
Submitted:
19 May 2025
Posted:
20 May 2025
You are already at the latest version
Abstract
Systemic light-chain (AL) amyloidosis is a challenging, complex and heterogeneous disease. AL amyloidosis is classified under the category of plasma cell neoplasms and other diseases with paraproteins in the fifth edition of the World Health Organization classification of lymphoid tumors. Epidemiological information is limited, largely due to its low incidence and the lack of a global network of population-based specific registries. Despite recent advances, AL amyloidosis is still considered an incurable disease. The presence of a precursor disease, particularly monoclonal gammopathy of uncertain sig-nificance, is the main consolidated risk factor. Limited knowledge about other risk factors precludes the possibility of establishing preventive measures. A relevant percentage of AL amyloidosis patients fulfill the current diagnostic criteria of multiple myeloma. In-cidence should be evaluated in the setting of population-based studies. On the one hand, incidence shows a slightly increasing pattern. On the other hand, survival is progres-sively increasing. Consequently, prevalence is also rising. Early mortality, commonly associated to advanced heart involvement, remains a serious drawback to improve the outcome. Epidemiology represents the first level of heterogeneity in AL amyloidosis. Both genomic and clinical epidemiological research in systemic AL amyloidosis have a crucial role in the global strategy to combat this multifaceted disease.
Keywords:
systemic light-chain amyloidosis
; epidemiology
; incidence
; prevalence
; survival
; mortality
; early mortality
; risk factors
; prevention
; population-based registry
1. Introduction
Amyloidoses are a heterogeneous group of diseases characterized by the presence of an extracellular material of fibrillar nature called “amyloid” [1]. The classification of amyloidoses is based on the precursor protein of the amyloid. Currently, 19 human amyloid fibril proteins have been associated with systemic deposition and 4 can led to localized or systemic deposits [2].
Transthyretin (ATTR) and immunoglobulin (Ig) light-chain (AL) amyloidosis are the two most frequent types of systemic amyloidosis. Systemic AL amyloidosis has been classically considered the most common type of systemic amyloidosis [1,3,4], but nowadays the landscape has changed and ATTR is the most prevalent form [5,6,7].
The precursor proteins of the amyloid fibrils in systemic AL amyloidosis are monoclonal Ig light chains (LCs), which are produced by a clonal plasma cell (cPC) proliferation usually located in the bone marrow (BM). The diagnosis of AL amyloidosis is based on the demonstration of amyloid in a biopsy, and the confirmation that the amyloid fibrils are LC-derived, by a typing procedure.
In most cases, AL amyloidosis is systemic, showing a clonal BMPC infiltration and the presence of amyloid in one or more organs. However, localized AL amyloidosis (ALL) constitutes approximately 5-7% of all AL patients [8,9]. These cases commonly involve the respiratory, gastrointestinal, head and neck, or genitourinary systems [10]. ALL has an excellent prognosis with overall survival (OS) similar to that of the general population [11], without requiring systemic treatment.
Systemic AL amyloidosis is still considered a rare, challenging and incurable disease, often misdiagnosed or underdiagnosed. Fatigue is the most common symptom, usually in the setting of heart failure, provided that the heart is the most frequently affected organ. Cardiac involvement represents the single most important prognostic marker [8,12], and cardiac amyloidosis (CA) is an intensive field of research in which cardiologists have a key role [13]. Remarkably, a timely diagnosis requires a high index of suspicion [14].
Systemic AL amyloidosis is included in the 5th edition of the World Health Organization (WHO) classification of lymphoid tumors [15] as “Ig-related AL amyloidosis”. It is placed on “Plasma cell neoplasms and other diseases with paraproteins” (category) and “Diseases with monoclonal Ig deposition” (family). From an epidemiological point of view, it is highly recommended the right use of the WHO classification which is linked with the International Classification of Diseases.
Building comprehensive clinical registries at different levels (single center, regional, national) is crucial for epidemiological research, particularly in the setting of rare diseases, such as AL amyloidosis. It is strongly encouraged the inclusion of this entity in population-based cancer registries (PBCR) international network.
The aim of this narrative review is to update and summarize the current knowledge about risk factors (RFs), incidence, prevalence, and mortality in systemic AL amyloidosis.
2. Risk Factors
Etiological research in rare diseases is challenging. Available information on this subject remains scarce in the setting of AL amyloidosis. On the contrary, the landscape of epidemiological research in multiple myeloma (MM) focusing on RFs is more advanced [16], in the global context of monoclonal gammopathies (MGs) [17].
Given the similarities between systemic AL amyloidosis and MM, and the relatively common scenario (AL/MM) in which patients fulfill criteria for both diseases [8], a certain parallelism in potential RFs is anticipated. Only epidemiological studies with a biopsy-proven amyloidosis and confirmed AL typing should be taken into account for the analysis of RFs. The current evidence on RFs for developing systemic AL amyloidosis is presented below and summarized in Figure 1.
2.1. Precursor Disease
Several entities can be considered as precursor diseases [17], mainly monoclonal gammopathy of uncertain significance (MGUS), monoclonal gammopathy of clinical significance (MGCS) [18], and smoldering multiple myeloma (SMM).
MGUS can progress to both MM, systemic AL amyloidosis and other entities. MM is the key precursor disease for MM and it is considered that virtually all MM patients had a previous MGUS, sometimes overlooked. The role of MGUS progression to AL amyloidosis is less known. However, a risk-based follow-up approach should be established in every patient with MGUS, including a high suspicion for signs or symptoms of AL amyloidosis. Assuming that MGUS can only progress to MM is a serious misconception [19]. Remarkably, the risk of progression of MGCS is higher than that of MGUS and the follow-up should be adapted accordingly [18].
2.2. Family History
The information about hereditary factors in AL amyloidosis is limited. In a large series from Boston [23] with 1621 AL patients, 44 probands (2.7%) were identified with 52 relatives affected. The most common entities in family members were MM (48%) and AL amyloidosis (18%).
Similarly, in a study from Mayo Clinic involving 2955 AL patients [24], 82 (2.8%) had a positive family history. Again, MM (83%) was the most frequent diagnosis. Interestingly, median overall survival (mOS) was significantly better in the group with a positive family history.
Regarding advances in this field, large genomic studies are warranted. Clinical epidemiology must be complemented with genomic epidemiology.
2.3. Genetic Susceptibility
Several genomic approaches has been used for the advancement in the knowledge of the genomic landscape of systemic AL amyloidosis. Over the past two decades, the use of genome-wide association studies reported that some single nucleotide polymorphisms associated with MM risk were also associated with AL risk, suggesting a shared genetic etiology for MM, AL and MGUS [25].
2.4. Age
2.5. Sex
2.6. Race
Few studies have focussed on race/ethnicity as a potential RF for systemic AL amyloidosis. Most of them come form USA or UK [31,32,33,34]. The main aim of these studies is to investigate the possible prognostic impact of key ethnic minorities instead of their etiological role. Regarding prognosis, race does not behaves as an independent prognostic factor after controlling for other prognosis-related variables.
Hispanics and non-Hispanic blacks are the most usually analyzed ethnic minorities [31]. In a large study [32] of 4028 AL patients (2010-2019), Black patients were more likely to have previous MGUS and higher AL-associated clinical burden, suggesting that the diagnostic delay was longer in this group. In another retrospective study [33] (2015-2018), Hispanics had a reduced risk of mortality compared to non-Hispanic whites and non-Hispanic blacks. In a prospective observational study from UK [34] involving 1387 patients, 10% of them comprised ethnic minorities (5% Black, 2% Asian, and 2% mixed). This group had poorer relative deprivation index.
In summary, other underlying RFs such as genomic and sociodemographic factors, or environmental exposures, may influence the potential impact of race in the development of systemic AL amyloidosis.
2.7. Obesity
Obesity is defined by a body mass index (BMI) of 30 Kg/m2 or higher. The evidence on the role of obesity as RF for MM is strong [16]. Despite obesity being the only currently modifiable RF for MM, controversy remains on its potential link with MGUS [35]. However, BMI has been associated with the risk of progression in MGUS [36], particularly among females.
Regarding systemic AL amyloidosis, only 13.7% had obesity as comorbidity [22]. To date, there are no studies identifying obesity as a RF for AL amyloidosis.
2.8. Other Risk Factors
There is growing evidence on the role of some sociodemographic factors (socioeconomic status, marital status, occupation, and others) as RFs for developing MM, but their role in the setting of AL amyloidosis remains to be determined.
3. Incidence and Prevalence
The estimated number of incident cases (NDAL) worldwide in 2018 was 74,000 in a systematic review [37]. The crude annual incidence rate was 10.44 cases per million population (PMP), with relevant heterogeneity between countries, ranging from 14.30 in Japan to 6.72 in Brazil.
The incidence of systemic AL amyloidosis should be analyzed ideally in the context of PBSs, including all incident cases emerging within a concrete population and geographical area during a determined period of time. Single centers and particularly referral centers are not suitable for assessing incidence due to a high risk of selection bias. Table 1 shows incidence rates in recent PBS.
Incidence rates can be described as crude incidence rates or age-standardized incidence rates, being the latter option preferable.
Epidemiological studies on CA should be assessed carefully, since not all systemic AL amyloidosis patients have cardiac involvement and patients with both AL and ATTR amyloidosis are commonly included.
A recent nationwide Swedish study [28] (2011-2019) demonstrated an increase in incidence over time from 10.5 PMP in 2011 to 15.1 PMP in 2019 (trend test: p < 0.001). Other smaller PBSs and RW studies are also showed a similar trend. Crude incidence rates ranged from 12.5 to 16.7 PMP [38,39,40,41], whereas age-standardized incidence rates showed lower values [28,29,42]. Table 1 summarizes AL incidence in key epidemiological studies.
The prevalence of AL amyloidosis is also increasing, mainly due the rise in the incidence and the improvement in OS.
The estimated worldwide prevalence in 2018 in the above systematic review [37] was 51 PMP, which was similar to the one reported in the PBS by Duhamel et al. [38] of 58 PMP. In the Swedish study [28], the prevalence increased from 32.0 in 2011 to 47.0 PMP in 2019 (trend test: p < 0.001). Similarly, the prevalence in the USA [39] increased significantly from 15.5 PMP in 2007 to 40.5 in 2015, with an annual percentage change of 12% (p < 0.001). Comparably, other retrospective study in USA [41] showed a prevalence increase from 22.7 to 69.1 PMP between 2017 and 2021. Moreover, a cross-sectional retrospective cohort study in the elderly population (≥ 65 years) of USA [43] also showed that the prevalence increased from 9.92 in 2018 to 14.01 in 2020.
Figure 2 highlights and summarizes basic epidemiological data.
4. Survival
Despite the lack of an efficient and timely access to new therapies, OS is steadily increasing in systemic AL amyloidosis, largely due to the use of new agents, the implementation of a multidisciplinary approach, improvement in supportive care and the application of selective criteria for autologous stem cell transplant.
In the Mayo Clinic, mOS was less than 18 months at the end of the past century [44]. Only 4.7% in a series of 841 patients reached long-term OS (ten o more years), whereas this percentage achieved 22% twenty years later [45]. In a large series from Boston (1980-2019, n = 2337), mOS was 55.2 months during the last era [46]. Comparable progress has been documented in Europe and Asia, highlighting mOS of 48.8 months in the European study (2004-2018, n = 4480) [30], 56 months in the Swedish study (2011-2019, n = 846) [28], 64 months in the National Clinical Registry in Germany (2018-2020, n = 527) [47], or 52.6 months in a Spanish single referral center study (2005- 2023, n = 134) [22]. Remarkably, the reported survival in PBSs is lower, which may be due to the analysis of an unselected population. Accordingly, mOS in the Italian study (2000-2019, n=281) was 22 months [48]. Regarding Asia, in a recent single-center study (2011-2021, n = 335) from China, a remarkable mOS of 77.5 months was achieved [49], whereas 42 months were reported in another single referral center from Korea (1995-2018, n = 302) [50]. However, in other referral centers of middle-income Latin America countries such as São Paulo in Brazil (2009-2020, n = 97), mOS was 18.5 months [51]. Finally, a similar result of 19 months was reported in a multicenter study from Chile (2010-2018, n = 42), (Peña et al. 2019) [52].
It is clear that there is still room for improvement, particularly in economically disadvantaged areas. Every effort should be made to avoid diagnostic delay and to ensure timely access to therapeutic advances.
5. Mortality
Mortality is closely associated to the heart involvement, which is the main prognostic factor in this entity [8]. Consequently, the main cause of death was disease-related in most studies [46]. The most common cause of death was organ failure (49%), mainly heart failure (HF), and the second cause (23%) was sudden unexpected death. In a French study (2010-2016, n = 187) death was of cardiovascular origin in 69% and infection was the most common non-cardiovascular cause [53]. In an American study (2009-2016, n = 194), the primary cause of death was determined to be cardiac in 82%, and noncardiac in 18%. For patients with cardiac cause of death, 68% died of advanced HF, and 32% died of sudden cardiac death [54]. In a study with 1341 patients, using nationally representative hospital discharge data (2017–2020), 8% experienced in-hospital death [55].
Again, mortality should ideally be assessed in the context of PBSs as an age- and sex-standardized mortality rate. This is the right way in which PBSs report mortality rates of other HMs such as MM [56]. However, most PBCR do not include systemic AL amyloidosis, resulting in a lack of high quality information about its adjusted mortality and trends over time. Similarly, data about the standardized cause of death in AL amyloidosis are lacking.
Overall, mortality in systemic AL amyloidosis is improving over time, but early mortality remains an unmet clinical need, particularly in advanced stages of AL-CA. The cut-off time for early mortality is not standardized, but it is commonly defined as the death rate at six-months of diagnosis. In the largest RW epidemiological study to date [30], early mortality within 3 months from start of therapy was 13.4% and it did not improve over time (11.4% in the pre-2010 and 14.4% in the post-2010 era). Early mortality within the first six months after diagnosis is decreasing over time in most studies: from 37% to 24% [57], and from 23% to 13% [46]. This is also true for series with a high rate of cardiac involvement (91%) in which a small reduction was also demonstrated, from 31% to 28.3% [22].
In a cross-sectional analysis of death certificates in US from 1999 to 2020 [58], a total of 40,288 amyloidosis-related deaths occurred among all age groups, of which 75.8% (30,524) occurred among adults aged ≥ 65 years. Age-adjusted mortality rates (AAMRs) were calculated by standardizing amyloidosis-related deaths to the year 2000 US population. AAMRs for amyloidosis increased from 2.7 (1999) to 5.6 (2020) /100,000 persons. AAMRs were higher in men (8.5 vs 3.5 in women in 2020), in Non-Hispanic Blacks (11.8), in large metropolitan areas (6.3 vs. 4.6 in rural areas), and in the Northeast region (6.9 vs 4.2 in the South). Overall, 68.8% (21,010) of deaths occurred due to CA, with AAMR rising from 1.7 to 4.2. These data highlight the need to improve the management of CA amyloidosis in the elderly population.
6. Conclusions
Systemic AL amyloidosis is a rare, complex, heterogeneous and challenging disease. Epidemiology plays a critical role to foster clinical, molecular and etiological research. Moreover, accurate data from PBS including incidence, prevalence, survival and mortality help to unveil the underlying real-world heterogeneity. Trends of epidemiological indicators over time underscore the importance of allocating resources to implement more efficient strategies to combat this disease.
The integration of efforts by different healthcare professionals in a collaborative approach is mandatory. In this regard, the inclusion of systemic AL amyloidosis in the international network of PBCR is strongly recommended.
The diversity in the clinical presentation and outcome of this multifaceted entity could be explained by the integration of several levels of heterogeneity. Remarkably, epidemiology should be considered the first level of heterogeneity and hence the relevance of performing a comprehensive approach including the analysis of healthcare system characteristics, sociodemographic factors, comorbidity, frailty, nutrition status, and other variables, besides genomics and standard clinical information.
Even today, in advanced countries with fully developed healthcare systems, this disease remains a challenge, considering it is mostly represented by a frail and elderly population, and that the global burden of the disease is increasing due to a well-documented rise in incidence and prevalence.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
To Ana Ríos Sánchez for the English revision of the manuscript.
Conflicts of Interest
The author declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AAMR | Age-adjusted mortality rates |
| AL | Light-chain amyloidosis |
| ATTR | Transthyretin amyloidosis |
| BM | Bone marrow |
| BMI | Body mass index |
| CA | Cardiac amyloidosis |
| cPC | Clonal plasma cell |
| HF | Heart failure |
| Ig | Immunoglobulin |
| LC | Light chains |
| MGs | Monoclonal gammopathies |
| MGCS | Monoclonal gammopathy of clinical significance |
| MGUS | Monoclonal gammopathy of uncertain significance |
| MM | Multiple myeloma |
| ND | Newly diagnosed |
| OS | Overall survival |
| PBCR | Population-based cancer registry |
| PBS | Population-based cancer registry |
| PMP | Cases per million population |
| RF | Risk factor |
| RW | Real world |
| SMM | Smoldering multiple myeloma |
| WHO | World Health Organization |
References
- Falk, R.H.; Comenzo, R.L.; Skinner, M. The systemic amyloidoses. N. Engl. J. Med. 1997, 337, 898-909. [CrossRef]
- Buxbaum, J.N.; Eisenberg, D.S.; Fändrich, M.; McPhail, E.D.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Westermark, P. Amyloid nomenclature 2024: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2024, 31, 249-256. [CrossRef]
- Pinney, J.H.; Smith, C.J.; Taube, J.B.; Lachmann, H.J.; Venner, C.P.; Gibbs, S.D.-J.; Dungu, J.; Banypersad, S.M.; Wechalekar, A.D.; Whelan, C.J.; et al. Systemic amyloidosis in England: an epidemiological study. Br. J. Haematol. 2013, 161, 525-532. [CrossRef]
- Ravichandran, S.; Lachmann, H.J.; Wechalekar, A. Epidemiologic and Survival Trends in Amyloidosis, 1987–2019. N. Engl. J. Med. 2020, 382,1567-1568. [CrossRef]
- Zampieri, M.; Nardi, G.; Del Monaco, G.; Allinovi, M.; Gabriele, M.; Zocchi, C.; Casagrande, S.; Fumagalli, C.; Di Mario, C.; Olivotto, I.; et al. Changes in the perceived epidemiology of amyloidosis: 20 year-experience from a Tertiary Referral Centre in Tuscany. Int. J. Cardiol. 2021, 335, 123-127. [CrossRef]
- Panichella, G.; Aimo, A.; Vergaro, G.; Castiglione, V.; Arzilli, C.; Giannoni, A.; Merlo, M.; Limongelli, G.; Emdin, M. Cardiac Amyloidosis How Its Epidemiology is Changing. Heart Fail. Clin. 2024, 20, e1–e10. [CrossRef]
- Yamaguchi, A.; Tasaki, M.; Ueda, M.; Ando, Y.; Naiki, H. 2024. Epidemiological study of the subtype frequency of systemic amyloidosis listed in the Annual of the Pathological Autopsy Cases in Japan. Pathol. Int. 2024, 74, 68–76. [CrossRef]
- Ríos-Tamayo, R.; Krsnik, I.; Gómez-Bueno, M.; Garcia-Pavia, P.; Segovia-Cubero, J.; Huerta, A.; Salas, C.; Silvestre, R.; Sánchez, A.; Manso, M.; et al. AL Amyloidosis and Multiple Myeloma: A Complex Scenario in Which Cardiac Involvement Remains the Key Prognostic Factor. Life 2023, 13, 1518. [CrossRef]
- Kourelis, T.V.; Kyle, R.A.; Dingli, D.; K Buadi, F.K.; Kumar, S.K.; Gertz, M.A.; Lacy, M.Q.; Kapoor, P.; Go, R.S.; Gonsalves, W.I.; et al. Presentation and Outcomes of Localized Immunoglobulin Light Chain Amyloidosis: The Mayo Clinic Experience. Mayo Clin. Proc. 2017, 92, 908-917. [CrossRef]
- Dima, D.; Goel, U.; Ullah, F.; Faiman, B.; Basali, D.; Mazzoni, S.; Williams, L.S.; Samaras, C.; Valent, J.; Anwer, F.; et al. Presentation and Outcomes of Localized Immunoglobulin Light Chain Amyloidosis: 14-Year Experience of an Academic Center. Hematol. Oncol. 2025, 43, e70082. [CrossRef]
- Martínez, J.C. and Lichtman, E.I. Localized light chain amyloidosis: A self-limited plasmacytic B-cell lymphoproliferative disorder. Front. Oncol. 2022, 12, 1002253. [CrossRef]
- Zanwar, S.; Gertz, M.A.; Muchtar, E. 2023. Immunoglobulin Light Chain Amyloidosis: Diagnosis and Risk Assessment. J. Natl. Compr. Canc. Netw. 2023, 21, 83–90. [CrossRef]
- Wechalekar, A.D.; Fontana, M.; Quarta, C.C.; Liedtke, M. AL Amyloidosis for Cardiologists: Awareness, Diagnosis, and Future Prospects: JACC: CardioOncology State-of-the-Art Review. J. Am. Coll. Cardiol. CardioOnc. 2022, 4, 427–441. [CrossRef]
- Stefani, G.; Kouvata, E.; Vassilopoulos, G. Light-Chain Amyloidosis: The Great Impostor. Life 2024, 14, 42. [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [CrossRef]
- Ríos-Tamayo, R.; Sánchez Rodríguez, D.; Chang-Chan, D.-Y.-L.; Sánchez Pérez, M.J. Epidemiology of Multiple Myeloma. In Update on Multiple Myeloma; Editor Khalid Ahmed Al-Anazi; Intech: London, UK, 2019; pp.13-33. [CrossRef]
- Ríos-Tamayo, R. Monoclonal Gammopathies. In Comprehensive Hematology and Stem Cell Research, 1st Edition; Editor Nima Rezaei; Elsevier: Amsterdam, Netherlands, 2024; pp. 358-374. [CrossRef]
- Ríos-Tamayo, R.; Paiva, B; Lahuerta, J.J.; Martínez López, J.; Duarte, R.F. Monoclonal Gammopathies of Clinical Significance: A Critical Appraisal. Cancers 2022, 14, 5247. [CrossRef]
- Gertz, M.A. Immunoglobulin light chain amyloidosis: 2024 update on diagnosis, prognosis, and treatment. Am. J. Hematol. 2024, 99, 309–324. [CrossRef]
- Sigurdardottir, E.E.; Turesson, I.; Lund, S.H.; Lindqvist, E.K.; Mailankody, S.; Korde, N.; Björkholm, M.; Landgren, O.; Kristinsson, S.Y. The role of diagnosis and clinical follow-up of monoclonal gammopathy of undetermined significacnce on survival in multiple myeloma. JAMA Oncol. 2015, 1, 168-174. [CrossRef]
- Ríos-Tamayo, R.; Sainz, J.; Jurado, M.; Puerta, J.M.; González, P.A.; Romero, A.; López, E.; Moratalla,L.; García de Veas, J.L.; Rodríguez, T.; et al. Multiple myeloma with prior precursor disease shows better outcome. Blood 2015, 126 (23), 1756. [CrossRef]
- Ríos-Tamayo, R.; Lecumberri, R.; Cibeira, M.T.; González-Calle, V.; Alonso, R.; Domingo-González, A.; Landete, E.; Encinas, C.; Iñigo, B.; Blanchard, M.J.; et al. A Simple Frailty Score Predicts Survival and Early Mortality in Systemic AL Amyloidosis. Cancers 2024, 16, 1689. [CrossRef]
- Staron, A.; Verma, K.; Sanchorawala, V. Prevalence of plasma cell and lymphoproliferative disorders among blood relatives of patients with light chain amyloidosis. Br. J. Haematol. 2022, 198, 861-865. [CrossRef]
- Visram, A.; Vachon, C.; Baughn, L.B.; Larson, D.; Smadbeck, J.; Dispenzieri, A.; Kapoor, P.; Lacy, M.Q.; Gertz, M.A.; Buadi, F.K.; et al. 2022. Family history of plasma cell disorders is associated with improved survival in MGUS, multiple myeloma, and systemic AL amyloidosis. Leukemia 2022, 36, 1058-1065. [CrossRef]
- Chattopadhyay, S.; Thomsen, H.; Weinhold, N.; Meziane, I.; Huhn, S.; da Silva Filho, M.I.; Vodicka,P.; Ludmila Vodickova, L.; Hoffmann, P.; Nöthen, M.M.; et al. Eight novel loci implicate shared genetic etiology in multiple myeloma, AL amyloidosis, and monoclonal gammopathy of unknown significance. Leukemia 2020, 34, 1187-1191. [CrossRef]
- Paiva, B.; Martinez-Lopez, J.; Corchete, L.A.; Sanchez-Vega, B.; Rapado, I.; Puig, N.; Barrio, S.; Sanchez, M.L.; Alignani, D.; Lasa, M.; et al. Phenotypic, transcriptomic, and genomic features of clonal plasma cells in light-chain amyloidosis. Blood 2016, 127, 3035-3039. [CrossRef]
- Kimura, K.; Tsukamoto, S.; Miyazaki, K.; Kawajiri-Manako, C.; Ishii, A.; Rahmutulla, B.; Fukuyo, M.; Oshima-Hasegawa, N.; Mitsukawaa, S.; Takeda, Y.; et al. 2021. Identification of clonal immunoglobulin λ light-chain gene rearrangements in AL amyloidosis using next-generation sequencing. Exp. Hematol. 2021, 101, 34-41. [CrossRef]
- Mellqvist, U.-H.; Cai, Q.; Hester, L.L.; Grövdal, M.; Börsum, J.; Rahman, I.; Ammann, E.M.; Hansson, M. Epidemiology and clinical outcomes of light-chain amyloidosis in Sweden: A nationwide population-based study. Eur. J. Hematol. 2023, 111, 697-705. [CrossRef]
- Kyle, R.A.; Larson, D.R.; Kurtin, P.J.; Kumar, S.; Cerhan, J.R.; Therneau, T.M.; Rajkumar, S.V.; Vachon, C.M.; Dispenzieri, A. 2019. Incidence of AL Amyloidosis in Olmsted County, Minnesota, 1990 through 2015. Mayo Clin. Proc.2019, 94, 465-471. [CrossRef]
- Palladini, G.; Schönland, S.; Merlini, G.; Milani, P.; Jaccard, A.; Bridoux, F.; Dimopoulos, M.A.; Ravichandran, S.; Hegenbart, U.; Roeloffzen, W.; et al. The management of light chain (AL) amyloidosis in Europe: clinical characteristics, treatment patterns, and efficacy outcomes between 2004 and 2018. Blood Cancer J. 2023, 13, 19. [CrossRef]
- Staron, A.; Connors, L.H.; Zheng, L.; Doros, G.; and Sanchorawala, V. Race/ethnicity in systemic AL amyloidosis: perspectives on disease and outcome disparities. Blood Cancer J. 2020, 10, 118. [CrossRef]
- D’Souza, A.; Pezzin, L.; Laud, P.; Singh, A. Racial disparities in patients diagnosed with light chain (AL) amyloidosis. Blood Cancer J. 2021, 11, 72. [CrossRef]
- Ream, S.; Ma, J.; Rodriguez, T.; Sarabia-Gonzalez, A.; Alvarado, L.A.; Dwivedi, A.K.; Mukherjee, D. 2023. Ethnic/racial differences in risk factors and clinical outcomes among patients with amyloidosis. Am. J. Med. Sci. 2023, 365, 232–241. [CrossRef]
- Khwaja, J.; Ravichandran, S.; Cohen, O.; Foard, D.; Martinez-Naharro, A.; Venneri, L.; Whelan, C.; Hawkins, P.N.; Gillmore, J.; Lachmann, H.J.; et al. Ethnicity in systemic AL amyloidosis may impact risk stratification. Haematologica 2025, 110, 1202-1206. [CrossRef]
- Castaneda-Avila, M.A.; Ulbricht, C.M.; Epstein, M.M. Risk factors for monoclonal gammopathy of undetermined significance: a systematic review. Ann. Hematol. 2021, 100, 855-863. [CrossRef]
- Kleinstern, G.; Larson, D.R.; Allmer, C.; Norman, A.D.; Muntifering, G.; Sinnwell, J.; Visram, A.; Rajkumar, V.; Dispenzieri, A.; Kyle, R.A.; et al. Body mass index associated with monoclonal gammopathy of undetermined significance (MGUS) progression in Olmsted County, Minnesota. Blood Cancer J. 2022, 12, 67. [CrossRef]
- Kumar, N.; Zhang, N.J.; Cherepanov, D.; Romanus, D.; Hughes, M.; Faller, D.V. Global epidemiology of amyloid light-chain amyloidosis. Orphanet J. Rare Di.s 2022, 17, 278. [CrossRef]
- Duhamel, S.; Mohty, D.; Magne, J.; Lavergne, D.; Bordessoule, D.; Aboyans, V.; Jaccard, A. Incidence and Prevalence of Light Chain Amyloidosis: A Population-Based Study. Blood 2017, 130 (Suppl_1), 5577. [CrossRef]
- Quock, T.P.; Yan, T.; Chang, E.; Guthrie, S.; Broder, M.S. Epidemiology of AL amyloidosis: a real-world study using US claims data. Blood Adv. 2018, 2, 1046-1053. [CrossRef]
- Zampieri, M.; Cappelli, F.; Allinovi, M.; Olivotto, I.; Antonioli, E.; Tassetti, L.; Zocchi, C.; Andrei, V.; Di Mario, C.; Nozzoli, C.; et al. Incidence of light chain amyloidosis in Florence metropolitan area, Italy: a population-based study. Amyloid 2021, 28, 211-212. [CrossRef]
- Laires, P.A., Evans, J.; Thompson, J.; Manwani, R.; Mudumby, P.; Field, M.; Fang, S. Prevalence, Incidence, and Characterization of Light Chain Amyloidosis in the USA: A Real-World Analysis Utilizing Electronic Health Records (EHR). Blood 2023, 142, 6767-6768. [CrossRef]
- Hou, H.-A.; Tang, C.-H.; Goh, C.H.; Shen, S.-P.; Huang, K.-C.; Qiu, H.; Siggins, S.; Rothwell, L.A.; Liu, Y. A population-based cohort study of the epidemiology of light-chain amyloidosis in Taiwan. Sci. Reports 2022, 12, 15736. [CrossRef]
- Bajaj, P.S.; Broder, M.S.; Das, A.K.; Chang, E.; Tarbox, M.H.; Conrad, A.; D’Souza, A. Increasing prevalence and incidence of AL amyloidosis among older adults in the US. XIX International Symposium on Amyloidosis Abstracts, Amyloid 2024, 31(sup1): S30-S31. [CrossRef]
- Kyle, R.A.; Gertz, M.A.; Greipp, P.R.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Therneau, T.M. Long-Term Survival (10 Years or More) in 30 Patients with Primary Amyloidosis. Blood 1999, 93, 1062-1066.
- Muchtar, E.; Gertz, M.A.; Lacy, M.Q.; Go, R.S.; Buadi, F.K.; Dingli, D.; Grogan, M.; AbouEzzeddine, O.F.; Hayman, S.R.; Kapoor, P.; et al. Ten-year survivors in AL amyloidosis: characteristics and treatment pattern. Br. J. Haematol. 2019, 187, 588–594. [CrossRef]
- Staron, A.; Zheng, L.; Doros, G.; Connors, L.H.; Mendelson, L.M.; Joshi, T.; Sanchorawala, V. Marked progress in AL amyloidosis survival: a 40-year longitudinal natural history study. Blood Cancer J. 2021, 11, 139. [CrossRef]
- Hegenbart, U.; aus dem Siepen, F.; Carpinteiro, A.; Hansen, T.; Kimmich, C.; Oldenburg, S.; Hofmann, E.; Fuhr, N.; Huber, L.; Ziehl, R.; et al. Long-term evaluation of amyloidosis diseases in Germany: National Clinical Amyloidosis Registry. XIX International Symposium on Amyloidosis Abstracts, Amyloid 2024, 31(sup1), S147. [CrossRef]
- Zampieri, M.; Nardi, G.; Del Monaco, G.; Allinovi, M.; Gabriele, M.; Zocchi, C.; Casagrande, S.; Fumagalli, C.; Di Mario, C.; Olivotto, I.; et al. Changes in the perceived epidemiology of amyloidosis: 20 year-experience from a Tertiary Referral Centre in Tuscany. Int. J. Cardiol. 2021, 335, 123-127. [CrossRef]
- Xie, W.; Wang, Q.; Zhou, F.; Wang, S.; Sun, Y.; Cen, X.; Ren, H.; Qiu, Z.; Dong, Y. Clinical characteristics and prognosis of a Chinese cohort with systemic light chain amyloidosis: a single-center study. Int. J. Hematol. 2023, 118, 231-241. [CrossRef]
- Yoon, S.E.; Kim, D.; Choi, J.-O.; Min, J.-H.; Kim, B.J.; Kim, J.-S.; Lee, J.E.; Choi, J.Y.; Jeon, E.-S.; Kim, S.J.; et al. A comprehensive overview of AL amyloidosis disease characteristics accumulated over two decades at a single referral center in Korea. Int. J. Hematol. 2023, 117, 706-717. [CrossRef]
- Shcolnik Szor, R.; Fernandes, F.; Martins Lino, A.M.; Oliveira Mendonça, L.; Salles Seguro, F.; Araujo Feitosa, V.; Bianchi Castelli, J.; Barbosa Jorge, L.; de Oliveira Alves, L.B.; Miranda de Menezes Neves, P.D.; et al. Systemic amyloidosis journey from diagnosis to outcomes: a twelve-year real-world experience of a single center in a middle-income country. Orphanet J. Rare Dis. 2022, 17,425. [CrossRef]
- Peña, C.; González, J.T.; López-Vidal, H.; Donoso, J.; Contreras, C.; Vergara, C.G.; Hojas, R.; Soto, P.; Correa, G.; Valjalo, R.; et al. AL amyloidosis in the Chilean public health system: a pending debt. Multicenter study of the Chilean Monoclonal Gammopathies Cooperative Group. Rev. Med. Chile 2019, 147, 1239-1246.
- Kharoubi, M.; Bodeza, D.; Bezard, M.; Zarouia, A.; Galat, A.; Guendouz, S.; Gendre, T.; Hittinger, L.; Attias, D.; Mohty, D.; et al. Describing mode of death in three major cardiac amyloidosis subtypes to improve management and survival. Amyloid 2022, 29, 79-91. [CrossRef]
- Barret, C.D.; Dobos, K.; Liedtke, M.; Tuzovic, M.; Haddad, F.; Kobayashi, Y.; Lafayette, R.; Fowler; M.B.; Arai, S.; Schrier, S.; et al. A Changing Landscape of Mortality for Systemic Light Chain Amyloidosis. J. Am. Coll. Cardiol. 2019, 7, 958–966. doi.org/10.1016/j.jchf.2019.07.007.
- Quock, T.P.; D’Souza, A.; Broder, M.S.; Bognar, K.; Chang, E.; Tarbox, M.H. In-hospital mortality in amyloid light chain amyloidosis: analysis of the Premier Healthcare Database. J. Comp. Eff. Res. 2023, 12, e220185. [CrossRef]
- Chang-Chan, D.-Y.-L.; Ríos-Tamayo, R.; Rodríguez Barranco, M.; Redondo-Sánchez, D.; González, Y.; Marcos-Gragera, R.; Sánchez, M.J. Trends of incidence, mortality and survival of multiple myeloma in Spain. A twenty-three-year population-based study. Clin. Transl. Oncol. 2021, 23, 1429-1439. [CrossRef]
- Muchtar, E.; Gertz, M.A.; Kumar, S.K.; Lacy, M.Q.; Dingli, D.; Buadi, F.K.; Grogan, M.; Hayman, S.R.; Kapoor, P.; Leung, N.; et al. Improved outcomes for newly diagnosed AL amyloidosis between 2000 and 2014: cracking the glass ceiling of early death. Blood 2017, 129, 2111-2119. [CrossRef]
- Dhaliwal, J.S.; Hussain, F.; Ahmed, H.; Khan, A.T.M.A.; Aslam Khan, A.; Asghar Memon, M.; Arshad, M.; Maisum Mehdi, S.; Toussf Hussain, A.; et al. Demographic and regional trends in systemic and cardiovascular amyloidosis-related mortality among older adults in the United States from 1999 to 2020. Intern. Emerg. Med. 2025. [Epub ahead of print]. [CrossRef]
Figure 1.
Risk factors associated with systemic AL amyloidosis. Gen Sus: Genetic susceptibility, Fam Hist: Family History, Prec Dis: Precursor disease.
Figure 1.
Risk factors associated with systemic AL amyloidosis. Gen Sus: Genetic susceptibility, Fam Hist: Family History, Prec Dis: Precursor disease.
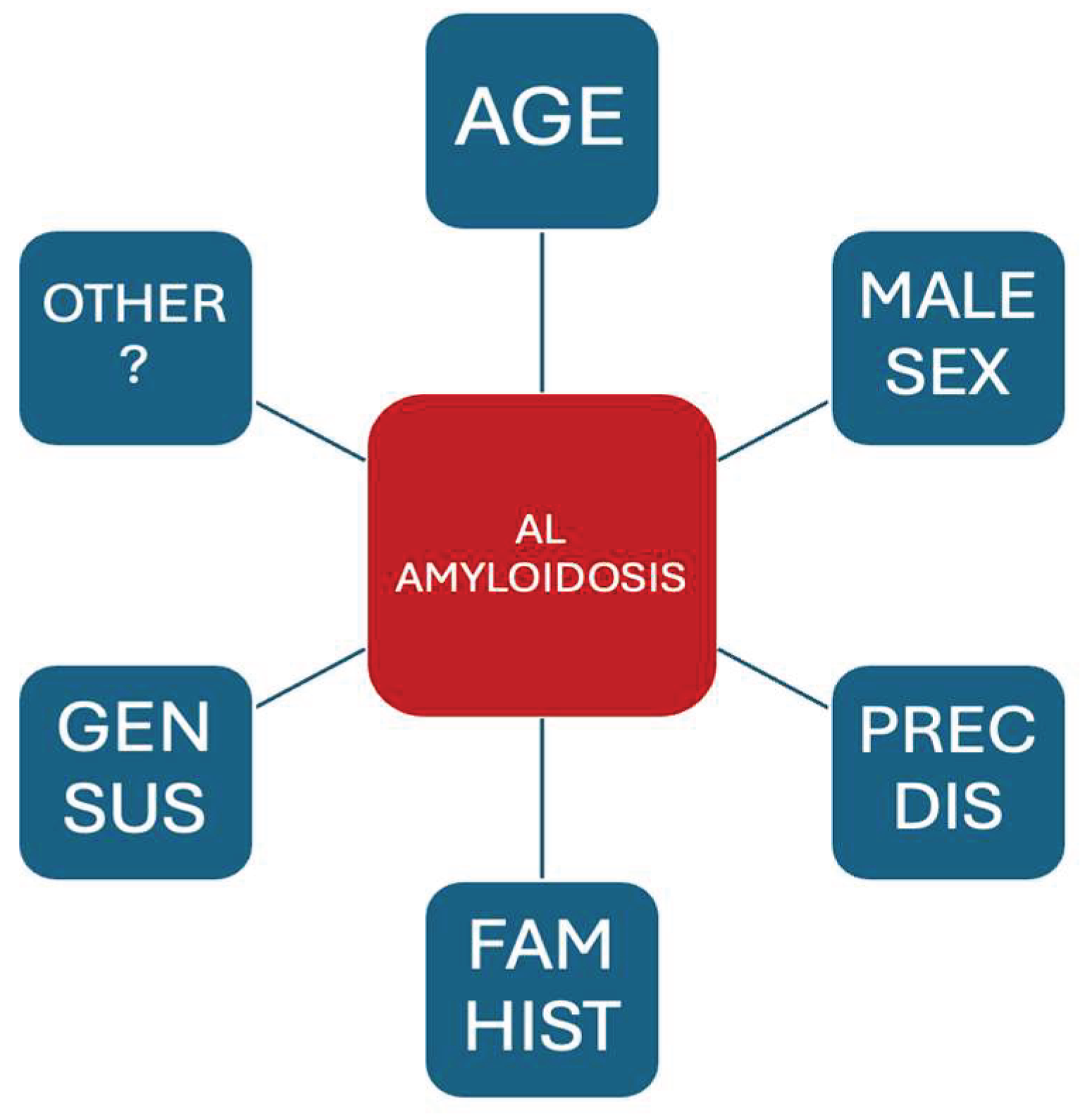
Figure 2.
Basic epidemiological information on systemic AL amyloidosis. EM: early mortality, F: female, INC: incidence, M: male, OS: overall survival, PMP: cases per million population, PREV: prevalence.
Figure 2.
Basic epidemiological information on systemic AL amyloidosis. EM: early mortality, F: female, INC: incidence, M: male, OS: overall survival, PMP: cases per million population, PREV: prevalence.
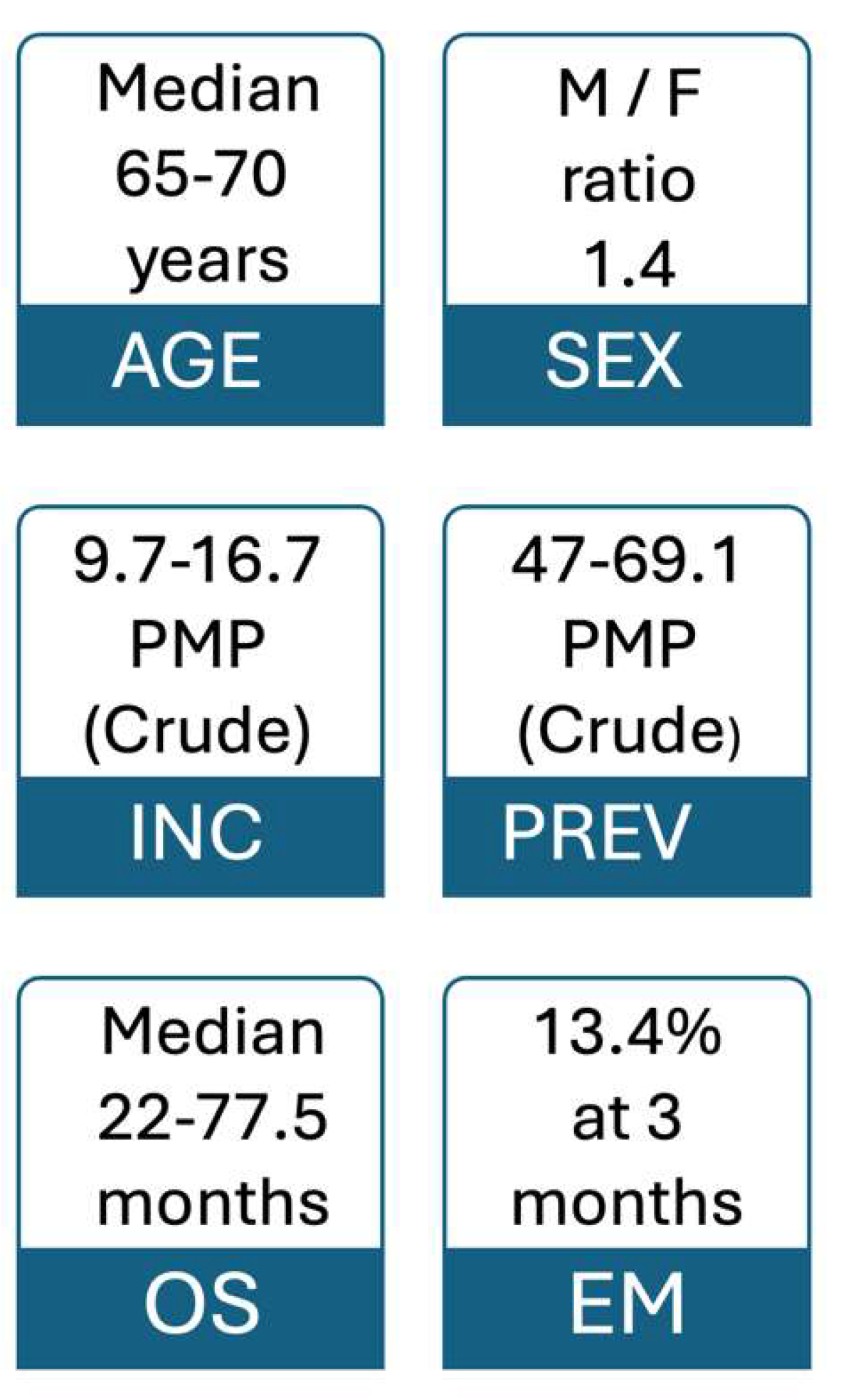

Table 1.
Incidence rates of systemic AL amyloidosis in population-based studies.
| Author/Year | Period | n | Incidence Rate |
|---|---|---|---|
| Duhamel, S. et al., 2017, France [38] | 2012-2016 | 46 | 12.5 c |
| Kyle, R.A. et al., 2019, USA [29] | 1990-2015 | 35 | 12 a |
| Zampieri, M. et al., 2021, Italy [40] | 2000-2019 | 281 | 9.2 nd |
| Hou, H.-A. et al., 2022, Taiwan [42] | 2016-2019 | 841 | 5.3-5.7 a |
| Mellqvist, U.-H. et al., 2023, Sweden [28] | 2011-2019 | 846 | 10.5-15.1 c |
A: adjusted, c: crude, n: number of patients, nd: not determined.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



