Submitted:
20 August 2025
Posted:
21 August 2025
You are already at the latest version
Abstract
Cardiovascular disease (CVD) remains the leading cause of morbidity and mortality in the elderly, driven not only by traditional risk factors but also by biological aging processes such as cellular senescence. Senescent cells accumulate in cardiovascular tissues with age and secrete a complex mix of pro-inflammatory cytokines, chemokines, proteases, and growth factors known as the senescence-associated secretory phenotype (SASP). While SASP may play beneficial roles in tissue repair, its chronic activity drives systemic inflammation, vascular remodeling, endothelial dysfunction, and myocardial fibrosis—all key features of age-related CVD. This review synthesizes the current understanding of SASP’s mechanistic contributions to vascular aging, atherosclerosis, heart failure, and arrhythmias in older adults. It highlights how SASP promotes arterial stiffness, plaque instability, cardiac remodeling, and electrical conduction abnormalities. Furthermore, the review explores emerging therapeutic strategies targeting SASP, including senolytics and senomorphics, and discusses their potential to mitigate age-related CVD. We also examine biomarker development and outline key knowledge gaps, emphasizing the need for longitudinal human studies to guide precision senescence-targeted therapies in elderly cardiovascular populations.
Keywords:
cellular senescence
; cardiovascular diseases
; inflammation
; aging
; vascular endothelium
Introduction
Aging is a strong independent risk factor for cardiovascular disease (CVD), which remains the leading cause of death globally, especially among older adults [1,2]. As populations continue to age worldwide, the healthcare burden of age-related CVDs—including heart failure (HF), atherosclerosis, cardiac arrhythmias, and vascular dysfunction—is expected to rise substantially. This trend highlights the urgent need to understand the biological underpinnings of aging that contribute to CV pathology [3,4,5]. One of the key cellular processes implicated in aging and age-related diseases is cellular senescence (i.e., a state of stable cell cycle arrest triggered by various stressors such as telomere shortening, DNA damage, and oxidative stress). Senescent cells, although initially protective, persist in tissues over time and secrete a broad array of bioactive molecules including pro-inflammatory cytokines, chemokines, growth factors, and proteases. Collectively, this pro-inflammatory phenotype is termed the senescence-associated secretory phenotype (SASP) [6,7]. While SASP can support tissue repair in acute contexts, its chronic expression drives tissue degeneration, systemic inflammation, and organ dysfunction—key features of aging [8,9]. Recent evidence has drawn direct connections between SASP and CV aging. Senescent cells accumulate in CV tissues such as the endothelium, myocardium, and vascular smooth muscle, where they disrupt tissue homeostasis through SASP-driven inflammation and extracellular matrix remodeling. These processes contribute to key pathological changes including arterial stiffness, endothelial dysfunction, and myocardial fibrosis, all of which are hallmarks of CVD in the elderly [10,11]. Given the expanding knowledge base, it is critical to synthesize our understanding of how SASP contributes to CV aging and to explore its potential as a therapeutic target. This review aims to explore the emerging role of SASP in age-related CVD, outline the molecular and cellular pathways involved, and evaluate therapeutic strategies targeting SASP to improve CV health in the elderly.
Cellular Senescence and SASP: Definitions and Mechanisms
Hallmarks of Cellular Senescence
Cellular senescence is a complex, highly conserved cellular state characterized by a permanent arrest in cell proliferation, typically triggered by stressors such as telomere attrition, DNA damage, oncogene activation, oxidative stress, and mitochondrial dysfunction [11]. Although senescence initially serves as a protective mechanism to prevent the replication of damaged or potentially oncogenic cells, the persistence of senescent cells in tissues over time can become detrimental, especially through their secretory activity [9]. Senescent cells remain metabolically active and undergo substantial alterations in gene expression and phenotype, most notably the development of SASP, a potent pro-inflammatory and tissue-modifying secretome that plays a significant role in aging and the pathogenesis of age-related diseases, including CVD [6,12,13]. One of the primary hallmarks of cellular senescence is a stable arrest of the cell cycle, often in the G1 phase. This arrest is regulated by key tumor suppressor pathways, particularly the p53/p21CIP1 and p16INK4a/Rb axes. These pathways act by inhibiting cyclin-dependent kinases (CDKs), ultimately preventing the phosphorylation of the retinoblastoma (Rb) protein, which is necessary for cell cycle progression [12]. This growth arrest is irreversible under normal physiological conditions and serves as the foundation of the senescent state. Chromatin remodeling is another prominent feature of senescence. Senescent cells often display the formation of senescence-associated heterochromatin foci (SAHF), which represent regions of condensed chromatin that contribute to the silencing of proliferation-promoting genes [14]. These epigenetic changes, including altered DNA methylation and histone modifications, help reinforce the permanent cell cycle arrest [15]. In parallel, senescent cells often exhibit a persistent DNA damage response (DDR), particularly marked by the presence of γ-H2AX foci, indicative of DNA double-strand breaks. This chronic DDR is especially prevalent at telomeres and acts as a continual signal maintaining the senescent phenotype [16,17]. Mitochondrial dysfunction is frequently observed in senescent cells, often accompanied by an increase in reactive oxygen species (ROS) production. Elevated ROS levels can promote oxidative damage and further reinforce senescence through positive feedback mechanisms involving DNA damage and inflammatory signaling. These dysfunctional mitochondria not only contribute to cellular stress but also play a role in triggering the development of SASP [18,19]. Among all the hallmarks, SASP is perhaps the most consequential in terms of impact on tissue and systemic physiology. These molecules can act in an autocrine manner to reinforce senescence, and in a paracrine manner to influence the behavior of neighboring cells, often promoting inflammation, matrix remodeling, and tissue degradation. In the context of aging tissues, especially the CV system, the chronic activity of SASP is implicated in driving many of the structural and functional changes that predispose older individuals to disease [20,21,22].
Composition and Regulation of SASP
SASP includes pro-inflammatory cytokines, chemokines, proteases, matrix-degrading enzymes, growth factors, bioactive lipids, and extracellular vesicles (Table 1). These components are not only instrumental in mediating the autocrine and paracrine effects of senescent cells but are also central to many age-related pathologies, including CVD [22,23]. The composition of SASP is context-dependent, varying by cell type, senescence-inducing stimulus, and the surrounding microenvironment. However, several factors consistently emerge across different models and tissues. Cytokines form the core of SASP, particularly interleukin (IL)-6, IL-1α, and tumor necrosis factor-alpha (TNF-α), all of which promote local and systemic inflammation [24]. IL-6, in particular, is a master regulator of chronic inflammation in aging and has been implicated in vascular dysfunction and the pathogenesis of atherosclerosis [25]. IL-1α acts upstream in the inflammatory cascade, often triggering further SASP component production via autocrine signaling loops [24]. These cytokines are critical not only in reinforcing the senescence growth arrest but also in creating a pro-inflammatory environment that disrupts tissue homeostasis [6,26]. Chemokines such as monocyte chemoattractant protein-1 (MCP-1)/C-C motif chemokine ligand 2 (CCL2), CXCL8 (IL-8), and CCL5 are also prevalent in SASP and play a crucial role in immune cell recruitment. These chemotactic signals attract various immune cells, including macrophages, neutrophils, and T cells, to sites of senescence [27,28]. While this immune surveillance can facilitate the clearance of senescent cells under optimal conditions, chronic secretion of these chemokines contributes to persistent inflammation, immune cell infiltration, and tissue damage. In the CV system, this mechanism is believed to underlie the infiltration of immune cells into atherosclerotic plaques and fibrotic cardiac tissue [10,11,21].
Proteases, particularly matrix metalloproteinases (MMPs) such as MMP-1, MMP-3, and MMP-9, are another key SASP component. These enzymes degrade the extracellular matrix (ECM), contributing to tissue remodeling but also to pathological processes such as fibrosis and plaque destabilization. In the vasculature, overexpression of MMPs leads to the breakdown of elastin and collagen, weakening vessel walls and promoting aneurysm formation or plaque rupture in atherosclerosis. The imbalance between proteases and their inhibitors is therefore a central feature of SASP-driven tissue degeneration in aging [29,30]. Growth factors such as vascular endothelial growth factor (VEGF), transforming growth factor-beta (TGF-β), and hepatocyte growth factor (HGF) are variably expressed in SASP and contribute to angiogenesis, fibrosis, and tissue remodeling. While these factors may aid in tissue repair and regeneration during acute injury, their chronic expression in the context of senescence promotes pathological angiogenesis and fibrotic remodeling. For instance, persistent TGF-β signaling is associated with cardiac and vascular fibrosis, which impairs tissue compliance and function in elderly individuals [31,32,33]. The regulation of SASP is tightly controlled at multiple levels, including transcriptional, epigenetic, and post-transcriptional mechanisms. Key transcription factors involved include nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and C/EBPβ, which coordinate the expression of many SASP genes. DDR signaling, particularly via ATM and p38 MAPK, plays a pivotal role in initiating and sustaining SASP production. Additionally, mechanistic target of rapamycin (mTOR) and GATA4 pathways have been implicated in modulating the intensity and composition of SASP (Figure 1) [15,34,35]. Notably, SASP is not static; its composition can evolve over time and in response to feedback from the microenvironment. This dynamic regulation means that SASP can shift from being initially beneficial, for instance in tissue repair, to chronically deleterious, promoting inflammation and disease progression [36].
Triggers and Amplifiers of SASP
The activation of SASP is intricately tied to a variety of cellular stressors and damage signals that induce and sustain cellular senescence. These triggers are not only responsible for initiating the senescent state but also play key roles in modulating the intensity and composition of SASP. Among the most well-characterized initiators are persistent DNA damage, telomere attrition, and mitochondrial dysfunction—each of which contributes to a chronic intracellular stress response that culminates in SASP expression [18,37]. DNA damage is a central trigger of SASP and is primarily sensed through the DDR pathway (Figure 1). Persistent double-strand DNA breaks, especially those at telomeres or induced by genotoxic stress, activate DDR kinases such as ATM and ATR. These kinases, in turn, stabilize and activate the p53 tumor suppressor protein, initiating a cascade that leads to cell cycle arrest via p21 and promotes the senescent phenotype. Crucially, even after cell cycle arrest is established, ongoing DDR signaling maintains the expression of SASP components, particularly through activation of the NF-κB and C/EBPβ transcription factors. The sustained DDR not only reinforces the senescence program but also amplifies the pro-inflammatory nature of SASP, contributing to tissue-wide inflammation and degeneration observed in aging organisms [16,38,39]. Telomere attrition (Figure 2) represents a more specific but equally potent driver of senescence and SASP activation. In somatic cells, telomeres progressively shorten with each round of replication due to the end-replication problem. Critically shortened telomeres are perceived by the cell as DNA double-strand breaks, triggering a DDR that leads to replicative senescence. This telomere-driven senescence is particularly relevant in aging tissues with high cellular turnover, such as the vascular endothelium and immune system. The resulting SASP not only halts further cellular proliferation but also induces inflammatory signaling that alters the tissue microenvironment, disrupts homeostasis, and can contribute to diseases such as atherosclerosis and myocardial fibrosis in the elderly [40,41].
Mitochondrial dysfunction is another crucial trigger of SASP and is often both a cause and consequence of senescence. Senescent cells typically exhibit dysfunctional mitochondria characterized by impaired oxidative phosphorylation, altered mitochondrial dynamics, and increased production of ROS. Elevated ROS levels induce oxidative damage to DNA, proteins, and lipids, further activating the DDR and reinforcing senescence. Importantly, ROS also serve as signaling molecules that enhance the expression of pro-inflammatory cytokines, such as IL-6 and IL-8, through redox-sensitive transcription factors like NF-κB. In this way, mitochondrial dysfunction acts as a feedback loop that maintains and amplifies SASP activity. This phenomenon has been particularly well-documented in the context of CV aging, where oxidative stress from senescent endothelial or myocardial cells accelerates vascular inflammation, arterial stiffening, and fibrosis [18,42]. In addition to these primary triggers, several intracellular signaling pathways function as amplifiers of SASP once it has been initiated. Notably, the mTOR pathway has been shown to regulate the translation of SASP components, while the cGAS-STING pathway senses cytosolic DNA fragments and activates type I interferon signaling that further enhances inflammatory SASP expression [43,44]. These amplifying mechanisms demonstrate that SASP is not merely a passive consequence of senescence but a tightly regulated and dynamically sustained program with systemic physiological impact [36].
Distinction Between Transient vs Chronic SASP
A critical conceptual distinction has emerged between transient and chronic SASP, which reflects differences in both the physiological roles and pathological consequences of senescent cells (Table 2). Transient SASP is typically observed during acute cellular senescence, such as that which occurs in response to tissue injury, wound healing, or during embryonic development [45]. In these settings, senescence is activated as a tightly regulated and time-limited process. The transient expression of SASP factors facilitates tissue repair by modulating inflammation, remodeling the ECM, and promoting the clearance of damaged or dysfunctional cells through immune-mediated mechanisms. This controlled release of SASP molecules such as ILs, MMPs, and growth factors plays a beneficial role in restoring tissue homeostasis. Importantly, the transient nature of SASP in this context is often terminated by the efficient removal of senescent cells by the immune system or through programmed cell death. Thus, transient SASP contributes to a regenerative and adaptive response that is essential for maintaining organismal health [9,23,45]. In contrast, chronic SASP emerges when senescent cells persist in tissues due to a failure in immune surveillance or an inability to undergo apoptosis. This is particularly common in aging tissues, where the accumulation of senescent cells leads to sustained and dysregulated SASP expression. Chronic SASP maintains a persistent pro-inflammatory milieu, characterized by continued secretion of cytokines such as IL-6 and IL-1β, chemokines, growth factors, and matrix-degrading enzymes [23,46]. Unlike the transient SASP, which is temporally limited and spatially confined, chronic SASP spreads its influence over time and affects surrounding tissues by promoting fibrosis, altering stem cell niches, and encouraging the senescence of neighboring cells through paracrine signaling [47]. This reinforces tissue dysfunction and is a key contributor to the progression of age-related diseases, including CV disease, where chronic inflammation and remodeling play central roles in disease pathogenesis [20]. Molecularly, chronic SASP is maintained by ongoing activation of key signaling pathways such as the DDR, NF-κB, p38 MAPK, and mTOR, which drive transcriptional and translational regulation of SASP components. Additionally, cytoplasmic chromatin fragments and activation of the cGAS-STING pathway have been implicated in reinforcing SASP expression in persistent senescent cells [38,44]. These pathways are often upregulated in senescent cells residing in aged or diseased tissues, further perpetuating the chronic nature of SASP. In the CV system, this persistent SASP expression can drive endothelial dysfunction, vascular remodeling, and myocardial fibrosis, contributing to the pathophysiology of conditions including atherosclerosis, HF, and hypertension (Table 3) [30,48].
SASP-Induced Chronic Low-Grade Inflammation ("Inflammaging")
Chronic low-grade inflammation, commonly referred to as “inflammaging,” represents a defining feature of biological aging and a fundamental contributor to CV pathology in elderly individuals. Inflammaging is characterized by a persistent, systemic inflammatory state that exists in the absence of acute infection and is marked by modest elevations in circulating pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β, and C-reactive protein (CRP). A principal driver of this phenomenon is SASP. As senescent cells persist in the absence of efficient immune clearance, they function as chronic sources of these inflammatory signals, establishing a systemic environment that promotes immune dysregulation, tissue degeneration, and heightened susceptibility to age-associated CVDs [49]. Within the CV system, the systemic propagation of SASP-induced inflammatory mediators contributes to endothelial dysfunction, vascular stiffening, and the promotion of atherosclerosis. Elevated levels of IL-6 and TNF-α in circulation activate endothelial cells, reduce nitric oxide (NO) bioavailability, and upregulate adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), facilitating leukocyte adhesion and transmigration into the vascular intima. These processes promote a pro-atherogenic milieu even in the absence of overt plaque formation and are particularly detrimental in aged vessels where endothelial resilience is diminished [50]. Moreover, SASP-induced inflammation promotes the polarization of monocytes and macrophages toward pro-inflammatory M1 phenotypes, which secrete additional cytokines and proteases that perpetuate vascular inflammation and matrix degradation. This cascade significantly contributes to the development of vulnerable plaques and impairs vascular elasticity, increasing pulse wave velocity and systolic blood pressure—hallmarks of vascular aging [51,52].
In addition to its effects on the vasculature, SASP-driven inflammaging has profound consequences for cardiac tissue integrity and function. Inflammatory cytokines produced systemically by senescent cells in peripheral tissues—including adipose tissue, bone marrow, and skeletal muscle—can reach the myocardium and contribute to cardiac remodeling. This systemic inflammatory burden is associated with increased myocardial infiltration of immune cells, activation of fibroblasts, and deposition of ECM components, which collectively promote interstitial fibrosis. Chronic exposure to inflammatory mediators also affects cardiomyocyte function directly by impairing calcium handling, altering β-adrenergic signaling, and promoting mitochondrial dysfunction, all of which contribute to contractile impairment and the development of HF phenotypes such as HFpEF [53,54]. Furthermore, systemic SASP-mediated inflammation is implicated in the dysfunction of vascular progenitor and stem cells, reducing their regenerative capacity and impairing vascular repair mechanisms. Inflammaging-induced disruption of endothelial progenitor cell function is particularly relevant in elderly individuals with impaired angiogenic responses, contributing to delayed wound healing and impaired collateral vessel formation in ischemic conditions [55]. These defects are compounded by the pro-coagulant effects of SASP, which include increased expression of tissue factor and plasminogen activator inhibitor-1 (PAI-1), thereby increasing the risk of thrombotic events in aging populations [56,57].
SASP, Endothelial Dysfunction, and Vascular Aging
Effects of SASP on Vascular Endothelial Cells
The aging of the vascular system is characterized by structural and functional changes that impair blood vessel integrity and regulation. A major contributor to this process is the accumulation of senescent cells within the vascular endothelium, which promotes chronic inflammation and disrupts endothelial function [58]. Endothelial cells, which line the interior of blood vessels, are vital for maintaining vascular homeostasis. They regulate vascular tone, inhibit thrombosis, and control immune cell trafficking. A key protective function of endothelial cells is the synthesis of NO by endothelial nitric oxide synthase (eNOS). NO promotes vasodilation, prevents platelet aggregation, and reduces leukocyte adhesion. However, exposure to SASP factors—including IL-6, TNF-α, and ROS—impairs eNOS expression and activity, significantly reducing NO bioavailability. This leads to impaired vasodilation and fosters a pro-inflammatory and pro-thrombotic endothelial phenotype [48,50,58]. SASP-induced dysfunction initiates a self-perpetuating cycle of oxidative stress and inflammation. SASP components activate NADPH oxidase and mitochondrial ROS production, which further decrease NO levels through direct scavenging and by oxidizing tetrahydrobiopterin (BH4), a critical eNOS cofactor. This results in eNOS uncoupling, where the enzyme generates superoxide instead of NO, exacerbating oxidative stress [50,59].
Additionally, SASP-related pro-inflammatory cytokines activate NF-κB and p38 MAPK signaling pathways in endothelial cells, promoting the expression of adhesion molecules such as VCAM-1 and ICAM-1. These molecules enhance leukocyte adhesion and infiltration, aggravating endothelial injury and driving vascular remodeling and stiffness [60,61]. Simultaneously, ROS can induce further cellular senescence in both an autocrine and paracrine manner and oxidize key biomolecules like lipids, proteins, and DNA [50,59]. This oxidative stress also activates redox-sensitive transcription factors such as activator protein-1 (AP-1), which boost the transcription of inflammatory SASP components, perpetuating the inflammatory loop [62,63]. SASP further contributes to vascular aging by stimulating the proliferation and phenotypic switching of vascular smooth muscle cells (VSMCs) into pro-inflammatory and osteogenic states, promoting vascular calcification and rigidity. The altered immune microenvironment also impairs the clearance of senescent cells, allowing them to persist and amplify inflammatory signaling [20,64,65]. As aging progresses, the balance between endothelial repair and damage becomes increasingly skewed due to declining progenitor cell function and rising senescent cell burden. SASP-induced endothelial dysfunction not only mirrors but accelerates vascular aging, contributing to arterial stiffness, plaque formation, and reduced vascular reactivity [30]. Importantly, experimental studies have shown that clearing senescent cells or inhibiting SASP factors can restore endothelial function and enhance NO bioavailability, underscoring the causal role of SASP in vascular aging [66,67].
Role of SASP in Arterial Stiffness
Cellular senescence contributes to arterial stiffness not merely through the cessation of cellular proliferation but more critically through the chronic paracrine signaling mediated by SASP. In aged vessels, the accumulation of senescent endothelial cells, VSMCs, and adventitial fibroblasts leads to a continuous release of SASP factors, which remodel the vascular ECM, compromise vascular tone, and initiate atherogenic signaling cascades [68,69]. Arterial stiffness arises from structural and functional alterations in the vascular wall, notably the breakdown of elastin fibers, increased collagen deposition, and cross-linking of ECM proteins. These changes reduce arterial compliance and impair the ability of vessels to buffer pulsatile blood flow. SASP factors play a central role in these alterations by increasing the expression and activation of MMPs such as MMP-2 and MMP-9, which degrade elastin and other ECM components [70,71]. Simultaneously, SASP components such as TGF-β and connective tissue growth factor (CTGF) drive fibroblast activation and collagen synthesis. In senescent VSMCs, SASP-induced phenotypic switching to a secretory and osteogenic state further promotes arterial calcification, exacerbating vessel rigidity. Moreover, chronic inflammation maintained by SASP cytokines such as IL-6 and TNF-α activates NF-κB and STAT3 pathways in vascular cells, which enhance the fibrotic response and suppress elastin repair mechanisms, compounding arterial stiffening over time [33,72]. Experimental clearance of senescent cells using senolytics has been shown to reduce arterial stiffness and improve endothelial function in aged animal models, providing compelling evidence of a causal role for SASP in vascular pathology [69,73,74]. These findings suggest that targeting SASP components or the senescent cells producing them could offer therapeutic benefit in attenuating arterial stiffness, thereby mitigating CV risk in the elderly.
SASP and Atherosclerotic Heart Disease
Contribution of Senescent Cells and SASP in Atherogenesis
Atherosclerosis is a complex, multifactorial disease characterized by the accumulation of lipids, inflammatory cells, and fibrous elements in the arterial wall, leading to plaque formation, vascular stiffening, and luminal obstruction. An emerging body of evidence implicates cellular senescence and the accompanying SASP as significant contributors to the initiation, progression, and destabilization of atherosclerotic plaques. A central and pathogenic feature of SASP in the context of atherosclerotic plaque development is its capacity to recruit immune cells into the vascular wall. In early atherogenesis, SASP factors derived from senescent endothelial cells and VSMCs initiate an inflammatory cascade by secreting high levels of chemokines, such as CCL2 (MCP-1), CXCL8 (IL-8), and CCL5 (RANTES). These chemokines bind to cognate receptors on circulating monocytes, neutrophils, and T lymphocytes, facilitating their adhesion to the endothelium and subsequent transmigration into the intima. This process is further enhanced by SASP-induced upregulation of adhesion molecules including ICAM-1, VCAM-1, and E-selectin on endothelial cells, which establish the necessary interface for leukocyte tethering and diapedesis. Once in the subendothelial space, recruited immune cells encounter a microenvironment rich in inflammatory cytokines—such as IL-1β, IL-6, and TNF-α—sustained by SASP. This milieu not only maintains immune cell activation but also amplifies local cytokine production, forming a self-sustaining inflammatory loop that drives lesion expansion [30,51,75]. Senescent macrophages, VSMCs, and endothelial cells actively secrete SASP factors that exacerbate the inflammatory milieu, promote ECM remodeling, and propagate further cellular dysfunction in a paracrine fashion, thereby facilitating atherogenesis and plaque evolution [30,76,77].
Senescent macrophages represent a central immune component of atherosclerotic lesions and contribute directly to plaque inflammation. These cells, initially recruited to clear oxidized low-density lipoprotein (oxLDL), undergo phenotypic changes under chronic stress and lipid overload. They transform into foam cells upon ingesting oxLDL, forming the lipid-rich necrotic core of the plaque. In the presence of persistent SASP signaling, these macrophages fail to undergo effective efferocytosis, leading to secondary necrosis and enlargement of the necrotic core [78,79]. In addition to impaired phagocytic capacity, senescent macrophages is also a sustained source of pro-inflammatory SASP components, including IL-1β, IL-6, TNF-α, and MMPs. These factors not only maintain a pro-atherogenic environment but also inhibit the resolution of inflammation and clearance of necrotic debris, further contributing to the growth of the lipid-rich necrotic core. Furthermore, SASP factors from senescent macrophages drive the recruitment and activation of additional immune cells, creating a self-reinforcing loop of inflammation within the plaque [80,81]. SASP also modulates the polarization of infiltrating macrophages and T cells. The pro-inflammatory cytokines produced by SASP-secreting cells favor a skewing toward M1 macrophages and Th1/Th17 T cell subsets, both of which contribute to enhanced inflammation and cytotoxicity. At the same time, SASP can suppress regulatory T cells and anti-inflammatory M2 macrophages, impairing the resolution of inflammation. This immunological imbalance favors continued immune cell infiltration, local tissue destruction, and increased oxidative stress, all of which converge to accelerate plaque progression and destabilization [51,82,83].
Senescent VSMCs also undergo functional reprogramming in atherosclerosis. Under conditions of oxidative stress and exposure to SASP cytokines, VSMCs switch from a contractile to a synthetic phenotype, and many enter a state of senescence [84]. These senescent VSMCs lose their ability to maintain ECM integrity and instead secrete MMPs that degrade collagen and elastin within the fibrous cap of the plaque. In addition to proteolytic activity, senescent VSMCs contribute to calcification processes by adopting osteochondrogenic features, which further stiffen the arterial wall and increase the risk of plaque rupture [85]. Critically, SASP derived from these VSMCs perpetuates local inflammation and promotes further VSMC and macrophage senescence, amplifying the pathogenic cascade within the lesion [86,87]. Senescent endothelial cells also play an equally vital role in the early stages of atherogenesis. These cells form the inner lining of arteries and are responsible for maintaining vascular homeostasis by regulating permeability, leukocyte adhesion, and vasodilation. Senescence in endothelial cells, driven by telomere shortening, disturbed flow, and ROS exposure, results in the loss of barrier integrity and decreased NO production. This dysfunction facilitates the transendothelial migration of monocytes and the retention of lipids in the subendothelial space [77,88,89]. Moreover, SASP factors released by senescent endothelial cells, including chemokines such as CCL2 and CXCL8, enhance leukocyte recruitment and amplify local inflammation [77,90,91].
SASP and Plaque Instability
The sustained immune cell infiltration driven by SASP factors has significant implications for plaque stability. Senescent macrophages themselves develop a SASP-like profile, secreting pro-inflammatory mediators and proteolytic enzymes that destabilize the plaque structure. MMPs, especially MMP-1, MMP-3, and MMP-9, are upregulated in response to chronic SASP exposure and degrade key ECM components such as collagen and elastin within the fibrous cap. The degradation of the ECM weakens the cap and increases the risk of rupture, an event that precipitates thrombosis and acute coronary syndromes [51,77,92]. Another important aspect of SASP-mediated plaque destabilization is its effect on VSMCs. While VSMCs in early plaque formation are involved in fibrous cap synthesis, those exposed to sustained SASP signaling undergo senescence and phenotypic switching, losing their contractile properties and matrix-producing functions. Instead, these senescent VSMCs secrete pro-inflammatory cytokines and MMPs, further contributing to ECM degradation. Moreover, SASP exposure induces VSMC apoptosis, depleting the cellular population necessary for cap maintenance. The combined effect of reduced matrix synthesis, increased matrix degradation, and VSMC loss leads to a thin, rupture-prone fibrous cap—hallmarks of vulnerable plaques that are prone to hemorrhage and thrombosis [93,94]. The pro-thrombotic state induced by SASP further contributes to plaque instability by increasing endothelial expression of tissue factor and reducing anti-coagulant molecules like thrombomodulin [77,90,91].
Evidence from Animal Models and Human Studies
The pathogenic role of SASP in the development, progression, instability, and rupture of atherosclerotic plaques has been substantiated through a growing body of evidence from both animal models and human studies. In vivo studies utilizing genetically engineered mouse models have been particularly instrumental in demonstrating the causal role of senescent cells and their SASP in vascular pathology [95,96]. In one of the seminal experiments using the INK-ATTAC transgenic mouse model, in which p16INK4a+ senescent cells can be selectively ablated, the clearance of senescent cells led to a significant reduction in atherosclerotic burden, preservation of vascular function, and increased fibrous cap stability [97,98]. These effects were accompanied by a notable decrease in SASP cytokines, reduced infiltration of pro-inflammatory macrophages, and diminished MMP activity in the aortic wall. These findings underscore the mechanistic link between senescent cell accumulation, SASP-driven inflammation, and structural degradation of the atherosclerotic plaque [99]. Further experimental validation comes from studies employing pharmacological senolytics such as dasatinib and quercetin (D+Q). In apolipoprotein E-deficient (ApoE-/-) mice, a widely used model of atherosclerosis, treatment with these agents resulted in the elimination of senescent cells within plaques, attenuation of SASP factor expression, and a corresponding reduction in plaque size and necrotic core area. This senolytic intervention also improved markers of plaque stability, including increased collagen deposition and decreased MMP activity. The restoration of a more stable plaque phenotype in these models supports the hypothesis that SASP not only exacerbates inflammation but also directly contributes to fibrous cap weakening and the risk of rupture [100,101,102]. However, an animal study evaluated the impact of the senolytic agent ABT-263 (Navitoclax) on advanced atherosclerotic lesions, focusing on its effects on smooth muscle cells (SMCs) and endothelial cells, which together constitute the majority of α-SMA+ cells in the stabilizing fibrous cap. Using lineage-traced ApoE−/− mice fed a Western diet for 18 weeks, followed by ABT-263 treatment at two dosing regimens, they found that ABT-263 did not reduce lesion size or alter the lumen area of the brachiocephalic artery. However, it significantly depleted SMCs by 90% and increased endothelial cell contributions to lesions via endothelial-to-mesenchymal transition (EndoMT) by 60%. These cellular changes were associated with a 60% reduction in fibrous cap thickness and an increase in mortality by over 50%. As such, ABT-263 treatment led to multiple detrimental outcomes, including features indicative of plaque destabilization and increased mortality, highlighting potential risks of senolytic therapy in the context of advanced atherosclerosis [103].
Human studies also have provided some insights, confirming the presence and pathogenic role of senescent cells and SASP components in advanced atherosclerotic lesions. Analyses of human carotid and coronary artery plaques have consistently demonstrated increased expression of senescence markers such as p16INK4a, p21CIP1, and senescence-associated β-galactosidase (SA-β-gal), particularly in areas of plaque vulnerability such as the fibrous cap and necrotic core [104]. These regions also show high levels of SASP factors, including IL-6, TNF-α, and MMPs. Importantly, SASP expression in human plaques correlates with clinical indices of plaque instability, such as reduced fibrous cap thickness, large lipid cores, and increased intraplaque hemorrhage, which are all predictors of rupture and acute thrombotic events. In addition, endothelial and smooth muscle cells within unstable plaques display transcriptional profiles consistent with a senescent phenotype, including the upregulation of SASP-related inflammatory and proteolytic genes [30]. Furthermore, studies in aged human populations have shown that individuals with higher circulating levels of SASP-associated cytokines—such as IL-1β and IL-6—are at increased risk for CV events, independent of traditional risk factors. These systemic markers reflect not only local vascular inflammation but also a systemic pro-inflammatory state driven by the widespread accumulation of senescent cells in aging tissues [26,105]. Histological examinations of post-mortem coronary arteries in patients who died of acute myocardial infarction have revealed dense infiltrates of senescent macrophages and T cells in ruptured plaques, suggesting a final common pathway of SASP-mediated inflammation and tissue degradation culminating in rupture [106]. Taken together, both experimental and clinical studies provide compelling evidence that SASP is not merely a bystander but an active driver of atherosclerotic plaque progression and destabilization. By orchestrating chronic inflammation, ECM degradation, immune cell recruitment, and VSMC dysfunction, SASP-producing senescent cells establish a microenvironment conducive to plaque rupture and acute vascular events.
SASP, Myocardial Remodeling, and Heart Failure
Senescence in Cardiomyocytes and Fibroblasts
Senescence in cardiomyocytes and cardiac fibroblasts plays a central role in the maladaptive remodeling processes that contribute to the development and progression of HF in aged individuals [107]. Although cardiomyocytes are traditionally considered terminally differentiated and largely post-mitotic, accumulating evidence demonstrates that these cells are nonetheless susceptible to senescence-inducing stimuli, including oxidative stress, mitochondrial dysfunction, telomere attrition, and genotoxic insults [108]. Senescent cardiomyocytes exhibit classical senescence markers such as increased expression of p16INK4a and p21CIP1, DNA damage foci marked by γ-H2AX, and senescence-associated β-gal activity. Critically, while these cells no longer proliferate, they remain metabolically active and contribute to pathological cardiac remodeling through the secretion of SASP, which includes pro-inflammatory cytokines, chemokines, MMPs, and extracellular vesicles [109,110,111]. SASP derived from senescent cardiomyocytes has significant downstream effects on the cardiac microenvironment. It acts in a paracrine manner to induce senescence in neighboring cells, propagate inflammation, and disrupt myocardial structural integrity. Chronic exposure to SASP components promotes leukocyte infiltration, endothelial dysfunction, and interstitial fibrosis, all of which impair cardiac compliance and diastolic function. Moreover, SASP factors such as IL-6, TNF-α, and TGF-β alter calcium handling and excitation-contraction coupling in adjacent non-senescent cardiomyocytes, contributing to contractile dysfunction. These mechanisms are particularly relevant in the pathophysiology of HF with preserved ejection fraction (HFpEF), a condition prevalent in the elderly, in which cardiac hypertrophy and fibrosis are prominent, despite preserved systolic function [112,113].
In parallel, cardiac fibroblasts—critical mediators of ECM homeostasis—undergo profound changes upon senescence. Senescent fibroblasts accumulate in the aging myocardium and express SASP factors that contribute to ECM remodeling and fibrosis. Unlike transiently activated fibroblasts that participate in reparative fibrosis following myocardial injury, senescent fibroblasts secrete sustained levels of MMPs, fibronectin, and TGF-β, leading to an imbalance between matrix synthesis and degradation. This results in diffuse interstitial fibrosis, increased ventricular stiffness, and impaired relaxation, further driving HFpEF pathophysiology. Additionally, SASP from senescent fibroblasts inhibits cardiac regeneration by impairing the function of cardiac progenitor cells and promoting their senescence, thereby reducing the regenerative capacity of the myocardium [20,114]. Evidence from aged murine models supports these findings. Mice with cardiac-specific accumulation of senescent cells demonstrate left ventricular hypertrophy, impaired relaxation, and elevated levels of inflammatory and fibrotic markers in cardiac tissue [115]. Conversely, genetic or pharmacological clearance of senescent cardiomyocytes and fibroblasts leads to reduced myocardial fibrosis, improved diastolic function, and enhanced cardiac output in aged animals. These interventions also attenuate the myocardial expression of SASP components, supporting the concept that the secretory profile of senescent cells drives adverse cardiac remodeling [116,117]. In human studies, myocardial biopsies from elderly patients with HF reveal elevated expression of senescence markers and SASP-related cytokines in both cardiomyocytes and fibroblasts, reinforcing the translational relevance of preclinical findings [118,119,120].
SASP-Driven Fibrosis, Hypertrophy, and Impaired Contractility
Myocardial remodeling in the context of aging and HF is characterized by pathological alterations including interstitial fibrosis, cardiomyocyte hypertrophy, and contractile dysfunction. SASP has emerged as a central mediator of these remodeling processes, primarily through its chronic paracrine signaling effects on cardiac cells and the ECM. In both experimental models and human tissues, persistent SASP activity from senescent cardiomyocytes, fibroblasts, and infiltrating immune cells has been shown to drive fibrotic and hypertrophic responses that culminate in impaired cardiac mechanics and progression to HF [114,121]. Fibrosis is a cardinal feature of aging myocardium and is exacerbated by SASP through both stimulation of myofibroblast activation and dysregulation of ECM turnover. SASP factors such as TGF-β1, IL-6, and MMPs secreted by senescent fibroblasts and cardiomyocytes create a profibrotic environment by inducing fibroblast-to-myofibroblast transition and increasing collagen I and III synthesis. Simultaneously, MMPs contribute to maladaptive ECM remodeling by degrading structural matrix components, thereby disturbing the mechanical architecture of the myocardium. This disruption leads to increased ventricular stiffness, reduced compliance, and diastolic dysfunction—pathological hallmarks of HFpEF, which disproportionately affects elderly patients [114,121,122].
Cardiomyocyte hypertrophy, another critical aspect of myocardial remodeling, is similarly influenced by chronic SASP exposure [20,123]. Pro-inflammatory cytokines such as IL-1β and TNF-α released by senescent cells activate hypertrophic signaling pathways in adjacent cardiomyocytes, notably the NF-κB and MAPK cascades. These pathways upregulate genes involved in sarcomeric reorganization and cellular growth, including atrial natriuretic peptide (ANP) and β-myosin heavy chain, resulting in increased cardiomyocyte size and altered contractile protein expression [124,125]. The hypertrophic response, while initially compensatory, ultimately becomes maladaptive by increasing myocardial oxygen demand, impairing energy efficiency, and reducing diastolic filling capacity. This hypertrophy is not accompanied by proportional vascular growth, leading to relative ischemia and further cardiomyocyte stress, which may perpetuate cellular senescence and SASP signaling in a self-reinforcing feedback loop [126]. SASP signaling also contributes to impaired myocardial contractility through several interrelated mechanisms. Chronic inflammatory mediators such as TNF-α and IL-6 disrupt calcium homeostasis by altering the expression and activity of calcium-handling proteins including sarco/endoplasmic reticulum calcium ATPase 2a (SERCA2a) and the ryanodine receptor (RyR). These disruptions lead to impaired excitation-contraction coupling, reduced calcium reuptake into the sarcoplasmic reticulum, and diastolic calcium overload. The resulting cytosolic calcium dysregulation contributes to contractile inefficiency and increased myocardial stiffness [127]. Furthermore, oxidative stress associated with SASP exacerbates mitochondrial dysfunction, reducing ATP availability for contractile processes and enhancing ROS production, which damages cellular components and exacerbates senescence. These effects collectively impair systolic and diastolic function and accelerate the transition from compensated hypertrophy to overt HF [128,129,130]. Human studies support these mechanistic insights. In myocardial biopsies from patients with HFpEF and HF with reduced ejection fraction (HFrEF), elevated expression of senescence markers and SASP-related cytokines have been observed, particularly in fibrotic and hypertrophic regions. Additionally, circulating levels of IL-6, TNF-α, and TGF-β—key SASP components—correlate with worse clinical outcomes and echocardiographic indices of impaired function [120,131]. These findings, combined with data from animal models demonstrating improved myocardial structure and function following senescent cell clearance.
Relevance in HFpEF in Elderly
HFpEF has emerged as the most prevalent form of HF among elderly populations, accounting for over half of all HF cases in individuals over 65 years old. Unlike HFrEF, HFpEF is characterized by diastolic dysfunction, myocardial stiffness, and impaired ventricular relaxation despite normal systolic contractility. This distinct phenotype is increasingly recognized not as a single disease but as a complex syndrome driven by systemic and myocardial inflammation, endothelial dysfunction, fibrosis, and altered cardiomyocyte mechanics [132,133]. Recent advances in aging biology have implicated cellular senescence and SASP as pivotal contributors to the pathophysiological mechanisms underlying HFpEF, particularly in the aging myocardium and vasculature [134,135]. The myocardial phenotype in HFpEF is marked by interstitial fibrosis, hypertrophy, and increased ventricular stiffness, all of which are influenced by senescent cardiomyocytes and fibroblasts through their SASP output. Senescent fibroblasts, which accumulate in the aging heart, secrete pro-fibrotic and pro-inflammatory mediators such as TGF-β, IL-6, and MMPs, which stimulate excessive collagen deposition and maladaptive ECM remodeling. This fibrotic burden impairs myocardial compliance and contributes to the increased filling pressures and pulmonary congestion seen in HFpEF patients. Concurrently, senescent cardiomyocytes release SASP components that further promote fibroblast activation and enhance the inflammatory burden of the cardiac interstitium. These interactions create a feedback loop that reinforces cellular senescence and SASP expression, amplifying structural remodeling and functional impairment [136,137].
Endothelial cell senescence and the associated SASP also contribute critically to the pathophysiology of HFpEF. Aging-associated endothelial dysfunction impairs NO bioavailability, which is essential for maintaining vascular tone, modulating cardiomyocyte stiffness, and promoting myocardial perfusion [134]. SASP factors such as IL-1β and TNF-α from senescent endothelial cells activate oxidative and inflammatory signaling pathways that suppress eNOS activity and promote vascular rarefaction and stiffness [138]. These vascular abnormalities exacerbate left ventricular stiffening and contribute to impaired myocardial relaxation. Furthermore, systemic inflammation driven by SASP-producing senescent cells in peripheral tissues such as adipose tissue and bone marrow is thought to contribute to the "inflammatory HFpEF phenotype," where comorbidities including obesity, diabetes, and hypertension converge with cardiac aging to precipitate HF [53]. Emerging evidence from both preclinical and clinical studies reinforces the relevance of SASP in HFpEF. Animal models of diastolic dysfunction in aged mice have demonstrated that senolytic therapies targeting p16INK4a+ senescent cells improve cardiac compliance, reduce fibrosis, and restore ventricular relaxation. These effects are mediated by attenuation of SASP-associated inflammatory and fibrotic signaling [139]. In human studies, myocardial biopsies from elderly HFpEF patients show enrichment of senescence markers in cardiomyocytes and fibroblasts, as well as elevated levels of SASP cytokines within myocardial and circulating compartments. Furthermore, serum levels of IL-6, growth differentiation factor 15 (GDF-15), and other SASP-linked biomarkers correlate with left ventricular stiffness and reduced exercise capacity in HFpEF patients, suggesting systemic SASP burden may reflect and drive disease severity [140,141,142]. Together, these findings position SASP not only as a biomarker of HFpEF in the elderly but also as a mechanistic driver of myocardial dysfunction. By promoting inflammation, fibrosis, endothelial dysfunction, and cardiomyocyte remodeling, SASP plays a pivotal role in the initiation and maintenance of HFpEF pathophysiology.
SASP and Cardiac Arrhythmias
SASP plays a pivotal role in linking inflammaging to the development and progression of cardiac arrhythmias in elderly individuals [143,144]. Cellular senescence, particularly within CV tissues and immunometabolically active organs such as visceral adipose tissue and the bone marrow, contributes to the sustained release of SASP factors including IL-6, TNF-α, IL-1β, and ROS. These pro-inflammatory mediators do not remain confined to the local tissue environment but circulate systemically, exerting paracrine and endocrine-like effects on the electrophysiological properties of the myocardium and the conduction system. The cumulative exposure of cardiac tissue to SASP-induced inflammation disrupts ion channel expression, gap junction integrity, and calcium handling—all of which are key pathophysiological mechanisms in age-related arrhythmogenesis [145,146,147]. One of the principal mechanisms by which SASP promotes arrhythmias is through its deleterious effects on cardiac conduction and electrical coupling. Chronic elevation of SASP cytokines alters the expression and function of connexins, particularly connexin-43 (Cx43), the predominant gap junction protein in ventricular myocardium. TNF-α and IL-1β have been shown to reduce Cx43 expression and promote its internalization from intercalated discs, resulting in impaired electrical conduction and increased dispersion of repolarization [148]. These changes create a substrate for reentry arrhythmias, which are especially dangerous in the setting of myocardial fibrosis—a common feature in aging hearts. Furthermore, SASP-induced MMP activation contributes to ECM remodeling and fibrotic deposition, exacerbating the structural heterogeneity of cardiac tissue and facilitating arrhythmic conduction blocks [149,150]. SASP also affects intracellular calcium homeostasis, a key determinant of both contractile function and arrhythmia susceptibility. IL-6 and TNF-α have been shown to modulate the expression and function of RyR2 and SERCA2a, leading to calcium leak and impaired reuptake, respectively. These abnormalities increase the likelihood of delayed afterdepolarizations (DADs) and triggered activity, especially under conditions of sympathetic stimulation or oxidative stress [151,152]. Additionally, elevated ROS levels induced by the SASP can oxidize calcium-handling proteins and mitochondrial membranes, further destabilizing cardiomyocyte electrophysiology [153]. This is particularly relevant in atrial tissue, where calcium dysregulation and inflammation synergize to promote the development of atrial fibrillation (AF), the most prevalent arrhythmia in the elderly and a major contributor to stroke and HF [154,155].
Another contributing factor is the pro-arrhythmic effect of SASP-mediated autonomic remodeling. Chronic systemic inflammation leads to sympathovagal imbalance by promoting sympathetic nerve sprouting and suppressing vagal tone. Pro-inflammatory cytokines increase norepinephrine release from sympathetic nerve terminals and upregulate β-adrenergic signaling pathways in cardiomyocytes, enhancing the propensity for arrhythmic triggers. Furthermore, senescent cardiac autonomic neurons themselves may contribute to dysregulated neurotransmission and impaired heart rate variability, which are well-established risk factors for ventricular tachyarrhythmias and sudden cardiac death in aging populations. These alterations in autonomic regulation, compounded by SASP-induced electrical and structural remodeling, result in a multifaceted arrhythmogenic substrate [156,157]. Clinical observations lend support to these mechanistic insights. Elevated serum levels of IL-6 and TNF-α in elderly patients have been independently associated with increased risk of AF, ventricular ectopy, and sudden cardiac arrest [158,159]. Moreover, atrial biopsies from patients with chronic AF exhibit increased expression of senescence markers such as p16INK4a and elevated SASP components, suggesting a direct link between local senescence burden and arrhythmogenic remodeling [154]. Interventional studies have shown that therapies targeting inflammation, such as IL-1 antagonists or statins, can reduce AF burden, although their effects may be limited by the persistence of senescent cells [160,161]. These findings underscore the potential of senescence- and SASP-targeted therapies, such as senolytics and senomorphics, as novel anti-arrhythmic strategies, particularly in elderly individuals with elevated inflammatory burden and structural heart disease [145,162]. In sum, SASP acts as a central mediator connecting inflammaging to age-related cardiac arrhythmias by disrupting electrical conduction, promoting fibrosis, impairing calcium handling, and altering autonomic tone. These multifactorial effects underscore the importance of targeting senescence and its secretory phenotype to mitigate arrhythmic risk in the elderly.
Biomarkers and Diagnostic Potential
Potential Biomarkers for SASP and Senescent Cell Burden
The identification of reliable and specific biomarkers for SASP and senescent cell burden (Table 4) is critical for advancing the clinical utility of senescence-targeting therapies. Currently, the lack of standardized, non-invasive biomarkers remains a significant barrier in the translational pathway, particularly for patient stratification, real-time monitoring of therapeutic efficacy, and risk prediction in CVD [163]. Senescence is a heterogeneous and context-dependent process, and SASP exhibits considerable variability depending on the cell type, senescence trigger, and tissue microenvironment [164,165]. Therefore, a biomarker strategy must encompass both cell-intrinsic markers of senescence and extracellular signatures reflective of SASP activity and systemic inflammatory burden. Canonical intracellular markers of senescence include increased expression of CDK inhibitors p16INK4a and p21CIP1, persistent DNA damage foci such as γ-H2AX, and SA-β-gal activity. These markers are commonly used in ex vivo tissue analyses, but their diagnostic applicability in clinical settings is limited due to their requirement for invasive biopsy and lack of specificity in complex tissues [166,167]. To circumvent these limitations, recent studies have focused on circulating biomarkers that reflect the secretory profile of senescent cells. Among the most consistently elevated SASP factors in aging and CVD are IL-6, IL-1β, TNF-α, MMPs (i.e., MMP-1, MMP-3, MMP-9), and chemokines such as CCL2 and CXCL8. These molecules contribute to systemic inflammation, endothelial dysfunction, and myocardial remodeling, and their levels correlate with age-related CV risk factors and disease severity [168]. In CV cohorts, elevated IL-6 and TNF-α have been associated with arterial stiffness, endothelial dysfunction, and increased incidence of myocardial infarction and HF [25,169]. Moreover, the composite inflammatory index known as the “SASP score,” derived from multiplex assays of circulating cytokines and proteases, has shown promise as a surrogate for systemic senescence burden [170]. These findings suggest that panels of SASP components, rather than single markers, may offer more robust diagnostic utility. The use of high-sensitivity multiplex immunoassays or mass spectrometry-based proteomics may further enhance the detection of low-abundance SASP proteins in plasma, improving both sensitivity and specificity.
Beyond protein biomarkers, cell-free nucleic acid-based markers have also emerged as potential tools for assessing senescence. Senescent cells can release DNA fragments into the circulation, often as part of the cytoplasmic chromatin fragments that activate the cGAS-STING pathway and sustain SASP expression. Quantification of cell-free nuclear and mitochondrial DNA with senescence-specific methylation signatures could provide a minimally invasive measure of senescent cell burden [171]. In addition, recent transcriptomic analyses of peripheral blood mononuclear cells (PBMCs) from elderly individuals have identified upregulation of senescence-related gene signatures, including increased expression of p16INK4a and SASP cytokines, which correlate with frailty, reduced physical performance, and CV comorbidities [172,173]. Advanced imaging modalities are also being developed to visualize senescent cell populations in vivo. Positron emission tomography (PET) tracers targeting β-gal or other senescence-associated enzymes have shown potential in preclinical models for non-invasively tracking senescent cell distribution and therapeutic clearance following senolytic administration [174]. Integration of imaging biomarkers with circulating SASP profiles may offer a comprehensive approach to assessing spatial and systemic senescence dynamics, particularly in the CV system, where focal accumulation of senescent cells contributes to localized pathology such as atherosclerotic plaques and fibrotic myocardial segments. In conclusion, multiplexed panels of SASP proteins, cell-free nucleic acids, and senescence-associated transcriptomic signatures, potentially coupled with advanced imaging, represent a promising multi-modal strategy to quantify senescence burden and guide therapeutic decision-making in age-related CVD.
Use in Risk Stratification and Monitoring Therapy in Elderly CVD Patients
The integration of senescence-associated biomarkers into clinical practice holds substantial promise for advancing risk stratification and therapeutic monitoring in elderly patients with CVD. As the aging population grows, traditional risk models—largely based on chronological age and conventional factors such as hypertension, dyslipidemia, and diabetes—are increasingly insufficient for capturing the biological heterogeneity and the complexity of CV aging. Senescence biomarkers, particularly those reflecting the burden and activity of SASP, offer a mechanistically grounded approach to identify patients at higher risk for adverse CV outcomes due to underlying molecular and cellular senescence processes [168]. Elevated levels of circulating SASP components such as IL-6, IL-1β, TNF-α, and MMPs (e.g., MMP-9) have been consistently associated with worse CV outcomes in elderly cohorts [175]. These biomarkers are not only indicative of systemic inflammation but also correlate with subclinical vascular dysfunction, arterial stiffness, and endothelial senescence. Longitudinal studies have demonstrated that higher baseline levels of IL-6 and TNF-α predict increased incidence of myocardial infarction, HF, and CV mortality in older adults, independent of traditional risk factors. This suggests that inclusion of SASP biomarkers could enhance existing risk prediction models by providing insight into the molecular drivers of CV aging and vulnerability. Moreover, the dynamic measurement of senescence biomarkers offers a valuable tool for monitoring therapeutic responses to senescence-targeting interventions [176]. In early studies, changes in circulating SASP factors have been used to assess the biological activity of senolytic treatments such as dasatinib and quercetin (D+Q), as well as senomorphic agents like metformin and rapamycin. For example, reductions in IL-6, MCP-1, and MMPs following senolytic therapy have correlated with improvements in endothelial function, physical performance, and systemic inflammation, suggesting that these markers can serve as pharmacodynamic indicators of target engagement and therapeutic efficacy [177,178]. This is particularly relevant in elderly patients with HFpEF, where senescence and inflammation play central pathogenic roles, and traditional biomarkers such as natriuretic peptides offer limited mechanistic information.
Risk stratification using senescence biomarkers could also facilitate personalized medicine approaches in CV care. For instance, older individuals exhibiting high levels of p16INK4a expression in peripheral blood T cells or elevated SASP profiles might be identified as candidates for early initiation of senomorphic or senolytic therapy [179]. This approach could be particularly beneficial in patients with multimorbidity or borderline CV risk, where traditional models may not justify aggressive intervention but where biologically driven senescence may predispose to rapid functional decline. Conversely, patients with low senescence burden might be spared from unnecessary exposure to potentially toxic agents, improving therapeutic risk-benefit ratios. The implementation of senescence biomarkers for monitoring therapy also opens the door to adaptive therapeutic regimens. For example, intermittent senolytic dosing schedules could be guided by real-time changes in SASP marker levels, allowing for personalized treatment cycles aimed at maintaining low inflammatory and senescence burden without continuous drug exposure [179,180]. Additionally, early biomarker shifts may signal impending therapeutic resistance or incomplete response, prompting escalation or combination therapy with anti-inflammatory or antioxidant agents.
Therapeutic Strategies Targeting SASP
Senolytics and Their CV Impact
Senolytic therapies, which selectively eliminate senescent cells, have emerged as a promising strategy to counteract the deleterious effects of SASP on the CV system (Table 5). Senolytics directly target the survival pathways that senescent cells rely on to resist apoptosis, thereby reducing their abundance and systemic SASP burden. Given the causal role of senescent cells in driving inflammation, fibrosis, endothelial dysfunction, and myocardial remodeling, the therapeutic application of senolytics offers the potential to reverse age-related CV decline and prevent the progression of diseases such as atherosclerosis, HF, and hypertension [181,182]. Preclinical studies in murine models have provided compelling evidence of the CV benefits of senolytic interventions. In aged mice treated with senolytic combinations such as D+Q, there is a marked reduction in senescent cell burden within the vasculature and myocardium, accompanied by attenuation of SASP components including IL-6, TNF-α, and MMPs [183,184]. In models of atherosclerosis, D+Q treatment reduces plaque size and inflammation, preserves fibrous cap integrity, and decreases necrotic core area, thereby improving plaque stability and reducing the risk of rupture. These effects are partly mediated by improved immune clearance and remodeling of the ECM following senescent cell ablation [185,186,187]. Senolytic therapies have also demonstrated efficacy in improving cardiac function in the context of age-related myocardial remodeling. In aged murine models and models of HFpEF, senescent cell clearance leads to significant improvements in diastolic relaxation, myocardial compliance, and ventricular stiffness. These benefits are associated with reduced myocardial fibrosis, diminished hypertrophic signaling, and improved calcium handling in cardiomyocytes [117,188]. Furthermore, senolytic treatment in aged mice restores endothelial NO bioavailability and reduces vascular stiffness, indicating systemic benefits beyond the myocardium. Notably, these improvements occur without adverse effects on ejection fraction, suggesting that senolytics preserve contractile function while alleviating age-associated structural remodeling [69].
Emerging clinical studies are beginning to translate these preclinical findings into human applications. A pilot clinical trial involving patients with diabetic kidney disease (DKD) treated with intermittent dosing of D+Q demonstrated a reduction in senescent cell biomarkers and circulating SASP cytokines [177]. The first-in-human open-label Phase 1 study enrolled nine older adults with DKD and administered oral dasatinib (100 mg) and quercetin (1000 mg) daily for three consecutive days. They collected blood, adipose tissue, and skin biopsies before treatment and 11 days after the final dose. Using SA-β-gal staining and immunohistochemistry, they measured the expression of senescence markers such as p16INK4A and p21CIP1, while also quantifying circulating SASP factors including IL-1α, IL-6, MMP-9, and MMP-12. As the results, there was a significant reduction in senescent cells in both adipose and skin tissues, evidenced by decreased expression of key senescence markers and reduced β-gal activity. Adipose tissue also showed fewer macrophage-rich crown-like structures, indicating a reduction in local inflammation. In addition, there was a marked decrease in systemic inflammatory SASP factors, which are known contributors to endothelial activation, vascular stiffening, and plaque instability in CVD [177]. Although CV endpoints were not the primary focus of this study, the reduction in systemic inflammation suggests a plausible mechanism for CV benefit.
Senomorphics and Their CV Impact
Senomorphics, or senescence-modulating agents, represent a therapeutic class aimed at attenuating the deleterious effects of SASP without eliminating senescent cells themselves (Table 6). Unlike senolytics, which induce apoptosis of senescent cells, senomorphics target the regulatory pathways that control SASP expression, thereby reducing the inflammatory, profibrotic, and tissue-degenerative signaling that contributes to CV aging. These agents are particularly advantageous in contexts where complete removal of senescent cells may be undesirable due to their physiological roles in tissue repair and tumor suppression. By modulating SASP, senomorphics aim to restore tissue homeostasis and reduce chronic inflammation, oxidative stress, and immune dysfunction associated with age-related CVD [189,190]. A key mechanistic target for senomorphic therapy is the NF-κB signaling pathway, a central transcriptional regulator of many SASP components including IL-6, IL-1β, and TNF-α. Pharmacological inhibition of NF-κB using compounds such as metformin or sodium salicylate has been shown to suppress SASP expression and reduce vascular inflammation in preclinical models of aging [191]. Metformin, in particular, has demonstrated pleiotropic CV benefits in both diabetic and non-diabetic populations, including improved endothelial function, reduced arterial stiffness, and attenuation of left ventricular hypertrophy. These effects are partly attributed to suppression of SASP-related inflammation and mitochondrial ROS generation via AMP-activated protein kinase (AMPK) activation and inhibition of mTOR signaling, both of which are upstream regulators of SASP expression [192,193].
Another major class of senomorphics includes mTOR inhibitors such as rapamycin and its analogs (rapalogs), which suppress translation of SASP components by modulating mTORC1 activity [194]. In aged rodent models, rapamycin has been shown to reduce arterial stiffness, improve endothelial NO bioavailability, and attenuate cardiac hypertrophy, effects closely tied to reduced expression of pro-inflammatory and pro-fibrotic SASP factors [195,196]. In the context of atherosclerosis, mTOR inhibition stabilizes plaques by decreasing macrophage infiltration and MMP activity, both of which are exacerbated by SASP signaling. Furthermore, intermittent rapamycin dosing has been proposed to mitigate the potential immunosuppressive effects of chronic treatment while retaining its CV and anti-SASP benefits [197,198]. Flavonoids, a class of naturally occurring polyphenols, have also demonstrated senomorphic properties by modulating key SASP pathways, including NF-κB, p38 MAPK, and the NLRP3 inflammasome [199]. Compounds such as apigenin, quercetin (when used in sub-senescent doses), and fisetin reduce SASP secretion and exhibit anti-inflammatory, antioxidant, and vasodilatory properties. These compounds have shown promise in improving endothelial function, reducing vascular stiffness, and mitigating myocardial remodeling in animal models of aging and metabolic syndrome [200,201]. Their intricate effects on both senescent and non-senescent cells make them attractive candidates for long-term CV prevention, particularly in populations with heightened inflammatory tone and comorbid metabolic dysfunction. In clinical contexts, senomorphics offer a feasible and safer approach for elderly individuals who may not tolerate the cytotoxic or off-target effects of senolytics. For example, in observational studies and randomized controlled trials, metformin use in elderly diabetic patients has been associated with reduced CV mortality and incidence of HF, effects that extend beyond glycemic control and likely involve attenuation of systemic SASP-mediated inflammation [202,203,204]. Ongoing trials are now investigating the use of mTOR inhibitors and flavonoids as geroprotective agents in non-diabetic populations to assess their capacity to modulate biological aging and CV risk.
Limitations of Currently Available Senolytics and Senomorphics
Despite the promising therapeutic potential of senolytics and senomorphics in targeting the deleterious effects of SASP, several critical limitations constrain their translational application in CVD. These limitations arise from biological, pharmacological, and clinical complexities that underscore the need for refined strategies, improved specificity, and a deeper understanding of senescence biology across tissues and disease contexts. One of the principal limitations of current senolytics is their lack of cell-type and context specificity. Most senolytic agents, such as the D+Q combination, target broadly expressed survival pathways including BCL-2 family proteins, PI3K/AKT, or FOXO signaling, which are not exclusive to senescent cells. As a result, these agents may induce off-target cytotoxicity in non-senescent but metabolically stressed or proliferatively active cells, especially in tissues with high turnover or in patients with underlying comorbidities [183,205]. For example, dasatinib is a tyrosine kinase inhibitor (TKI) with known hematologic and hepatic toxicities, which raises concerns about chronic or repeated use in elderly patients with pre-existing frailty or polypharmacy [206]. Additionally, the heterogeneity of senescent cells within and across tissues means that a single senolytic agent may not be universally effective, necessitating personalized approaches or combination therapies to achieve comprehensive clearance [164,207,208]. Another critical concern is the physiological role of senescent cells in tissue repair, tumor suppression, and wound healing. Acute senescence, particularly in transient settings such as after injury or during embryonic development, plays essential roles in promoting regeneration and modulating immune responses. Indiscriminate clearance of senescent cells through senolytics could disrupt these beneficial processes, especially if therapeutic timing is not precisely calibrated. This is particularly relevant in the CV system, where cardiomyocyte and endothelial senescence may initially limit damage by arresting the proliferation of damaged cells or modulating the reparative response. The risk of impairing these adaptive functions necessitates the development of biomarkers that distinguish deleterious chronic senescence from beneficial transient states, a distinction that remains technically challenging and poorly defined in clinical settings [209].
Senomorphics, while generally safer and more targeted in their mechanism of action, also face important limitations. One of the primary challenges is their reliance on continuous administration to suppress SASP signaling. Because senomorphics do not remove senescent cells, their effects are reversible and dependent on ongoing drug exposure. This necessitates long-term treatment, which increases the risk of cumulative side effects, drug resistance, and pharmacokinetic variability in aging populations [210]. Furthermore, the specificity of senomorphic agents for SASP suppression is often limited. For instance, metformin and rapamycin modulate multiple cellular pathways, including those involved in metabolism, autophagy, and protein synthesis, making it difficult to disentangle their SASP-targeting effects from broader systemic actions. These pleiotropic effects may be advantageous in some contexts but also raise concerns about unintended consequences, particularly when used chronically in elderly patients with multimorbidity [211]. Another limitation is the variability in SASP composition depending on the senescence inducer, cell type, and tissue microenvironment. This heterogeneity implies that senomorphic therapies may only partially suppress SASP or may inadvertently promote compensatory inflammatory pathways. For example, inhibition of mTOR may reduce IL-6 secretion but have minimal impact on MMPs or TGF-β. Such partial suppression may be insufficient to reverse disease phenotypes, especially in advanced CV pathology where multiple SASP components act synergistically to promote fibrosis, hypertrophy, and inflammation. Consequently, there is a pressing need for combinatorial approaches that can simultaneously modulate multiple SASP pathways while minimizing systemic toxicity [165,212]. Finally, a significant translational hurdle for both senolytics and senomorphics is the lack of robust biomarkers to monitor senescence burden and treatment efficacy in vivo. Current methods rely heavily on tissue biopsies or ex vivo assays, which are impractical for routine clinical use. The development of non-invasive imaging techniques or circulating biomarkers that accurately reflect senescent cell load and SASP activity is essential for patient stratification, therapeutic monitoring, and outcome prediction. Without such tools, clinical trials may face difficulties in demonstrating efficacy and optimizing dosing strategies [163].
Knowledge Gaps and Future Research Directions
Heterogeneity of Senescent Cells Across Tissues
A critical and unresolved challenge in the field of senescence biology is the pronounced heterogeneity of senescent cells across different tissues, which complicates both mechanistic understanding and therapeutic targeting. Senescent cells are not a uniform population; rather, they exhibit substantial variation in their molecular signatures, phenotypic characteristics, SASP composition, and functional outcomes depending on their cellular origin, tissue context, and the nature of the senescence-inducing stimulus. This heterogeneity presents a major barrier to developing universal biomarkers and pan-senescent therapeutic strategies, particularly in complex organ systems such as the CV system where multiple cell types—endothelial cells, VSMCs, fibroblasts, cardiomyocytes, and immune cells—undergo senescence and contribute differentially to disease progression [165]. The transcriptional and proteomic profiles of senescent cells induced by replicative exhaustion, oxidative stress, DNA damage, oncogene activation, or mitochondrial dysfunction can differ markedly, even within the same tissue type [164]. For example, endothelial cells rendered senescent by shear stress-induced DNA damage produce a SASP rich in chemokines and adhesion molecules that promote leukocyte recruitment and vascular inflammation, whereas senescent VSMCs adopt a matrix-degrading, calcifying phenotype with upregulation of MMPs, osteogenic factors, and pro-calcific SASP components [87,213]. These context-specific differences have been demonstrated in both human vascular tissues and murine models, where senescent VSMCs contribute more to plaque instability while senescent endothelial cells primarily mediate early inflammatory signaling and endothelial dysfunction.
This diversity is further complicated by cell-intrinsic epigenetic modifications and microenvironmental cues that modulate SASP and the cellular response to senescence. Epigenetic landscapes shaped by chromatin accessibility, histone modifications, and non-coding RNAs can influence whether a senescent cell adopts a highly inflammatory SASP or a more reparative, immunomodulatory secretome. For instance, senescent cardiac fibroblasts in a fibrotic heart may exhibit a profibrotic SASP rich in TGF-β and fibronectin, while senescent cardiomyocytes may produce cytokines that impair excitation-contraction coupling and promote paracrine senescence in neighboring cells. These distinctions are not merely academic; they have significant therapeutic implications, as senolytics or senomorphics that effectively target one senescent cell type or SASP profile may fail to affect—or may even exacerbate—senescence-driven pathology in another context [15,214]. Moreover, the temporal dynamics of senescence add another layer of complexity. The molecular markers that differentiate these senescent states are not fully elucidated, and the inability to distinguish between functionally distinct senescent populations impedes the development of precision senotherapeutics [215,216]. To address these challenges, future research must focus on high-resolution single-cell transcriptomic and proteomic analyses to delineate senescent cell populations across tissues and disease states. Integrative approaches that combine spatial transcriptomics, lineage tracing, and functional assays will be essential to map the origins, trajectories, and paracrine effects of senescent cells in situ. Additionally, improved animal models that more accurately reflect human senescence heterogeneity are needed, including tissue-specific and inducible senescence reporters that can capture the dynamics of senescent cell emergence and clearance under physiological and pathological conditions. These tools will be vital not only for understanding the context-specific roles of senescence in CVD but also for informing the design of targeted interventions that can modulate senescence with greater precision.
Need for Longitudinal Human Studies
Another significant barrier to the full clinical translation of senescence-targeted therapies and the integration of senescence biology into CVD management is the paucity of longitudinal human studies. While experimental models and cross-sectional human investigations have provided compelling evidence for the role of cellular senescence and SASP in CV aging [11,21], these designs are inherently limited in their ability to infer causality, track temporal changes in senescent cell burden, or evaluate the long-term effects of therapeutic intervention. Longitudinal studies are urgently needed to characterize the natural history of cellular senescence in humans, delineate its temporal relationship with CV pathogenesis, and establish senescence and SASP biomarkers as predictive or prognostic tools in clinical practice. Current knowledge regarding the accumulation of senescent cells in CV tissues largely derives from autopsy studies or surgical specimens obtained from patients with advanced disease [217,218]. These analyses are informative but static, capturing senescence at a single time point and typically under end-stage pathological conditions. This makes it difficult to determine whether senescence is a driver or merely a byproduct of disease progression. Moreover, individual variability in the onset and burden of senescent cells, influenced by genetic background, environmental exposures, and comorbidities, further necessitates longitudinal tracking in diverse human populations. Only through repeated measures across time can we understand the dynamics of senescence induction, SASP evolution, and their modulation by therapeutic or lifestyle interventions.
In the context of CV aging, longitudinal cohort studies incorporating senescence-specific endpoints could profoundly enhance risk stratification models. Serial measurement of circulating SASP biomarkers, such as IL-6, TNF-α, and MMPs, in conjunction with imaging-derived indices of vascular stiffness, myocardial fibrosis, or plaque instability, could elucidate how changes in senescence burden correlate with subclinical CVD progression and major adverse CV events. In addition, integration of epigenetic markers of senescence—such as DNA methylation clocks or p16INK4a expression levels in peripheral blood cells—into longitudinal designs could enable the tracking of biological aging trajectories and identify individuals who exhibit accelerated senescence-associated CV decline [179]. This approach may be particularly informative in high-risk groups such as patients with diabetes, chronic kidney disease, or HFpEF, where traditional risk factors inadequately explain clinical outcomes. Longitudinal human studies will also be essential for validating the efficacy and safety of senolytic and senomorphic therapies over extended periods. Most existing clinical trials in this domain have short follow-up durations and primarily focus on pharmacodynamic endpoints or surrogate biomarkers. While early data on intermittent dosing of senolytics such as D+Q are encouraging, long-term studies are required to assess whether these interventions can sustainably reduce senescence burden, delay CV events, and improve survival. Additionally, the potential for senescence re-emergence, compensatory SASP upregulation, or off-target effects needs to be systematically evaluated in real-world settings through prolonged observation. Such data are indispensable for informing dosing regimens, identifying appropriate treatment windows, and establishing monitoring protocols in aging patients with CVD. Moreover, longitudinal designs will facilitate the identification of senescence-modifying behaviors and environmental exposures. Lifestyle interventions such as caloric restriction, physical activity, or dietary supplementation may modulate senescence dynamics, but their effects remain poorly defined in human populations. Capturing longitudinal changes in senescence markers before and after lifestyle modification or pharmacologic treatment could yield critical insights into the plasticity of cellular senescence and inform non-pharmacological strategies to mitigate SASP-associated CV risks.
Interaction of SASP with Other Aging Hallmarks in CVD
Understanding the intersection between SASP and other established hallmarks of aging is vital for elucidating the multifactorial pathogenesis of CVD in the elderly. Aging is a systemic, progressive process characterized by cumulative damage and decline in cellular and organ function, driven by a constellation of interrelated biological mechanisms [219]. Among these, cellular senescence and its SASP play a pivotal but interconnected role, influencing and being influenced by other aging hallmarks such as mitochondrial dysfunction, genomic instability, telomere attrition, epigenetic alterations, proteostasis imbalance, and deregulated nutrient sensing. These processes do not occur in isolation; rather, they converge within cardiovascular tissues, exacerbating structural deterioration, functional decline, and inflammatory signaling, thereby accelerating disease progression. A critical area of interplay exists between SASP and mitochondrial dysfunction, which reciprocally reinforce each other in a deleterious feedback loop. Mitochondrial dysfunction is both a trigger and a consequence of cellular senescence. Impaired oxidative phosphorylation and elevated mitochondrial ROS levels lead to DNA damage and activation of DDR, one of the primary inducers of senescence. Senescent cells, in turn, exhibit altered mitochondrial dynamics and bioenergetics, with a metabolic shift toward glycolysis and increased mitochondrial mass but reduced function. These changes exacerbate ROS generation, perpetuating genotoxic stress and further enhancing SASP secretion via activation of redox-sensitive transcription factors such as NF-κB and p38 MAPK [220,221,222]. In cardiac and vascular tissues, this interaction promotes oxidative stress, inflammation, and endothelial dysfunction, which are central to atherosclerosis, arterial stiffening, and HFpEF [223,224].
SASP also interacts closely with genomic instability and telomere attrition, two fundamental drivers of senescence. Persistent DNA damage, whether from replication stress, oxidative lesions, or telomere erosion, activates p53 and p16INK4a pathways, initiating senescence and SASP expression. In CV cells, particularly endothelial cells and smooth muscle cells, telomere dysfunction has been strongly associated with increased senescence burden and pro-inflammatory SASP profiles [225]. The resulting SASP perpetuates DNA damage in surrounding cells via paracrine signaling, a phenomenon known as “bystander senescence.” This process accelerates clonal expansion of dysfunctional cells in atherosclerotic plaques and impairs the regenerative capacity of progenitor cells, further compromising vascular repair mechanisms. Moreover, the continuous activation of DDR pathways in senescent cells maintains the SASP in a sustained pro-inflammatory state, reinforcing local and systemic inflammation in elderly individuals with CVD [226,227]. Epigenetic alterations, another hallmark of aging, also influence SASP regulation and senescence phenotype heterogeneity. Age-related epigenetic drift, including global DNA hypomethylation, site-specific hypermethylation, histone modifications, and chromatin remodeling, affects the transcriptional landscape of senescent cells. These changes modulate the expression of SASP genes by altering the accessibility of enhancers and promoters to transcription factors such as C/EBPβ and NF-κB. In CV tissues, these epigenetic alterations contribute to persistent SASP activation and the establishment of a chronic inflammatory milieu. In addition, SASP factors such as IL-6 and TNF-α can feed back to induce epigenetic reprogramming in adjacent cells, promoting phenotypic shifts that favor fibrosis, calcification, or apoptosis, depending on cell context. This bidirectional interaction between SASP and epigenetic dysregulation creates a maladaptive epigenomic memory that underlies long-term pathological remodeling in the heart and vasculature [15,214,228].
Another key point of convergence lies with deregulated nutrient sensing, particularly the insulin/IGF-1 and mTOR pathways, which are intimately linked to both aging and SASP expression [229]. Activation of mTOR enhances translation of SASP factors and is a critical regulator of the pro-inflammatory state of senescent cells. In the context of cardiovascular aging, hyperactivation of mTOR not only promotes SASP activity but also drives cardiac hypertrophy, vascular remodeling, and metabolic dysfunction. Caloric restriction and pharmacologic mTOR inhibition (e.g., rapamycin) suppress SASP expression and ameliorate cardiovascular aging phenotypes, highlighting the therapeutic potential of targeting nutrient-sensing pathways to modulate SASP and its pathological consequences [230]. In conclusion, the SASP intersects with multiple hallmarks of aging to drive CV pathology through complex and reciprocal mechanisms. These interactions reinforce chronic inflammation, oxidative stress, genomic instability, and tissue remodeling, contributing to the pathogenesis of atherosclerosis, myocardial fibrosis, and diastolic dysfunction in the elderly. Future research must dissect these multifactorial interactions at cellular and systemic levels using integrative omics approaches and systems biology models. Understanding the crosstalk between SASP and other aging pathways will be essential for designing combinatorial interventions that simultaneously modulate multiple hallmarks to prevent or reverse age-associated CVD.
Conclusion
SASP has emerged as a critical molecular nexus linking cellular senescence to the pathogenesis of CVD in the aging population. Accumulating evidence demonstrates that SASP mediates a wide array of deleterious effects across CV tissues, including endothelial dysfunction, vascular inflammation, arterial stiffening, atherosclerotic plaque instability, myocardial fibrosis, impaired contractility, and arrhythmogenesis. These effects are driven by the chronic secretion of pro-inflammatory cytokines, chemokines, proteases, and growth factors by senescent cells, which act both locally and systemically to propagate tissue remodeling and inflammaging. The complex nature of SASP underscores its extensive crosstalk with other aging hallmarks—such as mitochondrial dysfunction, genomic instability, and epigenetic alteration—resulting in a complex and self-reinforcing cycle of cardiovascular deterioration. While senolytic and senomorphic therapies have shown promise in preclinical and early clinical studies, substantial challenges remain, including cell-type specificity, therapeutic safety, biomarker validation, and the heterogeneity of senescence phenotypes across tissues. Longitudinal human studies and integrated biomarker platforms are urgently needed to track senescence dynamics, personalize intervention strategies, and evaluate long-term outcomes. Ultimately, targeting SASP offers a paradigm-shifting approach to mitigate age-related CVD by intervening at a fundamental driver of cellular dysfunction, inflammation, and tissue degeneration.
Acknowledgement: None.
Author Contributions
Henry Sutanto: Conceptualization, Methodology, Formal analysis, Investigation, Resources, Writing - Original Draft, Writing - Review & Editing, Visualization. Deasy Fetarayani: Conceptualization, Writing - Original Draft, Writing - Review & Editing, Supervision. Rosyid Narendra: Writing - Original Draft, Writing - Review & Editing. Sally Aman Nasution: Writing - Original Draft, Writing - Review & Editing, Supervision.
Funding
This research receives no external funding.
Conflict of Interest
Henry Sutanto, Deasy Fetarayani, M. Rosyid Narendra, and Sally Aman Nasution have no conflicts of interest to declare.
References
- Rodgers, J.L.; Jones, J.; Bolleddu, S.I.; Vanthenapalli, S.; Rodgers, L.E.; Shah, K.; Karia, K.; Panguluri, S.K. Cardiovascular Risks Associated with Gender and Aging. J Cardiovasc Dev Dis 2019, 6, 19. [Google Scholar] [CrossRef]
- Di Cesare, M.; Perel, P.; Taylor, S.; Kabudula, C.; Bixby, H.; Gaziano, T.A.; McGhie, D.V.; Mwangi, J.; Pervan, B.; Narula, J.; et al. The Heart of the World. Glob Heart 2024, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, D.J.; Goldstein, D.R. Ageing and Atherosclerosis: Vascular Intrinsic and Extrinsic Factors and Potential Role of IL-6. Nat Rev Cardiol 2021, 18, 58–68. [Google Scholar] [CrossRef]
- Boccardi, V.; Mecocci, P. The Importance of Cellular Senescence in Frailty and Cardiovascular Diseases. In Frailty and Cardiovascular Diseases. In Frailty and Cardiovascular Diseases; Veronese, N., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, 2020; ISBN 978-3-030-33329-4. [Google Scholar]
- Triposkiadis, F.; Xanthopoulos, A.; Butler, J. Cardiovascular Aging and Heart Failure: JACC Review Topic of the Week. Journal of the American College of Cardiology 2019, 74, 804–813. [Google Scholar] [CrossRef]
- Olivieri, F.; Procopio, A.D.; Rippo, M.R. Cellular Senescence and Senescence-Associated Secretory Phenotype (SASP) in Aging Process. In Human Aging; Elsevier, 2021; pp. 75–88 ISBN 978-0-12-822569-1.
- Birch, J.; Gil, J. Senescence and the SASP: Many Therapeutic Avenues. Genes Dev 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- LeBrasseur, N.K.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and the Biology of Aging, Disease, and Frailty. In Nestlé Nutrition Institute Workshop Series; Fielding, R.A., Sieber, C., Vellas, B., Eds.; S. Karger AG, 2015; Vol. 83, pp. 11–18 ISBN 978-3-318-05477-4.
- Wilkinson, H.N.; Hardman, M.J. Senescence in Wound Repair: Emerging Strategies to Target Chronic Healing Wounds. Front. Cell Dev. Biol. 2020, 8, 773. [Google Scholar] [CrossRef]
- Suda, M.; Paul, K.H.; Minamino, T.; Miller, J.D.; Lerman, A.; Ellison-Hughes, G.M.; Tchkonia, T.; Kirkland, J.L. Senescent Cells: A Therapeutic Target in Cardiovascular Diseases. Cells 2023, 12, 1296. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Zhang, X.; Teng, T.; Ma, Z.-G.; Tang, Q.-Z. Cellular Senescence in Cardiovascular Diseases: A Systematic Review. Aging Dis 2022, 13, 103–128. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Zhu, Y.; Armstrong, J.L.; Tchkonia, T.; Kirkland, J.L. Cellular Senescence and the Senescent Secretory Phenotype in Age-Related Chronic Diseases. Current Opinion in Clinical Nutrition and Metabolic Care 2014, 17, 324–328. [Google Scholar] [CrossRef]
- Olan, I.; Handa, T.; Narita, M. Beyond SAHF: An Integrative View of Chromatin Compartmentalization during Senescence. Current Opinion in Cell Biology 2023, 83, 102206. [Google Scholar] [CrossRef]
- Dasgupta, N.; Arnold, R.; Equey, A.; Gandhi, A.; Adams, P.D. The Role of the Dynamic Epigenetic Landscape in Senescence: Orchestrating SASP Expression. NPJ Aging 2024, 10, 48. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a Break: Cellular Senescence as a DNA-Damage Response. Nat Rev Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Collins, P.L.; Purman, C.; Porter, S.I.; Nganga, V.; Saini, A.; Hayer, K.E.; Gurewitz, G.L.; Sleckman, B.P.; Bednarski, J.J.; Bassing, C.H.; et al. DNA Double-Strand Breaks Induce H2Ax Phosphorylation Domains in a Contact-Dependent Manner. Nat Commun 2020, 11, 3158. [Google Scholar] [CrossRef]
- Miwa, S.; Kashyap, S.; Chini, E.; von Zglinicki, T. Mitochondrial Dysfunction in Cell Senescence and Aging. J Clin Invest 2022, 132, e158447. [Google Scholar] [CrossRef]
- Martini, H.; Passos, J.F. Cellular Senescence: All Roads Lead to Mitochondria. The FEBS Journal 2023, 290, 1186–1202. [Google Scholar] [CrossRef] [PubMed]
- Khavinson, V.; Linkova, N.; Dyatlova, A.; Kantemirova, R.; Kozlov, K. Senescence-Associated Secretory Phenotype of Cardiovascular System Cells and Inflammaging: Perspectives of Peptide Regulation. Cells 2022, 12, 106. [Google Scholar] [CrossRef]
- Luan, Y.; Zhu, X.; Jiao, Y.; Liu, H.; Huang, Z.; Pei, J.; Xu, Y.; Yang, Y.; Ren, K. Cardiac Cell Senescence: Molecular Mechanisms, Key Proteins and Therapeutic Targets. Cell Death Discov. 2024, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Kotla, S.; Reddy Velatooru, L.; Abe, R.J.; Davis, E.A.; Cooke, J.P.; Schadler, K.; Deswal, A.; Herrmann, J.; Lin, S.H.; et al. Senescence-Associated Secretory Phenotype as a Hinge Between Cardiovascular Diseases and Cancer. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Saito, Y.; Yamamoto, S.; Chikenji, T.S. Role of Cellular Senescence in Inflammation and Regeneration. Inflammation and Regeneration 2024, 44, 28. [Google Scholar] [CrossRef]
- Frisch, S.M. Interleukin-1α: Novel Functions in Cell Senescence and Antiviral Response. Cytokine 2022, 154, 155875. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rane, M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circulation Research 2021, 128, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Tylutka, A.; Walas, Ł.; Zembron-Lacny, A. Level of IL-6, TNF, and IL-1β and Age-Related Diseases: A Systematic Review and Meta-Analysis. Front. Immunol. 2024, 15. [Google Scholar] [CrossRef]
- Yue, Z.; Nie, L.; Zhao, P.; Ji, N.; Liao, G.; Wang, Q. Senescence-Associated Secretory Phenotype and Its Impact on Oral Immune Homeostasis. Front. Immunol. 2022, 13. [Google Scholar] [CrossRef]
- Chaudhary, J.K.; Danga, A.K.; Kumari, A.; Bhardwaj, A.; Rath, P.C. Role of Chemokines in Aging and Age-Related Diseases. Mechanisms of Ageing and Development 2025, 223, 112009. [Google Scholar] [CrossRef]
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The Role of Matrix Metalloproteinases in Aging: Tissue Remodeling and Beyond. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2017, 1864, 2015–2025. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Liu, T.; Zhu, X.; Pan, X. The Multifaceted Role of the SASP in Atherosclerosis: From Mechanisms to Therapeutic Opportunities. Cell Biosci 2022, 12, 74. [Google Scholar] [CrossRef]
- Wang, B.; Han, J.; Elisseeff, J.H.; Demaria, M. The Senescence-Associated Secretory Phenotype and Its Physiological and Pathological Implications. Nat Rev Mol Cell Biol 2024, 25, 958–978. [Google Scholar] [CrossRef] [PubMed]
- Soeki, T.; Tamura, Y.; Shinohara, H.; Tanaka, H.; Bando, K.; Fukuda, N. Role of Circulating Vascular Endothelial Growth Factor and Hepatocyte Growth Factor in Patients with Coronary Artery Disease. Heart Vessels 2000, 15, 105–111. [Google Scholar] [CrossRef]
- Ren, L.-L.; Miao, H.; Wang, Y.-N.; Liu, F.; Li, P.; Zhao, Y.-Y. TGF-β as A Master Regulator of Aging-Associated Tissue Fibrosis. Aging Dis 2023, 14, 1633–1650. [Google Scholar] [CrossRef]
- Hao, X.; Wang, C.; Zhang, R. Chromatin Basis of the Senescence-Associated Secretory Phenotype. Trends Cell Biol 2022, 32, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Salotti, J.; Johnson, P.F. Regulation of Senescence and the SASP by the Transcription Factor C/EBPβ. Exp Gerontol 2019, 128, 110752. [Google Scholar] [CrossRef] [PubMed]
- Giroud, J.; Bouriez, I.; Paulus, H.; Pourtier, A.; Debacq-Chainiaux, F.; Pluquet, O. Exploring the Communication of the SASP: Dynamic, Interactive, and Adaptive Effects on the Microenvironment. International Journal of Molecular Sciences 2023, 24, 10788. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Yu, X.; Zhang, C.; Wang, Y.; Sun, Y.; Sun, H.; Zhang, H.; Shi, Y.; He, X. Telomeres and Mitochondrial Metabolism: Implications for Cellular Senescence and Age-Related Diseases. Stem Cell Rev Rep 2022, 18, 2315–2327. [Google Scholar] [CrossRef]
- Schumacher, B.; Pothof, J.; Vijg, J.; Hoeijmakers, J.H.J. The Central Role of DNA Damage in the Ageing Process. Nature 2021, 592, 695–703. [Google Scholar] [CrossRef]
- Shreeya, T.; Ansari, M.S.; Kumar, P.; Saifi, M.; Shati, A.A.; Alfaifi, M.Y.; Elbehairi, S.E.I. Senescence: A DNA Damage Response and Its Role in Aging and Neurodegenerative Diseases. Front Aging 2023, 4, 1292053. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence - Size Matters Not. eBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef]
- Rossiello, F.; Jurk, D.; Passos, J.F.; d’Adda di Fagagna, F. Telomere Dysfunction in Ageing and Age-Related Diseases. Nat Cell Biol 2022, 24, 135–147. [Google Scholar] [CrossRef]
- Dai, D.-F.; Rabinovitch, P.S.; Ungvari, Z. Mitochondria and Cardiovascular Aging. Circ Res 2012, 110, 10.1161–CIRCRESAHA.111. [Google Scholar] [CrossRef]
- Cayo, A.; Segovia, R.; Venturini, W.; Moore-Carrasco, R.; Valenzuela, C.; Brown, N. mTOR Activity and Autophagy in Senescent Cells, a Complex Partnership. Int J Mol Sci 2021, 22, 8149. [Google Scholar] [CrossRef]
- Schmitz, C.R.R.; Maurmann, R.M.; Guma, F.T.C.R.; Bauer, M.E.; Barbé-Tuana, F.M. cGAS-STING Pathway as a Potential Trigger of Immunosenescence and Inflammaging. Front Immunol 2023, 14, 1132653. [Google Scholar] [CrossRef]
- Thanapaul, R.J.R.S.; Shvedova, M.; Shin, G.H.; Roh, D.S. An Insight into Aging, Senescence, and Their Impacts on Wound Healing. Adv Geriatr Med Res 2021, 3, e210017. [Google Scholar] [CrossRef]
- Olivieri, F.; Prattichizzo, F.; Grillari, J.; Balistreri, C.R. Cellular Senescence and Inflammaging in Age-Related Diseases. Mediators Inflamm 2018, 2018, 9076485. [Google Scholar] [CrossRef]
- Ohtani, N. The Roles and Mechanisms of Senescence-Associated Secretory Phenotype (SASP): Can It Be Controlled by Senolysis? Inflammation and Regeneration 2022, 42, 11. [Google Scholar] [CrossRef]
- Picos, A.; Seoane, N.; Campos-Toimil, M.; Viña, D. Vascular Senescence and Aging: Mechanisms, Clinical Implications, and Therapeutic Prospects. Biogerontology 2025, 26, 118. [Google Scholar] [CrossRef] [PubMed]
- Barcena, M.L.; Aslam, M.; Pozdniakova, S.; Norman, K.; Ladilov, Y. Cardiovascular Inflammaging: Mechanisms and Translational Aspects. Cells 2022, 11, 1010. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-M.; Huang, A.; Kaley, G.; Sun, D. eNOS Uncoupling and Endothelial Dysfunction in Aged Vessels. American Journal of Physiology-Heart and Circulatory Physiology 2009, 297, H1829–H1836. [Google Scholar] [CrossRef]
- Vellasamy, D.M.; Lee, S.-J.; Goh, K.W.; Goh, B.-H.; Tang, Y.-Q.; Ming, L.C.; Yap, W.H. Targeting Immune Senescence in Atherosclerosis. International Journal of Molecular Sciences 2022, 23, 13059. [Google Scholar] [CrossRef]
- Molnár, A.Á.; Pásztor, D.T.; Tarcza, Z.; Merkely, B. Cells in Atherosclerosis: Focus on Cellular Senescence from Basic Science to Clinical Practice. International Journal of Molecular Sciences 2023, 24, 17129. [Google Scholar] [CrossRef]
- Mesquita, T.; Lin, Y.-N.; Ibrahim, A. Chronic Low-Grade Inflammation in Heart Failure with Preserved Ejection Fraction. Aging Cell 2021, 20, e13453. [Google Scholar] [CrossRef] [PubMed]
- Aranda, J.F.; Ramírez, C.M.; Mittelbrunn, M. Inflammageing, a Targetable Pathway for Preventing Cardiovascular Diseases. Cardiovascular Research 2024, cvae240. [Google Scholar] [CrossRef]
- Shimizu, S.; Iba, T.; Naito, H.; Rahmawati, F.N.; Konishi, H.; Jia, W.; Muramatsu, F.; Takakura, N. Aging Impairs the Ability of Vascular Endothelial Stem Cells to Generate Endothelial Cells in Mice. Angiogenesis 2023, 26, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Akrivou, D.; Perlepe, G.; Kirgou, P.; Gourgoulianis, K.I.; Malli, F. Pathophysiological Aspects of Aging in Venous Thromboembolism: An Update. Medicina 2022, 58, 1078. [Google Scholar] [CrossRef]
- Vaughan, D.E.; Rai, R.; Khan, S.S.; Eren, M.; Ghosh, A.K. PAI-1 Is a Marker and a Mediator of Senescence. Arterioscler Thromb Vasc Biol 2017, 37, 1446–1452. [Google Scholar] [CrossRef]
- Han, Y.; Kim, S.Y. Endothelial Senescence in Vascular Diseases: Current Understanding and Future Opportunities in Senotherapeutics. Exp Mol Med 2023, 55, 1–12. [Google Scholar] [CrossRef]
- Hernandez-Navarro, I.; Botana, L.; Diez-Mata, J.; Tesoro, L.; Jimenez-Guirado, B.; Gonzalez-Cucharero, C.; Alcharani, N.; Zamorano, J.L.; Saura, M.; Zaragoza, C. Replicative Endothelial Cell Senescence May Lead to Endothelial Dysfunction by Increasing the BH2/BH4 Ratio Induced by Oxidative Stress, Reducing BH4 Availability, and Decreasing the Expression of eNOS. International Journal of Molecular Sciences 2024, 25, 9890. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging Role of NF-κB Signaling in the Induction of Senescence-Associated Secretory Phenotype (SASP). Cellular Signalling 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Kułdo, J.M.; Westra, J.; Ásgeirsdóttir, S.A.; Kok, R.J.; Oosterhuis, K.; Rots, M.G.; Schouten, J.P.; Limburg, P.C.; Molema, G. Differential Effects of NF-κB and P38 MAPK Inhibitors and Combinations Thereof on TNF-α- and IL-1β-Induced Proinflammatory Status of Endothelial Cells in Vitro. American Journal of Physiology-Cell Physiology 2005, 289, C1229–C1239. [Google Scholar] [CrossRef]
- Kim, Y.-W.; West, X.Z.; Byzova, T.V. Inflammation and Oxidative Stress in Angiogenesis and Vascular Disease. J Mol Med 2013, 91, 323–328. [Google Scholar] [CrossRef] [PubMed]
- El Hadri, K.; Smith, R.; Duplus, E.; El Amri, C. Inflammation, Oxidative Stress, Senescence in Atherosclerosis: Thioredoxine-1 as an Emerging Therapeutic Target. International Journal of Molecular Sciences 2022, 23, 77. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Touyz, R.M. Oxidative Stress, Inflammation, and Vascular Aging in Hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-Silva, J.C.; Nolasco, P.; Krieger, J.E.; Miyakawa, A.A. Dynamic Crosstalk between Vascular Smooth Muscle Cells and the Aged Extracellular Matrix. International Journal of Molecular Sciences 2021, 22, 10175. [Google Scholar] [CrossRef]
- Li, Y.; Kračun, D.; Dustin, C.M.; El Massry, M.; Yuan, S.; Goossen, C.J.; DeVallance, E.R.; Sahoo, S.; St Hilaire, C.; Gurkar, A.U.; et al. Forestalling Age-Impaired Angiogenesis and Blood Flow by Targeting NOX: Interplay of NOX1, IL-6, and SASP in Propagating Cell Senescence. Proc Natl Acad Sci U S A 2021, 118, e2015666118. [Google Scholar] [CrossRef]
- Dookun, E.; Walaszczyk, A.; Redgrave, R.; Palmowski, P.; Tual-Chalot, S.; Suwana, A.; Chapman, J.; Jirkovsky, E.; Donastorg Sosa, L.; Gill, E.; et al. Clearance of Senescent Cells during Cardiac Ischemia–Reperfusion Injury Improves Recovery. Aging Cell 2020, 19, e13249. [Google Scholar] [CrossRef]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiological Reviews 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Clayton, Z.S.; Rossman, M.J.; Mahoney, S.A.; Venkatasubramanian, R.; Maurer, G.S.; Hutton, D.A.; VanDongen, N.S.; Greenberg, N.T.; Longtine, A.G.; Ludwig, K.R.; et al. Cellular Senescence Contributes to Large Elastic Artery Stiffening and Endothelial Dysfunction With Aging: Amelioration With Senolytic Treatment. Hypertension 2023, 80, 2072–2087. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z. Aging, Arterial Stiffness, and Hypertension. Hypertension 2015, 65, 252–256. [Google Scholar] [CrossRef]
- Yasmin; Wallace, S. ; McEniery, C.M.; Dakham, Z.; Pusalkar, P.; Maki-Petaja, K.; Ashby, M.J.; Cockcroft, J.R.; Wilkinson, I.B. Matrix Metalloproteinase-9 (MMP-9), MMP-2, and Serum Elastase Activity Are Associated With Systolic Hypertension and Arterial Stiffness. Arteriosclerosis, Thrombosis, and Vascular Biology 2005, 25, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Dutzmann, J.; Daniel, J.-M.; Bauersachs, J.; Hilfiker-Kleiner, D.; Sedding, D.G. Emerging Translational Approaches to Target STAT3 Signalling and Its Impact on Vascular Disease. Cardiovascular Research 2015, 106, 365–374. [Google Scholar] [CrossRef]
- Tuday, E.; Islam, T.; Kim, J.; Lesniewski, L.A.; Donato, A.J. Abstract 13452: Age-Related Increases in Arterial Stiffness Is Attenuated With Long-Term Senolytic Therapy in Mice. Circulation 2022, 146, A13452–A13452. [Google Scholar] [CrossRef]
- Mahoney, S.; Hutton, D.; Rossman, M.; Brunt, V.; VanDongen, N.; Casso, A.; Greenberg, N.; Ziemba, B.; Melov, S.; Campisi, J.; et al. Late-Life Treatment with the Senolytic ABT-263 Reverses Aortic Stiffening and Improves Endothelial Function with Aging. The FASEB Journal 2021, 35. [Google Scholar] [CrossRef]
- Shang, D.; Liu, H.; Tu, Z. Pro-Inflammatory Cytokines Mediating Senescence of Vascular Endothelial Cells in Atherosclerosis. Fundamental & Clinical Pharmacology 2023, 37, 928–936. [Google Scholar] [CrossRef]
- Costa-Beber, L.C.; Guma, F.T.C.R. The Macrophage Senescence Hypothesis: The Role of Poor Heat Shock Response in Pulmonary Inflammation and Endothelial Dysfunction Following Chronic Exposure to Air Pollution. Inflamm. Res. 2022, 71, 1433–1448. [Google Scholar] [CrossRef]
- Suzuki, K.; Susaki, E.A.; Nagaoka, I. Lipopolysaccharides and Cellular Senescence: Involvement in Atherosclerosis. International Journal of Molecular Sciences 2022, 23, 11148. [Google Scholar] [CrossRef]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. The Journal of Immunology 2017, 198, 1387–1394. [Google Scholar] [CrossRef]
- Adkar, S.S.; Leeper, N.J. Efferocytosis in Atherosclerosis. Nature Reviews Cardiology 2024, 21, 762–779. [Google Scholar] [CrossRef]
- Luo, G.; Xiang, L.; Xiao, L. Quercetin Alleviates Atherosclerosis by Suppressing Oxidized LDL-Induced Senescence in Plaque Macrophage via Inhibiting the p38MAPK/P16 Pathway. The Journal of Nutritional Biochemistry 2023, 116, 109314. [Google Scholar] [CrossRef]
- Wu, C.-M.; Zheng, L.; Wang, Q.; Hu, Y.-W. The Emerging Role of Cell Senescence in Atherosclerosis. Clinical Chemistry and Laboratory Medicine (CCLM) 2021, 59, 27–38. [Google Scholar] [CrossRef]
- Hou, P.; Fang, J.; Liu, Z.; Shi, Y.; Agostini, M.; Bernassola, F.; Bove, P.; Candi, E.; Rovella, V.; Sica, G.; et al. Macrophage Polarization and Metabolism in Atherosclerosis. Cell Death Dis 2023, 14, 1–14. [Google Scholar] [CrossRef]
- Snijckers, R.P.M.; Foks, A.C. Adaptive Immunity and Atherosclerosis: Aging at Its Crossroads. Front. Immunol. 2024, 15. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Xuan, X.; Hu, J.; Zhang, R.; Jin, H.; Dong, H. How Vascular Smooth Muscle Cell Phenotype Switching Contributes to Vascular Disease. Cell Commun Signal 2022, 20, 180. [Google Scholar] [CrossRef]
- Tang, H.-Y.; Chen, A.-Q.; Zhang, H.; Gao, X.-F.; Kong, X.-Q.; Zhang, J.-J. Vascular Smooth Muscle Cells Phenotypic Switching in Cardiovascular Diseases. Cells 2022, 11, 4060. [Google Scholar] [CrossRef]
- Uryga, A.K.; Bennett, M.R. Ageing Induced Vascular Smooth Muscle Cell Senescence in Atherosclerosis. The Journal of Physiology 2016, 594, 2115–2124. [Google Scholar] [CrossRef]
- Grootaert, M.O.J.; Moulis, M.; Roth, L.; Martinet, W.; Vindis, C.; Bennett, M.R.; De Meyer, G.R.Y. Vascular Smooth Muscle Cell Death, Autophagy and Senescence in Atherosclerosis. Cardiovascular Research 2018, 114, 622–634. [Google Scholar] [CrossRef]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial Cell Senescence in Human Atherosclerosis. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed]
- Voghel, G.; Thorin-Trescases, N.; Farhat, N.; Nguyen, A.; Villeneuve, L.; Mamarbachi, A.M.; Fortier, A.; Perrault, L.P.; Carrier, M.; Thorin, E. Cellular Senescence in Endothelial Cells from Atherosclerotic Patients Is Accelerated by Oxidative Stress Associated with Cardiovascular Risk Factors. Mechanisms of Ageing and Development 2007, 128, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Nagar, N.; Naidu, G.; Panda, S.K.; Gulati, K.; Singh, R.P.; Poluri, K.M. Elucidating the Role of Chemokines in Inflammaging Associated Atherosclerotic Cardiovascular Diseases. Mechanisms of Ageing and Development 2024, 220, 111944. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Xie, X.; Yuan, R.; Xin, Q.; Miao, Y.; Leng, S.X.; Chen, K.; Cong, W. Vascular Aging and Atherosclerosis: A Perspective on Aging. Aging Dis 2024, 16, 33–48. [Google Scholar] [CrossRef]
- Ali, I.; Zhang, H.; Zaidi, S.A.A.; Zhou, G. Understanding the Intricacies of Cellular Senescence in Atherosclerosis: Mechanisms and Therapeutic Implications. Ageing Research Reviews 2024, 96, 102273. [Google Scholar] [CrossRef]
- Harman, J.L.; Jørgensen, H.F. The Role of Smooth Muscle Cells in Plaque Stability: Therapeutic Targeting Potential. British Journal of Pharmacology 2019, 176, 3741–3753. [Google Scholar] [CrossRef]
- Zha, Y.; Zhuang, W.; Yang, Y.; Zhou, Y.; Li, H.; Liang, J. Senescence in Vascular Smooth Muscle Cells and Atherosclerosis. Front. Cardiovasc. Med. 2022, 9. [Google Scholar] [CrossRef]
- Fulop, G.A.; Kiss, T.; Tarantini, S.; Balasubramanian, P.; Yabluchanskiy, A.; Farkas, E.; Bari, F.; Ungvari, Z.; Csiszar, A. Nrf2 Deficiency in Aged Mice Exacerbates Cellular Senescence Promoting Cerebrovascular Inflammation. GeroScience 2018, 40, 513–521. [Google Scholar] [CrossRef]
- Zuccolo, E.; Badi, I.; Scavello, F.; Gambuzza, I.; Mancinelli, L.; Macrì, F.; Tedesco, C.C.; Veglia, F.; Bonfigli, A.R.; Olivieri, F.; et al. The microRNA-34a-Induced Senescence-Associated Secretory Phenotype (SASP) Favors Vascular Smooth Muscle Cells Calcification. International Journal of Molecular Sciences 2020, 21, 4454. [Google Scholar] [CrossRef]
- Childs, B.G.; Zhang, C.; Shuja, F.; Sturmlechner, I.; Trewartha, S.; Velasco, R.F.; Baker, D.J.; Li, H.; van Deursen, J.M. Senescent Cells Suppress Innate Smooth Muscle Cell Repair Functions in Atherosclerosis. Nat Aging 2021, 1, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-Positive Senescent Cells Delays Ageing-Associated Disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Schmidt, M.O.; Kallakury, B.; Jain, S.; Mehdikhani, S.; Levi, M.; Mendonca, M.; Welch, W.; Riegel, A.T.; Wilcox, C.S.; et al. Low Dose Chronic Angiotensin II Induces Selective Senescence of Kidney Endothelial Cells. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Dagher, O.; Mury, P.; Thorin-Trescases, N.; Noly, P.E.; Thorin, E.; Carrier, M. Therapeutic Potential of Quercetin to Alleviate Endothelial Dysfunction in Age-Related Cardiovascular Diseases. Front. Cardiovasc. Med. 2021, 8. [Google Scholar] [CrossRef]
- Nieto, M.; Konigsberg, M.; Silva-Palacios, A. Quercetin and Dasatinib, Two Powerful Senolytics in Age-Related Cardiovascular Disease. Biogerontology 2024, 25, 71–82. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic Senolytic Treatment Alleviates Established Vasomotor Dysfunction in Aged or Atherosclerotic Mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Karnewar, S.; Karnewar, V.; Shankman, L.S.; Owens, G.K. Treatment of Advanced Atherosclerotic Mice with ABT-263 Reduced Indices of Plaque Stability and Increased Mortality. JCI Insight 2024, 9, e173863. [Google Scholar] [CrossRef]
- Chen, J.; Huang, X.; Halicka, D.; Brodsky, S.; Avram, A.; Eskander, J.; Bloomgarden, N.A.; Darzynkiewicz, Z.; Goligorsky, M.S. Contribution of p16INK4a and p21CIP1 Pathways to Induction of Premature Senescence of Human Endothelial Cells: Permissive Role of P53. American Journal of Physiology-Heart and Circulatory Physiology 2006, 290, H1575–H1586. [Google Scholar] [CrossRef] [PubMed]
- Katkenov, N.; Mukhatayev, Z.; Kozhakhmetov, S.; Sailybayeva, A.; Bekbossynova, M.; Kushugulova, A. Systematic Review on the Role of IL-6 and IL-1β in Cardiovascular Diseases. Journal of Cardiovascular Development and Disease 2024, 11, 206. [Google Scholar] [CrossRef]
- Machado-Oliveira, G.; Ramos, C.; Marques, A.R.A.; Vieira, O.V. Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Cells 2020, 9, 2146. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, R.E.; Dookun, E.; Booth, L.K.; Camacho Encina, M.; Folaranmi, O.; Tual-Chalot, S.; Gill, J.H.; Owens, W.A.; Spyridopoulos, I.; Passos, J.F.; et al. Senescent Cardiomyocytes Contribute to Cardiac Dysfunction Following Myocardial Infarction. npj Aging 2023, 9, 1–7. [Google Scholar] [CrossRef]
- Tang, X.; Li, P.-H.; Chen, H.-Z. Cardiomyocyte Senescence and Cellular Communications Within Myocardial Microenvironments. Front. Endocrinol. 2020, 11. [Google Scholar] [CrossRef]
- Cianflone, E.; Torella, M.; Biamonte, F.; De Angelis, A.; Urbanek, K.; Costanzo, F.S.; Rota, M.; Ellison-Hughes, G.M.; Torella, D. Targeting Cardiac Stem Cell Senescence to Treat Cardiac Aging and Disease. Cells 2020, 9, 1558. [Google Scholar] [CrossRef]
- Zhai, P.; Sadoshima, J. Cardiomyocyte Senescence and the Potential Therapeutic Role of Senolytics in the Heart. J Cardiovasc Aging 2024, 4, 18. [Google Scholar] [CrossRef]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. JACC 2016, 67, 2018–2028. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Shakeri, H.; Leloup, A.J.; Van Hove, C.E.; De Meyer, G.R.Y.; Vrints, C.J.; Lemmens, K.; Van Craenenbroeck, E.M. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circulation: Heart Failure 2017, 10, e003806. [Google Scholar] [CrossRef] [PubMed]
- Gharagozloo, K.; Mehdizadeh, M.; Heckman, G.; Rose, R.A.; Howlett, J.; Howlett, S.E.; Nattel, S. Heart Failure With Preserved Ejection Fraction in the Elderly Population: Basic Mechanisms and Clinical Considerations. Canadian Journal of Cardiology 2024, 40, 1424–1444. [Google Scholar] [CrossRef] [PubMed]
- Mehdizadeh, M.; Aguilar, M.; Thorin, E.; Ferbeyre, G.; Nattel, S. The Role of Cellular Senescence in Cardiac Disease: Basic Biology and Clinical Relevance. Nat Rev Cardiol 2022, 19, 250–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Larson, D.F.; Watson, R. Age-Related Left Ventricular Function in the Mouse: Analysis Based on in Vivo Pressure-Volume Relationships. American Journal of Physiology-Heart and Circulatory Physiology 1999, 277, H1906–H1913. [Google Scholar] [CrossRef] [PubMed]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological Clearance of Senescent Cells Improves Survival and Recovery in Aged Mice Following Acute Myocardial Infarction. Aging Cell 2019, 18, e12945. [Google Scholar] [CrossRef]
- Salerno, N.; Marino, F.; Scalise, M.; Salerno, L.; Molinaro, C.; Filardo, A.; Chiefalo, A.; Panuccio, G.; De Angelis, A.; Urbanek, K.; et al. Pharmacological Clearance of Senescent Cells Improves Cardiac Remodeling and Function after Myocardial Infarction in Female Aged Mice. Mechanisms of Ageing and Development 2022, 208, 111740. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.J.; Zhang, X.; Kumar, A.; Atkinson, E.J.; Zhu, Y.; Jachim, S.; Mazula, D.L.; Brown, A.K.; Berning, M.; Aversa, Z.; et al. The Senescence-Associated Secretome as an Indicator of Age and Medical Risk. JCI Insight 2020, 5, e133668. [Google Scholar] [CrossRef]
- Salman, O.; Zamani, P.; Zhao, L.; Dib, M.J.; Gan, S.; Azzo, J.D.; Pourmussa, B.; Richards, A.M.; Javaheri, A.; Mann, D.L.; et al. Prognostic Significance and Biologic Associations of Senescence-Associated Secretory Phenotype Biomarkers in Heart Failure. Journal of the American Heart Association 2024, 13, e033675. [Google Scholar] [CrossRef]
- Qian, C.; Zamani, P.; Salman, O.; Jehangir, Q.; Cohen, J.; Gunawardhana, K.; Greenawalt, D.; Zhao, L.; Chang, C.-P.; Gordon, D.A.; et al. Abstract 11642: Biologic Associations of the Senescence-Associated Secretory Phenotype (SASP) in Heart Failure (HF): Insights From Plasma Proteomics. Circulation 2022, 146, A11642–A11642. [Google Scholar] [CrossRef]
- Ma, Y.; Chiao, Y.A.; Clark, R.; Flynn, E.R.; Yabluchanskiy, A.; Ghasemi, O.; Zouein, F.; Lindsey, M.L.; Jin, Y.-F. Deriving a Cardiac Ageing Signature to Reveal MMP-9-Dependent Inflammatory Signalling in Senescence. Cardiovascular Research 2015, 106, 421–431. [Google Scholar] [CrossRef]
- Osorio, J.M.; Espinoza-Pérez, C.; Rimassa-Taré, C.; Machuca, V.; Bustos, J.O.; Vallejos, M.; Vargas, H.; Díaz-Araya, G. Senescent Cardiac Fibroblasts: A Key Role in Cardiac Fibrosis. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2023, 1869, 166642. [Google Scholar] [CrossRef]
- Mehdizadeh, M.; Mackasey, M.; Tardif, J.-C.; Naud, P.; Ferbeyre, G.; Sirois, M.; Thorin, E.; Tanguay, J.-F.; Nattel, S. Abstract 11965: Cellular Senescence Produces Diastolic Dysfunction and Cardiac Hypertrophy With Aging in Mice. Circulation 2022, 146, A11965–A11965. [Google Scholar] [CrossRef]
- Martinez, P.F.; Bonomo, C.; Guizoni, D.M.; Junior, S.A.O.; Damatto, R.L.; Cezar, M.D.M.; Lima, A.R.R.; Pagan, L.U.; Seiva, F.R.; Bueno, R.T.; et al. Modulation of MAPK and NF-κB Signaling Pathways by Antioxidant Therapy in Skeletal Muscle of Heart Failure Rats. Cellular Physiology and Biochemistry 2016, 39, 371–384. [Google Scholar] [CrossRef]
- Li, Y.; Ha, T.; Gao, X.; Kelley, J.; Williams, D.L.; Browder, I.W.; Kao, R.L.; Li, C. NF-κB Activation Is Required for the Development of Cardiac Hypertrophy in Vivo. American Journal of Physiology-Heart and Circulatory Physiology 2004, 287, H1712–H1720. [Google Scholar] [CrossRef]
- Tanai, E.; Frantz, S. Pathophysiology of Heart Failure. Comprehensive Physiology 2016, 6, 187–214. [Google Scholar] [CrossRef]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L. Inflammation in Heart Failure: JACC State-of-the-Art Review. Journal of the American College of Cardiology 2020, 75, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.; Zhao, Y.; Lu, X. Cellular Senescence as a Key Player in Chronic Heart Failure Pathogenesis: Unraveling Mechanisms and Therapeutic Opportunities. Progress in Biophysics and Molecular Biology 2025, 196, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K. Cardiac Senescence, Heart Failure, and Frailty: A Triangle in Elderly People. The Keio Journal of Medicine 2016, 65, 25–32. [Google Scholar] [CrossRef]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative Stress and Heart Failure. Am J Physiol Heart Circ Physiol 2011, 301, H2181–2190. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, A.; Giugni, F.; Lamberson, V.; Yang, Y.; Boerwinkle, E.; Fornage, M.; Giannarelli, C.; Grams, M.; Windham, B.G.; Mosley, T.; et al. Abstract 4144054: Circulating Senescence-Associated Secretory Phenotype (SASP) Proteins Are Associated with Risk of Late Life Heart Failure: The ARIC Study. Circulation 2024, 150, A4144054–A4144054. [Google Scholar] [CrossRef]
- Ma, C.; Luo, H.; Fan, L.; Liu, X.; Gao, C. Heart Failure with Preserved Ejection Fraction: An Update on Pathophysiology, Diagnosis, Treatment, and Prognosis. Braz J Med Biol Res 2020, 53, e9646. [Google Scholar] [CrossRef]
- Stoicescu, L.; Crişan, D.; Morgovan, C.; Avram, L.; Ghibu, S. Heart Failure with Preserved Ejection Fraction: The Pathophysiological Mechanisms behind the Clinical Phenotypes and the Therapeutic Approach. International Journal of Molecular Sciences 2024, 25, 794. [Google Scholar] [CrossRef]
- Gevaert, A.B.; Shakeri, H.; Leloup, A.J.; Van Hove, C.E.; De Meyer, G.R.Y.; Vrints, C.J.; Lemmens, K.; Van Craenenbroeck, E.M. Endothelial Senescence Contributes to Heart Failure With Preserved Ejection Fraction in an Aging Mouse Model. Circ Heart Fail 2017, 10, e003806. [Google Scholar] [CrossRef]
- Kumar, M.; Yan, P.; Kuchel, G.A.; Xu, M. Cellular Senescence as a Targetable Risk Factor for Cardiovascular Diseases: Therapeutic Implications: JACC Family Series. JACC: Basic to Translational Science 2024, 9, 522–534. [Google Scholar] [CrossRef]
- Sanhueza-Olivares, F.; Troncoso, M.F.; Pino-de la Fuente, F.; Martinez-Bilbao, J.; Riquelme, J.A.; Norambuena-Soto, I.; Villa, M.; Lavandero, S.; Castro, P.F.; Chiong, M. A Potential Role of Autophagy-Mediated Vascular Senescence in the Pathophysiology of HFpEF. Front Endocrinol (Lausanne) 2022, 13, 1057349. [Google Scholar] [CrossRef]
- Grootaert, M.O.J. Cell Senescence in Cardiometabolic Diseases. npj Aging 2024, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Aroor, A.R.; Jia, C.; Sowers, J.R. Endothelial Cell Senescence in Aging-Related Vascular Dysfunction. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2019, 1865, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Tuday, E.; Islam, T.; Kim, J.; Lesniewski, L.A.; Donato, A. Abstract 11782: Senolytic Therapy Improves Diastolic Cardiac Function in Aged Mice. Circulation 2022, 146, A11782–A11782. [Google Scholar] [CrossRef]
- Vrabie, A.-M.; Totolici, S.; Delcea, C.; Badila, E. Biomarkers in Heart Failure with Preserved Ejection Fraction: A Perpetually Evolving Frontier. Journal of Clinical Medicine 2024, 13, 4627. [Google Scholar] [CrossRef]
- Hage, C.; Michaëlsson, E.; Linde, C.; Donal, E.; Daubert, J.-C.; Gan, L.-M.; Lund, L.H. Inflammatory Biomarkers Predict Heart Failure Severity and Prognosis in Patients With Heart Failure With Preserved Ejection Fraction. Circulation: Cardiovascular Genetics 2017, 10, e001633. [Google Scholar] [CrossRef]
- Daou, D.; Gillette, T.G.; Hill, J.A. Inflammatory Mechanisms in Heart Failure with Preserved Ejection Fraction. Physiology (Bethesda) 2023, 38, 217–230. [Google Scholar] [CrossRef]
- Zhao, S.; Johnston, A.M.; Yiu, C.H.K.; Moreira, L.M.; Reilly, S.; Wehrens, X.H.T. Aging-Associated Mechanisms of Atrial Fibrillation Progression and Their Therapeutic Potential. jca 2024, 4, N. [Google Scholar] [CrossRef]
- Taghdiri, A. Inflammation and Arrhythmogenesis: A Narrative Review of the Complex Relationship. International Journal of Arrhythmia 2024, 25, 4. [Google Scholar] [CrossRef]
- Sengun, E.; Baggett, B.; Murphy, K.; Lu, Y.; Kim, T.-Y.; Terentyev, D.; Choi, B.-R.; Sedivy, J.; Turan, N.N.; Koren, G. Manipulating Senescence of Cardiac Fibroblasts In Vivo and In Vitro to Investigate Arrhythmogenic Effect of Senescence. The FASEB Journal 2020, 34, 1–1. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Laghi-Pasini, F.; Boutjdir, M.; Capecchi, P.L. Inflammatory Cytokines and Cardiac Arrhythmias: The Lesson from COVID-19. Nat Rev Immunol 2022, 22, 270–272. [Google Scholar] [CrossRef]
- Lazzerini, P.E.; Abbate, A.; Boutjdir, M.; Capecchi, P.L. Fir(e)Ing the Rhythm: Inflammatory Cytokines and Cardiac Arrhythmias. JACC: Basic to Translational Science 2023, 8, 728–750. [Google Scholar] [CrossRef] [PubMed]
- De Jesus, N.M.; Wang, L.; Herren, A.W.; Wang, J.; Shenasa, F.; Bers, D.M.; Lindsey, M.L.; Ripplinger, C.M. Atherosclerosis Exacerbates Arrhythmia Following Myocardial Infarction: Role of Myocardial Inflammation. Heart Rhythm 2015, 12, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Baggett, B.C.; Murphy, K.R.; Sengun, E.; Mi, E.; Cao, Y.; Turan, N.N.; Lu, Y.; Schofield, L.; Kim, T.Y.; Kabakov, A.Y.; et al. Myofibroblast Senescence Promotes Arrhythmogenic Remodeling in the Aged Infarcted Rabbit Heart. eLife 2023, 12, e84088. [Google Scholar] [CrossRef] [PubMed]
- Ihara, K.; Sasano, T. Role of Inflammation in the Pathogenesis of Atrial Fibrillation. Front. Physiol. 2022, 13. [Google Scholar] [CrossRef]
- Alí, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, Inflammation, and Arrhythmias: Role for Interleukin-6 Molecular Mechanisms. Front. Physiol. 2019, 9. [Google Scholar] [CrossRef]
- Heijman, J.; Muna, A.P.; Veleva, T.; Molina, C.E.; Sutanto, H.; Tekook, M.; Wang, Q.; Abu-Taha, I.H.; Gorka, M.; Künzel, S.; et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ Res 2020, 127, 1036–1055. [Google Scholar] [CrossRef]
- Mondragon, R.R.; Wang, S.; Stevenson, M.D.; Lozhkin, A.; Vendrov, A.E.; Isom, L.L.; Runge, M.S.; Madamanchi, N.R. NOX4-Driven Mitochondrial Oxidative Stress in Aging Promotes Myocardial Remodeling and Increases Susceptibility to Ventricular Tachyarrhythmia. Free Radical Biology and Medicine 2025, 235, 294–305. [Google Scholar] [CrossRef]
- Xie, J.; Chen, Y.; Hu, C.; Pan, Q.; Wang, B.; Li, X.; Geng, J.; Xu, B. Premature Senescence of Cardiac Fibroblasts and Atrial Fibrosis in Patients with Atrial Fibrillation. Oncotarget 2017, 8, 57981–57990. [Google Scholar] [CrossRef] [PubMed]
- Pfenniger, A.; Yoo, S.; Arora, R. Oxidative Stress and Atrial Fibrillation. Journal of Molecular and Cellular Cardiology 2024, 196, 141–151. [Google Scholar] [CrossRef]
- Lei, Q.; Jiang, Z.; Shao, Y.; Liu, X.; Li, X. Stellate Ganglion, Inflammation, and Arrhythmias: A New Perspective on Neuroimmune Regulation. Front. Cardiovasc. Med. 2024, 11. [Google Scholar] [CrossRef]
- Manolis, A.A.; Manolis, T.A.; Apostolopoulos, E.J.; Apostolaki, N.E.; Melita, H.; Manolis, A.S. The Role of the Autonomic Nervous System in Cardiac Arrhythmias: The Neuro-Cardiac Axis, More Foe than Friend? Trends in Cardiovascular Medicine 2021, 31, 290–302. [Google Scholar] [CrossRef]
- Marcus, G.M.; Whooley, M.A.; Glidden, D.V.; Pawlikowska, L.; Zaroff, J.G.; Olgin, J.E. Interleukin 6 and Atrial Fibrillation in Patients with Coronary Artery Disease: Data from the Heart and Soul Study. Am Heart J 2008, 155, 303–309. [Google Scholar] [CrossRef]
- Bai, W.; Liu, Z.-Q.; He, P.-Y. ; Muhuyati The Role of IL-6, IL-10, TNF-α and PD-1 Expression on CD4 T Cells in Atrial Fibrillation. Heliyon 2023, 9, e18818. [Google Scholar] [CrossRef]
- Hadi, H.A.; Mahmeed, W.A.; Suwaidi, J.A.; Ellahham, S. Pleiotropic Effects of Statins in Atrial Fibrillation Patients: The Evidence. Vasc Health Risk Manag 2009, 5, 533–551. [Google Scholar] [CrossRef] [PubMed]
- Saljic, A.; Heijman, J.; Dobrev, D. Emerging Antiarrhythmic Drugs for Atrial Fibrillation. Int J Mol Sci 2022, 23, 4096. [Google Scholar] [CrossRef] [PubMed]
- Angendohr, S.; Zoehner, C.; Duplessis, A.; Amin, E.; Polzin, A.; Dannenberg, L.; Schmitt, J.; Srivastava, T.; Bejinariu, A.; Rana, O.; et al. Senolysis Ameliorates Increased Susceptibility to Ventricular Arrhythmias in Prediabetes. Europace 2024, 26, euae102.624. [Google Scholar] [CrossRef]
- Muthamil, S.; Kim, H.-Y.; Jang, H.-J.; Lyu, J.-H.; Shin, U.C.; Go, Y.; Park, S.-H.; Lee, H.G.; Park, J.H. Biomarkers of Cellular Senescence and Aging: Current State-of-the-Art, Challenges and Future Perspectives. Advanced Biology 2024, 8, 2400079. [Google Scholar] [CrossRef]
- Cohn, R.L.; Gasek, N.S.; Kuchel, G.A.; Xu, M. The Heterogeneity of Cellular Senescence: Insights at the Single-Cell Level. Trends in Cell Biology 2023, 33, 9–17. [Google Scholar] [CrossRef]
- Bitencourt, T.C.; Vargas, J.E.; Silva, A.O.; Fraga, L.R.; Filippi-Chiela, E. Subcellular Structure, Heterogeneity, and Plasticity of Senescent Cells. Aging Cell 2024, 23, e14154. [Google Scholar] [CrossRef]
- Kudlova, N.; De Sanctis, J.B.; Hajduch, M. Cellular Senescence: Molecular Targets, Biomarkers, and Senolytic Drugs. Int J Mol Sci 2022, 23, 4168. [Google Scholar] [CrossRef]
- Sorokina, A.G.; Orlova, Y.A.; Grigorieva, O.A.; Novoseletskaya, E.S.; Basalova, N.A.; Alexandrushkina, N.A.; Vigovskiy, M.A.; Kirillova, K.I.; Balatsky, A.V.; Samokhodskaya, L.M.; et al. Correlations between Biomarkers of Senescent Cell Accumulation at the Systemic, Tissue and Cellular Levels in Elderly Patients. Experimental Gerontology 2023, 177, 112176. [Google Scholar] [CrossRef]
- Cummings, S.R.; Lui, L.-Y.; Zaira, A.; Mau, T.; Fielding, R.A.; Atkinson, E.J.; Patel, S.; LeBrasseur, N. Biomarkers of Cellular Senescence and Major Health Outcomes in Older Adults. Geroscience 2024. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int J Mol Sci 2021, 22, 3850. [Google Scholar] [CrossRef]
- Farr, J.N.; Monroe, D.G.; Atkinson, E.J.; Froemming, M.N.; Ruan, M.; LeBrasseur, N.K.; Khosla, S. Characterization of Human Senescent Cell Biomarkers for Clinical Trials. Aging Cell 2025, 24, e14489. [Google Scholar] [CrossRef]
- González-Gualda, E.; Baker, A.G.; Fruk, L.; Muñoz-Espín, D. A Guide to Assessing Cellular Senescence in Vitro and in Vivo. The FEBS Journal 2021, 288, 56–80. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Zamudio, R.I.; Dewald, H.K.; Vasilopoulos, T.; Gittens-Williams, L.; Fitzgerald-Bocarsly, P.; Herbig, U. Senescence-associated Β-galactosidase Reveals the Abundance of Senescent CD8+ T Cells in Aging Humans. Aging Cell 2021, 20, e13344. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, C. Transcriptomic Analysis of Cellular Senescence: One Step Closer to Senescence Atlas. Mol Cells 2021, 44, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Dong, C.; Zhou, L.; Liu, J.; Rabinowitz, Z.M.; Zhang, Y.; Guo, H.; He, F.; Chen, X.; Wang, Y.; et al. Novel PET Imaging Probe for Quantitative Detection of Senescence In Vivo. J Med Chem 2024, 67, 5924–5934. [Google Scholar] [CrossRef]
- Katkenov, N.; Mukhatayev, Z.; Kozhakhmetov, S.; Sailybayeva, A.; Bekbossynova, M.; Kushugulova, A. Systematic Review on the Role of IL-6 and IL-1β in Cardiovascular Diseases. J Cardiovasc Dev Dis 2024, 11, 206. [Google Scholar] [CrossRef]
- Wang, J.; Hong, D.; Li, J.; Wang, L.; Xie, Y.; Da, J.; Liu, Y. Activatable Multiplexed 19F NMR Probes for Dynamic Monitoring of Biomarkers Associated with Cellular Senescence. ACS Meas. Sci. Au 2024, 4, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics Decrease Senescent Cells in Humans: Preliminary Report from a Clinical Trial of Dasatinib plus Quercetin in Individuals with Diabetic Kidney Disease. eBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Garbarino, V.R.; Palavicini, J.P.; Melendez, J.; Barthelemy, N.; He, Y.; Kautz, T.F.; Lopez-Cruzan, M.; Mathews, J.J.; Xu, P.; Zhan, B.; et al. Evaluation of Exploratory Fluid Biomarker Results from a Phase 1 Senolytic Trial in Mild Alzheimer’s Disease. Res Sq, 3994. [Google Scholar] [CrossRef]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16INK4a in Peripheral Blood T-Cells Is a Biomarker of Human Aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in Idiopathic Pulmonary Fibrosis: Results from a First-in-Human, Open-Label, Pilot Study. eBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Owens, W.A.; Walaszczyk, A.; Spyridopoulos, I.; Dookun, E.; Richardson, G.D. Senescence and Senolytics in Cardiovascular Disease: Promise and Potential Pitfalls. Mech Ageing Dev 2021, 198, 111540. [Google Scholar] [CrossRef]
- Sweeney, M.; Cook, S.A.; Gil, J. Therapeutic Opportunities for Senolysis in Cardiovascular Disease. The FEBS Journal 2023, 290, 1235–1255. [Google Scholar] [CrossRef]
- Gadecka, A.; Nowak, N.; Bulanda, E.; Janiszewska, D.; Dudkowska, M.; Sikora, E.; Bielak-Zmijewska, A. The Senolytic Cocktail, Dasatinib and Quercetin, Impacts the Chromatin Structure of Both Young and Senescent Vascular Smooth Muscle Cells. GeroScience 2025. [Google Scholar] [CrossRef] [PubMed]
- Kadoumi, N.; Zatarah, G.; Stokar, J.; Shivam, P.; Gurt, I.; Dresner-Pollak, R. 12489 Senolytics Dasatinib And Quercetin Alleviate Type 1 Diabetes-Related Cardiac Fibrosis In Male Akita Ins-/+Mice. Journal of the Endocrine Society 2024, 8, bvae163.927. [Google Scholar] [CrossRef]
- Takaba, M.; Iwaki, T.; Arakawa, T.; Ono, T.; Maekawa, Y.; Umemura, K. Dasatinib Suppresses Atherosclerotic Lesions by Suppressing Cholesterol Uptake in a Mouse Model of Hypercholesterolemia. Journal of Pharmacological Sciences 2022, 149, 158–165. [Google Scholar] [CrossRef]
- Lu, X.-L.; Zhao, C.-H.; Yao, X.-L.; Zhang, H. Quercetin Attenuates High Fructose Feeding-Induced Atherosclerosis by Suppressing Inflammation and Apoptosis via ROS-Regulated PI3K/AKT Signaling Pathway. Biomedicine & Pharmacotherapy 2017, 85, 658–671. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic Senolytic Treatment Alleviates Established Vasomotor Dysfunction in Aged or Atherosclerotic Mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Silva, E.; Tome, I.; Vasques-Novoa, F.; Silva, A.; Conceicao, G.; Miranda-Silva, D.; Pitrez, P.; Barros, A.; Leite-Moreira, A.; Pinto-Do-O, P.; et al. Pharmacological Targeting of Senescence with ABT-263 in Experimental Heart Failure with Preserved Ejection Fraction. Cardiovascular Research 2022, 118, cvac066.107. [Google Scholar] [CrossRef]
- Suda, M.; Katsuumi, G.; Tchkonia, T.; Kirkland, J.L.; Minamino, T. Potential Clinical Implications of Senotherapies for Cardiovascular Disease. Circ J 2024, 88, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Stojanović, S.D.; Thum, T.; Bauersachs, J. Anti-Senescence Therapies: A New Concept to Address Cardiovascular Disease. Cardiovascular Research 2025, 121, 730–747. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Pitcher, L.E.; Prahalad, V.; Niedernhofer, L.J.; Robbins, P.D. Targeting Cellular Senescence with Senotherapeutics: Senolytics and Senomorphics. The FEBS Journal 2023, 290, 1362–1383. [Google Scholar] [CrossRef]
- Abdelgawad, I.Y.; Agostinucci, K.; Sadaf, B.; Grant, M.K.O.; Zordoky, B.N. Metformin Mitigates SASP Secretion and LPS-Triggered Hyper-Inflammation in Doxorubicin-Induced Senescent Endothelial Cells. Front. Aging 2023, 4. [Google Scholar] [CrossRef]
- Le Pelletier, L.; Mantecon, M.; Gorwood, J.; Auclair, M.; Foresti, R.; Motterlini, R.; Laforge, M.; Atlan, M.; Fève, B.; Capeau, J.; et al. Metformin Alleviates Stress-Induced Cellular Senescence of Aging Human Adipose Stromal Cells and the Ensuing Adipocyte Dysfunction. eLife 2021, 10, e62635. [Google Scholar] [CrossRef]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin Inhibits the Secretory Phenotype of Senescent Cells by a Nrf2-Independent Mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Lesniewski, L.A.; Seals, D.R.; Walker, A.E.; Henson, G.D.; Blimline, M.W.; Trott, D.W.; Bosshardt, G.C.; LaRocca, T.J.; Lawson, B.R.; Zigler, M.C.; et al. Dietary Rapamycin Supplementation Reverses Age-Related Vascular Dysfunction and Oxidative Stress, While Modulating Nutrient-Sensing, Cell Cycle, and Senescence Pathways. Aging Cell 2017, 16, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Reihl, K.D.; Seals, D.R.; Henson, G.D.; LaRocca, T.J.; Magerko, K.; Bosshardt, G.C.; Lesniewski, L.A.; Donato, A.J. Dietary Rapamycin Selectively Improves Arterial Function in Old Mice. The FASEB Journal 2013, 27, 1194.17–1194.17. [Google Scholar] [CrossRef]
- Kang, N.; Kim, J.; Kwon, M.; Son, Y.; Eo, S.-K.; Baryawno, N.; Kim, B.S.; Yoon, S.; Oh, S.-O.; Lee, D.; et al. Blockade of mTORC1 via Rapamycin Suppresses 27-Hydroxycholestrol-Induced Inflammatory Responses. International Journal of Molecular Sciences 2024, 25, 10381. [Google Scholar] [CrossRef] [PubMed]
- Van Skike, C.E.; DeRosa, N.; Galvan, V.; Hussong, S.A. Rapamycin Restores Peripheral Blood Flow in Aged Mice and in Mouse Models of Atherosclerosis and Alzheimer’s Disease. Geroscience 2023, 45, 1987–1996. [Google Scholar] [CrossRef]
- Mbara, K.C.; Devnarain, N.; Owira, P.M.O. Potential Role of Polyphenolic Flavonoids as Senotherapeutic Agents in Degenerative Diseases and Geroprotection. Pharmaceut Med 2022, 36, 331–352. [Google Scholar] [CrossRef]
- Mahoney, S.A.; Venkatasubramanian, R.; Darrah, M.A.; Ludwig, K.R.; VanDongen, N.S.; Greenberg, N.T.; Longtine, A.G.; Hutton, D.A.; Brunt, V.E.; Campisi, J.; et al. Intermittent Supplementation with Fisetin Improves Arterial Function in Old Mice by Decreasing Cellular Senescence. Aging Cell 2024, 23, e14060. [Google Scholar] [CrossRef]
- Sun, Y.; Li, Q.; Kirkland, J.L. Targeting Senescent Cells for a Healthier Longevity: The Roadmap for an Era of Global Aging. Life Med 2022, 1, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Chun, K.-H.; Oh, J.; Lee, C.J.; Park, J.J.; Lee, S.E.; Kim, M.-S.; Cho, H.-J.; Choi, J.-O.; Lee, H.-Y.; Hwang, K.-K.; et al. Metformin Treatment Is Associated with Improved Survival in Diabetic Patients Hospitalized with Acute Heart Failure: A Prospective Observational Study Using the Korean Acute Heart Failure Registry Data. Diabetes & Metabolism 2024, 50, 101504. [Google Scholar] [CrossRef]
- Andersson, C.; Olesen, J.B.; Hansen, P.R.; Weeke, P.; Norgaard, M.L.; Jørgensen, C.H.; Lange, T.; Abildstrøm, S.Z.; Schramm, T.K.; Vaag, A.; et al. Metformin Treatment Is Associated with a Low Risk of Mortality in Diabetic Patients with Heart Failure: A Retrospective Nationwide Cohort Study. Diabetologia 2010, 53, 2546–2553. [Google Scholar] [CrossRef]
- Han, Y.; Xie, H.; Liu, Y.; Gao, P.; Yang, X.; Shen, Z. Effect of Metformin on All-Cause and Cardiovascular Mortality in Patients with Coronary Artery Diseases: A Systematic Review and an Updated Meta-Analysis. Cardiovasc Diabetol 2019, 18, 96. [Google Scholar] [CrossRef]
- Rad, A.N.; Grillari, J. Current Senolytics: Mode of Action, Efficacy and Limitations, and Their Future. Mechanisms of Ageing and Development 2024, 217, 111888. [Google Scholar] [CrossRef]
- Costa, A.; Abruzzese, E.; Latagliata, R.; Mulas, O.; Carmosino, I.; Scalzulli, E.; Bisegna, M.L.; Ielo, C.; Martelli, M.; Caocci, G.; et al. Safety and Efficacy of TKIs in Very Elderly Patients (≥75 Years) with Chronic Myeloid Leukemia. Journal of Clinical Medicine 2024, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. An Updated Landscape of Cellular Senescence Heterogeneity: Mechanisms, Technologies and Senotherapies. Translational Medicine of Aging 2023, 7, 46–51. [Google Scholar] [CrossRef]
- Neri, F.; Zheng, S.; Watson, M.; Desprez, P.-Y.; Gerencser, A.A.; Campisi, J.; Wirtz, D.; Wu, P.-H.; Schilling, B. Senescent Cell Heterogeneity and Responses to Senolytic Treatment Are Related to Cell Cycle Status during Cell Growth Arrest. bioRxiv 2024. [Google Scholar] [CrossRef]
- Garrido, A.M.; Kaistha, A.; Uryga, A.K.; Oc, S.; Foote, K.; Shah, A.; Finigan, A.; Figg, N.; Dobnikar, L.; Jørgensen, H.; et al. Efficacy and Limitations of Senolysis in Atherosclerosis. Cardiovasc Res 2021, 118, 1713–1727. [Google Scholar] [CrossRef]
- Suda, M.; Paul, K.H.; Tripathi, U.; Minamino, T.; Tchkonia, T.; Kirkland, J.L. Targeting Cell Senescence and Senolytics: Novel Interventions for Age-Related Endocrine Dysfunction. Endocrine Reviews 2024, 45, 655–675. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhou, L.; Makarczyk, M.J.; Feng, P.; Zhang, J. The Anti-Aging Mechanism of Metformin: From Molecular Insights to Clinical Applications. Molecules 2025, 30, 816. [Google Scholar] [CrossRef] [PubMed]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLOS Biology 2020, 18, e3000599. [Google Scholar] [CrossRef]
- Bloom, S.I.; Islam, M.T.; Lesniewski, L.A.; Donato, A.J. Mechanisms and Consequences of Endothelial Cell Senescence. Nat Rev Cardiol 2023, 20, 38–51. [Google Scholar] [CrossRef]
- Crouch, J.; Shvedova, M.; Thanapaul, R.J.R.S.; Botchkarev, V.; Roh, D. Epigenetic Regulation of Cellular Senescence. Cells 2022, 11, 672. [Google Scholar] [CrossRef]
- Safwan-Zaiter, H.; Wagner, N.; Michiels, J.-F.; Wagner, K.-D. Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging. Cells 2022, 11, 541. [Google Scholar] [CrossRef]
- Chandrasegaran, S.; Sluka, J.P.; Shanley, D. Modelling the Spatiotemporal Dynamics of Senescent Cells in Wound Healing, Chronic Wounds, and Fibrosis. PLOS Computational Biology 2025, 21, e1012298. [Google Scholar] [CrossRef]
- Savić, R.; Yang, J.; Koplev, S.; An, M.C.; Patel, P.L.; O’Brien, R.N.; Dubose, B.N.; Dodatko, T.; Rogatsky, E.; Sukhavasi, K.; et al. Integration of Transcriptomes of Senescent Cell Models with Multi-Tissue Patient Samples Reveals Reduced COL6A3 as an Inducer of Senescence. Cell Rep 2023, 42, 113371. [Google Scholar] [CrossRef]
- Tuttle, C.S.L.; Luesken, S.W.M.; Waaijer, M.E.C.; Maier, A.B. Senescence in Tissue Samples of Humans with Age-Related Diseases: A Systematic Review. Ageing Research Reviews 2021, 68, 101334. [Google Scholar] [CrossRef]
- Flint, B.; Tadi, P. Physiology, Aging. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2025. [Google Scholar]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes (Basel) 2017, 8, 398. [Google Scholar] [CrossRef]
- Somasundaram, I.; Jain, S.M.; Blot-Chabaud, M.; Pathak, S.; Banerjee, A.; Rawat, S.; Sharma, N.R.; Duttaroy, A.K. Mitochondrial Dysfunction and Its Association with Age-Related Disorders. Front Physiol 2024, 15, 1384966. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and Metabolic Dysfunction in Ageing and Age-Related Diseases. Nat Rev Endocrinol 2022, 18, 243–258. [Google Scholar] [CrossRef]
- De Luca, M.; Crisci, G.; Armentaro, G.; Cicco, S.; Talerico, G.; Bobbio, E.; Lanzafame, L.; Green, C.G.; McLellan, A.G.; Debiec, R.; et al. Endothelial Dysfunction and Heart Failure with Preserved Ejection Fraction—An Updated Review of the Literature. Life (Basel) 2023, 14, 30. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Pagliaro, P. Endothelial Dysfunction: Redox Imbalance, NLRP3 Inflammasome, and Inflammatory Responses in Cardiovascular Diseases. Antioxidants 2025, 14, 256. [Google Scholar] [CrossRef]
- Yeh, J.-K.; Wang, C.-Y. Telomeres and Telomerase in Cardiovascular Diseases. Genes (Basel) 2016, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Martini, H.; Lefevre, L.; Sayir, S.; Itier, R.; Maggiorani, D.; Dutaur, M.; Marsal, D.J.; Roncalli, J.; Pizzinat, N.; Cussac, D.; et al. Selective Cardiomyocyte Oxidative Stress Leads to Bystander Senescence of Cardiac Stromal Cells. Int J Mol Sci 2021, 22, 2245. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A Senescent Cell Bystander Effect: Senescence-Induced Senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Sidler, C.; Kovalchuk, O.; Kovalchuk, I. Epigenetic Regulation of Cellular Senescence and Aging. Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C. Nutrient Sensing, Signaling and Ageing: The Role of IGF-1 and mTOR in Ageing and Age-Related Disease. Subcell Biochem 2018, 90, 49–97. [Google Scholar] [CrossRef]
- Daneshgar, N.; Rabinovitch, P.S.; Dai, D.-F. TOR Signaling Pathway in Cardiac Aging and Heart Failure. Biomolecules 2021, 11, 168. [Google Scholar] [CrossRef]
Figure 1.
DNA damage-induced senescence and SASP via ATM/ATR-mediated molecular cascades. DNA double-strand breaks activate the DNA damage response (DDR), primarily through ATM/ATR kinases, initiating downstream signaling cascades that lead to cellular senescence and SASP. DDR signaling results in activation of CHK2, p53, GATA4, and NEMO. p53 upregulates p21CIP1, which inhibits CDK2/Cyclin E, enforcing G1/S cell cycle arrest. Independently, increased expression of p16INK4a inhibits CDK4/6, maintaining Rb in a hypophosphorylated state and contributing to senescence. DNA damage also leads to cytoplasmic chromatin fragment (CCF) formation, activating the cGAS–STING–TBK1 pathway, which induces IRF3 and NF-κB activation. NF-κB, in cooperation with C/EBPβ and GATA4, drives the transcription of pro-inflammatory SASP factors. Together, these pathways establish irreversible growth arrest and promote SASP expression in senescent cells.
Figure 1.
DNA damage-induced senescence and SASP via ATM/ATR-mediated molecular cascades. DNA double-strand breaks activate the DNA damage response (DDR), primarily through ATM/ATR kinases, initiating downstream signaling cascades that lead to cellular senescence and SASP. DDR signaling results in activation of CHK2, p53, GATA4, and NEMO. p53 upregulates p21CIP1, which inhibits CDK2/Cyclin E, enforcing G1/S cell cycle arrest. Independently, increased expression of p16INK4a inhibits CDK4/6, maintaining Rb in a hypophosphorylated state and contributing to senescence. DNA damage also leads to cytoplasmic chromatin fragment (CCF) formation, activating the cGAS–STING–TBK1 pathway, which induces IRF3 and NF-κB activation. NF-κB, in cooperation with C/EBPβ and GATA4, drives the transcription of pro-inflammatory SASP factors. Together, these pathways establish irreversible growth arrest and promote SASP expression in senescent cells.
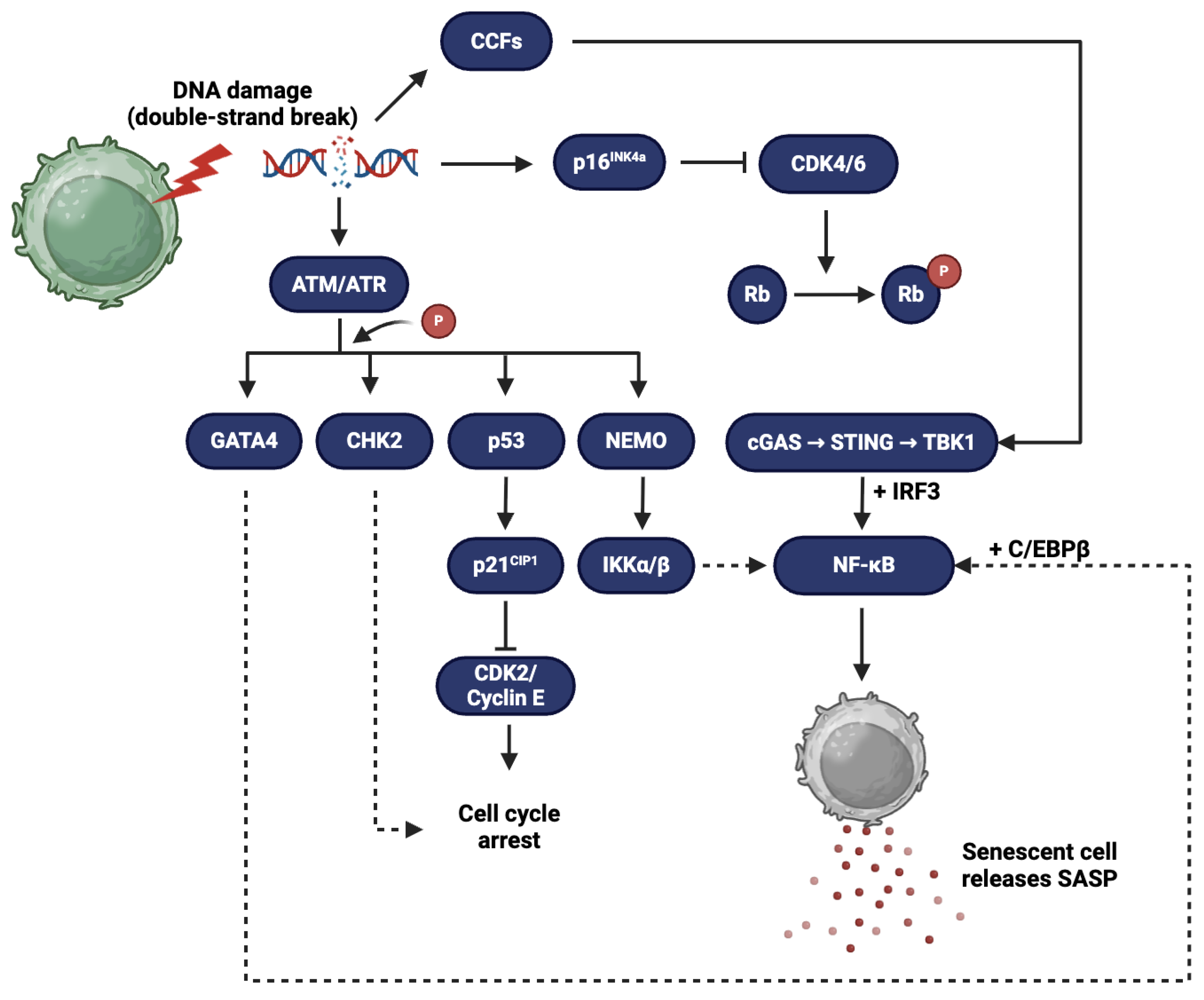
Figure 2.
Telomere shortening and its role in cellular senescence and SASP. Telomeres are repetitive nucleotide sequences (e.g., TTAGGG) located at the ends of chromosomes that protect genomic DNA. With each round of cell division, telomeres progressively shorten due to the end-replication problem, ultimately leading to critically short telomeres. When telomeres become too short, cells enter a state of irreversible growth arrest known as cellular senescence.
Figure 2.
Telomere shortening and its role in cellular senescence and SASP. Telomeres are repetitive nucleotide sequences (e.g., TTAGGG) located at the ends of chromosomes that protect genomic DNA. With each round of cell division, telomeres progressively shorten due to the end-replication problem, ultimately leading to critically short telomeres. When telomeres become too short, cells enter a state of irreversible growth arrest known as cellular senescence.
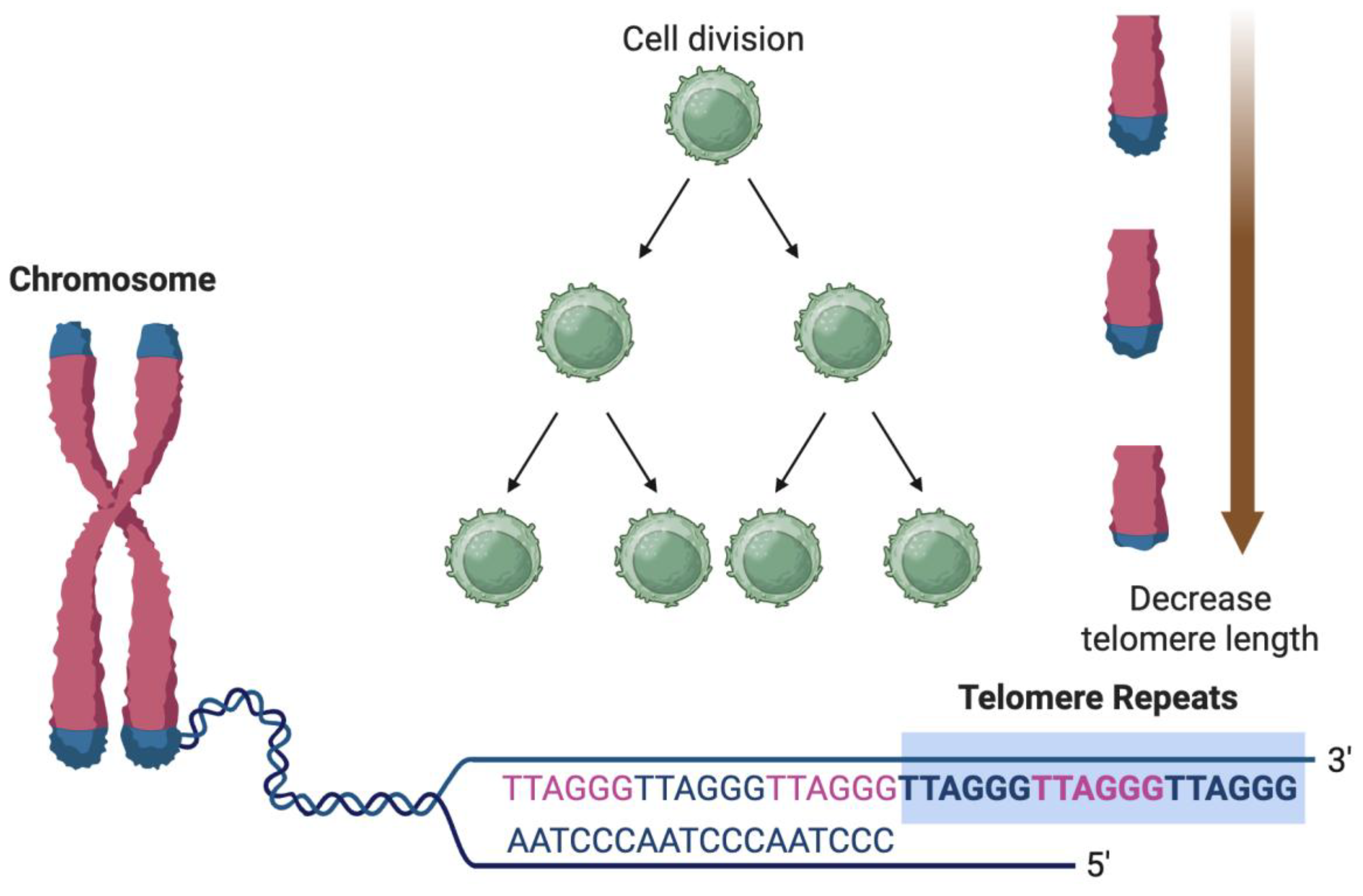

Table 1.
Key senescence-associated secretory phenotype (SASP) molecules and their functional roles in cellular senescence.
Table 1.
Key senescence-associated secretory phenotype (SASP) molecules and their functional roles in cellular senescence.
| SASP Molecule | Molecular Class | Primary Role in Senescence and Tissue Pathophysiology |
|---|---|---|
| IL-6 | Cytokine | Central mediator of chronic inflammation; promotes paracrine senescence via JAK/STAT pathway; impairs endothelial function and insulin signaling. |
| IL-1α / IL-1β | Cytokine | Early SASP inducers; amplify NF-κB signaling; enhance MMP expression and leukocyte recruitment; activate inflammasome. |
| TNF-α | Cytokine | Pro-inflammatory cytokine; disrupts cell-cell junctions; enhances ROS production and apoptosis in neighboring cells. |
| CXCL8 (IL-8) | Chemokine | Chemoattractant for neutrophils; promotes angiogenesis and inflammatory cell infiltration; contributes to plaque instability. |
| CCL2 (MCP-1) | Chemokine | Recruits monocytes/macrophages to sites of tissue injury or senescence; drives chronic inflammation and foam cell formation in atherosclerosis. |
| CCL5 (RANTES) | Chemokine | Attracts T cells and monocytes; involved in autoimmune and chronic inflammatory responses in vascular aging. |
| MMP-1, MMP-3, MMP-9 | Matrix Metalloproteinases | Degrade extracellular matrix; facilitate tissue remodeling, fibrosis, and plaque rupture; destabilize fibrous caps in atherosclerosis. |
| GM-CSF | Growth Factor / Cytokine | Promotes myeloid lineage differentiation; enhances pro-inflammatory macrophage polarization and activation. |
| TGF-β | Growth Factor | Dual role in senescence and fibrosis; induces ECM deposition; contributes to cardiac and vascular fibrosis and stiffening. |
| VEGF | Growth Factor | Promotes angiogenesis; contributes to neovascularization of plaques; involved in tissue repair and endothelial permeability. |
| IGFBP-3 / IGFBP-7 | Insulin-like Growth Factor Binding Proteins | Modulate IGF signaling; induce growth arrest; IGFBP-7 implicated in paracrine senescence and cardiac hypertrophy. |
| PAI-1 (SERPINE1) | Serine Protease Inhibitor | Mediator of TGF-β-induced senescence; inhibits fibrinolysis; associated with thrombosis and vascular remodeling in aged tissues. |
| Galectin-3 | Lectin | Promotes inflammation, fibrosis, and immune cell recruitment; elevated in heart failure and linked to SASP signaling. |
| HMGB1 | Alarmin / DAMP | Released during cellular stress or necrosis; triggers TLR4 signaling and promotes inflammatory SASP amplification. |
| SAA1 / SAA2 | Acute-Phase Proteins | Amplify inflammatory responses; stimulate chemokine production and monocyte recruitment. |
| IL-10 | Anti-inflammatory Cytokine | Context-dependent; may limit excessive inflammation but also contribute to immune evasion by senescent cells. |
| ROS / NO | Reactive Species | Promote DNA damage, mitochondrial dysfunction, and SASP induction; disrupt redox-sensitive ion channels and gap junctions. |
| Extracellular vesicles (EVs) | Nanoparticle-derived SASP | Transfer of microRNAs, damaged DNA, and inflammatory proteins to nearby cells; propagate senescence and SASP signaling systemically. |
Note: ROS = reactive oxygen species;NO = nitric oxide; IL-6 = interleukin-6; IL-1α = interleukin-1 alpha; IL-1β = interleukin-1 beta; IL-8 = interleukin-8 (also known as CXCL8); TNF-α = tumor necrosis factor-alpha; CXCL8 = C-X-C motif chemokine ligand 8; CCL2 = C-C motif chemokine ligand 2 (also known as MCP-1); MCP-1 = monocyte chemoattractant protein-1; CCL5 = C-C motif chemokine ligand 5 (also known as RANTES); RANTES = regulated on activation, normal T cell expressed and secreted; MMP-1 = matrix metalloproteinase-1; MMP-3 = matrix metalloproteinase-3; MMP-9 = matrix metalloproteinase-9; GM-CSF = granulocyte-macrophage colony-stimulating factor; TGF-β = transforming growth factor-beta; VEGF = vascular endothelial growth factor; IGFBP-3 = insulin-like growth factor binding protein 3; IGFBP-7 = insulin-like growth factor binding protein 7; PAI-1 = plasminogen activator inhibitor-1 (also known as SERPINE1); SERPINE1 = serpin family E member 1; HMGB1 = high mobility group box 1; SAA1 = serum amyloid A1; SAA2 = serum amyloid A2; IL-10 = interleukin-10; EVs = extracellular vesicles).
Table 2.
Key distinctions between transient and chronic SASP.
| Feature | Transient SASP | Chronic SASP |
|---|---|---|
| Triggering Context | Acute stress (e.g., wound healing, development, tumor suppression) | Persistent stress or damage (e.g., telomere attrition, DNA damage, oxidative stress) |
| Duration of SASP Expression | Short-lived; resolves within days to weeks | Long-lasting; persists over weeks to months or indefinitely |
| Senescent Cell Fate | Often cleared by immune surveillance or undergoes apoptosis | Resistant to apoptosis; persists and accumulates with age |
| Key SASP Components | Controlled expression of IL-6, IL-8, TGF-β, limited MMPs | Broad, high-level expression of pro-inflammatory cytokines (IL-6, IL-1β, TNF-α), MMPs, chemokines |
| Immune Modulation | Immunostimulatory; facilitates immune cell recruitment and clearance of senescent cells | Immunosuppressive or immunoevasive; leads to immune cell exhaustion and chronic inflammation |
| Paracrine Effects | Promotes transient proliferation, differentiation, or repair in neighboring cells | Induces paracrine senescence, inflammation, and tissue degradation in surrounding cells |
| Tissue Remodeling | Supports tissue regeneration and fibrosis resolution | Promotes fibrosis, ECM degradation, calcification, and loss of tissue integrity |
| NF-κB / p38MAPK Activation | Mild, transient activation | Sustained, amplified activation driving long-term SASP transcription |
| DNA Damage Response (DDR) | Temporary DDR activation | Persistent DDR signaling (e.g., γ-H2AX, ATM/ATR activation) |
| Epigenetic Landscape | Reversible chromatin changes (transient enhancer activation) | Stable heterochromatin formation (SAHF), long-term enhancer remodeling of SASP genes |
| Metabolic Signature | Mild metabolic reprogramming | Elevated glycolysis, mitochondrial dysfunction, increased ROS production |
| Relevance to CVD | Protective in early repair post-injury (e.g., myocardial infarction) | Pathogenic in aging-associated diseases: atherosclerosis, HFpEF, vascular calcification |
| Resolution Mechanism | Clearance by immune cells (NK, macrophages) | Immune evasion or exhaustion prevents clearance, leading to accumulation |
| Therapeutic Implications | May be beneficial and should be preserved | Targeted for removal or suppression using senolytics or senomorphics |
Table 3.
Role of SASP in cardiovascular and peripheral vascular diseases.
| Disease/Condition | Key SASP-Mediated Mechanisms | Prominent SASP Components | Pathophysiological Consequences |
|---|---|---|---|
| Hypertension | SASP-induced endothelial dysfunction reduces nitric oxide (NO); vascular senescence increases stiffness and tone | IL-6, TNF-α, TGF-β, ROS, MCP-1, MMPs, PAI-1 | Arterial stiffness, impaired vasodilation, increased systemic vascular resistance, pressure overload |
| Peripheral Vascular Disease (PVD) | Chronic inflammation in peripheral arteries; endothelial senescence impairs angiogenesis and perfusion | IL-6, IL-1β, TNF-α, MMP-9, CCL2 | Capillary rarefaction, impaired collateral vessel formation, limb ischemia, tissue necrosis |
| Atherosclerosis | Senescent endothelial and smooth muscle cells promote lipid accumulation, inflammation, and matrix degradation | IL-1β, IL-6, IL-8, MCP-1, TNF-α, MMP-1/3/9, TGF-β | Plaque growth, immune cell recruitment, necrotic core expansion, and fibrous cap weakening |
| Coronary Artery Disease (CAD) | SASP enhances leukocyte adhesion and endothelial dysfunction in coronary vessels; promotes thrombosis | IL-6, TNF-α, VCAM-1, ICAM-1, PAI-1, ROS | Reduced coronary perfusion, enhanced vasoconstriction, increased risk of plaque rupture and thrombus |
| Myocardial Infarction (MI) | Post-MI, SASP from senescent fibroblasts and immune cells sustains inflammation and impairs reparative remodeling | IL-1β, IL-6, TGF-β, MMPs, HMGB1 | Excessive fibrosis, impaired scar formation, ventricular dysfunction, higher risk of rupture |
| Heart Failure (HF) | Persistent SASP drives myocardial fibrosis, hypertrophy, and impaired cardiomyocyte contractility | TGF-β, IL-6, TNF-α, MCP-1, MMPs, IGFBP-3 | Left ventricular stiffening, diastolic dysfunction, progression of HFpEF and HFrEF |
| Cardiac Arrhythmias | SASP-induced fibrosis and inflammation disrupt electrical conduction and calcium homeostasis | IL-1β, TNF-α, IL-6, ROS, MMP-9, TGF-β | Gap junction uncoupling, Ca²⁺ handling defects, increased risk of atrial fibrillation and VT/VF |
Note: SASP factors drive chronic low-grade inflammation (inflammaging), ECM remodeling, oxidative stress, and paracrine senescence—all of which are central to vascular and myocardial aging. In atherosclerosis and CAD, SASP amplifies immune cell recruitment and matrix degradation, while in heart failure and MI, it contributes to maladaptive remodeling and fibrosis. In arrhythmias, especially atrial fibrillation, SASP disrupts ion channel expression, gap junctions, and calcium cycling, creating a pro-arrhythmic substrate.
Table 4.
Potential biomarkers for SASP and senescent cell burden.
| Biomarker | Biomarker Class | Source | Biological Relevance | Advantages / Limitations | Utility in CVD |
|---|---|---|---|---|---|
| IL-6 | SASP cytokine | Plasma / serum | Key pro-inflammatory SASP cytokine; drives systemic inflammaging via JAK/STAT pathway | Readily measurable; non-specific; elevated in many chronic conditions | Correlates with endothelial dysfunction, heart failure progression, and increased CVD mortality |
| IL-1β | SASP cytokine | Plasma / serum | Early SASP mediator; activates NF-κB and inflammasome | Reflects acute and chronic inflammation; overlap with infection signals | Linked to myocardial remodeling, atherosclerosis, and atrial fibrillation |
| TNF-α | SASP cytokine | Plasma / serum | Pro-inflammatory cytokine that promotes apoptosis, insulin resistance | Robust inflammatory marker; limited specificity | Predicts heart failure risk, myocardial fibrosis, and vascular dysfunction |
| MMP-1 / MMP-3 / MMP-9 | SASP protease | Plasma / tissue biopsies | Mediate ECM degradation, fibrosis, plaque rupture | Specific to tissue remodeling; can reflect active SASP | Associated with atherosclerosis progression, plaque instability, and myocardial fibrosis |
| PAI-1 (SERPINE1) | SASP effector / ECM regulator | Plasma / endothelial cells | Mediator of TGF-β-induced senescence; pro-thrombotic and fibrotic | Strongly associated with senescence; modulated by metabolic status | Marker of vascular senescence, pro-coagulant state, and hypertensive remodeling |
| CXCL8 (IL-8) | SASP chemokine | Plasma / PBMCs | Neutrophil chemoattractant; promotes leukocyte adhesion and chronic inflammation | Reflects SASP-driven immune cell recruitment; overlaps with acute inflammation markers | Elevated in unstable atherosclerosis and heart failure |
| HMGB1 | DAMP / Alarmin | Plasma / extracellular vesicles | Released from stressed or senescent cells; promotes sterile inflammation | Non-specific damage signal; also elevated in necrosis | Linked to post-MI inflammation, cardiac injury, and chronic heart failure |
| p16INK4a mRNA | Senescence marker | PBMCs / tissue biopsy | CDK inhibitor upregulated in senescent cells; cell-cycle arrest marker | Correlates with biological age; requires RNA quantification | Elevated in vascular and myocardial senescence; correlates with frailty and arterial aging |
| p21CIP1 | Senescence marker | PBMCs / tissue | DNA damage-induced CDK inhibitor; involved in early senescence | Reflects stress-induced senescence; overlap with quiescence | Associated with endothelial and fibroblast senescence in CVD |
| γ-H2AX foci | DNA damage marker | PBMCs / tissue nuclei | Histone mark of DNA double-strand breaks; persistent in senescent cells | Requires microscopy or imaging; gold-standard for DNA damage | Identifies cells with persistent DDR driving SASP in vascular and cardiac tissues |
| SA-β-Gal activity | Senescence enzymatic marker | Tissue biopsy / ex vivo cultures | Classic marker of lysosomal β-galactosidase upregulation in senescence | Requires fresh tissue or cells; limited in vivo applicability | Used in histological assessment of vascular and myocardial senescence |
| SASP Proteomic Panels | Multi-analyte | Plasma / serum | Combines IL-6, IL-8, MMPs, IGFBPs, TGF-β, PAI-1, etc. | Captures SASP complexity; multiplexed detection; requires standardization | Correlates with disease burden, biological age, and therapeutic response to senolytics |
| Circulating microRNAs | Non-coding RNA biomarkers | Plasma / extracellular vesicles | miRNAs involved in senescence regulation and secreted via EVs (e.g., miR-34a, miR-146a) | Non-invasive; still under investigation; may reflect specific SASP profiles | Linked to cardiac remodeling, fibrosis, and endothelial senescence |
| Cell-free mitochondrial DNA (cf-mtDNA) | Cell stress marker | Plasma | Released from damaged or senescent cells; activates cGAS-STING pathway | Reflects systemic mitochondrial dysfunction and cellular stress | Elevated in patients with HFpEF, MI, and vascular inflammation |
| Epigenetic Clocks (e.g., DNAm Age) | DNA methylation signature | Blood / saliva / tissues | Measures biological age acceleration via methylation at age-related CpG sites | Requires sequencing; validated in aging studies | Associated with vascular aging, arterial stiffness, and CVD risk beyond chronological age |
Table 5.
Available and emerging senolytics, and their potential roles in CVD.
| Senolytic Agent | Mechanism of Action | Senescent Cell Targets | Evidence in CVD Models | Current Status |
|---|---|---|---|---|
| Dasatinib (D) | Inhibits multiple tyrosine kinases (e.g., Src, EphA) required for senescent cell survival | Senescent preadipocytes, endothelial cells, VSMCs, macrophages | Reduces atherosclerosis and improves cardiac function in aged and hyperlipidemic mice | Clinical trials (e.g., diabetic kidney disease) |
| Quercetin (Q) | Inhibits PI3K, serpins, and BCL-2 family proteins; antioxidant properties | Endothelial cells, VSMCs | Improves endothelial function and reduces arterial stiffness | Used in combination with D (D+Q); ongoing trials |
| Navitoclax (ABT-263) | BCL-2/BCL-xL inhibitor; induces apoptosis in cells relying on anti-apoptotic signaling | Senescent endothelial cells, VSMCs, cardiomyocytes | Reduces myocardial fibrosis and improves cardiac function in aged mice | Preclinical; dose-limiting thrombocytopenia |
| Fisetin | Flavonoid with senolytic and antioxidant activity; modulates PI3K/AKT/mTOR signaling | Multiple senescent cell types | Reduces SASP expression, vascular inflammation, and improves survival in aged mice | Clinical trials in aging and frailty |
| UBX1325 / UBX0101 | Small molecules targeting pro-survival pathways (e.g., FOXO4-p53 interaction) | Chondrocytes, fibroblasts, possibly cardiac fibroblasts | UBX0101 shown to clear senescent fibroblasts in OA models; cardiovascular applications under investigation | Phase 1/2 trials for other diseases |
| FOXO4-DRI peptide | Disrupts FOXO4–p53 interaction, restoring p53-mediated apoptosis in senescent cells | Fibroblasts, endothelial cells | Restores cardiac function and decreases senescent cell burden in progeroid mouse models | Preclinical; limited by delivery challenges |
| Piperlongumine | Increases ROS selectively in senescent cells, triggering apoptosis | Broad senescent cell activity | Potential anti-fibrotic effects in cardiac remodeling; requires further CVD-specific validation | Preclinical |
| BPTES (Glutaminase inhibitor) | Inhibits glutamine metabolism, selectively killing glutamine-addicted senescent cells | Cancer-associated fibroblasts, senescent endothelial cells | May reduce metabolic-driven vascular senescence; under investigation | Experimental stage |
| HSP90 inhibitors (e.g., 17-AAG) | Destabilize survival proteins (e.g., AKT, HIF1α) in senescent cells | Multiple senescent types | Shown to deplete senescent cells in vitro and attenuate stress-induced vascular damage | Preclinical; toxicity concerns |
| Emerging PROTAC-based senolytics | Use proteolysis-targeting chimeras to degrade senescence-specific survival proteins | Customizable based on target (e.g., p16INK4a+, p21CIP1+ cells) | Potential for tissue- and cell-specific clearance of senescent cardiovascular cells | Early-stage development |
Table 6.
Available and emerging senomorphics.
| Senomorphic Agent | Mechanism of Action | Target Pathways | Effects on SASP / Senescence | Potential Role in CVD | Clinical Status |
|---|---|---|---|---|---|
| Rapamycin (Sirolimus) | Inhibits mTORC1 signaling | mTOR/PI3K/AKT pathway | Reduces translation of SASP mRNAs, suppresses IL-6, IL-1β, MMPs | Attenuates vascular aging, cardiac hypertrophy, fibrosis; improves diastolic function | Clinical trials in aging and HFpEF |
| Metformin | Activates AMPK; inhibits mitochondrial complex I and mTOR | AMPK, NF-κB, mTOR | Suppresses SASP cytokines, enhances autophagy, reduces oxidative stress | Reduces endothelial senescence, improves vascular reactivity, lowers inflammatory markers in elderly | Widely used; repurposing trials ongoing |
| Resveratrol | Activates SIRT1; modulates NF-κB and NLRP3 inflammasome | SIRT1, AMPK, NF-κB | Reduces IL-6, TNF-α, and ROS; enhances mitochondrial function | Protects against endothelial dysfunction, reduces arterial stiffness and inflammation | Nutraceutical; variable bioavailability |
| Curcumin | Inhibits NF-κB and p38 MAPK; antioxidant | NF-κB, p38 MAPK | Reduces SASP cytokine output, MMPs, and ROS | Improves vascular reactivity; anti-fibrotic effects in myocardium | Preclinical and nutraceutical use |
| Apigenin | Flavonoid; inhibits NF-κB and cyclin-dependent kinases | CDK1/2, NF-κB | Suppresses pro-inflammatory SASP components and DNA damage signaling | Reduces oxidative stress and inflammation in vascular tissues | Preclinical |
| Nicotinamide Riboside (NR) | NAD⁺ precursor; enhances mitochondrial function | SIRT1, PARP, mitochondrial pathways | Reduces mitochondrial-derived SASP signaling; promotes DNA repair | Reverses age-associated endothelial dysfunction; improves arterial compliance | In trials for metabolic and cardiac aging |
| JAK inhibitors (e.g., Ruxolitinib) | Inhibit JAK/STAT signaling downstream of SASP cytokines | JAK/STAT pathway | Reduces IL-6–driven inflammatory SASP output | Mitigates cardiac inflammation and hypertrophy in aged hearts; potential use in HFpEF | Approved for myelofibrosis; trials in aging |
| Glycyrrhizin | Inhibits HMGB1–TLR4 signaling | DAMP sensing (TLRs) | Suppresses SASP-induced sterile inflammation | Reduces inflammatory cardiomyopathy; protects against ischemia–reperfusion injury | Experimental |
| Bay 11-7082 | Irreversible inhibitor of IκB kinase (IKK) | NF-κB signaling | Suppresses NF-κB-dependent SASP gene expression | Reduces vascular inflammation and remodeling | Preclinical |
| Melatonin | Antioxidant and chronobiotic; modulates NF-κB, NLRP3, SIRT1 | NLRP3, SIRT1, ROS | Reduces pro-inflammatory SASP expression; promotes anti-inflammatory cytokines | Protects cardiac tissue from oxidative damage and fibrotic remodeling | Clinical use for sleep; trials ongoing in aging |
| Spermidine | Enhances autophagy and mitochondrial proteostasis | Autophagy, mTOR, AMPK | Suppresses pro-inflammatory and metabolic SASP profiles | Improves cardiac diastolic function and mitochondrial function in aging myocardium | Trials in cardiovascular aging |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



