
Submitted:
13 September 2025
Posted:
16 September 2025
You are already at the latest version
Abstract
Atypical parkinsonian disorders—progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and multiple system atrophy (MSA)—are rare, rapidly progressive neurodegenerative syndromes with complex clinical presentations, distinct neuropathological signatures, and limited therapeutic options. Accurate diagnosis is challenging, particularly in early stages, due to overlap with Parkinson’s disease and among themselves, phenotypic heterogeneity, and the lack of reliable stand-alone biomarkers. Misclassification is a frequent occurrence, with significant consequences for prognosis, patient care, and the design of clinical trials. This review synthesizes current evidence on clinical, neuroimaging, and biomarker-based diagnosis of PSP, CBD, and MSA. We describe core and variant phenotypes, provide epidemiological updates, and present neuropathological correlates, along with structured diagnostic approaches. Advances in MRI morphometry, tau- and α–synuclein–sensitive PET, quantitative oculomotor and autonomic testing, and fluid biomarkers—particularly neurofilament light chain—are critically evaluated for their diagnostic value and limitations. We identify ten persistent challenges to early and accurate diagnosis, including phenotypic mimicry, non-specific imaging, limited biomarker specificity, and inequitable access to advanced diagnostics. We propose tiered, multimodal algorithms that integrate standardized clinical phenotyping with quantitative imaging, molecular diagnostics, and pathology-linked validation. This approach may reduce diagnostic delays, enhance trial enrichment, and facilitate earlier, personalized interventions in these disorders.
Keywords:
progressive supranuclear palsy
; corticobasal degeneration
; multiple system atrophy
; atypical parkinsonism
; biomarkers
; diagnostic algorithms
1. Introduction
Parkinsonism is a clinical syndrome defined by bradykinesia with additional motor signs—rigidity, tremor, and postural instability—but its causes are heterogeneous. The most frequent is Parkinson's disease (PD); nevertheless, a substantial proportion reflects atypical parkinsonian disorders (APDs), notably progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and multiple system atrophy (MSA). These entities carry distinct biology—4-repeat tau in PSP and CBD, α-synuclein pathology in MSA—with different trajectories, complications, and treatment priorities. Early and accurate separation from PD and one another is therefore central to prognosis, counseling, and trial enrollment [1,2].
Over the past decade, diagnostic frameworks have evolved from purely clinicopathological descriptions to structured, phenotype-based criteria supported by imaging and fluid biomarkers. The 2017 MDS-PSP criteria recognize variant phenotypes beyond Richardson's syndrome, emphasizing ocular motor dysfunction and early postural instability, and have improved case capture across the phenotypic spectrum [1,3]. In MSA, the 2022 MDS criteria formalize "clinically established," "clinically probable," and "possible prodromal" categories, integrating autonomic failure, parkinsonism and/or cerebellar signs, and specific MRI markers—such as putaminal and middle cerebellar peduncle changes or the "hot cross bun" sign—to enhance early-stage accuracy [2]. CBD remains diagnostically challenging because corticobasal syndrome (CBS) is pathologically pleomorphic; contemporary reviews stress asymmetric limb apraxia or dystonia with cortical sensory loss while acknowledging that Alzheimer's disease (AD) and PSP frequently underlie CBS at autopsy [4,5].
Supportive investigations now carry greater weight. Quantitative MRI measures, such as the MR Parkinsonism Index 2.0 (MRPI 2.0), enhance the separation of PSP—particularly PSP-parkinsonism—from PD, with automated pipelines becoming increasingly available [6,7,8]. Cardiac 123I-MIBG scintigraphy often remains normal in MSA but is typically reduced in PD, aiding synucleinopathy subtyping when interpreted alongside autonomic testing [9]. Fluid biomarkers add complementary value: neurofilament light chain (NfL) levels in cerebrospinal fluid or serum are generally higher in APDs than PD and correlate with faster progression, supporting triage and prognostication [10]. Seed-amplification assays for misfolded α-synuclein show high sensitivity in PD and dementia with Lewy bodies. However, they are variably negative in MSA, a biologically informative distinction shaping trial stratification rather than serving as a stand-alone rule-in test for MSA [11,12].
This review updates and operationalizes diagnostic algorithms for PSP, CBD, and MSA by integrating these criteria and biomarkers with careful longitudinal re-evaluation—an approach reflected in the stepwise schemas provided (initial clinical screening; targeted ocular, cortical, cerebellar, and autonomic assessments; response to levodopa; MRI and selected functional imaging; and ongoing diagnostic revision) [6]. The goal is pragmatic: to reduce early misdiagnosis, direct patients to appropriate symptomatic care, and accelerate entry into disease-modifying trials. The field now requires a paradigm shift toward biology-led diagnosis, which couples phenotyping with standardized imaging and fluid panels, implemented within multidisciplinary services [11,13,14].
2. Progressive Supranuclear Palsy (PSP)
PSP is a primary 4-repeat (4R) tauopathy with characteristic neuronal and glial tau pathology—globose neurofibrillary tangles, coiled bodies, and tufted astrocytes—predominantly affecting brainstem, basal ganglia, and related white-matter tracts [6,15,16]. Clinically, the core syndrome (PSP–Richardson’s) combines early postural instability with backwards falls, vertical supranuclear gaze impairment, axial-predominant rigidity, dysarthria/dysphagia, and a dysexecutive, subcortical cognitive profile [1,3]. Contemporary epidemiology indicates a pooled prevalence of ~7 per 100,000 and an incidence of up to 2.6 per 100,000 person-years, with a typical onset in the early to mid-60s and a median survival of 5–7 years [17,18,19]. Diagnostic delays of 3–5 years remain common, underscoring the need for algorithmic approaches that integrate phenotype, oculomotor testing, imaging, and biomarkers [20].
2.1. Etiology and genetics
Most PSP is sporadic, but genetic susceptibility is substantial. Beyond the MAPT H1 haplotype, genome-wide studies replicate risk at MOBP, EIF2AK3, and STX6, and nominate additional loci (e.g., SLCO1A2, DUSP10, RUNX2; occasionally LRRK2 signals), with recent large-scale analyses also implicating complement C4A and glial-activation pathways [21,22,23,24,25]. Rare familial cases with pathogenic or likely pathogenic MAPT variants broaden the phenotypic spectrum but are the exception [26,27]. Mitochondrial and oxidative stress mechanisms are frequently discussed, but, to date, genetic and molecular evidence most consistently converges on tau biology, proteostasis, and neuroinflammation [22,28].
2.2. Diagnostic approach
Early PSP is frequently misclassified as PD because both share bradykinesia and postural instability in the initial years. However, PSP usually presents with axial-predominant rigidity, symmetric parkinsonism, poor and unsustained levodopa benefit, paucity of rest tremor, and the progression from vertical saccadic slowing to supranuclear gaze palsy with preserved vestibulo-ocular reflex [1,29]. Pseudobulbar features such as dysarthria and dysphagia, together with early executive dysfunction, also distinguish PSP from PD.
Ocular motor examination is pivotal. Quantitative video-oculography confirms that vertical saccade slowing and increased square-wave jerks have high specificity for PSP, with combined amplitude–velocity metrics outperforming single parameters for PSP vs PD discrimination [30,31,32]. These findings are incorporated into the 2017 MDS-PSP criteria, which stratify diagnostic certainty across PSP-Richardson’s syndrome (PSP-RS) and its variants [1,3].
To reduce misdiagnosis, a stepwise diagnostic algorithm (Figure 1) integrates clinical, ocular motor, imaging, and biomarker assessments:
- Initial clinical evaluation: Symmetric, axial-predominant parkinsonism with early postural instability, vertical gaze impairment, dysarthria/dysphagia, and dysexecutive profile should raise early suspicion of PSP, contrasting with the asymmetric onset, rest tremor, and levodopa responsiveness typical of PD.
- Ocular motor testing: Slowing of vertical saccades progressing to gaze palsy, together with square-wave jerks, is highly specific; video-oculography offers objective quantification.
- Phenotype classification: Applying the 2017 MDS-PSP criteria allows early recognition not only of PSP-RS but also of variant phenotypes (e.g., PSP-parkinsonism, PSP with predominant gait freezing, PSP-CBS, PSP-FTD).
- Neuroimaging: Midbrain atrophy with pontine sparing (“hummingbird sign”) is supportive, but MRPI 2.0, computed via automated tools, provides superior discrimination, especially between PSP-parkinsonism and PD/MSA.
- Supportive imaging: DAT-SPECT confirms presynaptic nigrostriatal degeneration but lacks nosological resolution. In contrast, cardiac 123I-MIBG scintigraphy is typically normal in PSP but reduced in Lewy body disorders, supporting differential diagnosis.
- Fluid biomarkers: Cerebrospinal fluid (CSF) or plasma NfL levels are consistently higher in PSP than in PD, with area under the receiver operating characteristic curve (AUC) values greater than 0.90 for PD compared to atypical parkinsonism. Although not diagnostic in isolation, NfL has prognostic and triage value, particularly when combined with MRI measures.
- Longitudinal reassessment: Diagnostic certainty increases as the disease evolves. Repeated clinical, ocular motor, and imaging assessments capture the progression from subtle ocular motor slowing to frank gaze palsy, thereby consolidating diagnostic accuracy.
This algorithm highlights the importance of multimodal integration: clinical red flags initiate suspicion, ocular motor examination provides high specificity, MRI morphometry and quantitative metrics improve diagnostic yield, and fluid biomarkers add prognostic value. Together, these steps embody the shift from descriptive syndromic diagnosis to biology-anchored diagnostic certainty in PSP.
2.3. Phenotypic spectrum
Beyond PSP-Richardson’s, clinically recognized variants include PSP-parkinsonism (PSP-P), PSP with predominant gait freezing (PSP-PGF), PSP-corticobasal syndrome (PSP-CBS), PSP-frontotemporal dementia (PSP-FTD), and progressive non-fluent aphasia variants. Phenotypes can evolve, demanding longitudinal re-evaluation rather than single-timepoint labelling [1,29].
2.4. Investigations and biomarkers
2.4.1. MRI
Midbrain atrophy with relative pontine preservation (“hummingbird” profile) is supportive but not sufficient. Planimetric metrics markedly improve performance. The MR Parkinsonism Index 2.0 (MRPI 2.0) distinguishes PSP—especially PSP-P—from PD and can be computed via validated automated pipelines, enhancing standardization across centers and supporting trials [6,7,8].
2.4.2. Tau PET
Second-generation tracers show promise for 4R-tau imaging. Multicenter data with [18F]PI-2620 and [18F]florzolotau (APN-1607/PM-PBB3) demonstrate uptake patterns in the pallidum, subthalamus, and midbrain that complement MRI and may aid in early diagnosis and monitoring, although harmonized thresholds and head-to-head comparisons across 4R tauopathies remain priorities [27,33,34,35].
2.4.3. Dopaminergic imaging and autonomic tracers
DAT-SPECT confirms presynaptic nigrostriatal denervation but does not reliably distinguish between PSP, PD, and MSA. Postsynaptic D2/3 imaging (e.g., 123I-IBZM) is no longer recommended for routine differential diagnosis due to limited added value [36]. Cardiac 123I-MIBG scintigraphy is typically normal in PSP and reduced in Lewy body disorders (PD/dementia with Lewy bodies); thus, it aids in the exclusion of PD when interpreted in the context of clinical findings, including newer protocols that incorporate salivary-gland uptake [37,38].
2.4.4. Fluid biomarkers
Neurofilament light chain in CSF or plasma is consistently higher in atypical parkinsonism (including PSP) than in PD and correlates with progression; recent comparative studies show AUCs >0.90 for PD vs APD, supporting triage and prognostication rather than stand-alone diagnosis [39,40]. Combining NfL with quantitative MRI (e.g., midbrain measures, quantitative susceptibility mapping) improves classification in research settings [41].
2.5. Pathology
2.6. Treatment and management
No disease-modifying therapy has shown efficacy to date. Randomized trials of coenzyme Q10 and the GSK-3β inhibitor tideglusib were negative for clinical endpoints, despite showing imaging signals in early studies [42,43]. Multiple anti-tau monoclonal antibodies—gosuranemab and tilavonemab—failed to slow progression in phase 2 trials [44]. Ongoing programs (e.g., bepranemab/UCB0107) focus on safety and target engagement, but clinical benefit remains unproven.
Management is multidisciplinary and symptomatic: cautious levodopa trials for parkinsonism (often limited benefit), amantadine for gait/freezing in select cases, botulinum toxin for blepharospasm or eyelid-opening apraxia, early speech-language therapy with proactive dysphagia strategies (including timely PEG discussion), tailored physiotherapy for balance and falls, occupational therapy for executive dysfunction, and targeted treatment of mood, sleep, and pseudobulbar affect [45]. The integration of structured diagnostic algorithms, anchored in MDS-PSP criteria, quantitative oculomotor testing, MRPI 2.0, and tiered biomarker panels, can reduce misdiagnosis and accelerate enrollment into trials that increasingly require biologically characterized cohorts [1,7,18].
3. Corticobasal Degeneration (CBD)
Corticobasal degeneration is an uncommon primary 4R tauopathy characterized by distinctive neuropathological features, including astrocytic plaques, ballooned neurons, and thread-like tau inclusions, which most prominently affect the frontoparietal cortex, basal ganglia, and substantia nigra [46,47]. Clinically, it is characterized by progressive, levodopa-resistant, asymmetric Parkinson's disease with cortical signs, including limb apraxia, cortical sensory deficits, and alien limb phenomena [46,48]. Population-based prevalence estimates are scarce but consistently lower than for PSP or MSA, ranging from 0.8 to 25 per 100,000, with incidence rates between 0.03 and 0.8 per 100,000 person-years [18]; onset typically occurs in the early 60s, and median survival is approximately 6–8 years from symptom onset [47].
3.1. Etiology and genetics
Most CBD cases are sporadic, but shared genetic susceptibility with other primary tauopathies is increasingly recognized. The MAPT H1 haplotype, a major risk factor for PSP, has also been associated with CBD [49]. Genome-wide association studies suggest overlapping genetic architecture between CBD, PSP, and certain forms of frontotemporal lobar degeneration (FTLD), implicating genes involved in microtubule stabilization, vesicle trafficking, and glial activation [50]. While familial CBD is rare, pathogenic MAPT mutations occasionally produce clinicopathological CBD, expanding the phenotypic range [51]. Mechanistically, CBD pathology converges on the abnormal aggregation of 4R tau, dysregulation of proteostasis, and microglial activation; however, the molecular triggers remain unclear [52].
3.2. Diagnostic approach
CBD most often manifests as corticobasal syndrome, a clinico-anatomical construct characterized by asymmetric akinetic-rigid parkinsonism, limb dystonia, and stimulus-sensitive myoclonus. Patients may perceive the affected limb as “dead” or “foreign,” producing alien limb phenomena with autonomous, purposeful-appearing movements [46,53]. Unlike PSP, ocular motor abnormalities in CBD usually present as difficulty initiating voluntary saccades or subtle gaze apraxia, rather than vertical saccadic slowing [54].
Cortical sensory deficits are frequent and may include impaired two-point discrimination, astereognosis, and limb-kinetic apraxia [55]. Cognitive impairment typically involves executive and visuospatial dysfunction, while episodic memory is often relatively preserved until late stages, a valuable clinical clue against AD [48,55].
The Armstrong criteria (2013) remain the standard framework for defining probable and possible CBD, but their sensitivity and specificity are modest in clinicopathological studies [46]. Importantly, CBS is often underpinned by non-CBD pathologies, most frequently AD, PSP, or FTLD with TPD-43-immunoreactive pathology (FTLD-TDP), underscoring the role of neuropathological examination as the definitive diagnostic standard [4,47,48,51].
To operationalize early recognition and reduce misclassification, a stepwise diagnostic algorithm (Figure 2) integrates structured clinical assessment with neuroimaging, biomarkers, and longitudinal reassessment:
- Initial presentation: Asymmetric, levodopa-resistant parkinsonism with cortical signs such as apraxia, cortical sensory loss, or alien limb phenomena is the core entry point for CBS and the most common clinical phenotype of CBD.
- Application of the Armstrong criteria: Differentiating between probable and possible CBD provides a standardized clinical framework, although with limited sensitivity and specificity in practice.
- Neuropsychological evaluation: Formal testing of executive, visuospatial, and language domains is crucial. A non-fluent/agrammatic language profile supports CBD, while early episodic memory loss points toward AD-related CBS.
- MRI assessment: Characteristically reveals asymmetric frontoparietal atrophy contralateral to the most affected limb, sometimes with basal ganglia atrophy. Although not specific, it strengthens the clinico-anatomical correlation.
- Functional imaging, such as [18F]Fluorodeoxyglucose positron emission tomography (FDG-PET) or perfusion SPECT, typically demonstrates asymmetric hypometabolism or hypoperfusion in the frontoparietal cortex and basal ganglia, with relative sparing of the midbrain and cerebellum. These features help separate CBD from PSP/MSA but overlap with AD-related CBS.
- Tau PET: New-generation tracers ([18F]PI-2620, [18F]florzolotau) show asymmetric uptake in perirolandic and basal ganglia regions in pathologically confirmed CBD, complementing MRI for early detection of 4R tau pathology.
- Fluid biomarkers: Elevated NfL is a common finding and correlates with disease progression. Combined with imaging and neuropsychological testing, it improves discrimination between CBS due to CBD and CBS from AD or other pathologies.
- Longitudinal reassessment: Phenotypes may evolve toward PSP-like Richardson’s syndrome, behavioral variant FTD, non-fluent aphasia, or posterior cortical atrophy. Ongoing clinical review is essential, as early labels often shift with disease progression.
This algorithm underscores that CBD diagnosis is inherently probabilistic, given the pleomorphic nature of CBS. Multimodal integration—anchoring clinical suspicion with structured criteria, neuropsychological profiling, advanced neuroimaging, and supportive biomarkers—maximizes diagnostic confidence and accuracy. Ultimately, however, pathology remains the gold standard, and diagnostic frameworks should be applied dynamically, with recognition that phenotypes may shift across the disease course.
3.3. Phenotypic spectrum
Pathologically confirmed CBD can present with diverse phenotypes beyond CBS, including behavioral variant FTD, PSP-like Richardson syndrome, non-fluent/agrammatic primary progressive aphasia, and posterior cortical atrophy [47,48,55]. Conversely, CBS may be produced by AD, PSP, FTLD-TDP, or mixed pathologies [47,48,55]. Phenotypic evolution over time is common, underscoring the importance of longitudinal reassessment rather than fixed diagnostic labelling [56].
3.4. Investigations and biomarkers
3.4.1. MRI
CBD typically shows asymmetric frontoparietal atrophy contralateral to the most affected limb, sometimes with basal ganglia atrophy [47,56]. Advanced morphometric analyses, such as voxel-based morphometry and cortical thickness mapping, may detect changes earlier but lack specificity at the individual level [6].
3.4.2. FDG-PET and perfusion SPECT
These modalities often reveal asymmetric hypometabolism or hypoperfusion in the frontoparietal cortex and basal ganglia, extending to the supplementary motor area, with relative sparing of the midbrain and cerebellum. This pattern supports differentiation from PSP and MSA but overlaps with AD-related CBS [48].
3.4.3. Tau PET
First-generation tracers such as [18F]flortaucipir have shown variable binding in CBS/CBD and lower sensitivity for pure 4R tau compared to AD-type mixed 3R/4R tau. Second-generation tracers ([18F]PI-2620, [18F]florzolotau) display higher affinity for 4R tau and, in pathologically confirmed CBD, show asymmetric uptake in perirolandic and basal ganglia regions that complement MRI findings [33].
3.4.4. Fluid biomarkers
No single CSF or plasma biomarker reliably distinguishes CBD from other tauopathies. NfL is often elevated, correlating with disease severity and rate of progression, and may aid in differentiating CBS due to CBD from CBS due to AD when combined with structural imaging and neuropsychological testing focused on language and executive function [39].
3.5. Pathology
Neuropathological examination reveals widespread deposition of hyperphosphorylated 4R tau in neurons and glia. Astrocytic plaques, particularly in the cortex and striatum, are a defining feature that distinguishes CBD from PSP and other tauopathies. Coiled bodies in oligodendrocytes and ballooned neurons further characterize the pathology. Lesion distribution corresponds to the asymmetric cortical and extrapyramidal signs observed clinically [47,48].
3.6. Treatment and management
There is no therapy proven to alter the underlying pathology of CBD. Management is symptomatic and multidisciplinary. Trials of levodopa may be undertaken for parkinsonism, though responses are typically modest and short-lived [46]. Amantadine can be considered for gait freezing and other parkinsonian features [56]. Botulinum toxin injections may reduce focal dystonia or rigidity that interferes with function [57]. Myoclonus may respond to valproate or levetiracetam. Supportive measures, including early speech-language therapy for dysarthria and dysphagia, occupational therapy to address apraxia, and physiotherapy with mobility aids to prevent falls, are critical [4].
Given the phenotypic overlap with other tauopathies and AD, a multimodal diagnostic approach—integrating detailed clinical characterization, longitudinal follow-up, structural and functional imaging, and targeted biomarker analysis—is essential for early and accurate recognition [6]. Such an approach not only guides prognosis and counselling but also facilitates enrolment into clinical trials testing tau-targeted disease-modifying therapies [33,39].
4. Multiple System Atrophy (MSA)
MSA is a rapidly progressive α-synucleinopathy defined by the combination of autonomic failure and motor features—parkinsonism and/or cerebellar ataxia—in the absence of an alternative diagnosis [11]. Two motor phenotypes are recognized: the parkinsonian form (MSA-P), in which striatonigral degeneration predominates and parkinsonism is the major motor presentation, and the cerebellar form (MSA-C), characterized by olivopontocerebellar atrophy and limb/gait ataxia [58]. Phenotypic evolution is common, with many individuals acquiring mixed features as the disease progresses [2,59]. Contemporary prevalence estimates are approximately 4 per 100,000, with onset typically occurring between the ages of 53 and 55 years, a roughly equal sex distribution, and a median survival of between 6 and 10 years [59,60]. Onset before age 30 is sporadic. Although rare, sporadic familial clusters have been reported, most notably in association with COQ2 mutations, particularly in East Asian cohorts [61,62].
4.1. Etiology and genetics
MSA is considered a primary oligodendrogliopathy, characterized by α-synuclein aggregation within glial cytoplasmic inclusions (Papp–Lantos bodies), which serves as the neuropathological hallmark [11]. The molecular basis remains incompletely understood. Rare pathogenic or risk variants in COQ2, a gene encoding a key enzyme in coenzyme Q10 biosynthesis, have been identified in Japanese and other Asian populations, particularly in MSA-C, and are associated with reduced tissue and plasma coenzyme Q10 levels [61,62]. Additional genetic studies have explored SNCA variants and other mitochondrial or lysosomal genes; however, no robust, population-wide associations have been established outside of COQ2 [59].
4.2. Diagnostic approach
The diagnosis of MSA requires the integration of autonomic, motor, and supportive features, as outlined in the 2022 MDS diagnostic criteria [2]. In MSA-P, parkinsonism is typically symmetric, poorly or transiently responsive to levodopa, and lacks the classic pill-rolling rest tremor of Parkinson’s disease. Jerky postural or action tremor, often associated with cortical myoclonus, is a common occurrence. In MSA-C, gait and limb ataxia predominate, accompanied by dysarthria and oculomotor abnormalities, with parkinsonism emerging later in the course.
Autonomic failure is a defining hallmark and may precede motor symptoms by several years. It encompasses neurogenic orthostatic hypotension, urinary urgency or retention with large post-void residuals, and erectile dysfunction in men [60]. Respiratory manifestations, notably nocturnal inspiratory stridor due to laryngeal abductor paresis, may appear early and are associated with worse survival [63,64]. Supportive clinical features include REM sleep behavior disorder, early postural instability, orofacial dystonia, and severe gait freezing, whereas early dementia or prominent hallucinations argue against MSA [2,59].
A structured stepwise diagnostic algorithm (Figure 3) supports this multimodal evaluation:
- Initial presentation: Suspect MSA when symmetric parkinsonism (MSA-P) or cerebellar syndrome (MSA-C) co-occurs with autonomic dysfunction. Autonomic features frequently precede motor involvement and are diagnostically pivotal.
- Autonomic assessment: Comprehensive evaluation—including tilt-table testing, Valsalva maneuver, and urodynamics—documents cardiovascular and urogenital dysfunction, confirming autonomic failure as a diagnostic cornerstone.
- Motor examination: Poor or transient levodopa response, jerky cortical myoclonus, or focal dystonia support MSA over PD, in which sustained levodopa benefit and classic rest tremor are typical.
- MRI assessment: Characteristic signs include the “hot cross bun” sign in the pons, putaminal atrophy or hypointensity, and middle cerebellar peduncle (MCP) hyperintensity/atrophy. Quantitative morphometry of MCP width and pons-to-MCP ratios enhances early diagnostic accuracy.
- Dopaminergic imaging: DAT-SPECT confirms presynaptic dopaminergic loss but does not differentiate MSA from PD or PSP, serving only to establish neurodegeneration.
- Cardiac 123I-MIBG scintigraphy: Normal uptake in MSA versus markedly reduced uptake in PD/dementia with Lewy bodies helps distinguish between synucleinopathies.
- FDG-PET: Hypometabolism in the putamen, pons, and cerebellum, with cortical sparing, supports MSA and helps separate it from PSP and PD.
- Pathology: The definitive diagnosis rests on the demonstration of α–synuclein–positive glial cytoplasmic inclusions in oligodendrocytes, but this is typically confirmed post-mortem.
- Longitudinal reassessment: As motor, autonomic, and cerebellar features evolve, ongoing clinical review is critical to ensure accurate classification, prognosis, and trial eligibility.
This algorithm emphasizes that early recognition of autonomic dysfunction, combined with supportive motor and imaging features, is central to diagnosing MSA. While classical MRI signs (e.g., “hot cross bun”) are highly supportive in the correct context, specificity is strengthened when combined with quantitative imaging, FDG-PET, and biomarker panels. Longitudinal reassessment remains essential, as phenotypic evolution—particularly from isolated autonomic failure to combined motor syndromes—often clarifies the diagnosis over time.
4.3. Phenotypic spectrum
Although MSA-P and MSA-C are the two recognized motor subtypes, patients often develop features of both forms as the disease progresses. MSA-P may later exhibit cerebellar signs, whereas MSA-C often evolves to include bradykinesia and rigidity [2,59]. The relative frequency of subtypes varies geographically, with MSA-C predominating in East Asia and MSA-P being more common in Western populations [2,59,65]. A prodromal phase, characterized by isolated autonomic failure, polysomnography-confirmed REM sleep behavior disorder, and subtle motor abnormalities, is increasingly recognized and is now a research category in the MDS criteria [2,59].
4.4. Investigations and biomarkers
4.4.1. MRI
Conventional imaging may reveal the “hot cross bun” sign in the pons, reflecting cruciform T2 hyperintensity due to selective pontocerebellar tract degeneration, as well as hyperintensity in the MCP with associated atrophy. In MSA-P, putaminal atrophy and posterolateral T2 hyperintensity with hypointense signal (“slit” sign) are common. Quantitative MRI morphometry, including MCP width and pons-to-MCP area ratios, improves early detection and reproducibility across centers [6].
4.4.2. Functional imaging
FDG-PET reveals hypometabolism in the putamen, pons, and cerebellum, with relative preservation of cortical metabolism—patterns that help distinguish MSA from PSP and Parkinson’s disease [66,67]. DAT-SPECT confirms presynaptic dopaminergic loss but cannot distinguish MSA from other degenerative parkinsonisms [68]. Cardiac 123I-MIBG scintigraphy is usually normal or only mildly reduced in MSA, in contrast to the marked reduction seen in PD and dementia with Lewy bodies, aiding differential diagnosis in appropriate contexts [69].
4.4.3. Autonomic testing
Formal autonomic evaluation, including tilt-table testing, Valsalva maneuver, and quantitative sudomotor axon reflex testing, documents the severity and pattern of dysfunction. Urodynamic studies often reveal detrusor overactivity with impaired contractility or detrusor–sphincter dyssynergia [70,71].
4.4.4. Fluid and molecular biomarkers
NfL concentrations in plasma and CSF are consistently higher in MSA than in PD and correlate with disease severity, rate of progression, and survival [72,73]. α-Synuclein seed amplification assays, while highly sensitive for Lewy body disorders, are less sensitive in MSA due to the distinct α-synuclein strains. However, ongoing refinements and alternative tissue sampling (e.g., skin) are improving sensitivity [11,74,75]. α-Synuclein PET tracers such as [18F]ACI-12589 have shown promising in vivo binding patterns in MSA and may enable pathology-specific diagnosis and staging in future clinical practice [76].
4.5. Pathology
Neuropathology reveals widespread oligodendroglial cytoplasmic inclusions composed of misfolded α-synuclein, accompanied by neuronal loss and gliosis in the striatonigral and olivopontocerebellar systems [77]. Lesion distribution explains the distinct motor phenotypes and associated imaging signatures. Emerging work highlights oligodendroglial vulnerability, myelin dysfunction, and innate immune activation as convergent disease mechanisms [59,75].
4.6. Treatment and management
There is no proven disease-modifying therapy for MSA. Parkinsonism may exhibit limited and transient benefit from levodopa, often constrained by the exacerbation of orthostatic hypotension. Management of neurogenic orthostatic hypotension involves fluid and salt supplementation, compression garments, head-up sleeping, and pharmacological agents such as midodrine, droxidopa, and, selectively, fludrocortisone [60]. Bladder dysfunction requires tailored interventions based on urodynamic findings, ranging from behavioral strategies to intermittent catheterization and intradetrusor botulinum toxin. Erectile dysfunction may respond to phosphodiesterase-5 inhibitors, provided that blood pressure is carefully monitored. Stridor is managed with early otolaryngology assessment, continuous positive airway pressure, or tracheostomy in severe cases, the latter associated with improved survival in selected patients [64]. REM sleep behavior disorder can be treated with melatonin or clonazepam. Multidisciplinary rehabilitation—including physiotherapy, occupational therapy, speech-language therapy, and nutritional support—should be initiated early to address gait instability, dysarthria, and dysphagia.
Several disease-modifying strategies are in clinical development. High-dose ubiquinol has shown modest slowing of motor decline in a phase 2 trial, supporting a possible role for coenzyme Q10 supplementation in genetically susceptible subgroups [61]. α-Synuclein immunotherapies, such as amlenotug (Lu AF82422), have demonstrated safety and target engagement in early-phase studies and are advancing to phase 3 evaluation [78]. The metal-binding modulator ATH434 has reported positive topline phase 2 results, with confirmatory trials underway [79]. As these therapies progress, the integration of the MDS diagnostic framework, quantitative imaging, and fluid biomarkers into recruitment strategies is becoming standard, allowing for earlier and biologically confirmed trial enrollment [2,59].
5. Discussion
Despite notable advances in clinical criteria, imaging, and biomarkers, the diagnosis of atypical parkinsonian disorders—PSP, CBD, and MSA—remains challenging, particularly during the early and prodromal phases when intervention might be most effective [1,2,46]. A comparative summary of the core clinical, pathological, and imaging features distinguishing PSP, CBS/CBD, MSA, and PD is provided in Table 1. This overview highlights the main elements that underlie diagnostic uncertainty and frame the subsequent discussion. The 2017 MDS-PSP and 2022 MDS-MSA criteria have expanded the phenotypic capture; however, clinicopathological correlation studies indicate that misclassification persists, with sensitivity and specificity varying across disease stages [1,2]. Moving from syndromic, probabilistic diagnosis to biologically anchored certainty will require the integration of multimodal evidence—disciplined clinical phenotyping, standardized oculomotor and autonomic testing, quantitative structural and molecular imaging, and fluid biomarkers—into harmonized diagnostic algorithms.
First, there is the problem of phenotypic overlap in the early phase. APDs often present with parkinsonism indistinguishable from Parkinson’s disease during the first years. Anchoring bias at the initial consultation, combined with the slow accrual of defining signs, leads to prolonged diagnostic latency [80]. This limitation underscores the need for longitudinal re-evaluation built into care pathways, with predefined review points and triggers for ancillary testing.
Second, the trade-off between sensitivity and specificity in diagnostic criteria remains unresolved. The broadened MDS-PSP criteria capture variant phenotypes (PSP-parkinsonism, PSP-CBS, and PSP-frontotemporal presentations) but show reduced specificity in the early stages of the disease [1]. The MDS-MSA framework defines “clinically probable” and “possible prodromal” categories, but validation in prospective, pathology-confirmed cohorts is incomplete [2]. Dynamic “living criteria” that incorporate imaging and biomarker thresholds, periodically recalibrated with autopsy-verified data, are needed.
Third, pathological pleomorphism—particularly in CBS—complicates clinicopathological correlation. While CBS is the most common presentation of CBD, it frequently results from PSP, AD, FTLD-TDP, or mixed pathologies [5]. Without amyloid and phospho-tau testing, or 4R tau–sensitive PET, distinguishing these entities remains unreliable. Incorporating such markers early could refine etiological diagnosis, stratify trial populations, and prevent the inclusion of inappropriate trial participants.
Fourth, imaging biomarkers are variably applied and not sufficiently standardized. Classical MRI signs—the hummingbird sign in PSP, the hot-cross-bun in MSA, asymmetric frontoparietal atrophy in CBD—remain supportive but nonspecific. Quantitative indices, such as MRPI 2.0, improve discrimination, particularly between PSP-parkinsonism and PD [6,7,8]. However, multicenter calibration, reproducible cut-offs, and open-source automated pipelines are needed for routine adoption.
Fifth, molecular imaging shows promise but lacks definitive clinical integration. Second-generation tau PET tracers such as [18F]PI-2620 and [18F]florzolotau have revealed characteristic uptake patterns in PSP/CBD, but require harmonization of acquisition protocols and disease-specific thresholds [33,34]. For MSA, α-synuclein PET ([18F]ACI-12589) offers an unprecedented pathology-specific signal [33,34,76]; however, larger studies must address confounders such as co-pathology and off-target binding.
Sixth, dopaminergic imaging has limited nosological resolution. DAT-SPECT confirms presynaptic nigrostriatal degeneration but cannot reliably distinguish PD from PSP or MSA [36]. Over-reliance on this modality risks overdiagnosis; it should be reserved for resolving the binary question of degenerative versus non-degenerative parkinsonism.
Seventh, autonomic and peripheral sympathetic markers remain underused. In the hands of experienced practitioners, cardiac 123I-MIBG scintigraphy supports the differentiation between Lewy body disorders and MSA/PSP [37], but its diagnostic accuracy is context-dependent. Comprehensive autonomic testing—utilizing tilt-table, Valsalva maneuver, and quantitative sudomotor axon reflex testing—should be incorporated into the early work-up where available.
Eighth, fluid biomarkers are supportive but not yet definitive. Plasma and CSF NfL levels are consistently elevated in APDs compared with PD and correlate with disease progression [72,73], but lack pathology specificity. Seed amplification assays for α-synuclein can identify Lewy body pathology but are often negative in MSA due to strain differences [81]. Multiplex biomarker panels integrating NfL with astrocytic and microglial markers could enhance discriminatory power.
Ninth, verification bias and scarcity of pathology-proven datasets undermine the external validity of diagnostic algorithms. Most validation studies rely on clinical diagnosis without neuropathological confirmation, perpetuating circular reasoning. Autopsy-linked, longitudinal cohorts, with interim biospecimen anchors, are needed to generate accurate diagnostic accuracy metrics.
Tenth, implementation barriers and inequitable access to advanced diagnostics create geographic and socioeconomic disparities in early diagnosis and trial enrolment. Resource-limited settings often lack access to quantitative MRI, tau PET, or autonomic laboratories. Hub-and-spoke referral models, shared-reading networks for advanced imaging, and the deployment of cost-effective minimal diagnostic panels could address these gaps. Machine learning applied to harmonized multimodal datasets may further enhance early-stage classification, provided that rigorous external validation is conducted and bias mitigation is explicitly addressed.
Future directions should prioritize integrating multimodal evidence into tiered algorithms, anchored in MDS criteria but refined with imaging, oculomotor, autonomic, and biomarker data. For PSP and CBD, combining MRPI 2.0 with 4R tau–sensitive PET and plasma/CSF NfL could raise diagnostic confidence years before classical signs emerge. For MSA, the combination of preserved cardiac 123I-MIBG uptake, characteristic MRI changes, high NfL, and emerging α-syn PET could achieve similar gains. CBS should routinely be screened for AD co-pathology using amyloid and p-tau markers, with 4R tau PET reserved for suspected primary tauopathies.
Ultimately, the shift from descriptive syndromes to biologically anchored diagnoses will depend on collaborative, standardized, and equity-conscious implementation of these tools, embedded in prospective, autopsy-verified cohorts. This approach offers the best prospect of shortening diagnostic delays, enriching disease-modifying trials with precisely characterized participants, and delivering earlier, more personalized care.
Author Contributions
Conceptualization, O.A.-C.; methodology, O.A.-C.; investigation, O.A.-C., E.O.-R. and E.R.-G.; resources, O.A.-C.; data curation, O.A.-C.; writing—original draft preparation, O.A.-C.; writing—review and editing, O.A.-C., E.O.-R. and E.R.-G.; visualization, E.O.-R. and E.R.-G.; supervision, O.A.-C.; project administration, O.A.-C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgements
The authors thank all colleagues and collaborators who contributed to this work. To those who consistently demonstrated the power of doing nothing — we did not wait.”
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| α-syn | Alpha-synuclein |
| 123I-IBZM | [123I]Iodobenzamide |
| 123I-MIBG | [123I]Metaiodobenzylguanidine |
| 4R tau | 4-repeat tauopathy |
| AD | Alzheimer’s disease |
| APD | Atypical parkinsonian disorder |
| AUC | Area under the receiver operating characteristic curve |
| CBS | Corticobasal syndrome |
| CBD | Corticobasal degeneration |
| CSF | Cerebrospinal fluid |
| DAT-SPECT | Dopamine transporter single-photon emission computed tomography |
| FDG-PET | [18F]Fluorodeoxyglucose positron emission tomography |
| FTD | Frontotemporal dementia |
| FTLD | Frontotemporal lobar degeneration |
| FTLD-TDP | FTLD with TPD-43-immunoreactive pathology |
| GSK-3β | Glycogen synthase kinase-3 beta |
| MCP | Middle cerebellar peduncle |
| MDS | Movement Disorder Society |
| MRI | Magnetic resonance imaging |
| MRPI 2.0 | Magnetic Resonance Parkinsonism Index 2.0 |
| MSA | Multiple system atrophy |
| MSA-C | Multiple system atrophy, cerebellar phenotype |
| MSA-P | Multiple system atrophy, parkinsonian phenotype |
| NfL | Neurofilament light chain |
| PD | Parkinson’s disease |
| PET | Positron emission tomography |
| PSP | Progressive supranuclear palsy |
| PSP-CBS | PSP with corticobasal syndrome |
| PSP-FTD | PSP with frontotemporal dementia |
| PSP-P | PSP-parkinsonism |
| PSP-PGF | PSP with predominant gait freezing |
| PSP-RS | PSP–Richardson’s syndrome |
References
- Hoglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Muller, U.; Nilsson, C.; Whitwell, J.L., et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord 2017, 32, 853-864. [CrossRef]
- Wenning, G.K.; Stankovic, I.; Vignatelli, L.; Fanciulli, A.; Calandra-Buonaura, G.; Seppi, K.; Palma, J.A.; Meissner, W.G.; Krismer, F.; Berg, D., et al. The Movement Disorder Society Criteria for the Diagnosis of Multiple System Atrophy. Mov Disord 2022, 37, 1131-1148. [CrossRef]
- Iankova, V.; Respondek, G.; Saranza, G.; Painous, C.; Camara, A.; Compta, Y.; Aiba, I.; Balint, B.; Giagkou, N.; Josephs, K.A., et al. Video-tutorial for the Movement Disorder Society criteria for progressive supranuclear palsy. Parkinsonism Relat Disord 2020, 78, 200-203. [CrossRef]
- Wilson, D.; Le Heron, C.; Anderson, T. Corticobasal syndrome: a practical guide. Practical Neurology 2021, 21, 276-285. [CrossRef]
- Nouh, C.D.; Younes, K. Diagnosis and Management of Progressive Corticobasal Syndrome. Curr. Treat. Options Neurol. 2024, 26, 319-338. [CrossRef]
- Ortega-Robles, E.; de Celis Alonso, B.; Cantillo-Negrete, J.; Carino-Escobar, R.I.; Arias-Carrion, O. Advanced Magnetic Resonance Imaging for Early Diagnosis and Monitoring of Movement Disorders. Brain Sci 2025, 15. [CrossRef]
- Quattrone, A.; Bianco, M.G.; Antonini, A.; Vaillancourt, D.E.; Seppi, K.; Ceravolo, R.; Strafella, A.P.; Tedeschi, G.; Tessitore, A.; Cilia, R., et al. Development and Validation of Automated Magnetic Resonance Parkinsonism Index 2.0 to Distinguish Progressive Supranuclear Palsy-Parkinsonism From Parkinson's Disease. Mov Disord 2022, 37, 1272-1281. [CrossRef]
- Quattrone, A.; Morelli, M.; Nigro, S.; Quattrone, A.; Vescio, B.; Arabia, G.; Nicoletti, G.; Nistico, R.; Salsone, M.; Novellino, F., et al. A new MR imaging index for differentiation of progressive supranuclear palsy-parkinsonism from Parkinson's disease. Parkinsonism Relat Disord 2018, 54, 3-8. [CrossRef]
- Catalan, M.; Dore, F.; Polverino, P.; Bertolotti, C.; Sartori, A.; Antonutti, L.; Cucca, A.; Furlanis, G.; Capitanio, S.; Manganotti, P. (123)I-Metaiodobenzylguanidine Myocardial Scintigraphy in Discriminating Degenerative Parkinsonisms. Mov. Disord. Clin. Pract. 2021, 8, 717-724. [CrossRef]
- Demiri, S.; Veltsista, D.; Siokas, V.; Spiliopoulos, K.C.; Tsika, A.; Stamati, P.; Chroni, E.; Dardiotis, E.; Liampas, I. Neurofilament Light Chain in Cerebrospinal Fluid and Blood in Multiple System Atrophy: A Systematic Review and Meta-Analysis. Brain Sci 2025, 15. [CrossRef]
- Arias-Carrion, O.; Guerra-Crespo, M.; Padilla-Godinez, F.J.; Soto-Rojas, L.O.; Manjarrez, E. alpha-Synuclein Pathology in Synucleinopathies: Mechanisms, Biomarkers, and Therapeutic Challenges. Int J Mol Sci 2025, 26. [CrossRef]
- Fernandes Gomes, B.; Farris, C.M.; Ma, Y.; Concha-Marambio, L.; Lebovitz, R.; Nellgard, B.; Dalla, K.; Constantinescu, J.; Constantinescu, R.; Gobom, J., et al. alpha-Synuclein seed amplification assay as a diagnostic tool for parkinsonian disorders. Parkinsonism Relat Disord 2023, 117, 105807. [CrossRef]
- Giannakis, A.; Konitsiotis, S.; Sioka, C. Differentiating Progressive Supranuclear Palsy and Corticobasal Syndrome: Insights from Cerebrospinal Fluid Biomarkers-A Narrative Review. Medicina (Kaunas) 2025, 61. [CrossRef]
- Gogola, A.; Lopresti, B.J.; Minhas, D.S.; Lopez, O.; Cohen, A.; Villemagne, V.L. Tau Imaging: Use and Implementation in New Diagnostic and Therapeutic Paradigms for Alzheimer's Disease. Geriatrics (Basel) 2025, 10. [CrossRef]
- Mena, A.M.; Strafella, A.P. Imaging pathological tau in atypical parkinsonisms: A review. Clin Park Relat Disord 2022, 7, 100155. [CrossRef]
- Stamelou, M.; Respondek, G.; Giagkou, N.; Whitwell, J.L.; Kovacs, G.G.; Hoglinger, G.U. Evolving concepts in progressive supranuclear palsy and other 4-repeat tauopathies. Nat. Rev. Neurol. 2021, 17, 601-620. [CrossRef]
- Barer, Y.; Chodick, G.; Cohen, R.; Grabarnik-John, M.; Ye, X.; Zamudio, J.; Gurevich, T. Epidemiology of Progressive Supranuclear Palsy: Real World Data from the Second Largest Health Plan in Israel. Brain Sci 2022, 12. [CrossRef]
- Lyons, S.; Trepel, D.; Lynch, T.; Walsh, R.; O'Dowd, S. The prevalence and incidence of progressive supranuclear palsy and corticobasal syndrome: a systematic review and meta-analysis. J. Neurol. 2023, 270, 4451-4465. [CrossRef]
- Street, D.; Bevan-Jones, W.R.; Malpetti, M.; Jones, P.S.; Passamonti, L.; Ghosh, B.C.; Rittman, T.; Coyle-Gilchrist, I.T.; Allinson, K.; Dawson, C.E., et al. Structural correlates of survival in progressive supranuclear palsy. Parkinsonism Relat Disord 2023, 116, 105866. [CrossRef]
- Nysetvold, E.; Lopez, L.N.; Cogell, A.N.; Fryk, H.; Pace, N.D.; Taylor, S.S.; Rhoden, J.; Nichols, C.A.; Pillas, D.; Klein, A., et al. Progressive Supranuclear palsy (PSP) disease progression, management, and healthcare resource utilization: a retrospective observational study in the US and Canada. Orphanet J. Rare Dis. 2024, 19, 215. [CrossRef]
- Chen, J.A.; Chen, Z.; Won, H.; Huang, A.Y.; Lowe, J.K.; Wojta, K.; Yokoyama, J.S.; Bensimon, G.; Leigh, P.N.; Payan, C., et al. Joint genome-wide association study of progressive supranuclear palsy identifies novel susceptibility loci and genetic correlation to neurodegenerative diseases. Mol. Neurodegener. 2018, 13, 41. [CrossRef]
- Farrell, K.; Humphrey, J.; Chang, T.; Zhao, Y.; Leung, Y.Y.; Kuksa, P.P.; Patil, V.; Lee, W.P.; Kuzma, A.B.; Valladares, O., et al. Genetic, transcriptomic, histological, and biochemical analysis of progressive supranuclear palsy implicates glial activation and novel risk genes. Nat. Commun. 2024, 15, 7880. [CrossRef]
- Hoglinger, G.U.; Melhem, N.M.; Dickson, D.W.; Sleiman, P.M.; Wang, L.S.; Klei, L.; Rademakers, R.; de Silva, R.; Litvan, I.; Riley, D.E., et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat. Genet. 2011, 43, 699-705. [CrossRef]
- Sanchez-Contreras, M.Y.; Kouri, N.; Cook, C.N.; Serie, D.J.; Heckman, M.G.; Finch, N.A.; Caselli, R.J.; Uitti, R.J.; Wszolek, Z.K.; Graff-Radford, N., et al. Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol. Neurodegener. 2018, 13, 37. [CrossRef]
- Wang, H.; Chang, T.S.; Dombroski, B.A.; Cheng, P.L.; Patil, V.; Valiente-Banuet, L.; Farrell, K.; McLean, C.; Molina-Porcel, L.; Rajput, A., et al. Whole-genome sequencing analysis reveals new susceptibility loci and structural variants associated with progressive supranuclear palsy. Mol. Neurodegener. 2024, 19, 61. [CrossRef]
- Cullinane, P.W.; Fumi, R.; Theilmann Jensen, M.; Jabbari, E.; Warner, T.T.; Revesz, T.; Morris, H.R.; Rohrer, J.D.; Jaunmuktane, Z. MAPT-Associated Familial Progressive Supranuclear Palsy with Typical Corticobasal Degeneration Neuropathology: A Clinicopathological Report. Mov. Disord. Clin. Pract. 2023, 10, 691-694. [CrossRef]
- Liu, F.T.; Lu, J.Y.; Li, X.Y.; Liang, X.N.; Jiao, F.Y.; Ge, J.J.; Wu, P.; Li, G.; Shen, B.; Wu, B., et al. (18)F-Florzolotau PET imaging captures the distribution patterns and regional vulnerability of tau pathology in progressive supranuclear palsy. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 1395-1405. [CrossRef]
- Chung, D.C.; Roemer, S.; Petrucelli, L.; Dickson, D.W. Cellular and pathological heterogeneity of primary tauopathies. Mol. Neurodegener. 2021, 16, 57. [CrossRef]
- Ichikawa-Escamilla, E.; Velasco-Martinez, R.A.; Adalid-Peralta, L. Progressive Supranuclear Palsy Syndrome: An Overview. IBRO Neurosci Rep 2024, 16, 598-608. [CrossRef]
- Chovatiya, H.; Pillai, K.; Reddy, C.; Thalakkattu, A.; Avarachan, A.; Chacko, M.; Kishore, A. Video-Oculography for Enhancing the Diagnostic Accuracy of Early Oculomotor Dysfunction in Progressive Supranuclear Palsy. J. Mov. Disord. 2025, 18, 77-86. [CrossRef]
- Facchin, A.; Buonocore, J.; Crasa, M.; Quattrone, A.; Quattrone, A. Systematic assessment of square-wave jerks in progressive supranuclear palsy: a video-oculographic study. J. Neurol. 2024, 271, 6639-6646. [CrossRef]
- Wunderlich, J.; Behler, A.; Dreyhaupt, J.; Ludolph, A.C.; Pinkhardt, E.H.; Kassubek, J. Diagnostic value of video-oculography in progressive supranuclear palsy: a controlled study in 100 patients. J. Neurol. 2021, 268, 3467-3475. [CrossRef]
- Brendel, M.; Barthel, H.; van Eimeren, T.; Marek, K.; Beyer, L.; Song, M.; Palleis, C.; Gehmeyr, M.; Fietzek, U.; Respondek, G., et al. Assessment of 18F-PI-2620 as a Biomarker in Progressive Supranuclear Palsy. JAMA Neurol. 2020, 77, 1408-1419. [CrossRef]
- Hong, J.; Lu, J.; Liu, F.; Wang, M.; Li, X.; Clement, C.; Lopes, L.; Brendel, M.; Rominger, A.; Yen, T.C., et al. Uncovering distinct progression patterns of tau deposition in progressive supranuclear palsy using [(18)F]Florzolotau PET imaging and subtype/stage inference algorithm. EBioMedicine 2023, 97, 104835. [CrossRef]
- Messerschmidt, K.; Barthel, H.; Brendel, M.; Scherlach, C.; Hoffmann, K.T.; Rauchmann, B.S.; Rullmann, M.; Marek, K.; Villemagne, V.L.; Rumpf, J.J., et al. (18)F-PI-2620 Tau PET Improves the Imaging Diagnosis of Progressive Supranuclear Palsy. J. Nucl. Med. 2022, 63, 1754-1760. [CrossRef]
- Wallert, E.D.; van de Giessen, E.; Knol, R.J.J.; Beudel, M.; de Bie, R.M.A.; Booij, J. Imaging Dopaminergic Neurotransmission in Neurodegenerative Disorders. J. Nucl. Med. 2022, 63, 27S-32S. [CrossRef]
- Ebina, J.; Mizumura, S.; Shibukawa, M.; Morioka, H.; Nagasawa, J.; Yanagihashi, M.; Hirayama, T.; Ishii, N.; Kobayashi, Y.; Inaba, A., et al. Comparison of MIBG uptake in the major salivary glands between Lewy body disease and progressive supranuclear palsy. Clin Park Relat Disord 2024, 11, 100287. [CrossRef]
- Pitton Rissardo, J.; Fornari Caprara, A.L. Cardiac 123I-Metaiodobenzylguanidine (MIBG) Scintigraphy in Parkinson's Disease: A Comprehensive Review. Brain Sci 2023, 13. [CrossRef]
- Baiardi, S.; Rossi, M.; Giannini, G.; Mammana, A.; Polischi, B.; Sambati, L.; Mastrangelo, A.; Magliocchetti, F.; Cortelli, P.; Capellari, S., et al. Head-to-head comparison of four cerebrospinal fluid and three plasma neurofilament light chain assays in Parkinsonism. NPJ Parkinsons Dis 2025, 11, 98. [CrossRef]
- Wang, S.Y.; Chen, W.; Xu, W.; Li, J.Q.; Hou, X.H.; Ou, Y.N.; Yu, J.T.; Tan, L. Neurofilament Light Chain in Cerebrospinal Fluid and Blood as a Biomarker for Neurodegenerative Diseases: A Systematic Review and Meta-Analysis. J. Alzheimers Dis. 2019, 72, 1353-1361. [CrossRef]
- Zhang, P.; Chen, J.; Cai, T.; He, C.; Li, Y.; Li, X.; Chen, Z.; Wang, L.; Zhang, Y. Quantitative susceptibility mapping and blood neurofilament light chain differentiate between parkinsonian disorders. Front Aging Neurosci 2022, 14, 909552. [CrossRef]
- Apetauerova, D.; Scala, S.A.; Hamill, R.W.; Simon, D.K.; Pathak, S.; Ruthazer, R.; Standaert, D.G.; Yacoubian, T.A. CoQ10 in progressive supranuclear palsy: A randomized, placebo-controlled, double-blind trial. Neurol Neuroimmunol Neuroinflamm 2016, 3, e266. [CrossRef]
- Tolosa, E.; Litvan, I.; Hoglinger, G.U.; Burn, D.; Lees, A.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; Del Ser, T.; Investigators, T. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord 2014, 29, 470-478. [CrossRef]
- Hoglinger, G.U.; Litvan, I.; Mendonca, N.; Wang, D.; Zheng, H.; Rendenbach-Mueller, B.; Lon, H.K.; Jin, Z.; Fisseha, N.; Budur, K., et al. Safety and efficacy of tilavonemab in progressive supranuclear palsy: a phase 2, randomized, placebo-controlled trial. Lancet Neurol 2021, 20, 182-192. [CrossRef]
- Rowe, J.B.; Holland, N.; Rittman, T. Progressive supranuclear palsy: diagnosis and management. Pract Neurol 2021, 21, 376-383. [CrossRef]
- Armstrong, M.J.; Litvan, I.; Lang, A.E.; Bak, T.H.; Bhatia, K.P.; Borroni, B.; Boxer, A.L.; Dickson, D.W.; Grossman, M.; Hallett, M., et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013, 80, 496-503. [CrossRef]
- Lee, S.E.; Rabinovici, G.D.; Mayo, M.C.; Wilson, S.M.; Seeley, W.W.; DeArmond, S.J.; Huang, E.J.; Trojanowski, J.Q.; Growdon, M.E.; Jang, J.Y., et al. Clinicopathological correlations in corticobasal degeneration. Ann. Neurol. 2011, 70, 327-340. [CrossRef]
- Aiba, I.; Hayashi, Y.; Shimohata, T.; Yoshida, M.; Saito, Y.; Wakabayashi, K.; Komori, T.; Hasegawa, M.; Ikeuchi, T.; Tokumaru, A.M., et al. Clinical course of pathologically confirmed corticobasal degeneration and corticobasal syndrome. Brain Commun 2023, 5, fcad296. [CrossRef]
- Valentino, R.R.; Koga, S.; Walton, R.L.; Soto-Beasley, A.I.; Kouri, N.; DeTure, M.A.; Murray, M.E.; Johnson, P.W.; Petersen, R.C.; Boeve, B.F., et al. MAPT subhaplotypes in corticobasal degeneration: assessing associations with disease risk, severity of tau pathology, and clinical features. Acta Neuropathol. Commun. 2020, 8, 218. [CrossRef]
- Yokoyama, J.S.; Karch, C.M.; Fan, C.C.; Bonham, L.W.; Kouri, N.; Ross, O.A.; Rademakers, R.; Kim, J.; Wang, Y.; Hoglinger, G.U., et al. Shared genetic risk between corticobasal degeneration, progressive supranuclear palsy, and frontotemporal dementia. Acta Neuropathol 2017, 133, 825-837. [CrossRef]
- Constantinides, V.C.; Paraskevas, G.P.; Paraskevas, P.G.; Stefanis, L.; Kapaki, E. Corticobasal degeneration and corticobasal syndrome: A review. Clin Park Relat Disord 2019, 1, 66-71. [CrossRef]
- Mimuro, M.; Iwasaki, Y. Age-Related Pathology in Corticobasal Degeneration. Int J Mol Sci 2024, 25. [CrossRef]
- Wenning, G.K.; Litvan, I.; Jankovic, J.; Granata, R.; Mangone, C.A.; McKee, A.; Poewe, W.; Jellinger, K.; Ray Chaudhuri, K.; D'Olhaberriague, L., et al. Natural history and survival of 14 patients with corticobasal degeneration confirmed at postmortem examination. J Neurol Neurosurg Psychiatry 1998, 64, 184-189. [CrossRef]
- Armstrong, R.A. Visual signs and symptoms of corticobasal degeneration. Clin Exp Optom 2016, 99, 498-506. [CrossRef]
- Jellinger, K.A. The Spectrum of Cognitive Impairment in Atypical Parkinsonism Syndromes: A Comprehensive Review of Current Understanding and Research. Diseases 2025, 13. [CrossRef]
- Mathew, R.; Bak, T.H.; Hodges, J.R. Diagnostic criteria for corticobasal syndrome: a comparative study. J Neurol Neurosurg Psychiatry 2012, 83, 405-410. [CrossRef]
- Unti, E.; Mazzucchi, S.; Calabrese, R.; Palermo, G.; Del Prete, E.; Bonuccelli, U.; Ceravolo, R. Botulinum toxin for the treatment of dystonia and pain in corticobasal syndrome. Brain Behav. 2019, 9, e01182. [CrossRef]
- Chelban, V.; Catereniuc, D.; Aftene, D.; Gasnas, A.; Vichayanrat, E.; Iodice, V.; Groppa, S.; Houlden, H. An update on MSA: premotor and non-motor features open a window of opportunities for early diagnosis and intervention. J. Neurol. 2020, 267, 2754-2770. [CrossRef]
- Krismer, F.; Wenning, G.K. Multiple system atrophy: insights into a rare and debilitating movement disorder. Nat. Rev. Neurol. 2017, 13, 232-243. [CrossRef]
- Fanciulli, A.; Wenning, G.K. Multiple-system atrophy. N. Engl. J. Med. 2015, 372, 249-263. [CrossRef]
- Mitsui, J.; Tsuji, S. Plasma Coenzyme Q10 Levels and Multiple System Atrophy-Reply. JAMA Neurol. 2016, 73, 1499-1500. [CrossRef]
- Porto, K.J.; Hirano, M.; Mitsui, J.; Chikada, A.; Matsukawa, T.; Ishiura, H.; Japan Multiple System Atrophy Registry, C.; Toda, T.; Kusunoki, S.; Tsuji, S. COQ2 V393A confers high risk susceptibility for multiple system atrophy in East Asian population. J. Neurol. Sci. 2021, 429, 117623. [CrossRef]
- Cortelli, P.; Calandra-Buonaura, G.; Benarroch, E.E.; Giannini, G.; Iranzo, A.; Low, P.A.; Martinelli, P.; Provini, F.; Quinn, N.; Tolosa, E., et al. Stridor in multiple system atrophy: Consensus statement on diagnosis, prognosis, and treatment. Neurology 2019, 93, 630-639. [CrossRef]
- Giannini, G.; Provini, F.; Cani, I.; Cecere, A.; Mignani, F.; Guaraldi, P.; Di Mirto, C.V.F.; Cortelli, P.; Calandra-Buonaura, G. Tracheostomy is associated with increased survival in Multiple System Atrophy patients with stridor. Eur. J. Neurol. 2022, 29, 2232-2240. [CrossRef]
- Krismer, F.; Fanciulli, A.; Meissner, W.G.; Coon, E.A.; Wenning, G.K. Multiple system atrophy: advances in pathophysiology, diagnosis, and treatment. Lancet Neurol 2024, 23, 1252-1266. [CrossRef]
- Arnone, A.; Allocca, M.; Di Dato, R.; Puccini, G.; Laghai, I.; Rubino, F.; Nerattini, M.; Ramat, S.; Lombardi, G.; Ferrari, C., et al. FDG PET in the differential diagnosis of degenerative parkinsonian disorders: usefulness of voxel-based analysis in clinical practice. Neurol. Sci. 2022, 43, 5333-5341. [CrossRef]
- Du, X.; Zhao, H.; Li, Y.; Dai, Y.; Gao, L.; Li, Y.; Fan, K.; Sun, Z.; Zhang, Y. The value of PET/CT in the diagnosis and differential diagnosis of Parkinson's disease: a dual-tracer study. NPJ Parkinsons Dis 2024, 10, 171. [CrossRef]
- Di Luca, D.G.; Perlmutter, J.S. Time for Clinical Dopamine Transporter Scans in Parkinsonism?: Not DAT Yet. Neurology 2024, 102, e209558. [CrossRef]
- Miyamoto, T.; Miyamoto, M. Reduced cardiac (123)I-MIBG uptake is a robust biomarker of Lewy body disease in isolated rapid eye movement sleep behaviour disorder. Brain Commun 2024, 6, fcae148. [CrossRef]
- Kimpinski, K.; Iodice, V.; Burton, D.D.; Camilleri, M.; Mullan, B.P.; Lipp, A.; Sandroni, P.; Gehrking, T.L.; Sletten, D.M.; Ahlskog, J.E., et al. The role of autonomic testing in the differentiation of Parkinson's disease from multiple system atrophy. J. Neurol. Sci. 2012, 317, 92-96. [CrossRef]
- Pena-Zelayeta, L.; Delgado-Minjares, K.M.; Villegas-Rojas, M.M.; Leon-Arcia, K.; Santiago-Balmaseda, A.; Andrade-Guerrero, J.; Perez-Segura, I.; Ortega-Robles, E.; Soto-Rojas, L.O.; Arias-Carrion, O. Redefining Non-Motor Symptoms in Parkinson's Disease. J. Pers. Med. 2025, 15. [CrossRef]
- Chelban, V.; Nikram, E.; Perez-Soriano, A.; Wilke, C.; Foubert-Samier, A.; Vijiaratnam, N.; Guo, T.; Jabbari, E.; Olufodun, S.; Gonzalez, M., et al. Neurofilament light levels predict clinical progression and death in multiple system atrophy. Brain 2022, 145, 4398-4408. [CrossRef]
- Singer, W.; Schmeichel, A.M.; Sletten, D.M.; Gehrking, T.L.; Gehrking, J.A.; Trejo-Lopez, J.; Suarez, M.D.; Anderson, J.K.; Bass, P.H.; Lesnick, T.G., et al. Neurofilament light chain in spinal fluid and plasma in multiple system atrophy: a prospective, longitudinal biomarker study. Clin. Auton. Res. 2023, 33, 635-645. [CrossRef]
- Gibbons, C.; Wang, N.; Rajan, S.; Kern, D.; Palma, J.A.; Kaufmann, H.; Freeman, R. Cutaneous alpha-Synuclein Signatures in Patients With Multiple System Atrophy and Parkinson Disease. Neurology 2023, 100, e1529-e1539. [CrossRef]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G., et al. Discriminating alpha-synuclein strains in Parkinson's disease and multiple system atrophy. Nature 2020, 578, 273-277. [CrossRef]
- Smith, R.; Capotosti, F.; Schain, M.; Ohlsson, T.; Vokali, E.; Molette, J.; Touilloux, T.; Hliva, V.; Dimitrakopoulos, I.K.; Puschmann, A., et al. The alpha-synuclein PET tracer [18F] ACI-12589 distinguishes multiple system atrophy from other neurodegenerative diseases. Nat. Commun. 2023, 14, 6750. [CrossRef]
- Valera, E.; Masliah, E. The neuropathology of multiple system atrophy and its therapeutic implications. Auton. Neurosci. 2018, 211, 1-6. [CrossRef]
- Buur, L.; Wiedemann, J.; Larsen, F.; Ben Alaya-Fourati, F.; Kallunki, P.; Ditlevsen, D.K.; Sorensen, M.H.; Meulien, D. Randomized Phase I Trial of the alpha-Synuclein Antibody Lu AF82422. Mov Disord 2024, 39, 936-944. [CrossRef]
- Alterity Therapeutics. Alterity Therapeutics Reports Positive Topline Data from Open-Label Phase 2 Clinical Trial of ATH434 in Multiple System Atrophy. Availabe online: https://www.globenewswire.com/news-release/2025/07/28/3122359/0/en/Alterity-Therapeutics-Reports-Positive-Topline-Data-from-Open-Label-Phase-2-Clinical-Trial-of-ATH434-in-Multiple-System-Atrophy.html (accessed on 2025/09/08).
- Respondek, G.; Kurz, C.; Arzberger, T.; Compta, Y.; Englund, E.; Ferguson, L.W.; Gelpi, E.; Giese, A.; Irwin, D.J.; Meissner, W.G., et al. Which ante mortem clinical features predict progressive supranuclear palsy pathology? Mov Disord 2017, 32, 995-1005. [CrossRef]
- Concha-Marambio, L.; Pritzkow, S.; Shahnawaz, M.; Farris, C.M.; Soto, C. Seed amplification assay for the detection of pathologic alpha-synuclein aggregates in cerebrospinal fluid. Nat. Protoc. 2023, 18, 1179-1196. [CrossRef]
Figure 1.
Diagnostic flowchart for progressive supranuclear palsy (PSP), based on the Movement Disorder Society (MDS) criteria, outlining inclusion criteria, core clinical domains, supportive features, and longitudinal review to classify cases as probable or possible.
Figure 1.
Diagnostic flowchart for progressive supranuclear palsy (PSP), based on the Movement Disorder Society (MDS) criteria, outlining inclusion criteria, core clinical domains, supportive features, and longitudinal review to classify cases as probable or possible.
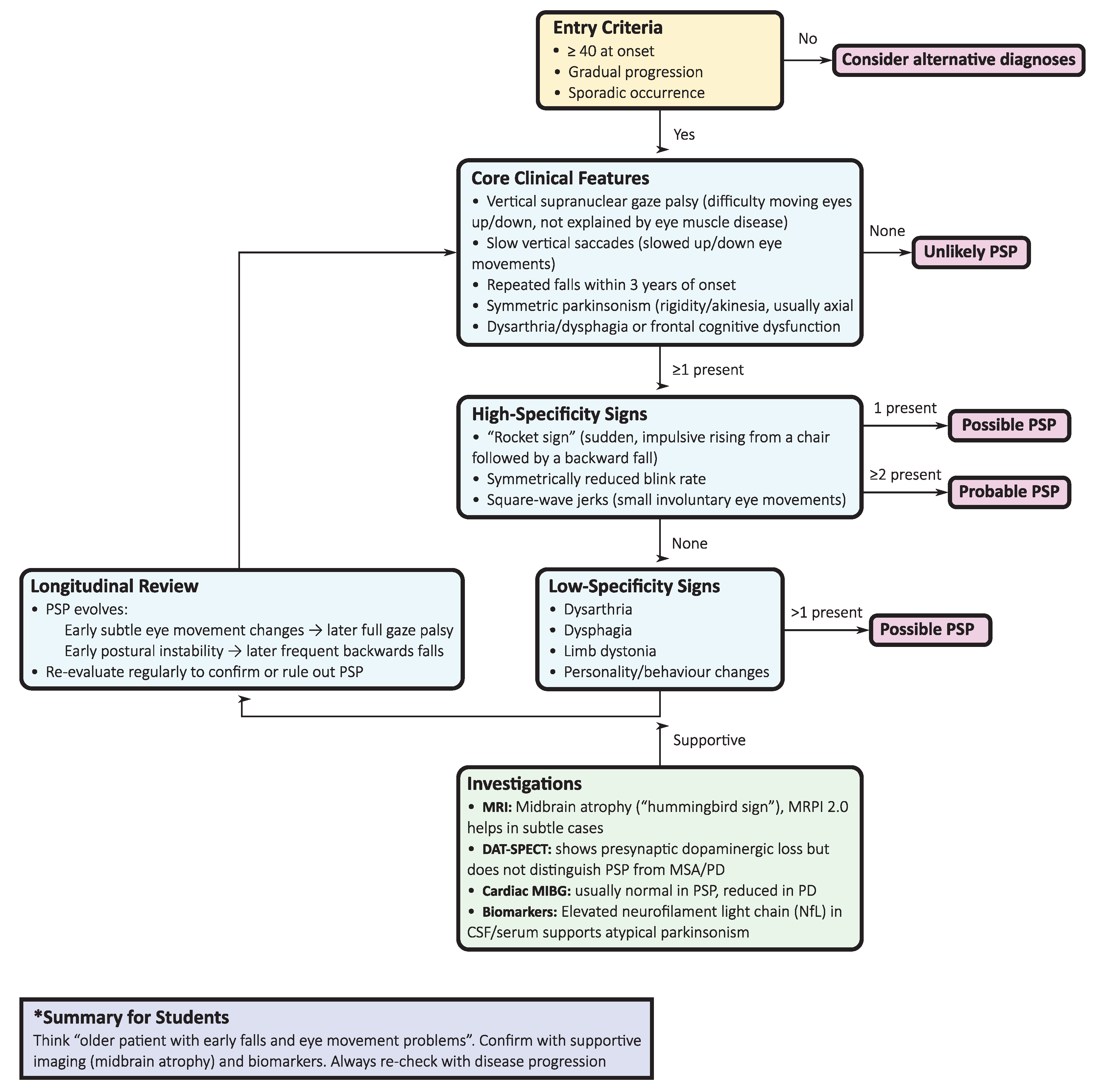
Figure 2.
Diagnostic flowchart for corticobasal syndrome (CBS). The algorithm summarizes clinical entry criteria, core motor and cortical features, diagnostic certainty (probable vs. possible corticobasal degeneration), supportive findings, exclusion red flags, and the need for longitudinal reassessment.
Figure 2.
Diagnostic flowchart for corticobasal syndrome (CBS). The algorithm summarizes clinical entry criteria, core motor and cortical features, diagnostic certainty (probable vs. possible corticobasal degeneration), supportive findings, exclusion red flags, and the need for longitudinal reassessment.
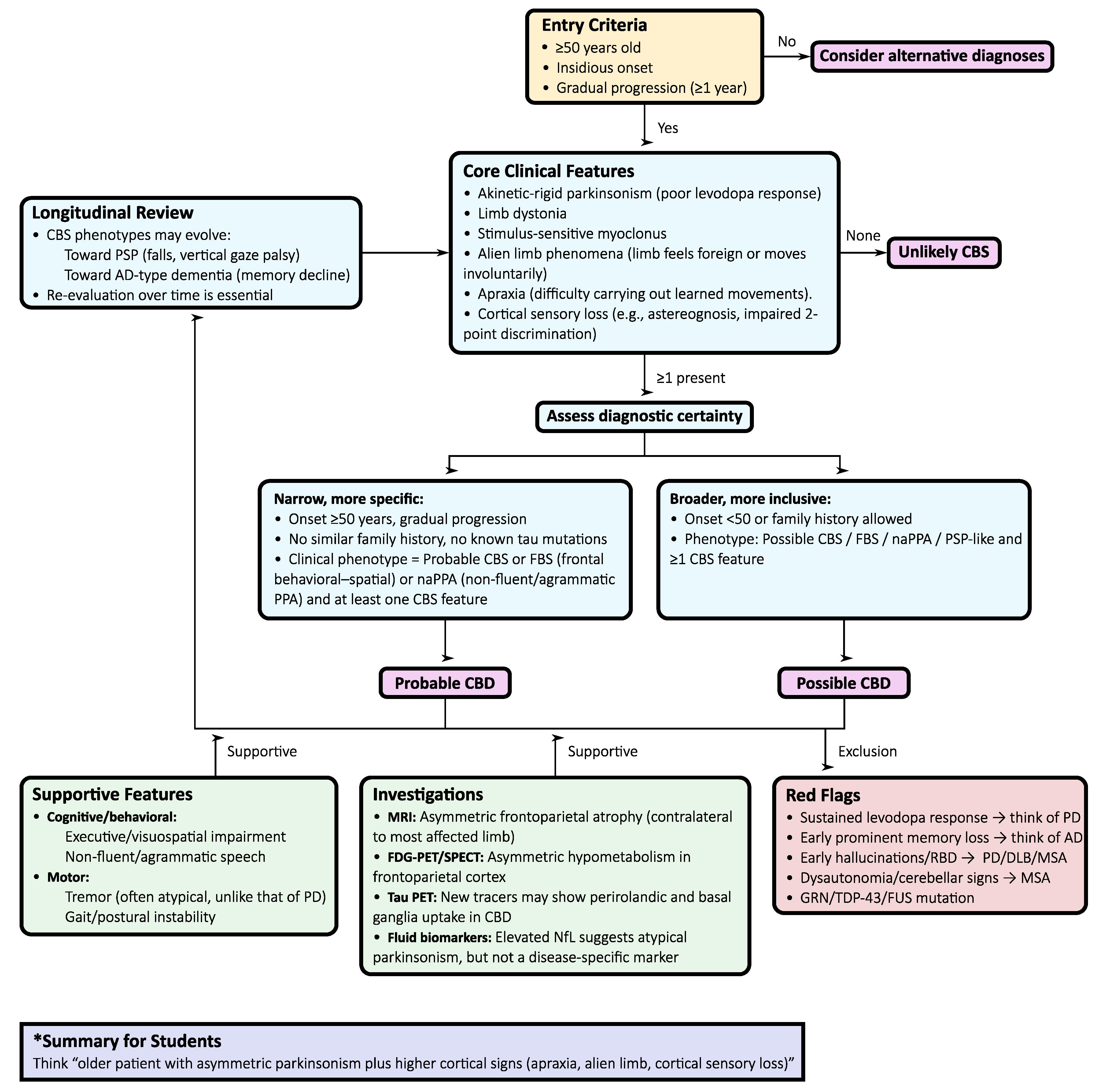
Figure 3.
Diagnostic flowchart for multiple system atrophy (MSA) according to the 2022 Movement Disorder Society criteria. The flowchart summarizes the stepwise evaluation of entry features, core clinical domains, supportive findings, MRI markers, and exclusion criteria to differentiate clinically established, clinically probable, and possible prodromal MSA.
Figure 3.
Diagnostic flowchart for multiple system atrophy (MSA) according to the 2022 Movement Disorder Society criteria. The flowchart summarizes the stepwise evaluation of entry features, core clinical domains, supportive findings, MRI markers, and exclusion criteria to differentiate clinically established, clinically probable, and possible prodromal MSA.
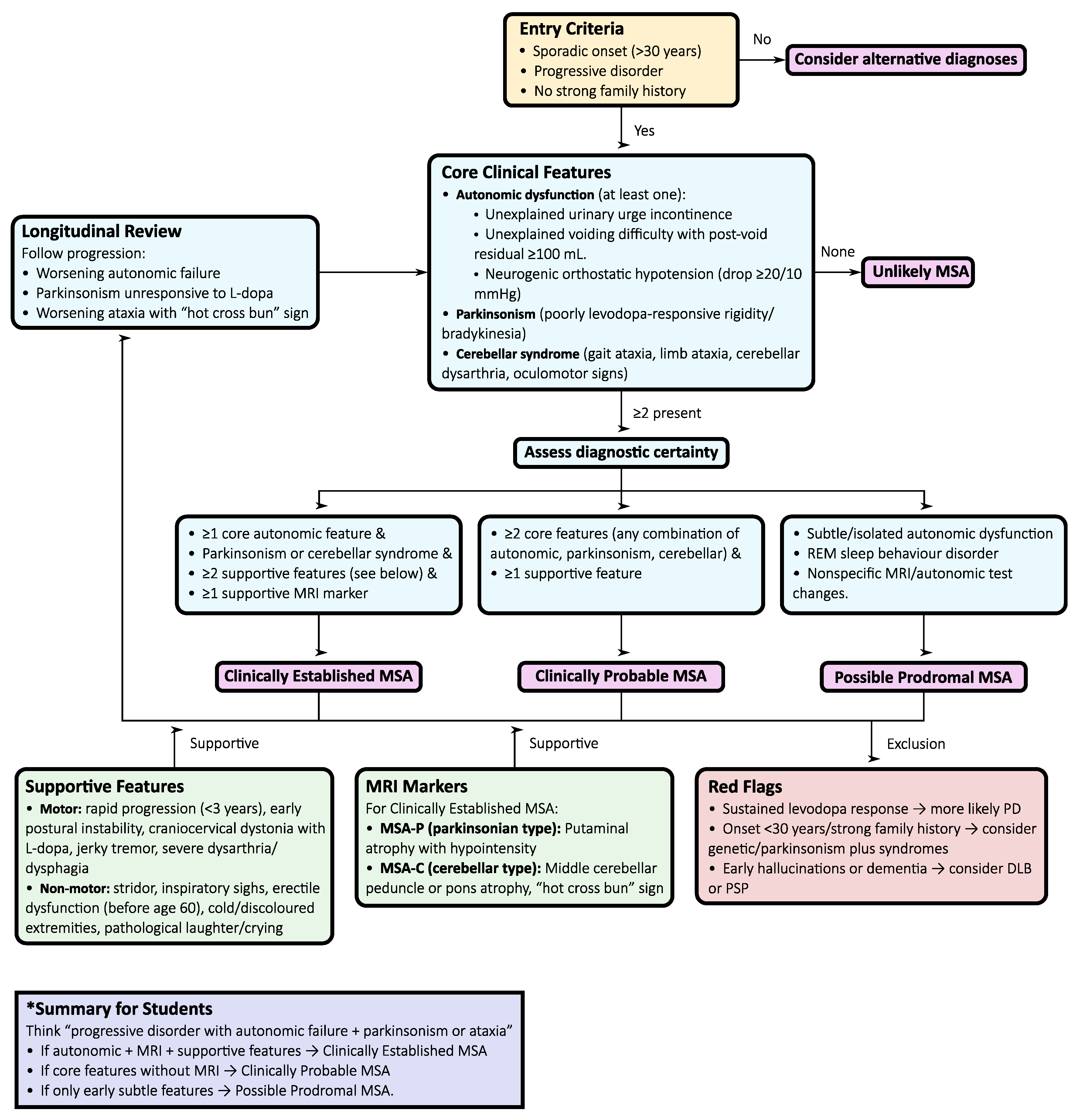

Table 1.
Differential Diagnosis: Parkinson’s Disease vs Atypical Parkinsonisms.
| Feature | Progressive Supranuclear Palsy (PSP) | Corticobasal Syndrome (CBS/CBD) | Multiple System Atrophy (MSA) | Parkinson’s Disease (PD) |
|---|---|---|---|---|
| Prevalence | ~7 per 100,000 | ~4 per 100,000 | ~4 per 100,000 | Much higher than atypical parkinsonisms |
| Onset Age | ~63 years | >60 years, variable | 53–55 years | Typically >60 years |
| Motor Symptoms | Symmetric parkinsonism, early axial rigidity, backwards falls | Asymmetric rigidity, dystonia, myoclonus, apraxia | MSA-P: symmetric parkinsonism (poor levodopa response); MSA-C: cerebellar signs (ataxia, dysarthria) | Asymmetric parkinsonism, classic rest tremor, shuffling gait |
| Ocular Signs | Vertical supranuclear gaze palsy, slow vertical saccades | Difficulty initiating voluntary saccades, gaze apraxia | Rare, nonspecific ocular signs | Rare ocular involvement |
| Cognitive Profile | Subcortical dementia (executive dysfunction, slowed processing) | Frontal-executive and parietal dysfunction; alien limb; cortical sensory loss | Cognitive impairment may occur, but not early or prominent | Cognitive impairment usually occurs late (dementia in advanced PD) |
| Autonomic Dysfunction | Not prominent early; may appear late | Rare or mild | Prominent: orthostatic hypotension, urinary incontinence/retention, erectile dysfunction, constipation | Mild compared with MSA |
| Key Pathology | 4R-tauopathy (globose tangles, tufted astrocytes) | 4R-tauopathy (astrocytic plaques, ballooned neurons) | α-synucleinopathy (glial cytoplasmic inclusions) | α-synucleinopathy (Lewy bodies) |
| Imaging | Midbrain atrophy (“hummingbird sign”); ↑MRPI 2.0 | Asymmetric frontoparietal atrophy (contralateral to the affected limb) | “Hot cross bun” sign (pons), putaminal atrophy/hypointensity | Often, normal or nonspecific changes |
| Levodopa Response | Poor, transient at best | Poor or absent | Poor or transient (rare sustained response) | Good, especially early |
| Other Key Features | Early falls (<3 yrs), pseudobulbar palsy, reduced blink, square-wave jerks | Alien limb, cortical sensory loss, asymmetric apraxia | Early stridor, rapid progression, cold extremities, REM sleep behavior disorder | Classic pill-rolling tremor, clear honeymoon response to levodopa |
| Teaching tip for students | Falls and eye movement problems early | Asymmetric parkinsonism with cortical signs (apraxia, alien limb phenomenon). | Autonomic failure plus parkinsonism/ataxia | Asymmetric, tremor-dominant, levodopa-responsive |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



