
Submitted:
27 June 2025
Posted:
30 June 2025
You are already at the latest version
Abstract
The dynamic phosphorylation of the human RNA Pol II CTD establishes a code applicable to all eukaryotic transcription processes. However, the ability of these specific post-translational modifications to convey molecular signals through structural changes remains unclear. We previously explained that each gene can be modeled as a combination of n circuits connected in parallel. RNA Pol II accesses these circuits and, through a series of pulses, matches the resonance frequency of the DNA qubits, enabling it to extract genetic information and quantum teleport it. Negatively charged phosphates react under RNA Pol II catalysis, which increases the electron density on the deoxyribose acceptor carbon ( 2'C in the DNA sugar backbone). The phosphorylation effect on the stability of a carbon radical connects tyrosine to the nitrogenous base, while the subsequent pulses link the protein to molecular water through hydrogen bonds. The selective activation of inert C(sp3)–H bonds begins by reading the quantum information stored in the nitrogenous bases. The coupling of hydrogen proton transfer with electron transfer in water generates a supercurrent, which is explained by the correlation of pairs of the same type of fermions exchanging a boson. All these changes lead to the formation of a molecular protein-DNA-water transcriptional condensate.
Keywords:
phosphorylation
; transcription processes
; hydrogen proton transfer
; protein-DNA-water condensate
1. Introduction
Phosphate esters and anhydrides dominate the living world. The main reservoirs of biochemical energy—adenosine triphosphate (ATP), creatine phosphate, and phosphoenolpyruvate—are phosphates 1. Unlike synthetic organic chemistry, nature has favored phosphates, as the leaving group is typically phosphate or pyrophosphate (PPi) in most natural biochemical and metabolic reactions. No other residue appears to fulfill the numerous roles of phosphate in biochemistry, particularly as an energy source. The importance of protein phosphorylation in life is evident in nearly every process. The human proteome contains around 100,000 potential phosphorylation sites 2. In cellular signaling, one-third of all proteins in the cell are phosphorylated at any given moment 3,4.
While the hydrophobic nitrogenous bases form complementary Hydrogen (H)-bonded pairs (A-T and G-C) within the macromolecule to minimize contact with water molecules, negatively charged phosphate groups in the backbone remain exposed to the solution 5. Under physiological conditions, water molecules surrounding DNA create a dynamic network of H-bonds that interact with the structure and dynamics of the double helix. DNA maintains a specific B-form with a hydration shell of 30-50 water molecules per nucleotide pair 6. These water molecules establish a dynamic network of H-bonds akin to bulk water. Water molecules that interact directly with the DNA atoms make up the most structured part of the hydration shell, particularly within the grooves of the double helix; these water complexes are the most ordered 7,8.
Specifically, within the DNA minor groove, a single water molecule can bridge two atoms of different nucleotide bases, forming a chain of structured water molecules known as the DNA hydration spine 6,9.
RNA polymerase II (Pol II)-driven transcription is a unidirectional multistep process. Each cycle can be divided into phases: initiation, elongation, and termination. Thousands of unique transcription cycles occur with the concerted action of transcriptional cyclin-dependent kinases, their cognate cyclins, and the opposing activity of transcriptional phosphatases 10. The dynamic phosphorylation of multiple residues within the Pol II complex carboxy-terminal domain (CTD) of the RPB1 subunit is central to controlling Pol II-driven transcription. In humans, this region comprises 52 heptad repeats of Tyrosine (Tyr), Serine (Ser), Proline (Pro), and Threonine (Thr) (Tyr1Ser2Pro3Thr4Ser5Pro6Ser7) 11. A growing list of proteins specifically recognizes post-translationally modified CTD residues, integrating additional transcriptional and co-transcriptional activities. DNA-binding transcriptional regulators are the most essential components of these regulatory systems 12.
The proper function of the genome depends on the spatial organization of DNA. The assembly of the Pol II complex occurs at the gene promoter 13–15. Protein compartmentalization, driven by multivalent interactions and liquid-liquid phase separation, provides a mechanism to regulate protein functions spatially 16–20. An emerging model for the transcription of protein-coding genes suggests that distinct transient condensates form at gene promoters and within gene bodies, concentrating the factors necessary for each transcription step. RNA pol II may shuttle between these condensates in a phosphorylation-dependent manner 21–23.
We recently described a quantum model of transcription. In this work, every gene in DNA was modeled as a combination of -circuits using Quantum Electrodynamics 10. Based on establishing DNA qubits (with the central H-bond acting like a Josephson Junction) as Radio Frequency Superconducting Quantum Interference Devices (RF SQUIDs) with intrinsically quantum behavior and the sugar-phosphate backbone as a Finite Transmission Line with Distributed Parameters (Figure 1A). DNA is unwound in the transcription bubble condensate before the RNA pol II catalytic site. The growing RNA is attached to the active center cleft with its 3′ end, forming the nine-bps hybrid duplex with the DNA template strand. The high Elongation Complex stability depends on the DNA-RNA tight binding, which facilitates transcription processivity 24. RNA Pol II changes the external magnetic flux through a phosphorylate mechanism with the exact pulse parameters times to read the DNA qubits. This generates a phase difference. A pulse is a finite time series of complex values (a pulse amplitude at a specific time). However, a pulse was defined in DNA as a finite time series of phosphorylation reactions 10.
The intrinsic multiscale nature of the DNA molecule necessitates chemistry and quantum physics to understand its structure, and quantum informatics to explain its function. Here, after characterizing the transcription process and DNA as a quantum computer, we describe why phosphates constitute the pulse emitted by RNA pol II to produce decoherence in the system and transmit genetic information. Furthermore, while there is ongoing discussion about the formation of molecular condensates during transcription, this article establishes, for the first time, the relationship between the proton released during the phosphorylation series of pulses, conductivity, and the structural formation of water associated with DNA, enabling the system to operate in a compartmentalized manner. Using valence theory and analyzing the orbitals, we establish how the proton jump is carried out, ruling out the creation of a pure proton current, as suggested in previous studies. We introduce the concept of a supercurrent of electrons induced by the movement of a proton and propose a physical-mathematical model. The establishment of resonant quantum states in which electron pairing occurs through boson exchange is a novel physical model, first analyzed by our group to explain aromaticity in benzene and, in this work, to elucidate the superconductivity of water during the transcription process. All this allows us to suggest that, in addition to all the qualities previously described concerning phosphates, their essential role as the pulse, the transfer of energy, and the source of protons that ensures the formation of the molecular condensate, without which transcription cannot occur, is now highlighted.
2. Material and Methods
This work presents a theoretical approach to describe the role of phosphates during the transcription cycles using concepts from physics and chemistry. The outline of this paper is as follows: Section 2.1 introduces phosphates as driving forces for RNA Pol II. Section 2.2 presents the theoretical model of phosphorylation-inducing interactions between RNA Pol II and DNA. Section 3 highlights the role of water molecules in DNA and intrinsically disordered proteins (IDPs). Section 3.1 proposes a physical model of phosphorylation mediating H+ proton transfer to explain superconductivity in water in Section 3.2 and 3.3. Section 4 discusses the theoretical background that underlies our model definition. All figures were created and edited using Adobe Illustrator (AI) software 2025. For the mathematical analysis, we used the Word Office 365 Equation Editor.
3. Results
3.1:. Characterization of the pulse during the transcription process
3.1.1: Phosphates as the driving force
The dynamic phosphorylation of Tyr-Ser-Thr-Pro in the Pol II CTD establishes a “phosphorylation code,” which acts as a signaling mechanism in all eukaryotic transcriptional processes. However, the ability of these specific post-translational modifications to convey molecular signals through structural changes remains unresolved. Another challenge is that most eukaryotic protein kinases share structural similarities 3. Our group has begun to address this question through theoretical analysis and modulation via local unconventional non-covalent interactions to examine the conformations of helical Tyr, Ser, Thr, and Pro residues in RNA Pol II CTD during transcription.
In general, a driving force is necessary for every chemical reaction. In phosphorylations, the phosphoryl group (PO3−) is transferred, for instance, from ATP to various substrates. Nucleotides transfer from ATP or guanosine triphosphate (GTP) during the synthesis of ribonucleic acids or deoxyribonucleic acids utilizes the favorable properties of phosphoric anhydrides 25,26. Living systems must exhibit longevity, particularly regarding genetic material. The stability of the phosphate ester bond renders it an ideal switch in information and energy transfer processes. The negative charge of the phosphate reduces the rate of nucleophilic attack on the ester, making it relatively resistant to hydrolysis and thereby enhancing DNA stability (Figure 1A). This is why phosphates persist in an aqueous environment. Moreover, they are thermodynamically unstable and can drive chemical processes to completion in the presence of a suitable catalyst (enzyme) 27.
In PO3−-transfer reactions, the nucleophile aligns with the phosphorus atom and the leaving group for an in-line attack on phosphorus. Enzyme interactions can position the nucleophile in this reactive geometry 25. In the loose transition state, the leaving group develops a substantial negative charge relative to the ground state, resulting in the near-complete formation of metaphosphate. The monomeric metaphosphate ion becomes almost free, if not entirely so 25. Thus, the transferred PO3− undergoes electrostatic and geometric changes 28. Unlike the simple intermediary metabolite phosphorylation, the group that leaves during nucleotide polymerization is PPi. Similarly, the activation of amino acids occurs through adenylation. Only nucleoside triphosphates can release PPi while retaining a phosphate for the sugar-phosphate backbone of RNA and DNA or amino acid activation.
Critical biological processes tend to occur within quantum molecular condensates 19,29,30. These condensates can be closed, exchanging energy, open, exchanging mass and energy, or isolated when they don´t exchange. Most ionized molecules are insoluble in lipids, keeping them within membranes. Phosphates, conversely, are ionized at physiological pH and, therefore, are trapped within cells. Moreover, the electrostatic interaction of positive and negative charges constitutes the most straightforward and possibly primitive interaction between molecules. The negative charges on phosphates are essential in binding coenzymes to enzymes and "packaging" nucleic acids.
In eukaryotic cells, phosphorylation primarily occurs on three hydroxyl (OH)-containing amino acids: Ser, Thr, and Tyr, with Ser being the predominant target. Proteomic analysis has shown that phosphoserine (pSer), phosphothreonine (pThr), and phosphotyrosine (pTyr) represent 86.4%, 11.8%, and 1.8%, respectively. The fully sequenced human genome is believed to contain 518 putative protein kinases that can be classified into two families: 90 Tyr kinases and 428 Ser/Thr kinases 3,4.
3.1.2: Model of phosphorylation inducing interactions between RNA pol II and DNA
Protein kinase enzymes catalyze the transphosphorylation of OH-containing substrates by converting ATP to adenosine diphosphate (ADP), a process facilitated by a divalent cation (typically Mg2+) in a substitution reaction centered around phosphorus 31. CTD Pol II is flexible and highly disordered. Most phosphorylation sites in proteins are located in their disordered or dynamic regions. Tyr phosphorylation has emerged as a key theme in cellular regulation, primarily due to the finding that activating a Tyr kinase is the initial event in many signaling pathways. The oncogenes of various retroviruses encode Tyr kinases, which promote unregulated Tyr kinase activities that transform cells 26,32,33. The effects of Tyr kinases are partly mediated through a substantial quantitative increase in the phosphorylation of aliphatic hydroxy-amino acids such as Ser and Thr 31. Tyr kinases indirectly activate Ser/Thr kinases through phospholipid-derived second messengers 26. This may explain why Tyr-phosphorylated proteins remain a small minority.
Tyr's phenol moiety contains a hydrophilic OH group that can act as an H-bond donor or acceptor and a hydrophobic benzene ring. Thus, it exhibits a bilateral mode of interaction. Tyr aromatic ring can form C−H⋅⋅⋅ interactions, changing the potential dispersion 34. Non-covalent interactions involving the Tyr phenol and other side-chain aromatic rings of amino acids have also been reported 35. The structure-stability relationship of the aromatic ring stacking and the effect of the OH group phosphorylation on the stacking is an example of the “stacked form-unstacked form” equilibrium regulation 36. It may serve as a chemical model of the biological processes involving Tyr phosphorylation.
In the stacked structure, the carbons ortho to the OH group of Tyr-OH conformation would allow the electron flow with the OH group, increasing the ring electron density. The stacking between rings with similar electron densities should have a weaker stabilizing effect 36. Radicals play an essential role in many chemical reactions because of their unpaired single electron, which makes them highly reactive, thus quickly attacking the substrate, destroying the stable electronic structure of the substrate by snatching electrons, and making them easily reaction-activated 37,38. Radicals are even more critical for highly polymerized organic molecules because their chain structure may provide continuous electron transfer (ET) for the reaction in thermodynamics and chemistry.
An approach to the interaction through the OH leads us to the ET theory. ET represents the tunneling transition process of an electron between two localized states on the donor and acceptor molecular groups 39. Biological ET is efficient, specific, and frequently reversible. It has evolved from the early ideas of bridge-mediated electron tunneling in the 1960s to a quantum mechanical process 38. Yet, any analytic ET theory results from some simplifying assumptions. A single-ET (SET) reaction has been proposed for the ligand-protein interaction between some molecules and Tyr residue 40,41. The proton-coupled electron transfer (PCET) and H atom transfer (HAT) are the other two mechanisms by which the OH radical initiates the interaction with bimolecular compounds 42.
Based on these findings, we theorize that Tyr1 in the RNA Pol II CTD initiates reading the quantum information contained in the first base pair during transcription. The primary catalytic apparatus consists of a conserved Tyr1 and deoxyribose. The conserved Tyr1 residue performs a nucleophilic attack on the ATP phosphate moiety to release H+ and form a covalent pTyr-nitrogenous base ribose complex. Non-covalent interactions may influence the formation of coordination compounds 43. The OH radical in Tyr1 targets the gamma-phosphorus group in the ATP chain. Then, it is transferred from ATP to Tyr1, enhancing the reactivity of the acceptor molecule (Figure 1B).
Figure 2 focuses on phosphorus-centered radicals. It explains that the first step is to induce a reaction in which the pTyr1-PO4 radical attaches to the H atom at the 2' carbon (C) position. A deoxyribose residue in the DNA backbone has seven H atoms attached to C that are, in principle, available for abstraction by an oxidizing agent. Although DNA presents a conformational challenge, geometry optimization and orientation suggest that 2'C-H is the most probable option. After removing the deoxyribose's H atom, the unpaired single electron becomes localized in the 2'C, increasing the electron spin density at this atom. As a result of the ET processes, the hybridization and bonding characteristics of the C atom will also change accordingly. In this scenario, the single bond between the single-electron 2'C atom and its adjacent C (1'C) atom tends to form a double bond through an electron rearrangement, accompanied by bond breaking. Thus, the single electron is transferred along the chain until the nitrogenous base is reached. The ET-specific mechanism can also be manifested as a spin-transfer reaction. The negatively charged phosphoryl group is attached to atom 2'C of the ribose. The presence of a single electron activates the C–C bond. Additionally, we hypothesize that the geometry adopted by Tyr facilitates non-covalent interactions between Tyr's benzene ring and the aromatic ring of the nitrogenous base attached to the sugar.
The Ser2 residue then carries out a second nucleophilic attack. Several proteins have α-helical segments containing Ser and Thr residues. These sequences are abundant in the human proteome, particularly in IDPs/ intrinsically disordered regions (IDRs) (IDPs/IDRs). Ser and Thr contain polar OH groups, with their side chains differing only by an additional methyl group for Thr. They also play similar roles in biology by forming H-bonding networks, participating in enzymatic active sites, and acting as phosphorylation targets. Because of their H-bonding potential, these residues might form H-bonding to the carbonyl oxygen in the preceding turn of the α-helix, changing its conformation 44. xn-H2O fragment ions occur exclusively at Ser and Thr residues 45. Neutral water loss by Radical-directed dissociation (RDD) is common in both amino acids, behaving similarly and facilitating nearly identical dissociation products 46. RDD is a fragmentation technique that utilizes the collisional activation of a radical precursor to access fragmentation pathways 46.
Eukaryotic proteins have notable linker regions that are often significantly disordered. Ser and Pro are among the most disorder-promoting residues. Specifically, Ser is prevalent in IDRs and ranks as the third most disorder-promoting residue 47. Pro is also familiar with linker regions that connect domains. On average, eukaryotic proteins consist of 450 residues, with 32% classified as disordered, amounting to an average of 145 disordered residues 48. This suggests that disordered eukaryotic protein residues contribute to increased linker regions' disorders. When interacting with specific binding partners, IDPs/IDRs undergo a disorder-to-order conformational transition 49. Pro residues are crucial in modulating local and global protein structures, as Pro is the only canonical amino acid capable of forming tertiary amide peptide bonds 50,51. In the absence of amide hydrogen, Pro can induce disorder by disrupting H-bonded secondary structures and promoting order. The trans/cis-amide isomerism at Pro creates significant pivots in the peptide backbone. Phosphorylation of Ser-Pro stabilizes the ground state of both trans- and cis-Pro amide conformations. It is proposed that phosphorylation of Ser-Thr-Pro sequences stabilizes both Pro amide isomers, although this typically occurs only in the trans-Pro conformation 52. Furthermore, Pro is unique in non-covalent interactions; its cyclic structure results in intense inductive polarization of its C-H bonds. These bonds are potent electron acceptors and have consistently been observed to engage in C-H/O interactions in various small-molecule and protein crystal structures 53.
Biomolecular condensates formed through the phase separation of IDPs/IDRs and nucleic acids are associated with water. Water makes up about 60–70% of the condensate volume and is believed to influence the interactions between chains and chain solvent, modulating the dynamics of the condensates 54. The active sites of many redox-active proteins are situated sufficiently close to highly polar water, allowing for a significant water component in the medium reorganization energy 39. Electron hopping in chains of biological redox cofactors often occurs with nearly zero reaction-free energy. The influence of water molecules on the mobility and reactivity of OH radicals presents a crucial knowledge gap in our current understanding of essential reactions. However, the role of water in condensates remains unclear, particularly concerning the key functions of protein hydration water in driving phase separation. Specifically, the relevant molecular mechanisms linked to the mobility of OH radicals and the potential for diffusion in water via an H-transfer reaction remain unresolved questions 55. Dehydrogenative transformations via a SET produce more functionalized molecules by forming C-heteroatom and heteroatom–heteroatom bonds. These reactions are characterized by chemical bond formation, including C–C, C–N, C–O, and other C-heteroatom bond formations 56.
In Figure 2, pTyr joins the CTD IDR chain with the DNA nitrogenous base through a non-covalent interaction with the deoxyribose. In contrast, the phosphorylation of Ser/Thr joins the Tyr-nitrogenous base complex with water molecules. In the next section, we will address the relationship between phosphorylation during transcription and the water associated with DNA in forming the biomolecular condensate, facilitating the quantum transmission of the genetically encoded information.
3.2:. Water molecules in DNA and intrinsically disordered proteins
Hydration significantly influences DNA binding with proteins and ligands and the intercalation of foreign molecules. Water molecules create hydration layers around DNA, stabilizing an entangled structure 57, though they may not uniformly cover its surface. Some water molecules, particularly those in the inner hydration shells of the minor and major grooves of DNA, are tightly bound to the DNA surface. In contrast, those found in the outer shells or interacting with Na+ counterions are only loosely bound. The counterions neutralize the DNA’s negative charge and affect the structural network of H-bonds around DNA atomic groups, influencing the dynamic characteristics of water molecules. Water molecules exhibit more restrictions in the minor and major grooves than in free water. This phenomenon has been described as a chiral helix of water molecules in the DNA minor groove 58,59.
The dynamics of water molecules are typically described in terms of residence time, reorientation time, and vibrational frequency 60. Water positioned near the DNA surface displays high frequencies. The changes in the characteristic residence time depend on their localization around the double helix. In the inner regions, it increases several times, reaching approximately 100 ps in the major groove and around one ns in the minor groove 60. The H-bond reorientation between different partner atoms occurs within this time range.
Additionally, water molecules can experience translational vibrations during residence times before reorienting and transitioning to another equilibrium state. These vibrations are prominent in the low-frequency spectral range. Six modes describe the intermolecular vibrations of water molecules. DNA's low-frequency spectral range includes the double helix's conformational vibrations 59. These modes consist of the stretching vibrations of H-bonds in nucleotide pairs, inter-nucleoside vibrations (near 80 cm−1), and backbone vibrations (near 15 cm−1). Translational vibrations of the hydration spine in the minor groove of the double helix occur within the frequency range of 160-210 cm−1 and are significantly influenced by the nucleotide sequence. The dynamics of counterions, which neutralize the negatively charged phosphate groups of the DNA backbone, are characterized by specific modes of ion-phosphate vibrations in the range of 100 to 180 cm−1, depending on the type of counterion 6. The conformational vibrations of DNA and counterions are coupled with the vibrational dynamics of water molecules surrounding the double helix.
Compared to the folded globular proteins involved in processes like folding, molecular recognition, and enzyme function, the properties of water molecules on the surfaces of IDPs/IDRs remain poorly understood. Research indicates that water motions are more restricted, and the coupling between the protein and its hydration water molecules is stronger for IDPs/IDRs than for folded proteins 61. Additionally, studies show that the diffusive water dynamics on the surfaces of IDPs/IDRs are more heterogeneous. Furthermore, the relationship between the translational diffusion of water molecules on protein surfaces and the temperature-dependent activation of large-scale protein motions is independent of the protein-folding state 62,63. Strong electrostatic repulsions, arising from a higher net charge and a lack of compaction-inducing force due to low mean hydrophobicity, are the primary reasons for the unfolded, extended structure of IDPs/IDRs.
Having IDRs rich in amino acids with OH groups susceptible to phosphorylation as a post-translational modification offers advantages compared to the globular structure of proteins. The general phosphorylation reaction scheme is represented in:
MgATP− + substrate-O: H → substrate-O: PO32− + MgADP + H+
Our model illustrating the formation of the tetrahedral structure of water is presented in Figure 3. The release of a proton of H+ during the phosphorylation pulse modifies the nearby water molecules. The kinetic energy the proton gains as it is transformed into a free particle is employed to attract one water molecule to form the hydronium ion (H3O+). The H3O+ positive charge attracts two additional water molecules and forms the tetrahedral structure H7O3. It features a bifurcated H-bond that links to two neighboring water molecules and is distinctive, representing its minimum energy structure. The predominance of positive charges binds the water molecules together, probably contributing to what has been observed in other studies about the helical structure of water. This will be addressed later in the Discussion section.
3.2.1:. Physical model of phosphorylation mediating H+ proton transfer
This section will address the physical model of phosphorylation that induces the tetrahedral water structure through H+ proton transfer, as illustrated in Figure 3. The released proton acts as a free particle, and its kinetic energy is calculated by solving the Schrödinger equation:
Where is the wave function of a free particle:
Where , and , because it is a free particle. Then, equation (1):
Then, replace (2) in (3):
We obtain that:
Considering Water is a complicated quantum problem of eight electrons and three nuclei (two of H and one of oxygen). The Hamiltonian of a water molecule would be expressed:
When the proton approaches the H2O molecule, the H3O+ is formed, which is electropositive with a Hamiltonian:
The kinetic energy of the proton pulls an electron from the lone pair of one oxygen, forming the H3O+. This leaves a hole in the orbital of oxygen.
The H3O+ has a trigonal pyramidal molecular geometry with the oxygen atom at its apex.
Water H3O+ attracts two additional water molecules, H2O, to form a regular tetrahedron with the following Hamiltonian:
Now, the oxygen of the H3O+ shares a lone pair electron with the proton, leaving a hole in the orbital. If an electron located in a 2p orbital moves to a orbital, energy is released:
This energy is used to correlate electron pairs in the orbitals of the oxygens, resulting in a superconducting current inside the tetrahedron.
3.2.2:. Superconductor character of water induced by H+ proton transfer
The movement of an H+ proton during phosphorylation reactions induces an electric current of electrons into the water clusters. In this work, we explain that the conductivity of water is not precisely due to the PT theory. But to the supercurrent created by the delocalized motion of electrons moving in resonant quantum states. We prefer to call this phenomenon a supercurrent induced by a proton. The kinetic energy that the proton brings when it becomes a free particle is used to attract a water molecule. Still, when the H3O+ is formed, an electron from the lone pair of a orbital of oxygen jumps to the orbital of H+, it releases a Boson of quantum energy, inducing the subsequent electron pairs correlation and movement.
Previously, we described the general oscillatory resonant quantum states process between electron and hole pairs forming in -orbitals. The overlap of two z orbitals brings two electrons with equal spin closer, allowing them to experiment with two interactions: the Coulombian repulsion and the electron-vibrational energy-electron. The Coulombian repulsion forced electron one () to occupy the position of hole one . During the translation, emits a quantum of energy to electron two (). The energy absorbed is used by to occupy the hole two () position. In the second half of the oscillation, and exchange to return to their original positions. The energy difference between the electrons is the same as 64. That is the condition for the electron and hole pairs, one below and one above the Fermi level with opposite momentum, to oscillate. The electrons cannot occupy other states, so they do not interact with other atoms in the molecule. The resistance or dispersion energy is canceled.
In Figure 4A, we explain the process of superconducting water based on these same principles. When the H3O+ cation is formed, the kinetic energy of the H+ proton attracts an electron from a lone pair located in a free orbital of an oxygen (O1) atom to the empty orbital, leaving a hole. The displacement of this electron then releases energy and leaves a hole in its position. This is now used by another electron located in the orbital of the second oxygen (O2) to move to the new hole left.
3.2.3:. Application of the physical-mathematical model for the correlation of pairs of the same type of fermions (two electrons) that exchange a boson to explain superconductivity in water
In Figure 4B, the correlation of pairs of electrons that exchange a boson explains the superconductivity of water. In the first part, the electron with down spin () moves to the empty orbital of H+. In the second, the processes of formation of correlated pairs are represented when the electron that moves to the orbital of H+ is the one that has spin up (). The electrons that correlate to form pairs are and because they have opposite momentum, spin, and different energies. Their energy difference equals that of the boson (photon) they exchange. Electrons and can’t pair because when exchanging the boson (photon), they do not have a permissible space (hole) for the oscillatory movement to occur in the resonant quantum states. Electrons and also can’t oscillate in resonant quantum states because they involve transitions of electrons within the same orbital.
Two processes are required to form electron pairs. Process one involves a half oscillation. In this half oscillation, we go from state a to state b. In process two, the fermion pairs exchange the boson, performing a second oscillation that defines the oscillatory resonant quantum states. In this second-half oscillation, the system goes from state b to c, where the initial state is reached. The internal Hamiltonian ( of the system in the state a () is given by:
Where is the Colombian repulsion potential between electrons and and is the interaction potential between that emits a boson and that absorbs it.
The of the system in the state b () is given by:
Where is the Colombian repulsion potential between electrons and , equal to and . Then, . is the interaction potential between , which emits a boson, and , which absorbs it. Then, .
The total Hamiltonian would be:
Considering:
, the correlation energy of the electron pair,
and
Then, we obtain the Classical Scattering Law of Correlated Pair Formation in Pair Formation in Superconductivity:
Here, the existence of superconductivity in water is demonstrated.
4. Discussion
Chemical reactions involving the formation or cleavage of P-O bonds in phosphate esters are ubiquitous in biological systems. This work made a theoretical analysis and modulation via local unconventional non-covalent interactions to explain how the RNA Pol II CTD utilizes the 52 repeats of the sequence Tyr1Ser2Pro3Thr4Ser5Pro6Ser7 to interpret the genetic information encoded quantumly in the nitrogenous bases. In a prior study based on Circuit Quantum Electrodynamics theory, our group described the fundamental structure of DNA genes as a combination of non-perturbative circuits. By considering each base pair as a RF-SQUID (A-T, T-A, G-C, C-G), the dispersive detuning regime enables the quantum measurement of the qubit state and its manipulation. A sequential series of phosphorylation reactions was proposed as the driving pulse for transcription. Irradiating at the qubit frequency coherently controls the qubit, while irradiating at the resonator frequency introduces photons into the cavity that become entangled with the qubit states (Riera Aroche, Ortiz García, Sánchez Moreno, et al., 2024). The coupling between n-phosphorylation pulses and a specific qubit could be selectively achieved through frequency matching 10
Traditionally, non-covalent interactions, such as H-bonds, hydrophobic effects, electrostatic interactions, and van der Waals forces, are fundamental for protein folding, structural stability, and function 65. The -moiety-involved interactions, such as - stacking, CH-, cation/anion-, n → *, and XH- interactions (XH = NH, OH, SH), are equally crucial non-bonded interaction forces 4,66,67. These non-covalent interactions govern photophysical properties that are sensitive toward the microenvironment. Thus, by altering these interactions, selective sensing for a particular analyte can be achieved 68.
It has been reported that Tyr can form - stacking interactions with aromatic rings, while the side chains of Ser and Thr can create OH- and NH- interactions 4. Compared to H-bonding interactions, the dispersion forces that dominate CH- and - stacking interactions are relatively weak. However, dehydration incurs a much lower (or zero) energy cost. As a result, significant interaction strengths persist in the aqueous phase for both CH- and - stacking interactions 69,70.
Here, we found that the phosphate group in pTyr produces H-abstraction at 2'C of deoxyribose. The distribution of the unpaired electron via SET activates the C–C bond, supported by a readjustment of the electronic configuration. Living systems use proximity to regulate biochemical processes 71, and some authors have characterized Tyr's defining trait as a stronger adhesive 72. Ligand-protein and protein-protein interactions via weak and transient connections are essential for regulating all dynamic biological phenomena.
Various molecular drivers influence water's H-bonding network changes during homotypic and heterotypic phase separations 54. Molecular characteristics, such as affinity, valence, and the competition between intermolecular and intramolecular contacts, also contribute to phase separation 73. Phase equilibria are established by balancing species-specific chemical potentials with osmotic pressure across the coexisting phases 74 75,76. Aromatic residues and other polar moieties play a role in the phase transitions of IPDs 76,77. Identifying these critical and dynamic interactions remains a challenge.
The OH radical in aqueous environments plays a significant role in initiating specific reactions. In one study, the OH radical extracts an H atom from the polyacrylamide main chain. This action simultaneously triggers the SET process on the same chain, as shown by the spin-density transfer, which leads to a reduction in the dissociation energy barrier of the C–C bond from approximately 90 to about 20 kcal/mol 37. It was found that the SET process is crucial to the mechanism initiated by the attack of radicals on specific polymers 78. Modeling studies have predicted the presence of H-bonding and hydrophobic interactions mediated by the amino acid residues of Ser and Tyr, which elucidate odorant receptors and binding sites 34. Based on these interactions, various heterobifunctional small molecules have recently been developed to enhance the proximity of two or more proteins, triggering proximity-dependent modifications 79.
The conversion of the Tyr phenol OH group to the phosphoester group through phosphorylation significantly decreases stability due to the hydrophilic nature of the negatively charged phosphoester moiety. Specific biological processes are regulated by modifying the Tyr side group through phosphorylation, with its observed effect on intermolecular stacking interactions suggested as the basis for understanding molecular recognition and the biological regulation of reactions 79.
Interactions between aromatic rings are crucial in protein and protein-DNA systems for stabilizing proteins and facilitating various regulatory processes. Insights into DNA interactions involving the side chains of amino acid residues from transcription factors and the nucleobases of DNA have significantly advanced our understanding of the roles played by weak interactions during the initial stages of DNA transcription 79. Additionally, ring substituents affect the stability of stacked structures. One study analyzed the transcription factor HcaR from Acinetobacter sp. and four HcaR-ligand complex structures. Each ligand was bound to the same deep cavity, surrounded by multiple electron-rich aromatic and hydrophobic residues, including Tyr. Several hydrophilic residues, such as Ser and Thr, could also form direct or water-mediated hydrogen bonds with the ligand at the dimer interface within the cavity 80. Intermolecular stacking interactions have been shown for nucleotides and complexes with aromatic ligands in both solution and solid states. As expected, these interactions depend on the structure of the aromatic rings and can be interpreted as resulting from the interaction between molecular orbitals 79.
C–H/O interactions occur between neutral or anionic oxygen lone pairs and highly polarized C–H bonds. These interactions are often described in electrostatic terms related to charge attraction, similar to H-bonds. While there is an electrostatic aspect to C–H/O interactions, experimental and computational analyses have shown that they are primarily stereoelectronic, characterized by the delocalization of oxygen lone pair electrons into an alkyl H antibonding orbital 81. Due to their significant stereoelectronic component, it has also been noted that the strength of C–H/O interactions is mainly independent of electrostatics in polar environments, such as water.
The mechanism of protons moving through water is central to acid-base chemistry reactions. Developing a molecular basis for these phenomena is significant in energy conversion applications, such as designing efficient fuel cells. For a successful transfer, the model requires solvent reorganization around the proton-receiving species to establish a coordination pattern similar to the species it will convert to, a process known as pre-solvation, 57,82. Despite various characterizations of the Grotthuss mechanism, the role of the connectivity of the water network has not been emphasized.
Introduced in 1806 by Grotthuss, the concept of proton hopping is still regarded as the most efficient PT mechanism, typically implying the PT between chains of host molecules through elementary reactions involving H-bonds 83–85. Popov et al. noted that PT between molecules in pure and 85% aqueous phosphoric acid occurs via surprisingly short proton jump lengths (0.5-0.7 Å), accompanied by a reduction in conductivity, contrary to the expected enhancement typically associated with the Grotthuss mechanism 86. Agreeing with this result, in our work, we argue that the conductivity of water is not solely due to proton movement but rather to the supercurrent generated by the delocalized motion of electrons in resonant quantum states. The kinetic energy the proton gains when it becomes a free particle is exclusively used to attract a water molecule. An electron supercurrent is created induced by the transfer of a proton. The mutual neutralization of H3O+ and hydroxide OH− ions is primarily attributed to an ET mechanism, for which a minor contribution is linked to PT 87.
H3O+ is the only molecular cation that forms in water with H+ ions. These cations do not exist freely; they are highly reactive and are quickly solvated by surrounding water molecules 87. Among the over 1,000 crystal structures of hydronium ions recognized by the Cambridge Crystallographic Database, the H3O+ ion with a bifurcated H-bond connecting to two adjacent water molecules is unique, representing its minimum energy structure. Huang et al. demonstrate that this optimized geometry aligns with an unusual transition state characterized by a single vibrational frequency associated with protonated water structures 88. Some solvation structures can be pretty significant: the ion H3O+(H2O)20, often referred to as "magic" due to its more excellent stability compared to other structures with a similar number of molecules 89, would trap the proton inside a dodecahedral formation.
Water features a helical component in its structure, primarily because of the proton-hopping properties of the H3O+. The foundation for the proposed water structure consists of tetrahedral substructures that, when combined, form long helitetrahedral chains shaped like Boerdijk–Coxeter (BC) helices. The BC helix represents a linear arrangement of regular tetrahedron 90.
PT has been considered to involve the simultaneous jumps of multiple protons 91. In this context, protons can delocalize over extensive H-bonded networks. This phenomenon occurs when water molecules form isolated chains in confined environments such as proteins. Rapid PT occurs through transient water wires forming during acid-base reactions in ice and water. When the H3O+ and OH- ions come within approximately 6 Å of each other, a water wire continuously connecting them experiences collective compression during their recombination 86. This event leads to the synchronized movement of three protons on a timescale of tens of femtoseconds. The formation of polarized water wires appears to be a crucial precursor to correlated PT events. This raises the question of whether wire-like structures can exist in liquid water. Charge migration includes bursts of activity along proton wires within the network, characterized by the coordinated movement of several protons and resting periods longer than expected, reminiscent of a jump-like diffusion mechanism. This remarkable dynamic activity is partly driven by the proton wires' ability to undergo collective compressions 92. Previous studies have highlighted that a wide distribution of closed rings characterizes liquid water 93.
We divide PT through water molecules into consecutive steps between adjacent multicopy donor-acceptor pairs. One proton donor is marked at the beginning, creating multiple copies. A sphere is drawn around this donor, generating numerous copies for all possible acceptors within this sphere and potential additional donors. Protons hop between the multicopy donors and acceptors. If a group of acceptor copies becomes protonated, this group is marked as the new donor for the next transport step. All non-protonated copies of this acceptor must take up a proton from one of the remaining donor copies within a specified timeframe. Subsequently, the newly marked donor becomes the center of a sphere containing multicopy groups for the next transport step. Protonated water clusters also play a crucial role in the confined spaces and cavities of proteins and biomolecules.
H+-transfer can be a very rapid process involving water and is significantly affected by structural fluctuations. A rough estimate of the total activation energy for the proton exchange reaction involving OH− has previously been assessed at 3 kcal/mol 94. Early electron movement is noticeable compared to the position of the H+ proton 95. This suggests a hybrid mechanism incorporating electron−PT elements. A weak H-bond is formed between the OH- H and the H-bond-accepting water in the transition state. Consequently, the radical center relocates to a new site within the H-bond network 55.
We also hypothesize that the movement of protons and the transformations they trigger in the surrounding biological water alter the frequency during the pulses of sequential phosphorylations. The coherent vibrational character of the coupled solute and solvent arises when strong H-bonds exist between the solute and the water molecules 96. The ability of Ser/Thr to form H-bonds links the CTD sequence Tyr1Ser2Pro3Thr4Ser5Pro6Ser7 repeats to water molecules, creating a nitrogenous base-deoxyribose-CTD-water condensate. As a result, a revision has been proposed for the typical depiction of the interaction between DNA proteins and the solvating water molecules, treating their interaction as a perturbation. Especially in the presence of strong H-bonding between the solute and water molecules, water vibrations tend to be delocalized 97, suggesting that the vibrational eigenstates associated with the solute in water may also be delocalized, with correlations extending throughout its solvation shell. Thus, the extent of the coherent vibrational character of the coupled solute and solvent determines the distance scale and mechanism over which concerted bimolecular chemical reactions, such as PT, can occur. In aqueous chemical reactions, these vibrational motions and dissipation would also act as the source of the detuning processes.
5. Conclusions
Ser-Thr-Pro sequences are plentiful in the human proteome, particularly in IDPs/IDRs. This abundance highlights their biological significance as dynamic sites for structural modulation. The human RNA Pol II CTD comprises 52 disordered Tyr1Ser2Pro3Thr4Ser5Pro6Ser7 heptad repeats. The dynamic phosphorylation of Ser2, Thr4, and/or Ser5 within the Pol II CTD establishes a “phosphorylation code” as a signaling mechanism in all eukaryotic transcriptional elongation and termination activities.
Protein residues may be essential components of PT in H-bond networks. Negatively charged phosphates react under the catalysis of enzymes that couple PT with electron transfer. Partially replacing H in deoxyribose with electron-rich PO3- enhances strong electronic interactions between Tyr and the sugar, linking the aromatic ring of Tyr with the aromatic nucleobases of DNA. Subsequently, the phosphorylation of Ser-Thr-Pro sequences alters the secondary structure and establishes H-bond connections with water molecules. The results suggest that this strategy can significantly increase the vacancy formation energy and the migration energy barrier of PO3-.
Meanwhile, the H+ ion utilizes its free energy to form H7O3 water clusters. The vibrational dynamics of water in aqueous chemical reactivity indicate that this reaction system likely involves an H-bond between the water molecule and the solute (CTD sequence Tyr1Ser2Pro3Thr4Ser5Pro6Ser7). The concerted reaction establishes a framework of DNA-protein-water interactions. Additionally, we anticipate that the system exhibits coherent vibrational motions between the coupled solute and solvent, reflected in the modulation of H during the concerted reaction, as the reaction system involves oscillations in proton donor-acceptor distances.
This natural electronic confinement-restrained strategy ensures the optimal temperature and pressure conditions needed to teleport the quantum information of each base pair. Utilizing the confinement effect of electron interactions offers new perspectives for exploiting and overcoming high-energy-density materials. Additionally, incorporating PO3- increased the number of lone pairs of electrons and optimized the band structure, benefiting from suitable conduction bands, sufficient electrons, and effective ET. The system limited the formation of intermediate products and enhanced catalytic efficiency. Multi-electron reduction achieved in electron-rich catalysts with high local electron concentrations will reduce the production of undesirable intermediates. With the active involvement of kinases and phosphatases, the process is reversibly converted to facilitate the reversible participation of multiple electrons in the electrochemical transcriptional process.
Also, in this work, we describe the superconducting character of water through the correlation of two electrons that exchange a boson. The coexistence of empty orbitals and outer lone pairs is beneficial, as the empty orbitals can accept electrons acting like holes. We have developed this theoretical model more extensively in our previous works, and it is a universal model for explaining superconductivity. But unlike these earlier works, the initial energy here is provided by the movement of a free H+, creating a proton-driven supercurrent of electrons. Nature chose Phosphates as transcription machinery-driven forces because of their multi-ET capabilities and high energy efficiency.
References
- Tian, T.; Chu, X.-Y.; Yang, Y.; Zhang, X.; Liu, Y.-M.; Gao, J.; Ma, B.-G.; Zhang, H.-Y. Phosphates as Energy Sources to Expand Metabolic Networks. Life 2019, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Yaron, T.M.; Huntsman, E.M.; Kerelsky, A.; Song, J.; Regev, A.; Lin, T.-Y.; Liberatore, K.; Cizin, D.M.; Cohen, B.M.; Vasan, N.; Ma, Y.; Krismer, K.; Robles, J.T.; van de Kooij, B.; van Vlimmeren, A.E.; Andrée-Busch, N.; Käufer, N.F.; Dorovkov, M.V.; Ryazanov, A.G.; Takagi, Y.; Kastenhuber, E.R.; Goncalves, M.D.; Hopkins, B.D.; Elemento, O.; Taatjes, D.J.; Maucuer, A.; Yamashita, A.; Degterev, A.; Uduman, M.; Lu, J.; Landry, S.D.; Zhang, B.; Cossentino, I.; Linding, R.; Blenis, J.; Hornbeck, P.V.; Turk, B.E.; Yaffe, M.B.; Cantley, L.C. An Atlas of Substrate Specificities for the Human Serine/Threonine Kinome. Nature 2023, 613, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Ubersax, J.A.; Ferrell Jr, J.E. Mechanisms of Specificity in Protein Phosphorylation. Nat Rev Mol Cell Biol 2007, 8, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Alqahtani, S.; Hu, X. Aromatic Rings as Molecular Determinants for the Molecular Recognition of Protein Kinase Inhibitors. Molecules 2021, 26, 1776. [Google Scholar] [CrossRef]
- Bubon, T.L.; Perepelytsya, S.M. Low-Frequency Vibrations of Water Molecules in DNA Minor Groove. The European Physical Journal E 2021, 44, 84. [Google Scholar] [CrossRef] [PubMed]
- Bubon, T.; Zdorevskyi, O.; Perepelytsya, S. Molecular Dynamics Study of Collective Water Vibrations in a DNA Hydration Shell. European Biophysics Journal 2023, 52, 69–79. [Google Scholar] [CrossRef]
- Dragan, A.I.; Read, C.M.; Crane-Robinson, C. Enthalpy–Entropy Compensation: The Role of Solvation. European Biophysics Journal 2017, 46, 301–308. [Google Scholar] [CrossRef]
- Duboué-Dijon, E.; Fogarty, A.C.; Hynes, J.T.; Laage, D. Dynamical Disorder in the DNA Hydration Shell. J Am Chem Soc 2016, 138, 7610–7620. [Google Scholar] [CrossRef]
- Tereshko, V.; Minasov, G.; Egli, M. A “Hydrat-Ion” Spine in a B-DNA Minor Groove. J Am Chem Soc 1999, 121, 3590–3595. [Google Scholar] [CrossRef]
- Riera Aroche, R.; Ortiz García, Y.M.; Sánchez Moreno, E.C.; Enriquez Cervantes, J.S.; Machado Sulbaran, A.C.; Riera Leal, A. DNA Gene’s Basic Structure as a Nonperturbative Circuit Quantum Electrodynamics: Is RNA Polymerase II the Quantum Bus of Transcription? Curr Issues Mol Biol 2024, 46, 12152–12173. [Google Scholar] [CrossRef]
- Rodríguez-Molina, J.B.; West, S.; Passmore, L.A. Knowing When to Stop: Transcription Termination on Protein-Coding Genes by Eukaryotic RNAPII. Mol Cell 2023, 83, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Vervoort, S.J.; Devlin, J.R.; Kwiatkowski, N.; Teng, M.; Gray, N.S.; Johnstone, R.W. Targeting Transcription Cycles in Cancer. Nat Rev Cancer 2022, 22, 5–24. [Google Scholar] [CrossRef] [PubMed]
- Patikoglou, G.A.; Kim, J.L.; Sun, L.; Yang, S.-H.; Kodadek, T.; Burley, S.K. TATA Element Recognition by the TATA Box-Binding Protein Has Been Conserved throughout Evolution. Genes Dev 1999, 13, 3217–3230. [Google Scholar] [CrossRef] [PubMed]
- Savinkova, L.K.; Sharypova, E.B.; Kolchanov, N.A. On the Role of TATA Boxes and TATA-Binding Protein in Arabidopsis Thaliana. Plants 2023, 12, 1000. [Google Scholar] [CrossRef]
- Vanaja, A.; Yella, V.R. Delineation of the DNA Structural Features of Eukaryotic Core Promoter Classes. ACS Omega 2022, 7, 5657–5669. [Google Scholar] [CrossRef]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; Li, C.H.; Guo, Y.E.; Day, D.S.; Schuijers, J.; Vasile, E.; Malik, S.; Hnisz, D.; Lee, T.I.; Cisse, I.I.; Roeder, R.G.; Sharp, P.A.; Chakraborty, A.K.; Young, R.A. Coactivator Condensation at Super-Enhancers Links Phase Separation and Gene Control. Science (1979) 2018, 361. [Google Scholar] [CrossRef]
- Yasuhara, T.; Xing, Y.-H.; Bauer, N.C.; Lee, L.; Dong, R.; Yadav, T.; Soberman, R.J.; Rivera, M.N.; Zou, L. Condensates Induced by Transcription Inhibition Localize Active Chromatin to Nucleoli. Mol Cell 2022, 82, 2738–2753.e6. [Google Scholar] [CrossRef]
- Cho, W.-K.; Spille, J.-H.; Hecht, M.; Lee, C.; Li, C.; Grube, V.; Cisse, I.I. Mediator and RNA Polymerase II Clusters Associate in Transcription-Dependent Condensates. Science (1979) 2018, 361, 412–415. [Google Scholar] [CrossRef]
- Pei, G.; Lyons, H.; Li, P.; Sabari, B.R. Transcription Regulation by Biomolecular Condensates. Nat Rev Mol Cell Biol 2024. [Google Scholar] [CrossRef]
- Gui, T.; Fleming, C.; Manzato, C.; Bourgeois, B.; Sirati, N.; Heuer, J.; Papadionysiou, I.; Montfort, D.I. van; Gijzen, M. van; Smits, L.M.M.; Burgering, B.M.T.; Madl, T.; Schuijers, J. Targeted Perturbation of Signaling-Driven Condensates. Mol Cell 2023, 83, 4141–4157.e11. [Google Scholar] [CrossRef]
- Cramer, P. Organization and Regulation of Gene Transcription. Nature 2019, 573, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Henninger, J.E.; Oksuz, O.; Shrinivas, K.; Sagi, I.; LeRoy, G.; Zheng, M.M.; Andrews, J.O.; Zamudio, A.V.; Lazaris, C.; Hannett, N.M.; Lee, T.I.; Sharp, P.A.; Cissé, I.I.; Chakraborty, A.K.; Young, R.A. RNA-Mediated Feedback Control of Transcriptional Condensates. Cell 2021, 184, 207–225.e24. [Google Scholar] [CrossRef] [PubMed]
- Lyons, H.; Veettil, R.T.; Pradhan, P.; Fornero, C.; De La Cruz, N.; Ito, K.; Eppert, M.; Roeder, R.G.; Sabari, B.R. Functional Partitioning of Transcriptional Regulators by Patterned Charge Blocks. Cell 2023, 186, 327–345.e28. [Google Scholar] [CrossRef] [PubMed]
- Ehara, H.; Yokoyama, T.; Shigematsu, H.; Yokoyama, S.; Shirouzu, M.; Sekine, S. Structure of the Complete Elongation Complex of RNA Polymerase II with Basal Factors. Science (1979) 2017, 357, 921–924. [Google Scholar] [CrossRef]
- Lassila, J.K.; Zalatan, J.G.; Herschlag, D. Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Annu Rev Biochem 2011, 80, 669–702. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Wang, Y.; Lin, C.; Zhang, D.; Chen, J.; Ouyang, L.; Wu, F.; Zhang, J.; Chen, L. Recent Progress on Vascular Endothelial Growth Factor Receptor Inhibitors with Dual Targeting Capabilities for Tumor Therapy. J Hematol Oncol 2022, 15, 89. [Google Scholar] [CrossRef]
- Kamerlin, S.C.L.; Sharma, P.K.; Prasad, R.B.; Warshel, A. Why Nature Really Chose Phosphate. Q Rev Biophys 2013, 46, 1–132. [Google Scholar] [CrossRef]
- Robertson, A.J.; Wilson, A.L.; Burn, M.J.; Cliff, M.J.; Popelier, P.L.A.; Waltho, J.P. The Relationship between Enzyme Conformational Change, Proton Transfer, and Phosphoryl Transfer in β-Phosphoglucomutase. ACS Catal 2021, 11, 12840–12849. [Google Scholar] [CrossRef]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat Rev Mol Cell Biol 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Visser, B.S.; Lipiński, W.P.; Spruijt, E. The Role of Biomolecular Condensates in Protein Aggregation. Nat Rev Chem 2024, 8, 686–700. [Google Scholar] [CrossRef]
- Kaufman, N.E.M.; Dhingra, S.; Jois, S.D.; Vicente, M. da G. H. Molecular Targeting of Epidermal Growth Factor Receptor (EGFR) and Vascular Endothelial Growth Factor Receptor (VEGFR). Molecules 2021, 26, 1076. [Google Scholar] [CrossRef] [PubMed]
- Dopkins, N.; Nixon, D.F. Activation of Human Endogenous Retroviruses and Its Physiological Consequences. Nat Rev Mol Cell Biol 2024, 25, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.; Rizzo, S.; Paz, W.E.; Kresinsky, A.; Thévenin, D.; Müller, J.P. Disrupting PTPRJ Transmembrane-Mediated Oligomerization Counteracts Oncogenic Receptor Tyrosine Kinase FLT3 ITD. Front Oncol 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Burevschi, E.; Alonso, E.R.; Sanz, M.E. Binding Site Switch by Dispersion Interactions: Rotational Signatures of Fenchone–Phenol and Fenchone–Benzene Complexes. Chemistry – A European Journal 2020, 26, 11327–11333. [Google Scholar] [CrossRef]
- Gibbs, C.A.; Weber, D.S.; Warren, J.J. Clustering of Aromatic Amino Acid Residues around Methionine in Proteins. Biomolecules 2021, 12, 6. [Google Scholar] [CrossRef]
- Yamauchi, O.; Odani, A. Structure-Stability Relationship in Ternary Copper(II) Complexes Involving Aromatic Amines and Tyrosine or Related Amino Acids. Intramolecular Aromatic Ring Stacking and Its Regulation through Tyrosine Phosphorylation. J Am Chem Soc 1985, 107, 5938–5945. [Google Scholar] [CrossRef]
- Zhang, J.; Hao, M.; Zhang, D.; Zhang, X.; Guo, S.; Wang, B.; Xiao, J.; Gao, Y.; Li, X. Enhanced Polyacrylamide Degradation via OH Radical-Initiated Single-Electron Transfer. ACS Omega 2023, 8, 46589–46597. [Google Scholar] [CrossRef]
- Blumberger, J. Recent Advances in the Theory and Molecular Simulation of Biological Electron Transfer Reactions. Chem Rev 2015, 115, 11191–11238. [Google Scholar] [CrossRef]
- Matyushov, D.V. Reorganization Energy of Electron Transfer. Physical Chemistry Chemical Physics 2023, 25, 7589–7610. [Google Scholar] [CrossRef]
- Nakane, K.; Nagasawa, H.; Fujimura, C.; Koyanagi, E.; Tomoshige, S.; Ishikawa, M.; Sato, S. Switching of Photocatalytic Tyrosine/Histidine Labeling and Application to Photocatalytic Proximity Labeling. Int J Mol Sci 2022, 23, 11622. [Google Scholar] [CrossRef]
- Qiao, H.; Xia, M.; Cheng, Y.; Zhou, J.; Zheng, L.; Li, W.; Wang, J.; Fang, P. Tyrosine-Targeted Covalent Inhibition of a TRNA Synthetase Aided by Zinc Ion. Commun Biol 2023, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Hazra, M.K. Impact of OH Radical-Initiated H2CO3 Degradation in the Earth’s Atmosphere via Proton-Coupled Electron Transfer Mechanism. J Phys Chem A 2016, 120, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Mahmudov, K.T.; Kopylovich, M.N.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Non-Covalent Interactions in the Synthesis of Coordination Compounds: Recent Advances. Coord Chem Rev 2017, 345, 54–72. [Google Scholar] [CrossRef]
- Gray, T.M.; Matthews, B.W. Intrahelical Hydrogen Bonding of Serine, Threonine and Cysteine Residues within α-Helices and Its Relevance to Membrane-Bound Proteins. J Mol Biol 1984, 175, 75–81. [Google Scholar] [CrossRef]
- Hubbard, E.E.; Julian, R.R. Unravelling the Favorability of Radical-Directed Xn-H2O Dissociation at Serine and Threonine. Int J Mass Spectrom 2025, 507, 117363. [Google Scholar] [CrossRef]
- Zhang, L.; Reilly, J.P. Radical-Driven Dissociation of Odd-Electron Peptide Radical Ions Produced in 157 Nm Photodissociation. J Am Soc Mass Spectrom 2009, 20, 1378–1390. [Google Scholar] [CrossRef]
- Uversky, V.N. The Intrinsic Disorder Alphabet. III. Dual Personality of Serine. Intrinsically Disord Proteins 2015, 3, e1027032. [Google Scholar] [CrossRef]
- Basile, W.; Salvatore, M.; Bassot, C.; Elofsson, A. Why Do Eukaryotic Proteins Contain More Intrinsically Disordered Regions? PLoS Comput Biol 2019, 15, e1007186. [Google Scholar] [CrossRef]
- Trivedi, R.; Nagarajaram, H.A. Intrinsically Disordered Proteins: An Overview. Int J Mol Sci 2022, 23, 14050. [Google Scholar] [CrossRef]
- Roy, S.; Lessing, J.; Meisl, G.; Ganim, Z.; Tokmakoff, A.; Knoester, J.; Jansen, T.L.C. Solvent and Conformation Dependence of Amide I Vibrations in Peptides and Proteins Containing Proline. J Chem Phys 2011, 135. [Google Scholar] [CrossRef]
- Kubyshkin, V.; Rubini, M. Proline Analogues. Chem Rev 2024, 124, 8130–8232. [Google Scholar] [CrossRef] [PubMed]
- Byun, B.J.; Kang, Y.K. Conformational Preferences and Prolyl Cis-Trans Isomerization of Phosphorylated Ser/Thr-Pro Motifs. Biopolymers 2010, 93, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Kubyshkin, V.; Mykhailiuk, P.K. Proline Analogues in Drug Design: Current Trends and Future Prospects. J Med Chem 2024, 67, 20022–20055. [Google Scholar] [CrossRef]
- Joshi, A.; Avni, A.; Walimbe, A.; Rai, S.K.; Sarkar, S.; Mukhopadhyay, S. Hydrogen-Bonded Network of Water in Phase-Separated Biomolecular Condensates. J Phys Chem Lett 2024, 15, 7724–7734. [Google Scholar] [CrossRef]
- Codorniu-Hernández, E.; Kusalik, P.G. Mobility Mechanism of Hydroxyl Radicals in Aqueous Solution via Hydrogen Transfer. J Am Chem Soc 2012, 134, 532–538. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, C.; Jiao, N. Recent Advances in Copper-Catalyzed Dehydrogenative Functionalization via a Single Electron Transfer (SET) Process. Chem Soc Rev 2012, 41, 3464. [Google Scholar] [CrossRef]
- Singh, A.K.; Wen, C.; Cheng, S.; Vinh, N.Q. Long-Range DNA-Water Interactions. Biophys J 2021, 120, 4966–4979. [Google Scholar] [CrossRef]
- McDermott, M.L.; Vanselous, H.; Corcelli, S.A.; Petersen, P.B. DNA’s Chiral Spine of Hydration. ACS Cent Sci 2017, 3, 708–714. [Google Scholar] [CrossRef]
- Perets, E.A.; Konstantinovsky, D.; Santiago, T.; Videla, P.E.; Tremblay, M.; Velarde, L.; Batista, V.S.; Hammes-Schiffer, S.; Yan, E.C.Y. Beyond the “Spine of Hydration”: Chiral SFG Spectroscopy Detects DNA First Hydration Shell and Base Pair Structures. J Chem Phys 2024, 161. [Google Scholar] [CrossRef]
- Mukherjee, S.; Mondal, S.; Acharya, S.; Bagchi, B. DNA Solvation Dynamics. J Phys Chem B 2018, 122, 11743–11761. [Google Scholar] [CrossRef]
- Gavrilov, Y.; Leuchter, J.D.; Levy, Y. On the Coupling between the Dynamics of Protein and Water. Physical Chemistry Chemical Physics 2017, 19, 8243–8257. [Google Scholar] [CrossRef] [PubMed]
- Conti Nibali, V.; D’Angelo, G.; Paciaroni, A.; Tobias, D.J.; Tarek, M. On the Coupling between the Collective Dynamics of Proteins and Their Hydration Water. J Phys Chem Lett 2014, 5, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.; Plazanet, M.; Gabel, F.; Kessler, B.; Oesterhelt, D.; Tobias, D.J.; Zaccai, G.; Weik, M. Coupling of Protein and Hydration-Water Dynamics in Biological Membranes. Proceedings of the National Academy of Sciences 2007, 104, 18049–18054. [Google Scholar] [CrossRef] [PubMed]
- Riera Aroche, R.; Ortiz García, Y.M.; Martínez Arellano, M.A.; Riera Leal, A. DNA as a Perfect Quantum Computer Based on the Quantum Physics Principles. Sci Rep 2024, 14, 11636. [Google Scholar] [CrossRef]
- Adhav, V.A.; Shelke, S.S.; Balanarayan, P.; Saikrishnan, K. Sulfur-Mediated Chalcogen versus Hydrogen Bonds in Proteins: A See-Saw Effect in the Conformational Space. QRB Discov 2023, 4, e5. [Google Scholar] [CrossRef]
- Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M.S.; Chakrabarti, P. CH–π Hydrogen Bonds in Biological Macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. [Google Scholar] [CrossRef]
- Mohan, N.; Vijayalakshmi, K.P.; Koga, N.; Suresh, C.H. Comparison of Aromatic NH···π, OH···π, and CH···π Interactions of Alanine Using MP2, CCSD, and DFT Methods. J Comput Chem 2010, 31, 2874–2882. [Google Scholar] [CrossRef]
- Singh, A.; Raj, P.; Dubowski, J.J.; Singh, N. ATP Induced Modulation in π–π Stacking Interactions in Pyrene Based Zinc Complexes: Chemosensor Study and Quantitative Investigation of Apyrase Activity. Cryst Growth Des 2018, 18, 4320–4333. [Google Scholar] [CrossRef]
- Brandl, M.; Weiss, M.S.; Jabs, A.; Sühnel, J.; Hilgenfeld, R. C-H…π-Interactions in Proteins. J Mol Biol 2001, 307, 357–377. [Google Scholar] [CrossRef]
- Krone, M.W.; Travis, C.R.; Lee, G.Y.; Eckvahl, H.J.; Houk, K.N.; Waters, M.L. More Than π–π–π Stacking: Contribution of Amide−π and CH−π Interactions to Crotonyllysine Binding by the AF9 YEATS Domain. J Am Chem Soc 2020, 142, 17048–17056. [Google Scholar] [CrossRef]
- Singh, S.; Tian, W.; Severance, Z.C.; Chaudhary, S.K.; Anokhina, V.; Mondal, B.; Pergu, R.; Singh, P.; Dhawa, U.; Singha, S.; Choudhary, A. Proximity-Inducing Modalities: The Past, Present, and Future. Chem Soc Rev 2023, 52, 5485–5515. [Google Scholar] [CrossRef] [PubMed]
- Bremer, A.; Farag, M.; Borcherds, W.M.; Peran, I.; Martin, E.W.; Pappu, R.V.; Mittag, T. Deciphering How Naturally Occurring Sequence Features Impact the Phase Behaviours of Disordered Prion-like Domains. Nat Chem 2022, 14, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.A.; Heidenreich, M.; Levy, E.D. Molecular and Environmental Determinants of Biomolecular Condensate Formation. Nat Chem Biol 2022, 18, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.V.; Cohen, S.R.; Dar, F.; Farag, M.; Kar, M. Phase Transitions of Associative Biomacromolecules. Chem Rev 2023, 123, 8945–8987. [Google Scholar] [CrossRef]
- Martin, E.W.; Holehouse, A.S.; Peran, I.; Farag, M.; Incicco, J.J.; Bremer, A.; Grace, C.R.; Soranno, A.; Pappu, R.V.; Mittag, T. Valence and Patterning of Aromatic Residues Determine the Phase Behavior of Prion-like Domains. Science (1979) 2020, 367, 694–699. [Google Scholar] [CrossRef]
- Vernon, R.M.; Forman-Kay, J.D. First-Generation Predictors of Biological Protein Phase Separation. Curr Opin Struct Biol 2019, 58, 88–96. [Google Scholar] [CrossRef]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; Baldwin, A.J. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol Cell 2015, 57, 936–947. [Google Scholar] [CrossRef]
- Gutowsk, M.; Kowalczyk, S. A Study of Free Radical Chemistry: Their Role and Pathophysiological Significance. Acta Biochim Pol 2016, 60, 1–16. [Google Scholar] [CrossRef]
- Meng, X.; Qi, J. Manipulating Tyrosine Phosphorylation by Heterobifunctional Small Molecules. ACS Cent Sci 2023, 9, 1512–1514. [Google Scholar] [CrossRef]
- Kim, Y.; Joachimiak, G.; Bigelow, L.; Babnigg, G.; Joachimiak, A. How Aromatic Compounds Block DNA Binding of HcaR Catabolite Regulator. Journal of Biological Chemistry 2016, 291, 13243–13256. [Google Scholar] [CrossRef]
- Czernek, J.; Brus, J.; Czerneková, V.; Kobera, L. Quantifying the Intrinsic Strength of C–H…O Intermolecular Interactions. Molecules 2023, 28, 4478. [Google Scholar] [CrossRef] [PubMed]
- Biswas, R.; Tse, Y.-L. S.; Tokmakoff, A.; Voth, G.A. Role of Presolvation and Anharmonicity in Aqueous Phase Hydrated Proton Solvation and Transport. J Phys Chem B 2016, 120, 1793–1804. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.; Lazaridis, T. Classical Models of Hydroxide for Proton Hopping Simulations. J Phys Chem B 2024, 128, 12161–12170. [Google Scholar] [CrossRef] [PubMed]
- Kier, L.B. Proton Hopping in Living Systems. Curr Comput Aided Drug Des 2021, 17, 333–336. [Google Scholar] [CrossRef]
- Farrell, K.M.; Zanni, M.T. Observing Aqueous Proton Transfer Dynamics. ACS Cent Sci 2019, 5, 1114–1116. [Google Scholar] [CrossRef]
- Popov, I.; Zhu, Z.; Young-Gonzales, A.R.; Sacci, R.L.; Mamontov, E.; Gainaru, C.; Paddison, S.J.; Sokolov, A.P. Search for a Grotthuss Mechanism through the Observation of Proton Transfer. Commun Chem 2023, 6, 77. [Google Scholar] [CrossRef]
- Bogot, A.; Poline, M.; Ji, M.; Dochain, A.; Simonsson, A.; Rosén, S.; Zettergren, H.; Schmidt, H.T.; Thomas, R.D.; Strasser, D. The Mutual Neutralization of Hydronium and Hydroxide. Science (1979) 2024, 383, 285–289. [Google Scholar] [CrossRef]
- Huang, L.; Matta, C.F.; Wallace, S.; Massa, L.; Bernal, I. A Unique Trapping by Crystal Forces of a Hydronium Cation Displaying a Transition State Structure. Comptes Rendus. Chimie 2015, 18, 511–515. [Google Scholar] [CrossRef]
- Yu, Q.; Bowman, J.M. Tracking Hydronium/Water Stretches in Magic H3O+(H2O)20 Clusters through High-Level Quantum VSCF/VCI Calculations. J Phys Chem A 2020, 124, 1167–1175. [Google Scholar] [CrossRef]
- Onaka, S. Periodic Arrangements of Tetrahedra Having Appearances Similar to That of the Boerdijk–Coxeter Helix. Sci Rep 2024, 14, 18260. [Google Scholar] [CrossRef]
- Limbach, H.-H. NMR Studies of Elementary Steps of Multiple Proton and Deuteron Transfers in Liquids, Crystals, and Organic Glasses. In Intermolecular Forces; Springer Berlin Heidelberg: Berlin, Heidelberg, 1991. [Google Scholar] [CrossRef]
- Hassanali, A.; Giberti, F.; Cuny, J.; Kühne, T.D.; Parrinello, M. Proton Transfer through the Water Gossamer. Proceedings of the National Academy of Sciences 2013, 110, 13723–13728. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Fang, H.; Ni, K.; Feng, Y. Water Clusters and Density Fluctuations in Liquid Water Based on Extended Hierarchical Clustering Methods. Sci Rep 2022, 12, 8036. [Google Scholar] [CrossRef] [PubMed]
- Frutos-Puerto, S.; Colín, M.J.; Corchado, J.C.; Sánchez, M.L.; Martín, M.E.; Aguilar, M.A. Photophysical and Photochemical Properties of 3-Hydroxyflavone in Ethanol Solution: Implicit vs Explicit Solvent Models. J Mol Liq 2023, 381, 121783. [Google Scholar] [CrossRef]
- Tyburski, R.; Liu, T.; Glover, S.D.; Hammarström, L. Proton-Coupled Electron Transfer Guidelines, Fair and Square. J Am Chem Soc 2021, 143, 560–576. [Google Scholar] [CrossRef] [PubMed]
- Dubey, V.; Kumar, N.; Daschakraborty, S. Importance of Solvents’ Translational–Rotational Coupling for Translational Jump of a Small Hydrophobic Solute in Supercooled Water. J Phys Chem B 2018, 122, 7569–7583. [Google Scholar] [CrossRef]
- Yu, C.-C.; Chiang, K.-Y.; Okuno, M.; Seki, T.; Ohto, T.; Yu, X.; Korepanov, V.; Hamaguchi, H.; Bonn, M.; Hunger, J.; Nagata, Y. Vibrational Couplings and Energy Transfer Pathways of Water’s Bending Mode. Nat Commun 2020, 11, 5977. [Google Scholar] [CrossRef]
Figure 1.
The figure illustrates the fundamental structure of DNA and ATP. A) The primary structure of DNA is examined as a combination of quantum circuits. Each base pair serves as a qubit, functioning like an RF-SQUID. The sugar-phosphate backbone resembles a transmission line with distributed parameters 10. B) Chemical description of ATP. The amino acid tyrosine contains a phenol group in its side chain that can undergo phosphorylation as a post-translational modification. The transphosphorylation of OH-containing substrates is a substitution reaction centered around phosphorus and releases a hydrogen proton. The beta and gamma phosphate groups are transferred from ATP to tyrosine.
Figure 1.
The figure illustrates the fundamental structure of DNA and ATP. A) The primary structure of DNA is examined as a combination of quantum circuits. Each base pair serves as a qubit, functioning like an RF-SQUID. The sugar-phosphate backbone resembles a transmission line with distributed parameters 10. B) Chemical description of ATP. The amino acid tyrosine contains a phenol group in its side chain that can undergo phosphorylation as a post-translational modification. The transphosphorylation of OH-containing substrates is a substitution reaction centered around phosphorus and releases a hydrogen proton. The beta and gamma phosphate groups are transferred from ATP to tyrosine.
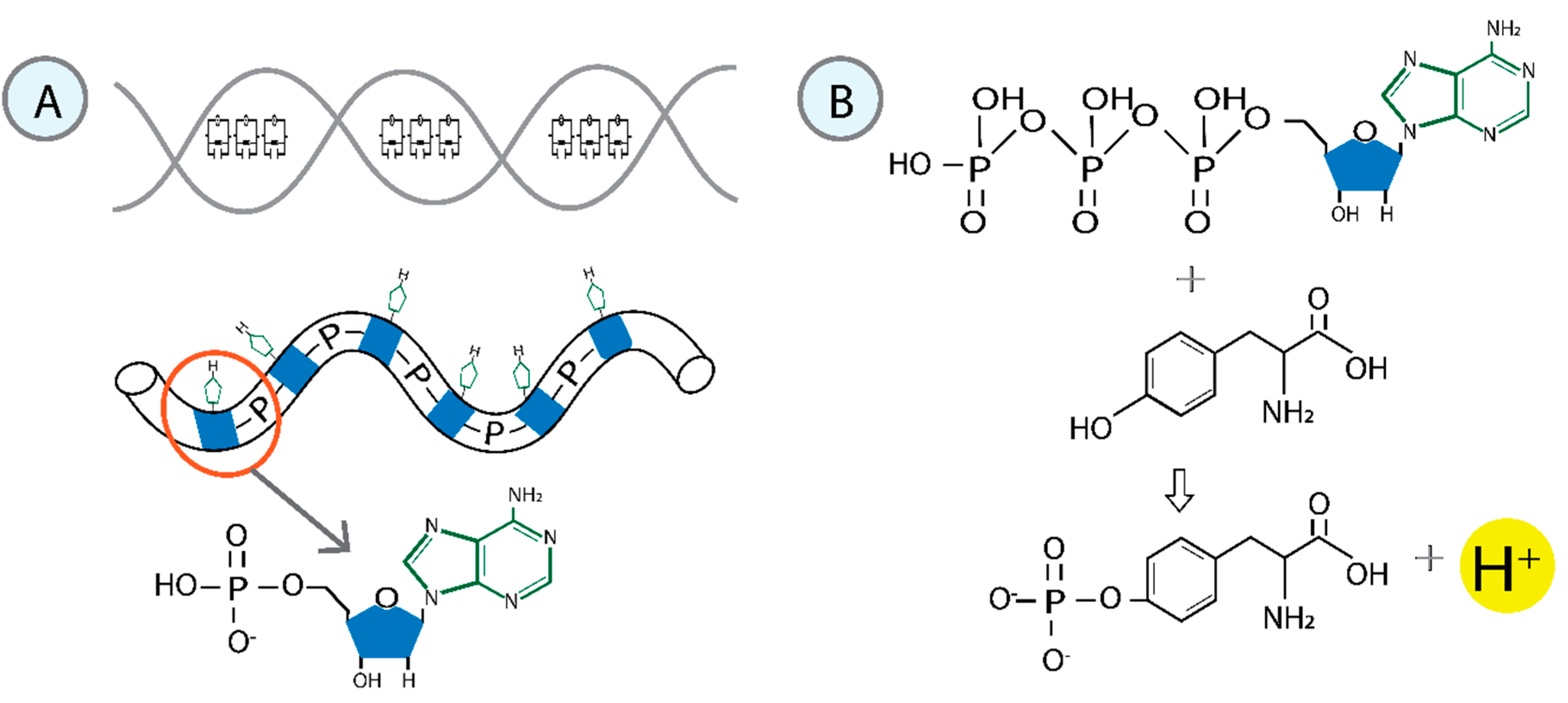
Figure 2.
Formation of the RNA polymerase II-DNA-water molecular condensate. The initial step involves inducing a reaction that attaches the phosphorylated tyrosine to the sugar of a nitrogenous base. After removing the hydrogen atom from deoxyribose, the unpaired single electron becomes localized at the corresponding carbon (2'C). The geometry adopted by tyrosine promotes non-covalent interactions between the protein and the nitrogenous base. A sequential series of phosphorylations is initiated, modifying the structure of the water associated with the molecular condensate. These non-covalent interactions govern the photophysical properties of the system.
Figure 2.
Formation of the RNA polymerase II-DNA-water molecular condensate. The initial step involves inducing a reaction that attaches the phosphorylated tyrosine to the sugar of a nitrogenous base. After removing the hydrogen atom from deoxyribose, the unpaired single electron becomes localized at the corresponding carbon (2'C). The geometry adopted by tyrosine promotes non-covalent interactions between the protein and the nitrogenous base. A sequential series of phosphorylations is initiated, modifying the structure of the water associated with the molecular condensate. These non-covalent interactions govern the photophysical properties of the system.

Figure 3.
The process of forming the tetrahedral structure of water. 1) The released proton (H+) functions as a free particle. 2) The kinetic energy generates the H3O+ cation from one water molecule. 3) H3O+ is highly reactive and is quickly solvated by neighboring water molecules. 4) The H3O+ ion, linked by a bifurcated H-bond to two adjacent water molecules, represents the minimum energy structure.
Figure 3.
The process of forming the tetrahedral structure of water. 1) The released proton (H+) functions as a free particle. 2) The kinetic energy generates the H3O+ cation from one water molecule. 3) H3O+ is highly reactive and is quickly solvated by neighboring water molecules. 4) The H3O+ ion, linked by a bifurcated H-bond to two adjacent water molecules, represents the minimum energy structure.
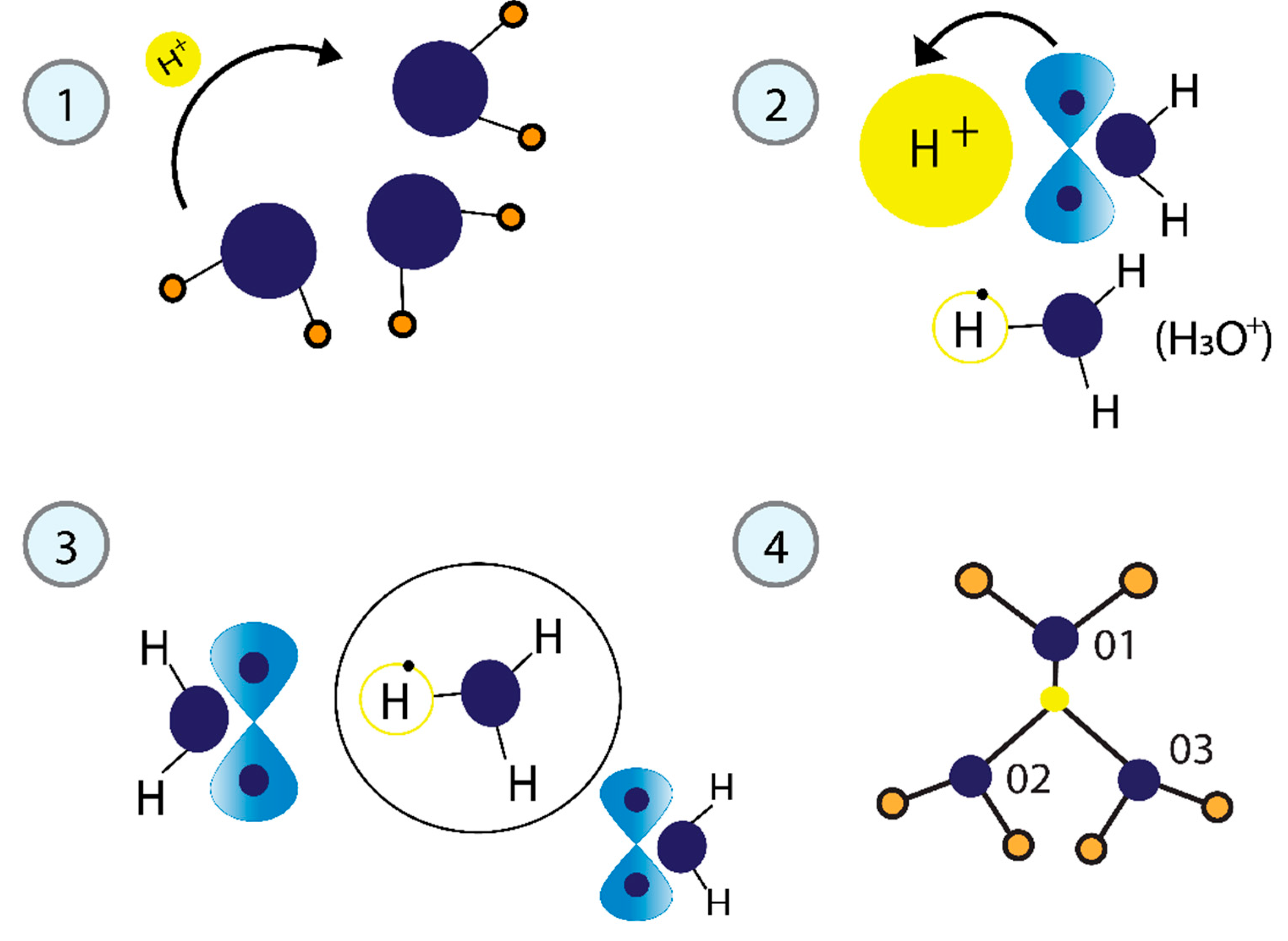
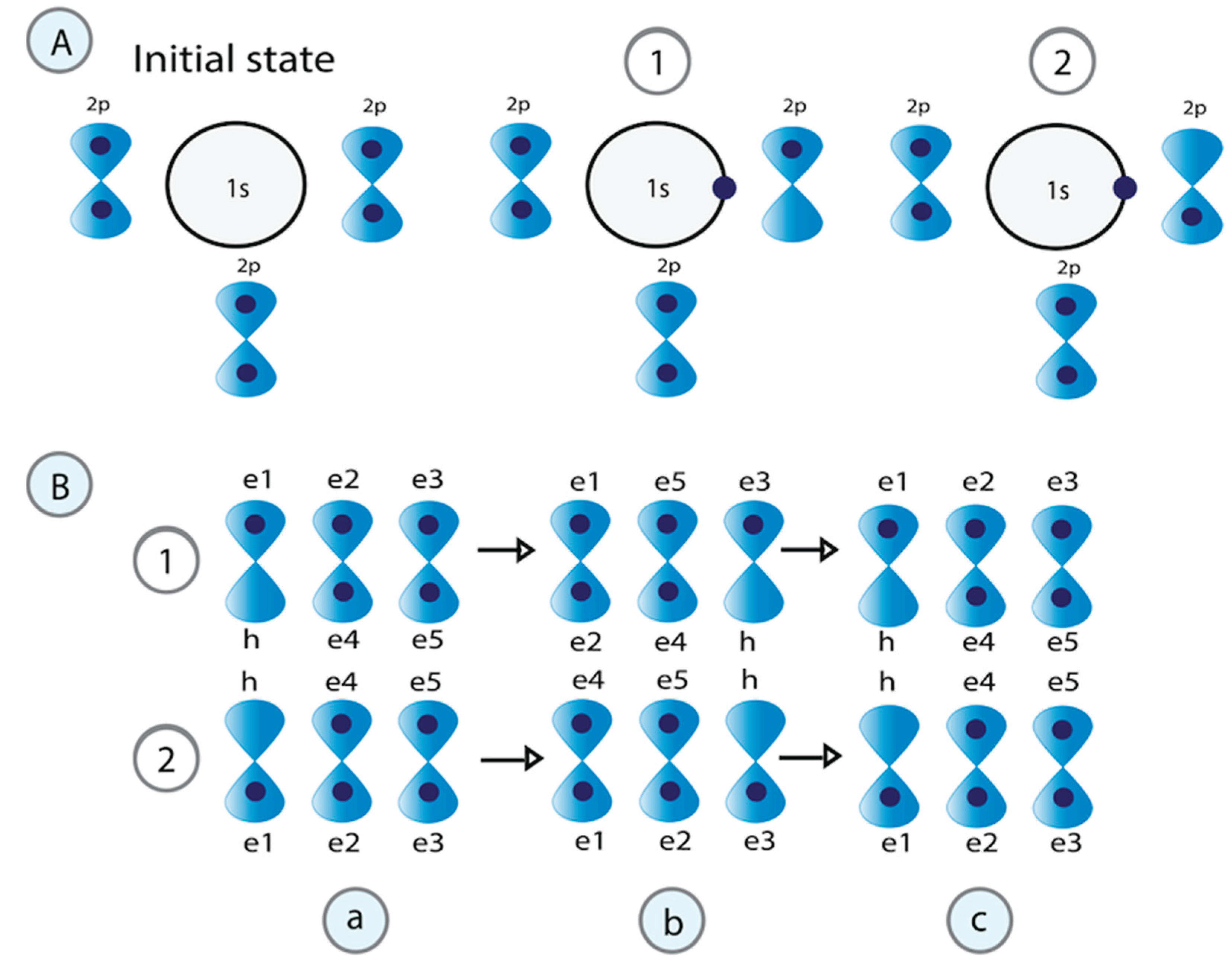
Figure 4.
Structure of water associated with DNA in the molecular condensate. A) During the formation of the H3O+ cation, one electron from a lone pair in the oxygen orbital moves to the empty H+ orbital, creating a vacancy. The released energy () is used to form correlated pairs of electrons. The electrons that correlate to form pairs should have opposite momentum, spin, and different energies. Their energy difference equals that . Two processes are required to create electron pairs. Processes a to b involve a half oscillation. In processes b to c, the fermion pairs exchange , a second oscillation defines the oscillatory resonant quantum states.
Figure 4.
Structure of water associated with DNA in the molecular condensate. A) During the formation of the H3O+ cation, one electron from a lone pair in the oxygen orbital moves to the empty H+ orbital, creating a vacancy. The released energy () is used to form correlated pairs of electrons. The electrons that correlate to form pairs should have opposite momentum, spin, and different energies. Their energy difference equals that . Two processes are required to create electron pairs. Processes a to b involve a half oscillation. In processes b to c, the fermion pairs exchange , a second oscillation defines the oscillatory resonant quantum states.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



