Submitted:
21 September 2025
Posted:
23 September 2025
You are already at the latest version
Abstract
Abstract
The OX40 and OX40L signaling axis represents a key costimulatory pathway in the TNF and TNFR superfamilies. In tumor immunotherapy, OX40L binds to OX40 on activated T cells to promote T cell expansion and cytokine production while inhibiting the inhibitory activity of Tregs and amplifying antitumor immune responses. Clinical trials have shown that OX40 agonists inhibit tumor progression and induce long-lasting systemic immune memory. Several OX40 agonist-based antibodies are currently in early phase I/II clinical studies and have shown good safety and tolerability and great therapeutic potential. This article reviews the OX40/OX40L pathway-related molecular structure, signaling mechanism, regulatory role in the tumor microenvironment and targeted therapy strategies. This article also summarizes some clinical trials using OX40 agonists, analyzes the existing problems and limitations of current therapies, proposes solutions to solve them, and proposes future development directions.
Keywords:
OX40
; OX40L
; tumor immunity
; targeted therapy
1. Introduction
In the field of oncology, immunotherapy has rapidly evolved into a paradigm-shifting therapeutic modality that potentiates host antitumor immunity to eradicate malignant cells within the tumor microenvironment [1]. Although surgery, radiotherapy, and chemotherapy remain the cornerstones of oncologic management, their therapeutic efficacy is often restricted in advanced or relapsed/refractory malignancies and is accompanied by considerable toxicity. In recent years, immunotherapy has achieved precise and durable tumor cell elimination by reinvigorating host immunity, thereby redefining the therapeutic paradigm for numerous solid and hematologic neoplasms [2,3]. Among the immunotherapeutic strategies, the OX40/OX40L costimulatory axis is pivotal in initiating and amplifying immune responses and has become a focal point of translational research. Immune checkpoints encompass stimulatory or inhibitory signals mediated by membrane-bound receptor–ligand pairs, intracellular enzymes, or secreted molecules that preserve the dynamic equilibrium between pathogen clearance and prevention of autoimmunity [4,5]. Tumors exploit this regulatory circuitry by upregulating checkpoint molecules, resulting in immune escape [6,7]. Checkpoint inhibition, as illustrated by monoclonal antibodies directed against programmed cell death protein-1/programmed death-ligand 1 (PD-1/PD-L1) and cytotoxic T-lymphocyte antigen-4 (CTLA-4), has markedly extended overall survival among individuals with advanced-stage malignancies [8]. Nevertheless, many tumors remain refractory owing to scarce T cell infiltration, underscoring the imperative for innovative strategies to reactivate antitumor immunity. The OX40/OX40L costimulatory axis delivers signals complementary to the inhibitory checkpoints. Upon T-cell receptor (TCR) engagement, OX40 is transcriptionally upregulated; its ligation to OX40L orchestrates multiple signaling cascades that enhance proliferation, cytokine secretion, and anti-apoptotic signaling in both CD4+ and CD8+ T cells, thereby fostering memory T-cell generation and mitigating tumor recurrence [9,10,11]. OX40 signaling attenuates Treg-mediated immunosuppression and modulates the functional programs of B cells, dendritic cells, tumor-associated macrophages, and natural killer cells to counteract tumor progression and reprogram the immunosuppressive microenvironment. By employing genetic engineering to control OX40L expression, investigators can substantially enhance the antitumor potency of both conventional T lymphocytes and chimeric antigen receptor–modified T (CAR-T) cells. Several OX40 agonist antibodies have been developed and have demonstrated promising activity in experimental animal systems and initial human studies [12]. However, there is no unified standard for the optimal timing and dose of administration, and the best combination of treatments has not been found. Studies on the same dosing strategy in different patients are lacking. This results in highly variable dosing patterns in clinical trials, which are difficult to compare.
2. Biological Characteristics and Functions of OX40/OX40L
OX40, a glycoprotein of the type I transmembrane class, forms part of the TNFR superfamily and has an estimated molecular mass of 48–50 kDa. The encoding gene spans roughly 1.4 kb and resides on chromosome 1, clustered with other TNFR members at 1p36 [13]. The initial identification of OX40 occurred on activated CD4+ T cells in mice and was subsequently detected in human cells. Current evidence indicates that OX40 is broadly expressed on activated CD4+ and CD8+ T cells and is detectable in dendritic cells (DC), natural killer (NK) cells, and epithelial cells [14,15].OX40L is classified within the TNF superfamily and functions as a type II transmembrane glycoprotein. OX40L spans approximately 1 kb, is tightly linked to TNFSF6, and is located on chromosome 1q25. OX40L is a 183 amino acids polypeptide with a molecular mass of approximately 34–40 kDa. It is mainly expressed in professional antigen-presenting cells and is present in tissues including the heart, bone, and lung. Activated T cells additionally express OX40L at low levels [16].
The OX40/OX40L axis promotes dendritic cell maturation and differentiation, enhances intercellular adhesion, amplifies T cell function, facilitates helper T cell (Th) polarization, and sustains T cell activity and persistence [10,17]. Professional antigen-presenting cells (APC) provide the primary signal via the major histocompatibility complex–peptide engagement with the T cell receptor (TCR) and the second signal through B7–CD28 interactions. Activated T cells upregulate OX40 and TNFSF5 (CD40L), which engages TNFRSF5 (CD40) on APC, increasing APC OX40L expression and modulating the axis. OX40/OX40L action proceeds sequentially: (a) in lymph nodes, guided by this signal, CD4+ T cells that have undergone activation relocate into B-cell follicles, where they facilitate germinal-center development and antibody secretion; (b) OX40-mediated co-stimulation licenses activated CD4+ T lymphocytes to egress into the circulation and subsequently home to sites of inflammation; (c) tissue-derived OX40L+ APC mediates local inflammatory effects of CD4+ T cells in peripheral tissues [18,19,20,21].
3. Overview of Signaling Pathways
The core mechanism of OX40/OX40L signaling involves sequential kinase cascades and transcription factor activation, ultimately modulating T cells by regulating the expression of genes associated with cell survival, metabolism, and differentiation. Figure 1 illustrates the signaling pathway of OX40/OX40L.
3.1. PI3K/AKT Pathway
Recruits tumor necrosis factor receptor-associated factor 2 and 5 (TRAF2/5) adaptor proteins following OX40 engagement, which initiates a signaling cascade. This process activates phosphatidylinositol 3-kinase (PI3K) and subsequently induces phosphorylation of protein kinase B (AKT). Activated AKT significantly enhances T cell survival and amplifies effector responses through the transcriptional repression of pro-apoptotic factors Bim and Bad, coupled with the upregulation of anti-apoptotic proteins Bcl-2 and Bcl-xL [22]. Through its activation of mechanistic target of rapamycin complex 1, AKT promotes metabolic reprogramming and enhances the proliferation of antigen-specific T-cell clones [23].
3.2. The NF-κB Pathway
OX40 signaling activates both the canonical and noncanonical nuclear factor kappa B (NF-κB) pathways through TRAF-dependent mechanisms. Within the canonical cascade, phosphorylation of NF-κB inhibitory protein α (IκBα) by the IκB kinase complex liberates NF-κB heterodimers, which then migrate into the nucleus to initiate the expression of proinflammatory mediators including interleukin-2 (IL-2) and interferon-γ (IFN-γ) [24]. Activation of the RelB–p52 heterodimer in the noncanonical cascade depends upon NF-κB-inducing kinase, thereby contributing to memory T cell formation [25,26].
3.3. MAPK Pathway
Ligation of OX40 to OX40L triggers simultaneous activation of p38 mitogen-activated protein kinase (p38 MAPK) and c-Jun N-terminal kinase, leading to enhanced activator protein 1 transcriptional activity. p38 MAPK not only stabilizes mRNA encoding type 2 helper T cell (Th2) cytokines such as interleukin-4 and interleukin-5, but also phosphorylates MAPK-interacting kinase 1 to facilitate eukaryotic initiation factor 4E-mediated protein translation, thereby promoting the differentiation of both Th2 and type 17 helper T cell (Th17) subsets [19,27].
3.4. Calcium Ion Signaling and PKC Activation
Upon OX40 engagement, phospholipase C is activated and cleaves phosphatidylinositol-4,5-bisphosphate into inositol-1,4,5-trisphosphate and diacylglycerol. Inositol-1,4,5-trisphosphate triggers endoplasmic reticulum Ca2+ release, elevates intracellular free Ca2+ levels, activates calcineurin, and consequently drives the nuclear import of nuclear factor of activated T cells [28,29]. Diacylglycerol subsequently triggers protein kinase C (PKC), particularly PKCβ2, which enhances NF-κB signaling through phosphorylation of IκBα and facilitates T cell migration to inflammatory sites [30].
4. Role of OX40 Pathway in Tumor Immunity
4.1. Enhanced T Cell Activity and Persistence
OX40/OX40L exerts direct stimulatory effects on stimulated CD4+ and CD8+ T cells, orchestrating activation, multiplication and functional persistence via convergent signaling cascades [31,32]. Through canonical NF-κB and PI3K–Akt modules, the axis amplifies TCR-mediated activation while upregulating anti-apoptotic Bcl-2 family proteins, thereby preventing activation-induced cell death and prolonging T cell viability [33,34]. OX40L potentiates CD8+ cytotoxic T lymphocyte effector functions by inducing IL-2 and IFN-γ secretion, which augments tumor-antigen recognition and clonal burst size [35,36]. Metabolic reprogramming within the tumor microenvironment (TME) is evidenced by elevated glycolytic flux and oxidative phosphorylation, enhanced mitochondrial biogenesis, improved respiratory efficiency, diminished reactive oxygen species burden and delayed exhaustion [37,38]. This axis preserves metabolic homeostasis and proliferative capacity through upregulation of amino-acid transporters and increased import of essential substrates [39]. Furthermore, OX40L mitigates the suppressive effects imposed by PD-1/PD-L1, CTLA-4 and T cell immunoglobulin and mucin-domain containing-3 (TIM-3), thereby antagonizing these inhibitory checkpoints. it mitigates PD-1-driven exhaustion, attenuates Treg-mediated suppression and promotes secretion of granzyme B and IFN-γ, thereby potentiating cytotoxicity and restoring effector T cell competence [40,41,42]. Positive feedback loops established via upregulated costimulatory receptors further lower the activation threshold [43]. OX40L also programs transcriptional networks that promote the conversion of effector T cells into long-lived central memory T and stem-cell-like memory T subsets. These memory subsets demonstrate superior self-renewal, long-term persistence, rapid clonal expansion and robust effector cytokine production upon antigen re-encounter, thereby sustaining durable immune surveillance [44,45]. Experimental data demonstrate that concurrent OX40L agonism and PD-1 pathway inhibition significantly augments intratumoral memory T cell frequency and prolongs antitumor immunity [46].
4.2. Regulates the Action of Immune Cells Against Tumors
4.2.1. B Cells
B cells engage T cells via OX40L to orchestrate follicular helper T cell (Tfh) differentiation, thereby supporting germinal center reactions and high-affinity antibody generation [18,47]. In the unique microenvironment of B cell malignancies such as follicular lymphoma, the OX40 axis undergoes functional inversion; by attenuating Tfh-mediated support to neoplastic B cells, it indirectly restrains tumor growth [48]. OX40 signaling also reprograms tumor-infiltrating B cells, particularly immunosuppressive regulatory B cells. These Bregs frequently exert protumoral effects [49], yet OX40 activation skews B cell differentiation, selectively reducing intra-tumoral Breg frequency and relieving immunosuppression while potentiating antitumor humoral immunity [50,51]. Beyond this, the axis markedly enhances B cell activation and plasma cell transition, augments tumor-specific antibody production [52], upregulates the costimulatory molecule CD40 to strengthen B cell–T cell crosstalk, and amplifies T cell activation and expansion. Additionally, it tunes the metabolic state of B cells toward an antitumor phenotype [53].
4.2.2. Dendritic Cells
DCs are indispensable for initiating and sustaining antitumor immunity and constitute a principal cellular reservoir of OX40L.By enhancing antigen presentation and T cell priming, OX40L-expressing DCs engage naïve T cells within lymph nodes and deliver potent costimulatory signals that drive antitumor responses [54]. Within the TME, metabolic perturbations such as hypoxia, lactate accumulation, and immunosuppressive cues frequently impair DC antigen presentation [55]. The OX40/OX40L axis restores DC functionality and re-establishes their capacity to activate CD8+ T cells through multifaceted mechanisms [56]. OX40 signaling activates the NF-κB pathway in DCs, partially reversing metabolic suppression and promoting proinflammatory cytokine secretion [57]. Additionally, this signaling cascade fosters DC maturation and migratory competence, facilitating efficient capture and presentation of tumor antigens [58,59]. OX40 engagement further upregulates costimulatory molecules on DCs, thereby intensifying DC–T cell interactions and amplifying T cell activation and proliferation. Moreover, OX40 signaling reprograms the metabolic state of DCs to favor proinflammatory immune responses [16]. Batf3-derived dendritic cells leverage OX40L together with ancillary cues to amplify antitumor T cell activity under PD-1/PD-L1 checkpoint inhibition, rendering them essential for optimal therapeutic efficacy [60].
4.2.3. Natural Killer Cells
As an essential constituent of innate immunity, NK cells exert a critical antineoplastic function. Studies have shown that activation of OX40 signaling promotes NK cell activation and proliferation, thereby enhancing the release of IFN-γ and TNF-α, key proinflammatory mediators [61]. The upregulation of activation receptors on NK cells augments their tumoricidal function, a process augmented by this signaling cascade [62]. Furthermore, OX40 signaling improves NK cell survival within the tumor microenvironment by modulating their metabolic state [63]. In the experiment, the combination of an OX40 agonist with chemotherapeutic agents induces a marked increase in intratumoral NK cell density, augments their IFN-γ production, enhances NK cell antitumor activity, and significantly suppresses tumor growth [64,65].
4.2.4. Tumor-Associated Macrophages
Within neoplastic tissues, tumor-associated macrophages (TAMs) exhibit pronounced phenotypic plasticity, and the OX40/OX40L signaling axis markedly influences their polarization state and functional properties [66]. Studies indicate that this pathway can direct TAMs toward either the proinflammatory M1 or the anti-inflammatory M2 phenotype in response to microenvironmental cues, and that manipulating TAM polarization through this axis enhances their antitumor activity [67]. Upon OX40 activation, TAMs are skewed toward the M1 phenotype, resulting in increased secretion of the proinflammatory cytokines IL-12 and TNF-α while suppressing M2-associated immunosuppressive functions. This reprogramming is primarily mediated through metabolic rewiring, which shifts TAMs from an immunosuppressive to an antitumor state. Concurrently, OX40 signaling augments TAM antigen uptake and presentation, indirectly priming the adaptive immunity. Upregulation of costimulatory molecules in tumor-associated macrophages through this pathway can enhance their interaction with T cells [68,69]. This highlights the essential function of the OX40/OX40L pathway in reprogramming immunosuppressive macrophages in the tumor microenvironment [70,71].
5. Targeted Therapy Strategies
5.1. Agonist Therapy
OX40 agonists are immunostimulatory monoclonal antibodies or derivatives that target OX40 and belong to the TNFR family. By binding to OX40 on T cells, these agonists mimic or potentiate the signal delivered by the natural ligand OX40L, thereby amplifying antitumor immunity [72]. Stimulation of OX40 with agonist antibodies initiates NF-κB signaling via tumor necrosis factor receptor–associated factors. This activation potentiates T cell expansion, longevity, and effector functions, including cytokine secretion and a Th1-polarized response marked by IFN-γ production [32]. During antitumor immune responses, OX40 stimulation augments TCR signal duration. This effect promotes the enrichment of CD8+ T cells within tumors along with the clonal expansion of high-affinity, tumor antigen-specific populations [73]. Notably, OX40 agonists do not directly abrogate regulatory T cell suppression; rather, they indirectly expand Treg and conventional T cell (Tconv) populations by enhancing IL-2 production from Tconv cells [74]. Thus, the core mechanism is driven primarily by the stimulation and clonal proliferation of T cells, not by the direct suppression of immunosuppressive functions. OX40 agonistic antibodies are currently available as full-length IgG, Fab/scFv fragments, Fc fusion proteins, multivalent/bispecific constructs, and multimers. In clinical studies, fully human IgG1 monoclonal antibodies constitute the dominant form, representing intact antibodies rather than fragments, fusion proteins, or multivalent constructs [75,76]. Clinically, these agents are principally investigated for cancer immunotherapy; nevertheless, studies have also demonstrated their efficacy against cutaneous inflammation and leukemia [77,78]. Multiple major pharmaceutical companies are developing OX40 agonists, most of which have entered clinical trials (Table 1).
5.2. Genetic Engineering and Cell Therapy
Genetic engineering can enhance T cell therapy by modulating OX40L expression and serves to optimize the costimulatory function of the OX40/OX40L axis, fine-tune T cell subset activity, and integrate with gene editing technologies [79]. Conceptually, OX40L overexpression can be delivered into antigen-presenting cells or engineered T cells via viral or nonviral vectors, thereby intensifying OX40 engagement [80,81]. Autocrine or paracrine OX40L signaling subsequently cooperates to activate T cells and augment antitumor activity [82]. Targeted delivery systems can further restrict OX40L gene transfer to APCs or T cells within the tumor microenvironment, locally reinforcing the OX40/OX40L signal [83]. The bidirectional regulatory capacity of OX40 signaling also permits precise control of T cell subset balance. OX40L overexpression expands CD4+ and CD8+ effector T cells, diminishes Treg suppressive function, and prolongs systemic T cell persistence by fostering memory T cell formation, thereby conferring long-term immunological protection [31,84]. Chimeric antigen receptor T cells are genetically engineered autologous or allogeneic T cell products that express synthetic receptors, endowing them with the capacity to recognize, activate, expand, and eradicate tumors [85]. The specific interaction between OX40 and OX40L optimizes CAR-T cell functionality across multiple dimensions, including cell survival, functional differentiation, and TME modulation [86]. Activation of the OX40/OX40L axis markedly enhances CAR-T cell survival and proliferation by triggering downstream NF-κB and PI3K-Akt signaling, suppressing pro-apoptotic proteins, and thus extending persistence and antitumor efficacy within the TME [87]. In addition, OX40 costimulation can promote the transformation of CAR-T cells to an effector cell phenotype, and down-regulate the expression of exhaustion markers TIM-3 and PD-1, which effectively reverses the dysfunction of T cells and improves the killing ability of solid tumors [88]. By reprogramming transcription factors, the OX40/OX40L axis orchestrates the differentiation of CAR-T cells into central memory T cells or stem cell–like memory T cells populations. This process fosters long-lasting antitumor immunity, enhances their sustained persistence in vivo [89]. OX40 signaling also ameliorates the immunosuppressive TME via a dual mechanism: it inhibits the production of IL-10 and transforming growth factor-β by Tregs. Concurrently, it potentiates the expression of IFN-γ and IL-2 in the CAR-T cells. This altered cytokine milieu culminates in the engagement of innate immune cells; key populations including macrophages and dendritic cells are recruited and activated to mount a synergistic attack against tumors [90,91,92]. Evidence further indicates that OX40 signaling enhances CAR-T cell vascular penetration and tumor infiltration efficiency [88]. The cooperative action of OX40L therefore offers additional solutions to overcome limitations in CAR-T cell therapy, including insufficient persistence, TME-mediated suppression, and toxicity risks; nevertheless, optimal activation strategies and dosing regimens require further investigation. Figure 2 illustrates the rationale for gene therapy and CAR-T cell therapies.
5.3. Combination Treatment Strategies
5.3.1. Combined with Immune Checkpoint Inhibitors
Although the OX40/OX40L signaling axis potentiates antitumor immunity, its efficacy is modulated by the complex tumor microenvironment. Tumor cells can secrete specific factors or alter metabolic states to impair OX40 signal transduction [92,93]. This has led to the development of combination therapies as a novel approach in cancer immunotherapy [94]. The inhibitors of PD-1 and CTLA-4 have been widely used in clinical practice and have achieved encouraging results; concurrent or sequential administration with OX40 agonists is expected to yield superior therapeutic efficacy [95,96]. Mechanistically, PD-1/PD-L1 blockade releases T cells from inhibitory signaling, yet multiple suppressive factors persist in some TMEs. The addition of OX40 agonists delivers a positive costimulatory signal, thereby achieving a dual effect of stimulation and inhibition [40]. The PD-1 immune checkpoint attenuates T cell activation via the suppression of TCR-induced phosphotyrosine signaling, whereas OX40 costimulation induces PI3K/AKT/mTOR activation without CD28 involvement, thereby rescuing T cell proliferative potential. A synergistic blockade of OX40 and PD-1 potently enhances CD8+ T cell functionality, marked by increased secretion of Granzyme B and IFN-γ while concurrently depleting Tregs in the tumor, thereby inducing significant immunomodulation [97].
5.3.2. Combination with Radiotherapy or Chemotherapy
Radiotherapy produces synergistic antitumor activity with OX40 agonists through local tumor reduction and immunomodulation. Irradiation causes DNA damage that potentiates the presentation of tumor-associated antigens (TAAs) and damage-associated molecular patterns (DAMPs). As a result, DCs become activated and efficiently prime T cells [98]. Concurrently, radiotherapy upregulates major histocompatibility complex class I and costimulatory molecules on tumor cells, thereby improving T cell recognition and cytotoxicity [99]. OX40 agonists amplify the systemic antitumor immunity elicited by radiotherapy, potentiating the abscopal effect and suppressing the growth of distant metastases [100,101].
Chemotherapeutic agents further synergize with OX40 agonists by inducing immunogenic cell death, which liberates TAAs and DAMPs [102]. Clinical studies have shown that combining OX40 agonists with chemotherapy enhances antitumor responses, chemotherapy increases antigen availability and presentation, and OX40 signaling heightens T cell recognition of tumor antigens and reverses immune suppression within the tumor microenvironment [103]. Chemotherapy also reduces the frequency of myeloid-derived suppressor cells (MDSCs) and Tregs, and co-administration with OX40 agonists further alleviates TME immunosuppression [104]. Chemotherapy induces antigen release, radiotherapy enhances local immune activation, and OX40 agonists amplify systemic T cell responses. Chemotherapy and radiotherapy decrease Treg and MDSC populations, while OX40 agonists block Treg function and augment effector T cell activity.
6. Research Progress
6.1. Preclinical Studies
At present, the most extensively studied OX40L-targeted strategy is OX40 agonist therapy. Preclinical studies demonstrate that OX40 agonists, whether used as monotherapy or in combination, elicit robust antitumor activity in murine tumor models. Ma and colleagues reported that pancreatic ductal adenocarcinoma appears to evade immunity by inducing suppressive T cells. Compared with controls, mice receiving antiOX40 monotherapy exhibited improved survival; Coadministration of antiOX40 and antiPD-1 led to Treg depletion concurrent with an expansion in CD4+ and CD8+ T cell populations, resulting in complete tumor eradication [46]. In a bladder cancer model, an OX40 agonist antibody enhanced CpG-mediated antitumor activity and prolonged overall survival [105]. In colon cancer models, Zhang et al. showed that the OX40 agonist SHR-1806 markedly increased IFN-γ secretion, promoted antitumor T cell responses, and preserved both antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity to eliminate Tregs and achieve tumor regression [106]. In another colon cancer study, Jiang et al. employed the OX40 agonist antibody BGB-A445 and demonstrated that it activated T cells without compromising NK cell function, while antibody-dependent cellular cytotoxicity depleted Tregs in vitro and in vivo, resulting in superior immunostimulation and antitumor activity [17]. In melanoma models, an OX40 agonist antibody enhanced CD8+ T cell cytotoxicity, fostered tumor-specific memory formation, delayed tumor progression, and extended survival [93]. In a glioma study, combination therapy with an OX40 agonist elevated the proportion of Th1 CD4+ T lymphocytes, reversed intracerebral T cell exhaustion, decreased PD-1 expression, and augmented Th1-mediated antitumor immunity [107]. Guo and colleagues conducted the first study to combine antiOX40 with antiPD-1 in an ovarian cancer model; the regimen expanded CD4+ and CD8+ T cells, reduced Treg and MDSC numbers, induced a local immunostimulatory milieu, and markedly inhibited tumor development [108]. In mice with breast cancer, co-administration of Sm16 and antiOX40 elicited potent antitumor effects, attenuated tumor growth and metastasis, and reinforced resistance to rechallenge, accompanied by robust tumor-specific peripheral memory IFN-γ responses [109]. Collectively, these preclinical studies demonstrate the strong antitumor activity of OX40 agonist antibodies, providing a solid foundation for their clinical translation. The progress of related clinical investigations is presented in the following section.
6.2. Clinical Trials of OX40 Agonist Monotherapy in Tumor Intervention
Table 2 provides an overview of some clinical trials of OX40 agonist antibodies, both as monotherapy and in combination. The safety and efficacy of BAT6026(a humanized IgG1 monoclonal antibody)were assessed in a phase I study (NCT05105971) [110] involving individuals with advanced solid tumors. Escalating doses of 0.01 to 10 mg/kg were administered intravenously to 30 enrolled patients on day 1 of each 21-day cycle. During the expansion phase, a cohort of 6 participants was treated at a dose of 10 mg/kg. The antiOX40 agent demonstrated a favorable safety profile and was well tolerated. No dose-limiting toxicities were reported and the maximum tolerated dose was not attained in this trial. Regarding antitumor activity, neither complete nor partial responses were observed among participants. Stable disease (SD) was achieved in 10 subjects, yielding a disease control rate (DCR) of 38.5%.
To evaluate the safety and feasibility of neoadjuvant antiOX40 therapy, patients diagnosed with head and neck squamous cell carcinoma (HNSCC) received preoperative administration of an OX40 agonist antibody MEDI6469(a humanized IgG2a monoclonal antibody) in a clinical trial (NCT02274155) [111]. A total of 17 participants were treated with the antiOX40 agent at a dosage of 0.4 mg/kg on days 1, 3, and 5. The therapy exhibited a favorable safety profile and was well tolerated, with only grade 1 or 2 adverse events observed. After a median follow-up of 39 months, the overall survival (OS) and disease-free survival (DFS) rates were 82% and 71%, respectively. Immunological profiling indicated an elevation in CD4+ and CD8+ T cell counts after treatment. Furthermore, expansion of CD103+CD39+CD8+ tumor-infiltrating lymphocytes was noted in a subset of patients. This increase was correlated with restrained tumor progression and did not elevate the risk of postponing surgery.
The safety and antitumor activity of the OX40 agonist antibody MEDI0562(a humanized IgG1 monoclonal antibody) administered as a single agent were investigated in a phase I/II study (NCT02318394) [112] involving patients with advanced solid malignancies. The participants were administered intravenous infusions of the antibody biweekly, at dose levels escalating from 0.01 to 10 mg/kg. The therapy demonstrated a manageable safety profile and was well-tolerated. Among 55 evaluable subjects, an objective response rate (ORR) of 4% was observed, including two partial responses. Immunological analyses revealed a marked elevation in Ki67-positive CD4+ and CD8+ memory T cell populations after treatment. This immunostimulatory effect was concomitant with a decrease in OX40+Foxp3+ Tregs in the tumor microenvironment.
In patients presenting with advanced solid malignancies, the therapeutic efficacy of INCAGN01949(a human IgG4 monoclonal antibody) designed to activate OX40, was assessed in a phase I/II clinical study (NCT02923349) [113]. A dose-escalation study of the antibody as monotherapy was conducted over 14-day cycles, with doses ranging from 7 to 1400 mg. Among the 87 patients, the ORR was 1.5%, with one patient achieving a partial response. Twenty-three patients attained SD, with one patient maintaining SD for over six months. No safety concerns were identified with monotherapy; however, the treatment did not enhance T-cell proliferation or reduce the number of Tregs.
The safety, tolerability, and antitumor efficacy of the OX40 agonist antibody MOXR0916(a humanized IgG1 monoclonal antibody) were assessed in a phase I clinical trial (NCT02219724) [114] involving individuals with advanced solid malignancies. The participants were administered the antibody intravenously in a three-week cycle, with doses escalating from 0.2 to 1200 mg. A favorable safety and tolerability profile was observed, wherein most treatment-emergent adverse events were grade 1 or 2. Among the 172 treated participants, the ORR was 1.2%, which consisted of two partial responses. Immunological analyses demonstrated an elevation in CD8+ T cell numbers and increased cytokine concentrations in a subgroup of participants.
6.3. Combination Therapy Trials
Preliminary data from a phase I/II clinical study (NCT04215978) [115] involving subjects with advanced solid malignancies were disclosed at the annual conference of American Society of Clinical Oncology. The investigation assessed the safety and antitumor activity of BGB A445(a humanized IgG1 monoclonal antibody), administered both as a single-agent therapy and in combination with an anti PD 1 antibody. The data revealed that no severe adverse events occurred in the enrolled participants. Therapeutic interventions were generally well tolerated, and no dose limiting toxicities were detected. Among the 50 patients receiving monotherapy, the ORR was 4%, with two partial responses. In the combination therapy cohort, 30 patients received BGB-A445 plus 200 mg Tislelizumab (a humanized antiPD-1 IgG4 monoclonal antibody), resulting in an ORR of 23% with seven partial responses.
The clinical efficacy of the OX40 agonist antibody GSK3174998(a humanized IgG1 monoclonal antibody) was examined in a phase I study (NCT02528357) [116] involving patients with advanced solid tumors, evaluating both single-agent and combination regimens with immune checkpoint blocking agents. NO clear dose-toxicity relationship was identified, and the study did not reach the maximum tolerated dose. Adverse effects were predominantly grade 1 or 2 in severity. In Part 1 (monotherapy), which included 45 patients, the ORR was 0% and the DCR was 9%. In Part 2 (combination therapy), 96 patients received GSK3174998 in combination with 200 mg pembrolizumab (a humanized antiPD-1 IgG4 monoclonal antibody), achieving an ORR of 8%, which included two complete responses and four partial responses.
A phase I/II clinical study (NCT02737475) [117] evaluated the OX40 agonist antibody BMS986178(a fully human monoclonal antibody) as a monotherapy and in combination with other agents in patients with advanced solid tumors. The maximum tolerated dose was not achieved. The treatment demonstrated a favorable safety profile; most adverse events were grade 1 or 2 in severity, and grade 3 or 4 events occurred in a minority of patients. The ORR was 0% for monotherapy and ranged from 0% to 13% across combination therapy cohorts.
The safety and efficacy of a combination regimen containing the OX40 agonist antibody PF-04518600(a humanized IgG2 monoclonal antibody), nivolumab(a fully human antiPD-1 IgG4 monoclonal antibody), and ipilimumab (a human antiCTLA-4 IgG1 monoclonal antibody) were evaluated in a phase I trial (NCT02315600) [118] involving patients with advanced solid tumors. The trial employed a dose escalation design. The patient tolerated the combination therapy well. Grade 3-4 adverse events occurred in 28 of the 57 assessable patients. Partial responses were achieved in two melanoma patients, and stable disease was documented in 18 patients. A DCR of 35.1% was observed across all the dose cohorts.
A phase I study (NCT02317747) [119] assessed a triple therapy regimen comprising the OX40 agonist PF-04518600 in combination with avelumab (a fully human antiPD-L1 IgG1 monoclonal antibody) and utomilumab(a fully human anti4-1BB agonist IgG2 monoclonal antibody) in individuals with gynecologic cancers. This combination therapy resulted in a favorable safety profile. Only grade 1 or 2 adverse events were documented, with no dose-limiting toxicities, and the regimen was generally well tolerated. DCR was 78% in patients receiving avelumab and utomilumab alone. In a group of nine subjects administered avelumab plus utomilumab, an ORR of 11% was achieved, including one partial response. Among the 35 participants treated with the triple antibody combination, the ORR was 2.9% with a DCR of 37.1%.
7. Discussion
Although OX40/OX40L targeted therapy has shown great potential in preclinical experiments. However, clinical trials have not shown the same efficacy. To date, no OX40 agonist clinical trials have successfully progressed beyond phase II. And there are many problems and research gaps in clinical trials. OX40 agonist monotherapy has limited efficacy and low objective response rates, and although its combination with other immune checkpoint inhibitors improves response rates, it does not show a significant advantage. In addition, inadequate activation of receptor signaling can affect its efficacy. The optimized downstream signal of the OX40 signaling pathway requires multivalent activation, while the traditional bivalent OX40 agonist cannot effectively induce receptor clustering, and the effect of activating T cells is not good, so it cannot achieve the expected antitumor effect [122,123,124]. In the future, it is necessary to develop new, more efficient agonist molecular structures that can activate signaling pathways at lower doses. There is a mismatch between biological activity and clinical benefit, with receptor and T-cell activation signals detected in the peripheral blood and tumors in multiple trials, but without inhibition of tumor growth or survival benefit. This is because OX40 expression is upregulated approximately 3 days after the full activation of T cells. However, many trials use biweekly or triweekly dosing regimens, which may miss the optimal window for signal costimulation [125,126]. To this end, we can match the time window of T cell activation by precise dosing times rather than fixed long intervals. OX40 agonists have had limited clinical benefits as monotherapy, but combining them with other immune stimuli, such as radiotherapy and chemotherapy, can create a larger activation window and optimize drug delivery. In addition, the drug escalation strategy was used inappropriately, and the dosing schedule did not match the immunokinetics. In clinical trials, researchers tend to push agonists to the maximum tolerated dose. After signal activation, T cells expand rapidly and easily enter the activation exhaustion state, which reduces the therapeutic potential [127]. We should avoid sustained high-dose therapy in favor of immunodynamics-based dose exploration to identify the lowest effective dose that activates T cells while avoiding exhaustion. The optimal combination with other treatments has not been determined. In clinical trials, OX40 agonists are often combined with immune checkpoint inhibitors or other drugs; however, the combination regimen is not optimized based on OX40 biological mechanisms, which may also lead to poor synergistic effects [128]. To explore the optimal therapeutic combination, combination design can be carried out according to the mechanism, and the timing and sequence of administration can be optimized to achieve the best efficacy.
There are differences between patients; most of the patients participating in phase II clinical trials have advanced solid tumors, patients are not screened, and only a few patients may respond to treatment [129]. No study to date has screened patients with high OX40 expression for OX40 agonist therapy; therefore, this study could be performed to maximize efficacy. Clinical trials have shown that OX40 agonists combined with chemoradiotherapy have preliminary efficacy [100,104], but there are no studies on the combination of the three, which may lead to better efficacy.
8. Conclusion
OX40 agonist antibodies have been used to optimize the host antitumor immune response at multiple levels, inhibit tumor growth, and prolong patient survival. Although the safety window is wide, toxicity limits dose escalation. Most trials have not yet reached the maximum tolerated dose, but different degrees of adverse reactions have been reported. Therefore, further studies are needed to expand the sample size, adjust the dosing regimen, and prolong observation times. In the future, OX40 agonist therapy will shift from simple immune activation to precise, safe, and efficient activation. Molecular designs are moving toward multispecificity, and delivery strategies will evolve from systemic intravenous administration to more effective targeted delivery systems designed to limit costimulatory signals in the tumor microenvironment while minimizing systemic toxicity.
Author Contributions
Zixuan Yuan: Conceptualization, Methodology, Data curation, formal analysis, writing – original draft, writing – review and editing, and visualization. Yutong Pan: Data curation, formal analysis, and visualization. Aihua Gong: Conceptualization, Writing – review and editing, supervision. All authors have and approved the final manuscript.
Data Availability Statement
All experimental data analyzed in this study were obtained from publicly available articles and trial registries accessed via PubMed and ClinicalTrials.gov. The original datasets are listed in the reference list. No new data were generated or deposited in connection with the study.
Acknowledgments
I sincerely thank those who guided and helped me in the writing of this review. Their professional guidance meant a lot to me, and it was only because of your cooperation that this task was made possible. Thank you for your contributions to this article. we thank to BioRender for the license (CC-BY 4.0).
Conflicts of Interest
The authors did not receive support from any organization for the submitted work. The authors have no competing interests to declare relevant to the content of this article.
References
- Chen, Q.; Liu, Y.; Chen, Q.; Li, M.; Xu, L.; Lin, B.; et al. DNA Nanostructures: Advancing Cancer Immunotherapy. Small (Weinheim an der Bergstrasse, Germany). 2024, 20, e2405231. [Google Scholar] [CrossRef]
- Schaller, J.; Agudo, J. Metastatic Colonization: Escaping Immune Surveillance. Cancers 2020, 12. [Google Scholar] [CrossRef]
- Deng, H.; Zhang, Z. The application of nanotechnology in immune checkpoint blockade for cancer treatment. Journal of controlled release : official journal of the Controlled Release Society. 2018, 290, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer. 2012, 12, 252–64. [Google Scholar] [CrossRef] [PubMed]
- Najafi-Hajivar, S.; Zakeri-Milani, P.; Mohammadi, H.; Niazi, M.; Soleymani-Goloujeh, M.; Baradaran, B.; et al. Overview on experimental models of interactions between nanoparticles and the immune system. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2016, 83, 1365–78. [Google Scholar]
- Fu, J.; Yu, A.; Xiao, X.; Tang, J.; Zu, X.; Chen, W.; et al. CD4(+) T cell exhaustion leads to adoptive transfer therapy failure which can be prevented by immune checkpoint blockade. American journal of cancer research. 2020, 10, 4234–50. [Google Scholar]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018, 24, 4175–86. [Google Scholar] [CrossRef]
- Kim, A.; Lee, E.J.; Han, J.H.; Chung, H.S. Caryophylli Cortex Suppress PD-L1 Expression in Cancer Cells and Potentiates Anti-Tumor Immunity in a Humanized PD-1/PD-L1 Knock-In MC-38 Colon Cancer Mouse Model. Nutrients 2024, 16. [Google Scholar] [CrossRef]
- Yang, Z.; Hai, L.; Chen, X.; Wu, S.; Lv, Y.; Cui, D.; et al. OX40 ligand promotes follicular helper T cell differentiation and development in mice with immune thrombocytopenia. Journal of Zhejiang University Science B. 2025, 26, 240–53. [Google Scholar] [CrossRef]
- Sato, A.; Nagai, H.; Suzuki, A.; Ito, A.; Matsuyama, S.; Shibui, N.; et al. Generation and characterization of OX40-ligand fusion protein that agonizes OX40 on T-Lymphocytes. Frontiers in immunology. 2024, 15, 1473815. [Google Scholar] [CrossRef]
- Alharshawi, K.; Marinelarena, A.; Kumar, P.; El-Sayed, O.; Bhattacharya, P.; Sun, Z.; et al. PKC-ѳ is dispensable for OX40L-induced TCR-independent Treg proliferation but contributes by enabling IL-2 production from effector T-cells. Scientific reports. 2017, 7, 6594. [Google Scholar] [CrossRef]
- Oberst, M.D.; Auge, C.; Morris, C.; Kentner, S.; Mulgrew, K.; McGlinchey, K.; et al. Potent Immune Modulation by MEDI6383, an Engineered Human OX40 Ligand IgG4P Fc Fusion Protein. Molecular cancer therapeutics. 2018, 17, 1024–38. [Google Scholar] [CrossRef]
- Sadrolashrafi, K.; Guo, L.; Kikuchi, R.; Hao, A.; Yamamoto, R.K.; Tolson, H.C.; et al. An OX-Tra’Ordinary Tale: The Role of OX40 and OX40L in Atopic Dermatitis. Cells 2024, 13. [Google Scholar] [CrossRef]
- Fouladi, S.; Masjedi, M.; Ghasemi, R. ; MGH; Eskandari, N. The In Vitro Impact of Glycyrrhizic Acid on CD4+ T Lymphocytes through OX40 Receptor in the Patients with Allergic Rhinitis. Inflammation. 2018, 41, 1690–701. [Google Scholar] [CrossRef] [PubMed]
- Imianowski, C.J.; Kuo, P.; Whiteside, S.K.; von Linde, T.; Wesolowski, A.J.; Conti, A.G.; et al. IFNgamma Production by Functionally Reprogrammed Tregs Promotes Antitumor Efficacy of OX40/CD137 Bispecific Agonist Therapy. Cancer research communications. 2024, 4, 2045–57. [Google Scholar] [CrossRef] [PubMed]
- Gajdasik, D.W.; Gaspal, F.; Halford, E.E.; Fiancette, R.; Dutton, E.E.; Willis, C.; et al. Th1 responses in vivo require cell-specific provision of OX40L dictated by environmental cues. Nature communications. 2020, 11, 3421. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhang, T.; Deng, M.; Jin, W.; Hong, Y.; Chen, X.; et al. BGB-A445, a novel non-ligand-blocking agonistic anti-OX40 antibody, exhibits superior immune activation and antitumor effects in preclinical models. Frontiers of medicine. 2023, 17, 1170–85. [Google Scholar] [CrossRef]
- Tahiliani, V.; Hutchinson, T.E.; Abboud, G.; Croft, M.; Salek-Ardakani, S. OX40 Cooperates with ICOS To Amplify Follicular Th Cell Development and Germinal Center Reactions during Infection. Journal of immunology (Baltimore, Md : 1950). 2017, 198, 218–28. [Google Scholar] [CrossRef]
- Huang, L.; Wang, M.; Yan, Y.; Gu, W.; Zhang, X.; Tan, J.; et al. OX40L induces helper T cell differentiation during cell immunity of asthma through PI3K/AKT and P38 MAPK signaling pathway. Journal of translational medicine. 2018, 16, 74. [Google Scholar] [CrossRef]
- Liu, B.; Yu, H.; Sun, G.; Sun, X.; Jin, H.; Zhang, C.; et al. OX40 promotes obesity-induced adipose inflammation and insulin resistance. Cellular and molecular life sciences : CMLS. 2017, 74, 3827–40. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Croft, M.; Geng, B.; Rynkiewicz, N.; Lucchesi, D.; Peakman, M.; et al. The role of OX40 ligand/OX40 axis signalling in atopic dermatitis. The British journal of dermatology. 2024, 191, 488–96. [Google Scholar] [CrossRef]
- So, T.; Choi, H.; Croft, M. OX40 complexes with phosphoinositide 3-kinase and protein kinase B (PKB) to augment TCR-dependent PKB signaling. Journal of immunology (Baltimore, Md : 1950). 2011, 186, 3547–55. [Google Scholar] [CrossRef]
- Croft, M.; So, T.; Duan, W.; Soroosh, P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunological reviews. 2009, 229, 173–91. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhang, C.; Sun, C.; Zhao, X.; Tian, D.; Shi, W.; et al. OX40 expression in neutrophils promotes hepatic ischemia/reperfusion injury. JCI insight. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Sun, X.; Li, W.; Liu, K.; Tian, D.; Dong, Y.; et al. Critical role of OX40 in the expansion and survival of CD4 T-cell-derived double-negative T cells. Cell death & disease. 2018, 9, 616. [Google Scholar]
- Kawamata, S.; Hori, T.; Imura, A.; Takaori-Kondo, A.; Uchiyama, T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. The Journal of biological chemistry. 1998, 273, 5808–14. [Google Scholar] [CrossRef]
- Mestas, J.; Crampton, S.P.; Hori, T.; Hughes, C.C. Endothelial cell co-stimulation through OX40 augments and prolongs T cell cytokine synthesis by stabilization of cytokine mRNA. International immunology. 2005, 17, 737–47. [Google Scholar] [CrossRef]
- Yan, J.; Wang, C.; Du, R.; Liu, P.; Chen, G. OX40-OX40 ligand interaction may activate phospholipase C signal transduction pathway in human umbilical vein endothelial cells. Chemico-biological interactions. 2009, 180, 460–4. [Google Scholar] [CrossRef]
- Lv, Y.W.; Chen, Y.; Lv, H.T.; Li, X.; Tang, Y.J.; Qian, W.G.; et al. Kawasaki disease OX40-OX40L axis acts as an upstream regulator of NFAT signaling pathway. Pediatric research. 2019, 85, 835–40. [Google Scholar] [CrossRef]
- Burgess, J.K.; Carlin, S.; Pack, R.A.; Arndt, G.M.; Au, W.W.; Johnson, P.R.; et al. Detection and characterization of OX40 ligand expression in human airway smooth muscle cells: a possible role in asthma? The Journal of allergy and clinical immunology. 2004, 113, 683–9. [Google Scholar] [CrossRef]
- Weinberg, A.D.; Rivera, M.M.; Prell, R.; Morris, A.; Ramstad, T.; Vetto, J.T.; et al. Engagement of the OX-40 receptor in vivo enhances antitumor immunity. Journal of immunology (Baltimore, Md : 1950). 2000, 164, 2160–9. [Google Scholar] [CrossRef]
- Kjaergaard, J.; Tanaka, J.; Kim, J.A.; Rothchild, K.; Weinberg, A.; Shu, S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer research. 2000, 60, 5514–21. [Google Scholar]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nature reviews Immunology. 2015, 15, 486–99. [Google Scholar] [CrossRef]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends in immunology. 2015, 36, 265–76. [Google Scholar] [CrossRef]
- Fromm, G.; de Silva, S.; Giffin, L.; Xu, X.; Rose, J.; Schreiber, T.H. Gp96-Ig/Costimulator (OX40L, ICOSL, or 4-1BBL) Combination Vaccine Improves T-cell Priming and Enhances Immunity, Memory, and Tumor Elimination. Cancer immunology research. 2016, 4, 766–78. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.M.; Alejo, A.; Avia, J.M.; Rodriguez-Martin, D.; Sanchez, C.; Alcami, A.; et al. Activation of OX40 and CD27 Costimulatory Signalling in Sheep through Recombinant Ovine Ligands. Vaccines 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jia, W.; Zhou, X.; Ma, Z.; Liu, J.; Lan, P. CBX4 suppresses CD8(+) T cell antitumor immunity by reprogramming glycolytic metabolism. Theranostics. 2024, 14, 3793–809. [Google Scholar] [CrossRef]
- Lin, M.C.; Moller, S.H.; Ho, P.C. Re-”Formate” T-cell Antitumor Responses. Cancer discovery. 2023, 13, 2507–9. [Google Scholar] [CrossRef]
- Ren, W.; Liu, G.; Yin, J.; Tan, B.; Wu, G.; Bazer, F.W.; et al. Amino-acid transporters in T-cell activation and differentiation. Cell death & disease. 2017, 8, e2655. [Google Scholar]
- Polesso, F.; Weinberg, A.D.; Moran, A.E. Late-Stage Tumor Regression after PD-L1 Blockade Plus a Concurrent OX40 Agonist. Cancer immunology research. 2019, 7, 269–81. [Google Scholar] [CrossRef]
- Yu, J.; Cui, J.; Zhang, X.; Xu, H.; Chen, Z.; Li, Y.; et al. The OX40-TRAF6 axis promotes CTLA-4 degradation to augment antitumor CD8(+) T-cell immunity. Cellular & molecular immunology. 2023, 20, 1445–56. [Google Scholar]
- Deng, Z.; Tian, Y.; Wang, J.; Xu, Y.; Liu, Z.; Xiao, Z.; et al. Enhanced Antitumor Immunity Through T Cell Activation with Optimized Tandem Double-OX40L mRNAs. International journal of nanomedicine. 2025, 20, 3607–21. [Google Scholar] [CrossRef] [PubMed]
- Akiba, H.; Oshima, H.; Takeda, K.; Atsuta, M.; Nakano, H.; Nakajima, A.; et al. CD28-independent costimulation of T cells by OX40 ligand and CD70 on activated B cells. Journal of immunology (Baltimore, Md : 1950). 1999, 162, 7058–66. [Google Scholar] [CrossRef] [PubMed]
- Luoma, A.M.; Suo, S.; Wang, Y.; Gunasti, L.; Porter, C.B.M.; Nabilsi, N.; et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell. 2022, 185, 2918–35. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, D.T.; Delpoux, A.; Wieland, D.; Huang, X.; Lai, C.Y.; Hofmann, M.; et al. Active Maintenance of T Cell Memory in Acute and Chronic Viral Infection Depends on Continuous Expression of FOXO1. Cell reports. 2018, 22, 3454–67. [Google Scholar] [CrossRef]
- Ma, Y.; Li, J.; Wang, H.; Chiu, Y.; Kingsley, C.V.; Fry, D.; et al. Combination of PD-1 Inhibitor and OX40 Agonist Induces Tumor Rejection and Immune Memory in Mouse Models of Pancreatic Cancer. Gastroenterology. 2020, 159, 306–19. [Google Scholar] [CrossRef]
- Cortini, A.; Ellinghaus, U.; Malik, T.H.; Cunninghame Graham, D.S.; Botto, M.; Vyse, T.J. B cell OX40L supports T follicular helper cell development and contributes to SLE pathogenesis. Annals of the rheumatic diseases. 2017, 76, 2095–103. [Google Scholar] [CrossRef]
- Zhao, J.; Li, L.; Feng, X.; Yin, H.; Fan, X.; Gao, C.; et al. Blockade of OX40/OX40L signaling using anti-OX40L alleviates murine lupus nephritis. European journal of immunology. 2024, 54, e2350915. [Google Scholar] [CrossRef]
- Zhang, X.; Xiao, X.; Lan, P.; Li, J.; Dou, Y.; Chen, W.; et al. OX40 Costimulation Inhibits Foxp3 Expression and Treg Induction via BATF3-Dependent and Independent Mechanisms. Cell reports. 2018, 24, 607–18. [Google Scholar] [CrossRef]
- Du, X.; Zhu, Y.; Lu, W.; Fu, N.; Wang, Q.; Shi, B. Regulation of the Function of T Follicular Helper Cells and B Cells in Type 1 Diabetes Mellitus by the OX40/OX40L Axis. The Journal of clinical endocrinology and metabolism. 2024, 109, 2823–30. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, L.; Sui, B.; Luo, Z.; Zhang, Y.; Wang, Y. Recombinant Rabies Virus Overexpressing OX40-Ligand Enhances Humoral Immune Responses by Increasing T Follicular Helper Cells and Germinal Center B Cells. Vaccines 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Chtanova, T.; Tangye, S.G.; Newton, R.; Frank, N.; Hodge, M.R.; Rolph, M.S.; et al. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. Journal of immunology (Baltimore, Md : 1950). 2004, 173, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Wei, Y.; Hu, B.; Liao, Y.; Wang, X.; Wan, W.H.; et al. c-Myc-driven glycolysis polarizes functional regulatory B cells that trigger pathogenic inflammatory responses. Signal transduction and targeted therapy. 2022, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Giladi, A.; Barboy, O.; Hamon, P.; Li, B.; Zada, M.; et al. The interaction of CD4(+) helper T cells with dendritic cells shapes the tumor microenvironment and immune checkpoint blockade response. Nature cancer. 2022, 3, 303–17. [Google Scholar] [CrossRef]
- Zhu, S.; Yang, N.; Wu, J.; Wang, X.; Wang, W.; Liu, Y.J.; et al. Tumor microenvironment-related dendritic cell deficiency: a target to enhance tumor immunotherapy. Pharmacological research. 2020, 159, 104980. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, W.; Liu, M.; Jia, R.; Wang, J.; Wang, F.; et al. OX40L enhances the immunogenicity of dendritic cells and inhibits tumor metastasis in mice. Microbiology and immunology. 2023, 67, 79–89. [Google Scholar] [CrossRef]
- Chen, S.; Fang, L.; Guo, W.; Zhou, Y.; Yu, G.; Li, W.; et al. Control of T(reg) cell homeostasis and immune equilibrium by Lkb1 in dendritic cells. Nature communications. 2018, 9, 5298. [Google Scholar] [CrossRef]
- Poropatich, K.; Dominguez, D.; Chan, W.C.; Andrade, J.; Zha, Y.; Wray, B.; et al. OX40+ plasmacytoid dendritic cells in the tumor microenvironment promote antitumor immunity. The Journal of clinical investigation. 2020, 130, 3528–42. [Google Scholar] [CrossRef]
- Badillo-Godinez, O.; Niemi, J.; Helfridsson, L.; Karimi, S.; Ramachandran, M.; Mangukiya, H.B.; et al. Brain tumors induce immunoregulatory dendritic cells in draining lymph nodes that can be targeted by OX40 agonist treatment. Journal for immunotherapy of cancer 2025, 13. [Google Scholar] [CrossRef]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer cell. 2017, 31, 711–23. [Google Scholar] [CrossRef]
- Nuebling, T.; Schumacher, C.E.; Hofmann, M.; Hagelstein, I.; Schmiedel, B.J.; Maurer, S.; et al. The Immune Checkpoint Modulator OX40 and Its Ligand OX40L in NK-Cell Immunosurveillance and Acute Myeloid Leukemia. Cancer immunology research. 2018, 6, 209–21. [Google Scholar] [CrossRef]
- Zaini, J.; Andarini, S.; Tahara, M.; Saijo, Y.; Ishii, N.; Kawakami, K.; et al. OX40 ligand expressed by DCs costimulates NKT and CD4+ Th cell antitumor immunity in mice. The Journal of clinical investigation. 2007, 117, 3330–8. [Google Scholar] [CrossRef]
- Kweon, S.; Phan, M.T.; Chun, S.; Yu, H.; Kim, J.; Kim, S.; et al. Expansion of Human NK Cells Using K562 Cells Expressing OX40 Ligand and Short Exposure to IL-21. Frontiers in immunology. 2019, 10, 879. [Google Scholar] [CrossRef]
- Niknam, S.; Barsoumian, H.B.; Schoenhals, J.E.; Jackson, H.L.; Yanamandra, N.; Caetano, M.S.; et al. Radiation Followed by OX40 Stimulation Drives Local and Abscopal Antitumor Effects in an Anti-PD1-Resistant Lung Tumor Model. Clinical cancer research : an official journal of the American Association for Cancer Research. 2018, 24, 5735–43. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Lin, D.; Yan, N.; Li, H.; Li, J.; Huang, Z. Oxaliplatin facilitates tumor-infiltration of T cells and natural-killer cells for enhanced tumor immunotherapy in lung cancer model. Anti-cancer drugs. 2022, 33, 117–23. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, T.; Ou, M.; Luo, R.; Chen, H.; Ren, H.; et al. OX40L-expressing M1-like macrophage exosomes for cancer immunotherapy. Journal of controlled release : official journal of the Controlled Release Society. 2024, 365, 469–79. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Xu, L.; Wu, H.; Liao, H.; Luo, L.; Liao, M.; et al. OX40 expression in hepatocellular carcinoma is associated with a distinct immune microenvironment, specific mutation signature, and poor prognosis. Oncoimmunology. 2018, 7, e1404214. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, Q.; Yang, K.; Zhang, D.; Li, F.; Chen, J.; et al. Reprogramming macrophage by targeting VEGF and CD40 potentiates OX40 immunotherapy. Biochemical and biophysical research communications. 2024, 698, 149546. [Google Scholar] [CrossRef]
- Badeaux, M.D.; Rolig, A.S.; Agnello, G.; Enzler, D.; Kasiewicz, M.J.; Priddy, L.; et al. Arginase Therapy Combines Effectively with Immune Checkpoint Blockade or Agonist Anti-OX40 Immunotherapy to Control Tumor Growth. Cancer immunology research. 2021, 9, 415–29. [Google Scholar] [CrossRef]
- Chua, C.Y.X.; Jain, P.; Susnjar, A.; Rhudy, J.; Folci, M.; Ballerini, A.; et al. Nanofluidic drug-eluting seed for sustained intratumoral immunotherapy in triple negative breast cancer. Journal of controlled release : official journal of the Controlled Release Society. 2018, 285, 23–34. [Google Scholar] [CrossRef]
- Perry, C.J.; Munoz-Rojas, A.R.; Meeth, K.M.; Kellman, L.N.; Amezquita, R.A.; Thakral, D.; et al. Myeloid-targeted immunotherapies act in synergy to induce inflammation and antitumor immunity. The Journal of experimental medicine. 2018, 215, 877–93. [Google Scholar] [CrossRef]
- Fu, Y.; Lin, Q.; Zhang, Z.; Zhang, L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta pharmaceutica Sinica B. 2020, 10, 414–33. [Google Scholar] [CrossRef]
- Moran, A.E.; Polesso, F.; Weinberg, A.D. Immunotherapy Expands and Maintains the Function of High-Affinity Tumor-Infiltrating CD8 T Cells In Situ. Journal of immunology (Baltimore, Md : 1950). 2016, 197, 2509–21. [Google Scholar] [CrossRef]
- Polesso, F.; Sarker, M.; Weinberg, A.D.; Murray, S.E.; Moran, A.E. OX40 Agonist Tumor Immunotherapy Does Not Impact Regulatory T Cell Suppressive Function. Journal of immunology (Baltimore, Md : 1950). 2019, 203, 2011–9. [Google Scholar] [CrossRef]
- Kuang, Z.; Jing, H.; Wu, Z.; Wang, J.; Li, Y.; Ni, H.; et al. Development and characterization of a novel anti-OX40 antibody for potent immune activation. Cancer immunology, immunotherapy : CII. 2020, 69, 939–50. [Google Scholar] [CrossRef]
- Papp, K.A.; Gooderham, M.J.; Girard, G.; Raman, M.; Strout, V. Phase I randomized study of KHK4083, an anti-OX40 monoclonal antibody, in patients with mild to moderate plaque psoriasis. Journal of the European Academy of Dermatology and Venereology : JEADV. 2017, 31, 1324–32. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Simpson, E.L.; Reich, K.; Kabashima, K.; Igawa, K.; Suzuki, T.; et al. An anti-OX40 antibody to treat moderate-to-severe atopic dermatitis: a multicentre, double-blind, placebo-controlled phase 2b study. Lancet (London, England). 2023, 401, 204–14. [Google Scholar] [CrossRef]
- Short, N.J.; Borthakur, G.; Pemmaraju, N.; Dinardo, C.D.; Kadia, T.M.; Jabbour, E.; et al. A multi-arm phase Ib/II study designed for rapid, parallel evaluation of novel immunotherapy combinations in relapsed/refractory acute myeloid leukemia. Leukemia & lymphoma. 2022, 63, 2161–70. [Google Scholar]
- Schober, K.; Muller, T.R.; Busch, D.H. Orthotopic T-Cell Receptor Replacement-An “Enabler” for TCR-Based Therapies. Cells 2020, 9. [Google Scholar] [CrossRef]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nature reviews Immunology. 2012, 12, 269–81. [Google Scholar] [CrossRef]
- Reichenbach, P.; Giordano Attianese, G.M.P.; Ouchen, K.; Cribioli, E.; Triboulet, M.; Ash, S.; et al. A lentiviral vector for the production of T cells with an inducible transgene and a constitutively expressed tumour-targeting receptor. Nature biomedical engineering. 2023, 7, 1063–80. [Google Scholar] [CrossRef]
- Rui, X.; Alvarez Calderon, F.; Wobma, H.; Gerdemann, U.; Albanese, A.; Cagnin, L.; et al. Human OX40L-CAR-T(regs) target activated antigen-presenting cells and control T cell alloreactivity. Science translational medicine. 2024, 16, eadj9331. [Google Scholar] [CrossRef] [PubMed]
- Ye, K.; Li, F.; Wang, R.; Cen, T.; Liu, S.; Zhao, Z.; et al. An armed oncolytic virus enhances the efficacy of tumor-infiltrating lymphocyte therapy by converting tumors to artificial antigen-presenting cells in situ. Molecular therapy : the journal of the American Society of Gene Therapy. 2022, 30, 3658–76. [Google Scholar] [CrossRef] [PubMed]
- Flynn, S.; Toellner, K.M.; Raykundalia, C.; Goodall, M.; Lane, P. CD4 T cell cytokine differentiation: the B cell activation molecule, OX40 ligand, instructs CD4 T cells to express interleukin 4 and upregulates expression of the chemokine receptor, Blr-1. The Journal of experimental medicine. 1998, 188, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Yoon, S.E.; Kim, W.S. Current Challenges in Chimeric Antigen Receptor T-cell Therapy in Patients With B-cell Lymphoid Malignancies. Annals of laboratory medicine. 2024, 44, 210–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, F.; Cao, J.; Wang, X.; Cheng, H.; Qi, K.; et al. A chimeric antigen receptor with antigen-independent OX40 signaling mediates potent antitumor activity. Science translational medicine 2021, 13. [Google Scholar] [CrossRef]
- Moreno-Cortes, E.; Franco-Fuquen, P.; Garcia-Robledo, J.E.; Forero, J.; Booth, N.; Castro, J.E. ICOS and OX40 tandem co-stimulation enhances CAR T-cell cytotoxicity and promotes T-cell persistence phenotype. Frontiers in oncology. 2023, 13, 1200914. [Google Scholar] [CrossRef]
- Zhang, H.; Zhong, R.; Wang, W.; Huang, Y.; Li, F.; Liang, J.; et al. OX40-heparan sulfate binding facilitates CAR T cell penetration into solid tumors in mice. Science translational medicine. 2025, 17, eadr2151. [Google Scholar] [CrossRef]
- Tan, J.; Jia, Y.; Zhou, M.; Fu, C.; Tuhin, I.J.; Ye, J.; et al. Chimeric antigen receptors containing the OX40 signalling domain enhance the persistence of T cells even under repeated stimulation with multiple myeloma target cells. Journal of hematology & oncology. 2022, 15, 39. [Google Scholar]
- Rittig, S.M.; Lutz, M.S.; Clar, K.L.; Zhou, Y.; Kropp, K.N.; Koch, A.; et al. Controversial Role of the Immune Checkpoint OX40L Expression on Platelets in Breast Cancer Progression. Frontiers in oncology. 2022, 12, 917834. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, A.; An, Z.; Huang, S.; Zhang, C.; Zhong, X.; et al. B cells enhance EphA2 chimeric antigen receptor T cells cytotoxicity against glioblastoma via improving persistence. Human immunology. 2024, 85, 111093. [Google Scholar] [CrossRef]
- Huang, X.; Guo, J.; Li, T.; Jia, L.; Tang, X.; Zhu, J.; et al. c-Met-targeted chimeric antigen receptor T cells inhibit hepatocellular carcinoma cells in vitro and in vivo. Journal of biomedical research. 2021, 36, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Williams, L.J.; Xu, C.; Melendez, B.; McKenzie, J.A.; Chen, Y.; et al. Anti-OX40 Antibody Directly Enhances The Function of Tumor-Reactive CD8(+) T Cells and Synergizes with PI3Kbeta Inhibition in PTEN Loss Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2019, 25, 6406–16. [Google Scholar] [CrossRef] [PubMed]
- Shrimali, R.K.; Ahmad, S.; Verma, V.; Zeng, P.; Ananth, S.; Gaur, P.; et al. Concurrent PD-1 Blockade Negates the Effects of OX40 Agonist Antibody in Combination Immunotherapy through Inducing T-cell Apoptosis. Cancer immunology research. 2017, 5, 755–66. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Huang, Y.; Li, P.; Wang, L.; Tang, Z.; Liu, X.; et al. Physical- and Chemical-Dually ROS-Responsive Nano-in-Gel Platforms with Sequential Release of OX40 Agonist and PD-1 Inhibitor for Augmented Combination Immunotherapy. Nano letters. 2023, 23, 1424–34. [Google Scholar] [CrossRef]
- Yadav, R.; Redmond, W.L. Current Clinical Trial Landscape of OX40 Agonists. Current oncology reports. 2022, 24, 951–60. [Google Scholar] [CrossRef]
- Hoffmann, P.R.; Hoffmann, F.W.; Premeaux, T.A.; Fujita, T.; Soprana, E.; Panigada, M.; et al. Multi-antigen Vaccination With Simultaneous Engagement of the OX40 Receptor Delays Malignant Mesothelioma Growth and Increases Survival in Animal Models. Frontiers in oncology. 2019, 9, 720. [Google Scholar] [CrossRef]
- Schoenfeld, J.D.; Giobbie-Hurder, A.; Ranasinghe, S.; Kao, K.Z.; Lako, A.; Tsuji, J.; et al. Durvalumab plus tremelimumab alone or in combination with low-dose or hypofractionated radiotherapy in metastatic non-small-cell lung cancer refractory to previous PD(L)-1 therapy: an open-label, multicentre, randomised, phase 2 trial. The Lancet Oncology. 2022, 23, 279–91. [Google Scholar] [CrossRef]
- Yang, Y.J.; Cao, L.; Li, Z.W.; Zhao, L.; Wu, H.F.; Yue, D.; et al. Fluorouracil-based neoadjuvant chemoradiotherapy with or without oxaliplatin for treatment of locally advanced rectal cancer: An updated systematic review and meta-analysis. Oncotarget. 2016, 7, 45513–24. [Google Scholar] [CrossRef]
- Han, M.G.; Wee, C.W.; Kang, M.H.; Kim, M.J.; Jeon, S.H.; Kim, I.A. Combination of OX40 Co-Stimulation, Radiotherapy, and PD-1 Inhibition in a Syngeneic Murine Triple-Negative Breast Cancer Model. Cancers 2022, 14. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Zhang, J.; Peng, Q.; Wang, X.; Xiao, X.; et al. Nanomaterial-mediated modulation of the cGAS-STING signaling pathway for enhanced cancer immunotherapy. Acta biomaterialia. 2024, 176, 51–76. [Google Scholar] [CrossRef]
- Ramser, M.; Eichelberger, S.; Daster, S.; Weixler, B.; Kraljevic, M.; Mechera, R.; et al. High OX40 expression in recurrent ovarian carcinoma is indicative for response to repeated chemotherapy. BMC cancer. 2018, 18, 425. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, Y.; Yu, J.; Meng, S.; Bi, C.; Guan, Q.; et al. OX40 shapes an inflamed tumor immune microenvironment and predicts response to immunochemotherapy in diffuse large B-cell lymphoma. Clinical immunology (Orlando, Fla). 2023, 251, 109637. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn-Cymerman, D.; Rizzuto, G.A.; Merghoub, T.; Cohen, A.D.; Avogadri, F.; Lesokhin, A.M.; et al. OX40 engagement and chemotherapy combination provides potent antitumor immunity with concomitant regulatory T cell apoptosis. The Journal of experimental medicine. 2009, 206, 1103–16. [Google Scholar] [CrossRef]
- Gulyás, D.; Kovács, G.; Jankovics, I.; Mészáros, L.; Lőrincz, M.; Dénes, B. Effects of the combination of a monoclonal agonistic mouse anti-OX40 antibody and toll-like receptor agonists: Unmethylated CpG and LPS on an MB49 bladder cancer cell line in a mouse model. PloS one. 2022, 17, e0270802. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, L.; Sun, X.; Lin, Y.; Yuan, J.; Yang, C.; et al. SHR-1806, a robust OX40 agonist to promote T cell-mediated antitumor immunity. Cancer biology & therapy. 2024, 25, 2426305. [Google Scholar]
- Jahan, N.; Talat, H.; Curry, W.T. Agonist OX40 immunotherapy improves survival in glioma-bearing mice and is complementary with vaccination with irradiated GM-CSF-expressing tumor cells. Neuro-oncology. 2018, 20, 44–54. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, X.; Cheng, D.; Xia, Z.; Luan, M.; Zhang, S. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PloS one. 2014, 9, e89350. [Google Scholar] [CrossRef]
- Garrison, K.; Hahn, T.; Lee, W.C.; Ling, L.E.; Weinberg, A.D.; Akporiaye, E.T. The small molecule TGF-β signaling inhibitor SM16 synergizes with agonistic OX40 antibody to suppress established mammary tumors and reduce spontaneous metastasis. Cancer immunology, immunotherapy : CII. 2012, 61, 511–21. [Google Scholar] [CrossRef]
- Zhou, H.; Ma, Y.; Li, Y.; Tang, L.; Guo, Y.; Yuan, G.; et al. Anti-OX40 antibody BAT6026 in patients with advanced solid tumors: A multi-center phase I study. iScience. 2025, 28, 112270. [Google Scholar] [CrossRef]
- Duhen, R.; Ballesteros-Merino, C.; Frye, A.K.; Tran, E.; Rajamanickam, V.; Chang, S.C.; et al. Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells. Nature communications. 2021, 12, 1047. [Google Scholar] [CrossRef] [PubMed]
- Glisson, B.S.; Leidner, R.S.; Ferris, R.L.; Powderly, J.; Rizvi, N.A.; Keam, B.; et al. Safety and Clinical Activity of MEDI0562, a Humanized OX40 Agonist Monoclonal Antibody, in Adult Patients with Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2020, 26, 5358–67. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.J.; Martin-Liberal, J.; Kristeleit, R.; Cho, D.C.; Blagden, S.P.; Berthold, D.; et al. First-in-human phase I/II, open-label study of the anti-OX40 agonist INCAGN01949 in patients with advanced solid tumors. Journal for immunotherapy of cancer 2022, 10. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Burris, H.A.; de Miguel Luken, M.J.; Pishvaian, M.J.; Bang, Y.J.; Gordon, M.; et al. First-In-Human Phase I Study of the OX40 Agonist MOXR0916 in Patients with Advanced Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2022, 28, 3452–63. [Google Scholar] [CrossRef]
- Fracp, J.D. A phase 1 study of the OX40 agonist BGB-A445, with or without tislelizumab, an anti-PD-1 monoclonal antibody, in patients with advanced solid tumors American Society of Clinical Oncology (ASCO) Annual Meeting 2023, June 2-6, 2023, Chicago, IL, USA2023.
- Postel-Vinay, S.; Lam, V.K.; Ros, W.; Bauer, T.M.; Hansen, A.R.; Cho, D.C.; et al. First-in-human phase I study of the OX40 agonist GSK3174998 with or without pembrolizumab in patients with selected advanced solid tumors (ENGAGE-1). Journal for immunotherapy of cancer 2023, 11. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; et al. Efficacy and Safety of Nivolumab Plus Ipilimumab in Patients With Advanced Hepatocellular Carcinoma Previously Treated With Sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA oncology. 2020, 6, e204564. [Google Scholar] [CrossRef]
- Hamid, O.; Chiappori, A.A.; Thompson, J.A.; Doi, T.; Hu-Lieskovan, S.; Eskens, F.; et al. First-in-human study of an OX40 (ivuxolimab) and 4-1BB (utomilumab) agonistic antibody combination in patients with advanced solid tumors. Journal for immunotherapy of cancer. 2022, 10(10).
- Knisely, A.; Ahmed, J.; Stephen, B.; Piha-Paul, S.A.; Karp, D.; Zarifa, A.; et al. Phase 1/2 trial of avelumab combined with utomilumab (4-1BB agonist), PF-04518600 (OX40 agonist), or radiotherapy in patients with advanced gynecologic malignancies. Cancer. 2024, 130, 400–9. [Google Scholar] [CrossRef]
- Shree, T.; Czerwinski, D.; Haebe, S.; Sathe, A.; Grimes, S.; Martin, B.; et al. A Phase I Clinical Trial Adding OX40 Agonism to In Situ Therapeutic Cancer Vaccination in Patients with Low-Grade B-cell Lymphoma Highlights Challenges in Translation from Mouse to Human Studies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2025, 31, 868–80. [Google Scholar] [CrossRef]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer research. 2013, 73, 7189–98. [Google Scholar] [CrossRef]
- Redmond, WL. Challenges and opportunities in the development of combination immunotherapy with OX40 agonists. Expert opinion on biological therapy. 2023, 23, 901–12. [Google Scholar] [CrossRef]
- Kuang, Z.; Pu, P.; Wu, M.; Wu, Z.; Wang, L.; Li, Y.; et al. A Novel Bispecific Antibody with PD-L1-assisted OX40 Activation for Cancer Treatment. Molecular cancer therapeutics. 2020, 19, 2564–74. [Google Scholar] [CrossRef] [PubMed]
- Holay, N.; Yadav, R.; Ahn, S.J.; Kasiewicz, M.J.; Polovina, A.; Rolig, A.S.; et al. INBRX-106: a hexavalent OX40 agonist that drives superior antitumor responses via optimized receptor clustering. Journal for immunotherapy of cancer 2025, 13. [Google Scholar] [CrossRef]
- Moiseyenko, A.; Muggia, F.; Condamine, T.; Pulini, J.; Janik, J.E.; Cho, D.C. Sequential therapy with INCAGN01949 followed by ipilimumab and nivolumab in two patients with advanced ovarian carcinoma. Gynecologic oncology reports. 2020, 34, 100655. [Google Scholar] [CrossRef]
- Sanchez, S.; Dangi, T.; Awakoaiye, B.; Lew, M.H.; Irani, N.; Fourati, S.; et al. Delayed reinforcement of costimulation improves the efficacy of mRNA vaccines in mice. The Journal of clinical investigation 2024, 134. [Google Scholar] [CrossRef]
- Jiang, T.; Yang, Z.; Su, Q.; Fang, L.; Xiang, Q.; Tian, C.; et al. Bivalent OX40 Aptamer and CpG as Dual Agonists for Cancer Immunotherapy. ACS applied materials & interfaces. 2025, 17, 7353–62. [Google Scholar]
- He, B.; Zhao, R.; Zhang, B.; Pan, H.; Liu, J.; Huang, L.; et al. Endothelial OX40 activation facilitates tumor cell escape from T cell surveillance through S1P/YAP-mediated angiogenesis. The Journal of clinical investigation 2025, 135. [Google Scholar] [CrossRef]
- Thapa, B.; Kato, S.; Nishizaki, D.; Miyashita, H.; Lee, S.; Nesline, M.K.; et al. OX40/OX40 ligand and its role in precision immune oncology. Cancer metastasis reviews. 2024, 43, 1001–13. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The OX40/OX40L interaction and down-stream signaling pro-cesses lead to the prolif-eration and survival of T cells. It also inhibited the transcription of FoxP3.
Figure 1.
The OX40/OX40L interaction and down-stream signaling pro-cesses lead to the prolif-eration and survival of T cells. It also inhibited the transcription of FoxP3.
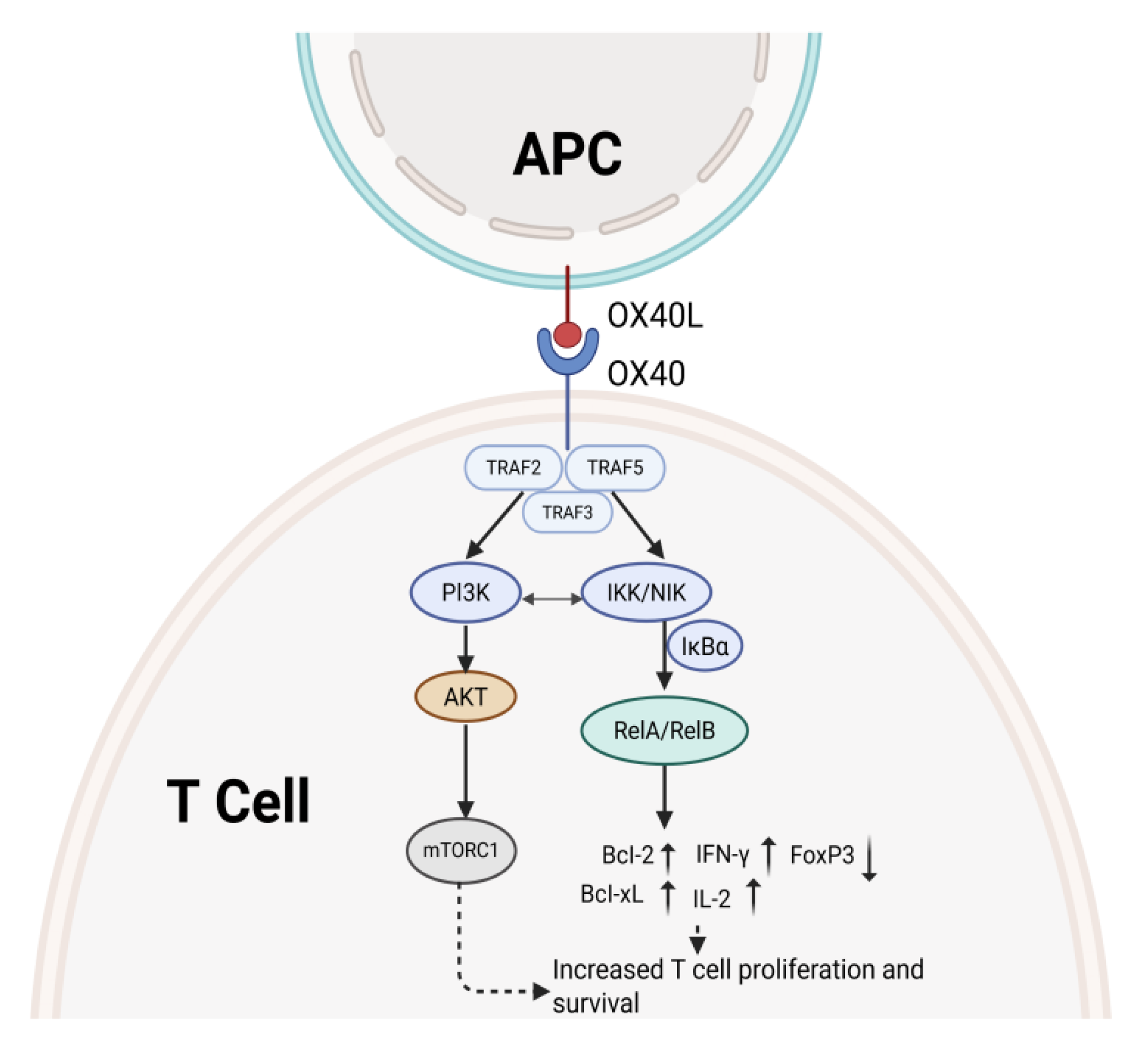
Figure 2.
illustrates the basic principles of gene therapy and CAR-T cell therapy.
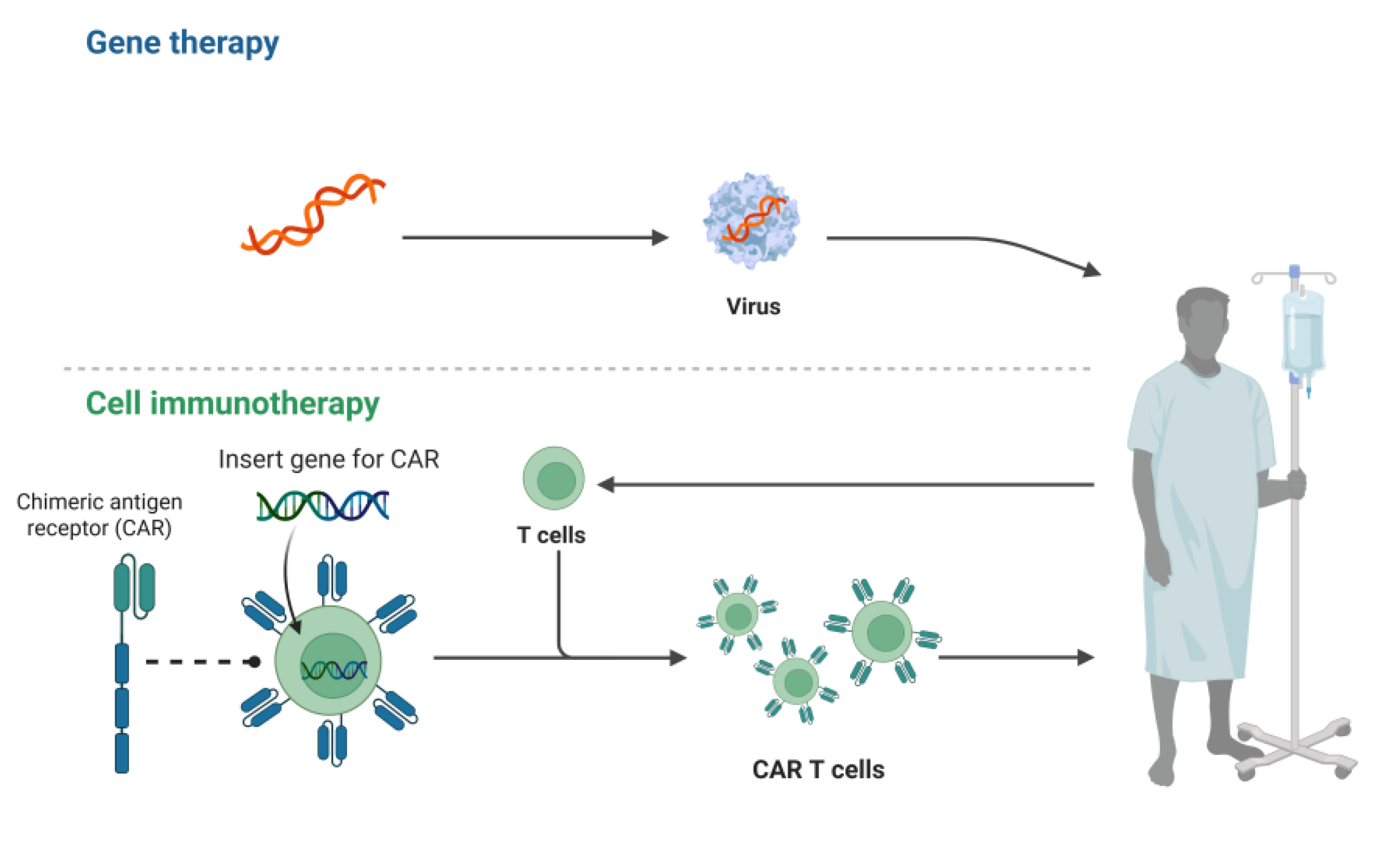

Table 1.
Examples of ongoing clinical trials targeting monoclonal antibodies against OX40.
| Clinical ID | Format | Phase Status | Name | Tumor Type | information by |
|---|---|---|---|---|---|
| NCT01862900 | IgG1 | I/II | MEDI6469 | Metastatic breast cancer | Providence Health Services |
| NCT02274155 | IgG1 | I | MEDI0562 | Head and neck cancer/Solid tumors/Ovarian cancer | Providence Health Services |
| NCT02315066 | IgG2 | I/II | PF-04518600 | Metastatic carcinoma/malignancies | Pfizer |
| NCT02410512 | IgG1 | I | MOXR0916 | Metastatic Solid tumors | Genentech |
| NCT02737475 | IgG1 | I/II | BMS-986178 | Solid tumors | Bristol-Myers Squibb |
| NCT03410901 | IgG1 | I | BMS-986178 | Low-grade B-Cell non-Hodg-kin lymphomas | Ronald Levy Stanford university |
| NCT04387071 | IgG4 | I/II | INCAGN01949 | Pancreatic cancer | University of southern california |
| NCT03758001 | IgG4 | I | IBI101 | Solid tumors | Innovent Biologics |
| NCT05229601 | IgG1 | I | HFB301001 | Solid tumors | HiFiBio Therapeutics |
| NCT03092856 | IgG2 | I/II | PF-04581600 | Renal Cell Carcinoma | Pfizer |
| NCT01775631 | IgG2 | I/II | BMS-663513 | B-Cell non-Hodg-kin lymphomas | Bristol-Myers Squibb |
| NCT02598960 | IgG2 | I/II | BMS-986156 | Advanced-stage Solid tumors | Bristol-Myers Squibb |
| NCT02221960 | IgG2 | I/II | MEDI6383 | Recurrent or metastatic solid tumors | MedImmune LLC |
| NCT04215978 | IgG1 | I/II | BGB-A445 | Advanced solid tumors | BeiGene |
| NCT02274155 | IgG2 | I/II | 9B12 | Head and neck cancer | Providence Health Services |
Table 2.
Examples of OX40agonistic monospecific antibodies in clinical studies.
| Name | Phase | Combination |
Type of mouse tumor | Type of OX40 therapy | Effect | References |
|---|---|---|---|---|---|---|
| BAT6026 | I | Single agent | Advanced cancer | Anti-OX40,0.01-10mg/kg, IV, Q3W. | N=30, ORR 0%, DCR 38.5% | [110] |
| MEDI6469 | I/II | Single agent | HNSCC | Anti-OX40,0.4mg/kg, IV, day1.3.5 | n=17, OS 82%; PFS 71% at 3 years | [111] |
| MEDI0562 | I | Single agent | Advanced Solid tumors | Anti-OX40,0.03-10mg/kg, IV, Q2W. | N=55, ORR 4%; PR1, DCR 22% | [112] |
| INCAGN01949 | I/II | Single agent | Advanced Solid tumors | Anti-OX40,7-1400mg, IV, Q2W. | N=87, ORR 1.5%; PR1 | [113] |
| MOXR0916 | I | Single agent | Advanced Solid tumors | Anti-OX40,0.2-1200mg, IV, Q3W. | N=172, ORR 1.2% PR2, DCR66% | [114] |
| BGB-A445 | Ib | Tislelizumab | Advanced Solid tumors | Anti-OX40,0.03-40mg/kg, IV, Q3W. Anti-PD-1,200mg, IV, Q3W. | N=30, ORR 23%; PR 7 | [115] |
| GSK3174998 | I | Pembrolizumab | Advanced Solid tumors | Anti-OX40,0.03-10mg/kg, IV, Q3W. Anti-PD-1,200mg, IV, Q3W. | N=138, Single agent ORR0%; Combination with aPD-1, ORR 8%; CR 2; PR 4 | [116] |
| PF-04518600 | I/II | Nivolumab Ipilimumab |
Advanced Solid tumors | Anti-OX40,0.1-3mg/kg, IV, Q2W. Anti-4-1BB,20/100mg, IV, Q4W. | N=57, ORR 3.5%; PR 2; DCR 35% | [118] |
| BMS986178 | I | SD-101 Radiation |
LG-B-NHL | Anti-OX40,0.3mg/kg, IV, Day8.15.22 SD-101,1-8mg, IT, Day8.15.22.29.36 RT, Day1. |
N=29, ORR 31%; CR 4; PR 5 | [120] |
| BMS986178 | I/II | Nivolumab Ipilimumab |
Advanced Solid tumors | Anti-OX40,20-320mg, IV, Q2W Anti-PD-1,240-480mg, IV, Q2W Anti-CTLA-4,1-3mg/kg, IV, Q3W |
N=165, Single agent ORR 0%(n=20), Combination therapy ORR 0-13%(n=145) |
[117] |
| PF-04518600 | I/II | Avelumab UtomilumabRadiation |
Advanced gynecologic malignancies | Anti-OX40,0.3mg/kg, IV, Q2W Anti-PD-L1,10mg/kg, IV, Q2W Anti-4-1BB,20/100mg, IV, Q4W |
N=35,Avelumab+Utomilumab:ORR11%;PR1(n=9),Avelumab+Utomilumab+PF04518600:ORR2.9%,DCR37.1%,(n=35) | [119] |
| 9B12 | I/II | Single agent | Advanced cancer | Anti-OX40,0.1-2mg/kg, IV, Day1,3,5 | N=30, ORR 0%, tumors remission | [121] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



