Submitted:
27 May 2025
Posted:
28 May 2025
You are already at the latest version
Abstract
Since the advent of recombinant DNA technologies and leading up to the clinical approval of T cell engager blinatumomab, the modular design of therapeutic antibodies has enabled the fusion of antibody fragments with proteins of various functionalities. This has resulted in an expansive array of possible mechanisms of actions and has given birth to fragment-based antibodies (fbAbs) with immune cell engager modalities. In searchable databases, the preclinical development of these antibodies has shown promises; however, clinical outcomes and restructuring efforts involving these agents have produced mixed results and uncertainties. Amid budgetary cuts in both academia and industry, critical planning and evaluation of drug R&D would be more essential than ever before. While many reviews have provided outstanding summaries of preclinical phase fbAbs and cataloged relevant clinical trials, to date, very few of the articles in searchable databases have comprehensively reviewed the details clinical outcomes along with the underlying reasons or potential explanations for the success and failures of these fbAb drug products. To fill the gap, in this review, we seek to provide the readers with clinically driven insights, in accompany with translational and mechanistic studies, on the current landscape of fragment-based immune cell engager antibodies in treating cancer, infectious and autoimmune diseases.
Keywords:
fragment-based antibody
; immune cell engagers
; T cell engagers
; NK cell engagers
; myeloid cell engagers
; B cell engagers
; immune checkpoint
; antibody engineering
; antibody translational medicine
; antibody clinical development
1. Introduction
Antibody is one of the most essential classes of drug in the contemporary pharmaceutical field. Despite its success as a class of drug, research and development of antibodies can face challenges due to many factors: 1) lack of clinical efficacy, 2) excessive toxicity, 3) poor pharmacokinetic profile, and 4) high immunogenicity. During periods of budgetary surplus, these factors might be considered “tolerable” in clinical development and receive approval. However, during periods of economic downturn as well as budget cuts, more frequent termination of drug development could occur when the above-mentioned factors occur. Given the availability of literature documenting clinical results of successful and unsuccessful drug candidates, an understanding of these valuable cases could help to provide insights for designing and polishing the drug candidate to meet clinical and commercial needs.
With the advancement of recombinant technologies in synthetic biology, antibodies with countless structural varieties could be crafted out of blueprints on computer software. Fragment-based antibodies (fbAbs) are a relatively general engineering approach for formatting recombinant antibodies. Compared to other chimeric IgG formats (such as chimeric bispecifics) or IgG-like structures that retain the main framework of the IgG, fbAbs strive to utilize the lowest molecular weight possible to achieve the desired therapeutic functionality as needed, by minimally drawing upon the fragments of IgG, such as scFv or Fc, fused with other molecules of interest. Due to their versatileness, fragment-based antibodies are frequently the blueprints for constructing immune cell engagers. fbAbs were investigated in cancer, infectious disease, and autoimmune diseases, with more than 60 designs and more than 50 clinical trials on record [1,2], highlighting the attention attracted by their flexibility to meet therapeutic needs.
A key advantage of the multidomain antibodies is their outstanding capability to be modularized, where each module (or domain) is assigned to bind an arbitrary target. With the aid of bioinformatics, an understanding of pharmacology gives rise to demands for platform of tailorable targeted therapies. Besides targeting antigens to recruit immune cells and complement complex, designers of modular multivalent antibodies also have the liberty to simultaneously target multiple types of pathological antigens [3], in order to elicit synergistic effects [4], to target human serum albumin for extended half-life [5], to incorporate an immune cell-modulating moieties to boost immune cell function [6], and to potentially neutralize target antigens like viral particles [7,8,9,10,11,12]. In terms of the mechanisms of actions, those fbAb that entered clinical trials had these roles or mechanisms of action: 1) Immune cell engagement, 2) immune checkpoint modulation, 3) cytokine and chemokine modulation, 4) disease target binding for inhibition, activation, or neutralization, 5) payload toxicity [13]. Readers who are interested may read this article [14]. Key parameters for engineering successful fbAb include: 1) persistence (pharmacokinetics), 2) affinity modulation [15], 3) conditional activation, 4) antigen selectivity, 5) co-stimulation of the immune cell targets. The basic building blocks of these multivalent antibodies are usually derived from these two components: VHH and scFv. For those readers who are interested in more detailed comparisons between the structural activities of VHH and scFv, this review paper can be helpful [16]. In addition, protein subunits, such as Fc [17], PD-1, CD40L, cytokines, and immunostimulatory small molecules, are also embedded into the structure of the antibody to achieve enhanced pharmacokinetic and/or pharmacological properties (Figure 1).
2. Harness the Incidence of Anti-Drug Antibody and Pharmacokinetics
2.1. Mechanism for the Incidence of Anti-Drug Antibody (ADA)
Immunogenicity of antibodies pose significant threat to the success of the treatment, as it introduces the binding of anti-drug antibody (ADA) towards the antibody drug of interest. While pre-existing antibodies are existing in the patients due to prior treatment or patient-specific immune response, treatment-emergent ADA is induced due to the treatment with the antibody. When the antibody is required for repeated dosing, the presence of ADA can cause the antibody drug of interest to be rapidly neutralized or cleared from the body, thus jeopardizing efficacy.
Immunogenicity of therapeutic antibodies in general depends on several factors. From the side of the patient, it could be due to genetic background (e.g., HLA polymorphisms), baseline immunogenic status (medication and disease condition). From the product itself, its amino acid sequence, mechanism of action, dosing route, and manufacturing process can contribute to the incidence of ADA [18,19,20]. ADA could also induce various types of adverse effects, including but not limited to: type I hypersensitivity reactions and anaphylaxis (IgE ADA-mediated), type III hypersensitivity (inflammation caused by immune complexes, which can lead to fever, rash, hematuria, proteinuria) [21], alongside with reduced treatment efficacy and an increased potential risk of reaction against a similar class of drugs.
Amino acid sequence is the primary mechanism leading to the generation of ADA. Upon internalization by antigen presenting cells (APCs, such as dendritic cells), antibody is digested into peptides, being displayed to helper T cells via APC by MHC-II-TCR (T cell receptor) interactions on the APC surface. Subsequently, the activated T cell, capable of identifying the peptide sequence of the antibody, can activate and maturate B cells with complementary BCR (B cell receptor), which are responsible for producing ADAs. In cases of T cell-independent pathways, the antibody can directly crosslink with the BCR, stimulating the differentiation of B cells into plasma cells to produce ADAs [19].
The extent of humanization is also a factor that is frequently being discussed. Yet, it may not serve as a definitive contributing factor for immunogenicity. According to two systematic/meta-analysis [22,23], the incidence of ADA for fully human antibodies range from 1.12% to 7.61% (1.12% for tezepelumab, 3.8% for golimumab, 7.5% for adalimumab, 7.61% for dupilumab), whereas the incidence of ADA for humanized antibodies ranges from 0%-28%: (0% for omalizumab, 4.39% for reslizumab, 3.63% for mepolizumab, 8.35% for benralizumab, 10.9% for certolizumab, 16.0% for natalizumab and 28.0% for infliximab), indicating an overlap of ADA rates with or without humanization. Therefore, a fully human antibody may not always have lower ADA incidence rate than humanized antibodies, and vice versa.
The mechanism of action of the antibody itself may also facilitate the generation of ADAs. Multivalent antibodies, capable of crosslinking more than one of the same antigens, may have higher chances of forming immune complexes, especially when the antigen is soluble and/or containing more than one epitope. Immune complexes, a gigantic scaffold of crosslinked matrix of the antibody and the antigen, can be more easily captured and digested by APCs, serving as reservoir for generating more ADAs. For instance, the anti-TNF antibody has an ADA incidence rate ranging from 28% in dinutuximab to 66.7% in infliximab [19]. Aglycosylation for silencing the Fc is another main contributor of ADA, which can cause aggregation of the antibody in around 30-48% of the patients [20,24]. Anti-B cell antibodies, such as blinatumomab (≤2%), glofitamab (0%), and teclistamab (0-0.46%), could have lower ADA incidence rates [19], due to their mechanism of action to eliminate B cells that serve as the source for producing ADA.
Last but not least, dosing route is an important factor for influencing the incidence of ADA. Subcutaneous (SC) route forces the antibody (since molecules >20 kDa are too bulky to be able to directly enter capillary circulation) to traffic through interstitial matrix and reach peripheral lymphatic tissue, where the antibody is phagocytosed by antigen-presenting cells (APCs), and the immunogenic epitopes of the antibody may be presented to T cells, potentially eliciting an eventual ADA response [18], especially when the drug has to be dosed more frequently due to PK issues.
For instance, in phase I study of the BiTE AMG212 [25], 96.7% (n=31) of SC dosed patients developed ADA (93.3% being neutralizing), whereas none (n=16) of the patients dosed with continuous intravenous infusion (CIV) developed ADA. Those emergent ADA were characterized by early onset (cycle 2 day 1), resulting in increased drug clearance level, contributing to rebound PSA levels and reducing drug efficacy. Topically applied glucocorticoid cream, which could theoretically inhibit dendritic cell-mediated ADA induction, could not prevent or mitigate ADA development. In more detailed analysis of peptide epitopes, it was found that three peptide fragments from the epitope of AMG212 were immunogenic to CD4+ T cells in a clinical memory recall assay to PBMCs from patients who had been treated with AMG160 (which has 98.4% sequence homology with AMG212), as evidenced by elevated interferon-gamma levels. These observations also raise concerns for patients who need to be treated with analogous drugs. On the other hand, antibody dosed via intravenous route can still possess immunogenicity, as seen in the case of BiTE AMG160 with 20% rate of incidence for ADA [18].
2.2. Pharmacokinetic Challenges and Solutions
When an fbAb enters the human body, it can be consumed through the following ways: 1) renal filtration (especially for protein less than 70 kDa), 2) liver absorption, 3) target or receptor-mediated endocytosis, and 4) biochemical degradation [17]. The total number of half-life extension drugs published by FDA had reached to more than 50 by the year 2024 [17], highlighting the significance of these strategies. Clinically speaking, fbAb with very low half-life and no half-life extending strategies may need frequent dosing and even extended periods of infusion, such as Blinatumomab, which may place a burden on the patient. Last but not the least, presence or availability of immune cells at the site of treatment is as important as the availability of immune cell engagers at the site of treatment. PBPK model has been developed and suggested that the conditions as well as availability of the immune cells in the microenvironment can affect the treatment outcome of solid tumor [26,27]. With similar mechanism of actions, depleting tissue-resident B cells in autoimmune disease as well as pathogen infected tissue-resident cells by immune cell engager should follow similar roles.
2.2.1. Molecular Weight: Balancing the Tissue Penetration, Half-Life, & “Switch”
When it comes to antibody engineering or targeted therapy engineering, especially in oncology, arguably, the most important difference to note in categorizing different types would be whether the cancer is hematological origin or solid tumor origin. Intuitively, in blood cancers such as leukemia and lymphoma, malignant cells are mostly in circulation within bloodstream. Such accessibility can contribute to the success of hematological targeted therapies, including antibodies and CAR-T [28]. When it comes to solid tumors and deep tissue (which can serve as sanctuary for autoreactive B cells [29] and viral infected cells [29]), the dense and complex structures within the stroma, composed of a heterogenous mixture of cells and extracellular matrix (ECM), play roles as a physical barrier that limits antibody penetration and binding to the target cell of interest [30].
To gain access to core regions of solid tumors and tissues, antibody engineers have devised to design for smaller antibodies. A popular theory [31] claims that when it comes to molecular weight (MW), a trade-off often exists between tumor/tissue penetration and tumor retention in antibody engineering. As the MW of the antibody increases, its penetration is reduced but its retention is increased, and vice versa. Antibodies with very low MW face challenges if their MW is below renal threshold limit, which is a cutoff for antibodies around 70 kDa [31]; this leads to significantly reduced half-life due to rapid renal clearance. On the other hand, although many may believe in the enhanced permeability retention (EPR) effect, which suggests penetration of high MW protein through blood vessels and accumulation in tumor, the advantage of EPR is not as prominent in clinical settings [30], as demonstrated by the lack of clinical success of payload nanoparticles in oncology research [32]. Potential explanation for such clinical challenges involves blood coagulation from the production of vascular permeability factors [33].
The short half-life of fbAb like blinatumomab gives rise to its continuous dosing regimen to make up for its rapid clearance [34,35]. Like yin and yang, despite the dosing regimen being less convenient, it possesses the safety feature of allowing rapid stop of treatment, which is in stark contrast with CAR-T, as CAR-T cannot be easily turned off in short time even with engineered off-switch designs that may need more than 12 hours to days [36,37,38].
2.2.2. Strategy to Improve Half-Life: Fc Fusion
To achieve a balance between permeability and retention in tissues, besides altering the sizes of the antibodies, one could opt to append an Fc domain, which can significantly extend the tumor uptake rate of the fbAb. Fc can bind to FcRn, which stands for neonatal Fc receptor. The binding occurs in a pH-dependent manner, allowing the antibody tagged with Fc to be taken up by cells into acidic endosomes. In the process, the antibody is protected from lysosomal degradation and is recycled and released back into the bloodstream [39]. In an investigation [40] for HER2 targeted multivalent antibodies based on the 4D5 clone, it was demonstrated that the (scFv)2-Fc has significantly longer half-life than (scFv)2 (12.2% uptake at 21-hour timepoint vs 1.37-2.1% update at 24-hour time point). In comparison, the full-length IgG antibody has an 18.05% update at 24-hour time point, highlighting the essential contribution of Fc made in promoting overall tumor uptake.
Nevertheless, wildtype Fc possesses the capacity to interact with Fcγ, which can activate immune cells and complement pathways [41]. Such activation could be unwanted, especially when the antibody has a secondary immune cell engaging subdomain, and in such case, the antibody could cause Fc mediated attack on immune cells recruited by the secondary immune cell-engaging subdomain. Therefore, researchers have engineered various Fc variants to inactivate their Fcγ-binding capacity while retaining the FcRn binding capacity. Examples of such engineered or mutated Fc include: L234A/L235A (also known as LALA), L234A/L235A/P329G (LALA-PG), as well as endosomal pH-depending binding variants, such as M252Y/S254T/T256E (YTE) or Q311R/M428L/N434W (REW) [17].
2.2.3. Strategy to Improve Half-Life: Targeting Serum Albumin
For enhancement of circulation time in bloodstream, attaching a serum albumin-binding domain to the antibody design can draw on the presence of serum albumin, analogous to a hitchhiking effect, which increases the bulkiness of the antibody and thereby slow down renal clearance. For instance, in a study [42] that designed a tandem anti-DR5 VHH fbAb possessing a domain binding to serum albumin, compared to the constructs without an anti-serum albumin domain (NbDR54, which has four tandem repeats targeting DR5), the pNbDR54-NbSA exhibited at least several hundred-fold higher area-under-the-curve (AUC). Nevertheless, the in vivo data from the study show that NbDR54-NbSA exhibited significantly lower efficacy than NbDR54. One possible explanation is that the anti-serum albumin subunit serves as a sink, which reduces the availability of the drug at the tumor site. The drug may also become rapidly internalized into the tumor and therefore impair its ability to activate death receptor 5 for inducing apoptosis of cancer cells. For the next step, the researchers may opt to design a serum albumin binder that exhibits weaker binding affinity, which may strike a balance between extending half-life and reducing the impact of sinking or internalization.
3. Features of fbAb (Fragment-Based Antibody) Immune Cell Engager Platforms
BiTE (bispecific T-cell engager) platform, developed by Amgen, with the well-known example of blinatumomab, has been a hotspot for clinical development ever since the approval of blinatumomab by the FDA. From a structural standpoint, BiTE relies solely on a flexible peptide linker between its anti-CD19 and anti-CD3 scFvs, which provides a minimalist structural requirement for bispecificity. Blinatumomab has a low incidence of ADA of 2%, and among the 2% patients, 78% (7/9) of them had developed neutralizing antibodies [35]. In the clinical trials of tarlatamab [43], 9.3% of patients (n=420) were found to be positive for ADA. In the case of PSMA x CD3 pasotuxizumab [44], the incidence of ADA is 0 out of 16. Despite the promising prospect of the BiTE platform, not all BiTE constructs exhibits equally low immunogenicity. In a phase I study of the CD3 x PSMA (prostate-specific membrane antigen) BiTE AMG212 [25], treatment-induced ADA was observed in 96.7% of subjects (n=31), with 93.3% of the subjects possessing neutralizing antibodies, across a dose range from 0.5ug/d to 144 ug/d, despite none of the subject having pre-existing ADAs. In a trial for AMG211/MEDI-565 (CD3 x CEA), ADAs were observed in all patients receiving more than 3.2mg of the antibody [45]. BiTE has a half-life of 1.41-2.10 h for blinatumomab [35,46]. Half-life could be boosted to 5.7 days with Fc fusion in BiTE tarlatamab (AMG757) [47,48].
DART (dual affinity re-targeting) platform, developed by MacroGenics, with the example of CD3 x CD123 Flotetuzumab, incorporates an interchain disulfide bond at the C-terminus of its two scFvs, a feature absent in blinatumomab's BiTE structure [49,50]. This covalent linkage stabilizes the heterodimeric configuration, preventing dissociation of the CD123 and CD3-binding domains under physiological conditions. The DART platform, due to the necessity of VH-VL pairing, is believed to reduce aggregation and enhance stability [49]. In the case of flotetuzumab, the incidence of ADA is around 0.9% (n=88) after treatment cycles or at the end of treatment visit [51]. Although clinical pharmacokinetic data are lacking, the half-life of unmodified DART is expected to be similar to that of BiTE due to their structural similarities. By fusing DART to an Fc, the half-life of unmodified DART could reach as long as 26.6-45.8 h as could be seen in the case of PF-06671008 [52,53], or even better, up to 11-12 days as seen in the case of MGD013 and MGD019 [54,55,56,57].
ImmTAC (immune-mobilizing monoclonal T-cell receptor against cancer) platform, developed by Immunocore, is composed of an engineered TCR that can recognize MHC-peptide complexes and is linked via a peptide linker to an anti-CD3 domain (made of scFv in tebentafusp). A notable example is the FDA-approved tebentafusp (CD3 x MHC-gp100) [58]. The modality of the antibody is to mimic the recognition of MHC-peptide complex by TCR, followed by activation of the CD3 complex. Since the natural binding affinity between the TCR of cytotoxic CD8+ T cell for self-antigen-presented MHC is low due to self-tolerance mechanism, the design of ImmTAC enables the possibility to target tumor associated antigens using an artificial TCR [58]. In addition, ImmTAV (immune-mobilizing monoclonal T-cell receptor against virus), a similar fbAb product that targets viral antigen instead, is also being developed to treat HIV [59]. ADA incidence rate for tebentafusp was 29% and 33% across two clinical studies, with a median onset time of ADA around 6-9 weeks after tebentafusp-tebn treatment. Patients with higher titer ADA are shown to have higher antibody clearance rate, suggesting the presence of neutralizing antibody in some patients. Exploratory analysis with limited data suggests the formation of ADA does not appear to have clinically significant effect on frequency or severity of hypersensitivity related adverse reactions and no observed impact of survival [60]. Tebentafusp has a half-life of 7.5h [47,48], which is shorter than that of IMCnyeso, a CD3 x NY-ESO-1 ImmTAC with an half-life of 25 h [61], suggesting potential influence from target-mediated drug disposition (TMDD).
Fab derivatives are derived from fusing two fab fragments or from fusing Fab and other proteins of interest. F(ab’)2, composed of two Fabs linked an immunoglobin hinge region [58], has been gaining interest as an antivenom (e.g., for Black Widow spider venom) [62], antiviral (anti-COVID-19 [63,64]), antibacterial [65], antitumor drug [66]. Preexisting ADA could be a concern for F(ab’)2 due to its exposed hinge region as well as its equine origin, as seen in cynomolgus monkey [67]. Due to this feature, F(ab’)2 antibodies are mostly investigated for purpose of short-term therapeutic purpose. For instance, F(ab’)2 like FDA-approved Anavip is used as an antivenom, where the transient dosing nature renders ADA less concerning. As an alternative approach, one or two Fab fragments can be fused with other proteins of interest. IMB071703 is an F(ab’)2 derivative, fused to a CD40L protein cluster in order to target CD40. Clinical study was conducted with IMB071703 [68] but no data for antidrug antibody was available. IMB071703 has a half-life of 2.45 hours [68]. Given that the molecular weight of a Fab fragment is around 50 kDa and that of CD40L trimer is around 54-60 kDa, the total molecular weight of IMB071703 should be close to 100 kDa, which is much lower than the renal filtration threshold of 70 kDa [31]. Moreover, its half-life is also significantly lower than that of tebentafusp, which has an lower molecular weight around 77 kDa but higher half-life of 7.5h [47,48], indicating the need for further investigating the reason and for further enhancing its pharmacokinetic profile.
Fab fragment can also be combined with other antibody fragments, as can be seen in the case of TriNKET (trispecific NK cell engager therapeutics; Dragonfly therapeutics and Merck) and ANKET (antibody-based NK cell engager therapeutics; Innate Pharma). TriNKET is a class of tri and tetra-specific fbab, with scFv, Fab, and Fc components, targeting CD16, NKp46, cancer antigen, and/or potentially the beta chain of IL-2 receptor [69,70]. DF1001 (CD16, NKp46, HER2) has shown efficacy in relapsed/refractory solid tumors [71]. Data for the incidence of ADA and half-life for the platforms cannot be found. ANKET platform antibody is composed of two Fab, each recognizing NK cell receptor or tumor-associated antigens, and one Fc domain. Preliminary clinical results for ANKET format antibody SAR443579/IPH6101 was announced by Innate Pharma [72], although no data for incidence of ADA or half-life were available.
TriTAC (tri-specific T cell activating construct) platform, developed by Harpoon therapeutics (acquired by Merck/MSD), is a type of trispecific T-cell engager antibody, tandemly connecting humanized scFv/VHH that bind a pathogenic antigen, CD3, and human serum albumin, effectively achieving T cell engagement of the pathogen as well as half-life extension [73]. Although clinical data is unavailable for incidence of ADA, in study with cynomolgus monkey for TriTAC HPN328 (DLL3 x CD3 x albumin) [74], ADA was detected in 7 out of 10 animals at 1mg/kg/dose group and 3 out of 10 animals in the 10mg/kg/dose group. TriTAC-XR is an prodrug version of TriTAC containing masked antigen-binding region [75], with potentially enhanced safety and pharmacokinetic profile.
TandAb (tandem diabody), developed by Affimed, is a bispecific or tetraspecific antibody format with tandemly linked scFvs. TandAb also serves as part of the ROCK platform (redirected optimized cell killing), with clinical examples including AFM13 (acimtamig), and AFM28 [76]. The CD30 x CD16A AFM13 has completed phase II trial [77], where ADA was detected in 17.6% patients (n=108), and out of the 45 ADA-positive samples, 38 samples were determined to be neutralizing. Currently, Fc-fusion version of TandAb AFM24 (CD16A x CD123) is in clinical trials [76]. The half-life of TandAb ranges from 18.4 to 22.9 h for AFM11 [78,79] and 20.6h for AFM13 [80].
DARPin (designed ankyrin repeat protein) is based on naturally occurring ankyrin repeat domains, with roughly a total of 404 types of ankyrin repeats coded in human genes, which are typically the building blocks of human proteins in various functions [81]. For example, the tetradomain DARPin, MP0250, contains five repeats, with domains recognizing human serum albumin, VEGF, and HGF [81]. DARPin platform includes AMG 506 (MP0310) [82], MP0274 [83]. MP0250 has ADA incidence in 20 of 42 (47.6%), and the incidence of ADA did not affect the PK of the MP0250 [84]. The half-life of MP0317 is 70.5 h [85,86] whereas that of MP0250 is 15-16 d due to its ability to bind to serum albumin, allowing convenient dosing intervals of every 2, 3, or even 4 weeks [84,87].
TriKE (tri-specific killer cell engager) is a class of trispecific fbab, targeting NK cell antigen CD16, cancer/viral antigen, and includes tandemly linked scFv or VHH with an inserted IL-15 domain to stimulate NK cells [88,89,90]. While the CD16 x IL-15 x CD33 GTB-3550 has shown promising clinical results [91,92], no data regarding the incidence of ADA is available. The half-life of GTB-3550 was claimed to be “short as predicted” [71], while the corporate presentation of GT biopharma has shown that the half-life of GTB-3550 ranges from 2.2 h to 2.52 h [93], consistent with fragment-based antibody at similar molecular weight. Future clinical development of the antibody will involve replacement of the antibody component with VHH [93].
The (VHH)n-Fc format antibody links VHH and Fc domains altogether, mimicking the Y-shaped IgG format, with the exception that one arm of the antibody can host multiple tandemly linked VHH, providing multiple options in designing the antibody. At present, erfonrilimab (KN046) is the most clinically advanced example for fragment-based immune cell engager incorporating multiple VHHs, with an incidence of ADA at 67.9% as seen in a phase 2 multicenter trial [94]. KN046 has a half-life of 111.0-137.4 h, likely due to the insertion of Fc [95].
ISAC (immune-stimulating antibody conjugate) is a type of immune cell engaging antibody, utilizing conjugation of immune cell receptor ligands to antibody fragment to achieve immune cell engagement. Even though ISAC frequently targets APCs, such as the TLR7/8 receptors, the incidence of ADA varies significantly. For instance, the HER2 x TLR7/8 BDC-1001 ISAC demonstrated no ADA incidence in 118 patients with 16 tumor types [96], whereas the HER2 x TLR7 NJH395 showed a 100% incidence of ADA in all 14 patients [97]. The half-life of BDC-1001 is 4.8 d, potentially due to its Fc and molecular weight [96,98,99].
4. T cell Engagers (TCE)
T cell engagers (TCEs) are a class of drugs that bind to activating receptors on T cells, such as CD3. Since CD3+ T cell play major roles in immune memory (in addition to B cells), they have become a focus in the field of targeted immunotherapy. Because of the complexity of T cell immunology, their roles in cancer, infection, and autoimmunity are very different. In terms of the FDA-approved fbAbs (fragment-based antibodies), there are examples of blinatumomab, abciximab, ranibizumab, certolizumab pegol, emicizumab, caplacizumab, tebentafusp [100]. Among these drugs, blinatumomab and tebentafusp are used to treat cancer. In this section, the roles of T cells in these diseases will be examined based on the disease type.
TCE works by crosslinking T cells and the target cells, facilitating the formation of immune synapses, a process involving the rearrangement of cellular surface proteins that can bridge the T cell and the target cell [101]. The target cell presenting antigens of interest could be “healthy cells” that are responsible for autoimmune disease, cells infected with intracellular pathogens, and cancer cells. By binding to T cell activating receptor (such as CD3) and the antigen/target on the target cell, TCE can transiently connect T cell and the target cell, forming a cytolytic immune synapse. The signal from TCE binding then causes the T cell to release pore-forming protein perforin, which disrupts the membrane of the target cell, and apoptosis-inducer, granzyme, to activate cell death [102,103]. Moreover, the formation of the synapse can also promote release of pro-inflammatory cytokines (IFN-γ and TNF-α) and proliferation of T-cells [101]. Essentially, all cytotoxic T-cells can be engaged by this mechanism of action, including CD8+ T cells, CD4+ T cells, gamma-delta T cells, and NKT cells [102].
Compared to IgG and IgG-like TCE antibodies with larger size, fbAbs with smaller sizes, are capable of creating tight junctions (close distance binding or membrane proximity) between the T cell and the target cell, thereby creating an artificial immune synapse analogous to the ones observed during binding of antigen peptide-loaded HLA complexes and T cell receptor (TCR) [104,105]. Therefore, by exploring the size advantage of fbAb, TCE could have promising application as preferable structural blueprints for designing TCE.
Simple as it sounds, the complexity of designing an appropriate T cell engager necessitates a significant amount of attention to details, in the whole picture of drug development. Particularly, the minute details in CD3 modalities are the most studied finely tuned ones. Anti-CD3 domain is so far the most popular component in T cell engager design, as is seen in the fbAbs, such as blinatumomab and tebentafusp. One prominent difference is the affinity of the TCE against CD3. The affinity of CD3 in FDA-approved T cell engagers ranges from the lowest 260 nM for the CD3 x CD19 blinatumomab [106] to the highest of 4.73 nM for the CD3 x CD20 Epcoritamab [107]. This diversity in affinity suggests the potential for optimizing CD3 binding affinity to maximize TCE efficacy. Pre-clinical studies suggest that weak binding affinity (KD≥600 nM) could still be sufficient to avoid impacting the potency of the antibody if the density of the antigen on the target cell (such as TSA, tumor associated antigen) is high. However, if TSA density is low, both the affinity towards TSA and the affinity towards CD3 can significantly alter the outcome [15,104]. Therefore, when targeting cells expressing high levels of the antigen of interest, the engineer may choose to reduce the affinity of the antibody against CD3, so as to balance out adverse effects, such as CRS [15].
Selection of the epitope(s) or the different subdomain(s) of CD3 by TCE can also significantly alter the pharmacology of the drug, in terms of the efficacy and safety. The signaling patterns of their activation profile can be biologically different, as discussed in this review article [108]. Preclinical studies [109] have shown that co-targeting CD3δε and CD3γε could help to reduce dose-dependent, activation-induced cell death compared to target CD3δε alone. Studies in CAR-T has also noticed different functionalities of CAR-T when engineered with different CD3 subdomains [110].
It is widely known that TCE and CAR-T are usually compared and contrasted with each other, specifically when it comes to CD19-targeted therapies (applicable for cancers and autoimmune diseases), where arguments have been made favoring CAR-T or [111] TCE (BiTE, bispecific T cell engager) [112], over the other. The favoring view [112] suggests the following: off-the-shelf (availability), lower cost, almost no manufacturability issues (comparatively), no need for lymphodepletion, significantly lower incidence of ≥grade 3 adverse effects in many categories, around one fifth the cost of CAR-T, less risk of antigen loss, flexibility in targeting multiple antigens (e.g., as cocktails), and easier approaches to contain T cell exhaustion. The opposing view [111] claims that compared to blinatumomab, CAR-T has higher complete response rate (CR) in adults (blinatumomab=36%-44%; CAR-T=67%-100%), superior trafficking to treat disease site in CNS and extramedullary region, and better performance under higher tumor burden.
While the purpose of this review does not include a comparison between CAR-T and TCE, the shortcomings of TCE will be discussed, so as to shed light on its improvement. One key difference between blinatumomab and CAR-T is that both FDA-approved CAR-Ts (Tisagenlecleucel and Axicabtagene ciloleucel), are second generation CAR-T products that are equipped with both CD3 and a costimulatory domain, namely either CD28 or 4-1BB. The presence of costimulatory domain in CAR-T may put CAR-T at an advantage over blinatumomab by itself, given that the first-generation CAR-T (with only an CD3ζ domain for stimulation), achieved much less efficacy than blinatumomab [113]. Moreover, a recent phase 1b trial [114] has shown that BiTE is able to achieve 92.3% (n=13) of CR/CRh in oncology, if administered through the subcutaneous route, highlighting the potential to enhance the efficacy of fbAb TCE through improving dosing regimen.
One key advantage of TCE is their capacity to benefit patients who had undergone CAR-T therapy or even antibody therapy targeting the same antigen. For instance, in treating relapsed or refractory large B cell lymphoma in a phase I/II trial [115], Epcoritamab, an CD3 x CD20, TCE, has demonstrated higher efficacy for patients who had relapsed from anti-CD20 therapy (but lower efficacy from disease refractory to anti-CD20). Similarly, those patients who are refractory to CAR-T would benefit less [115]. This trial represents the synergistic potential for combining conventional IgG, fbAb TCE, and CAR-T in treating cancer, which can provide a broader coverage of the tumor associated antigen and avoid antigen escape. For instance, there are pediatric regimens which combine mini-hyper-CVD (cyclophosphamide, vincristine, and dexamethasone) with inotuzumab ozogamicin (INO), rituximab, and blinatumomab, resulting in relapsed/refractory B-cell ALL, with an overall response rate of 75% [116]. The mini-hyper-CVD regimen with INO, rituximab, and blinatumomab in adult patients yielded an overall response rate of 80% [116].
Last but not the least, TCE is beneficial for patients who have immunodeficiency, such AIDS, where a clinical case report [117] has shown that a patient with refractory multiple myeloma and HIV could benefit from BCMA x CD3 teclistamab, where no HIV rebound was observed. In contrast, HIV-infection could render the patient ineligible for CAR-T therapy due to the fact that detection of HIV (PCR, ≥13.2 copies/mL), is a violation of GMP standard [117].
4.1. T Cell Engagers in Cancer
As published in numerous pieces of literatures, T cell engagers have gained popularity in recent years in oncology, as evidenced by the FDA approval of mosunetuzumab (CD3 x CD20), Glofitamab (CD3 x CD20), Epcoritamab (CD3 x CD20), Teclistamab (CD3 x BCMA), Elranatamab (CD3 x BCMA), Talquetamab (CD3 x GPRC5D), Tarlatamab (CD3 x DLL3), Tebentafusp (CD3 x gp100-MHC), and last but not the least, blinatumomab (CD3 x CD19) [50,118]. The following section will discuss the clinical outcomes of fbAb in hematological malignancies and solid tumors, as well as some insights from their preclinical studies.
4.1.1. fbAb TCE for Treating Hematological Malignancies
Blinatumomab is the most well-studied fbAb so far, where the pros and cons from the roadmap of developing the drug serve as the best beacon for clinical and preclinical studies on fbAbs, consisting of an scFv-scFv fusion. The drug is approved to treat CD19+ acute lymphoblastic leukemia (ALL), and Philadelphia chromosome-positive B-cell precursor ALL that has relapsed or is refractory to treatment, A meta-analysis [46] of 18 studies involving 1373 patients treated with blinatumomab has shown a complete remission (CR) rate of 54% (95%CI:44%-64%). In randomized controlled trials [119], blinatumomab-treated group showed significantly better outcomes compared to the chemotherapy group, with a 12% improvement in OS (overall survival), and an 2.16-fold higher event-free survival rate.
Compared to the safety profile of CAR-T (in the same meta-analysis with 446 patients [120]), in terms of ≥grade 3 AEs, blinatumomab has lower neurological toxicity (18% vs 7%), lower CRS (19% vs 3%), and a similar level of neutropenia (38% vs 31%). One of the reasons for its relatively lower CRS and neurological toxicity is its dosing regimen, which, if designed properly, can mitigate toxicity, improve T cell functionality, and ultimately enhance efficacy. According to the latest drug label revised in 2018, blinatumomab is administered as a continuous infusion over 4 weeks, with a dose escalation of 0.5–90 μg/m2/d, followed by a 14-day or 56-day treatment-free interval or TFI [34,35]. The dose escalation allows clinicians to monitor the patients’ reactions from the patient and to alter the dose or provide additional support as needed.
The treatment-free interval (TFI) is crucial for maintaining the efficacy of the drug, as it could give some break for the T cells to prevent exhaustion. Analysis of the T cells from patients who underwent continuous treatment with blinatumomab has shown a reduction in cytotoxicity and IFN-γ level [121]. The same study [121] has also shown that continuous exposure of T cells to a half-life extended CD3 T cell engager antibody, AMG 562, can induce expression of T cell exhaustion markers (PD-1+Tim-3+LAG-3+), a reduction in T cell proliferation, cytotoxicity, release of granzyme B and IL-2. Furthermore, the adoption of a treatment free interval (TFI) or dasatinib (which can turn off T cell activation signaling), can also enhance treatment efficacy in terms of T cell proliferation, cytotoxicity, granzyme releasing, and the ratio of PD-1+Tim-3+LAG-3+ T cells [121].
While the pharmacokinetic challenges of blinatumomab posed significant challenges for its clinical development, efforts have been made to address this issue. Duvortuxizumab (MGD011) is a DART-Fc format CD19 x CD3 antibody intended to treat B cell malignancies [45], where the Fc domain could take advantage of the FcRn pathway. While the clinical results have not been published, an announcement from its parental company, MacroGenics, indicates that while multiple objective responses were observed, a number of patients experienced treatment-related neurological toxicities, even though these toxicities are similar to those seen in other CD19-targeted T cell engagers. Due to commercial competition, the development of duvortuxizumab is no longer attractive, highlighting the increasing commercial demand for higher standards of efficacy and safety for developing similar targeted TCEs. Similarly, during a phase I trial of AFM11, a CD19 x CD3 TandAb [78], development was halted due to a lack of efficacy and a significantly high incidence of severe adverse effects, including dose-limiting toxicities. As a result, neurotoxicity and limited efficacy led to the termination of the drug’s clinical development [45].
Amgen, in addition to developing blinatumomab, has also been developing many other BiTE drugs for treating hematological malignancies. One key lesson learned from these examples is the CD3 x BCMA AMG420 (also known as BI836909) for Relapsed and/or Refractory Multiple Myeloma (RRMM). During its phase I trial [122], it achieved an ORR of 31%, and a response rate of 70% and 50% of MRD-negative complete response in the maximum tolerated dose (400 μg/d) group [123]. Serious AEs were observed in 48% of patients (n=20), with the most prominent being infection (n=14), and polyneuropathy (n=2). While the efficacy of AMG420 is promising, it was surpassed by its analog drug, the FDA-approved CD3 x BCMA antibody elranatamab, which achieved an ORR of 63.6% and CR of 38.2% in phase 1 trial (n=55) [124]. Although elranatamab outperformed AMG 420 in terms of efficacy, AMG420 still achieved a 70% response rate at its maximum tolerable dose (400μg/d; 4-week infusion/6-week cycle) [122]. In comparison, Elranatamab showed its best response (ORR=83.3%) at a dose of 1000 μg/kg administered subcutaneously [124]. Moreover, both elranatamab and AMG 420 demonstrated dose-dependent efficacy profiles, highlighting the need for balancing pharmacokinetics with toxicity in the pursuit of high efficacy. This may explain why Amgen incorporated Fc domain in their subsequent product designs. AMG 330, a CD3 x CD33 BiTE, achieved a fair ORR of 14.2% but a serious AE incidence rate of 66% [125]. Due to lack of efficacy and toxicity, AMG330 clinical development has been terminated according to NIH website (NCT04478695) [45].
Besides BiTE, Flotetuzumab (MGD006), a CD3 x CD123 DART (dual-affinity retargeting) fbAb for treating acute myeloid leukemia, had achieved a promising outcome with a complete response of 11.7% for all groups and 26.7% in the patient group at the recommended phase 2 dose (RP2D) in the phase I/II trial [51], while the AEs are mostly grade 1-2 and manageable with appropriate interventions [51].
A preclinical research paper on Flotetuzumab (MGD006) suggests that the drug can modestly upregulate the expression of PD-1, but the exhaustion marker, TIM-3, remained low, with concurrent presence of high T-cell cytotoxicity following prolonged exposure to MGD006 suggests that the antibody has a lower propensity to induce T-cell exhaustion. [126]. Despite the promising efficacy, flotetuzumab has been discontinued from development. As explained by the CEO of MacroGenics to the investors [127], Fc-incorporation, along with a next generation of CD3 component in MGD024, the next generation CD123 x CD3 drug intended to replace MGD006, is able to extend half-life and significantly reduce cytokine release. MGD024 is currently under clinical investigation [128].
4.1.2. fbAb TCE for Treating Solid Tumor
Tarlatamab (AMG757), an CD3 x DDL3 BiTE antibody, has been approved by the FDA for treating extensive stage small cell lung cancer (SCLC) and therefore serves as the first BiTE approved to treat solid tumors. In its phase II trial [47], an ORR rate of 40% and 32% were observed in the 10-mg and 100-mg groups, respectively. Such efficacy is promising, as limited options are available for pretreated small-cell lung cancer patients. Compared to blinatumomab, its predecessor BiTE molecule with a half-life of 2.1 hours [129], tarlatamab, by fusing with an Fc domain [130,131], is able to achieve a clinical terminal elimination half-life of 5.7 days [132]. Therefore, while blinatumomab requires continuous infusion on a daily basis, tarlatamab only requires one-hour infusion on a biweekly basis, significantly reducing the burden on patients. (DLL3 is a Notch ligand that) plays a role in neuroendocrine differentiation and SCLC tumorigenesis [130,131]. Tarlatamab is able to recognize cells with low levels of DLL3 expression with less than 1000 DLL3 molecules per cell, demonstrating efficacy against patient-derived xenografts and orthotopic mouse model [130,131]. Compared to blinatumomab, tarlatamab has a binding affinity of 3 nM towards CD3 and 3.1 nM towards DLL3, which is much higher than that of blinatumomab (260 nM towards CD3 [106]). As the result, likely due to the stronger binding and activation, PD-L1 expression was significantly upregulated after co-incubating T cells, cancer cells, and tarlatamab [130,131]. Such observations, also observed in the study of AMG562 [121], are potential signs of T cells exhaustion, supporting the rationale for treatment-free intervals or combination with immune checkpoint blockers (ICBs).
The BiTE platform has provided successful examples. Although some business reorganization and strategic decisions may contribute to termination of BiTE development, from insights in business development [133], the BiTE platform from Amgen’s oncology devision stll has 4 active BiTE pipelines (excluding the two marketed drugs, blinatumomab and tarlatamab) and 14 inactivated BiTE molecules that were terminated after phase I. Except portfolio prioritization, clinical and scientific reasons, including toxicity, need for infusion, and immunogenicity, were major reasons why these BiTEs were terminated [133].
Similar to TriTAC HPN424, the PSMA x CD3 BiTE pasotuxizumab (BAY 2010122/AMG212) was clinically tested to treat metastatic castration-resistant prostate cancer [44]. While some efficacies were noted (≥50% PSA decrement in 3 patients, n=16; one patient showed complete regression of soft-tissue metastasis), 81% of patients have experienced at least one ≥grade 3 AEs. BiTE AMG211 (MT111/MEDI-565), an CEA x CD3 BiTE [134], has shown high immunogenicity so that the therapeutic window could not be defined in clinical trial, accompanied by insufficient exposure to establish objective response [45]. BiTE AMG 596 (also known as etevritamab), an EGFRvIII x CD3 BiTE [135], has been used to treat EGFRvIII+ recurrent glioblastoma (RGBM). Serious adverse effects were observed in 50% of patients (n=14), and 1 out of 8 patients had a partial response.
While the clinical landscape of BiTE has ups and downs, a similar trend could be observed in the TriTAC plaform. Similar to BiTE tarlatamab, HPN328, an CD3 x DLL3 x albumin TriTAC fbAb, also achieved promising clinical efficacy, exhibiting antitumor activity against relapsed/refractory metastatic SCLC (confirmed ORR=50%), neuroendocrine cancer (NEC, other than prostate neuroendocrine cancer; confirmed ORR=44%), while achieving linear PK and a median half-life of 71h [136]. Other TriTAC format antibodies, such as CD3 x PSMA x albumin HPN424 [137] and CD3 x mesothelin x albumin HPN536 (NCT03872206), are in clinical phases. HPN424 [137] has a well-tolerated safety profile, but disease progression (metastatic castration-resistant prostate cancer) has led to discontinuation of the treatment. PSA level declined in 21% of patients (n=63) with follow-up at 24 weeks. Heavy pretreatment (<2 prior systematic therapies), and the presence of antigen-escape or DLL3-negative tumor subtype in prostate cancer patients [138], could also be contributing factors. Last but not the least, in preclinical studies, compared to other peer TriTACs, HPN424 [73] and HPN536 [139], which are configured in a tandem domain format of anti-CD3-anti-albumin-anti-tumor target configuration (also known as the C:A:T format), HPN328 is configured in an anti-tumor target-anti-albumin-anti-CD3 configuration (T:A:C format) [74]. It was noted by the authors [74] that the C:A:T format HPN328 is 3.1-fold more potent than its format in T:A:C in in vitro, suggesting that geometric influence the binding between the anti-DLL3 domain and DLL3.
4.1.3. Improving and Examining the Safety of TCE in Treating Solid Tumor
As mentioned in the previous section, incorporation of Fc domain into the design of antibody could facilitate its half-life. Nevertheless, if the incorporated Fc is unsilenced, it could potentially cause issues by unintentionally depleting immune cells of interest. Antibody with unsilenced Fc and anti-CD3 domain can crosslink Fc-effector cells and T-cells, leading to mutual attacks by the immune cells [140], as is evidenced by catumaxomab, which was withdrawn in 2017 [140]. As shown in preclinical studies [141,142], TCE with active Fc and anti-CD3 domain not only failed to direct T cell to tumor but also induced T cell depletion and aggregation of T cells in the lung (for anti-HER2 TCE). Meanwhile, Fc-silenced TCEs do not sequester T cells in lungs but are able to induce T cell infiltration in tumors. Mutations like N297A and K322A can reduce ADCC or CDC towards T cells, incidence of CRS, while preserving the cytotoxicity of T cell and the consistency of pharmacokinetic profile [140,141,142].
One example is PF-06671008, a P- cadherin x CD3 DART with an extra Fc domain for extended half-life to ~4.4 days [53]. PF-06671008 was tested in a phase I clinical trial (N=27), and its further development was trial terminated due to lack of efficacy [52]. The clinical trial also shown that ≥grade 3 AEs (adverse events) occurred in 62.9% patients, with an incidence of ≥grade 3 CRS at 18.5%, ≥grade 3 lymphopenia at 33.3%, and ≥grade 3 Hypophosphatemia at 18.5%. Pre-clinical research on PF-06671008 [53] suggests the antibody has good tolerability at even a dose of 10 μg/kg in hPBMC/hFcRn-humanized mice, indicating that preclinical mouse model may not detect immune cell depletion by Fc-mediated mechanisms without cellular characterization. The Fc domain of the PF-06671008 DART antibody includes only knob-in-hole and disulfide bond engineering [53], but lacks modifications that deactivate Fc gamma receptor binding [143,144], posing potential threat to its efficacy.
When investigating the impact of off-tumor on-target toxicities using animal models, three approaches are generally employed. The first approach would be to use genetically modified animal expressing human gene encoding the target of interest, and the second approach would be to use non-genetically engineered animal with “animalized” version of the drug of interest (i.e., an antibody engineered to precisely target the animal homolog of the human protein target), and the third approach would be to test human drugs in non-human primates, like cynomolgus monkeys. Although all three approaches have their own advantages, their disadvantages are profound. The first approach may not recapitulate the interaction of the knock-in target protein with other proteins in the animal in physiological context, the second approach may not be able to recreate a drug analog that targets the animal target with a sufficiently similar binding modality, and the third approach can overlook subtle differences between human and non-human primate.
The abovementioned first and third approaches were utilized in the development of solitomab (AMG110 or MT110), an EpCAM x CD3 BiTE. Development of the drug terminated, due to on-target dose-limiting toxicity seen in clinical trial. 95% of patients have ≥grade 3 treatment-related adverse effect, with major TRAEs observed in lung, liver, and GI tract. Only one unconfirmed partial response was observed among sixty-five patients [131,145], yielding a maximum tolerable dose below potentially therapeutic dose level. Dosing selection was made from 1 μg to 96 μg /day, and it was determined that the maximum tolerated dose (MTD) is 24 μg /day. IHC shows CD3+ T cell infiltration of EpCAM+ duodenal epithelium as well as damage of crypt structure with villus collapse by H&E staining [131,145].
The preclinical testing of solitomab highlights the limitations of animal model for prediction of clinical toxicity for immune cell engagers and the necessity for more critical experimental design and data interpretation. When treating mouse (Female BALB/c mice (Janvier, Le Genest-Saint-Isle, France) 7 weeks old) for two days with 50 μg/kg/day of muS110, the murine version of AMG110, weight loss, hypoactivity, and diarrhea were observed [145]. Similar to results in clinical biopsy, mouse necropsy shows significance damage to vacuolated enterocytes in duodenum tissue, accompanied by granzyme B-expressing CD4+ lymphocyte infiltration. Interestingly, in early preclinical study of muS110 BiTE [146], muS110 was well-tolerated up to 50 μg/kg, while efficacy could be achieved as low as 5 μg/kg, both of which concentrations are significantly higher than those seen in later in vivo study or clinical study (24 μg/day) [131,145]. No significant weight loss was observed at 12.5 μg/kg until reaching the 12.5-400 μg/kg range in a BALB/c CT-26 IV tail vein xenograft model. Moreover, PBMC-humanized mice didn’t seem to display toxicity as well [146]. It could be noted that application of IHC could have been helpful to identify the toxicity profile of the drug, but still, the MTD in mouse and human are drastically different.
Importantly, the difference of MTDs of solitomab in mouse study and human study could not be explained by the binding affinity (for EpCAM, KD muS110 = 21 nM, KD MT110 = 13 nM), (for CD3, KD muS110 = 2.9 nM, KD MT110 = 100 nM) [146]. Despite comparable anti-EpCAM affinity and lower anti-CD3 affinity in MT110, solitomab could even be administered to cynomolgus monkey at a high concentration for 1mg/kg for a period of 6-10 day in pharmacokinetic study [147], suggesting a potentially safer profile in cynomolgus monkey than in mice.
These preclinical findings underscore the limited translatability of preclinical antibody safety research to clinical setting in solid tumor, where on-target off-tumor effect can frequently damage orangs and tissues. Organ-on-chip models [148] could be an effective method to assay on-target off-tumor toxicity of immune cell engagers. It was seen that lung-chip and intestine-chip, as well as their organoids, could help to reproduce and help to predict target-dependent toxicity of immune cell engager. For instance, in a study investigating anti-FOLR1 and anti-CEA therapy, under the presence of PBMC, signs of on-target off-tumor toxicity was observed, as evidenced by elevated levels of IFN-γ, granzyme B, and IL-6 by PBMC, by anti-FOLR1 engager against healthy lung and kidney cells, and by anti-CEA antibody against healthy duodenum cells in the chip [148].
Another methodology for designing safer T-cell engager is by targeting tumor-specific antigens displayed by the major histocompatibility complex (MHC), through engineering a TCE that incorporates a T cell receptor (TCR) that can recognize the tumor antigen-MHC complex. At present, such antibody design has been achieved to target tumor-associated antigen, with arguably the best-known example being the FDA-approved drug tebentafusp (in the ImmTAC format), containing an anti-CD3 scFv and a TCR domain that recognizes the complex of MHC-I (HLA-A*02:01) and the proteosome-digested fragment of gp100. From a meta-analysis [149], it has a partial response rate of 8% and a stable disease rate of 36% in treating metastatic uveal melanoma, a rare form of solid tumor originated in the eye. Other therapies, such as the anti-PD-1/PD-L1 immune checkpoint blockers, have limited efficacy due to the lack of PD-L1 expression in the primary tumor and liver metastasis. In a propensity score-weighted analysis [150], the 1-year OS rates for tebentafusp and pembrolizumab were 73% and 59% in a randomized, phase III trial (IMCgp100-202; N = 378, respectively, while in the single-arm GEM1402 (N = 52), ) in untreated metastatic uveal melanoma (mUM) the 1-year OS rate for nivolumab plus ipilimumab (N+I) in mUM was 52%. According to the phase 2 trial result of tebentafusp [151], among the 59% of patients with ≥grade 3 AEs, the breakdown of the most common AEs was as follows: rash (16%), hypotension (8%), CRS (4%), and pruritus (4%). Tebentafusp has an affinity of KD=38 nM towards CD3 [152] compared to that of blinatumomab [106]. Since the target of the antibody gp100, is abundantly presented on uveal melanoma cells, the resulting digested form of the peptide displayed by the MHC-complex gp100 has a decent amount of display on the cancer cells [153]. However, given that healthy melanocytic tissue also express gp100 [154], rashes caused by T cell and macrophage activation are also prominent adverse effects seen in clinical trial [150].
Tebentafusp not only serves as the harbinger for fbAb and TCE development in treating solid tumors but also paved the way for developing antibody therapies against MHC-peptide complexes as well as neoantigen therapy, a promising avenue for curing even the most recalcitrant types of cancer. Because MHC is able to display peptide fragments of intracellular origin, the antigen repository accessible to antibodies targeting MHC is more diverse than that of antibodies. Moreover, MHC-peptide display can present tumor-specific antigen (TSA), which can theoretically eliminate the risk of on-target off-tumor toxicity. Such methodology is promising in immuno-oncology. One of the most famous clinical studies [155] of neoantigen therapy involved a vaccine composed of up to 20 neoantigens plus atezolizumab, in treating pancreatic ductal adenocarcinoma (PDAC).
Among the 50% of patients who responded in the study, the recurrence-free survival was not reached at a median follow-up of 3.2 years and that of the non-responders was 11.0 months. In the study, the antigens were formulated as mRNA in lipoplex nanoparticles. Each of the mRNA encoded up to 10 MHC-I and MHC-II neoepitopes, and 11% of antigen epitopes were able to induce a T cell response sufficient to be detectable via ex vivo IFN-γ ELISpot. Besides neoantigen vaccines, engineered autologous T cell receptor (TCR) therapies recognizing specific MHC-peptide complexes have also shown promises, such as the FDA-approved Afamitresgene autoleucel, which can recognize the MAGE-A4 peptide presented by HLA-A*02 for treatment of advanced synovial sarcoma [156]. Despite the promising aspects of neoantigen vaccines and TCR cell therapy, the manufacture time and treatment cost are still major roadblocks due to the need for individualization, and these disadvantages can be addressed by off-the-shelf fbAbs like tebentafusp. Antibody and TCR-based fbAbs against MHC-peptide complex for mutant p53 [157,158,159] or KRAS [160,161], have been developed (see one review here for more details [162]), serving as arsenals for antibody engineers to generate optimized versions of fbAbs.
Similarly, besides tebentafusp, other formats of ImmTACs, like IMC-C103C (CD3 x MAGE-A4), and IMC-F106C (CD3 x PRAME) [1], are also being tested in clinical trials. A clinical trial of IMC-F106C in cutaneous melanoma (CM) for patients who had been treated with immune checkpoint inhibitor (ICI) has shown a combined partial response (PR) plus stable disease (SD) of 61%, with most common AE being grade 1/2 CRS in 50% of patients [163]. On the other hand, despite showing promise in clinical trials, IMC-C103C has been discontinued from development by Roche, which does not seem due to lack of safety or efficacy but rather due to business reasons [164], likely due to potential competition from the newly FDA-approved anti-MAGE-A4 TCR Afamitresgene autoleucel [156].
4.1.4. Targeting 4-1BB and CD28 in TCE Design
Targeting T cell costimulatory receptors, such as 4-1BB and CD28, by fbAb, has also been investigated for clinical efficacy. NM21-1480, an scMATCH3 (scFv3) format fbAb with tri-specificity for PD-L1, 4-1BB, and human serum albumin (HSA), has also been tested for clinical safety and efficacy and serves as a notable example for discussion. The antibody marks both an example of a trispecific fbAb, demonstrating successful half-life extension and controllable 4-1BB-medaited T cell activation [165,166]. The 4-1BB-targeting scFv of NM21-1480 binds to a membrane-distal epitope of 4-1BB. Since clustering of at least three 4-1BB molecules is required for stimulation of 4-1BB signaling [167,168], the activation of 4-1BB can be achieved once the antibody is bound to a target cell expressing PD-L1. The ultrahigh binding affinity of NM21-1480 towards PD-L1 allows the antibody to serve as an anchor on the cancer cell for T cells, transforming the cancer cell into a platform for 4-1BB clustering to enable tumor-specific activation. The lower affinity of NM21-1480 for 4-1BB prevents it from effectively clustering 4-1BB [165,166]. This design of NM21-1480 also suggests that multivalent antibody targeting more than one 4-1BB domain may be less desirable compared to a monovalent approach for targeting 4-1BB. A preclinical study [165] shows that NM21-1480 can lead to tumor regression in 60%-80% of animals across multiple cancer types, including non-small cell lung cancer (NSCLC), breast adenocarcinoma, and colorectal adenocarcinoma. The study also shows no detectable systematic cytokine release, confirming localized activity. A phase I trial (N=26) [166] for unresectable solid tumors, has shown that NM21-1480 exhibits a disease control rate of 57% in the 24 mg-800 mg dose range groups, where 62% of patients had received anti-PD-(L)1 therapy. The trial also shows that NM21-1480 has no maximum tolerated dose, with no incidence of grade >3 AE, endorsing the safety of the fbAb platform. Moreover, the drug is dosed every two weeks [166], in alignment with its preclinical profile for a half-life of 14 days in cynomolgus monkeys [165], indicating a major improvement of pharmacokinetics and dosing regimen compared to the non-half-life-extended blinatumomab. Other similar 4-1BB targeted therapies are being developed, such as GEN1046 (4-1BB x PD-L1, [169]), and TJ-CD4B/ABL111 (4-1BB x Claudin 18.2, [170]).
Like targeting 4-1BB, targeting CD28 has also been investigated. CD28-tageted therapy, unlike CD3-targeted therapy, provides direct co-stimulation independent of TCR signaling, enabling activation even in environments with suboptimal antigen presentation [171]. CD28 bispecifics can also counteract T cell exhaustion by delivering strong co-stimulatory signals that restore metabolic competence in T cells, as evidenced by increased mitochondrial biogenesis and enhanced glycolytic capacity in treated cells [171].
4.2. fbAb TCE in Treating Infectious Disease
As will be discussed in this section, T cell engagers have been studied and clinically tested for viral infections [172,173,174,175,176], the efficacy and toxicity of these agents have been examined. Activation of T-cell by TCE engager can usually leads to the release of substantial amount of IFN-γ and TNF. In combating viral infection, IFN-γ can downregulate the expression of viral entry receptors (e.g., claudin-1 for HCV and CD4 for HIV), inhibit RNA synthesis, block protein synthesis, induce RNA degradation for HBV, disrupt capsid assembly for HBV, and destabilize cccDNA (covalently enclosed circular DNA) for HBV [177]. IFN-γ can also upregulate MHC processing and presentation, which can facilitate the generation of adaptive immune responses [178,179]. TNF can induce RNA degradation (in SARS-CoV-2, EMCV, and influenza), help activate NK cells and M1 macrophages for pathogen clearance, and synergize with IFN- γ [180].
Moreover, TCE-activated T cell can release a substantial amount of cytokines contributing to CRS. For instance, COVID infection and TCE activation are both characterized by elevated levels of IL-6, IL-10, TNF- α, and GM-CSF [181,182,183,184,185,186]. While CRS can be helpful for combating infection, TCE and CAR-T therapies designed to treat COVID must account for the risk of CRS [183,184], which, when compounded with the CRS resulting from COVID infection [185,186], may cause severe adverse effects and even potentially death of patient. For instance, an analysis has shown that B-cell depleting TCE can significantly increase the mortality of COVID patients compared to the canonical IgG-based B-cell depletion drug rituximab (57,1% vs 12.5%, p=0.002), yielding an adjusted odds ratio of 7.08 (95% CI = [1.29-38.76]) [176]. Therefore, both the safety and efficacy of the antibody should be considered during the design process.
4.2.1. fbAb TCE in Treating SARS-CoV-2 and Influenza (Pre-Clinical)
SARS-CoV2-infected cells display spike proteins on the cell surface. After SARS-CoV-2 viral infection, the viral spike protein is synthesized in the endoplasmic reticulum, and is substantially transported and integrated into host cell membrane, serving as anchorage points for adhering to and infecting neighboring cells [187,188,189]. Considering this feature, two fbAbs with the CD3 x ACE2 and CD3 x spike protein [183,190] BiTE designs can achieve efficient outcomes in pre-clinical studies. Research [190] for S-BiTE has shown that the binding region of S-BiTE within the RBD is evolutionarily conserved across variants (including Omicron BA.1/BA.2/BA.4/BA.5). S-BiTE can also induce activation of IFN-γ and TNF when added to co-cultures of T cells and spike-expressing cells.
Influenza, while targetable by vaccinations, can still be potentially treated by T cell engagers. Recent research [191] has identified a broadly cross-reactive TCR from CD8+ T cells capable of targeting 9-12 influenza virus variants spanning from 1918 to 2024, where the targeted epitope is highly variable but remains immunodominant. Such epitopes can be used for screening in display platforms, while platforms such as ImmTAV [172,173,174,175], which utilizes TCR as part of its structure, can directly draw on the availability of the TCR to rapidly develop TCE.
4.2.2. fbAb TCE in Treating HIV
A key challenge for HIV treatment lies in the presence of a latent viral reservoir, in which a population of CD4+ T cells harbors replication-competent provirus that persists despite antiretroviral therapy (ART) [192]. These cells (also known as viral “reservoirs”) can evade immune detection due to minimal viral protein expression, posing challenges for treating HIV with antibodies and T-cell based therapies. Latency-reversing agents (LRAs), such as histone deacetylase (HDAC) inhibitors like vorinostat and romidepsin, have shown limited success in reactivating latent HIV and inducing re-expression of viral antigen [192]. Theoretically, polyclonal or multispecific antibodies could exploit multiple epitopes or different viral antigens, whereas ultrahigh-affinity antibodies may be able overcome the scarcity of cell surface viral antigen density.
One of the best examples of the ultrahigh affinity approach is the ImmTAV fbAb platform, with the clinical stage example of IMC-M113V and IMC-I109V. The platform utilizes anti-CD3 scFv as well as a picomolar-affinity TCR to bind to HLA (HLA-A*02:01) on HIV-infected cells that are displaying gag peptide [174,175]. Due to the ultrahigh-affinity nature of the fbAb, it is capable of eliminating HIV-infected CD4+ T cells from ART-treated patients, and the efficacy is correlated with HIV gag expression [174]. The study also showed some limitations, where immune dysfunction (PD-L1+TIM-3+) in T cells from ART-treated patient was associated with reduced efficacy compared to T cells from healthy sources, suggesting a potential needs to combine immune checkpoint inhibitor and/or interleukins for correcting patients T cells for maximum efficacy. In addition, in phase 1/2 trial update from the company [175], IMC-M113V demonstrated good tolerability (no serious adverse events), dose-dependent cytokine increase, as well as dose-dependent viral control after interruption of antiretroviral treatment (ART). Three patients with evidence of viral control were suspended from ART, for analytical treatment interruption as specified in the protocol of the clinical trial for up to twelve weeks. The three patients experienced viral rebound, followed by viral reduction to approximately 200 c/mL, which occurs in less than 1% of all patients living with HIV in general [193], suggesting the presence of an immune response against the virus [175].
Besides ImmTAV, the DART-Fc format antibody, MGD014, which directly binds to gp120 of HIV (CD3 x gp120), was also clinically examined, showing good tolerability in a phase I trial [194]. Nevertheless, when used as standalone therapy, neither MGD014, nor the DART-Fc MGD020 (CD3 x gp41), were able to decrease persistent viral infection biomarkers, such as cell-associated viral RNA or time to rebound of viremia after ART interruption [194], highlighting the need to apply combinatory approach to enhance the efficacy.
Simultaneously reactivating latent HIV and targeting viral antigen has also been attempted, as evidenced by preclinical studies of a trispecific TCE targeting CD3/CD28 x gp120 [195,196]. The platform utilizes an antibody fragment that targets a conserved sequence in the CD4-binding region of gp120, allowing identification of viral antigen, whereas binding of CD3 could stimulate latent infected-T cell (ACH2, J1.1 and OM10) and cause reactivation of latent HIV. Overall, binding of gp120 and CD3/CD28 can cause latent T cells to be reactivated [197], expressing HIV and HIV-related proteins, forcing the reactivated HIV-infected T cell to be immunogenic for the attack by the gp120 x CD3 TCE, ultimately achieving both in vitro (HIV patient cells) and in vivo efficacy in rhesus macaques [195,196].
4.2.3. fbAb TCE in Treating HBV
Immunotherapy for HBV faces challenges in curing HBV infection, such as difficulty in eliminating cccDNA reservoir, suppression of pathogen recognition receptors (PPPs), and the exhaustion of CD8+ T cells from chronic infection [198]. Unlike the usage of T cell-based therapies in treating HIV, controlled activation is crucial in treating HBV, since HBV can hijack healthy liver cells. T cells can cause hepatocyte apoptosis and liver damage, which can be exacerbated by the pre-existing inflammatory microenvironment [198]. Moreover, the clonal diversity of HBV should also be addressed to avoid mutant escape.
The MHC-restricted targeting approach could be helpful for limiting the toxicity of TCE. Similar to IMC-M113V, IMC-I109V [172,173] is also part of the ImmTAV variant fbAb utilizing a TCR-based method that can recognize a conserved region of HBV Env protein. In a clinical trial, IMC-I109V displayed good tolerability, with no reports of serious adverse effects, and no association with CRS as seen in preliminary clinical result. A rise of IL-6 and alanine aminotransferase level was also observed, indicating a response consistent with the mechanism of action of the antibody [172,173]. Efficacy from the prospective phase 2 trial will need to be confirmed, as in the phase 1 trial, a decline in serum viral surface antigen was observed in a low dose group, albeit with a transient response duration [172,173]. Moreover, a preclinical study has demonstrated that ImmTAV can also be used to target HBV Pol, Core, and Env, by activating T cells and promoting the release of inflammatory cytokines [199].
Similarly, preclinical studies of TCE fbAbs with various formats directly targeting CD3/CD28 x HBV antigens have been developed [200]. The study compared BiMAb (four scFv connected to an Fc-delta by a G4 or G4S3 linker) and FabMAb (Fab connected to an scFv with an G4S3 linker). Each BiMAb or FabMAb binds to only CD3 or CD28. It was observed that FabMAb and BiMAb performed similarly in activating CD8+ T cells (LAMP1+CD25+) when equipped to bind to CD3, whereas when binding to CD28, only FabMAb exhibited activation but not BiMAb. Co-treatment with both CD3- and CD28-fbAb TCEs can achieve a synergistic effect in T cell activation. Interestingly, during prolonged co-culture of the fbAbs (mixture of CD3 and CD28-TCE) with PBMC and HBV infected HepG2-NTCP cells, from day 10-12 in the medium, FabMAb achieved superior control of BHeAg, compared to BiMAb.
4.3. fbAb TCE in Autoimmune Disease
Efforts have been made to deplete B cells with the aim of resetting B cell immunity to eliminate the production of autoantibodies by B cells and activation of autoreactive T cells observed in many autoimmune diseases, such as systematic lupus erythematosus (SLE), rheumatoid arthritis (RA), and systematic sclerosis. Studies utilizing rituximab in B cell depletion therapy for treating SLE has been shown a significant reduction in the levels of anti-dsDNA [201,202,203]. Nevertheless, rituximab cannot reduce the level of anti-histone, anti-RNP autoantibodies [204]. It is believed that long-term presence of high serological levels of anti-Sm/RNP and Ro/La autoantibodies is likely due to germinal center (GC)-derived long-lived plasma cells as well as memory B cells, whereas the transitory presence of anti-dsDNA autoantibodies is likely an indication of presence of short-lived plasma cells [203]. Therefore, targeting both short-lived and long-lived plasma cells is needed to address the therapeutic need of resetting the B cell immunity. Since CD20 is a marker for less mature B cells while CD19 is a marker for more differentiated B cells [205], CD19 targeted therapies are believed to have more comprehensive coverage of B cells lineages fall under different growth stages and therefore offer greater potential to cure autoimmune diseases. Indeed, CD19 profiling of SLE patients who had received CD19 CAR-T therapy after three months showed that autoantibodies against dsDNA, Ro/La, and histone were reduced in five out of six patients [206], indicating a likelihood of the removal of autoreactive long-lived B cells [203]. In another clinical study [207], all five patients receiving CD19 CAR-T therapy achieved remission for SLE according to the DORIS criteria.
In contrast to CAR-T, TCE can also be utilized in a short-term manner with an option of stop treatment as needed. Compared to traditional IgG antibodies, TCE has the advantage of being able to leverage the T cells in germinal center and tertiary lymphoid tissue, and this advantage could be further reinforced by fbAbs with even lower smaller molecular weight designs [208]. Clinical results [209,210] for compassionate use of blinatumomab for rheumatoid arthritis (RA) have shown decreased disease activity and reduced autoantibody levels. Improved synovitis was confirmed by both ultrasound and FAPI-PET-C. Flow cytometry showed a reset with depletion of activated memory B cells, which were replaced by none-class-switched IgD-positive naïve B cells. Treatment was safe, characterized by a brief increase in body temperature and no signs of CRS. [209,210].
Success has also been seen in a case report describing treating a patient with severe systemic sclerosis in a case report [211]. In the case report, a patient with severe systemic sclerosis experienced significant improvement of symptom, characterized by a regained ability to move freely and resolution of burning skin sensations. By the end of the treatment, the patient had significant improvement in their condition. Interestingly, the treatment did not lead to an increased risk to infections. Serum IgG levels and specific antibody titers against infectious agents (haemophilus influenzae, pneumococcus) or toxins (tetanus anddiphtheria) were not affected by the therapy [211], resembling the clinical observation of anti-CD19 CAR-T in treating SLE [207].
In alignment with targeting CD19, CLN-978, a CD3 x CD19 x serum albumin fbAb, is being investigated for clinical trial in hematological malignancy [212] and SLE (NCT06613360), which has shown promising result as announced by the company and is being investigated for phase 1b trial for SLE [213]. Results in oncology [214] has shown that CLN-978 can effectively deplete B cells in B-non-Hodgkin Lymphoma, with a peripheral B cell depletion rate ranging from 93%-98% within 96 hours after a 30 μg subcutaneous dosing.
5. fbAb NK Cell Engagers (NKCE)
Natural killer cell engager are specialized antibodies connecting with activating receptors, such as CD16a NKG2D, NKp30, NKp46, NKG2C, as well as inhibitory receptors such as Siglec-7 [215], and are capable of inducing antibody-dependent cellular cytotoxicity (ADCC). Conventional antibodies, which activates interaction of NK cells, exhibits weaker and more transient interaction with CD16a through its Fc domain [215,216].
Receptors on natural killer cells play crucial roles in the immune responses against tumors and viral infections. For instance, CD16a mediates binding to Fc of IgG format antibodies and significantly contributes to the efficacy of FDA-approved oncology antibodies, including rituximab, trastuzumab, and cetuximab. NKG2D pairs with CD94 to respond to cytomegalovirus (CMV) infection by counteracting inhibitory signal of NKG2A. NKp46 binds to CD3ζ or FcRγ to mediate the lysis of target cells. NKp30 induces NK activation via interacting with B7-H6 [215,216]. Similar to T cells, NK can form immunological synapse with the target cell via the bridging of antibody, subsequently releasing cytotoxic molecules, such as granzyme B, perforin, IFN and TNF [215,216]. NK cells have two subsets: CD16+CD56dim and CD16-CD56bright subsets, where the former is primarily cytotoxic and the latter is associated with helper functions similar to CD4+ helper cells, and sometimes acts as regulatory NK cells by producing IL-10 [217].
Unlike non-engineered CD8+ T cell, which require recognition of peptide displayed on MHC-I molecules to perform cytotoxic functional, NK cells would kill MHC-I-negative cells, who are considered to be “non-self”. This feature serves as a complementary immune surveillance mechanism to T cells, allowing detection of cancer cells or viral-infected cells that downregulate or inhibit the expression of MHC-I molecule to evade MHC-TCR-based T cell detection [218].
5.1. fbAb NKCE in Treating Cancer
NKCEs have gained popularity in oncology in both fbAb format (AFM28, AFM24, AFM13, GTB-3550, SAR443579/ IPH6101) and IgG-like format (AFM26/RO7297089, AFVT-2101, TriNKET). While T cell engager clearly demonstrated efficacy in numerus trials, NKCE also possess similar potential, if not superior efficacy and safety in certain cases. AFM28 (TandAb-Fc, CD16A x CD123) has achieved an composite complete response rate of 30% (n=12) at dose levels of 250 mg and 300 mg against relapsed/refractory acute malignant leukemia (R/R AML) in a phase 1 trial [216, 76]. Grade ≥ 3 treatment-related adverse effect (TRAE) were observed in 13% of patients. In comparison, phase I results of CD3 x CD123 T cell engager vibecotamab (XmAb14045) [219] showed an ORR of 11.5% in efficacy-evaluable group with 0.75 μg/kg or higher dose group (n=87), with 87.5% (n=120) of grade ≥ 3 TRAEs. Addition, combined NKCE and TCE therapies also hold promise in reducing cytokine secretion by T cells, as shown in preclinical study targeting BCMA [220].
Moreover, NKCE can also be combined with immune checkpoint blockers for synergistic effects. AFM24 (TandAb-Fc, CD16A x EGFR), when combined with atezolizumab (480 mg AFM24 + 840 mg ate via infusion), achieved an ORR of 27% (4/15; 1 CR + 3 PR) in patients with WT-EGFR+ metastatic NSCLC with at least one prior therapy [221]. This efficacy is significantly higher compared to AFM24 monotherapy (1/10 of PR) [222] or atezolizumab monotherapy (ORR=15%, [223]), suggesting a synergistic potential. Similar success was observed with AFM13 (TandAb, CD16A x CD30) when combined with pembrolizumab (ORR = 64.2%-73.9%, [224]), achieving an ORR of 88% (n=30) in relapsed/refractory Hodgkin lymphoma. However, the incidence rate of infusion-related reaction increased, necessitating the use of acetaminophen and corticosteroid [80]. AFM13, has also demonstrated monotherapy efficacy in relapsed/refractory peripheral T cell lymphoma (ORR=32.4%, serious TRAE=8%, n=108 [225]), and in relapse/refractory classical Hodgkin lymphoma (R/R HL) (ORR=16.7%, n=25, [226]. When combined with pembrolizumab, AFM13 achieves an ORR of 83% in relapsed/refractory Hodgkin lymphoma [80]. Given that cancer patients may undergo intensive pretreatments that compromise or exhaust conditions of NK cells, adoptive NK cell therapy is being clinically explored in combination NKCE. For instance, AFM13 has been combined with allogenic NK cells (ab-101) in treating R/R HL [227], and AFM24 (CD16 x EGFR) with autologous SNK01 NK cells, which do not require lymphodepletion [228].
TriKE is a class of trispecific fbab, with a first domain targeting the NK cell antigen CD16, a second cancer or viral antigen, and a third domain composed of an inserted IL-15 fragment to stimulate NK cells [88,89,90]. The CD16 x IL-15 x CD33 TriK, GTB-3550, was evaluated in a phase I trial and had shown to be able to control the disease progression (ORR=0, disease control rate =2/3, n=3), with no clinically significant toxicity observed [91,92]. Researchers observed that the number of activated NK cells (CD69+) increased beginning on day 3 and remained above baseline through day 15 and day 22, until treatment completion [91,92]. To enhance therapeutic outcomes, GT Biopharama is developing a second-generation version of the drug, GTB-3650, which replaces the scFv domain of the drug with a camelid VHH. GTB-3650 has received FDA IND clearance, with a clinical study set to begin on January 1, 2025 [229].
Co-targeting CD16A and NKp46 is another approach to further enhance NK cell toxicity, as exemplified by the TriNKET as well as the ANKET platforms. TriNKET is a multispecific NK cell engager, targeting CD16a, NKp46, tumor associated antigens, and in certain cases, the beta chain of IL-2 receptor [70]. Unlike multivalent CD38-CD38 crosslinking by daratumumab, which may cause fratricide [230], preclinical studies have shown that CD16A-CD16A [230], or CD16-NKp46 crosslinking [69], does not cause fratricide. The safety profile of TriNKET DF1001 (CD16, NKp46, HER2) was favorable (incidence of grade 3 TRAE = 14%), and its efficacy (ORR=13.9%, n=36) suggests potentials use in combination therapy for relapsed/refractory solid tumors [71]. Similarly, ANKET format antibody developed by Innate Pharma and Sanofi has demonstrated promising preliminary clinical results. According to the company’s updated clinical result, the trispecific ANKET SAR443579/IPH6101 (CD16 x NKp46 x CD123), induced complete response in 5 patients at 1mg/kg dose in a phase 1/2 hematological malignancy trial [72].
5.2. fbAb NKCE in Treating Infectious Disease (Preclinical)
While no clinical-phase research antibody are current available, preclinical studies of NKCE have shown promise. NK cells are natural defenders against viral infection, capable of detecting MHC-downregulated target cells and engaging them via neutralizing antibodies via ADCC [231]. The BiKE CD16a x gp160/gp140/gp120 antibody [232] is constructed by fusing a single-domain soluble CD4 to an scFv. This antibody can bind to either coated gp140sc (gp140, soluble and cleaved), gp160sc-expressing 293T cells, or gp160-expressing CHO cells. gp160 is cleaved into gp120 and gp41, whereas gp140 is an engineered version of gp160 that lacks gp41 so that it is more soluble and easier to study for experimental purpose [233]. These antibodies can induce NK cell degranulation and killing of Env-expressing cells, as well as the elimination of both chronically and acutely HIV-infected T cells by PBMC. Due to their small size, these antibodies may also be capable of crossing the blood-brain barrier (BBB) and achieve deep tissue removal of HIV-infected cells.
5.3. fbAb NKCE in Treating Autoimmune Disease
The current literature suggests a limited number of clinical phases NKCE developed for the treatment of autoimmune disease is available. Nevertheless, insights from allogenic and engineered NK (CAR-NK) can provide valuable clinical prospectives. Analogous to the concept of CD19 CAR-T, the allogenic CD19 CAR-NK, named NKX019, when combined with rituximab or obinutuzumab, can synergistic deplete pathogenic B cells [234]. As the result, NKX019 is currently being evaluating in phase I trial for systemic lupus erythematosus (SLE) requiring for lymphodepletion [235]. As treatment with immune cell engagers do not require lymphodepletion, it is possible that NKCE poses a competitive edge over CAR-NK for treating autoimmune disease.
This observation suggests not only the feasibility of combining CD19 and CD20 as combination targets for treating lupus, but also that immune cell engagers may potentially be combined with CAR-NK for enhanced therapeutic outcomes. Preclinical research has shown that combining CD19-CAR-NK and rituximab (anti-CD20) can help to overcome CD19- antigen, preferentially sparing CD19+ healthy B cells (versus cancer cells), and boost CAR-NK degranulation and cytokine production capacity [236]. In addition, in vitro study has shown that combining CD19 antibody with CD19 CAR-NK or CAR-T can enhance serial killing capacity of these immune cells by facilitating rapid detachment of these CAR effector cells from target cell by reducing trogocytosis [237], further testifying the validity of combined B-cell depleting immunotherapy in treating autoimmune disease. Similarly, iPSC-derived CD19-CAR-NK, such as FT522, is armed with features such as anti-CD19 CAR, non-cleavable CD16 (binding to therapeutic antibody), and IL-15/IL-15 receptor, CD38 knockout to increase mitochondrial respiratory capacity. FT522 have demonstrated B cell-depleting capacity in oncology when combined with rituximab [238].
6. Myeloid Cell Engagers (MCE) and B Cell Engager (BCE)
Besides T cells and NK cells, targeting myeloid cells a can not only address the specific therapeutic needs but also potentially creating synergistic effects with other immunotherapies. Due to the overlapping of some cell surface receptors between myeloid and B cells, such as CD40 and TLRs, myeloid cell engagers can sometimes activate both cell types, leading to additive efficacy. As of 2024, there are eight MCE in clinical trial, although five MCE have been discontinued from further clinical development, highlighting the opportunities and challenges in this field [101,216]. Numerous macrophage and neutrophil engagers (FcγRI, FcαRI, SIRPα), dendritic cell engagers (CD40), granulocyte engagers, and B-cell engagers, which are identified, studied and put for clinical trial [101,216].
Macrophages, derived from circulating monocytes, perform phagocytosis, engulfing targets such as cell debris, cancer cells, or pathogens. Phagocytosis can be facilitated through antibody bridging, a process known as antibody-dependent cellular phagocytosis (ADCP) [101,216]. Neutrophils are usually recruited to sites of infection or tumorigenesis, where they perform degranulation, phagocytosis, and trogocytosis. Both macrophages and neutrophils can exhibit proinflammatory/antitumor phenotype or immunosuppressive/protumorigenic phenotype [101,216]. B cells, in contrast, produce antibodies and coordinate with T cells, which can play therapeutic roles in targeting pathogens and tumors or pathogenic roles in generating autoreactive immunity.
The discontinuation of development of many myeloid cell engagers (MCEs) was not due to the drug themselves. Many clinical trials for these MCEs were performed in the early 2000s, when limitations in clinical understanding and biomedical technologies hindered the development of these agents. At that time, the lack of modern cytokine release syndrome (CRS) management protocols and anti-IL-6 drugs like tocilizumab (approved in 2017 [239]) prevented the developers from effective mitigating safety concerns. In addition, early clinical trials often lack biomarker-driven patient selection. For instance, the phase II trial of MDX-H210 [240] included patients with varying HER2 expression levels, and the trial for MDX-447 [241] also did not require EGFR positivity from in patient. Moreover, the immature state of recombinant technologies and the high cost of synthetic biological production at the time presented challenges for manufacturing multispecific antibodies. The rise of immune checkpoint inhibitor drugs has diverted corporate resources away from MCEs, leading to pipeline discontinuation. Macrophages, which express all classes of FcγR receptors and FcαR receptor can execute ADCP, ADCC, and mediation of inflammation and cytokine release [101,216]. With these earlier roadblocks now largely overcome, MCE R&D may experience a resurgence.
Many clinical phase MCEs were examined in clinical trials [101,216], including but not limited to: MDX-210/MDX-H210 (FcγRI/CD64 x HER-2 [240,242,243]), MDX-447 (FcγRI x EGFR [241]), H22xKi4 (FcγRI x CD30 [244]), BDC-1001 (TLR7/8 x HER2 [96,245]), NJH395 (TLR7 x HER2 [97,246]), TAC-001 (TLR9 x CD22 [247,248]), BR105 (SIRPα with silenced Fc [249]), IMB071703 (XFab-α4-1BB/CD40L [68]), MGD010 (DART-Fc, CD32B x CD79B [250]), and MP0317 (the only clinical phase DARPin fbAb, CD40 x FAP [85,86]). These antibodies incorporate scFvs to bind to the antigens or employ covalent fusion to protein ligands or small-molecule agonists, collective referred to as immune-stimulating antibody conjugates (ISACs), resulting in a diverse spectrum of myeloid cell engagers.
6.1. MCE and BCE in Treating Cancer
FcγRI/CD64-based, antibodies, such as MDX-210/MDX-H210, MDX-447, and H22xKi4, can activate macrophage, monocytes, dendritic cells, and cytokine-primed neutrophils by binding to IgG, thereby initiating ADCC and ADCP [101,216]. Since these drugs were tested during clinical trials of early 2000s, their limitations were highlighted by difficulties in managing drug-related symptoms. Overall, moderate or no efficacies and only transient responses were reported in these trials [240,241,242,243,244]. One of the challenges in targeting FcγRI is the that serum IgG can compete for CD64 binding, thereby reducing efficacy through competitive inhibition [251]. Engineering of these antibodies to target different epitopes may resolve this issue. Alternatively, co-activating both FcγRI and FcαRI [252] could further enhance macrophage potency and coordinate cytotoxicity with NK cells.
CD40-targeted therapies, including IMB071703 (XFab-α 4-1BB/CD40L [68]) and the DARPin format fbAb MP0317 (CD40 x FAP [85,86]), function by orchestrating interactions between T cells and antigen-presenting cells (APCs). IMB071703 targets 4-1BB on T cell and CD40 on APCs (including B cells), particularly dendritic cells, promoting immune synapse formation and facilitating crosstalk between the cells to activate antigen specific adaptive immune response. However, clinical trial showed no objective response [68], in contrast to promising outcomes observed in preclinical study [253]. The dosing regimen, which involved intratumoral injection, may not present optimal pharmacokinetic profile as have indicated by its clinical half-life of 2.45 hours [85,86]. Infusion or subcutaneous route (as discussed in the previous sections for T cell engagers), may offer more sustained stimulation and a higher cumulative chance of eliciting adaptive immunity.
MP0317, on the other hand, activates CD40+ APCs via its anti-FAP arm, which recognizes fibroblasts in the tumor microenvironment, therefore limiting toxicity to fibroblast-rich tumors. Phase I studies [85,86] demonstrated well tolerability in all dose groups (0.01-10mg/kg). Tumor biopsies confirmed successful co-localization of MP0317 with FAP and CD40. Genes associated with B cell trafficking (CXCL3, CXCR5, CCR6, and CCL20) and IFN-γ downstream chemokines (CXCL9, CXCL10) were upregulated, while pro-inflammatory genes (IL-2, IL-6, IL-8) were not, indicating tumor microenvironment remodeling [85,86].
TLR7/8 and TLR/9 are expressed on myeloid cells and B cells [101]. These receptors play key roles in pathogen recognition, particularly of ssRNA and CpG DNA. Due to their small molecular size, TLR ligands can be covalently linked to fbAb to create anti-TLR functionality. TLR7 promotes production of type I interferons (IFN-α/β) and dendritic cell maturation [254], while TLR8 can activate activates NF-κB and induces proinflammatory cytokines (e.g., TNF-α, IL-12, IL-1β) that recruits neutrophils and macrophages [255,256]. TLR9 triggers interferon regulatory factors and NF-κB pathways, regulating B cell tolerance and potentially suppress TLR7 driven autoantibodies in lupus models [255,257].
Recent clinical development of TLR-targeting antibodies differs from early MCE trials by taking advantage of combinatory therapy. For example, the phase I/II trial of BDC-1001 (TLR7/8 x HER2) included co-treatment with nivolumab [96], demonstrating good tolerability and efficacy in heavily pretreated patients. NJH395 (TLR7 x HER2), a first-in-class ISAC [258] showed a manageable safety profile in clinical trials, although neuroinflammation was observed at high doses, suggesting potential interaction with CNS-resident myeloid cells. Anti-drug antibodies were detected in all tested patients, potentially limiting repeat dosing. Non-linear pharmacokinetics indicated target-mediated drug disposition. While both BDC-1001 and NJH395 targets HER2 and TLR, NJH395 specifically targets TLR7, thereby inducing IFN-α/β without IL-1β induction via TLR8, which may improve efficacy at the cost of increased systemic toxicity [259]. Due insufficient efficacy, both BDC-1001 [98] and NJH395 [260] were terminated for further development.
TAC-001 is a TLR9 x CD22 antibody that significantly activates both B and T cells, including immunoglobin secretion, and is currently under investigation in a phase I/II trial [247,248]. In addition to its oncology application, TAC-001 has also shown promises in enhancing vaccination efficacy in preclinical studies involving elderly animal model [261]. For reference, this review [259] provides a comprehensive summary of TLR-based drug clinical trials in cancer therapy.
Both macrophages and neutrophils express the SIRPα receptor, which binds to CD47, a receptor that can inhibit phagocytosis (known as the “don’t eat me signal”) [259]. Thus, blocking SIRPα-CD47 interactions may promote phagocytosis, leading to both tumor cell clearance and enhanced antigen presentation. BR105 is an anti-SIRPα antibody with silenced Fc region [249]. Phase I clinical results show no dose-limiting toxicities, although all patients experienced treatment-related adverse events. However, no treatment-related discontinuation or deaths occurred. Pre-clinical studies [262] showed that BR105 binds to various SIRPα variants, synergizes with therapeutic antibodies to promote phagocytosis, and inhibiting tumor growth in vivo.
Activation of STING (stimulator of interferon genes) can trigger interferon expression, enhancing the efficacy of cancer therapies including immune checkpoint and radiation therapy, by sensitizing myeloid cells [263]. TAK-500 [263], a STING agonist, activates CCR2+ myeloid cells in the tumor microenvironment and converts immune suppressive “cold tumor” into “hot tumor” characterized by CD8+ T cell proliferation and activation [263,264]. TAK-500 is currently being evaluate in combination with pembrolizumab in a clinical trials [264].
6.2. MCE and BCE in Treating Infectious Disease
MDX-240 (FcγRI x gp41) is a clinical stage antibody aimed to target HIV gp41, and its clinical development was terminated, with very limited data available for reference [101]. While the reasons behind the termination of clinical trials might be similar to those other myeloid cell engagers tested from the 1990s to 2000s for cancer treatment (see the previous section), preclinical data indicate that MDX-240 could reduce (but not eliminate) reverse transcriptase activity in cell culture, even in macrophage with active infection, demonstrating efficacy. However, the antibody alone is not likely to be sufficient for treating HIV. Cocktail approaches, involving cytotoxicity and coordination of multiple immune cells are most likely required. A research [265] has shown that antibodies from AIDS patients who are HIV controller and untreated progressors exhibited enhanced phagocytic activity compared to treated patients. In these patients, antibodies demonstrated a potential to bind to FcγR2a (CD32a), an activating receptor. Blocking the inhibitory FcγR2a receptor has no effect on the phagocytosis of HIV antibody from chronically infected patient but increased phagocytosis from antibodies derived from controllers, indicates that antibodies from patient at different pathological stages can target different Fc gamma receptors, and that specifically targeting activating receptors of myeloid cells can potentially improve treatment efficacy.
While engineering high affinity FcγR-targeting antibody may be considered as advantageous for combating viral infection, a phenomenon known as antibody-dependent enhancement (ADE) is frequently hijacked by virus to facilitate entry into and infection of immune cells through crosslinking of the virus and the immune cell by the antibody. Dengue virus has been shown to exhibit greater severity at lower serum antibody concentration, and reducing antibody binding affinity to FcγRs may help to mitigate this issue [266]. Myeloid cells expressing FcγR, especially macrophages and dendritic cells, are particularly susceptible to this process [267,268]. Pathogens like dengue virus, Zika virus, coronavirus, HIV, and RSV are known to induce ADE, although the mechanism of actions differ [268]. For instance, ADE caused by dengue virus arises due to inability of the antibody to neutralize heterologous serotype, resulting in binding without neutralization. Coronavirus-induced ADE is attributed to conformational changes in the spike protein, facilitating virial entry via canonical receptor-dependent pathways involving the receptor binding domain. HIV can exploit the antibodies to the surface of target cells, promoting membrane fusion via FcRs and complement receptors. In some cases, IgA from HIV-infected patient can mediate and promote IV infection of primary human monocytes [268,269,270].
6.3. MCE and BCE in Treating Autoimmune Disease
Crosstalk between APC cells and B cells and T cells play a key role in inducing autoimmunity. APCs can present endogenous antigens to helper T cells, leading to the activation and proliferation of autoreactive T cell clones. Blocking the interaction between CD28 and CD80/CD86 may reduce T cell activation. One of the mostly studied antibodies is abatacept, an FDA approved anti-CD80/CD86 antibody, which functions by inhibiting the binding between T cells and APCs. It has been investigated for the treatment of RA, juvenile idiopathic arthritis, psoriatic arthritis, and prophylaxis of acute graft-versus-host diseases. Clinical data suggest that abatacept can not only treats patient with rheumatoid arthritis [271], but also helps reduce the progression of high risk patient into rheumatoid arthritis in high-risk individuals [272]. In addition, although clinical trial with shorter treatment duration for Sjögren’s syndrome (SS) have shown limited efficacy [273], longer treatment duration demonstrated significant therapeutic benefit [274].
In addition to targeting myeloid cells, targeting CD40-CD40L interaction between T cells, B cells, and APC cells has proven efficacious in treating autoimmune disease. CD40L is expressed on activated T cells and platelets, whereas CD40 is expressed on myeloid cells and B cells. Disrupting this crosstalk reduces the ability of B cells to mount antigenic responses to endogenous antigens, and it hinders T cell-APC interactions, holding promise for reducing the generation of adaptive autoimmunity [275].
The CD40L agonist dapirolizumab pegol (DZP), a pegylated Fab antibody, is being developed to treat SLE. A preclinical study [275] of DZP (knowns as CDP7657) demonstrated that DZP could inhibit antigen-specific immune responses to tetanus toxoid in cynomolgus monkey, indicating DZP’s ability to reduce antigenic response that contributes to autoimmunity. Importantly, the antibody did not induce thrombotic complications, a key safety consideration, as CD40L is also expressed on platelet [275]. DZP has a high affinity of 7.9 pM for CD40L, enabling engaging with its ligand [276]. Although DZP shows efficacy, its phase 2 study [276] revealed that withdraw of the drug can lead to worsening of disease symptoms. In its phase 3 results [277], DZP combined with the standard of care (SOC) lead to a 60.1% (n=208) response rate at at week 48 according to SRI-4, compared to 41.1% in the placebo + SOC group (n=107; p=0.0014). (Severe BILAG flares occurred in 11.6% of patients receiving DZP + SOC, versus 23.4% in the placebo + SOC group by week 48 (p=0.0257).) Patients also experienced reductions in corticosteroid doses following DZP treatment, and serious adverse events were less frequent in the DZP group compared to placebo (9.9% vs 14.8%).
Inhibiting B cell is another strategy to modulate autoimmune disease. By blocking B cell signaling activation, it is possible to inhibit downstream pathways responsible autoantibody production. MGD010 is a DART-Fc antibody that targets CD32B and CD79B. CD32B is an inhibitory receptor for B cell, and CD79B is a component of the B cell receptor complex; crosslinking these two receptors can suppress B cell activity without depleting the cells. Clinical evidence suggests that MGD010 could reduce B cell activation, as indicated by decreased BCR-induced Ca2+ mobilization and sustained downregulation of surface BCRs on circulating CD27+ memory B cells [250]. The safety profile of MGD010 has also confirmed, with all adverse events being below grade 3 and no serve adverse effects reported [250].
7. Immune Checkpoint Blocker (ICB) and Immune Checkpoint Agonist (ICA)
As shown by FDA approvals, immune checkpoint blockers target PD-1 (Nivolumab, pembrolizumab, cemiplimab), PD-L1 (atezolizumab, avelumab, durvalumab), and CTLA-4 (ipilimumab). CTLA-4 can negatively regulate T-cell activation via competitive binding to the ligands of CD28, which are CD80 and CD86. Anti-CTLA-4, by preserving CD28 binding free of CTLA-4 interference, serves as a preserver for CD28 signaling in T cells [278]. Moreover, if equipped with Fc, such as ipilimumab, such antibody can mediate ADCC to lyse CTLA-4+ regulatory T cells [279], which can be beneficial for treating cancer and infectious diseases but potentially harmful for autoimmune conditions. PD-L1 is expressed on macrophages, dendritic cells, B cells, and T cells, whereas PD-L1 and PD-L2 are expressed on both antigen-presenting cells and cancer cells [278]. Activation of the PD-1/PD-L1 axis can blunt T cell inflammatory responses and restrict their proliferation after activation [278]. While both anti-PD-1 and anti-PD-L1 antibodies can block the PD-1/PD-L1 interactions, anti-PD-L1 possesses the additional feature of freeing CD80 from PD-L1 binding, thereby freeing additional CD80+ dendritic cells [278]. In terms of biological differences, CTLA-4 is expressed early during T cell activation and primes T cells in lymphoid tissue, whereas PD-1 is largely expressed at later stages of T cell activation and functions predominantly in peripheral tissues to limit T cell activity in inflammation or infection. Besides targeting PD(L)1 and CTLA-4, ICBs that target LAG-3, TIGIT, and TIM-3 are also under clinical development [280]. Fragment-based immune cell engagers can also circumvent the need for Fc silencing, which can otherwise lead to phagocytosis of T cells by macrophages via Fc-PD-1 mediated binding [281].
Similar to treating cancer, the aim of ICB in treating infectious diseases has been strategically focused on reinvigorating or preventing the occurrence of exhausted immune cells, as well as counteracting evasion or inhibition of immunity by pathogens [282,283]. Chronic infections, such as HIV, HBV, HCV malaria, and tuberculosis (TB) can lead to upregulation of immune checkpoint marker expression, as they can induce T cell exhaustion [284].
As CD4+T cells with exhaustion markers (PD-1, LAG-3, and TIGIT) have been shown to contain HIV DNA at higher frequency, these targets of ICBs are also biomarkers, serving as good targets for eliminating viral reservoirs [280]. In HIV treatment, the effect of immune checkpoint blocker displayed mixed outcomes and should be considered with caution. Both reversal of viral latency/reactivation of virus due to pembrolizumab (PD-1) treatment [285], and suppression of viral rebound from budigalimab (PD-1) treatment [286], have been reported. Such conflicting results highlight a need to investigate the mechanisms of ICB in virology research.
For the case of budigalimab, delayed rebound of HIV RNA was observed [286]. Oncological clinical studies of budigalimab [287] have shown that budigalimab exhibits similar efficacy (ORR=13% for HNSCC and 19% for the NSCLC) compared with pembrolizumab (ORR=18% for NSCLC). Budigalimab has the L234A/L235A (LALA) IgG1 mutation in the Fc domain, whereas pembrolizumab contains an IgG4 S228P mutation in the Fc [288]. It has been shown that while the S228P mutation poses negligible impact on binding to Fc gamma receptors, the L234A/L235A mutation can significantly reduce binding to Fc gamma receptors [289], suggesting potential impact from Fc-dependent modalities. For pembrolizumab, a study [290] utilizing patient CD4+T cells with latent HIV infection has shown that PD-1 signaling is able to suppress T-cell receptor (TCR) induced P-TEFb pathway, a key regulator for HIV transcription. Inhibition of PD-1 with pembrolizumab can remove such inhibitory signal, restoring the function of P-TEFb as well as promoting the transcription of HIV RNA. Increased unspliced HIV RNA and plasma viremia, but not HIV specific T cell responses to clear reactivated cells, were induced by pembrolizumab, even while ART was not interrupted [285].
Similarly, HBV, HCV, and EBV-infected patients have upregulated PD-1 levels, whereas in the acute HBV PD-1 is upregulated, in chronic HBV condition, PD-L1, TIM-3, and CTLA-4 are also upregulated [283]. In parasitic malaria infectiosn, due to the recurrence of infection where multiple infections are needed for conferring immunity, continuous antigenic exposure can cause immunosuppression, through upregulation of PD-L1, LAG-3, CTLA-4, OX40, and TIM-3 [282,283]. Mice lacking PD-1 can clear infection of a usually chronic strain of malaria [291]. In bacterial infection from M. tuberculosis, H. pylori, and L. donovani, PD-1 levels are also increased [282,283]. However, mouse studies also suggest that mice lacking PD-1 can suffer from dramatically higher mortality rate from TB, while other cases suggest that PD-1 can reduce interferon production to avoid immune-mediated tissue damage, suggesting protective roles of PD-1 in some diseases [283].
Clinical observations of oncological treatments with ICBs have led to interest in applying ICBs in treating autoimmune diseases. Incidence of immune-related adverse events (irAEs) is a prominent side effect of ICBs, caused by overriding self-tolerance signals, such as PD-1, that are expressed on healthy tissues and organs [292]. While ICBs used in oncology primarily serve to remove inhibitory signals, in autoimmune diseases, immune checkpoint agonists (ICAs) could be used to suppress pathogenic autoimmunity [293]. Various PD-1 and CTLA-4-targeting antibodies are being evaluated placed for clinical trials. Phase I trials like anti-PD-1 rosnilimab [294] have shown to induce a higher Treg to Teff (effector T cell) ratio as well asa reduction in interferon-gamma levels. According to the company’s announcement [295], rosnilimab has achieved significant responses by week 14 for moderate to severe rheumatoid arthritis based on ACR20 (American College of Rheumatology 20 standard or 20% improvement from baseline of disease) and ACR50 standard. Phase II trial of anti-PD-1 peresolimab [296] has shown improvement of patients with rheumatoid arthritis (RA) according to the ACR20 standard at 700 mg dose group.
To maximizing the efficacy from blocking immune checkpoints, co-targeting multiple immune checkpoints are also very promising strategy. Specifically, a multi-targeting approach could more selectively target immune cell populations that express two or more of the desired checkpoint targets. Interestingly, multispecific immune checkpoint antibodies have the potential to induce fewer adverse effects compared to combinations of multiple monospecific immune checkpoint blockers. For instance, according to a metanalysis [297,298], combined monotherapies regimens of CTLA-4 (lpilimumab) and PD-1/PD (pembrolizumab or nivolumab) can cause incidence rate of irAEs for 35-47% (≥grade 3: 13-15%) with sequential dosing, and irAEs of 60-83% (≥grade 3: 27.5-36%) for simultaneous dosing. In comparison, for bispecific PD-1/CTLA-4 antibody AK104 in a phase II trial [299], the rate of irAE is 53.3% (≥grade 3: 17.8%), which is comparable to the irAE of sequential dosing and less toxic than that of simultaneous dosing.
Similarly, the clinical progress for other co-stimulatory antibodies, such as the CTLA-4 x PD-1 AK104, MEDI5752, XmAb20717, as well as the PD-1 x LAG-3 tebotelimab (DART-Fc, also known as MGD013), PD-L1 x CTLA-4 lorigerlimab (DART-Fc, also known as MGD019) and PD-1 x TIM-3 RO7121661 [56,297], has been encouraging. In its phase I trial against both solid tumor and hematological malignancies [54], tebotelimab induced tumor shrinkage in 34% of patients, including patients with PD-1 refractory disease, LAG3+ non-Hodgkin’s lymphoma, and CAR-T refractory disease. Clinical responses were also observed in patients who were unresponsive to anti-HER2 and anti-PD-1 combination therapy. Lorigerlimab achieved an ORR of 25.7%, with 32.3% of irAEs and 7.9% of ≥grade 3 irAE in the treatment of metastatic castration-resistant prostate cancer (mCRPC) [56].
The VHH-Fc-based antibody PD-L1 x CTLA-4 erfonrilimab (KN046) has achieved promising result for clinical trial, as monotherapy or combination therapy with chemotherapy or targeted therapy [94,95,300,301,302], It has been tested as first-line therapy for metastatic triple-negative breast cancer as first line therapy in combination with nab-paclitaxel (ORR=44%) [95], hepatocellular carcinoma in combination with lenvatinib (ORR=51.9%) [301], pancreatic cancer as combination with nab-paclitaxel and gemcitabine [303] (ORR=45.2%), and nasopharyngeal carcinoma (ORR=12.5%) [300]. Interestingly, the design of erfonrilimab preserves the Fc function, allowing Fc gamma receptor-mediated effector function such as ADCC and CDC, which in turn regulatory T cells in the tumor microenvironment [300].
Importantly, erfonrilimab serves as an example demonstrating that a multispecificity fashion of design immune checkpoint blocker is capable of exerting higher efficacy than combination therapies. In the treatment of metastatic non-small cell lung cancer, erfonrilimab with pemetrexed as first-line therapy achieved an ORR=46% and a median OS of 26.6 months [94]. In comparison to other cocktail of Anti-PD-L1 with CTLA-4 trial, combining nivolumab plus ipilimumab yielded an ORR of 33.4% with median OS of 17.1 months in the CheckMate 227 trial and an ORR of 38.2% and median OS of 15.6 months in the CheckMate 9LA trial, while co-treating durvalumab, tremelimumab, and chemotherapy yielded an ORR of 38.8% and median OS of 14 months in the POSEIDON trial [94].
8. Conclusion
In the new era of immunotherapy, pharmacogenomics and precision medicine can assist the physician to make more effective and more personalized treatment plans for the patients. Therapies like CAR-T and neoantigen vaccines, represent major therapeutic alternatives to immune cell engagers. The versatility of engineering methods in CAR designs provides numerous strategies to engineer CAR immune cells, including logic gates (AND/OR/IF-THEN), multiple antigen targeting, safety off-switches, genetically incorporated tonic signaling modulation, checkpoint regulation, which are described in detail in another review [304]. These methodologies, while they might appear to “compete” from the viewpoints of antibody engineers, also suggest that combination therapy and multivalency strategies used in CAR can be applied to the development of antibody and immune cell engager therapy regimens, such as by creating cocktails that combine multiple immune cell engagers with the standard of care.
Insights from designing neoantigen cancer vaccine suggest a tumor specific approach that can simultaneously target multiple antigens, thus exponentially reducing the chance of antigen escape. While targeting cancer specific antigens can be carefully selected with display methodologies, simultaneous targeting multiple antigens remain a challenge, as currently available drug candidates are generally mono or bi valent towards cancer antigens. In clinical practice, with increased availability of approved immune cell engagers, clinicians many soon have access to a robust arsenal of therapies. Hopefully, within the first half of twenty-first century, immune cell engager could turn more diseases from incurable to curable.
Cancer, infectious disease, and autoimmune disorders, mark three of the primary area of focus in immunotherapy. The crosstalk of pathways and strategies for treating these diseases motivate researchers to investigate and develop more efficacious drugs. Recent advancement in AI, machine learning, multi-omics, ex vivo organoid systems, and high throughput methods, have significantly accelerated the pace of drug discovery and development. While these tools can miraculously shorten the time required to obtain results, the difficulty for critical evaluation could increase as the volume of data researchers must process continues to grow. The translatability of drug research is complex, as highlighted by challenges such as anti-drug antibody formation, pharmacokinetics, efficacy, and unexpected toxicities (Table 1). These issues underscore the importance of examining such traits during preclinical phase and comparing them with clinical-stage or approved drug products, to prudently assess their competitive edge from a business and humanitarian perspective.
The global economy has remained at a nadir since the onset of COVID-19. Reduced availability of grant and funding has compelled both industry and academia to adopt stricter criteria for approving or terminating of drug R&D efforts. Thus, the ability to critically evaluate the clinical potential of drug candidates is more essential than ever. As more clinical trial results become available, learning from the past and anticipating issues in in future clinical phases can serve as a guiding light for navigating the future of immune cell engagers in this era of uncertainty.
Author Contributions
Conceptualization, G.Y. and M.M.; methodology, G.Y.; software, G.Y. and M.M.; investigation, G.Y.; resources, G.Y. and M.M.; data curation, G.Y.; writing—original draft preparation, G.Y.; writing—review and editing, G.Y. and M.M.; visualization, M.M.; supervision, M.M.; project administration, G.Y. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
The research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of interest: The authors declare no conflicts of interest.
References
- Elshiaty M, Schindler H, Christopoulos P. Principles and Current Clinical Landscape of Multispecific Antibodies against Cancer. Int J Mol Sci. 2021;22(11).
- Wang J, Kang G, Yuan H, Cao X, Huang H, de Marco A. Research Progress and Applications of Multivalent, Multispecific and Modified Nanobodies for Disease Treatment. Front Immunol. 2021;12:838082.
- Ding L, Tian C, Feng S, Fida G, Zhang C, Ma Y, et al. Small sized EGFR1 and HER2 specific bifunctional antibody for targeted cancer therapy. Theranostics. 2015;5(4):378-98.
- Larbouret C, Robert B, Navarro-Teulon I, Thèzenas S, Ladjemi MZ, Morisseau S, et al. In vivo therapeutic synergism of anti-epidermal growth factor receptor and anti-HER2 monoclonal antibodies against pancreatic carcinomas. Clin Cancer Res. 2007;13(11):3356-62.
- Xu Z, Qiu C, Wen B, Wang S, Zhu L, Zhao L, et al. A bispecific nanobody targeting the dimerization interface of epidermal growth factor receptor: Evidence for tumor suppressive actions in vitro and in vivo. Biochem Biophys Res Commun. 2021;548:78-83.
- Vallera DA, Oh F, Kodal B, Hinderlie P, Geller MA, Miller JS, et al. A HER2 Tri-Specific NK Cell Engager Mediates Efficient Targeting of Human Ovarian Cancer. Cancers (Basel). 2021;13(16).
- Lu Y, Li Q, Fan H, Liao C, Zhang J, Hu H, et al. A Multivalent and Thermostable Nanobody Neutralizing SARS-CoV-2 Omicron (B.1.1.529). Int J Nanomedicine. 2023;18:353-67.
- Henry KA, MacKenzie CR. Antigen recognition by single-domain antibodies: structural latitudes and constraints. MAbs. 2018;10(6):815-26.
- Schriek AI, van Haaren MM, Poniman M, Dekkers G, Bentlage AEH, Grobben M, et al. Anti-HIV-1 Nanobody-IgG1 Constructs With Improved Neutralization Potency and the Ability to Mediate Fc Effector Functions. Front Immunol. 2022;13:893648.
- Terryn S, Francart A, Rommelaere H, Stortelers C, Van Gucht S. Post-exposure Treatment with Anti-rabies VHH and Vaccine Significantly Improves Protection of Mice from Lethal Rabies Infection. PLoS Negl Trop Dis. 2016;10(8):e0004902.
- Bessalah S, Jebahi S, Mejri N, Salhi I, Khorchani T, Hammadi M. Perspective on therapeutic and diagnostic potential of camel nanobodies for coronavirus disease-19 (COVID-19). 3 Biotech. 2021;11(2):89.
- Xu J, Xu K, Jung S, Conte A, Lieberman J, Muecksch F, et al. Nanobodies from camelid mice and llamas neutralize SARS-CoV-2 variants. Nature. 2021;595(7866):278-82.
- Jin S, Sun Y, Liang X, Gu X, Ning J, Xu Y, et al. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduction and Targeted Therapy. 2022;7(1):39.
- Ai Z, Wang B, Song Y, Cheng P, Liu X, Sun P. Prodrug-based bispecific antibodies for cancer therapy: advances and future directions. Front Immunol. 2025;16:1523693.
- Haber L, Olson K, Kelly MP, Crawford A, DiLillo DJ, Tavaré R, et al. Generation of T-cell-redirecting bispecific antibodies with differentiated profiles of cytokine release and biodistribution by CD3 affinity tuning. Sci Rep. 2021;11(1):14397.
- Asaadi Y, Jouneghani FF, Janani S, Rahbarizadeh F. A comprehensive comparison between camelid nanobodies and single chain variable fragments. Biomark Res. 2021;9(1):87.
- Binder U, Skerra A. Strategies for extending the half-life of biotherapeutics: successes and complications. Expert Opinion on Biological Therapy. 2025;25(1):93-118.
- Jones GB, Collins DS, Harrison MW, Thyagarajapuram NR, Wright JM. Subcutaneous drug delivery: An evolving enterprise. Sci Transl Med. 2017;9(405).
- Vaisman-Mentesh A, Gutierrez-Gonzalez M, DeKosky BJ, Wine Y. The Molecular Mechanisms That Underlie the Immune Biology of Anti-drug Antibody Formation Following Treatment With Monoclonal Antibodies. Front Immunol. 2020;11:1951.
- Davda J, Declerck P, Hu-Lieskovan S, Hickling TP, Jacobs IA, Chou J, et al. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J Immunother Cancer. 2019;7(1):105.
- Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. Aaps j. 2014;16(4):658-73.
- Bots SJ, Parker CE, Brandse JF, Löwenberg M, Feagan BG, Sandborn WJ, et al. Anti-Drug Antibody Formation Against Biologic Agents in Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. BioDrugs. 2021;35(6):715-33.
- Chen ML, Nopsopon T, Akenroye A. Incidence of Anti-Drug Antibodies to Monoclonal Antibodies in Asthma: A Systematic Review and Meta-Analysis. J Allergy Clin Immunol Pract. 2023;11(5):1475-84.e20.
- Li M, Zhao R, Chen J, Tian W, Xia C, Liu X, et al. Next generation of anti-PD-L1 Atezolizumab with enhanced anti-tumor efficacy in vivo. Sci Rep. 2021;11(1):5774.
- Penny HL, Hainline K, Theoharis N, Wu B, Brandl C, Webhofer C, et al. Characterization and root cause analysis of immunogenicity to pasotuxizumab (AMG 212), a prostate-specific membrane antigen-targeting bispecific T-cell engager therapy. Front Immunol. 2023;14:1261070.
- Trivedi A, Stienen S, Zhu M, Li H, Yuraszeck T, Gibbs J, et al. Clinical Pharmacology and Translational Aspects of Bispecific Antibodies. Clin Transl Sci. 2017;10(3):147-62.
- Friedrich SW, Lin SC, Stoll BR, Baxter LT, Munn LL, Jain RK. Antibody-directed effector cell therapy of tumors: analysis and optimization using a physiologically based pharmacokinetic model. Neoplasia. 2002;4(5):449-63.
- Peng L, Sferruzza G, Yang L, Zhou L, Chen S. CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors. Cellular & Molecular Immunology. 2024;21(10):1089-108.
- Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nature Reviews Drug Discovery. 2021;20(3):179-99.
- Matsumura, Y. Barriers to antibody therapy in solid tumors, and their solutions. Cancer Science. 2021;112(8):2939-47.
- Cuesta AM, Sainz-Pastor N, Bonet J, Oliva B, Alvarez-Vallina L. Multivalent antibodies: when design surpasses evolution. Trends Biotechnol. 2010;28(7):355-62.
- Matsumura Y, Gotoh M, Muro K, Yamada Y, Shirao K, Shimada Y, et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Annals of Oncology. 2004;15(3):517-25.
- Matsumura, Y. Cancer stromal targeting therapy to overcome the pitfall of EPR effect. Adv Drug Deliv Rev. 2020;154-155:142-50.
- Sigmund AM, Sahasrabudhe KD, Bhatnagar B. Evaluating Blinatumomab for the Treatment of Relapsed/Refractory ALL: Design, Development, and Place in Therapy. Blood Lymphat Cancer. 2020;10:7-20.
- Administration USFaD. BLINCYTO® (blinatumomab) for injection, for intravenous use Initial U.S. Approval: 2014. Revised: 6/2024.
- Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, et al. Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS One. 2013;8(12):e82742.
- Tey SK, Dotti G, Rooney CM, Heslop HE, Brenner MK. Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant. 2007;13(8):913-24.
- Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160-70.
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nature Reviews Immunology. 2007;7(9):715-25.
- Deyev SM, Lebedenko EN. Multivalency: the hallmark of antibodies used for optimization of tumor targeting by design. Bioessays. 2008;30(9):904-18.
- Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nature Reviews Immunology. 2020;20(10):633-43.
- Vermeire G, De Smidt E, Casteels P, Geukens N, Declerck P, Hollevoet K. DNA-based delivery of anti-DR5 Nanobodies improves exposure and anti-tumor efficacy over protein-based administration. Cancer Gene Therapy. 2021;28(7):828-38.
- (FDA) USFaDA. NDA/BLA Multi-disciplinary Review and Evaluation: IMDELLTRA - Tarlatamab (BLA 761344). 2024.
- Hummel H-D, Kufer P, Grüllich C, Deschler-Baier B, Chatterjee M, Goebeler M-E, et al. Phase 1 study of pasotuxizumab (BAY 2010112), a PSMA-targeting Bispecific T cell Engager (BiTE) immunotherapy for metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2019;37(15_suppl):5034-.
- Ahamadi-Fesharaki R, Fateh A, Vaziri F, Solgi G, Siadat SD, Mahboudi F, et al. Single-Chain Variable Fragment-Based Bispecific Antibodies: Hitting Two Targets with One Sophisticated Arrow. Mol Ther Oncolytics. 2019;14:38-56.
- Liu H, Xi R, Mao D, Zhao X, Wu T. Efficacy and Safety of Blinatumomab for the Treatment of Relapsed/Refractory Acute Lymphoblastic Leukemia: A Systemic Review and Meta-Analysis. Clin Lymphoma Myeloma Leuk. 2023;23(3):e139-e49.
- Ahn M-J, Cho BC, Felip E, Korantzis I, Ohashi K, Majem M, et al. Tarlatamab for Patients with Previously Treated Small-Cell Lung Cancer. New England Journal of Medicine. 2023;389(22):2063-75.
- Tang D, Kang R. Tarlatamab: the promising immunotherapy on its way from the lab to the clinic. Transl Lung Cancer Res. 2023;12(6):1355-7.
- Kang J, Sun T, Zhang Y. Immunotherapeutic progress and application of bispecific antibody in cancer. Front Immunol. 2022;13:1020003.
- Cech P, Skórka K, Dziki L, Giannopoulos K. T-Cell Engagers-The Structure and Functional Principle and Application in Hematological Malignancies. Cancers (Basel). 2024;16(8).
- Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 2021;137(6):751-62.
- Harding JJ, Garrido-Laguna I, Chen X, Basu C, Dowlati A, Forgie A, et al. A Phase 1 Dose-Escalation Study of PF-06671008, a Bispecific T-Cell-Engaging Therapy Targeting P-Cadherin in Patients With Advanced Solid Tumors. Front Immunol. 2022;13:845417.
- Root AR, Cao W, Li B, LaPan P, Meade C, Sanford J, et al. Development of PF-06671008, a Highly Potent Anti-P-cadherin/Anti-CD3 Bispecific DART Molecule with Extended Half-Life for the Treatment of Cancer. Antibodies (Basel). 2016;5(1).
- Luke JJ, Patel MR, Blumenschein GR, Hamilton E, Chmielowski B, Ulahannan SV, et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nature Medicine. 2023;29(11):2814-24.
- Luke JJ, Patel MR, Hamilton EP, Chmielowski B, Ulahannan SV, Kindler HL, et al. A phase I, first-in-human, open-label, dose-escalation study of MGD013, a bispecific DART molecule binding PD-1 and LAG-3, in patients with unresectable or metastatic neoplasms. Journal of Clinical Oncology. 2020;38(15_suppl):3004-.
- Luke JJ, Sharma M, Chandana SR, Lugowska IA, Szczylik C, Zolnierek J, et al. Lorigerlimab, a bispecific PD-1×CTLA-4 DART molecule in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): A phase 1 expansion (exp) cohort. Journal of Clinical Oncology. 2023;41(6_suppl):155-.
- Sharma M, Sanborn RE, Cote GM, Bendell JC, Kaul S, Chen F, et al. 1020O A phase I, first-in-human, open-label, dose escalation study of MGD019, an investigational bispecific PD-1 x CTLA-4 DART® molecule in patients with advanced solid tumours. Annals of Oncology. 2020;31:S704-S5.
- Bates A, Power CA. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies (Basel). 2019;8(2).
- Zaia, JA. A New Agent in the Strategy to Cure AIDS. Molecular Therapy. 2016;24(11):1894-6.
- Administration FaD. KIMMTRAK (tebentafusp-tebn) injection, for intravenous use: Highlights of prescribing information. 2022(Initial U.S. Approval: 2022).
- Lopez JS, Milhem M, Butler MO, Thistlethwaite F, Van Tine BA, D'Angelo SP, et al. Phase 1 study of IMCnyeso, a T cell receptor bispecific ImmTAC targeting NY-ESO-1-expressing malignancies. Cell Rep Med. 2025;6(4):101994.
- Dart RC, Bush SP, Heard K, Arnold TC, Sutter M, Campagne D, et al. The Efficacy of Antivenin Latrodectus (Black Widow) Equine Immune F(ab')(2) Versus Placebo in the Treatment of Latrodectism: A Randomized, Double-Blind, Placebo-Controlled, Clinical Trial. Ann Emerg Med. 2019;74(3):439-49.
- Lopardo G, Belloso WH, Nannini E, Colonna M, Sanguineti S, Zylberman V, et al. RBD-specific polyclonal F(ab´)2 fragments of equine antibodies in patients with moderate to severe COVID-19 disease: A randomized, multicenter, double-blind, placebo-controlled, adaptive phase 2/3 clinical trial. eClinicalMedicine 2021, 34. [Google Scholar]
- Gupta D, Ahmed F, Tandel D, Parthasarathy H, Vedagiri D, Sah V, et al. Equine immunoglobulin fragment F(ab')(2) displays high neutralizing capability against multiple SARS-CoV-2 variants. Clin Immunol. 2022;237:108981.
- Akiyoshi DE, Sheoran AS, Rich CM, Richard L, Chapman-Bonofiglio S, Tzipori S. Evaluation of Fab and F(ab')2 fragments and isotype variants of a recombinant human monoclonal antibody against Shiga toxin 2. Infect Immun. 2010;78(3):1376-82.
- Buist MR, Kenemans P, den Hollander W, Vermorken JB, Molthoff CJM, Burger CW, et al. Kinetics and Tissue Distribution of the Radiolabeled Chimeric Monoclonal Antibody MOv18 IgG and F(ab′)2 Fragments in Ovarian Carcinoma Patients1. Cancer Res. 1993;53(22):5413-8.
- Ruppel J, Brady A, Elliott R, Leddy C, Palencia M, Coleman D, et al. Preexisting Antibodies to an F(ab')2 Antibody Therapeutic and Novel Method for Immunogenicity Assessment. J Immunol Res. 2016;2016:2921758.
- Xue J, Ge X, Li Q, Xue L, Zhao W, Lin F, et al. A phase Ia, dose-escalation study of IMB071703 injection in patients (pts) with recurrent or metastatic, advanced solid tumors. Journal of Clinical Oncology. 2024;42(16_suppl):e14502-e.
- Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell. 2019;177(7):1701-13.e16.
- Demaria O, Habif G, Le Floch F, Chiossone L, Remark R, Vetizou M, et al. IPH6501 Is a Novel NKp46-Targeting Tetraspecific Antibody-Based Natural Killer Cell Engager Therapeutic (ANKET) Armed with a Non-Alpha IL-2 Variant and Developed for the Treatment of CD20-Positive Malignancies. Blood. 2022;140(Supplement 1):11559-.
- Safran H, Cassier PA, Vicier C, Forget F, Gomez-Roca CA, Penel N, et al. Phase 1/2 Study of DF1001, a novel tri-specific, NK cell engager therapy targeting HER2, in patients with advanced solid tumors: Phase 1 DF1001 monotherapy dose-escalation results. Journal of Clinical Oncology. 2023;41(16_suppl):2508-.
- Pharma, I. Innate Pharma shares updated results from the Sanofi developed blood cancer Phase 1/2 SAR443579/IPH6101 trial. 2024.
- Austin RJ, Lemon BD, Aaron WH, Barath M, Culp PA, DuBridge RB, et al. TriTACs, a Novel Class of T-Cell–Engaging Protein Constructs Designed for the Treatment of Solid Tumors. Molecular Cancer Therapeutics. 2021;20(1):109-20.
- Molloy ME, Aaron WH, Barath M, Bush MC, Callihan EC, Carlin K, et al. HPN328, a Trispecific T Cell–Activating Protein Construct Targeting DLL3-Expressing Solid Tumors. Molecular Cancer Therapeutics. 2024;23(9):1294-304.
- Kwant KS, Rocha SS, Yu T, Stephenson K, Banzon RR, Vollhardt S, et al. Abstract 2861: TriTAC-XR: An extended-release T cell engager platform designed to minimize cytokine release syndrome. Cancer Res. 2022;82(12_Supplement):2861-.
- Montesinos P, Arnan M, De Botton S, Calbacho M, Rodriguez Veiga R, Bories P, et al. Engaging Innate Immunity By AFM28, an Innate Cell Engager (ICE®) Targeting CD123-Positive Leukemic Cells in Patients with Relapsed/Refractory Acute Myeloid Leukemia: Safety and Efficacy Results of a First-in-Human Phase 1 Study. Blood. 2024;144(Supplement 1):738-.
- Kim WS, Shortt J, Zinzani PL, Mikhailova N, Radeski D, Ribrag V, et al. A Phase II Study of Acimtamig (AFM13) in Patients with CD30-Positive, Relapsed, or Refractory Peripheral T-cell Lymphomas. Clin Cancer Res. 2025;31(1):65-73.
- Topp M, Dlugosz-Danecka M, Skotnicki AB, Salogub G, Viardot A, Klein AK, et al. Safety of AFM11 in the treatment of patients with B-cell malignancies: findings from two phase 1 studies. Trials. 2023;24(1):4.
- Reusch U, Duell J, Ellwanger K, Herbrecht C, Knackmuss SH, Fucek I, et al. A tetravalent bispecific TandAb (CD19/CD3), AFM11, efficiently recruits T cells for the potent lysis of CD19(+) tumor cells. MAbs. 2015;7(3):584-604.
- Bartlett NL, Herrera AF, Domingo-Domenech E, Mehta A, Forero-Torres A, Garcia-Sanz R, et al. A phase 1b study of AFM13 in combination with pembrolizumab in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2020;136(21):2401-9.
- Stumpp MT, Dawson KM, Binz HK. Beyond Antibodies: The DARPin(®) Drug Platform. BioDrugs. 2020;34(4):423-33.
- Link A, Juglair L, Poulet H, Lemaillet G, Reichen C, Schildknecht P, et al. Abstract 2273: Selection of first-in-human clinical dose range for the tumor-targeted 4-1BB agonist MP0310 (AMG 506) using a pharmacokinetic/pharmacodynamics modeling approach. Cancer Res. 2020;80(16_Supplement):2273-.
- Baird R, Omlin A, Kiemle-Kallee J, Fiedler U, Zitt C, Feurstein D, et al. Abstract OT1-03-02: MP0274-CP101: A phase 1, first-in-human, single-arm, multi-center, open-label, dose escalation study to assess safety, tolerability, and pharmacokinetics of MP0274 in patients with advanced HER2-positive solid tumors. Cancer Res. 2018;78(4_Supplement):OT1-03-2-OT1--2.
- Baird RD, Linossi C, Middleton M, Lord S, Harris A, Rodón J, et al. First-in-Human Phase I Study of MP0250, a First-in-Class DARPin Drug Candidate Targeting VEGF and HGF, in Patients With Advanced Solid Tumors. J Clin Oncol. 2021;39(2):145-54.
- Gomez-Roca CA, Steeghs N, Gort EH, Winter HAMD, Fernandez E, Stavropoulou V, et al. Phase I study of MP0317, a FAP-dependent DARPin, for tumor-localized CD40 activation in patients with advanced solid tumors. Journal of Clinical Oncology. 2023;41(16_suppl):2584-.
- Steeghs N, Gomez-Roca CA, Korakis I, Gort EH, Winter HAMD, Stojcheva N, et al. Effect of MP0317, a FAP x CD40 DARPin, on safety profile and tumor-localized CD40 activation in a phase 1 study in patients with advanced solid tumors. Journal of Clinical Oncology. 2024;42(16_suppl):2573-.
- Knop S, Szarejko M, Grząśko N, Bringhen S, Trautmann-Grill K, Jurczyszyn A, et al. A phase 1b/2 study evaluating efficacy and safety of MP0250, a designed ankyrin repeat protein (DARPin) simultaneously targeting vascular endothelial growth factor (VEGF) and hepatocyte growth factor (HGF), in combination with bortezomib and dexamethasone, in patients with relapsed or refractory multiple myeloma. eJHaem. 2024;5(5):940-50.
- Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clinical Cancer Research. 2016;22(14):3440-50.
- Felices M, Lenvik TR, Kodal B, Lenvik AJ, Hinderlie P, Bendzick LE, et al. Potent Cytolytic Activity and Specific IL15 Delivery in a Second-Generation Trispecific Killer Engager. Cancer Immunol Res. 2020;8(9):1139-49.
- Sarhan D, Brandt L, Felices M, Guldevall K, Lenvik T, Hinderlie P, et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018;2(12):1459-69.
- Felices M, Warlick E, Juckett M, Weisdorf D, Vallera D, Miller S, et al. 444 GTB-3550 tri-specific killer engager TriKE™ drives NK cells expansion and cytotoxicity in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients. Journal for ImmunoTherapy of Cancer. 2021;9(Suppl 2):A473-A.
- Warlick ED, Weisdorf DJ, Vallera DA, Wangen R, Lewis D, Knox J, et al. GTB-3550 TriKE™ for the Treatment of High-Risk Myelodysplastic Syndromes (MDS) and Refractory/Relapsed Acute Myeloid Leukemia (AML) Safely Drives Natural Killer (NK) Cell Proliferation At Initial Dose Cohorts. Blood. 2020;136(Supplement 1):7-8.
- GT Biopharma, I. Corporate presentation describing TriKE® platform, pipeline, and clinical data as of November 2022. GT Biopharma Corporate Presentation. 2022.
- Zhao Y, Chen G, Li X, Wu J, Chang B, Hu S, et al. KN046, a bispecific antibody against PD-L1 and CTLA-4, plus chemotherapy as first-line treatment for metastatic NSCLC: A multicenter phase 2 trial. Cell Rep Med. 2024;5(3):101470.
- Li Q, Liu J, Zhang Q, Ouyang Q, Zhang Y, Liu Q, et al. The anti-PD-L1/CTLA-4 bispecific antibody KN046 in combination with nab-paclitaxel in first-line treatment of metastatic triple-negative breast cancer: a multicenter phase II trial. Nat Commun. 2024;15(1):1015.
- Li BT, Pegram MD, Lee K-W, Sharma M, Lee J, Spira AI, et al. A phase 1/2 study of a first-in-human immune-stimulating antibody conjugate (ISAC) BDC-1001 in patients with advanced HER2-expressing solid tumors. Journal of Clinical Oncology. 2023;41(16_suppl):2538-.
- Janku F, Han S-W, Doi T, Ajani J, Kuboki Y, Mahling P, et al. 378 A first in-human, multicenter, open-label, dose-finding phase 1 study of the immune stimulator antibody conjugate NJH395 in patients with nonbreast HER2+ advanced malignancies. Journal for ImmunoTherapy of Cancer. 2020;8(Suppl 3):A230-A.
- Biotherapeutics, B. Bolt Biotherapeutics Reports First Quarter 2024 Results, Announces Strategic Pipeline Prioritization and Changes to Leadership Team. 2024.
- Li BT, Lee KW, Pegram M, Sharma MR, Lee J, Spira AI, et al. 657MO Recommended phase II dose (RP2D) selection and pharmacodynamic (PD) data of the first-in-human immune-stimulating antibody conjugate (ISAC) BDC-1001 in patients (pts) with advanced HER2-expressing solid tumors. Annals of Oncology. 2023;34:S462-S3.
- André AS, Moutinho I, Dias JNR, Aires-da-Silva F. In vivo Phage Display: A promising selection strategy for the improvement of antibody targeting and drug delivery properties. Front Microbiol. 2022;13:962124.
- Rolin C, Zimmer J, Seguin-Devaux C. Bridging the gap with multispecific immune cell engagers in cancer and infectious diseases. Cell Mol Immunol. 2024;21(7):643-61.
- Baeuerle PA, Wesche H. T-cell-engaging antibodies for the treatment of solid tumors: challenges and opportunities. Curr Opin Oncol. 2022;34(5):552-8.
- Dustin, ML. The immunological synapse. Cancer Immunol Res. 2014;2(11):1023-33.
- Poussin M, Sereno A, Wu X, Huang F, Manro J, Cao S, et al. Dichotomous impact of affinity on the function of T cell engaging bispecific antibodies. J Immunother Cancer. 2021;9(7).
- Singh K, Hotchkiss KM, Mohan AA, Reedy JL, Sampson JH, Khasraw M. For whom the T cells troll? Bispecific T-cell engagers in glioblastoma. Journal for ImmunoTherapy of Cancer. 2021;9(11):e003679.
- Dreier T, Lorenczewski G, Brandl C, Hoffmann P, Syring U, Hanakam F, et al. Extremely potent, rapid and costimulation-independent cytotoxic T-cell response against lymphoma cells catalyzed by a single-chain bispecific antibody. International Journal of Cancer. 2002;100(6):690-7.
- Administration FaD. 761324Orig1s000 Multi-Discipline Review (Epcoritamab, BLA 761324). 2023.
- Menon AP, Moreno B, Meraviglia-Crivelli D, Nonatelli F, Villanueva H, Barainka M, et al. Modulating T Cell Responses by Targeting CD3. Cancers (Basel). 2023;15(4).
- Trinklein ND, Pham D, Schellenberger U, Buelow B, Boudreau A, Choudhry P, et al. Efficient tumor killing and minimal cytokine release with novel T-cell agonist bispecific antibodies. MAbs. 2019;11(4):639-52.
- Velasco Cárdenas RM, Brandl SM, Meléndez AV, Schlaak AE, Buschky A, Peters T, et al. Harnessing CD3 diversity to optimize CAR T cells. Nat Immunol. 2023;24(12):2135-49.
- Molina JC, Shah NN. CAR T cells better than BiTEs. Blood Advances. 2021;5(2):602-6.
- Subklewe, M. BiTEs better than CAR T cells. Blood Adv. 2021;5(2):607-12.
- M E, R G, D R, D B, N K, J H, et al. - First generation anti-CD19 chimeric antigen receptor-modified T cells for management of B cell malignances: initial analysis of an ongoing Phase I clinical trial. J Immunother Cancer. 2014;2(Suppl 3):12.
- Jabbour E, Zugmaier G, Agrawal V, Martínez-Sánchez P, Rifón Roca JJ, Cassaday RD, et al. Single agent subcutaneous blinatumomab for advanced acute lymphoblastic leukemia. Am J Hematol. 2024;99(4):586-95.
- Thieblemont C, Phillips T, Ghesquieres H, Cheah CY, Clausen MR, Cunningham D, et al. Epcoritamab, a Novel, Subcutaneous CD3xCD20 Bispecific T-Cell-Engaging Antibody, in Relapsed or Refractory Large B-Cell Lymphoma: Dose Expansion in a Phase I/II Trial. J Clin Oncol. 2023;41(12):2238-47.
- Gibson A, Nunez C, Robusto L, Kammerer B, Garcia M, Roth M, et al. Combination low-intensity chemotherapy plus inotuzumab ozogamicin, blinatumomab and rituximab for pediatric patients with relapsed/refractory B-cell acute lymphoblastic leukemia. Haematologica. 2024;109(9):3042-7.
- Cords L, Schaefers C, Kamili A, Hoffmann C, Cichutek S, Haag F, et al. BCMA x CD3 T-cell engager in a patient with pentarefractory multiple myeloma and HIV: a clinical and immunological report. Haematologica. 2024;109(9):3071-7.
- Géraud A, Hueso T, Laparra A, Bige N, Ouali K, Cauquil C, et al. Reactions and adverse events induced by T-cell engagers as anti-cancer immunotherapies, a comprehensive review. European Journal of Cancer. 2024;205:114075.
- Chen B, Zou Z, Zhang Q, Chen K, Zhang X, Xiao D, et al. Efficacy and safety of blinatumomab in children with relapsed/refractory B cell acute lymphoblastic leukemia: A systematic review and meta-analysis. Frontiers in Pharmacology. 2023;13.
- Aamir S, Anwar MY, Khalid F, Khan SI, Ali MA, Khattak ZE. Systematic Review and Meta-analysis of CD19-Specific CAR-T Cell Therapy in Relapsed/Refractory Acute Lymphoblastic Leukemia in the Pediatric and Young Adult Population: Safety and Efficacy Outcomes. Clin Lymphoma Myeloma Leuk. 2021;21(4):e334-e47.
- Philipp N, Kazerani M, Nicholls A, Vick B, Wulf J, Straub T, et al. T-cell exhaustion induced by continuous bispecific molecule exposure is ameliorated by treatment-free intervals. Blood. 2022;140(10):1104-18.
- Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Anti–B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. Journal of Clinical Oncology. 2020;38(8):775-83.
- Topp MS, Duell J, Zugmaier G, Attal M, Moreau P, Langer C, et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J Clin Oncol. 2020;38(8):775-83.
- Bahlis NJ, Costello CL, Raje NS, Levy MY, Dholaria B, Solh M, et al. Elranatamab in relapsed or refractory multiple myeloma: the MagnetisMM-1 phase 1 trial. Nat Med. 2023;29(10):2570-6.
- Ravandi F, Stein AS, Kantarjian HM, Walter RB, Paschka P, Jongen-Lavrencic M, et al. A Phase 1 First-in-Human Study of AMG 330, an Anti-CD33 Bispecific T-Cell Engager (BiTE®) Antibody Construct, in Relapsed/Refractory Acute Myeloid Leukemia (R/R AML). Blood. 2018;132(Supplement 1):25-.
- Chichili GR, Huang L, Li H, Burke S, He L, Tang Q, et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci Transl Med. 2015;7(289):289ra82.
- Biotech, F. MacroGenics switches horses in CD123 race, canning flotetuzumab in favor of next-gen successor. 2022.
- Winer ES, Maris M, Sharma MR, Kaminker P, Zhao E, Ward A, et al. A Phase 1, First-in-Human, Dose-Escalation Study of MGD024, a CD123 x CD3 Bispecific Dart® Molecule, in Patients with Relapsed or Refractory CD123-Positive (+) Hematologic Malignancies. Blood. 2022;140(Supplement 1):11753-4.
- Hoffman LM, Gore L. Blinatumomab, a Bi-Specific Anti-CD19/CD3 BiTE(®) Antibody for the Treatment of Acute Lymphoblastic Leukemia: Perspectives and Current Pediatric Applications. Front Oncol. 2014;4:63.
- Chou J, Egusa EA, Wang S, Badura ML, Lee F, Bidkar AP, et al. Immunotherapeutic Targeting and PET Imaging of DLL3 in Small-Cell Neuroendocrine Prostate Cancer. Cancer Res. 2023;83(2):301-15.
- Giffin MJ, Cooke K, Lobenhofer EK, Estrada J, Zhan J, Deegen P, et al. AMG 757, a Half-Life Extended, DLL3-Targeted Bispecific T-Cell Engager, Shows High Potency and Sensitivity in Preclinical Models of Small-Cell Lung Cancer. Clin Cancer Res. 2021;27(5):1526-37.
- Paz-Ares L, Champiat S, Lai WV, Izumi H, Govindan R, Boyer M, et al. Tarlatamab, a First-in-Class DLL3-Targeted Bispecific T-Cell Engager, in Recurrent Small-Cell Lung Cancer: An Open-Label, Phase I Study. J Clin Oncol. 2023;41(16):2893-903.
- Plieth, J. Reality Bites again for Amgen. OncologyPipeline (ApexOnco).
- Oberst MD, Fuhrmann S, Mulgrew K, Amann M, Cheng L, Lutterbuese P, et al. CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. MAbs. 2014;6(6):1571-84.
- Rosenthal MA, Balana C, van Linde ME, Sayehli C, Fiedler WM, Wermke M, et al. ATIM-49 (LTBK-01). AMG 596, A NOVEL ANTI-EGFRVIII BISPECIFIC T CELL ENGAGER (BITE(®)) MOLECULE FOR THE TREATMENT OF GLIOBLASTOMA (GBM): PLANNED INTERIM ANALYSIS IN RECURRENT GBM (RGBM). Neuro Oncol. 2019;21(Suppl 6):vi283.
- Beltran H, Johnson ML, Jain P, Schenk EL, Sanborn RE, Thompson JR, et al. Updated results from a phase 1/2 study of HPN328, a tri-specific, half-life (T1/2) extended DLL3-targeting T-cell engager in patients (pts) with small cell lung cancer (SCLC) and other neuroendocrine cancers (NEC). Journal of Clinical Oncology. 2024;42(16_suppl):8090-.
- Bono JSD, Fong L, Beer TM, Gao X, Geynisman DM, III HAB, et al. Results of an ongoing phase 1/2a dose escalation study of HPN424, a tri-specific half-life extended PSMA-targeting T-cell engager, in patients with metastatic castration-resistant prostate cancer (mCRPC). Journal of Clinical Oncology. 2021;39(15_suppl):5013-.
- Gorges TM, Riethdorf S, von Ahsen O, Nastał YP, Röck K, Boede M, et al. Heterogeneous PSMA expression on circulating tumor cells: a potential basis for stratification and monitoring of PSMA-directed therapies in prostate cancer. Oncotarget. 2016;7(23):34930-41.
- Molloy ME, Austin RJ, Lemon BD, Aaron WH, Ganti V, Jones A, et al. Preclinical Characterization of HPN536, a Trispecific, T-Cell–Activating Protein Construct for the Treatment of Mesothelin-Expressing Solid Tumors. Clinical Cancer Research. 2021;27(5):1452-62.
- Middelburg J, Kemper K, Engelberts P, Labrijn AF, Schuurman J, van Hall T. Overcoming Challenges for CD3-Bispecific Antibody Therapy in Solid Tumors. Cancers (Basel). 2021;13(2).
- Wang L, Hoseini SS, Xu H, Ponomarev V, Cheung NK. Silencing Fc Domains in T cell-Engaging Bispecific Antibodies Improves T-cell Trafficking and Antitumor Potency. Cancer Immunol Res. 2019;7(12):2013-24.
- Labrijn AF, Meesters JI, Bunce M, Armstrong AA, Somani S, Nesspor TC, et al. Efficient Generation of Bispecific Murine Antibodies for Pre-Clinical Investigations in Syngeneic Rodent Models. Sci Rep. 2017;7(1):2476.
- Vaks L, Litvak-Greenfeld D, Dror S, Shefet-Carasso L, Matatov G, Nahary L, et al. Design Principles for Bispecific IgGs, Opportunities and Pitfalls of Artificial Disulfide Bonds. Antibodies. 2018;7(3):27.
- Kang TH, Jung ST. Boosting therapeutic potency of antibodies by taming Fc domain functions. Experimental & Molecular Medicine. 2019;51(11):1-9.
- Kebenko M, Goebeler ME, Wolf M, Hasenburg A, Seggewiss-Bernhardt R, Ritter B, et al. A multicenter phase 1 study of solitomab (MT110, AMG 110), a bispecific EpCAM/CD3 T-cell engager (BiTE®) antibody construct, in patients with refractory solid tumors. Oncoimmunology. 2018;7(8):e1450710.
- Amann M, Brischwein K, Lutterbuese P, Parr L, Petersen L, Lorenczewski G, et al. Therapeutic Window of MuS110, a Single-Chain Antibody Construct Bispecific for Murine EpCAM and Murine CD3. Cancer Res. 2008;68(1):143-51.
- Schlereth B, Lorenczewski G, Friedrich M, Lutterbuese P, Lutterbuese R, Kischel R, et al. Feasibility of repeated subcutaneous delivery supports a new route of administration for treating cancer patients with EpCAM-specific BiTE antibody MT110. Cancer Res. 2008;68(9_Supplement):2403-.
- Kerns SJ, Belgur C, Petropolis D, Kanellias M, Barrile R, Sam J, et al. Human immunocompetent Organ-on-Chip platforms allow safety profiling of tumor-targeted T-cell bispecific antibodies. Elife. 2021;10.
- Dian Y, Liu Y, Zeng F, Sun Y, Deng G. Efficacy and safety of tebentafusp in patients with metastatic uveal melanoma: A systematic review and meta-analysis. Hum Vaccin Immunother. 2024;20(1):2374647.
- Piulats JM, Watkins C, Costa-García M, Del Carpio L, Piperno-Neumann S, Rutkowski P, et al. Overall survival from tebentafusp versus nivolumab plus ipilimumab in first-line metastatic uveal melanoma: a propensity score-weighted analysis. Ann Oncol. 2024;35(3):317-26.
- Carvajal RD, Butler MO, Shoushtari AN, Hassel JC, Ikeguchi A, Hernandez-Aya L, et al. Clinical and molecular response to tebentafusp in previously treated patients with metastatic uveal melanoma: a phase 2 trial. Nature Medicine. 2022;28(11):2364-73.
- Administration USFaD. (tebentafusp ) NDA/BLA Multi-Disciplinary Review and Evaluation for Application Number 761228Orig1s000. 2022.
- Chen LN, Carvajal RD. Tebentafusp for the treatment of HLA-A*02:01-positive adult patients with unresectable or metastatic uveal melanoma. Expert Rev Anticancer Ther. 2022;22(10):1017-27.
- Martinez-Perez D, Viñal D, Solares I, Espinosa E, Feliu J. Gp-100 as a Novel Therapeutic Target in Uveal Melanoma. Cancers (Basel). 2021;13(23).
- Rojas LA, Sethna Z, Soares KC, Olcese C, Pang N, Patterson E, et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature. 2023;618(7963):144-50.
- D'Angelo SP, Araujo DM, Abdul Razak AR, Agulnik M, Attia S, Blay J-Y, et al. Afamitresgene autoleucel for advanced synovial sarcoma and myxoid round cell liposarcoma (SPEARHEAD-1): an international, open-label, phase 2 trial. The Lancet. 2024;403(10435):1460-71.
- Low L, Goh A, Koh J, Lim S, Wang CI. Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat Commun. 2019;10(1):5382.
- Hsiue EH, Wright KM, Douglass J, Hwang MS, Mog BJ, Pearlman AH, et al. Targeting a neoantigen derived from a common TP53 mutation. Science. 2021;371(6533).
- Chai D, Wang J, Fan C, Lim JM, Wang X, Neeli P, et al. Remodeling of anti-tumor immunity with antibodies targeting a p53 mutant. J Hematol Oncol. 2024;17(1):45.
- Du J, Tang W, Jiao X, Zhao L, Du P, Zhang Y, et al. Abstract 6717: Targeting mutant KRAS proteins with novel TCR-mimic fully human antibodies. Cancer Res. 2024;84(6_Supplement):6717-.
- Du J, Tang W, Jiao X, Zhao L, Du P, Zhang Y, et al. 1168 Identification of fully human TCR-mimic antibodies targeting the KRAS G12V/HLA complex generated in HLA-transgenic RenMabTM mice. Journal for ImmunoTherapy of Cancer. 2023;11(Suppl 1):A1287-A.
- Duan Z, Ho M. Targeting the cancer neoantigens p53 and KRAS with TCR mimic antibodies. Antibody Therapeutics. 2021;4(4):208-11.
- Hamid O, Williams A, Lopez JS, Olson D, Sato T, Shaw HM, et al. Phase 1 safety and efficacy of IMC-F106C, a PRAME × CD3 ImmTAC bispecific, in post-checkpoint cutaneous melanoma (CM). Journal of Clinical Oncology. 2024;42(16_suppl):9507-.
- N, T. Roche, seeing MAGE magic wane, punts pair of solid tumor bispecifics, makes $490M bets disappear. Fierce Biotech [Internet]. 2023.
- Warmuth S, Gunde T, Snell D, Brock M, Weinert C, Simonin A, et al. Engineering of a trispecific tumor-targeted immunotherapy incorporating 4-1BB co-stimulation and PD-L1 blockade. Oncoimmunology. 2021;10(1):2004661.
- Luke J, Johnson M, Gadgeel S, Spira A, Yang J, Johnson J, et al. 732 First-in-human trial to evaluate safety, PK/PD and initial clinical activity of NM21–1480, an affinity-balanced PD-L1x4–1BBxHSA trispecific antibody: results of phase 1 dose escalation. Journal for ImmunoTherapy of Cancer. 2022;10(Suppl 2):A764-A.
- Chin SM, Kimberlin CR, Roe-Zurz Z, Zhang P, Xu A, Liao-Chan S, et al. Structure of the 4-1BB/4-1BBL complex and distinct binding and functional properties of utomilumab and urelumab. Nat Commun. 2018;9(1):4679.
- Vanamee É S, Faustman DL. Structural principles of tumor necrosis factor superfamily signaling. Sci Signal. 2018;11(511).
- Muik A, Garralda E, Altintas I, Gieseke F, Geva R, Ben-Ami E, et al. Preclinical Characterization and Phase I Trial Results of a Bispecific Antibody Targeting PD-L1 and 4-1BB (GEN1046) in Patients with Advanced Refractory Solid Tumors. Cancer Discov. 2022;12(5):1248-65.
- Gao J, Wang Z, Jiang W, Zhang Y, Meng Z, Niu Y, et al. CLDN18.2 and 4-1BB bispecific antibody givastomig exerts antitumor activity through CLDN18.2-expressing tumor-directed T-cell activation. Journal for ImmunoTherapy of Cancer. 2023;11(6):e006704.
- Correnti CE, Laszlo GS, de van der Schueren WJ, Godwin CD, Bandaranayake A, Busch MA, et al. Simultaneous multiple interaction T-cell engaging (SMITE) bispecific antibodies overcome bispecific T-cell engager (BiTE) resistance via CD28 co-stimulation. Leukemia. 2018;32(5):1239-43.
- Bourgeois S, Lim YS, Gane EJ, Lee HW, Cheng W, Heo J, et al. IMC-I109V, a novel T cell receptor (TCR) bispecific (ENVxCD3) designed to eliminate HBV-infected hepatocytes in chronic HBV patients: initial data from a first-in-human study. Poster Presentation (SAT437). The International Liver Congress 2022, 22- Journal of Hepatology2022. p. S872. 26 June.
- Zheng J-R, Wang Z-L, Feng B. Hepatitis B functional cure and immune response. Frontiers in Immunology. 2022;13.
- Yang H, Buisson S, Bossi G, Wallace Z, Hancock G, So C, et al. Elimination of Latently HIV-infected Cells from Antiretroviral Therapy-suppressed Subjects by Engineered Immune-mobilizing T-cell Receptors. Molecular Therapy. 2016;24(11):1913-25.
- Immunocore. Immunocore announces initial Phase 1 safety and pharmacodynamic activity data with first soluble TCR therapy for people living with HIV. 2023.
- Lee CM, Choe PG, Kang CK, Jo HJ, Kim NJ, Yoon SS, et al. Impact of T-Cell Engagers on COVID-19-Related Mortality in B-Cell Lymphoma Patients Receiving B-Cell Depleting Therapy. Cancer Res Treat. 2024;56(1):324-33.
- Kang S, Brown HM, Hwang S. Direct Antiviral Mechanisms of Interferon-Gamma. Immune Netw. 2018;18(5):e33.
- Zhou, F. Molecular mechanisms of IFN-gamma to up-regulate MHC class I antigen processing and presentation. Int Rev Immunol. 2009;28(3-4):239-60.
- Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front Immunol. 2021;12:636568.
- Mihaescu G, Chifiriuc MC, Filip R, Bleotu C, Ditu LM, Constantin M, et al. Role of interferons in the antiviral battle: from virus-host crosstalk to prophylactic and therapeutic potential in SARS-CoV-2 infection. Front Immunol. 2023;14:1273604.
- Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121(26):5154-7.
- Chen LY, Kothari J. Supportive care measures for bispecific T-cell engager therapies in haematological malignancies. Current Opinion in Supportive and Palliative Care. 2024;18(2):92-9.
- Dogan M, Kozhaya L, Placek L, Karabacak F, Yigit M, Unutmaz D. Targeting SARS-CoV-2 infection through CAR-T-like bispecific T cell engagers incorporating ACE2. Clinical & Translational Immunology. 2022;11(10):e1421.
- Busca A, Salmanton-García J, Corradini P, Marchesi F, Cabirta A, Di Blasi R, et al. COVID-19 and CAR T cells: a report on current challenges and future directions from the EPICOVIDEHA survey by EHA-IDWP. Blood Adv. 2022;6(7):2427-33.
- Hong R, Zhao H, Wang Y, Chen Y, Cai H, Hu Y, et al. Clinical characterization and risk factors associated with cytokine release syndrome induced by COVID-19 and chimeric antigen receptor T-cell therapy. Bone Marrow Transplantation. 2021;56(3):570-80.
- Wang W, Liu X, Wu S, Chen S, Li Y, Nong L, et al. Definition and Risks of Cytokine Release Syndrome in 11 Critically Ill COVID-19 Patients With Pneumonia: Analysis of Disease Characteristics. J Infect Dis. 2020;222(9):1444-51.
- Desai, PJ. Expression and fusogenic activity of SARS CoV-2 Spike protein displayed in the HSV-1 Virion. bioRxiv. 2023.
- Pizzato M, Baraldi C, Boscato Sopetto G, Finozzi D, Gentile C, Gentile MD, et al. SARS-CoV-2 and the Host Cell: A Tale of Interactions. Frontiers in Virology. 2022;1.
- Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nature Reviews Molecular Cell Biology. 2022;23(1):3-20.
- Li F, Xu W, Zhang X, Wang W, Su S, Han P, et al. A spike-targeting bispecific T cell engager strategy provides dual layer protection against SARS-CoV-2 infection in vivo. Communications Biology. 2023;6(1):592.
- Quiñones-Parra SM, Gras S, Nguyen THO, Farenc C, Szeto C, Rowntree LC, et al. Molecular determinants of cross-strain influenza A virus recognition by αβ T cell receptors. Science Immunology. 2025;10(104):eadn3805.
- Deeks SG, Archin N, Cannon P, Collins S, Jones RB, de Jong MAWP, et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nature Medicine. 2021;27(12):2085-98.
- Gunst JD, Gohil J, Li JZ, Bosch RJ, White CSA, Chun T-W, et al. Time to HIV viral rebound and frequency of post-treatment control after analytical interruption of antiretroviral therapy: an individual data-based meta-analysis of 24 prospective studies. Nature Communications. 2025;16(1):906.
- Nordstrom JL, Ferrari G, Margolis DM. Bispecific antibody-derived molecules to target persistent HIV infection. J Virus Erad. 2022;8(3):100083.
- Promsote W, Xu L, Hataye J, Fabozzi G, March K, Almasri CG, et al. Trispecific antibody targeting HIV-1 and T cells activates and eliminates latently-infected cells in HIV/SHIV infections. Nature Communications. 2023;14(1):3719.
- Pegu A, Asokan M, Wu L, Wang K, Hataye J, Casazza JP, et al. Activation and lysis of human CD4 cells latently infected with HIV-1. Nature Communications. 2015;6(1):8447.
- Bosque A, Planelles V. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood. 2009;113(1):58-65.
- Bertoletti A, Le Bert N. Immunotherapy for Chronic Hepatitis B Virus Infection. Gut Liver. 2018;12(5):497-507.
- Fergusson JR, Wallace Z, Connolly MM, Woon AP, Suckling RJ, Hine DW, et al. Immune-Mobilizing Monoclonal T Cell Receptors Mediate Specific and Rapid Elimination of Hepatitis B-Infected Cells. Hepatology. 2020;72(5):1528-40.
- Quitt O, Luo S, Meyer M, Xie Z, Golsaz-Shirazi F, Loffredo-Verde E, et al. T-cell engager antibodies enable T cells to control HBV infection and to target HBsAg-positive hepatoma in mice. J Hepatol. 2021;75(5):1058-71.
- Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012;64(4):1215-26.
- Anolik JH, Barnard J, Cappione A, Pugh-Bernard AE, Felgar RE, Looney RJ, et al. Rituximab improves peripheral B cell abnormalities in human systemic lupus erythematosus. Arthritis Rheum. 2004;50(11):3580-90.
- Han S, Zhuang H, Shumyak S, Yang L, Reeves WH. Mechanisms of Autoantibody Production in Systemic Lupus Erythematosus. Frontiers in Immunology. 2015;6.
- Cambridge G, Leandro MJ, Teodorescu M, Manson J, Rahman A, Isenberg DA, et al. B cell depletion therapy in systemic lupus erythematosus: effect on autoantibody and antimicrobial antibody profiles. Arthritis Rheum. 2006;54(11):3612-22.
- Schett G, Nagy G, Krönke G, Mielenz D. B-cell depletion in autoimmune diseases. Ann Rheum Dis. 2024;83(11):1409-20.
- Nunez D, Patel D, Volkov J, Wong S, Vorndran Z, Müller F, et al. Cytokine and reactivity profiles in SLE patients following anti-CD19 CART therapy. Molecular Therapy Methods & Clinical Development. 2023;31.
- Mackensen A, Müller F, Mougiakakos D, Böltz S, Wilhelm A, Aigner M, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nature Medicine. 2022;28(10):2124-32.
- Shah K, Leandro M, Cragg M, Kollert F, Schuler F, Klein C, et al. Disrupting B and T-cell collaboration in autoimmune disease: T-cell engagers versus CAR T-cell therapy? Clinical and Experimental Immunology. 2024;217(1):15-30.
- McHugh, J. BiTEing refractory RA. Nature Reviews Rheumatology. 2024;20(7):395-.
- Bucci L, Hagen M, Rothe T, Raimondo MG, Fagni F, Tur C, et al. Bispecific T cell engager therapy for refractory rheumatoid arthritis. Nature Medicine. 2024;30(6):1593-601.
- Subklewe M, Magno G, Gebhardt C, Bücklein V, Szelinski F, Arévalo HJR, et al. Application of blinatumomab, a bispecific anti-CD3/CD19 T-cell engager, in treating severe systemic sclerosis: A case study. Eur J Cancer. 2024;204:114071.
- Shouse GP, Blum KA, Haydu JE, Abramson JS, Narkhede M, Ip A, et al. A Phase 1, Open-Label, Dose Escalation and Dose Expansion Study of CLN-978 (CD19XCD3XHSA) in Patients with Relapsed/Refractory (R/R) B-Cell Non-Hodgkin Lymphoma (B-NHL). Blood. 2023;142(Supplement 1):3142-.
- Cullinan Therapeutics, I. Cullinan Therapeutics Announces Preclinical Data for CLN-978, a CD19-directed T Cell Engager, to be Presented at ACR Convergence 2024. Press release announcing preclinical data for CLN-978 T cell engager in autoimmune diseases Includes details of Phase 1b SLE trial design and clinical development plans. 2024.
- Máire, F. Quigley JSM, Jeffrey Jones, Farrukh T. Awan, Geoffrey P. Shouse, Yue Zhang, Todd Shearer, Judy Inumerable, Irina M. Shapiro, Patrick A. Baeuerle and Stephen Wax. CLN-978, a CD19-directed T Cell Engager (TCE), Leads to Rapid and Deep B Cell Depletion and Has Broad Potential for Development in Autoimmune Diseases. ACR Convergence 2024. 2024(Abstract Number: 0003).
- Nikkhoi SK, Li G, Hatefi A. Natural killer cell engagers for cancer immunotherapy. Front Oncol. 2024;14:1483884.
- Fenis A, Demaria O, Gauthier L, Vivier E, Narni-Mancinelli E. New immune cell engagers for cancer immunotherapy. Nature Reviews Immunology. 2024;24(7):471-86.
- Kucuksezer UC, Aktas Cetin E, Esen F, Tahrali I, Akdeniz N, Gelmez MY, et al. The Role of Natural Killer Cells in Autoimmune Diseases. Front Immunol. 2021;12:622306.
- Rosenberg J, Huang J. CD8(+) T Cells and NK Cells: Parallel and Complementary Soldiers of Immunotherapy. Curr Opin Chem Eng. 2018;19:9-20.
- Ravandi F, Bashey A, Foran J, Stock W, Mawad R, Short N, et al. Phase 1 study of vibecotamab identifies an optimized dose for treatment of relapsed/refractory acute myeloid leukemia. Blood Adv. 2023;7(21):6492-505.
- Xiao X, Cheng Y, Zheng X, Fang Y, Zhang Y, Sun R, et al. Bispecific NK-cell engager targeting BCMA elicits stronger antitumor effects and produces less proinflammatory cytokines than T-cell engager. Front Immunol. 2023;14:1113303.
- Kim HR, Saavedra O, Cervantes A, Lugowska IA, Oberoi A, El-Khoueiry AB, et al. Preliminary results from the phase 2 study of AFM24 in combination with atezolizumab in patients with EGFR wild-type (EGFR-WT) non-small cell lung cancer (NSCLC). Journal of Clinical Oncology. 2024;42(16_suppl):2522-.
- El-Khoueiry AB, Rivas D, Lee S-H, Thomas JS, Kim YJ, Cervantes A, et al. Leveraging innate immunity with AFM24, a novel CD16A and epidermal growth factor receptor (EGFR) bispecific innate cell engager: Interim results for the non-small cell lung cancer (NSCLC) cohort. Journal of Clinical Oncology. 2023;41(16_suppl):2533-.
- Kontić M, Marković F, Nikolić N, Samardžić N, Stojanović G, Simurdić P, et al. Efficacy of Atezolizumab in Subsequent Lines of Therapy for NSCLC Patients: Insights from Real-World Data. Cancers. 2024;16(21):3696.
- Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P, et al. Phase II Study of the Efficacy and Safety of Pembrolizumab for Relapsed/Refractory Classic Hodgkin Lymphoma. Journal of Clinical Oncology. 2017;35(19):2125-32.
- Kim WS, Shortt J, Zinzani PL, Mikhaylova N, Marin-Niebla A, Radeski D, et al. Abstract CT024: REDIRECT: A Phase 2 study of AFM13 in patients with CD30-positive relapsed or refractory (R/R) peripheral T cell lymphoma (PTCL). Cancer Res. 2023;83(8_Supplement):CT024-CT.
- Sasse S, Bröckelmann PJ, Momotow J, Plütschow A, Hüttmann A, Basara N, et al. AFM13 in patients with relapsed or refractory classical Hodgkin lymphoma: final results of an open-label, randomized, multicenter phase II trial. Leuk Lymphoma. 2022;63(8):1871-8.
- Moskowitz A, Harstrick A, Emig M, Overesch A, Pinto S, Ravenstijn P, et al. AFM13 in Combination with Allogeneic Natural Killer Cells (AB-101) in Relapsed or Refractory Hodgkin Lymphoma and CD30 + Peripheral T-Cell Lymphoma: A Phase 2 Study (LuminICE). Blood. 2023;142(Supplement 1):4855-.
- Inc. ANVaNA. Affimed and NKMax America to Study the Combination of AFM24, an EGFR-Targeted Innate Cell Engager, with SNK01 Natural Killer Cell Therapy. 2020.
- GT Biopharma, I. Product Pipeline Overview. 2025.
- Yang G, Nikkhoi SK, Owji H, Li G, Massumi M, Cervelli J, et al. A Novel Tetravalent Bispecific Immune Cell Engager Activates Natural Killer Cells to Kill Cancer Cells without Mediating Fratricide. Antibodies. 2024;13(3):75.
- Bernard NF, Alsulami K, Pavey E, Dupuy FP. NK Cells in Protection from HIV Infection. Viruses. 2022;14(6).
- Li W, Wu Y, Kong D, Yang H, Wang Y, Shao J, et al. One-domain CD4 Fused to Human Anti-CD16 Antibody Domain Mediates Effective Killing of HIV-1-Infected Cells. Sci Rep. 2017;7(1):9130.
- Harris A, Borgnia MJ, Shi D, Bartesaghi A, He H, Pejchal R, et al. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc Natl Acad Sci U S A. 2011;108(28):11440-5.
- Tohmé M, Davis T, Zhang M, Lemar H, Duong B, Tan J, et al. 902 NKX019, an off-the-shelf CD19 CAR-NK cell, mediates improved anti-tumor activity and persistence in combination with CD20-directed therapeutic mAbs. Journal for ImmunoTherapy of Cancer. 2022;10(Suppl 2):A940-A.
- Askanase A, Khalili L, Chang C, Blaus A, Gip P, Karis E, et al. A Phase 1 Study of NKX019, an Allogeneic Chimeric Antigen Receptor Natural Killer (CAR-NK) Cell Therapy in Patients with Systemic Lupus Erythematosus. Blood. 2024;144:4846.1.
- Cichocki F, Goodridge JP, Bjordahl R, Mahmood S, Davis ZB, Gaidarova S, et al. Dual antigen-targeted off-the-shelf NK cells show durable response and prevent antigen escape in lymphoma and leukemia. Blood. 2022;140(23):2451-62.
- Koh SK, Kim H, Han B, Jo H, Doh J, Park J, et al. Anti-CD19 antibody cotreatment enhances serial killing activity of anti-CD19 CAR-T/-NK cells and reduces trogocytosis. Blood. 2025;145(9):956-69.
- Bachanova V, Deol A, Al-Juhaishi TMS, Lulla PD, Byrne MT, Wong C, et al. Safety and Efficacy of FT522, a First-in-Class, Multi-Antigen Targeted, Off-the-Shelf, iPSC-Derived CD19 CAR NK Cell Therapy with Alloimmune Defense Receptor (ADR) in Relapsed/Refractory B-Cell Lymphoma. Blood. 2024;144(Supplement 1):6543-.
- Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, et al. FDA Approval Summary: Tocilizumab for Treatment of Chimeric Antigen Receptor T Cell-Induced Severe or Life-Threatening Cytokine Release Syndrome. Oncologist. 2018;23(8):943-7.
- James ND, Atherton PJ, Jones J, Howie AJ, Tchekmedyian S, Curnow RT. A phase II study of the bispecific antibody MDX-H210 (anti-HER2 x CD64) with GM-CSF in HER2+ advanced prostate cancer. Br J Cancer. 2001;85(2):152-6.
- Fury MG, Lipton A, Smith KM, Winston CB, Pfister DG. A phase-I trial of the epidermal growth factor receptor directed bispecific antibody MDX-447 without and with recombinant human granulocyte-colony stimulating factor in patients with advanced solid tumors. Cancer Immunol Immunother. 2008;57(2):155-63.
- Repp R, van Ojik HH, Valerius T, Groenewegen G, Wieland G, Oetzel C, et al. Phase I clinical trial of the bispecific antibody MDX-H210 (anti-FcγRI × anti-HER-2/neu) in combination with Filgrastim (G-CSF) for treatment of advanced breast cancer. British Journal of Cancer. 2003;89(12):2234-43.
- Valone FH, Kaufman PA, Guyre PM, Lewis LD, Memoli V, Deo Y, et al. Phase Ia/Ib trial of bispecific antibody MDX-210 in patients with advanced breast or ovarian cancer that overexpresses the proto-oncogene HER-2/neu. Journal of Clinical Oncology. 1995;13(9):2281-92.
- Borchmann P, Schnell R, Fuss I, Manzke O, Davis T, Lewis LD, et al. Phase 1 trial of the novel bispecific molecule H22xKi-4 in patients with refractory Hodgkin lymphoma. Blood. 2002;100(9):3101-7.
- Pegram MD, Calfa C, Chen C, Salgado AC, Heeke AL, Kang I, et al. Phase 2 study of novel HER2-targeting, TLR7/8 immune-stimulating antibody conjugate (ISAC) BDC-1001 (trastuzumab imbotolimod) +/- pertuzumab (P) in patients (pts) with HER2-positive metastatic breast cancer (MBC) previously treated with trastuzumab deruxtecan (T-DXd). Journal of Clinical Oncology. 2024;42(16_suppl):TPS1121-TPS.
- Janku F, Han SW, Doi T, Amatu A, Ajani JA, Kuboki Y, et al. Preclinical Characterization and Phase I Study of an Anti-HER2-TLR7 Immune-Stimulator Antibody Conjugate in Patients with HER2+ Malignancies. Cancer Immunol Res. 2022;10(12):1441-61.
- Perez C, Henry J, Kim S, El-Khoueiry A, Gutierrez M, Pavlick A, et al. 743 INCLINE-101: preliminary safety, tolerability, pharmacokinetics (PK), and pharmacodynamics (PD) of TAC-001 (TLR9 agonist conjugated to a CD22 mAb) in patients with advanced or metastatic solid tumors. Journal for ImmunoTherapy of Cancer. 2023;11(Suppl 1):A838-A.
- Kuo TC, Harrabi O, Chen A, Sangalang ER, Doyle L, Fontaine D, et al. Abstract 1721: TAC-001, a toll-like receptor 9 (TLR9) agonist antibody conjugate targeting B cells, promotes anti-tumor immunity and favorable safety profile following systemic administration in preclinical models. Cancer Res. 2021;81(13_Supplement):1721-.
- Lin N, Du T, Zhou H, Li Y, Wang L, Dong X, et al. BR105 Is a Novel Anti-Sirpα Monoclonal Antibody That Demonstrates a Favorable Safety Profile and Potent Anti-Tumor Efficacy in Patients with Relapsed/Refractory Lymphoid Malignancies: Preliminary Results from a Phase I Study. Blood. 2024;144(Supplement 1):4175-.
- Pandya N, Chen W, Lohr J, Yao X-T, Burns R, Li H, et al. OP0201 Safety, Tolerability, and Functional Activity of MGD010, A Dart® Molecule Targeting CD32B and CD79B, Following A Single Dose Administration in Healthy Volunteers. Annals of the Rheumatic Diseases. 2016;75(Suppl 2):132-3.
- Holtrop T, Budding K, Brandsma AM, Leusen JHW. Targeting the high affinity receptor, FcγRI, in autoimmune disease, neuropathy, and cancer. Immunother Adv. 2022;2(1):ltac011.
- Heemskerk N, Gruijs M, Temming AR, Heineke MH, Gout DY, Hellingman T, et al. Augmented antibody-based anticancer therapeutics boost neutrophil cytotoxicity. J Clin Invest. 2021;131(6).
- Wang B, Liu Y, Yuan R, Dou X, Qian N, Pan X, et al. XFab-α4-1BB/CD40L fusion protein activates dendritic cells, improves expansion of antigen-specific T cells, and exhibits antitumour efficacy in multiple solid tumour models. Cancer Immunol Immunother. 2023;72(12):4015-30.
- Bender AT, Tzvetkov E, Pereira A, Wu Y, Kasar S, Przetak MM, et al. TLR7 and TLR8 Differentially Activate the IRF and NF-κB Pathways in Specific Cell Types to Promote Inflammation. ImmunoHorizons. 2020;4(2):93-107.
- Desnues B, Macedo AB, Roussel-Queval A, Bonnardel J, Henri S, Demaria O, et al. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc Natl Acad Sci U S A. 2014;111(4):1497-502.
- Sun K, Salmon SL, Lotz SA, Metzger DW. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect Immun. 2007;75(3):1196-202.
- Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, et al. TLR9 Regulates TLR7- and MyD88-Dependent Autoantibody Production and Disease in a Murine Model of Lupus. The Journal of Immunology. 2010;184(4):1840-8.
- Janku F, Han S-W, Doi T, Amatu A, Ajani JA, Kuboki Y, et al. Preclinical Characterization and Phase I Study of an Anti–HER2-TLR7 Immune-Stimulator Antibody Conjugate in Patients with HER2+ Malignancies. Cancer Immunology Research. 2022;10(12):1441-61.
- Le Naour J, Kroemer G. Trial watch: Toll-like receptor ligands in cancer therapy. Oncoimmunology. 2023;12(1):2180237.
- AxisPharm. ADC: All You Need to Know About Targeting, Termination, Optimization, and Phase II/III Clinical Trials.
- Bonacorsi MZ, Chen A, Harrabi O, Li M, Sangalang ER, Fontaine D, et al. Abstract 6746: A B-cell targeted TLR9 agonist antibody conjugate potentiates cancer vaccine efficacy and rejuvenates vaccine responses in the elderly. Cancer Res. 2024;84(6_Supplement):6746-.
- Wu ZH, Li N, Mei XF, Chen J, Wang XZ, Guo TT, et al. Preclinical characterization of the novel anti-SIRPα antibody BR105 that targets the myeloid immune checkpoint. J Immunother Cancer. 2022;10(3).
- Appleman V, Matsuda A, Ganno M, Lopez AM, Rosentrater E, Christensen C, et al. 1153 Preclinical activity of C-C chemokine receptor 2 (CCR2)-targeted immune stimulating antibody conjugate (ISAC), motivating clinical testing of TAK-500. Journal for ImmunoTherapy of Cancer. 2022;10(Suppl 2):A1196-A.
- Schalper KA, Matsuda A, Ganno-Sherwood M, Maldonado-Lopez AE, Rosentrater E, Porciuncula A, et al. Abstract 1841: TAK-500 is a clinical stage immune-cell directed antibody drug conjugate (iADC) inducing STING activation in CCR2-expressing intratumor myeloid cells and favorable immunomodulation. Cancer Res. 2023;83(7_Supplement):1841-.
- Ackerman ME, Dugast AS, McAndrew EG, Tsoukas S, Licht AF, Irvine DJ, et al. Enhanced phagocytic activity of HIV-specific antibodies correlates with natural production of immunoglobulins with skewed affinity for FcγR2a and FcγR2b. J Virol. 2013;87(10):5468-76.
- Pantaleo G, Correia B, Fenwick C, Joo VS, Perez L. Antibodies to combat viral infections: development strategies and progress. Nature Reviews Drug Discovery. 2022;21(9):676-96.
- Flipse J, Diosa-Toro MA, Hoornweg TE, van de Pol DPI, Urcuqui-Inchima S, Smit JM. Antibody-Dependent Enhancement of Dengue Virus Infection in Primary Human Macrophages; Balancing Higher Fusion against Antiviral Responses. Sci Rep. 2016;6(1):29201.
- Yang X, Zhang X, Zhao X, Yuan M, Zhang K, Dai J, et al. Antibody-Dependent Enhancement: ″Evil″ Antibodies Favorable for Viral Infections. Viruses. 2022;14(8).
- Janoff EN, Wahl SM, Thomas K, Smith PD. Modulation of human immunodeficiency virus type 1 infection of human monocytes by IgA. J Infect Dis. 1995;172(3):855-8.
- Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J Virol. 2020;94(5).
- Genovese MC, Becker J-C, Schiff M, Luggen M, Sherrer Y, Kremer J, et al. Abatacept for Rheumatoid Arthritis Refractory to Tumor Necrosis Factor α Inhibition. New England Journal of Medicine. 2005;353(11):1114-23.
- Cope AP, Jasenecova M, Vasconcelos JC, Filer A, Raza K, Qureshi S, et al. Abatacept in individuals at high risk of rheumatoid arthritis (APIPPRA): a randomised, double-blind, multicentre, parallel, placebo-controlled, phase 2b clinical trial. The Lancet. 2024;403(10429):838-49.
- Baer AN, Gottenberg J-E, St Clair EW, Sumida T, Takeuchi T, Seror R, et al. Efficacy and safety of abatacept in active primary Sjögren’s syndrome: results of a phase III, randomised, placebo-controlled trial. Annals of the Rheumatic Diseases. 2021;80(3):339-48.
- de Wolff L, van Nimwegen JF, Mossel E, van Zuiden GS, Stel AJ, Majoor KI, et al. Long-term abatacept treatment for 48 weeks in patients with primary Sjögren's syndrome: The open-label extension phase of the ASAP-III trial. Seminars in Arthritis and Rheumatism. 2022;53:151955.
- Shock A, Burkly L, Wakefield I, Peters C, Garber E, Ferrant J, et al. CDP7657, an anti-CD40L antibody lacking an Fc domain, inhibits CD40L-dependent immune responses without thrombotic complications: an in vivo study. Arthritis Res Ther. 2015;17(1):234.
- Furie RA, Bruce IN, Dörner T, Leon MG, Leszczyński P, Urowitz M, et al. Phase 2, randomized, placebo-controlled trial of dapirolizumab pegol in patients with moderate-to-severe active systemic lupus erythematosus. Rheumatology (Oxford). 2021;60(11):5397-407.
- Clowse M ID, Merrill J, Dörner T, Petri M, Vital E, Morand E, Jimenez T, Brookes S, Gaiha-Rohrbach J, Martin C, Nelde A, Stach C. Dapirolizumab Pegol Demonstrated Significant Improvement in Systemic Lupus Erythematosus Disease Activity: Efficacy and Safety Results of a Phase 3 Trial [abstract]. Arthritis Rheumatology. 2024;76(suppl 9).
- Ackerman SE, Pearson CI, Gregorio JD, Gonzalez JC, Kenkel JA, Hartmann FJ, et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nature Cancer. 2021;2(1):18-33.
- Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci U S A. 2015;112(19):6140-5.
- Gubser C, Chiu C, Lewin SR, Rasmussen TA. Immune checkpoint blockade in HIV. eBioMedicine. 2022;76.
- Moreno-Vicente J, Willoughby JE, Taylor MC, Booth SG, English VL, Williams EL, et al. Fc-null anti-PD-1 monoclonal antibodies deliver optimal checkpoint blockade in diverse immune environments. J Immunother Cancer. 2022;10(1).
- Papadakis M, Karniadakis I, Mazonakis N, Akinosoglou K, Tsioutis C, Spernovasilis N. Immune Checkpoint Inhibitors and Infection: What Is the Interplay? In Vivo. 2023;37(6):2409-20.
- King HAD, Lewin SR. Immune checkpoint inhibitors in infectious disease. Immunological Reviews. 2024;328(1):350-71.
- Wykes MN, Lewin SR. Immune checkpoint blockade in infectious diseases. Nature Reviews Immunology. 2018;18(2):91-104.
- Uldrick TS, Adams SV, Fromentin R, Roche M, Fling SP, Gonçalves PH, et al. Pembrolizumab induces HIV latency reversal in people living with HIV and cancer on antiretroviral therapy. Sci Transl Med. 2022;14(629):eabl3836.
- Safety, pharmacokinetics, and exploratory efficacy of the PD-1 inhibitor budigalimab in antiretroviral treatment-suppressed people living with HIV-1: preliminary analysis of 2 Phase 1b studies including an analytical treatment interruption. 19th European AIDS Conference (EACS 2023.
- Italiano A, Cassier PA, Lin CC, Alanko T, Peltola KJ, Gazzah A, et al. First-in-human phase 1 study of budigalimab, an anti-PD-1 inhibitor, in patients with non-small cell lung cancer and head and neck squamous cell carcinoma. Cancer Immunol Immunother. 2022;71(2):417-31.
- Scapin G, Yang X, Prosise WW, McCoy M, Reichert P, Johnston JM, et al. Structure of full-length human anti-PD1 therapeutic IgG4 antibody pembrolizumab. Nat Struct Mol Biol. 2015;22(12):953-8.
- Hale G, De Vos J, Davy AD, Sandra K, Wilkinson I. Systematic analysis of Fc mutations designed to reduce binding to Fc-gamma receptors. MAbs. 2024;16(1):2402701.
- Fromentin R, DaFonseca S, Costiniuk CT, El-Far M, Procopio FA, Hecht FM, et al. PD-1 blockade potentiates HIV latency reversal ex vivo in CD4(+) T cells from ART-suppressed individuals. Nat Commun. 2019;10(1):814.
- Karunarathne DS, Horne-Debets JM, Huang JX, Faleiro R, Leow CY, Amante F, et al. Programmed Death-1 Ligand 2-Mediated Regulation of the PD-L1 to PD-1 Axis Is Essential for Establishing CD4(+) T Cell Immunity. Immunity. 2016;45(2):333-45.
- Esfahani K, Meti N, Miller WH, Jr., Hudson M. Adverse events associated with immune checkpoint inhibitor treatment for cancer. Cmaj. 2019;191(2):E40-e6.
- Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front Immunol. 2018;9:2306.
- Luu K, Dahl M, Hare E, Sibley C, Lizzul P, Randazzo B. DOP81 Rosnilimab, a novel PD-1 agonist monoclonal antibody, reduces T cell proliferation, inflammatory cytokine secretion, and PD-1high expressing CD4 and CD8 T cells: Results from a Phase 1 healthy volunteer clinical trial. Journal of Crohn's and Colitis. 2024;18(Supplement_1):i226-i.
- AnaptysBio, I. naptys announces rosnilimab achieved positive results in RA Phase 2b trial and highest ever reported CDAI LDA response over 6 months. 2025.
- Tuttle J, Drescher E, Simón-Campos JA, Emery P, Greenwald M, Kivitz A, et al. A Phase 2 Trial of Peresolimab for Adults with Rheumatoid Arthritis. New England Journal of Medicine. 2023;388(20):1853-62.
- Farhangnia P, Ghomi SM, Akbarpour M, Delbandi AA. Bispecific antibodies targeting CTLA-4: game-changer troopers in cancer immunotherapy. Front Immunol. 2023;14:1155778.
- Almutairi AR, McBride A, Slack M, Erstad BL, Abraham I. Potential Immune-Related Adverse Events Associated With Monotherapy and Combination Therapy of Ipilimumab, Nivolumab, and Pembrolizumab for Advanced Melanoma: A Systematic Review and Meta-Analysis. Front Oncol. 2020;10:91.
- Wang J, Lou H, Cai H-B, Huang X, Li G, Wang L, et al. A study of AK104 (an anti-PD1 and anti-CTLA4 bispecific antibody) combined with standard therapy for the first-line treatment of persistent, recurrent, or metastatic cervical cancer (R/M CC). Journal of Clinical Oncology. 2022;40(16_suppl):106-.
- Ma Y, Xue J, Zhao Y, Zhang Y, Huang Y, Yang Y, et al. Phase I trial of KN046, a novel bispecific antibody targeting PD-L1 and CTLA-4 in patients with advanced solid tumors. Journal for ImmunoTherapy of Cancer. 2023;11(6):e006654.
- Xing B, Da X, Zhang Y, Ma Y. A phase II study combining KN046 (an anti-PD-L1/CTLA-4 bispecific antibody) and lenvatinib in the treatment for advanced unresectable or metastatic hepatocellular carcinoma (HCC): Updated efficacy and safety results. Journal of Clinical Oncology. 2022;40(16_suppl):4115-.
- Jin G, Guo S, Xu J, Liu R, Liang Q, Yang Y, et al. A multicenter, randomized, double-blind phase III clinical study to evaluate the efficacy and safety of KN046 combined with nab-paclitaxel and gemcitabine versus placebo combined with nab-paclitaxel and gemcitabine in patients with advanced pancreatic cancer (ENREACH-PDAC-01). Journal of Clinical Oncology. 2022;40(16_suppl):TPS4189-TPS.
- Gang J, Guo S, Zhang Y, Ma Y, Guo X, Zhou X, et al. A phase II study of KN046 monotherapy as 2nd line and above treatment for unresectable locally advanced or metastatic pancreatic ductal adenocarcinoma (PDAC). Journal of Clinical Oncology. 2022;40(16_suppl):e16305-e.
- Labanieh L, Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. 2023;614(7949):635-48.
- Immunocore Holdings plc. Immunocore presents initial multiple ascending dose data for HIV functional cure candidate in an oral presentation at CROI 2025. 2025.
- Bourgeois S LY, Gane E, Lee HW, Cheng W, Heo J, Kim W, Buti Ferret M, Thompson A, Matthews G, Janczewska E, Ryder S, Andrade RJ, Yuan Y, Benlahrech A, Wustner J, Dorrell L, Yuen MF. Title: IMC-I109V, a novel soluble T cell receptor (TCR) bispecific (ENVxCD3) designed to eliminate HBV-infected hepatocytes in chronic HBV patients: initial data from a first-in-human study. The International Liver Congress (ILC) 2022. 2022.
- Chamberlain C, Colman PJ, Ranger AM, Burkly LC, Johnston GI, Otoul C, et al. Repeated administration of dapirolizumab pegol in a randomised phase I study is well tolerated and accompanied by improvements in several composite measures of systemic lupus erythematosus disease activity and changes in whole blood transcriptomic profiles. Ann Rheum Dis. 2017;76(11):1837-44.
- Webster JA, Luskin MR, Rimando J, Blackford A, Zeidan AM, Sharon E, et al. Blinatumomab in Combination with Immune Checkpoint Inhibitors (ICIs) of PD-1 and CTLA-4 in Adult Patients with Relapsed/Refractory (R/R) CD19 Positive B-Cell Acute Lymphoblastic Leukemia (ALL): Results of a Phase I Study. Blood. 2023;142(Supplement 1):966-.
- Institute, NC. A Dose Escalation Study of Duvortuxizumab in Participants With Relapsed or Refractory B-cell Malignancies (NCT02454270). US National Library of Medicine.
- Liu L, Lam C-YK, Long V, Widjaja L, Yang Y, Li H, et al. MGD011, A CD19 x CD3 Dual-Affinity Retargeting Bi-specific Molecule Incorporating Extended Circulating Half-life for the Treatment of B-Cell Malignancies. Clinical Cancer Research. 2017;23(6):1506-18.
- MacroGenics, I. MacroGenics Announces Termination of Duvortuxizumab Collaboration and License Agreement with Janssen. 2017.
- Howlett S, Carter TJ, Shaw HM, Nathan PD. Tebentafusp: a first-in-class treatment for metastatic uveal melanoma. Ther Adv Med Oncol. 2023;15:17588359231160140.
- Hamid O, Hassel JC, Shoushtari AN, Meier F, Bauer TM, Salama AKS, et al. Tebentafusp in combination with durvalumab and/or tremelimumab in patients with metastatic cutaneous melanoma: a phase 1 study. J Immunother Cancer. 2023;11(6).
- Ravandi F, Subklewe M, Walter RB, Vachhani P, Ossenkoppele G, Buecklein V, et al. Safety and tolerability of AMG 330 in adults with relapsed/refractory AML: a phase 1a dose-escalation study. Leuk Lymphoma. 2024;65(9):1281-91.
- NIH. A Study of AMG 330 With Pembrolizumab in Adult Patients With Relapsed/Refractory Acute Myeloid Leukemia.ClinicalTrials.gov identifier: NCT04478695.
- Johnson ML, Dy GK, Mamdani H, Dowlati A, Schoenfeld AJ, Pacheco JM, et al. Interim results of an ongoing phase 1/2a study of HPN328, a tri-specific, half-life extended, DLL3-targeting, T-cell engager, in patients with small cell lung cancer and other neuroendocrine cancers. Journal of Clinical Oncology. 2022;40(16_suppl):8566-.
- Moek KL, Waaijer SJH, Kok IC, Suurs FV, Brouwers AH, Menke-van der Houven van Oordt CW, et al. (89)Zr-labeled Bispecific T-cell Engager AMG 211 PET Shows AMG 211 Accumulation in CD3-rich Tissues and Clear, Heterogeneous Tumor Uptake. Clin Cancer Res. 2019;25(12):3517-27.
- E, DV. Continuous intravenous administration of AMG 211 (CEA CD3 BiTE®) in patients with relapsed/refractory gastrointestinal adenocarcinomas. 2025 ASCO Annual Meeting. 2025;Abstract 148667.
- Amgen) LDeA. Summary of clinical trials and development status for AMG 596 (etevritamab), an anti-EGFRvIII bispecific T cell engager (BiTE) for glioblastoma, including trial outcomes and preclinical data. 2024.
- El-Khoueiry A, Saavedra O, Thomas J, Livings C, Garralda E, Hintzen G, et al. First-in-Human Phase I Study of a CD16A Bispecific Innate Cell Engager, AFM24, Targeting EGFR-Expressing Solid Tumors. Clin Cancer Res. 2025;31(7):1257-67.
Figure 1.
structural diversity of fragment-based immune cell engagers discussed in this review.
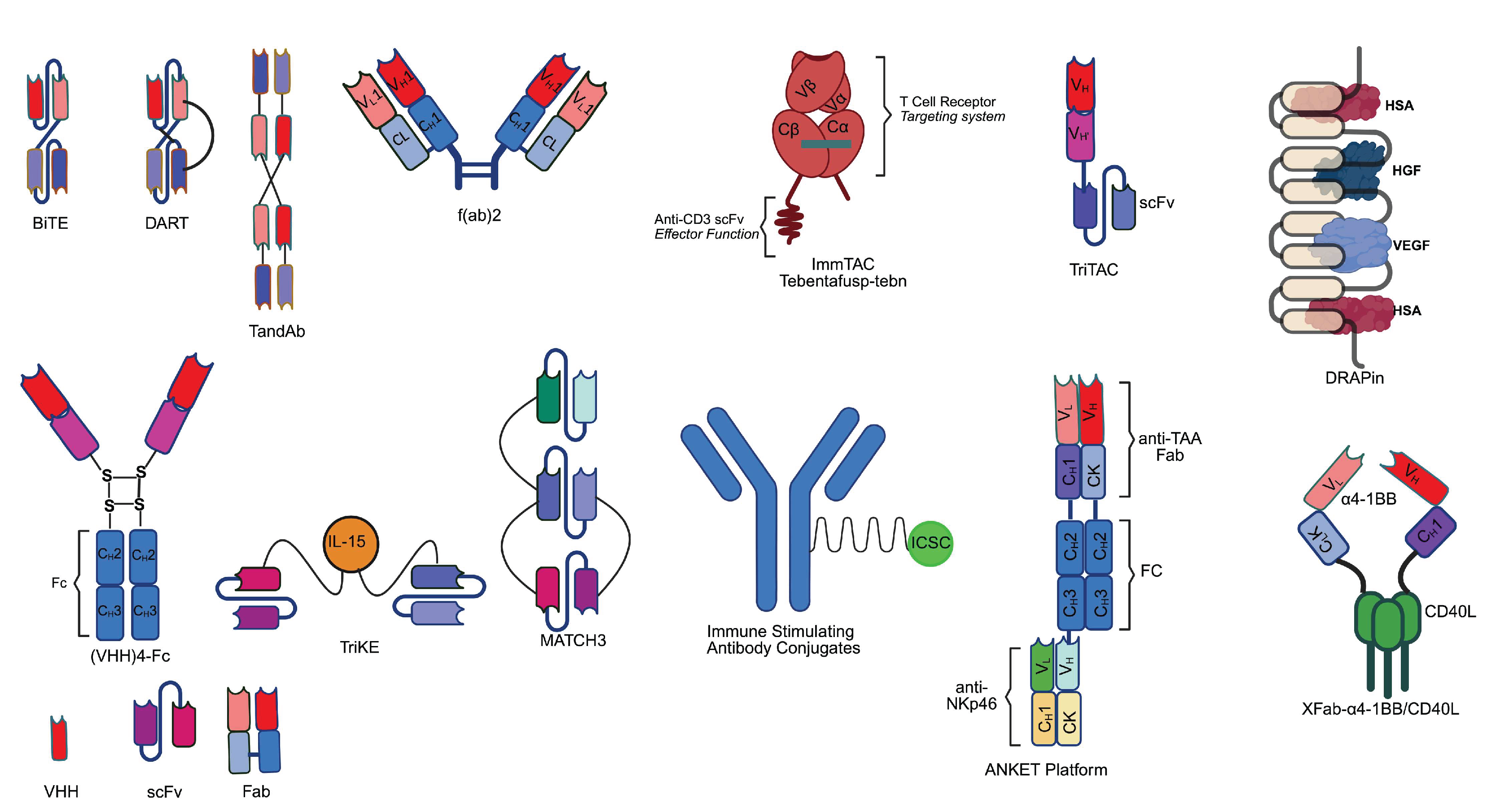

Table 1.
summary of clinical results for fragment-based immune cell engagers discussed in this review. Abbreviations: ADA: anti-drug antibody, HLA: human leukocyte antigen, irAE: immune-related adverse events, TEAE: treatment-emergent adverse events, TRAE: treatment-related adverse events, SAE: serious adverse events, AE: adverse events. CR: complete response rate, ORR: overall response, PK: pharmacokinetics, SC: subcutaneous (route), cIV: continuous IV infusion, PR: partial response, SD: stable disease, AML: acute myeloid leukemia, ALL: acute lymphoblastic leukemia, MDS: myelodysplastic syndromes, SCLC: small cell lung cancer, NSCLC: non-small cell lung cancer, NEC: neuroendocrine carcinoma, SRI-4: reduction in the SLE Disease Activity Index (SLEDAI) by ≥ 4 points. Note: Data for incidence of ADA and half-life may come from a different source of trial, rather than the source of trial for efficacy and adverse events (with patient size indicated).
Table 1.
summary of clinical results for fragment-based immune cell engagers discussed in this review. Abbreviations: ADA: anti-drug antibody, HLA: human leukocyte antigen, irAE: immune-related adverse events, TEAE: treatment-emergent adverse events, TRAE: treatment-related adverse events, SAE: serious adverse events, AE: adverse events. CR: complete response rate, ORR: overall response, PK: pharmacokinetics, SC: subcutaneous (route), cIV: continuous IV infusion, PR: partial response, SD: stable disease, AML: acute myeloid leukemia, ALL: acute lymphoblastic leukemia, MDS: myelodysplastic syndromes, SCLC: small cell lung cancer, NSCLC: non-small cell lung cancer, NEC: neuroendocrine carcinoma, SRI-4: reduction in the SLE Disease Activity Index (SLEDAI) by ≥ 4 points. Note: Data for incidence of ADA and half-life may come from a different source of trial, rather than the source of trial for efficacy and adverse events (with patient size indicated).
| antibody characteristic | clinical result | reference | |||||||||||||||||||||
| antibody name(s) | format | half-life | incidence of ADA | disease | target(s) | development stage | prospect | patient sample size (N) | Efficacy/Outcome | ≥grade 3 events | reference | ||||||||||||
| T cell engager (Infectious disease) | |||||||||||||||||||||||
| IMC-M113V | ImmTAV | N/A | N/A | AIDS/HIV | CD3 x gag (HLA-A*02:01) | phase 1/2 on going | on going development | 16 | Dose-dependent delayed viral rebound and/or viremia control (18.8%) | "no serious adverse events" | [305] | ||||||||||||
| IMC-I109V | ImmTAV | N/A | N/A | Chronic HBV infection | CD3 x env (HBV; HLA-A*02:01) | phase 1 ongoing | on going development | 21 | decline of serum viral surface antigen level (HBsAg); elevation in ALT and IL-6 | 0% | [306] | ||||||||||||
| MGD014 | DART-Fc | 12 d | 8.30% | AIDS/HIV | CD3 x gp120 | phase I completed | on going development | 24 | N/A | "well tolerated" | [194] | ||||||||||||
| Myeloid cell engager and B cell engager (infectious disease) | |||||||||||||||||||||||
| MDX-240 | bispecific | N/A | N/A | AIDS/HIV | FcγRI x gp41 | Clinical phase | development terminated | N/A | efficacy unknown; reduce infectivity in human monocyte-derived macrophages | N/A | [101] | ||||||||||||
| T cell engager (autoimmune disease) | |||||||||||||||||||||||
| Blinatumomab | BiTE | N/A | N/A | multidrug-resistant rheumatoid arthritis (RA) | CD3 x CD19 | clinical (compassionate use) | ongoing development | 6 | Sustained remission at month3; reduced autoantibody level | "no serious adverse events" | [209,210] | ||||||||||||
| Blinatumomab | BiTE | N/A | N/A | severe systemic sclerosis | CD3 x CD19 | case report | possible development | 1 | "significant improvement of symptom, regained ability to move" | "well tolerated" | [211] | ||||||||||||
| CLN-978 | BiTE-albumin | N/A | N/A | systemic lupus erythematosus (SLE) | CD3 x CD19 x albumin | phase 1b ongoing | ongoing development | 2 | N/A | N/A | [214] | ||||||||||||
| Myeloid cell engager and B cell engager (autoimmune disease) | |||||||||||||||||||||||
| MGD010 | DART-Fc | 8 d | N/A | autoimmunity | CD32B x CD79B | phase I completed | Collaborative development terminated | 8 | "decreased B cell activation with no signs of B-cell depletion" | 0% | [250] | ||||||||||||
| dapirolizumab pegol | Fab (PEG) | N/A | N/A | autoimmunity | CD40L | phase 3 completed | ongoing development | 208 | 60.1% SRI-4 vs control (41.1%) | 9.9% (TEAE) | [277,307] | ||||||||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



