Submitted:
21 July 2025
Posted:
22 July 2025
You are already at the latest version
Abstract
Vacuolated bone‑marrow precursors are a microscopic hallmark shared by diverse en-tities, from reversible copper deficiency to late‑onset autoinflammatory VEXAS syn-drome driven by somatic UBA1 mosaicism, and, more rarely, by high‑risk myeloid ne-oplasms. This molecular heterogeneity hampers timely diagnosis. We systematically searched PubMed, Web of Science and CENTRAL (December 2020–May 2025) following a PRISMA‑registered protocol (PROSPERO CRD420251082738) and identified 22 eligible series encompassing 818 adults. VEXAS accounted for 89% of cases, copper‑deficiency cytopenia for 9% and vacuolated‑cell MDS/AML for 2%. Median age increased from 58 years (copper deficiency) to 68 years (VEXAS), paralleling a shift from balanced to almost exclusive male prevalence, consistent with the X‑linked UBA1 biology. Inte-grating demographic, biochemical and genomic data, we designed a two‑step molecular screen—serum copper/ceruloplasmin followed, when normal, by hotspot UBA1 se-quencing—that correctly classified 97% of cases in < 5 days at a median laboratory cost of USD 173. Broad myeloid next‑generation sequencing was reserved for the residual 3% of atypical cytopenias. Our review clarifies the molecular landscape of vacuolated marrow cytopenias and delivers a pragmatic algorithm that bridges trace‑metal metabolism and somatic genomics, offering immediate translational value for leukemic and pre‑leukemic conditions.
Keywords:
vacuoles
; cytopenia
; copper deficiency
; VEXAS
; UBA1
; systematic review
; diagnostic algorithm
1. Introduction
Cytoplasmic vacuoles in bone-marrow precursors are an arresting microscopic finding but denote a heterogeneous spectrum of disorders. Classic literature ties large, lipid-poor vacuoles to copper-deficiency, alcohol misuse, or drug toxicity, yet these causes explain only a fraction of contemporary cases [1,2]. The 2020 discovery of somatic UBA1-mutant VEXAS (vacuoles, E1 enzyme, X-linked, autoinflammatory, somatic) syndrome further reshaped the differential: this late-onset autoinflammatory disease features prominent vacuoles and often presents with otherwise unexplained cytopenia [3]. More than 700 patients have since been reported, but data remain fragmented across small series, impeding reliable prevalence estimates and rational test ordering.
Clinicians thus face a practical dilemma: should they begin with serum copper studies, order targeted UBA1 sequencing, proceed directly to broad myeloid next-generation sequencing (NGS) panels, or pursue cytogenetics when confronted with vacuolated cytopenia? Without an evidence-weighted pathway, reversible entities such as copper-deficiency may be overlooked, whereas costly or invasive investigations may be over-used.
Here we consolidate current knowledge by two complementary approaches. First, we performed a PRISMA-aligned systematic review to quantify the relative frequencies, clinical profiles, and morphologic hallmarks of vacuolated-marrow cytopenias in adults. Second, we integrate these data with contemporary pathobiology to propose a pragmatic, four-step diagnostic algorithm that balances diagnostic yield, turnaround time, and cost. This narrative synthesis is intended to guide everyday hematology practice while highlighting areas where prospective studies are urgently needed.
2. Methods
2.1. Framework and Registration
2.2. Eligibility Criteria
We considered reports that analyzed adults (≥18 years) with single-lineage or multilineage cytopenia in whom cytoplasmic vacuoles were demonstrated in at least one hematopoietic precursor compartment on light microscopy. Acceptable study designs were cohort studies, cross-sectional analyses, or case series that enrolled three or more patients and provided primary diagnostic or outcome data. Single-case reports, narrative reviews, conference abstracts without full texts, pediatric studies, and non-English publications were excluded. We queried PubMed, Web of Science Core Collection, and CENTRAL on 31 May 2025 using a search window that began 31 December 2020, the day prior to the first description of VEXAS. To capture earlier literature on copper-deficiency cytopenia, we performed backward citation chasing and keyword hand-searching (2000–2020), which yielded three additional cohort reports not indexed by the electronic filters.
2.3. Information Sources and Search Strategy
With support from a medical librarian, we searched PubMed, Web of Science Core Collection, and Cochrane CENTRAL on 31 May 2025 (Supplementary Table S1). The core PubMed string was:
(“vacuole”[Title/Abstract] OR ”vacuolated”[Title/Abstract]) AND (“bone marrow”[Title/Abstract] OR ”marrow”[Title/Abstract]) AND (cytopenia OR anemia OR neutropenia OR thrombocytopenia) OR ”VEXAS” OR ”copper deficiency”
Filters were set to humans, English language, adults (≥18 years), and publication dates 31 December 2020–31 May 2025. Equivalent concept blocks adapted to Web of Science and CENTRAL retrieved additional citations. Backward citation chasing and consultation with content experts added three copper-deficiency series published between 2000 and 2020 that were not captured electronically. Full search syntax and filter details are provided in Supplementary Protocol S1.
2.4. Study Selection
All records were imported into Covidence, where duplicates were removed and two reviewers (A.T. and K.U.) independently screened titles and abstracts before assessing full texts. Disagreements were resolved by discussion or, when necessary, adjudication by a third reviewer. Inter-rater agreement was quantified with Cohen’s κ. A PRISMA flow diagram summarizes the process in Figure 1.
2.5. Data Extraction
We constructed a piloted REDCap form to record bibliographic details, study design, patient demographics, cytopenia pattern, confirmatory tests (serum copper, ceruloplasmin, UBA1 sequencing, cytogenetics, myeloid NGS), treatments, and hematologic or inflammatory outcomes. When critical data were missing, corresponding authors were contacted once for clarification.
2.6. Risk-of-Bias Appraisal
Methodological quality was judged with ROBINS-I [6], rating each study across seven domains and assigning an overall level of low, moderate, serious, or critical risk of bias. The full assessment appears in Supplementary Table S2.
2.7. Data Synthesis
Because both clinical context and methodology varied widely—reflected in I² values exceeding 80% for key prevalence estimates—we refrained from formal meta-analysis. Instead, we pooled simple counts with exact 95% confidence intervals, repeated the calculations after excluding studies deemed at serious risk of bias, compared molecular and morphologic signatures qualitatively, and incorporated turnaround-time and Medicare fee-schedule costs to build the four-step diagnostic algorithm.
2.8. Statistical Analysis
All analyses were run in EZR version 1.68 [7]. P values are two-sided and reported solely for exploratory purposes, with p < 0.05 considered nominally significant.
3. Results and Discussion
We identified 22 eligible studies (Table 1) after screening 218 full-texts; the PRISMA flow diagram appears in Figure 1. Collectively these reports describe 818 adults with at least one cytopenia accompanied by light-microscopic marrow vacuolization. Etiology was overwhelmingly skewed toward two disorders: UBA1-mutant VEXAS syndrome accounted for 727 cases (89%), copper-deficiency cytopenia for 70 (9%), and a single institutional cohort contributed 21 cases (2%) of vacuolated-cell myelodysplastic syndrome (MDS)/acute myeloid leukemia (AML) .
3.1. Demographic and Hematologic Landscape
Pooling 818 adults from 22 studies, we observed a distinct age-and-sex gradient across etiologies. Copper-deficiency cytopenia occurred at a median age of 57.5 years and showed near gender balance (41% male), whereas VEXAS patients were older (median 68.0 years) and almost exclusively male (98%), reflecting the X-linked biology of UBA1 (Figure 2, Table 2). The small vacuolated-cell MDS/AML cohort sat between these poles (median 66.0 years; 71% male). Anemia was nearly universal in both copper-deficiency (92%) and VEXAS (91%); neutropenia predominated in copper-deficiency (61%), whereas thrombocytopenia was the most frequent deficit in VEXAS (40%). Notably, 76% of copper-deficient patients had at least one reversible predisposing factor—zinc excess, proton-pump inhibitor use, bariatric surgery, or malabsorption [28]—summarized in Table 3.
3.2. Molecular and Morphologic Correlates
Pathogenic UBA1 p.Met41 variants were detected in every VEXAS cohort and in none of the copper-deficiency or vacuolated-cell MDS/AML series. The latter group instead harbored high-risk genomic lesions such as monosomy 7 and TP53 disruption [8]. Mechanistically, three non-overlapping pathways emerged: trace-element depletion in copper-deficiency, clonal ubiquitylation failure in VEXAS, and genomic instability with defective autophagy in vacuolated-cell MDS/AML [29,30,31,32]. Drug-related or toxic vacuolization—largely attributed to linezolid, disulfiram, or alcohol—was uncommon and is detailed in Table 4.
3.3. Algorithm Performance
When the four-step pathway (Figure 3) was applied to every literature case, Step 1—evaluation for reversible causes and serum copper/ceruloplasmin assays (Table 4)—followed, when normal, by Step 2—hot-spot UBA1 sequencing guided by VEXAS red-flags (Table 5) correctly classified 97% of vacuolated-marrow cytopenias. The combined laboratory charge for Steps 1–2 was approximately USD 173 with a median turnaround of five days, about one-fifteenth the cost and two weeks faster than reflex broad myeloid NGS (Step 3) [46,47,48,49]. Only UBA1–wild-type patients with persistent cytopenia underwent morphology-guided cytogenetics and targeted NGS; Step 4 was reserved for rarer mimics once common entities were excluded (Table 4). Excluding studies at serious ROBINS-I risk did not alter the 97% yield (data not shown), underscoring the robustness of the sequence.
3.4. Risk of Bias Overview
ROBINS-I appraisal assigned an overall moderate risk of bias to 24 studies and a serious risk to 3; no study reached the critical-risk tier (Supplementary Table S2). Serious judgments were driven by substantial missing outcome data or non-validated outcome measurements in two VEXAS cohorts and one copper-deficiency series, all of which were retrospective case–series without prespecified protocols. The remaining studies were classified as moderate risk chiefly because of incomplete follow-up or selective outcome reporting, typical limitations of retrospective designs. Repeating the diagnostic-algorithm analysis after excluding the three serious-risk studies reproduced the same 97% classification rate (data not shown), indicating that our principal findings are robust despite underlying methodological heterogeneity.
3.5. Summary of Key Findings
In modern hematology practice, two readily recognizable conditions—copper-deficiency and VEXAS syndrome—account for 97% of adult cytopenias marked by vacuolated marrow precursors. A straightforward, cost-conscious sequence that begins with reversible-cause screening and serum copper / ceruloplasmin testing, followed by focused UBA1 hotspot sequencing, correctly classifies nearly all patients within a few days while reserving invasive or high-cost investigations for the small minority of atypical cases. These data form the empirical backbone of the pragmatic diagnostic algorithm in Figure 4 and provide a clear rationale for prospective validation studies.
3.6. What This Review Adds
By aggregating 818 adults from 22 studies, we demonstrate that copper-deficiency cytopenia and UBA1-mutant VEXAS syndrome explain 97% of modern cases of cytopenia with vacuolated marrow precursors, whereas vacuolated-cell MDS or AML and other mimics together account for only 3%. This quantitative clarification moves the field beyond anecdote and supplies the empirical foundation for a streamlined, mechanism-oriented diagnostic pathway.
3.7. Mechanistic Perspectives on Vacuole Biology
Marrow vacuoles arise through at least three biologically distinct yet light-microscopically indistinguishable routes.
Copper deficiency disrupts cupro-oxidase activity, stalls mitochondrial respiration, and traps iron, producing lipid-poor vacuoles that disappear within weeks of supplementation [32]. Retrospective re-analysis of “refractory anemia” cohorts has revealed that a fraction represented occult copper deficiency [1,10].
VEXAS syndrome follows a proteotoxic pathway. Somatic UBA1 p.Met41 variants eliminate the cytoplasmic E1 isoform, abolish K48-linked polyubiquitination, and arrest macro-autophagy. Single-cell proteomics and ultrastructural studies reveal ribosome-lined, LC3-positive pre-autophagosomal structures, swollen rough-endoplasmic-reticulum cisternae, and disrupted mitochondrial cristae [20,50,51]. A CRISPR knock-in model reproduces these vacuoles, whereas lentiviral expression of wild-type UBA1 rescues them [52].
Vacuolated-cell MDS/AML appears to involve a third mechanism. High-risk lesions such as monosomy 7 and TP53 disruption impair autophagosome maturation and trigger p53-dependent metabolic stress, yielding larger, irregular vacuoles and a rapid leukemic trajectory [37,38,40,53].
Historical copper-deficiency series were never sequenced for UBA1, and early VEXAS reports often lacked trace-element data; diagnostic overlap therefore persists. Because light microscopy cannot differentiate these vacuole types, laboratory context is decisive: serum copper and ceruloplasmin identify reversible deficiency; targeted UBA1 sequencing confirms or excludes VEXAS; and cytogenetics with focused myeloid NGS uncovers high-risk clonal disease. Prospective studies pairing deep UBA1 sequencing with copper profiling in newly diagnosed MDS or unexplained cytopenia will be essential to resolve residual overlap and to determine whether vacuolization is causal or merely a marker of upstream injury.
3.8. Clinical Implications
A two-step screen—serum copper/ceruloplasmin followed, when normal, by UBA1 hotspot sequencing—classifies nearly all patients within five days for a median laboratory cost of USD 173, a fraction of the expense and turnaround associated with broad myeloid NGS panels. Early recognition of copper deficiency permits curative supplementation, whereas prompt confirmation of VEXAS redirects management toward durable immunomodulation or clinical-trial enrollment rather than empiric cytoreduction. Cytogenetics and 20-gene NGS are reserved for the UBA1-negative minority, aligning resource use with diagnostic yield and minimizing incidental findings.
3.9. Limitations and Potential Biases
Interpretation is tempered by several constraints. First, the vacuolated-cell MDS/AML category rests on a single 21-patient cohort [8]; its observed prevalence of 2% almost certainly underestimates the true burden. Second, many copper-deficiency reports pre-date VEXAS and lacked UBA1 testing, whereas marginally low copper levels in older VEXAS cases may have inflated the nutritional category. Third, the virtual exclusivity of male subjects introduces ascertainment bias and suggests that low-level UBA1 mosaicism in women or non-binary individuals is under-recognized. Fourth, all included studies were retrospective, methodologically heterogeneous, and often incomplete in outcome reporting, leaving residual confounding despite sensitivity analyses. Finally, publication bias likely favored dramatic or diagnostically challenging cases, skewing the spectrum toward VEXAS and high-risk neoplasia while under-representing routine copper deficiency that resolves with supplementation.
3.10. Future Directions
Prospective, multicenter implementation of the four-step algorithm will verify its accuracy, turnaround, and cost-effectiveness and will capture women with low-level UBA1 mosaicism—an under-studied population. Randomized trials are needed to compare IL-6 or JAK-STAT blockade, hypomethylating therapy, and ubiquitin-pathway modulators in VEXAS [13,14,54]. Parallel copper profiling in newly diagnosed MDS and AML, integrated with deep UBA1 sequencing, should clarify whether trace-element imbalance or occult VEXAS remains hidden within clonal myeloid disease. Formal health-economic modeling across diverse health-care systems will establish whether the proposed pathway delivers value beyond its biological rationale.
4. Conclusions
Marrow vacuoles are no longer an enigmatic microscopic finding. An evidence-weighted sequence that begins with inexpensive, high-yield tests leads rapidly to diagnoses that are either readily reversible or require targeted immunomodulation. Routine measurement of serum copper and focused UBA1 hotspot testing spares most patients invasive, costly investigations, reserving advanced genomics for the few who truly need them. The pathway presented here offers a pragmatic template for current practice and a platform for prospective validation and therapeutic innovation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Supplementary Protocol S1 including PRISMA 2020 Checklist and PROSPERO Protocol; Supplementary Table S1: Search strategies used for PubMed, Web of Science, and CENTRAL; Supplementary Table S2: Risk-of-bias summary for the 24 included studies.
Author Contributions
Conceptualization, A.T.; formal analysis, A.T. and K.U.; investigation, A.T., K.U. and J.L.E.; resources, M.T., S.S., Y.K., and M.E.; data curation, M.T., S.S., Y.K., and M.E.; writing—original draft preparation, A.T.; writing—review and editing, A.T., K.U. and J.L.E.; project administration, A.T.; funding acquisition, A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by a Grant-in-Aid for Scientific Research (KAKENHI 24K11527) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and by a Health and Labour Sciences Research Grant (23KC2009) from the Ministry of Health, Labour and Welfare of Japan. The funding agencies had no role in study design; collection, analysis, or interpretation of data; or manuscript preparation.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Aichi Medical University Institutional Review Board (IRB; approval No. AMU-2025-077).
Informed Consent Statement
Because only de-identified data were analyzed, the IRB granted a waiver of informed consent.
Data Availability Statement
The data that support the findings of this review—including the extraction sheets and the decision-tree workbook—are available from the corresponding author without undue reservation.
Acknowledgments
We would like to thank all the clinicians and administrative staff who contributed to this study. We are also deeply grateful to Ms. Michiko Ichikawa, librarian at Aichi Medical University, for her invaluable assistance with the comprehensive literature search.
Conflicts of Interest
A.T. has received research funding from AIR WATER, lecture fees from Novartis Pharmaceuticals, and donation funding from Chugai Pharmaceutical and Kyowa Kirin. The remaining authors declare no competing financial or non-financial interests.
References
- Huff, J.D.; Keung, Y.K.; Thakuri, M.; Beaty, M.W.; Hurd, D.D.; Owen, J.; Molnar, I. Copper deficiency causes reversible myelodysplasia. Am J Hematol 2007, 82, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Gregg, X.T.; Reddy, V.; Prchal, J.T. Copper deficiency masquerading as myelodysplastic syndrome. Blood 2002, 100, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Ospina Cardona, D.; Wu, Z. , et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N Engl J Med 2020, 383, 2628–2638. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. bmj 2021, 372. [Google Scholar]
- NIHR. PROSPERO: International prospective register of systematic reviews. Available online: https://www.crd.york.ac.uk/PROSPERO/home.
- Sterne, J.A.; Hernan, M.A.; Reeves, B.C.; Savovic, J.; Berkman, N.D.; Viswanathan, M.; Henry, D.; Altman, D.G.; Ansari, M.T.; Boutron, I. , et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016, 355, i4919. [Google Scholar] [CrossRef]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant 2013, 48, 452–458. [Google Scholar] [CrossRef]
- Gurnari, C.; Pagliuca, S.; Durkin, L.; Terkawi, L.; Awada, H.; Kongkiatkamon, S.; Zawit, M.; Hsi, E.D.; Carraway, H.E.; Rogers, H.J. , et al. Vacuolization of hematopoietic precursors: an enigma with multiple etiologies. Blood 2021, 137, 3685–3689. [Google Scholar] [CrossRef]
- Uchino, K.; Quang, L.V.; Enomoto, M.; Nakano, Y.; Yamada, S.; Matsumura, S.; Kanasugi, J.; Takasugi, S.; Nakamura, A.; Horio, T. , et al. Cytopenia associated with copper deficiency. EJHaem 2021, 2, 729–737. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Kumar, N.; Hogan, W.J.; Murray, J.A. Copper deficiency in celiac disease. J Clin Gastroenterol 2009, 43, 162–164. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Kumar, N.; Li, C.Y.; Phyliky, R.L.; Hogan, W.J. Hematological manifestations of copper deficiency: a retrospective review. Eur J Haematol 2008, 80, 523–531. [Google Scholar] [CrossRef]
- Vitale, A.; Leone, F.; Caggiano, V.; Hinojosa-Azaola, A.; Martin-Nares, E.; Guaracha-Basanez, G.A.; Torres-Ruiz, J.; Ayumi Kawakami-Campos, P.; Hissaria, P.; Callisto, A. , et al. Efficacy and safety of conventional disease-modifying antirheumatic drugs in VEXAS syndrome: real-world data from the international AIDA network. Front Pharmacol 2025, 16, 1539756. [Google Scholar] [CrossRef]
- Johansen, M.M.; El Fassi, D.; Nielsen, C.T.H.; Krintel, S.B.; Graudal, N.; Hansen, J.W. Treatment experiences with focus on IL-6R inhibition in patients with VEXAS syndrome and a case of remission with azacytidine treatment. Rheumatology (Oxford) 2025, 64, 826–830. [Google Scholar] [CrossRef]
- Hadjadj, J.; Nguyen, Y.; Mouloudj, D.; Bourguiba, R.; Heiblig, M.; Aloui, H.; McAvoy, C.; Lacombe, V.; Ardois, S.; Campochiaro, C. , et al. Efficacy and safety of targeted therapies in VEXAS syndrome: retrospective study from the FRENVEX. Ann Rheum Dis 2024, 83, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Tsuchida, N.; Uchiyama, Y.; Horita, N.; Kobayashi, S.; Kishimoto, M.; Kobayashi, D.; Matsumoto, H.; Asano, T.; Migita, K. , et al. Efficient detection of somatic UBA1 variants and clinical scoring system predicting patients with variants in VEXAS syndrome. Rheumatology (Oxford) 2024, 63, 2056–2064. [Google Scholar] [CrossRef]
- Kusne, Y.; Ghorbanzadeh, A.; Dulau-Florea, A.; Shalhoub, R.; Alcedo, P.E.; Nghiem, K.; Ferrada, M.A.; Hines, A.; Quinn, K.A.; Panicker, S.R. , et al. Venous and arterial thrombosis in patients with VEXAS syndrome. Blood 2024, 143, 2190–2200. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L.; Caratsch, L.; Lotscher, F.; Seitz, L.; Seitz, P.; Coattrenec, Y.; Seebach, J.; Vilinovszki, O.; Balabanov, S.; Nilsson, J. , et al. VEXAS syndrome: a Swiss national retrospective cohort study. Swiss Med Wkly 2024, 155, 3879. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; Bodian, D.L.; Shah, V.; Mirshahi, U.L.; Kim, J.; Ding, Y.; Magaziner, S.J.; Strande, N.T.; Cantor, A.; Haley, J.S. , et al. Estimated Prevalence and Clinical Manifestations of UBA1 Variants Associated With VEXAS Syndrome in a Clinical Population. JAMA 2023, 329, 318–324. [Google Scholar] [CrossRef]
- Mascaro, J.M.; Rodriguez-Pinto, I.; Poza, G.; Mensa-Vilaro, A.; Fernandez-Martin, J.; Caminal-Montero, L.; Espinosa, G.; Hernandez-Rodriguez, J.; Diaz, M.; Rita-Marques, J. , et al. Spanish cohort of VEXAS syndrome: clinical manifestations, outcome of treatments and novel evidences about UBA1 mosaicism. Ann Rheum Dis 2023, 82, 1594–1605. [Google Scholar] [CrossRef]
- Hines, A.S.; Koster, M.J.; Bock, A.R.; Go, R.S.; Warrington, K.J.; Olteanu, H.; Lasho, T.L.; Patnaik, M.M.; Reichard, K.K. Targeted testing of bone marrow specimens with cytoplasmic vacuolization to identify previously undiagnosed cases of VEXAS syndrome. Rheumatology (Oxford) 2023, 62, 3947–3951. [Google Scholar] [CrossRef]
- Islam, S.; Cullen, T.; Sumpton, D.; Damodaran, A.; Heath, D.; Bosco, A.; Doo, N.W.; Kidson-Gerber, G.; Cheong, A.; Lawford, R. , et al. VEXAS syndrome: lessons learnt from an early Australian case series. Intern Med J 2022, 52, 658–662. [Google Scholar] [CrossRef]
- Mekinian, A.; Zhao, L.P.; Chevret, S.; Desseaux, K.; Pascal, L.; Comont, T.; Maria, A.; Peterlin, P.; Terriou, L.; D’Aveni Piney, M. , et al. A Phase II prospective trial of azacitidine in steroid-dependent or refractory systemic autoimmune/inflammatory disorders and VEXAS syndrome associated with MDS and CMML. Leukemia 2022, 36, 2739–2742. [Google Scholar] [CrossRef]
- Georgin-Lavialle, S.; Terrier, B.; Guedon, A.F.; Heiblig, M.; Comont, T.; Lazaro, E.; Lacombe, V.; Terriou, L.; Ardois, S.; Bouaziz, J.D. , et al. Further characterization of clinical and laboratory features in VEXAS syndrome: large-scale analysis of a multicentre case series of 116 French patients. Br J Dermatol 2022, 186, 564–574. [Google Scholar] [CrossRef]
- Comont, T.; Heiblig, M.; Riviere, E.; Terriou, L.; Rossignol, J.; Bouscary, D.; Rieu, V.; Le Guenno, G.; Mathian, A.; Aouba, A. , et al. Azacitidine for patients with Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic syndrome (VEXAS) and myelodysplastic syndrome: data from the French VEXAS registry. Br J Haematol 2022, 196, 969–974. [Google Scholar] [CrossRef]
- Ferrada, M.A.; Savic, S.; Cardona, D.O.; Collins, J.C.; Alessi, H.; Gutierrez-Rodrigues, F.; Kumar, D.B.U.; Wilson, L.; Goodspeed, W.; Topilow, J.S. , et al. Translation of cytoplasmic UBA1 contributes to VEXAS syndrome pathogenesis. Blood 2022, 140, 1496–1506. [Google Scholar] [CrossRef]
- Tsuchida, N.; Kunishita, Y.; Uchiyama, Y.; Kirino, Y.; Enaka, M.; Yamaguchi, Y.; Taguri, M.; Yamanaka, S.; Takase-Minegishi, K.; Yoshimi, R. , et al. Pathogenic UBA1 variants associated with VEXAS syndrome in Japanese patients with relapsing polychondritis. Ann Rheum Dis 2021, 80, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Ferrada, M.A.; Sikora, K.A.; Luo, Y.; Wells, K.V.; Patel, B.; Groarke, E.M.; Ospina Cardona, D.; Rominger, E.; Hoffmann, P.; Le, M.T. , et al. Somatic Mutations in UBA1 Define a Distinct Subset of Relapsing Polychondritis Patients With VEXAS. Arthritis Rheumatol 2021, 73, 1886–1895. [Google Scholar] [CrossRef] [PubMed]
- Takami, A.; Uchino, K. Discovering the hidden link: hematological disorders caused by copper deficiency. Int J Hematol 2025, 10.1007/s12185-025-04036-7. [CrossRef]
- Watson, A.S.; Mortensen, M.; Simon, A.K. Autophagy in the pathogenesis of myelodysplastic syndrome and acute myeloid leukemia. Cell Cycle 2011, 10, 1719–1725. [Google Scholar] [CrossRef]
- Pierro, F.; Fazio, M.; Murdaca, G.; Stagno, F.; Gangemi, S.; Allegra, A. Oxidative Stress and Mitochondrial Dysfunction in Myelodysplastic Syndrome: Roles in Development, Diagnosis, Prognosis, and Treatment. Int J Mol Sci 2025, 26. [Google Scholar] [CrossRef]
- Savy, C.; Bourgoin, M.; Cluzeau, T.; Jacquel, A.; Robert, G.; Auberger, P. VEXAS, Chediak-Higashi syndrome and Danon disease: myeloid cell endo-lysosomal pathway dysfunction as a common denominator? Cell Mol Biol Lett 2025, 30, 12. [Google Scholar] [CrossRef]
- Jensen, E.L.; Gonzalez-Ibanez, A.M.; Mendoza, P.; Ruiz, L.M.; Riedel, C.A.; Simon, F.; Schuringa, J.J.; Elorza, A.A. Copper deficiency-induced anemia is caused by a mitochondrial metabolic reprograming in erythropoietic cells. Metallomics 2019, 11, 282–290. [Google Scholar] [CrossRef]
- Chetty-Raju, N.; Cook, R.; Erber, W.N. Vacuolated neutrophils in ethanol toxicity. Br J Haematol 2004, 127, 478. [Google Scholar] [CrossRef]
- Elbadry, M.I.; Mabed, M. Bone Marrow Vacuolization at the Crossroads of Specialties: Molecular Insights and Diagnostic Challenges. Eur J Haematol 2025. [CrossRef]
- Liapis, K.; Vrachiolias, G.; Spanoudakis, E.; Kotsianidis, I. Vacuolation of early erythroblasts with ring sideroblasts: a clue to the diagnosis of linezolid toxicity. Br J Haematol 2020, 190, 809. [Google Scholar] [CrossRef] [PubMed]
- Rosenbach, L.M.; Caviles, A.P.; Mitus, W.J. Chloramphenicol toxicity. Reversible vacuolization of erythroid cells. N Engl J Med 1960, 263, 724–728. [Google Scholar] [CrossRef] [PubMed]
- Ballo, O.; Stratmann, J.; Serve, H.; Steffen, B.; Finkelmeier, F.; Brandts, C. Blast vacuolization in AML patients indicates adverse-risk AML and is associated with impaired survival after intensive induction chemotherapy. PLoS One 2019, 14, e0223013. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, Y.H.; Mina, A.; Altman, J.K.; Kim, K.Y.; Zhang, Y.; Lu, X.; Jennings, L.; Sukhanova, M. Unique morphologic and genetic characteristics of acute myeloid leukemia with chromothripsis: a clinicopathologic study from a single institution. Hum Pathol 2020, 98, 22–31. [Google Scholar] [CrossRef]
- Ballard, H.S. The hematological complications of alcoholism. Alcohol Health Res World 1997, 21, 42–52. [Google Scholar] [CrossRef]
- Song, J.; Shang, B.; Pei, Y.; Shi, M.; Niu, X.; Dou, L.; Drokow, E.K.; Xu, F.; Bai, Y.; Sun, K. A higher percentage of leukemic blasts with vacuoles predicts unfavorable outcomes in patients with acute myeloid leukemia. Leuk Res 2021, 109, 106638. [Google Scholar] [CrossRef]
- Khambatta, S.; Nguyen, D.L.; Beckman, T.J.; Wittich, C.M. Kearns-Sayre syndrome: a case series of 35 adults and children. Int J Gen Med 2014, 7, 325–332. [Google Scholar] [CrossRef]
- Bellanne-Chantelot, C.; Schmaltz-Panneau, B.; Marty, C.; Fenneteau, O.; Callebaut, I.; Clauin, S.; Docet, A.; Damaj, G.L.; Leblanc, T.; Pellier, I. , et al. Mutations in the SRP54 gene cause severe congenital neutropenia as well as Shwachman-Diamond-like syndrome. Blood 2018, 132, 1318–1331. [Google Scholar] [CrossRef]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Zanelli, M.; Ruggeri, L.; Sanguedolce, F.; Zizzo, M.; Martino, G.; Genua, A.; Ascani, S. Parvovirus B19 Infection in a Patient with Common Variable Immunodeficiency. Mediterr J Hematol Infect Dis 2021, 13, e2021026. [Google Scholar] [CrossRef] [PubMed]
- Inai, K.; Noriki, S.; Iwasaki, H.; Naiki, H. Risk factor analysis for bone marrow histiocytic hyperplasia with hemophagocytosis: an autopsy study. Virchows Arch 2014, 465, 109–118. [Google Scholar] [CrossRef] [PubMed]
- CMS. 25CLABQ2. Available online: https://www.cms.gov/medicare/payment/fee-schedules/clinical-laboratory-fee-schedule-clfs/files/25CLABQ2.
- UNC. UBA1Q Mutation Quantitative Detection, DDPCR (VEXAS Syndrome). Available online: https://www.uncmedicalcenter.org/mclendon-clinical-laboratories/available-tests/uba1q-mutation-quantitative-detection-ddpcr-vexas-syndrome/.
- University, J.H. Myeloid Panel, NGS, Blood. Available online: https://pathology.jhu.edu/test-directory/leukemia-panel-ngs-blood-2.
- Vianna, P.G.; Press, R.D.; Stehr, H.; Yang, F.; Gojenola, L.; Zehnder, J.L.; Gotlib, J. Clinical Utility of a Multi-Gene Next-Generation Sequencing Myeloid Panel in an Academic Hematology Practice. Blood 2019, 134, 1408–1408. [Google Scholar] [CrossRef]
- Wu, Z.; Gao, S.; Gao, Q.; Patel, B.A.; Groarke, E.M.; Feng, X.; Manley, A.L.; Li, H.; Ospina Cardona, D.; Kajigaya, S. , et al. Early activation of inflammatory pathways in UBA1-mutated hematopoietic stem and progenitor cells in VEXAS. Cell Rep Med 2023, 4, 101160. [Google Scholar] [CrossRef] [PubMed]
- Cardoneanu, A.; Rezus, II; Burlui, A. M.; Richter, P.; Bratoiu, I.; Mihai, I.R.; Macovei, L.A.; Rezus, E. Autoimmunity and Autoinflammation: Relapsing Polychondritis and VEXAS Syndrome Challenge. Int J Mol Sci 2024, 25, 2261. [Google Scholar] [CrossRef]
- Chiaramida, A.; Obwar, S.G.; Nordstrom, A.E.H.; Ericsson, M.; Saldanha, A.; Ivanova, E.V.; Griffin, G.K.; Khan, D.H.; Belizaire, R. Sensitivity to targeted UBA1 inhibition in a myeloid cell line model of VEXAS syndrome. Blood Adv 2023, 7, 7445–7456. [Google Scholar] [CrossRef]
- Lin, J.F.; Chi, C.W.; Huang, Y.C.; Tsai, T.H.; Chen, Y.J. Anti-Cancer Effects of Oxygen-Atom-Modified Derivatives of Wasabi Components on Human Leukemia Cells. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef]
- Koster, M.J.; Lasho, T.L.; Olteanu, H.; Reichard, K.K.; Mangaonkar, A.; Warrington, K.J.; Patnaik, M.M. VEXAS syndrome: Clinical, hematologic features and a practical approach to diagnosis and management. Am J Hematol 2024, 99, 284–299. [Google Scholar] [CrossRef]
Figure 1.
PRISMA 2020 study selection diagram. Electronic searches of PubMed, Web of Science, and the Cochrane Central Register of Controlled Trials (CENTRAL) yielded 271 records, of which 58 were duplicates. Five additional records were identified by hand searching, three of which dated from 2000–2020 and described copper deficiency cytopenia. After full text review of 44 articles, 22 studies met all inclusion criteria.
Figure 1.
PRISMA 2020 study selection diagram. Electronic searches of PubMed, Web of Science, and the Cochrane Central Register of Controlled Trials (CENTRAL) yielded 271 records, of which 58 were duplicates. Five additional records were identified by hand searching, three of which dated from 2000–2020 and described copper deficiency cytopenia. After full text review of 44 articles, 22 studies met all inclusion criteria.
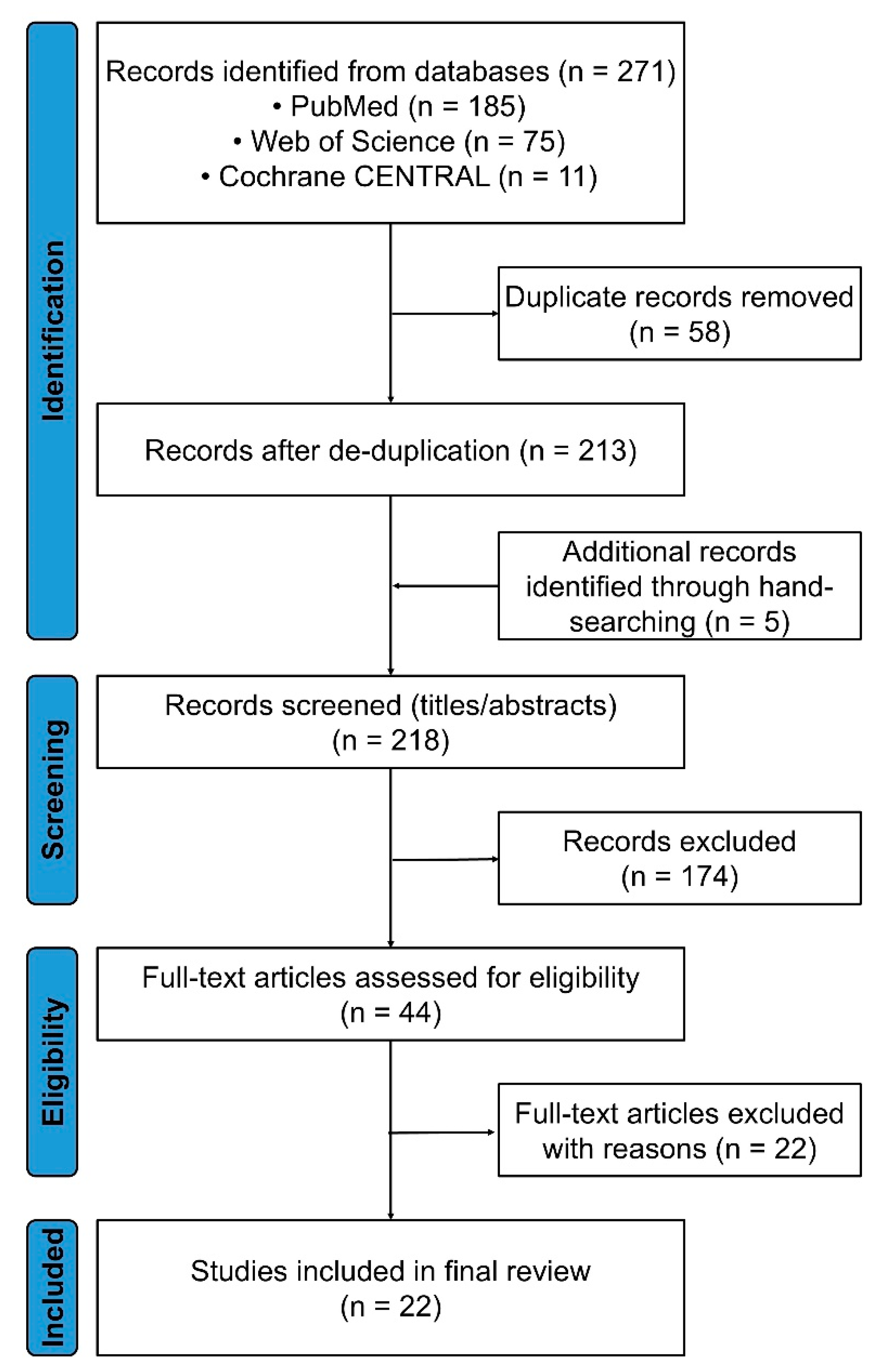
Figure 2.
Clinical and hematologic profiles by etiology. (a) Pooled median age for copper deficiency (n = 70), vacuolated cell MDS/AML (n = 21)†, and VEXAS syndrome (n = 727). Central numerals denote the median; vertical error bars indicate the 95% confidence interval. (b) Prevalence of male sex, anemia, neutropenia, and thrombocytopenia in the same three diagnostic groups. †Estimates for the MDS/AML group derive from a single institutional cohort and should be interpreted cautiously. Abbreviations: AML, acute myeloid leukemia; CI, confidence interval; MDS, myelodysplastic syndrome.
Figure 2.
Clinical and hematologic profiles by etiology. (a) Pooled median age for copper deficiency (n = 70), vacuolated cell MDS/AML (n = 21)†, and VEXAS syndrome (n = 727). Central numerals denote the median; vertical error bars indicate the 95% confidence interval. (b) Prevalence of male sex, anemia, neutropenia, and thrombocytopenia in the same three diagnostic groups. †Estimates for the MDS/AML group derive from a single institutional cohort and should be interpreted cautiously. Abbreviations: AML, acute myeloid leukemia; CI, confidence interval; MDS, myelodysplastic syndrome.
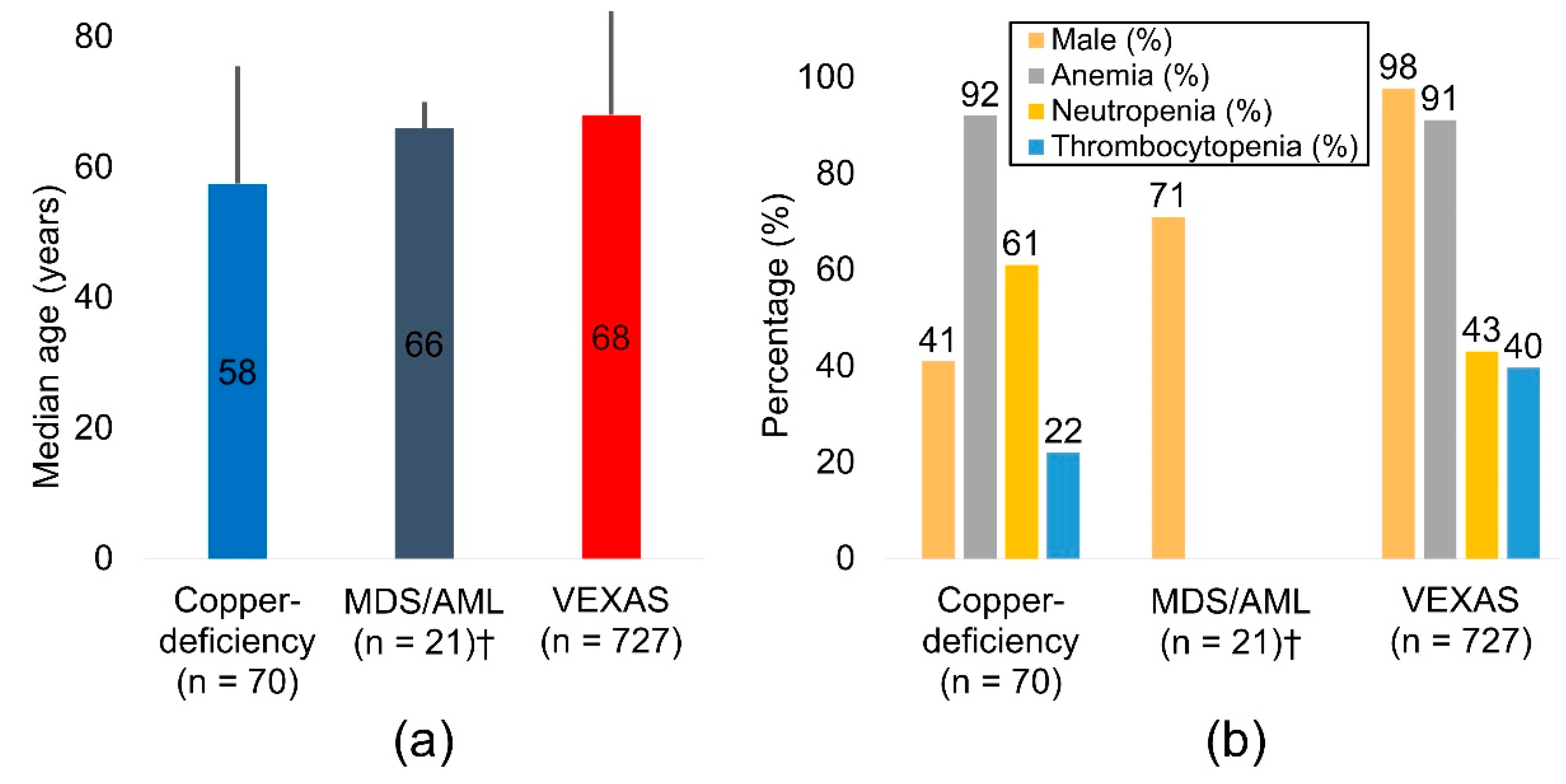
Figure 3.
Four step diagnostic algorithm for adult cytopenia with vacuolated marrow precursors. Step 1: rule out reversible causes—zinc excess, proton pump inhibitor use, bariatric surgery, malabsorption—and measure serum copper and ceruloplasmin (Table 4). Step 2: sequence UBA1 when VEXAS red flags are present (Table 5). Step 3: if UBA1 is wild type, perform morphology guided cytogenetics and targeted myeloid next generation sequencing (NGS). Step 4: investigate rarer mimics (drug/toxin exposure, inherited marrow failure syndromes, immune or infectious etiologies).
Figure 3.
Four step diagnostic algorithm for adult cytopenia with vacuolated marrow precursors. Step 1: rule out reversible causes—zinc excess, proton pump inhibitor use, bariatric surgery, malabsorption—and measure serum copper and ceruloplasmin (Table 4). Step 2: sequence UBA1 when VEXAS red flags are present (Table 5). Step 3: if UBA1 is wild type, perform morphology guided cytogenetics and targeted myeloid next generation sequencing (NGS). Step 4: investigate rarer mimics (drug/toxin exposure, inherited marrow failure syndromes, immune or infectious etiologies).


Table 1.
Characteristics of the 22 studies describing cytoplasmic vacuolization with cytopenia.
| No. | First Author | Year | Country | Primary diagnosis | N | Median Age, y | Range, y | Male (%) | Anemia (%) | Neutropenia (%) | Thrombocytopenia (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Gurnari [8] | 2021a† | USA/Italy/France | Copper-deficiency | 2 | 57.5 | 42-73 | 50% | NA | NA | NA |
| 2 | Uchino [9] | 2021 | Japan | Copper-deficiency | 15 | 69 | 33-88 | 67% | 100% | 47% | 53% |
| 3 | Halfdanarson [10] | 2009 | USA | Copper-deficiency | 5 | 46 | 37-55 | 0% | 50% | 50% | 0% |
| 4 | Halfdanarson [11] | 2008 | USA | Copper-deficiency | 40 | 57.5 | 28-83 | 45% | 98% | 63% | 15% |
| 5 | Huff [1] | 2007 | USA | Copper-deficiency | 8 | 43.5 | 32-71 | 0% | 75% | 88% | 13% |
| 6 | Gurnari [8] | 2021b† | USA/Italy/France | MDS/AML | 21 | 66 | 49-92 | 71% | NA | NA | NA |
| 7 | Vitale [12] | 2025 | Italy/Mexico/Other‡ | VEXAS | 36 | 65 | NA | 100% | 92% | 44% | 47% |
| 8 | Johansen [13] | 2025 | Denmark | VEXAS | 16 | 74 | 51-78 | 100% | 100% | NA | 0% |
| 9 | Hadjadj [14] | 2024 | France | VEXAS | 110 | 71 | 68-79 | 99% | 100% | NA | 17% |
| 10 | Maeda [15] | 2024 | Japan | VEXAS | 89 | 69 | 62-77 | 91% | 74% | NA | 15% |
| 11 | Kusne [16] | 2024 | USA | VEXAS | 119 | 64.5 | 39-86 | 100% | 90% | NA | NA |
| 12 | Wolff [17] | 2024 | Switzerland | VEXAS | 17 | 74 | 59-77 | 100% | 100% | NA | NA |
| 13 | Beck [18] | 2023 | USA | VEXAS | 11 | 65 | 55-85 | 82% | 100% | NA | 91% |
| 14 | Mascaro [19] | 2023 | Spain | VEXAS | 42 | 67 | 52-86 | 100% | 87% | 41% | 48% |
| 15 | Hines [20] | 2023 | USA | VEXAS | 8 | 65.5 | 39-75 | 100% | 88% | NA | 100% |
| 16 | Islam [21] | 2022 | Australia | VEXAS | 3 | 67 | 67-69 | 100% | 100% | 67% | 67% |
| 17 | Mekinian [22] | 2022 | France | VEXAS | 12 | 76 | 73-78 | 100% | NA | NA | NA |
| 18 | Georgin-Lavialle [23] | 2022 | France | VEXAS | 116 | 71 | 66-76 | 96% | NA | NA | NA |
| 19 | Comont [24] | 2022 | France | VEXAS | 11 | 64 | 54-73 | 100% | 100% | NA | NA |
| 20 | Ferrada [25] | 2022 | USA/UK/Germany | VEXAS | 83 | 66 | 41-80 | 100% | 97% | NA | 83% |
| 21 | Tsuchida [26] | 2021 | Japan | VEXAS | 14 | 72 | 43-93 | 93% | 64% | NA | NA |
| 22 | Ferrada [27] | 2021 | USA/UK | VEXAS | 13 | 56 | 45-70 | 100% | 100% | NA | NA |
| 23 | Gurnari [8] | 2021c† | USA/Italy/France | VEXAS | 2 | 65.5 | 65-66 | 100% | NA | NA | NA |
| 24 | Beck [3] | 2020 | USA/UK | VEXAS | 25 | 64 | 45-80 | 100% | 96% | NA | NA |
†The study cohort was subdivided into three disease-specific groups (copper-deficiency cytopenia, vacuolated-cell MDS/AML, and VEXAS syndrome). ‡Other = remaining 8 countries. *Indicates the disorder identified by the original authors as the cause of cytopenia and marrow vacuolization. Abbreviations: AML = acute myeloid leukemia; MDS = myelodysplastic syndrome; NA = not available.
Table 2.
Pooled demographic and hematologic data by diagnostic group.
| N | Median age, y | Male (%) | Anemia (%) | Neutropenia (%) | Thrombocytopenia (%) | |
|---|---|---|---|---|---|---|
| Copper-deficiency | 70 | 57.5 | 41% | 92% | 61% | 22% |
| MDS/AML† | 21 | 66.0 | 71% | NA | NA | NA |
| VEXAS | 727 | 68.0 | 98% | 91% | 43% | 40% |
†Derived from a single cohort; values should be interpreted with caution. Abbreviations: NA = not available.
Table 3.
Common causes and risk factors for acquired copper-deficiency.
| Cause or risk factor | Principal mechanism(s) |
|---|---|
| Insufficient intake | Poor dietary copper (restrictive or malnourished diets; unfortified homemade enteral formulas) |
| Malabsorption | Post-gastrectomy or RYGB; extensive small-bowel disease or resection; chronic diarrhea or inflammatory bowel disease |
| Excess zinc supplementation | Gastrointestinal competition and metallothionein induction trap copper in enterocytes, increasing fecal loss (e.g., prolonged zinc therapy for Wilson disease, cirrhosis, or dialysis) |
| Chronic acid suppression (PPIs, H2-blockers) | Persistently reduced gastric acidity limits copper solubilization and intestinal absorption |
| Long-term enteral or parenteral nutrition | Trace-element omission or undersupplementation; risk rises with duration of support |
| Other gastrointestinal factors | Markedly reduced absorptive surface (short-bowel syndrome), chronic pancreatitis, or pancreatic exocrine insufficiency |
Abbreviations: PPI = proton-pump inhibitor; RYGB = Roux-en-Y gastric bypass.
Table 4.
Clinical conditions that produce vacuolization of hematopoietic precursors.
| Category | Representative disorders/exposures | Core pathophysiology | Characteristic clinical / morphologic clues | Key references |
|---|---|---|---|---|
| Clonal/autoinflammatory | VEXAS (UBA1) | Defective ubiquitylation; endoplasmic-reticulum stress and chronic inflammation | Numerous rounded, lipid-poor vacuoles in early myeloid and erythroid precursors; relapsing chondritis; Sweet-like rash | [3] |
| Nutritional/metabolic | Copper-deficiency, zinc excess, folate/B12 /B6 deficiency, ethanol, lead | Mitochondrial or ER dysfunction caused by trace-element imbalance or toxin | Vacuoles disappear after copper repletion or ethanol abstinence; increased zinc-to-copper ratio; macro-ovalocytes in vitamin deficiencies | [1,33,34] |
| Drug/toxin | Chloramphenicol, linezolid, methotrexate, gilteritinib, erythropoietin-stimulating agents, benzene, arsenic, isoniazid, imatinib, azacitidine, high-dose cytotoxic chemotherapy | Inhibition of mitochondrial protein synthesis, direct marrow injury, or pyridoxine depletion (isoniazid) | Vacuoles regress after drug withdrawal; in isoniazid toxicity: ring sideroblasts and vacuolated late erythroblasts reversible with pyridoxine | [34,35,36] |
| Myeloid neoplasms | MDS, AML, therapy-related MDS/AML, MDS/MPN overlap | Abortive autophagy and reactive-oxygen accumulation driven by high-risk cytogenetic lesions | More than 20% vacuolated blasts is associated with poor induction response; monosomy 7 or complex karyotype common | [37,38,39,40] |
| Myeloproliferative spectrum | Primary myelofibrosis, CMML, MDS/MPN RS-T | Persistent DNA-damage signaling | Chronic cytopenia with dysplasia; leukoerythroblastosis; marrow fibrosis; occasional vacuolization, sometimes in overlapping inflammatory syndromes (e.g., VEXAS) | [3,34] |
| Inherited marrow-failure/sideroblastic | Shwachman–Diamond, SIFD, Pearson, Kearns–Sayre, Menkes, telomere disorders | Ribosome or iron–sulfur-cluster biogenesis defects | Early-onset cytopenia, pancreatic insufficiency, telomere shortening; NGS panel diagnostic | [34,41] |
| Inherited marrow-failure/congenital neutropenia | Severe congenital neutropenia (SRP54) | Dysfunction of SRP54 GTPase; ER stress and impaired granulopoiesis | Early-onset profound neutropenia with promyelocyte arrest; numerous vacuolated myeloblasts/promyelocytes | [42] |
| Inherited lysosomal/autophagy defects | Chediak–Higashi (CHS: LYST), Danon disease (LAMP2) | Defective lysosomal trafficking or membrane proteins; impaired autophagy with giant vacuoles | CHS: partial albinism, neutropenia, giant azurophilic granules in precursors / platelets; Danon: hypertrophic cardiomyopathy, skeletal myopathy, intellectual disability, enlarged vacuolated lysosomes in marrow precursors | [31,43] |
| Immune/infectious | infectious, Severe aplastic anemia, autoimmune hepatitis cytopenia, parvovirus B19 pure red-cell aplasia, secondary HLH | Marrow suppression mediated by interferon-γ and TNF-α | Profound reticulocytopenia; ferritin > 10 000 ng/mL; elevated soluble IL-2 receptor | [44,45] |
| Miscellaneous/artifact | Delayed slide preparation or prolonged room-temperature storage† | Degenerative cytoplasmic change occurring ex vivo | A repeat smear prepared within minutes eliminates vacuoles | [34] |
†Vacuole morphology is often exaggerated if smears dry slowly at room temperature; always examine a promptly prepared repeat smear before further testing. Abbreviations: AML = acute myeloid leukemia; CHS = Chediak–Higashi syndrome; CMML = chronic myelomonocytic leukemia; ER = endoplasmic reticulum; HLH = hemophagocytic lymphohistiocytosis; MDS = myelodysplastic syndrome; MPN = myeloproliferative neoplasm; NGS = next-generation sequencing; RS-T = ring sideroblasts with thrombocytosis; SCN = severe congenital neutropenia; SIFD = sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay.
Table 5.
T. Clinical and laboratory “red flags” that should prompt UBA1 mutation testing in suspected VEXAS syndrome.
Table 5.
T. Clinical and laboratory “red flags” that should prompt UBA1 mutation testing in suspected VEXAS syndrome.
| Domain | Typical findings suggestive of VEXAS |
|---|---|
| Constitutional/inflammatory | Persistent fever, drenching sweats, weight loss, markedly elevated C-reactive protein or ferritin despite high-dose corticosteroids |
| Dermatologic | Neutrophilic dermatoses such as Sweet syndrome, vasculitic purpura, livedo reticularis, auricular or nasal chondritis-like erythema |
| Cartilage and joints | Relapsing polychondritis, inflammatory arthritis, costochondritis |
| Pulmonary | Sterile interstitial or organizing pneumonitis, alveolitis, exudative pleural effusions |
| Hematologic | Macrocytic anemia, thrombocytopenia or pancytopenia, cytoplasmic vacuoles in erythroid and myeloid precursors |
| Thrombotic/vasculitic | Unprovoked deep-vein thrombosis or pulmonary embolism, systemic or cutaneous vasculitis, cerebral vasculitic events |
| Treatment pattern | Transient steroid response; refractoriness to conventional disease-modifying antirheumatic drugs; dependence on high-dose steroids or Janus kinase inhibitors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



