
Submitted:
31 July 2025
Posted:
04 August 2025
You are already at the latest version
Abstract
Over recent years, epitranscriptomic research has provided a new layer of gene regulation during hematopoietic development and aberrant hematopoiesis. Among the 170 identified RNA chemical marks, N6-methyladenosine (m6A) is the most abundant in eukaryotic cells and plays a critical role in various biological processes. This dynamic modification is regulated by a series of methyltransferases, demethylases and m6A binding proteins, known as writers, erasers and readers, respectively. Emerging evidence suggests that m6A modification and its regulators are involved in every aspect of normal hematopoietic development, from the emergence of hematopoietic stem cell to the generation of mature blood cells. Also, it has been established that abnormal expression of m6A regulators is implicated in the initiation of blood diseases. In this review, we summarize the latest findings refarding the role of m6A in erythropoiesis and highlight its implication in the pathophysiology of hemoglobin disorders.
Keywords:
epitranscriptomics
; m6A modification
; m6A regulators
; hematopoietic stem cells
; erythropoiesis
; ineffective erythropoiesis
; thalassemia
; hemoglobin H disease
1. Introduction
1.1. The Biochemistry of N6-methyladenosine RNA Modification
During the last few decades, advances in epigenetic research have provided novel insights into the mechanisms that control either normal hematopoiesis or lead to blood disorders. The production and development of hematopoietic stem cells (HSCs) is a complex process regulated at multiple levels by epigenetic and epitranscriptomic factors, including DNA methylation, histone modification, chromatin modification, and RNA methylation [1]. So far, a significant number of post-transcriptional modifications of mRNA have been identified, among which the nitrogen-6-methyladenosine (m6A) is the most common. m6A is a dynamic RNA modification abundant in endogenous eukaryotic messenger RNAs [2]. Although m6A was first discovered in the early 1970s, only the recent advances in sequencing technologies have allowed mapping the m6A modified sites in mammalian cells and tissues, and thus have begun deciphering its role in physiological processes and disease. Marks of m6A occurring in adenines of RRACH sequence (R=G or A, H=A, C or U) are in abundance in the 3’ untranslated regions (3’ UTRs), in long internal exons, and around stop codons [3]. These marks are reversibly installed in the nucleus by the m6A methyltransferase (MTase) complex (writers), consisting of METTL3, which is the catalytic subunit, METTL14, WTAP, and other cofactors, while demethylases FTO and ALKBH5 (erasers) reduce the global m6A levels. Finally, m6A binding proteins, known as readers, are located both in the nucleus and in the cytoplasm, and determine the fate of the m6A-modified transcripts, by affecting mRNA nuclear export, splicing, stability and translation efficiency, as depicted in Figure 1. The best-described readers belong to the YT521-B homology (YTH) domain family, which includes the YTHDF1, YTHDF2, YTHDF3, YTHDC1, and YTHDC2 proteins. Most recently, insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) and proteins of the heterogeneous nuclear ribonucleoprotein (HNRNP) family. were found to impact the stability and translation of targeted mRNAs, as well as the alternative splicing and microRNA processing, respectively [4]. Regarding the biological role of m6A, accumulating evidence suggests that it is involved in a broad spectrum of biological processes, such as tissue development, circadian rhythm, apoptosis, autophagy, and tumorigenesis [5]. Recent studies revealed that the N6A-methyladenosine RNA modification plays a crucial role in hematopoietic cell fate decisions, by regulating both self-renewal and HSC differentiation [6].
2. The Role of m6A RNA Methylation in Normal Hematopoiesis and in Disease
2.1. HSCs and Endothelial-to-Hematopoietic Transition
The blood system is comprised of more than ten different blood cell types, termed lineages, with numerous functions, from immune surveillance and defense to O2 transport and wound healing. All blood cell types are generated by hematopoietic stem cells (HSCs) that reside mostly in the bone marrow, a major site of adult hematopoiesis [7]. The primary stages of embryonic development of blood tissue literally take place in the absence of any blood cells. The sites of the de novo hematopoietic stem and progenitor cell (HSPC) generation, as well as the maintenance and expansion are constantly changing in the developing embryo. The first hematopoietic cells are generated in the extraembryonic yolk sac, before the first heartbeat and later in the allantois and placenta [8]. All adult and many of the later embryonic blood cells, are generated from HSCs. However, there is a transient population of primitive blood cells in the early embryo, generated from endothelium through a highly conserved process known as endothelial-to-hematopoietic transition (EHT). Molecular signaling by Notch ligands, hedgehog, bone morphogenetic protein (BMP), vascular endothelial growth factor (VEGF) and nitric oxide signaling, play a crucial role in the induction of the hemogenic program in endothelial cells [8].
During EHT, cells undergo significant morphological changes, including the breaking of tight connections with surrounding endothelial cells and rounding up, before escaping into the bloodstream [9]. A complex interplay of critical transcription factors and signaling pathways, as well as inputs from adjacent tissues, coordinate the entire process [9]. Accumulating evidence so far, suggests that m6A modification plays also a key role in the successful generation of HSCs. Zhang and colleagues [10]. showed that reduced METTL3 protein, and hence reduced m6A levels in zebrafish embryos, leads to EHT disruption and eventually to complete loss of specialized blood cells. Interestingly, epitranscriptome mapping revealed that the lack of HSC emergence was caused by reduced m6A mRNA methylation of Notch1 and of other arterial endothelial genes, which are normally under the YTHDF2-mediated degradation [10]. Also, lower levels of the HSPC markers runx1 and cmyb were detected. Analogous results were obtained by ythdf2 zebrafish morphants [10]. Additionally, the m6A implication on EHT regulation was confirmed in Mettl3-knockdown hematopoietic stem and progenitor cells, acquired from the aorta-gonad-mesonephros region (AGM) of mice [10]. Employment of m6A-RNA immunoprecipitation (RIP)-qPCR assays and qPCR analysis, revealed that Notch1 is m6A-targeted, and its expression is enhanced after Mettl3-knockdown. Furthermore, in vivo experiments using the Cre/LoxP system to knockout the expression of Mettl3 specifically in endothelial cells of the mouse AGM region, provided consistent results [10]. Collectively, METTL3 and YTHDF2 operate synergistically to suppress the Notch signaling pathway and thus affect the EHT process [10]. Similarly, a study by Lv et al [11], showed that METTL3 depletion in vascular endothelial cells of mouse AGM, leads to a significant suppression of HSPC function. The above findings clearly show that m6A modification plays an essential role during vertebrate definitive hematopoiesis. Finally, there seems to be an interplay between epigenetic and epitranscriptomic mechanisms that are involved in EHT, such as miRNAs, known to affect HSC production and control m6A levels via modulating METTL3 binding to mRNAs [1].
2.2. HSC Self-Renewal and Expansion
Hematopoietic stem cells are multipotent precursors that have the dual ability for self-renewal and for giving rise to myeloid, lymphoid, and erythroid cell lineages throughout life [7]. This equilibrium is maintained by cell fate decisions made during cellular division [7]. It has not been fully elucidated yet, which signals determine whether a cellular division will result in lineage commitment and differentiation or in self-renewal [7,12]. According to recent studies, m6A-related regulators, seem to be key participants in HSC homeostasis [13,14,15,16].
Specifically, the reader protein YTHDF2, which mediates targeted m6A-mRNA degradation, was reported to uniquely influence HSC regeneration and expansion. Recent studies showed that the loss of YTHDF2, leads to increased proliferation of HSC without altering lineage bias [13]. Particularly, Wang et al [13], exploited Cre/LoxP technology to create hematopoietic-specific Ythdf2-knockout mice. According to their study, Ythdf2 expression in HSCs expedites the decay of m6A-marked mRNAs of Wnt target genes, leading to suppression of Wnt signaling at steady state. Given the fact that aberrant activation of Wnt signaling leads to an increase in the number of HSCs with lower functional response to hematological stressors, YTHDF2 probably has a protective role. Interestingly, upon YTHDF2 loss, not only was the number of HSCs increased, but so was the regenerative capacity of HSCs under stress circumstances [13]. Furthermore, Mapperley et al [14], showed that YTHDF2 protects HSCs from proinflammatory signals, which cause excessive proliferation and promote myeloid-biased differentiation. These data also suggest that Ythdf2 deletion results in the expansion of HSCs and multipotent progenitors (MPPs) [14]. Similar observations were made by Li et al [15], after the depletion of YTHDF2 in mouse HSPCs and human umbilical cord blood HSCs. Data obtained by Ythdf2 knockout mice indicated that YTHDF2 normally suppresses HSC self-renewal through RNA degradation. Furthermore, m6A mapping revealed that mRNAs of transcription factors important for stem cell self-renewal, such as Tal1 and Gata2, are m6A-labeled and recognized by YTHDF2, which drives them to decay sites [15]. Moreover, Ythdf2 knockdown in human umbilical cord blood HSCs, led to decreased apoptotic rate and enhanced expansion ex vivo, providing possible new strategies for future stem cell-based treatments [15]. The nuclear m6A reader YTHDC1 is also essential for normal hematopoiesis and HSPCs maintenance according to in vivo studies [16]. In line with the previous studies, Yin et al [17], proposed that m6A could be considered as a quality control system for the preservation of HSC integrity. Their findings suggest that the reader protein IGF2BP2. plays a critical role in maintaining HSC stemness by regulating the expression state of m6A-methylated mRNAs as well as the mitochondrial activity of HSCs.
Other m6A regulators whose role has been studied in mammalian hematopoietic development, include METTL3 and METTL14. Their role in the control of HSC self-renewal, was assessed in adult mouse bone marrow, utilizing Cre/LoxP technology to create Mettl3-/Mettl14- deficient mice [18]. Deletion of these genes led to the accumulation of HSCs and reduction of their self-renewal capacity. Specifically, it was shown that METTL3 enhances the expression of genes related to HSC quiescence, maintenance and self-renewal, such as Nr4a2, p21, Bmi-1, and Prdm16. Notably, qPCR analysis showed no difference in the expression of genes related to HSC differentiation such as Ikaros, PU.1, and Mef2c [18]. Consistent with these results, deletion of Mettl3 in adult mouse HSCs, and therefore lack of m6A, leads to an accumulation of HSCs in the bone marrow, while having no important effect on HSC self-renewal and quiescence [19]. On the contrary, a most recent study demonstrated that the previously identified accumulated HSCs, are actually blocked HSC-multipotent progenitor-like cells, who fail to differentiate due to loss of Myc, which controls HSC symmetric commitment and is normally under m6A regulation [20]. Moreover, it was shown that Mettl3 knock out HSCs, are less quiescent and have increased metabolism, as well as mitochondrial activity, indicating that m6A is essential for HSC quiescence and self-renewal [20]. In addition, loss of METTL3-mediated m6A modification caused enhanced formation of endogenous double-stranded RNAs and abnormal stimulation of MDA5-RIG-I, PKR-eIF2α, and OAS-RNase L pathways in HSPCs, leading to accumulation of dysfunctional immature cells in vitro and in vivo [21].
2.3. Erythropoiesis
Hematopoietic stem cells replenish all blood cell types on a continuous basis through a series of differentiation stages and recurrent cellular divisions that entail the production of lineage-committed progenitors. Daily, the erythroid progenitors expand enormously to generate 200 billion red blood cells in the bone marrow, during a stepwise maturation process, known as erythropoiesis [22]. Erythropoiesis is controlled by a network of factors, such as oxygen sensors, cytokines, and regulators of iron metabolism [23]. Any abnormalities in critical components of this process can result in severe disorders such as anemia or myelodysplastic syndromes [23]. Briefly, HSCs initially give rise to multipotent progenitors (MPPs), which in turn give rise either to common myeloid precursors (CMPs) or common lymphoid precursors (CLPs), through cytokine signaling and activation of several transcription factors. MPPs differentiate into CLPs that originate lymphocytes and natural killer cells that rely mainly on activation by PU.1, Ikaros and GATA-3 transcription factors [24]. MPP fate-decision differentiation into CMP, generates granulocyte-macrophage (GMP) and megakaryocyte-erythrocyte progenitors (MEPs), and is modulated by PU.1 and GATA-1 [24]. The differentiation of CMP is relied on secretion of granulocyte-macrophage-colony stimulating factor (GM-CSF), and subsequently on macrophage-colony stimulating factor (M-CSF), which regulates the differentiation of monocytes/macrophages, while granulocyte-colony stimulating factor (G-CSF), regulates the differentiation of neutrophils, basophils, and eosinophils [24]. During erythropoiesis, MEP differentiates into burst-forming unit-erythroid (BFU-E) and, finally, into colony-forming unit-erythroid (CFU-E) [23]. This process is modulated by soluble factors such as erythropoietin (EPO), stem cell factor (SCF), IL-3 and IL-6, as well as by the activation of GATA-1, STAT-5, and Krüppel-like factor-1 (KLF-1) pathways [25].
Despite the extended knowledge in this field, many of the regulatory features that control HSC differentiation, as well as production, and maturation of erythrocytes remain still unknown. Numerous studies have shown that m6A RNA methylation plays a pivotal role in normal and pathological erythropoiesis [6]. Epitranscriptomic sequencing either with m6A-seq or by MeRIP-seq technology, has enabled the mapping of m6A sites on the mRNA molecules of mammalian cells. Notably, transcripts of transcription factors that coordinate erythroid differentiation, such as KLF1, GATA1, GATA2, FLI1, and MPL, and of genes involved in erythroid disorders, were found to be enriched in m6A RNA methylation [26]. KLF1 and GATA1 are known to work together, as well as with other cofactors and chromatin modifiers to drive the erythroid transcriptional program [26]. Low levels or complete loss of GATA1 have been linked with impaired terminal erythroid differentiation and anemia development [27]. KLF1 is a principal regulator of erythropoiesis that directly regulates the expression of genes involved in α and β-globin and iron-bound heme biosynthesis [28]. Particularly, KLF1 has been demonstrated to activate the expression of β-globin and regulate the expression of genes that participate in the production of hemoglobin molecules, such as BCL11A, which is known to control the production of fetal γ-globin [27]. Erythropoiesis is substantially hampered upon loss of KLF1. As a result, reduction or absence of β-globin chain production, leads to the accumulation of excess of α-globin chains, which precipitate. forming insoluble aggregates in the red blood cell precursors, leading to apoptosis at the polychromatophilic erythroblast stage [29].
CRISPR-Cas9 whole-genome screening in human erythroleukemia (HEL) cells was employed to identify regulatory molecules affecting erythropoiesis [26]. Specifically, transduction of HEL cells with lentiviral sgRNA reagents, followed by sorting of CD235- cells out of the population of the outgrown HEL cells, disclosed that METTL3, METTL14, and WTAP were among the genes required for normal erythroid differentiation. Next, CRISPR-Cas9 gene targeting was carried out to knock out each of the main components of the MTase complex, and thus to elicit a reduction of global m6A levels. The data documented that elimination of the m6A MTase complex, results in erythroid differentiation blockage, without affecting the development of other non-lymphoid cell lineages. Further analysis showed that m6A marking enhanced the translation of genes involved in erythroblast differentiation and heme biosynthesis, as well as of genes that constitute the H3K4 MTase network, e.g., SETD1A, SETS1B, and KMT2D [26]. Intriguingly, METTL3 and WTAP knockdown. led to the loss of H3K4me3 marking in KLF1-bound promoters, leading to decreased binding capacity [26]. Similar results were obtained following m6A loss in human adult bone marrow HSPCs. Knockdown of MTase complex components revealed that their activity is required for early erythropoiesis but not for megakaryocytic differentiation [26]. Also, it was revealed that WTAP enhances the translation of PABPC1 and PABPC4, which promote the expression of β-globin in human HSPCs. Taken together, the data suggest that m6A modification is implicated in the regulation of erythropoiesis by maintaining the H3K4me3 marking at erythroid gene targets and promoting erythroid lineage development [26].
Interestingly, transcripts of c-MYC, BCL-2, and PTEN are also enriched in m6A marks [30]. Specifically, METTL3-mediated m6A targeting of these transcripts, promotes their translation and subsequently blocks differentiation of HSPCs [30]. MYC, a proto-oncogenic protein, is known to control the balance between HSC self-renewal and differentiation. During terminal erythroid differentiation, its protein levels are rapidly decreased [30]. Furthermore, key components of the MTase complex, seem to regulate Myc RNA stability and expression in HSPCs [20]. Specifically, siRNA depletion of Mettl3 in normal murine HSPCs, resulted in decreased MYC protein levels and blockage of HSC symmetric commitment, while in Mettl3 knockout mice, accumulation of immature erythroblasts, leading spleen enlargement and disruption of its architecture, were observed. Moreover, it was found that the expression of METTL14 and FTO is significantly lower in mature hematopoietic cells. No significant expression pattern was found for METTL3, WTAP, and ALKBH5. Consistent with these data, recent studies showed that ALKBH5 is not required for maintaining the function of normal HSCs [31,32]. Moreover, METTL14 knockdown in normal CD34+ cells, resulted in enhanced myeloid differentiation, colony formation inhibition, and decreased expression of the oncogenic transcription factors MYB and MYC [20]. Normally, METTL14 and FTO were found to promote stability of Myc mRNA via IGF2BP-mediated regulation of its expression [20]. Both YTHDF2 and IGF2BPs act as regulators of mRNA stability. However, contrary to the mRNA degradation- promoting function of YTHDF2, IGF2BPs were reported to promote mRNA stability [33]. In vivo models of murine acute myeloid leukemia (AML) and analysis of primary samples from patients with AML, revealed that the RNA-binding protein YBX1 affects the stability of m6A-enriched transcripts of apoptotic genes, such as Bcl2 and c-Myc, by interacting with IGF2BPs [34]. However, two independent studies demonstrated that YBX1 does not affect normal hematopoiesis or the function of HSCs [34,35]. It is also noteworthy, that a recent study in cancer cells, documented that the m6A reader protein YTHDF1, affects the transferrin receptor-mediated iron metabolism [36].
Analysis of the ENCODE Chip-Seq data in ChIPBase, revealed that METTL14 is negatively regulated by the SPI1 transcription factor, both in normal and pathological conditions, such as acute myeloid leukemia [37]. For the first time, Weng and coworkers [37], described the signaling axis SPI1-METTL14-MYB/MYC in normal myelopoiesis and leukemogenesis, and proposed that METTL14 could be a new therapeutic target to treat AML. This view is further supported by the fact that upon its deletion, HSCs are expanded, while AML malignant stem cells are selectively compromised [38]. Moreover, c-Myc expression is regulated by the RNA-binding motif protein 15 (RBM15), which was recently identified as a component of the MTase complex [39]. RBM15 was found to be highly expressed in the hematopoietic system [39,40]. Particularly, in vitro studies of cultured AML cells, showed that knock down of RBM15, leads to impaired differentiation and increased apoptosis rate. Also, knockout mouse studies, revealed that RBM15 is implicated in HSC expansion and differentiation, probably by affecting Notch signaling and c-Myc expression, respectively [40].
2.4. Ineffective Erythropoiesis and Apoptosis
Apoptosis and autophagy are cell death machineries widely associated with ineffective erythropoiesis and blood cancers. Notably, transcripts of pro-apoptotic and apoptotic genes were found to be enriched in m6A marks [41]. Several studies have shown that METTL3, which is the core component of the MTase complex, plays an important role in apoptosis, mainly by regulating the expression of Bcl-2 [42]. METTL3 can either inhibit or trigger cell death in different tissues by modulating Bcl-2, SHH/GLI, and ZNF750/FGF14 signaling pathways [42]. Additionally, the demethylases FTO and ALKBH5 primarily function to prevent apoptosis. FTO-knockdown in AML cell lines led to increased m6A levels in MYC mRNA, promoting its degradation and thus, enhancing apoptosis [43]. The m6A regulators are also involved in the autophagy pathway [44,45]. Specifically, it has been demonstrated that m6A RNA methylation controls the expression of autophagy-related gene (ATG) transcripts [44,45]. The m6A marked ATG transcripts are under YTHDF2-mediated degradation and therefore, autophagy is inhibited [44]. Further research is needed to decipher how the relationship between m6A and cell death could be implicated in the pathophysiology of hematologic diseases.
2.5. The Role of m6A Modification in Thalassemia
Recent studies revealed that m6A RNA modification affects the expression of genes involved in hemoglobin production, heme biosynthesis, iron metabolism and cell death mechanisms, such as apoptosis and autophagy [26,36,43]. These findings imply that m6A modification could be also a key participant in the pathogenesis of thalassemic hematopoiesis. Recently, Ruan et al [46], showed for the first time that m6A modification is linked to the phenotype of the Hemoglobin H Constant Spring variant, a severe form of α-thalassemia, caused by mutations of α-genes (--/αCSα) that affect the production of α-globin chains. For their study, immature erythrocytes were derived from peripheral blood of 16 thalassemia patients and 15 healthy individuals. Data acquired from qRT-PCR and m6A mRNA and lncRNA epitranscriptomic microarrays, revealed that the expression pattern of m6A-related factors, is significantly different between the two groups. Specifically, they discovered that the expression of the MTase complex components METTL16, WTAP, CBLL1, RBM15B, and ZC3H13 was lower in thalassemia, whereas the eraser protein ALKBH5, as well as the readers IGF2BP2 and YTHDF3, were overexpressed. Furthermore, they used m6A mapping to identify the differentially hypo-m6A-methylated transcripts in the two cohorts. Interestingly, the transcript of the anti-apoptotic protein BCL2A1 was downregulated in thalassemia patients. Therefore, in HbH Constant Spring thalassemia, apoptosis remains uninhibited as a result of the decreased expression of the BCL2A1, eventually leading to hemolytic anemia. According to their findings, the ALKBH5-mediated alteration of the m6A methylation status of BCL2A1, contributes to the pathogenesis of thalassemia. However, the exact mechanism by which m6A modification precisely regulates BCL2A1 expression remains to be elucidated.
Further experimental evidence for the role of METTL16 in the pathogenesis of hemoglobin H disease, was provided recently by studies [47], where the METTL16, YTHDF3, and SLC5A3 mRNAs were downregulated in HbH patients. Modification of the METTL16 levels in human erythroleukemia K562 cells, provided evidence that METTL16 affects the expression of hemoglobin via the IGF2BP3 reader, which regulates the phenotype og HbH disease [47].
3. Conclusions and Future Perspectives
In conclusion, a significant number of studies have confirmed the importance of m6A RNA methylation in normal hematopoiesis, and its implication in erythroid disorders. As discussed above. almost every step of HSC development and erythroid differentiation is controlled by m6A and its regulators. To our knowledge, the role of m6A RNA methylation in major clonal hematological malignancies, such as AML, has been well established. However, there is limited knowledge regarding the role of m6A modification in anemia-related blood disorders, such as the thalassemia syndromes. To date, there are only two studies [46,47], as discussed above, demonstrating the significant role of m6A modification in α-thalassemia. Therefore, it would be interesting to explore whether the m6A modification is implicated in the pathogenesis of other forms of thalassemia, which can be further exploited to attenuate the effects of ineffective erythropoiesis and improve the stability of globin mRNAs.
In addition, major questions regarding the molecular basis of m6A are yet to be addressed, including m6A stoichiometry and topology, as well as regulation of the MTase complex [48]. Further research is needed to elucidate the exact function of each component of the m6A regulatory network in governing cell fate decisions, and to explore the underlying mechanisms by which altered m6A levels affect gene expression. Indeed, recent studies have documented that m6A modification is a potent inducer of ribosome stalling, leading to ribosome collisions at the m6A sites, which in turn is followed by recruitment of the YTHDFs reader proteins, promoting RNA degradation [49]. The distribution of m6A marks is very consistent among tissues, probably due to the conserved gene architecture [48]. For that reason, any detected differentially regulated m6A sites, could be used to explore the role of other molecular pathways to m6A formation [48,50].
Looking to the future, targeting the m6A pathway could be exploited as a new therapeutic tool for treating hematopoietic disorders. However, focus should be given on developing therapeutic strategies that do not have any adverse effects on healthy cells [51]. Lastly, among the remaining major challenges to be addressed, is to determine experimentally whether these key m6A regulators or the newly discovered coding sequence-m6A decay (CMD) pathway [50], could be efficiently targeted by small molecule inhibitors [52], before moving the translation of such findings to the clinical stage.
Author Contributions
A-GV and EK wrote the manuscript. ED prepared the Figure. NPA and ED critically read the manuscript. All authors contributed to the article and approved the submitted version.
Funding
A-GV was supported by a Graduate Scholarship from the Bodossaki Foundation, Athens, Greece.
Acknowledgments
The Figure was created with Biorender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Kasper DM, Nicoli S. Epigenetic and epitranscriptomic factors make a mark on hematopoietic stem cell development. Curr Stem Cell Rep. 2018; 4(1):22-32. [CrossRef]
- Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015; 29(13):1343-1355. [CrossRef]
- Zhang Y, Geng X, Li Q, et al. m6A modification in RNA: biogenesis, functions and roles in gliomas. J Exp Clin Cancer Res. 2020; 39:192. [CrossRef]
- Jiang X, Liu B, Nie Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021; 6(1):74. [CrossRef]
- Zhu ZM, Huo FC, Pei DS. Function and evolution of RNA N6-methyladenosine modification. Int J Biol Sci. 2020; 16(11):1929-1940. [CrossRef]
- VU P, Cheng Y, Kharas G. The biology of m6A RNA methylation in normal and malignant hematopoiesis. Cancer Discov. 2019; 9(1):25-33/.
- Rieger MA, Schroeder T. Hematopoiesis. Cold Spring Harb Perspect Biol. 2012; 4(12):a008250.
- Gekas C, Rhodes KE, Van Handel B, Chhabra A, Ueno M, Mikkola HK. Hematopoietic stem cell development in the placenta. Int J Dev Biol. 2010; 54(6-7):1089-98. [CrossRef]
- Ottersbach K. Endothelial-to-haematopoietic transition: an update on the process of making blood. Biochem Soc Trans. 2019; 47(2):591-601. [CrossRef]
- Zhang C, Chen Y, Sun B, et al. m6A modulates haematopoietic stem and progenitor cell specification. Nature. 2017; 549(7671):273-276. [CrossRef]
- Lv J, Zhang Y, Gao S et al. Endothelial-specific m6A modulates mouse hematopoietic stem and progenitor cell development via Notch signaling. Cell Res. 2018; 28:249–252. [CrossRef]
- Grinenko T, Eugster A, Thielecke L, et al. Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nat Commun. 2018; 9:1898. [CrossRef]
- Wang H, Zuo H, Liu J, et al. Loss of YTHDF2-mediated m6A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. Cell Res. 2018; 28(10):1035-1038. [CrossRef]
- Mapperley C, van de Lagemaat LN, Lawson H, et al. The mRNA m6A reader YTHDF2 suppresses proinflammatory pathways and sustains hematopoietic stem cell function. J Exp Med. 2021; 218(3):e20200829. [CrossRef]
- Li Z, Qian P, Shao W, et al. Suppression of m6A reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018; 28(9):904-917. [CrossRef]
- Sheng Y, Wei J, Yu F, et al. A critical role of nuclear m6A reader YTHDC1 in Leukemogenesis by Regulating MCM Complex-Mediated DNA Replication Blood. 2021; blood.2021011707. [CrossRef]
- Yin R, Chang J, Li Y, et al. Differential m6A RNA landscapes across hematopoiesis reveal a role for IGF2BP2 in preserving hematopoietic stem cell function. Cell Stem Cell. 2022;29(1):149-159. [CrossRef]
- Yao QJ, Sang L, Lin M et al. Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res. 2018; 28(9):952-954. [CrossRef]
- Lee H, Bao S, Qian Y, et al. Stage-specific requirement for Mettl3-dependent m6A mRNA methylation during haematopoietic stem cell differentiation. Nat Cell Biol. 2019; 21(6):700-709. [CrossRef]
- Cheng Y, Luo H, Izzo F, et al. m6A RNA methylation maintains hematopoietic stem cell identity and symmetric commitment. Cell Rep. 2019; 28(7):1703-1716. [CrossRef]
- Gao Y, Vasic R, Song Y, et al. m6A modification prevents Formation of endogenous double-Stranded RNAs and deleterious innate immune responses during hematopoietic development. Immunity. 2020; 16; 52(6):1007-1021. [CrossRef]
- Dzierzak E, Philipsen S. Erythropoiesis: development and differentiation. Cold Spring Harb Perspect Med. 2013; 3(4):a011601.
- Valent P, Büsche G, Theurl I, et al. Normal and pathological erythropoiesis in adults: from gene regulation to targeted treatment concepts. Haematologica. 2018; 103(10):1593-1603. [CrossRef]
- Cheng H, Zheng Z, Cheng T. New paradigms on hematopoietic stem cell differentiation. Protein Cell. 2020; 11(1):34-44. [CrossRef]
- Scharf P, Broering MF, Oliveira da Rocha GH, Farsky SHP. Cellular and molecular mechanisms of environmental pollutants on hematopoiesis. Int JMol Sci. 2020; 21(19):6996. [CrossRef]
- Kuppers DA, Arora S, Lim Y, et al. N6-methyladenosine mRNA marking promotes selective translation of regulons required for human erythropoiesis. Nat Commun. 2019; 10:4596. [CrossRef]
- Barbarani G, Fugazza C, Strouboulis J, Ronchi AE. The pleiotropic effects of GATA1 and KLF1 in physiological erythropoiesis and in dyserythropoietic disorders. Front Physiol. 2019; 10:91. [CrossRef]
- Tallack MR, Perkins AC. KLF1 directly coordinates almost all aspects of terminal erythroid differentiation. IUBMB Life. 2010; 62(12):886-890. [CrossRef]
- Lithanatudom P, Leecharoenkiat A, Wannatung T, et al. A mechanism of ineffective erythropoiesis in β-thalassemia/HbE disease. Haematologica. 2010; 95(5):716-723.
- Vu LP, Pickering BF, Cheng Y, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017; 23(11):1369-1376. [CrossRef]
- Shen C, Sheng Y, Zhu AC, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell. 2020; 27(1):64-80. [CrossRef]
- Wang J, Li Y, Wang P, et al. Leukemogenic chromatin alterations promote AML leukemia stem cells via a KDM4C-ALKBH5-AXL signaling axis. Cell Stem Cell. 2020; 27(1):81-97.
- Huang H, Weng H, Sun W, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018; 20(9):1098.
- Feng M, Xie X, Han G, et al. YBX1 is required for maintaining myeloid leukemia cell survival by regulating BCL2 stability in an m6A-dependent manner. Blood. 2021; 138(1):71-85. [CrossRef]
- Jayavelu AK, Schnöder TM, Perner F, et al. Splicing factor YBX1 mediates persistence of JAK2-mutated neoplasms. Nature. 2020; 588: 157–163. [CrossRef]
- Ye J, Wang Z, Chen X, et al. YTHDF1-enhanced iron metabolism depends on TFRC m6A methylation. Theranostics. 2020; 10(26):12072-12089. [CrossRef]
- Weng H, Huang H, Wu H, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. 2018; 22(2):191-205.
- Paris J, Morgan M, Campos J, et al. Targeting the RNA m6A reader YTHDF2 selectively compromises cancer stem cells in acute myeloid leukemia. Cell Stem Cell. 2019; 25(1):137-148. [CrossRef]
- Raffel GD, Mercher T, Shigematsu H, et al. Ott1 (Rbm15) has pleiotropic roles in hematopoietic development. Proc Natl Acad Sci USA. 2007; 104:6001–6006.
- Niu C, Zhang J, Breslin P, et al. c-Myc is a target of RNA-binding motif protein 15 in the regulation of adult hematopoietic stem cell and megakaryocyte development. Blood. 2009; 114 (10): 2087–2096. [CrossRef]
- Chen J, Wang C, Fei W, Fang X, Hu X. Epitranscriptomic m6A modification in the stem cell field and its effects on cell death and survival. Am J Cancer Res. 2019; 9(4):752-764.
- Liu S, Zhuo L, Wang J, et al. METTL3 plays multiple functions in biological processes. Am J Cancer Res. 2020; 10(6):1631-1646.
- Xu L, Zhang C, Yin H, et al. RNA modifications act as regulators of cell death. RNA Biol. 2021; 1-11. [CrossRef]
- Chen X, Wang J, Tahir M, et al. Current insights into the implications of m6A RNA methylation and autophagy interaction in human diseases. Cell Biosci. 2021; 11:147. [CrossRef]
- Liu S, Li Q, Li G, et al. The mechanism of m6A methyltransferase METTL3- mediated autophagy in reversing gefitinib resistance in NSCLC cells by β-elemene. Cell Death Dis. 2020; 11:969. [CrossRef]
- Ruan H, Yang F, Deng L, et al. Human m6A-mRNA and lncRNA epitranscriptomic microarray reveal function of RNA methylation in hemoglobin H-Constant Spring disease. Sci Rep. 2021; 11:20478. [CrossRef]
- Liao Y, Zhang F, Yang F, et al. MRTTL16 participates in hemoglobin H disease through m6A modification. PLoS ONE 2024; 19(8):e0306043.
- Murakami S, Jaffrey S. Hidden codes in mRNA: Control of gene expression by m6A. Mol Cell. 2022; 82(12):2236-2251. [CrossRef]
- Murakami S, Olarenin-George AO, Liu JF, et al. m6A alters ribosome dynamics to initiate mRNA degradation. Cell, 2025; 188(14):3728-3743. [CrossRef]
- Corovic M, Hoch-Kraft P, Zhou Y, et al. m6A in the coding sequence: linking deposition, translation and decay. Trends Genet. 2025; S0168-9525(25)00132-5. [CrossRef]
- Sommerkamp P, Brown JA, Haltalli MLR, Mercier FE, Vu LP, Kranc KR. m6A RNA modifications: Key regulators of normal and malignant hematopoiesis. Exp Hematol. 2022;111:25-31. [CrossRef]
- Feng G, Wu Y, Hu Y, et al. Small molecule inhibitors targeting m6A regulators. J Hematol Oncol. 2024; 17:30. [CrossRef]
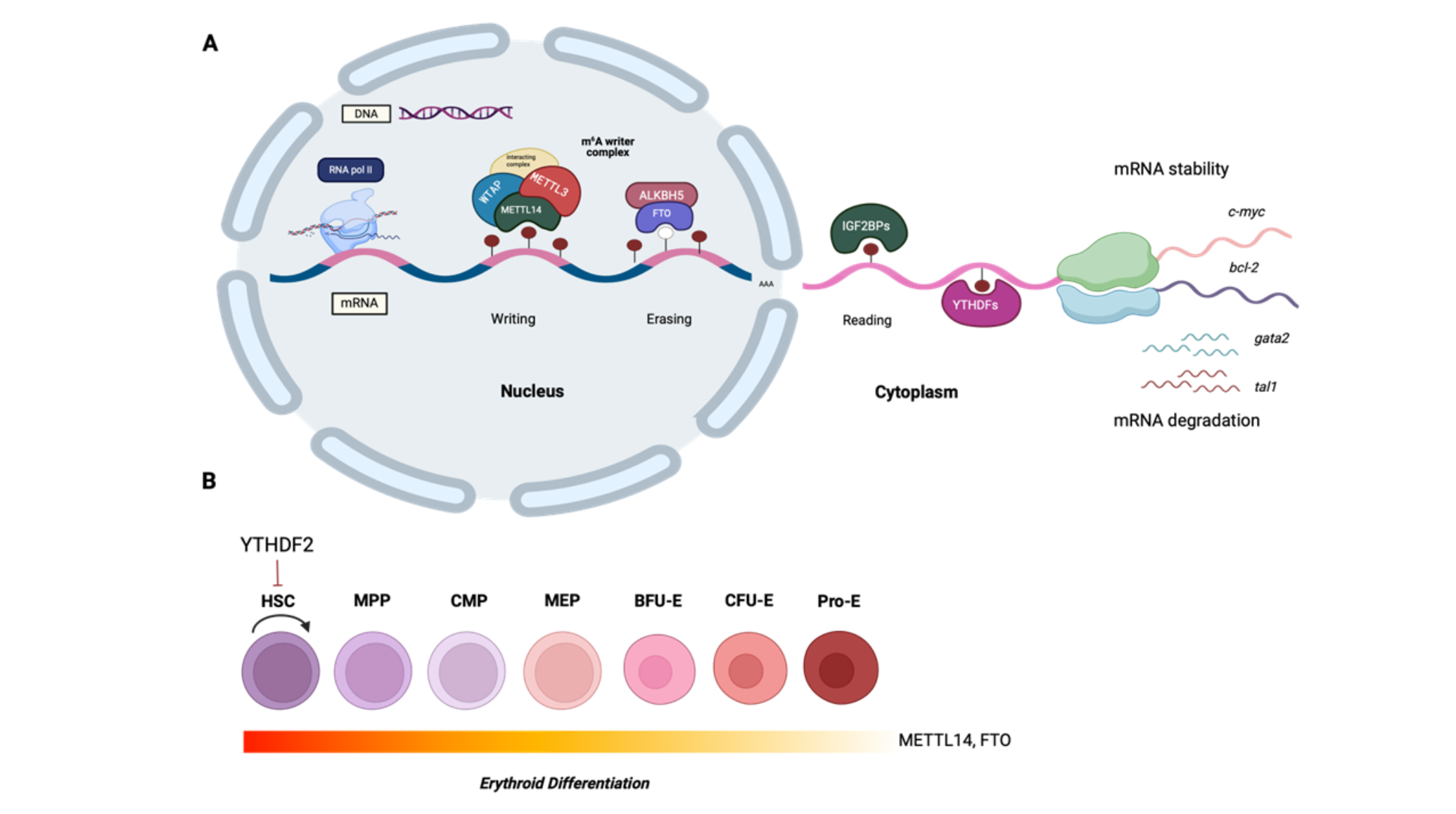
Figure 1.
Features of the m6A RNA modification and its role on hematopoietic stem cell (HSC) development. (A). The m6A methylation occurs in the nucleus by the MTase complex (writers), consisting of METTL3, which is the catalytic subunit, METTL14, WTAP, and other cofactors, and is erased by the demethylases ALKBH5 and FTO (erasers). The fate of the m6A-tagged mRNAs is determined by the m6A-binding proteins known as readers (e.g. YTH family of proteins, IGF2BP1, 2, and 3, HNRNPA2B1), whose actions can confer either mRNA stability, or induce mRNA degradation (B). Recent findings suggest that the m6A-regulators are key players during the hematopoietic stem cell (HSC) development and erythroid differentiation. For instance, YTHDF2 suppresses the capacity of HSCs for self-renewal, while METTL14 and FTO are active primarily in the early stages of normal erythroid differentiation.
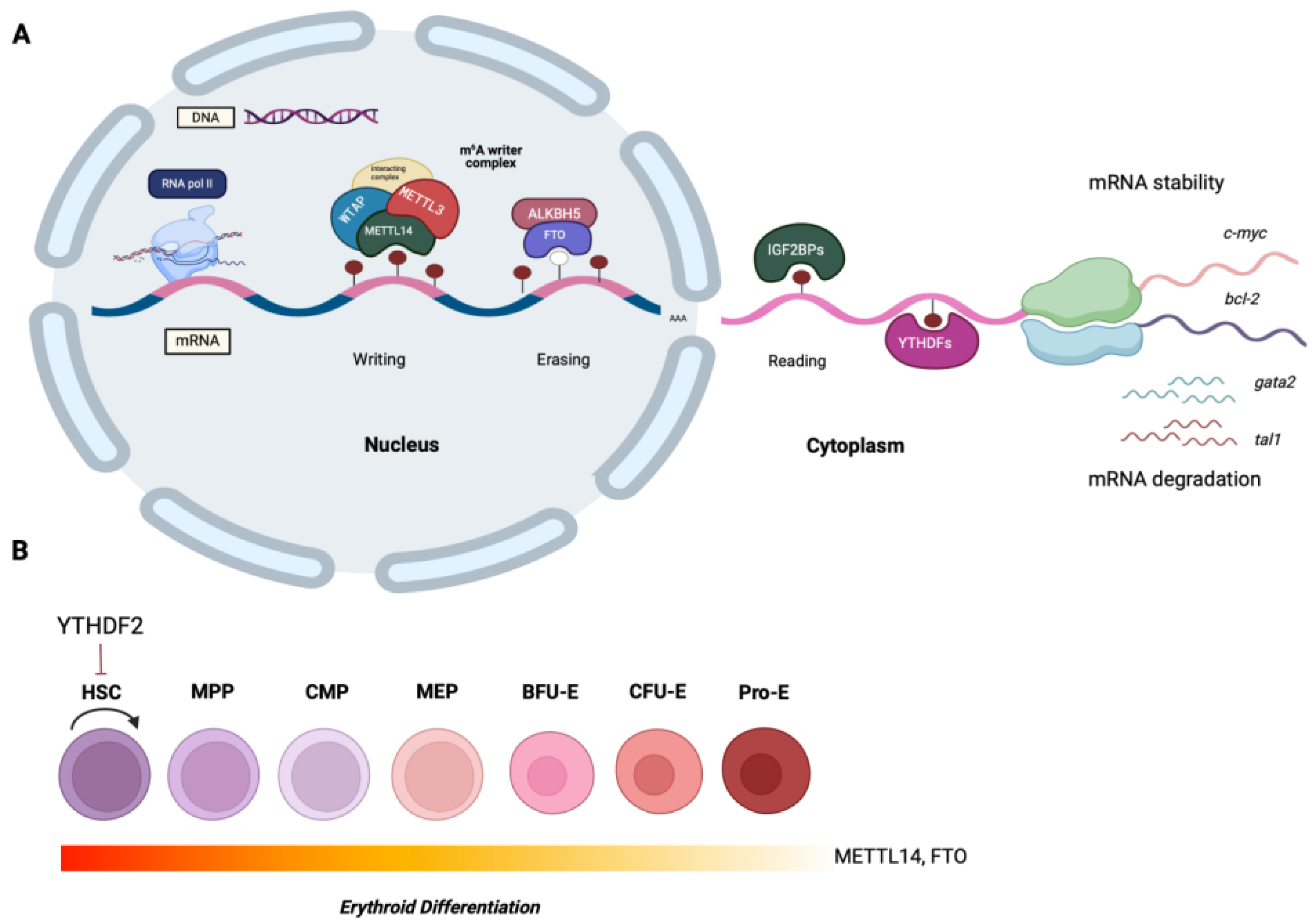
Figure 1.
Features of the m6A RNA modification and its role on hematopoietic stem cell (HSC) development. (A). The m6A methylation occurs in the nucleus by the MTase complex (writers), consisting of METTL3, which is the catalytic subunit, METTL14, WTAP, and other cofactors, and is erased by the demethylases ALKBH5 and FTO (erasers). The fate of the m6A-tagged mRNAs is determined by the m6A-binding proteins known as readers (e.g. YTH family of proteins, IGF2BP1, 2, and 3, HNRNPA2B1), whose actions can confer either mRNA stability, or induce mRNA degradation (B). Recent findings suggest that the m6A-regulators are key players during the hematopoietic stem cell (HSC) development and erythroid differentiation. For instance, YTHDF2 suppresses the capacity of HSCs for self-renewal, while METTL14 and FTO are active primarily in the early stages of normal erythroid differentiation.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



