucosamine-6-phosphate deaminase: (B) glucosamine-6-phosphate. Amino acid deaminases: (C) neutral amino acids, (D) 1-pyrroline-4-hydroxy-2-carboxylate, (E) 2-aminomuconate, (F) 1-aminocyclopropane-1-carboxylic acid, (G) serine [R=H] and threonine [R=CH3] derived imino acids. dUTPase superfamily: (H) dCTP. Both Cytidine Deaminase Superfamily (CDAS) and Amidohydrolase Superfamily (AHS) deaminases: (I) guanine, (J) cytosine, (K) ammeline. AHS: (L) adenine, (M) adenosine, (N) 5′-methylthioadenosine, (O) dAMP, (P) N6-Methyl-AMP, (Q) aminofutalosine, (R) pterin-6-carboxylate, (S) isoxanthopterin, (T) 8-oxoguanine, (U) ammelide. CDAS: (V) cytidine, (W) blasticidin S, (X) DARIPP [2,5-diamino-6-ribitylamino-4(3H)-pyrimidinone 5’-phosphate], (Y) DAROPP [2.5-diamino-6-ribosylamino-4-(3H)-pyrimidinone 5’-phosphate], (Z) dCMP, (AA) isoguanine, (AB) 5-azacytosine, (AC) acetoguanide, (AD) deoxy/cytidine-polynucleotide [R2=H/OH], (AE) adenosine-polynucleotide, (AF) guanosine.
Figure 2.
Deaminase substrates by protein groups. The deaminase substrates across porphobilinogen deaminases (orange), glucosamine-6-phosphate deaminase (grey), amino acid deaminases (green), dUTPase superfamily deaminases (olive), amidohydrolase superfamily (red), cytidine deaminase superfamily (blue), and substrates of both AHS and CDA superfamilies (purple). Porphobilinogen deaminase: (A) porphobilinogen. Glucosamine-6-phosphate deaminase: (B) glucosamine-6-phosphate. Amino acid deaminases: (C) neutral amino acids, (D) 1-pyrroline-4-hydroxy-2-carboxylate, (E) 2-aminomuconate, (F) 1-aminocyclopropane-1-carboxylic acid, (G) serine [R=H] and threonine [R=CH3] derived imino acids. dUTPase superfamily: (H) dCTP. Both Cytidine Deaminase Superfamily (CDAS) and Amidohydrolase Superfamily (AHS) deaminases: (I) guanine, (J) cytosine, (K) ammeline. AHS: (L) adenine, (M) adenosine, (N) 5′-methylthioadenosine, (O) dAMP, (P) N6-Methyl-AMP, (Q) aminofutalosine, (R) pterin-6-carboxylate, (S) isoxanthopterin, (T) 8-oxoguanine, (U) ammelide. CDAS: (V) cytidine, (W) blasticidin S, (X) DARIPP [2,5-diamino-6-ribitylamino-4(3H)-pyrimidinone 5’-phosphate], (Y) DAROPP [2.5-diamino-6-ribosylamino-4-(3H)-pyrimidinone 5’-phosphate], (Z) dCMP, (AA) isoguanine, (AB) 5-azacytosine, (AC) acetoguanide, (AD) deoxy/cytidine-polynucleotide [R2=H/OH], (AE) adenosine-polynucleotide, (AF) guanosine.
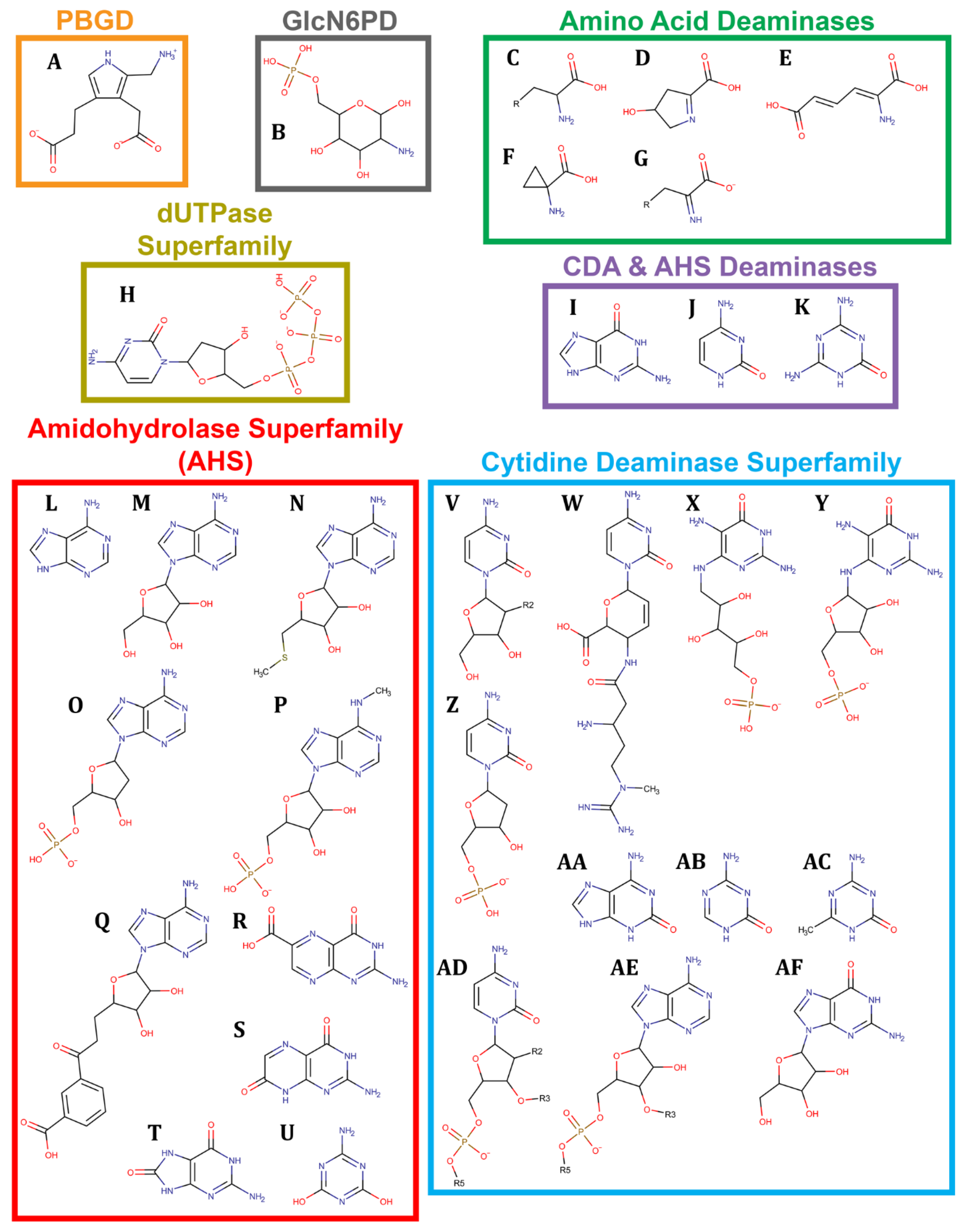
Scheme 2.
Reaction of porphobilinogen to hydroxymethylbilane.

Figure 3.
Porphobilinogen deaminase (PBGD) structures. The catalytic cysteine is in skyblue connected to DPM in purple bound to 2 substrate pyrroles in pink, and 1 unbound substrate in light pink. Protein cartoon (A) and surface (B) with domain 1 in blue, domain 2 in green, and domain 3 in red. A transparent view (C) of the catalytic pocket with the cartoon view coloured by secondary structure while the surface arginines are shown in blue and the polar residues in orange. All structures from PDB;7CD0 [6].
Figure 3.
Porphobilinogen deaminase (PBGD) structures. The catalytic cysteine is in skyblue connected to DPM in purple bound to 2 substrate pyrroles in pink, and 1 unbound substrate in light pink. Protein cartoon (A) and surface (B) with domain 1 in blue, domain 2 in green, and domain 3 in red. A transparent view (C) of the catalytic pocket with the cartoon view coloured by secondary structure while the surface arginines are shown in blue and the polar residues in orange. All structures from PDB;7CD0 [6].
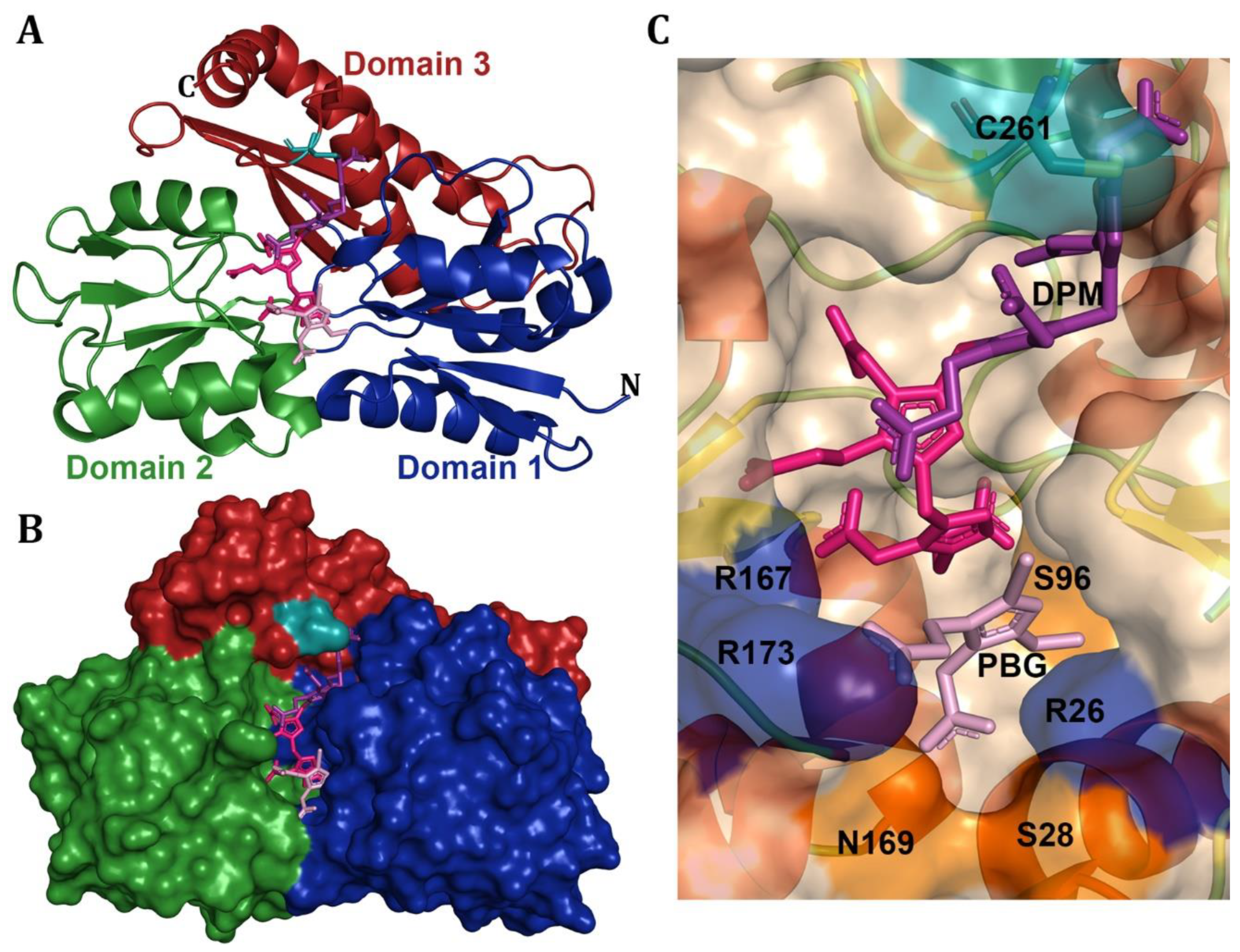
Scheme 3.
Reaction of glucosamine-6-phosphate to fructose-6-phosphate.
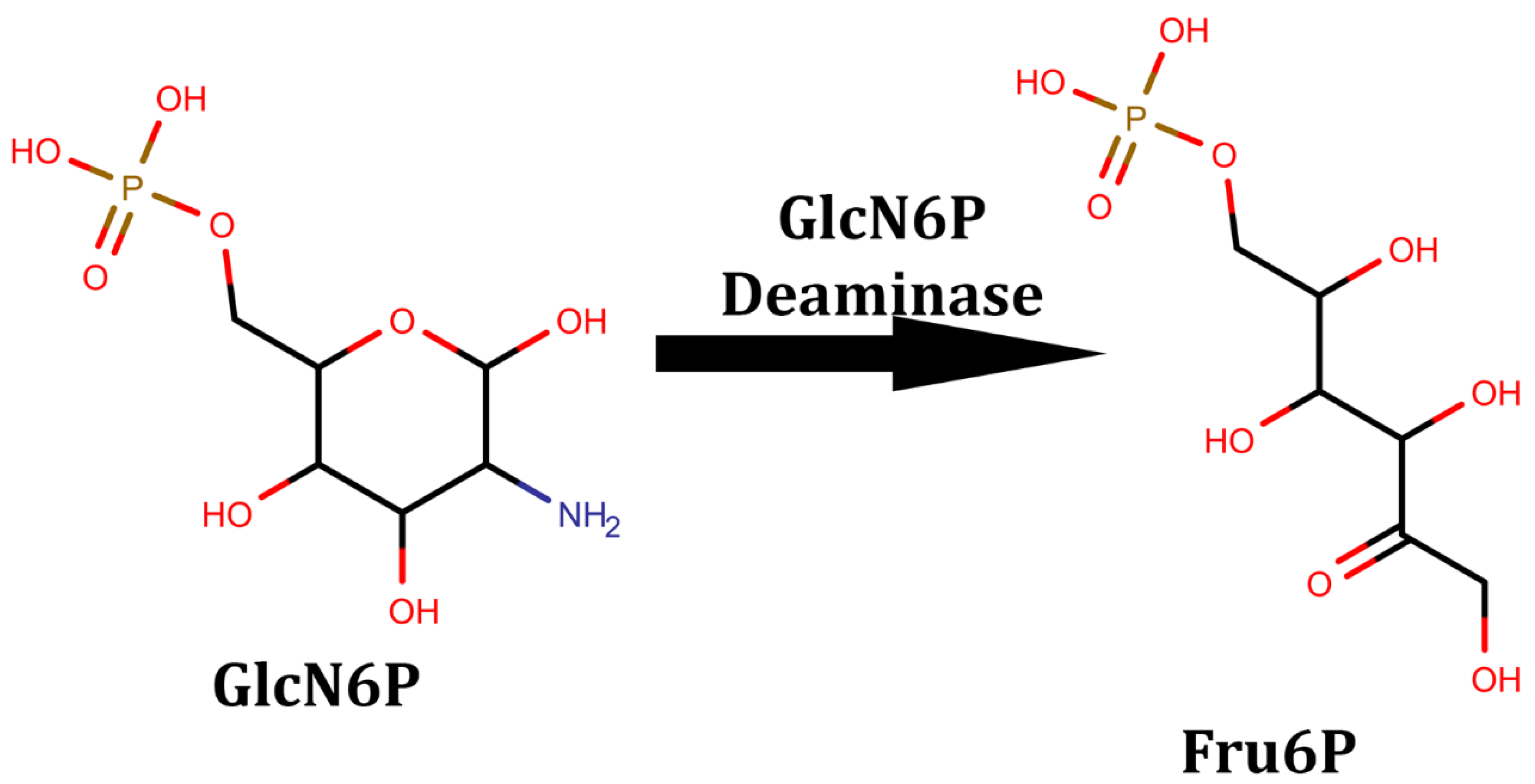
Figure 4.
Glucosamine-6-phosphate (GlcN6P) structures. Glucosamine-6-phosphate deaminase cartoon structure (A) coloured by N-term to C-term spectrum from blue to red, and the T-state (B) and R-state (C) with transparent surface showing cartoon coloured by secondary structure and surface coloured with the lid in green, and the dual-proton-shuttle and stabilizing aspartate in blue. Fructose-6-phosphate sticks are in purple in the R-state (C). Hyperthermophilic glucosamine-6-phosphate dimer cartoon (D) shown with a spectrum from blue to red with the opposing monomer in sand, as well as shown by secondary structure (E) with the catalytic lysine and glutamate in deepteal and the coordinating histidine on the opposing sand monomer in blue. Structures are from (A,C) 1FQO [44], (B) 1FSF [44], and (D,E) 2CB0 [56].
Figure 4.
Glucosamine-6-phosphate (GlcN6P) structures. Glucosamine-6-phosphate deaminase cartoon structure (A) coloured by N-term to C-term spectrum from blue to red, and the T-state (B) and R-state (C) with transparent surface showing cartoon coloured by secondary structure and surface coloured with the lid in green, and the dual-proton-shuttle and stabilizing aspartate in blue. Fructose-6-phosphate sticks are in purple in the R-state (C). Hyperthermophilic glucosamine-6-phosphate dimer cartoon (D) shown with a spectrum from blue to red with the opposing monomer in sand, as well as shown by secondary structure (E) with the catalytic lysine and glutamate in deepteal and the coordinating histidine on the opposing sand monomer in blue. Structures are from (A,C) 1FQO [44], (B) 1FSF [44], and (D,E) 2CB0 [56].
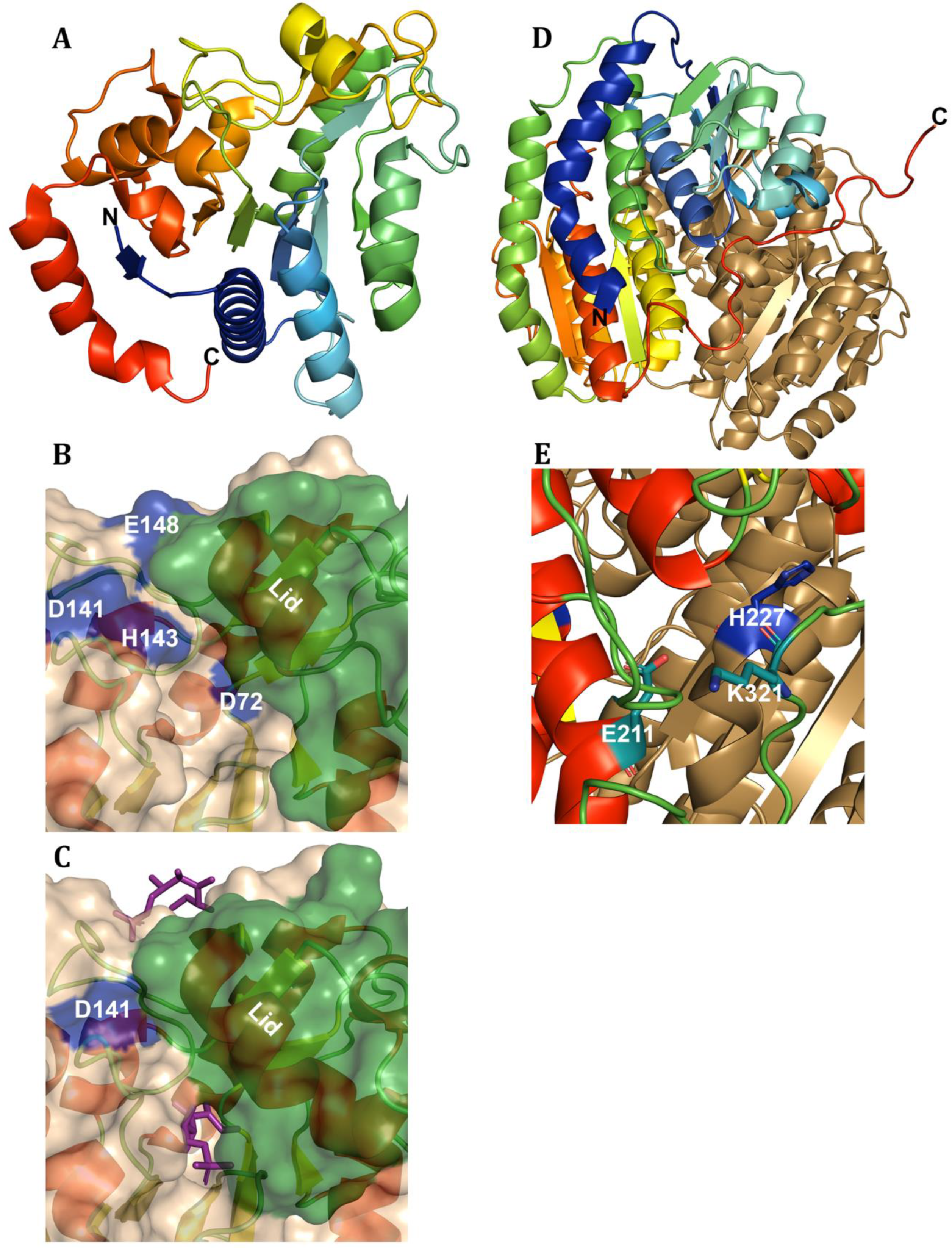
Scheme 4.
Amino acid and imino acid deamination.
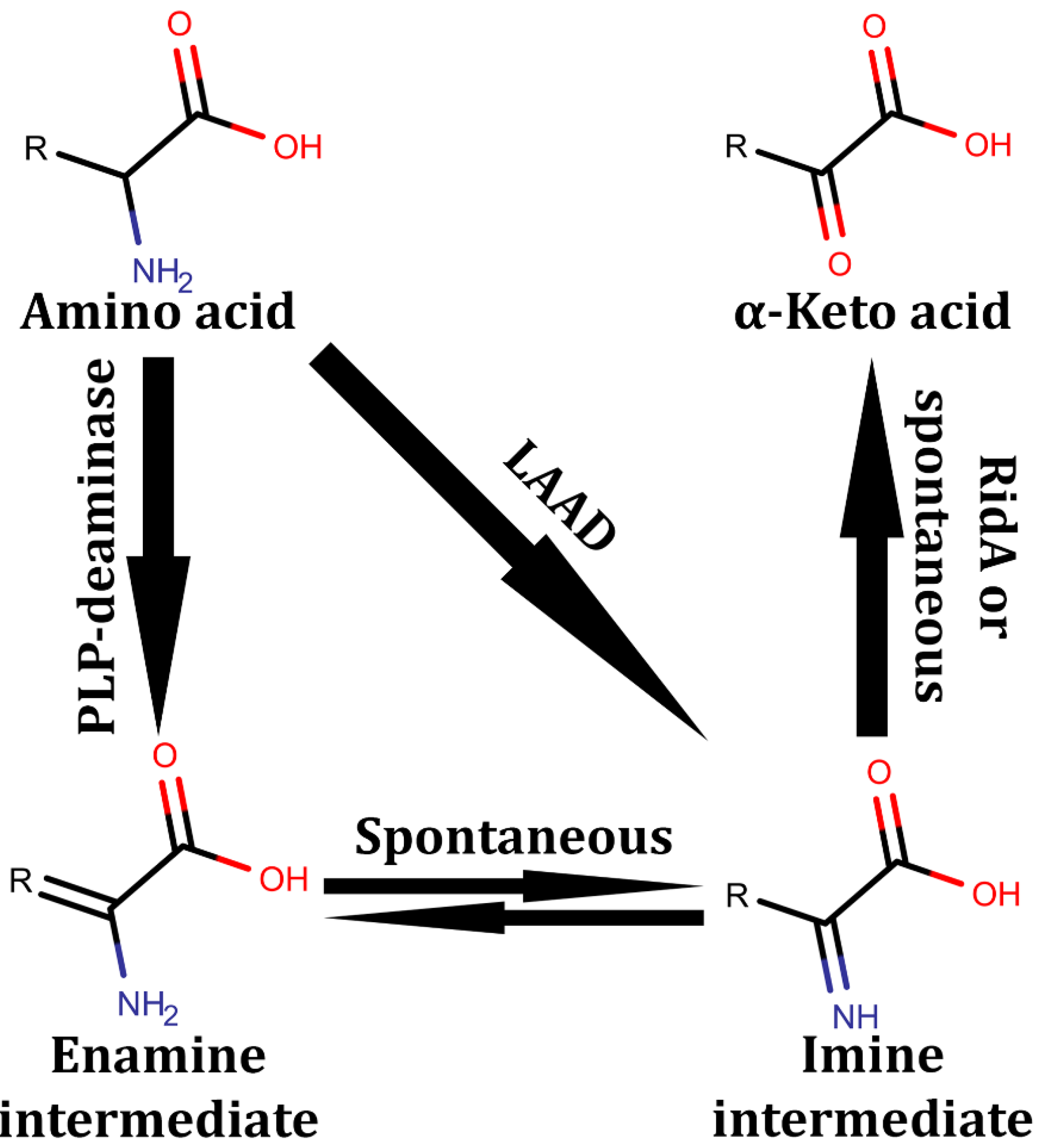
Figure 5.
Amino acid deaminase structures. Cartoons of amino acid deaminases with sticks of purple substrates, (A-G) cyan PLP, and orange catalytic lysine. PLP deaminases include (A) serine deaminase domains shown as deepteal and forest with the cartoon pocket shown by secondary structure with (B) lysine binding PLP and (C) PLP-binding serine, (D) chicken serine deaminase, (E) pocket of 1-aminocyclopropane-1-carboxylic acid deaminase, (F) MppP [L-arginine deaminase], and (G) aromatic amino acid deaminase cartoon dimer with one coloured by secondary structure and the other in deepteal. Non-PLP amino acid deaminases include (H) L-amino acid deaminase with the membrane insertion module in deepteal, substrate-binding domain in red, and FAD-binding domain in green, with FAD as purple stick. The (I) RidA trimer shown in red, deepteal, and green with purple substrate, and (J) HPC deaminase coloured by secondary structure with orange lysine and purple substrate. Structures are from (A,B) 1P5J [64], (C) 1PWH [23], (D) 3AWO [60], (E) 1J0D [2], (F) 5DJ3 [25], (G) 6EEI [59], (H) 5FJM [73], (I) 5HP8 [76], and (J) 4O0K [86].
Figure 5.
Amino acid deaminase structures. Cartoons of amino acid deaminases with sticks of purple substrates, (A-G) cyan PLP, and orange catalytic lysine. PLP deaminases include (A) serine deaminase domains shown as deepteal and forest with the cartoon pocket shown by secondary structure with (B) lysine binding PLP and (C) PLP-binding serine, (D) chicken serine deaminase, (E) pocket of 1-aminocyclopropane-1-carboxylic acid deaminase, (F) MppP [L-arginine deaminase], and (G) aromatic amino acid deaminase cartoon dimer with one coloured by secondary structure and the other in deepteal. Non-PLP amino acid deaminases include (H) L-amino acid deaminase with the membrane insertion module in deepteal, substrate-binding domain in red, and FAD-binding domain in green, with FAD as purple stick. The (I) RidA trimer shown in red, deepteal, and green with purple substrate, and (J) HPC deaminase coloured by secondary structure with orange lysine and purple substrate. Structures are from (A,B) 1P5J [64], (C) 1PWH [23], (D) 3AWO [60], (E) 1J0D [2], (F) 5DJ3 [25], (G) 6EEI [59], (H) 5FJM [73], (I) 5HP8 [76], and (J) 4O0K [86].
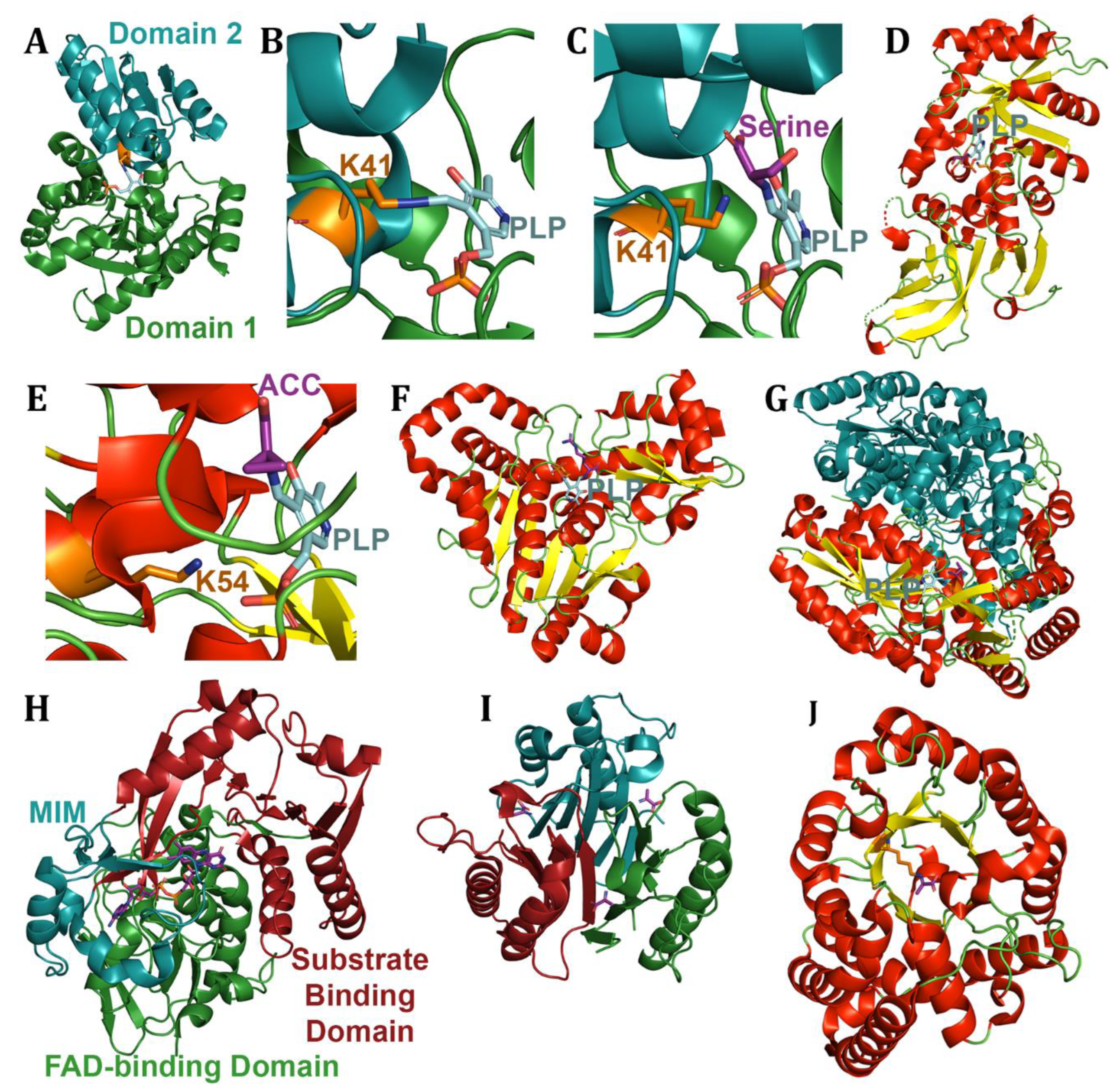
Scheme 5.
Guanine and guanosine deamination. (A) Guanine and (B) guanosine deamination reactions.
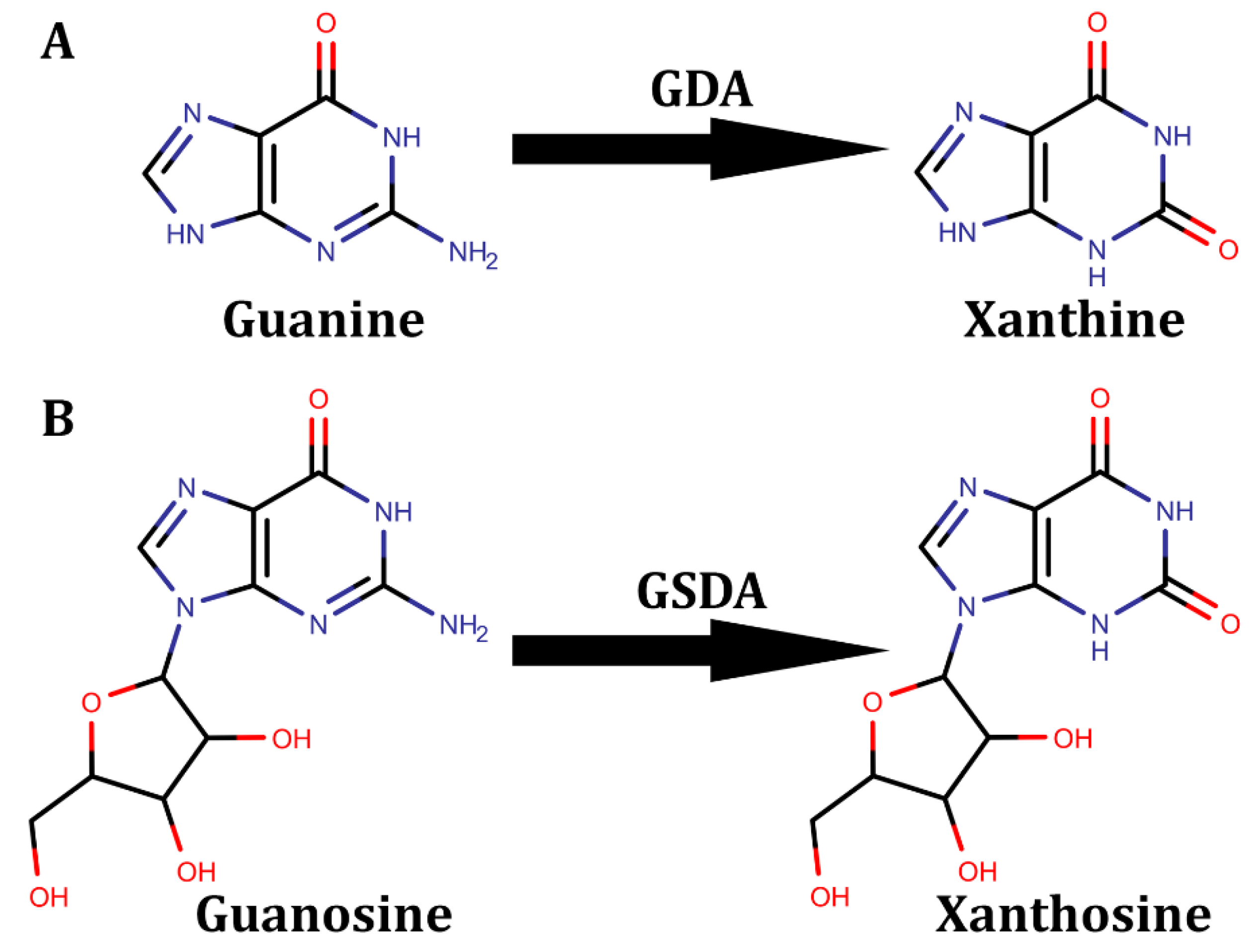
Figure 6.
Guanine derivative deaminases. The (A-C) AHS guanine deaminase shown (A) as a cartoon spectrum from N-term blue to C-term red, (B) a cartoon with the TIM β-barrel domain in deepteal and PDZ domain in red, and (C) pocket view coloured by secondary structure with the substrate purple, coordinating histidines in skyblue and dual proton shuttle in orange. The CDAS (D-G) guanine and (H) guanosine deaminases shown as cartoon dimers with the second subunit shown as sand. Guanine deaminase of (D) N. europaea and (E) B. subtilis shown as a full structure with cartoon coloured as a spectrum from blue to red. The (F) dimeric interface of GDA N. europaea shown in deepteal. The catalytic pockets of (G) GDA B. subtilis and (H) GSDA coloured by secondary structure with sticks of the ligand in purple, with the coordinating residues in skyblue, the proton shuttle in orange, and the lid shown in pink. Structures are from (A-C) 6OHB [94], (D,F) 4LC5 [17], (E,G) 1WKQ [97], and (H) 7DOW [18].
Figure 6.
Guanine derivative deaminases. The (A-C) AHS guanine deaminase shown (A) as a cartoon spectrum from N-term blue to C-term red, (B) a cartoon with the TIM β-barrel domain in deepteal and PDZ domain in red, and (C) pocket view coloured by secondary structure with the substrate purple, coordinating histidines in skyblue and dual proton shuttle in orange. The CDAS (D-G) guanine and (H) guanosine deaminases shown as cartoon dimers with the second subunit shown as sand. Guanine deaminase of (D) N. europaea and (E) B. subtilis shown as a full structure with cartoon coloured as a spectrum from blue to red. The (F) dimeric interface of GDA N. europaea shown in deepteal. The catalytic pockets of (G) GDA B. subtilis and (H) GSDA coloured by secondary structure with sticks of the ligand in purple, with the coordinating residues in skyblue, the proton shuttle in orange, and the lid shown in pink. Structures are from (A-C) 6OHB [94], (D,F) 4LC5 [17], (E,G) 1WKQ [97], and (H) 7DOW [18].
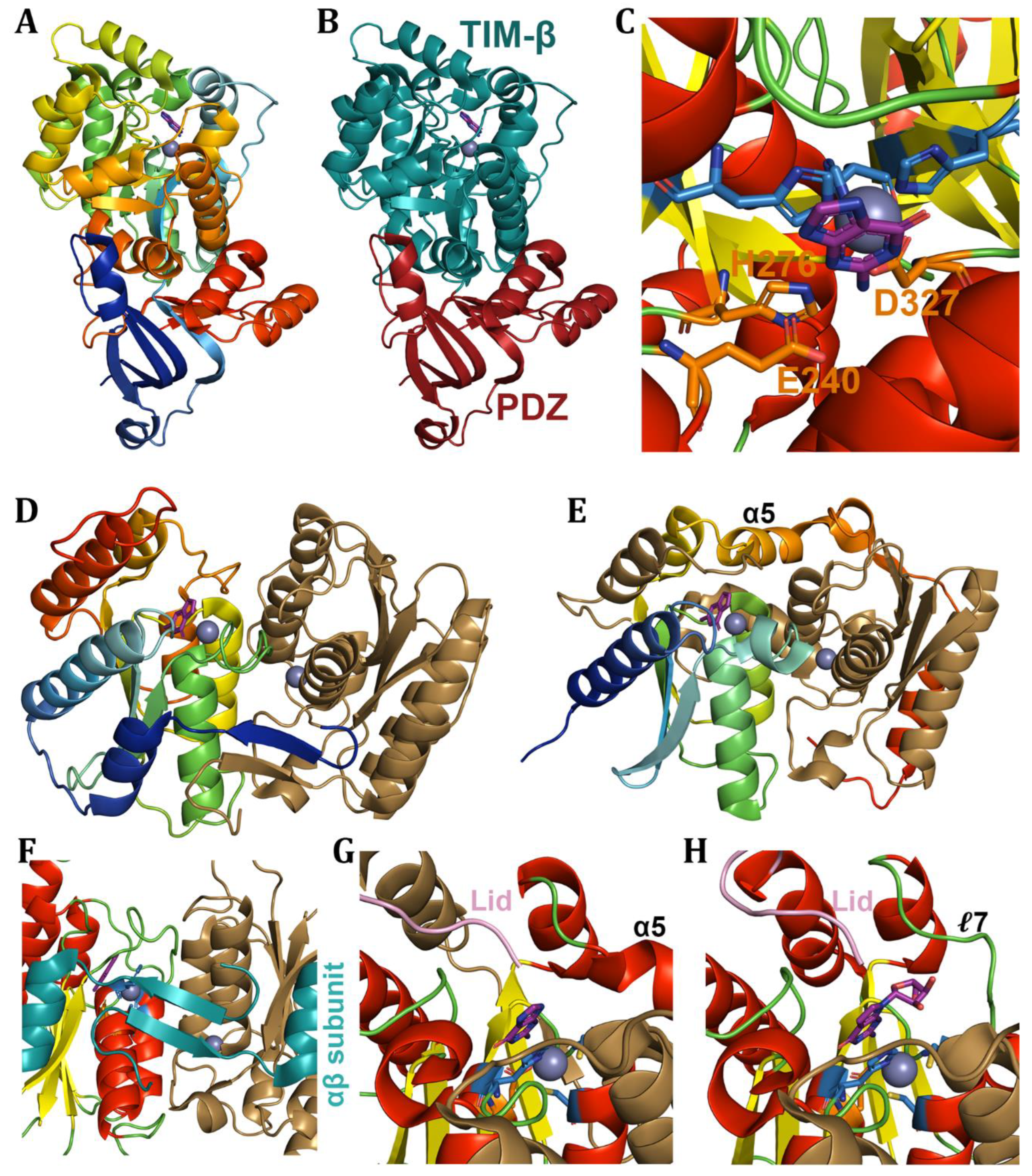
Scheme 6.
Adenine and adenosine based-deamination. (A) Adenine, (B) adenosine, and (C) N6-methyl-AMP deamination reactions.
Scheme 6.
Adenine and adenosine based-deamination. (A) Adenine, (B) adenosine, and (C) N6-methyl-AMP deamination reactions.
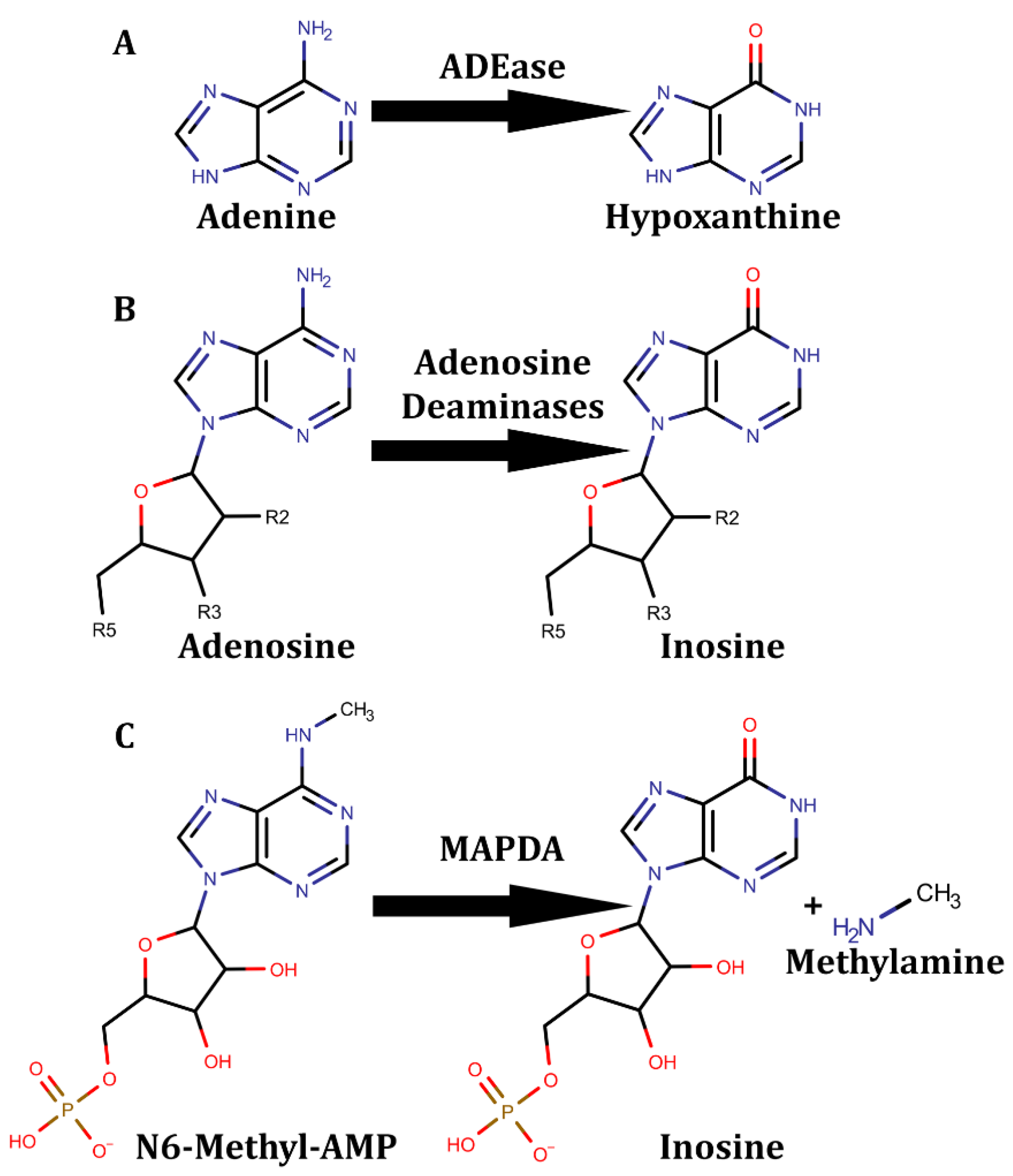
Figure 7.
Adenine derivative deaminases / free nucleobase/nucleoside deaminases (AHS ADA/ADEase). Deaminases shown as (A,C-F) cartoons coloured via secondary structures with skyblue coordination residues, orange proton shuttle and purple substrate. Structures shown are (A) pocket of dual divalent cationic ADEase, (B) A. fabrum triple domain ADEase shown with the PDZ domain in red along with the TIM β-barrel domain in deepteal and the domain of unknown function in green, with a (C) pocket view, the (D) full structure of ADA, with a (E) pocket view, and a (F) pocket view of MAPDA. Structures are from (A) 2ICS [105], (B-C) 3T81 [101], (D-E) 1ADD [102], and (F) 6IJN [122].
Figure 7.
Adenine derivative deaminases / free nucleobase/nucleoside deaminases (AHS ADA/ADEase). Deaminases shown as (A,C-F) cartoons coloured via secondary structures with skyblue coordination residues, orange proton shuttle and purple substrate. Structures shown are (A) pocket of dual divalent cationic ADEase, (B) A. fabrum triple domain ADEase shown with the PDZ domain in red along with the TIM β-barrel domain in deepteal and the domain of unknown function in green, with a (C) pocket view, the (D) full structure of ADA, with a (E) pocket view, and a (F) pocket view of MAPDA. Structures are from (A) 2ICS [105], (B-C) 3T81 [101], (D-E) 1ADD [102], and (F) 6IJN [122].
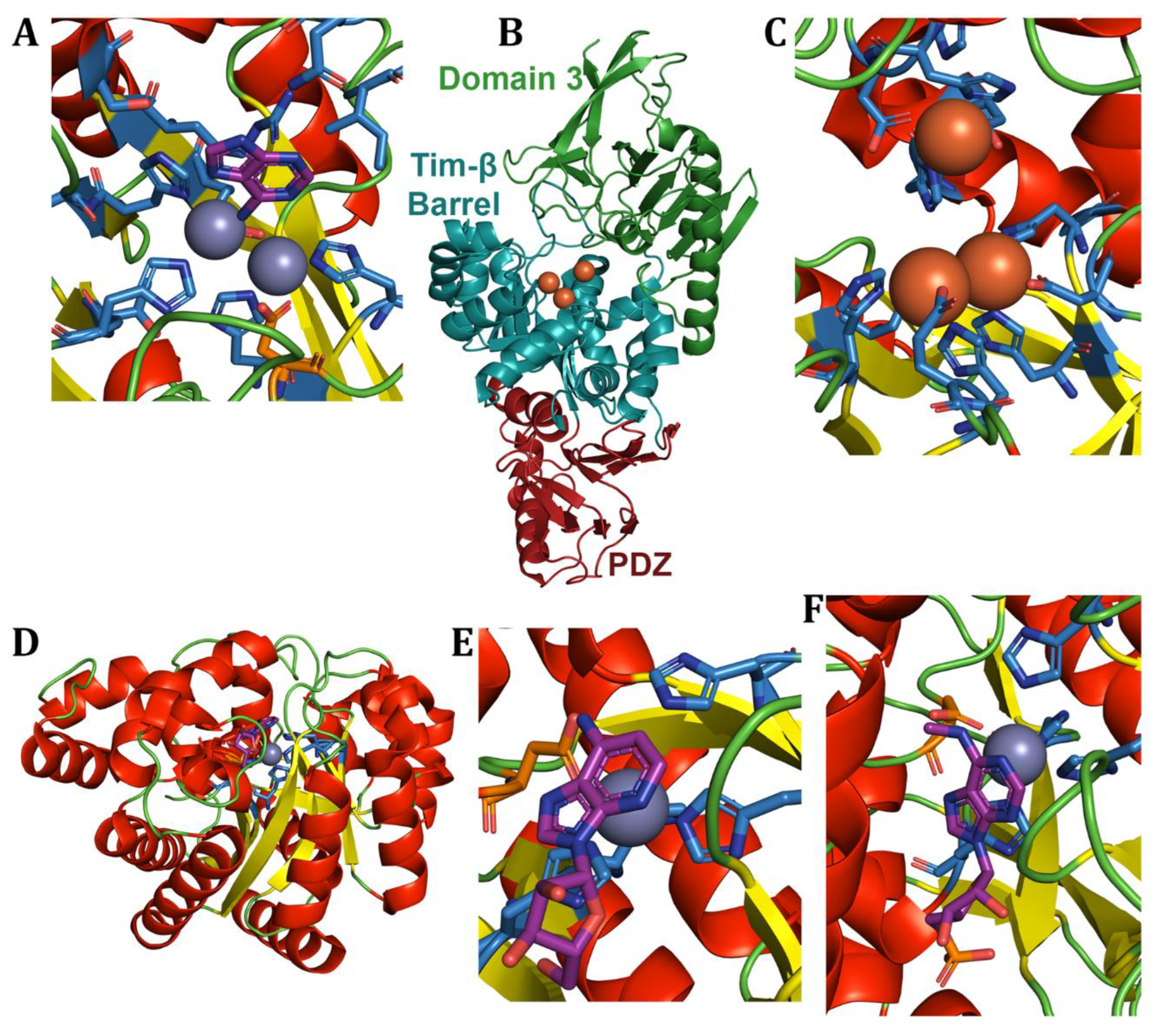
Figure 8.
Adenine derivative deaminases / tRNA deaminases (CDA superfamily). (A,B) TadA dimer coloured by secondary structure on one subunit and coloured sand on the other with the substrate in purple, coordinating residues in skyblue, proton shuttle in orange, and FFxxxR motif in cyan. ADAT2/3 shown by (C) domain with ADAT2 in deepteal, ADAT3-LOBE in green and ADAT3-PCD in red, as well as shown coloured by (D) secondary structure, both with substrate in purple. Structures are from (A-B) 2B3J and (C-D) 8AW3 [19].
Figure 8.
Adenine derivative deaminases / tRNA deaminases (CDA superfamily). (A,B) TadA dimer coloured by secondary structure on one subunit and coloured sand on the other with the substrate in purple, coordinating residues in skyblue, proton shuttle in orange, and FFxxxR motif in cyan. ADAT2/3 shown by (C) domain with ADAT2 in deepteal, ADAT3-LOBE in green and ADAT3-PCD in red, as well as shown coloured by (D) secondary structure, both with substrate in purple. Structures are from (A-B) 2B3J and (C-D) 8AW3 [19].
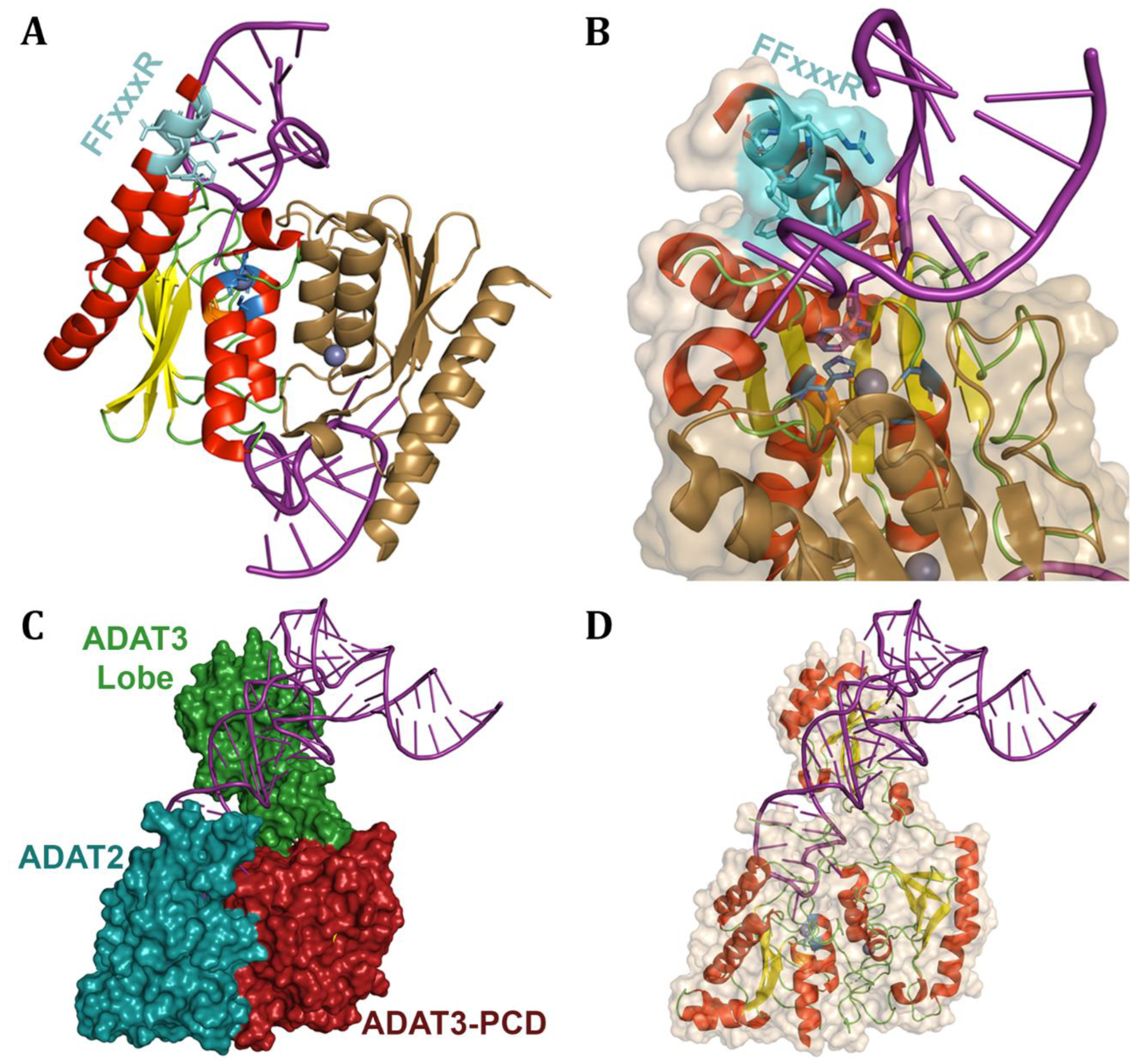
Figure 9.
Adenine derivative deaminases / dsRNA deaminases (CDA superfamily). ADAR proteins coloured by (A-B,D-E) secondary structure or by (C) domain with all substrates in purple. Shown are the (A) Z-DNA-binding domain and (B) dsRNA-binding motif, (C) dimerization of 2 ADAR catalytic domains in green and deepteal with dsRBM in red, (D) full catalytic domain of ADAR, and the catalytic pocket with coordinating residues in skyblue, proton shuttle in orange and E488 in green flipped into the dsRNA. Structures are from (A) 5ZU1 [156], (B) 2L2K [162], (C) 6VFF [148], and (D-E) 5HP2 [164].
Figure 9.
Adenine derivative deaminases / dsRNA deaminases (CDA superfamily). ADAR proteins coloured by (A-B,D-E) secondary structure or by (C) domain with all substrates in purple. Shown are the (A) Z-DNA-binding domain and (B) dsRNA-binding motif, (C) dimerization of 2 ADAR catalytic domains in green and deepteal with dsRBM in red, (D) full catalytic domain of ADAR, and the catalytic pocket with coordinating residues in skyblue, proton shuttle in orange and E488 in green flipped into the dsRNA. Structures are from (A) 5ZU1 [156], (B) 2L2K [162], (C) 6VFF [148], and (D-E) 5HP2 [164].
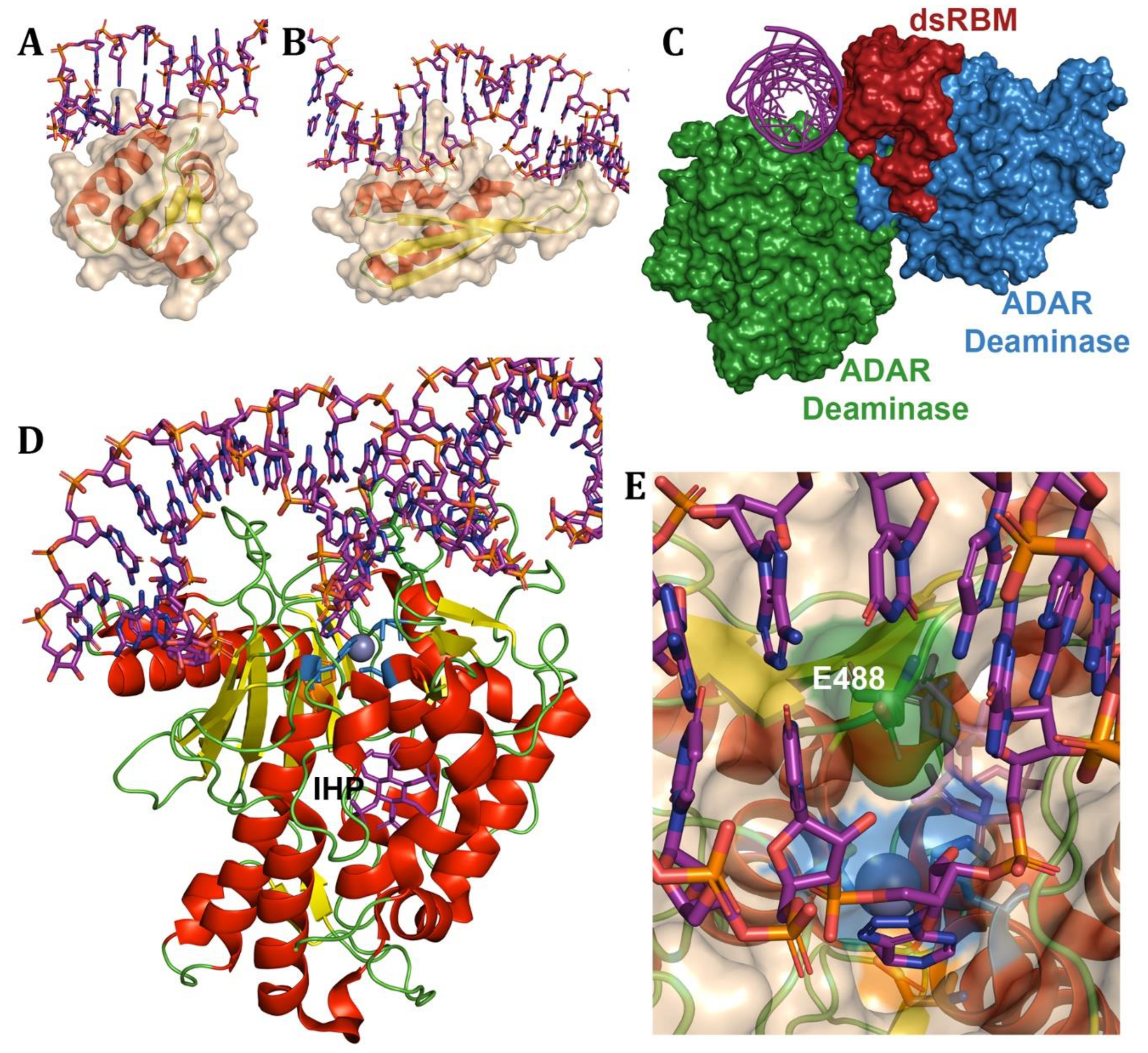
Scheme 7.
Cytosine and derivative deamination. (A) Cytosine and s-triazine scaffold based (B) 5-azacytosine deamination reactions.
Scheme 7.
Cytosine and derivative deamination. (A) Cytosine and s-triazine scaffold based (B) 5-azacytosine deamination reactions.
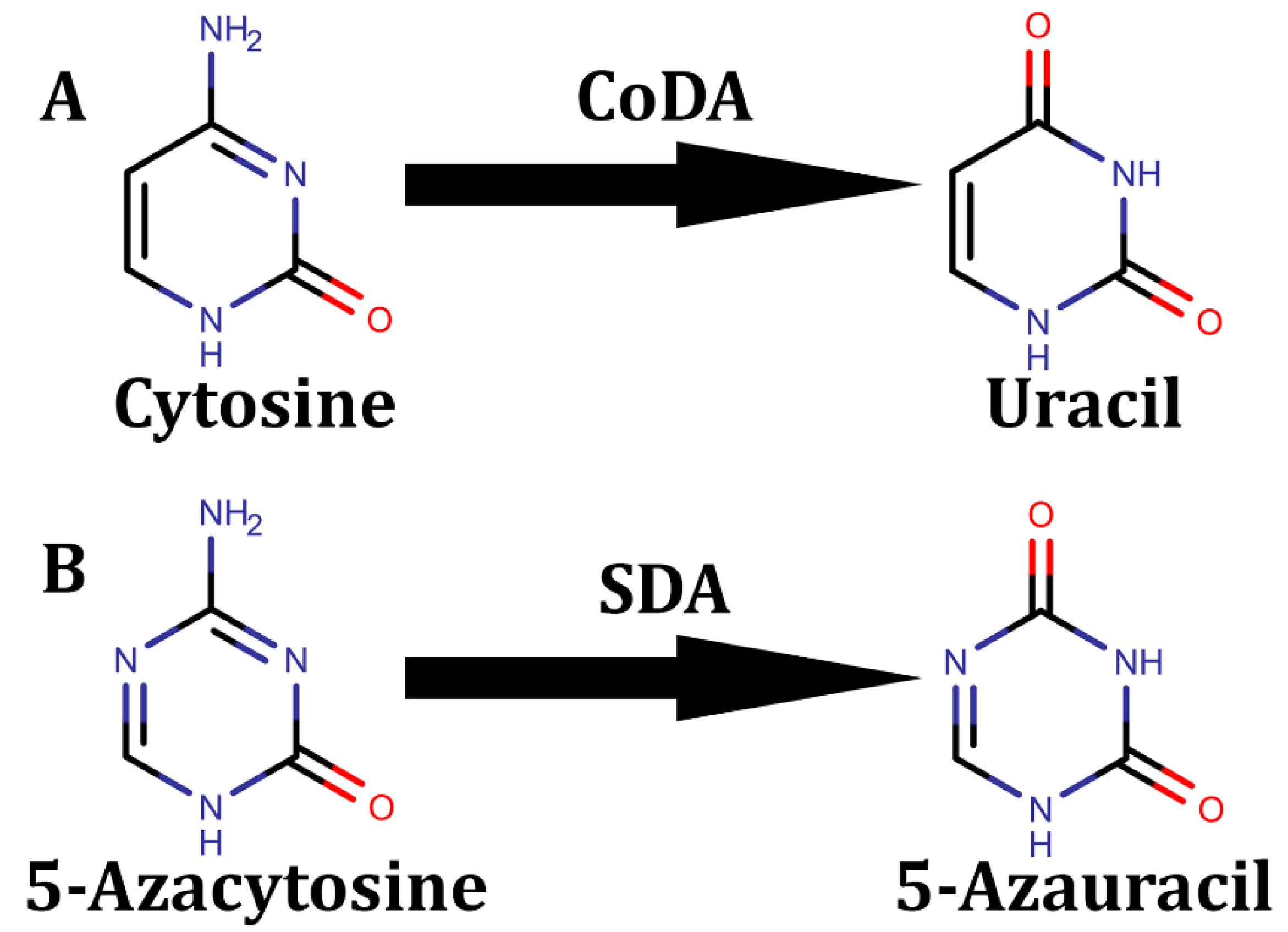
Figure 10.
Cytosine free nucleobase deaminase and derivatives. Cartoons coloured by secondary structure of catalytic pockets of (A) bCoDA, (B) yCoDA, and (C) SDA. Ligands are purple, coordinating residues skyblue, and proton shuttles orange. (B,C) opposing monomer in dimer coloured sand, with (B) lid coloured pink. Structures are from (A) 1K70 [168], (B) 1P6O [169], and (C) 5XKP [178].
Figure 10.
Cytosine free nucleobase deaminase and derivatives. Cartoons coloured by secondary structure of catalytic pockets of (A) bCoDA, (B) yCoDA, and (C) SDA. Ligands are purple, coordinating residues skyblue, and proton shuttles orange. (B,C) opposing monomer in dimer coloured sand, with (B) lid coloured pink. Structures are from (A) 1K70 [168], (B) 1P6O [169], and (C) 5XKP [178].
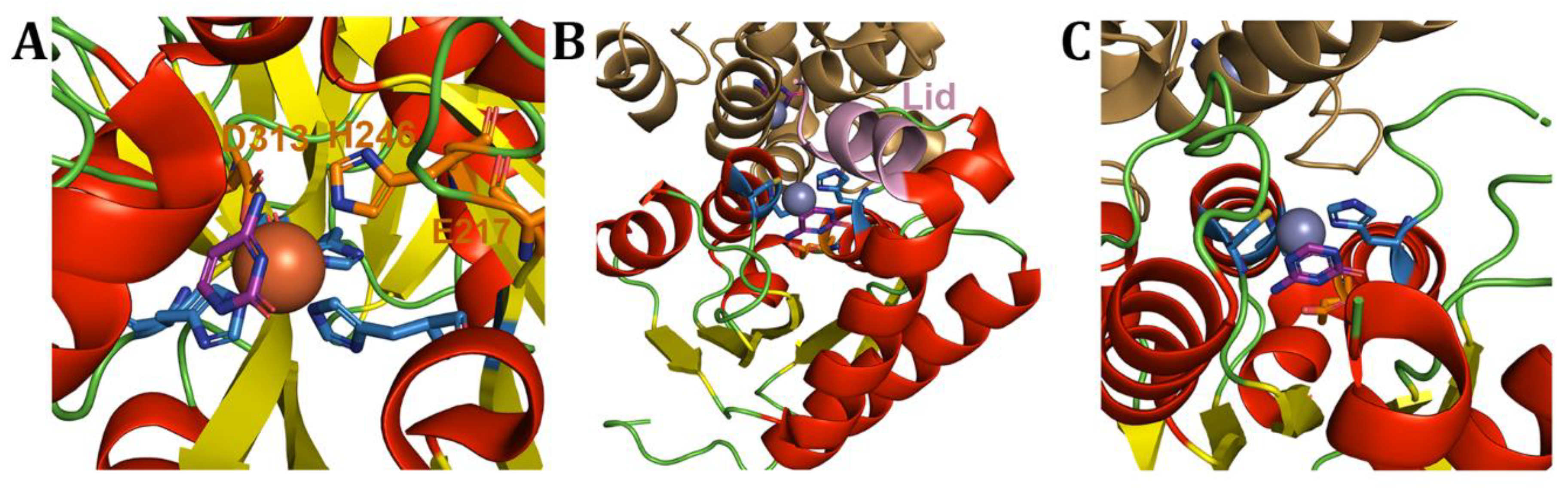
Scheme 8.
Cytidine derived deamination. (A) Cytidine, and (B) blasticidine S deamination reactions.
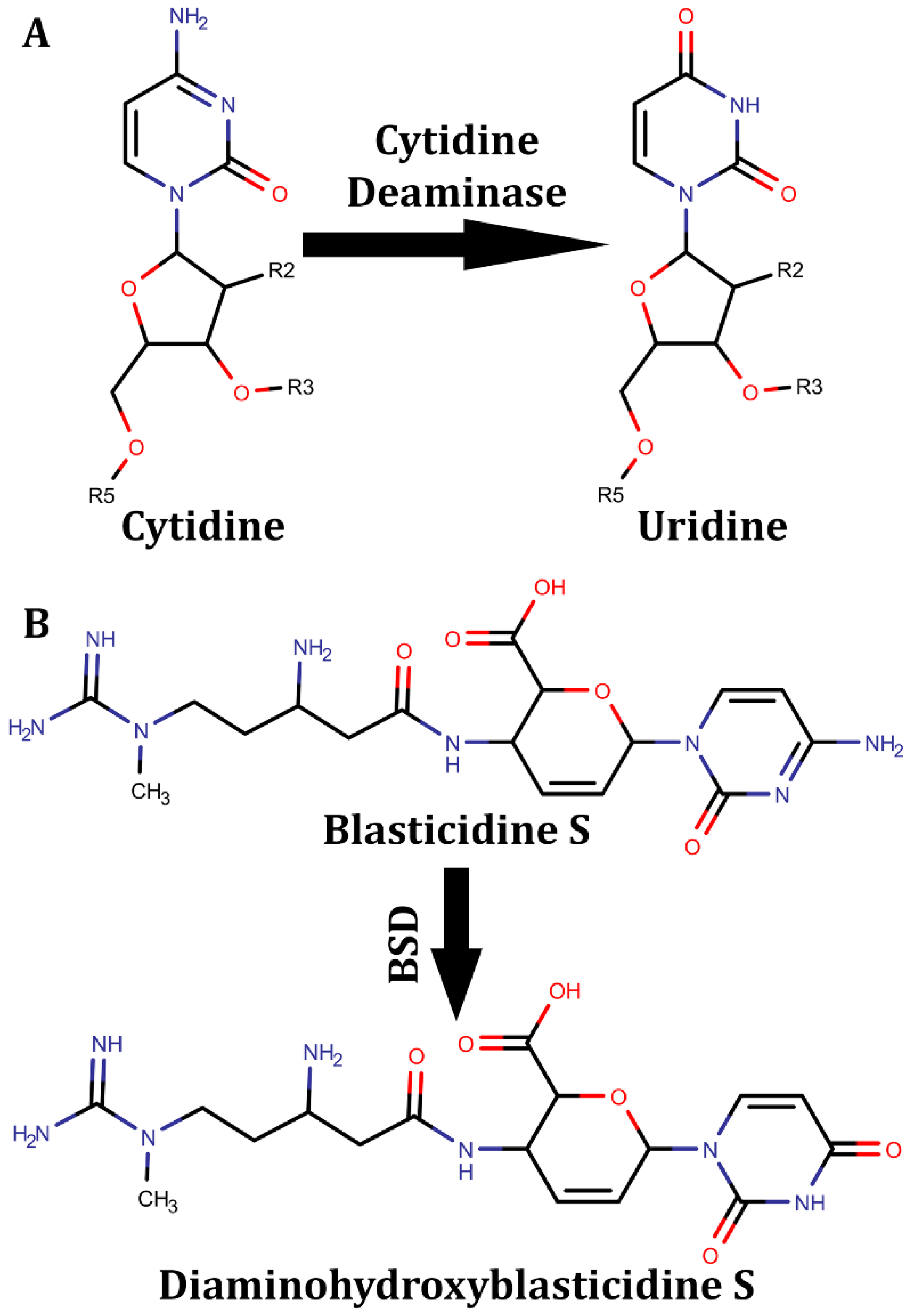
Figure 11.
Cytidine free nucleoside deaminases structures. Structures of (A-B) T-CDA, (C-D) BSD, and (E-F) D-CDA. The (A,C) T-CDA and BSD tetramers are shown in different colours and D-CDA shown with catalytic domains in deepteal and wheat while pseudocatalytic domains in red and green with the (A,E) lid of Chain B [deepteal] highlighted in pink over the T-CDA’s catalytic pocket. (B,D,F) Catalytic pockets with coordinating residues in skyblue, proton shuttle in orange, and substrates in purple. (B) Cartoons from other domains with lid shown in pink and coordinating arginine in cyan. (D) residues coordinating chlorine in cyan. Structures are from (A-B) 2FR6 [15], (C-D) 2Z3I [8], (E-F) 4EG2 [194].
Figure 11.
Cytidine free nucleoside deaminases structures. Structures of (A-B) T-CDA, (C-D) BSD, and (E-F) D-CDA. The (A,C) T-CDA and BSD tetramers are shown in different colours and D-CDA shown with catalytic domains in deepteal and wheat while pseudocatalytic domains in red and green with the (A,E) lid of Chain B [deepteal] highlighted in pink over the T-CDA’s catalytic pocket. (B,D,F) Catalytic pockets with coordinating residues in skyblue, proton shuttle in orange, and substrates in purple. (B) Cartoons from other domains with lid shown in pink and coordinating arginine in cyan. (D) residues coordinating chlorine in cyan. Structures are from (A-B) 2FR6 [15], (C-D) 2Z3I [8], (E-F) 4EG2 [194].
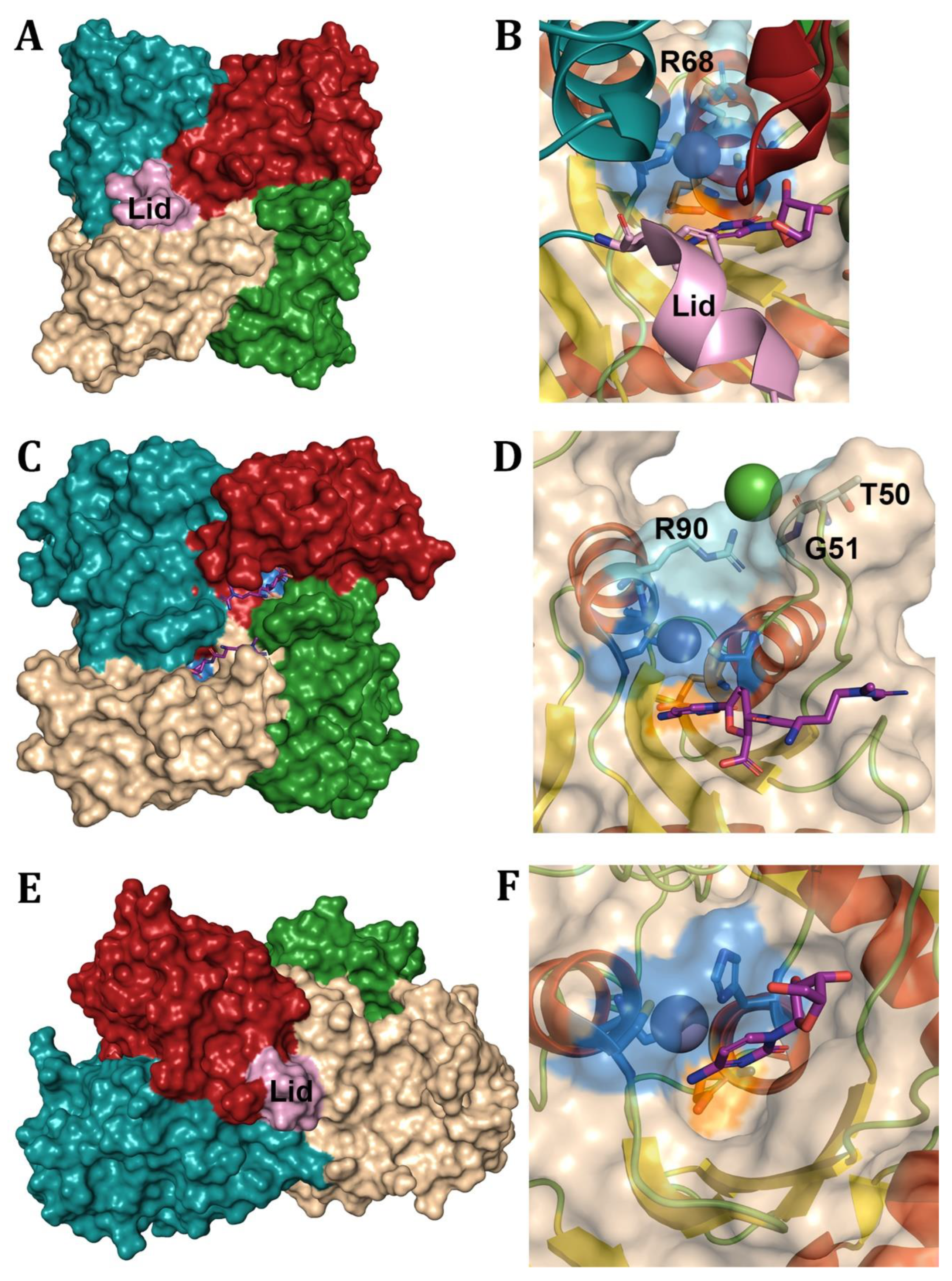
Figure 12.
Cytidine free nucleotide deaminase structures. Trimeric (A,B) dCTPD and (C) DCD:DUT with each subunit coloured differently and the ligand in purple. The (B) pocket of dCTPD is shown with the catalytic residues in orange with other stabilizing residues in cyan. The (D) hexameric form of dCMPD from cyanophage S-TIM5 coloured by subunit binding dCMP. View of the variant structures next to the catalytic pocket of (E) T4 Bacteriophage’s zinc and coordinating residues in cyan, and the (F) cyanophage S-TIM5 with the disulfide bond of cysteines in cyan. Surface view of cyanophage S-TIM5 highlighting coordinating residues in skyblue, binding (G) dCMP and (H) dTMP, forming an active and inactive pocket respectively. Structures are from (A-B) 4A6A [213], (C) 4XJC [200], (D,G) 4P9C [211], (E) 1VQ2 [195], and (F,H) 4P9D [211].
Figure 12.
Cytidine free nucleotide deaminase structures. Trimeric (A,B) dCTPD and (C) DCD:DUT with each subunit coloured differently and the ligand in purple. The (B) pocket of dCTPD is shown with the catalytic residues in orange with other stabilizing residues in cyan. The (D) hexameric form of dCMPD from cyanophage S-TIM5 coloured by subunit binding dCMP. View of the variant structures next to the catalytic pocket of (E) T4 Bacteriophage’s zinc and coordinating residues in cyan, and the (F) cyanophage S-TIM5 with the disulfide bond of cysteines in cyan. Surface view of cyanophage S-TIM5 highlighting coordinating residues in skyblue, binding (G) dCMP and (H) dTMP, forming an active and inactive pocket respectively. Structures are from (A-B) 4A6A [213], (C) 4XJC [200], (D,G) 4P9C [211], (E) 1VQ2 [195], and (F,H) 4P9D [211].
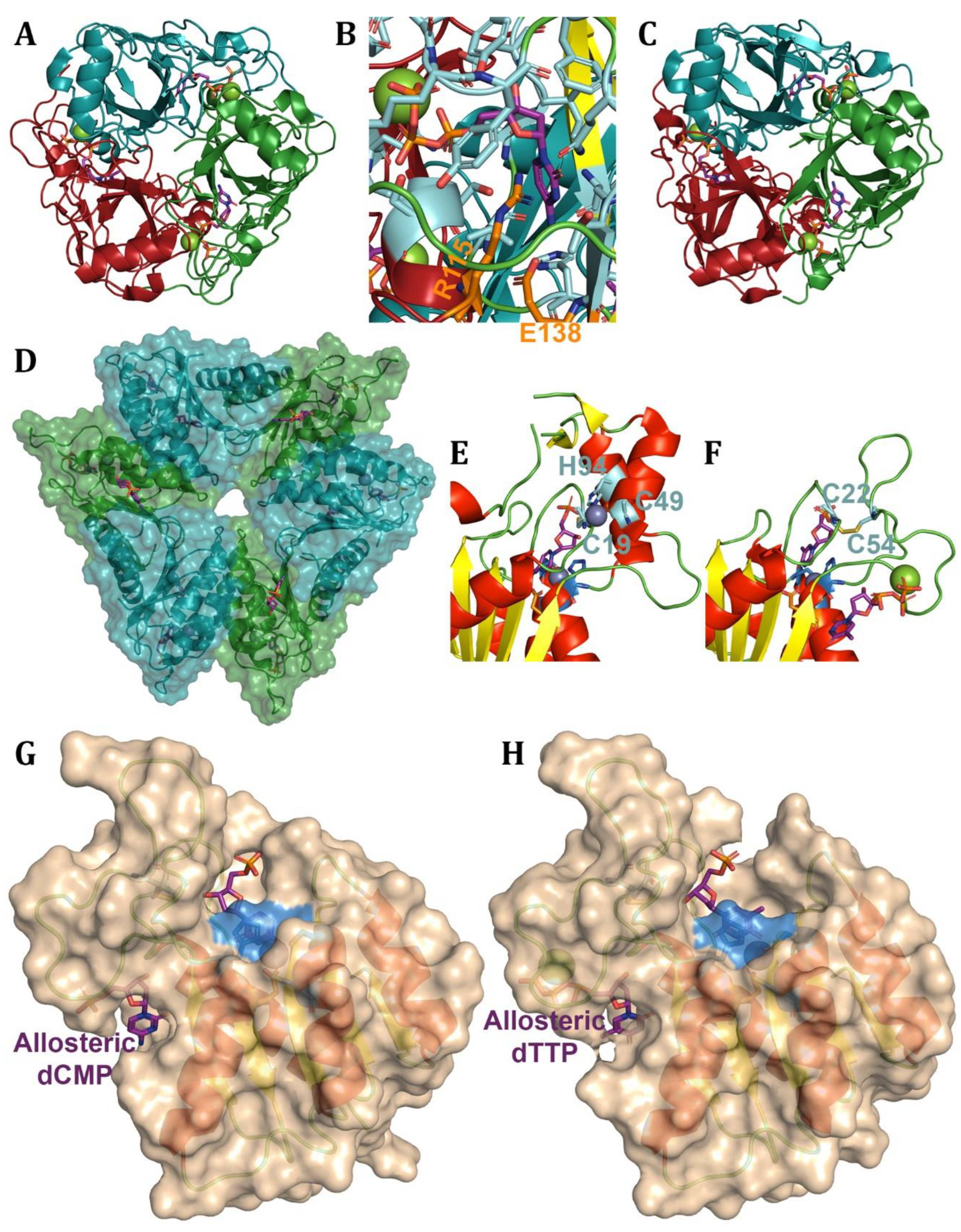
Scheme 9.
Vitamin B2 pathway.
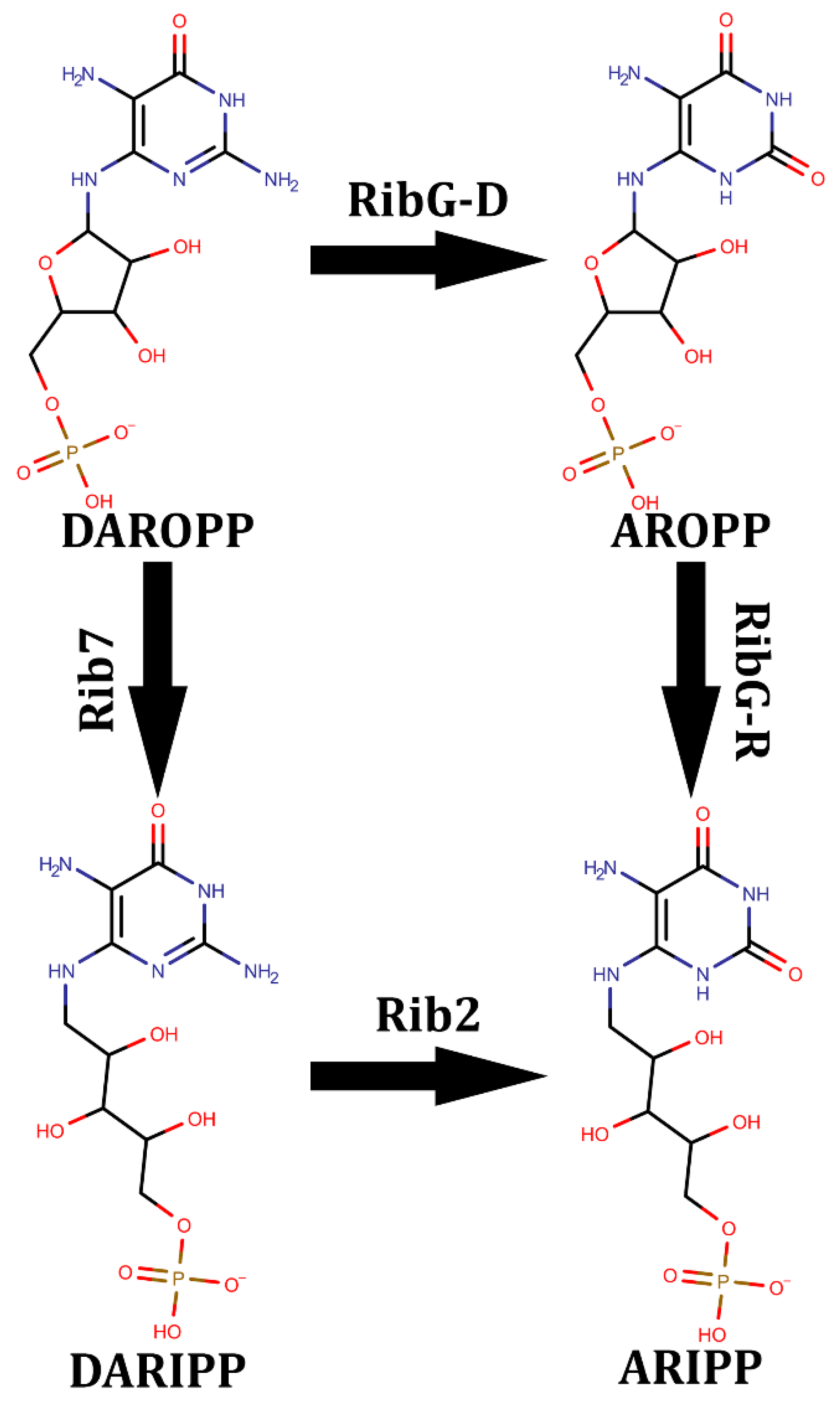
Figure 13.
RibG and Rib2 structures. Cartoon of (A) RibG-D and (B) Rib2 coloured by secondary structure with coordinating residues in skyblue, proton shuttle in orange, substrate in purple, and other important residues in cyan. (C) B. subtilis RibG tetrameric ring structure with chain A coloured by secondary structure and the 3 other chains coloured by subunit. Structures are from (A,C) 3EX8 [215], (B) 7DS1 [218].
Figure 13.
RibG and Rib2 structures. Cartoon of (A) RibG-D and (B) Rib2 coloured by secondary structure with coordinating residues in skyblue, proton shuttle in orange, substrate in purple, and other important residues in cyan. (C) B. subtilis RibG tetrameric ring structure with chain A coloured by secondary structure and the 3 other chains coloured by subunit. Structures are from (A,C) 3EX8 [215], (B) 7DS1 [218].
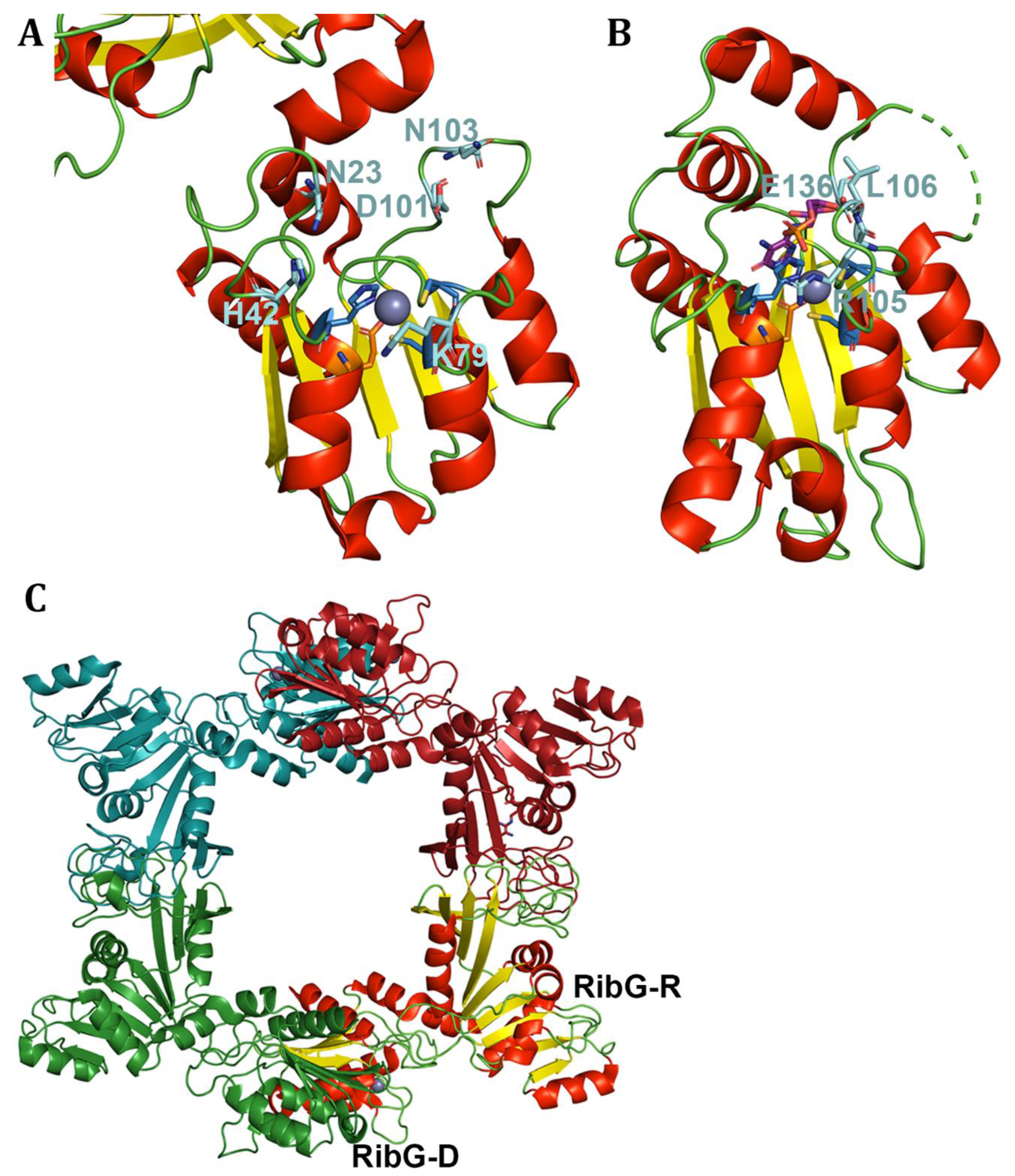
Figure 14.
Cytidine RNA deaminases. Structures of (A-C) CDAT8 and the (D-F) DYW domain. Cartoons are coloured by (A,C,E-F) secondary structure with skyblue coordinating residues and orange proton shuttle. The surface of CDAT8 is coloured by (A) domain with green deaminase domain, skyblue FLD domain, and red THUMP domain. The surfaces of CDAT8 subunits dimerized are coloured in (B) skyblue and red, and wheat and sand, respectively. The DYW (D) subdomains are coloured skyblue for the gate, green for the deaminase and the DYW motif in red. The (E) closed gate and (F) open gate are shown with the DYW motif in sand, the lysine in pink, and the R895 contacting D922 or D872 in cyan, respectively. Structures are from (A-C) 3G8Q [224], (D,F) 7O4F [223], (E) 7O4E [223].
Figure 14.
Cytidine RNA deaminases. Structures of (A-C) CDAT8 and the (D-F) DYW domain. Cartoons are coloured by (A,C,E-F) secondary structure with skyblue coordinating residues and orange proton shuttle. The surface of CDAT8 is coloured by (A) domain with green deaminase domain, skyblue FLD domain, and red THUMP domain. The surfaces of CDAT8 subunits dimerized are coloured in (B) skyblue and red, and wheat and sand, respectively. The DYW (D) subdomains are coloured skyblue for the gate, green for the deaminase and the DYW motif in red. The (E) closed gate and (F) open gate are shown with the DYW motif in sand, the lysine in pink, and the R895 contacting D922 or D872 in cyan, respectively. Structures are from (A-C) 3G8Q [224], (D,F) 7O4F [223], (E) 7O4E [223].
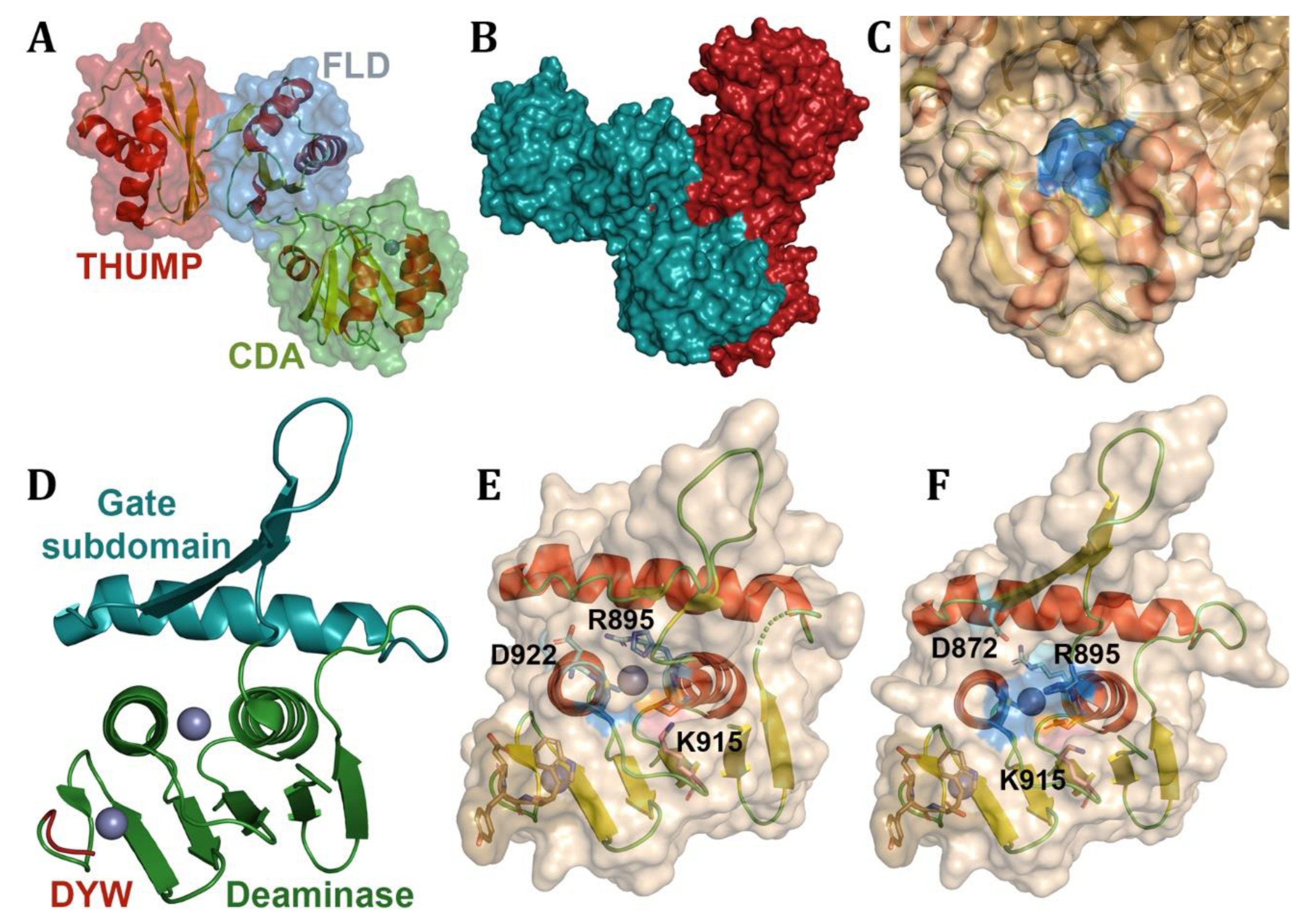
Figure 15.
Interbacterial DNA toxin cytidine deaminases. Cartoons of (A,C-D) DddA and (B,E) SsdA are coloured by secondary structure. The coordinating residues are coloured skyblue, proton shuttle orange, the region of the (A, B) SWG highlighted in cyan, and (A, C) phenylalanine in green. The (C) substrate is in purple and pink to differentiate the double strands interacting with DddA. The inhibitors (D) DddAi and (E) SsdA are shown in deepteal. Structures are from (A,C) 8E5E [227], (B,E) 7JTU [226], and (D) 6U08 [10].
Figure 15.
Interbacterial DNA toxin cytidine deaminases. Cartoons of (A,C-D) DddA and (B,E) SsdA are coloured by secondary structure. The coordinating residues are coloured skyblue, proton shuttle orange, the region of the (A, B) SWG highlighted in cyan, and (A, C) phenylalanine in green. The (C) substrate is in purple and pink to differentiate the double strands interacting with DddA. The inhibitors (D) DddAi and (E) SsdA are shown in deepteal. Structures are from (A,C) 8E5E [227], (B,E) 7JTU [226], and (D) 6U08 [10].
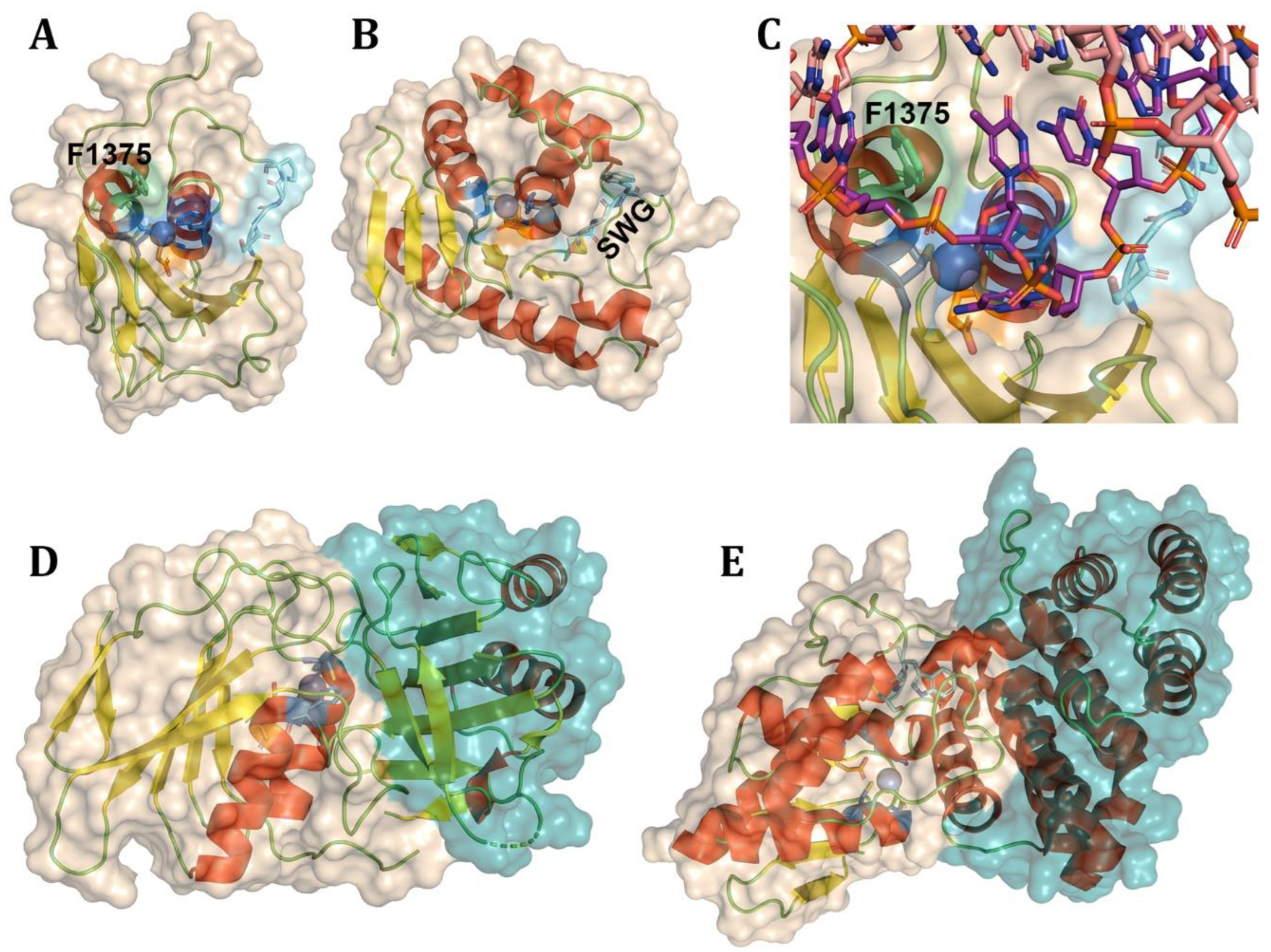
Figure 16.
AID/APOBEC family deaminases across evolution. Evolution of AID/APOBECs across clades of life shown through labelling of different AID/APOBECs where colours denote continuation of lineage, while white box within section denotes absence across the clade. (A) Shows the evolution of single domain AID/APOBECs through A3Z domain subtypes and their lineages in primates. (B) Shows the evolution of A3Z subtypes across non-primates.
Figure 16.
AID/APOBEC family deaminases across evolution. Evolution of AID/APOBECs across clades of life shown through labelling of different AID/APOBECs where colours denote continuation of lineage, while white box within section denotes absence across the clade. (A) Shows the evolution of single domain AID/APOBECs through A3Z domain subtypes and their lineages in primates. (B) Shows the evolution of A3Z subtypes across non-primates.
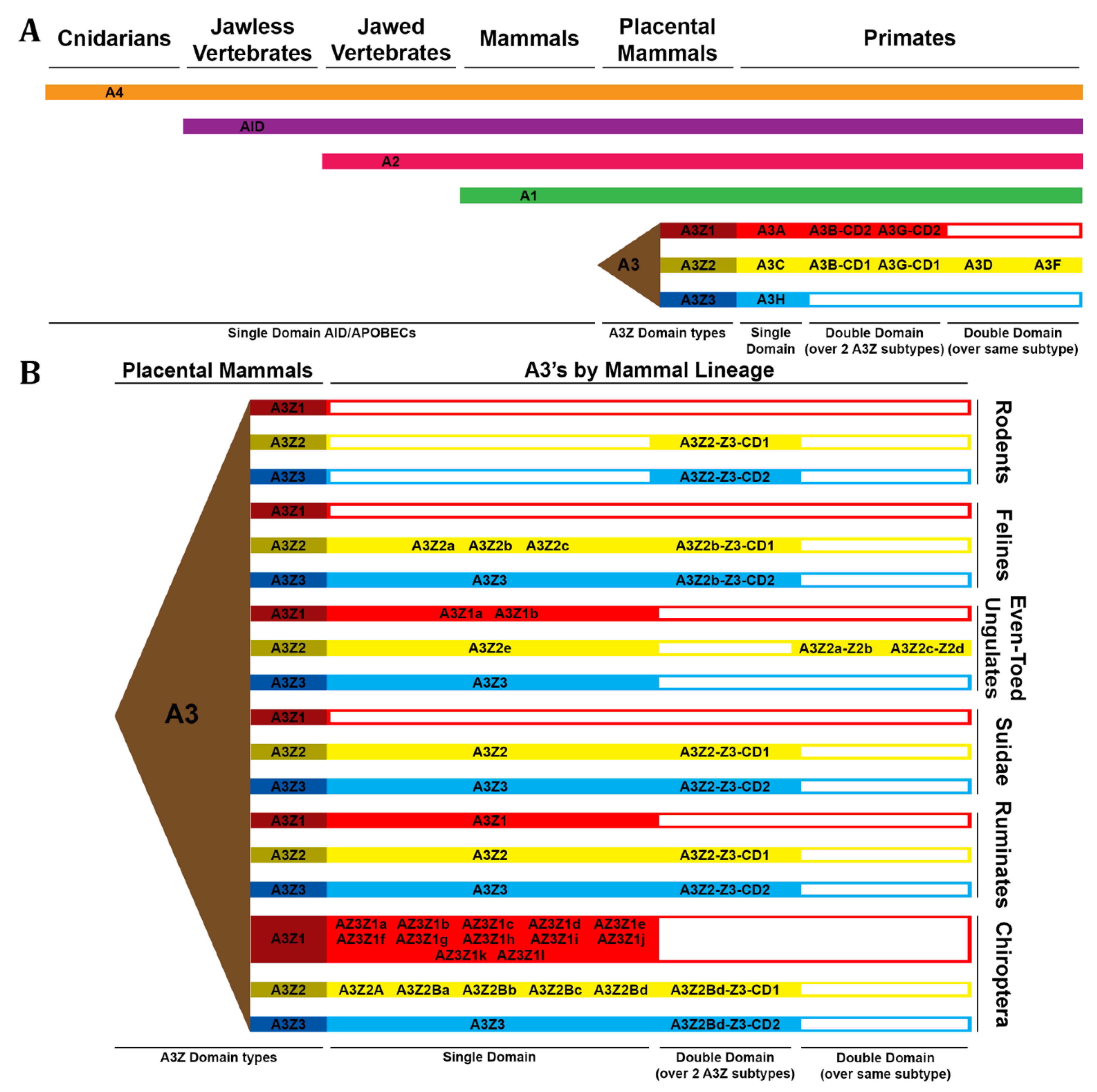
Figure 17.
APOBEC3 viral VIF/CBF-β interactions. Structures of A3s and Vif/CBF-β across primates. (A) Cartoon and surface structure of Vif in wheat, and CBF-β in deepteal with the binding site of A3C, A3D, and A3F in orange, binding site of A3G in blue, and A3H haplotype 2 in purple. Structures of (B) A3C, (C) A3D-CD2, (D) A3F-CD2, (E) A3G-CD1, and (F) A3H haplotype 2 with Vif-binding site in firebrick and CBF-β-binding site on A3C, A3D-CD2 and A3G-CD1 in deepteal. Structure of (G) A3F-CD2 and (H) A3G in forest with DNA in purple interacting with Vif in firebrick and CBF-β in deepteal. Structures are from (A,G) 6NIL [271], (B) 3VM8 [301], (C) was created in AlphaFold2, (D) 5ZVB [265], (E) 5K82 [273], (F) 5W3V [235], and (H) 8CX2 [302].
Figure 17.
APOBEC3 viral VIF/CBF-β interactions. Structures of A3s and Vif/CBF-β across primates. (A) Cartoon and surface structure of Vif in wheat, and CBF-β in deepteal with the binding site of A3C, A3D, and A3F in orange, binding site of A3G in blue, and A3H haplotype 2 in purple. Structures of (B) A3C, (C) A3D-CD2, (D) A3F-CD2, (E) A3G-CD1, and (F) A3H haplotype 2 with Vif-binding site in firebrick and CBF-β-binding site on A3C, A3D-CD2 and A3G-CD1 in deepteal. Structure of (G) A3F-CD2 and (H) A3G in forest with DNA in purple interacting with Vif in firebrick and CBF-β in deepteal. Structures are from (A,G) 6NIL [271], (B) 3VM8 [301], (C) was created in AlphaFold2, (D) 5ZVB [265], (E) 5K82 [273], (F) 5W3V [235], and (H) 8CX2 [302].
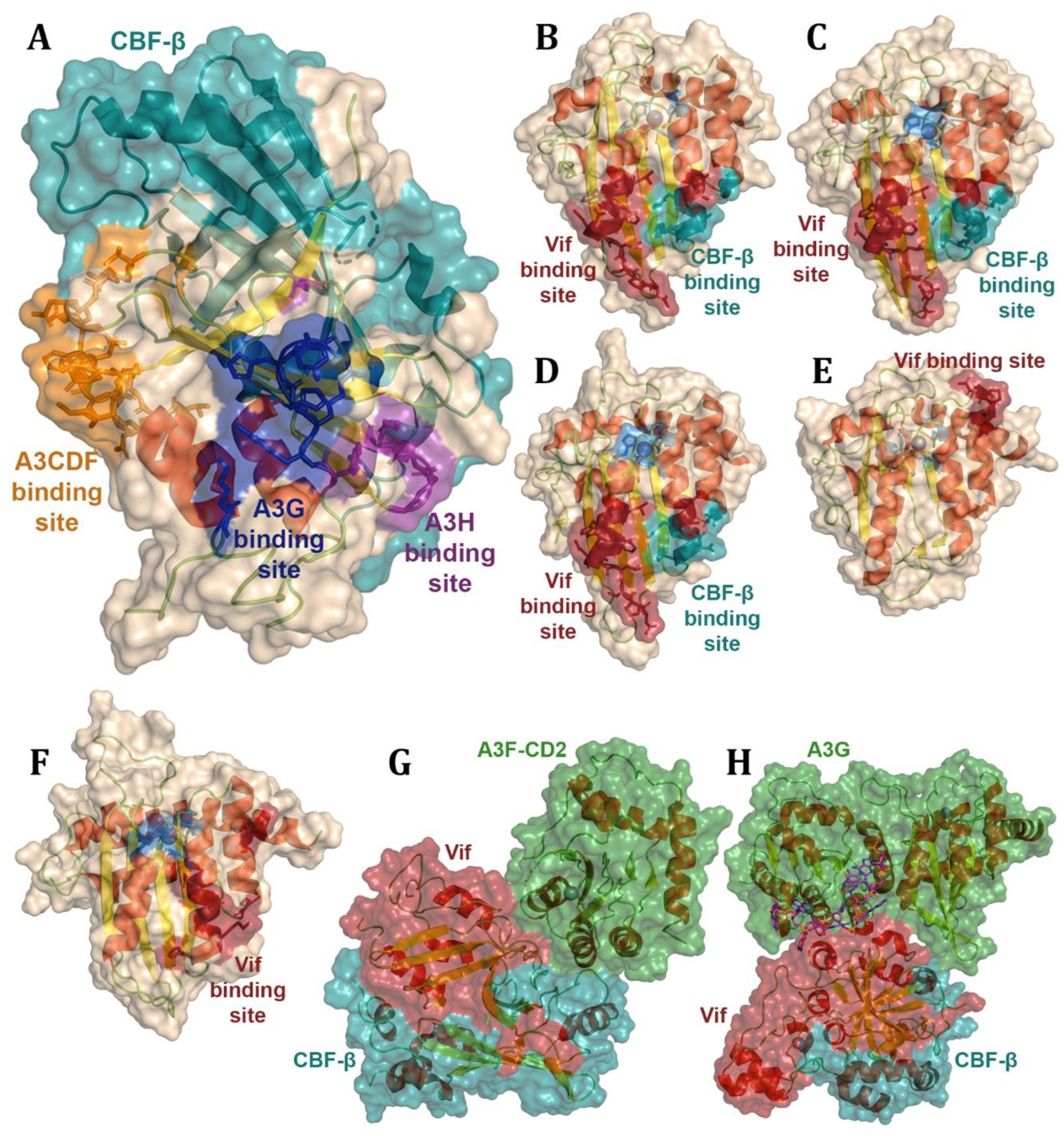
Figure 18.
AID/APOBEC family deaminases base structure. AID/APOBEC structures with cartoons coloured by secondary structure, with skyblue coordinating residues, orange proton shuttle, and purple substrates. AID/APOBEC fold shown with an example of (A) AID as a cartoon, as well as (B) surface structure with light green loop 1 highlighting the gatekeeper residue in forest green, pink loop 3, red loop 5, and blue loop 7. (C) AID cartoon shown with different α7 positions in deepteal covering the assistant patch and blue by the β-sheet. (D) APOBEC1 cartoon with the HD domain in deepteal. (E) A3B-CD2 bound to DNA coloured by loop (as in B). A3G shown in (F) “dumbbell” and (G) “globular” forms. (H) Apobec1 dimerization with catalytic domain in wheat and red, with HD domain in sand and green. Dimerization of (I) A3A and (J) Apobec2 shown in wheat and deepteal. (K) A3H dimerization in wheat and sand with dsRNA coloured in purple and hot pink. Structures are from (A-B) 5W1C [275], (D,H) 6X91 [230], (E) 5TD5 [252], (F) 6P40 [234], (G) 8EDJ [267], (I) 4XXO [248], (J) 2NYT [198], and (K) 5W3V [235].
Figure 18.
AID/APOBEC family deaminases base structure. AID/APOBEC structures with cartoons coloured by secondary structure, with skyblue coordinating residues, orange proton shuttle, and purple substrates. AID/APOBEC fold shown with an example of (A) AID as a cartoon, as well as (B) surface structure with light green loop 1 highlighting the gatekeeper residue in forest green, pink loop 3, red loop 5, and blue loop 7. (C) AID cartoon shown with different α7 positions in deepteal covering the assistant patch and blue by the β-sheet. (D) APOBEC1 cartoon with the HD domain in deepteal. (E) A3B-CD2 bound to DNA coloured by loop (as in B). A3G shown in (F) “dumbbell” and (G) “globular” forms. (H) Apobec1 dimerization with catalytic domain in wheat and red, with HD domain in sand and green. Dimerization of (I) A3A and (J) Apobec2 shown in wheat and deepteal. (K) A3H dimerization in wheat and sand with dsRNA coloured in purple and hot pink. Structures are from (A-B) 5W1C [275], (D,H) 6X91 [230], (E) 5TD5 [252], (F) 6P40 [234], (G) 8EDJ [267], (I) 4XXO [248], (J) 2NYT [198], and (K) 5W3V [235].

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



