Submitted:
10 September 2025
Posted:
10 September 2025
You are already at the latest version
Abstract
Background/Objectives: Despite decades of focus on the Somatic Mutation Theory (SMT), survival outcomes for cancer remain modest, with most therapies offering only marginal benefits. Mounting evidence suggests that mitochondrial dysfunction, rather than random mutations, represents the central initiating event in carcinogenesis. Methods: We applied a systems-based Root Cause Analysis (RCA) framework, adapted from engineering and healthcare, to trace modifiable upstream biological drivers of mitochondrial collapse. Literature spanning environmental, nutritional, metabolic, infectious, and hormonal domains was reviewed to identify initiating contributors. Results: We propose a three-layer model of cancer initiation: (1) upstream initiating drivers (toxins, nutrient deficiencies, chronic infections, hormonal disruption, psychosocial stress, and iatrogenic factors); (2) central bioenergetic collapse via mitochondrial dysfunction, oxidative stress, and metabolic instability; and (3) downstream oncogenic phenotype, encompassing genomic instability, inflammation, immune evasion, and tumor progression. RCA organizes these drivers into a coherent sequence, highlighting prevention and intervention leverage points.
Keywords:
integrative cancer metabolic therapy
; mitochondrial dysfunction
; root cause analysis
; orthomolecular medicine
; metabolic reprogramming
; oxidative stress
; high-dose intravenous vitamin C (HDIVC)
1. Introduction
1.1. Cancer: A Growing Global Burden and Therapeutic Challenge
Despite decades of intensive research and the approval of hundreds of cancer drugs, cancer remains a major global health challenge. Between 2000 and 2020, global cancer incidence has steadily increased, with projections indicating 28.4 million new cases annually by 2040—driven by aging populations and increased exposure to environmental carcinogens, especially in low- and middle-income countries [1,2,3].
Yet the most striking concern is not merely the rising incidence, but the disappointing real-world benefit of most modern cancer treatments. A growing body of large-scale analyses across multiple countries reveals a sobering reality: the mutation-centric model has produced negligible survival gains despite decades of dominance, reflecting a systemic failure of paradigm rather than a limitation of technology.
Key findings from major reviews include:
- FDA (2000–2016): Among 90 approved cancer drugs, the median overall survival (OS) benefit was only 2.4 months, with most approvals based on tumor shrinkage—not life extension [4].
- UK Cancer Drugs Fund: Despite £1.3 billion in expenditures, most drugs showed no meaningful benefit in survival or quality of life [5].
- China (2005–2020): Among 68 approved drugs, 50% showed no survival benefit, and the rest extended life by only ~4 months, often with high toxicity [6].
- Nature (2017): Review of 277 global oncology trials found that 85% of drugs had no clinical benefit; the most expensive drugs often performed worst [7].
- Australia–US (2004): Cytotoxic chemotherapy contributed only ~2% to 5-year survival in adult solid tumors [8].
More recent reviews confirm this trend:
- Lancet Oncology (2024): Of 223 FDA approvals based on immature survival data, only 32% showed statistically significant OS benefit upon follow-up [9].
- Global Meta-Analysis (2023): Across nearly 400 approved drugs, only ~1/3 improved survival, while ~2/3 showed no meaningful benefit [10].
- Pooled RCTs (2023): Median OS benefit from 234 modern trials was just 2.8 months, reinforcing earlier findings [11].
These findings highlight a persistent mismatch between therapeutic promise and patient outcomes. Despite unprecedented financial investment and technological progress, most cancer drugs offer limited benefit—especially in terms of quality of life and long-term survival.
This widening disconnect suggests a critical flaw in the prevailing framework. Decades of emphasis on downstream molecular targets and somatic mutations have not translated into substantial progress. It is increasingly clear that cancer must be re-evaluated not as a genetic end-point, but as a biological systems failure rooted in earlier upstream disruptions—notably at the mitochondrial and metabolic levels [12].
Thomas Seyfried’s seminal work, Cancer as a Metabolic Disease [13,14], revives Otto Warburg’s foundational insight [15] that the defining hallmark of cancer is not genomic mutation, but mitochondrial dysfunction—a shift in energy metabolism that precedes genetic instability. Building on this foundation, we propose a systems-organizing model—Root Cause Analysis (RCA)—to help trace the upstream biological stressors and initiating drivers that impair mitochondrial function. Rather than replacing existing models, RCA serves as a complementary framework that seeks to redirect research, clinical strategy, and public health policy toward earlier, more effective, and preventive interventions.
1.2. The Hallmarks of Cancer: Describing What Cancer Does, Not How It Begins
In 2000 and again in 2011, Hanahan and Weinberg proposed the influential “Hallmarks of Cancer” framework [16,17], which conceptualizes cancer not as a singular genetic defect but as a set of acquired functional capabilities—such as sustained proliferative signaling, resistance to cell death, induction of angiogenesis, and immune evasion. These hallmarks have become a foundational reference in oncology, shaping both research and therapeutic development strategies over the past two decades.
However, while the Hallmarks framework provides a comprehensive description of how cancer behaves, it offers limited insight into how cancer originates. It catalogs the downstream phenotypic traits of malignant cells but does not address the upstream biological disruptions that give rise to these behaviors.
In this paper, we extend the Hallmarks framework by introducing a Root Cause Analysis (RCA) approach to explore how these hallmark traits emerge—tracing them to mitochondrial dysfunction as the central initiating mechanism, and further upstream to the environmental, nutritional, and inflammatory stressors that disrupt cellular energy metabolism.
This systems-level perspective does not challenge the existence of molecular mutations or hallmarks but rather seeks to organize and integrate them within a broader causal network—redirecting the focus from symptom management to early intervention on modifiable biological disruptions.
2. Conceptual Framework: Applying Root Cause Analysis (RCA) to Carcinogenesis
We applied a structured Root Cause Analysis (RCA) framework to clarify the layered contributors to cancer initiation, particularly those upstream of mitochondrial dysfunction. RCA, widely used in engineering and healthcare to identify preventable system failures, is here adapted to biological complexity. The purpose is not to redefine causality in absolute terms but to organize known contributors by temporal sequence, mechanistic impact, and clinical modifiability.
In this framework:
-
Upstream biological drivers include
- Environmental & Occupational Toxins;
- Dietary & Metabolic Stressors;
- Micronutrient Deficiencies;
- Chronic Infections & Immune Dysregulation;
- Hormonal Imbalance & Endocrine Disruption;
- Lifestyle & Behavioral Risk Factors;
- Psychosocial & Emotional Stress;
- Developmental & Early-Life Programming;
- Genetic & Epigenetic Susceptibility;
- Medical Iatrogenesis
- Central initiating mechanism is mitochondrial dysfunction, characterized by impaired oxidative phosphorylation, excess reactive oxygen species (ROS), metabolic destabilization, and immune dysregulation.
- Downstream clinical expression is cancer (malignancy), manifesting as histological transformation, uncontrolled proliferation, invasion, and tumor progression.
This layered model builds upon and complements the metabolic theory of cancer, which identifies mitochondrial dysfunction as the proximate cellular trigger of carcinogenesis [14]. Upstream factors—such as toxins, deficiencies, and infections—are best understood as modifiable stressors rather than immutable primary causes.
By integrating root-level drivers and metabolic intermediaries, this RCA-based approach provides a structured framework for patient-centered prevention, therapeutic strategies, and public health planning. A schematic overview is presented in Figure 1, and a complementary tabular summary in Table 1.
This framework builds upon and complements the metabolic theory of cancer, which posits mitochondrial dysfunction as the initiating event in carcinogenesis [14]. The upstream factors we examine—such as toxins, deficiencies, and infections—are better understood as contributory stressors rather than ontological primary causes. While they initiate damage, mitochondrial dysfunction remains the proximate cellular trigger, and these stressors represent modifiable drivers within the causal cascade.
By integrating both root-level triggers and metabolic intermediaries, this approach supports patient-centered strategies for cancer prevention, treatment, and public health planning—while remaining fully compatible with the core tenets of metabolic oncology.
2.1. Scientific Justification of RCA in Complex, Nonlinear Systems like Cancer
Some critics have questioned whether RCA—originally developed for engineered systems—can be validly applied to complex biological processes such as carcinogenesis. This skepticism often stems from a misunderstanding of RCA’s intent. Its core function is not to replace molecular mechanisms, but to map upstream contributors that precede and often precipitate system failure [18,19,20].
In nonlinear, adaptive systems like human biology, cancer arises from interactions among genetic, metabolic, immune, and environmental variables over time. Linear causality is often insufficient to explain emergent disease behavior [21,22,23].
Systems biology and complexity science support this layered causality:
- Convergent failure modes (e.g., mitochondrial collapse) can arise from diverse upstream inputs [23];
While the metabolic theory places mitochondrial dysfunction as the primary cause of cancer initiation, RCA does not contradict this view. Rather, it provides a contextual framework to trace what causes mitochondrial dysfunction—a layer of analysis that may include modifiable factors such as toxic exposures, nutrient deficiencies, infections, or hormonal disruption. These factors may not be "causes" in a strict ontological sense, but they often precede and precipitate the core metabolic collapse.
Thus, RCA:
- Complements reductionist models by embedding them in a systems logic;
- Supports mechanistic mapping from exposure to phenotype;
- Offers practical, testable hypotheses at molecular, cellular, and population levels.
Our model positions RCA not as a deterministic algorithm, but as a heuristic tool for identifying intervention points—especially those modifiable in the clinical or public health context. By doing so, it bridges the gap between the mechanistic insights of metabolic oncology and the complex, modifiable exposures that shape individual cancer risk.
3. Overview of Major Theories of Carcinogenesis
For over a century, researchers have proposed various frameworks to explain the origins of cancer. While diverse in mechanisms, most theories focus on observable downstream abnormalities such as uncontrolled proliferation, genetic instability, immune evasion, and metabolic shifts. However, few models attempt to systematically trace these abnormalities back to their upstream biological initiators—a gap this paper seeks to address by applying Root Cause Analysis (RCA) as a systems-organizing lens.
Below is a concise overview of the major competing and complementary theories that have shaped our understanding of cancer biology and influenced treatment paradigms:
3.1. Somatic Mutation Theory (SMT) – The Prevailing Paradigm
- Has dominated cancer research, drug development, and public messaging since the 1970s.
-
- Many tumors lack clear driver mutations.
- Fails to account for non-genetic causes of transformation.
- Targeted therapies based on this model often show limited durability and modest survival benefit.
- SMT has not only failed to account for the majority of cancers, but its dominance has actively delayed the exploration of more plausible upstream metabolic explanations.
RCA insight: SMT may describe cancer’s phenotype but fails to identify its initiating biological stressors or primary cellular event.
3.2. Viral and Infectious Theories
- Examples: HPV (cervical cancer), EBV (nasopharyngeal carcinoma), H. pylori (gastric cancer), HBV/HCV (hepatocellular carcinoma).
- Estimated to contribute to ~15–20% of global cancer burden, especially in low-income regions [28].
RCA insight: Chronic infections act as upstream biological stressors, impairing immune surveillance and destabilizing mitochondrial function, thus contributing to carcinogenesis.
3.3. Epigenetic Dysregulation
- Attributes cancer to reversible changes in gene expression—including DNA methylation, histone modification, and chromatin remodeling—rather than irreversible mutations [31].
-
Explains key cancer features:
- ○
- Cell plasticity
- ○
- Phenotypic heterogeneity
- ○
- Therapy resistance [32].
- Enabled therapeutic approaches such as HDAC inhibitors and DNA methyltransferase inhibitors [33].
Limitation: Epigenetic changes are often secondary responses to upstream stressors—such as environmental toxins, nutrient deficiencies, or inflammation—rather than initiating events [34,35].
RCA insight: Epigenetic dysregulation functions as an intermediary mechanism, not an initiating driver of malignancy.
3.4. Cancer Stem Cell (CSC) Theory
- Proposes that a subpopulation of stem-like tumor-initiating cells drives cancer initiation, progression, and recurrence [36].
-
CSCs exhibit:
- ○
- Self-renewal
- ○
- Metabolic flexibility
- ○
- Chemoresistance and radioresistance [37].
-
Notably, CSCs show metabolic traits consistent with the mitochondrial model:
- Their behavior is shaped by a permissive tumor microenvironment involving hypoxia, inflammation, and immune suppression [40].
RCA insight: CSC traits likely emerge in response to upstream metabolic collapse, with mitochondrial dysfunction serving as the central initiating mechanism.
3.5. Immune Surveillance and Immune Escape Theories
-
Forms the theoretical foundation for immunotherapies, including checkpoint inhibitors [16].Limitation: Immune dysfunction is often a consequence of upstream issues such as:
RCA insight: Immune evasion is a downstream effect; restoring immune surveillance requires correcting underlying metabolic and redox imbalances.
3.6. Mitochondrial Metabolic Theory (Warburg–Seyfried Model)
- Warburg first observed a metabolic shift from oxidative phosphorylation to aerobic glycolysis—known as the Warburg effect [15].
-
Seyfried extended the model by demonstrating:
- ○
- Mitochondrial damage precedes genetic instability
- ○
- Restoring mitochondrial function suppresses tumorigenesis—even in cells with nuclear mutations.
- Cytoplasmic–nuclear transfer experiments further show that healthy mitochondria can reverse tumorigenic potential.
- The ketogenic diet exploits this vulnerability—targeting cancer cells’ dependence on glucose and glutamine—but it does not fully address the upstream initiating drivers of cancer. Without simultaneous attention to toxins, infections, nutrient deficiencies, and hormonal disruption, the root causes of mitochondrial collapse remain uncorrected. Thus, diet should be seen as a critical but partial tool within a broader framework.
- While the MMT correctly centers mitochondrial dysfunction, it often risks being interpreted in isolation. This limitation sets the stage for expansion: mitochondria are not ultimate causes, but vulnerable sentinels shaped by upstream biological stressors.
Key strength: Unifies multiple cancer traits—including mutations, inflammation, immune evasion, and epigenetic drift—under a primary energy failure model driven by mitochondrial collapse.
3.7. Chromosomal Instability and Aneuploidy Theory
-
Aneuploidy induces:
Limitations:
- Poor predictive power in early tumor development.
- Fails to explain what initiates aneuploidy [47].
RCA insight: Mitochondrial dysfunction and oxidative stress can induce chromosomal instability, positioning aneuploidy as a secondary effect, not an initiating event [23].
3.8. Synthesis and Transition: Toward a Systems-Based RCA Framework
Each of the above models contributes valuable mechanistic insights. However, most focus on downstream abnormalities without accounting for the upstream initiating drivers that give rise to these disruptions.
By integrating these perspectives into a systems-organizing RCA framework, and positioning mitochondrial dysfunction as the primary cellular event, we propose a more coherent model that connects disparate theories. This approach redirects attention toward modifiable, real-world biological stressors—including environmental toxins, micronutrient insufficiencies, chronic inflammation, and infectious burden—that compromise mitochondrial function and initiate carcinogenesis.
This complementary framework does not reject existing models but instead aims to organize them along a unified biological progression, enabling earlier and more effective interventions—both clinically and in public health.
4. From Mutation to Metabolism—and Beyond
Cancer has long been conceptualized as a genetic disease, driven by the accumulation of somatic mutations. This mutation-centric view has dominated research for decades, yet survival benefits from gene-targeted therapies remain modest. The Warburg–Seyfried model corrects this by positioning mitochondrial dysfunction as the initiating event. However, it stops short of fully addressing what causes mitochondria to fail in the first place.
Our contribution is to extend the MMT using Root Cause Analysis (RCA), a systems-based framework for tracing initiating drivers. In this model, mitochondria are not ultimate causes but vulnerable sentinels responding to diverse upstream stressors. These initiating drivers include environmental pollutants, nutrient-depleted diets, chronic infections, psychosocial stressors, and iatrogenic medical interventions. RCA integrates these factors into a structured progression: initiating stressors → mitochondrial collapse → downstream hallmarks of malignancy.
This expanded framework not only clarifies the biological sequence but also highlights actionable prevention strategies grounded in lifestyle, nutrition, and environmental health. By shifting attention from mutation suppression to initiating-driver mitigation, we offer a more comprehensive and prevention-oriented model—one that is scientifically grounded, scalable, and aligned with public health goals.
5. Initiating Drivers of Mitochondrial Dysfunction: A Systems-Based Overview
A Root Cause Analysis (RCA) framework, when applied as a systems-organizing tool, urges us to look upstream of mitochondrial dysfunction to identify the biological stressors that initiate its decline. These initiating drivers compromise mitochondrial integrity, energy metabolism, and redox balance—setting the stage for the downstream hallmarks of cancer. Below, we organize these initiating contributors into ten modifiable domains relevant to both clinical intervention and public health strategy.
5.1. Environmental & Occupational Toxins
Action: Identify high-risk exposures, reduce contact, and support detoxification.
Environmental toxins are among the most pervasive and modifiable initiating stressors in carcinogenesis. They impair mitochondrial function, generate oxidative stress, damage DNA, and trigger chronic inflammation—creating a cellular environment conducive to malignant transformation.
Key contributors include:
- Nanoparticles & microplastics: industrial and food-chain exposure [50]
Systems insight: These exposures act as initiating biological stressors that compromise mitochondrial stability and immune function, often decades before clinical cancer appears.
5.2. Dietary & Metabolic Stressors
Action: Promote nutrient-dense, anti-inflammatory, low-carbohydrate diets to restore mitochondrial efficiency and metabolic flexibility.
Modern dietary patterns—dominated by ultra-processed foods, refined sugars, industrial seed oils, and overconsumption—are potent initiating drivers of cancer. They disrupt mitochondrial bioenergetics, increase oxidative damage, and drive chronic hyperinsulinemia and inflammation.
Key contributors include:
Systems insight: These metabolic disruptions are early and modifiable stressors that impair mitochondrial function—well before cancer is detectable.
5.3. Micronutrient Deficiencies
Action: Assess and correct subclinical deficiencies to restore mitochondrial respiration, redox capacity, immune integrity, and DNA repair.
Micronutrient insufficiencies are widespread—even in affluent populations—and represent a silent yet profound initiating influence on cancer risk. Deficiencies impair core mitochondrial functions and immune surveillance.
Common cancer-relevant deficiencies:
Systems insight: Chronic nutrient depletion primes the body for mitochondrial instability and immune failure—conditions that favor malignant progression.
5.4. Chronic Infections & Immune Dysregulation
Action: Screen for persistent infections, reduce immune suppression, and restore mitochondrial-immune homeostasis.
Persistent infections and microbial imbalance are often overlooked initiating drivers of carcinogenesis. Pathogens such as HPV, EBV, H. pylori, and stealth species promote inflammation, immune tolerance, and mitochondrial stress [28,75,76].
Key contributors:
-
Oncoviruses:
-
Bacterial infections:
-
Chronic fungal and parasitic infections:
-
Immune exhaustion and dysregulation:
Systems insight: These infections are initiating stressors, not just cofactors. They impair mitochondrial function and immune clearance mechanisms that otherwise suppress tumor development.
5.5. Hormonal Imbalance & Endocrine Disruption
Action: Reduce exposure to endocrine-disrupting chemicals (EDCs) and restore hormonal-metabolic balance.
Hormonal dysregulation—endogenous or environmental—can initiate mitochondrial destabilization and drive carcinogenesis, particularly in hormone-sensitive tissues.
Key contributors include:
- Estrogen dominance & low progesterone: linked to breast and endometrial cancer via proliferative signaling and impaired apoptosis [85].
- Insulin resistance & hyperinsulinemia: activate IGF-1 and mTOR pathways, promoting anabolic, pro-cancer metabolism [86].
- Thyroid dysfunction: impairs mitochondrial oxygen use and ATP production [87].
- Cortisol dysregulation & HPA axis stress: suppress immune surveillance and promote chronic inflammation [88].
-
Endocrine-disrupting chemicals (EDCs):
Systems insight: Hormones regulate mitochondrial biogenesis, apoptosis, and immune tone. Hormonal imbalance functions as a high-level initiating influence in many cancers.
5.6. Lifestyle & Behavioral Risk Factors
Action:
Support movement, sleep, sun exposure, and reduce behavioral stressors to enhance cellular resilience.
Sedentary lifestyles, poor sleep, low sunlight, and harmful substance use collectively disrupt mitochondrial and immune health.
Key contributors:
- High-risk sexual behavior: increases exposure to oncogenic viruses (e.g., HPV)
Systems insight: Lifestyle patterns shape the terrain in which initiating biological stressors either manifest or are mitigated.
5.7. Psychosocial & Emotional Stress
Action: Support trauma healing, circadian balance, and community connection to reduce stress-related carcinogenic signaling.
Chronic psychological stress and emotional trauma are increasingly recognized as biological disruptors that elevate cancer risk. Stress dysregulates the HPA axis, elevates cortisol and catecholamines, and drives systemic inflammation. These changes suppress immunity, impair mitochondrial function, promote DNA damage, and weaken cellular resilience against malignant transformation.
Key contributing factors include:
- Sleep deprivation and circadian disruption: Disturbances in daylight exposure, shift work, and poor sleep quality suppress melatonin (an oncostatic hormone), impair DNA repair, and disturb antioxidant cycling. Night shift work is classified as "probably carcinogenic" by IARC (Group 2A) due to these mechanisms [108,109].
- Social isolation & emotional loneliness: Meta-analyses find that chronic loneliness and isolation are associated with increased all-cause and cancer-specific mortality—likely through immune dysfunction and elevated inflammatory markers [110].
Systems insight: Emotional stress is not merely psychosomatic—it functions as a systemic initiating driver that reshapes neuroendocrine-immune balance and mitochondrial function.
5.8. Developmental & Early-Life Programming
Action: Protect maternal–child health and reduce prenatal/early-life exposure to epigenetic and mitochondrial disruptors.
The developmental period—from conception through early childhood—is one of the most critical windows of vulnerability to cancer-inducing insults. Exposures and deficiencies during this time can cause lasting epigenetic changes, alter immune and hormonal programming, and impair mitochondrial function—establishing a biological foundation for cancer risk decades later.
Key contributors include:
- Gestational metabolic stress: maternal obesity, insulin resistance, and gestational diabetes increase childhood cancer risk [117]
Systems insight: These exposures initiate epigenetic programming and mitochondrial fragility, raising long-term cancer risk without early clinical signs.
5.9. Genetic & Epigenetic Susceptibility
Action: Use genomic tools to guide personalized prevention by identifying vulnerabilities to upstream stressors.
Most cancers are not directly caused by genes but by how genetic predispositions interact with upstream stressors.
Key contributors:
- Family history of cancer in high-risk environments: reflects both shared genes and shared exposures [134]
Systems insight: Genetic susceptibility amplifies the effects of upstream initiators. Prevention should prioritize reducing stressor load, not just genetic screening.
5.10. Medical Iatrogenesis
Action: Minimize unnecessary exposure to radiation, immunosuppression, and mitochondrial-toxic treatments.
While life-saving, many modern interventions can initiate or accelerate mitochondrial dysfunction, especially when overused or unbalanced.
Key contributors:
- Excessive antibiotic use: disrupts the gut microbiome, weakens immune defenses, and promotes inflammation
Systems insight: Iatrogenic stressors must be factored into the full map of upstream biological disruptions that can initiate or exacerbate cancer.
6. From Mechanism to Policy: Implications for Public Health
Conventional cancer policy remains largely focused on downstream indicators—such as BRCA mutations, PSA levels, and late-stage interventions (e.g., surgery, chemotherapy, radiation). While these tools have value, they arrive too late in the biological timeline to meaningfully alter cancer incidence on a population scale.
A systems-based framework—centered on mitochondrial dysfunction as the central initiating mechanism—calls for upstream reform. By applying Root Cause Analysis (RCA) as a systems-organizing tool, we can trace policy relevance back to the initiating biological stressors that undermine mitochondrial and immune integrity. Below are strategic domains for upstream intervention.
6.1. Environmental Regulation
Recommended Actions:
- Ban or restrict high-risk exposures such as glyphosate, PFAS, endocrine-disrupting chemicals, and EMF-emitting infrastructure in residential zones.
- Enforce industrial detoxification mandates and environmental remediation.
- Mandate transparency and labeling of known carcinogenic or mitochondrial-disrupting chemicals.
Rationale: Environmental toxins are well-established initiating stressors in cancer etiology. Mitigating these exposures is foundational to upstream prevention.
6.2. Nutritional Policy
Recommended Actions:
- Eliminate subsidies for refined grains, added sugars, and industrial seed oils.
- Prioritize nutrient-dense, whole-food dietary models (e.g., low-carbohydrate, anti-inflammatory) in public nutrition programs.
- Implement targeted micronutrient fortification based on population-level deficiency data.
Rationale: Nutritional deprivation and metabolic stress are central initiating drivers of mitochondrial decline. Nutritional policy must shift from caloric sufficiency to mitochondrial sufficiency.
6.3. Infection Control
Recommended Actions:
- Screen for chronic latent infections (e.g., HPV, EBV, H. pylori) in high-risk groups.
- Integrate immune-nutritional interventions (vitamins C, D, zinc, selenium) into preventive protocols.
- Reassess vaccine formulations with regard to mitochondrial safety and adjuvant toxicity.
Rationale: Chronic infections act as biological initiators of mitochondrial dysfunction, immune suppression, and tumor microenvironment dysregulation.
6.4. Medical Reform
Recommended Actions:
- Shift clinical care from pharma-driven symptom suppression to nutritional, metabolic, and detoxification therapies.
- Redirect research funding toward prevention, environmental detoxification, and metabolic oncology.
- Incorporate systems-based RCA training into medical education, emphasizing initiating drivers and early mitochondrial disruption.
Rationale: Downstream pharmacological interventions must give way to upstream, patient-centered strategies that address the true drivers of cancer biology.
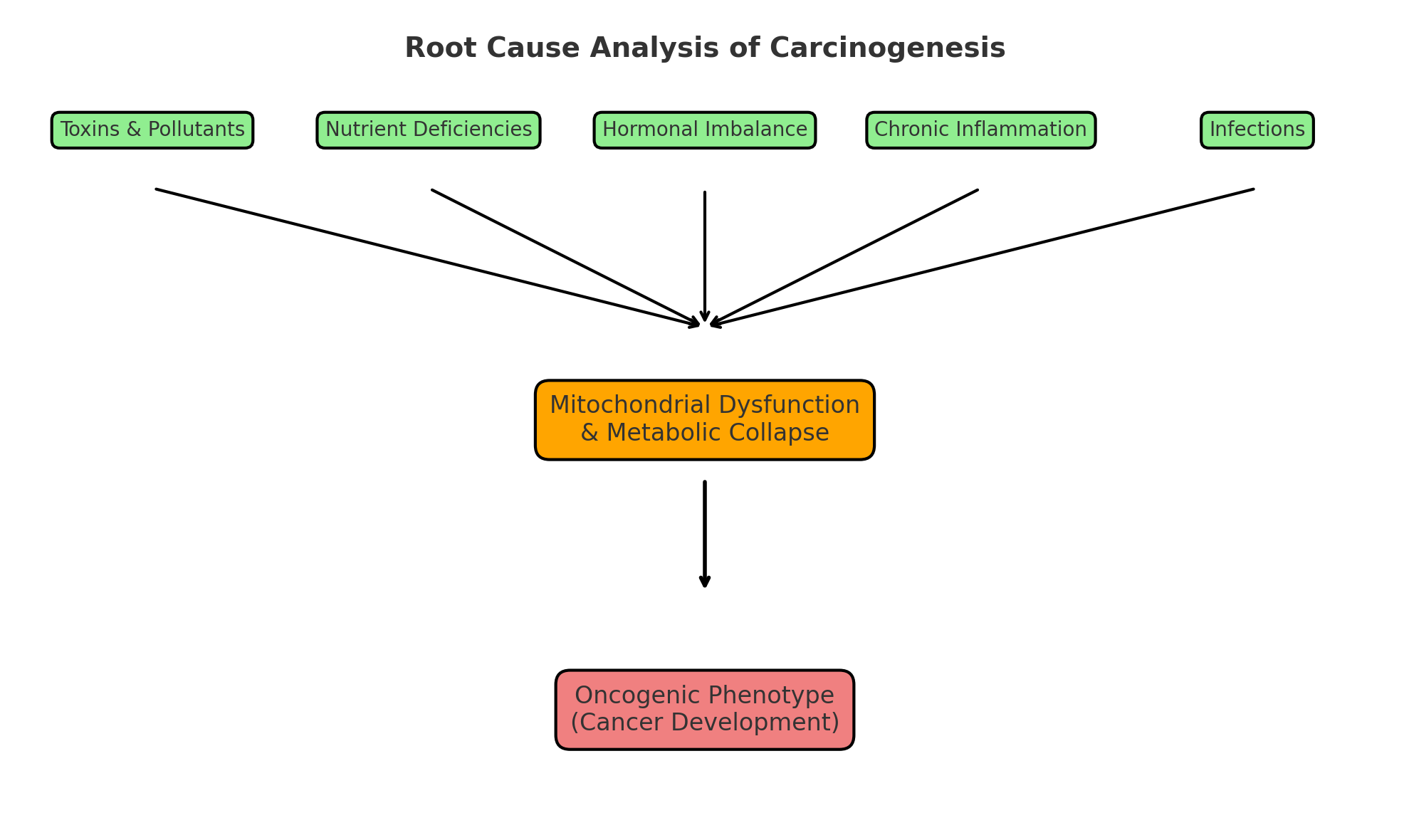
The figure illustrates a three-layer model of cancer initiation and progression. Layer 1 represents upstream initiating drivers, including environmental toxins, dietary carcinogens, micronutrient deficiencies, chronic infections, hormonal disruption, psychosocial stress, developmental insults, and medical iatrogenesis. Layer 2 shows mitochondrial collapse and metabolic destabilization as the central bioenergetic failure that links initiating stressors to disease expression. Layer 3 depicts the downstream oncogenic phenotype, including genomic instability, inflammation, immune evasion, and epigenetic drift. Together, the RCA framework highlights how diverse upstream drivers converge on mitochondrial dysfunction to produce the hallmarks of cancer.
This table contrasts the dominant Somatic Mutation Theory (SMT), the Mitochondrial Metabolic Theory (MMT/Warburg–Seyfried model), and the proposed Root Cause Analysis (RCA) framework. SMT emphasizes nuclear mutations as initiating events, but explains survival outcomes poorly and offers limited preventive strategies. MMT centers on mitochondrial dysfunction as the driver of malignant transformation, integrating evidence from metabolic profiling and nuclear–cytoplasmic transfer experiments, but often underemphasizes the upstream stressors that destabilize mitochondria. The RCA framework expands on MMT by systematically tracing modifiable initiating drivers, positioning mitochondria as vulnerable sentinels rather than ultimate causes, and emphasizing prevention-oriented, systems-level interventions.
7. Discussion: Toward an IOM Framework for Cancer Prevention and Reversal
The escalating global cancer burden cannot be meaningfully reduced through downstream, symptom-suppressing interventions alone. Integrative Orthomolecular Medicine (IOM) offers a paradigm shift—one that targets the upstream biological stressors that initiate carcinogenesis, including mitochondrial dysfunction as the central initiating mechanism, along with micronutrient depletion, chronic inflammation, toxin overload, and hormonal and metabolic imbalance.
We must abandon the illusion that a gene edit or pharmaceutical pill can reverse decades of cumulative exposure to initiating biological stressors. Instead, the true path forward is rebuilding the cellular foundations of health through prevention-focused, systems-based care.
The IOM Model Emphasizes:
- Cellular health as the foundation of systemic health
- Safety, accessibility, and sustainability over drug-centric interventions
- Prevention-first strategies, grounded in nutrition, detoxification, and metabolic repair
This is not simply a shift in treatment preference—it is a call for a public health transformation guided by biological fidelity, not technological novelty. Lasting reversal of the cancer epidemic requires:
- Nutrient-dense, anti-inflammatory diets
- Mitochondrial support and metabolic flexibility
- Toxin exposure reduction and detoxification support
- Circadian alignment and lifestyle rhythm restoration
This integrative approach does not replace existing models but complements them, offering a systems-organizing framework that clarifies the sequence from initiating stressors to mitochondrial collapse, and ultimately to malignant transformation.
The somatic mutation theory is no longer just scientifically inadequate—it is a public health limitation that diverts resources from more promising strategies.
By restoring upstream biological integrity, IOM provides a more practical, cost-effective, and sustainable path forward—for both individuals and health systems.
As an example, high-dose intravenous vitamin C (HDIVC) has been investigated as a metabolic therapy that directly engages oxidative stress pathways, illustrating how root-cause analysis may guide future therapeutic innovation without being limited to mutation-centric strategies.
8. Conclusion: Reframing Cancer as a Systems-Initiated, Mitochondrial Disease
Cancer is not merely the result of genetic errors; it is a systemic biological failure initiated by upstream stressors and culminating in mitochondrial dysfunction—the primary cellular event in carcinogenesis. This metabolic collapse is driven and sustained by a convergence of environmental toxins, nutritional deficiencies, chronic infections, hormonal disruptions, and iatrogenic medical stressors.
To reverse the trajectory of the cancer epidemic, both public health and clinical medicine must adopt a fundamentally different orientation:
- From reaction to proactive prevention
- From genetic determinism to metabolic resilience
- From symptom suppression to initiating driver mitigation
This paradigm shift—rooted in the principles of Integrative Orthomolecular Medicine—offers a complementary, systems-level roadmap for cancer prevention, improved treatment outcomes, and long-term healthspan extension.
The future of oncology lies not only in new drugs or targeted therapies, but in the restoration of cellular integrity through upstream intervention. Only by addressing the initiating biological stressors that undermine mitochondrial and immune function can we truly change the course of chronic disease and cancer.
Cancer research must move beyond the dead-end of mutation-centrism. Our RCA framework reframes cancer as a systems-initiated, mitochondria-mediated disease, providing a practical roadmap for prevention and reversal.
Supplementary Materials
Not applicable.
Author Contributions
The author is solely responsible for all aspects of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
The author gratefully acknowledges colleagues in the international orthomolecular medicine community for their insights and discussions that helped shape this manuscript. The author also serves as Editor-in-Chief of the Orthomolecular Medicine News Service (OMNS). The author specifically thanks Dr. Thomas N. Seyfried (Boston College) for his valuable comments on earlier drafts of this manuscript.
Abbreviations
The following abbreviations are used in this manuscript:
| RCA | Root Cause Analysis |
| SMT | Somatic Mutation Theory |
| MMT | Mitochondrial Metabolic Theory |
| IOM | Integrative Orthomolecular Medicine |
| ROS | Reactive Oxygen Species |
| OS | Overall Survival |
| FDA | U.S. Food and Drug Administration |
| RCT | Randomized Controlled Trial |
References
- Zarocostas, J. Global Cancer Cases and Deaths Are Set to Rise by 70% in next 20 Years. BMJ 2010, 340, c3041. [Google Scholar] [CrossRef]
- Pilleron, S.; Sarfati, D.; Janssen-Heijnen, M.; Vignat, J.; Ferlay, J.; Bray, F.; Soerjomataram, I. Global Cancer Incidence in Older Adults, 2012 and 2035: A Population-Based Study. Int J Cancer 2019, 144, 49–58. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ladanie, A.; Schmitt, A.M.; Speich, B.; Naudet, F.; Agarwal, A.; Pereira, T.V.; Sclafani, F.; Herbrand, A.K.; Briel, M.; Martin-Liberal, J.; et al. Clinical Trial Evidence Supporting US Food and Drug Administration Approval of Novel Cancer Therapies Between 2000 and 2016. JAMA Netw Open 2020, 3, e2024406. [Google Scholar] [CrossRef]
- Aggarwal, A.; Fojo, T.; Chamberlain, C.; Davis, C.; Sullivan, R. Do Patient Access Schemes for High-Cost Cancer Drugs Deliver Value to Society?-Lessons from the NHS Cancer Drugs Fund. Annals of oncology : official journal of the European Society for Medical Oncology 2017, 28, 1738–1750. [Google Scholar] [CrossRef]
- Zhang, Y.; Naci, H.; Wagner, A.K.; Xu, Z.; Yang, Y.; Zhu, J.; Ji, J.; Shi, L.; Guan, X. Overall Survival Benefits of Cancer Drugs Approved in China From 2005 to 2020. JAMA Netw Open 2022, 5, e2225973. [Google Scholar] [CrossRef]
- Sullivan, R.; Pramesh, C.S.; Booth, C.M. Cancer Patients Need Better Care, Not Just More Technology. Nature 2017, 549, 325–328. [Google Scholar] [CrossRef]
- Morgan, G.; Ward, R.; Barton, M. The Contribution of Cytotoxic Chemotherapy to 5-Year Survival in Adult Malignancies. Clinical oncology (Royal College of Radiologists (Great Britain)) 2004, 16, 549–560. [Google Scholar] [CrossRef]
- Naci, H.; Zhang, Y.; Woloshin, S.; Guan, X.; Xu, Z.; Wagner, A.K. Overall Survival Benefits of Cancer Drugs Initially Approved by the US Food and Drug Administration on the Basis of Immature Survival Data: A Retrospective Analysis. Lancet Oncol 2024, 25, 760–769. [Google Scholar] [CrossRef]
- Elbaz, J.; Haslam, A.; Prasad, V. An Empirical Analysis of Overall Survival in Drug Approvals by the US FDA (2006-2023). Cancer Med 2024, 13, e7190. [Google Scholar] [CrossRef]
- Michaeli, D.T.; Michaeli, J.C.; Michaeli, T. Advances in Cancer Therapy: Clinical Benefit of New Cancer Drugs. Aging (Albany NY) 2023, 15, 5232–5234. [Google Scholar] [CrossRef]
- Maimets, T. Cancer Research and the Mainstream of Biology. Front Cell Dev Biol 2025, 13, 1623849. [Google Scholar] [CrossRef]
- Seyfried, T.N. Cancer as a Metabolic Disease: On the Origin, Management, and Prevention of Cancer, 1st ed.; Wiley, 2012; ISBN 0-470-58492-0. [Google Scholar]
- Seyfried, T.N.; Shelton, L.M. Cancer as a Metabolic Disease. Nutrition & Metabolism 2010, 7, 7. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Singh, G.; Patel, R.H.; Vaqar, S.; Boster, J. Root Cause Analysis and Medical Error Prevention. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2025. [Google Scholar]
- Peerally, M.F.; Carr, S.; Waring, J.; Dixon-Woods, M. The Problem with Root Cause Analysis. BMJ Qual Saf 2017, 26, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Dinicola, S.; D’Anselmi, F.; Pasqualato, A.; Proietti, S.; Lisi, E.; Cucina, A.; Bizzarri, M. A Systems Biology Approach to Cancer: Fractals, Attractors, and Nonlinear Dynamics. OMICS 2011, 15, 93–104. [Google Scholar] [CrossRef]
- Soto, A.M.; Sonnenschein, C. The Somatic Mutation Theory of Cancer: Growing Problems with the Paradigm? Bioessays 2004, 26, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Fardet, A.; Rock, E. Perspective: Reductionist Nutrition Research Has Meaning Only within the Framework of Holistic and Ethical Thinking. Adv Nutr 2018, 9, 655–670. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and Cancer. Nat Rev Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P.A. Why Most Published Research Findings Are False. PLoS Med 2005, 2, e124. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer Genes and the Pathways They Control. Nat Med 2004, 10, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a Metabolic Disease: Implications for Novel Therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- zur Hausen, H. Papillomaviruses in the Causation of Human Cancers - a Brief Historical Account. Virology 2009, 384, 260–265. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global Burden of Cancer Attributable to Infections in 2018: A Worldwide Incidence Analysis. Lancet Glob Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Plummer, M.; de Martel, C.; Vignat, J.; Ferlay, J.; Bray, F.; Franceschi, S. Global Burden of Cancers Attributable to Infections in 2012: A Synthetic Analysis. Lancet Glob Health 2016, 4, e609–616. [Google Scholar] [CrossRef]
- Damania, B.; Münz, C. Immunodeficiencies That Predispose to Pathologies by Human Oncogenic γ-Herpesviruses. FEMS Microbiology Reviews 2019, 43, 181–192. [Google Scholar] [CrossRef]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Tsai, H.-C.; Baylin, S.B. Cancer Epigenetics: Tumor Heterogeneity, Plasticity of Stem-like States, and Drug Resistance. Mol. Cell 2014, 54, 716–727. [Google Scholar] [CrossRef]
- Kelly, T.K.; De Carvalho, D.D.; Jones, P.A. Epigenetic Modifications as Therapeutic Targets. Nat Biotechnol 2010, 28, 1069–1078. [Google Scholar] [CrossRef]
- Feil, R.; Fraga, M.F. Epigenetics and the Environment: Emerging Patterns and Implications. Nat Rev Genet 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Lillycrop, K.A.; Burdge, G.C. Epigenetic Changes in Early Life and Future Risk of Obesity. Int J Obes (Lond) 2011, 35, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem Cells, Cancer, and Cancer Stem Cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour Stem Cells and Drug Resistance. Nat Rev Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc Natl Acad Sci U S A 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of Cancer Stem Cell Metabolism. Br J Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef]
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu Rev Pathol 2016, 11, 47–76. [Google Scholar] [CrossRef]
- Burnet, F.M. The Concept of Immunological Surveillance. Prog Exp Tumor Res 1970, 13, 1–27. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef]
- Cruzat, V.; Macedo Rogero, M.; Noel Keane, K.; Curi, R.; Newsholme, P. Glutamine: Metabolism and Immune Function, Supplementation and Clinical Translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef]
- Miller, M.D.; Marty, M.A. Impact of Environmental Chemicals on Lung Development. Environ Health Perspect 2010, 118, 1155–1164. [Google Scholar] [CrossRef]
- Duesberg, P.; Li, R.; Fabarius, A.; Hehlmann, R. The Chromosomal Basis of Cancer. Cell Oncol 2005, 27, 293–318. [Google Scholar] [CrossRef]
- Duesberg, P.; Mandrioli, D.; McCormack, A.; Nicholson, J.M. Is Carcinogenesis a Form of Speciation? Cell Cycle 2011, 10, 2100–2114. [Google Scholar] [CrossRef]
- Rasnick, D. Aneuploidy Theory Explains Tumor Formation, the Absence of Immune Surveillance, and the Failure of Chemotherapy. Cancer Genet Cytogenet 2002, 136, 66–72. [Google Scholar] [CrossRef]
- Flora, G.; Gupta, D.; Tiwari, A. Toxicity of Lead: A Review with Recent Updates. Interdisciplinary Toxicology 2012, 5, 47–58. [Google Scholar] [CrossRef]
- Duarte-Hospital, C.; Tête, A.; Brial, F.; Benoit, L.; Koual, M.; Tomkiewicz, C.; Kim, M.J.; Blanc, E.B.; Coumoul, X.; Bortoli, S. Mitochondrial Dysfunction as a Hallmark of Environmental Injury. Cells 2021, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- Reddam, A.; McLarnan, S.; Kupsco, A. Environmental Chemical Exposures and Mitochondrial Dysfunction: A Review of Recent Literature. Curr Environ Health Rep 2022, 9, 631–649. [Google Scholar] [CrossRef]
- Biswas, G.; Srinivasan, S.; Anandatheerthavarada, H.K.; Avadhani, N.G. Dioxin-Mediated Tumor Progression through Activation of Mitochondria-to-Nucleus Stress Signaling. Proc Natl Acad Sci U S A 2008, 105, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Fiolet, T.; Srour, B.; Sellem, L.; Kesse-Guyot, E.; Allès, B.; Méjean, C.; Deschasaux, M.; Fassier, P.; Latino-Martel, P.; Beslay, M.; et al. Consumption of Ultra-Processed Foods and Cancer Risk: Results from NutriNet-Santé Prospective Cohort. BMJ 2018, 360, k322. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, C.A.; Cannon, G.; Levy, R.B.; Moubarac, J.-C.; Louzada, M.L.; Rauber, F.; Khandpur, N.; Cediel, G.; Neri, D.; Martinez-Steele, E.; et al. Ultra-Processed Foods: What They Are and How to Identify Them. Public Health Nutr 2019, 22, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Shafiu, M.; Sundaram, S.; Le, M.; Ishimoto, T.; Sautin, Y.Y.; Lanaspa, M.A. Sugar, Uric Acid, and the Etiology of Diabetes and Obesity. Diabetes 2013, 62, 3307–3315. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sun, T.; Liu, H.; Li, J.; Tian, L. Carbohydrate Quality vs Quantity on Cancer Risk: Perspective of Microbiome Mechanisms. Journal of Functional Foods 2024, 118, 106246. [Google Scholar] [CrossRef]
- Cosmin Stan, M.; Paul, D. Diabetes and Cancer: A Twisted Bond. Oncol Rev 2024, 18, 1354549. [Google Scholar] [CrossRef]
- Ramsden, C.E.; Zamora, D.; Leelarthaepin, B.; Majchrzak-Hong, S.F.; Faurot, K.R.; Suchindran, C.M.; Ringel, A.; Davis, J.M.; Hibbeln, J.R. Use of Dietary Linoleic Acid for Secondary Prevention of Coronary Heart Disease and Death: Evaluation of Recovered Data from the Sydney Diet Heart Study and Updated Meta-Analysis. BMJ 2013, 346, e8707. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Nara, T.Y. Structure, Function, and Dietary Regulation of Delta6, Delta5, and Delta9 Desaturases. Annu Rev Nutr 2004, 24, 345–376. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Woodruff, S.; Goodman, S.; Cai, W.; Chen, X.; Pyzik, R.; Yong, A.; Striker, G.E.; Vlassara, H. Advanced Glycation End Products in Foods and a Practical Guide to Their Reduction in the Diet. J Am Diet Assoc 2010, 110, 911–916.e12. [Google Scholar] [CrossRef]
- Hung, C.-H.; Lin, Y.-C.; Tsai, Y.-G.; Lin, Y.-C.; Kuo, C.-H.; Tsai, M.-L.; Kuo, C.-H.; Liao, W.-T. Acrylamide Induces Mitophagy and Alters Macrophage Phenotype via Reactive Oxygen Species Generation. Int J Mol Sci 2021, 22, 1683. [Google Scholar] [CrossRef]
- Smith, R.L.; Soeters, M.R.; Wüst, R.C.I.; Houtkooper, R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr Rev 2018, 39, 489–517. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Wang, J.; Guo, X.; Song, Y.; Fu, K.; Gao, Z.; Liu, D.; He, W.; Yang, L.-L. Energy Metabolism in Health and Diseases. Sig Transduct Target Ther 2025, 10, 69. [Google Scholar] [CrossRef]
- Lovell, D.I.; Stuelcken, M.; Eagles, A. Exercise Testing for Metabolic Flexibility: Time for Protocol Standardization. Sports Med Open 2025, 11, 31. [Google Scholar] [CrossRef]
- Grant, W.B.; Wimalawansa, S.J.; Pludowski, P.; Cheng, R.Z. Vitamin D: Evidence-Based Health Benefits and Recommendations for Population Guidelines. Nutrients 2025, 17, 277. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D Deficiency. N Engl J Med 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.F.; Garland, F.C.; Gorham, E.D.; Lipkin, M.; Newmark, H.; Mohr, S.B.; Holick, M.F. The Role of Vitamin D in Cancer Prevention. Am J Public Health 2006, 96, 252–261. [Google Scholar] [CrossRef]
- Naidu, K.A. Vitamin C in Human Health and Disease Is Still a Mystery? An Overview. Nutr J 2003, 2, 7. [Google Scholar] [CrossRef]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C Pharmacokinetics in Healthy Volunteers: Evidence for a Recommended Dietary Allowance. Proc Natl Acad Sci U S A 1996, 93, 3704–3709. [Google Scholar] [CrossRef]
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate Deficiency Causes Uracil Misincorporation into Human DNA and Chromosome Breakage: Implications for Cancer and Neuronal Damage. Proc Natl Acad Sci U S A 1997, 94, 3290–3295. [Google Scholar] [CrossRef] [PubMed]
- Lucock, M. Folic Acid: Nutritional Biochemistry, Molecular Biology, and Role in Disease Processes. Mol Genet Metab 2000, 71, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M.; Veronese, N.; Dominguez, L.J. Magnesium in Aging, Health and Diseases. Nutrients 2021, 13, 463. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, R. Magnesium Metabolism and Its Disorders. Clin Biochem Rev 2003, 24, 47–66. [Google Scholar] [PubMed]
- Rayman, M.P. Selenium and Human Health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Maret, W.; Sandstead, H.H. Zinc Requirements and the Risks and Benefits of Zinc Supplementation. J Trace Elem Med Biol 2006, 20, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. The Search for Infectious Causes of Human Cancers: Where and Why. Virology 2009, 392, 1–10. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Bosch, F.X.; Lorincz, A.; Muñoz, N.; Meijer, C.J.L.M.; Shah, K.V. The Causal Relation between Human Papillomavirus and Cervical Cancer. J Clin Pathol 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Rickinson, A.B. Epstein-Barr Virus: 40 Years On. Nat Rev Cancer 2004, 4, 757–768. [Google Scholar] [CrossRef]
- Yu, C.; He, S.; Zhu, W.; Ru, P.; Ge, X.; Govindasamy, K. Human Cytomegalovirus in Cancer: The Mechanism of HCMV-Induced Carcinogenesis and Its Therapeutic Potential. Front Cell Infect Microbiol 2023, 13, 1202138. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular Carcinoma and Hepatitis C in the United States. Hepatology 2002, 36, S74–83. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M.; Wilson, K.T. Helicobacter Pylori and Gastric Cancer: Factors That Modulate Disease Risk. Clin Microbiol Rev 2010, 23, 713–739. [Google Scholar] [CrossRef]
- Yacoub, E.; Saed Abdul-Wahab, O.M.; Al-Shyarba, M.H.; Ben Abdelmoumen Mardassi, B. The Relationship between Mycoplasmas and Cancer: Is It Fact or Fiction ? Narrative Review and Update on the Situation. J Oncol 2021, 2021, 9986550. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the Microbiota in Immunity and Inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef]
- Yager, J.D.; Davidson, N.E. Estrogen Carcinogenesis in Breast Cancer. N Engl J Med 2006, 354, 270–282. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and Insulin-like Growth Factor Signalling in Neoplasia. Nat Rev Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Wrutniak-Cabello, C.; Casas, F.; Cabello, G. Thyroid Hormone Action in Mitochondria. J Mol Endocrinol 2001, 26, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Reiche, E.M.V.; Nunes, S.O.V.; Morimoto, H.K. Stress, Depression, the Immune System, and Cancer. Lancet Oncol 2004, 5, 617–625. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E.; Bourguignon, J.-P.; Giudice, L.C.; Hauser, R.; Prins, G.S.; Soto, A.M.; Zoeller, R.T.; Gore, A.C. Endocrine-Disrupting Chemicals: An Endocrine Society Scientific Statement. Endocrine Reviews 2009, 30, 293–342. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Blumberg, B.; Cave, M.; Machtinger, R.; Mantovani, A.; Mendez, M.A.; Nadal, A.; Palanza, P.; Panzica, G.; Sargis, R.; et al. Metabolism Disrupting Chemicals and Metabolic Disorders. Reprod Toxicol 2017, 68, 3–33. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I. The Key Role of Mitochondrial Function in Health and Disease. Antioxidants 2023, 12, 782. [Google Scholar] [CrossRef] [PubMed]
- Clemente-Suárez, V.J.; Martín-Rodríguez, A.; Redondo-Flórez, L.; Ruisoto, P.; Navarro-Jiménez, E.; Ramos-Campo, D.J.; Tornero-Aguilera, J.F. Metabolic Health, Mitochondrial Fitness, Physical Activity, and Cancer. Cancers (Basel) 2023, 15, 814. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Sammy, M.J.; Ballinger, S.W. Mitochondrial Toxicity of Tobacco Smoke and Air Pollution. Toxicology 2017, 391, 18–33. [Google Scholar] [CrossRef]
- Yang, Z.; Harrison, C.M.; Chuang, G.C.; Ballinger, S.W. The Role of Tobacco Smoke Induced Mitochondrial Damage in Vascular Dysfunction and Atherosclerosis. Mutat Res 2007, 621, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an Underestimated Risk Factor for Cancer Development: Role of Genetics in Ethanol Metabolism. Genes Nutr 2010, 5, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Becker, P. Alcohol Metabolism and Cancer Risk. Alcohol Res Health 2007, 30, 38–41. [Google Scholar] [PubMed]
- Alcohol and Cancer Risk Fact Sheet - NCI. Available online: https://www.cancer.gov/about-cancer/causes-prevention/risk/alcohol/alcohol-fact-sheet (accessed on 1 August 2025).
- Rodríguez-Santana, C.; Florido, J.; Martínez-Ruiz, L.; López-Rodríguez, A.; Acuña-Castroviejo, D.; Escames, G. Role of Melatonin in Cancer: Effect on Clock Genes. Int J Mol Sci 2023, 24, 1919. [Google Scholar] [CrossRef]
- Szataniak, I.; Packi, K. Melatonin as the Missing Link Between Sleep Deprivation and Immune Dysregulation: A Narrative Review. Int J Mol Sci 2025, 26, 6731. [Google Scholar] [CrossRef]
- Holick, M.F. Cancer, Sunlight and Vitamin D. J Clin Transl Endocrinol 2014, 1, 179–186. [Google Scholar] [CrossRef]
- Raymond-Lezman, J.R.; Riskin, S.I. Benefits and Risks of Sun Exposure to Maintain Adequate Vitamin D Levels. Cureus 2023, 15, e38578. [Google Scholar] [CrossRef]
- Anwar, M.J.; Alenezi, S.K.; Alhowail, A.H. Molecular Insights into the Pathogenic Impact of Vitamin D Deficiency in Neurological Disorders. Biomedicine & Pharmacotherapy 2023, 162, 114718. [Google Scholar] [CrossRef]
- Liu, Z.; Lei, M.; Bai, Y. Chronic Stress Mediates Inflammatory Cytokines Alterations and Its Role in Tumorigenesis. J Inflamm Res 2025, 18, 1067–1090. [Google Scholar] [CrossRef]
- Vignjević Petrinović, S.; Milošević, M.S.; Marković, D.; Momčilović, S. Interplay between Stress and Cancer-A Focus on Inflammation. Front Physiol 2023, 14, 1119095. [Google Scholar] [CrossRef]
- Sarkar, S.; Jackson, B.; Manzo, L.L.; Jeon, S.; Poghosyan, H. Association between Adverse Childhood Experiences and Self-Reported Health-Risk Behaviors among Cancer Survivors: A Population-Based Study. PLoS One 2024, 19, e0299918. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Thacker, L.R.; Cohen, S.A. Association between Adverse Childhood Experiences and Diagnosis of Cancer. PLOS ONE 2013, 8, e65524. [Google Scholar] [CrossRef]
- Yan, J.; Chen, Y.; Luo, M.; Hu, X.; Li, H.; Liu, Q.; Zou, Z. Chronic Stress in Solid Tumor Development: From Mechanisms to Interventions. J Biomed Sci 2023, 30, 8. [Google Scholar] [CrossRef]
- Samuelsson, L.B.; Bovbjerg, D.H.; Roecklein, K.A.; Hall, M.H. Sleep and Circadian Disruption and Incident Breast Cancer Risk: An Evidence-Based and Theoretical Review. Neurosci Biobehav Rev 2018, 84, 35–48. [Google Scholar] [CrossRef]
- Zeng, Y.; Guo, Z.; Wu, M.; Chen, F.; Chen, L. Circadian Rhythm Regulates the Function of Immune Cells and Participates in the Development of Tumors. Cell Death Discov. 2024, 10, 199. [Google Scholar] [CrossRef]
- Wheldon, C.W.; Shahsavar, Y.; Choudhury, A.; McCormick, B.P.; Albertorio-Díaz, J.R. Loneliness among Adult Cancer Survivors in the United States: Prevalence and Correlates. Sci Rep 2025, 15, 3914. [Google Scholar] [CrossRef]
- Bommarito, P.A.; Martin, E.; Fry, R.C. Effects of Prenatal Exposure to Endocrine Disruptors and Toxic Metals on the Fetal Epigenome. Epigenomics 2017, 9, 333–350. [Google Scholar] [CrossRef]
- Chen, C.; Ma, C.; Li, Q.; Hang, J.G.; Shen, J.; Nakayama, S.F.; Kido, T.; Lin, Y.; Feng, H.; Jung, C.-R.; et al. Prenatal Exposure to Heavy Metals and Adverse Birth Outcomes: Evidence From an E-Waste Area in China. Geohealth 2023, 7, e2023GH000897. [Google Scholar] [CrossRef] [PubMed]
- Gernand, A.D.; Schulze, K.J.; Stewart, C.P.; West, K.P.; Christian, P. Micronutrient Deficiencies in Pregnancy Worldwide: Health Effects and Prevention. Nat Rev Endocrinol 2016, 12, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, M.; Fernández de Cossío, L.; Chakravarty, M.M.; Tremblay, M.-È. From Maternal Diet to Neurodevelopmental Disorders: A Story of Neuroinflammation. Front. Cell. Neurosci. 2021, 14. [Google Scholar] [CrossRef]
- Russo, F. The Impact of Nutritional Deficiencies During Pregnancy: A Comprehensive Overview. Maternal and Pediatric Nutrition 2024, 9, 1–2. [Google Scholar] [CrossRef]
- Prentice, S. They Are What You Eat: Can Nutritional Factors during Gestation and Early Infancy Modulate the Neonatal Immune Response? Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Marley, A.R.; Domingues, A.; Ghosh, T.; Turcotte, L.M.; Spector, L.G. Maternal Body Mass Index, Diabetes, and Gestational Weight Gain and Risk for Pediatric Cancer in Offspring: A Systematic Review and Meta-Analysis. JNCI Cancer Spectr 2022, 6, pkac020. [Google Scholar] [CrossRef]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10. [Google Scholar] [CrossRef]
- Elliott, R.L.; Jiang, X.P.; Baucom, C.C. Antibiotic Overusage Causes Mitochondrial Dysfunction Which May Promote Tumorigenesis. J. Cancer Treat. Res. 2017, 5, 62–65. [Google Scholar] [CrossRef]
- Antibiotics-Induced Obesity: A Mitochondrial Perspective | Public Health Genomics | Karger Publishers. Available online: https://karger.com/phg/article-abstract/20/5/257/272852/Antibiotics-Induced-Obesity-A-Mitochondrial?redirectedFrom=fulltext (accessed on 1 August 2025).
- Monzer, N.; Hartmann, M.; Buckert, M.; Wolff, K.; Nawroth, P.; Kopf, S.; Kender, Z.; Friederich, H.-C.; Wild, B. Associations of Childhood Neglect With the ACTH and Plasma Cortisol Stress Response in Patients With Type 2 Diabetes. Front Psychiatry 2021, 12, 679693. [Google Scholar] [CrossRef] [PubMed]
- Kuhlman, K.R.; Vargas, I.; Geiss, E.G.; Lopez-Duran, N.L. Age of Trauma Onset and HPA Axis Dysregulation Among Trauma-Exposed Youth. J Trauma Stress 2015, 28, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Amavizca, B.E.; Orozco-Castellanos, R.; Ortíz-Orozco, R.; Padilla-Gutiérrez, J.; Valle, Y.; Gutiérrez-Gutiérrez, N.; García-García, G.; Gallegos-Arreola, M.; Figuera, L.E. Contribution of GSTM1, GSTT1, and MTHFR Polymorphisms to End-Stage Renal Disease of Unknown Etiology in Mexicans. Indian J Nephrol 2013, 23, 438–443. [Google Scholar] [CrossRef]
- Goldman, G.S.; Cheng, R.Z. The Immature Infant Liver: Cytochrome P450 Enzymes and Their Relevance to Vaccine Safety and SIDS Research. Int J Med Sci 2025, 22, 2434–2445. [Google Scholar] [CrossRef]
- Aronica, L.; Ordovas, J.M.; Volkov, A.; Lamb, J.J.; Stone, P.M.; Minich, D.; Leary, M.; Class, M.; Metti, D.; Larson, I.A.; et al. Genetic Biomarkers of Metabolic Detoxification for Personalized Lifestyle Medicine. Nutrients 2022, 14, 768. [Google Scholar] [CrossRef]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human Mitochondrial DNA: Roles of Inherited and Somatic Mutations. Nat Rev Genet 2012, 13, 878–890. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA Damage and Reactive Oxygen Species in Neurodegenerative Disease. FEBS Lett 2018, 592, 728–742. [Google Scholar] [CrossRef]
- Toprani, S.M.; Mordukhovich, I.; McNeely, E.; Nagel, Z.D. Suppressed DNA Repair Capacity in Flight Attendants after Air Travel. Sci Rep 2025, 15, 16513. [Google Scholar] [CrossRef]
- Frey, Y.; Haj, M.; Ziv, Y.; Elkon, R.; Shiloh, Y. Broad Repression of DNA Repair Genes in Senescent Cells Identified by Integration of Transcriptomic Data. Nucleic Acids Res 2025, 53, gkae1257. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA Damage Response in Cancer. Redox Biol 2019, 25, 101084. [Google Scholar] [CrossRef]
- Alegría-Torres, J.A.; Baccarelli, A.; Bollati, V. Epigenetics and Lifestyle. Epigenomics 2011, 3, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Bekdash, R.A. Epigenetics, Nutrition, and the Brain: Improving Mental Health through Diet. Int J Mol Sci 2024, 25, 4036. [Google Scholar] [CrossRef] [PubMed]
- Savic, B.; Savic, B.; Stanojlovic, S. Epigenetic DNA Methylation Under the Influence of Low-Dose Ionizing Radiation, and Supplementation with Vitamin B12 and Folic Acid: Harmful or Beneficial for Professionals? Epigenomes 2025, 9, 17. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Holm, N.V.; Verkasalo, P.K.; Iliadou, A.; Kaprio, J.; Koskenvuo, M.; Pukkala, E.; Skytthe, A.; Hemminki, K. Environmental and Heritable Factors in the Causation of Cancer--Analyses of Cohorts of Twins from Sweden, Denmark, and Finland. N Engl J Med 2000, 343, 78–85. [Google Scholar] [CrossRef]
- Dominic, A.; Hamilton, D.; Abe, J.-I. Mitochondria and Chronic Effects of Cancer Therapeutics: The Clinical Implications. J Thromb Thrombolysis 2021, 51, 884–889. [Google Scholar] [CrossRef]
- Bai, B.; Ma, Y.; Liu, D.; Zhang, Y.; Zhang, W.; Shi, R.; Zhou, Q. DNA Damage Caused by Chemotherapy Has Duality, and Traditional Chinese Medicine May Be a Better Choice to Reduce Its Toxicity. Front Pharmacol 2024, 15, 1483160. [Google Scholar] [CrossRef]
- van den Boogaard, W.M.C.; Komninos, D.S.J.; Vermeij, W.P. Chemotherapy Side-Effects: Not All DNA Damage Is Equal. Cancers (Basel) 2022, 14, 627. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.-X.; Zhou, P.-K. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Sig Transduct Target Ther 2020, 5, 60. [Google Scholar] [CrossRef]
- Second Cancers Related to Treatment. Available online: https://www.cancer.org/cancer/survivorship/long-term-health-concerns/second-cancers-in-adults/treatment-risks.html (accessed on 1 August 2025).
- Indari, O.; Ghosh, S.; Bal, A.S.; James, A.; Garg, M.; Mishra, A.; Karmodiya, K.; Jha, H.C. Awakening the Sleeping Giant: Epstein–Barr Virus Reactivation by Biological Agents. Pathogens and Disease 2024, 82, ftae002. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, F.; Rasizadeh, R.; Sharaflou, S.; Aghbash, P.S.; Shamekh, A.; Jafari-Sales, A.; Bannazadeh Baghi, H. Coinfection of EBV with Other Pathogens: A Narrative Review. Front. Virol. 2024, 4. [Google Scholar] [CrossRef]
- Hewavisenti, R.V.; Arena, J.; Ahlenstiel, C.L.; Sasson, S.C. Human Papillomavirus in the Setting of Immunodeficiency: Pathogenesis and the Emergence of next-Generation Therapies to Reduce the High Associated Cancer Risk. Front Immunol 2023, 14, 1112513. [Google Scholar] [CrossRef]
- Does Hormone Replacement Therapy (HRT) Increase the Risk of Cancer? Available online: https://www.cancerresearchuk.org/about-cancer/causes-of-cancer/hormones-and-cancer/does-hormone-replacement-therapy-increase-cancer-risk (accessed on 1 August 2025).
- Vinogradova, Y.; Coupland, C.; Hippisley-Cox, J. Use of Hormone Replacement Therapy and Risk of Breast Cancer: Nested Case-Control Studies Using the QResearch and CPRD Databases. BMJ 2020, 371, m3873. [Google Scholar] [CrossRef]
- Cao, C.-F.; Ma, K.-L.; Shan, H.; Liu, T.-F.; Zhao, S.-Q.; Wan, Y.; Jun-Zhang, null; Wang, H.-Q. CT Scans and Cancer Risks: A Systematic Review and Dose-Response Meta-Analysis. BMC Cancer 2022, 22, 1238. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, J.; Zhang, Q.; Zhang, J.; Dai, Q.; Zhang, D. Radiation Exposure in Recurrent Medical Imaging: Identifying Drivers and High-Risk Populations. Front. Public Health 2025, 13. [Google Scholar] [CrossRef]
Figure 1.
Root Cause Analysis (RCA) Framework of Cancer Initiation.
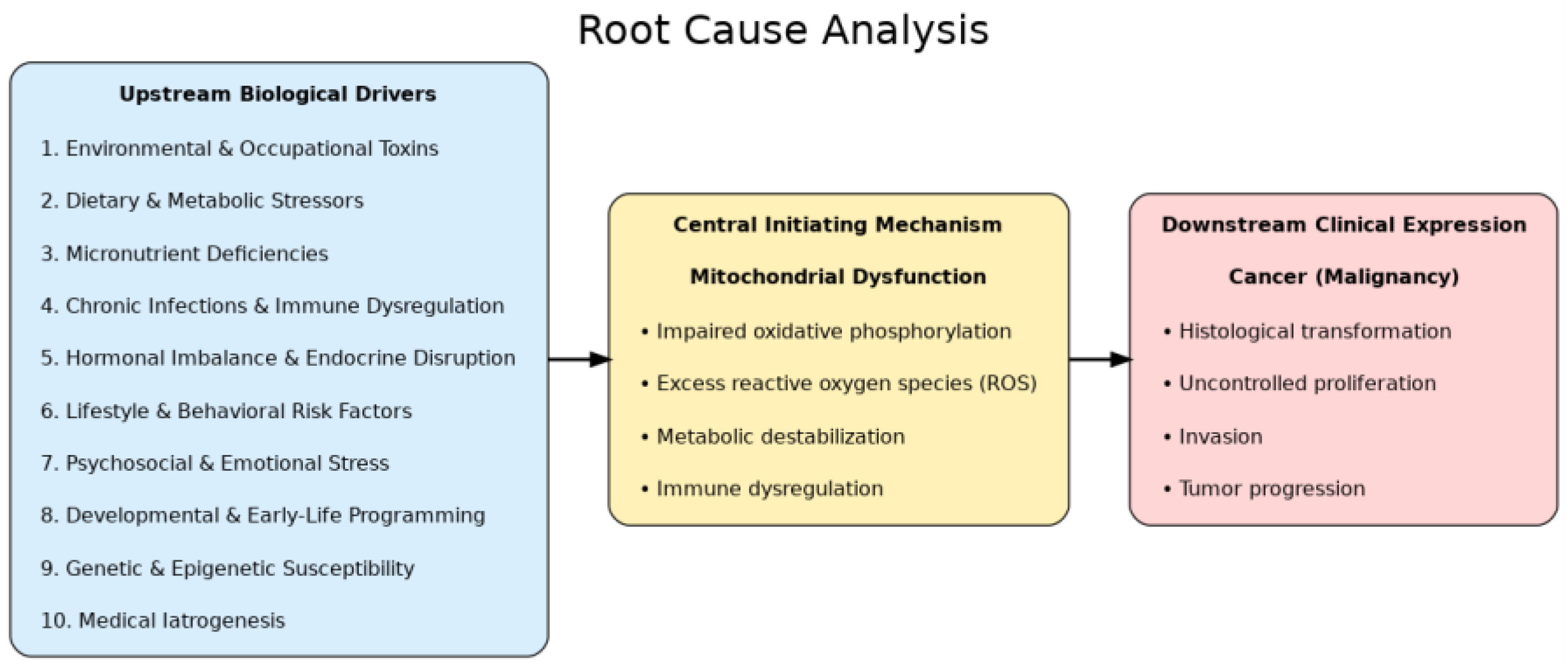

Table 1.
Comparison of Cancer Theories and Their Explanatory Scope.
| Layer | Definition | Representative Factors / Mechanisms | Clinical Relevance |
|---|---|---|---|
| 1. Upstream Biological Drivers | Modifiable exposures and conditions that destabilize cellular homeostasis and predispose to mitochondrial damage | Environmental & Occupational Toxins; Dietary & Metabolic Stressors; Micronutrient Deficiencies; Chronic Infections & Immune Dysregulation; Hormonal Imbalance & Endocrine Disruption; Lifestyle & Behavioral Risk Factors; Psychosocial & Emotional Stress; Developmental & Early-Life Programming; Genetic & Epigenetic Susceptibility; Medical Iatrogenesis | Identifies modifiable drivers for prevention and early intervention |
| 2. Central Initiating Mechanism: Mitochondrial Dysfunction | Collapse of oxidative phosphorylation and loss of metabolic stability that directly trigger malignant transformation | Impaired oxidative phosphorylation; excess reactive oxygen species (ROS) and oxidative stress; metabolic destabilization; immune dysregulation | Provides proximate target for therapeutic strategies (metabolic, nutritional, pharmacologic) |
| 3. Downstream Clinical Expression | Phenotypic outcomes of mitochondrial dysfunction manifesting as cancer | Histological transformation; uncontrolled proliferation; invasion; tumor progression | Guides clinical diagnosis, staging, and integrative disease management |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



