
Submitted:
19 May 2025
Posted:
20 May 2025
You are already at the latest version
Abstract
We present results of DFT calculations of electronic properties, i.e. band structure, density of states and energy gap for arrays of parallelly ordered infinitely long small-diameter carbon nanotubes (CNTs). It turns out that if the distance between CNTs is sufficiently large (of the order 7.5\AA or greater), the electronic properties of arrays are very close to isolated CNTs. However, when in arrays CNTs are forced to be at a distance of the order of 5 \AA, they undergo radical structural rearrangement, leading to completely different structures: graphene-like or intermediate between graphite and diamond. Electronic properties are also rebuilt in a drastic manner.
Keywords:
carbon nanotubes
; density functional theory
; band structure
; energy gap
1. Introduction
Carbon nanotubes (CNTs) – quasi-one-dimensional allotropes of carbon, being cylindrical tubes with nanometers in diameter and up to several millimeters in length – have attracted significant research interest ever since their discovery due to their outstanding properties: electronic, optic, mechanical and even supercoducting [1]. From authors’ personal point of view, particularly interesting are: electronic, optic and excitonic properties, due to possible applications of CNTs as active layer in solar cells [5,8]. Electronic properties of isolated CNTs have been subject of numerous theoretical studies, see for example [4,5,7].
However, CNTs in experiments rarely appear as independent entities – most often they form random array of disordered fibers, or are placed into “matrices” of external host. Obecnie coraz wieksza uwage przyciagaja arrays of parallelly aligned CNTs. Its potential applications obey, among others as adhesive films, blackbody absorbers, biological sensors, gas sensors, electron-emitting materials [2,3]. From this point of view, theoretical studies on arrays of parallelly oriented CNT’s are highly desirable.
In our paper, we present results for electronic properties of arrays of parallelly aligned CNTs.
They obey: band structure and densities of states (DOS), and in particular the gap. In our study, we present result for some small-diameter (of ’radius’ 2 and 3) CNTs . The general conclusion is that electronic structure of an array does not differ significantly from properties of isolated ones for distances between CNTs being sufficiently large. However, we observe that when they are sufficiently close each other, they undergo the structural metamorphosis to the graphene-like, or even more complicated structures.
2. Computational Methods
We undertook a systematic exploration of small-diameter carbon nanotubes to examine its electronic properties We have performed first-principles calculations within the framework of the density-functional theory (DFT) as implemented in SIESTA [9].
The exchange and correlation effects were accounted for by the generalized gradient approximation (GGA) with PBE exchange-correlation potential. The optimized atomic structures were obtained by fully relaxing both atomic positions as well as cell parameters by using conjugate gradients (CG) method. The optimization algorithm varied the atomic structure of the system until all forces were smaller than eV/Å. All parameters critical for convergence, such as the k-points mesh and the energy cutoff, were carefully tested to ensure the most accurate results.
3. Results
3.1. Isolated CNTs
CNTs are characterized by two parameters , where n is diameter, (so n is a natural number greater than 1) whereas m denotes the chirality parameter, which – for given n – take values from 0 to n. In our study, we compute properties of small-diameter CNTs: n take values 2 and 3. We took two conducting (i.e. zero-gap) and two insulating (non-zero gap)CNTs: diameter-2 series: (2,0) (non-zero gap) and (2,2) (zero gap); and diameter-3 series: (3,2) (non-zero gap) and (3,3) (zero gap). We display their structures on Figure 1
3.2. CNT Arrays
We repeated calculations for arrays of parallelly oriented CNTs. We considered nanotube systems distributed over a 2D triangular lattice (to achieve the ’close packing’) with nanotube distances of 15.0, 10.0, 7.5, and 5.0Å. The ’distance’ means the distance between middles of CNTs. The array structure is illustrated on Figure 4.
Results for band structure and DOSes are very similar for single (isolated) CNTs and their arrays, for intertube distances: 15Åand 10Å. When the distance is equal to 7.5Åone observes some changes, but not too significant. See Table 2. The geometric structures of CNTs in arrays are also similar to those for the single tube. But the situation changes drammatically when the distance is forced to be 5Åin all three cases: DOSes, band structure and gap, and the geometry of cristal lattice. We describe these changes in more details for every individual CNT.
(2,0) The band structure changes radically for distances less than 5Å. The structure of DOS and bands changes complitely and energy gap opens, so the system becomes an insulator. See Figure 5. It is accompanied by complete reorganization of crystal structure to graphene-like one. See Figure 8.
(2,2).
In the case of (2,2) CNTs, the structure rebuilds at the distance about 5 Å, resulting it the set of graphene-like planes. See Figure 9. The bands and DOS also undergo drammatic metamorphosis. See Figure 6.
(3,2).
In this case, we have no definite results as the optimization process does not converge even after large number of steps. Perhaps the optimized structure is very complicated and includes very large number of atoms.
(3,3) Here we present structures for initial distances 7.5Å and 5Å. It is seen that the structure changes completely, resulting in something intermediate between graphite and diamond. See Figure 10. What is not strange, also DOS and bands rebuild in a profound manner. See Figure 7.
Figure 7.
2x2 array DOS and band structure for (3,3) CNT with Å (top), Å (center) and Å(bottom). It is seen that the DOS and band changes drammatically for the distance 5Å; in particular, the energy gap opens
Figure 7.
2x2 array DOS and band structure for (3,3) CNT with Å (top), Å (center) and Å(bottom). It is seen that the DOS and band changes drammatically for the distance 5Å; in particular, the energy gap opens
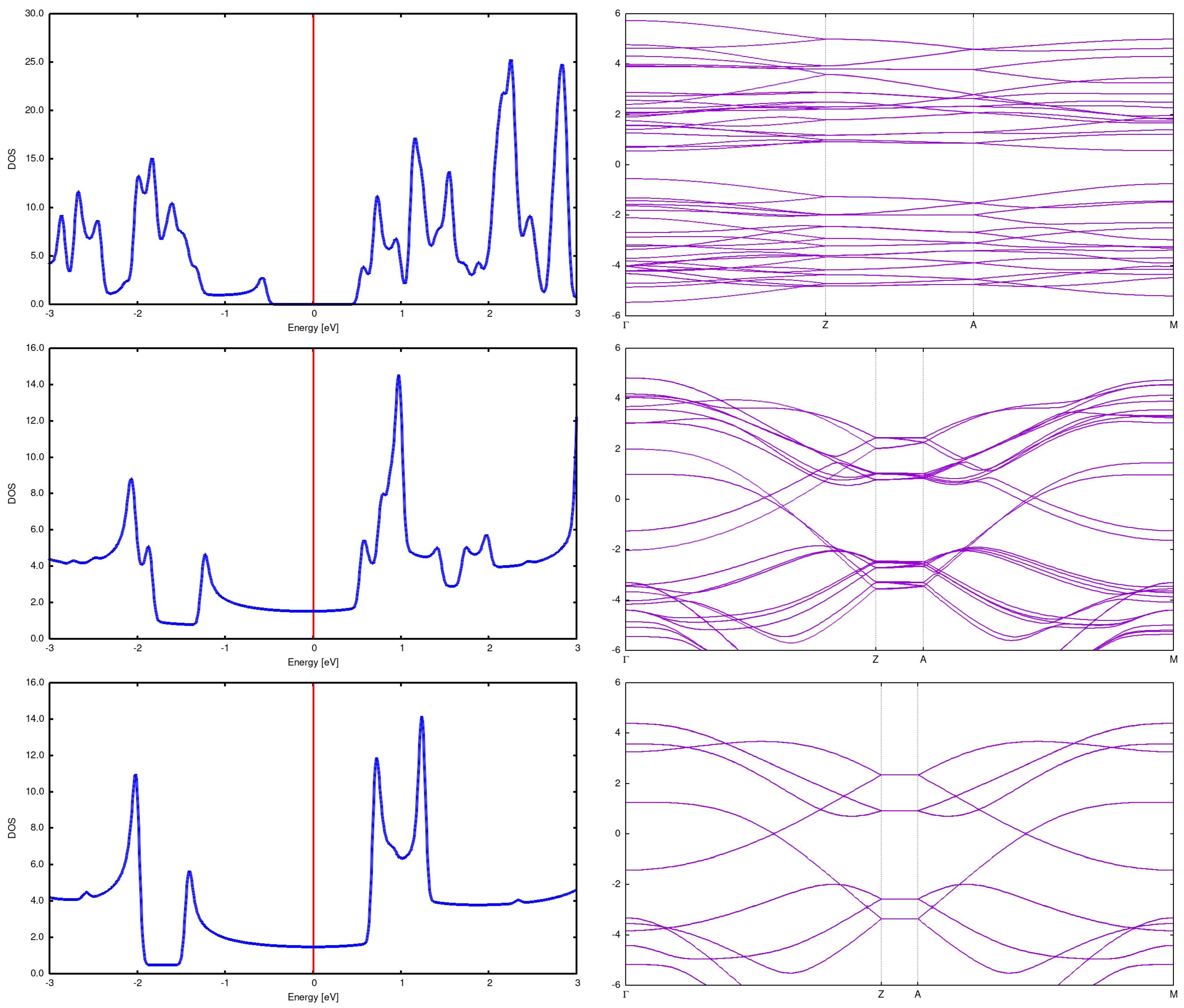
Figure 8.
Crystal structure after optimisation for arrays of (2,0)CNTs for intertube distances 5Å(left) and 4Å(right). It is seen that for 5Åthe ’tube-like’ structure is preserved, whereas for 4Åthe completely new structure appears – the planes which ’remembers’ the nanotubes.
Figure 8.
Crystal structure after optimisation for arrays of (2,0)CNTs for intertube distances 5Å(left) and 4Å(right). It is seen that for 5Åthe ’tube-like’ structure is preserved, whereas for 4Åthe completely new structure appears – the planes which ’remembers’ the nanotubes.

Figure 9.
(2,2) CNT optimized structures for initial distances 7.5Åand 5Å. It is seen that for 5Åthe graphene-like structure emerges.
Figure 9.
(2,2) CNT optimized structures for initial distances 7.5Åand 5Å. It is seen that for 5Åthe graphene-like structure emerges.
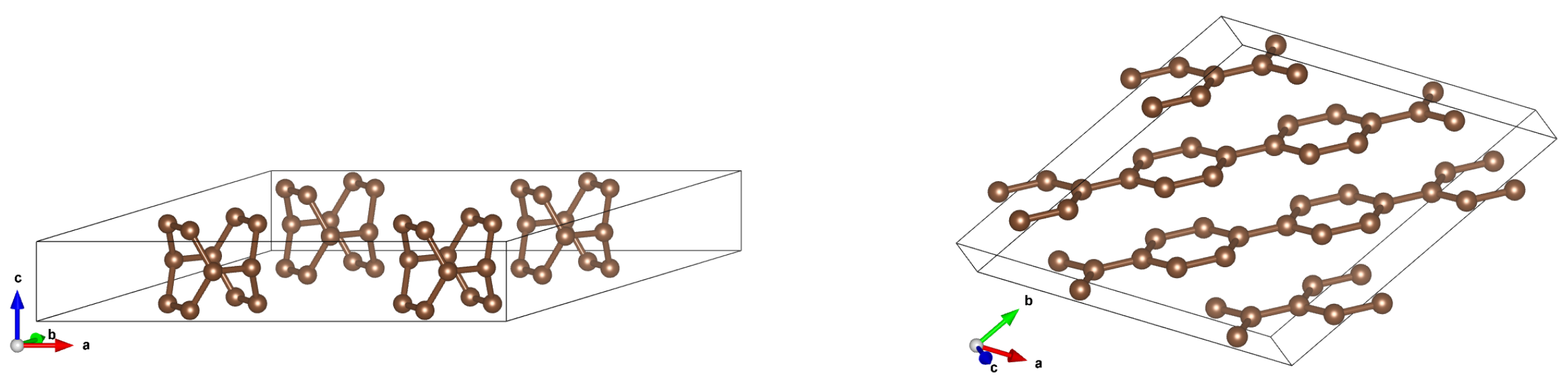
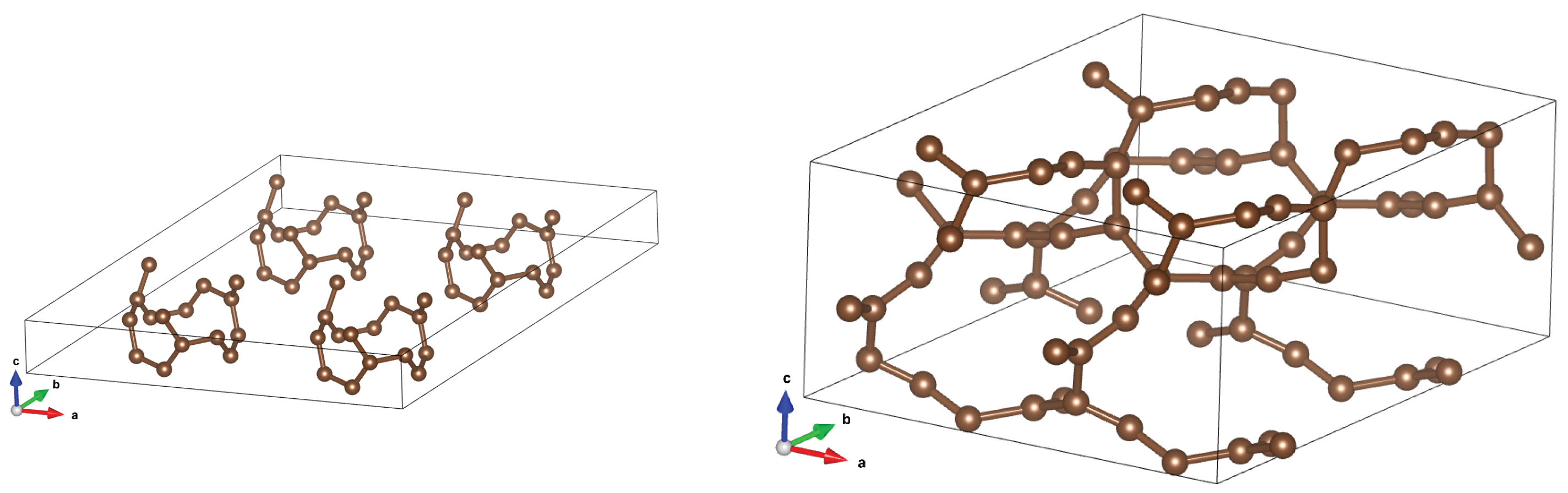
Figure 10.
CNT optimized structures for initial distances 7.5Åand 5Å. It is seen that something intermediate between the diamond and graphite appears.
Figure 10.
CNT optimized structures for initial distances 7.5Åand 5Å. It is seen that something intermediate between the diamond and graphite appears.
4. Summary, Perspectives of Further Investigations
We have calculated electronic properties of arrays of small-diameter carbon nanotubes. It turned out that for large intertube distances, the DOS and band structures does not differ from the case of isolated CNTs. But for smaller distances, the optimized structure differs radically from initial ones, which implies also radical change of DOS and band structures.
It would be very desirable to continue these calculations in order to:
- Know the equilibrium distance between CNTs. On the ground of present calculation, we conjecture that such arrays does not exist for small-diameter CNTs and are stable only for larger-diameter CNTs. One can guess that in this case the distance between walls of CNTs is the order of 3.5Å, like the distance between graphene layers in graphite. However, such a task is much more demanding from numerical point of view than present calculations.
- For equilibrium-distance arrays, one would know the relation between isolated tube properties and those in arrays: Geometry of CNTs; DOS and band structure; optical properties; exciton diffusion in arrays. All these properties are crucial for the idea of CNT-array-based solar cells. Unfortunately, the numerical demand seems to be enormous.
References
- A. Jorio, G. Dresselhaus, M. S. Dresselhaus: Carbon Nanotubes: Advanced Topics in the Synthesis, Structure, Properties, and Applications. Berlin, Springer Verlag, 2008.
- C. Zamora-Ledezma, Ch. Blanc, N. Puech, M. Maugey, C. Zakri, E. Anglaret, and P. Poulin: Conductivity anisotropy of assembled and oriented carbon nanotubes. Phys. Rev. E 85, 019901 (2012).
- Ding Wang, Pengcheng Song, Changhong Liu, Wei Wu and Shoushan Fan: Highly oriented carbon nanotube papers made of aligned carbon nanotubes. Nanotechnology 19 (2008) 075609.
- B. Brzostowski, A.P. Durajski, K.M. Gruszka, J. Wojtkiewicz: LNCS 13827 (2023) 392–402.
- J. Wojtkiewicz, M. Pilch: Comput. Theoret. Chem. 1216 (2022) 113846.
- C. Kamal and A. Chakrabarti: Comparison of electronic and geometric structures of nanotubes with subnanometer diameters: A density functional theory study. Phys. Rev. B 76, 075113 (2007).
- J. Yuan and Huang: Journal of Molecular Structure: THEOCHEM 942 (2010) 88-92.
- Wojtkiewicz, J. , Brzostowski B., Pilch M. (2020) Electronic and Optical Properties of Carbon Nanotubes Directed to Their Applications in Solar Cells. In: Wyrzykowski R., Deelman E., Dongarra J., Karczewski K. (eds) Parallel Processing and Applied Mathematics. PPAM 2019. Lecture Notes in Computer Science, vol. 12044. Springer, Cham. [CrossRef]
- J.M. Soler, E. Artacho, J.D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal: The SIESTA method for ab initio order-N materials simulation.Journal of Physics: Condensed Matter 14, 2745–2779 (2002).
Figure 1.
Elementary cells for optimized structure for: a) (2,0) b) (2,2) c) (3,3) d) (3,2) carbon nanotube, respectively
Figure 1.
Elementary cells for optimized structure for: a) (2,0) b) (2,2) c) (3,3) d) (3,2) carbon nanotube, respectively
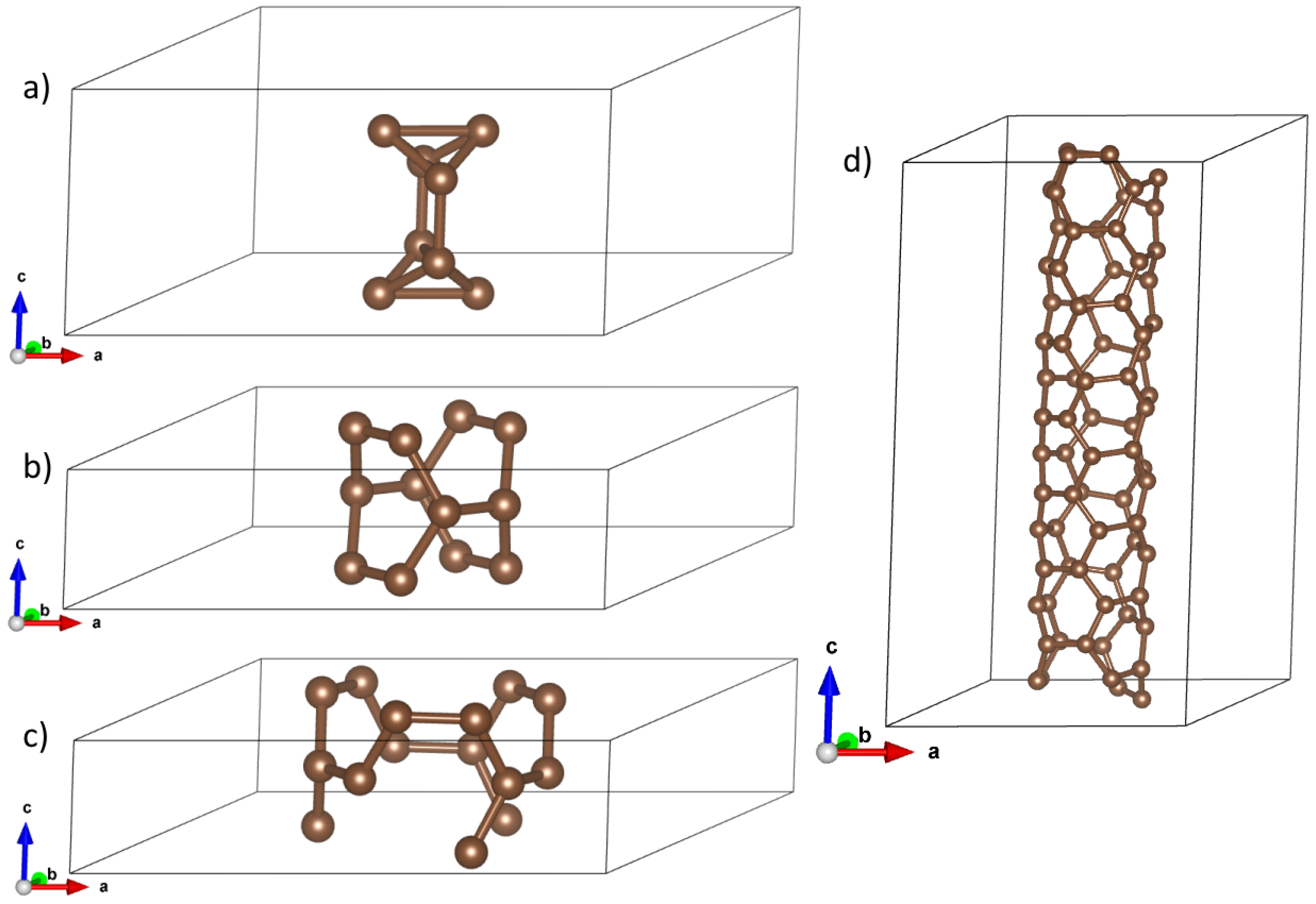
Figure 2.
DOS for single CNTs: (2,0) top left (2,2) bottom left (3,2) top right and (3,3) bottom right
Figure 2.
DOS for single CNTs: (2,0) top left (2,2) bottom left (3,2) top right and (3,3) bottom right
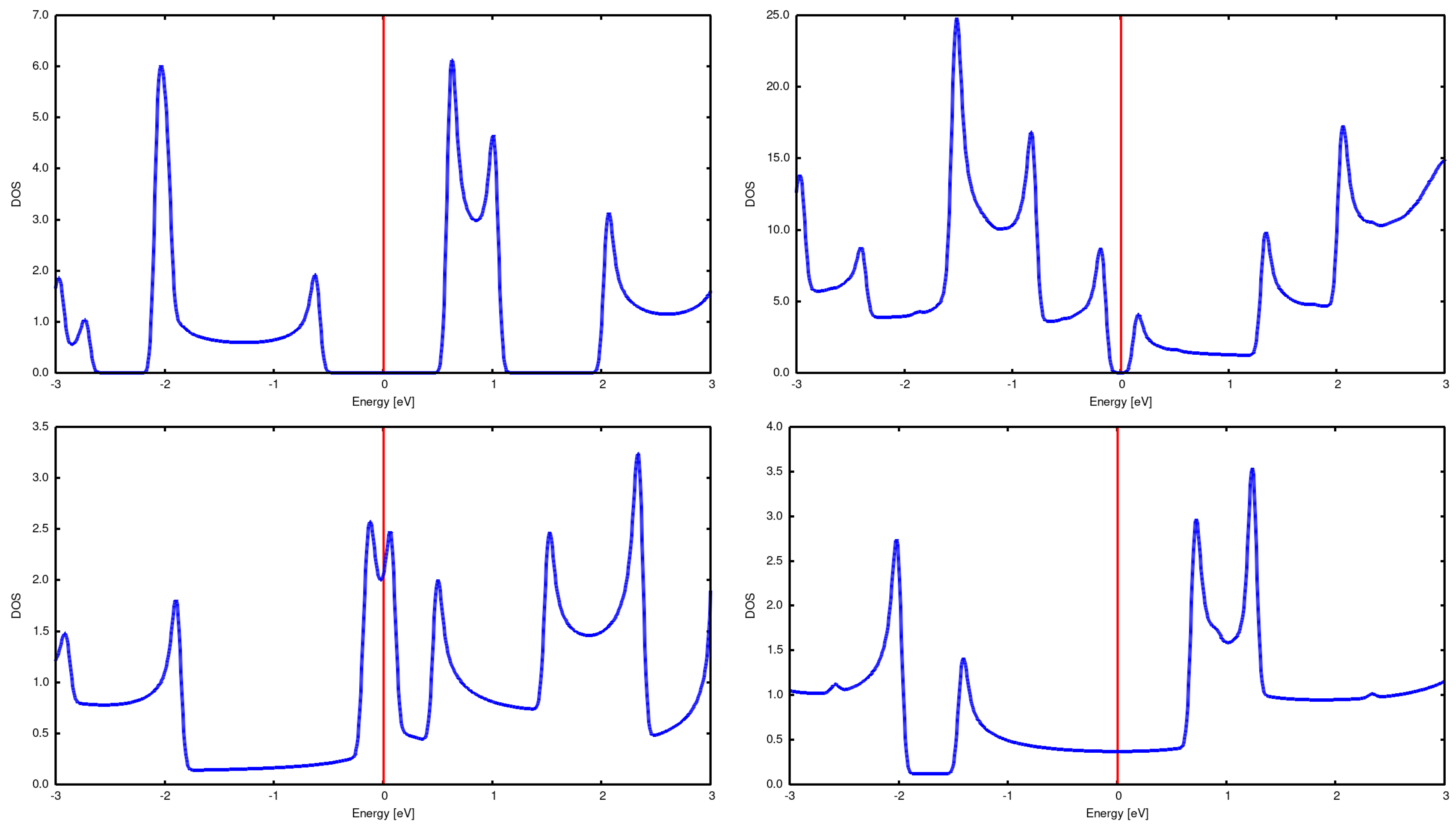
Figure 3.
Band structure for single CNTs: (2,0) top left (2,2) bottom left (3,2) top right and (3,3) bottom right
Figure 3.
Band structure for single CNTs: (2,0) top left (2,2) bottom left (3,2) top right and (3,3) bottom right
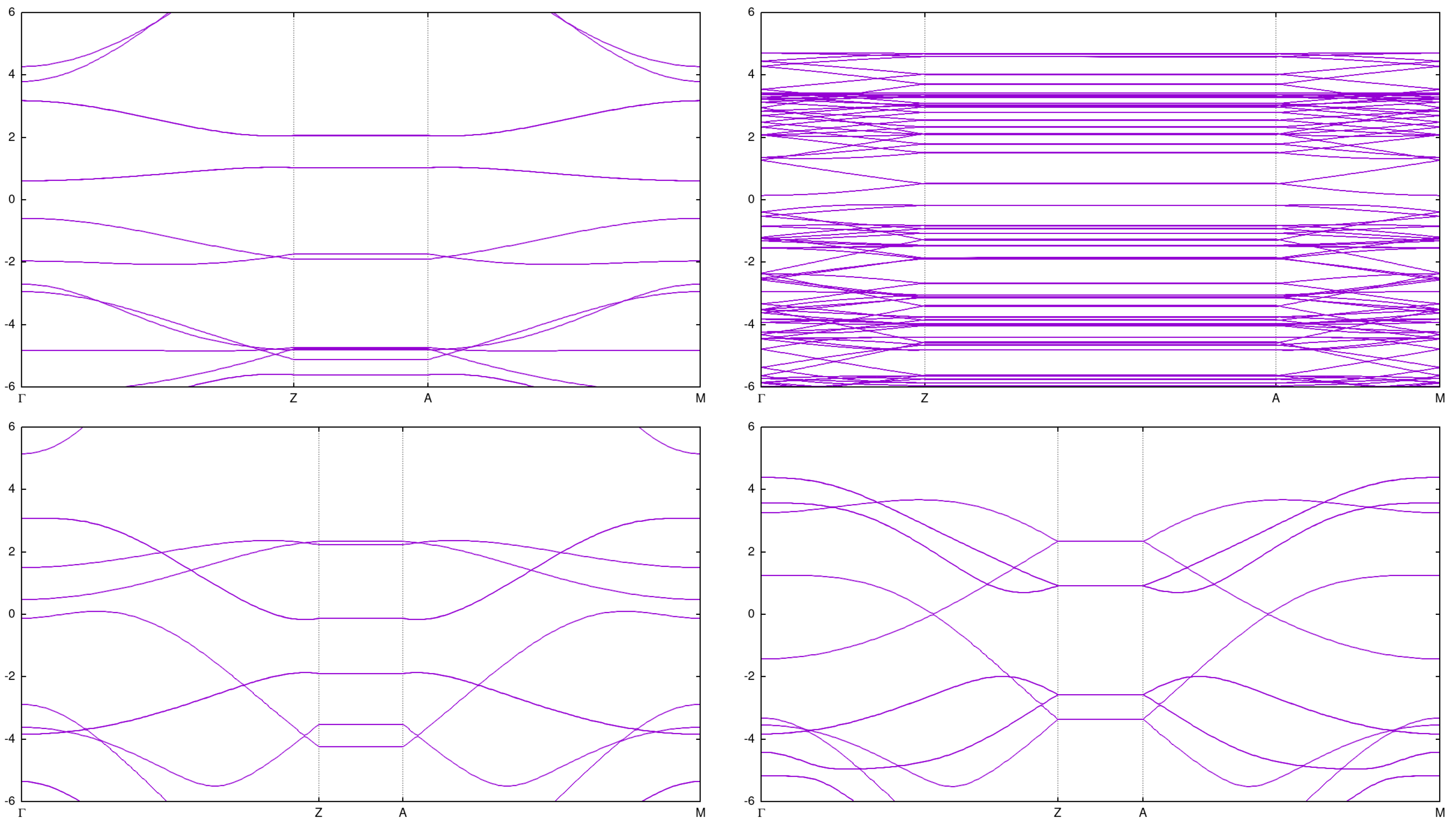
Figure 4.
Structure of arrays for (2,0), (2,2), (3,2) and (3,3) CNTs.
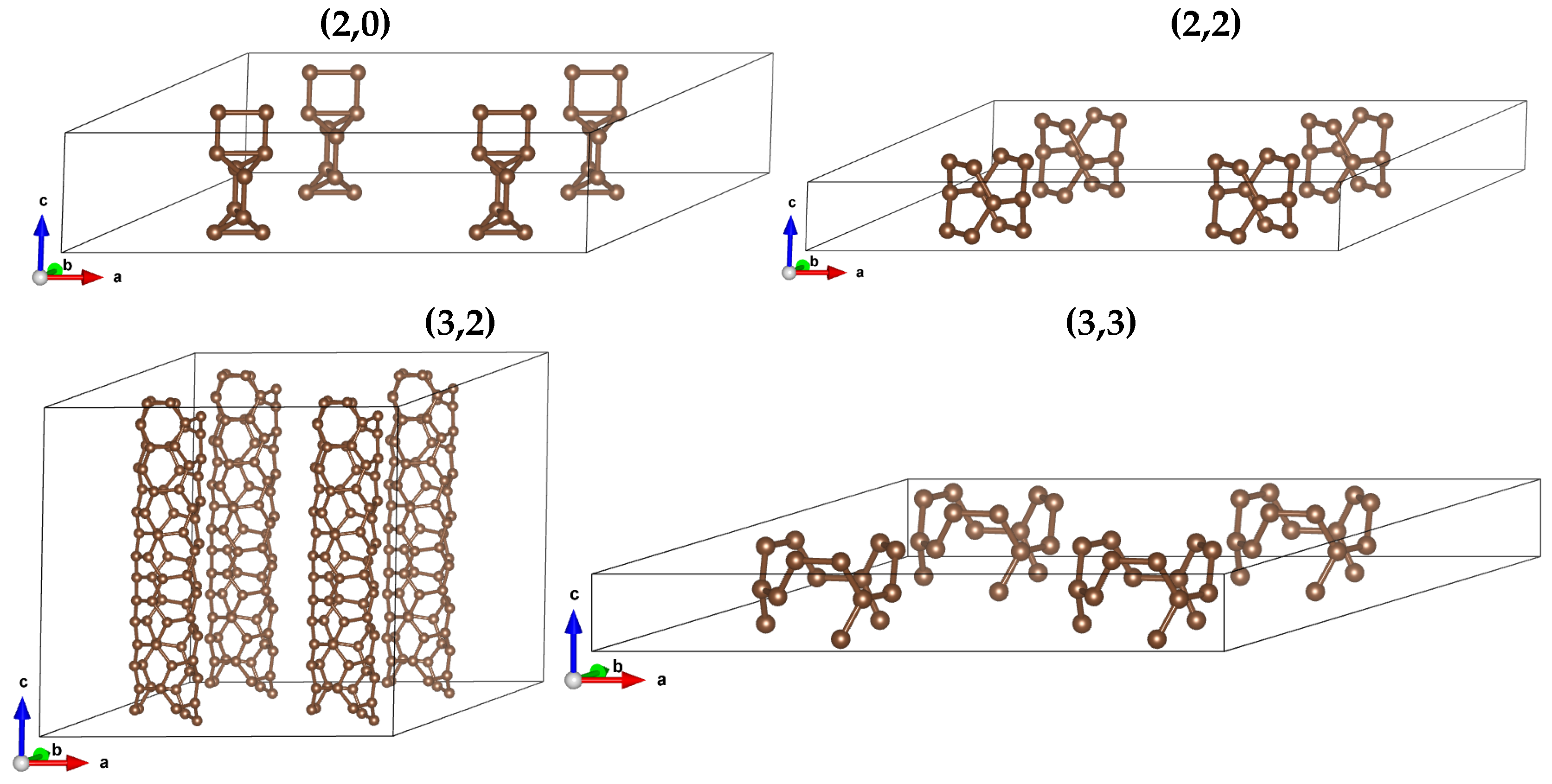
Figure 5.
2x2 array DOS and band structure for (2,0) CNT with Å (top), Å (center) and Å(bottom). For the distance equal to 5Å one still observes the nanotube structure, but they are very deformed in comparison to isolated case. Moreover, the energy gap closes.
Figure 5.
2x2 array DOS and band structure for (2,0) CNT with Å (top), Å (center) and Å(bottom). For the distance equal to 5Å one still observes the nanotube structure, but they are very deformed in comparison to isolated case. Moreover, the energy gap closes.
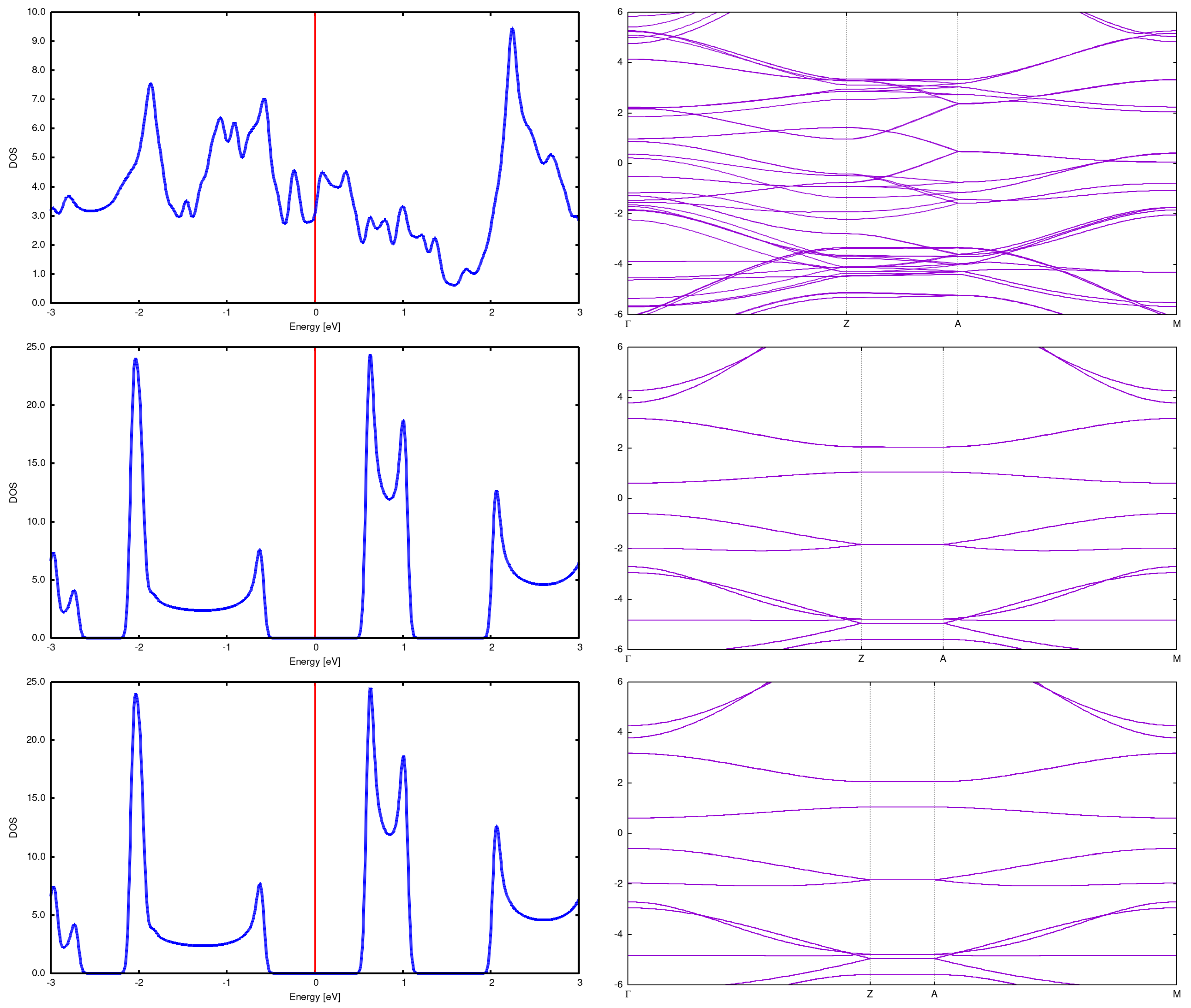
Figure 6.
2x2 array DOS and band structure for (2,2) CNT with Å (top), Å (center) and Å(bottom).
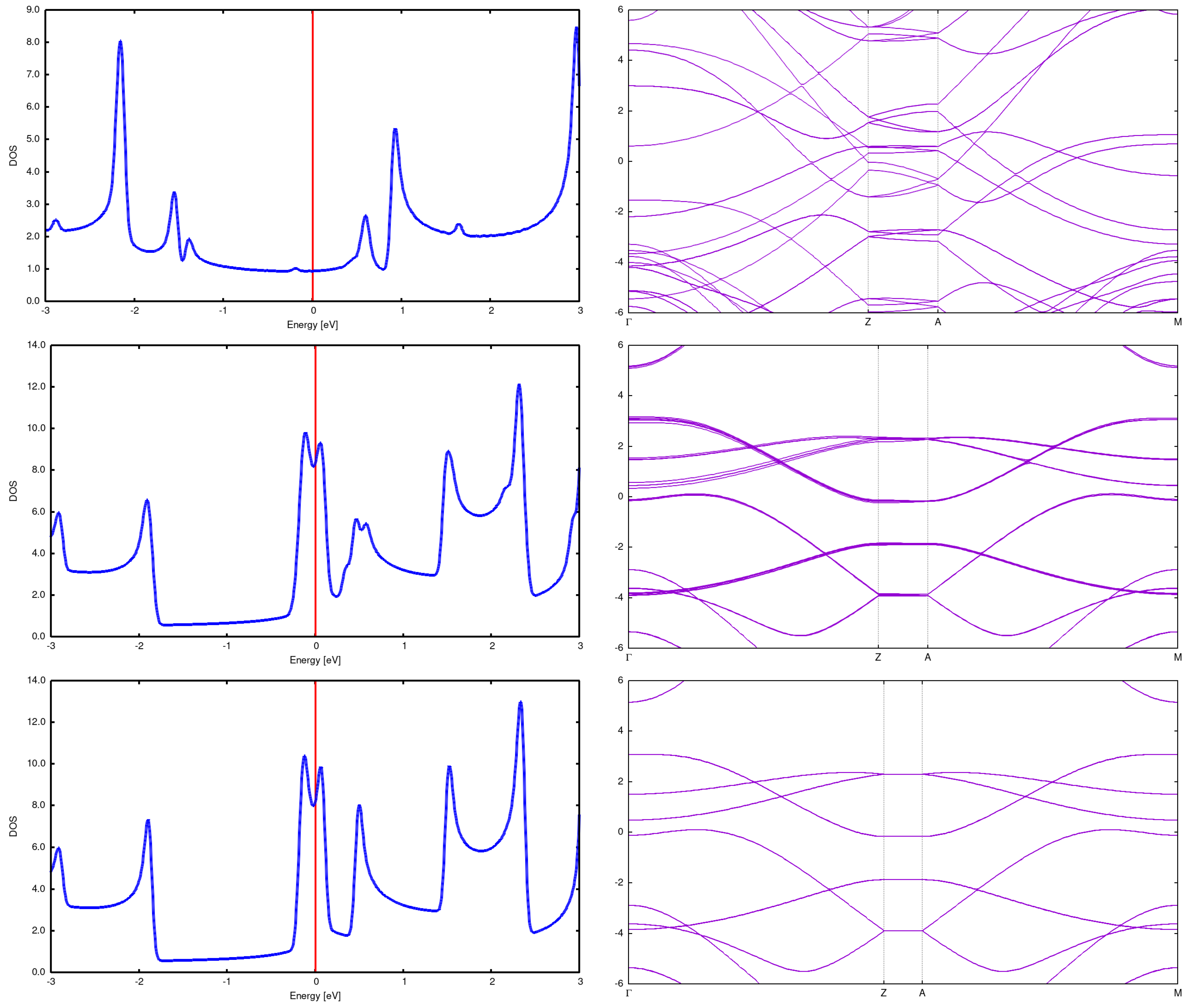

Table 1.
HOMO-LUMO gaps [eV] for isolated nanotubes.
| CNT | (2,0) | (2,2) | (3,2) | (3,3) |
|---|---|---|---|---|
| H-L gap | 1.20 | 0.00 | 0.29 | 0.00 |
Table 2.
HOMO-LUMO gap
| d [Å] | (2,0) | (2,0) | (3,3) | (3,2) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1x1 | 2x2 | 3x3 | 1x1 | 2x2 | 3x3 | 1x1 | 2x2 | 1x1 | 2x2 | |
| 5.0 | 1.58 | 0.00 | 0.445 | 0.01 | 0.01 | 0.00 | 2.15 | 1.10 | – | – |
| 7.5 | 1.20 | 1.2 | 1.20 | 0.00 | 0.00 | 0.00 | 0.02 | 0.00 | 0.55 | – |
| 10.0 | 1.20 | 1.20 | 1.20 | 0.00 | 0.01 | 0.01 | 0.00 | 0.00 | 0.29 | 0.29 |
| 15.0 | 1.20 | 1.20 | 1.20 | 0.00 | 0.01 | 0.01 | 0.00 | 0.00 | 0.29 | 0.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



