Submitted:
03 September 2025
Posted:
03 September 2025
You are already at the latest version
Abstract
Pancreatic neuroendocrine tumors (PNETs) represent a biologically heterogeneous group of neoplasms shaped by both intrinsic genomic alterations and dynamic interactions with the tumor microenvironment (TME). Conventional analytical approaches offered limited insight into these complex mechanisms. However, the emergence of multi-omics technologies including genomics, transcriptomics, proteomics, and spatial single-cell platforms dramatically expanded our understanding of tumor evolution, immune-stromal crosstalk, and phenotypic plasticity. In this review, we discuss the recent integrated omics findings to highlight co-evolutionary dynamics between tumor cells and the TME, uncover novel biomarkers, and explore therapeutic implications. We further propose that multi-omics analysis offers not only a descriptive snapshot of tumor composition but also a mechanistic framework for precision oncology. This paradigm enables more accurate patient stratification and the identification of actionable vulnerabilities that may guide targeted therapeutic strategies in PNETs.
Keywords:
pancreatic neuroendocrine tumors
; multi-omics profiling
; tumor microenvironment
; immune modulation
; genomic instability
; single-cell rna-seq
; spatial transcriptomics
1. Introduction
Pancreatic neuroendocrine tumors (PNETs) are a rare and biologically diverse class of neoplasms originating from the endocrine cells of the pancreas, distinguished by their complex pathological architecture and diverse clinical behavior [1,2,3,4,5]. Moreover, PNETs exhibit a high propensity for liver metastasis and postoperative recurrence, with the liver being the predominant site of disease relapse [6,7]. Based on hormone secretion, PNETs are broadly classified into functional and non-functional subtypes clinically. Functional PNETs (F-PNETs), such as insulinomas and gastrinomas, are typically diagnosed earlier due to hormone-related syndromes. In contrast, non-functional PNETs (NF-PNETs), which comprise the majority of cases, are often asymptomatic until late stages and indicate a wider range of malignant potential. The World Health Organization (WHO) further stratifies PNETs into grades G1, G2, and G3 based on mitotic count and Ki-67 proliferation index [8,9]. While G1 and G2 tumors are generally well-differentiated and may follow a more indolent course, G3 tumors encompass both well-differentiated high-grade PNETs and poorly differentiated pancreatic neuroendocrine carcinomas (PNECs), each with distinct molecular and clinical behavior. Such evolutionary progression is generally indicative of poor clinical outcomes [10,11,12,13]. Defining the evolutionary dynamics and progression characteristics of PanNETs holds potential for enhancing prognostic assessment.
While the WHO grading system classifies tumors according to proliferative indices, it overlooks the functional plasticity and microenvironmental reprogramming that often underlie aggressive phenotypes in histologically similar tumors. Mounting evidence suggests that the tumor microenvironment (TME), a dynamic assembly of stromal cells, immune infiltrates, vasculature, and extracellular matrix (ECM) plays a central role in dictating tumor progression, therapeutic responsiveness, and immune evasion [14,15]. In many instances, TME features shown stronger prognostic and predictive value than tumor-intrinsic factors alone, emphasizing the need to understand this intricate ecosystem [16,17,18]. A key unmet need is the development of integrative approaches that place the tumor microenvironment at the forefront, alongside tumor-intrinsic factors, to improve prognostic accuracy and optimize therapeutic strategies in PNETs.
Historically, limited resolution of analytic tools constrained mechanistic insight into TME–tumor interactions. However, recent advances in multi-omics technologies—including single-cell RNA sequencing (scRNA-seq), spatial transcriptomics, proteogenomics, and deconvolution of bulk RNA-Seq data—enabled comprehensive profiling of both tumor-intrinsic and extrinsic features. In initial studies, genomic analyses revealed that MEN1, DAXX, and ATRX are among the most frequently mutated genes in PNETs, often associated with poor clinical outcomes and disrupted chromatin regulation [5,19,20,21]. Beyond these earlier genomic profiling studies, the newer multi-omics platforms not only reconstruct the cellular architecture of PNETs but also unveil functional programs such as lineage plasticity, immune remodeling, metabolic rewiring, and tumor–stroma crosstalk that lead to disease progression. In this review, we integrate findings from cutting-edge multi-omics studies to reframe our understanding of PNET biology through the lens of TME co-evolution. We uncover core mechanisms such as genomic instability, stromal remodeling, immune modulation, and biomarker discovery, with a focus on their translational relevance for patient stratification and targeted therapeutic development.
2. Genomic and Evolutionary Remodeling
2.1. Genomic Instability as a Diver of Dedifferentiation and Transcriptomic Plasticity
The transition from well-differentiated, indolent tumors to poorly differentiated, high-grade PNETs is fundamentally driven by increasing genomic instability and transcriptomic reprogramming. Cutting-edge technologies enabled the mapping of this transformation across multiple molecular layers [22,23]. Low-grade tumors often present low copy number variation (CNV) and maintain endocrine identity through the stable expression of lineage-specific transcription factors and chromatin regulators. However, as tumors progress to higher grades, widespread CNVs, chromosomal instability, and altered DNA repair mechanisms emerge. These changes are accompanied by global shifts in gene expression, including downregulation of neuroendocrine markers and upregulation of genes associated with cell cycle progression, EMT, hypoxia, and inflammation [24].
Single-cell and bulk transcriptomic analyses stratified by WHO grade (G1–G3) uncover a correlation between tumor grade and increased proliferative signaling, loss of endocrine differentiation, and activation of epithelial–mesenchymal transition (EMT) and MYC-driven transcriptional programs in PNETs [25,26,27]. CNV inference from scRNA-seq (e.g., inferCNV) in NF-PNETs demonstrated that G3 tumors harbor subclonal populations marked by chromosomal instability and dedifferentiation [28]. These subclones preferentially localize to the invasive tumor margins and express markers of metastasis and cell cycle progression. Moreover, in CNV-high tumors, widespread chromosomal aberrations, elevated tumor mutational burden (TMB), and activation of DNA repair and oxidative stress pathways are observed. These molecular features are often accompanied by microenvironmental alterations, including expansion of myofibroblastic cancer-associated fibroblasts (CAFs), extracellular matrix (ECM) deposition, and immune exclusion [29,30].
In addition to gene-level mutations, large-scale chromosomal alterations are a hallmark of PNET progression. CNV profiling through whole-genome sequencing (WGS) revealed three major structural subtypes: CNV-low, CNV-altered, and CNV-recurrent [28]. CNV-low tumors typically retain endocrine differentiation and display stable karyotypes. In contrast, CNV-altered and CNV-recurrent tumors exhibit broad chromosomal gains and losses. For example, recurrent CNV profiling by proteomic analysis identified frequent arm-level and focal gains and losses across the NF-PNET genome, even in tumors classified as low or intermediate grade [31]. Common gains are observed on chromosomes 4p, 4q, 7p, 7q, 13q, 14q, 19p, 19q, 20p, and 20q, while losses frequently affect 11p and 11q. Notably, amplification of CDK6 (chromosome 7) was functionally linked to enhanced cell cycle progression and defines a subset of highly proliferative tumors [32]. Similarly, amplification of EZH2, a histone methyltransferase also located on chromosome 7, may contribute to immune modulation by influencing interferon signaling and epigenetic silencing [33]. Conversely, loss of MEN1 on chromosome 11—one of the most frequently deleted tumor suppressors in PanNETs—disrupts chromatin regulation and is considered a fundamental early event in tumor initiation [19,21]. Similarly, ATRX/DAXX mutations, often associated with alternative lengthening of telomeres (ALT), co-occur with chromosomal rearrangements involving chromosome 21q or 9p [34,35,36].
Transcriptomic data including bulk RNA sequencing, single-cell RNA-seq, Proteogenomic and spatial transcriptomics further illuminate this process. ScRNA-seq of primary tumors revealed enrichment of gene programs associated with angiogenesis and stemness [25]. Especially, transcription factor 4 (TCF4), which was found to involve in neural development and neuroendocrine differentiation [37,38], enhanced activity in primary site. However, the significantly enriched cellular functions in metastatic tumor were related to biosynthesis, mitosis, cell cycle, and cell proliferation. Proteogenomic profiling revealed that high-grade tumors downregulate these markers and upregulate genes associated with hypoxia responses, EMT and an inflammatory signature, which correlated with aggressive phenotype, such as invasion and metastasis [28,39]. Spatial transcriptomics implied that these proliferative, dedifferentiated states are enriched at invasive and hypoxic regions with dense CAF infiltration and limited immune surveillance [34]. In G3 PNETs, tumor cells and CAFs co-express EMT and invasion-associated genes (e.g., MMPs), while CAFs secrete paracrine inducers of dedifferentiation, notably TGFβ1 [31,34,40]. This reciprocal signaling loop reinforces malignant transformation and illustrates the coupled evolution of tumor and stroma.
A notable case study described a patient with a germline FANCD2 mutation whose PNET exhibited extensive chromosomal instability and a dedifferentiated transcriptional profile [41]. Whole exome sequencing of this tumor revealed marked germline mutation of WNT10A (a WNT signaling activator) and deletion of MUTYH (a connection with base excision repair deficiency) [42,43,44,45]. Moreover, due to FANCD2 germline mutation, impaired DNA damage repair and unprotected genome resulted in high mutation rate in PNETs [46].
Altogether, these data supported that genomic instability is not merely a consequence of tumor progression, but a foundational feature that reshapes cellular identity and primes tumors for invasion, metastasis, and therapy resistance.
2.2. Clonal Evolution and the Pre-Configuration of Metastatic Niches
Longitudinal and cross-sectional integrated omics studies using pseudotime trajectory inference and CNV profiling has uncovered dynamic clonal transitions during PNET progression. These transitions are marked by acquisition of metabolic plasticity, angiogenic signaling, and transcriptional programs associated with immune evasion and proliferative expansion [25,47]. These changes are not stochastic but reflect selective pressures imposed by evolving microenvironmental niches.
Metastatic lesions, particularly in the liver, are often populated by tumor clones enriched in oxidative phosphorylation, MYC signaling, and resistance to apoptosis depend on increased E2F family and BAX expression via scRNA-seq [25]. These environments are also characterized by pro-angiogenic endothelial cells and M1-like macrophages, forming niches conducive to metastatic colonization. Spatial transcriptomics demonstrated that dissemination is often preceded by the emergence of spatially confined, proliferation-primed subclones at the tumor margin, frequently associated with hypoxia-driven gene expression (e.g., MMP9) [34].
Proteogenomic profiling delineated that EMT-like subpopulations, enriched in high-grade and metastatic tumors, indicate TGF-β activation, glycolysis, MYC signaling, and hypoxia-responsive transcription [31,48]. Especially, hypoxia-induced EMT signature exhibits positive correlation with TGF-β signaling pathway [49]. ScRNA transcriptomics identified these metastatic clusters localize to invasive fronts with high CAF density and minimal CD8+ T cell infiltration [28]. These findings suggested that metastatic competency is not acquired de novo but preconfigured within transcriptionally primed subpopulations at the primary tumor margins. To define the metastatic potential in the high-grade tumors, scRNA transcriptome analysis in PNETs revealed the role of a gene signature with prominent emerge of two genes PCSK1 and SMOC1 [25,28].
In summary, genomic instability in PNETs promotes not only tumor-intrinsic transformation but also adaptation to selective pressures imposed by the microenvironment. Its co-occurrence with dedifferentiation, immune evasion, and stromal remodeling underscores its pivotal role in malignant progression (Figure 1). These findings collectively illustrate the need for therapeutic strategies that account for intratumoral heterogeneity and spatial dynamics to effectively target the evolving landscape of PNETs.
3.1. Integrated Omics Profiling of Tumor-Stroma Interactions
While tumor-intrinsic alterations are critical, a full understanding of PNET progression requires dissecting their dynamic interactions with the surrounding stroma and immune compartments. ScRNA-seq and spatial transcriptomics revolutionized our ability to deconstruct the cellular heterogeneity of the PNET microenvironment. These high-resolution technologies exposed a complex ecosystem composed of malignant neuroendocrine cells, cancer-associated fibroblasts (CAFs), endothelial cells, immune subsets including CD4+ and CD8+ T cells, B cells, dendritic cells, tumor-associated macrophages (TAMs), and mast cells each exhibiting context-dependent differentiation states and functional programs [25,28].
Spatial transcriptomic mapping shown that these cellular populations exhibit non-random, spatially organized distributions [34]. Proliferative tumor regions are typically bordered by dense fibroblast-rich stroma, which serves both as a physical barrier and a source of immunomodulatory cues. Proteogenomic profiling uncovered that hypoxic and fibrotic zones often co-localize with reduced immune infiltration, highlighting spatial regulation of immune exclusion and therapeutic resistance [31]. These spatially defined patterns provide critical insights into how microenvironmental architecture constrains or supports tumor progression.
3.2. CAF-Driven Niches Promoting Dedifferentiation and Immune Exclusion
Building upon transcriptomic findings, recent studies identified distinct CAF subtypes in PNETs with specialized roles in shaping functional tumor niches [25,28,34]. Spatial profiling of sorted α-SMA+ stromal cells show that CAFs evolve with tumor grade: G1/G2 tumors exhibit ECM-remodeling CAFs, while G3 tumors harbor immunoregulatory and pro-fibrotic CAFs expressing TGFB1, FN1 (fibronectin) and FGF8 (fibroblast growth factor 8). These changes contribute to immune exclusion, enhanced invasiveness, and hypoxia.
Single-cell transcriptomic and spatial data reported that dedifferentiated tumor cells localize to CAF-enriched fibrotic zones, where EMT and cell cycle genes (e.g., MMPs, MYC) are upregulated [28,34]. These data illustrate how functional state transitions—such as the loss of neuroendocrine identity and acquisition of mesenchymal or progenitor-like traits are not solely driven by intrinsic oncogenic signaling but are spatially reinforced by CAF-derived cues within the fibrotic tumor margins. Pseudotime trajectory analyses elucidated that proliferative, dedifferentiated subclones emerge along a continuum from endocrine to mesenchymal transcriptional states, particularly at hypoxic, fibrotic margins [25]. Moreover, spatial constraints imposed by the stroma also shape immune dynamics. Co-localization of CAFs with immunosuppressive cells (e.g., M2 macrophages, FOXP3+ Tregs) forms exclusionary zones where CD8+ T cells are scarce or dysfunctional, especially in patient with metastasis. Single-cell and proteomic profiling elucidated that tumor cells from PNET patients within these micro-niches frequently express exhaustion markers (e.g. PDCD1, LAG3, and CTLA4), further reinforcing immune evasion [25,28,31,48]. Importantly, these immune-excluded niches overlap with areas harboring chromosomally unstable subclones identified by inferCNV analysis, suggesting a convergence of structural instability, functional dedifferentiation, and immune suppression.
Together, these observations underscore a model in which tumor cell state plasticity, stromal remodeling, and immune exclusion co-evolve within spatially defined microenvironments. Dissecting these coupled networks at single-cell and spatial resolution is critical for identifying actionable targets and designing therapies that disrupt both malignant progression and its supportive niche.
4. Immune Landscape and Functional States
4.1. Immune Phenotypes in PNET Progression
To fully appreciate the immunologic consequences of stromal remodeling, we turn to the spatial and functional characterization of immune infiltration within the PNET microenvironment. The immune contexture of PNETs emerged as a key determinant of disease progression, metastatic potential, and therapeutic responsiveness. Multi-omics approaches including bulk RNA-Seq, scRNA-seq, and immune deconvolution—enabled classification of PNETs into distinct immunophenotypes: immune-cold, immune-active, and immune-suppressive [31,48,50]. Immune-cold tumors displayed sparse lymphocyte infiltration and diminished expression of antigen-presentation machinery, suggesting a state of immune ignorance or exclusion. Conversely, immune-active tumors are characterized by abundant CD8+ cytotoxic T cells and M1-like macrophages, elevated interferon-γ signaling, and upregulation of costimulatory molecules, features indicative of a partially activated or exhausted immune response. Immune-suppressive tumors display high infiltration of regulatory T cells (Tregs), M2-polarized macrophages, and elevated expression of immune checkpoint ligands such as PD-L1, IDO1, and TIM-3, along with TGF-β pathway activation. These immune phenotypes are strongly associated with tumor grade and microenvironmental features, providing critical insight into prognosis and therapeutic stratification. Moreover, multiplex spatial profiles validated that these immune profiles are spatially compartmentalized within the TME of PNETs [50]. The metastasis-like primary (MLP) subtype, defined by miRNA profiles [26,51], presented increased macrophages under hypoxia and necroptosis.
4.2. Spatial and Molecular Determinants of Immune Modulation
Spatially resolved transcriptomic and proteotranscriptomic profiling of PNETs further clarified how architectural and molecular features of the TME regulate immune infiltration and function [34,52]. Immune-active regions are often segregated by fibrotic stroma enriched in CAFs and immunosuppressive cytokines, which limit lymphocyte trafficking and activation. These barriers are maintained by signaling pathways such as TGF-β and Hippo-YAP/TAZ, which promote extracellular matrix deposition and immune exclusion. In contrast, immune-permissive zones display higher expression of MHC class I and II molecules, chemokines, and costimulatory ligands that facilitate effector T cell recruitment and retention. The juxtaposition of immune-active and immune-silent regions within the same tumor reflected the spatial heterogeneity of immunologic pressure and underscored the limitations of bulk profiling in capturing clinically relevant immune dynamics.
Together, these findings underscore the necessity of spatially integrated multi-omics approaches to fully resolve the immunologic landscape of PNETs. A deeper understanding of spatial immune organization may guide the rational design of combination therapies, particularly in overcoming immune suppression in stroma-rich, high-grade tumors.
5. Translational Implications and Biomarker Discovery
The integration of multi-omics technologies accelerated the identification of clinically relevant biomarkers embedded within the PNET microenvironment. These biomarkers hold significant promise for refining prognosis, predicting therapeutic response, and guiding personalized interventions. Among the emerging candidates, VCAN—a stromal proteoglycan enriched in mesenchymal subtypes was proposed as a non-invasive biomarker detectable in both tumor tissue and plasma of PNETs [48]. Transcriptional co-activators YAP1 and TAZ, central mediators of Hippo pathway signaling, are preferentially activated in stromal-rich, immune-excluded tumors and represent potential therapeutic targets for PNET patients [52]. From an immunologic perspective, the expression of immune checkpoint molecules including PD-L1, TIM-3, and IDO1 aligns with suppressive immune phenotypes and may identify patients most likely to benefit from immune checkpoint blockade [48]. Spatial and proteogenomic profiling further refines these associations by capturing context-specific expression patterns within distinct tumor–stroma compartments.
These findings nominate a combination therapeutic strategy for stroma-rich PNETs, incorporating YAP/TAZ inhibition, immune checkpoint blockade, and TGF-β pathway suppression [52]. Notably, the case study of a FANCD2 germline mutation-associated PNET demonstrated the feasibility of leveraging scRNA-seq for uncovering targetable features such as MYC activation and DNA damage response pathways, underscoring the utility of integrating rare-case multi-omics data into biomarker-driven strategies. These findings emphasize the value of multi-omics-informed stratification for personalized therapy.
6. Future Outlook and Conclusions
The advent of multi-omics technologies profoundly transformed our understanding of PNETs. Once classified primarily by morphology and proliferation indices, these tumors are now recognized as dynamic, spatially structured ecosystems shaped by genomic instability, transcriptional diversity, and microenvironmental remodeling (Figure 2). Despite recent progress, several critical questions remain unanswered. The cellular origins of PNET subtypes are still under investigation, and it remains unclear how treatment-naïve tumors differ from those with acquired resistance. Predictive markers of metastatic potential are urgently needed.
Understanding how specific signals guide the formation of immune niches and how tumor–stroma metabolic interactions evolve under therapy is essential for therapeutic reprogramming. Addressing spatial heterogeneity remains a major hurdle, as do the identification of reliable biomarkers and the development of physiologically relevant preclinical models. To move the field forward, several strategies are required. Advanced organoid and microfluidic systems will be essential for modeling tumor–immune–stroma interactions. Humanized mouse models that preserve immune function will be crucial for validating new therapies. Future clinical trials should integrate multi-omics endpoints and spatial profiling, supported by standardized methodologies to enable broader implementation in pathology workflows.
Looking ahead, the integration of basic discovery with translational research promises to usher in a new era of personalized oncology for PNET patients. Advancing precision oncology in PNETs will require continued investment in multi-omics discovery, spatial profiling, and collaborative clinical translation.
Author Contributions
Conceptualization, Y.W., A.S.A. and Y.S.; Methodology, Y.W. and Y.S.; Formal Analysis, Investigation and Resources, Y.W.; Writing – Original Draft Preparation, Y.W., Y.Z. and Y.S.; Writing – Review & Editing, A.S.A., P.A.P., H.C. and B.E.; Supervision, Project Administration and Funding Acquisition, A.S.A. and Y.S.
Funding
This research was partly supported by the NIH/NCI Cancer Center Support Grant P30CA022453, and the Cancer Research Horizons Fund Grant at the Karmanos Cancer Institute.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Reid, M.D., et al., Neuroendocrine tumors of the pancreas: current concepts and controversies. Endocr Pathol, 2014. 25(1): p. 65-79.
- Lawrence, B., et al., The epidemiology of gastroenteropancreatic neuroendocrine tumors. Endocrinol Metab Clin North Am, 2011. 40(1): p. 1-18, vii. [CrossRef]
- Cives, M. and J.R. Strosberg, Gastroenteropancreatic Neuroendocrine Tumors. CA Cancer J Clin, 2018. 68(6): p. 471-487.
- Simon, T., et al., DNA methylation reveals distinct cells of origin for pancreatic neuroendocrine carcinomas and pancreatic neuroendocrine tumors. Genome Med, 2022. 14(1): p. 24. [CrossRef]
- Chan, C.S., et al., ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat Commun, 2018. 9(1): p. 4158. [CrossRef]
- Garcia-Carbonero, R., et al., Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (GEP-NETs): results from the National Cancer Registry of Spain (RGETNE). Ann Oncol, 2010. 21(9): p. 1794-1803. [CrossRef]
- Singh, S., et al., Recurrence in Resected Gastroenteropancreatic Neuroendocrine Tumors. JAMA Oncol, 2018. 4(4): p. 583-585. [CrossRef]
- Inzani, F., G. Petrone, and G. Rindi, The New World Health Organization Classification for Pancreatic Neuroendocrine Neoplasia. Endocrinol Metab Clin North Am, 2018. 47(3): p. 463-470. [CrossRef]
- Nagtegaal, I.D., et al., The 2019 WHO classification of tumours of the digestive system. Histopathology, 2020. 76(2): p. 182-188. doi:10.1111/his.13975.
- Tang, L.H., et al., Well-Differentiated Neuroendocrine Tumors with a Morphologically Apparent High-Grade Component: A Pathway Distinct from Poorly Differentiated Neuroendocrine Carcinomas. Clin Cancer Res, 2016. 22(4): p. 1011-7. [CrossRef]
- Crona, J., et al., Multiple and Secondary Hormone Secretion in Patients With Metastatic Pancreatic Neuroendocrine Tumours. J Clin Endocrinol Metab, 2016. 101(2): p. 445-52. [CrossRef]
- de Mestier, L., et al., Metachronous hormonal syndromes in patients with pancreatic neuroendocrine tumors: a case-series study. Ann Intern Med, 2015. 162(10): p. 682-9.
- Botling, J., et al., High-Grade Progression Confers Poor Survival in Pancreatic Neuroendocrine Tumors. Neuroendocrinology, 2020. 110(11-12): p. 891-898. [CrossRef]
- de Visser, K.E. and J.A. Joyce, The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell, 2023. 41(3): p. 374-403.
- Xiao, Y. and D. Yu, Tumor microenvironment as a therapeutic target in cancer. Pharmacol Ther, 2021. 221: p. 107753. [CrossRef]
- Zhang, J., et al., Turning cold tumors hot: from molecular mechanisms to clinical applications. Trends Immunol, 2022. 43(7): p. 523-545. [CrossRef]
- Wang, L., et al., Hot and cold tumors: Immunological features and the therapeutic strategies. MedComm (2020), 2023. 4(5): p. e343. [CrossRef]
- Baghban, R., et al., Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal, 2020. 18(1): p. 59. [CrossRef]
- Scarpa, A., et al., Whole-genome landscape of pancreatic neuroendocrine tumours. Nature, 2017. 543(7643): p. 65-71.
- Jiao, Y., et al., DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science, 2011. 331(6021): p. 1199-203. [CrossRef]
- Hong, X., et al., Whole-genome sequencing reveals distinct genetic bases for insulinomas and non-functional pancreatic neuroendocrine tumours: leading to a new classification system. Gut, 2020. 69(5): p. 877-887. [CrossRef]
- Zhou, W., et al., Targeted deep sequencing reveals the genetic heterogeneity in well-differentiated pancreatic neuroendocrine tumors with liver metastasis. Hepatobiliary Surg Nutr, 2023. 12(3): p. 302-313. [CrossRef]
- Abbas, T., M.A. Keaton, and A. Dutta, Genomic instability in cancer. Cold Spring Harb Perspect Biol, 2013. 5(3): p. a012914.
- Ciriello, G., et al., Emerging landscape of oncogenic signatures across human cancers. Nat Genet, 2013. 45(10): p. 1127-33. [CrossRef]
- Zhou, Y., et al., Single-cell RNA sequencing reveals spatiotemporal heterogeneity and malignant progression in pancreatic neuroendocrine tumor. Int J Biol Sci, 2021. 17(14): p. 3760-3775. [CrossRef]
- Sadanandam, A., et al., A Cross-Species Analysis in Pancreatic Neuroendocrine Tumors Reveals Molecular Subtypes with Distinctive Clinical, Metastatic, Developmental, and Metabolic Characteristics. Cancer Discov, 2015. 5(12): p. 1296-313.
- Miki, M., et al., CLEC3A, MMP7, and LCN2 as novel markers for predicting recurrence in resected G1 and G2 pancreatic neuroendocrine tumors. Cancer Med, 2019. 8(8): p. 3748-3760. [CrossRef]
- Ye, Z., et al., Single-cell sequencing reveals the heterogeneity of pancreatic neuroendocrine tumors under genomic instability and histological grading. iScience, 2024. 27(9): p. 110836. [CrossRef]
- Zhao, Y., et al., Stromal cells in the tumor microenvironment: accomplices of tumor progression? Cell Death Dis, 2023. 14(9): p. 587. [CrossRef]
- Li, M., X. Gao, and X. Wang, Identification of tumor mutation burden-associated molecular and clinical features in cancer by analyzing multi-omics data. Front Immunol, 2023. 14: p. 1090838. [CrossRef]
- Tanaka, A., et al., Proteogenomic characterization of pancreatic neuroendocrine tumors uncovers hypoxia and immune signatures in clinically aggressive subtypes. iScience, 2024. 27(8): p. 110544. [CrossRef]
- Shi, Y., et al., Cell Cycle Protein Expression in Neuroendocrine Tumors: Association of CDK4/CDK6, CCND1, and Phosphorylated Retinoblastoma Protein With Proliferative Index. Pancreas, 2017. 46(10): p. 1347-1353.
- April-Monn, S.L., et al., EZH2 Inhibition as New Epigenetic Treatment Option for Pancreatic Neuroendocrine Neoplasms (PanNENs). Cancers (Basel), 2021. 13(19). [CrossRef]
- Niedra, H., et al., Tumor and alpha-SMA-expressing stromal cells in pancreatic neuroendocrine tumors have a distinct RNA profile depending on tumor grade. Mol Oncol, 2025. 19(3): p. 659-681. [CrossRef]
- Gisder, D.M., et al., DAXX, ATRX, and MSI in PanNET and Their Metastases: Correlation with Histopathological Data and Prognosis. Pathobiology, 2023. 90(2): p. 71-80. [CrossRef]
- Heaphy, C.M. and A.D. Singhi, Reprint of: The Diagnostic and Prognostic Utility of Incorporating DAXX, ATRX, and Alternative Lengthening of Telomeres (ALT) to the Evaluation of Pancreatic Neuroendocrine Tumors (PanNETs). Hum Pathol, 2023. 132: p. 1-11. [CrossRef]
- Lee, G.T., et al., TCF4 induces enzalutamide resistance via neuroendocrine differentiation in prostate cancer. PLoS One, 2019. 14(9): p. e0213488. [CrossRef]
- Forrest, M.P., et al., The emerging roles of TCF4 in disease and development. Trends Mol Med, 2014. 20(6): p. 322-31. [CrossRef]
- Ruan, K., G. Song, and G. Ouyang, Role of hypoxia in the hallmarks of human cancer. J Cell Biochem, 2009. 107(6): p. 1053-62. [CrossRef]
- Ye, Z., et al., The stromal microenvironment endows pancreatic neuroendocrine tumors with spatially specific invasive and metastatic phenotypes. Cancer Lett, 2024. 588: p. 216769. [CrossRef]
- Avsievich, E., et al., Pancreatic Neuroendocrine Tumor: The Case Report of a Patient with Germline FANCD2 Mutation and Tumor Analysis Using Single-Cell RNA Sequencing. J Clin Med, 2024. 13(24). [CrossRef]
- Pilati, C., et al., Mutational signature analysis identifies MUTYH deficiency in colorectal cancers and adrenocortical carcinomas. J Pathol, 2017. 242(1): p. 10-15. [CrossRef]
- Zhang, J.H., et al., Deficiency of Wnt10a causes female infertility via the beta-catenin/Cyp19a1 pathway in mice. Int J Med Sci, 2022. 19(4): p. 701-710. [CrossRef]
- Zhang, J.H., et al., Deletion of Wnt10a Is Implicated in Hippocampal Neurodegeneration in Mice. Biomedicines, 2022. 10(7). [CrossRef]
- Li, P., et al., Clinical significance and biological role of Wnt10a in ovarian cancer. Oncol Lett, 2017. 14(6): p. 6611-6617. [CrossRef]
- Liu, W., et al., FANCD2 and RAD51 recombinase directly inhibit DNA2 nuclease at stalled replication forks and FANCD2 acts as a novel RAD51 mediator in strand exchange to promote genome stability. Nucleic Acids Res, 2023. 51(17): p. 9144-9165. [CrossRef]
- Backman, S., et al., The evolutionary history of metastatic pancreatic neuroendocrine tumours reveals a therapy driven route to high-grade transformation. J Pathol, 2024. 264(4): p. 357-370. [CrossRef]
- Ji, S., et al., Proteogenomic characterization of non-functional pancreatic neuroendocrine tumors unravels clinically relevant subgroups. Cancer Cell, 2025. 43(4): p. 776-796 e14. [CrossRef]
- Deshmukh, A.P., et al., Identification of EMT signaling cross-talk and gene regulatory networks by single-cell RNA sequencing. Proc Natl Acad Sci U S A, 2021. 118(19). [CrossRef]
- Young, K., et al., Immune landscape, evolution, hypoxia-mediated viral mimicry pathways and therapeutic potential in molecular subtypes of pancreatic neuroendocrine tumours. Gut, 2021. 70(10): p. 1904-1913. [CrossRef]
- Olson, P., et al., MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev, 2009. 23(18): p. 2152-65. [CrossRef]
- Yang, K.C., et al., Proteotranscriptomic classification and characterization of pancreatic neuroendocrine neoplasms. Cell Rep, 2021. 37(2): p. 109817. [CrossRef]
Figure 1.
Figure 1. Genomic instability and TME co-evolution in PNETs: Summary of how increasing genomic instability drives dedifferentiation, EMT, and inflammatory programs, reinforced by CAF-rich, immune-excluded niches revealed through multi-omics profiling.3. Stromal and immune ecosystem.
Figure 1.
Figure 1. Genomic instability and TME co-evolution in PNETs: Summary of how increasing genomic instability drives dedifferentiation, EMT, and inflammatory programs, reinforced by CAF-rich, immune-excluded niches revealed through multi-omics profiling.3. Stromal and immune ecosystem.
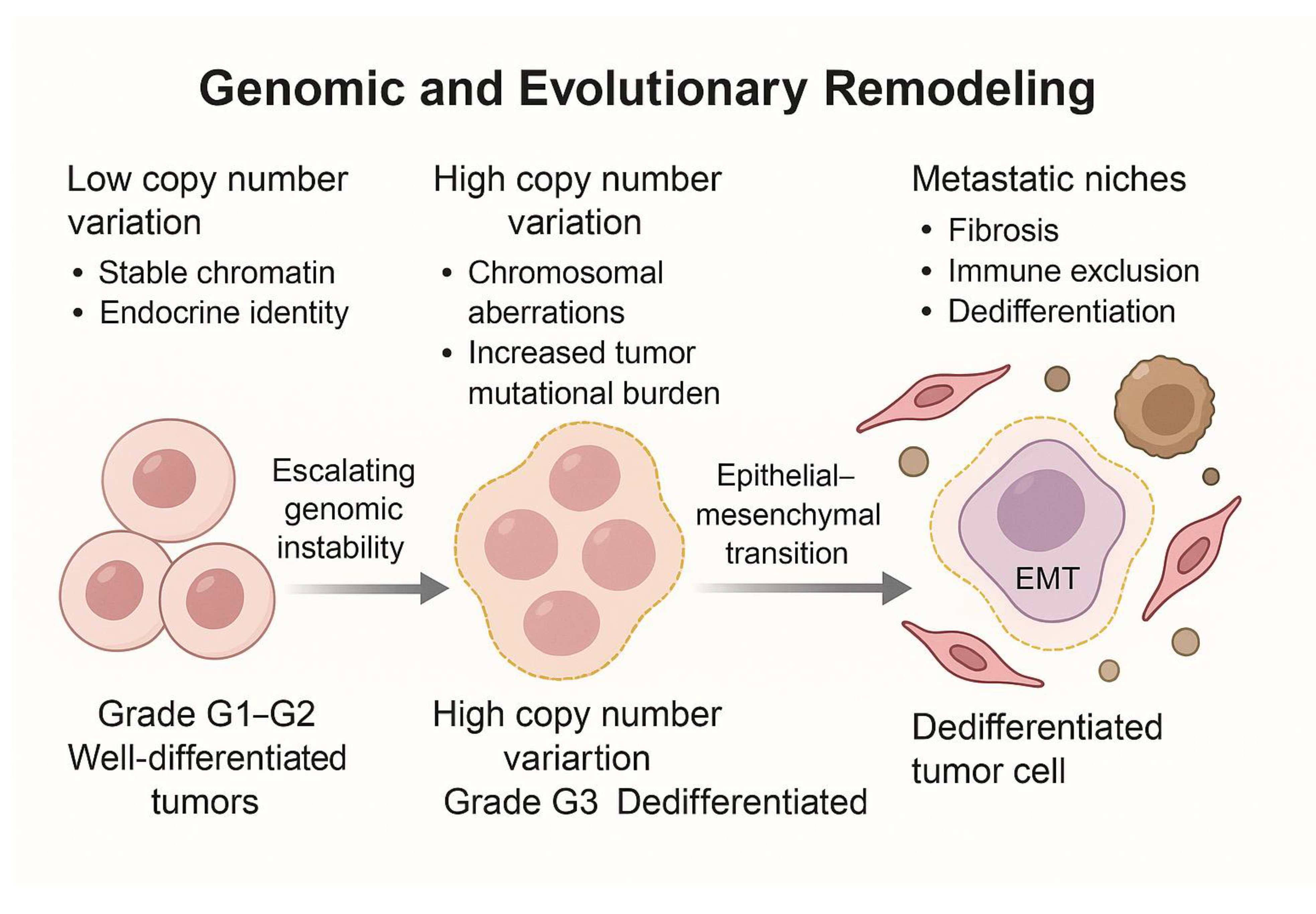
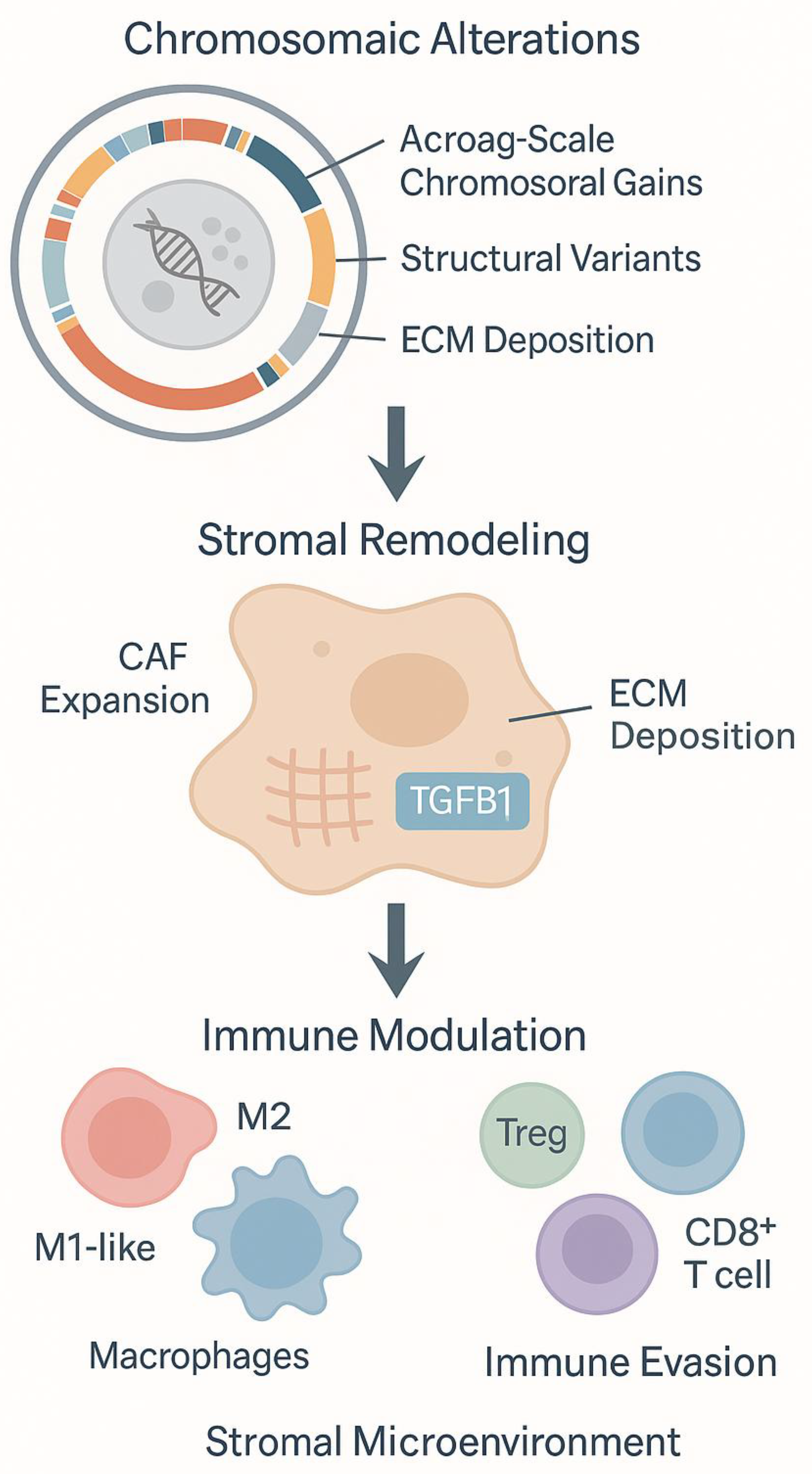
Figure 2.
Multi-omics dissection of the PNET ecosystem: Integrative genomics, transcriptomics, proteogenomics, and spatial profiling uncover tumor heterogeneity, stromal remodeling, immune landscapes, and biomarker opportunities for precision therapy.
Figure 2.
Multi-omics dissection of the PNET ecosystem: Integrative genomics, transcriptomics, proteogenomics, and spatial profiling uncover tumor heterogeneity, stromal remodeling, immune landscapes, and biomarker opportunities for precision therapy.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



