Submitted:
17 July 2025
Posted:
17 July 2025
You are already at the latest version
Abstract
Globally, Sub-Saharan Africa is experiencing the highest mortality rates for several cancer types. Exasperated by a one-size-fits-all, non-African-derived treatment strategies, Africa has largely been excluded from the genomic era and received little benefits from precision oncology. Through a thorough literature review, we identified five whole cancer genome databases that include patients from Sub-Saharan Africa. Irrespective of cancer type (breast, esophageal, prostate and Burkitt lymphoma), these studies report higher tumour genome instability, including African-specific cancer drivers and mutational signatures, suggesting unique contributory mechanisms at play. Reviewing bioinformatic tools applied to African databases, we provide a rationale for workflow selection that incorporates cohort-level data and integrates a scalable design achieving high-level parallelism through physical data or genomic interval chunking strategies. Furthermore, we provide rationale for improving variant calling accuracy for African data, including adopting more sequencing techniques and sourcing African-derived data to meet different priorities of applications. Together, these enhancements and genomic scaling, aim to facilitate early diagnosis and treatment strategies, and ultimately reduce the disparity gap in cancer mortality rates across Sub-Saharan Africa.
Keywords:
Africa
; computational workflow
; parallelism
; cancer genomics
; whole-genome sequencing
1. Introduction
Sub-Saharan Africa bears a disproportionate burden of many cancer types based on GLOBOCAN 2022 [1]. Besides cancers with a known viral aetiology, such as cervical uteri cancer and Kaposi sarcoma, Africa has the highest age-standardised mortality rates per 100,000 people per year for eight additional cancer types originating from the breast (19.2), prostate (17.3), non-Hodgkin lymphoma (3.3), thyroid (0.64), vulva (0.63), Hodgkin lymphoma (0.41), salivary glands (0.38), and vagina (0.24) [1]. Despite this burden, tailored clinical care remains restricted. This is largely attributed to limited tumour genome profiling and progression of precision medicine from related cancer studies [2,3,4,5], perpetuated by inadequate investment, resources and technical capacity [6].
Cancer whole-genome research conducted across high-income countries has inevitably been ancestrally biased [6]. While patients of European ancestry predominate, African ancestral representation is largely African American. Consequently, ancestral fractions are biased towards West African origins, with further masking through European–fractions (on average, 18%) [7], limiting cross-continental correlations. This African American focus applies not only to studies of a particular cancer type, taking prostate cancer (PCa) as an example [8,9,10,11,12,13,14,15,16], but also to pan-cancer studies. The largest of these, Pan-Cancer Analysis of Whole Genomes (PCAWG), is derived from merging efforts generated by the UK-led International Cancer Genome Consortium (ICGC) [17] and the US-led The Cancer Genome Atlas (TCGA) [18]. Among the 2,583 tumour-normal matched whole-genome sequences (WGS) across 38 cancer types, only 5% were of African ancestry (African ancestral fraction: median, 83.91%; range, 50.02 – 99.96%) [19]. Another study of 333,908 tumour-only samples across six cancer types included 9.8% of patients of African ancestry (unknown African ancestral fractions) [20]. As such, regions of Africa most impacted by cancer mortality, such as southern and central Africa for PCa and northern, eastern and southern Africa for non-Hodgkin lymphoma [1], are unlikely to fully benefit from targeted cancer care built on these datasets. Nevertheless, tumours derived from African American patients show molecular differences compared to European patients. For example, pan-cancer databases show African American derived tumours to have significantly elevated whole-genome duplication (WGD) [21], including PCAWG (n=1,293 P-value=0.034) and TCGA (n=8,060, P-value=0.022), while validated by the Memorial Sloan Kettering - Metastatic Events and Tropisms (MSK-MET) array-sequenced study (n=13,071, P-value=0.016). The increased WGD may be partly attributed to the higher frequencies of TP53 mutations and cyclin E (CCNE1) gain in African American patients (P-value=5.8e-7 and 2.5e-5, respectively) [21]. Additionally, TCGA through whole-exome sequencing reported a higher level of intratumor heterogeneity (ITH) in African American breast cancer (BRCA) patients (n=768) by 5.1 units calculated by mutant-allele ITH algorithm [22]. In contrast, lower frequencies of TMPRSS2-ERG gene fusions and PTEN losses have been noted in African American PCa patients (n=24; 21% versus 40-80%, 8% versus 40%, respectively) [14]. These differences highlight the importance of including regionally diverse populations across the African continent. Leveraging large-scale whole-genome tumour data, cancer discoveries can be extended to critical non-coding and more complex structural cancer drivers, mutational signatures, and molecular subtyping to reveal potential aetiologies specific to African patients.
Large-scale studies involving whole tumour genome interrogation face computational challenges in processing workflows. PCAWG reported a total of 10 million CPU-core hours used for their workflows [19]. Generating and analysing WGS data in Binary Alignment Map (BAM) format (each ~100 GB for a 30X genome) requires substantial computational resources, including large storage hardware, multiple CPUs, and high memory allocation. Such demands increase proportionally with cohort size, incurring additional computational costs associated with achieving greater accuracy. For example, to accurately identify short variants (single nucleotide variants (SNVs), insertion and deletion (indels) variants less than 50 bp), PCAWG employed six different algorithms to produce a consensus call set [19]. Notably, the Genome Analysis Toolkit (GATK) pipeline includes a joint-calling step to incorporate cohort-wide information [23,24]. Such resource requirements can only be met by high-performance computing (HPC) platforms [25,26], supporting job parallelism and allocating hundreds of CPUs and terabytes (TB) of random-access memory (RAM) in a single run. The scatter-gather approach proposed by GATK pipeline [23] enables several thousand parallel tasks in a cost-effective and fast manner. It is a higher level of parallelism than simply parallel-by-sample, allowing for simultaneous execution of multiple tasks divided from a single step of processing a sample. Integrating high-level parallelism for the interrogation of African WGS data using HPC platforms, would accelerate the pace of research and as such greatly contribute to closing the gap for African inclusion.
We first examine all publicly available WGS databases that include tumours and matched blood or normal derived tissue from cancer patients from Sub-Saharan Africa. We highlight ancestry-related molecular features and bioinformatic tools used across studies for genome alignment and variant calling. We show the scientific importance and computational demands of analysing African cancer genomes. To alleviate the computational burden and enhance the efficiency, we present a scalable bioinformatics workflow deployed on HPC infrastructure. Computationally intensive steps are enhanced by high-level parallelism through physical data or genomic interval chunking strategies. For these steps, we benchmarked the processing and tested the scalability of tools using African WGS data. Furthermore, we discussed the potential improvements and applications of introducing new genomic technologies.
2. WGS Data of African Patient-Derived Tumours
Through literature review using PubMed with the following search terms: ‘WGS’ or ‘whole-genome’, ‘cancer’ or ‘tumour’ or ‘carcinoma’, ‘Africa’ or ‘African’ or ‘African descent’ or ‘Sub-Saharan’, ‘patients’ or ‘cohort’, a total of 154 publications were identified at 3rd June 2025. After selecting studies with patients from any of the 43 countries across Sub-Saharan Africa, seven publications from five consortia interrogated WGS datasets derived from tumour-blood patient-matched samples, as summarised in Table 1 and Figure 1. For each consortium, we briefly reviewed (i) the cohort information, such as the countries and cohort size, (ii) reported biological findings, and (iii) and workflow for analysing WGS data.
2.1. Cohort Information of African Patients
The Southern African Prostate Cancer Study (SAPCS) [34] expanded from six Black South African patients [28] to include a total of 118 genetically-defined African ancestral PCa cases [27]. More recently, SAPCS has merged with partner studies as part of the Health Equity Research Outcomes and Improvement Consortia (HEROIC) Prostate Cancer Precision Health (PCaPH) Africa1K [35]. In East Africa, three studies have emerged, two focusing on esophageal squamous cell carcinoma (ESCC) [29,30] and one on Burkitt lymphoma (BL) [31]. The largest study cohort for Sub-Saharan Africa, Oesophageal Squamous Cell Carcinoma African Prevention Research (ESCCAPE), included 162 patients from Kenya, Malawi, and Tanzania [29]. The second ESCC study, as part of the African Esophageal Cancer Consortium (AfrECC), recruited 61 patients from Tanzania [30]. The Burkitt Lymphoma Genome Sequencing Project (BLGSP), including the Epidemiology of Burkitt’s Lymphoma in East African Children and Minors (EMBLEM) study, focused on the role of the Epstein-Barr virus associated with BL [31,32]. The study did WGS sequencing on samples from 74 patients and later expanded to 87 patients. In West Africa, the Nigerian Breast Cancer Study (NBCS) generated whole-genome data from 97 Nigerian women diagnosed BRCA [33].
2.2. Cancer Discoveries from African Genomic Studies
We further interrogated genomic features that are predominant in African patients and shared across cancer types, as summarised in Table 2. SAPCS and NBCS included clinicopathologically matched European ancestral patients to provide direct clinical, technical and informatic comparative analysis, while NBCS further included an African American cohort for comparison. In contrast, ESCCAPE, AfrECC and BLGSP did not include direct ancestral comparisons. For this reason, we compared the ESCCAPE and BLGSP results by the country, where the ancestries of patients were not determined, and compared the AfrECCE results referred to external publications. BLGSP reported that Epstein-Barr virus (EBV) infection in BL showed a higher mutational burden and more aberrant somatic hypermutation, which could also associate with ancestral or geographical factors (e.g. higher exposure to EBV) given that EBV-positive patients were largely Ugandan (68 out of 71, 96%). Overall, African-derived tumours presented with significantly more variants, ranging from short to structural variant types, with elevated frequencies and longer-tail of cancer driver mutations. In contrast, African-derived prostate tumours showed a diminished frequency for TMPRSS2-ERG, which is common to European patients, and ESCC tumours showed a decreased frequency for TP53 mutations, although it remained the top candidate driver.
As cancer drivers are implicated in promoting tumour initiation and progression, African patients presenting distinct mutational patterns may undergo a unique evolutionary trajectory triggered by previously unrecognised aetiologies. SPACS identified two African-specific mutational subtypes in PCa; one predominated by driver gene copy number (CN) gain and included enrichment for driver mutations in KMT2C, MTOR and TP53 among inferred tumour subclones, while the second demonstrated a combination of CN gain and hemizygous loss in cancer drivers. Further studies using SAPCS data found that the aggressive presentation of prostate tumours, defined as International Society of Urological Pathology (ISUP) 3, was significantly associated with other molecular features for African patients. This includes type-specific hyper-SV subtypes [39], shortened tumour telomere lengths against leucocyte-derived lengths [40], megabase impacting Y-chromosomal CN gains over losses [41], and the percentage of single base substitutions (SBS) signature 2 contributing to the focal hypermutation kataegis that links to APOBEC enzyme activity [42]. NBCS reported a molecular subtype of BRCA featured by an African-related cancer driver, GATA3 at the early clonal stage, with a 10.5-year early diagnosis, and a novel aetiology-unknown signature (INDEL-B) strongly associated with African ancestry. Investigated by ESCCAPE, smoking and alcohol consumption are known factors for ESCC, but their associated genomic signatures were not identified in patients from Africa. Likewise, AfrECC showed no association with smoking and African relevant RNA-derived subtypes. Together, these findings reveal a spectrum of African relevant mutational patterns largely lacking known aetiologies or established clinical implications. This highlights an urgent need for African-inclusive studies to investigate underlying risk factors with comprehensive clinical follow-up data and a sufficient cohort size to achieve statistical power.
2.3. Challenges of Analysing WGS Data of African Patients
African cancer study workflows described above have mostly followed the same pipeline architecture from raw read alignment to variant detection, with utilised tools listed in Table 3. For read alignment (or read mapping) to a known/reference human genome, all studies used the BWA-MEM aligner [43]. The aligned reads, stored in BAM file format, are used for subsequent variant detection, with studies employing different tools. The choice of variant calling tools is known to impact the sensitivity, accuracy, and reproducibility of the results [44], as well as computational resource requirements and scalability for large cohort consideration.
The computational challenges of analysing African-derived WGS data from large-cohort studies stem from three aspects: the large size of the WGS data, the methods adopted for variant calling with enhanced sensitivity and accuracy, and the elevated mutational burden of African-derived tumours. Firstly, WGS data of tumour and patient-matched normal samples typically require a minimum of 60X and 30X sequencing coverages, respectively, with an average size of 300 GB per patient in SAPCS. High coverage, demanding extensive time for alignment and analysis, benefits downstream analyses, such as clonality interrogation which is essential for studying cancer development. The SAPCS, for example, spent a total of 712,200 service/compute units in HPC servers to process 190 patients, of which 118 were African. Secondly, the computational burden is exacerbated by leveraging cohort-wide information and multiple-caller adoption for the sensitivity and accuracy of variant calling. For germline short variants, the HaplotypeCaller employed by SAPCS used 16,500 service units for joint calling, which exclusively allows for genotyping at the cohort level without any sample size restriction (a maximum of ten for Strelka2). Joint calling reduces false negatives by enhancing the detection of common variants within samples that may be affected by quality issues at each genomic position; reduces sequencing errors falsely called as variants by downgrading the confidence of calls in one sample that are invariant in all others; and provides genotype consistency which is difficult to attain when merging single-sample variant data. For somatic variants, SAPCS (40,700 service units used) and NBCS created a panel of normals (PoN) to filter out false positives caused by germline variants and artefacts raised from sequencing and data processing. Similar to the PoN strategy, BLGSP filtered out a set of SVs that were called in multiple samples. In addition, consensus call sets merged from several callers have been adopted for somatic short variants by ESCCAPE, BLGSP, and NBCS, as well as for SVs by SAPCS, BLGSP, and NBCS. Lastly, compute time is longer for African patients with higher genomic instability. Using SAPCS data, we found longer execution hours for African data than European data when performing GRIDSS for SV detection (median, 11 versus 9.6 hours; P-value=0.0002). These computational burdens are expected to be exacerbated with expanding cohort size, highlighting a need for scalable and well-optimised workflows.
3. Rapid and Scalable HPC Workflow for African Genomic Studies
Aiming to meet substantial computational demands while improving rapid WGS processing time and aligning resource usage with underlying computer hardware, SAPCS has reported adaptive pipelines for rapid and scalable processing on HPC platforms. Here, we provide a closer evaluation of the SAPCS workflow by briefly introducing (i) steps of processing WGS data, (ii) the parallelism strategies applied to computational-intensive steps, and (iii) describing more recent improvements.
3.1. SAPCS Workflow Overview
SAPCS applied a parallelism-integrated workflow [58,59,60] on African WGS data adapted to HPC infrastructure. Ideally, the workflow could finish processing any size of the cohort in two days, if ignoring the queue time of the HPC server and allowing enough computational resources. The modular workflow applies physical data chunking to the most compute-intensive phase of read mapping and genomic interval chunking to the GATK Best Practices workflows for germline and somatic variant detection [23,24]. The workflows consist of four pipelines from data pre-processing to variant identification, as presented it Figure 2. Analysis-ready BAM files are prepared in Pipeline 1 for variant discoveries, including short germline variants, short somatic variants and SVs, which are processed in Pipelines 2 to 4, respectively. Using real-world SAPCS data, we benchmarked the optimised resource configurations on Australia’s National Computational Infrastructure (NCI) Gadi HPC, with performance summarised in Supplementary Table 1 and determined the best batch-processing configuration for high-level parallelism steps, presented in Table 4.
Pipeline 1 processes raw WGS data from each blood and tumour sample in FASTQ format into analysis-ready data in BAM format, which contains information about the aligned coordinates of reads on the human reference genome. The ALT-aware function of BWA-MEM extends the mapping region from the primary human reference genome GRCh38 to a list of alternative haplotypes derived from broader populations, thereby expanding the investigation of immune regions among African patients. However, reads aligned with multiple regions may receive low mapping quality scores, requiring manual checking during variant calling. Without high-level parallelism, the read alignment required 15.1 hours for a 30X coverage WGS data (6-CPU, 24-GB RAM allocation). Therefore, the pipeline performs alignment through physical data chunking, enabling the parallelisation of large, multi-node jobs with reliably high CPU efficiency and predictable execution time. The generated scattered alignments are merged into one BAM file per sample utilising SAMBAMBA. The following refinement, which facilitates the utmost sensitivity and specificity of variant calling, includes masking duplicate reads (technical artefacts that may cause false positives) utilising SAMBLASTER and GATK base quality score recalibration (BQSR) to lessen the impact of systematic errors introduced during sequencing. The GATK package was overhauled with version 4 to replace multi-threading functionality for resource-intensive tasks with scatter-gather capability via the ‘intervals’ flag. The final quality control stage examines mapping quality, sample contamination, and tumour cellularity using Qualimap, Sequenza, and QSignature, respectively.
Pipeline 2 identifies germline short variants using BAM files from blood samples (or normal tissue) generated in Pipeline 1, employing variant calling, joint-calling, and filtration stages. Samples are first processed with GATK HaplotypeCaller by 3,200 genomic intervals, followed by the joint-calling stage to enhance variant detection sensitivity, as previously discussed. To reduce the memory demand of joint-calling, we utilised intervals enabling 3,200-fold parallelisation per cohort and the GenomicsDB format that deals with the cohort-wise variant data, followed by merging all intervals to one cohort-level variant file via GatherVcfs. The following filtration stage of variant quality score recalibration (VQSR) applies machine learning algorithms to assess the pattern of known validated variants (provided in the form of reference SNP and indel databases) from the cohort-level variant file, which estimates the trustworthiness of all variants. To ensure sufficient data for the model training, VQSR does not employ any chunking strategies.
Pipelines 3 and 4 identify somatic short and SVs, respectively. Somatic variants are those present only in a tumour sample and absent in the matched blood (or normal when applicable) sample, so the identification process takes BAM files from paired tumour and blood samples as inputs. Pipeline 3 first creates a PoN to enhance variant accuracy as previously described. Similar to the strategy applied in Pipeline 2, the PoN data are generated in 3,200 genomic intervals, transformed into GenomicsDB format, and merged into a single cohort-level file. The PoN is included in the variant calling stage performed by Mutect2. The Mutect2, with its improvement in detecting low-frequency variants, facilitates the investigation of cancer subclonal evolution. The last filtration stage with FilterMutectCalls excludes several types of artefacts fitted by models, such as those introduced by formalin fixation (although not relevant to the fresh tissue derived SAPCS).
Pipeline 4 is more complicated and computationally intensive than short variant detection. This is because SVs can involve thousands to millions of base pairs, span multiple chromosomes, and often have very complex forms, including deleted or inverted sequences, chromosomal translocations, or combinations of different SV types. Due to the inescapable fact that different types of SVs are called with varying accuracy using different algorithms [66], SAPCS adopted GRIDSS and Manta callers to find a consensus call set. These callers require access to the entire dataset of a sample (tumour and matched blood samples), so data or interval chunking is not possible but parallelised by the sample.
3.2. High-Level Parallelism
For the improved efficiency of WGS data processing for tumour-normal pairs, especially for those of African ancestry that often present elevated germline and somatic variants, the required workflows need to scale across hundreds of computational cores while preserving accuracy and resource efficiency. We have implemented two parallelisation strategies tailored to different computational profiles: physical data chunking for read alignment and genomic interval scatter-gather for variant calling.
3.2.1. Parallelism via Physical Data Chunking for Alignment
Using SAPCS African data, we show that alignment time is improved to less than two hours through physical data chunking (or sharding) of input reads while maintaining mapping accuracy due to the independent alignment of each sequencing read. The paralleled alignment stage is achieved by three steps: (i) splitting FASTQ inputs into small and independent files of homogenous size; (ii) mapping small files in parallel; and (iii) merging BAM alignment files, as shown in Figure 2. Around one hour was expected to split input FASTQ files into about 184 pairs (forward and reverse reads) of small files that each contained two million reads and took five minutes for alignment by BWA-MEM (6 CPUs). The BWA-MEM mapping showed high and consistent performance over 0.87 and CPU efficiency over 0.83 throughout the compute allocations from one to eight nodes (48 CPUs per node) as shown in Figure 3a. For an 80-node allocation job processing 20 blood samples (~30X coverage each; 3,840 parallel tasks expected) at once, the batch was completed in 0.53 hours (32 minutes) with high CPU efficiency (0.84). The outputs of mapping – scattered BAM files – took around 0.3 hours to merge per blood sample (~30X coverage) and twice the time for tumour samples attributed to a doubling of sequence coverage (~60X coverage), as shown in Table 4.
3.2.2. Parallelism via Genomic Interval Chunking
Genomic interval chunking, also known as scatter-gather by GATK, is a parallelism strategy developed particularly for bioinformatics analysis. The human reference genome should be partitioned into evenly sized, abutting intervals. Each interval is processed independently in parallel. The strategy is applied in Pipelines 1-3 with varying numbers of genomic intervals to optimise output accuracy, execution time, and computational load. For the BQSR stage in Pipeline 1, the recalibration implements machine learning models of known variants to estimate a variant’s quality, so the step was parallelised into 32 interval tasks to allow for adequate training data per interval for the recalibration model. The following step of applying the recalibration is not computationally intensive and is parallelised into 24 intervals. In contrast, the 3.2 billion nucleotide-long human genome was divided into 3,200 intervals to computationally intensive steps in Pipelines 2 and 3, such as the variant calling of local re-assembly of DNA haplotypes via HaplotypeCaller for germline variants and MuTect2 for somatic variants.
We assessed the scalability of HaplotypeCaller and Mutect2 using scaling tests and batch processing with African SAPCS data. HaplotypeCaller maintained performance over 0.80 and CPU efficiency over 0.77 when using one or two compute nodes, and the CPU efficiency decreased when allocating more than two nodes (3,200 parallel tasks for a 30X blood sample), as depicted in Figure 3b. The CPU efficiency was affected by idle CPUs caused by varying execution time of parallel tasks which depends on the local read depth and the unpredictable number of potential variants per interval. Scaling to process 20 blood samples at a single run, the batch job was completed in 1.8 hours and maintained a 0.98 CPU efficiency with a 20-node allocation. Additionally, MuTect2 performs two steps of the Pipeline 3 somatic variant discovery pipeline, including creating a PoN and performing variant calling. The PoN creation step showed good scalability, performance over 0.88 and CPU efficiency over 0.9 when allocated from one to six nodes (3,200 parallel tasks for a blood sample), as depicted in Figure 3c. Consistently high CPU efficacies of MuTect2 were also shown for the batch processing of PoN (20 blood samples, 64,000 parallel tasks) and variant calling steps (20 pairs of tumour and matched blood samples, 64,000 parallel tasks), which completed within one hour (0.58 and 0.81 hours, respectively).
3.3. Integration with Workflow Management Tools
While parallelisation methods dramatically improve the performance of computationally intensive processes within a workflow, the real-world implementation of large-scale WGS workflows also requires robust orchestration to manage thousands of tasks and ensure reproducibility. Manual submission of batch jobs in HPC environments with strict wall time limits and diverse job profiles introduces inefficiency. While steps employing high-level parallelism are optimised to have similar execution times for each parallel task by ensuring even input sizes or genomic intervals, other steps that batch-process multiple samples and utilise parallelism only at the sample level can suffer from idle CPU time due to the varying execution times between parallel tasks. Reducing manual manipulation and idle CPUs could be achieved simultaneously by introducing workflow management tools, such as Nextflow [67]. Instead of batch processing, Nextflow enables the independent processing of multiple samples which could decrease the idle CPU time by automatically assigning idle CPUs to tasks, as exemplified by FASTQ splitting and BAM merging steps in the alignment, as illustrated in Figure 4.
4. Emerging Technologies and Resources to be Integrated to African Genomic Studies
Although the diversification of African biospecimen collections is gaining attention, it is still drastically insufficient, while analytic methods and reference resources remain African-exclusive in genomic applications. The application of new genomic technologies brings promises of improvements in both cancer research and clinical implications, as summarised in Figure 5. The emerging long-range sequencing/non-sequencing technologies can facilitate SV detection. The optical genome mapping (OGM) or digital karyotyping method is designed to capture single molecules up to megabases [68], as presented in Figure 5a. OGM provides a cost-effective option to detect and visualise SVs requiring no bioinformatic skills due to the user-friendly Bionano platform. As such, OGM is suitable for clinical use, such as a quick preliminary screening for progressive tumours to determine the necessity of in-depth investigation of SVs. For cancer research, OGM can validate tumour DNA sequences impacted by SVs. Complementary to OGM, long-read sequencing enables base resolution with phasing information and allows for an extended search in low-complexity regions [69]. A recent study suggests a link between the early diagnosis of cervical cancer in African American women with YAP1 amplification and the YAP1-BIRC3-BIRC2 breakage-fusion-bridge cycle identified from cell lines using long-read sequencing [70].
Long-range sequencing technologies also contribute to a more comprehensive genome assembly. The growing recognition of diversity and inclusion in human genetics has led to widespread calls for improving methods for presenting global variation. Recently, using the DNA technologies described above to include gapless telomere-to-telomere (T2T) assemblies, a truly complete genome for an individual has been constructed, as illustrated in Figure 5b. For example, A complete hydatidiform mole (CHM13) has filled 8% gaps in GRCh38, but is biased towards European ancestry [71]. Following this, although not gapless, the Human Pangenome Reference Consortium (HPRC) released a pangenome draft built from 47 subjects, including four from Sierra Leone, three Nigerians, a Kenyan, and a Gambian [72]. The pangenome draft has reported the identification of African-specific SVs that are related to epigenetic features [72]. Subsequent bioinformatic methods [73] are urgently needed to refine each T2T assembly from genetically diverse individuals for the real-world use of this advanced pangenome reference concept. These population-aware efforts in previously under-ascertained regions of the human genome pave the way for generating practical and translational insights from cancer genomics studies in Africa, as illustrated in Figure 5c.
While OGM cannot exclude for sequencing, as it is unable to detect small variants, both OGM and long-read sequencing technologies are limited by their dependence on acquiring high or ultra-high molecular weight DNA. Infective for use on highly abundant, yet highly degraded formalin fixed tissue, these technologies require a higher quality of tissue source and abundance, as well as efficient laboratory skills to efficiently acquire intact kilo-to-megabase long DNA molecules. The latter highlighting the need for effective biobanking of fresh tissues across Africa to facilitate future application to these emerging technologies.
5. Conclusions and Challenges
Sub-Saharan African countries have demonstrated greater risks of many cancer types than countries of other continents. However, cancer genomic studies, especially large-scale studies, often lack representative data from Sub-Saharan Africa. We report published research on tumour WGS data derived from African cancer patients, revealing only five databases representing four cancer types. Reviewing genetic findings from these studies, unique molecular patterns within tumours derived from African patients have been observed when compared to European patients. These include higher genomic instability, varying frequencies for cancer drivers, and a diversity of tumour subtypes with unknown underlying mechanisms. Although limited by the small number of studies, the findings support a pressing need to strive for African-inclusive cancer research to facilitate equitable patient care and outcomes. Exploring the WGS bioinformatics workflow implemented in these limited African focused cancer studies, we describe computational barriers. Due to the challenges, we introduce a highly scalable, efficient and rapid workflow that outlines how modern computing techniques, combined with appropriate access to computing hardware, can meet the computational burden for large-scale African inclusive cancer studies. Beyond short-read WGS data, emerging genomic technologies offer more accurate options that could be applied in future research and clinical use, from generating African-representative T2T-finshed pangenome references to addressing the heightened genomic complexity observed across these limited African cancer genome studies. While technologies may reduce the need for high-skilled computational biologists, in turn, their requirement for high-quality intact DNA calls for concerted efforts for fresh tissue biobanking across Africa. With improved computing practices that scale efficiently to large cohorts and advanced technologies that provide unprecedented genomic resolution, these combined could help progress genomic applications in cancer diagnosis and treatment in Sub-Saharan Africa.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
V.M.H. curated the data. J.J., C.W., and T.C. performed the data analyses. J.J., G.S., C.W., V.M.H. and W.J. wrote and revised the paper. G.S., C.W., and T.C. developed the methods. G.S., C.W., V.M.H. and W.J. critically reviewed the paper. V.M.H. and W.J. supervised the project.
Funding
Genomic sequencing and analytics for the SAPCS was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia including 2018/GNT1165762, 2020/GNT2001098 and 2021/GNT2010551 to V.M.H.. Further analytics was supported by a U.S.A. Congressionally Directed Medical Research Programs (CDMRP) Prostate Cancer Research Program (PCRP) Idea Development Award PC200390 (TARGET Africa) to V.M.H. and HEROIC Consortium Award PC210168 and PC23067 (HEROIC PCaPH Africa1K) to V.M.H. (with co-Principal Investigators Professors Riana Bornman, University of Pretoria, South Africa; Gail Prins, University of Illinois at Chicago, U.S.A.; and Mungai Peter Ngugi, University of Nairobi, Kenya), a U.S.A. National Institute of Health (NIH) National Cancer Institute (NCI) Award 1R01CA285772-01 to V.M.H., as well as a NHMRC Ideas grant 2024/GNT2037298 to W.J.. J.J. is further supported by a U.S.A. Prostate Cancer Foundation (PCF) Scholarship as part of a 2023 Challenge Award 2023CHAL4150 to V.M.H., while V.M.H. is supported by the Petre Foundation via the University of Sydney Foundation, Australia.
Conflicts of Interest
The author declares no conflicts of interest.
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R. L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2024, 74 (3), 229-263. [CrossRef]
- Hayes, V. M.; Gong, T.; Mutambirwa, S. B.; Jaratlerdsiri, W.; Bornman, M. R. African inclusion in prostate cancer genomic studies provides the first glimpses into addressing health disparities through tailored clinical care. Clinical and Translational Medicine 2023, 13 (1), e1142. [CrossRef]
- Rubagumya, F.; Carson, L.; Mushonga, M.; Manirakiza, A.; Murenzi, G.; Abdihamid, O.; Athman, A.; Mungo, C.; Booth, C.; Hammad, N. An analysis of the African cancer research ecosystem: tackling disparities. BMJ Global Health 2023, 8 (2), e011338. [CrossRef]
- Drake, T. M.; Knight, S. R.; Harrison, E. M.; Søreide, K. Global inequities in precision medicine and molecular cancer research. Frontiers in Oncology 2018, 8, 346. [CrossRef]
- Pereira, L.; Mutesa, L.; Tindana, P.; Ramsay, M. African genetic diversity and adaptation inform a precision medicine agenda. Nature Reviews Genetics 2021, 22 (5), 284-306. [CrossRef]
- Omotoso, O.; Teibo, J. O.; Atiba, F. A.; Oladimeji, T.; Paimo, O. K.; Ataya, F. S.; Batiha, G. E.-S.; Alexiou, A. Addressing cancer care inequities in sub-Saharan Africa: current challenges and proposed solutions. International journal for equity in health 2023, 22 (1), 189. [CrossRef]
- Lawson, D. J.; Van Dorp, L.; Falush, D. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nature communications 2018, 9 (1), 3258. [CrossRef]
- Liu, W.; Zheng, S. L.; Na, R.; Wei, L.; Sun, J.; Gallagher, J.; Wei, J.; Resurreccion, W. K.; Ernst, S.; Sfanos, K. S. Distinct genomic alterations in prostate tumors derived from African American men. Molecular Cancer Research 2020, 18 (12), 1815-1824. [CrossRef]
- Kittles, R. A.; Baffoe-Bonnie, A. B.; Moses, T. Y.; Robbins, C. M.; Ahaghotu, C.; Huusko, P.; Pettaway, C.; Vijayakumar, S.; Bennett, J.; Hoke, G. A common nonsense mutation in EphB2 is associated with prostate cancer risk in African American men with a positive family history. Journal of medical genetics 2006, 43 (6), 507-511. [CrossRef]
- Khani, F.; Mosquera, J. M.; Park, K.; Blattner, M.; O'Reilly, C.; MacDonald, T. Y.; Chen, Z.; Srivastava, A.; Tewari, A. K.; Barbieri, C. E. Evidence for molecular differences in prostate cancer between African American and Caucasian men. Clinical Cancer Research 2014, 20 (18), 4925-4934. [CrossRef]
- Huang, F. W.; Mosquera, J. M.; Garofalo, A.; Oh, C.; Baco, M.; Amin-Mansour, A.; Rabasha, B.; Bahl, S.; Mullane, S. A.; Robinson, B. D. Exome sequencing of African-American prostate cancer reveals loss-of-function ERF mutations. Cancer discovery 2017, 7 (9), 973-983. [CrossRef]
- Blattner, M.; Lee, D. J.; O'Reilly, C.; Park, K.; MacDonald, T. Y.; Khani, F.; Turner, K. R.; Chiu, Y.-L.; Wild, P. J.; Dolgalev, I. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia 2014, 16 (1), 14-W10.
- Yuan, J.; Kensler, K. H.; Hu, Z.; Zhang, Y.; Zhang, T.; Jiang, J.; Xu, M.; Pan, Y.; Long, M.; Montone, K. T. Integrative comparison of the genomic and transcriptomic landscape between prostate cancer patients of predominantly African or European genetic ancestry. PLoS genetics 2020, 16 (2), e1008641. [CrossRef]
- Lindquist, K. J.; Paris, P. L.; Hoffmann, T. J.; Cardin, N. J.; Kazma, R.; Mefford, J. A.; Simko, J. P.; Ngo, V.; Chen, Y.; Levin, A. M. Mutational landscape of aggressive prostate tumors in African American men. Cancer research 2016, 76 (7), 1860-1868. [CrossRef]
- Xiao, Q.; Sun, Y.; Dobi, A.; Srivastava, S.; Wang, W.; Srivastava, S.; Ji, Y.; Hou, J.; Zhao, G.-P.; Li, Y. Systematic analysis reveals molecular characteristics of ERG-negative prostate cancer. Scientific reports 2018, 8 (1), 12868. [CrossRef]
- Petrovics, G.; Li, H.; Stümpel, T.; Tan, S.-H.; Young, D.; Katta, S.; Li, Q.; Ying, K.; Klocke, B.; Ravindranath, L. A novel genomic alteration of LSAMP associates with aggressive prostate cancer in African American men. EBioMedicine 2015, 2 (12), 1957-1964. [CrossRef]
- Consortium, I. C. G. International network of cancer genome projects. Nature 2010, 464 (7291), 993.
- Weinstein, J. N.; Collisson, E. A.; Mills, G. B.; Shaw, K. R.; Ozenberger, B. A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J. M. The cancer genome atlas pan-cancer analysis project. Nature genetics 2013, 45 (10), 1113-1120. [CrossRef]
- Aaltonen, L. A.; Abascal, F.; Abeshouse, A.; Aburatani, H.; Adams, D. J.; Agrawal, N.; Ahn, K. S.; Ahn, S.-M.; Aikata, H.; Akbani, R.; et al. Pan-cancer analysis of whole genomes. Nature 2020, 578 (7793), 82-93. [CrossRef]
- Jiagge, E.; Jin, D. X.; Newberg, J. Y.; Perea-Chamblee, T.; Pekala, K. R.; Fong, C.; Waters, M.; Ma, D.; Dei-Adomakoh, Y.; Erb, G. Tumor sequencing of African ancestry reveals differences in clinically relevant alterations across common cancers. Cancer cell 2023, 41 (11), 1963-1971. e1963. [CrossRef]
- Brown, L. M.; Hagenson, R. A.; Koklič, T.; Urbančič, I.; Qiao, L.; Strancar, J.; Sheltzer, J. M. An elevated rate of whole-genome duplications in cancers from Black patients. Nature Communications 2024, 15 (1), 8218. [CrossRef]
- Johnson, J. A.; Moore, B. J.; Syrnioti, G.; Eden, C. M.; Wright, D.; Newman, L. A. Landmark series: the cancer genome atlas and the study of breast cancer disparities. Annals of Surgical Oncology 2023, 30 (11), 6427-6440. [CrossRef]
- Van der Auwera, G. A.; Carneiro, M. O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Current protocols in bioinformatics 2013, 43 (1), 11.10. 11-11.10. 33. [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 2010, 20 (9), 1297-1303. [CrossRef]
- Huang, Z.; Rustagi, N.; Veeraraghavan, N.; Carroll, A.; Gibbs, R.; Boerwinkle, E.; Venkata, M. G.; Yu, F. A hybrid computational strategy to address WGS variant analysis in> 5000 samples. BMC bioinformatics 2016, 17, 1-12. ttps://doi.org/10.1186/s12859-016-1211-6.
- Meggendorfer, M.; Jobanputra, V.; Wrzeszczynski, K. O.; Roepman, P.; de Bruijn, E.; Cuppen, E.; Buttner, R.; Caldas, C.; Grimmond, S.; Mullighan, C. G. Analytical demands to use whole-genome sequencing in precision oncology. In Seminars in cancer biology, 2022; Elsevier: Vol. 84, pp 16-22. [CrossRef]
- Jaratlerdsiri, W.; Jiang, J.; Gong, T.; Patrick, S. M.; Willet, C.; Chew, T.; Lyons, R. J.; Haynes, A.-M.; Pasqualim, G.; Louw, M.; et al. African-specific molecular taxonomy of prostate cancer. Nature 2022, 609 (7927), 552-559. [CrossRef]
- Jaratlerdsiri, W.; Chan, E. K.; Gong, T.; Petersen, D. C.; Kalsbeek, A. M.; Venter, P. A.; Stricker, P. D.; Bornman, M. R.; Hayes, V. M. Whole-genome sequencing reveals elevated tumor mutational burden and initiating driver mutations in African men with treatment-naïve, high-risk prostate cancer. Cancer research 2018, 78 (24), 6736-6746.
- Moody, S.; Senkin, S.; Islam, S. M. A.; Wang, J.; Nasrollahzadeh, D.; Cortez Cardoso Penha, R.; Fitzgerald, S.; Bergstrom, E. N.; Atkins, J.; He, Y.; et al. Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidence. Nature Genetics 2021, 53 (11), 1553-1563. [CrossRef]
- Van Loon, K.; Mmbaga, E. J.; Mushi, B. P.; Selekwa, M.; Mwanga, A.; Akoko, L. O.; Mwaiselage, J.; Mosha, I.; Ng, D. L.; Wu, W. A Genomic Analysis of Esophageal Squamous Cell Carcinoma in Eastern Africa. Cancer Epidemiology, Biomarkers & Prevention 2023, 32 (10), 1411-1420. [CrossRef]
- Grande, B. M.; Gerhard, D. S.; Jiang, A.; Griner, N. B.; Abramson, J. S.; Alexander, T. B.; Allen, H.; Ayers, L. W.; Bethony, J. M.; Bhatia, K. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood, The Journal of the American Society of Hematology 2019, 133 (12), 1313-1324. [CrossRef]
- Thomas, N.; Dreval, K.; Gerhard, D. S.; Hilton, L. K.; Abramson, J. S.; Ambinder, R. F.; Barta, S.; Bartlett, N. L.; Bethony, J.; Bhatia, K. Genetic subgroups inform on pathobiology in adult and pediatric Burkitt lymphoma. Blood 2023, 141 (8), 904-916. [CrossRef]
- Ansari-Pour, N.; Zheng, Y.; Yoshimatsu, T. F.; Sanni, A.; Ajani, M.; Reynier, J.-B.; Tapinos, A.; Pitt, J. J.; Dentro, S.; Woodard, A. Whole-genome analysis of Nigerian patients with breast cancer reveals ethnic-driven somatic evolution and distinct genomic subtypes. Nature communications 2021, 12 (1), 6946. [CrossRef]
- Tindall, E. A.; Monare, L. R.; Petersen, D. C.; Van Zyl, S.; Hardie, R. A.; Segone, A. M.; Venter, P. A.; Bornman, M. R.; Hayes, V. M. Clinical presentation of prostate cancer in black South Africans. The Prostate 2014, 74 (8), 880-891. [CrossRef]
- Hayes, V. M.; Patrick, S. M.; Shirinde, J.; Jaratlerdsiri, W.; Nenzhelele, M.; Radzuma, M. B.; Gheybi, K.; Mokua, W.; Oyaro, M. O.; Moreira, D. M. Health equity research outcomes and improvement Consortium Prostate Cancer Health Precision Africa1K: closing the health equity gap through rural community inclusion. Journal of Urologic Oncology 2024, 22 (2), 144-149. [CrossRef]
- Zhang, R.; Li, C.; Wan, Z.; Qin, J.; Li, Y.; Wang, Z.; Zheng, Q.; Kang, X.; Chen, X.; Li, Y. Comparative genomic analysis of esophageal squamous cell carcinoma among different geographic regions. Frontiers in Oncology 2023, 12, 999424. [CrossRef]
- Li, M.; Zhang, Z.; Wang, Q.; Yi, Y.; Li, B. Integrated cohort of esophageal squamous cell cancer reveals genomic features underlying clinical characteristics. Nature Communications 2022, 13 (1), 5268. [CrossRef]
- Cui, Y.; Chen, H.; Xi, R.; Cui, H.; Zhao, Y.; Xu, E.; Yan, T.; Lu, X.; Huang, F.; Kong, P. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell research 2020, 30 (10), 902-913. [CrossRef]
- Gong, T.; Jaratlerdsiri, W.; Jiang, J.; Willet, C.; Chew, T.; Patrick, S. M.; Lyons, R. J.; Haynes, A.-M.; Pasqualim, G.; Brum, I. S. Genome-wide interrogation of structural variation reveals novel African-specific prostate cancer oncogenic drivers. Genome medicine 2022, 14 (1), 100. [CrossRef]
- Huang, R.; Bornman, M. R.; Stricker, P. D.; Simoni Brum, I.; Mutambirwa, S. B.; Jaratlerdsiri, W.; Hayes, V. M. The impact of telomere length on prostate cancer aggressiveness, genomic instability and health disparities. Scientific Reports 2024, 14 (1), 7706. [CrossRef]
- Soh, P. X.; Adams, A.; Bornman, M. R.; Jiang, J.; Stricker, P. D.; Mutambirwa, S. B.; Jaratlerdsiri, W.; Hayes, V. M. Y chromosome variation and prostate cancer ancestral disparities. iScience 2025, 28 (5). [CrossRef]
- Hayes, V.; Jiang, J.; Tapinos, A.; Huang, R.; Bornman, R.; Stricker, P.; Mutambirwa, S.; Wedge, D.; Jaratlerdsiri, W. Kataegis associated mutational processes linked to adverse prostate cancer presentation in African men. Research Square 2024, rs. 3. rs-4597464.
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 2013.
- Chen, Z.; Yuan, Y.; Chen, X.; Chen, J.; Lin, S.; Li, X.; Du, H. Systematic comparison of somatic variant calling performance among different sequencing depth and mutation frequency. Scientific Reports 2020, 10 (1), 3501. [CrossRef]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M. A.; Fennell, T. J.; Carneiro, M. O.; Van der Auwera, G. A.; Kling, D. E.; Gauthier, L. D.; Levy-Moonshine, A.; Roazen, D. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2017, 201178.
- Cibulskis, K.; Lawrence, M. S.; Carter, S. L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E. S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 2013, 31 (3), 213-219. [CrossRef]
- Cameron, D. L.; Baber, J.; Shale, C.; Valle-Inclan, J. E.; Besselink, N.; van Hoeck, A.; Janssen, R.; Cuppen, E.; Priestley, P.; Papenfuss, A. T. GRIDSS2: comprehensive characterisation of somatic structural variation using single breakend variants and structural variant phasing. Genome Biology 2021, 22, 1-25. [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A. J.; Kruglyak, S.; Saunders, C. T. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2015, 32 (8), 1220-1222. [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A. L.; Bekritsky, M. A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P. Strelka2: fast and accurate calling of germline and somatic variants. Nature methods 2018, 15 (8), 591-594. [CrossRef]
- Jones, D.; Raine, K. M.; Davies, H.; Tarpey, P. S.; Butler, A. P.; Teague, J. W.; Nik-Zainal, S.; Campbell, P. J. cgpCaVEManWrapper: simple execution of CaVEMan in order to detect somatic single nucleotide variants in NGS data. Current protocols in bioinformatics 2016, 56 (1), 15.10. 11-15.10. 18. [CrossRef]
- Raine, K. M.; Hinton, J.; Butler, A. P.; Teague, J. W.; Davies, H.; Tarpey, P.; Nik-Zainal, S.; Campbell, P. J. cgpPindel: identifying somatically acquired insertion and deletion events from paired end sequencing. Current protocols in bioinformatics 2015, 52 (1), 15.17. 11-15.17. 12. [CrossRef]
- Radenbaugh, A. J.; Ma, S.; Ewing, A.; Stuart, J. M.; Collisson, E. A.; Zhu, J.; Haussler, D. RADIA: RNA and DNA integrated analysis for somatic mutation detection. PloS one 2014, 9 (11), e111516. [CrossRef]
- Wilm, A.; Aw, P. P. K.; Bertrand, D.; Yeo, G. H. T.; Ong, S. H.; Wong, C. H.; Khor, C. C.; Petric, R.; Hibberd, M. L.; Nagarajan, N. LoFreq: a sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic acids research 2012, 40 (22), 11189-11201. [CrossRef]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S. R. F.; Wilkie, A. O. M.; McVean, G.; Lunter, G.; Consortium, W. G. S. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nature Genetics 2014, 46 (8), 912-918. [CrossRef]
- Saunders, C. T.; Wong, W. S.; Swamy, S.; Becq, J.; Murray, L. J.; Cheetham, R. K. Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 2012, 28 (14), 1811-1817. [CrossRef]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stütz, A. M.; Benes, V.; Korbel, J. O. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28 (18), i333-i339. [CrossRef]
- Layer, R. M.; Chiang, C.; Quinlan, A. R.; Hall, I. M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biology 2014, 15 (6), R84. [CrossRef]
- Sadsad, R., Samaha, G., & Chew, T. . Fastq-to-bam @ NCI-Gadi. WorkflowHub 2021. [CrossRef]
- Sadsad, R., Samaha, G., & Chew, T. . Germline-ShortV @ NCI-Gadi. WorkflowHub 2021. [CrossRef]
- Sadsad, R., & Chew, T. Somatic-ShortV @ NCI-Gadi. WorkflowHub 2021. [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34 (17), i884-i890. [CrossRef]
- Tarasov, A.; Vilella, A. J.; Cuppen, E.; Nijman, I. J.; Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 2015, 31 (12), 2032-2034. [CrossRef]
- Faust, G. G.; Hall, I. M. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 2014, 30 (17), 2503-2505. [CrossRef]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L. M.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T. F.; Conesa, A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics 2012, 28 (20), 2678-2679. [CrossRef]
- Favero, F.; Joshi, T.; Marquard, A. M.; Birkbak, N. J.; Krzystanek, M.; Li, Q.; Szallasi, Z.; Eklund, A. C. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Annals of Oncology 2015, 26 (1), 64-70. [CrossRef]
- Gong, T.; Hayes, V. M.; Chan, E. K. Detection of somatic structural variants from short-read next-generation sequencing data. Briefings in bioinformatics 2021, 22 (3), bbaa056. [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E. W.; Barja, P. P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nature biotechnology 2017, 35 (4), 316-319. [CrossRef]
- Chan, E. K.; Cameron, D. L.; Petersen, D. C.; Lyons, R. J.; Baldi, B. F.; Papenfuss, A. T.; Thomas, D. M.; Hayes, V. M. Optical mapping reveals a higher level of genomic architecture of chained fusions in cancer. Genome research 2018, 28 (5), 726-738. [CrossRef]
- Sakamoto, Y.; Sereewattanawoot, S.; Suzuki, A. A new era of long-read sequencing for cancer genomics. Journal of human genetics 2020, 65 (1), 3-10. [CrossRef]
- Rodriguez, I.; Rossi, N. M.; Keskus, A. G.; Xie, Y.; Ahmad, T.; Bryant, A.; Lou, H.; Paredes, J. G.; Milano, R.; Rao, N.; et al. Insights into the mechanisms and structure of breakage-fusion-bridge cycles in cervical cancer using long-read sequencing. Am J Hum Genet 2024, 111 (3), 544-561. [CrossRef]
- Nurk, S.; Koren, S.; Rhie, A.; Rautiainen, M.; Bzikadze, A. V.; Mikheenko, A.; Vollger, M. R.; Altemose, N.; Uralsky, L.; Gershman, A. The complete sequence of a human genome. Science 2022, 376 (6588), 44-53.
- Liao, W.-W.; Asri, M.; Ebler, J.; Doerr, D.; Haukness, M.; Hickey, G.; Lu, S.; Lucas, J. K.; Monlong, J.; Abel, H. J.; et al. A draft human pangenome reference. Nature 2023, 617 (7960), 312-324. [CrossRef]
- Rhie, A.; McCarthy, S. A.; Fedrigo, O.; Damas, J.; Formenti, G.; Koren, S.; Uliano-Silva, M.; Chow, W.; Fungtammasan, A.; Kim, J. Towards complete and error-free genome assemblies of all vertebrate species. Nature 2021, 592 (7856), 737-746. [CrossRef]
Figure 1.
Cancer genomic databases established in Sub-Saharan Africa. PCa, prostate cancer; ESCC, esophageal squamous cell carcinoma; BL, Burkitt lymphoma cancer; and BRCA, breast cancer. Note, (i) two databases have Tanzanian patients diagnosed with ESCC, and (ii) the BLGSP study cohort consists of subjects no more than 15 years old except for one at age 19 and is therefore defined as paediatric Burkitt Lymphoma.
Figure 1.
Cancer genomic databases established in Sub-Saharan Africa. PCa, prostate cancer; ESCC, esophageal squamous cell carcinoma; BL, Burkitt lymphoma cancer; and BRCA, breast cancer. Note, (i) two databases have Tanzanian patients diagnosed with ESCC, and (ii) the BLGSP study cohort consists of subjects no more than 15 years old except for one at age 19 and is therefore defined as paediatric Burkitt Lymphoma.
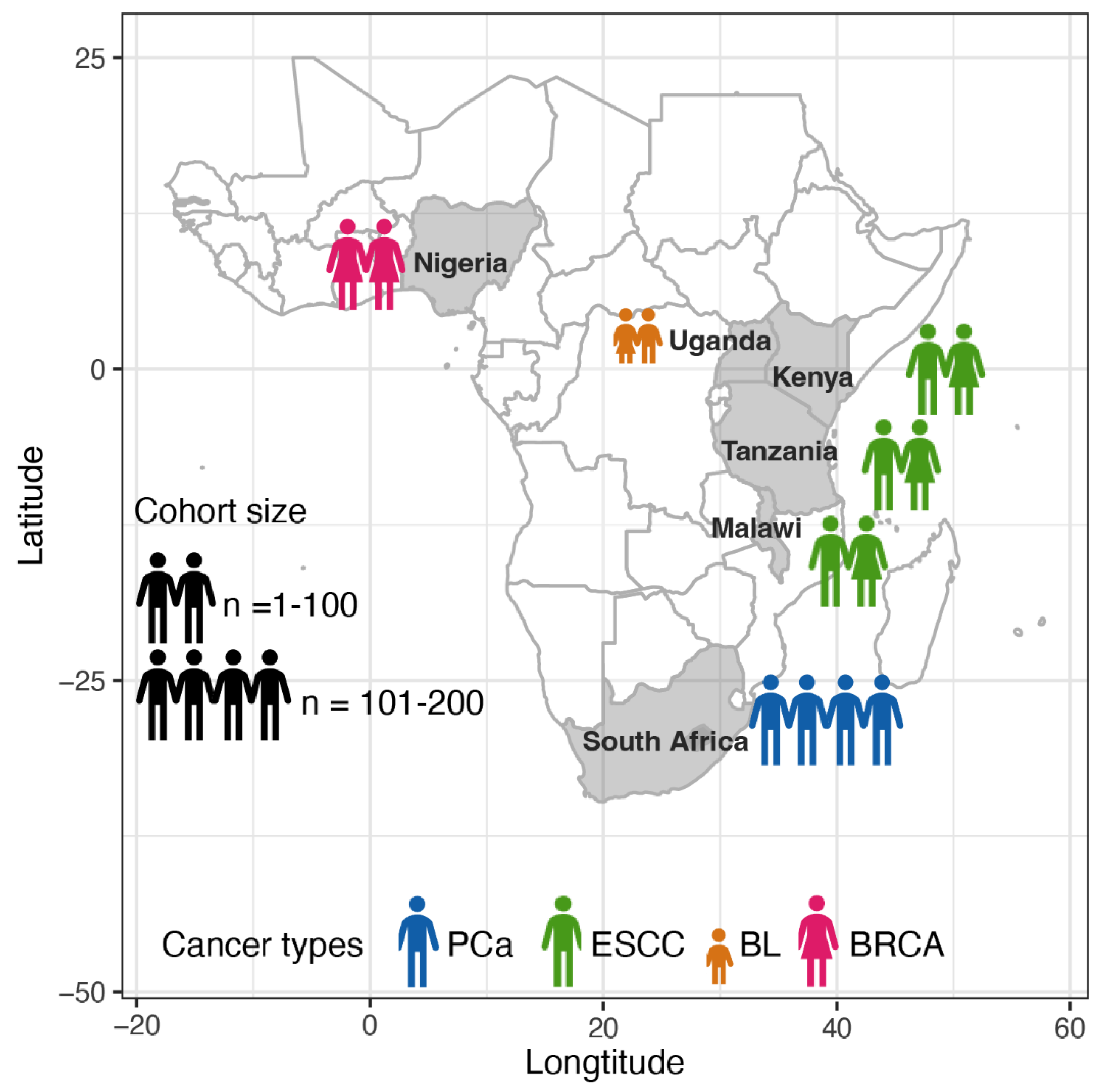
Figure 2.
Schematic SAPCS workflow for processing African-inclusive cancer genomics data from raw WGS data to variant calling. The workflow is broken down into four pipelines including 1, data processing and alignment; 2, germline short variant calling; 3, somatic short variant calling and 4, somatic structural variant calling.
Figure 2.
Schematic SAPCS workflow for processing African-inclusive cancer genomics data from raw WGS data to variant calling. The workflow is broken down into four pipelines including 1, data processing and alignment; 2, germline short variant calling; 3, somatic short variant calling and 4, somatic structural variant calling.
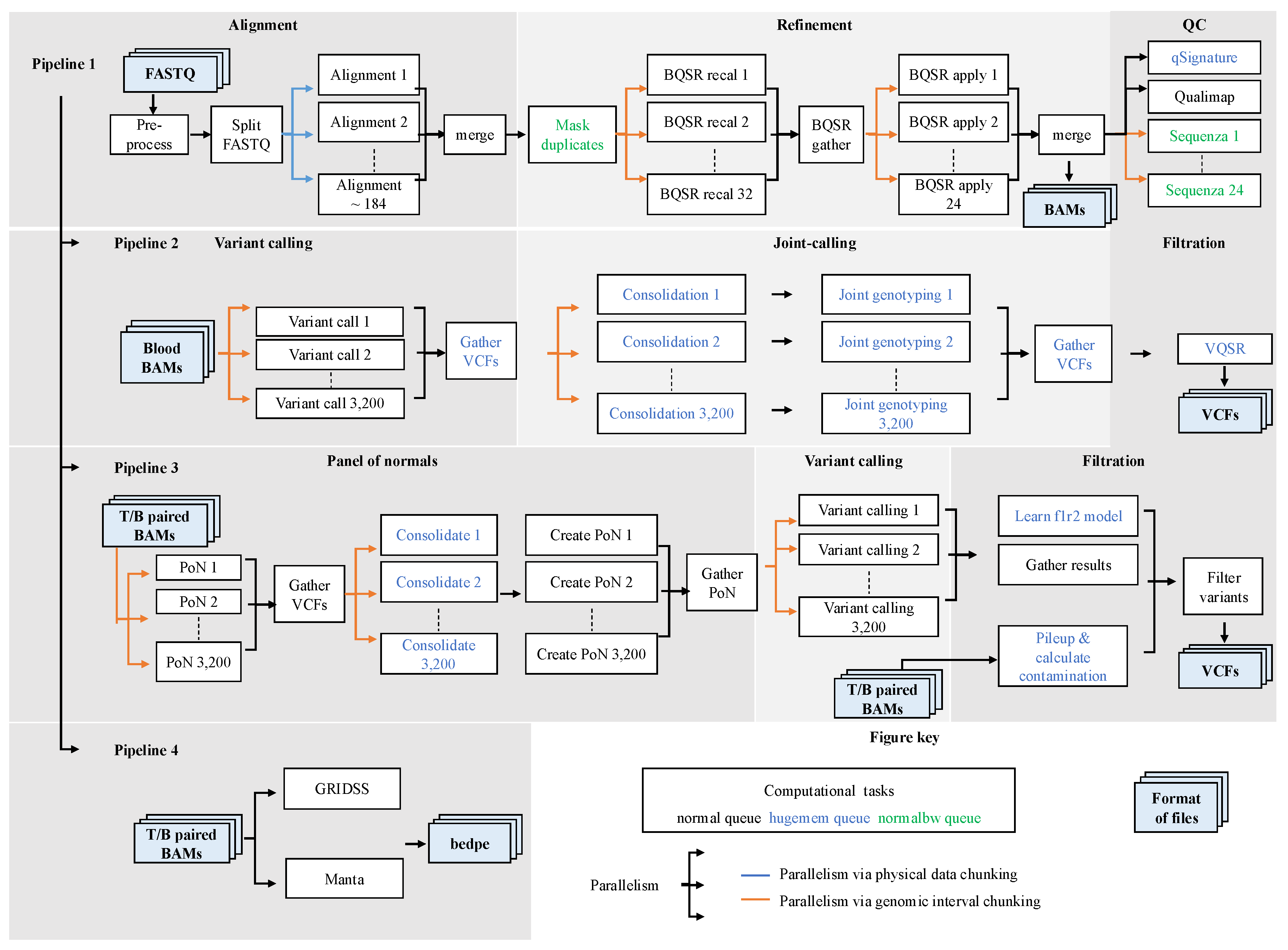
Figure 3.
Scaling tests on computationally intensive analyses using African data from SAPCS. a. The scalability of BWA-MEM was tested by aligning reads from a blood sample (30X coverage, 184 parallel tasks) with allocation of one to eight nodes (48 CPUs per node). Each parallel task was allocated six CPUs. b. HaplotypeCaller was tested to call germline variants of a blood sample (3,200 parallel tasks) with allocations from one to eight nodes. Each task is allocated one CPU. c. Mutect2 was tested to process a blood sample (3,200 parallel tasks) with allocations from one to six nodes. While the ideal speed-up scales linearly with the number of CPUs, the actual speed-up is defined as the product of execution time and CPU count for each process, compared to that of the process with the lowest CPU allocation. The lowest CPU allocation is six for BWA-MEM and eight for HaplotypeCaller and Mutect2. Performance is estimated as the inverse of the actual speed-up. CPU efficiency is an estimate of CPU time divided by the execution time and CPU count.
Figure 3.
Scaling tests on computationally intensive analyses using African data from SAPCS. a. The scalability of BWA-MEM was tested by aligning reads from a blood sample (30X coverage, 184 parallel tasks) with allocation of one to eight nodes (48 CPUs per node). Each parallel task was allocated six CPUs. b. HaplotypeCaller was tested to call germline variants of a blood sample (3,200 parallel tasks) with allocations from one to eight nodes. Each task is allocated one CPU. c. Mutect2 was tested to process a blood sample (3,200 parallel tasks) with allocations from one to six nodes. While the ideal speed-up scales linearly with the number of CPUs, the actual speed-up is defined as the product of execution time and CPU count for each process, compared to that of the process with the lowest CPU allocation. The lowest CPU allocation is six for BWA-MEM and eight for HaplotypeCaller and Mutect2. Performance is estimated as the inverse of the actual speed-up. CPU efficiency is an estimate of CPU time divided by the execution time and CPU count.
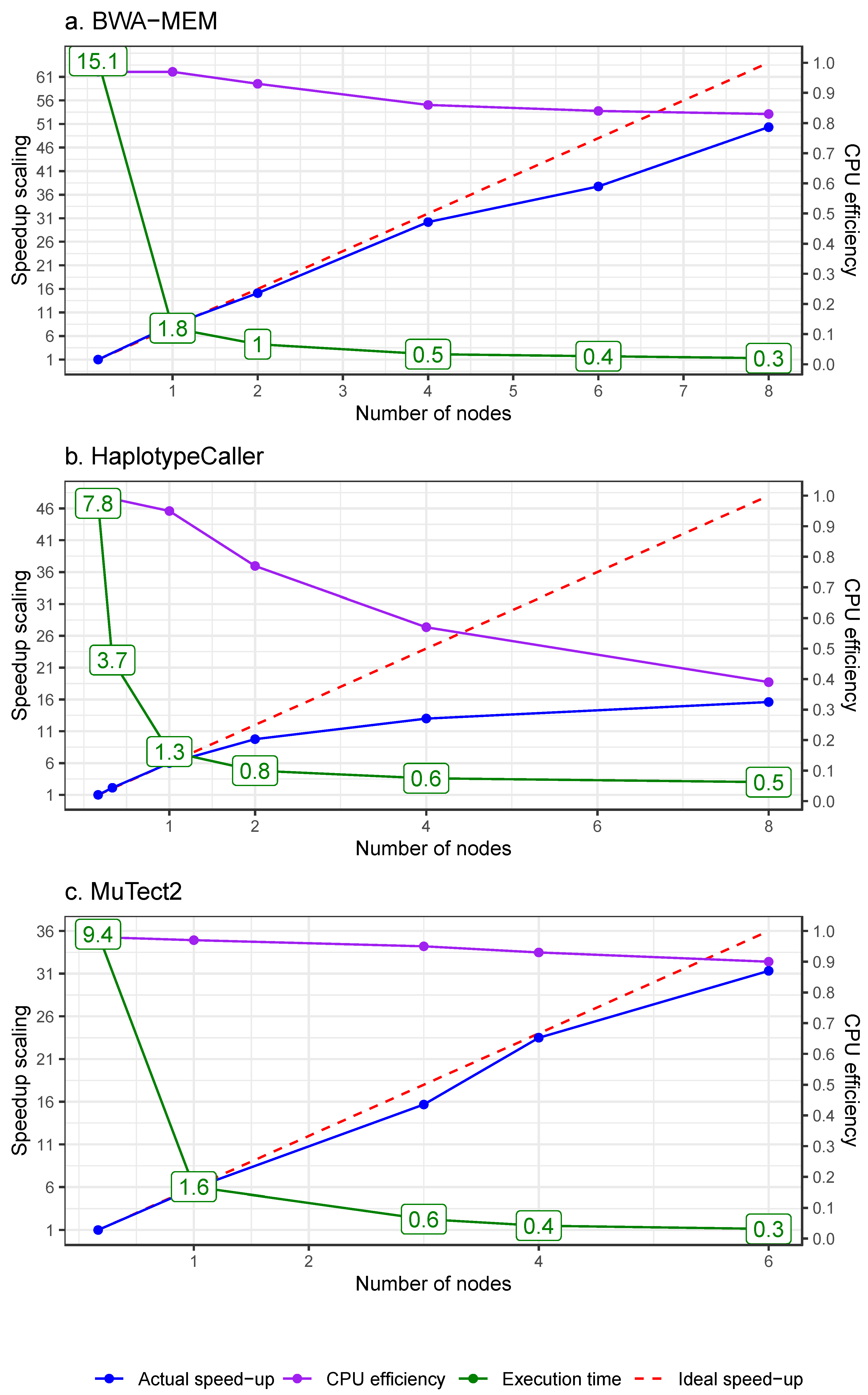
Figure 4.
Schematic comparisons of African data processing in the alignment step using sequential processing, simple parallelisation and automated workflow.
Figure 4.
Schematic comparisons of African data processing in the alignment step using sequential processing, simple parallelisation and automated workflow.
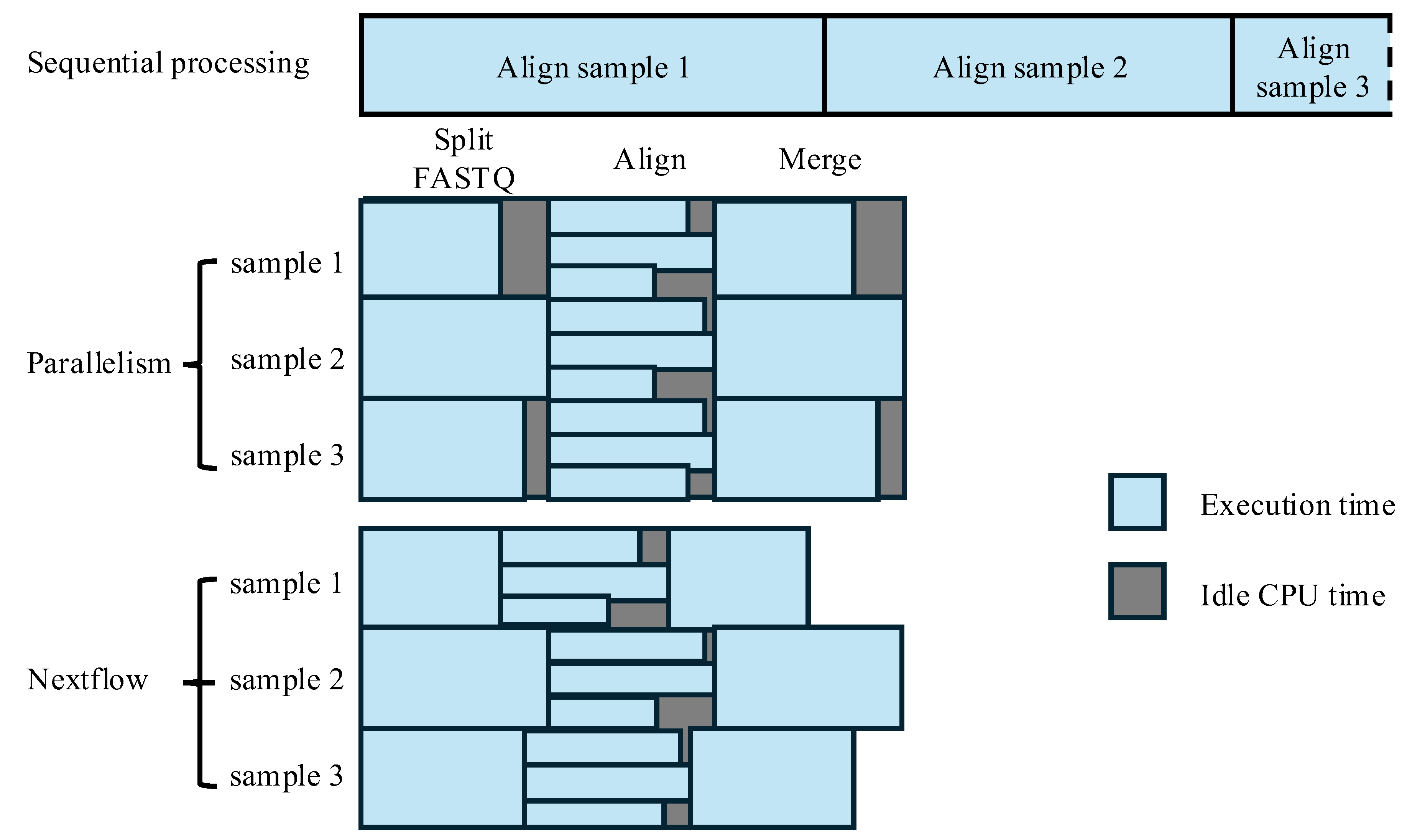
Figure 5.
Overarching schematic of cancer genomics with a focus on diversity and inclusion. a Starting with the global collection of biospecimens chosen with unique patterns of digital karyotyping as exemplified by Chan et al [68]. b an innovative reference-free cancer genomics pipeline can be built based on the foundation of long-read technologies and telomere-to-telomere assembled chromosomes, followed by c a new ecosystem of analysis tools for their use in clinical applications.
Figure 5.
Overarching schematic of cancer genomics with a focus on diversity and inclusion. a Starting with the global collection of biospecimens chosen with unique patterns of digital karyotyping as exemplified by Chan et al [68]. b an innovative reference-free cancer genomics pipeline can be built based on the foundation of long-read technologies and telomere-to-telomere assembled chromosomes, followed by c a new ecosystem of analysis tools for their use in clinical applications.
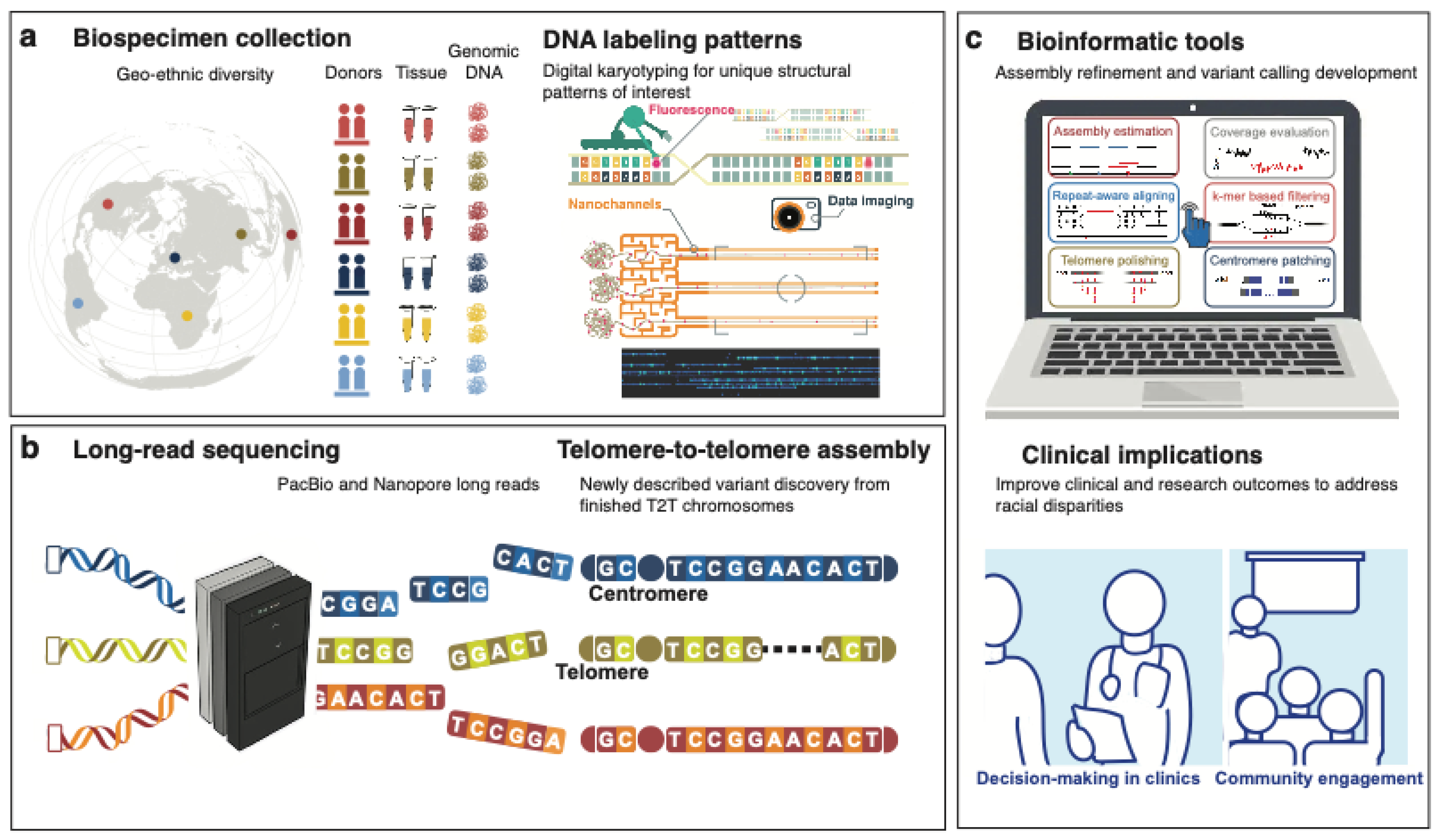

Table 1.
Cohort information of African WGS datasets of tumour and matched-normal tissue.
| Consortium or project | Cancer type | Country | Cohort size a | Tissue fixation b | Coverage of tumour, normal (median/mean) |
Recruitment time | Recruitment hospitals |
| SAPCS [27,28] | PCa | South Africa | 123 | FF | 88.69X, 44.3X (median) | 2013 – 2018 | Polokwane Urology Clinic, Limpopo; Tshilidzini Hospital, Limpopo; Pretoria's Steve Biko Academic Hospitals, Gauteng; Dr. George Mukhari Academic Hospitals, Gauteng; and Kalafong Academic Hospital, Gauteng |
| ESCCAPE [29] | ESCC | Kenya | 68 | FF | 49X, 26X (mean c) | 2014 - 2020 | Moi Teaching and Referral Hospital, Eldoret; |
| Malawi | 59 | Queen Elizabeth Central Hospital, Blantyre; | |||||
| Tanzania | 35 | Kilimanjaro Clinical Research Institute, Moshi | |||||
| AfrECC [30] | ESCC | Tanzania | 61 | FFPE | 60X, 30X (targeted coverage, de facto values unavailable) | 2016 - 2018 | Muhimbili National Hospital, Dar es Salaam, |
| BLGSP [31,32] | BL | Uganda | 87 | 83 FF, 4 FFPE | 82X, 41X (mean c); 72.6X (mean across sample types c) |
Unavailable | Uganda Cancer Institute, Kampala; St. Mary's Hospital, Gulu |
| NBCS [33] | BRCA | Nigeria | 97 | FPAX | 103.2X, 35.1X (mean) | 2013 - 2015 | Lagos State University Teaching Hospital, Lagos |
a cohort size: the number of cancer patients whose tumour and matched blood/normal samples underwent WGS, b FF: fresh frozen tissue; FPAX: Fresh PAXgene; FFPE: formalin-fixed paraffin-embedded tissue, c the mean coverage is calculated from the whole study cohort that include patients outside Africa.
Table 2.
Main ancestral-related findings from African WGS datasets.
| Cancer type | Measurement | Values or odds ratios | P-value | Comparison b |
| Short variants (nucleotide variants, insertion and deletion variants less than 50bp) | ||||
| PCa | Tumour mutational burden (TMB, mutations per Mb) | 1.197 versus 1.061 | 0.013 | EUR |
| PCa | Predicted damaging mutations (count) | 14 versus 11 | 0.022 | EUR |
| BRCA | Insertions and deletions (indels) | N/A | 6.510−5, 210−4 | EUR, AA |
| Driver genes | ||||
| BRCA | GATA3 | 6.3-fold | FDR=0.038 | EUR, AA |
| BRCA | Non-coding region, upstream of ZNF217 (frequency) | 42.3% versus 4.3% | FDR=0.037 | EUR, AA |
| BRCA | Non-coding region, spanning SYPL1 (frequency | 28.9% versus 0% | FDR=0.097 | EUR, AA |
| ESCC | TP53 (frequency) | 72% versus 74.8% - 87% [36,37,38] | EUR, AA | |
| BL | SIN3A (frequency) | 18.4% versus 9.1% | patients from the USA | |
| BL | HIST1H1E (frequency) | 9.2% versus 4.5% | ||
| BL | CHD8 (frequency) | 9.2% versus 4.5% | ||
| Somatic copy number alteration (SCNA) | ||||
| PCa | Percentage of genome alteration (PGA) | 7.26% versus 2.82% | 0.021 | EUR |
| BRCA | Whole-genome duplications (WGD) | 3-fold | FDR=0.02 | EUR, AA |
| Structural variants (SV) | ||||
| PCa | Duplication (relative frequency, count) [39] | 1.6-fold, 2.5-fold | EUR | |
| PCa | A single type hyper-SV frequency [39] a | 2-fold | EUR | |
| PCa | PCAT1 | 9.09-fold | 0.012 | EUR |
| PCa | TMPRSS2-ERG | 0.26-fold | 0.0004 | EUR |
| Several types of variants combined | ||||
| BRCA | intra-tumoral heterogeneity (ITH, increase %) | 3.4%, 5.7 % | 0.005, 0.00017 | EUR, AA |
| PCa | NCOA2 | 5.81-fold | 3.1410−6 | EUR |
| PCa | DDX11L1 | 4.17-fold | 0.0001 | EUR |
| PCa | STK19 | 4.65-fold | 0.004 | EUR |
| PCa | SETBP1 | 2.80-fold | 0.012 | EUR |
a A single-type hyper-SV is defined as a tumour with at least 100 SVs dominated by a single type, b EUR, AA means significant comparisons between African patients with European, and African American patients, respectively.
Table 3.
Bioinformatic tools applied to African WGS data.
| Consortium or project | Genome | Variant callers | ||
| Short variants | Structural variants | |||
| Germline | Somatic | |||
| SAPCS | GRCh38 | GATK HaplotypeCaller [45] | GATK MuTect2 [46] | GRIDSS [47], Manta [48] |
| ESCCAPE | GRCh37 | Strelka2 [49] | Strelka2, and cgpCaVEMan [50] for SNVs; cgpPindel [51] for INDELs | BRASSa |
| AfrECC | GRCh37 | - | RADIA [52] | - |
| BLGSP | GRCh38 | - | Strelka2, GATK Mutect2, Lofreq [53], and SAGEb | GRIDSS, Manta |
| NBCS | GRCh37 |
Platypus [54] | GATK MuTect and Strelka [55] | Manta, DELLY [56], and Lumpy [57] |
Table 4.
Configurations for SAPCS workflow compute jobs. Estimates of data processing with a batch of 20 pairs of tumour and matched-blood samples using National Computational Infrastructure (NCI) facilities.
Table 4.
Configurations for SAPCS workflow compute jobs. Estimates of data processing with a batch of 20 pairs of tumour and matched-blood samples using National Computational Infrastructure (NCI) facilities.
| Steps | Sample type a | CPU/task | Total tasks | Batches | CPUs/batch | Execution time (hr) | Main algorithm with version |
| Pipeline 1 Data pre-processing for variant discovery | 14.4 | ||||||
| Split FASTQ | Bood | 4 | 20 | 1 | 96 | 0.9 | fastp [61] v0.20.0 |
| Tumour | 4 | 20 | 1 | 96 | 1.8 | ||
| Alignment | Both | 6 | 11,040 | 3 | 3,840 | 0.5 | BWA-MEM v0.7.15 |
| Merge | Bood | 24 | 20 | 1 | 480 | 0.4 | SAMBAMBA [62] v0.7.1 |
| Tumour | 24 | 20 | 1 | 480 | 0.8 | ||
| Mask duplicate | Bood | 14 | 20 | 1 | 280 | 1.3 | SAMBLASTER [63] v0.1.24 |
| Tumour | 14 | 20 | 1 | 280 | 2.6 | ||
| BQSR recal | Bood | 1 | 640 | 1 | 640 | 0.2 | GATK v4.4.0.0 b BaseRecalibrator |
| Tumour | 1 | 640 | 1 | 640 | 0.3 | ||
| BQSR apply | Bood | 2 | 480 | 1 | 960 | 0.3 | GATK ApplyBQSR |
| Tumour | 2 | 480 | 1 | 960 | 0.6 | ||
| qSignature | Bood | 24 | 20 | 1 | 480 | 0.7 | QSignature c v0.1pre (75) |
| Tumour | 24 | 20 | 1 | 480 | 1.4 | ||
| Qualimap | Bood | 6 | 20 | 2 | 144 | 1.4 | Qualimap [64] v.2.2.1 |
| Tumour | 6 | 20 | 2 | 144 | 2.8 | ||
| Sequenza | Pair | 2 | 480 | 1 | 504 | 3.6 | Sequenza [65] v3.0.0 |
| Pipeline 2 Germline short variant discovery | 8.1 | ||||||
| Variant call | Bood | 1 | 64,000 | 1 | 480 | 1.8 | GATK HaplotypeCaller |
| Consolidation | Bood | 1 | 3,200 | 11 | 144 | 1.3 | GATK GenomicsDBImport |
| Joint genotyping | Bood | 1 | 3,200 | 1 | 144 | 2 | GATK GenotypeGVCFs |
| VQSR | Blood | 16 | 1 | 1 | 16 | 3 | GATK VariantFiltration, MakeSitesOnlyVcf, VariantRecalibrator, CollectVariantCallingMetrics, ApplyVQSR, CollectVariantCallingMetrics |
| Pipeline 3 Somatic short variant discovery | 3.3 | ||||||
| PoN | Bood | 1 | 64,000 | 1 | 2,880 | 0.6 | GATK Mutect2 |
| Consolidate | Blood | 2 | 3,200 | 1 | 96 | 0.3 | GATK GenomicsDBImport |
| Create PoN | Blood | 1 | 3,200 | 1 | 960 | 1.6 | GATK CreateSomaticPON |
| Variant call | Pair | 1 | 64,000 | 1 | 2,880 | 0.8 | GATK Mutect2 |
| Pipeline 4 Structural variant discovery | 23 | ||||||
| GRIDSS | Pair | 8 | 20 | 20 | 8 | Range, 10 - 20 | GRIDSS v2.8.3 |
| Manta | Pair | 24 | 20 | 2 | 48 | 3.0 | Manta v1.6.0 |
a Both means that tumour and blood samples are processed in one job but as separate tasks. Pair means that tumour and the matched blood are processed together in one task. Steps performing high-level parallelism are highlighted in grey. Small steps processing for a few minutes are omitted. b GATK tools are all v.4.4.0.0 version, c https://github.com/AdamaJava/adamajava/tree/master/qsignature.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



