Submitted:
07 August 2025
Posted:
08 August 2025
You are already at the latest version
Abstract
Glioblastoma multiforme (GBM) remains the most common and fatal primary brain tumour, with low survival rates due to its heterogeneous nature. Traditional diagnostic methods, such as magnetic resonance imaging (MRI), computed tomography (CT) scans, and invasive tissue biopsies for histopathological analysis, are often insufficient in differentiating treatment-related changes from tumour progression, leading to misdiagnosis and delays. Repeated biopsies are impractical for ongoing monitoring due to their invasive nature.
A promising alternative is liquid biopsy, a minimally invasive procedure that analyses biofluids like blood and cerebrospinal fluid (CSF) for tumour-related biomarkers. Tumours release a variety of components into the bloodstream, including circulating tumour cells (CTCs), circulating tumour DNA (ctDNA), microRNAs (miRNAs), extracellular vesicles (EVs), and proteins, which can cross the blood-brain barrier and provide a non-invasive means of assessing genetic, epigenetic, proteomic, and metabolomic markers associated with GBM. Liquid biopsy can be performed multiple times throughout the disease course, offering a rapid way to gather critical and changing tumour information.
However, challenges remain in the clinical implementation of liquid biopsies for GBM diagnosis, including low levels of circulating biomarkers and the lack of standardised assays for biofluid collection and analysis. Combining multiple biomarkers from various bio-elements, such as CTCs, ctDNA, miRNAs, and EVs, could enhance sensitivity and specificity, addressing some of these limitations. The integration of artificial intelligence (AI) for analysing liquid biopsy biomarkers holds great promise in overcoming these challenges. AI can enhance biomarker identification, improve diagnostic accuracy, and expand the clinical utility of liquid biopsy in GBM management. Further research, and large-scale clinical validation are needed to optimise these approaches.
This review explores the potential of GBM biomarkers found in blood and CSF samples, focusing on their applications in diagnosis, prediction, and prognosis, and highlights recent advancements, challenges, and future perspectives, including the integration of AI to improve outcomes in GBM management. Liquid biopsies bridge the gap between invasive methods and emerging technologies, offering transformative potential for GBM diagnosis and treatment.
Keywords:
glioblastoma multiforme
; liquid biopsies
; biomarkers
1. Introduction
Glioblastoma Multiforme (GBM) is the most common and aggressive primary brain tumour, with a median survival of around 15 months despite intensive treatments including surgery, radiation, and chemotherapy [1]. Its highly invasive nature, resistance to therapy, and recurrence make it one of the most challenging cancers to treat. GBM is also marked by substantial molecular heterogeneity, including gene mutations such as IDH-R132H, H3-K27M, and EGFR (EGFRvIII) amplification complicating treatment approaches and prognosis. Moreover, the blood-brain barrier (BBB) further limits the effectiveness of conventional therapies and monitoring methods [2,3,4].
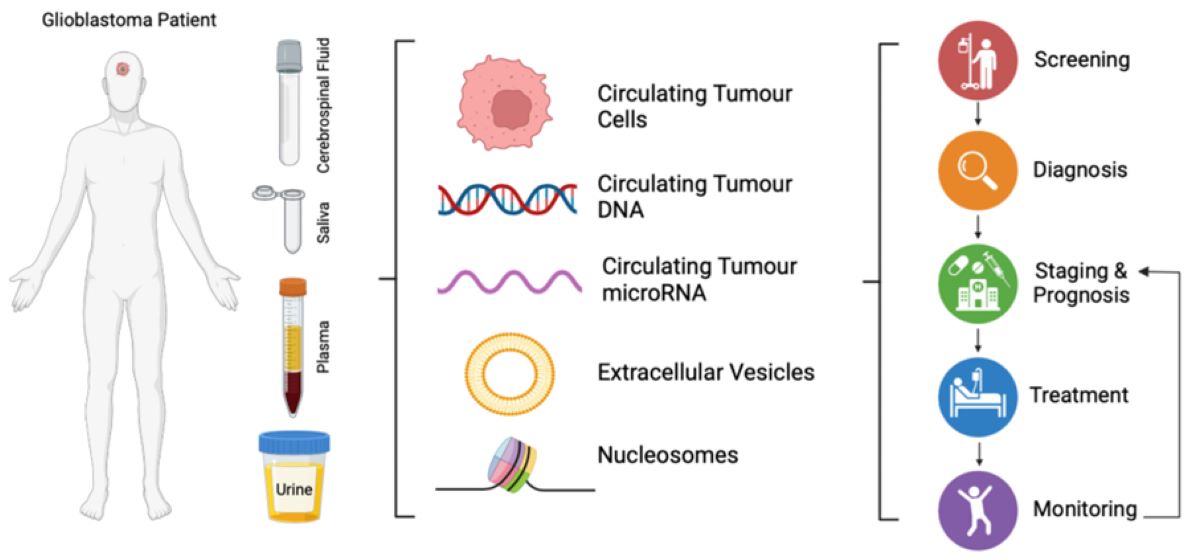
Diagnosing GBM typically includes neuroimaging, followed by histopathological and molecular analysis for confirmation[5,6,7]. However, both imaging and tissue-based methods have limitations, such as distinguishing between relapse and pseudo progression, which are lesions related to treatment simulating recurrence of GBM; a continuous challenge [8]. These challenges underscore the need for alternative, non-invasive diagnostic tools, such as liquid biopsy, which has surfaced as a promising approach to detect early recurrence for GBM patients, principally through blood tests, urine, or cerebrospinal fluid (CSF), providing real-time insights into tumour dynamics and treatment response[9,10]. Liquid biopsies comprise the detection and quantification of tumour related content in biofluids that is released by tumours, such as circulating tumour cells (CTCs), circulating tumour DNA (ctDNA), circulating tumour microRNA (miRNA), extracellular vesicles (EVs) and nucleosomes or in response to tumours, all of which can provide critical insights into GBM progression and recurrence [11].
Being minimally or non-invasive, liquid biopsy offers a valuable alternative to invasive tissue biopsies for detecting early recurrence, monitoring minimal residual disease (MRD), and guiding treatment strategies in GBM. While the BBB hinders the amount of tumour-derived entities that may be found in body fluids, ongoing research aims to increase sensitivity and specificity of liquid biopsy tests, making them a promising tool for advancing clinical management of GBM. This review will explore recent advancements in liquid biopsy technologies and their potential for enhancing the diagnosis and treatment of GBM.
2. Circulating Biomarkers of Glioblastoma from Liquid Biopsies
2.1. Circulating Tumour Cells
CTCs are individual cells or cell clusters that are released from primary or metastatic solid tumours into bodily fluids. When disseminated into the bloodstream and other body fluids, they may contribute to metastasis and can be used as biomarkers for cancer detection, prognosis, and therapeutic monitoring[9,12]. The analysis of CTCs emerged as a potential tool for monitoring MRD in cancer patients[13,14,15]. Higher levels of CTCs are directly related to severity and spread of multiple cancers[16]. Key methods for CTC enumeration, which quantify CTC numbers include sensitive assays like CellSearch® (Menarini Silicon Biosystems, Italy), which detects as few as one CTC per 10 mL of blood. However, CellSearch® uses antibody-mediated capture, mainly targeting epithelial cell adhesion molecule (EpCAM) on CTC membrane surfaces using a positive selection method[17,18,19,20,21].
EpCAM is not expressed in GBM tumours due to their predominantly mesenchymal phenotype[12,22]. Hence, alternative techniques for isolation of CTCs in GBM patients have been explored (Table 1). One such method is negative selection, which enriches for CTCs by eliminating leukocytes from whole blood by means of antibodies against leukocyte antigens[9,19,29,32]. Several different techniques of CTC isolation have been used which are dependent on the CTCs physical properties[23,24]. These include density-gradient centrifugation[27,28], spiral microfluidic technology[30,31] and chip technology[21]. Although promising, it is important to highlight how rare CTC detection in the peripheral circulation of GBM patients actually is. Reported detection rates vary widely, with some studies identifying CTCs in as few as 20-40% of GBM patients, depending on the sensitivity of the detection method and sample volume and timing (20.6% - 77% of patients with detectable CTCs, Table 1). Along with the complexity of isolating CTCs from peripheral blood, this rarity underscores the need for highly sensitive and specific detection technologies to validate their clinical utility as reliable biomarkers. It also further complicates standardisation efforts and underlines the need for optimisation and harmonisation of isolation protocols.
CTC analysis methods for GBM, other than enumeration, include, molecular profiling (e.g., next generation sequencing (NGS), Reverse Transcriptase-PCR (RT-PCR)), immunophenotyping using GBM-specific markers (such as GFAP, Nestin, Sox2, EGFR, GLAST)[21,27,29,30,31], single-cell sequencing[27], and epigenetic profiling[25], with CSF-based analysis often offering higher sensitivity than blood[26,27,28]. These methods enable the detection of clinically-relevant alterations, such as IDH1 mutations and MGMT promoter hypermethylation, which are important for prognosis and for guiding targeted therapy decisions in GBM[19].
Monitoring CTC kinetics over time can provide insights into the effectiveness of treatment and the risk of disease recurrence. Changes in CTC count, phenotype or genetic profile can act as early indicators of therapeutic response of tumour progression[21,27,28,29,31,32] (Table 1).
These studies demonstrate the potential of CTCs as a tool for cancer diagnosis and prognosis, however, there are still challenges that need to be addressed: (i) Low abundance: CTCs are present in very low numbers in the bloodstream being 1 CTC in 109 normal blood cells per 10 mL of blood, making their isolation and analysis challenging[9,18]. Low abundance can lead to false-negative results and reduce the sensitivity of CTC-based assays. (ii) Heterogeneity: CTCs show intra- and inter-patient heterogeneity and may differ in morphology, phenotype, and genetic profile. This heterogeneity complicates the identification and characterisation of CTCs, and it may require the use of multiple markers or assays to detect different subsets of CTCs[35,36]. (iii) Technical variability: The isolation and analysis of CTCs require specialised equipment and protocols, and it can affect the accuracy and reproducibility of CTC-based assays. The choice of isolation and analysis method, as well as the handling and storage of blood samples, can all impact the results of CTC assays[37,38]. (iv) Lack of standardisation: There is currently no standardised protocol for the isolation and analysis of CTCs, and different studies may use different methods or markers to detect CTCs. This lack of standardisation can make it difficult to compare results across studies and to establish the clinical utility of CTC-based assays[39]. (v) Cost and accessibility: CTC-based assays can be costly and require specialised equipment and expertise, which may limit their availability and accessibility in a clinical setting[40].
Overcoming these barriers will require continued innovation in sensitive and specific CTC isolation and analysis techniques, along with greater collaboration between researchers and clinicians in establishing the clinical utility of this approach. By integrating CTC-based assays with other clinical and pathological parameters, and rigorously validating these methods in clinical trials, the clinical utility and potential for earlier accurate cancer detection and monitoring tumour progression may be determined.
2.2. Circulating Tumour DNA
Cell-free DNA (cfDNA) are short DNA fragments of 140-170 base pairs (bp) present in body fluids. In cancer patients, a portion of cfDNA originates from tumour cells following apoptosis or necrosis, referred to as circulating tumour DNA (ctDNA)[9,10,18,41,42]. Representing a promising tool for non-invasive cancer diagnosis, particularly in GBM, ctDNA consists of shorter DNA fragments (< 145 bp) that carry the genetic and epigenetic signatures of the originating tumour[12,18]. A number of studies have validated that ctDNA reflects the genetic profile of the original tumour, showing uniformity with its mutational profile and that of the corresponding tumour tissue across various cancers, including GBM[9,43,44,45], thereby offering insights into tumour genetics and treatment response (see Table 2). Key detection methods include assessing ctDNA levels and known point mutations using polymerase chain reaction (PCR)-based method such as digital droplet PCR (ddPCR), and next-generation sequencing (NGS) or whole genome sequencing (WGS), method that detects novel or unknown mutations[46,47,48]. ddPCR is particularly sensitive for identifying low-frequency mutations such as IDH1, TERT promoter mutations, and EGFRvIII in plasma samples[48]. NGS enables comprehensive genomic profiling, detecting a broad spectrum of mutations and copy number alterations[49]. Methylation-specific PCR targets epigenetic modifications, like MGMT promoter methylation, which are relevant for GBM prognosis and therapy selection[47].
Detecting ctDNA in GBM is challenging due to tumour’s location and the BBB, which decreases yield of ctDNA in peripheral circulation. Despite these obstacles, significant work has been done towards ctDNA detection by employing highly sensitive molecular technologies as described in Table 2. It is noteworthy to mention the baseline level of cfDNA is greater in serum compared to plasma, which may be in result of contamination with DNA that is released from lysed immune cells, thus plasma samples are preferred for studying ctDNA[46,47,48]. While plasma ctDNA alone may not yet be sufficiently sensitive for reliable GBM diagnosis or monitoring, detectable levels offer promise for non-invasive molecular profiling and could help identify actionable mutations and resistance mechanisms, potentially improving treatment strategies. Studies have shown that ctDNA can be detected in a subset of GBM patients, though plasma detection rates are generally lower than in other cancers, likely due to the BBB limiting ctDNA release into the bloodstream[50]. Piccioni et al. found ctDNA mutations in the blood of 50% of brain tumour patients, with 55% of these being GBM cases[51]. However, dependent on patient cohort and cfDNA extraction method 75.0% sensitivity (95% CI: 64.1%–84.0%) and 88.7% specificity (95% CI: 77.0%–95.7%) has been achieved for detection of the IDH1 mutation R132H, and negativity for this mutation is a key classifier of GBM[52,53].
EGFRvIII ctDNA was detectable in plasma and can serve as a dynamic biomarker for preoperative and postoperative assessment, as well as for monitoring disease recurrence in GBM patients[54]. Zill et al. used next-generation sequencing of plasma ctDNA in 107 GBM patients to identify actionable mutations, supporting the use of ctDNA sequencing for personalised cancer treatment[55]. Liu et al., and Miller et al., further demonstrated that WGS of CSF ctDNA achieves high detection rates and can identify clinically relevant mutations, reinforcing its utility as a minimally invasive biomarker for GBM monitoring[56,57].
Furthermore, studies have demonstrated that CSF is a more reliable source for ctDNA in GBM patients compared to plasma, overcoming the limitations imposed by the BBB in regard to ctDNA release into the bloodstream[58,59,60]. For instance, ctDNA was detected in 61% of CSF samples compared to 37% in serum of 19 GBM patients using methylation-specific PCR[61], while nested PCR allowed for detection in 92% of CSF samples compared to only 8% in plasma samples of 38 GBM patients [62]. Another study using WGS on CSF samples of 11 GBM patients demonstrated a 100% ctDNA detection rate[63]. These findings highlight the superior sensitivity of CSF-based liquid biopsies, likely due to the proximity of CSF to the brain tumour and the reduced interference from background cfDNA found in blood[9,12,18,64]. However, CSF collection via lumbar puncture is invasive and may not be suitable for all patients[18]. As alternatives, less invasive fluids like nasal secretions or saliva may provide an alternative to CSF, offering a less invasive means to access brain-derived biomarkers but needed further investigation.
Urine has also emerged as a non-invasive source of ctDNA for GBM diagnosis, offering the advantage of easy and repeatable collection throughout treatment[10]. Following renal filtration, ctDNA can be detected as transrenal DNA (trDNA), enabling molecular diagnostics through urinary DNA analysis[10,65]. Several urinary biomarkers—including matrix metalloproteinases (MMP-2, MMP-9), neutrophil gelatinase-associated lipocalin (NGAL), and vascular endothelial growth factor (VEGF) have demonstrated high sensitivity (95.2%), specificity (95.7%), and accuracy (92.5%) in detecting primary brain tumours, including GBM (see Table 2). Notably, elevated levels of these urinary markers in GBM patients have been shown to correlate with their expression in tumour tissue and, in CSF (for MMP-9 expression)[65,66,67,68]. In addition, urine-based detection of ctDNA epigenetic alterations, such as DNA methylation, offers further potential. Methylation-specific PCR enables the quantification of tumour-specific methylation, such as O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation[69,70], a well-established predictor of GBM patient response to alkylating agents like temozolomide (TMZ)[71,72,73].
In conclusion, ctDNA analysis has shown significant promise as a tool for detecting and monitoring genetic alterations in brain cancers, although some challenges still need to be addressed. These include variability in ctDNA levels depending on the stage and anatomical location of cancer, as well as its rapid degradation in circulation and sensitivity to pre-analytical factors, which can complicate collection and analysis. Overall, detection of ctDNA in CSF and plasma provides valuable insights into tumour evolution, resistance mechanisms, and potential therapeutic targets, offering a non-invasive approach to guide personalised treatment for CNS cancers. Further advancements in ctDNA isolation, sequencing methods, and clinical validation will be crucial to unlock its full potential for early diagnosis, treatment monitoring, and personalised cancer management.
2.3. Nucleosomes
Chromatin structure was first described in 1974 as repeating nucleosome units. Each nucleosome consists of an octamer of two copies, each of the core histones H2A, H2B, H3 and H4, wrapped by 146-147 base pairs of DNA[80,81]. Nucleosomes are generated by DNA associated with proteins, including histone H1. Histones have a globular domain for histone-histone interactions and flexible, positively charged tails (20-35 amino acids) that undergo post-translational modifications (PTMs)[82]. As the basic units of chromatin, nucleosomes regulate key nuclear processes like transcription, replication and repair, primarily through PTMs[83]. These modifications influence gene expression on an additional level to DNA methylation, and activating oncogenes or silencing tumour suppressor genes may contribute to cancer development and progression[83,84]. Histone PTMs include methylation, acetylation, phosphorylation, ubiquitination, sumoylation, glycosylation, homocysteinylation, and crotonylation[85]. Methylation may activate or repress gene expression depending on the site and degree[83], while acetylation generally promotes transcription by neutralising lysine’s positive charge, reducing chromatin compaction and enhancing accessibility for transcription factors[85]. The interplay of these PTMs forms a “histone code” read by cellular proteins to control transcription, replication, and repair, with core histone variants adding further complexity[85,86].
Nucleosomes and their PTMs can be detected in plasma or serum using immunoassays like chemiluminescence immunoassay (ChLIA) or enzyme-linked immunosorbent assay (ELISA) (Table 3). These assays detect specific biomarkers from complex matrices, and can be applied on automated platforms which would allow faster and more reproducible results[87]. Cancer cell death, including that of GBM cells results in the release of nucleosomes into the bloodstream, which are transported as mono- or oligo- nucleosomes bound to ctDNA.
Cancer patients have a greater count of circulating nucleosomes due to the increase in cellular turnover in comparison to healthy controls and due to the cytotoxic effect of treatments which lead to cell death[83]. The high levels of nucleosomes are not explicit to cancer or GBM per se, since it is also linked to trauma, stroke and sepsis which limits its clinical use as a unique biomarker for GBM detection. The latest studies have however, reported that the PTMs found on circulating nucleosomes may have cancer specificity and are thus be investigated as biomarkers[88].
Enzymes that regulate PTMs are often dysregulated in GBM. Histone deacetylases and demethylases like lysine-specific histone demethylase 1, can alter the epigenetic status of brain cells, influencing cancer development and progression [88,89]. In paediatric high-grade gliomas, recurrent histone mutations are common. The H3K27M mutation, a lysine-to-methionine substitution at position 27 of H3.1 or H3.3 histone genes, is associated with childhood diffuse intrinsic pontine glioma, disrupting polycomb repressive complex 2 function, leading to global H3K27 methylation loss and altered gene expression[90]. H3G34R/V mutations, involving glycine 34 substitutions in H3.3 are seen in gliomas of the cerebral hemispheres in both children and adults causing redistribution of the activating H3K36 methylation mark and transcriptional dysregulation[88,90].
Another epigenetic marker dysregulated in GBM is histone 3 lysine 4 (H3K4), which is decreased in severe GBM cases, leading to gene repression[89]. Acetylation of histone 3 at lysine 18 (H3K18Ac), is another common PTMs with dysregulated expression seen in a number of cancers including GBM, where lower levels identify better prognosis for the patient[91]. Accordingly, a profile of PTMs of circulating nucleosome linked histone modifications has potential to be studied in GBM patients which would be in addition to MRI, a guide to diagnosing and monitoring tumour progression in GBM patients[9].
Nucleosomes are generally less useful for cancer diagnosis due to elevated levels in benign diseases. However, in gastrointestinal cancers, nucleosome levels correlate with tumour stage and metastasis. Nucleosomes are more valuable for therapy monitoring, as their levels decrease in remission and increase with disease progression during chemotherapy and radiotherapy, a pattern observed in lung, pancreatic, colorectal cancers, and haematological malignancies [92,93].In GBM, circulating nucleosomes in serum and CSF have shown potential for diagnosis and prognosis. Patients developing cerebral oedema post-surgery exhibited a ~200-fold rise in CSF nucleosomes. Monitoring these levels may help detect complications and track tumour progression and treatment response[94]. However, further research is needed to refine their specificity and clinical application, especially in distinguishing cancer from other conditions.
Collectively, circulating nucleosomes and their PTMs present a promising avenue for GBM detection and monitoring. While total nucleosome levels in plasma and CSF lack disease specificity, unique histone modifications, such as H3K27M and H3K18Ac, provide greater diagnostic potential. Emerging technologies, including immunoassays and single-molecule detection methods, have enabled precise profiling of nucleosome-bound epigenetic markers, distinguishing GBM patients from healthy individuals. These findings suggest that nucleosome-based liquid biopsies could serve as a valuable adjunct to MRI and conventional diagnostics, particularly in monitoring disease status and therapy response. Additionally, nucleosome levels may help identify postoperative complications, such as cerebral oedema, offering further insight into tumour progression and treatment outcomes. However, further validation in larger clinical studies is necessary to refine their specificity and clinical application, especially in differentiating cancer from other conditions.
2.4. Circulating Tumour microRNA
Circulating tumour microRNA (miRNA), is the most common small non-coding RNA, typically 21-23 nucleotides long, and regulates up to 30% of protein-coding genes[96,97]. It plays a key role in cancer by downregulating tumour suppressor genes. miRNAs are highly stable and detectable in bodily fluids such as blood, urine, and CSF, and are also present in extracellular vesicles (EVs)[12,98,99,100].
Altered expression of specific miRNAs have been found to distinguish between GBM patients and lower-grade glioma patients (Table 4). For instance, downregulation of miR-125b, miR-16, miR-497, miR-128, miR-342-3p, miR-205 and upregulation of miR-210, miR-182, miR-20a-5p, miR-454-3p, miR-106a-5p, miR-181b-5p have been linked to GBM pathophysiology[101,102,103,104,105,106,107,108]. Thus, miRNA dysregulation in GBM involves both upregulation of oncogenic miRNAs (“oncomirs”) and downregulation of tumour-suppressive miRNAs, each playing distinct roles in tumour biology.
Most miRNAs which show an increase in their expression level in GBM are oncogenic and are known as “oncomirs”[109]. Oncomirs usually promote tumour growth by inhibiting tumour suppressor genes and genes linked to cell differentiation or apoptosis. The first oncomir which showed significantly increased levels in GBM tissue samples compared to normal tissue was miR21. It can target several tumour suppressors such as PTEN[110]. The levels of miR-21 in GBM tissue and plasma samples was higher in grade II and III GBM samples compared to controls, yet there was no significant change between pre-surgery and post-surgery sample measures, while miR21 level was decreased after chemo irradiation therapy [101].
Several miRNAs function as tumour suppressors and are commonly downregulated in GBM patients[111]. Tumour-suppressive miRNA such as Let-7, which targets oncogenes like K-RAS and MYC inhibit GBM cell proliferation by interfering with histone methyltransferase EZH2[112,113,114]. miR-128 and miR-342-3p, significantly reduced in the plasma of GBM patients, have been shown to return to normal levels after surgery and chemo-radiation, indicating the patient’s response to treatment[102]. These miRNAs are glioma-specific, and their loss or downregulation may contribute to tumour progression including malignant transformation of meningioma and pituitary adenomas into GBM [115,116]. A meta-analysis demonstrated that serum miR-125b, markedly reduced in GBM patients, shows a 3.5-fold higher detection frequency compared to healthy controls, highlighting its potential as a screening biomarker[117]. Similarly, miR-205, another tumour suppressor, was significantly decreased in GBM patients, increased post-surgery, and declined again upon recurrence, suggesting its value as a dynamic biomarker[107]. Accordingly, the dysregulated expression of miRNA, shapes the prediction of cancer, early diagnosis, prognosis and histological classification[9,98]
Other studies have advocated that the BBB is not permeable for some miRNAs which increases the value of miRNA profiling of the CSF samples. As miR-10b is greatly overexpressed in GBM but absent in normal brain, it can thus be detected in CSF of majority of GBM patients (89%) but not in the CSF of healthy controls. Considering extracranial tissues express miR-10b, its absence in CSF of healthy controls designated that it may not pass the BBB under non-neoplastic conditions[98,118].
Several studies have identified specific miRNAs that are dysregulated in GBM patients, with both upregulated and downregulated miRNAs linked to tumour progression and prognosis. miRNAs such as miR-21, miR-128, and miR-342-3p have been highlighted for their potential as biomarkers for early GBM detection, offering high sensitivity and specificity, particularly in blood and urinary samples. These clinical data signify that the dysregulation of miRNAs, whether through upregulation or downregulation, can be used to diagnose or monitor GBM patients. The circulation biomarker: miRNA should serve as a non-invasive method for screening patients suspecting GBM. Though the results are thought-provoking, some limitations do exist, like the small size of regimens and the lack of a standard method for blood collection, RNA extraction and sequencing. Of note is that specificity of miRNA detection may be lower than that of ctDNA[9], but it may be considered complementary to ctDNA detection. Despite challenges, miRNA-based liquid biopsy offers a promising, less-invasive alternative to traditional tissue biopsies for GBM diagnosis, prognosis, and treatment monitoring.
2.5. Extracellular Vesicles
Extracellular vesicles (EVs), including exosomes and microvesicles (MVs) are small membrane enclosed particles released by normal and tumour cells. They serve as carriers of macromolecules in liquid biopsies of brain tumour patients[18]. EVs are heterogeneous in size, quantity and origin of molecular content and biological activity. Two main groups of EVs exist, which differ in origin and size. MVs (50 to 500 nm) bud from the cell membrane, while exosomes (50 to 150 nm) originate from the endosomal system[125,126]. EVs protect their cargo; mRNAs, miRNAs, lipids or proteins, which are specific to the cell origin[127] from enzymatic degradation via their lipid bilayer, enabling them to cross the BBB[125].
The significance of EVs is emphasised by the fact that their transcriptomic and proteomic profile is specific to the cell of origin and can differ in response to diverse stimuli[10]. Their imperative role in intercellular communications allows them to alter the phenotype of recipient cells by transferring genetic information and proteins[12,128]. These molecules often reflect malignant processes, underlining their importance as liquid biopsy tools in cancer, including GBM[127]. EV-mediated crosstalk has been demonstrated between GBM and its tumour microenvironment, fostering tumour progression[129]. Exosomes from hypoxic GBM cells overexpress VEGF-A, disrupting the BBB by downregulating occludin and claudin-5[130].
EV isolation typically involves immunoaffinity capture or differential centrifugation gradients[9]. Exosomes are characterised by transmission electron microscopy (TEM), nanoparticle-tracking analysis (NTA) and surface markers such as CD63, CD81, CD9, CD37, CD53, CD82, ICAM-1 and integrins, detectable by flow cytometry or Western blot[127].
Exosome-based liquid biopsies show promise in cancer diagnostics, with tests like ExoDx™ Lung (ALK) and ExoDx Prostate IntelliScore (EPI) already approved. For GBM, research is ongoing[131]. Emerging biomarkers include exosomal circular RNAs (circRNAs), EGFRvIII RNA for monitoring CAR-T cell therapy response, and MGMT methylation for predicting treatment outcomes. Despite promising findings (see Table 5), no exosome-based GBM test has yet reached clinical practice, pending further validation in larger cohorts [131,132,133,134,135].
Studies indicate that EVs from GBM stem cells exhibit increased adhesion-related proteins after TMZ treatment, potentially promoting tumour progression[136]. TMZ resistance may be associated with EVs carrying MGMT mRNA, as a potential marker of resistance, alongside adhesion proteins (e.g., TGM2, CD44 and CD133), and stemness markers like NESTIN[137,138,139]. These EV-associated molecules may serve as biomarkers to monitor drug resistance and treatment failure[10].
Overall, EVs, particularly exosomes, are promising biomarkers for GBM detection, monitoring, and prognosis. Exosomal RNA, proteins, and lipids include disease-specific molecular cargo, such as EGFRvIII RNA and syndecan-1 with high diagnostic potential. EV-derived molecular cargo may reflect or potentially even contribute to treatment resistance and tumour progression, though their mechanistic role remains under investigation. Although exosome-based liquid biopsy presents a minimally invasive alternative to traditional tissue biopsies, larger patient cohorts need to be analysed and standardised isolation and characterisation techniques are needed to validate their utility in GBM. Despite these limitations, ongoing research suggests that EV-based liquid biopsies could revolutionise GBM management, offering real-time insights into disease progression and therapeutic response.
3. Application of Machine Learning and Artificial Intelligence
The diagnosis of glioblastoma using signals derived from liquid biopsy samples remains particularly challenging, traditional statistical approaches frequently fall short in detecting the nonlinear patterns and subtle molecular variations that characterize GBM biology, increasing the risk of misdiagnosis or missed detection when relying solely on liquid biopsy data. To address these challenges, artificial intelligence (AI) and machine learning (ML) approaches are increasingly integrated into GBM liquid biopsy workflows. Novel ML algorithms can extract meaningful insights from noisy and heterogeneous datasets, enabling earlier and more accurate detection of GBM tumours, even when biomarker levels are extremely low in blood or CSF. Various liquid biopsy-based signatures have shown potential for tumour detection, including plasma denaturation profiles, CSF proteomic signatures, and serum miRNA signatures120, 160. In omics analysis for GBM, ML techniques uncover complex molecular signatures linked to tumour subtype classification, disease progression, and therapeutic response[163,164]. This allows the development of robust predictive models that outperform traditional biomarker-based approaches, advancing precision diagnostics and personalized treatment strategies for glioblastoma patients. Once trained, these models can be applied in clinical practice[163,164].
Fragmentation patterns and personalised cfDNA sequencing in urine and plasma have differentiated glioma patients from healthy individuals[165]. Tumour-educated platelets (TEPs), analysed with swarm algorithms, have identified spliced RNA biomarkers to distinguish false positives from true disease progression[166].
ML has also identified tumour-specific DNA methylation markers in blood and tumour-associated MRI features[167,168]. In several studies, ML algorithm identified MRI based tumour-associated features, while serum spectroscopy combined with MRI detected spectra variations between healthy and tumour-affected patients. Additionally, ML has been applied to investigate the relationship between brain tumour volume and liquid biopsy test performance. In a cohort of 177 patients (90 patients with high-grade glioma (GBM or anaplastic astrocytoma), or low-grade glioma (astrocytoma, oligoastrocytoma and oligodendroglioma) spectroscopic liquid biopsy approach detected small and low-grade gliomas, supporting early diagnosis[169,170].
AI enhances liquid biopsy applications and diagnostic performance by enabling rapid analysis of cancer-related circulating proteins and nucleic acids and enhancing imaging from subjective interpretation to quantitative analysis.[171,172,173] Multimodal AI models integrate proteomic and genomic data from liquid biopsies and quantitative imaging to create a more comprehensive diagnostic pathway[174,175]. This helps distinguish benign from malignant lesions, reduces unnecessary follow-ups and guides therapeutic decisions.
Despite its potential, the application of AI and ML in liquid biopsy and cancer diagnostics faces several challenges. One major hurdle is the need for large, high-quality datasets to train robust models, as variability in sample collection, processing, and sequencing methods introduces bias[176]. Additionally, the interpretability of AI-driven models remains a concern, as complex algorithms often function as "black boxes," making it difficult for clinicians to understand the reasoning behind predictions[164,174]. Regulatory approval and clinical validation also pose significant barriers, requiring extensive trials to ensure reliability and accuracy. Finally, integrating AI into existing healthcare systems demands substantial computational resources, infrastructure, and clinician training, which may limit widespread adoption[168,174,176,177,178].
4. Conclusions
In the coming years, clinical studies are expected to clarify which liquid biopsy techniques provide the most value for diagnosing, prognosing, and treating primary brain tumours, potentially surpassing traditional imaging and tissue-based methods. GBM liquid biopsy research is rapidly gaining traction. Analysis of body fluids, such as blood, or CSF, from GBM patients reveal various tumour-derived components, including CTCs, ctDNA, miRNAs, EVs, nucleosomes, and metabolites. These biomarkers may serve as alternatives to tissue samples and support genotype-guided therapies and personalised GBM management. They also show promise for monitoring tumour progression, treatment response, and guiding therapeutic choices. However, accessing tumour-derived material is challenging due to the BBB, which limits the release of biomarkers into the bloodstream—currently the most studied liquid biopsy. Consequently, tumour-derived nucleic acids in serum or plasma often occur at low levels, hindering routine clinical use. Improving sensitivity will require optimising sample volumes, technological platforms, and the use of artificial intelligence. Combining the measurement of multiple different liquid biopsy-based markers from the same biopsy will likely improve overall sensitivity of single modality testing but this idea needs to be further validated in the future.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This review received no external funding.
.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lah, T. T.; Novak, M.; Breznik, B. Brain malignancies: Glioblastoma and brain metastases. Semin Cancer Biol 2020, 60, 262–273. [Google Scholar] [CrossRef]
- Ostrom, Q. T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J. S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol 2018, 20 (suppl_4), iv1–iv86. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M. E.; Mason, W. P.; van den Bent, M. J.; Taphoorn, M. J.; Janzer, R. C.; Ludwin, S. K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Tan, A. C.; Ashley, D. M.; López, G. Y.; Malinzak, M.; Friedman, H. S.; Khasraw, M. Management of glioblastoma: State of the art and future directions. CA Cancer J Clin 2020, 70, 299–312. [Google Scholar] [CrossRef]
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol 2015, 129, 829–848. [Google Scholar] [CrossRef]
- Clarke, J. L.; Chang, S. M. Neuroimaging: diagnosis and response assessment in glioblastoma. Cancer J 2012, 18, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Tanaka, R.; Takeda, N. Magnetic resonance imaging and histopathology of cerebral gliomas. Neuroradiology 1992, 34, 463–469. [Google Scholar] [CrossRef]
- Delgado-López, P. D.; Riñones-Mena, E.; Corrales-García, E. M. Treatment-related changes in glioblastoma: a review on the controversies in response assessment criteria and the concepts of true progression, pseudoprogression, pseudoresponse and radionecrosis. Clin Transl Oncol 2018, 20, 939–953. [Google Scholar] [CrossRef]
- Ronvaux, L.; Riva, M.; Coosemans, A.; Herzog, M.; Rommelaere, G.; Donis, N.; D'Hondt, L.; Douxfils, J. Liquid Biopsy in Glioblastoma. Cancers (Basel) 2022, 14. [Google Scholar] [CrossRef]
- Saenz-Antoñanzas, A.; Auzmendi-Iriarte, J.; Carrasco-Garcia, E.; Moreno-Cugnon, L.; Ruiz, I.; Villanua, J.; Egaña, L.; Otaegui, D.; Samprón, N.; Matheu, A. Liquid Biopsy in Glioblastoma: Opportunities, Applications and Challenges. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Sareen, H.; Garrett, C.; Lynch, D.; Powter, B.; Brungs, D.; Cooper, A.; Po, J.; Koh, E. S.; Vessey, J. Y.; McKechnie, S.; et al. The Role of Liquid Biopsies in Detecting Molecular Tumor Biomarkers in Brain Cancer Patients. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Müller Bark, J.; Kulasinghe, A.; Chua, B.; Day, B. W.; Punyadeera, C. Circulating biomarkers in patients with glioblastoma. Br J Cancer 2020, 122, 295–305. [Google Scholar] [CrossRef]
- Qazi, M. A.; Salim, S. K.; Brown, K. R.; Mikolajewicz, N.; Savage, N.; Han, H.; Subapanditha, M. K.; Bakhshinyan, D.; Nixon, A.; Vora, P.; et al. Characterization of the minimal residual disease state reveals distinct evolutionary trajectories of human glioblastoma. Cell Rep 2022, 40, 111420. [Google Scholar] [CrossRef]
- Strati, A.; Markou, A.; Kyriakopoulou, E.; Lianidou, E. Detection and Molecular Characterization of Circulating Tumour Cells: Challenges for the Clinical Setting. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Lion, T. Minimal residual disease. Curr Opin Hematol 1999, 6, 406–411. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T. N. A.; Huang, P. S.; Chu, P. Y.; Hsieh, C. H.; Wu, M. H. Recent Progress in Enhanced Cancer Diagnosis, Prognosis, and Monitoring Using a Combined Analysis of the Number of Circulating Tumor Cells (CTCs) and Other Clinical Parameters. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M. M.; Ramani, V. C.; Jeffrey, S. S. Circulating tumor cell technologies. Mol Oncol 2016, 10, 374–394. [Google Scholar] [CrossRef]
- Shankar, G. M.; Balaj, L.; Stott, S. L.; Nahed, B.; Carter, B. S. Liquid biopsy for brain tumors. Expert Rev Mol Diagn 2017, 17, 943–947. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat Rev Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Allard, W. J.; Matera, J.; Miller, M. C.; Repollet, M.; Connelly, M. C.; Rao, C.; Tibbe, A. G.; Uhr, J. W.; Terstappen, L. W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Lynch, D. , Powter, B., Po, J. W., Cooper, A., Garrett, C., Koh, E.-S., Sheridan, M., van Gelder, J., Darwish, B., Mckechnie, S., Bazina, R., Jaeger, M., Roberts, T. L., de Souza, P., & Becker, T. M.. Isolation of Circulating Tumor Cells from Glioblastoma Patients by Direct Immunomagnetic Targeting. Applied Sciences 2020, 10. [Google Scholar]
- Sullivan, J. P.; Nahed, B. V.; Madden, M. W.; Oliveira, S. M.; Springer, S.; Bhere, D.; Chi, A. S.; Wakimoto, H.; Rothenberg, S. M.; Sequist, L. V.; et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov 2014, 4, 1299–1309. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Peng, Z.; Lin, S. C.; Geri, M.; Li, S.; Li, P.; Chen, Y.; Dao, M.; Suresh, S.; Huang, T. J. Cell separation using tilted-angle standing surface acoustic waves. Proc Natl Acad Sci U S A 2014, 111, 12992–12997. [Google Scholar] [CrossRef] [PubMed]
- Kulasinghe, A.; Wu, H.; Punyadeera, C.; Warkiani, M. E. The Use of Microfluidic Technology for Cancer Applications and Liquid Biopsy. Micromachines (Basel) 2018, 9. [Google Scholar] [CrossRef]
- Kolostova, K.; Pospisilova, E.; Pavlickova, V.; Bartos, R.; Sames, M.; Pawlak, I.; Bobek, V. Next generation sequencing of glioblastoma circulating tumor cells: non-invasive solution for disease monitoring. Am J Transl Res 2021, 13, 4489–4499. [Google Scholar]
- Valerius, A. R.; Webb, M. J.; Hammad, N.; Sener, U.; Malani, R. Cerebrospinal Fluid Liquid Biopsies in the Evaluation of Adult Gliomas. Curr Oncol Rep 2024, 26, 377–390. [Google Scholar] [CrossRef]
- Mathur, R.; Wang, Q.; Schupp, P. G.; Nikolic, A.; Hilz, S.; Hong, C.; Grishanina, N. R.; Kwok, D.; Stevers, N. O.; Jin, Q.; et al. Glioblastoma evolution and heterogeneity from a 3D whole-tumor perspective. Cell 2024, 187, 446–463.e416. [Google Scholar] [CrossRef]
- Berezovsky, A. D.; Poisson, L. M.; Cherba, D.; Webb, C. P.; Transou, A. D.; Lemke, N. W.; Hong, X.; Hasselbach, L. A.; Irtenkauf, S. M.; Mikkelsen, T.; et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia 2014, 16, 193–206, 206.e119-125. [Google Scholar] [CrossRef]
- Müller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A.; Matschke, J.; Langer-Freitag, S.; Gasch, C.; Stoupiec, M.; et al. Hematogenous dissemination of glioblastoma multiforme. Sci Transl Med 2014, 6, 247ra101. [Google Scholar] [CrossRef]
- Macarthur, K. M.; Kao, G. D.; Chandrasekaran, S.; Alonso-Basanta, M.; Chapman, C.; Lustig, R. A.; Wileyto, E. P.; Hahn, S. M.; Dorsey, J. F. Detection of brain tumor cells in the peripheral blood by a telomerase promoter-based assay. Cancer Res 2014, 74, 2152–2159. [Google Scholar] [CrossRef]
- Gao, F.; Cui, Y.; Jiang, H.; Sui, D.; Wang, Y.; Jiang, Z.; Zhao, J.; Lin, S. Circulating tumor cell is a common property of brain glioma and promotes the monitoring system. Oncotarget 2016, 7, 71330–71340. [Google Scholar] [CrossRef]
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B. M.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of circulating tumour cell clusters in human glioblastoma. Br J Cancer 2018, 119, 487–491. [Google Scholar] [CrossRef]
- Müller Bark, J.; Kulasinghe, A.; Hartel, G.; Leo, P.; Warkiani, M. E.; Jeffree, R. L.; Chua, B.; Day, B. W.; Punyadeera, C. Isolation of Circulating Tumour Cells in Patients With Glioblastoma Using Spiral Microfluidic Technology - A Pilot Study. Front Oncol 2021, 11, 681130. [Google Scholar] [CrossRef]
- Gao, F.; Zhao, W.; Li, M.; Ren, X.; Jiang, H.; Cui, Y.; Lin, S. Role of circulating tumor cell detection in differentiating tumor recurrence from treatment necrosis of brain gliomas. Biosci Trends 2021, 15, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Parker, N. R.; Khong, P.; Parkinson, J. F.; Howell, V. M.; Wheeler, H. R. Molecular heterogeneity in glioblastoma: potential clinical implications. Front Oncol 2015, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Menyailo, M. E.; Tretyakova, M. S.; Denisov, E. V. Heterogeneity of Circulating Tumor Cells in Breast Cancer: Identifying Metastatic Seeds. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Zhuang, R.; Long, M.; Pavlovic, M.; Kang, Y.; Ilyas, A.; Asghar, W. Circulating tumor cell isolation, culture, and downstream molecular analysis. Biotechnol Adv 2018, 36, 1063–1078. [Google Scholar] [CrossRef]
- Batth, I. S.; Mitra, A.; Rood, S.; Kopetz, S.; Menter, D.; Li, S. CTC analysis: an update on technological progress. Transl Res 2019, 212, 14–25. [Google Scholar] [CrossRef]
- Ju, S.; Chen, C.; Zhang, J.; Xu, L.; Zhang, X.; Li, Z.; Chen, Y.; Zhou, J.; Ji, F.; Wang, L. Detection of circulating tumor cells: opportunities and challenges. Biomark Res 2022, 10, 58. [Google Scholar] [CrossRef]
- Jan, Y. J.; Chen, J. F.; Zhu, Y.; Lu, Y. T.; Chen, S. H.; Chung, H.; Smalley, M.; Huang, Y. W.; Dong, J.; Chen, L. C.; et al. NanoVelcro rare-cell assays for detection and characterization of circulating tumor cells. Adv Drug Deliv Rev 2018, 125, 78–93. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F. O.; Hesch, R. D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res 2001, 61, 1659–1665. [Google Scholar]
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Tosoni, A.; Lodi, R.; Brandes, A. A. Liquid Biopsy in Glioblastoma Management: From Current Research to Future Perspectives. Oncologist 2021, 26, 865–878. [Google Scholar] [CrossRef]
- Murtaza, M.; Dawson, S. J.; Tsui, D. W.; Gale, D.; Forshew, T.; Piskorz, A. M.; Parkinson, C.; Chin, S. F.; Kingsbury, Z.; Wong, A. S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Leary, R. J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J. D.; Craig, D.; O'Shaughnessy, J.; Kinzler, K. W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med 2012, 4, 162ra154. [Google Scholar] [CrossRef]
- Lebofsky, R.; Decraene, C.; Bernard, V.; Kamal, M.; Blin, A.; Leroy, Q.; Rio Frio, T.; Pierron, G.; Callens, C.; Bieche, I.; et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol Oncol 2015, 9, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Sasmita, A. O.; Wong, Y. P.; Ling, A. P. K. Biomarkers and therapeutic advances in glioblastoma multiforme. Asia Pac J Clin Oncol 2018, 14, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Birkó, Z.; Nagy, B.; Klekner, Á.; Virga, J. Novel Molecular Markers in Glioblastoma-Benefits of Liquid Biopsy. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Wadden, J.; Ravi, K.; John, V.; Babila, C. M.; Koschmann, C. Cell-Free Tumor DNA (cf-tDNA) Liquid Biopsy: Current Methods and Use in Brain Tumor Immunotherapy. Front Immunol 2022, 13, 882452. [Google Scholar] [CrossRef] [PubMed]
- Parker, A. L.; Kavallaris, M.; McCarroll, J. A. Microtubules and their role in cellular stress in cancer. Front Oncol 2014, 4, 153. [Google Scholar] [CrossRef]
- Schwaederle, M.; Husain, H.; Fanta, P. T.; Piccioni, D. E.; Kesari, S.; Schwab, R. B.; Banks, K. C.; Lanman, R. B.; Talasaz, A.; Parker, B. A.; et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget 2016, 7, 9707–9717. [Google Scholar] [CrossRef]
- Piccioni, D. E.; Achrol, A. S.; Kiedrowski, L. A.; Banks, K. C.; Boucher, N.; Barkhoudarian, G.; Kelly, D. F.; Juarez, T.; Lanman, R. B.; Raymond, V. M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol 2019, 8, Cns34. [Google Scholar] [CrossRef]
- Batool, S. M.; Escobedo, A. K.; Hsia, T.; Ekanayake, E.; Khanna, S. K.; Gamblin, A. S.; Zheng, H.; Skog, J.; Miller, J. J.; Stemmer-Rachamimov, A. O.; et al. Clinical utility of a blood based assay for the detection of IDH1.R132H-mutant gliomas. Nat Commun 2024, 15, 7074. [Google Scholar] [CrossRef] [PubMed]
- Stoyanov, G. S.; Lyutfi, E.; Georgieva, R.; Georgiev, R.; Dzhenkov, D. L.; Petkova, L.; Ivanov, B. D.; Kaprelyan, A.; Ghenev, P. Reclassification of Glioblastoma Multiforme According to the 2021 World Health Organization Classification of Central Nervous System Tumors: A Single Institution Report and Practical Significance. Cureus 2022, 14, e21822. [Google Scholar] [CrossRef] [PubMed]
- Salkeni, M. A.; Zarzour, A.; Ansay, T. Y.; McPherson, C. M.; Warnick, R. E.; Rixe, O.; Bahassi el, M. Detection of EGFRvIII mutant DNA in the peripheral blood of brain tumor patients. J Neurooncol 2013, 115, 27–35. [Google Scholar] [CrossRef]
- Zill, O. A.; Banks, K. C.; Fairclough, S. R.; Mortimer, S. A.; Vowles, J. V.; Mokhtari, R.; Gandara, D. R.; Mack, P. C.; Odegaard, J. I.; Nagy, R. J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin Cancer Res 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [PubMed]
- Miller, A. M.; Shah, R. H.; Pentsova, E. I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S. A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef]
- Liu, A. P. Y.; Smith, K. S.; Kumar, R.; Robinson, G. W.; Northcott, P. A. Low-coverage whole-genome sequencing of cerebrospinal-fluid-derived cell-free DNA in brain tumor patients. STAR Protoc 2022, 3, 101292. [Google Scholar] [CrossRef]
- Bettegowda, C.; Sausen, M.; Leary, R. J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B. R.; Wang, H.; Luber, B.; Alani, R. M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med 2014, 6, 224ra224. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M. A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S. A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Yan, Y. Y.; Guo, Q. R.; Wang, F. H.; Adhikari, R.; Zhu, Z. Y.; Zhang, H. Y.; Zhou, W. M.; Yu, H.; Li, J. Q.; Zhang, J. Y. Cell-Free DNA: Hope and Potential Application in Cancer. Front Cell Dev Biol 2021, 9, 639233. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, W.; Wang, Y.; Guo, Y.; Cong, Z.; Du, F.; Song, B. MGMT promoter methylation in serum and cerebrospinal fluid as a tumor-specific biomarker of glioma. Biomed Rep 2015, 3, 543–548. [Google Scholar] [CrossRef]
- Juratli, T. A.; Stasik, S.; Zolal, A.; Schuster, C.; Richter, S.; Daubner, D.; Juratli, M. A.; Thowe, R.; Hennig, S.; Makina, M.; et al. TERT Promoter Mutation Detection in Cell-Free Tumor-Derived DNA in Patients with IDH Wild-Type Glioblastomas: A Pilot Prospective Study. Clin Cancer Res 2018, 24, 5282–5291. [Google Scholar] [CrossRef]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K. W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G. L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc Natl Acad Sci U S A 2015, 112, 9704–9709. [Google Scholar] [CrossRef]
- Jones, J.; Nguyen, H.; Drummond, K.; Morokoff, A. Circulating Biomarkers for Glioma: A Review. Neurosurgery 2021, 88, E221–e230. [Google Scholar] [CrossRef]
- Mair, R.; Mouliere, F.; Smith, C. G.; Chandrananda, D.; Gale, D.; Marass, F.; Tsui, D. W. Y.; Massie, C. E.; Wright, A. J.; Watts, C.; et al. Measurement of Plasma Cell-Free Mitochondrial Tumor DNA Improves Detection of Glioblastoma in Patient-Derived Orthotopic Xenograft Models. Cancer Res 2019, 79, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Krauze, A. V.; Won, M.; Graves, C.; Corn, B. W.; Muanza, T. M.; Howard, S. P.; Mahadevan, A.; Schultz, C. J.; Haas, M. L.; Mehta, M. P.; et al. Predictive value of tumor recurrence using urinary vascular endothelial factor levels in patients receiving radiation therapy for Glioblastoma Multiforme (GBM). Biomark Res 2013, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Rieger, J.; Bähr, O.; Maurer, G. D.; Hattingen, E.; Franz, K.; Brucker, D.; Walenta, S.; Kämmerer, U.; Coy, J. F.; Weller, M.; et al. ERGO: a pilot study of ketogenic diet in recurrent glioblastoma. Int J Oncol 2014, 44, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Ishikawa, E.; Nakai, K.; Matsuda, M.; Masumoto, T.; Yamamoto, T.; Matsumura, A. Bevacizumab in Japanese patients with malignant glioma: from basic research to clinical trial. Onco Targets Ther 2014, 7, 1551–1562. [Google Scholar] [CrossRef]
- Constâncio, V.; Nunes, S. P.; Henrique, R.; Jerónimo, C. DNA Methylation-Based Testing in Liquid Biopsies as Detection and Prognostic Biomarkers for the Four Major Cancer Types. Cells 2020, 9. [Google Scholar] [CrossRef]
- Lavon, I.; Refael, M.; Zelikovitch, B.; Shalom, E.; Siegal, T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro Oncol 2010, 12, 173–180. [Google Scholar] [CrossRef]
- Watson, N.; Grieu, F.; Morris, M.; Harvey, J.; Stewart, C.; Schofield, L.; Goldblatt, J.; Iacopetta, B. Heterogeneous staining for mismatch repair proteins during population-based prescreening for hereditary nonpolyposis colorectal cancer. J Mol Diagn 2007, 9, 472–478. [Google Scholar] [CrossRef] [PubMed]
- McCord, M.; Steffens, A.; Javier, R.; Kam, K. L.; McCortney, K.; Horbinski, C. The efficacy of DNA mismatch repair enzyme immunohistochemistry as a screening test for hypermutated gliomas. Acta Neuropathol Commun 2020, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Senhaji, N.; Squalli Houssaini, A.; Lamrabet, S.; Louati, S.; Bennis, S. Molecular and Circulating Biomarkers in Patients with Glioblastoma. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Mair, R.; Chandrananda, D.; Marass, F.; Smith, C. G.; Su, J.; Morris, J.; Watts, C.; Brindle, K. M.; Rosenfeld, N. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol Med 2018, 10. [Google Scholar] [CrossRef]
- Martínez-Ricarte, F.; Mayor, R.; Martínez-Sáez, E.; Rubio-Pérez, C.; Pineda, E.; Cordero, E.; Cicuéndez, M.; Poca, M. A.; López-Bigas, N.; Ramon, Y. C. S.; et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clin Cancer Res 2018, 24, 2812–2819. [Google Scholar] [CrossRef]
- Wu, J.; Liu, Z.; Huang, T.; Wang, Y.; Song, M. M.; Song, T.; Long, G.; Zhang, X.; Li, X.; Zhang, L. Cerebrospinal fluid circulating tumor DNA depicts profiling of brain metastasis in NSCLC. Mol Oncol 2023, 17, 810–824. [Google Scholar] [CrossRef]
- Hickman, R. A.; Miller, A. M.; Holle, B. M.; Jee, J.; Liu, S. Y.; Ross, D.; Yu, H.; Riely, G. J.; Ombres, C.; Gewirtz, A. N.; et al. Real-world experience with circulating tumor DNA in cerebrospinal fluid from patients with central nervous system tumors. Acta Neuropathol Commun 2024, 12, 151. [Google Scholar] [CrossRef]
- Guo, W.; Jin, L.; Liang, J.; Lin, G.; Zheng, J.; Zhou, D.; Zhan, S.; Sun, H.; Jiang, X. Detection of mutation profiles and tumor mutation burden of cerebrospinal fluid circulating DNA by a cancer genomic panel sequencing in glioma patients. Clin Chim Acta 2022, 534, 81–92. [Google Scholar] [CrossRef]
- Bruzek, A. K.; Ravi, K.; Muruganand, A.; Wadden, J.; Babila, C. M.; Cantor, E.; Tunkle, L.; Wierzbicki, K.; Stallard, S.; Dickson, R. P.; et al. Electronic DNA Analysis of CSF Cell-free Tumor DNA to Quantify Multi-gene Molecular Response in Pediatric High-grade Glioma. Clin Cancer Res 2020, 26, 6266–6276. [Google Scholar] [CrossRef]
- Kornberg, R. D. Chromatin structure: a repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Cutter, A. R.; Hayes, J. J. A brief review of nucleosome structure. FEBS Lett 2015, 589, 2914–2922. [Google Scholar] [CrossRef]
- Peterson, C. L.; Laniel, M. A. Histones and histone modifications. Curr Biol 2004, 14, R546–551. [Google Scholar] [CrossRef]
- Kim, Y. Z. Altered histone modifications in gliomas. Brain Tumor Res Treat 2014, 2, 7–21. [Google Scholar] [CrossRef]
- Was, H.; Krol, S. K.; Rotili, D.; Mai, A.; Wojtas, B.; Kaminska, B.; Maleszewska, M. Histone deacetylase inhibitors exert anti-tumor effects on human adherent and stem-like glioma cells. Clin Epigenetics 2019, 11, 11. [Google Scholar] [CrossRef]
- McAnena, P.; Brown, J. A.; Kerin, M. J. Circulating Nucleosomes and Nucleosome Modifications as Biomarkers in Cancer. Cancers (Basel) 2017, 9. [Google Scholar] [CrossRef]
- Kunadis, E.; Lakiotaki, E.; Korkolopoulou, P.; Piperi, C. Targeting post-translational histone modifying enzymes in glioblastoma. Pharmacol Ther 2021, 220, 107721. [Google Scholar] [CrossRef]
- Holdenrieder, S.; Stieber, P.; Bodenmüller, H.; Fertig, G.; Fürst, H.; Schmeller, N.; Untch, M.; Seidel, D. Nucleosomes in serum as a marker for cell death. Clin Chem Lab Med 2001, 39, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Romani, M.; Pistillo, M. P.; Banelli, B. Epigenetic Targeting of Glioblastoma. Front Oncol 2018, 8, 448. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Huang, B. S.; Xiong, Y. Y.; Yang, L. J.; Wu, L. X. 4,5-Dimethoxycanthin-6-one is a novel LSD1 inhibitor that inhibits proliferation of glioblastoma cells and induces apoptosis and pyroptosis. Cancer Cell Int 2022, 22, 32. [Google Scholar] [CrossRef] [PubMed]
- Lowe, B. R.; Maxham, L. A.; Hamey, J. J.; Wilkins, M. R.; Partridge, J. F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Liu, B. L.; Cheng, J. X.; Zhang, X.; Wang, R.; Zhang, W.; Lin, H.; Xiao, X.; Cai, S.; Chen, X. Y.; Cheng, H. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol Biomarkers Prev 2010, 19, 2888–2896. [Google Scholar] [CrossRef]
- Gahan, P. B. Circulating nucleic acids in plasma and serum: diagnosis and prognosis in cancer. Epma j 2010, 1, 503–512. [Google Scholar] [CrossRef]
- Holdenrieder, S. , Spuler, A., Tischinger, M., Nagel, D., Stieber, P. Presence of Nucleosomes in Cerebrospinal Fluid of Glioblastoma Patients – Potential for Therapy Monitoring. In Springer Nature, Gahan; 2010.
- Andreas Spuler, M. T. , Dorothea Nagel and Petra Stieber Presence of Nucleosomes in Cerebrospinal Fluid of Glioblastoma Patients – Potential for Therapy Monitoring; 2010.
- Erez, N.; Furth, N.; Fedyuk, V.; Wadden, J.; Aittaleb, R.; Schwark, K.; Niculcea, M.; Miclea, M.; Mody, R.; Franson, A.; et al. Single-molecule systems for detection and monitoring of plasma circulating nucleosomes and oncoproteins in Diffuse Midline Glioma. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Lee, R. C.; Feinbaum, R. L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Felekkis, K.; Touvana, E.; Stefanou, C.; Deltas, C. microRNAs: a newly described class of encoded molecules that play a role in health and disease. Hippokratia 2010, 14, 236–240. [Google Scholar]
- Klekner, Á.; Szivos, L.; Virga, J.; Árkosy, P.; Bognár, L.; Birkó, Z.; Nagy, B. Significance of liquid biopsy in glioblastoma - A review. J Biotechnol 2019, 298, 82–87. [Google Scholar] [CrossRef]
- Ebrahimkhani, S.; Vafaee, F.; Hallal, S.; Wei, H.; Lee, M. Y. T.; Young, P. E.; Satgunaseelan, L.; Beadnall, H.; Barnett, M. H.; Shivalingam, B.; et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. NPJ Precis Oncol 2018, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Hallal, S.; Ebrahim Khani, S.; Wei, H.; Lee, M. Y. T.; Sim, H. W.; Sy, J.; Shivalingam, B.; Buckland, M. E.; Alexander-Kaufman, K. L. Deep Sequencing of Small RNAs from Neurosurgical Extracellular Vesicles Substantiates miR-486-3p as a Circulating Biomarker that Distinguishes Glioblastoma from Lower-Grade Astrocytoma Patients. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Wang, Q.; Li, P.; Li, A.; Jiang, W.; Wang, H.; Wang, J.; Xie, K. Plasma specific miRNAs as predictive biomarkers for diagnosis and prognosis of glioma. J Exp Clin Cancer Res 2012, 31, 97. [Google Scholar] [CrossRef]
- Sun, J.; Liao, K.; Wu, X.; Huang, J.; Zhang, S.; Lu, X. Serum microRNA-128 as a biomarker for diagnosis of glioma. Int J Clin Exp Med 2015, 8, 456–463. [Google Scholar]
- Lai, N. S.; Wu, D. G.; Fang, X. G.; Lin, Y. C.; Chen, S. S.; Li, Z. B.; Xu, S. S. Serum microRNA-210 as a potential noninvasive biomarker for the diagnosis and prognosis of glioma. Br J Cancer 2015, 112, 1241–1246. [Google Scholar] [CrossRef]
- Shao, N.; Wang, L.; Xue, L.; Wang, R.; Lan, Q. Plasma miR-454-3p as a potential prognostic indicator in human glioma. Neurol Sci 2015, 36, 309–313. [Google Scholar] [CrossRef]
- Regazzo, G.; Terrenato, I.; Spagnuolo, M.; Carosi, M.; Cognetti, G.; Cicchillitti, L.; Sperati, F.; Villani, V.; Carapella, C.; Piaggio, G.; et al. A restricted signature of serum miRNAs distinguishes glioblastoma from lower grade gliomas. J Exp Clin Cancer Res 2016, 35, 124. [Google Scholar] [CrossRef]
- Xiao, Y.; Zhang, L.; Song, Z.; Guo, C.; Zhu, J.; Li, Z.; Zhu, S. Potential Diagnostic and Prognostic Value of Plasma Circulating MicroRNA-182 in Human Glioma. Med Sci Monit 2016, 22, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lan, F.; Hu, M.; Pan, Q.; Wang, Q.; Wang, J. Downregulation of serum microRNA-205 as a potential diagnostic and prognostic biomarker for human glioma. J Neurosurg 2016, 124, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Zhi, F.; Shao, N.; Wang, R.; Deng, D.; Xue, L.; Wang, Q.; Zhang, Y.; Shi, Y.; Xia, X.; Wang, S.; et al. Identification of 9 serum microRNAs as potential noninvasive biomarkers of human astrocytoma. Neuro Oncol 2015, 17, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Chan, J. A.; Krichevsky, A. M.; Kosik, K. S. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Meng, F.; Henson, R.; Wehbe-Janek, H.; Ghoshal, K.; Jacob, S. T.; Patel, T. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E. A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B. L.; Mak, R. H.; Ferrando, A. A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Johnson, S. M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K. L.; Brown, D.; Slack, F. J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Sampson, V. B.; Rong, N. H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N. J.; Dunn, S. P.; Krueger, L. J. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res 2007, 67, 9762–9770. [Google Scholar] [CrossRef]
- Lee, S. T.; Chu, K.; Oh, H. J.; Im, W. S.; Lim, J. Y.; Kim, S. K.; Park, C. K.; Jung, K. H.; Lee, S. K.; Kim, M.; et al. Let-7 microRNA inhibits the proliferation of human glioblastoma cells. J Neurooncol 2011, 102, 19–24. [Google Scholar] [CrossRef]
- Papagiannakopoulos, T.; Friedmann-Morvinski, D.; Neveu, P.; Dugas, J. C.; Gill, R. M.; Huillard, E.; Liu, C.; Zong, H.; Rowitch, D. H.; Barres, B. A.; et al. Pro-neural miR-128 is a glioma tumor suppressor that targets mitogenic kinases. Oncogene 2012, 31, 1884–1895. [Google Scholar] [CrossRef]
- Shi, L.; Wan, Y.; Sun, G.; Gu, X.; Qian, C.; Yan, W.; Zhang, S.; Pan, T.; Wang, Z.; You, Y. Functional differences of miR-125b on the invasion of primary glioblastoma CD133-negative cells and CD133-positive cells. Neuromolecular Med 2012, 14, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Chen, D.; Lv, T.; Li, G.; Qu, S. Serum MicroRNA-125b as a Potential Biomarker for Glioma Diagnosis. Mol Neurobiol 2016, 53, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Teplyuk, N. M.; Mollenhauer, B.; Gabriely, G.; Giese, A.; Kim, E.; Smolsky, M.; Kim, R. Y.; Saria, M. G.; Pastorino, S.; Kesari, S.; et al. MicroRNAs in cerebrospinal fluid identify glioblastoma and metastatic brain cancers and reflect disease activity. Neuro Oncol 2012, 14, 689–700. [Google Scholar] [CrossRef] [PubMed]
- Swellam, M.; Ezz El Arab, L.; Al-Posttany, A. S.; S, B. S. Clinical impact of circulating oncogenic MiRNA-221 and MiRNA-222 in glioblastoma multiform. J Neurooncol 2019, 144, 545–551. [Google Scholar] [CrossRef]
- Morokoff, A.; Jones, J.; Nguyen, H.; Ma, C.; Lasocki, A.; Gaillard, F.; Bennett, I.; Luwor, R.; Stylli, S.; Paradiso, L.; et al. Serum microRNA is a biomarker for post-operative monitoring in glioma. J Neurooncol 2020, 149, 391–400. [Google Scholar] [CrossRef]
- Muhtadi, R.; Bernhardt, D.; Multhoff, G.; Hönikl, L.; Combs, S. E.; Krieg, S. M.; Gempt, J.; Meyer, B.; Barsegian, V.; Lindemann, M.; et al. Liquid Biopsy in Whole Blood for Identification of Gene Expression Patterns (mRNA and miRNA) Associated with Recurrence of Glioblastoma WHO CNS Grade 4. Cancers (Basel) 2024, 16. [Google Scholar] [CrossRef]
- Kitano, Y.; Aoki, K.; Ohka, F.; Yamazaki, S.; Motomura, K.; Tanahashi, K.; Hirano, M.; Naganawa, T.; Iida, M.; Shiraki, Y.; et al. Urinary MicroRNA-Based Diagnostic Model for Central Nervous System Tumors Using Nanowire Scaffolds. ACS Appl Mater Interfaces 2021, 13, 17316–17329. [Google Scholar] [CrossRef]
- Akers, J. C.; Hua, W.; Li, H.; Ramakrishnan, V.; Yang, Z.; Quan, K.; Zhu, W.; Li, J.; Figueroa, J.; Hirshman, B. R.; et al. A cerebrospinal fluid microRNA signature as biomarker for glioblastoma. Oncotarget 2017, 8, 68769–68779. [Google Scholar] [CrossRef]
- Drusco, A.; Bottoni, A.; Laganà, A.; Acunzo, M.; Fassan, M.; Cascione, L.; Antenucci, A.; Kumchala, P.; Vicentini, C.; Gardiman, M. P.; et al. A differentially expressed set of microRNAs in cerebro-spinal fluid (CSF) can diagnose CNS malignancies. Oncotarget 2015, 6, 20829–20839. [Google Scholar] [CrossRef]
- Cordonnier, M.; Chanteloup, G.; Isambert, N.; Seigneuric, R.; Fumoleau, P.; Garrido, C.; Gobbo, J. Exosomes in cancer theranostic: Diamonds in the rough. Cell Adh Migr 2017, 11, 151–163. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; D'Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D. W.; Simpson, R. J. Extracellular vesicles in cancer - implications for future improvements in cancer care. Nat Rev Clin Oncol 2018, 15, 617–638. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Kumari Singh, D.; Panda, S.; Shiras, A. Extracellular Vesicles As Modulators of Tumor Microenvironment and Disease Progression in Glioma. Front Oncol 2017, 7, 144. [Google Scholar] [CrossRef]
- Zhao, C.; Wang, H.; Xiong, C.; Liu, Y. Hypoxic glioblastoma release exosomal VEGF-A induce the permeability of blood-brain barrier. Biochem Biophys Res Commun 2018, 502, 324–331. [Google Scholar] [CrossRef]
- Yu, D.; Li, Y.; Wang, M.; Gu, J.; Xu, W.; Cai, H.; Fang, X.; Zhang, X. Exosomes as a new frontier of cancer liquid biopsy. Mol Cancer 2022, 21, 56. [Google Scholar] [CrossRef]
- Wu, X.; Shi, M.; Lian, Y.; Zhang, H. Exosomal circRNAs as promising liquid biopsy biomarkers for glioma. Front Immunol 2023, 14, 1039084. [Google Scholar] [CrossRef]
- Rosas-Alonso, R.; Colmenarejo-Fernández, J.; Pernía, O.; Burdiel, M.; Rodríguez-Antolín, C.; Losantos-García, I.; Rubio, T.; Moreno-Velasco, R.; Esteban-Rodríguez, I.; Martínez-Marín, V.; et al. Evaluation of the clinical use of MGMT methylation in extracellular vesicle-based liquid biopsy as a tool for glioblastoma patient management. Sci Rep 2024, 14, 11398. [Google Scholar] [CrossRef]
- Indira Chandran, V.; Gopala, S.; Venkat, E. H.; Kjolby, M.; Nejsum, P. Extracellular vesicles in glioblastoma: a challenge and an opportunity. NPJ Precis Oncol 2024, 8, 103. [Google Scholar] [CrossRef]
- Karami Fath, M.; Azami, J.; Masoudi, A.; Mosaddeghi Heris, R.; Rahmani, E.; Alavi, F.; Alagheband Bahrami, A.; Payandeh, Z.; Khalesi, B.; Dadkhah, M.; et al. Exosome-based strategies for diagnosis and therapy of glioma cancer. Cancer Cell Int 2022, 22, 262. [Google Scholar] [CrossRef]
- André-Grégoire, G.; Bidère, N.; Gavard, J. Temozolomide affects Extracellular Vesicles Released by Glioblastoma Cells. Biochimie 2018, 155, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, V.; Kushwaha, D.; Koay, D. C.; Reddy, H.; Mao, Y.; Zhou, L.; Ng, K.; Zinn, P.; Carter, B.; Chen, C. C. Post-transcriptional regulation of O(6)-methylguanine-DNA methyltransferase MGMT in glioblastomas. Cancer Biomark 2011, 10, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Garnier, D.; Meehan, B.; Kislinger, T.; Daniel, P.; Sinha, A.; Abdulkarim, B.; Nakano, I.; Rak, J. Divergent evolution of temozolomide resistance in glioblastoma stem cells is reflected in extracellular vesicles and coupled with radiosensitization. Neuro Oncol 2018, 20, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Akers, J. C.; Ramakrishnan, V.; Kim, R.; Skog, J.; Nakano, I.; Pingle, S.; Kalinina, J.; Hua, W.; Kesari, S.; Mao, Y.; et al. MiR-21 in the extracellular vesicles (EVs) of cerebrospinal fluid (CSF): a platform for glioblastoma biomarker development. PLoS One 2013, 8, e78115. [Google Scholar] [CrossRef]
- Koch, C. J.; Lustig, R. A.; Yang, X. Y.; Jenkins, W. T.; Wolf, R. L.; Martinez-Lage, M.; Desai, A.; Williams, D.; Evans, S. M. Microvesicles as a Biomarker for Tumor Progression versus Treatment Effect in Radiation/Temozolomide-Treated Glioblastoma Patients. Transl Oncol 2014, 7, 752–758. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D. H.; Gainche, L.; Sena-Esteves, M.; Curry, W. T., Jr.; Carter, B. S.; Krichevsky, A. M.; Breakefield, X. O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Evans, S. M.; Putt, M.; Yang, X. Y.; Lustig, R. A.; Martinez-Lage, M.; Williams, D.; Desai, A.; Wolf, R.; Brem, S.; Koch, C. J. Initial evidence that blood-borne microvesicles are biomarkers for recurrence and survival in newly diagnosed glioblastoma patients. J Neurooncol 2016, 127, 391–400. [Google Scholar] [CrossRef]
- Osti, D.; Del Bene, M.; Rappa, G.; Santos, M.; Matafora, V.; Richichi, C.; Faletti, S.; Beznoussenko, G. V.; Mironov, A.; Bachi, A.; et al. Clinical Significance of Extracellular Vesicles in Plasma from Glioblastoma Patients. Clin Cancer Res 2019, 25, 266–276. [Google Scholar] [CrossRef]
- Manda, S. V.; Kataria, Y.; Tatireddy, B. R.; Ramakrishnan, B.; Ratnam, B. G.; Lath, R.; Ranjan, A.; Ray, A. Exosomes as a biomarker platform for detecting epidermal growth factor receptor-positive high-grade gliomas. J Neurosurg 2018, 128, 1091–1101. [Google Scholar] [CrossRef]
- Lan, F.; Qing, Q.; Pan, Q.; Hu, M.; Yu, H.; Yue, X. Serum exosomal miR-301a as a potential diagnostic and prognostic biomarker for human glioma. Cell Oncol (Dordr) 2018, 41, 25–33. [Google Scholar] [CrossRef]
- Manterola, L.; Guruceaga, E.; Gállego Pérez-Larraya, J.; González-Huarriz, M.; Jauregui, P.; Tejada, S.; Diez-Valle, R.; Segura, V.; Samprón, N.; Barrena, C.; et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro Oncol 2014, 16, 520–527. [Google Scholar] [CrossRef]
- Santangelo, A.; Imbrucè, P.; Gardenghi, B.; Belli, L.; Agushi, R.; Tamanini, A.; Munari, S.; Bossi, A. M.; Scambi, I.; Benati, D.; et al. A microRNA signature from serum exosomes of patients with glioma as complementary diagnostic biomarker. J Neurooncol 2018, 136, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Indira Chandran, V.; Welinder, C.; Månsson, A. S.; Offer, S.; Freyhult, E.; Pernemalm, M.; Lund, S. M.; Pedersen, S.; Lehtiö, J.; Marko-Varga, G.; et al. Ultrasensitive Immunoprofiling of Plasma Extracellular Vesicles Identifies Syndecan-1 as a Potential Tool for Minimally Invasive Diagnosis of Glioma. Clin Cancer Res 2019, 25, 3115–3127. [Google Scholar] [CrossRef]
- Tan, S. K.; Pastori, C.; Penas, C.; Komotar, R. J.; Ivan, M. E.; Wahlestedt, C.; Ayad, N. G. Serum long noncoding RNA HOTAIR as a novel diagnostic and prognostic biomarker in glioblastoma multiforme. Mol Cancer 2018, 17, 74. [Google Scholar] [CrossRef] [PubMed]
- de Mooij, T.; Peterson, T. E.; Evans, J.; McCutcheon, B.; Parney, I. F. Short non-coding RNA sequencing of glioblastoma extracellular vesicles. J Neurooncol 2020, 146, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ye, L.; Wang, L.; Quan, R.; Zhou, Y.; Li, X. Identification of miRNA signatures in serum exosomes as a potential biomarker after radiotherapy treatment in glioma patients. Ann Diagn Pathol 2020, 44, 151436. [Google Scholar] [CrossRef]
- Maire, C. L.; Fuh, M. M.; Kaulich, K.; Fita, K. D.; Stevic, I.; Heiland, D. H.; Welsh, J. A.; Jones, J. C.; Görgens, A.; Ricklefs, T.; et al. Genome-wide methylation profiling of glioblastoma cell-derived extracellular vesicle DNA allows tumor classification. Neuro Oncol 2021, 23, 1087–1099. [Google Scholar] [CrossRef]
- Tzaridis, T.; Reiners, K. S.; Weller, J.; Bachurski, D.; Schäfer, N.; Schaub, C.; Hallek, M.; Scheffler, B.; Glas, M.; Herrlinger, U.; et al. Analysis of Serum miRNA in Glioblastoma Patients: CD44-Based Enrichment of Extracellular Vesicles Enhances Specificity for the Prognostic Signature. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Dobra, G.; Bukva, M.; Szabo, Z.; Bruszel, B.; Harmati, M.; Gyukity-Sebestyen, E.; Jenei, A.; Szucs, M.; Horvath, P.; Biro, T.; et al. Small Extracellular Vesicles Isolated from Serum May Serve as Signal-Enhancers for the Monitoring of CNS Tumors. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Chen, W. W.; Balaj, L.; Liau, L. M.; Samuels, M. L.; Kotsopoulos, S. K.; Maguire, C. A.; Loguidice, L.; Soto, H.; Garrett, M.; Zhu, L. D.; et al. BEAMing and Droplet Digital PCR Analysis of Mutant IDH1 mRNA in Glioma Patient Serum and Cerebrospinal Fluid Extracellular Vesicles. Mol Ther Nucleic Acids 2013, 2, e109. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, J. M.; Skog, J.; Akers, J.; Li, H.; Komotar, R.; Jensen, R.; Ringel, F.; Yang, I.; Kalkanis, S.; Thompson, R.; et al. Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro Oncol 2017, 19, 1494–1502. [Google Scholar] [CrossRef] [PubMed]
- Ricklefs, F. L.; Alayo, Q.; Krenzlin, H.; Mahmoud, A. B.; Speranza, M. C.; Nakashima, H.; Hayes, J. L.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune evasion mediated by PD-L1 on glioblastoma-derived extracellular vesicles. Sci Adv 2018, 4, eaar2766. [Google Scholar] [CrossRef]
- Shao, H.; Chung, J.; Balaj, L.; Charest, A.; Bigner, D. D.; Carter, B. S.; Hochberg, F. H.; Breakefield, X. O.; Weissleder, R.; Lee, H. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat Med 2012, 18, 1835–1840. [Google Scholar] [CrossRef]
- Müller Bark, J., Paul Leo7, Rosalind L. Jeffree, Benjamin Chua, Bryan W. Day, Chamindie Punyadeera. Isolation and characterization of exosomes from plasma and saliva of glioblastoma patients. Metro North Health QLD, 2020.
- Mikolajewicz, N.; Khan, S.; Trifoi, M.; Skakdoub, A.; Ignatchenko, V.; Mansouri, S.; Zuccatto, J.; Zacharia, B. E.; Glantz, M.; Zadeh, G.; et al. Leveraging the CSF proteome toward minimally-invasive diagnostics surveillance of brain malignancies. Neurooncol Adv 2022, 4, vdac161. [Google Scholar] [CrossRef]
- Buehler, M.; Yi, X.; Ge, W.; Blattmann, P.; Rushing, E.; Reifenberger, G.; Felsberg, J.; Yeh, C.; Corn, J. E.; Regli, L.; et al. Quantitative proteomic landscapes of primary and recurrent glioblastoma reveal a protumorigeneic role for FBXO2-dependent glioma-microenvironment interactions. Neuro Oncol 2023, 25, 290–302. [Google Scholar] [CrossRef]
- Soylemez, B.; Bulut, Z.; Şahin-Bölükbaşı, S. Investigating the Potential of Lipids for Use as Biomarkers for Glioblastoma via an Untargeted Lipidomics Approach. J Korean Neurosurg Soc 2022. [Google Scholar] [CrossRef]
- Tsvetkov, P. O.; Eyraud, R.; Ayache, S.; Bougaev, A. A.; Malesinski, S.; Benazha, H.; Gorokhova, S.; Buffat, C.; Dehais, C.; Sanson, M.; et al. An AI-Powered Blood Test to Detect Cancer Using NanoDSF. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Eledkawy, A.; Hamza, T.; El-Metwally, S. Precision cancer classification using liquid biopsy and advanced machine learning techniques. Sci Rep 2024, 14, 5841. [Google Scholar] [CrossRef]
- Mouliere, F.; Smith, C. G.; Heider, K.; Su, J.; van der Pol, Y.; Thompson, M.; Morris, J.; Wan, J. C. M.; Chandrananda, D.; Hadfield, J.; et al. Fragmentation patterns and personalized sequencing of cell-free DNA in urine and plasma of glioma patients. EMBO Mol Med 2021, 13, e12881. [Google Scholar] [CrossRef]
- Sol, N.; In 't Veld, S.; Vancura, A.; Tjerkstra, M.; Leurs, C.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Zwaan, K.; et al. Tumor-Educated Platelet RNA for the Detection and (Pseudo)progression Monitoring of Glioblastoma. Cell Rep Med 2020, 1, 100101. [Google Scholar] [CrossRef] [PubMed]
- Herrgott, G. A.; Asmaro, K. P.; Wells, M.; Sabedot, T. S.; Malta, T. M.; Mosella, M. S.; Nelson, K.; Scarpace, L.; Barnholtz-Sloan, J. S.; Sloan, A. E.; et al. Detection of tumor-specific DNA methylation markers in the blood of patients with pituitary neuroendocrine tumors. Neuro Oncol 2022, 24, 1126–1139. [Google Scholar] [CrossRef]
- Menna, G.; Piaser Guerrato, G.; Bilgin, L.; Ceccarelli, G. M.; Olivi, A.; Della Pepa, G. M. Is There a Role for Machine Learning in Liquid Biopsy for Brain Tumors? A Systematic Review. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Theakstone, A. G.; Brennan, P. M.; Jenkinson, M. D.; Mills, S. J.; Syed, K.; Rinaldi, C.; Xu, Y.; Goodacre, R.; Butler, H. J.; Palmer, D. S.; et al. Rapid Spectroscopic Liquid Biopsy for the Universal Detection of Brain Tumours. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Bukva, M.; Dobra, G.; Gomez-Perez, J.; Koos, K.; Harmati, M.; Gyukity-Sebestyen, E.; Biro, T.; Jenei, A.; Kormondi, S.; Horvath, P.; et al. Raman Spectral Signatures of Serum-Derived Extracellular Vesicle-Enriched Isolates May Support the Diagnosis of CNS Tumors. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Aaes, T. L.; Vandenabeele, P. The intrinsic immunogenic properties of cancer cell lines, immunogenic cell death, and how these influence host antitumor immune responses. Cell Death Differ 2021, 28, 843–860. [Google Scholar] [CrossRef]
- Ginghina, O.; Hudita, A.; Zamfir, M.; Spanu, A.; Mardare, M.; Bondoc, I.; Buburuzan, L.; Georgescu, S. E.; Costache, M.; Negrei, C.; et al. Liquid Biopsy and Artificial Intelligence as Tools to Detect Signatures of Colorectal Malignancies: A Modern Approach in Patient's Stratification. Front Oncol 2022, 12, 856575. [Google Scholar] [CrossRef]
- Tiwari, A.; Mishra, S.; Kuo, T. R. Current AI technologies in cancer diagnostics and treatment. Mol Cancer 2025, 24, 159. [Google Scholar] [CrossRef]
- Foser, S.; Maiese, K.; Digumarthy, S. R.; Puig-Butille, J. A.; Rebhan, C. Looking to the Future of Early Detection in Cancer: Liquid Biopsies, Imaging, and Artificial Intelligence. Clin Chem 2024, 70, 27–32. [Google Scholar] [CrossRef]
- Parvin, N.; Joo, S. W.; Jung, J. H.; Mandal, T. K. Multimodal AI in Biomedicine: Pioneering the Future of Biomaterials, Diagnostics, and Personalized Healthcare. Nanomaterials (Basel) 2025, 15. [Google Scholar] [CrossRef]
- Ma, L.; Guo, H.; Zhao, Y.; Liu, Z.; Wang, C.; Bu, J.; Sun, T.; Wei, J. Liquid biopsy in cancer current: status, challenges and future prospects. Signal Transduct Target Ther 2024, 9, 336. [Google Scholar] [CrossRef]
- Cheung, H. M. C.; Rubin, D. Challenges and opportunities for artificial intelligence in oncological imaging. Clin Radiol 2021, 76, 728–736. [Google Scholar] [CrossRef]
- Bi, W. L.; Hosny, A.; Schabath, M. B.; Giger, M. L.; Birkbak, N. J.; Mehrtash, A.; Allison, T.; Arnaout, O.; Abbosh, C.; Dunn, I. F.; et al. Artificial intelligence in cancer imaging: Clinical challenges and applications. CA Cancer J Clin 2019, 69, 127–157. [Google Scholar] [CrossRef]

Table 1.
CTC isolation and analysis in patients with GBM.
| Isolation method | Patient numbers | Markers used to verify GBM origin | Findings | References and publication date |
|---|---|---|---|---|
| CTC-iChip microfluidic technology. Leukocyte depletion using magnetically-tagged anti-CD45 and anti-CD16 antibodies. | 33 | SOX-2, EGFR, c-MET, A2B5 tubulin β-3 | Isolated CTCs from peripheral blood of 39% (13 of 33) GBM patients. Greater CTC counts in patients with progressive disease relative to stable disease. CTCs have a mesenchymal phenotype. |
[22] (2014) |
| Density gradient centrifugation | 141 | Single-cell genomics for common GBM mutations, GFAP staining, tumour specific anomalies like amplification of EGFR gene and gains and losses in chromosomes 7 and 10 genomic regions by chromosomal and array CGH on whole genome amplification. | Isolated CTCs from 20.6% (29/141) of patients. A convenient diagnostic tool for identifying patients with extracranial tumour cell spread and indicates that CTCs could be used to monitor the progression of glioblastoma. | [29] (2014) |
| Density gradient centrifugation using the OncoQuick® system. | 11 | Telomerase-based test was used to identify CTCs. |
CTCs detected in 72% (8 of 11) of patients before radiotherapy but dropped to 8% (1 of 8) in post-radiotherapy patients. This suggests that CTCs may be useful to monitor the progression of cancers before and after therapies. | [30] (2014) |
| Immunoaffinity-based methods. CTC separation with a matrix and negative depletion of white blood cells using immunomagnetic beads. |
31 | Polyploidy chromosome-8-positive detection was employed as a positive measure for CTCs using subtraction enrichment and immunostaining-fluorescence in situ hybridization (SE-iFISH), in addition to GFAP-positive or GFAP-negative cells and CD45-negative cells grading to confirm cell origin. | CTCs were detected in 77% (24 of 31) of GBM patients. Monitoring treatment using CTCs was slightly better than MRI in distinguishing radionecrosis from recurrence of glioma. CTCs can dynamically monitor the microenvironment of gliomas which is a significant complement to radiographic imaging. |
[31] (2016) |
| Immunotargeted enrichment of MSP & MCAM expressing cells. | 13 | CTCs were isolated based on cell surface MSP and MCAM and identified by probing for GLAST and/or GFAP expression. | ≥ 1 CTCs were detected in 69% of patients (9/13), using the combination of 2 isolation and 2 identification markers increased CTC detection. | [21] (2020) |
| Immunopheno-typing. CTCs were isolated by size separation using a Parsortix® microfluidic cassette. | 13 | CTCs were isolated based on no expression of CD45, while expressing EGFR, Ki67, and EB1 microtubule associated protein. For confirmation, the GBM CTC clusters and a biopsy from the primary tumour of the patient were stained with GBM marker SOX-2. |
CTC clusters identified in 53.8% of 13 GBM patients. GBM markers validated that multicellular CTC clusters can be formed and pass the BBB in patients with GBM to reach peripheral circulation and be used for monitoring. |
[32] (2018) |
| Spiral microfluidic technology | 20 | CTCs exhibited characteristic molecular features of GBM, such as EGFR amplification and mutations in TP53 and IDH1. | The study found that CTCs could be isolated from both early- and late-stage GBM patients, highlighting the potential of this technique for non-invasive monitoring of tumour progression. | [33] (2021) |
| CTC Subtraction enrichment/depletion with magnetic immunoaffinity beads and immunostaining-FISH | 22 | Detection of CTCs to differentiate between treatment-induced necrosis and tumour recurrence in brain gliomas. CTC detection outperformed both DSC-MRP and MET-PET in diagnostic accuracy. Additionally, it showed potential for predicting recurrence in one patient. |
The mean CTC count was significantly higher in patients with tumour recurrence (6.10 ± 3.28) compared to those with treatment necrosis (1.08 ± 2.54). A threshold CTC count of 2 provided 100% sensitivity and 91.2% specificity (AUC = 0.933) for identifying tumour recurrence. CTC detection could be a valuable tool for distinguishing tumour recurrence from necrosis, warranting further validation in larger clinical studies. | [34] (2021) |
Table 2.
ctDNA isolation and analysis in patients with GBM.
| Isolation method | Patient numbers | Markers used to verify GBM origin | Findings | References and publication date |
|---|---|---|---|---|
| Guardant360® and digital NGS | 171 | NGS targeting 54 cancer related genes, including assessments for copy number variants in EGFR, ERBB2, and MET. | Of the 33 patients diagnosed with GBM, 73% had unaltered ctDNA, 24% had one alteration and 3% had two or more alterations. | [50] (2016) |
| Guardant360® and digital NGS | 222 | Single nucleotide variants were detected in 61 genes, with amplifications detected in ERBB2, MET, EGFR. | ctDNA mutations were detected in blood samples from 55% of GBM patients. | [51] (2019) |
| Illustra triplePrep Kit (GE healthcare BioSciences Corp) and WGS | 13 | EGFRvIII mutation characterised by a deletion on exons 2 through 7. | EGFRvIII mutant DNA detected in the plasma of GBM patients, with its presence correlating with the mutation in tumour tissue. | [54] (2013) |
| DNA extraction and NGS | 107 | Tumour-specific mutations such as EGFRvIII | ctDNA detection rate was 51% Genomic alteration in the ctDNA of patients highlight the potential of guiding personalised cancer treatment. |
[55] (2018) |
| DNA extraction and methylation specific PCR assay | 19 | MGMT promoter methylation GBM patients serum and cerebrospinal fluid CSF samples for ctDNA detection using methylation specific PCR assay, |
Detected 37% ctDNA in serum and 61% ctDNA in CSF. MGMT promoter methylation was detected with higher sensitivity in CSF (72.0%) compared to serum (41.7%), suggesting CSF as a promising tool for early diagnosis, treatment monitoring, and recurrence detection. |
[61] (2015) |
| DNA extraction and nested PCR-based assays | 38 | ctDNA was analysed for TERT promoter mutations (C228T and C250T) and IDH hotspot mutations. | Detected 8% (3 of 38 patients) ctDNA in plasma and 92% (35 of 38 patients) ctDNA in CSF. | [62] (2018) |
| DNA extraction and WGS | 11 | Tumour-specific mutations in the ctDNA extracted from CSF. | 100% ctDNA detection rate in CSF. | [63] (2015) |
| DNA extraction and WGS | 13 | Copy number alterations in ctDNA. | 50% ctDNA detection rate in CSF. Identified copy number alterations in the ctDNA, which closely reflected tumour genetic profile. Fragmentation patterns in CSF-derived ctDNA Copy number alterations in ctDNA were consistent with tumour tissue. |
[74] (2018) |
| DNA extraction and NGS | 16 | IDH1/2 and 1p/19q codeletion. | ctDNA detected in 49.4% of CSF samples. Genetic alterations detected closely matched those found in tumour biopsies. |
[75] (2018) |
| DNA extraction and NGS | NA | IDH1 (R132H variant), TERT promoter (C228T mutation), TP53, ATRX, H3F3A and HIST1H3B. | CSF ctDNA more accurately reflected BM mutations, detecting all mutations in 83.33% of cases versus 27.78% for plasma ctDNA. CSF ctDNA more accurately reflected BM mutations, detecting all mutations in 83.33% of cases versus 27.78% for plasma ctDNA. Mutant allele frequency (MAF) in CSF ctDNA strongly correlated with BM tumour size (r = 0.95) and was higher than in plasma ctDNA (38.05% vs. 4.57%). MAF and tumour mutational burden in CSF ctDNA closely matched BM values (r = 0.96 and 0.97, respectively). CSF ctDNA exhibited superior concordance with BM (99.33%) compared to plasma ctDNA (67.44%), improving the identification of clinically relevant mutations. However, for multiple BM, plasma ctDNA performed well, achieving a 93.01% concordance, comparable to CSF ctDNA. |
[76] (2023) |
| DNA extraction and MSK-IMPACT™, a NGS assay | 711 | The distribution of clinically actionable somatic alterations was consistent with tumour-type specific alterations across the AACR GENIE cohort. | Genetic alterations were detected in 53% (489/922) of CSF samples from patients with confirmed CNS tumours, while none of the 85 samples from patients without CNS tumours contained detectable ctDNA. The identified mutations aligned with tumour-type-specific alterations observed in the AACR GENIE cohort. Repeated ctDNA testing revealed clonal evolution and resistance mechanisms, and the presence of ctDNA linked to reduced overall survival after CSF collection. |
[77] (2024) |
| QIAamp Circulating Nucleic Acids kit (QIAGEN), Western blot, histopathology, and immunochemistry, digital PCR, and shallow whole genome sequencing were utilised | 64 | Patient-derived orthotopically implanted xenograft models of GBM. | Analysis of fragment length profiles of host (rat) and tumour (human) ctDNA identified a peak at 145 bp in the human DNA fragments, indicating a difference in the origin or processing of the ctDNA. The concentration of ctDNA correlated with cell death only after treatment with TMZ and radiotherapy. ddPCR detection of plasma tumour mitochondrial DNA (tmtDNA), an alternative to detection of nuclear ctDNA, improved plasma DNA detection rate (82% versus 24%) and allowed detection in CSF and urine. The total amount of tmtDNA in the plasma (558 copies) is ~13 times higher than in the CSF (43 copies), showing therefore that the BBB does not prevent significant amounts of tumour DNA |
[65] (2019) |
| QIAamp DNA micro kit (Qiagen) and amplicon sequencing | 20 | DH1, IDH2, TP53, TERT, ATRX, H3F3A, and HIST1H3B gene mutations | The genomic analysis in CSF extracted ctDNA facilitates the diagnosis of diffuse gliomas into subtypes to support the surgical and clinical management of these patients. | [75] (2018) |
| QIAamp Circulating Nucleic Acid kit and NGS | 26 | Cancer genomic panel sequencing on the CSF-derived ctDNA. | A high detection rate of ctDNA (24/26, 92.3%) was observed in CSF. ctDNA mutations had high concordance rates with tumour DNA, especially in non-copy number variations and in GBM. CSF ctDNA TMB also exhibited a strong correlation with tumour DNA TMB (R2 = 0.879, P < 0.001), particularly in GBM (R2 = 0.992, P < 0.001). |
[78] (2022) |
| DNeasy Blood and Tissue Kit (Qiagen), and targeted DNA sequencing by Oxford Nanopore Technology MinION device. | 12 paediatric high-grade glioma patients and 6 controls | Analysis of ctDNA with a handheld platform (Oxford Nanopore MinION) to quantify patient-specific CSF ctDNA variant allele fraction (VAF) | Nanopore demonstrated 85% sensitivity and 100% specificity in CSF samples (n = 127 replicates) with 0.1 femtomole DNA limit of detection and 12 h results, all of which compared favourably with NGS. Multiplexed analysis provided concurrent analysis of H3.3A (H3F3A) and H3C2 (HIST1H3B) mutations in a non-biopsied patient and results were confirmed by ddPCR. Serial CSF ctDNA sequencing by Nanopore demonstrated correlation of radiological response on a clinical trial, with one patient showing dramatic multi-gene molecular response that predicted long-term clinical response. |
[79] (2020) |
Table 3.
Nucleosome isolation and analysis in patients with GBM.
| Isolation method | Patient numbers | Markers used to verify GBM origin | Findings | References |
|---|---|---|---|---|
| Cell Death Detection ELISA Plus kit | 10 | NA | Pre-therapeutic nucleosome levels in both serum and CSF did not significantly differ among GBM patients and control groups. Postoperative Increase: In GBM patients, nucleosome levels in serum and CSF increased moderately during the week following surgery and intracavitary chemotherapy. Cerebral Oedema Correlation: Three out of ten GBM patients developed cerebral oedema post-surgery. In these patients, CSF nucleosome levels increased almost 200-fold, peaking on day 3 postoperatively. In contrast, the seven patients without oedema exhibited only slight increases in nucleosome levels. Clinical Implication: Monitoring CSF nucleosome levels may serve as an indicator for postoperative complications such as cerebral oedema in GBM patients. |
[94] |
| Single-molecule technology to detect and monitor plasma-circulating nucleosomes | NA | H3-K27M mutation and mutant p53 | The single-molecule analysis revealed epigenetic patterns unique to diffuse midline glioma, enabling differentiation from healthy individuals and patients with other cancer types. This approach profiles multiple histone modifications on individual nucleosomes from less than 1 mL of plasma, revealing epigenetic patterns unique to glioma that significantly differentiate these patients from healthy individuals and those with other cancer types. The detection strategy demonstrated a correlation with MRI measurements and ddPCR assessments of ctDNA, highlighting its potential utility in non-invasive treatment monitoring. Suitable for paediatric patients and scenarios where sample volume is limited. |
[95] |
Table 4.
miRNA isolation and analysis in patients with GBM.
| Isolation method | Patient numbers | Markers used to verify GBM origin | Findings | Reference and publication date |
|---|---|---|---|---|
| miRNA extraction and qPCR | 20 | miR-221 and miR-222 | Both miR-221 and miR-222 were significantly upregulated in the plasma of GBM patients compared to healthy controls. miR-221 demonstrated 90% sensitivity and 100% specificity and miR-222 demonstrated 85% sensitivity and 100% specificity for GBM detection. Expression levels of miR-221 and miR-222 decreased following treatment |
[119] (2019) |
| miRNA profiling performed using the Nanostring® platform | 91 | miR-223 and miR-320e, IDH mutation status and1p/19q co-deletion. | Dynamic changes of miR-320e were linked to tumour volume of GBM patients. A 9-miRNA signature was established, distinguishing glioma patients from healthy controls with 99.8% accuracy. miRNA levels did not increase in cases of pseudo-progression. This supports their use in distinguishing true progression from treatment effects and in post-operative monitoring |
[120] (2020) |
| mirVana™ miRNA Isolation Kit and qRT-PCR | 50 | miR-21, miR-128, and miR-342-3p |
miR-21 was significantly upregulated, while miR-128 and miR-342-3p were markedly downregulated in glioma patients compared to healthy controls Notably, miR-21 demonstrated a high diagnostic performance with 90% sensitivity, and 100% specificity. |
[101] (2012) |
| RNA isolation and NGS, including mRNA-seq and small RNA-seq | 7 | mRNA and miRNA candidates | The study identified differentially expressed genes in individual patients, with up to 93 mRNA and 19 miRNA candidates linked to GBM recurrence. | [121] (2024) |
| Urinary microRNA-based diagnostic model for CNS tumours using nanowire scaffolds. | 119 | Differential miRNA expression profiles | The study reported high diagnostic performance, with sensitivity and specificity values of 100% and 97%, respectively, for detecting early-stage CNS tumours. Non-invasive method holds promise for early detection and monitoring of CNS tumours through urine-based liquid biopsy. |
[122] (2021) |
| miRCURY RNA Isolation Kit and qRT-PCR | 10 | miR-21, miR-218, miR-193b, miR- 331, and miR-374a, miR- 548c, miR-520f, miR-27b, and miR-130b | Sampling of CSF from the lumbar region to extract 9 signature miRNA for GBM. The overexpressed signatures were miR-21, miR-218, miR-193b, miR- 331, and miR-374a, while the down regulated were miR- 548c, miR-520f, miR-27b, and miR-130b in GBM CSF The study compared the diagnostic performance of miRNA detection between CSF obtained from the cisternal and lumbar regions. The cisternal CSF samples demonstrated a sensitivity of 80% and specificity of 67% for GBM detection, whereas the lumbar CSF samples showed a sensitivity of 28% and specificity of 95%. |
[123] (2017) |
| TaqMan Low Density Array platform for miRNA profiling and qRT-PCR | 16 GBM and 9 healthy patients | miR-451, miR-711, miR-935, miR-223 | Showed that miRNA from CSF can differentiate between tumour and non-tumour diseases states. Identified distinct miRNA signature in CSF that can distinguish CNS malignancies (GBM) from non-tumour controls. |
[124] (2015) |
Table 5.
EV isolation and analysis in patients with GBM.
| Isolation method | Patient numbers | Markers used to verify GBM origin | Findings | References and publication date |
|---|---|---|---|---|
| Differential centrifugation and flow cytometry | 11 | GFAP | Concentration and composition of circulating MVs in patient plasma correlated with tumour progression. Elevated levels of tumour-derived MVs were associated with true tumour progression, while lower levels were indicative of treatment-related changes or pseudoprogression. |
[140] (2014) |
| Differential centrifugation and ultracentrifugation, filtration and flotation density gradient centrifugation. | 25 | Tumour-specific EGFRvIII mRNA within the vesicles | EVs contain functional RNA and proteins that may influence the tumour microenvironment and serve as diagnostic tools GBM cells release MVs containing mRNA, miRNA, and angiogenic proteins. Detection of tumour-specific EGFRvIII mRNA in serum-derived MVs supports their potential as non-invasive biomarkers for GBM diagnosis and monitoring |
[141] (2008) |
| Serial centrifugation and flow cytometry | 16 | Annexin V, CD41, CD235 and Anti-EGFR | An increase in Annexin V-positive MV levels during chemoradiation therapy was associated with earlier tumour recurrence and shorter overall survival Patients with higher levels of MVs had > 4-fold increase in the hazard ratio for recurrence compared to those with lower levels The study provided initial evidence that monitoring blood-borne MV levels could serve as a non-invasive method to predict disease progression and patient outcomes in newly diagnosed GBM patients. |
[142] (2016) |
| Ultrafiltration and ultracentrifugation and NTA | 43 | GFAP |
Plasma EV concentration was higher in GBM compared with healthy controls (p=0.0099). Average size of GBM and healthy EVs were similar in discovery (p=0.548) and validation cohort (p=0.075). The amount of circulating EV was not affected by tumour size (p=0.318) However, the extent of necrosis influenced the degree of secretion (p=0.045): higher necrosis in GBM samples (grade 3) substantially reduced EV secretion. Elevated EV levels in GBM plasma decrease post-surgery and rose at recurrence. |
[143] (2019) |
| Precipitation using ThermoFisher kit and Semi quantitative RT-PCR | 96 | EGFRvIII mRNA within the vesicles | EGFRvIII prevalence in the data set was 39.58%. The sensitivity and specificity of serum EV analysis for EGFRvIII was 81.58% (95% CI 65.67%–92.96%) and 79.31% (95% CI 66.65%–88.83%), respectively |
[144] (2018) |
| Differential centrifugation and qRT-PCR | 60 | miR-301a | Serum exosomal miR-301a levels were significantly elevated in glioma patients compared to healthy controls. Higher miR-301a levels were associated with higher tumour grades and lower Karnofsky Performance Status (KPS) scores. Post-surgical samples showed a significant reduction in miR-301a levels, which increased again during tumour recurrence, suggesting its potential as a marker for disease monitoring. Kaplan-Meier survival analysis indicated that patients with higher serum exosomal miR-301a levels had shorter overall survival. |
[145] (2018) |
| Ultracentrifugation and TEM | 42 | miR-320, miR-574-3p and RNU6-1 | RNU6-1 identified in serum exosomes could effectively distinguish GBM patients from healthy individuals. The elevated levels of RNU6-1 in GBM patients' exosomes suggest its potential as a non-invasive diagnostic biomarker. |
[146] (2014) |
| Differential centrifugation and qPCR | 12 | miR-182-5p, miR-328-3p, miR-339-5p, miR-340-5p, miR-485-3p, miR-486-5p and miR-543 | The identified miRNA signature in serum exosomes could effectively distinguish GBM patients from healthy individuals and those with lower-grade gliomas. | [99] (2018) |
| Density gradient ultracentrifugation, using OptiPrep™ Density Gradient Medium and sequencing followed by differential expression analysis | 12 GBM (astrocytoma grade IV) and 5 astrocytoma grade II-III | Analysis of CUSA (cavitron ultrasonic surgical aspirate) EV and serum EV miRNA and piRNA | Seven miRNA species were determined to be the most stable classifiers for GBM (miR-182-5p, 382-3p, 339-5p, 340-5p, 485-3p, 486-3p and 543) with an overall predictive power of 91.7%; within multivariate models, six iterations of these markers were capable of distinguishing GBM patients from healthy controls with 100% accuracy. | [100] (2020) |
| Chemical precipitation using ExoQuick-TC and qPCR | 100 patients with glioma, 11 with metastatic brain tumours, 30 healthy patients | Expression of 3 miRNAs: miR-21, miR-222 and miR-124-3p, in serum exosomes. | Exosome miR-21, 222 and 124-3p were robust for discriminating patients with GBM from healthy controls, with an area under curve of 0.84 (95% CI 0.7538–0.9371, p < 0.001), 0.80 (95% CI 0.6967–0.8980, p < 0.001) and 0.78 (95% CI 0.6732–0.8904, p < 0.001), respectively. miR-21 alone appears to be the best predictor for distinguishing patients with High grade glioma from those with low grade glioma, displaying the highest AUC of 0.83 (95% CI 0.7395–0.9398, p < 0.001 |
[147] (2018) |
| Ultracentrifugation, NTA and TEM for plEV isolation with proximity extension assay–based ultrasensitive immunoprofiling. | 82 | Syndecan-1 |
SDC1 in plEVs could discriminate between GBM and low-grade glioma with a sensitivity of 71%, and specificity of 91%. The findings support the concept of circulating plEVs as a tool for non-invasive diagnosis and monitoring of gliomas. |
[148] (2019) |
| Total Exosome Isolation reagent followed by ultracentrifugation and qRT-PCR | 43 GBM, 23 other brain tumour patients and 40 healthy individuals | Serum analysed for presence of EVs and HOTAIR biomarker | HOTAIR can be used as a biomarker for Dx and progression of some brain tumours including GBM with sensitivity and specificity of HOTAIR 86.1% and 87.5% respectively. | [149] (2018) |
| Serial ultracentrifugation and short non-coding RNA sequencing using the OASIS-2.0 platform. | 5 | Isolate EV’s from human differentiated GBM cells in vitro and perform short, non-coding RNA sequencing to determine expression pattern | Small genome sequencing revealed a total of 712 non-coding RNA sequences most of which have not been associated with GBM EV’s previously, including the let-7 miRNA family, miR-3182, miR-4448, miR-100-5p and miR-27-3p. Furthermore, multiple non-microRNA short non-coding RNA species were identified including piRNA, snRNA, snoRNA, and yRNA. |
[150] (2020) |
| qPCR | 5 Glioma patients before and after radiotherapy | Expression signature of miRNAs in glioma patients before and after radiotherapy | Identified 18 up-regulated differentially expressed (DE) miRNAs and 16 down-regulated DE miRNAs and the target genes of DE miRNAs were predicted based on multiple miRNA-target databases. | [151] (2020) |
| Differential centrifugation and DNA analysis through methylation array analysis Proteome analysis with differential quantitative proteomics |
Unspecified number of glioma patients and non-tumorous temporal tissue from patients undergoing epilepsy surgery | DNA and protein analysis of Glioma and non-tumorous EVs | Tumour-specific mutations and copy number variations were detected in EV-DNA with high accuracy. Proteome analysis did not allow specific tumour identification or classification |
[152] (2021) |
| Size-exclusion chromatography (by using qEV columns from IZON®), followed by immunoprecipitation with CD44-conjugated beads and qRT-PCR | 55 | Suitability of novel EV isolation procedure and analysis of serum EVs for miRNA biomarkers and their correlation with prognosis. | Four serum biomarkers were identified to be predictive of prognosis, miR-15b-3p, miR-21-3p, and miR-328-3p exhibiting a negative correlation (high levels were associated with an inferior prognosis) and miR-106a-5p a positive correlation (high levels were associated with a better prognosis prediction of GBM prognosis |
[153] (2020) |
| Differential centrifugation and NTA | 96 total patients, 24 GBM, 24 meningioma, 24 BM from NSCLC and 24 controls from patients with benign disk herniation. | Proteomic analysis of serum and serum derived small EVs | Identified 10- and 17-membered protein panel for whole serum and sEV samples respectively. While none of these proteins appeared to be able to distinguish between the patient groups individually, their combination was found to reliably discriminate between the different patient groups suggesting that instead of a few candidates, a specific protein panel is required for a perfect differentiation between various tumour types. | [154] (2020) |
| Ultracentrifugation and RNA extraction from EVs, ddPCR and flow cytometry for rare mutation detection. | 14 glioma patients (4 with GBM) | Mutant IDH1 G395A | CSF is a viable bio fluid to examine contents of EVs. Mutant IDH1 G395A identified in CSF EVs with a sensitivity of 63% and specificity of 100%. CSF EVs generally contained higher levels of mutant mRNA than serum EVs. EVs carry tumour-specific RNA signatures that can be detected non-invasively, supporting the potential of EVs as liquid biopsy tools for glioma diagnosis and monitoring. |
[155] (2013) |
| Ultracentrifugation and purification with sucrose cushion for EV isolation. qRT-PCR for mRNA transcript detection. | 25 GBM, 5 low-grade glioma and 4 healthy patients | EGFRvIII mutant mRNA | EGFRvIII oncogene, in EV RNA can be accomplished with a sensitivity of 60% and 98% specificity in comparison to the gold standard qPCR of EGFRvIII transcript from brain tumour tissue. | [156] (2017) |
| Ultracentrifugation and. TEM for EV isolation. Western blotting for exosomal markers. qRT-PCR for mRNA transcript quantification. |
9 GBM and 5 healthy patients | Exosomal markers (CD63 and TSG101) miR-21 |
Showed that miR-21 from CSF EVs can differentiate between tumour and non-tumour diseases states. |
[139] (2013) |
| Ultracentrifugation and OptiPrep™ density gradient ultracentrifugation for further purification. NTA, flow cytometry and western blotting (to detect PD-L1 on EV surface). | 10 | Patient derived GBM stem cells. | PD-L1 expression on the surface of GBM derived EVs, can prevent T-cell activation and proliferation upon binding directly to PD-L1. This indicates PD-L1 expression on EVs can be an immune-escape mechanism for GBM |
[157] (2018) |
| Differential centrifugation of MVs and microfluidic chip-based immunomagnetic technique called μNMR (miniaturized nuclear magnetic resonance) for protein typing of EVs/MVs. Also used magnetic nanoparticles (MNPs) conjugated to antibodies to detect specific tumour-associated proteins on circulating MVs. | 15 | EGFRvIII mutant, PDPN and IDH1 |
Tumour-derived MVs carrying EGFR/EGFRvIII proteins were successfully detected in patient plasma. Showed four protein panels of EV surface proteins can be used to discriminate GBM patients from healthy controls using novel antibody capture method. The system enabled real-time monitoring of tumour progression and treatment response by tracking changes in circulating tumour-derived MV profiles. |
[158] (2012) |
| Ultracentrifugation for exosome isolation and characterisation by western blotting for expression protein markers, TEM for morphology and NTA for size and concentration. | 10 | CD63, CD81 and CD9 | Saliva and plasma were used as liquid biopsies to isolate exosomes before and after surgery. GBM patients exhibited smaller but more abundant exosomes compared to healthy donors. EVs from GBM plasma showed globally reduced levels of cytokines and co-stimulatory molecules, but PD-L1 presence was similar to healthy donors. |
[159] (2020) |
| Proteomics using mass spectrometry | 22 | MGMT and IDH statuses and GAP43 | CSF proteomics using mass spectrometry enables GBM biomarker discovery from small volumes (~30 μL). Mikolajewics et al., identified 755 unique proteins in 73 CSF samples (22 GBM), with MGMT and IDH statuses accurately detected at 94.1% and 33.3%, respectively. Single-cell RNA sequencing confirmed GAP43 as GBM-specific, while TFF3 and CACNA2D2 were specific to BM and CNS lymphoma. |
[160] (2022) |
| Proteomics with sequential window acquisition of all theoretical fragment ion spectra mass spectrometry (SWATH-MS) and immunohistochemistry for validation. | 134 | BCAS1, INF1, and FBXO2 | Identified overexpressed proteins CSF proteomics in recurrent GBM, to quantify the proteomes of newly diagnosed and recurrent GBM patients and validated the markers using immunohistochemistry. | [161] (2023) |
| Lipidomics using Quadrupole time-of-flight liquid mass spectrometer Q-TOF LCMS/MS | 14 GBM and 14 healthy patients | NA; based on statistically significant differences in blood lipid species between GBM and controls | Lipidomics, also holds potential Identified differential lipid species including fatty acids, saccharolipid, sphingolipid, glycerolipid and sterol lipid from blood samples. |
[162] (2022) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



