Submitted:
26 June 2025
Posted:
30 June 2025
You are already at the latest version
Abstract
Cardiac physiology and pathology have been extensively explored at the transcriptional level, but are less understood at the translational level. Translation of mRNA to protein is the final step in the central dogma for protein synthesis. Translation machinery includes a family of essential “housekeeping” factors and enzymes required for mRNA translation. These translation factors ensure the accurate processing of mRNA to protein according to the genetic code and maintain the optimal quality and quantity of cellular proteins for normal cardiac function. Translation factors also control the efficiency, speed, and fidelity of protein production and participate in cardiac pathological remodeling under stress conditions. This review summarizes discoveries of the pathophysiological function and molecular mechanism of translational control in cardiac health and disease. Translational control has extensive crosstalk to other processes such as transcriptional regulation, mi-tochondrial metabolism, and sarcomere homeostasis. We discuss the translational regu-lation directed by specific regulatory factors in cardiac physiology and the etiology of heart disease when they undergo genetic mutation, expression dysregulation, or func-tional alteration. Because transcript-specific translational regulation of pathological and protective proteins occurs in heart disease, target-selective translation inhibitors and enhancers can be developed. These inhibitors and enhancers provide valuable insights into novel therapeutic targets and RNA-based drug development for heart disease treatment.
Keywords:
cardiac regeneration
; cardiomyocyte
; congenital heart disease
; fibroblast
; fibrosis
; heart failure
; hypertrophy
; mRNA translation
; RNA-binding protein
; RNA therapeutics
1. Introduction
The heart is the organ responsible for pumping blood to supply oxygen and nutrients to other organs throughout the body. Cardiomyocytes (CMs) and cardiac fibroblasts (CFs) are the two major cell types in the heart. CMs handle the contractile function of the heart, while CFs stabilize the cardiac structure and repair potential cardiac damage. Cardiovascular disease is the leading cause of morbidity and mortality worldwide. Heart failure (HF), a major manifestation of cardiovascular disease, often results from cardiac ischemic events such as myocardial infarction (MI). About 64 million people worldwide are affected by HF, and half of these patients will die within five years of diagnosis (1). An MI triggers proliferation and activation of cardiac fibroblasts; if uncontrolled, this can lead to excessive cardiac fibrosis, reducing heart function and leading to HF. HF is categorized based on ejection fraction (EF), which measures the percentage of blood pumped out of the heart with each beat: heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF) (2). HFrEF occurs when the left ventricle cannot contract properly and ejects less oxygen-rich blood after MI. Conversely, HFpEF maintains an EF above 50% but has impaired diastolic function and a reduced ability to fill the left ventricle with oxygenated blood, often developing after long-term hypertension, obesity, or diabetes. Chronic HF remains the leading cause of hospitalization among patients over 65 and poses significant clinical challenges and economic burdens both in the U.S. and globally.
The central dogma of molecular biology states that DNA is transcribed into mRNA (transcription), and mRNA is then translated into proteins (translation). Although transcriptional regulation has been studied extensively, fewer investigations have focused on understanding cardiac health and disease through translational control. Translation is crucial in producing functional proteins following DNA transcription to mRNA in all biological processes (Figure 1), including cardiac cell proliferation, differentiation, hypertrophy, and fibrosis. While increased protein synthesis of pro-hypertrophic and pro-fibrotic genes has been observed during cardiac remodeling, transcriptional regulation alone does not explain all the increases. Transcription factors and microRNAs (miRNAs) are sequence-specific regulators of transcription and post-transcriptional processes (mRNA stability and translatability), respectively. The roles and mechanisms of transcription factors and miRNAs in cardiac biology have been extensively studied over the past twenty years. However, little is known about the molecular mechanisms of miRNA-independent translational control and the therapeutic potential of targeting the general translation process or transcript-specific translational regulation in heart disease treatment. Understanding the abnormal translation control mechanisms that promote pro-hypertrophic and pro-fibrotic mRNA translation in cardiac cells is essential.
Figure 1.
Overview of mammalian mRNA translation.
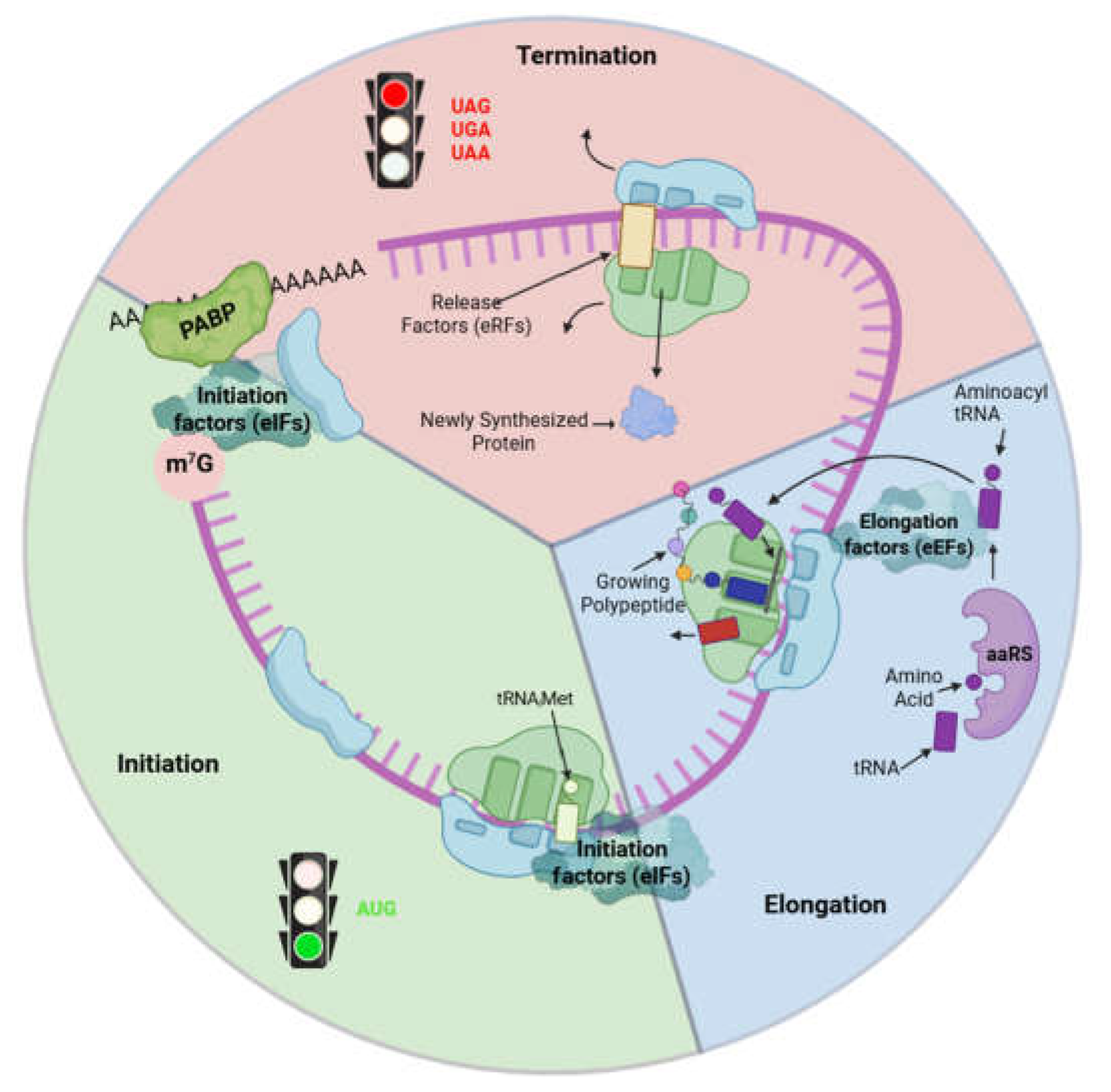
In any eukaryotic cell, two sets of translation machinery exist: one in the cytoplasm and one in the mitochondria. Both translation machines have four major components: aminoacyl-tRNA synthetases (ARSs), translation factors, ribosomes, and translation-regulatory RNA-binding proteins (RBPs) (Figure 1). ARSs are a family of evolutionarily conserved housekeeping enzymes ubiquitously present in the three major domains of life. ARSs catalyze the ligation of amino acids to the 3'-terminus of cognate tRNAs bearing the correct anticodon triplet to ensure accurate “reading” of mRNA to protein according to the genetic code (3, 4). Mammals have 20 cytoplasmic ARSs and 17 nuclear-encoded mitochondrial ARSs (3 ARSs are common in both cellular compartments). ARSs generally contain catalytic aminoacylation and tRNA anticodon recognition functions in separate domains. Several ARSs, including LARS1, IARS1, VARS1, AARS1, TARS1, FARS1, and EPRS1, contain a separate editing domain for hydrolyzing mis-aminoacylated products to maintain translation fidelity (5, 6). Translation factors such as initiation, elongation, and termination factors are required for uploading, translocating, and disassembling ribosome subunits, respectively. Cytoplasmic ribosomes comprise a 40S small subunit and a 60S large subunit, while mitochondrial ribosomes comprise a 28S small subunit and a 39S large subunit. All ribosome subunits consist of numerous ribosome proteins associated with ribosomal RNAs and are central for protein biosynthesis. RNA-binding proteins and miRNAs bind to sequence- or structure-specific cis-acting elements and regulate the translation efficiency of target mRNAs. In any disease state, phenotypic changes are driven by the alteration of pathogenic proteins produced by the translation machinery. However, due to translational control, the protein expression often does not correlate with mRNA abundance (7, 8).
Prior reviews have extensively discussed RNA-binding proteins and mRNA metabolism at the posttranscriptional level (9, 10). This review will focus on understanding translational control in cardiac biology and heart disease. We first introduce various techniques that can be used to study translational control: biochemical methods, deep sequencing, and imaging-based techniques. We then discuss translational control in cardiac development and congenital heart disease. We follow this with an overview of translational control in adult cardiac disease. Finally, we discuss therapies that target translational control mechanisms and what we believe are future areas of interest for investigations and therapies based on translational control in the heart.
2. Techniques and Methods to Investigate mRNA Translation in the Heart
2.1. Biochemical methods for studying translational control
2.1.1. RNA-binding protein immunoprecipitation (RIP) to identify bound target RNAs
This method uses immunoprecipitation via specific antibodies to apply the pulldown of a specific target protein and its respective negative control (11, 12). Briefly, healthy growing cells or tissues (such as cardiac cells or the heart) undergo lysis with appropriate RNase and protease inhibitors, and cell or tissue lysates are prepared for immunoprecipitation. An immunoprecipitation-graded antibody for a target protein and a specific IgG isotype control are used for the RIP experiment. Before immunoprecipitation, blocking and pre-clearing steps can be performed to minimize nonspecifically interacting proteins pulled down. mRNAs and interacting proteins can be extracted separately from the precipitated protein-RNA complexes. The isolated RNAs are used for quantitative (q)PCR or next-generation deep sequencing (RIP-seq) to check for RNA association or determine the global RNA targets. The retrieved proteins are subjected to immunoblot or mass spectrometry to confirm protein interaction or unbiasedly identify the interactome of the target protein of interest.
2.1.2. Crosslinking and immunoprecipitation (CLIP) to map RBP-binding sites on RNAs
This method examines the direct physical association between RNAs and their interacting proteins (13, 14). CLIP applies UV or formaldehyde crosslinking of live cells or tissues, which forms covalent bonds between RNAs and proteins in proximity (15, 16). Cell or tissue lysates are then prepared and subjected to partial fragmentation by a selected ribonuclease (e.g., RNase A or T1), followed by immunoprecipitation using a validated antibody for the target protein of interest and an IgG isotype control antibody. RNA fragments from the precipitated protein-RNA complexes are ligated with DNA adaptors on 3' and 5' ends (for individual-nucleotide resolution CLIP (iCLIP), DNA adaptors can be ligated on 5' ends after reverse transcription) and purified by SDS-PAGE with size selection. The isolated products are subjected to proteinase K digestion and reverse transcription to cDNA and amplified by appropriate cycles of PCR reaction. Following the library construction, next-generation deep sequencing (CLIP-seq) is carried out to map transcriptome-wide protein binding sites on RNAs. If an immunoprecipitation-competent antibody for the target protein is unavailable, recombinant tagged RNA-binding proteins can be overexpressed, or transgenic tagging of endogenous RNA-binding proteins can be used for CLIP in a specific cell type.
2.1.3. In vitro pulldown of interacting proteins of biotinylated RNA
This technique uses a cell-free system to study RBP and RNA interactions. Briefly, RNA can be in vitro transcribed with internal biotin (biotinylated CTP) or 5' biotin modification and incubated with cardiac cell or tissue lysates to capture interactions with specific RNA-binding proteins. The respective control oligo (scrambled or antisense oligo) and only beads are taken as negative controls, and an immunoblotting experiment is undertaken to decipher any possible interactions (17). Alternatively, mass spectrometry analysis can be conducted to identify novel RNA-interacting proteins as an unbiased approach, followed by immunoblot confirmation.
2.1.4. Proximity ligation assay associated with immunoblot or mass spectrometry
This technique applies selective biotinylating of proteins adjacent to a target protein of interest and can be used to study protein-protein interactions (18). In brief, a biotin ligase is fused to a target protein of interest and expressed in living cells, which biotinylates proteins that potentially interact with the target protein in the presence of external biotin added to culture media. The biotinylated proteins can be isolated using streptavidin beads and coupled with mass spectrometry or immunoblot analysis to examine protein-protein interactions and draw the interactome atlas of the target protein of interest. The mass spectrometry-based, label-free quantitative proteomics data (e.g., spectral count or intensity) can be scored using the SAINT (Significance Analysis of INTeractome) software package to identify high-confidence protein-protein interactions (SAINT score > 0.95) (19).
2.1.5. Puromycin incorporation assay to assess global translation efficiency
This is the most commonly used technique for studying the protein synthesis status of cultured primary cells in vitro and animal tissues in vivo (20). Cells are subjected to puromycin treatment for 15-20 minutes at a 37ºC incubator, followed by harvesting with a cell lysis buffer containing an appropriate protease inhibitor for protein isolation. The cellular protein lysates undergo immunoblotting analysis. Untreated cells are used as a negative control. In vivo puromycin incorporation allows imaging of nascent proteins and evaluating the regulation of translation spatially and temporally in whole organisms (21). An alkyne analog of puromycin, O-propargyl-puromycin (OP-puro), can form covalent conjugates with nascent polypeptide chains and label and visualize nascent proteins in the target organ of interest. This method broadly applies to imaging protein synthesis under physiological and pathological conditions in vitro and in vivo.
2.2. Deep sequencing-based translatome profiling in cells and animals
2.2.1. Polysome profiling-sequencing (polysome-seq)
Total cell or tissue lysates are subject to sucrose gradient solution and ultracentrifugation to separate different translation fractions, including free mRNP (mRNA-ribonucleoprotein complex), 40S ribosome small subunit, 60S ribosome large subunit, 80S monosome, and polysomes (disome, trisome, and multiple ribosomes). Actively translated mRNAs bound by polysomes can be determined by next-generation deep sequencing to evaluate the translation efficiency of individual mRNAs as normalized by total RNA-seq signal (22). Also, multiple pools can be collected, including non-polysome, monosome, and polysome, and their associated RNAs can be sequenced to calculate the distribution of specific mRNAs among different pools or individual fractions as translation efficiency, depending on the cost efficiency and demand of the resolution.
2.2.2. Translating ribosome affinity purification sequencing (TRAP-seq)
Genetic engineering of endogenous ribosome protein-coding genes in the mouse genome introduces a peptide or protein tag to the gene for subsequent affinity pulldown of ribosomes in vivo. Currently, three conditional inducible knock-in mouse models are available, including RPL22-3xHA, RPL10-EGFP, and mRPL62-FLAG (23-25). In the former two cases, cytoplasmic ribosomes can be affinity-purified using HA or EGFP antibodies from tissue lysates to capture ribosomes from a specific cell type by immunoprecipitation of the large ribosome subunit proteins RPL22 or RPL10 based on the use of the Cre recombinase transgenic mouse model. The latter targets the mitochondrial large ribosome subunit protein mRPL62 for purifying ribosomes in mitochondria. RNA-seq following the translating ribosome affinity purification (TRAP-seq) quantitatively measures the translation efficiency in a specific cell type across various organs in vivo.
2.2.3. Translational landscape in human and mouse heart failure determined by ribosome profiling (Ribo-seq)
Ribosome profiling, also known as Ribo-seq, is a technique that measures the activity of ribosomes in translating mRNA into protein in a cell at a specific time. Ribo-seq uses deep sequencing to analyze ribosome-protected mRNA fragments after digesting the unprotected RNA around the ribosome footprints with ribonucleases, like micrococcal nuclease or RNase I. The sequenced ribosome-protected fragments can be used to determine the positions of the translating ribosomes to define open reading frames (ORF), determine the protein synthesis rate, map translation start sites, examine translational control, and identify ribosome stalling sites. Ribosome profiling provides a snapshot of ribosome density on individual mRNAs and translation activity.
Our understanding of translational control of gene expression in human diseases has always been sparse and less systematic due to the diversity of global and specific regulation machinery. A recent study by van Heesch, S., et al. has attempted to solve this problem and provide a comprehensive landscape of translational regulation related to heart failure (26) by combining RNA-seq, ribosome profiling (Ribo-seq), and mass spectrometry. Results were generated from 80 human hearts, including 65 failing and 15 non-failing hearts. The investigators provided a detailed assessment of translational control in the human heart, including regulation of translation by diverse cis- or trans-elements. By comparing RNA-seq and Ribo-seq results, the investigators revealed translational downregulation of 327 genes and upregulation of 474 genes, including many cardiac disease marker genes, such as the extracellular matrix components genes associated with cardiac fibrosis. They also revealed specific cis-acting elements on mRNA correlated with translational regulation. For example, transcripts with a 5' terminal oligopyrimidine (TOP) motif are significantly translationally upregulated, which agrees with previous studies about mTOR activation during heart disease. Apart from the 5' TOP motif, upstream open reading frames (uORFs), another cis-acting element known to affect translation, have also been detected. They suggest no detectable correlation between the translation efficiency of the main ORF and the efficiency of uORF for most of the proteins. However, several proteins, such as eIF4G2, show an anti-correlation between the translation of the main ORF versus uORF, indicating a selective negative regulation of specific mRNAs via uORF in the diseased human hearts at the endpoint. Alternatively, the correlation analysis using the whole hearts at the endpoint of heart failure does not reveal the causative relationship between the translations of uORF and mORF due to the lack of temporal and spatial resolutions, regarding early versus late stages of disease status and averaging effects from multiple cell types and anatomic locations.
They also detected a correlation between naturally occurring genetic variations and translation. They monitored the influence of single-nucleotide variants, insertions, and deletions on mRNA abundance, ribosome occupancy, and translation efficiency. Although most variations do not correlate with ribosome occupancy or translation efficiency, genetic association with translation is detected for around 100 genes. Intriguingly, nonsense genetic mutations do not efficiently induce nonsense-mediated mRNA decay (NMD) since many protein-truncated variants (PTVs) are detected at the mRNA level. Ribosome occupancy of these PTVs shows little difference upstream or downstream of the premature stop codon, indicating inefficient termination or efficient re-initiation. The presence of these PTVs provides a possible cause of cardiac disease. Titin-truncating variants (TTNtvs) are detected in 13 dilated cardiomyopathy patients with different constitutive exons. No evidence of efficient NMD has been found, and significant ribosome occupancy downstream of the premature stop codon is detected in some cases, indicating that different TTNtvs may be translated differently and affect cardiac function in various ways. This observation partially explains the incomplete penetrance of heart disease occurrence in human patients with the same genetic mutation of TTNtvs. One possibility is additional expression and activity changes in eukaryotic release factors that may cause ribosome readthrough of premature termination codons but not native stop codons.
To complement Ribo-seq in whole human hearts, cell-type-specific translatomic analysis has been conducted. For instance, although post-transcriptional regulators such as IL-11 have been identified as related to cardiac fibrosis (27), the post-transcriptional mechanisms underlying myofibroblast transformation remained unexplored until 2019, when Chothani, S., et al. monitored the global changes of transcriptome and translatome during human cardiac fibroblast activation and transformation to myofibroblast using RNA-seq and ribosome profiling (28). TGFβ was used to stimulate fibrosis in primary cardiac fibroblasts isolated from atrial biopsies. Then, they performed RNA-seq and Ribo-seq at baseline and in a time-course ranging from 45 minutes to 24 hours after TGFβ stimulation to capture a dynamic picture of the transcriptional and posttranscriptional changes underlying the transition of quiescent fibroblasts into myofibroblasts.
They identified dynamically transcribed genes from RNA-seq and determined dynamically translated mRNAs from the integrated analysis of both RNA-seq and Ribo-seq results. A total of 1691 dynamically translated mRNAs were captured during the fibrotic response, of which translational changes alone were enough to cause changes in protein abundance. Sixty-seven dynamically translated mRNAs showed instant translational changes 45 minutes after TGFβ stimulation. The most enriched gene ontology term of those dynamically translated mRNAs was "transcription regulator activity," suggesting that instant translational changes may modulate subsequent transcriptional changes. The impact of translation then gradually decreases at later time points while the effect of transcription gradually increases, indicating a possible shift from translational regulation to transcriptional regulation. However, although plenty of dynamically transcribed genes were detected, about 29% of the transcriptional changes are buffered by translational regulation, implying that translation efficiency may be upregulated on genes with limited transcripts or vice versa.
Chothani S. et al. suggest that over one-third of all gene expression changes detected in myofibroblast transformation involve translational regulation, which may be carried out through RNA-binding proteins (RBPs). During myofibroblast transformation, fifty-three differentially expressed RBPs were detected. The targets of these RBPs were predominantly enriched in dynamically translated genes, but not in dynamically transcribed genes, highlighting the possibility that these RBPs shape the fibrotic response. However, loss-of-function and gain-of-function validation studies for individual RBPs are required to form the causative relationship between these RBPs and the fibrotic response.
2.3. Imaging-based techniques for evaluating translation efficiency and localized translation in cardiomyocytes
Puromycin, an unnatural analog of tyrosyl-tRNA, can be incorporated into the aminoacyl (A) site of translating ribosomes, leading to truncated protein production terminated at this residue. Treatment of cultured cardiac cells with puromycin can evaluate the global protein synthesis rate using immunoblot. Moreover, injecting puromycin into animals can allow direct imaging of the translation events in vivo. This method can provide information on the location of translation in organs and allow quantification of the translation efficiency by comparing control and genetically modified or disease-triggering animal models. One example is the visualization of translation machinery colocalized with sarcomere protein network in mouse cardiomyocytes in vivo (29). Macromolecular protein complexes, such as the sarcomere, the basic contractile macromolecular complex of cardiomyocytes, are maintained with proper localization and fixed subunit stoichiometry. Single-cell analysis of cardiomyocytes using mRNA and protein synthesis imaging demonstrates three different but related mechanisms for retaining the sarcomere (29): i) Mature mRNAs encoding sarcomere component proteins are localized to the sarcomere where their protein products are assembled. ii) Translation machinery, such as ribosomes, is located at the sarcomere with localized translation of sarcomere protein-coding mRNAs. iii) A specific localized E3 ubiquitin ligase allows rapid and efficient degradation of excess unincorporated sarcomere component proteins. These three mechanisms are distinct and required. Cooperation of the mechanisms is essential to ensure appropriate spatial localization of sarcomere proteins and to buffer the variability in mRNA expression levels of these proteins. Cardiomyocytes maintain their sarcomeres using localized translation at high rates and continuous proteasomal degradation to remove excess proteins and maintain the homeostatic stoichiometric ratio of different component proteins in the sarcomere protein network. Therefore, tightly regulated localization of mRNA transcripts, translation, and protein degradation controls the organization of sarcomere assembly and maintains the spatiotemporal features.
During cardiac hypertrophy, stress-induced signal transduction enhances CM mRNA translation, adding new contractile sarcomere units to enlarge cell size and enhance contractility. In this process, microtubules are required for cardiomyocyte growth via spatiotemporal control of the translation machinery (30). In particular, Scarborough et al. show that microtubule motor protein Kinesin-1 localizes ribosomes and mRNAs along microtubule tracks to different regions within the cell. Microtubules normally deliver mRNAs and translation machinery to specific sites to promote local translation and cardiomyocyte contractile unit assembly. This microtubule network is disrupted upon hypertrophic stress stimulus (phenylephrine treatment), causing mRNA and ribosome collapse around the nucleus, leading to mislocalized protein synthesis and rapid degradation of newly synthesized proteins. Consequently, cardiomyocytes fail to grow despite increased translation rates, suggesting that properly localized translation, not just the translation rate, is a key determinant of cardiac hypertrophy.
Synthesis of sarcolemma and sarcoplasmic reticulum membrane-associated proteins in cardiomyocytes was assumed to follow the general secretory pathway with localized mRNA translation in perinuclear areas, followed by protein trafficking and delivery to the functional sites. However, limited experimental evidence was provided. Using a single-molecule level visualization and a proximity-ligated in situ hybridization approach, researchers visualized ribosome-associated mRNAs for ion channel-related proteins, such as SERCA2A and SCN5A, providing detailed information on the localized translation sites within the cell (31). The translation machinery for membrane-associated protein synthesis occurs throughout the cardiomyocyte and enables the distributed synthesis of specific transmembrane proteins within specific sub-cellular locations. In these niches, the localized ribosomes synthesize proteins on local mRNA pools trafficked from in the nucleus by association with the microtubules and cytoskeleton network. As an evolutionarily conserved mechanism from mouse to human, membrane protein mRNAs are widely distributed across the cardiomyocyte in normal and failing human heart tissues. These findings confirm that local protein synthesis in cardiomyocytes regulates cardiac structure and function. At the pathophysiological level, arrhythmias and sudden death can occur with a mild imbalance between inward sodium and outward potassium currents. One new paradigm provides insights into the mechanisms of maintaining this critical balance. Electrophysiological and single-molecule fluorescence imaging analysis reveal that two mRNAs encoding SCN5A (INa) and hERG (IKr) channels are associated and coordinated in defined discrete complexes, namely, “micro-translatomes”, during protein translation (32). About half of the hERG-translating complexes contain SCN5A mRNA transcripts. Moreover, both mRNA transcripts are regulated at co-translational levels, and consequently, this regulation alters the expression of both functional ion channels localized at the cytoplasmic membrane.
3. Translational Control in Cardiac Development and Congenital Heart Disease
3.1. Human genetic mutations in translation machinery and congenital heart disease
3.1.1. Diamond Blackfan Anemia and other heart disease-causing mutations in cytoplasmic translation factors
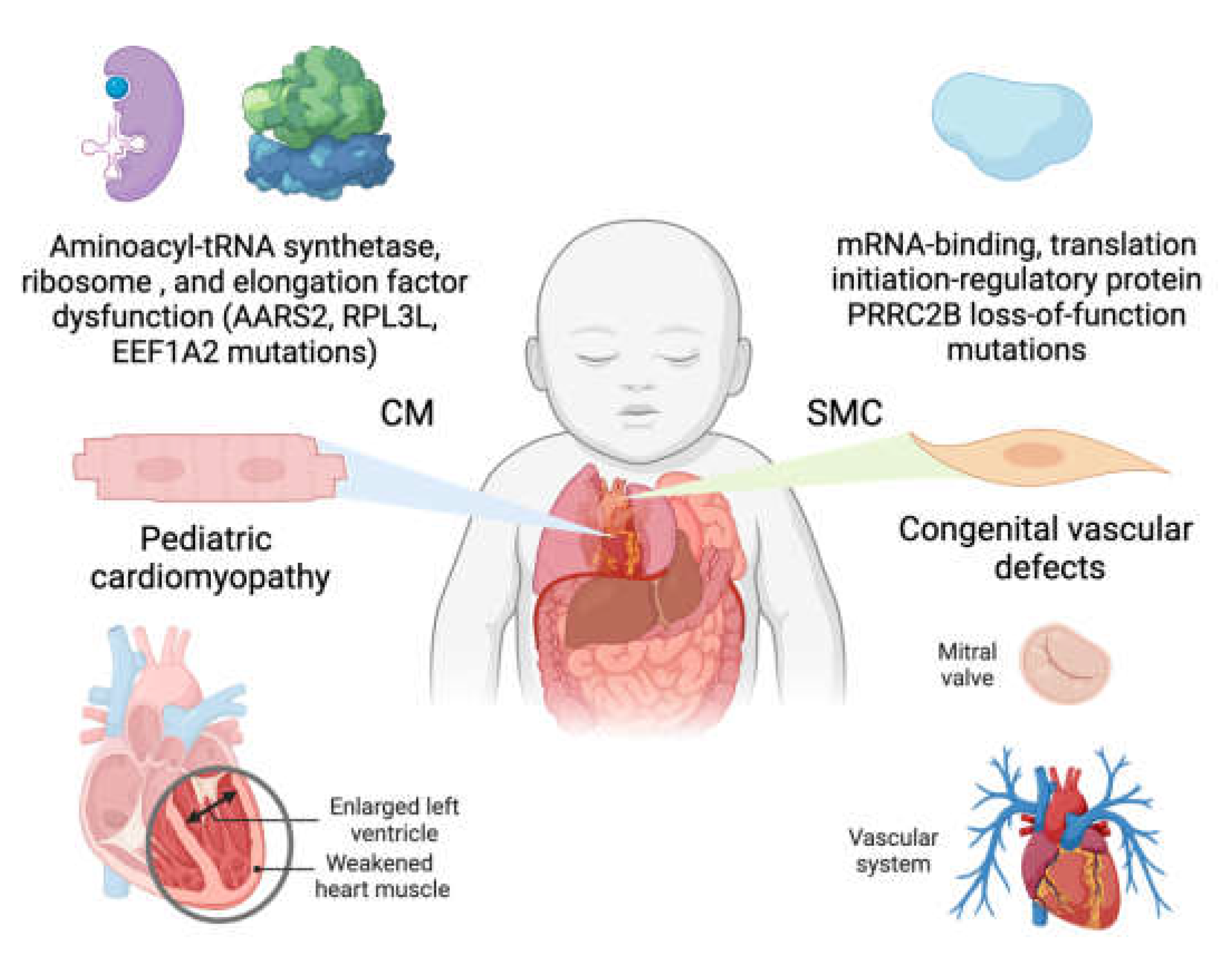
Human genetic mutations in cytoplasmic large ribosomal subunit protein 5 (RPL5), among many other cytoplasmic ribosome protein-coding genes, lead to Diamond Blackfan Anemia with congenital cardiac developmental defects (33-35), suggesting that loss-of-function of general house-keeping translation factors can result in cell-type- and organ-specific disorders. This observation has been well recapitulated in mouse and zebrafish genetic models with ribosome protein mutations (36). Most of these ribosome proteins are expressed ubiquitously across organs and cell types, such as RPL5 and RPL3 (large ribosomal subunit protein 3). Intriguingly, multiple ribosomal protein paralogs are expressed in a tissue-specific manner. It is under debate how these proteins influence translation in specific organs and affect the development and function, such as the heart. Large ribosomal subunit protein 3-like (RPL3L), a paralog of RPL3, is specifically expressed in cardiomyocytes and skeletal muscle cells of the heart and skeletal muscle, respectively, and modulates the dynamics of the translation elongation process. Genetic mutations of RPL3L in humans are associated with pediatric cardiomyopathy and age-related atrial fibrillation (37) (Figure 2). To recapitulate the genetic defects in human patients, a homozygous Rpl3l knockout mouse model has been established in multiple labs. Shiraishi’s group reported that a deficiency of RPL3L-bearing ribosomes in Rpl3l global knockout mice (CRISPR-Cas9-mediated deletion of the exon 2) caused reduced cardiac contractility (38). Transcriptome-wide ribosome profiling assay showed that ribosome occupancy at mRNA genetic codons was changed in the Rpl3l-null heart, and the changes were negatively correlated with those observed in myoblast cells with RPL3L overexpression. Compared with RPL3-bearing canonical ribosomes, RPL3L-bearing ribosomes were less prone to ribosome collisions on the mRNA. The reduction of RPL3L-containing ribosomes reprograms the translation elongation dynamics for the global transcriptome. Intriguingly, this translation-altering effect is most significant for mRNA transcripts encoding proteins related to cardiac muscle contraction and dilated cardiomyopathy, with the quantity of these proteins being decreased due to repressed translation. Thus, RPL3L-bearing ribosomes are essential to maintain the translation elongation dynamics required for normal cardiac function. This finding provides insights into the mechanisms of tissue-specific ribosome protein-mediated translational regulation with physiological and pathological relevance in human patients.
In contrast, Milenkovic’s group recently demonstrated a dynamic interplay between RPL3- and RPL3L-bearing ribosomes that regulates mitochondrial activity and ATP production in the mammalian heart as a translation-independent noncanonical mechanism. Different cell types possess distinct types of ribosomes with specialized ribosome proteins like RPL3L. This phenomenon is defined as ribosome heterogeneity. However, whether this ribosome heterogeneity results in functionally diverse “specialized ribosomes” in canonical mRNA translation or noncanonical functions remains controversial. Milenkovic’s group used a similar but different Rpl3l knockout mouse strain (CRISPR-Cas9-mediated deletion of a 13-bp DNA fragment in the exon 5) to uncover a rescue mechanism in which compensatory induction of RPL3 is triggered upon RPL3L inactivation, accumulating RPL3-bearing ribosomes instead of RPL3L-bearing ribosomes that are uniquely present in cardiomyocytes (39). In contrast to Shiraishi’s group’s findings, using ribosome profiling and a novel approach of ribosome pulldown coupled with nanopore RNA-seq (Nano-TRAP), RPL3L is found to modulate neither translational efficiency nor ribosome affinity towards a specific subset of mRNA transcripts. Interestingly, knockout of Rpl3l leads to enhanced ribosome–mitochondria interactions and a significant increase in mitochondrial activity and subsequent ATP synthesis in CMs. This study suggests that tissue-specific ribosome protein paralogues may not regulate the translation of specific mRNA transcripts. Alternatively, the presence or absence of RPL3L alters the expression of RPL3, which changes the subcellular localization of cytoplasmic ribosomes and modifies the mitochondrial activity. What factors cause the discrepancy between these two studies using similar approaches remains unclear. One of the most reproducible findings suggest that unrelated genetic deletion distinct from Rpl3l loss-of-function, such as conditional knockout of a cytosolic gene, glutamyl-prolyl-tRNA synthetase (Erps1), or a nuclear-encoded mitochondrial gene family with sequence similarity 210 member A (Fam210a) in CMs, caused heart failure and exhibited simultaneous increase in RPL3 and decrease in RPL3L mRNA and protein expression levels (22, 40). Cryo-electron microscopy visualization of RPL3- and RPL3L-bearing ribosome structures and comparative bioinformatic re-analysis of independent Ribo-seq data from both groups will provide critical insights into better deciphering the functional and mechanistic divergence between the two specialized ribosomes in the CMs.
A recent report from Molkentin's group showed that mouse cardiac ventricles express RPL3 during the neonatal stage (37). RPL3 is then replaced by RPL3L in adulthood but is re-expressed under cardiac hypertrophy and remodeling. This follows a similar expression pattern of the fetal gene program, such as the key transcription factors and sarcomere proteins. Intriguingly, Rpl3l−/− mice (CRISPR-Cas9 mediated deletion and frameshifting in the exon 5) showed no overt changes in cardiac structure or function at baseline or after transverse aortic constriction surgery-based, pressure overload-induced hypertrophy. Possibly, loss-of-function of RPL3L could be compensated by RPL3, as the expression of the latter was persistently upregulated in the adult heart (37). Transcriptomic profiling analysis and polysome profiling assay show little differences between Rpl3l knockout and wild-type control hearts from adult mice. Moreover, in adult Rpl3l knockout cardiomyocytes, no changes were found in cellular localization of the ribosome, cardiac tissue ultrastructure, or mitochondrial function compared to wild-type control cells. Adeno-associated virus-9 (AAV9)-mediated overexpression of either RPL3 or RPL3L in the hearts failed to cause pathogenesis in mice. Rpl3l null mice had significantly smaller hearts during cardiac aging than wild-type controls at 18 months after birth. Unlike the other two groups, Molkentin's lab demonstrates that Rpl3l knockout can be fully compensated by RPL3, although Rpl3l deletion leads to a slight but significant reduction in heart weight. More replicated studies are required to reproduce and confirm any of these findings because the different genetic knockout strategies from the three labs could partially contribute to the contradictory conclusions and distinct phenotypes and mechanisms. Ribo-seq mapping can provide detailed information about subtle changes in translation elongation dynamics that polysome-seq cannot. To resolve this controversy, genetic knock-in of the specific human mutation in the mouse genome must be performed to recapitulate the heart disease phenotype observed in human patients. If no spontaneous phenotype is triggered, it indicates the possibility of human mutations as an accompanying or modifier variant, which may not directly cause heart disease symptoms.
Intriguingly, a preprint manuscript from Wu’s lab provided further mechanistic insights into how PRL3L mutant proteins lead to dilated cardiomyopathy 2D (CMD2D; OMIM # 619371) due to a gain of toxic function in humans (41) (Figure 2).
Figure 2.
Translation machinery defects in early-onset human heart disease.
They identified new, rare yet highly pathogenic heterozygous hotspot mutations D308V/N and G27D in the RPL3L gene. These mutations were co-initiated with known loss-of-function mutations (frameshift, missense mutation, alternative splicing) or low-pathogenic missense mutations. Despite carrying autosomal recessive alleles, such patients do not exhibit severe heart failure. The authors observed a decrease in 28S rRNA but not 18S rRNA in the tissue of these patients. Moreover, neither RPL3 mRNA nor protein levels were found to be upregulated as a compensatory response. To evaluate the impact of each mutation, the authors developed an AC16 human cardiomyocyte cell model with inducible RPL3 and different variants of RPL3L to assess the effect of each mutation. D308V/N and G27D variants of RPL3L showed a reduction in 28S rRNA and 60S ribosomal subunits, resulting in the loss of 80S monosomes and polysomes. Being heterozygous and recessive, the mutations confer a gain of toxic function rather than a loss of function. The D308V/N and G27D RPL3L proteins were detected solely in the nucleus. Interactome research revealed their interaction with proteins involved in 28S rRNA processing and 60S ribosome subunit biogenesis. The nuclear-localized mutant RPL3L exhibited the highest binding affinity with the ribosomal chaperones and biogenesis factors GRWD1 and C7ORF50, as indicated by HA-tagged RPL3L overexpression followed by immunoprecipitation and mass spectrometry analysis. Thus, D308N and G27D proteins may sequester ribosomal biogenesis factors, ultimately diminishing overall translation. Additionally, the authors found that the R161W and T189M mutants of RPL3L induced a compensatory effect on RPL3, mirroring the upregulation of Rpl3 mRNA in the Rpl3l knockout mouse model. These two mutations enhance RPL3 mRNA stability but do not affect its transcription. However, the compensation from increased RPL3 was inadequate to rescue the failure caused by the gain-of-function mutation allele of RPL3L. Publicly available human genome sequencing databases reveal more than ten RPL3L homozygous knockout humans with no reported heart diseases. This supports the idea of a gain of toxic function of the RPL3L mutations. Noticeably, they used lentiviral overexpression of mutant RPL3L with shRPL3 knockdown in an immortalized human AC16 ventricular cardiomyocyte cell line fused with human fibroblast cells. This experimental system may not necessarily recapitulate the gene expression and protein localization in vivo in the human hearts. Genetic knock-in mouse model for the human RPL3L mutations and human iPSC-CM after maturation need to be exploited to validate their findings in the AC16 cell line. Also, the mechanism underlying this RPL3 mRNA stabilization by mutant RPL3L protein remains unclear and warrants future thorough biochemical characterization. As an example, a gain-of-toxic-function human mutation S637G in RBM20 (RNA binding motif protein 20) leads to mislocalization of this protein in the cytoplasm, formation of RBM20-ribonucleoprotein granules, thereby causing severe dilated cardiomyopathy via inhibiting mRNA translation most likely. The severe heart failure symptoms and high mortality rate are faithfully recapitulated in a Rbm20S639G knock-in mouse model, but not in the Rbm20 knockout model with a mild heart disease phenotype(42).
Unbiased animal genome- and transcriptome-wide associated studies (GAWS and TWAS) and subsequent expression quantitative trait locus (eQTL), as well as translation efficiency quantitative trait locus (teQTL) provide a complementary approach to human GWAS, TWAS, eQTL, and subsequent characterization of the causative genetic variants of diseases. One such study discovered a trans locus that causes ribosomopathy in hypertrophic hearts that modulates mRNA translation in a protein length-dependent manner using teQTL analysis in rats (43). This study investigated the influence of trans-acting genetic variation in distant genetic loci on the mRNA translation efficiency. It defined their contribution to developing complex disease phenotypes within a panel of rat inbred lines. One of the tissue-specific master regulatory loci, associated explicitly with hypertrophic hearts, drives a transcriptome-wide protein length-dependent regulation in mRNA translation efficiency, altering the stoichiometric translation rates of many sarcomeres protein-coding mRNAs. Mechanistically, significant differences in global polysome profiles and dysregulation of the small nucleolar RNA SNORA48 influence ribosome biogenesis and activity, leading to a translation machinery defect. Reproducible protein length-dependent shifts in translational efficiency were observed as an evolutionarily conserved trait of translation machinery mutants across multiple species from yeast to humans, including ribosomopathy-causing translation machinery component protein mutants. Mutations in different trans-acting factors can reduce or enhance a negative correlation between protein length and translation rates. This effect is potentially caused by transcript-specific translation initiation and re-initiation rate imbalances.
Another ribosome-related translation component is eukaryotic elongation factor 1A (eEF1A). eEF1A mediates aminoacyl-tRNA recruitment to the aminoacyl-tRNA site (A-site) of the eukaryotic cytoplasmic 80S ribosome. Therefore, eEF1A is crucial to the translation elongation process on mRNAs. Two different isoforms exist, eEF1A1 and eEF1A2, though antibodies to differentiate these isoforms have only recently become available. eEF1A1 is ubiquitously expressed through embryonic and neonatal mice and is then reduced and replaced by eEF1A2 in the heart, brain, spinal cord neurons, and skeletal muscle. Mice with a 15.8 kilobase deletion in the Eef1a2 gene have a “wasted” phenotype, experiencing muscle wasting, neurodegeneration, and death at postnatal day 28 (44). Humans with a Pro333-to-Leu mutation in the EEF1A2 gene have dilated cardiomyopathy, developmental delay, epilepsy, and early death (45) (Figure 2). Xmlc2-Cre+ driven cardiomyocyte-specific conditional knockout of Eef1a2 and knock-in of Pro333-to-Leu mutation in mice resulted in left ventricular chamber dilation and systolic dysfunction, followed by full penetrance of death at around 8-17 weeks (46), suggesting an essential role of eEF1A2 in heart development and functional maintenance. Like RPL3L and RPL3, it is crucial to dissect the functional redundancy and uniqueness between eEF1A1 and eEF1A2 as a future direction.
3.1.2. Human mutations in mitochondrial translation machinery lead to genetic cardiomyopathy
Numerous human mutations in mitochondrial translation machinery have been reported to cause spontaneous early-onset dilated or hypertrophic cardiomyopathy. These cardiomyopathy-related mitochondrial translation machinery mutations include mitochondrial aminoacyl-tRNA synthetases and tRNAs, translation factors, mitoribosomal proteins, and mitochondrial RNA-binding proteins (47). An illustration of this is a mutation in the mitochondrial alanyl-tRNA synthetase, AARS2, resulting in infant-onset cardiomyopathy (48-50) (Figure 2). This implies that AARS2-dependent translation is necessary for normal cardiac development. Another example of mitochondrial translation affecting cardiomyopathy was shown in experiments by Rudler et al (51). They generated a knockout mouse model of mtIF3, a mitochondrial translation initiation factor. A global knockout mouse model of Mtif3fl/fl with a global Cre transgene was embryonic lethal. In contrast, a heart and skeletal muscle knockout mouse model with Ckmm-Cre resulted in dilated cardiomyopathy. Evidence of mutations in mitochondrial translation elements resulting in cardiomyopathy highlights the importance of normal mitochondrial translation in maintaining mitochondrial integrity and cardiac function.
3.1.3. Loss-of-function of PRRC2B-mediated translation initiation regulation causes congenital cardiovascular defect in humans and mice
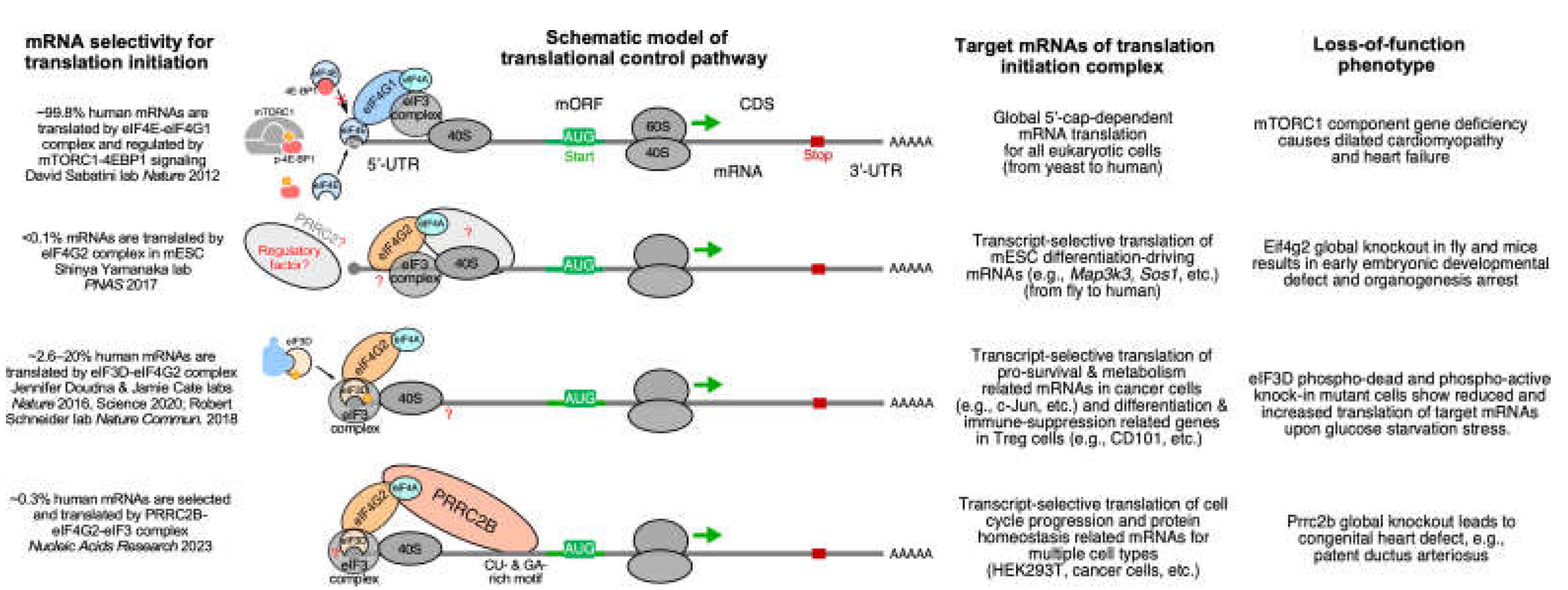
Posttranscriptional control of gene expression, including RNA splicing, transport, modification, translation, and degradation, is primarily mediated by RBPs and their interaction with target mRNAs (52). Recently, our group characterized the function of a novel RBP, Proline-rich coiled-coil 2B (PRRC2B) (13) (Figure 2). Transcriptome-wide CU- or GA-rich RNA regions were identified as PRRC2B binding sites near the translation initiation codon on a specific cohort of mRNAs by photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation and sequencing (PAR-CLIP-seq) in human cells. These PRRC2B-bound mRNAs, including oncogenes, protein homeostasis-related factors, and cell cycle regulators such as cyclin D2 (CCND2), showed decreased translation efficiency upon inducible PRRC2B knockdown, leading to compromised G1/S phase transition and cell proliferation. Antisense oligonucleotides (ASOs) masking PRRC2B interacting sites within CCND2 mRNA 5' UTR reduced its translation and inhibited cell cycle progression and proliferation. Mechanistically, PRRC2B interacts with eukaryotic translation initiation factors 4G2 (eIF4G2) and eIF3 in an RNA-independent manner to form a ribonucleoprotein complex together with target mRNAs. The interaction between PRRC2B and eIF4G2 is essential for sufficient translation initiation of CCND2 mRNA. Therefore, PRRC2B is a critical translation regulatory factor for the efficient expression of a selective cohort of proteins (~0.3% of human genes) required for cell cycle progression and proliferation (Figure 3). We confirmed that the shRNA-mediated knockdown of PRRC2B inhibits the translation of a selective cohort of endogenous mRNAs in human cells, such as CCND2, among others (13). Interestingly, CCND2 protein was reported to promote the proliferation of cardiomyocytes when overexpressed in porcine hearts as a potential factor to enhance heart regeneration in large mammals (53), implying that PRRC2B might be involved in cardiomyocyte proliferation at the translational level. In line with this idea, Teleman’s group reported the critical role of PRRC2 family proteins, PRRC2A, PRRC2B, and PRRC2C, in the cell proliferation of human cancer cells (54).
Our recent work highlighted a novel function of PRRC2B associated with congenital heart disease (55). We identified two alternatively spliced isoforms of PRRC2B and confirmed their conservation in human and mouse hearts and HEK293T cell lines. Kimchi’s group recently reported the same alternative spliced isoforms in multiple additional human cancer cell lines, such as HeLa, HCT116, A549, among others (56). Interestingly, our in vivo exon-16-containing premature termination codon knock-in mouse model for full-length PRRC2B did not show any severe cardiac phenotype, suggesting a possible compensation either by the alternate spliced isoform (exclusion of exon 16, as termed ΔE16) or possibly by PRRC2A or PRRC2C. However, global knockout of both full-length and ΔE16 Prrc2b mRNA isoforms (genetic deletion of exon 4 shared by both isoforms) causes a high penetrance of neonatal lethality in mice by triggering patent ductus arteriosus (PDA), a genetic disorder with cardiovascular developmental defects observed in humans. Bulk and single-nucleus RNA-seq data identify a decrease in smooth muscle cell number and expression of smooth muscle-specific genes upon global Prrc2b deletion. Moreover, polysome-seq, RNA-seq, and mass spectrometry analysis from CRISPR-Cas9-mediated PRRC2B knockout HEK293T cells suggest a significant reduction of genes involved in cell proliferation, heart and vascular development, indicating a possible regulation of PRRC2B in vascular smooth muscle cell proliferation and contraction. Two heterozygous loss-of-function mutations of the PRRC2B gene in patients with congenital heart disease are reported to manifest symptoms of pulmonary vein atresia and mitral regurgitation and stenosis, underscoring the connection between PRRC2B and cardiovascular development and disorders. Therefore, PRRC2B has been hypothesized to regulate protein translation of specific proteins in cardiac cells, such as smooth muscle cells, to maintain normal heart morphogenesis, and loss-of-function of PRRC2B causes aberrant gene expression and congenital heart defects. Our recent findings indicate that the knockdown of PRRC2B in primary human aorta-derived smooth muscle cells inhibited cell proliferation and migration (55) (Figure 3). PRRC2B is a promising RNA-binding protein that could be pivotal in cardiovascular disease development and a future therapeutic target.
Figure 3.
eIF4G2-PRRC2B complex-mediated translation initiation regulatory pathway for cell proliferation and cardiac developmental integrity. Bottom row compared to eIF4G1-eIF4E-driven, cap-dependent canonical translation initiation (top row), eIF4G2-mediated cap-independent (second row), and eIF3D-mediated cap-dependent (third row) noncanonical translation initiation pathways.
Figure 3.
eIF4G2-PRRC2B complex-mediated translation initiation regulatory pathway for cell proliferation and cardiac developmental integrity. Bottom row compared to eIF4G1-eIF4E-driven, cap-dependent canonical translation initiation (top row), eIF4G2-mediated cap-independent (second row), and eIF3D-mediated cap-dependent (third row) noncanonical translation initiation pathways.
3.1.4. eIF4E1C regulates cardiomyocyte metabolism and proliferation during heart regeneration in zebrafish
In eukaryotes, the eIF4E family of translation initiation factors, such as canonical eIF4E1A, bind 5' methylated guanosine caps of mRNAs as a limiting step for mRNA translation. Another family member, eIF4E1C, present in aquatic vertebrates but lost in terrestrial species, plays a vital role during cardiac development and heart regeneration in zebrafish (57). eIF4E1C is broadly expressed across multiple cell types and organs in fish. Genetic deletion of the eif4e1c gene in zebrafish caused growth defects and reduced juvenile survival. The knockout zebrafish surviving to adulthood had a significantly decreased number of cardiomyocytes and compromised proliferation in response to cardiac injury compared to wild-type controls. Translatome-wide Ribo-seq analysis of eif4e1c-null hearts reveals changes in the translation efficiency of mRNAs encoding proteins that regulate cardiomyocyte cell proliferation. Disruption of eIF4E1C function in the mutant fish had the most pronounced impact on the heart at juvenile stages. This suggests a specialized requirement for fine-tuning the control of translation initiation via a unique eIF4E paralog for heart regeneration and development in fish. Identifying a similar translation factor and regulatory mechanism in terrestrial species (e.g., mammals) is the key to generalizing this concept in evolution.
3.2. Translational control in mitochondrial cardiomyopathy
Many genetic mutations have been reported in the genes encoding mitochondrial translation machinery that cause spontaneous cardiomyopathy with mitochondrial dysfunction, including mitochondrial aminoacyl-tRNA synthetases and tRNAs, mitochondrial translation initiation and elongation factors, mitoribosome proteins, and other translation regulatory factors or RNA-binding proteins localized in the mitochondria (47). Persistent activation of an evolutionarily conserved central cytosolic translational control pathway, namely, integrated stress response (ISR), is considered a common shared feature with multiple types of mitochondrial cardiomyopathy and other mitochondrial dysfunction-related diseases caused by these mitochondrial protein-coding gene mutations (58, 59).
Translational regulation inside mitochondria is still underexplored, and more research is needed to discover novel therapeutic targets in the translational process to treat mitochondrial diseases. Prior genome-wide association studies in humans revealed that FAM210A (family with sequence similarity 210 member A) gene mutations were associated with skeletal muscle disorder and bone fractures (60, 61). We recently showed that the Fam210a cardiomyocyte-specific genetic knockout mouse model exhibited progressive mitochondrial cardiomyopathy and heart failure (62). Multi-omics analyses, including RNA-seq, Ribo-seq, proteomic mass spectrometry, and metabolomics, revealed a reduction of mitochondrial encoded mRNA translation elongation and consequent activation of ISR in Fam210a knockout hearts. Chronic and persistent ISR activation inhibits cap-dependent translation initiation in the cytoplasm, leading to translational reprogramming and disrupted protein homeostasis. Interestingly, FAM210A protein expression is reduced in diseased hearts from ischemic heart failure patients and mice with myocardial infarction surgery. This discovery demonstrates a novel crosstalk mechanism between mitochondrial and cytosolic translation processes via FAM210A-mediated regulation of mitochondrial translation elongation. Ribo-seq proved that mitochondrial translation elongation is compromised upon genetic knockout of Fam210a in cardiomyocytes in mice, as indicated by increased ribosome footprints of mRNAs transcribed from mitochondrial-encoded genes. Adeno-associated virus (AAV9)-mediated overexpression of FAM210A can significantly enhance mitochondrial translation and protect the heart from ischemia stress-induced cardiac dysfunction (62) as a potential gene therapy (Figure 4).
Figure 4.
FAM210A-EF-Tu complex regulates mitochondrial translation elongation and cardiac mitochondrial homeostasis in hypertensive cardiomyopathy and ischemic heart failure. Left panel: miR-574-5p and miR-574-3p downregulate FAM210A expression and limit cardiac hypertrophy in transverse aortic constriction-induced hypertensive cardiomyopathy. Right panel: FAM210A expression is reduced in left anterior descending coronary artery ligation-induced myocardial infarction, leading to pronounced mitochondrial dysfunction and contributing to heart failure.
Figure 4.
FAM210A-EF-Tu complex regulates mitochondrial translation elongation and cardiac mitochondrial homeostasis in hypertensive cardiomyopathy and ischemic heart failure. Left panel: miR-574-5p and miR-574-3p downregulate FAM210A expression and limit cardiac hypertrophy in transverse aortic constriction-induced hypertensive cardiomyopathy. Right panel: FAM210A expression is reduced in left anterior descending coronary artery ligation-induced myocardial infarction, leading to pronounced mitochondrial dysfunction and contributing to heart failure.
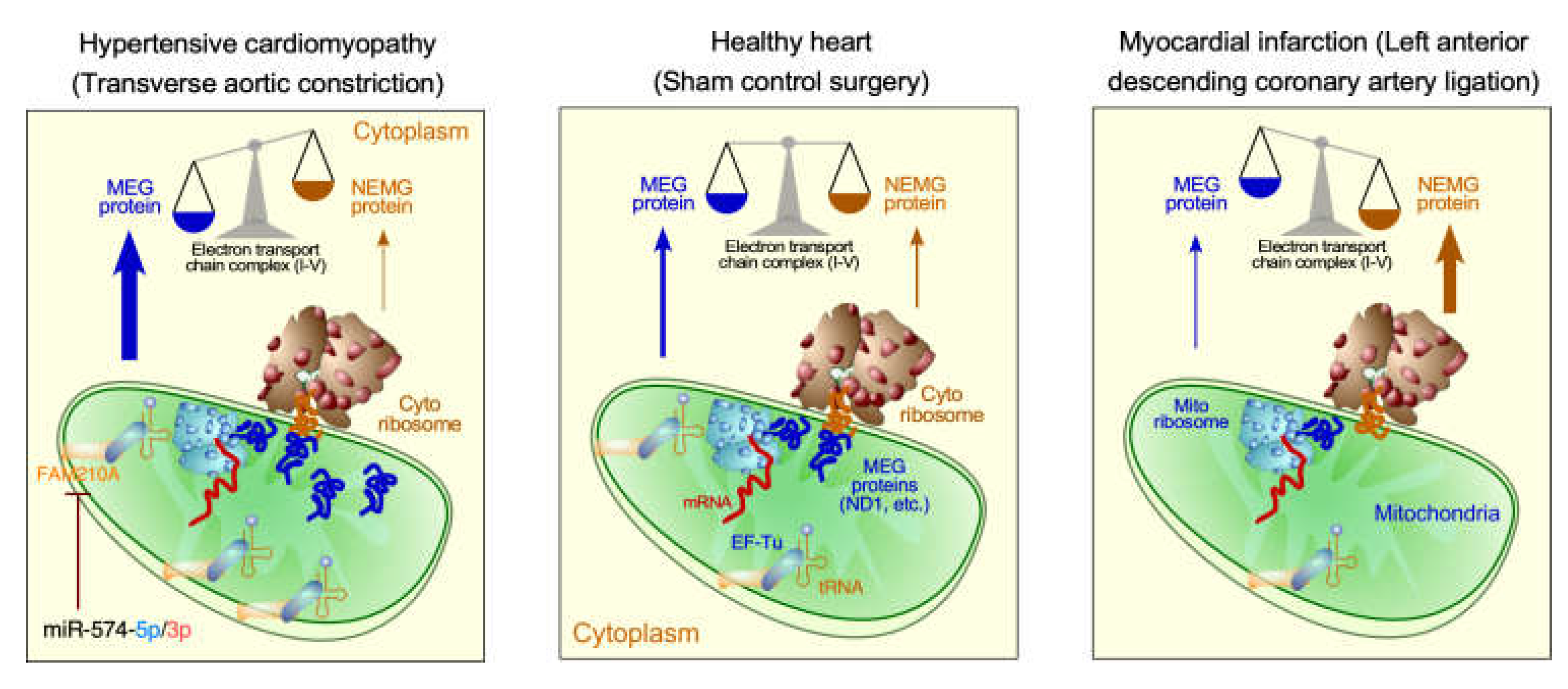
Following a prior miRNA expression screen for MI models (63), we identified mammalian miR-574 (including guide and passenger strands miR-574-5p and miR-574-3p) as involved in cardiac hypertrophy and pathological remodeling. We discovered that dual-strand miRNAs, miR-574-5p and miR-574-3p, are induced in human and mouse non-ischemic failing hearts compared to healthy hearts. Using the miR-574 genetic knockout mouse model and RNA-seq, we found that miR-574-5p and miR-574-3p target Fam210a mRNA in mouse cardiomyocytes and cardiac fibroblasts, thereby maintaining mitochondrial homeostasis and preventing cardiac hypertrophy and ventricular remodeling in a pressure overload-induced hypertensive cardiomyopathy model (64). This work demonstrates that the miR-574-FAM210A axis regulates mitochondrial translation for maintaining optimal expression of mitochondrial electron transport chain complex genes in non-ischemic heart disease (64) (Figure 4). More importantly, miR-574 delivered in the hypertensive cardiomyopathy mouse models via nanoparticles can be a potential therapeutic tool for antagonizing cardiac pathological remodeling (64). Based on these findings, we proposed “normalizing” mitochondrial translation to maintain the homeostatic balance with cytosolic translation to protect the heart from progressive pathological remodeling and heart failure.
To characterize the biochemical and biophysical properties, the protein structure of FAM210A and its complex with EF-Tu or other interacting partners needs to be resolved. This was highly challenging as FAM210A is a mitochondrial transmembrane protein. We overexpressed human FAM210A with a truncated mitochondria-targeting signal peptide at the N-terminus in bacteria and purified the recombinant protein from E. coli (65). Interestingly, bacteria-derived translation elongation factor EF-Tu is co-purified with human FAM210A, which recapitulates the formation of FAM210A-EF-Tu complex in cardiac mitochondria as seen by immunoprecipitation-mass spectrometry (64). Consistently, recombinant human FAM210A protein is localized in the plasma membrane of E. coli, like the localization in the inner mitochondrial membrane in CMs.
3.3. Translational regulation of cardiac cell proliferation and differentiation
Congenital heart disease (CHD) is the leading cause of birth defect-related death and can lead to severe adult heart disease, such as heart failure. The severity of CHD emphasizes the importance of the normal developmental program of the cardiovascular system, which depends on precise spatial and temporal control of the expression of genes encoding structural proteins, transcription factors, and cell cycle-related proteins. The gene expression programs of developing organs are established at both transcription and translation levels at the early stages of embryonic development. Transcriptional and epigenetic mechanisms are well-known for initiating and arranging developmental time courses for organogenesis. It is well-established that multiple transcription factors and cell cycle-related proteins control the development of the cardiovascular system (66-69). However, the regulatory mechanism upstream of these factors at the translational level remains largely unexplored. Translational regulation is competent for determining the fate of human embryonic stem cells (hESCs) towards cardiac differentiation by a specific mRNA translation regulatory pathway directed by RBPMS (RNA binding protein with multiple splicing) (70). Under cardiac cell differentiation conditions, RBPMS is associated with actively translating ribosomes in hESCs to activate the translation of a specific cohort of key factors required for initiating a cardiac commitment program, such as the Wingless/Integrated (WNT) signaling. As a result, the loss-of-function of RBPMS profoundly impairs cardiac mesoderm specification, thereby leading to profound morphogenetic and tissue patterning defects in cultured human cardiac organoids. RBPMS acts in translational control via two separate and related molecular mechanisms, including selectively binding to the 3'-UTR of specific target mRNAs and globally promoting translation initiation for enhancing protein synthesis. RBPMS depletion leads to inhibition of translation initiation, indicated by abnormal eIF3 complex retention and eIF5A drop-off on mRNAs, thereby blocking ribosome recruitment and elongation during protein synthesis.
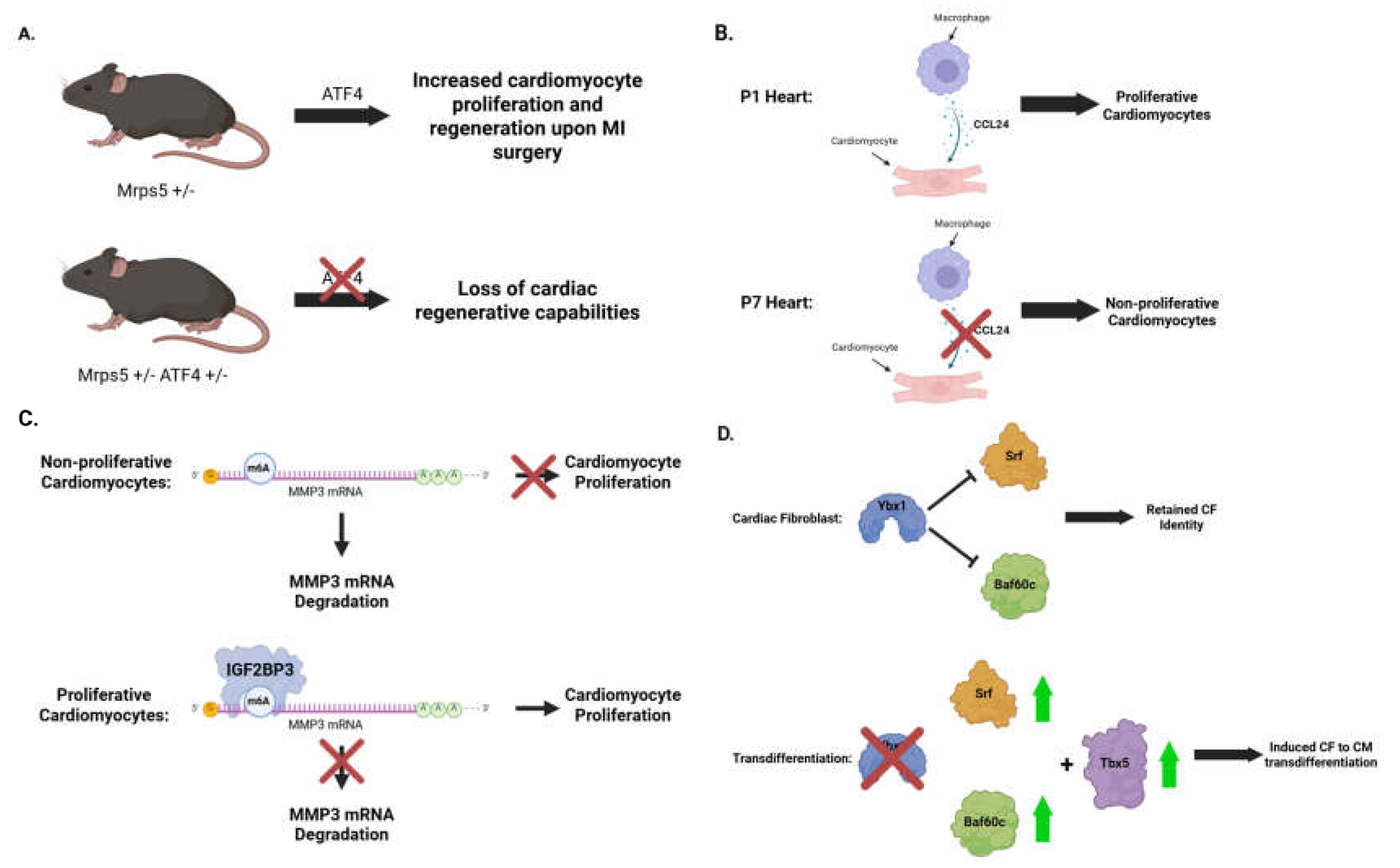
Mitochondria play essential roles in maintaining normal cardiac function and preventing heart disease. Generally, reduced mitochondrial translation causes a mitochondrial-nuclear proteomic imbalance and leads to changes in the activity of the electron-transporter chain complex. Intriguingly, deleting a single allele of mitochondrial small ribosomal subunit protein 5 (Mrps5) in mice enhances cardiomyocyte proliferation and cardiac regeneration in a surgical myocardial infarction mouse model (71) (Figure 5A). Cardiac function after MI surgery is significantly improved in mice with haploinsufficiency of Mrps5. Activating transcription factor 4 (ATF4) is a critical regulator of the mitochondrial stress response in cardiomyocytes from heterozygous Mrps5 knockout mice. Mechanistically, ATF4 regulates KNL1 (kinetochore scaffold 1) expression, increasing cytokinesis during cardiomyocyte proliferation. Doxycycline-mediated inhibition of mitochondrial translation counterintuitively promoted cardiomyocyte proliferation. Cardiomyocyte proliferation of Mrps5+/− can be attenuated when one allele of Atf4 is also genetically deleted (Mrps5+/− Atf4+/−), resulting in a loss of the cardiac regenerative capacity. MRPS5 reduction and doxycycline treatment activate an evolutionarily conserved regulatory mechanism that increases the proliferation of human induced pluripotent stem cells (hiPSC)-derived cardiomyocytes, providing a new approach for treating cardiac injury and activating heart regeneration.
Figure 5.
Translational regulatory mechanisms driving cardiomyocyte proliferation and cardiac fibroblast-to-myocyte transdifferentiation. A. Mild mitochondrial translational defects activate integrated stress response and cardiomyocyte proliferation. B. Paracrine secretion of CCL24 from macrophages stimulates cardiomyocyte proliferation. C. The molecular mechanism of IGF2BP3-mediated binding and stabilization of m6A-modified MMP3 mRNA contributes to cardiomyocyte proliferation. D. Loss-of-function of YBX1 promotes cardiac fibroblast-to-myocyte transdifferentiation with TBX5 overexpression.
Figure 5.
Translational regulatory mechanisms driving cardiomyocyte proliferation and cardiac fibroblast-to-myocyte transdifferentiation. A. Mild mitochondrial translational defects activate integrated stress response and cardiomyocyte proliferation. B. Paracrine secretion of CCL24 from macrophages stimulates cardiomyocyte proliferation. C. The molecular mechanism of IGF2BP3-mediated binding and stabilization of m6A-modified MMP3 mRNA contributes to cardiomyocyte proliferation. D. Loss-of-function of YBX1 promotes cardiac fibroblast-to-myocyte transdifferentiation with TBX5 overexpression.
In addition to the stoichiometry of ribosome proteins influencing cardiomyocyte proliferation, the posttranslational modifications contribute to cardiomyocyte differentiation. 2-oxoglutarate and iron-dependent oxygenase domain-containing protein 1 (OGFOD1), a ribosomal prolyl-hydroxylase, catalyzes the posttranslational hydroxylation of proline 62 in the small ribosomal protein S23 (RPS23). Genetic deletion of OGFOD1 in an in vitro cell culture model of human cardiomyocytes decreases the translation of specific proteins, such as RNA-binding proteins, which may, in turn, regulate translation and alternative splicing as a secondary effect (72). Loss of OGFOD1 causes alterations in protein translation and reprograms the cardiac proteome, thereby increasing the synthesis of sarcomere proteins, including cardiac troponins, titin, and cardiac myosin-binding protein C. Consistent with these translational changes, OGFOD1 expression is reduced during cardiomyocyte differentiation.
Mammalian cardiomyocytes exit the cell cycle shortly after birth. The adult heart cannot regenerate in response to injury, whereas the neonatal heart can efficiently regenerate following MI surgery, but this capacity is lost by postnatal day (P)7 (73). RNA-seq analysis for regenerative (P1) and nonregenerative (P8) mouse hearts after MI surgery revealed that the transcriptome of post-MI regenerative hearts reverts rapidly to a baseline pattern compared to uninjured control hearts. In contrast, post-MI nonregenerative hearts exhibited a distinct gene expression pattern (74). Integrated with active chromatin landscapes, genes and biological processes activated in injured hearts were identified, among which the immune response and embryonic developmental gene programs were strikingly divergent between regenerative and nonregenerative hearts. The macrophage-mediated innate immune response has been reported to play a critical role in neonatal heart regeneration (75). Acute activation of immune-related genes in regenerative hearts is evident through the deposition of histone H3 lysine-27 acetylation (H3K27ac), which marks active enhancers and promoters, serving as key steps in triggering regeneration; notably, the injury-induced immune factor CCL24 secreted from P1 macrophages promoted CM proliferation during neonatal heart regeneration (Figure 5B). Moreover, the regenerative P1 heart retained developmental and cell-cycle gene programs, within which an RNA-binding protein, IGF2BP3 (insulin-like growth factor 2 mRNA-binding protein), was identified to promote CM proliferation and restore cardiac morphology and function after MI, possibly driven by translational activation of CM regenerative factor mRNAs such as IGF2 (insulin-like growth factor 2) through the binding of 3'-UTR.
Understanding the molecular mechanisms underlying the reactivation of CM proliferation and heart regeneration is essential for inducing cardiac repair in response to injury. IGF2BP3 belongs to a family of N6-methyladenosine (m6A) readers that recognize the consensus GG(m6A)C sequence and promote the stability of thousands of m6A-modified mRNA targets (76). IGF2BP3 expression progressively declines in mouse hearts during postnatal development and is nearly undetectable by P28. While MI induces IGF2BP3 upregulation in neonatal hearts, its expression remains barely detectable in adult hearts (77). Overexpression of IG2BP3 in P7 CMs promotes mitosis and cell-cycle progression, while IGF2BP3 knockdown decreases CM proliferation (Figure 5C). In vivo, adeno-associated virus AAV9-mediated IGF2BP3 overexpression in the left ventricular myocardium of P1 mice, followed by MI surgery, increases proliferative CMs and results in improved cardiac hypertrophy, reduced myocardial infarction size, decreased myocardial fibrosis, and preserved left ventricular ejection fraction. Among the most enriched mRNAs pulled down by IGF2BP3, MMP3 mRNA was identified as a target of IGF2BP3-mediated post-translational regulation. Mechanistically, the KH3 and KH4 domains of IGF2BP3 directly bind to the MMP3 mRNA and stabilize it through m6A modification, increasing MMP3 protein translation. Functionally, MMP3 acts as a downstream target of IGF2BP3 to promote heart regeneration and improve cardiac function after myocardial infarction.
Cardiomyocytes lose their proliferative ability shortly after their differentiation and maturation. This does not allow an adult mammal heart to regenerate after damage. Heart-specific triggers and pathways responsible for proliferation remain enigmatic. Understanding the mRNA expression signature in proliferating cardiomyocytes is one of the keys to studying heart regeneration. A recent unbiased comparative study used artificial intelligence (AI)-based tools to find such signatures among two pre-existing in vivo (mice and pigs) and one in vitro (human induced pluripotent stem cell-to-cardiomyocyte, hiPSC-CM) proliferating CM model. In vivo mouse and pig models provide single-nucleus (sn)RNA-seq on different days after induced MI. MI was also performed on P28 in pigs alone or combined with another apical resection surgery on P1. The in vitro model includes bulk RNA-seq of the hiPSC-CM cells on different days after differentiation. The AI tool identifies clusters of proliferative cells based on RNA-seq analysis of hiPSC-CM 16 days after differentiation, when these cells maintain their proliferative ability, compared to hiPSC-CM 140 days after differentiation, when cells stop proliferating. Upregulated and downregulated mRNA for each model and each cluster were found. Many upregulated genes are related to mitochondrial metabolism, protein biosynthesis, and mRNA modifications or processing. The investigators identified twenty-one overlapping up-regulated genes among all models across three species. Nine coded proteins are associated with ribosomes, including a well-established CM regenerative factor IGF2BP3, HSPA5, DHX9, and BLM, among others. Three genes code classic RNA-binding proteins (DHX9, PTBP3, and IGF2BP3); others are metabolic enzymes, cytoskeleton maintenance, and heat shock proteins, which have been identified as noncanonical RBPs (78). Immunohistochemistry in hiPSC-CM proved overexpression of multiple proteins in pig hearts with proliferating CMs, such as PTBP3, DHX9, DDX6, and HNRNPUL1.
In addition to hESC/hiPSC-CM differentiation and reactivating cardiomyocyte proliferation, direct reprogramming of cardiac fibroblasts into induced cardiomyocytes (iCMs) is another promising strategy for heart regeneration (79, 80). Recent studies reveal new insights into the translational landscape underlying the CF-to-iCM trans-differentiation and reprogramming process through integrative translatomic and transcriptomic profiling (81). A gene-specific targeted loss-of-function screening for translational regulatory factors identified an RNA-binding protein, Y-box binding protein 1 (YBX1), as a critical barrier to iCM trans-differentiation (Figure 5D). In a mouse myocardial infarction-induced heart failure model, reducing Ybx1 expression enhanced CF-to-iCM reprogramming efficiency in vivo, resulting in improved cardiac function and decreased fibrosis and scar size. Ybx1 removal activates the translation of its direct mRNA targets Srf and Baf60c, which directs the effect of Ybx1 depletion on facilitating iCM induction. Depletion of Ybx1 combined with overexpression of a single transcription factor, Tbx5, is sufficient in mediating CF-to-iCM conversion. This strategy simplifies the well-established overexpression of the transcription factor “cocktail”, including GATA4, MEF2C, and TBX5, with a specific stoichiometric ratio (82).
4. Translational Control in Adult Cardiac Disease
4.1. Translational control in cardiomyocyte hypertrophy
The human translation machinery is comprised of three main parts: ribosomes (ribosome proteins: RPs; ribosome RNA [rRNA]), translation factors (initiation and elongation factors), and ARSs and substrate transfer RNAs (tRNAs) (83) (Figure 1). Elevated global mRNA translation and protein synthesis are common features in cardiac hypertrophy (84-86). Hypertrophic stimuli activate mTOR (mammalian target of rapamycin) signaling (87-89), which promotes the synthesis of ribosome proteins and activation of translation factors (90). In hypertrophic CMs, ribosome proteins and rRNAs are markedly increased to support the increase of protein mass and CM size (84, 91-93). Therefore, both the capacity and efficiency of translation are increased in response to hypertrophic stimuli. A few specific translation factors required for cardiac hypertrophy have been defined.
4.1.1. Role of translation initiation factors in cardiac hypertrophy
Theoretically, translation efficiency can be regulated in three steps (i.e., initiation, elongation, and termination). In mammals, translation initiation is a critical control point in most cellular responses (94). Translation initiation is the process in which the assembly of elongation-competent 80S ribosomes occurs at the initiation codon, allowing further elongation to synthesize a full-length protein. Generally, the initiation process comprises two steps: forming 48S initiation complexes and joining 48S complexes with 60S large ribosomal subunits. Translation initiation happens canonically at the 5' end of mRNAs harboring a 7-methylguanosine (m7G) cap, i.e., cap-dependent translation initiation, which requires the cooperation of multiple translation initiation factors (94, 95) (Figure 3). Alterations in translation initiation are a crucial feature of cancer, viral infection, and cardiac hypertrophy (96-98).
The changes in protein synthesis rate in cardiomyocytes during stress-induced cardiac hypertrophy have been shown to correlate with changes in the activity of translation initiation factors, which modulate the rate of translation initiation (99-101). For instance, the activity of translation initiation factor eIF4E is increased upon hypertrophic stimuli in cardiomyocytes in a pressure overload model (98, 102) (Figure 6A). eIF4E is one component of the eIF4F complex that interacts with the m7G cap. When activated, eIF4E facilitates loading the 43S initiation complex onto mRNA to form 48S initiation complexes and promotes ribosome scanning in the 5'-UTR. The activity of eIF4E depends on its phosphorylation (103) (Figure 3). Expression of eIF3E has been shown to increase during both electrical pacing- or α1-adrenoceptor-induced hypertrophy of quiescent neonatal rat CMs (102). Phosphorylation of eIF4E is increased in adult feline CMs in culture during electrical pacing and in canine CMs in vivo after imposition of pressure overload (98). An increase in translation efficiency was concomitant with an increase in eIF4E phosphorylation in hypertrophied CMs in vivo (101). Overexpression of an inactive form of eIF4E slows down protein synthesis and reduces CM hypertrophy (104). However, increased expression or phosphorylation of eIF4E alone cannot induce hypertrophy in non-stimulated CMs (104). Thus, increased eIF4E activity is required for the accelerated rate of protein synthesis upon pressure overload, but increased eIF4E activity alone is insufficient for hypertrophy.
Besides eIF4E, modulated eIF2 activity has also been found after hypertrophic stimuli and other cardiovascular stress conditions (59, 105, 106). eIF2 is essential for forming the eIF2-GTP-Met-tRNAMet ternary complex during translation initiation. It cycles between GTP-bound active and GDP-bound inactive forms through phosphorylation. Phosphorylation of eIF2 locks eIF2 in the GDP-bound inactive form, reducing translation initiation (107). Glycogen synthase kinase 3b (GSK-3β), a kinase for eIF2Bε-S535 phosphorylation, is inhibited in isoproterenol-induced hypertrophied neonatal rat CMs, resulting in decreased eIF2 phosphorylation and accelerated translation initiation. Decreases in eIF2Bε-S535 phosphorylation were also observed in a rat model of cardiac hypertrophy in vivo (105). Overexpression of a consistently phosphor-active eIF2Bε mutant causes hypertrophy growth like isoproterenol treatment. In contrast, a phosphor-inactive eIF2Bε mutant blocks the effect of isoproterenol treatment (105). Therefore, unlike eIF4E, activated eIF2 alone is sufficient to cause CM hypertrophy (Figure 6B). Furthermore, the modulation of phosphorylation of eIF2α has also been found to be related to cardiovascular stress (59, 106). Gcn2−/− mice lacking a key eIF2α kinase were less prone to ventricular dysfunction, myocardial apoptosis, and fibrosis when subjected to transverse aortic constriction (106). A dephosphorylation inhibitor, salubrinal, has been shown to attenuate pressure-overload-induced cardiac hypertrophy (108). Although modulation of translation initiation factor activity correlates with cardiac disease, the global effect and high conservation of these factors among different cell types make it difficult to propose a treatment specific to cardiac disease without affecting other essential biological processes.
In addition to the translation initiation rate, the fidelity of translation initiation is also associated with cardiac function and disease (51). An example of this comes from mitochondrial translation, which synthesizes 13 essential electron transport chain complex component proteins to assemble the oxidative phosphorylation (OXPHOS) system required for energy production. One mitochondrial translation initiation factor, MTIF3, is required for molecular proofreading during the mitochondrial translation process. Loss of MTIF3 will increase the protein synthesis rate at the expense of reduced translation initiation fidelity, resulting in uncoordinated translation of the electron transport chain complex proteins, reducing the correct assembly of the OXPHOS system and ATP production (51). Heart-specific depletion of MTIF3 in mice causes spontaneous dilated cardiomyopathy, probably because of mitochondrial dysfunction in cardiac cells (51).
Apart from the canonical cap-dependent initiation, translation initiation can also happen in an alternative manner at internal ribosome entry sites (IRES) (109, 110). Cap-independent translation initiation is associated with many diseases, including cardiac diseases (109, 111, 112). One example of an IRES is Connexin 43 (Cx43) and its truncated isoform GJA1-20k (113, 114). Cx43 is the most widely expressed gap junction protein translated from an IRES in the 5' uORF (115) , while GJA1-20k is a truncated protein isoform generated from an IRES within the mORF of Cx43 (114). Levels of GJA1-20k regulate the formation of Cx43 gap junctions, which in cardiomyocytes are necessary for the electrical conduction that facilitates heartbeat (111). The IRES activity of GJA1-20k is increased in response to hypoxic conditions in cardiomyocytes (114). Ectopic expression of GJA1-20k has been found to rescue gap junction loss during acute ischemia, proving that modulating alternative translation initiation may protect against loss of electrical coupling, particularly in heart disease (111). Translation of GJA1-20k may be more complicated than “normal” IRES-driven translation since the evidence shows that m7G cap and ribosome scanning are also required (116). The non-canonical translation initiation of GJA1-20k exemplifies the role of alternative translation initiation related to cardiac disease. The mechanism needs to be elucidated regarding regulating the stoichiometric ratio of GJA1-20k to the full-length GJA1 at the translational level under baseline and cardiac stress conditions.
4.1.2. Role of translation elongation factors in cardiac hypertrophy
Although translation initiation has been established as the primary step for translational regulation (94), it is not surprising that translation elongation is also highly regulated since it is the most energy-consuming step during protein synthesis (117). Translation elongation is the process by which ribosomes decode mRNAs by bringing in the proper amino acids carried by aminoacyl-transfer RNAs (aa-tRNA) to form peptides (Figure 1). This process includes the selection of aa-tRNA according to mRNA codon in the ribosome aminoacyl (A) site, peptide bond formation, and movement of both tRNAs and the mRNA through the ribosome (118). A complex set of factors individually or cooperatively affects either the rate or the precision of translation elongation (117). The translation elongation step is affected by modification, abundance, and aminoacylation status of tRNAs, as well as the activity of elongation factors (117). Understanding the regulation of translation elongation is essential since it has been established that dysregulation of elongation is related to human disease (e.g., heart disease (119), tumors (120), neurodegeneration (121)). Evidence showing the correlation between the regulation of translation elongation and cardiac disease is seen by changes in the phosphorylation status of the eukaryotic elongation factor 2 (eEF2) induced by hypertrophic stimuli (122-125) (Figure 6B). eEF2 is a GTPase catalyzing the GTP-dependent ribosome translocation step during translation elongation. eEF2 enters the A-site and induces a conformational change of the ribosome through GTP hydrolysis, allowing the newly formed A-site-bound peptidyl-tRNA and P-site-bound deacylated tRNA to move to the P and E sites. The ribosome can then recruit a new aminoacyl-tRNA into the A-site and continue peptide elongation (126). The activity of eEF2 largely depends on its phosphorylation status (126). Dephosphorylated eEF2 results in eEF2 activation and increases elongation rate, while phosphorylation of eEF2 at Thr56 blocks translation elongation (123). The growth rate of CMs also correlates closely with eEF2 phosphorylation (122). Hypertrophic growth of neonatal CMs induced by angiotensin II is associated with mitogen-activated protein kinase-dependent decreases in eEF2 phosphorylation (123). Decreased eEF2 can be detected within 30 minutes after angiotensin II treatment in isolated neonatal CMs, together with increased protein synthesis, which is believed to be one of the primary causes of cardiac hypertrophy (123). Other hypertrophic agonists, including endothelin-1, phenylephrine, and insulin, are also reported to facilitate dephosphorylation of eEF2 in CMs (124), suggesting that hypertrophy correlates with increased translation elongation caused by eEF2 phosphorylation. However, in another model where isoproterenol was used to induce hypertrophy in isolated adult rat ventricular myocytes, increased eEF2 phosphorylation was observed as early as 1 minute after isoproterenol treatment and remained elevated for 20 minutes (125). The inconsistent results of different models indicate the possibility that modulation of translation elongation during hypertrophy is complex. Isolated pharmaceutical hypertrophy models in vitro may not reflect what happens in vivo. In vivo mouse studies have shown that changes in eEF2 phosphorylation remain undetectable 1 week after isoproterenol injection into adult mice, while eEF2 phosphorylation is significantly increased after transverse aortic constriction surgery (122).
Another example showing the correlation between elongation regulation and hypertrophy comes from eukaryotic elongation factor 1A (eEF1A) (127). eEF1A promotes the GTP-dependent binding of aminoacyl-tRNA into the A-site of ribosomes, allowing downstream interactions to add one amino acid to the elongating peptide. The activity of eEF1A depends on its essential cofactor, eEF1B2 (128). TIP30 has been identified as an endogenous translation elongation inhibitor that interacts with eEF1A to prevent its association with eEF1B2. Tip30+/− and Tip30−/− mice are prone to developing cardiac hypertrophy and heart failure after transverse aortic constriction surgery. Overexpression of Tip30 in neonatal rat CMs restricts hypertrophy induced by hypertrophic growth factors, phenylephrine, or endothelin-1 (127).
Besides cytosolic translation elongation, mitochondrial translation elongation is highly regulated and relevant to cardiac disease. One example of this is the correlation between mitochondrial translation and ischemia (129). Mitochondrial dysfunction is a well-characterized feature of ischemic myocardial injury (130). Translation in the mitochondria produces 13 peptides essential for the electron transport chain and OXPHOS (131). Ischemic stimuli partially affect OXPHOS and energy production by regulating mitochondrial translation elongation. Mechanistically, ischemia increases the phosphorylation of the mitochondrial translational elongation factor Tu (EF-Tu) (129). Unphosphorylated EF-Tu forms a ternary complex with GTP and mitochondrial aminoacyl-tRNAs, facilitating the recruitment of aminoacyl-tRNAs to the A-site of the elongating ribosomes (131). Phosphorylation on Thr382 of EF-Tu inhibits this process by preventing ternary complex formation, thus inhibiting protein synthesis (132). Increased phosphorylation of EF-Tu has been found in mouse hearts subjected to ischemia followed by reperfusion (129). Incubation of isolated non-ischemic mitochondria in cytosol from ischemic hearts increases EF-Tu phosphorylation. In contrast, incubation of mitochondria in cytosol from healthy hearts decreases EF-Tu phosphorylation compared with the absence of cytosol, suggesting a mechanism in the cytosol to prevent EF-Tu phosphorylation and maintain the normal function of mitochondria is inhibited under ischemia conditions (129).
Apart from elongation factors, the rate of protein synthesis is also regulated by the availability of ribosomes, which affects the capacity of translation elongation (117, 119). It has been demonstrated that increased ribosome content is required for the hypertrophic growth of CMs under phenylephrine stimulation (133). Increased transcription of the 45S rDNA (encoding rRNA for ribosome assembly) has been observed in cardiac hypertrophy induced by phenylephrine, endothelin-1, phorbol myristate acetate, angiotensin II, and contraction, partially through increasing RNA polymerase I activity (134-136). Ribosome proteins are also increased to coordinate with the increased rRNAs through enhanced translation. S6K1, a mitogen-stimulated protein kinase downstream of mTOR, has been shown to account for the increased translation of ribosomal proteins (137). S6K1 phosphorylates and activates the 40S ribosomal protein S6, facilitating the translation of multiple ribosome protein mRNAs harboring a 5' polypyrimidine tract (137). Phosphoinositide 3 kinase (PI3K), upstream of S6K1 but downstream of mTOR, has also been shown to promote hypertrophic growth of CMs in a transgenic mouse model (138).
4.1.3. Genetic loss-of-function of mTORC1 causes heart failure in mice
The mammalian target of rapamycin C1 (mTORC1) is a master regulator of cell growth and survival. Activation of mTORC1 induces protein synthesis in response to physiological and pathological stimuli via increased cellular translational activity. Notable dysregulation of translational events indicates the acute cellular response incurred during cardiac stress and remodeling, which ultimately culminates in cardiac diseases or heart failure if it persists (139, 140).
For mTORC1-mediated translational initiation, mTORC1 activation induces the phosphorylation of its major downstream effectors, including ribosomal S6 kinase 1 (S6K1) and eIF4E-binding protein-1 (4E-BP1) (141). S6K1 is required in ribosome biogenesis primarily via positive regulation of pyrimidine biosynthesis and rDNA transcription. Thus, mTORC1 activation through S6K1 can direct rRNA production to synthesize more ribosomes, impacting the protein translational activity (142-144). Whereas 4E-BP1 can bind to the cap-binding protein eIF4E and prevent it from interacting with other translation initiation factors to form the eIF4F complex, which is essential during the formation of the 48S initiation complex in translation initiation (145). In other words, mTORC1 activation increases phosphorylated 4E-BP1, which relieves the inhibitory constraint on eIF4E, culminating in augmented translation initiation.
Figure 6.
Translational control mechanisms in cardiomyocyte hypertrophy. A. Increased expression of eukaryotic initiation factor 4E (eIF4E) promotes global translation to support cardiomyocyte hypertrophy. B. Increased expression of eukaryotic elongation factor 2 (eEF2) enhances global translation to promote cardiac hypertrophy. C. Activation of mTORC1 kinase activity phosphorylates 4EBP1 and S6K1 and enhance eIF4E-mediated cap-dependent translation initiation, rRNA transcription, and ribosome biogenesis, thereby enhancing global mRNA translation and cardiomyocyte hypertrophy. D. Poly(A)-binding protein (PABPC1) upregulation increases global mRNA translation and cardiac hypertrophy.
Figure 6.
Translational control mechanisms in cardiomyocyte hypertrophy. A. Increased expression of eukaryotic initiation factor 4E (eIF4E) promotes global translation to support cardiomyocyte hypertrophy. B. Increased expression of eukaryotic elongation factor 2 (eEF2) enhances global translation to promote cardiac hypertrophy. C. Activation of mTORC1 kinase activity phosphorylates 4EBP1 and S6K1 and enhance eIF4E-mediated cap-dependent translation initiation, rRNA transcription, and ribosome biogenesis, thereby enhancing global mRNA translation and cardiomyocyte hypertrophy. D. Poly(A)-binding protein (PABPC1) upregulation increases global mRNA translation and cardiac hypertrophy.
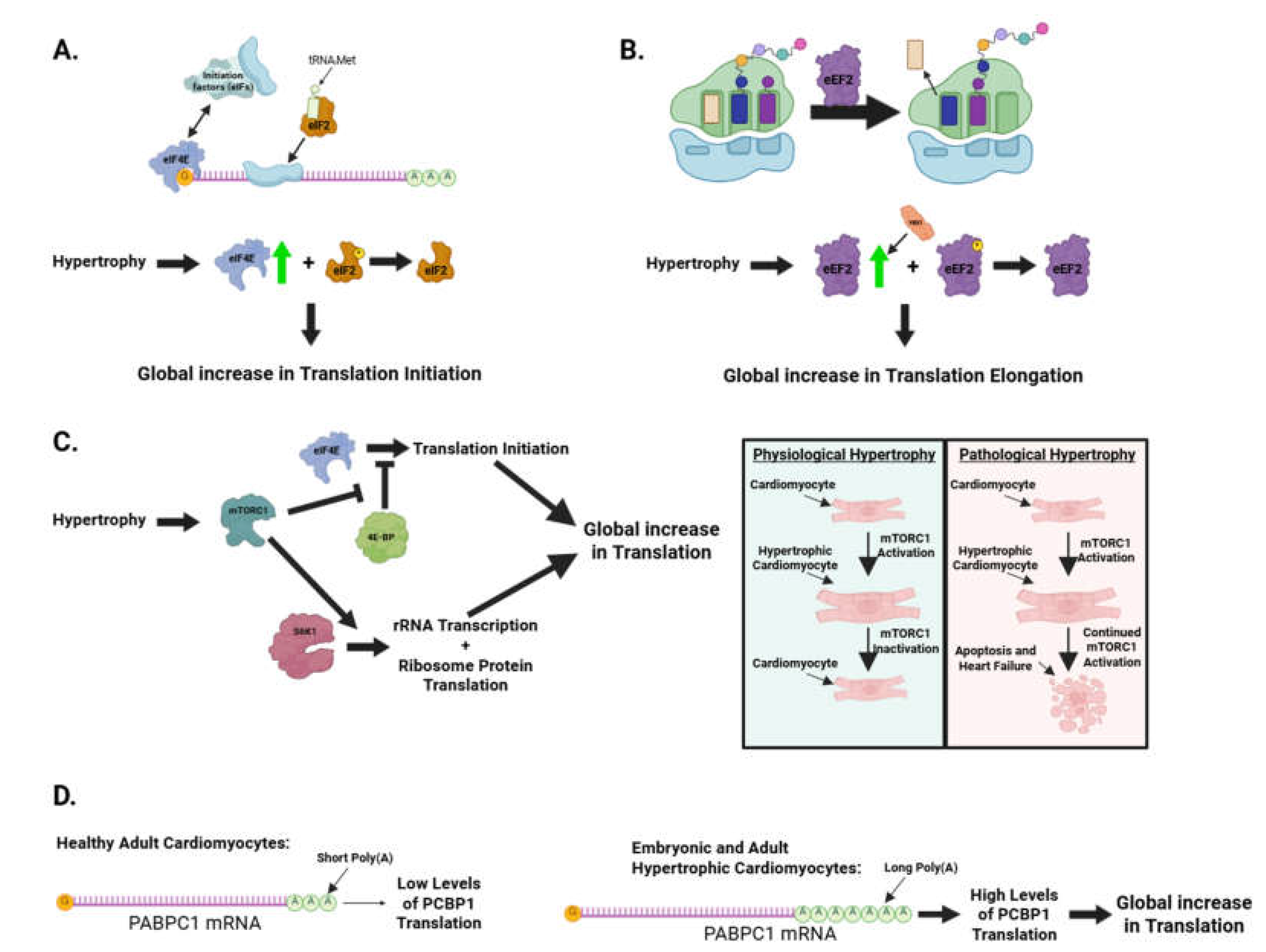
mTORC1 is essential in normal cardiac physiology (Figure 6C). mTORC1 plays a crucial role in CM proliferation and growth during murine heart development (146, 147) despite its initial low expression in the heart at embryonic stages. In adult mice, mTORC1 deficiency mediated by inducible cardiac-specific Raptor and Mtor ablation leads to lethal dilated cardiomyopathy. Raptor ablation in the myocardium results in severe heart failure within 6 weeks of gene deletion. Re-expression of the fetal gene program precedes the cardiac phenotypes, indicating cardiac stress response is incurred by the overexpression of ANP, BNP, and b-MHC in the myocardium (148). Mtor-ablated myocardium demonstrated enhanced apoptosis and impaired mitochondrial function (149). Thus, a negative causal relationship is evident between mTORC1 loss-of-function and cardiac dysfunction. Under pathological stimuli, mTORC1 deficiency impairs the myocardial response to cardiac stress at postnatal stages. Upon pressure overload, reduced mTORC1 activity in Mtor ablated mice hampers the hypertrophic response and accelerates heart failure progression (149). Compared to wild-type mice, Raptor-ablated mice show no cardiac hypertrophy at the organ or tissue level after aortic constriction (148). This supports the hypothesis that mTORC1 inactivation attenuates the translation activity required to induce an adaptive hypertrophic response to preserve cardiac function and integrity upon pressure overload or hemodynamic stress. However, the adaptive hypertrophic response, which is beneficial to cardiac function that requires mTORC1, converts to pathological hypertrophy if the pathological stimuli are sustained. Therefore, selective and partial inhibition of mTORC1 is cardioprotective in response to persistent pathological hypertrophy (140, 150-152). Rapamycin treatment attenuates load-induced cardiac hypertrophy, associated with reduced left ventricular diastolic and systolic diameters and improved cardiac contractile function (150, 151). Rheb1 deletion inhibits mTORC1 activation, conferring this cardio-protective effect in myocardial infarction and transverse aortic constriction mouse models (152). Furthermore, overexpression of PRAS40, an mTORC1 inhibitor, suppresses pathological hypertrophic growth in CMs. PRAS40 gain-of-function mice exhibited decreased heart size, reduced perivascular fibrosis, and preserved systolic function compared to control mice. mTORC1 inhibition mediated by PRAS40 overexpression or rapamycin treatment can effectively regress the established pathological remodeling and hypertrophy in mice with pressure overload (140, 151). Collectively, mTORC1 inhibition may serve as a therapeutic strategy to prevent heart failure progression, and rapamycin is currently tested in clinical trials for treating HFpEF (Figure 6C).
4.1.4. PABPC1-mediated translational control of physiological and pathological cardiac hypertrophy
Poly(A)-binding protein C1 (PABPC1) interacts with the 3' poly(A) tail of mRNAs and the eukaryotic translation initiation factor 4G (eIF4G) to promote mRNA circularization and translation initiation efficiency. PABPC1 protein expression is high in the embryonic heart but becomes undetectable in the adult heart, with no difference at the level of Pabpc1 mRNA in mice (85) (Figure 6D). The Pabpc1 mRNA has a substantially shorter poly(A) tail in the adult heart, leading to extremely low translation efficiency. In contrast, the poly(A) tail length on the Pabpc1 mRNA and the expression level of the PABPC1 protein are restored in adult hearts during cardiac hypertrophy. This mechanism is conserved in both exercise-triggered physiological hypertrophy and heart disease-caused pathological hypertrophy, with the common feature of enhanced protein production in cardiomyocytes. Transgenic overexpression of PABPC1 in adult hearts is sufficient to drive global protein synthesis and cardiac hypertrophy in mice (85).
4.1.5. Translational control of Ybx1 expression regulates cardiac function in response to pressure overload in vivo
YBX1, an RNA-binding protein, is known to regulate translation by binding to target mRNAs. In the hearts undergoing pathological remodeling, the mTORC1 signaling pathway activates the translation of Ybx1 mRNA without altering the transcription level (153). RNA immunoprecipitation (RIP)-seq uncovered Eef2 (eukaryotic translation elongation factor 2) mRNA as a critical target of YBX1. Eef2 mRNA contains a mTORC1-regulated terminal oligopyrimidine motif in the 5'-UTR. eEF2 protein, upregulated during cardiac pathological remodeling, enhances global protein synthesis and promotes cardiac hypertrophy. AAV9-mediated expression of Ybx1 shRNA reduced cardiomyocyte hypertrophy and restored left ventricular contractile function. Therefore, YBX1 is considered a promising therapeutic target, as inhibiting or reducing YBX1 expression in cardiac fibroblasts and myocytes compromises cardiac fibrosis (81) and hypertrophy (153).
4.1.6. m6A-dependent translational control in maintaining normal cardiac function
Chemical modification of N6-methyladenosine (m6A) in mRNAs regulates cardiomyocyte function by controlling mRNA stability or translatability. Increased global m6A levels have been identified as a shared feature in heart failure with different etiologies. m6A reader proteins orchestrate gene expression in cardiac pathological remodeling. For instance, a key m6A reader protein, Ythdf2, controls cardiac function. Genetic deletion of Ythdf2 in cardiomyocytes in mice causes mild cardiac hypertrophy, increased fibrosis, and reduced heart function upon pressure overload stress and during cardiac aging (154). The eukaryotic elongation factor 2 (eEF2) is post-transcriptionally regulated by YTHDF2 as indicated by cell-type-specific ribosome profiling analysis. Another study identifies that m6A methylation is directly associated with the progression of heart failure in both humans and mice. In this study, they identified a transcription-independent mechanism of mRNA translation regulation, where alteration of m6A methylation on mRNAs results in differential polysome occupancy and changes in protein expression. Furthermore, RNA demethylase Fto knockout in cardiomyocytes in mice shows impaired heart function, suggesting mRNA m6A methylation as a potential target for therapeutic interventions (155).
4.2. Translational control in cardiac fibroblast activation during fibrosis
4.2.1. Translational regulation in human TGFβ-activated cardiac fibroblasts
Fibrosis is a substantial symptom in multiple types of heart diseases and many other organ disorders. It can occur in inherited cardiomyopathy, ischemic heart disease, HFpEF, diabetes, and aging (156, 157). Cardiac fibrosis is primarily caused by excessive extracellular matrix accumulation secreted by highly specialized myofibroblast cells (157). Myofibroblasts are generated through a process called myofibroblast transformation, wherein quiescent fibroblasts in the myocardium transdifferentiate into active myofibroblasts upon profibrotic stimuli such as transforming growth factor β (TGFβ), angiotensin II, endothelin-1, or serum response factor (157). TGFβ has been identified as a primary and potent mediator of myofibroblast transformation. The induction of myofibroblast transformation by TGFβ appears to be a universal phenomenon across different organs (27, 157, 158). Expression of TGFβ is upregulated upon injury, and the protein is subsequently secreted into the ECM, where it binds to either type I or type II TGFβ receptors on the surface of fibroblasts (157, 158). The promotion of myofibroblast transformation by TGFβ primarily depends on the activation of profibrotic gene transcription either through phosphorylation of transcription factor SMAD2/3 (canonical signaling) or mitogen-activated protein kinase (MAPK) signaling (non-canonical signaling) (157). The significant effector downstream of TGFβ, interleukin-11 (IL-11), is related to the translational regulation of profibrotic gene expression required for myofibroblast transformation (27).
IL-11 is a hematopoietic IL-6 family cytokine with pleiotropic effects. Obana, M. et al. first demonstrated its correlation with cardiac fibrosis by showing that IL-11 was upregulated at the mRNA level in a mouse model 1 day after MI (159). They also suggested administering recombinant human IL-11 to mice after an MI operation, which reduced cardiac fibrosis at day 14 (159). However, Schafer S. et al. strongly challenged this opinion, suggesting mice cannot respond to human IL-11 (27). In contrast, Schafer S. et al. suggested that IL-11 contributes to cardiac fibrosis downstream of TGFβ. In their studies, IL-11 increased the expression of profibrotic proteins without changing the mRNA level, indicating translational or at least posttranscriptional regulation. IL-11 was also knocked down or overexpressed in a mouse model undergoing MI to show its induction of fibrosis. However, they did not elucidate the mechanism of profibrotic protein translation induced by IL-11. Although there are still arguments supporting the inhibiting effect of IL-11 on fibrosis, IL-11 is more accepted as a contributor to cardiac fibrosis and other organ aging processes with the potential to become a drug target, according to recent pre-clinical studies (27, 160).
Since human TGFβ plays a pleiotropic role in inducing various pathological conditions, unraveling the downstream key regulator in driving the pathogenesis of cardiac fibrosis is important. A comprehensive computational study (161) using RNA-seq and Ribo-seq databases of human heart samples is consistent with previous observations (162), wherein the incurred cardiac fibrotic response is reflected by the modulation of about a third of all genes at the translational level. Hence, understanding the molecular mechanism of action underlying fibroblast activation into myofibroblasts is of great interest for more effective and specific anti-fibrotic therapy development.
During pro-fibrotic response induced by TGFβ in primary human atrial cardiac fibroblasts, ribosome profiling reveals 1691 differentially translated mRNAs (139). Specifically, the translating ribosome is augmented in mRNAs of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1), while it is less pronounced in integrin subunit alpha 3 (ITGA3), although the transcript abundance remains unchanged. Intriguingly, the effect of pro-fibrotic response on translational dynamics is the most prominent at 24 hours despite a gradual decrease at 2 and 6 hours after TGFβ induction. Differentially transcribed genes and translated mRNAs imply that the profibrotic response induced by TGFβ alters mRNA abundance and translation efficiency differently. Hence, these can be further categorized into several distinct gene expression regulations: forwarded, exclusive, buffered, and intensified. Forwarded genes have unchanged ribosome occupancy without mRNA level alteration. Exclusive genes have altered ribosome occupancy but unchanged mRNA levels. Both ribosome occupancy and mRNA levels are modified in buffered and intensified genes. For example, gene expression regulation of transcription factor HES1 is known as an intensified gene due to even more robust protein upregulation than would be expected by qPCR or RNA sequencing data alone. The upregulation effect of the translational level culminates in a prominent increase in the HES1 protein level beyond the transcriptional surge.
4.2.2. EPRS1 promotes cardiac fibrosis by enhancing proline-rich extracellular matrix protein translation
Cytoplasmic glutamyl-prolyl-tRNA synthetase (EPRS1), the only bi-functional ARS in higher eukaryotic organisms (from C. elegans to humans), catalyzes the attachment of two amino acids, glutamic acid (E) and proline (P), to their cognate tRNAs for protein synthesis. In addition to EPRS1’s canonical aminoacylation function in translation, our previous work and others’ studies have shown that EPRS1 exerts a noncanonical function in translational silencing of inflammation-related mRNAs (e.g., VEGFA) in monocytes or macrophages as an anti-inflammatory response (12, 163-166). Hypoactive mutations in the PRS (prolyl-tRNA synthetase) domain of EPRS1 lead to hypomyelinating leukodystrophy (167) without causing cardiac dysfunction in patients, implying that reduced PRS enzymatic activity does not adversely affect hearts.
The role of cardiac EPRS1 in the heart has been recently elucidated. EPRS1 is the sole common ARS gene, containing single nucleotide polymorphisms (SNPs) associated with congenital heart disease in humans (168), TGFβ-induced ARSs in human cardiac fibroblasts (8), mouse GWAS of isoproterenol-induced cardiomyopathy model (169), and isoproterenol-induced ARSs in mouse failing hearts (170). Our work shows that EPRS1 is induced in cardiac fibrosis triggered by multiple pro-fibrotic and pro-hypertrophic stimuli (e.g., isoproterenol, TGFβ, IL-11) as an integrated node for translational control in human and mouse cardiac remodeling and fibrosis (22). EPRS1 activation contributes to elevated translation of proline-rich mRNAs via enhanced translation elongation in cardiac fibroblasts (Figure 7A).
Figure 7.
The mechanisms of dysfunctional aminoacyl-tRNA synthetases in cardiac fibrosis. A. Increased glutamyl-prolyl-tRNA synthetase (EPRS1) and hyperactive eIF5A are essential for proline-rich pro-fibrotic extracellular matrix protein translation elongation. B. Editing domain-inactivating mutation C723A in alanyl-tRNA synthetase (AARS1) causes mis-incorporation of incorrect serine amino acid to alanine codons in the proteome in the cardiac muscle, leading to severe cardiac fibrosis.
Figure 7.
The mechanisms of dysfunctional aminoacyl-tRNA synthetases in cardiac fibrosis. A. Increased glutamyl-prolyl-tRNA synthetase (EPRS1) and hyperactive eIF5A are essential for proline-rich pro-fibrotic extracellular matrix protein translation elongation. B. Editing domain-inactivating mutation C723A in alanyl-tRNA synthetase (AARS1) causes mis-incorporation of incorrect serine amino acid to alanine codons in the proteome in the cardiac muscle, leading to severe cardiac fibrosis.
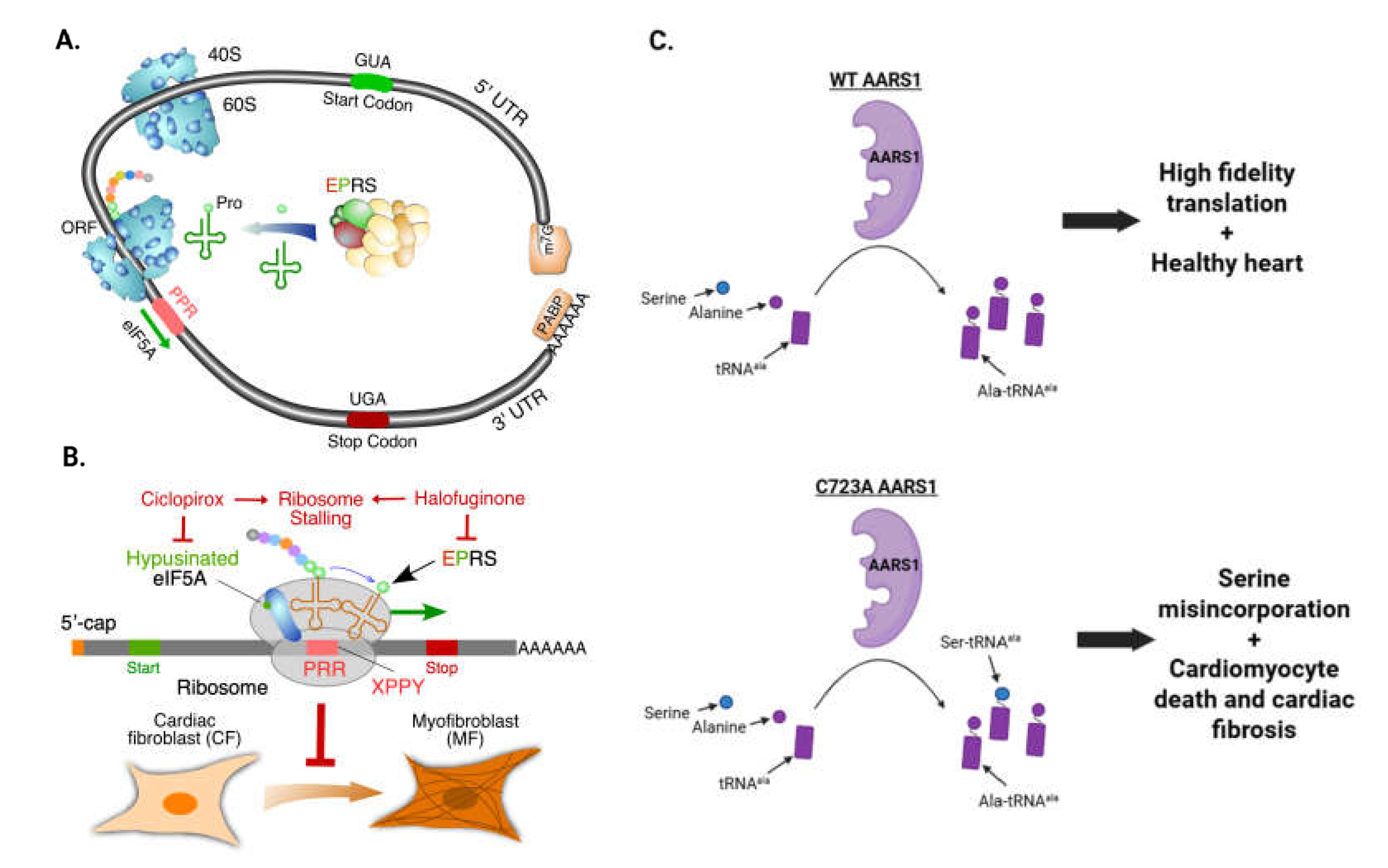
EPRS1 expression is correlated with the progression of cardiac fibrosis (22). Elevated EPRS1 expression was observed in human hearts with dilated cardiomyopathy and ischemic heart failure. A similar elevation in EPRS1 was found in multiple HF mouse models, including isoproterenol-driven neurohormonal stimulation, transverse aortic constriction triggered pressure overload, and myocardial infarction with ischemic and inflammatory stresses. A reduced EPRS1 level in haploinsufficient (Eprs1+/−) mice and tamoxifen-inducible, myofibroblast-specific Eprs1 conditional knockout mice reduces cardiac fibrosis and partially restores left ventricular contractile function in vivo. Using the PRS-specific inhibitor halofuginone, EPRS1 was shown to be essential for the translation of many proline-rich mRNAs in cardiac fibroblasts, including many collagen proteins and secretory signaling proteins (e.g, LTBP2 and SULF1), among other profibrotic ligands, receptors, and enzymes (Figure 7B). These genes are considered novel anti-fibrotic targets for developing therapeutic approaches to treat heart failure with severe cardiac fibrosis. Interestingly, multiple pro-fibrotic (e.g., TGFβ and IL-11) and pro-hypertrophic agents (e.g., isoproterenol and angiotensin II) induce transcription of EPRS1 mRNA. Possibly, extracellular signals activate the EPRS1 expression and promote pro-fibrotic protein expression and cardiac scar formation. The reduced level of charged Pro-tRNAPro following the reduction of EPRS1 activity or amount might, in turn, cause ribosome stalling and collisions on proline-rich codons and enhance no-go mRNA decay due to reduced ribosome occupancy (171). Although EPRS1 is ubiquitous in all cells, proline-rich transcripts will likely vary in different cell types. Thus, halofuginone is likely to cause mRNA-selective and cell-type-specific translational repression dependent on the heterogeneity of proline-rich proteome across distinct cell types, such as cardiomyocytes (172).
4.2.3. eIF5A: an anti-fibrosis target translation elongation factor
The biosynthesis of poly-proline peptide-containing proteins tends to cause ribosome stalling and translation arrest because of slow peptide bond formation (173, 174). A specialized translation elongation factor P (EF-P) overcomes this ribosome stalling effect in bacteria (173). As a homolog of EF-P, eIF5A (eukaryotic initiation factor 5A, it is actually “elongation factor 5, EF5”) binds to the stalled ribosome between the peptidyl-tRNA binding and the tRNA-exiting sites, and facilitates peptidyl-transferase activity, thus resuming translation at poly-proline codons among other proline-rich codon motifs (174). Eleven proline-proline dipeptide-containing motifs (XPPY) are among the top motifs for eIF5A-mediated ribosome readthrough in eukaryotic cells (174). Hypusination of an evolutionarily conserved lysine residue in EF-P or eIF5A is essential for its biological function (175). eIF5A inhibitors have also been tested in pre-clinical models for treating cancer and metabolic syndrome (176, 177). eIF5A may serve as a novel anti-fibrotic therapeutic target, and eIF5A inhibitors can be used to inhibit organ fibrosis in the future. For example, an antibiotic, ciclopirox, was repurposed to reduce cardiac fibrosis by inhibiting eIF5A activity in animal models of heart failure, following myocardial infarction (178) (Figure 7B).
4.2.4. Editing-defective Aars1 mouse shows spontaneous cardiac proteinopathy and fibrosis
In the previous sections, we discussed how quantitative changes in the global proteome or specific fibrotic proteins may affect cardiac function and cause heart disease. In addition to protein abundance, protein sequence and folding accuracy are also essential. Mistranslated or misfolded proteins are an emerging hallmark of multiple human diseases, including cardiac disease (179, 180). Accumulation of misfolded proteins results in protein aggregation, leading to cell dysfunction and death. In most cases, misfolded protein comes from genetic mutations wherein missense mutations in the exon of one specific gene will lead to the misfolded product of that protein. However, misfolded proteins can also be generated by inaccurate translation (181-183).
In eukaryotic cells, translation is strictly supervised to ensure fidelity. The correct translation of proteins requires the addition of the correct amino acid to their tRNA by a specific aminoacyl-tRNA synthetase and selecting the correct aminoacyl-tRNA according to the elongation cycle. Adding the proper amino acid to the tRNA largely depends on the editing activity of aminoacyl-tRNA synthetases (ARSs), which ensures accurate cognate tRNA aminoacylation (181, 184). Although errors in aminoacylation are relatively low, even small decreases in the editing activity of ARS dramatically affect cell survival (185-188).
Deficiency in the editing activity of ARS has been found to produce misfolded proteins associated with several diseases, including heart disease. One example of this comes from the editing-defective alanyl-tRNA synthetase (AARS1), which is caused by the introduction of missense mutations in the editing domain of AARS1 (182) (Figure 7C). Cys723, an essential amino acid in the editing domain of AARS1, was mutated into Ala, leading to excessive production of mischarged Ser-tRNAAla instead of Ala-tRNAAla, as demonstrated in vitro. When introduced in vivo, this editing-defective mutation resulted in the production of misfolded protein and increased cell death. Furthermore, Aars1C732A/C732A mice were found to be embryonic lethal, while Aars1C732A/+ mice showed extensive age-related cardiac fibrosis and CM death. Although the mechanism behind these phenotypes is not fully understood, and no human editing function mutations have been identified, these animal studies suggest that editing activity is essential for the survival of multicellular organisms and provide evidence for the correlation between translation fidelity and cardiac disease in rodents.
5. Translation-Manipulating Therapeutics for Heart Disease Treatment
5.1. Translation-targeted medicines for cardiac disorders
Cardioprotective roles of mild translational inhibition in heart failure: Since elevated global mRNA translation was observed in cardiac remodeling (84, 86, 91), scientists have taken multiple measures to block cardiac protein synthesis for heart failure treatment. Contrary to the expectation, rabbits treated with puromycin, a global translation inhibitor, or fed with a protein-free diet manifested accelerated heart failure progression in a heart disease model (189). These detrimental effects probably result from the robust translational inhibitory effect of puromycin and complete amino acid starvation, indicating a need for mild and selective translation reduction strategies. Consistent with this concept, rapamycin, a small molecule inhibitor of mTORC1, can attenuate transverse aortic constriction-induced cardiac hypertrophy in mouse models through S6K suppression (150), which implies that mild translational inhibition is a viable therapeutic strategy to antagonize pathogenic cardiac remodeling. However, inhibition of the mTORC1 pathway causes adverse immune-suppressant effects and may not be a safe approach to long-term anti-fibrotic treatment. Nevertheless, rapamycin has been tested in a phase I clinical trial to treat patients with heart failure with preserved ejection fraction (ClinicalTrials.gov ID: NCT04996719).
A prolyl-tRNA synthetase-specific inhibitor, halofuginone (190, 191), is derived from an anti-malaria Chinese herbal medicine, Chang Shan. Halofuginone blocks the binding of proline and tRNAPro to (E)PRS and prevents the ligation of the amino acid to its cognate tRNA (190, 191). Halofuginone has been shown to reduce collagen expression (190) and skeletal muscle fibrosis in Duchenne Muscular Dystrophy (DMD) (HT-100, delayed-release halofuginone from Akashi Therapeutics) in phase II clinical trials. Halofuginone protects the heart from multiple cardiac stresses in pre-clinical mouse heart failure models of angiotensin II infusion, transverse aortic constriction-induced pressure overload, and ischemia-reperfusion (192). Halofuginone has two significant cardioprotective effects: 1) it promotes cardiomyocyte autophagy and cell survival, and 2) it reduces extracellular matrix protein expression and inhibits cardiac fibrosis (192). The therapeutic mechanism of halofuginone has been studied at the transcriptional level to trigger an amino acid starvation response by inhibiting the incorporation of proline into the protein synthetic chain (190, 192, 193). Our group studied its direct impact on the translation of pro-fibrotic genes and anti-fibrotic effects in vivo (22). General translation factors are well-established antibiotic targets for eradicating infectious microorganisms (163). We proposed that mild translational inhibition using an EPRS1-specific inhibitor and genetic knockout of one allele of the Eprs1 gene can effectively reduce cardiac remodeling and fibrosis by specifically inhibiting proline-rich mRNA translation, while preserving global mRNA translation (non-proline-rich genes). This new therapeutic approach targeting evolutionarily conserved, ubiquitous translation factors can be used to treat cardiac fibrosis of multiple etiologies and is generalizable to treating fibrosis from various organs.
In addition, the integrated stress response inhibitor (ISRIB), an eIF2B complex inhibitor that inhibits ISR, can protect hearts from ischemic stress and post-infarct atrial fibrillation (194). In another study, ISRIB suppressed hyperlipidemia-induced inflammasome activation and inflammation, thereby reducing atherosclerosis (195). Intriguingly, the mechanism of action for ISRIB is opposite to halofuginone in terms of activation of the ISR. Rigorous clinical studies are required to evaluate any potential beneficial effects of ISRIB in heart disease patients and whether the cardioprotective benefit results from inhibition of ISR.
5.2. RNA secondary structure as a therapeutic target for ASO treatment of cardiac hypertrophy
The underlying molecular mechanisms governing uORF regulation within cells remain enigmatic. Our lab has identified a double-stranded RNA (dsRNA) structure within the GATA4 5' uORF, enhancing uORF translation while concurrently inhibiting mORF translation (11) (Figure 8A, B). We demonstrated that antisense oligonucleotides (ASOs) designed to disrupt this dsRNA structure effectively promoted mORF translation. Conversely, ASOs designed to base-pair immediately downstream, forming a bimolecular double-stranded region of either the uORF or mORF start codon, enhanced the translation of their respective sequences (Figure 8B). In human CMs, employing GATA4 uORF-enhancing ASOs reduced levels of cardiac GATA4 protein and increased resistance to CM hypertrophy (Figure 8, 9). Conversely, the inhibitory ASOs induced cellular shrinkage, in line with the observed GATA4 loss-of-function phenotypes. Our findings extend beyond GATA4, demonstrating the broad applicability of uORF-dsRNA- or mORF-targeting ASOs in regulating translation for other mRNAs. This research unveils a novel regulatory paradigm that governs translational regulation and offers a valuable strategy for modulating protein expression and cellular phenotypes by selectively disrupting or forming dsRNA structures downstream of uORF or mORF start codons.
Figure 8.
uORF-dsRNA modulates mORF translation of GATA4 mRNA and cardiomyocyte hypertrophy. A. Double-stranded RNA downstream of uAUG or mGUG activates GATA4 uORF translation and mORF translation of eIF4G2. B. Left panel: cis- and trans-acting molecular mechanisms regulating the translational balance between GATA4 uORF and mORF. Middle panel: physiological function of uORF in human induced embryonic pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) and mice (using CRISPR-Cas9-mediated gene editing to introduce ATG-to-TTG knock-in mutation). Right panel: therapeutic intervention of cardiac hypertrophy by nanoparticle embedding antisense oligonucleotides (ASOs) targeting GATA4 uORF-dsRNA. uORF translation is enhanced while mORF translation is inhibited, thereby GATA4 protein level decreases, and transverse aortic constriction surgery-induced cardiac hypertrophy is compromised.
Figure 8.
uORF-dsRNA modulates mORF translation of GATA4 mRNA and cardiomyocyte hypertrophy. A. Double-stranded RNA downstream of uAUG or mGUG activates GATA4 uORF translation and mORF translation of eIF4G2. B. Left panel: cis- and trans-acting molecular mechanisms regulating the translational balance between GATA4 uORF and mORF. Middle panel: physiological function of uORF in human induced embryonic pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) and mice (using CRISPR-Cas9-mediated gene editing to introduce ATG-to-TTG knock-in mutation). Right panel: therapeutic intervention of cardiac hypertrophy by nanoparticle embedding antisense oligonucleotides (ASOs) targeting GATA4 uORF-dsRNA. uORF translation is enhanced while mORF translation is inhibited, thereby GATA4 protein level decreases, and transverse aortic constriction surgery-induced cardiac hypertrophy is compromised.
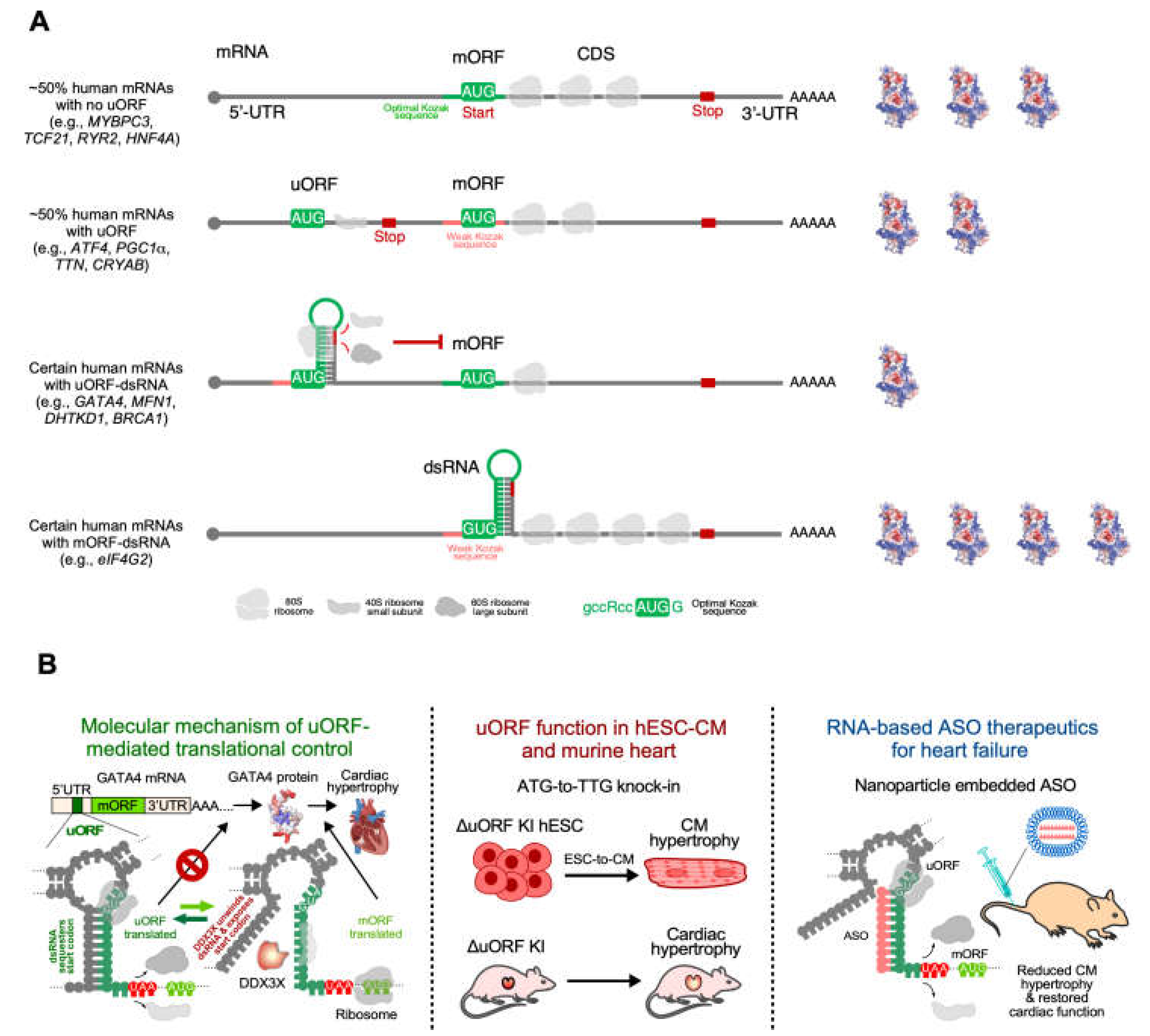
In the future, several remaining questions need to be addressed: 1) What distance and length rules govern the structural and functional features of uORF-dsRNA elements? 3) What trans-acting factors and pathological stress stimuli regulate uORF-dsRNA activity? 4) What is the in vivo significance of uORF and dsRNA elements in translating GATA4 mRNA and cardiac pathophysiology? Answers to these questions will help us understand how uORFs are involved in downstream mORF translational control and how translational control of GATA4 (or other proteins) is vital to normal development or pathogenic processes. Our recent findings reveal that a CRISPR-Cas9-mediated ATG-to-TTG followed by a PAM (protospacer adjacent motif) mutation (CTG-to-CTC) global knock-in mouse model shows spontaneous cardiac hypertrophy without significant fibrosis after 6-7 months after birth. Single-nucleus RNA-seq and ATAC-seq (Assay for Transposase-Accessible Chromatin) dual-omics analysis confirms the upregulation of GATA4 target gene transcription, including Ttn, Tmx1, Myh6, Actc1, and Mybpc3 (196).
Interestingly, a contemporary study showed a similar uORF-dsRNA-mediated translation regulatory mechanism acting in plants and human cancer cells to control the protein synthesis of stress-responsive factors and tumor suppressor genes, respectively (197). As an evolutionarily conserved molecular mechanism, similar observations were reported for alternative upstream translation initiation at near-cognate start codons in yeast (198) and repeat-associated non-AUG (RAN) translation of the GC-rich dsRNA in human diseased brains with genetic expansion of gain-of-function toxic hexanucleotide GGGGCC repeats (199).
5.3. Chemically modified mRNA-based CAR-T-mediated therapeutics for cardiac fibrosis
Cardiac fibrosis, indicated by stiffening and scarring of heart tissues, is a hallmark of most heart diseases and is considered a primary cause of mortality. Cardiac fibroblasts undergo proliferation upon heart injury, such as acute myocardial infarction and chronic hypertension, and are activated to differentiate into myofibroblasts. Myofibroblasts secrete and deposit excessive amounts of extracellular matrix proteins, leading to increased stiffness and reduced compliance of the cardiac tissue. Prolonged cardiac fibrosis is a key contributory factor in the progression of various forms of cardiac disease and consequent heart failure. Currently, limited effective clinical interventions and therapies exist to target fibrosis. A recent conceptual advance demonstrates the efficiency of reprogrammed T-cell immunotherapy to target pathological cardiac fibrosis in mice specifically (200). Engineered mouse cardiac fibroblasts expressing a xenogeneic antigen protein can be effectively targeted and eradicated by transferring antigen-specific CD8-positive T cells. An endogenous target of cardiac fibroblasts, fibroblast activation protein (FAP), was discovered by transcriptome profiling-based gene signature analysis of the heart tissues obtained from healthy control, dilated cardiomyopathy, and hypertrophic cardiomyopathy patients. Adoptive transfer of T cells expressing a chimeric antigen receptor (CAR-T) against FAP results in a substantial reduction in cardiac fibrosis and restoration of left ventricle contractile function in mice treated with angiotensin II and epinephrine. These findings provide an innovative approach to developing immune therapies for treating heart disease.
The researchers can generate therapeutic CAR-T cells in vivo by introducing CD5-targeted lipid nanoparticles bearing pre-designed instructive mRNAs required to reprogram T lymphocytes. Preclinical testing in a heart disease mouse model demonstrated that the approach successfully reduced cardiac fibrosis and restored heart function. The potential to generate CAR-T cells in vivo using modified synthetic mRNAs may have many therapeutic applications. However, one of the caveats lies in the long-term side effects of CAR-T attacking physiological fibrosis processes such as wound healing and skeletal regeneration. To avoid the potential systematic off-target effects and related toxicity, the same group of researchers developed an improved immunotherapy strategy to produce transient anti-fibrotic CAR-T cells that can recognize the cardiac fibroblasts in the mouse hearts by delivering chemically modified mRNA in T cell-targeted lipid nanoparticles (201) (Figure 9). The efficacy of the reprogrammed CAR-T cells was evaluated in vivo by injecting CD5-targeted lipid nanoparticles into a neurohumoral agonist-induced heart failure mouse model. Efficient lipid nanoparticle-directed delivery of modified mRNA encoding the CAR to T lymphocytes generated transient but effective CAR T cells in animals. These CAR-T cells promoted trogocytosis and retained the specific target antigen protein as they accumulated in the spleen. Treatment with the engineered mRNA-targeted lipid nanoparticles reduced fibrosis and partially improved cardiac function in mouse heart failure models. As a generalizable therapeutic platform, this method of in vivo generation of CAR-T cells is promising for treating various human diseases related to organ fibrosis. At the technical level, more strategies need to be developed to increase the translation efficiency further and allow minimal mRNA to produce the maximal amount of therapeutic proteins, such as multi-capped mRNA technology (202).
5.4. Chemically modified mRNA-based therapeutics for ischemic heart disease
mRNA carries genetic codon-based instructions transcribed by RNA polymerases from DNA, which ribosomes then translate to produce proteins. Proteins are the primary bioactive molecules directing various biochemical and biological functions in cells. Pfizer and Moderna’s pioneering mRNA therapeutics are designed to utilize cellular translation machinery to produce specific medicinal proteins (Figure 9), such as the VEGF-A protein. Vascular endothelial growth factor A (VEGF-A) is a paracrine cytokine required for new blood vessel formation. It can stimulate the proliferation of epicardial progenitor cells, which differentiate into endothelial cells that facilitate heart repair by promoting angiogenesis and blood flow. Therefore, VEGF-A has therapeutic effects on the cardiovascular system, but insufficient delivery is a significant challenge that impedes clinical development. AstraZeneca and Moderna have developed VEGF-A mRNA therapeutics for treating myocardial infarction patients. A Phase II clinical study, EPICCURE, was completed using modified mRNA encoding VEGF-A (AZD8601) without lipid encapsulation for injection into the hearts of patients with reduced left ventricular ejection fraction and coronary artery bypass surgery (203). AZD8601 met the primary endpoint of safety and tolerability in heart failure patients with no arrhythmias, mortality, and mRNA-triggered immune responses. Positive outcomes suggest improvement in left-ventricular ejection fraction and pro-B-type natriuretic peptide levels. However, this study is limited in size, and future clinical trials are required to confirm efficacy and safety with a larger patient cohort.
Figure 9.
RNA-based translation-manipulating therapeutics for treating heart disease.
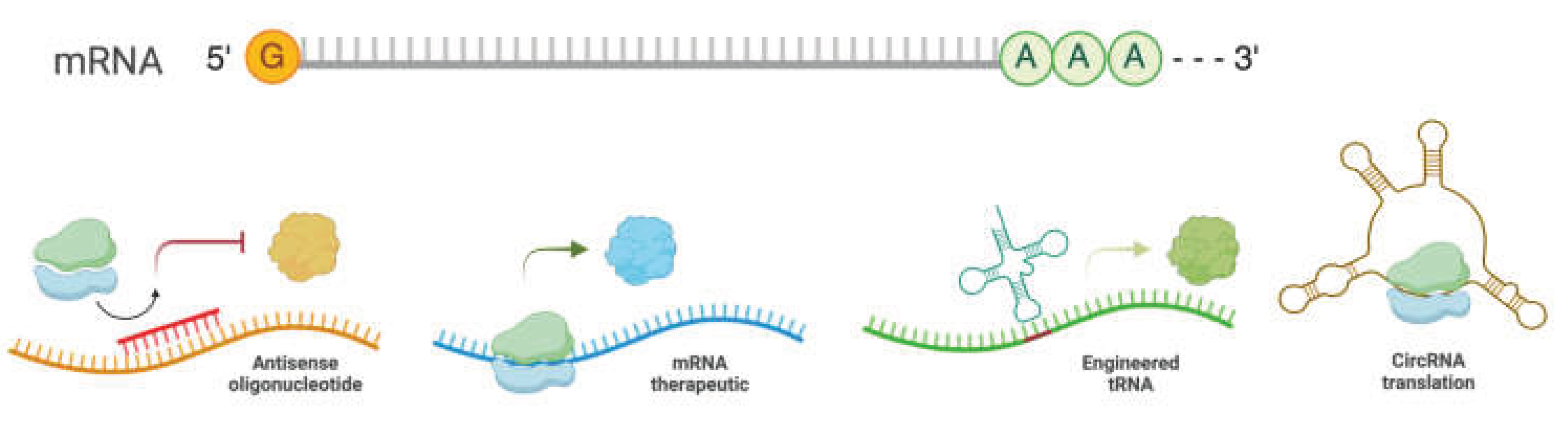
Cardiomyocyte-specific overexpression of CCND2 (cyclin D2) has been reported to promote recovery from cardiac injury in mammalian models of heart failure triggered by myocardial infarction. Modified mRNA of CCND2 was recently reported to activate the cell cycle of CMs in hearts under myocardial infarction injury in small and large mammalian hearts, such as mice and pigs (53). The CM-specific modified CCND2 mRNA translation system (CM SMRTs) contains two modRNA modules. One modRNA encodes CCND2 protein and bears an L7Ae-binding site. The other modRNA encodes L7Ae (an archaea large ribosome protein that recognizes and binds to kink-turn motifs in RNA) and possesses the specific recognition elements for the CM-specific microRNAs miR-208 and miR-1. Therefore, L7Ae is silenced by highly abundant endogenous miR-1 and miR-208 in CMs, while it inhibits CCND2 mRNA translation in non-CM cells. This bidirectional regulatory effect from the modRNA drives CM-specific CCND2 expression. Intramyocardial injections of the CM CCND2 SMRTs significantly enhanced CM proliferation and reduced infarction in both MI models of mice and pigs. They increased the left ventricular ejection fraction in animals compared to the control group with myocardial infarction surgery. The long-term goal is to advance mRNA technology and the ability to engineer different mRNAs as payloads to produce therapeutic proteins for disease treatment.
6. Concluding Remarks and Future Perspective
Along with transcription and epigenetic regulation, signal transducers, sarcomere network, protein modifications and degradation, and translational control are crucial for cardiac health and disease. Hyperactive translation at the transcriptome level or in a specific cohort of mRNAs encoding pathogenic proteins in cardiomyocytes and cardiac fibroblasts promotes cardiac hypertrophy and fibrosis, which drive the progression of pathological cardiac remodeling and heart failure. Conversely, hypoactive translation activity in the fetal heart leads to decreased cardiac cell proliferation and differentiation, causing developmental defects during embryonic stages and resulting in congenital heart disease. Understanding how translational control operates in cardiac pathological remodeling has significant implications for developing new treatments to combat heart diseases.
Global translation inhibition, such as with mTOR inhibitors, can reduce cardiomyocyte hypertrophy and fibrosis by silencing the production of pro-hypertrophic and pro-fibrotic proteins involved in sarcomere and extracellular matrix formation. However, potent inhibition of protein synthesis may lead to unpredictable cytotoxicity and side effects. To achieve a safer therapeutic approach, it is important to target more specific translation factors or RBPs to suppress the translation of pathogenic factors or optimize dosage and treatment duration. Alternatively, transcript-specific translational regulation can be targeted for manipulation of protein synthesis for one or multiple selected mRNAs. This method can further reduce unintended off-target effects. Using ASO that bind precisely to a unique site on the mRNA prevents specific RBPs from interacting with the target mRNA. This approach modulates the translation efficiency of the targeted mRNA for therapeutic purposes (13).
Among more than a thousand classical RNA-binding proteins, specific cohorts of RBPs can bind to either single-stranded RNA (e.g., linear motif) or double-stranded RNA (e.g., structural element). Secondary and tertiary structures formed by dsRNA elements are critical in processing, stability, translation, and interaction with RBPs. Understanding the structure of RNA can provide valuable insights into RNA-protein interactions and RBP-mediated mechanisms of translational regulation. It can also guide the design, screening, and development of RNA-based therapeutic compounds, including ASOs, siRNAs, miRNAs, and mRNAs. To determine RNA structures, a chemical probing method, SHAPE-MaP (Selective 2'-Hydroxyl Acylation analyzed by Primer Extension and Mutational Profiling), can be employed to measure RNA flexibility and conformation at a single-nucleotide resolution. In single-stranded regions, 2-methylnicotinic acid imidazolide (NAI) forms an adduct with free 2'-OH groups on the RNA sugar backbone. Adduct formation occurs at a 1-3% rate and further induces cDNA mutations during reverse transcription. Mutation rates above DMSO control are calculated into reactivity scores that guide RNA folding using publicly available web tools, such as RNAStructure or RNAFold. Thus, a comprehensive atlas of RNA secondary structures can be drawn as a blueprint for searching target sites for drug targeting to manipulate mRNA translation. For instance, transcriptome-wide uORF-dsRNA elements need to be precisely mapped in human or mouse cells to uncover novel therapeutic targets for ASO or siRNA. Based on Ionis’ pioneering work and our studies (11, 204), novel translation-manipulating ASO-based therapeutics can be developed for treating diseases across multiple organs, including the heart and blood vessels. A significant hurdle for RNA-based treatment of cardiovascular disease is the cardiac cell type-specific delivery of RNA therapeutics. There is an urgent need to invent novel RNA drug delivery tools with high uptake efficiency, cell-type specificity, low immunogenicity, and minimal interference with RNA drug efficacy, such as lipid nanoparticles and RNA aptamers (205-207). In the long term, we hope to generalize and apply translation-manipulating ASOs in different cell types, organs, and disease models.
In addition to ASO (11, 204), multiple promising RNA-based translation-modulating therapeutic strategies are emerging, including tRNA (208), circRNA (circular RNA) (209), lncRNA (long noncoding RNA) (210), SINE-up element (211), among others (Figure 9). Anticodon-engineered tRNA enables ribosome recoding of premature termination codons to treat genetic diseases caused by nonsense mutations (208). Further mechanistic studies are needed to confirm the safety of tRNA-mediated readthrough of premature termination codons across the transcriptome, while avoiding native stop codons, to prevent the production of potentially harmful C-terminal extended proteins. CircRNA is currently in preclinical testing as a more effective protein expression platform than linear mRNAs, due to its high stability, long half-life in serum and cells (lacking 5'-cap or 3'-polyA tail, which are targeted by exoribonucleases), and its potential for endless translation—possibly bypassing ribosome recycling once loaded through an engineered internal ribosome entry site (start and stop codons are close together for continuous ribosome reinitiation after translation termination without drop-off) (209). However, it remains unclear whether the internal ribosome entry site structure remains stable upon delivery into target cells and whether it can efficiently recruit ribosomes to initiate translation adequately to produce therapeutic proteins compared to a linear mRNA.
Intracellular protein homeostasis is crucial for maintaining optimal cellular function, and disruptions can lead to pathological conditions. Maintaining protein homeostasis involves a balanced process of protein synthesis, post-translational processing, and protein degradation. Accumulating evidence indicates that coordinating translational control with protein degradation pathways—such as ribosome-associated quality control (RQC) (212), eIF2α phosphorylation-dependent autophagy (213), and mTORC1-dependent regulation of translation and autophagy (214)—is essential for cardiac protein homeostasis. Ribosome stalling caused by abnormal translation elongation produces truncated proteins and activates RQC. Although research increasingly links RQC dysfunction to neurological disorders, cancer, developmental disorders, and metabolic diseases (215), its role in heart biology and disease remains largely unexplored. A loss-of-function mutation in the ribosomal protein RPL3L, expressed specifically in heart and skeletal muscle, has been shown to slow the translation of specific codons for proline and alanine, causing ribosomes to collide along the mRNA. These collisions lead to misfolded cardiac muscle contraction-related proteins, which are then degraded by RQC. Functionally, RPL3L deficiency results in impaired heart contractility and neonatal dilated cardiomyopathy (38). As we advance, studying RQC and related E3-ubiquitin ligases, such as RNF10—responsible for RPS3 ubiquitination and the degradation of stalled 40S ribosomal subunits—will be important for better understanding heart diseases (216, 217).
During ribosome stalling and collision, de novo synthesized polypeptide chains can misfold and aggregate, resulting in unfolded protein stress. Autophagy degrades misfolded proteins and aberrant protein aggregates to reduce proteotoxic stress and provide metabolic reserves. Phosphorylation of eIF2α by protein kinase R-like ER kinase (PERK) inhibits eIF2α-GDP recycling to eIF2α-GTP, thereby suppressing overall translation; however, this mechanism selectively promotes the translation of autophagy-associated activating transcription factor 4 (ATF4) via a uORF-dependent mechanism(218). Thrombospondin 1 (THBS1), which activates ER stress, mediates PERK-eIF2α-ATF4-induced autophagy, leading to cardiac atrophy and lethal cardiomyopathy(219). Melatonin reduces the interaction between vascular endothelial growth factor-B (VEGF-B) and glucose-regulated protein 78 (GRP78), as well as between GRP78 and PERK, which increases PERK phosphorylation and induces PERK-eIF2α-ATF4-mediated autophagy. As a result, melatonin alleviates diabetic cardiomyopathy, reduces cardiac hypertrophy and fibrosis(220), and decreases CM apoptosis while protecting the heart from MI injury by increasing eIF2α phosphorylation(221). This highlights the critical role of eIF2α phosphorylation in managing misfolded and unfolded protein stress in the heart. Besides its function in protein synthesis, mTORC1 negatively regulates autophagy by modulating downstream autophagy-initiating kinase 1 (ULK1) and ATG13, orchestrating the delicate balance between anabolic and catabolic pathways necessary for maintaining cardiac protein homeostasis. AMP-activated protein kinase (AMPK) directly phosphorylates ULK1 to promote autophagy(222). Activation of AMPK has been shown to enhance autophagy in hearts under pressure overload, resulting in delayed cardiac hypertrophy and improved cardiac function(223). AMPK also activates autophagy and protects the heart against diabetes and ischemic injury(224, 225). Similar effects were observed with the use of metformin, a commonly prescribed anti-diabetic drug and AMPK activator(226, 227).
As another future research direction, a deeper understanding of how translational control interfaces with protein degradation pathways may uncover new regulatory circuits vital for cardiac protein homeostasis and disease mechanisms, potentially aiding in developing more effective and precise therapies for cardiac conditions like HFpEF. Li, C et al provides an exemplary mechanism of a noncanonical IRE1 (Inositol-requiring enzyme 1)-eIF4G1-eIF3 translation initiation complex involved in transcript-selective translational control of epidermal growth factor receptor through binding of a 5'-UTR terminal oligopyrimidine motif. This work indicates a connection among unfolded protein response, protein degradation, and mRNA translation(228). Investigating translational control mechanisms in the development and progression of metabolic syndrome, diabetic cardiomyopathy, and HFpEF is essential to identify novel therapeutic targets and develop innovative treatment strategies. Additionally, exploring the translation landscape across different cardiac and vascular cell types (e.g., endothelial cells, smooth muscle cells, and immune cells) and during crosstalk among various organs (such as the heart, lung, kidney, liver, and brain) will emerge as a key area for multidisciplinary research and systematic investigation, facilitated by extensive collaborations within a diverse scientific community sharing common interests and diverse expertise.
Author Contributions
Conceptualization, X.X. and Y.Y.; methodology, X.X.; software, X.X.; validation, X.X., Y.Y. and Z.Z.; formal analysis, X.X.; investigation, X.X.; resources, X.X.; data curation, X.X.; writing—original draft preparation, X.X.; writing—review and editing, X.X.; visualization, X.X.; supervision, X.X.; project administration, X.X.; funding acquisition, Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health grants R01 HL132899, R01 HL147954, R01 HL164584, R01 HL169432, American Heart Association Established Investigator Award 24EIA1255341, and the Harold S. Geneen Charitable Trust Awards Program for Coronary Heart Disease Research (to P.Y.), American Heart Association postdoctoral fellowship 25POST1371944 (to D.D.), and NIH T32 predoctoral fellowship GM135134 (to L.W.; this publication was made possible by Grant Number T32 GM135134 from Institutional Ruth L. Kirschstein National Research Service Award).
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Martin SS, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation. 2024;149(8):e347-e913.
- Simmonds SJ, Cuijpers I, Heymans S, Jones EAV. Cellular and Molecular Differences between HFpEF and HFrEF: A Step Ahead in an Improved Pathological Understanding. Cells. 2020;9(1).
- Schimmel, P. Aminoacyl tRNA synthetases: general scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annu Rev Biochem. 1987;56:125-58.
- Ibba M, Söll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617-50.
- Yadavalli SS, Ibba M. Quality control in aminoacyl-tRNA synthesis its role in translational fidelity. Adv Protein Chem Struct Biol. 2012;86:1-43.
- Ling J, Reynolds N, Ibba M. Aminoacyl-tRNA synthesis and translational quality control. Annu Rev Microbiol. 2009;63:61-78.
- Liu Y, Beyer A, Aebersold R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell. 2016;165(3):535-50.
- Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552(7683):110-5.
- Gao C, Wang Y. mRNA Metabolism in Cardiac Development and Disease: Life After Transcription. Physiol Rev. 2020;100(2):673-94.
- Volkers M, Preiss T, Hentze MW. RNA-binding proteins in cardiovascular biology and disease: the beat goes on. Nat Rev Cardiol. 2024.
- Hedaya OM, Venkata Subbaiah KC, Jiang F, Xie LH, Wu J, Khor ES, et al. Secondary structures that regulate mRNA translation provide insights for ASO-mediated modulation of cardiac hypertrophy. Nat Commun. 2023;14(1):6166.
- Yao P, Potdar AA, Arif A, Ray PS, Mukhopadhyay R, Willard B, et al. Coding Region Polyadenylation Generates a Truncated tRNA Synthetase that Counters Translation Repression. Cell. 2012;149(1):88-100.
- Jiang F, Hedaya OM, Khor E, Wu J, Auguste M, Yao P. RNA binding protein PRRC2B mediates translation of specific mRNAs and regulates cell cycle progression. Nucleic Acids Res. 2023;51(11):5831-46.
- Anastasakis DG, Apostolidi M, Garman KA, Polash AH, Umar MI, Meng Q, et al. Nuclear PKM2 binds pre-mRNA at folded G-quadruplexes and reveals their gene regulatory role. Mol Cell. 2024;84(19):3775-89 e6.
- Kim B, Kim VN. fCLIP-seq for transcriptomic footprinting of dsRNA-binding proteins: Lessons from DROSHA. Methods. 2019;152:3-11.
- Danan C, Manickavel S, Hafner M. PAR-CLIP: A Method for Transcriptome-Wide Identification of RNA Binding Protein Interaction Sites. Methods Mol Biol. 2016;1358:153-73.
- Panda AC, Martindale JL, Gorospe M. Affinity Pulldown of Biotinylated RNA for Detection of Protein-RNA Complexes. Bio Protoc. 2016;6(24).
- Sears RM, May DG, Roux KJ. BioID as a Tool for Protein-Proximity Labeling in Living Cells. Methods Mol Biol. 2019;2012:299-313.
- Mellacheruvu D, Wright Z, Couzens AL, Lambert JP, St-Denis NA, Li T, et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods. 2013;10(8):730-6.
- de la Parra C, Ernlund A, Alard A, Ruggles K, Ueberheide B, Schneider RJ. A widespread alternate form of cap-dependent mRNA translation initiation. Nat Commun. 2018;9(1):3068.
- Liakath-Ali K, Mills EW, Sequeira I, Lichtenberger BM, Pisco AO, Sipila KH, et al. An evolutionarily conserved ribosome-rescue pathway maintains epidermal homeostasis. Nature. 2018;556(7701):376-80.
- Wu J, Xie HL, Jiang F, Mickelson DJ, Myers JR, Tang WH, et al. EPRS regulates proline-rich pro-fibrotic protein synthesis during cardiac fibrosis. bioRxiv. 2019.
- Zhou P, Zhang Y, Ma Q, Gu F, Day DS, He A, et al. Interrogating translational efficiency and lineage-specific transcriptomes using ribosome affinity purification. Proc Natl Acad Sci U S A. 2013;110(38):15395-400.
- Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A. 2009;106(33):13939-44.
- Busch JD, Cipullo M, Atanassov I, Bratic A, Silva Ramos E, Schondorf T, et al. MitoRibo-Tag Mice Provide a Tool for In Vivo Studies of Mitoribosome Composition. Cell Rep. 2019;29(6):1728-38 e9.
- van Heesch S, Witte F, Schneider-Lunitz V, Schulz JF, Adami E, Faber AB, et al. The Translational Landscape of the Human Heart. Cell. 2019;178(1):242-60.e29.
- Schafer S, Viswanathan S, Widjaja AA, Lim W-W, Moreno-Moral A, DeLaughter DM, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature. 2017;552(7683):110-5.
- Chothani S, Schäfer S, Adami E, Viswanathan S, Widjaja AA, Langley SR, et al. Widespread Translational Control of Fibrosis in the Human Heart by RNA-Binding Proteins. Circulation. 2019;140(11):937-51.
- Lewis YE, Moskovitz A, Mutlak M, Heineke J, Caspi LH, Kehat I. Localization of transcripts, translation, and degradation for spatiotemporal sarcomere maintenance. J Mol Cell Cardiol. 2018;116:16-28.
- Scarborough EA, Uchida K, Vogel M, Erlitzki N, Iyer M, Phyo SA, et al. Microtubules orchestrate local translation to enable cardiac growth. Nat Commun. 2021;12(1):1547.
- Bogdanov V, Soltisz AM, Moise N, Sakuta G, Orengo BH, Janssen PML, et al. Distributed synthesis of sarcolemmal and sarcoplasmic reticulum membrane proteins in cardiac myocytes. Basic Res Cardiol. 2021;116(1):63.
- Eichel CA, Rios-Perez EB, Liu F, Jameson MB, Jones DK, Knickelbine JJ, et al. A microtranslatome coordinately regulates sodium and potassium currents in the human heart. Elife. 2019;8.
- Tominaga M, Hamanoue S, Goto H, Saito T, Nagai JI, Masuno M, et al. Diamond-Blackfan anemia caused by chromosome 1p22 deletion encompassing RPL5. Hum Genome Var. 2019;6:36.
- Ulirsch JC, Verboon JM, Kazerounian S, Guo MH, Yuan D, Ludwig LS, et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am J Hum Genet. 2019;104(2):356.
- Vlachos A, Osorio DS, Atsidaftos E, Kang J, Lababidi ML, Seiden HS, et al. Increased Prevalence of Congenital Heart Disease in Children With Diamond Blackfan Anemia Suggests Unrecognized Diamond Blackfan Anemia as a Cause of Congenital Heart Disease in the General Population: A Report of the Diamond Blackfan Anemia Registry. Circ Genom Precis Med. 2018;11(5):e002044.
- Liu YL, Shibuya A, Glader B, Wilkes MC, Barna M, Sakamoto KM. Animal models of Diamond-Blackfan anemia: updates and challenges. Haematologica. 2023;108(5):1222-31.
- Grimes KM, Prasad V, Huo J, Kuwabara Y, Vanhoutte D, Baldwin TA, et al. Rpl3l gene deletion in mice reduces heart weight over time. Front Physiol. 2023;14:1054169.
- Shiraishi C, Matsumoto A, Ichihara K, Yamamoto T, Yokoyama T, Mizoo T, et al. RPL3L-containing ribosomes determine translation elongation dynamics required for cardiac function. Nat Commun. 2023;14(1):2131.
- Milenkovic I, Santos Vieira HG, Lucas MC, Ruiz-Orera J, Patone G, Kesteven S, et al. Dynamic interplay between RPL3- and RPL3L-containing ribosomes modulates mitochondrial activity in the mammalian heart. Nucleic Acids Res. 2023;51(11):5301-24.
- Wu J, Subbaiah KCV, Hedaya O, Chen S, Munger J, Tang WHW, et al. FAM210A regulates mitochondrial translation and maintains cardiac mitochondrial homeostasis. Cardiovasc Res. 2023;119(14):2441-57.
- Michael, R. Murphy MG, Teresa M. Lee, Joshua M. Fisher, Megha V. Patel, Parul Jayakar, Amanda Buchanan, Rajesh K. Soni, Yue Yin, Feiyue Yang, Muredach P. Reilly, Wendy K. Chung, Xuebing Wu. Recessive but damaging alleles of muscle-specific ribosomal protein gene RPL3L drive neonatal dilated cardiomyopathy. In: Columbia University Irving Medical Center, editor. BioRxiv2025.
- Methawasin M, Zhang Y, Gregorich ZR, He Y, Liu C, Muldoon J, et al. Reducing Granules Without Splicing Restoration Alleviates RBM20 Cardiomyopathy. Circ Res. 2025;136(10):1134-46.
- Witte F, Ruiz-Orera J, Mattioli CC, Blachut S, Adami E, Schulz JF, et al. A trans locus causes a ribosomopathy in hypertrophic hearts that affects mRNA translation in a protein length-dependent fashion. Genome Biol. 2021;22(1):191.
- Khalyfa A, Bourbeau D, Chen E, Petroulakis E, Pan J, Xu S, et al. Characterization of elongation factor-1A (eEF1A-1) and eEF1A-2/S1 protein expression in normal and wasted mice. J Biol Chem. 2001;276(25):22915-22.
- Cao S, Smith LL, Padilla-Lopez SR, Guida BS, Blume E, Shi J, et al. Homozygous EEF1A2 mutation causes dilated cardiomyopathy, failure to thrive, global developmental delay, epilepsy and early death. Human Molecular Genetics. 2017;26(18):3545-52.
- Feng W, Wang L, Veevers J, Liu C, Huang T, Chen J. Loss of eEF1A2 (Eukaryotic Elongation Factor 1 A2) in Murine Myocardium Results in Dilated Cardiomyopathy. Circ Heart Fail. 2021;14(10):e008665.
- Boczonadi V, Horvath R. Mitochondria: impaired mitochondrial translation in human disease. Int J Biochem Cell Biol. 2014;48(100):77-84.
- Euro L, Konovalova S, Asin-Cayuela J, Tulinius M, Griffin H, Horvath R, et al. Structural modeling of tissue-specific mitochondrial alanyl-tRNA synthetase (AARS2) defects predicts differential effects on aminoacylation. Frontiers in Genetics. 2015;6.
- Götz A, Tyynismaa H, Euro L, Ellonen P, Hyötyläinen T, Ojala T, et al. Exome Sequencing Identifies Mitochondrial Alanyl-tRNA Synthetase Mutations in Infantile Mitochondrial Cardiomyopathy. The American Journal of Human Genetics. 2011;88(5):635-42.
- Sommerville EW, Zhou X-L, Oláhová M, Jenkins J, Euro L, Konovalova S, et al. Instability of the mitochondrial alanyl-tRNA synthetase underlies fatal infantile-onset cardiomyopathy. Human Molecular Genetics. 2018;28(2):258-68.
- Rudler DL, Hughes LA, Perks KL, Richman TR, Kuznetsova I, Ermer JA, et al. Fidelity of translation initiation is required for coordinated respiratory complex assembly. Science Advances. 2019;5(12):eaay2118.
- Gebauer F, Schwarzl T, Valcarcel J, Hentze MW. RNA-binding proteins in human genetic disease. Nat Rev Genet. 2021;22(3):185-98.
- Sun J, Wang L, Matthews RC, Walcott GP, Lu YA, Wei Y, et al. CCND2 Modified mRNA Activates Cell Cycle of Cardiomyocytes in Hearts With Myocardial Infarction in Mice and Pigs. Circ Res. 2023;133(6):484-504.
- Bohlen J, Roiuk M, Neff M, Teleman AA. PRRC2 proteins impact translation initiation by promoting leaky scanning. Nucleic Acids Res. 2023;51(7):3391-409.
- Das D, Khor ES, Jiang F, He J, Kawakami Y, Wainwright L, et al. Loss-of-function of RNA-binding protein PRRC2B causes translational defects and congenital cardiovascular malformation. medRxiv. 2024.
- Goldberg N, Bril D, Eisenstein M, Olender T, Savidor A, Bialik S, et al. The RNA-binding protein PRRC2B preserves 5’ TOP mRNA during starvation to maintain ribosome biogenesis during nutrient recovery. bioRxiv. 2024:2024.12.04.626744.
- Rao A, Lyu B, Jahan I, Lubertozzi A, Zhou G, Tedeschi F, et al. The translation initiation factor homolog eif4e1c regulates cardiomyocyte metabolism and proliferation during heart regeneration. Development. 2023;150(20).
- Jobava R, Mao Y, Guan B-J, Hu D, Krokowski D, Chen C-W, et al. Adaptive translational pausing is a hallmark of the cellular response to severe environmental stress. Molecular Cell. 2021;81(20):4191-208.e8.
- Santos-Ribeiro D, Godinas L, Pilette C, Perros F. The integrated stress response system in cardiovascular disease. Drug Discovery Today. 2018;23(4):920-9.
- Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44(5):491-501.
- Tanaka K-i, Xue Y, Nguyen-Yamamoto L, Morris JA, Kanazawa I, Sugimoto T, et al. FAM210A is a novel determinant of bone and muscle structure and strength. Proceedings of the National Academy of Sciences. 2018;115(16):E3759-E68.
- Wu J, Subbaiah KCV, Hedaya O, Chen S, Munger J, Tang WHW, et al. FAM210A regulates mitochondrial translation and maintains cardiac mitochondrial homeostasis. Cardiovasc Res. 2023.
- van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105(35):13027-32.
- Wu J, Venkata Subbaiah KC, Jiang F, Hedaya O, Mohan A, Yang T, et al. MicroRNA-574 regulates FAM210A expression and influences pathological cardiac remodeling. EMBO Mol Med. 2020:e12710.
- Hollinger J, Wu J, Awayda KM, O'Connell MR, Yao P. Expression and purification of the mitochondrial transmembrane protein FAM210A in Escherichia coli. Protein Expr Purif. 2023;210:106322.
- Mohamed TMA, Ang YS, Radzinsky E, Zhou P, Huang Y, Elfenbein A, et al. Regulation of Cell Cycle to Stimulate Adult Cardiomyocyte Proliferation and Cardiac Regeneration. Cell. 2018;173(1):104-16 e12.
- Aballo TJ, Roberts DS, Bayne EF, Zhu W, Walcott G, Mahmoud AI, et al. Integrated proteomics reveals alterations in sarcomere composition and developmental processes during postnatal swine heart development. J Mol Cell Cardiol. 2023;176:33-40.
- Zhao M, Nakada Y, Wei Y, Bian W, Chu Y, Borovjagin AV, et al. Cyclin D2 Overexpression Enhances the Efficacy of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes for Myocardial Repair in a Swine Model of Myocardial Infarction. Circulation. 2021;144(3):210-28.
- Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332(6028):458-61.
- Bartsch D, Kalamkar K, Ahuja G, Lackmann JW, Hescheler J, Weber T, et al. mRNA translational specialization by RBPMS presets the competence for cardiac commitment in hESCs. Sci Adv. 2023;9(13):eade1792.
- Gao F, Liang T, Lu YW, Pu L, Fu X, Dong X, et al. Reduced Mitochondrial Protein Translation Promotes Cardiomyocyte Proliferation and Heart Regeneration. Circulation. 2023;148(23):1887-906.
- Stoehr A, Kennedy L, Yang Y, Patel S, Lin Y, Linask KL, et al. The ribosomal prolyl-hydroxylase OGFOD1 decreases during cardiac differentiation and modulates translation and splicing. JCI Insight. 2019;5(13).
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. 2011;331(6020):1078-80.
- Wang Z, Cui M, Shah AM, Ye W, Tan W, Min YL, et al. Mechanistic basis of neonatal heart regeneration revealed by transcriptome and histone modification profiling. Proc Natl Acad Sci U S A. 2019;116(37):18455-65.
- Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124(3):1382-92.
- Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285-95.
- Li S, Shen S, Xu H, Cai S, Yuan X, Wang C, et al. IGF2BP3 promotes adult myocardial regeneration by stabilizing MMP3 mRNA through interaction with m6A modification. Cell Death Discov. 2023;9(1):164.
- Caudron-Herger M, Jansen RE, Wassmer E, Diederichs S. RBP2GO: a comprehensive pan-species database on RNA-binding proteins, their interactions and functions. Nucleic Acids Res. 2021;49(D1):D425-D36.
- Nakano K, Sadahiro T, Fujita R, Isomi M, Abe Y, Yamada Y, et al. Development of adeno-associated viral vectors targeting cardiac fibroblasts for efficient in vivo cardiac reprogramming. Stem Cell Reports. 2024;19(10):1389-98.
- Tani H, Sadahiro T, Yamada Y, Isomi M, Yamakawa H, Fujita R, et al. Direct Reprogramming Improves Cardiac Function and Reverses Fibrosis in Chronic Myocardial Infarction. Circulation. 2023;147(3):223-38.
- Xie Y, Wang Q, Yang Y, Near D, Wang H, Colon M, et al. Translational landscape of direct cardiac reprogramming reveals a role of Ybx1 in repressing cardiac fate acquisition. Nat Cardiovasc Res. 2023;2(11):1060-77.
- Wang L, Liu Z, Yin C, Asfour H, Chen O, Li Y, et al. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ Res. 2015;116(2):237-44.
- Kim S, Coulombe PA. Emerging role for the cytoskeleton as an organizer and regulator of translation. Nat Rev Mol Cell Biol. 2010;11(1):75-81.
- Hannan RD, Jenkins A, Jenkins AK, Brandenburger Y. Cardiac hypertrophy: a matter of translation. Clin Exp Pharmacol Physiol. 2003;30(8):517-27.
- Chorghade S, Seimetz J, Emmons R, Yang J, Bresson SM, Lisio M, et al. Poly(A) tail length regulates PABPC1 expression to tune translation in the heart. Elife. 2017;6.
- Gudbjarnason S, Telerman M, Chiba C, Wolf PL, Bing RJ. Myocardial Protein Synthesis in Cardiac Hypertrophy. J Lab Clin Med. 1964;63:244-53.
- Gonzalez-Teran B, Lopez JA, Rodriguez E, Leiva L, Martinez-Martinez S, Bernal JA, et al. p38gamma and delta promote heart hypertrophy by targeting the mTOR-inhibitory protein DEPTOR for degradation. Nat Commun. 2016;7:10477.
- Dorn GW, 2nd, Force T. Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest. 2005;115(3):527-37.
- Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114(3):549-64.
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274-93.
- Brandenburger Y, Arthur JF, Woodcock EA, Du XJ, Gao XM, Autelitano DJ, et al. Cardiac hypertrophy in vivo is associated with increased expression of the ribosomal gene transcription factor UBF. FEBS Lett. 2003;548(1-3):79-84.
- Luyken J, Hannan RD, Cheung JY, Rothblum LI. Regulation of rDNA transcription during endothelin-1-induced hypertrophy of neonatal cardiomyocytes. Hyperphosphorylation of upstream binding factor, an rDNA transcription factor. Circ Res. 1996;78(3):354-61.
- Hannan RD, Luyken J, Rothblum LI. Regulation of ribosomal DNA transcription during contraction-induced hypertrophy of neonatal cardiomyocytes. J Biol Chem. 1996;271(6):3213-20.
- Pestova TV, Kolupaeva VG, Lomakin IB, Pilipenko EV, Shatsky IN, Agol VI, et al. Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci U S A. 2001;98(13):7029-36.
- Gebauer F, Hentze MW. Molecular mechanisms of translational control. Nat Rev Mol Cell Biol. 2004;5(10):827-35.
- Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5' cap. Nature. 1990;345(6275):544-7.
- Ventoso I, Blanco R, Perales C, Carrasco L. HIV-1 protease cleaves eukaryotic initiation factor 4G and inhibits cap-dependent translation. Proceedings of the National Academy of Sciences. 2001;98(23):12966.
- Wada H, Ivester CT, Carabello BA, Cooper G, McDermott PJ. Translational Initiation Factor eIF-4E: A LINK BETWEEN CARDIAC LOAD AND PROTEIN SYNTHESIS. Journal of Biological Chemistry. 1996;271(14):8359-64.
- Tuxworth WJ, Wada H, Ishibashi Y, McDermott PJ. Role of load in regulating eIF-4F complex formation in adult feline cardiocytes. American Journal of Physiology-Heart and Circulatory Physiology. 1999;277(4):H1273-H82.
- Makhlouf AA, Namboodiri AMS, McDermott PJ. Transcriptional regulation of the rat eIF4E gene in cardiac muscle cells: the role of specific elements in the promoter region. Gene. 2001;267(1):1-12.
- Nagatomo Y, Carabello BA, Hamawaki M, Nemoto S, Matsuo T, McDermott PJ. Translational mechanisms accelerate the rate of protein synthesis during canine pressure-overload hypertrophy. American Journal of Physiology-Heart and Circulatory Physiology. 1999;277(6):H2176-H84.
- Makhlouf AA, McDermott PJ. Increased expression of eukaryotic initiation factor 4E during growth of neonatal rat cardiocytes in vitro. American Journal of Physiology-Heart and Circulatory Physiology. 1998;274(6):H2133-H42.
- Gingras A-C, Raught B, Sonenberg N. eIF4 Initiation Factors: Effectors of mRNA Recruitment to Ribosomes and Regulators of Translation. Annual Review of Biochemistry. 1999;68(1):913-63.
- Saghir AN, Tuxworth WJ, Jr., Hagedorn CH, McDermott PJ. Modifications of eukaryotic initiation factor 4F (eIF4F) in adult cardiocytes by adenoviral gene transfer: differential effects on eIF4F activity and total protein synthesis rates. Biochem J. 2001;356(Pt 2):557-66.
- Hardt SE, Tomita H, Katus HA, Sadoshima J. Phosphorylation of Eukaryotic Translation Initiation Factor 2Bε by Glycogen Synthase Kinase-3β Regulates β-Adrenergic Cardiac Myocyte Hypertrophy. Circulation Research. 2004;94(7):926-35.
- Lu Z, Xu X, Fassett J, Kwak D, Liu X, Hu X, et al. Loss of the Eukaryotic Initiation Factor 2α Kinase General Control Nonderepressible 2 Kinase Protects Mice From Pressure Overload-Induced Congestive Heart Failure Without Affecting Ventricular Hypertrophy. Hypertension. 2013;63.
- Kimball, SR. Eukaryotic initiation factor eIF2. The International Journal of Biochemistry & Cell Biology. 1999;31(1):25-9.
- Rani S, Sreenivasaiah PK, Cho C, Kim DH. Salubrinal Alleviates Pressure Overload-Induced Cardiac Hypertrophy by Inhibiting Endoplasmic Reticulum Stress Pathway. Mol Cells. 2017;40(1):66-72.
- James CC, Smyth JW. Alternative mechanisms of translation initiation: An emerging dynamic regulator of the proteome in health and disease. Life Sci. 2018;212:138-44.
- Martínez-Salas E, Piñeiro D, fernandez sanchez N. Alternative Mechanisms to Initiate Translation in Eukaryotic mRNAs. Comparative and functional genomics. 2012;2012:391546.
- Basheer WA, Xiao S, Epifantseva I, Fu Y, Kleber AG, Hong T, et al. GJA1-20k Arranges Actin to Guide Cx43 Delivery to Cardiac Intercalated Discs. Circulation research. 2017;121(9):1069-80.
- Peters Nicholas S, Coromilas J, Severs Nicholas J, Wit Andrew L. Disturbed Connexin43 Gap Junction Distribution Correlates With the Location of Reentrant Circuits in the Epicardial Border Zone of Healing Canine Infarcts That Cause Ventricular Tachycardia. Circulation. 1997;95(4):988-96.
- Smyth JW, Shaw RM. Autoregulation of connexin43 gap junction formation by internally translated isoforms. Cell Rep. 2013;5(3):611-8.
- Ul-Hussain M, Olk S, Schoenebeck B, Wasielewski B, Meier C, Prochnow N, et al. Internal ribosomal entry site (IRES) activity generates endogenous carboxyl-terminal domains of Cx43 and is responsive to hypoxic conditions. The Journal of biological chemistry. 2014;289(30):20979-90.
- Schiavi A, Hudder A, Werner R. Connexin43 mRNA contains a functional internal ribosome entry site. FEBS Letters. 1999;464(3):118-22.
- Salat-Canela C, Sesé M, Peula C, Ramón y Cajal S, Aasen T. Internal translation of the connexin 43 transcript. Cell Communication and Signaling. 2014;12(1):31.
- Knight JRP, Garland G, Pöyry T, Mead E, Vlahov N, Sfakianos A, et al. Control of translation elongation in health and disease. Disease Models & Mechanisms. 2020;13(3):dmm043208.
- Voorhees RM, Ramakrishnan V. Structural Basis of the Translational Elongation Cycle. Annual Review of Biochemistry. 2013;82(1):203-36.
- Hannan R, Jenkins A, Jenkins A, Brandenburger Y. Cardiac hypertrophy: A matter of translation. Clinical and Experimental Pharmacology and Physiology. 2003;30(8):517-27.
- Faller WJ, Jackson TJ, Knight JRP, Ridgway RA, Jamieson T, Karim SA, et al. mTORC1-mediated translational elongation limits intestinal tumour initiation and growth. Nature. 2015;517(7535):497-500.
- Delaidelli A, Jan A, Herms J, Sorensen PH. Translational control in brain pathologies: biological significance and therapeutic opportunities. Acta Neuropathologica. 2019;137(4):535-55.
- Kameshima S, Okada M, Ikeda S, Watanabe Y, Yamawaki H. Coordination of changes in expression and phosphorylation of eukaryotic elongation factor 2 (eEF2) and eEF2 kinase in hypertrophied cardiomyocytes. Biochemistry and Biophysics Reports. 2016;7.
- Everett AD, Stoops TD, Nairn AC, Brautigan D. Angiotensin II regulates phosphorylation of translation elongation factor-2 in cardiac myocytes. American Journal of Physiology-Heart and Circulatory Physiology. 2001;281(1):H161-H7.
- Wang L, Proud CG. Regulation of the phosphorylation of elongation factor 2 by MEK-dependent signalling in adult rat cardiomyocytes. FEBS Letters. 2002;531(2):285-9.
- McLeod LE, Wang L, Proud CG. beta-Adrenergic agonists increase phosphorylation of elongation factor 2 in cardiomyocytes without eliciting calcium-independent eEF2 kinase activity. FEBS Letters. 2001;489(2-3):225-8.
- Kaul G, Pattan G, Rafeequi T. Eukaryotic elongation factor-2 (eEF2): its regulation and peptide chain elongation. Cell Biochemistry and Function. 2011;29(3):227-34.
- Grund A, Szaroszyk M, Korf-Klingebiel M, Malek Mohammadi M, Trogisch F, Schrameck U, et al. TIP 30 counteracts cardiac hypertrophy and failure by inhibiting translational elongation. EMBO Molecular Medicine. 2019;11.
- Pittman Y, Kandl K, Lewis M, Valente L, Kinzy T. Coordination of Eukaryotic Translation Elongation Factor 1A (eEF1A) Function in Actin Organization and Translation Elongation by the Guanine Nucleotide Exchange Factor eEF1B. The Journal of biological chemistry. 2009;284:4739-47.
- He H, Chen M, Scheffler NK, Gibson Bradford W, Spremulli Linda L, Gottlieb Roberta A. Phosphorylation of Mitochondrial Elongation Factor Tu in Ischemic Myocardium. Circulation Research. 2001;89(5):461-7.
- Borutaite V, Mildaziene V, Brown GC, Brand MD. Control and kinetic analysis of ischemia-damaged heart mitochondria: which parts of the oxidative phosphorylation system are affected by ischemia? Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1995;1272(3):154-8.
- Cai YC, Bullard J, Thompson N, Spremulli L. Interaction of Mitochondrial Elongation Factor Tu with Aminoacyl-tRNA and Elongation Factor Ts. The Journal of biological chemistry. 2000;275:20308-14.
- Lippmann C, Lindschau C, Buchner K, Erdmann VA, editors. Phosphorylation of Elongation Factor Tu in Vitro and in Vivo1991; Berlin, Heidelberg: Springer Berlin Heidelberg.
- Brandenburger Y, Jenkins A, Autelitano DJ, Hannan RD. Increased expression of UBF is a critical determinant for rRNA synthesis and hypertrophic growth of cardiac myocytes. The FASEB Journal. 2001;15(11):2051-3.
- Hannan RD, Luyken J, Rothblum LI. Regulation of rDNA Transcription Factors during Cardiomyocyte Hypertrophy Induced by Adrenergic Agents. Journal of Biological Chemistry. 1995;270(14):8290-7.
- Hannan RD, Luyken J, Rothblum LI. Regulation of Ribosomal DNA Transcription during Contraction-induced Hypertrophy of Neonatal Cardiomyocytes. Journal of Biological Chemistry. 1996;271(6):3213-20.
- Luyken J, Hannan RD, Cheung JY, Rothblum LI. Regulation of rDNA Transcription During Endothelin-1Induced Hypertrophy of Neonatal Cardiomyocytes. Circulation Research. 1996;78(3):354-61.
- Sadoshima J, Izumo S. Rapamycin Selectively Inhibits Angiotensin II–Induced Increase in Protein Synthesis in Cardiac Myocytes In Vitro. Circulation Research. 1995;77(6):1040-52.
- Oh H, Fujio Y, Kunisada K, Hirota H, Matsui H, Kishimoto T, et al. Activation of Phosphatidylinositol 3-Kinase through Glycoprotein 130 Induces Protein Kinase B and p70 S6 Kinase Phosphorylation in Cardiac Myocytes. Journal of Biological Chemistry. 1998;273(16):9703-10.
- Chothani S, Schafer S, Adami E, Viswanathan S, Widjaja AA, Langley SR, et al. Widespread Translational Control of Fibrosis in the Human Heart by RNA-Binding Proteins. Circulation. 2019;140(11):937-51.
- Volkers M, Toko H, Doroudgar S, Din S, Quijada P, Joyo AY, et al. Pathological hypertrophy amelioration by PRAS40-mediated inhibition of mTORC1. Proc Natl Acad Sci U S A. 2013;110(31):12661-6.
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10(5):307-18.
- Ben-Sahra I, Howell JJ, Asara JM, Manning BD. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science. 2013;339(6125):1323-8.
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science. 2013;339(6125):1320-3.
- Hannan KM, Brandenburger Y, Jenkins A, Sharkey K, Cavanaugh A, Rothblum L, et al. mTOR-dependent regulation of ribosomal gene transcription requires S6K1 and is mediated by phosphorylation of the carboxy-terminal activation domain of the nucleolar transcription factor UBF. Mol Cell Biol. 2003;23(23):8862-77.
- Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11(2):113-27.
- Zhu Y, Pires KM, Whitehead KJ, Olsen CD, Wayment B, Zhang YC, et al. Mechanistic target of rapamycin (Mtor) is essential for murine embryonic heart development and growth. PLoS One. 2013;8(1):e54221.
- Mazelin L, Panthu B, Nicot AS, Belotti E, Tintignac L, Teixeira G, et al. mTOR inactivation in myocardium from infant mice rapidly leads to dilated cardiomyopathy due to translation defects and p53/JNK-mediated apoptosis. J Mol Cell Cardiol. 2016;97:213-25.
- Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123(10):1073-82.
- Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120(8):2805-16.
- Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, et al. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107(12):1664-70.
- McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109(24):3050-5.
- Wu X, Cao Y, Nie J, Liu H, Lu S, Hu X, et al. Genetic and pharmacological inhibition of Rheb1-mTORC1 signaling exerts cardioprotection against adverse cardiac remodeling in mice. Am J Pathol. 2013;182(6):2005-14.
- Varma E, Burghaus J, Schwarzl T, Sekaran T, Gupta P, Gorska AA, et al. Translational control of Ybx1 expression regulates cardiac function in response to pressure overload in vivo. Basic Res Cardiol. 2023;118(1):25.
- Kmietczyk V, Oelschlager J, Gupta P, Varma E, Hartl S, Furkel J, et al. Ythdf2 regulates cardiac remodeling through its mRNA target transcripts. J Mol Cell Cardiol. 2023;181:57-66.
- Berulava T, Buchholz E, Elerdashvili V, Pena T, Islam MR, Lbik D, et al. Changes in m6A RNA methylation contribute to heart failure progression by modulating translation. Eur J Heart Fail. 2020;22(1):54-66.
- Rockey DC, Bell PD, Hill JA. Fibrosis--a common pathway to organ injury and failure. N Engl J Med. 2015;372(12):1138-49.
- Davis J, Molkentin JD. Myofibroblasts: Trust your heart and let fate decide. Journal of Molecular and Cellular Cardiology. 2014;70:9-18.
- Akhurst RJ, Hata A. Targeting the TGFβ signalling pathway in disease. Nature Reviews Drug Discovery. 2012;11(10):790-811.
- Obana M, Maeda M, Takeda K, Hayama A, Mohri T, Yamashita T, et al. Therapeutic Activation of Signal Transducer and Activator of Transcription 3 by Interleukin-11 Ameliorates Cardiac Fibrosis After Myocardial Infarction. Circulation. 2010;121(5):684-91.
- Widjaja AA, Lim WW, Viswanathan S, Chothani S, Corden B, Dasan CM, et al. Inhibition of IL-11 signalling extends mammalian healthspan and lifespan. Nature. 2024;632(8023):157-65.
- Schneider-Lunitz V, Ruiz-Orera J, Hubner N, van Heesch S. Multifunctional RNA-binding proteins influence mRNA abundance and translational efficiency of distinct sets of target genes. PLoS Comput Biol. 2021;17(12):e1009658.
- Doroudgar S, Hofmann C, Boileau E, Malone B, Riechert E, Gorska AA, et al. Monitoring Cell-Type-Specific Gene Expression Using Ribosome Profiling In Vivo During Cardiac Hemodynamic Stress. Circ Res. 2019;125(4):431-48.
- Yao P, Fox PL. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med. 2013;5(3):332-43.
- Sampath P, Mazumder B, Seshadri V, Gerber CA, Chavatte L, Kinter M, et al. Noncanonical function of glutamyl-prolyl-tRNA synthetase: gene-specific silencing of translation. Cell. 2004;119(2):195-208.
- Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL. A stress-responsive RNA switch regulates VEGFA expression. Nature. 2009;457(7231):915-9.
- Yao P, Potdar AA, Ray PS, Eswarappa SM, Flagg AC, Willard B, et al. The HILDA complex coordinates a conditional switch in the 3'-untranslated region of the VEGFA mRNA. PLoS Biol. 2013;11(8):e1001635.
- Mendes MI, Gutierrez Salazar M, Guerrero K, Thiffault I, Salomons GS, Gauquelin L, et al. Bi-allelic Mutations in EPRS, Encoding the Glutamyl-Prolyl-Aminoacyl-tRNA Synthetase, Cause a Hypomyelinating Leukodystrophy. Am J Hum Genet. 2018;102(4):676-84.
- Da M, Feng Y, Xu J, Hu Y, Lin Y, Ni B, et al. Association of aminoacyl-tRNA synthetases gene polymorphisms with the risk of congenital heart disease in the Chinese Han population. PLoS One. 2014;9(10):e110072.
- Rau CD, Romay MC, Tuteryan M, Wang JJ, Santolini M, Ren S, et al. Systems Genetics Approach Identifies Gene Pathways and Adamts2 as Drivers of Isoproterenol-Induced Cardiac Hypertrophy and Cardiomyopathy in Mice. Cell Syst. 2017;4(1):121-8 e4.
- Galindo CL, Skinner MA, Errami M, Olson LD, Watson DA, Li J, et al. Transcriptional profile of isoproterenol-induced cardiomyopathy and comparison to exercise-induced cardiac hypertrophy and human cardiac failure. BMC physiology. 2009;9:23.
- Harigaya Y, Parker R. No-go decay: a quality control mechanism for RNA in translation. Wiley Interdiscip Rev RNA. 2010;1(1):132-41.
- Wu J, Hollinger J, Bonanno E, Jiang F, Yao P. Cardiomyocyte-Specific Loss of Glutamyl-prolyl-tRNA Synthetase Leads to Disturbed Protein Homeostasis and Dilated Cardiomyopathy. Cells. 2023;13(1).
- Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV. EF-P is essential for rapid synthesis of proteins containing consecutive proline residues. Science. 2013;339(6115):85-8.
- Schuller AP, Wu CC, Dever TE, Buskirk AR, Green R. eIF5A Functions Globally in Translation Elongation and Termination. Mol Cell. 2017;66(2):194-205 e5.
- Lassak J, Wilson DN, Jung K. Stall no more at polyproline stretches with the translation elongation factors EF-P and IF-5A. Mol Microbiol. 2016;99(2):219-35.
- Nakanishi S, Cleveland JL. Targeting the polyamine-hypusine circuit for the prevention and treatment of cancer. Amino Acids. 2016;48(10):2353-62.
- Imam S, Mirmira RG, Jaume JC. Eukaryotic translation initiation factor 5A inhibition alters physiopathology and immune responses in a "humanized" transgenic mouse model of type 1 diabetes. Am J Physiol Endocrinol Metab. 2014;306(7):E791-8.
- Subbaiah KCV, Wu J, Tang WHW, Yao P. Ciclopirox Inhibition of eIF5A Hypusination Attenuates Fibroblast Activation and Cardiac Fibrosis. J Cardiovasc Dev Dis. 2023;10(2).
- Goldfarb L, Dalakas M. Tragedy in a heartbeat: malfunctioning desmin causes skeletal and cardiac muscle disease. The Journal of Clinical Investigation. 2011;121:455-.
- McLendon P, Robbins J. Desmin-related cardiomyopathy: An unfolding story. American journal of physiology Heart and circulatory physiology. 2011;301:H1220-8.
- Park SG, Schimmel P, Kim S. Aminoacyl tRNA synthetases and their connections to disease. Proceedings of the National Academy of Sciences. 2008;105(32):11043.
- Liu Y, Satz J, Vo M-N, Nangle L, Schimmel P, Ackerman S. Deficiencies in tRNA synthetase editing activity cause cardioproteinopathy. Proc Natl Acad Sci U S A. 2014;111.
- Ling J, Reynolds N, Ibba M. Aminoacyl-tRNA Synthesis and Translational Quality Control. Annual review of microbiology. 2009;63:61-78.
- Yao P, Zhu B, Jaeger S, Eriani G, Wang ED. Recognition of tRNALeu by Aquifex aeolicus leucyl-tRNA synthetase during the aminoacylation and editing steps. Nucleic Acids Res. 2008;36(8):2728-38.
- Beebe K, Pouplana L, Schimmel P. Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. The EMBO journal. 2003;22:668-75.
- Lee j-w, Beebe K, Nangle L, Jang J, Guess C, Cook S, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50-5.
- Nangle L, Motta C, Schimmel P. Global Effects of Mistranslation from an Editing Defect in Mammalian Cells. Chemistry & biology. 2006;13:1091-100.
- Huang Q, Yao P, Eriani G, Wang ED. In vivo identification of essential nucleotides in tRNALeu to its functions by using a constructed yeast tRNALeu knockout strain. Nucleic Acids Res. 2012;40(20):10463-77.
- Zuhlke V, Du Mesnil de R, Gudbjarnason S, Bing RJ. Inhibition of protein synthesis in cardiac hypertrophy and its relation to myocardial failure. Circ Res. 1966;18(5):558-72.
- Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol. 2012;8(3):311-7.
- Zhou H, Sun L, Yang XL, Schimmel P. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature. 2013;494(7435):121-4.
- Qin P, Arabacilar P, Bernard RE, Bao W, Olzinski AR, Guo Y, et al. Activation of the Amino Acid Response Pathway Blunts the Effects of Cardiac Stress. J Am Heart Assoc. 2017;6(5).
- Sundrud MS, Koralov SB, Feuerer M, Calado DP, Kozhaya AE, Rhule-Smith A, et al. Halofuginone inhibits TH17 cell differentiation by activating the amino acid starvation response. Science. 2009;324(5932):1334-8.
- Zhang T, Wu Y, Hu Z, Xing W, Kun LV, Wang D, et al. Small-Molecule Integrated Stress Response Inhibitor Reduces Susceptibility to Postinfarct Atrial Fibrillation in Rats via the Inhibition of Integrated Stress Responses. J Pharmacol Exp Ther. 2021;378(3):197-206.
- Onat UI, Yildirim AD, Tufanli O, Cimen I, Kocaturk B, Veli Z, et al. Intercepting the Lipid-Induced Integrated Stress Response Reduces Atherosclerosis. J Am Coll Cardiol. 2019;73(10):1149-69.
- Hedaya OM, Jiang F, Baliga U, Ivanov A, Chen S, Schwartz JL, et al. Upstream open reading frame inactivation augments GATA4 translation and cardiomyocyte hypertrophy in mice. bioRxiv. 2025.
- Xiang Y, Huang W, Tan L, Chen T, He Y, Irving PS, et al. Pervasive downstream RNA hairpins dynamically dictate start-codon selection. Nature. 2023;621(7978):423-30.
- Guenther UP, Weinberg DE, Zubradt MM, Tedeschi FA, Stawicki BN, Zagore LL, et al. The helicase Ded1p controls use of near-cognate translation initiation codons in 5' UTRs. Nature. 2018;559(7712):130-4.
- Cheng W, Wang S, Zhang Z, Morgens DW, Hayes LR, Lee S, et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron. 2019;104(5):885-98 e8.
- Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573(7774):430-3.
- Rurik JG, Tombacz I, Yadegari A, Mendez Fernandez PO, Shewale SV, Li L, et al. CAR T cells produced in vivo to treat cardiac injury. Science. 2022;375(6576):91-6.
- Chen H, Liu D, Aditham A, Guo J, Huang J, Kostas F, et al. Chemical and topological design of multicapped mRNA and capped circular RNA to augment translation. Nat Biotechnol. 2024.
- Anttila V, Saraste A, Knuuti J, Hedman M, Jaakkola P, Laugwitz KL, et al. Direct intramyocardial injection of VEGF mRNA in patients undergoing coronary artery bypass grafting. Mol Ther. 2023;31(3):866-74.
- Liang XH, Shen W, Sun H, Migawa MT, Vickers TA, Crooke ST. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat Biotechnol. 2016;34(8):875-80.
- Chen Q, Yu T, Gong J, Shan H. Advanced Nanomedicine Delivery Systems for Cardiovascular Diseases: Viral and Non-Viral Strategies in Targeted Therapy. Molecules. 2025;30(4).
- Philippou S, Mastroyiannopoulos NP, Tomazou M, Oulas A, Ackers-Johnson M, Foo RS, et al. Selective Delivery to Cardiac Muscle Cells Using Cell-Specific Aptamers. Pharmaceuticals (Basel). 2023;16(9).
- Narayan C, Lin LH, Barros MN, Gilbert TC, Brown CR, Reddin D, et al. Identification of In Vivo Internalizing Cardiac-Specific RNA Aptamers. bioRxiv. 2024.
- Albers S, Allen EC, Bharti N, Davyt M, Joshi D, Perez-Garcia CG, et al. Engineered tRNAs suppress nonsense mutations in cells and in vivo. Nature. 2023;618(7966):842-8.
- Chen R, Wang SK, Belk JA, Amaya L, Li Z, Cardenas A, et al. Engineering circular RNA for enhanced protein production. Nat Biotechnol. 2023;41(2):262-72.
- Carrieri C, Cimatti L, Biagioli M, Beugnet A, Zucchelli S, Fedele S, et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature. 2012;491(7424):454-7.
- Arnoldi M, Zarantonello G, Espinoza S, Gustincich S, Di Leva F, Biagioli M. Design and Delivery of SINEUP: A New Modular Tool to Increase Protein Translation. Methods Mol Biol. 2022;2434:63-87.
- Ford PW, Narasimhan M, Bennett EJ. Ubiquitin-dependent translation control mechanisms: Degradation and beyond. Cell Rep. 2024;43(12):115050.
- Santos-Ribeiro D, Godinas L, Pilette C, Perros F. The integrated stress response system in cardiovascular disease. Drug Discov Today. 2018;23(4):920-9.
- Sciarretta S, Forte M, Frati G, Sadoshima J. New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ Res. 2018;122(3):489-505.
- McGirr T, Onar O, Jafarnejad SM. Dysregulated ribosome quality control in human diseases. FEBS J. 2025;292(5):936-59.
- Ford PW, Garshott DM, Narasimhan M, Ge X, Jordahl EM, Subramanya S, et al. RNF10 and RIOK3 facilitate 40S ribosomal subunit degradation upon 60S biogenesis disruption or amino acid starvation. Cell Rep. 2025;44(3):115371.
- Garzia A, Meyer C, Tuschl T. The E3 ubiquitin ligase RNF10 modifies 40S ribosomal subunits of ribosomes compromised in translation. Cell Rep. 2021;36(5):109468.
- Clemens, MJ. Initiation factor eIF2 alpha phosphorylation in stress responses and apoptosis. Prog Mol Subcell Biol. 2001;27:57-89.
- Vanhoutte D, Schips TG, Vo A, Grimes KM, Baldwin TA, Brody MJ, et al. Thbs1 induces lethal cardiac atrophy through PERK-ATF4 regulated autophagy. Nat Commun. 2021;12(1):3928.
- Zhang S, Tian W, Duan X, Zhang Q, Cao L, Liu C, et al. Melatonin attenuates diabetic cardiomyopathy by increasing autophagy of cardiomyocytes via regulation of VEGF-B/GRP78/PERK signaling pathway. Cardiovasc Diabetol. 2024;23(1):19.
- Li RJ, He KL, Li X, Wang LL, Liu CL, He YY. Salubrinal protects cardiomyocytes against apoptosis in a rat myocardial infarction model via suppressing the dephosphorylation of eukaryotic translation initiation factor 2alpha. Mol Med Rep. 2015;12(1):1043-9.
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132-41.
- Li Y, Chen C, Yao F, Su Q, Liu D, Xue R, et al. AMPK inhibits cardiac hypertrophy by promoting autophagy via mTORC1. Arch Biochem Biophys. 2014;558:79-86.
- He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62(4):1270-81.
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914-22.
- Xie W, Du L. Diabetes is an inflammatory disease: evidence from traditional Chinese medicines. Diabetes Obes Metab. 2011;13(4):289-301.
- Kanamori H, Naruse G, Yoshida A, Minatoguchi S, Watanabe T, Kawaguchi T, et al. Metformin Enhances Autophagy and Provides Cardioprotection in delta-Sarcoglycan Deficiency-Induced Dilated Cardiomyopathy. Circ Heart Fail. 2019;12(4):e005418.
- Li C, Li S, Zhang G, Li Q, Song W, Wang X, et al. IRE1alpha Mediates the Hypertrophic Growth of Cardiomyocytes Through Facilitating the Formation of Initiation Complex to Promote the Translation of TOP-Motif Transcripts. Circulation. 2024;150(13):1010-29.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



