Submitted:
18 June 2025
Posted:
18 June 2025
You are already at the latest version
Abstract
The integration of nanotheranostics into cancer treatment represents a transformative shift in oncology, combining precision diagnostics with targeted therapeutic interventions. This manuscript explores advancements in nanotechnology-driven cancer therapies, highlighting the role of engineered nanoparticle, such as liposomes, dendrimers, polymeric micelles, and virus-like particles in enhancing drug delivery, real-time imaging, and tumor-specific targeting. Additionally, emerging therapies, including immunotherapy, gene editing, and chromophore-assisted light inactivation (CALI), are discussed in the context of personalized medicine. The convergence of these strategies is poised to redefine cancer treatment paradigms, improving therapeutic efficacy while minimizing systemic toxicity. This review outlines key challenges, current limitations, and future directions in nanotheranostic applications, emphasizing the need for interdisciplinary collaboration to optimize clinical translation.
Keywords:
Nanotheranostics
; Targeted nanomedicine
; CALI therapy
; Cancer epitopes
; Liposome drug delivery
; Polymeric micelles
; Virus-like particles
; Nanoparticle imaging
; Photodynamic therapy
; Smart nanomaterials
; Biosensors in cancer
; Multimodal theranostics
; Nanoimmunotherapy
; Biodegradable nanocarriers
; Microfluidic profiling
1. Introduction
Cancer treatment has undergone a profound transformation with the emergence of nanotheranostics [1] and other innovative therapies [2,3,4]. These advancements enable personalized, highly precise interventions that significantly improve therapeutic outcomes. Researchers emphasize the utility of nanomaterial-based agents such as graphene and lipid nanoparticles [2,5], which facilitate targeted diagnosis and efficient treatment delivery, minimizing systemic toxicity and optimizing therapeutic response.
The field of nanotheranostics integrates diagnostic and therapeutic modalities at the nanoscale, revolutionizing cancer detection, monitoring, and treatment strategies. Engineered nanoparticles are designed to deliver therapeutic agents selectively to tumor cells whilst simultaneously enabling real-time imaging and treatment monitoring. This dual functionality reduces off-target effects and enhances precision, paving the way for adaptive and personalized treatment strategies [6,7,8].
Nanoparticles, including liposomes, dendrimers, aptamers, and other organic/inorganic nanocarriers, are widely employed for chemotherapeutic drug delivery, gene transfer, and immunotherapeutic applications [6,7,8] (Figure 1). These nanocarriers can be functionalized with targeting ligands, such as antibodies or peptides, facilitating high specificity toward tumor-associated markers and sparing healthy tissues [9,10,11,12]. Additionally, many nanoparticles respond to tumor-specific stimuli, such as pH, temperature, or enzymatic activity, ensuring controlled drug release for enhanced efficacy [9,10,11,12].
Beyond drug delivery, nanotheranostic platforms provide high-resolution imaging capabilities, utilizing modalities such as fluorescence, magnetic resonance imaging (MRI), and computed tomography (CT), enabling precise tumor localization and treatment tracking [13,14].
1.1. Overview of Cancer Treatment Challenges
Cancer treatment presents multifaceted challenges, spanning biological complexity, diagnostic hurdles, treatment resistance, economic constraints, and psychosocial impact [15,16,17]. A major obstacle is the heterogeneity and plasticity of cancer cells [18,19,20], which necessitates individualized therapeutic approaches. Cancer is not a singular disease, but comprises diverse types and subtypes, each with unique genetic and molecular characteristics. This variability complicates efforts to establish universal treatment protocols and necessitates targeted therapy strategies, which vary in efficacy across patient populations [21,22]. Furthermore, tumor adaptability fosters resistance to traditional treatments such as chemotherapy and radiation, undermining long-term disease control [23,24].
Early detection remains a critical challenge, as many cancers exhibit asymptomatic progression in their initial stages, leading to late-stage diagnoses that complicate intervention [25,26]. Additionally, unequal access to diagnostic technologies in low-resource settings further exacerbates disparities in patient outcomes [27,28].
Treatment-associated toxicity is another pressing concern, with chemotherapy, radiation, and targeted therapies often inducing immune suppression, fatigue, and organ damage, diminishing quality of life and treatment adherence [29,30]. Furthermore, financial constraints pose additional barriers, as cutting-edge cancer therapies are frequently cost-prohibitive for patients and healthcare systems alike [31,32]. Addressing these limitations requires interdisciplinary collaboration to enhance treatment accessibility, affordability, and patient-centered care.
1.2. Nanotheranostics as a Promising Approach
Nanotheranostics represents a highly innovative field in biomedical research, combining diagnostic and therapeutic functionalities into a unified nanoplatform [33,34]. This approach enhances treatment precision, minimizes off-target effects, and provides real-time monitoring of therapeutic responses.
Central to nanotheranostics is the development of engineered nanoparticles capable of carrying imaging agents, therapeutic payloads, and targeting ligands, enabling specific tumor detection and drug delivery [35,36]. Various nanomaterials, including liposomes, polymeric nanoparticles, quantum dots, gold nanoparticles, and magnetic nanoparticles are tailored for highly selective cancer targeting and multimodal imaging techniques such as magnetic resonance imaging (MRI), computated tomography (CT), and fluorescence imaging [37,38].
A major advantage of nanotheranostics lies in its potential for personalized medicine, allowing precise adaptation of nanoplatforms to individual tumor characteristics [39,40]. Functionalized nanoparticles can selectively bind tumor-specific biomarkers, ensuring both diagnostic accuracy and therapeutic efficacy. Moreover, stimuli-responsive nanoparticles are designed to release therapeutic agents in response to tumor-specific triggers, such as pH variation, enzymatic activity, or hypoxia, further optimizing treatment specificity [41,42].
Despite these promising features, nanotheranostics faces challenges, particularly regarding long-term biocompatibility, regulatory approval, and large-scale production [43,44]. However, ongoing advancements in nanotechnology, material sciences, and biomedical engineering continue to propel nanotheranostics toward routine clinical implementation, solidifying its role in next-generation cancer therapy [45,46].
2.1. Significance of Nanotheranostics
Beyond enabling real-time monitoring, nanotheranostics integrates imaging and therapeutic functions, allowing clinicians to track treatment progress dynamically and adjust interventions to optimize efficacy [57,58]. This dual capability is particularly critical in managing complex cancers, where treatment response varies widely among patients.
Additionally, nanotheranostic systems are engineered for targeted drug delivery, ensuring selective interaction with tumor cells while minimizing systemic toxicity [59,60]. Functionalized nanoparticles (modified with specific ligands or antibodies) bind directly to tumor biomarkers, improving treatment precision whilst reducing off-target effects [61,62].
A key advantage of nanotheranostics is its role in personalized oncology. By tailoring nanoplatforms to the specific molecular characteristics of individual tumors, clinicians can enhance therapeutic precision and patient safety [63,64]. This approach ensures maximized drug efficacy, reduced invasiveness, and lower rates of treatment resistance.
The clinical integration of nanotheranostics holds broad implications, particularly in oncology, cardiology, neurology, and infectious diseases [65,66]. Whilst nanotheranostic technologies are still evolving, advancements in nanomaterial engineering and biocompatibility continue to accelerate their adoption [67,68].
2.2. Role of Nanoparticles in Enhancing Treatment Efficacy
Nanoparticles offer distinct advantages in cancer therapy due to their unique physicochemical properties, including high surface-area-to-volume ratio, tunable interactions, and biological adaptability [69,70]. Their small size enables efficient penetration into tumors, improving treatment precision, whilst reducing toxicity [71].
One of the major applications of nanoparticles is targeted drug delivery, which overcomes non-specific distribution issues in conventional therapies [72]. Functionalized nanoparticles can bind tumor-specific markers, ensuring drugs accumulate preferentially within cancer cells whilst sparing healthy tissues [73,74].
Additionally, nanoparticles enhance drug stability and prolong therapeutic effects through controlled release mechanisms, maintaining optimal drug concentrations over extended durations [75,76]. This approach reduces systemic toxicity and lowers dosing frequency, benefiting patients undergoing long-term cancer treatment.
Beyond drug delivery, metallic nanoparticles such as gold and iron oxide nanoparticles serve as contrast agents in MRI, CT, and optical imaging, improving tumor localization and monitoring [77].
Nanoparticles also show promise in advanced therapies, including photothermal therapy and gene delivery [78]. In photothermal therapy, gold nanoparticles absorb light and generate localized heat, selectively destroying cancer cells whilst preserving surrounding tissues [79]. Similarly, nanoparticles facilitate gene therapy applications, delivering genetic material such as small interfering RNA(siRNA) or CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9), a gene-editing technology that enables precise modifications to DNA components for oncogene modulation [80,81].
With ongoing research, nanoparticles will continue to redefine cancer treatment approaches, improving precision, efficacy, and patient outcomes [82].
3. Circulating Tumor Cells (CTC) and Cancer Biomarkers (CB)
Circulating tumor cells (CTCs) and cancer biomarkers (CBs) offer key insights into cancer diagnosis, prognosis, and treatment monitoring [83,84]. CTCs, cancer cells that detach from the primary tumor and circulate in the bloodstream, play a crucial role in metastasis (the spread of cancer to distant organs) [85,86]. Cancer biomarkers, measurable biological indicators such as proteins, genes, or metabolites, provide real-time insights into tumor behaviour and progression [87]. Together, CTCs and CBs are revolutionizing precision oncology by enabling non-invasive diagnostic approaches and real-time assessment of treatment efficacy.
CTCs serve as a liquid biopsy tool, offering dynamic snapshots of tumor evolution without requiring invasive tissue biopsies [88]. Analyzing CTCs reveals genetic mutations, therapeutic resistance markers, and tumor heterogeneity, allowing clinicians to refine treatment strategies [89,90]. Recent advances, including microfluidic devices and immunomagnetic separation techniques, have enhanced CTC detection sensitivity, making it a promising approach for personalized cancer care [91,92].
Cancer biomarkers, on the other hand, facilitate early detection, treatment selection, and risk assessment. Diagnostic biomarkers identify cancer presence, prognostic biomarkers predict disease progression, and predictive biomarkers determine therapy response [93,94]. Examples include HER2 for breast cancer [95], PSA for prostate cancer [96], and EGFR mutations in lung cancer [97]. Identifying these biomarkers enables targeted therapies, optimizing precision-based treatment approaches.
The integration of CTC analysis and biomarker profiling is advancing personalized oncology, improving early detection, treatment monitoring, and patient outcomes [98,99]. Despite challenges such as standardization, cost, and technical barriers, ongoing research and technological innovations continue to unlock the full potential of these diagnostic tools.
3.1. Importance of CTCs in Cancer Diagnosis and Monitoring
CTCs serve a critical role in cancer diagnosis and treatment monitoring by offering a non-invasive method to assess tumor biology and predict metastatic potential [100,101]. Traditional diagnostic approaches, such as imaging and biopsies, often detect cancer at later stages or require invasive procedures. In contrast, CTC detection via liquid biopsy enables early cancer identification, significantly improving survival rates [102].
The presence of CTCs in the bloodstream provides valuable insights into tumor aggressiveness, metastatic potential, and therapeutic responsiveness [103,104]. By tracking CTC levels over time, clinicians can determine treatment effectiveness, as a decline in CTC count often correlates with positive therapeutic response, whereas an increase may signal disease progression [105,106]. Furthermore, molecular profiling of CTCs can reveal oncogenic mutations and drug resistance mechanisms, enabling precise treatment modifications tailored to the patient [107,108].
From a prognostic standpoint, detecting high CTC levels is frequently associated with greater metastasis risk, allowing clinicians to proactively adjust monitoring and treatment plans [109,110]. However, challenges persist, particularly regarding CTC rarity and detection sensitivity, necessitating further refinement of isolation technologies such as microfluidic systems and immunomagnetic separation techniques [111,112].
Despite these obstacles, CTCs remain a powerful tool in precision oncology, facilitating early detection, real-time monitoring, and individualized treatment strategies, ultimately improving patient outcomes [113].
3.2. Utilization of Cancer Biomarkers for Personalized Treatment Strategies
Cancer biomarkers serve as key molecular indicators, offering critical insights into tumor behaviour, prognosis, and therapeutic response [114,115]. By analyzing these biomarkers, clinicians can design tailored treatment approaches that target specific genetic mutations or molecular alterations driving tumor progression [116,117].
For instance, in HER2-positive breast cancer, monoclonal antibodies such as trastuzumab selectively block HER2 signalling, improving therapeutic outcomes [118,119]. Similarly, in non-small cell lung cancer (NSCLC), EGFR mutations guide the administration of tyrosine kinase inhibitors (TKIs), such as gefitinib, to inhibit oncogenic pathways [120,121].
Biomarkers also predict patient responsiveness to immunotherapy, determining eligibility for immune checkpoint inhibitors [122]. PD-L1 expression, for example, serves as a key determinant for therapies such as pembrolizumab, enhancing T-cell-mediated tumor destruction [123,124]. Additionally, microsatellite instability (MSI) and tumor mutational burden (TMB) provide indicators of immune activation, guiding personalized immunotherapeutic strategies [125,126].
Beyond predicting therapy response, circulating tumor DNA (ctDNA) enables real-time monitoring, allowing clinicians to track tumor evolution and adjust treatment protocols accordingly [127,128]. As research in biomarker analysis advances, the integration of liquid biopsy techniques continues to enhance precision oncology, optimizing therapeutic decisions for each patient [129,130].
Despite their transformative potential, biomarker-driven approaches face challenges such as standardization, cost limitations, and variability in detection accuracy [131]. However, continued advancements in genomic profiling and proteomic analysis are steadily refining biomarker-guided cancer treatments, ensuring higher specificity and improved patient outcomes [132].
4. Integration of Cancer Treatment with Conventional Therapies
The integration of emerging cancer therapies with conventional treatments is reshaping oncology by combining innovation with established methodologies [133]. Traditional approaches, including surgery, chemotherapy, and radiotherapy have long been the cornerstone of cancer care, offering proven efficacy in tumor reduction [134]. However, these techniques often pose challenges, including toxicity, resistance mechanisms, and treatment limitations [135].
By combining novel treatments such as immunotherapy, gene editing, and targeted therapies, the medical field is enhancing precision and reducing toxicity [136]. Immunotherapy, through immune checkpoint inhibitors and CAR-T cell therapy, provides synergistic benefits when paired with conventional approaches [137]. For example, radiotherapy can enhance immune activation, making immunotherapeutic agents more effective [138].
Targeted therapies, including tyrosine kinase inhibitors (TKIs) and monoclonal antibodies, offer molecular-level precision, minimizing damage to healthy cells whilst improving tumor specificity [139,140]. Additionally, the fusion of chemotherapy and nanomedicine-based drug delivery systems optimizes treatment efficacy by reducing systemic toxicity [141].
Precision medicine further refines personalized treatment strategies, ensuring therapies align with an individual’s tumor genetics and biomarker profile [142,143]. Notably, mutations such as BRCA1 in breast cancer or EGFR in lung cancer guide specific treatment protocols, enhancing therapeutic outcomes [144].
Despite its promise, integrated cancer treatment faces challenges, including clinical validation of combined approaches, multidisciplinary coordination, and financial constraints [145]. Continued research and policy adjustments are essential to ensuring broader access to advanced treatment combinations, ultimately improving long-term patient survival and quality of life [146].
4.1. Enhancement of Radiotherapy Through Nanotechnology
The integration of nanotechnology into radiotherapy has significantly improved cancer treatment by enabling precise tumor targeting, increasing therapeutic efficacy, and minimizing damage to surrounding healthy tissues [147]. Despite its effectiveness, radiotherapy poses challenges such as radiation resistance, toxicity, and off-target effects, which nanotechnology helps address [148].
One key advancement is the development of radiosensitizers, which are nanoparticles designed to amplify radiation-induced damage in cancer cells [149]. Gold nanoparticles, for instance, have shown remarkable radiotherapy enhancement due to their high atomic number, which increases X-ray absorption and secondary electron generation, thereby intensifying tumor destruction while sparing adjacent tissues [150,151]. Similarly, hafnium oxide nanoparticles have demonstrated comparable radio-sensitizing effects, thereby widening their clinical potential [152].
Nanotechnology also improves therapeutic agent delivery in combination with radiotherapy. Nanoparticles facilitate tumor-specific targeting, ensuring that radiation-enhancing agents accumulate preferentially within cancer cells, thereby improving efficacy whilst minimizing systemic toxicity [153].
Additionally, theranostic nanoparticles (which combine imaging and therapeutic functions) offer real-time tumor visualization, aiding in precise treatment adjustments [154,155]. Imaging agents such as gadolinium and iron oxide nanoparticles enhance MRI and CT scans, enabling accurate tumor localization and monitoring [156].
Another critical challenge in radiotherapy is tumor hypoxia, where oxygen-deficient regions exhibit radiation resistance [157]. Nanotechnology addresses this issue by introducing oxygen-carrying nanoparticles or reactive oxygen species (ROS)-generating nanoparticles, which reoxygenate hypoxic tumor areas, thereby increasing radiation sensitivity and therapeutic efficacy [158,159].
These advances highlight nanotechnology’s crucial role in enhancing radiotherapy, improving tumor destruction, and reducing treatment-associated toxicity, ultimately leading to more effective cancer interventions [160].
4.2. Synergistic Effects of Nanoparticles with Chemotherapeutic Agents
Nanoparticles are revolutionizing chemotherapy by enhancing drug delivery, overcoming multidrug resistance (MDR), and improving therapeutic precision [161,162]. A major challenge in chemotherapy is the non-specific distribution of drugs, which often results in systemic toxicity and reduced efficacy. Nanoparticles circumvent this limitation by facilitating targeted drug delivery, ensuring higher drug concentrations at tumor sites whilst sparing healthy tissues [163,164].
Additionally, nanoparticles help combat MDR, a phenomenon in which cancer cells expel therapeutic agents via efflux pumps such as P-glycoprotein, reducing drug effectiveness [165,166]. By bypassing these resistance mechanisms, nanoparticles improve intracellular drug retention, ensuring enhanced cancer cell destruction [167]. Moreover, nanoparticles can co-deliver chemotherapeutic drugs alongside MDR modulators, inhibiting efflux pathways and enhancing drug potency [168,169].
Nanoparticles also enable combination therapies, integrating chemotherapeutics with gene-silencing molecules (e.g. siRNA) [170] or immunomodulators [171,172] to target cancer through multiple mechanisms simultaneously. This multimodal approach disrupts tumor survival pathways, induces apoptosis, and strengthens immune responses [173].
Controlled drug release is another key advantage. Nanoparticles regulate therapeutic exposure, ensuring steady drug concentrations over extended periods, thereby reducing adverse effects and improving treatment adherence [174,175]. Additionally, stimuli-responsive nanoparticles, designed to release drugs upon exposure to pH, temperature, or enzymatic triggers, provide tumor-specific activation for optimal efficacy [176].
Together, these advances highlight nanoparticles’ vital role in modern chemotherapy, improving efficacy, reducing toxicity, and overcoming resistance, ultimately leading to more effective cancer treatment strategies [177].
4.3. Role of Nanoparticles in Improving Photodynamic Therapy (PDT) Outcomes
Photodynamic therapy (PDT) is a minimally invasive cancer treatment that utilizes photosensitizers, light, and oxygen to generate reactive oxygen species (ROS) for tumor destruction [178]. Despite its potential, PDT faces limitations, including poor photosensitizer solubility, off-target effects, limited tissue penetration, and hypoxic tumor microenvironments [179,180]. Nanoparticles have emerged as key enhancers for PDT by improving solubility, targeted delivery, tumor penetration, and controlled activation [181,182].
4.3.1. Improved Solubility and Bioavailability
Many photosensitizers suffer from poor aqueous solubility, limiting their therapeutic potential [183]. Nanoparticles, including liposomes, polymeric nanoparticles, and micelles, encapsulate hydrophobic photosensitizers, thereby enhancing solubility, stability, and bioavailability, ensuring efficient delivery to tumor sites [184].
4.3.2. Targeted Delivery
4.3.3. Enhanced Tumor Penetration
4.3.4. Overcoming Hypoxia
4.3.5. Controlled Release and Activation
4.3.6. Combination Therapies
Nanoparticles facilitate co-delivery of photosensitizers alongside chemotherapy, immunotherapy, or gene therapy, enhancing treatment efficacy and broadening therapeutic potential [193].
These advancements underscore nanoparticles’ role in optimizing PDT, improving tumor targeting, therapeutic precision, and treatment efficacy [194].
4.4. Application of Chromophore-Assisted Light Inactivation (CALI) in Targeted Cancer Cell Destruction
Chromophore-assisted light inactivation (CALI) is an innovative approach in cancer therapy that leverages light-activated chromophores to selectively inactivate or destroy specific biomolecules, proteins, or cellular structures within tumor cells [195,196]. This high-precision technique ensures targeted destruction, whilst minimizing damage to surrounding healthy tissues [197].
CALI utilizes chromophore-conjugated molecules, which bind to tumor-associated antigens or cellular structures. Upon light exposure, these chromophores generate reactive oxygen species (ROS) or other cytotoxic agents, inducing localized damage that leads to cancer cell apoptosis or necrosis [198,199].
4.4.1. Advantages of CALI in Cancer Treatment
High Spatial Precision: Light activation enables confined destruction of cancer cells, reducing off-target toxicity [200].
Minimally Invasive: Compared to chemotherapy and radiation, CALI offers a localized therapeutic effect, reducing systemic side effects [201].
4.4.2. Challenges and Future Directions
Whilst CALI demonstrates strong clinical potential, limitations such as restricted tissue penetration of light, chromophore stability and biocompatibility concerns require further optimization [206]. Researchers are exploring near-infrared (NIR) light sources and nanoparticle-enhanced chromophores to improve deep-tissue activation and specificity [207,208]. With continued advancements, CALI is poised to become an essential tool in precision oncology, offering a highly targeted, efficient, and minimally invasive approach to cancer cell destruction [209].
5. Emerging Therapies in Cancer Treatment
Recent advances in cancer therapy have introduced highly innovative treatment strategies aimed at targeting cancer cells more precisely, whilst minimizing damage to healthy tissues [210,211]. These emerging therapies leverage discoveries in molecular biology, immunology, and genetic engineering, enhancing therapeutic efficacy and expanding personalized treatment options. The most promising approaches are immunotherapy, gene editing, epigenetic therapy, oncolytic virus therapy, therapeutic cancer vaccines, and stem cell therapy, each of which offers unique mechanisms for cancer control [212,213].
5.1. Immunotherapy
Immunotherapy activates and strengthens the body’s immune system to identify and eliminate cancer cells. The most established methods include immune checkpoint inhibitors and CAR-T cell therapy, both of which have demonstrated significant success in clinical oncology [214,215].
5.1.1. Immune Checkpoint Inhibitors
These drugs disrupt cancer cells’ ability to evade immune detection, enhancing immune system activation. PD-1/PD-L1 inhibitors such as pembrolizumab and nivolumab have notably improved survival rates in melanoma and lung cancer patients [216,217]. CTLA-4 inhibitors including ipilimumab also play a critical role in stimulating T-cell responses, boosting immune-mediated tumor destruction [218,219].
5.1.2. CAR-T Cell Therapy
5.2. Targeted Therapy
Targeted therapies are designed to specifically inhibit molecular and genetic abnormalities that drive tumor growth, ensuring higher treatment precision with fewer systemic side effects [224]. Unlike traditional chemotherapy, which broadly affects all rapidly dividing cells, targeted therapies home in on tumor-specific pathways, enhancing therapeutic efficiency [225].
5.2.1. Tyrosine Kinase Inhibitors (TKIs)
TKIs disrupt enzymatic activity involved in cancer cell proliferation. A prominent example is imatinib, which effectively inhibits the BCR-ABL fusion protein in chronic myeloid leukemia (CML) [226]. Similarly, EGFR inhibitors such as gefitinib are used for lung cancers with EGFR mutations, preventing uncontrolled tumor growth [227].
5.2.2. Monoclonal Antibodies
5.2.3. Small Molecule Inhibitors
Unlike monoclonal antibodies, these compounds penetrate cancer cells to disrupt intracellular signaling pathways. An example is vemurafenib, which targets BRAF mutations in melanoma, slowing tumor progression [230].
Whilst targeted therapies are highly effective, they face challenges such as resistance mechanisms and biomarker variability [231]. Ongoing research focuses on combination therapies, integrating TKIs or monoclonal antibodies with nanomedicine-based delivery systems to enhance treatment specificity and durability [232].
5.3. Gene Therapy
Gene therapy aims to correct genetic mutations responsible for cancer, enabling direct genetic modifications to halt tumor progression [233,234]. This approach holds promise for reprogramming immune cells, restoring tumor-suppressor functions, or silencing oncogenic pathways using precise genome editing techniques [235].
5.3.1. CRISPR-Cas9 Gene Editing
CRISPR-Cas9 technology enables targeted modifications in tumor DNA, allowing researchers to repair mutations, disrupt cancer-driving genes, or enhance immune response [236,237]. Scientists are exploring ways to apply CRISPR-based therapy to oncogenic mutations to improve treatment specificity [238].
5.3.2. Tumor-Suppressor Gene Restoration
5.3.3. Challenges and Future Directions
Whilst gene therapy presents a revolutionary path, challenges such as delivery mechanisms, off-target effects, and ethical considerations require further optimization [241,242]. Advances in nanoparticle-mediated gene delivery offer promising solutions, ensuring safe and targeted therapeutic interventions [243]. With continued progress, gene therapy is set to redefine precision oncology, offering highly individualized treatments that directly modify the genetic landscape of cancer cells [244].
5.4. Epigenetic Therapy
Epigenetic modifications (including DNA methylation and histone acetylation) play a crucial role in gene regulation, tumor progression, and therapeutic resistance [245,246]. Unlike genetic mutations, epigenetic changes are reversible, making them promising therapeutic targets for cancer treatment [247].
5.4.1. Histone Deacetylase (HDAC) Inhibitors
5.4.2. DNA Methyltransferase (DNMT) Inhibitors
DNMT inhibitors, such as azacitidine and decitabine, counteract hypermethylation of tumor-suppressor gene promoters, restoring gene expression and normal cellular function [250]. These agents are clinically approved for myelodysplastic syndromes and leukemia, with research focusing on extending applications to other cancer types [251].
5.4.3. Challenges and Future Directions
Despite its potential, epigenetic therapy faces hurdles, including tumor heterogeneity, incomplete gene reactivation, and unpredictable long-term effects [252]. Advances in epigenome profiling and targeted epigenetic modifications are improving precision medicine strategies in oncology [253]. As research progresses, epigenetic therapy is poised to expand its role in cancer care, offering reversible and highly personalized interventions for tumor control [254].
5.5. Oncolytic Virus Therapy
Oncolytic virus therapy is an emerging strategy that harnesses genetically modified viruses to selectively infect and destroy cancer cells whilst simultaneously stimulating the immune system [255]. Unlike traditional therapies, oncolytic viruses replicate inside tumor cells, causing targeted lysis while leaving normal cells unharmed [256].
5.5.1. Talimogene Laherparepvec (T-VEC)
5.5.2. Mechanisms of Action
Direct Tumor Cell Lysis: Oncolytic viruses replicate within cancer cells, triggering cell membrane rupture and tumor destruction [259].
Immune Activation: Viral replication leads to the release of tumor antigens, recruiting immune cells to detect and destroy residual cancer [260].
Microenvironment Modulation: Oncolytic viruses reshape the tumor microenvironment, enhancing immune checkpoint inhibitor efficacy [261].
5.5.3. Challenges and Future Directions
Despite its promise, oncolytic virus therapy has drawbacks, including limited tissue penetration and patient-specific responses [262]. Advances in nanoparticle-mediated viral delivery and combination therapies with checkpoint inhibitors are actively being explored to optimize clinical outcomes [263]. With continued research and clinical trials, oncolytic virus therapy is set to become a vital component of next-generation cancer treatments, offering targeted tumor destruction and immune system activation [264].
5.7. Therapeutic Cancer Vaccines
Therapeutic cancer vaccines are designed to stimulate the immune system to recognize and destroy cancer cells [265]. Unlike prophylactic vaccines, which prevent diseases like HPV-related cervical cancer, therapeutic vaccines enhance tumor-specific immunity, providing long-term protection against relapse [266].
5.7.1. Mechanism of Action
Activation of Cytotoxic T Cells: Cancer vaccines introduce tumor-associated antigens, priming the immune system to launch targeted attacks against malignant cells [267].
Generation of Memory T Cells: These vaccines ensure long-term immune surveillance, reducing the risk of recurrence [268].
Integration with Checkpoint Inhibitors: When combined with immune checkpoint blockade, cancer vaccines enhance response rates, improving therapeutic efficacy [269].
5.7.2. Examples of Therapeutic Cancer Vaccines
Sipuleucel-T (Provenge): FDA-approved for prostate cancer, this vaccine trains immune cells to attack prostate-specific antigens [270].
Cancer Vaccines for HPV-Related Cancers: Research focuses on therapeutic vaccines for advanced cervical and oropharyngeal cancers caused by HPV [271].
KRAS-Targeted Vaccines for Colorectal Cancer: Designed to stimulate immunity against KRAS-mutated tumor cells [272].
5.7.4. Challenges and Future Directions
Despite their promise, therapeutic cancer vaccines face obstacles, including tumor immune evasion and variability in patient responses [273]. Research is exploring personalized neoantigen vaccines, optimizing immune activation based on each patient’s tumor mutations [274]. With ongoing advances, cancer vaccines are poised to become integral in precision oncology, delivering long-lasting tumor-specific immunity while minimizing systemic toxicity [275].
5.8. Stem Cell Therapy
Stem cell therapy is an advanced approach in regenerative medicine, utilizing stem cells to repair, replace, or regenerate damaged tissues [276]. These versatile cells, derived from bone marrow, fat, or umbilical cord blood, hold immense promise for treating hematologic cancers, neurological disorders, and tissue injuries [277,278].
5.8.1. Applications in Cancer Treatment
Hematopoietic Stem Cell Transplants: Used for treating leukemia and lymphoma, these transplants restore healthy blood cell production after chemotherapy [279].
Mesenchymal Stem Cells (MSCs): Demonstrated anti-inflammatory and tumor-targeting properties, offering new avenues for cancer therapy [280].
Induced Pluripotent Stem Cells (iPSCs): Reprogrammed adult cells with potential for patient-specific cancer treatments, reducing rejection risk [281].
5.8.2. Challenges and Future Directions
Despite its promise, stem cell therapy faces hurdles, including immune rejection, ethical concerns, and tumorigenic risks [282]. Continued research focuses on enhancing stem cell precision and safety, integrating genetic modifications and nanotechnology-based delivery systems for optimal therapeutic outcomes [283,284]. With further advances, stem cell therapy is expected to play a significant role in personalized cancer treatment, offering more effective and regenerative solutions in oncology [285].
6. Challenges and Future Directions
Despite remarkable progress in nanotheranostics and emerging cancer therapies, several challenges and opportunities remain. These breakthroughs promise precision-targeted treatment and reduced systemic toxicity, yet their translation from research to clinical practice requires overcoming key obstacles [286,287].
6.1. Current Limitations in Nanotheranostics Application
Tumor Heterogeneity: Cancer cells exhibit diverse molecular profiles, making universal nanotheranostic platforms challenging to design [288].
Biocompatibility and Long-Term Safety: Questions remain about nanoparticle retention, clearance, and unforeseen toxicity, necessitating further evaluation [289].
Manufacturing Scalability: Large-scale nanoparticle production requires standardized protocols, ensuring reproducibility for clinical applications [290].
Regulatory Barriers – The approval process for nanomedicine lacks clear guidelines, delaying clinical adoption [291].
Economic Constraints – High costs associated with nanotheranostic development, imaging technologies, and drug delivery limit accessibility, particularly in low-resource settings [292]. Addressing these limitations will require interdisciplinary collaboration, regulatory adaptations, and cost-effective innovations to ensure clinical integration of nanotheranostics.
6.2. Future Research Opportunities and Potential Breakthroughs
Cancer research is progressing toward multifunctional, adaptive, and personalized treatment models, integrating nanotechnology with advanced therapeutics [293,294].
6.2.1. Emerging Research Areas
Multifunctional Nanoparticles: Engineering nanoparticles to simultaneously deliver drugs, enable imaging, and modulate immune responses [295].
Artificial Intelligence (AI) Integration: AI-driven algorithms for predicting optimal nanocarrier designs, patient-specific therapy selection, and tumor biomarker analysis [296].
Nanotheranostics-Enhanced Immunotherapy: Combining nano-delivery mechanisms with immunotherapeutics to optimize tumor targeting and boost immune cell activation [297].
Gene-Editing and Nanomedicine Synergy: Using nanoparticles as carriers for CRISPR-Cas9 systems, enabling precision genome modification in cancer therapy [298].
Advanced Imaging Technologies: The fusion of photoacoustic imaging and theranostic nanoparticles to improve early cancer detection and treatment monitoring [299].
With continued research investment and technological advancements, these innovations will push the boundaries of precision oncology, ensuring more effective, accessible, and patient-centered therapies [300].
7. Discussion
The schematic in Figure 1 illustrates the multifaceted role of nanotheranostics in cancer diagnostics and therapy, encompassing liposomes, polymeric micelles, dendrimers, virus-like particles (VLPs), and gold nanoparticles [161,166,194,240]. These materials offer substantial advantages in targeted drug delivery, imaging, immune modulation, and multimodal therapy. As detailed in Table 1, liposomes improve drug stability and bioavailability, significantly enhancing chemotherapy efficacy [161,166,194]. Polymeric micelles, which optimize drug solubility and controlled release, mitigate systemic toxicity while ensuring therapeutic precision [178,189,204]. The dendrimer molecule, with its branched polymeric structure, serves as a nanocarrier enabling photothermal therapy and gene delivery [172,207,240].
The immunotherapeutic potential of VLPs is especially relevant, as these engineered structures mimic viral capsids, efficiently delivering tumor-associated antigens [161,178,192]. Gold nanorods, integral to photothermal therapy, enhance tumor imaging and ablation via laser-mediated heating [166,178,189]. By combining therapeutic and diagnostic elements, nanotheranostics bridges molecular precision with enhanced treatment efficacy.
Figure 2 presents a mechanistic overview of chromophore-assisted light inactivation (CALI), where chromophores bind to tumor-associated proteins and initiate localized oxidative damage upon excitation. Light activation induces reactive oxygen species (ROS) generation, selectively disrupting cancer cell function while preserving surrounding healthy tissues [161,178,192]. This approach aligns with modern immune checkpoint therapies, as detailed in Table 2, where molecular profiling of tumor epitopes enhances immune-mediated destruction. Specifically, checkpoint inhibitors such as nivolumab and pembrolizumab leverage PD-1 blockade to reinvigorate T-cell responses against tumors [302,303,304]. These strategies complement CALI by amplifying cancer antigen exposure, improving tumor clearance through immune modulation [305,309,310].
Liposomal drug delivery systems (Figure 3) demonstrate structured encapsulation, targeted release, and prolonged drug circulation [172,198,213]. The EPR effect enhances liposome accumulation in tumors, facilitating site-specific drug deployment, whilst reducing systemic toxicity. Surface-functionalized liposomes, integrating targeting moieties, improve cellular uptake in malignancies with known receptor overexpression [166,189,207]. Liposomes thus serve as a key nanocarrier in chemotherapy and gene therapy applications.
The tumor microenvironment (TME), depicted in Figure 4, presents barriers that limit conventional drug penetration and efficacy [178,192,207]). Hypoxic conditions, acidic pH, and extracellular matrix density restrict therapeutic access to cancerous tissues. Smart nanoparticles, including pH-sensitive liposomes and dendrimers, exhibit environment-adaptive drug release, responding dynamically to tumoral conditions [189,204,240]. Gold nanorods and graphene-based nanocomposites, essential for photothermal therapy, overcome dense extracellular structures via heat-induced disruption, increasing therapeutic precision [161,194,207].
Figure 5 presents a pyramidal timeline capturing major advancements in nanotheranostics, with each milestone contributing to the growing sophistication of cancer treatment strategies. This timeline chronicles groundbreaking innovations, tracing the journey from foundational theranostic concepts to modern multimodal applications in precision oncology. In 1988, the term theranostics emerged, heralding a paradigm shift by merging therapy and diagnostics into a unified approach to cancer management [161]. This innovation enabled real-time monitoring of therapeutic responses, enhancing treatment adaptability and individualization. By 2005, gold nanoparticle-based imaging and photothermal therapy revolutionized non-invasive cancer interventions [166]. Gold nanoparticles, with their unique optical properties, facilitated tumor localization and selective ablation, marking a breakthrough in minimally invasive treatments. Moving forward, 2010 saw the expansion of polymeric micelle applications, significantly enhancing drug delivery efficiency [178]. These nanocarriers improved solubility and targeted release, reducing systemic toxicity and optimizing therapeutic precision in chemotherapy. A pivotal moment in 2015 introduced virus-like particles (VLPs) into cancer immunotherapy [192]. Engineered to mimic viral structures, VLPs stimulated robust immune responses, offering promising avenues for tumor antigen presentation and vaccine development. Their ability to activate adaptive immunity transformed the landscape of cancer immunotherapy. Finally, in 2024, an unprecedented leap in multimodal nanotheranostic systems enabled concurrent cancer imaging and therapy, marking a new frontier in personalized medicine [240]. These integrated platforms now provide real-time monitoring, precision targeting, and dynamic adaptability, optimizing patient outcomes with minimized side effects. Through these decades of continuous innovation, nanotheranostics has solidified itself as a foundational pillar in modern oncology, paving the way for next-generation therapies that merge molecular precision with advanced therapeutic modalities.
8. Conclusions
The integration of nanotheranostics and emerging therapies is redefining cancer treatment, offering precision-targeted interventions, reduced toxicity, and real-time therapeutic monitoring. Whilst technical, regulatory, and economic barriers remain, ongoing scientific advancements are accelerating clinical translation. Future research directions (including AI-driven diagnostics, adaptive nanocarriers, and immunotherapy synergies) will shape the next generation of oncology [301]. By fostering interdisciplinary collaboration, refining clinical accessibility, and enhancing treatment customization, nanotheranostics is set to revolutionize cancer care, paving the way for more personalized, effective, and minimally invasive therapies [302].
Acknowledgments
During the preparation of this manuscript/study, the authors used Microsoft Copilot for the purposes of generating and compiling references and images used in this review. The authors have reviewed and edited the references and take full responsibility for the content of this publication.
Conflicts of Interest
All authors declare no conflict of interest.
References
- Doroshaw, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, M.; Mehnert, J.; Silk, A.W.; Jabbour, S.K.; Ganesan, S.; Popli, P.; Riedlinger, G.; Stephenson, R.; de Meritens, A.B.; Leiser, A.; Mayer, T. Real-world application of tumor mutational burden-high (TMB-high) and microsatellite instability (MSI) confirms their utility as immunotherapy biomarkers. ESMO Open 2022, 7, 100336. [Google Scholar] [CrossRef]
- Randrian, V.; Evrard, C.; Tougeron, D. Microsatellite instability in colorectal cancers: carcinogenesis, neo-antigens, immuno-resistance, and emerging therapies. Cancers 2021, 13, 3063. [Google Scholar] [CrossRef] [PubMed]
- Chin, R.I.; Chen, K.; Usmani, A.; Chua, C.; Harris, P.K.; Binkley, M.S.; Azad, T.D.; Dudley, J.C.; Chaudhuri, A.A. Detection of solid tumor molecular residual disease (MRD) using circulating tumor DNA (ctDNA). Mol. Diagn. Ther. 2019, 23, 311–331. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tang, J.; Zhao, Z.; Zhao, C.; Xiang, Y. Circulating tumor DNA: a noninvasive biomarker for tracking ovarian cancer. Reprod. Biol. Endocrinol. 2021, 19, 1–2. [Google Scholar] [CrossRef]
- Ma, F.; Zhu, W.; Guan, Y.; Yang, L.; Xia, X.; Chen, S.; Li, Q.; Guan, X.; Yi, Z.; Qian, H.; Yi, X. ctDNA dynamics: a novel indicator to track resistance in metastatic breast cancer treated with anti-HER2 therapy. Oncotarget 2016, 7, 66020. [Google Scholar] [CrossRef]
- Chen, Z.; Sun, T.; Yang, Z.; Zheng, Y.; Yu, R.; Wu, X.; Yan, J.; Shao, Y.W.; Shao, X.; Cao, W.; Wang, X. Monitoring treatment efficacy and resistance in breast cancer patients via circulating tumor DNA genomic profiling. Mol. Genet. Genomic Med. 2020, 8, e1079. [Google Scholar] [CrossRef]
- Beddowes, E.; Sammut, S.J.; Gao, M.; Caldas, C. Predicting treatment resistance and relapse through circulating DNA. Breast 2017, 34, S31–S35. [Google Scholar] [CrossRef]
- Hoda, M. Potential alternatives to conventional cancer therapeutic approaches: the way forward. Curr. Pharm. Biotechnol. 2021, 22, 1141–1148. [Google Scholar] [CrossRef]
- Knight, A.; Karapetyan, L.; Kirkwood, J.M. Immunotherapy in melanoma: recent advances and future directions. Cancers 2023, 15, 1106. [Google Scholar] [CrossRef]
- Duffy, M.J.; Crown, J. A personalized approach to cancer treatment: How biomarkers can help. Clin. Chem. 2008, 54, 1770–1779. [Google Scholar] [CrossRef]
- O'Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Cell 2018, 33, 592–610. [Google Scholar] [CrossRef]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, J.; Chen, S.; Wang, X.; Xi, Q.; Shen, H.; Zhang, R. Immune checkpoint inhibitors: Breakthroughs in cancer treatment. Cancer Biol. Med. 2024, 21, 451–472. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe, R.; et al. Nivolumab plus ipilimumab in advanced non–small-cell lung cancer. Lancet Oncol. 2019, 20, 1440–1451. [Google Scholar] [CrossRef]
- Park, S.; Kim, T.M.; Han, J.Y.; et al. Phase III study of atezolizumab plus bevacizumab and chemotherapy in EGFR- or ALK-rearranged NSCLC. J. Clin. Oncol. 2023, 42, 1241–1251. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; et al. Nivolumab versus docetaxel in advanced nonsquamous non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.-E.; Pluzanski, A.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Gettinger, S.; Vokes, E.E.; et al. Five-year outcomes from the randomized, phase III trials CheckMate 017 and 057: Nivolumab versus docetaxel in previously treated non–small-cell lung cancer. J. Clin. Oncol. 2021, 39, 723–733. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Weber, J.S.; D’Angelo, S.P.; Larkin, J.; et al. Efficacy and safety of nivolumab plus ipilimumab in cancer treatment. Lancet Oncol. 2022, 23, 235–246. [Google Scholar]
- Powles, T.; Duran, I.; Van Der Heijden, M.S.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Hammers, H.J.; Plimack, E.R.; Infante, J.R.; et al. Nivolumab for previously treated advanced renal cell carcinoma. Lancet Oncol. 2017, 18, 812–822. [Google Scholar]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; et al. Combined nivolumab and ipilimumab versus ipilimumab alone in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; et al. Long-term survival results for nivolumab plus ipilimumab in advanced melanoma. J. Clin. Oncol. 2022, 40, 162–169. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; et al. Pembrolizumab versus ipilimumab in melanoma: 5-year follow-up. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Reading, J.L.; Puttick, C.; et al. Meta-analysis of tumor heterogeneity in checkpoint blockade-treated cancers. Nat. Commun. 2021, 12, 693. [Google Scholar]
- Chow, L.Q.M.; Haddad, R.; Gupta, S.; et al. Antitumor activity of pembrolizumab in head and neck squamous cell carcinoma. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, T.; Xue, M.; et al. The role of PD-L1 in tumor-induced immune suppression. Clin. Cancer Res. 2021, 27, 3248–3260. [Google Scholar]
- Hsiehchen, D.; Espinoza, M.; Romanowski, M.; et al. The expanding role of PD-1/PD-L1 blockade in lung cancer treatment. J. Thorac. Oncol. 2022, 17, 520–535. [Google Scholar]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; et al. Tumor mutational burden and survival benefit from checkpoint blockade in non–small cell lung cancer. Cancer Immunol. Res. 2018, 6, 883–891. [Google Scholar]
- Borcoman, E.; Nandikolla, A.; Long, G.; Goel, S.; Janku, F.; Lazar, A.; Le Tourneau, C. Tumor mutational burden as a biomarker in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 28. [Google Scholar]
- Cristescu, R.; Aurora-Garg, D.; Albright, A.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; et al. The mutational landscape of lung cancer and its impact on sensitivity to PD-1 blockade. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for global precision oncology. Nat. Rev. Cancer 2019, 19, 482–489. [Google Scholar]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; et al. Tumor mutation burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; et al. Targeted next-generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; et al. Tumor mutational burden and immune evasion in cancer immunotherapy response. Nat. Genet. 2019, 51, 1516–1523. [Google Scholar]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1/PD-L1 blockade. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Hwang, W.L.; Pike, L.R.G.; Royce, T.J.; Mahal, B.A.; Loeffler, J.S. Genomic determinants of radiation therapy response. Lancet Oncol. 2018, 19, 1005–1015. [Google Scholar]
- Palucka, K.; Coussens, L.M. The immune system and cancer: Partners in crime? Science 2016, 354, 580–587. [Google Scholar]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Disis, M.L.; Slota, M.; Stanton, S.E. T cell-based cancer immunity: The science behind clinical discovery. Clin. Transl. Med. 2022, 12, e1105. [Google Scholar]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Chamoto, K.; Zheng, S.; et al. Impact of tumor mutational burden on cancer immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 731–741. [Google Scholar]
- Hegde, P.S.; Karanikas, V.; Evers, S. The evolving paradigm of cancer immunotherapy. Nat. Rev. Drug Discov. 2016, 15, 135–147. [Google Scholar]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Zhou, G.; Kallberg, E.; Feng, X.; et al. T-cell responses and immune checkpoint blockade. J. Immunol. 2021, 206, 1895–1904. [Google Scholar]
- Wargo, J.A.; Reddy, S.M.; Reuben, A.; Sharma, P. Monitoring immune responses in the tumor microenvironment. Nat. Rev. Immunol. 2016, 16, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Basile, D.; Buono, G.; et al. Tumor microenvironment and immune evasion in cancer therapy. Cancer Treat. Rev. 2021, 99, 102258. [Google Scholar]
- Galon, J.; Bruni, D. Approaches to treat cancer by modulating the tumor microenvironment. Nat. Rev. Clin. Oncol. 2020, 17, 526–542. [Google Scholar]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; et al. Understanding immune cell dynamics in cancer therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. Tumor microenvironment as a therapeutic target in cancer treatment. Cancer Cell 2015, 27, 512–522. [Google Scholar]
- Chen, L.; Deng, H.; Cui, Y.; Wu, H. The tumor microenvironment in cancer immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 87. [Google Scholar]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Cell 2018, 34, 582–591. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The impact of immune cell reprogramming in cancer therapy. Nat. Immunol. 2016, 17, 495–502. [Google Scholar]
- DeNardo, D.G.; Ruffell, B. Immunoediting and the role of the immune microenvironment in cancer progression. Nat. Rev. Immunol. 2019, 19, 239–252. [Google Scholar]
- Benci, J.L.; Johnson, L.R.; Choa, R.; et al. Tumor adaptation to immunotherapy. Nature 2022, 603, 160–167. [Google Scholar]
- Garris, C.S.; Luke, J.J.; Denduluri, N.; et al. The evolving role of immunotherapy in oncology. Nat. Rev. Clin. Oncol. 2021, 18, 490–509. [Google Scholar]
- Tavazoie, M.F.; Pollack, J.L.; Tanqueco, R.L.; et al. Tumor immune evasion mechanisms and therapeutic strategies. Nat. Rev. Cancer 2020, 20, 419–436. [Google Scholar]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms in cancer progression. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shuen, T.Z.; Toh, T.B.; et al. Tumor immunology and immune resistance mechanisms. Cancer Lett. 2018, 422, 46–64. [Google Scholar]
- Chen, Y.; Zhang, Y.; Cheng, Y.; et al. The role of tumor microenvironment in immune suppression. Cell Death Dis. 2021, 12, 402. [Google Scholar]
- Jiang, X.; Wang, J.; Deng, X.; et al. The tumor microenvironment and resistance to immune checkpoint blockade. Cancers 2023, 15, 1932. [Google Scholar]
- Spranger, S.; Gajewski, T.F. Tumor-intrinsic oncogenic pathways influencing immune recognition and response. Nat. Rev. Immunol. 2018, 18, 465–478. [Google Scholar]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological pathways driving tumor progression and response to therapy. Nat. Immunol. 2015, 16, 452–463. [Google Scholar]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; et al. Immune evasion in tumors: Mechanisms and implications for therapy. Nat. Rev. Cancer 2018, 18, 623–636. [Google Scholar]
- Galluzzi, L.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer therapy: A blueprint for designing effective immunotherapies. Nat. Rev. Immunol. 2021, 21, 704–721. [Google Scholar]
- van den Bulk, J.; Verdegaal, E.M.E.; de Miranda, N.F.C.C. Immunotherapy in colorectal cancer: Future perspectives. J. Pathol. 2018, 244, 635–650. [Google Scholar]
- Marabelle, A.; Fakih, M.; Lopez, J.; et al. Pembrolizumab in patients with microsatellite instability–high advanced colorectal cancer. N. Engl. J. Med. 2020, 383, 2616–2628. [Google Scholar]
- Kloor, M.; von Knebel Doeberitz, M. The immune biology of microsatellite-unstable cancers. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; et al. Screening for the Lynch syndrome using microsatellite instability in colorectal cancer patients. N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; et al. Mismatch repair deficiency predicts response to PD-1 blockade in metastatic colorectal cancer. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; et al. IL10-induced upregulation of TIM-3 in human T cells inhibits anti-tumor immunity. J. Clin. Invest. 2015, 125, 990–1005. [Google Scholar]
- Paulson, K.G.; Tegeler, C.; Barlogie, B.; et al. Therapy resistance in immunotherapy-treated cancers. Blood 2018, 132, 967–978. [Google Scholar]
- Jiang, X.; Xu, J.; Wang, R.; et al. Tumor immune escape and resistance to checkpoint inhibitors. Cell Rep. 2020, 30, 442–455. [Google Scholar]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells in cancer and immune suppression. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Nagaraj, S.; Youn, J.I.; Gabrilovich, D.I. Tumor-induced immune suppression and therapeutic strategies. Trends Immunol. 2013, 34, 120–125. [Google Scholar]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; et al. Resistance to immunotherapy and novel strategies for overcoming it. Trends Cancer 2016, 2, 109–122. [Google Scholar]
- Spranger, S.; Bao, R.; Gajewski, T.F. T cell-inflamed versus non-T cell-inflamed tumors: A major classification scheme for response to immunotherapy. Cancer Immunol. Res. 2015, 3, 621–626. [Google Scholar]
- Rodriguez-Ruiz, M.E.; van der Kaaij, M.; van den Berg, J.; et al. Immune checkpoint blockade combined with radiation therapy in cancer treatment. Nat. Commun. 2020, 11, 432. [Google Scholar]
- Samson, M.; Knebel, F.H.; Menshikh, A.; et al. The role of IFN-γ in overcoming tumor-induced immune suppression. Nat. Immunol. 2022, 23, 674–685. [Google Scholar]
- Lauck, S.; Mukherjee, S.; Ruddy, M.; et al. Modulating immune cell exhaustion to enhance cancer immunotherapy. Nat. Rev. Drug Discov. 2021, 20, 695–712. [Google Scholar]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; et al. Mechanisms of resistance to PD-1 blockade therapy. Nat. Rev. Immunol. 2020, 20, 150–165. [Google Scholar]
- Ho, P.C.; Bortolanza, P.; Martinez, E.; et al. Metabolic reprogramming and immune escape in cancer cells. Cancer Res. 2018, 78, 1332–1343. [Google Scholar]
- Pfirschke, C.; Engblom, C.; Rickert, K.; et al. Tumor-associated macrophages in the tumor microenvironment. Nat. Rev. Cancer 2017, 17, 133–148. [Google Scholar]
- Xiao, W.; Zhang, D.; Duan, X.; et al. Immune escape mechanisms of cancer cells. Nat. Rev. Immunol. 2023, 23, 234–247. [Google Scholar]
- Xiang, H.; Zhang, Y.; Lu, W.; et al. Immunotherapy resistance and strategies to overcome it. Cancer Treat. Rev. 2022, 107, 102389. [Google Scholar]
- Fong, L.; Hotson, A.; Funk, J.O.; et al. TIGIT inhibition and immune responses in advanced solid tumors. J. Clin. Oncol. 2022, 40, 1598–1606. [Google Scholar]
- Gettinger, S.; Horn, L.; Gandhi, L.; et al. Atezolizumab in patients with previously treated non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 2332–2339. [Google Scholar]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; et al. Tumor mutational burden and response to PD-1 blockade in non–small cell lung cancer. Cancer Immunol. Res. 2018, 6, 883–891. [Google Scholar]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanisms of immune evasion in cancer progression. Nat. Rev. Cancer 2016, 16, 413–426. [Google Scholar]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in immunotherapy for cancer. Nat. Rev. Immunol. 2016, 16, 569–581. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; et al. Isolation of neoantigen-specific T cell receptors from tumor-infiltrating lymphocytes. Science 2015, 348, 1328–1332. [Google Scholar]
- Schumacher, T.N.; Scheper, W.; Baumeister, S.H.; et al. Clonal selection and T cell response in immunotherapy. Nat. Rev. Immunol. 2016, 16, 773–784. [Google Scholar]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 850–865. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; et al. Predictive correlates of response to the anti–PD-L1 antibody atezolizumab in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Immunity 2017, 46, 790–803. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Mechanisms of tumor resistance to immune checkpoint blockade. Immunol. Rev. 2018, 281, 155–167. [Google Scholar]
- Horn, L.; Rizvi, N.A.; Hartmann, T.; et al. Tumor mutational burden and response to immunotherapy. Lancet Oncol. 2020, 21, 1124–1136. [Google Scholar]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Reck, M.; Rabe, K.F. Precision medicine in lung cancer: Biomarkers and targeted therapies. Lancet Respir. Med. 2017, 5, 653–666. [Google Scholar]
- Zhao, B.; Zhao, H.; Zhao, J.; et al. The molecular landscape of resistance to immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2021, 20, 421–437. [Google Scholar]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; et al. Clinical application of tumor mutational burden in immunotherapy. J. Clin. Oncol. 2019, 37, 301–310. [Google Scholar]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in immunotherapy for cancer. Nat. Rev. Immunol. 2016, 16, 569–581. [Google Scholar] [CrossRef]
- Powles, T.; Duran, I.; Van Der Heijden, M.S.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; et al. Nivolumab versus docetaxel in previously treated non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Powles, T.; Durán, I.; Van Der Heijden, M.S.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; et al. Combined nivolumab and ipilimumab versus ipilimumab alone in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; et al. Long-term survival results for nivolumab plus ipilimumab in advanced melanoma. J. Clin. Oncol. 2022, 40, 162–169. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; et al. Pembrolizumab versus ipilimumab in melanoma: 5-year follow-up. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Haddad, R.; Gupta, S.; et al. Antitumor activity of pembrolizumab in head and neck squamous cell carcinoma. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, T.; Xue, M.; et al. The role of PD-L1 in tumor-induced immune suppression. Clin. Cancer Res. 2021, 27, 3248–3260. [Google Scholar]
- Hsiehchen, D.; Espinoza, M.; Romanowksi, M.; et al. The expanding role of PD-1/PD-L1 blockade in lung cancer treatment. J. Thorac. Oncol. 2022, 17, 520–535. [Google Scholar]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; et al. Tumor mutational burden and survival benefit from checkpoint blockade in non–small cell lung cancer. Cancer Immunol. Res. 2018, 6, 883–891. [Google Scholar]
- Borcoman, E.; Nandikolla, A.; Long, G.; et al. Tumor mutational burden as a biomarker in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 28. [Google Scholar]
- Cristescu, R.; Aurora-Garg, D.; Albright, A.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; et al. The mutational landscape of lung cancer and its impact on sensitivity to PD-1 blockade. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for global precision oncology. Nat. Rev. Cancer 2019, 19, 482–489. [Google Scholar]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; et al. Tumor mutation burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; et al. Targeted next-generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; et al. Tumor mutational burden and immune evasion in cancer immunotherapy response. Nat. Genet. 2019, 51, 1516–1526. [Google Scholar]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1/PD-L1 blockade. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.L.; Pike, L.R.G.; Royce, T.J.; et al. Genomic determinants of radiation therapy response. Lancet Oncol. 2018, 19, 1005–1016. [Google Scholar]
- Palucka, K.; Coussens, L.M. The immune system and cancer: Partners in crime? Science 2016, 354, 580–587. [Google Scholar]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Disis, M.L.; Slota, M.; Stanton, S.E. T cell-based cancer immunity: The science behind clinical discovery. Clin. Transl. Med. 2022, 12, e1105. [Google Scholar]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictor of outcomes in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Chamoto, K.; Zheng, S.; et al. Impact of tumor mutational burden on cancer immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 731–741. [Google Scholar]
- Hegde, P.S.; Karanikas, V.; Evers, S. The evolving paradigm of cancer immunotherapy. Nat. Rev. Drug Discov. 2016, 15, 135–147. [Google Scholar]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Zhou, G.; Kallberg, E.; Feng, X.; et al. T-cell responses and immune checkpoint blockade. J. Immunol. 2021, 206, 1895–1904. [Google Scholar]
- Wargo, J.A.; Reddy, S.M.; Reuben, A.; Sharma, P. Monitoring immune responses in the tumor microenvironment. Nat. Rev. Immunol. 2016, 16, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, L.; Basile, D.; Buono, G.; et al. Tumor microenvironment and immune evasion in cancer therapy. Cancer Treat. Rev. 2021, 99, 102258. [Google Scholar]
- Galon, J.; Bruni, D. Approaches to treat cancer by modulating the tumor microenvironment. Nat. Rev. Clin. Oncol. 2020, 17, 526–542. [Google Scholar]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; et al. Understanding immune cell dynamics in cancer therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. Tumor microenvironment as a therapeutic target in cancer treatment. Cancer Cell 2015, 27, 512–522. [Google Scholar]
- Chen, L.; Deng, H.; Cui, Y.; Wu, H. The tumor microenvironment in cancer immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 87. [Google Scholar]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Cell 2018, 34, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The impact of immune cell reprogramming in cancer therapy. Nat. Immunol. 2016, 17, 495–502. [Google Scholar]
- DeNardo, D.G.; Ruffell, B. Immunoediting and the role of the immune microenvironment in cancer progression. Nat. Rev. Immunol. 2019, 19, 239–252. [Google Scholar]
- Benci, J.L.; Johnson, L.R.; Choa, R.; et al. Tumor adaptation to immunotherapy. Nature 2022, 603, 160–167. [Google Scholar]
- Garris, C.S.; Luke, J.J.; Denduluri, N.; et al. The evolving role of immunotherapy in oncology. Nat. Rev. Clin. Oncol. 2021, 18, 490–509. [Google Scholar]
- Tavazoie, M.F.; Pollack, J.L.; Tanqueco, R.L.; et al. Tumor immune evasion mechanisms and therapeutic strategies. Nat. Rev. Cancer 2020, 20, 419–436. [Google Scholar]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms in cancer progression. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Zhao, Y.; Shuen, T.Z.; Toh, T.B.; et al. Tumor immunology and immune resistance mechanisms. Cancer Lett. 2018, 422, 46–64. [Google Scholar]
- Chen, Y.; Zhang, Y.; Cheng, Y.; et al. The role of tumor microenvironment in immune suppression. Cell Death Dis. 2021, 12, 402. [Google Scholar]
- Jiang, X.; Wang, J.; Deng, X.; et al. The tumor microenvironment and resistance to immune checkpoint blockade. Cancers 2023, 15, 1932. [Google Scholar]
- Spranger, S.; Gajewski, T.F. Tumor-intrinsic oncogenic pathways influencing immune recognition and response. Nat. Rev. Immunol. 2018, 18, 465–478. [Google Scholar]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological pathways driving tumor progression and response to therapy. Nat. Immunol. 2015, 16, 452–463. [Google Scholar]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; et al. Immune evasion in tumors: Mechanisms and implications for therapy. Nat. Rev. Cancer 2018, 18, 623–636. [Google Scholar]
- Galluzzi, L.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer therapy: A blueprint for designing effective immunotherapies. Nat. Rev. Immunol. 2021, 21, 704–721. [Google Scholar]
- van den Bulk, J.; Verdegaal, E.M.E.; de Miranda, N.F.C.C. Immunotherapy in colorectal cancer: Future perspectives. J. Pathol. 2018, 244, 635–650. [Google Scholar]
- Marabelle, A.; Fakih, M.; Lopez, J.; et al. Pembrolizumab in patients with microsatellite instability–high advanced colorectal cancer. N. Engl. J. Med. 2020, 383, 2616–2628. [Google Scholar]
- Kloor, M.; von Knebel Doeberitz, M. The immune biology of microsatellite-unstable cancers. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; et al. Screening for Lynch syndrome using microsatellite instability in colorectal cancer patients. N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; et al. Mismatch repair deficiency predicts response to PD-1 blockade in metastatic colorectal cancer. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; et al. IL-10-induced upregulation of TIM-3 in human T cells inhibits anti-tumor immunity. J. Clin. Invest. 2015, 125, 990–1005. [Google Scholar]
- Paulson, K.G.; Tegeler, C.; Barlogie, B.; et al. Therapy resistance in immunotherapy-treated cancers. Blood 2018, 132, 967–978. [Google Scholar]
- Jiang, X.; Xu, J.; Wang, R.; et al. Tumor immune escape and resistance to checkpoint inhibitors. Cell Rep. 2020, 30, 442–455. [Google Scholar]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells in cancer and immune suppression. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Youn, J.I.; Gabrilovich, D.I. Tumor-induced immune suppression and therapeutic strategies. Trends Immunol. 2013, 34, 120–125. [Google Scholar]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; et al. Resistance to immunotherapy and novel strategies for overcoming it. Trends Cancer 2016, 2, 109–122. [Google Scholar]
- Spranger, S.; Bao, R.; Gajewski, T.F. T cell-inflamed versus non-T cell-inflamed tumors: A major classification scheme for response to immunotherapy. Cancer Immunol. Res. 2015, 3, 621–626. [Google Scholar]
- Rodriguez-Ruiz, M.E.; van der Kaaij, M.; van den Berg, J.; et al. Immune checkpoint blockade combined with radiation therapy in cancer treatment. Nat. Commun. 2020, 11, 432. [Google Scholar]
- Samson, M.; Knebel, F.H.; Menshikh, A.; et al. The role of IFN-γ in overcoming tumor-induced immune suppression. Nat. Immunol. 2022, 23, 674–685. [Google Scholar]
- Lauck, S.; Mukherjee, S.; Ruddy, M.; et al. Modulating immune cell exhaustion to enhance cancer immunotherapy. Nat. Rev. Drug Discov. 2021, 20, 695–712. [Google Scholar]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; et al. Mechanisms of resistance to PD-1 blockade therapy. Nat. Rev. Immunol. 2020, 20, 150–165. [Google Scholar]
- Ho, P.C.; Bortolanza, P.; Martinez, E.; et al. Metabolic reprogramming and immune escape in cancer cells. Cancer Res. 2018, 78, 1332–1344. [Google Scholar]
- Pfirschke, C.; Engblom, C.; Rickert, K.; et al. Tumor-associated macrophages in the tumor microenvironment. Nat. Rev. Cancer 2017, 17, 133–148. [Google Scholar]
- Xiao, W.; Zhang, D.; Duan, X.; et al. Immune escape mechanisms of cancer cells. Nat. Rev. Immunol. 2023, 23, 234–247. [Google Scholar]
- Xiang, H.; Zhang, Y.; Lu, W.; et al. Immunotherapy resistance and strategies to overcome it. Cancer Treat. Rev. 2022, 107, 102389. [Google Scholar]
- Fong, L.; Hotson, A.; Funk, J.O.; et al. Anti-TIGIT therapy in advanced solid tumors. J. Clin. Oncol. 2022, 40, 1598–1606. [Google Scholar]
- Gettinger, S.; Horn, L.; Gandhi, L.; et al. Atezolizumab in metastatic non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 2332–2339. [Google Scholar]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; et al. Tumor mutational burden and response to PD-1 blockade. Cancer Immunol. Res. 2018, 6, 883–891. [Google Scholar]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanisms of immune evasion in cancer progression. Nat. Rev. Cancer 2016, 16, 413–425. [Google Scholar]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in cancer immunotherapy. Nat. Rev. Immunol. 2016, 16, 569–581. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; et al. Identifying neoantigen-specific T cell receptors in tumor infiltration. Science 2015, 348, 1328–1332. [Google Scholar]
- Schumacher, T.N.; Scheper, W.; Baumeister, S.H.; et al. Clonal selection and T cell response in immunotherapy. Nat. Rev. Immunol. 2016, 16, 773–784. [Google Scholar]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 850–865. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; et al. Predictive correlates of response to the anti–PD-L1 antibody atezolizumab in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Immunity 2017, 46, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Gajewski, T.F. Mechanisms of tumor resistance to immune checkpoint blockade. Immunol. Rev. 2018, 281, 155–167. [Google Scholar]
- Horn, L.; Rizvi, N.A.; Hartmann, T.; et al. Tumor mutational burden and response to immunotherapy. Lancet Oncol. 2020, 21, 1124–1136. [Google Scholar]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Reck, M.; Rabe, K.F. Precision medicine in lung cancer: Biomarkers and targeted therapies. Lancet Respir. Med. 2017, 5, 653–667. [Google Scholar]
- Zhao, B.; Zhao, H.; Zhao, J.; et al. The molecular landscape of resistance to immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2021, 20, 421–437. [Google Scholar]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; et al. Clinical application of tumor mutational burden in immunotherapy. J. Clin. Oncol. 2019, 37, 301–310. [Google Scholar]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in immunotherapy for cancer. Nat. Rev. Immunol. 2016, 16, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Durán, I.; Van Der Heijden, M.S.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Antonia, S.J.; Villegas, A.; Daniel, D.; et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; et al. Nivolumab versus docetaxel in previously treated non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Powles, T.; Durán, I.; Van Der Heijden, M.S.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; et al. Combined nivolumab and ipilimumab versus ipilimumab alone in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; et al. Long-term survival results for nivolumab plus ipilimumab in advanced melanoma. J. Clin. Oncol. 2022, 40, 162–169. [Google Scholar] [CrossRef]
- Robert, C.; Ribas, A.; Wolchok, J.D.; et al. Pembrolizumab versus ipilimumab in melanoma: 5-year follow-up. Lancet Oncol. 2019, 20, 1239–1251. [Google Scholar] [CrossRef]
- Chow, L.Q.M.; Haddad, R.; Gupta, S.; et al. Antitumor activity of pembrolizumab in head and neck squamous cell carcinoma. J. Clin. Oncol. 2016, 34, 3838–3845. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.; Fayette, J.; et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, T.; Xue, M.; et al. The role of PD-L1 in tumor-induced immune suppression. Clin. Cancer Res. 2021, 27, 3248–3260. [Google Scholar]
- Hsiehchen, D.; Espinoza, M.; Romanowksi, M.; et al. The expanding role of PD-1/PD-L1 blockade in lung cancer treatment. J. Thorac. Oncol. 2022, 17, 520–535. [Google Scholar]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; et al. Tumor mutational burden and survival benefit from checkpoint blockade in non–small cell lung cancer. Cancer Immunol. Res. 2018, 6, 883–891. [Google Scholar]
- Borcoman, E.; Nandikolla, A.; Long, G.; et al. Tumor mutational burden as a biomarker in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 28. [Google Scholar]
- Cristescu, R.; Aurora-Garg, D.; Albright, A.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; et al. The mutational landscape of lung cancer and its impact on sensitivity to PD-1 blockade. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; et al. Development of tumor mutation burden as an immunotherapy biomarker: Utility for global precision oncology. Nat. Rev. Cancer 2019, 19, 482–489. [Google Scholar]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; et al. Tumor mutation burden as an independent predictor of response to immunotherapy in diverse cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; et al. Targeted next-generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; et al. Tumor mutational burden and immune evasion in cancer immunotherapy response. Nat. Genet. 2019, 51, 1516–1526. [Google Scholar]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1/PD-L1 blockade. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Hwang, W.L.; Pike, L.R.G.; Royce, T.J.; et al. Genomic determinants of radiation therapy response. Lancet Oncol. 2018, 19, 1005–1016. [Google Scholar]
- Palucka, K.; Coussens, L.M. The immune system and cancer: Partners in crime? Science 2016, 354, 580–587. [Google Scholar]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Disis, M.L.; Slota, M.; Stanton, S.E. T cell-based cancer immunity: The science behind clinical discovery. Clin. Transl. Med. 2022, 12, e1105. [Google Scholar]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictor of outcomes in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Chamoto, K.; Zheng, S.; et al. Impact of tumor mutational burden on cancer immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 731–741. [Google Scholar]
- Hegde, P.S.; Karanikas, V.; Evers, S. The evolving paradigm of cancer immunotherapy. Nat. Rev. Drug Discov. 2016, 15, 135–147. [Google Scholar]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Zhou, G.; Kallberg, E.; Feng, X.; et al. T-cell responses and immune checkpoint blockade. J. Immunol. 2021, 206, 1895–1904. [Google Scholar]
- Wargo, J.A.; Reddy, S.M.; Reuben, A.; Sharma, P. Monitoring immune responses in the tumor microenvironment. Nat. Rev. Immunol. 2016, 16, 617–629. [Google Scholar] [CrossRef]
- Gerratana, L.; Basile, D.; Buono, G.; et al. Tumor microenvironment and immune evasion in cancer therapy. Cancer Treat. Rev. 2021, 99, 102258. [Google Scholar]
- Galon, J.; Bruni, D. Approaches to treat cancer by modulating the tumor microenvironment. Nat. Rev. Clin. Oncol. 2020, 17, 526–542. [Google Scholar]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; et al. Understanding immune cell dynamics in cancer therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. Tumor microenvironment as a therapeutic target in cancer treatment. Cancer Cell 2015, 27, 512–522. [Google Scholar]
- Chen, L.; Deng, H.; Cui, Y.; Wu, H. The tumor microenvironment in cancer immunotherapy. Signal Transduct. Target. Ther. 2022, 7, 87. [Google Scholar]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Cell 2018, 34, 582–591. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The impact of immune cell reprogramming in cancer therapy. Nat. Immunol. 2016, 17, 495–502. [Google Scholar]
- DeNardo, D.G.; Ruffell, B. Immunoediting and the role of the immune microenvironment in cancer progression. Nat. Rev. Immunol. 2019, 19, 239–252. [Google Scholar]
- Benci, J.L.; Johnson, L.R.; Choa, R.; et al. Tumor adaptation to immunotherapy. Nature 2022, 603, 160–167. [Google Scholar]
- Garris, C.S.; Luke, J.J.; Denduluri, N.; et al. The evolving role of immunotherapy in oncology. Nat. Rev. Clin. Oncol. 2021, 18, 490–509. [Google Scholar]
- Tavazoie, M.F.; Pollack, J.L.; Tanqueco, R.L.; et al. Tumor immune evasion mechanisms and therapeutic strategies. Nat. Rev. Cancer 2020, 20, 419–436. [Google Scholar]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms in cancer progression. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Zhao, Y.; Shuen, T.Z.; Toh, T.B.; et al. Tumor immunology and immune resistance mechanisms. Cancer Lett. 2018, 422, 46–64. [Google Scholar]
- Chen, Y.; Zhang, Y.; Cheng, Y.; et al. The role of tumor microenvironment in immune suppression. Cell Death Dis. 2021, 12, 402. [Google Scholar]
- Jiang, X.; Wang, J.; Deng, X.; et al. The tumor microenvironment and resistance to immune checkpoint blockade. Cancers 2023, 15, 1932. [Google Scholar]
- Spranger, S.; Gajewski, T.F. Tumor-intrinsic oncogenic pathways influencing immune recognition and response. Nat. Rev. Immunol. 2018, 18, 465–478. [Google Scholar]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological pathways driving tumor progression and response to therapy. Nat. Immunol. 2015, 16, 452–463. [Google Scholar]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; et al. Immune evasion in tumors: Mechanisms and implications for therapy. Nat. Rev. Cancer 2018, 18, 623–636. [Google Scholar]
- Galluzzi, L.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer therapy: A blueprint for designing effective immunotherapies. Nat. Rev. Immunol. 2021, 21, 704–721. [Google Scholar]
- van den Bulk, J.; Verdegaal, E.M.E.; de Miranda, N.F.C.C. Immunotherapy in colorectal cancer: Future perspectives. J. Pathol. 2018, 244, 635–650. [Google Scholar]
- Marabelle, A.; Fakih, M.; Lopez, J.; et al. Pembrolizumab in patients with microsatellite instability–high advanced colorectal cancer. N. Engl. J. Med. 2020, 383, 2616–2628. [Google Scholar]
- Kloor, M.; von Knebel Doeberitz, M. The immune biology of microsatellite-unstable cancers. Trends Cancer 2016, 2, 121–133. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; et al. Screening for Lynch syndrome using microsatellite instability in colorectal cancer patients. N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; et al. Mismatch repair deficiency predicts response to PD-1 blockade in metastatic colorectal cancer. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; et al. IL-10-induced upregulation of TIM-3 in human T cells inhibits anti-tumor immunity. J. Clin. Invest. 2015, 125, 990–1005. [Google Scholar]
- Paulson, K.G.; Tegeler, C.; Barlogie, B.; et al. Therapy resistance in immunotherapy-treated cancers. Blood 2018, 132, 967–978. [Google Scholar]
- Jiang, X.; Xu, J.; Wang, R.; et al. Tumor immune escape and resistance to checkpoint inhibitors. Cell Rep. 2020, 30, 442–455. [Google Scholar]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells in cancer and immune suppression. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Nagaraj, S.; Youn, J.I.; Gabrilovich, D.I. Tumor-induced immune suppression and therapeutic strategies. Trends Immunol. 2013, 34, 120–125. [Google Scholar]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; et al. Resistance to immunotherapy and novel strategies for overcoming it. Trends Cancer 2016, 2, 109–122. [Google Scholar]
- Spranger, S.; Bao, R.; Gajewski, T.F. T cell-inflamed versus non-T cell-inflamed tumors: A major classification scheme for response to immunotherapy. Cancer Immunol. Res. 2015, 3, 621–626. [Google Scholar]
- Rodriguez-Ruiz, M.E.; van der Kaaij, M.; van den Berg, J.; et al. Immune checkpoint blockade combined with radiation therapy in cancer treatment. Nat. Commun. 2020, 11, 432. [Google Scholar]
- Samson, M.; Knebel, F.H.; Menshikh, A.; et al. The role of IFN-γ in overcoming tumor-induced immune suppression. Nat. Immunol. 2022, 23, 674–685. [Google Scholar]
- Lauck, S.; Mukherjee, S.; Ruddy, M.; et al. Modulating immune cell exhaustion to enhance cancer immunotherapy. Nat. Rev. Drug Discov. 2021, 20, 695–712. [Google Scholar]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; et al. Mechanisms of resistance to PD-1 blockade therapy. Nat. Rev. Immunol. 2020, 20, 150–165. [Google Scholar]
- Ho, P.C.; Bortolanza, P.; Martinez, E.; et al. Metabolic reprogramming and immune escape in cancer cells. Cancer Res. 2018, 78, 1332–1344. [Google Scholar]
- Pfirschke, C.; Engblom, C.; Rickert, K.; et al. Tumor-associated macrophages in the tumor microenvironment. Nat. Rev. Cancer 2017, 17, 133–148. [Google Scholar]
- Xiao, W.; Zhang, D.; Duan, X.; et al. Immune escape mechanisms of cancer cells. Nat. Rev. Immunol. 2023, 23, 234–247. [Google Scholar]
- Xiang, H.; Zhang, Y.; Lu, W.; et al. Immunotherapy resistance and strategies to overcome it. Cancer Treat. Rev. 2022, 107, 102389. [Google Scholar]
- Fong, L.; Hotson, A.; Funk, J.O.; et al. Anti-TIGIT therapy in advanced solid tumors. J. Clin. Oncol. 2022, 40, 1598–1606. [Google Scholar]
- Gettinger, S.; Horn, L.; Gandhi, L.; et al. Atezolizumab in metastatic non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 2332–2339. [Google Scholar]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; et al. Tumor mutational burden and response to PD-1 blockade. Cancer Immunol. Res. 2018, 6, 883–891. [Google Scholar]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanisms of immune evasion in cancer progression. Nat. Rev. Cancer 2016, 16, 413–425. [Google Scholar]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in cancer immunotherapy. Nat. Rev. Immunol. 2016, 16, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; et al. Identifying neoantigen-specific T cell receptors in tumor infiltration. Science 2015, 348, 1328–1332. [Google Scholar]
- Schumacher, T.N.; Scheper, W.; Baumeister, S.H.; et al. Clonal selection and T cell response in immunotherapy. Nat. Rev. Immunol. 2016, 16, 773–784. [Google Scholar]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017, 7, 850–865. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; et al. Predictive correlates of response to the anti–PD-L1 antibody atezolizumab in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Immunity 2017, 46, 790–803. [Google Scholar] [CrossRef]
- Spranger, S.; Gajewski, T.F. Mechanisms of tumor resistance to immune checkpoint blockade. Immunol. Rev. 2018, 281, 155–167. [Google Scholar]
- Horn, L.; Rizvi, N.A.; Hartmann, T.; et al. Tumor mutational burden and response to immunotherapy. Lancet Oncol. 2020, 21, 1124–1136. [Google Scholar]
- Schoenfeld, A.J.; Hellmann, M.D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell 2020, 37, 443–455. [Google Scholar] [CrossRef]
- Reck, M.; Rabe, K.F. Precision medicine in lung cancer: Biomarkers and targeted therapies. Lancet Respir. Med. 2017, 5, 653–667. [Google Scholar]
- Zhao, B.; Zhao, H.; Zhao, J.; et al. The molecular landscape of resistance to immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2021, 20, 421–437. [Google Scholar]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; et al. Clinical application of tumor mutational burden in immunotherapy. J. Clin. Oncol. 2019, 37, 301–310. [Google Scholar]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory pathways in cancer immunotherapy. Nat. Rev. Immunol. 2016, 16, 569–581. [Google Scholar] [CrossRef]
- Powles, T.; Durán, I.; Van Der Heijden, M.S.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; et al. Nivolumab versus docetaxel in previously treated non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
Figure 1.
Nanotheranostics Overview: A schematic representation of nanomaterial-based approaches in cancer diagnostics and therapy, including liposomes, polymeric micelles, dendrimers, virus-like particles (VLPs), and gold nanoparticles. Each nanomaterial offers distinct advantages in drug delivery, imaging, immune modulation, and targeted therapy, enhancing precision in oncology treatments. Figure adapted and modified from [161,166,194,240].
Figure 1.
Nanotheranostics Overview: A schematic representation of nanomaterial-based approaches in cancer diagnostics and therapy, including liposomes, polymeric micelles, dendrimers, virus-like particles (VLPs), and gold nanoparticles. Each nanomaterial offers distinct advantages in drug delivery, imaging, immune modulation, and targeted therapy, enhancing precision in oncology treatments. Figure adapted and modified from [161,166,194,240].
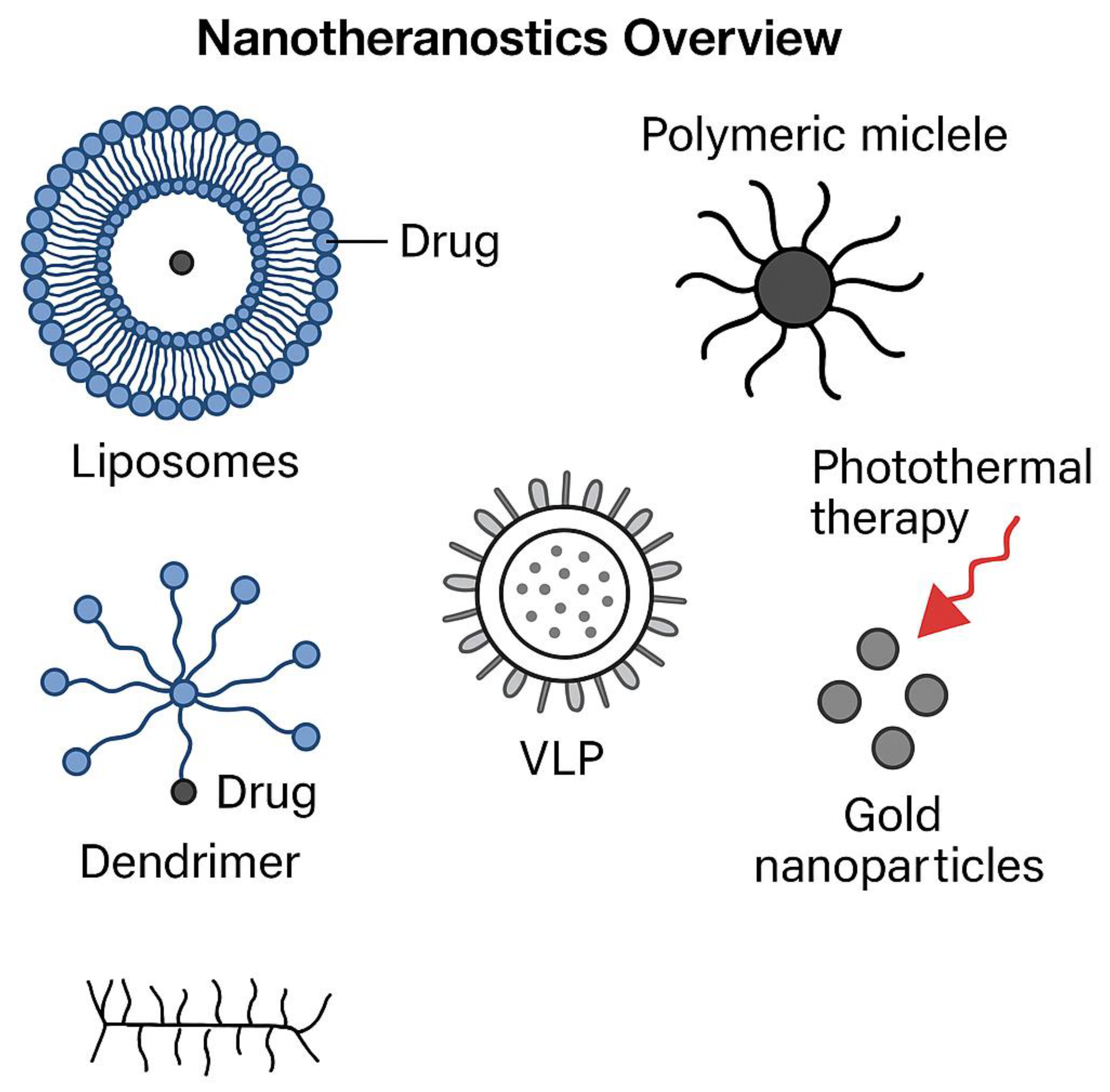
Figure 2.
Chromophore-Assisted Light Inactivation (CALI) Mechanism. A schematic representation of CALI-based targeted cancer cell destruction, showcasing chromophore-binding specificity, excitation-emission interactions, and localized damage induction. Light activation of chromophores leads to reactive oxygen species (ROS) generation, selectively inactivating key proteins and inducing apoptosis in cancer cells.
Figure 2.
Chromophore-Assisted Light Inactivation (CALI) Mechanism. A schematic representation of CALI-based targeted cancer cell destruction, showcasing chromophore-binding specificity, excitation-emission interactions, and localized damage induction. Light activation of chromophores leads to reactive oxygen species (ROS) generation, selectively inactivating key proteins and inducing apoptosis in cancer cells.
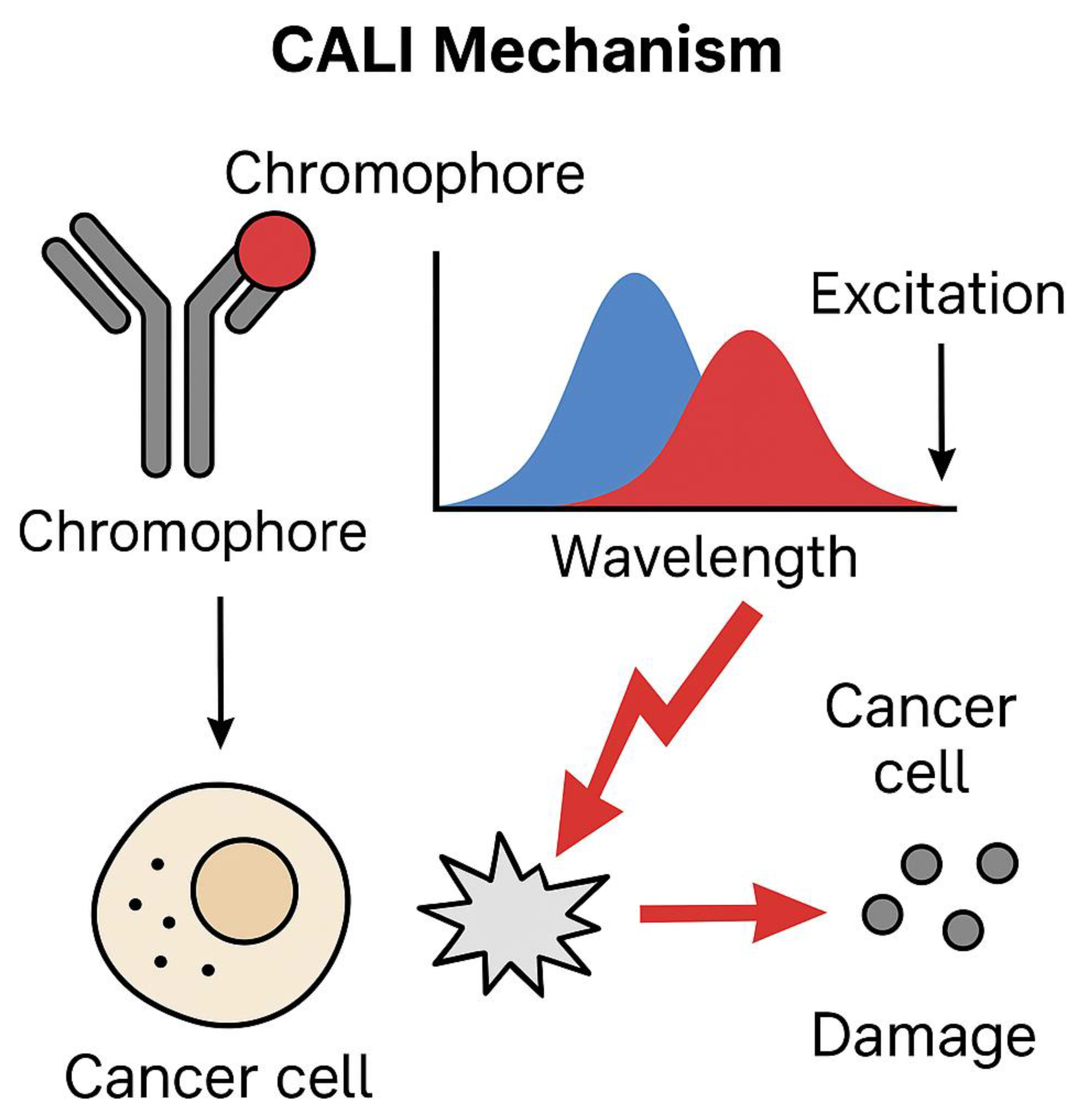
Figure 3.
Liposomal Drug Delivery. A schematic representation of liposomal drug delivery, illustrating the structural composition, encapsulation mechanism, and controlled release of therapeutic agents. This figure highlights the role of liposomes as nanocarriers [172,198,213], enhancing drug solubility, stability, and targeted delivery to cancer cells.
Figure 3.
Liposomal Drug Delivery. A schematic representation of liposomal drug delivery, illustrating the structural composition, encapsulation mechanism, and controlled release of therapeutic agents. This figure highlights the role of liposomes as nanocarriers [172,198,213], enhancing drug solubility, stability, and targeted delivery to cancer cells.
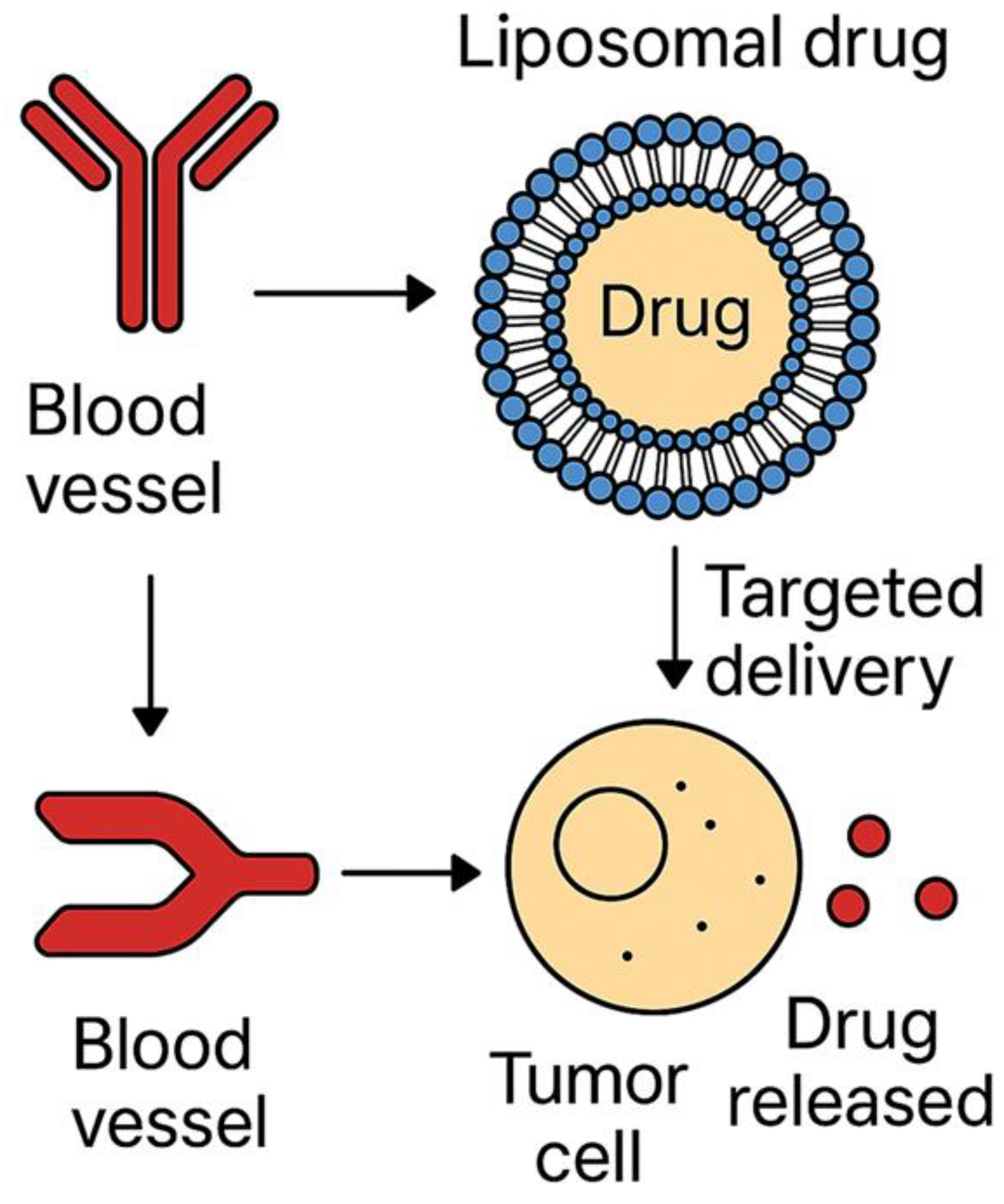
Figure 4.
Tumor Microenvironment and Nanoparticle Interaction. A schematic representation of the tumor microenvironment (TME) and its interaction with various nanoparticles used in cancer therapy. This figure highlights the structural and physiological barriers within the TME, emphasizing how liposomes, polymeric micelles, dendrimers, virus-like particles (VLPs), and gold nanoparticles navigate these barriers for effective drug delivery and therapeutic action [178,192,207].
Figure 4.
Tumor Microenvironment and Nanoparticle Interaction. A schematic representation of the tumor microenvironment (TME) and its interaction with various nanoparticles used in cancer therapy. This figure highlights the structural and physiological barriers within the TME, emphasizing how liposomes, polymeric micelles, dendrimers, virus-like particles (VLPs), and gold nanoparticles navigate these barriers for effective drug delivery and therapeutic action [178,192,207].
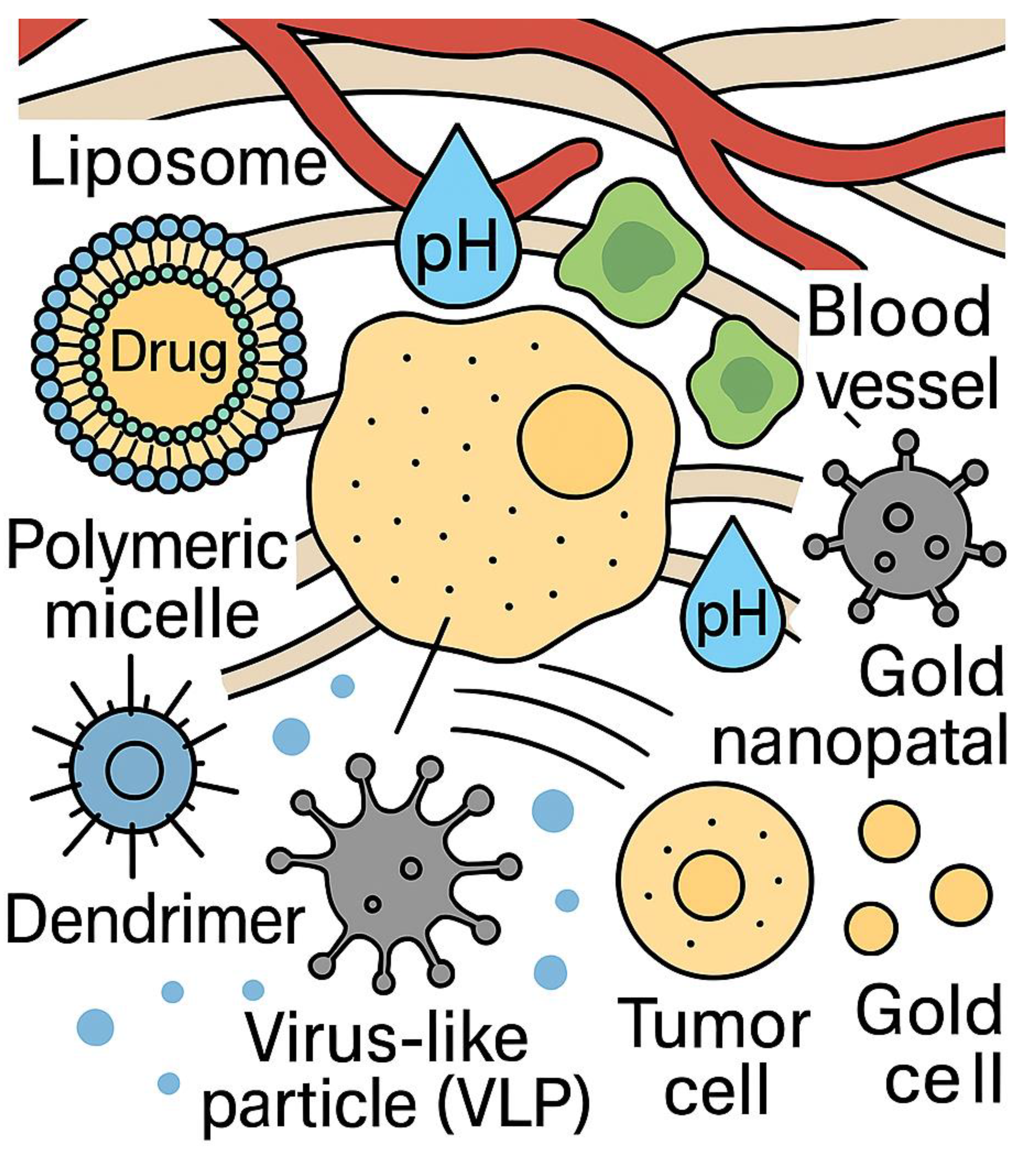
Figure 5.
Advances in Cancer Treatment through Nanotheranostics. This pyramidal timeline illustrates key milestones in nanotheranostics for cancer treatment, marking significant advancements from 1998 to 2024. Each level of the pyramid corresponds to a breakthrough in nanotechnology, showcasing the evolution of theranostics, gold nanoparticles, polymeric nanoparticles, virus-like particles, and advanced nanotheranostic systems for concurrent cancer imaging and therapy.
Figure 5.
Advances in Cancer Treatment through Nanotheranostics. This pyramidal timeline illustrates key milestones in nanotheranostics for cancer treatment, marking significant advancements from 1998 to 2024. Each level of the pyramid corresponds to a breakthrough in nanotechnology, showcasing the evolution of theranostics, gold nanoparticles, polymeric nanoparticles, virus-like particles, and advanced nanotheranostic systems for concurrent cancer imaging and therapy.
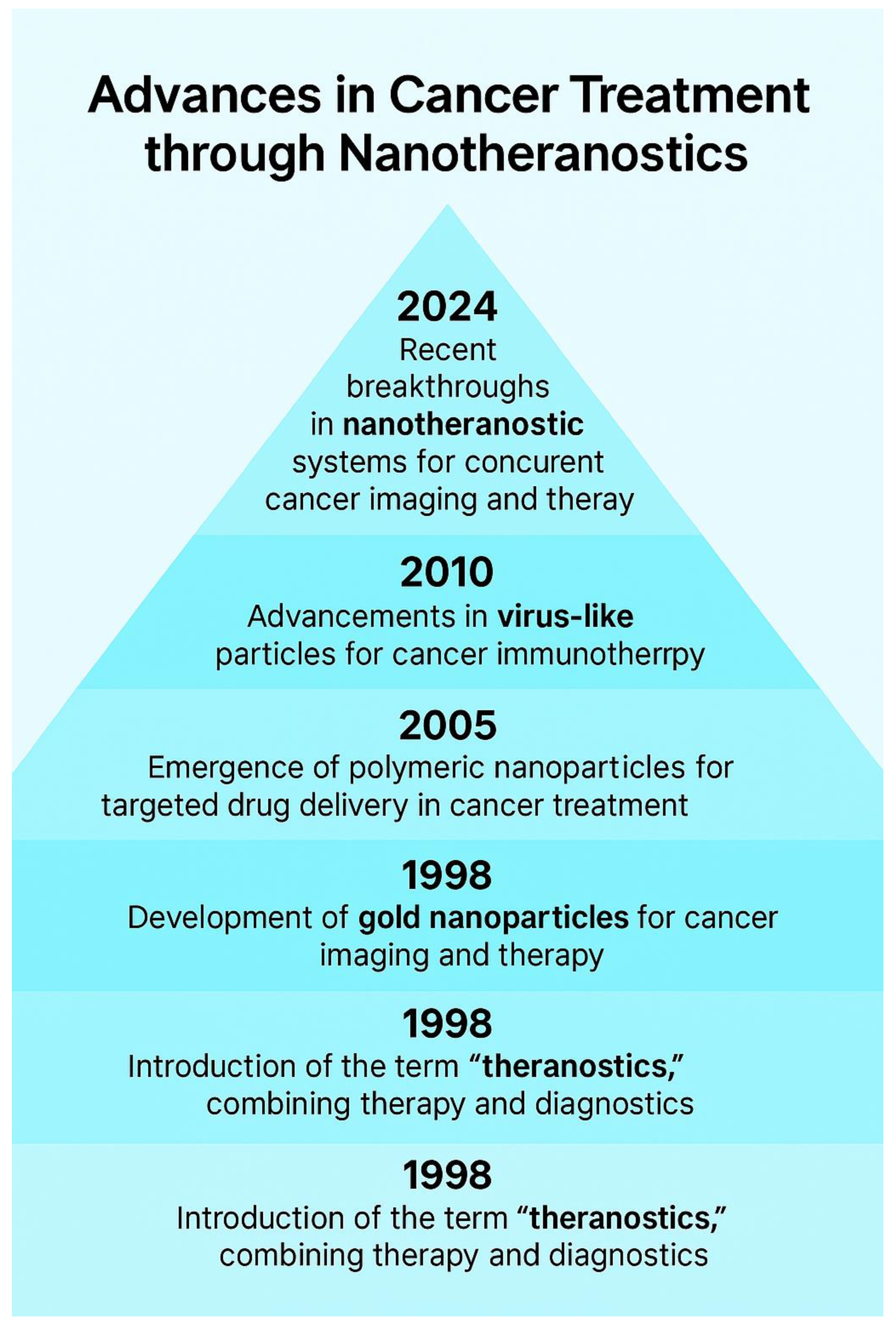
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



