Submitted:
22 April 2025
Posted:
23 April 2025
You are already at the latest version
Abstract
Obesity has a complex and incompletely understood pathogenesis, with an underlying interplay between our genetic architecture and obesogenic environment. The public understanding of the development of obesity is shrouded in myth with widespread societal misconceptions, often promoted by (or at least insufficiently countered by) the mass media. This includes ideas that weight gain and obesity stem from human vices and ‘sins’ such as greed, gluttony and sloth. Body Mass Index (BMI) is a highly heritable trait. However, despite reports from recent ‘genome-wide association studies’, only a small proportion of the overall heritability of BMI is known to be lurking within the human genome. Other non-genetic heritable traits may contribute towards BMI. The gut microbiome is an excellent candidate, with complex interlinks with hypothalamic control of appetite and metabolism through both direct and indirect routes that implicate entero-endocrine, autonomic, and neuro-humeral pathways. Furthermore, recent evidence promotes a trans-generational transmission of the gut microbiome from the mother to offspring via both vaginal delivery and breast-feeding. The latter implicates a maternal ‘entero-mammary pathway’ in which the maternal gut microbiota translocate across the gut wall and are transported endogenously to the breast milk. Although maternally derived gut microbiota tend to have a permanence within the gut, other microbiota manifest mutability that responds to changes in lifestyle and diet. We should all strive to optimise a healthy lifestyle and diet that is replete with varied and unprocessed plant-based foods to nurture our gut microbiome. Women of reproductive age should optimise their gut microbiome, particularly pre-conception, ante- and postnatally to enable the establishment of a healthy gut microbiome in their offspring from birth. Finally, we should redouble our efforts to educate the populace on the pathogenesis of obesity, and the role of heritable factors such as genetics and the gut microbiome. Such understanding and insights would help to promote the widespread adoption of healthy lifestyles and diet but also help in the transition from our current dispassionate and stigmatised societal approach to obesity, to one that is epitomised by understanding, support, and compassion towards people living with obesity.
Keywords:
gut microbiota
; BMI
; heritability
; appetite
; Metabolism
1. Introduction
The effective prevention and management of any disease usually requires deep knowledge and insight into its underlying aetiopathogenesis, and the design of effective preventive and therapeutic strategies that target key pathways and nodes within this pathogenic arena. In the case of monogenic conditions, the underlying aetiopathogenesis is often clearly understood, serving as a beacon for potential therapeutic strategies. Furthermore, with the emergence of genetically targeted therapies delivered by adenoviruses and the like, it is likely that in the future monogenic conditions as a group will be amongst the most treatable of diseases, even curable in some cases. At the other end of the pathogenic spectrum sit the many chronic diseases that typify our modern-day era, which far from originating from a single gene mutation, have complex and multifaceted causal pathways, implicating interplay between genetic and environmental components, and which in many cases are incompletely understood. Obesity is one such disease. Unfortunately, the complex and opaque aetiopathogenesis of conditions like obesity poses unique challenges in the search for effective preventive and management strategies.
Obesity is the most important health crisis of our time and is classified as a non-infectious global pandemic by the World Health Organisation [WHO]. The global prevalence of obesity exceeds 890 million, and overweight affects 2.5 billion adults [1]. The dysmetabolic sequelae of obesity stem from its association with insulin resistance [IR], particularly in the context of visceral adiposity and ectopic fat deposition [2]. There are >50 obesity-related conditions that include most notably Type 2 Diabetes Mellitus [T2D], and others like hypertension, dyslipidaemia, Polycystic Ovary Syndrome [PCOS] [3,4], Obstructive Sleep Apnoea [OSA] and Metabolic-Associated Fatty Liver Disease [MAFLD] [5,6]. Furthermore, obesity is an important risk factor for many malignancies [7,8], and confers a substantial health economic burden on humanity stemming from direct and indirect costs [9]. Obesity impairs work productivity [10], psychosocial functioning [11] and overall wellbeing and quality of life.
An unfortunate consequence of any common modern-day chronic disease with a complex and incompletely understood underlying pathogenesis, is that this provides an ideal opportunity for the generation and propagation of myths. This is perhaps illustrated by obesity more so than any other chronic disease. The media portray obesity resulting from our lifestyle choices: It is assumed, we all have the choice of what and when to eat, and how much to eat, and it is the unique will of each of us to control our own appetite. The corollary of this oversimplistic and frankly false message is that weight gain therefore results from an inability to control one’s own appetite through vices like greed and gluttony, and this is compounded perhaps by laziness and an unwillingness to engage in physical exercise and activity. This is a common myth that is widespread within our society and is even shared by some healthcare professionals. In the context of a complex and incompletely understood pathogenesis, it is easy to understand the popularization of such a myth. Furthermore, as with many conspiracy theories in our modern-day world, it is easy to see the attraction of such a myth, providing a simple explanation for weight gain and obesity that everyone can understand and relate to, that implicitly and conveniently apportions blame to the individual living with obesity, and which is intellectually lazy and requires little effort to understand. As humans, we are generally averse to uncertainty and unknowns, and we seem much happier living with a simple explanation for phenomena such as obesity, even if this explanation is clearly false. [Human history is littered with such examples!].
It is our view that the widely held misconception and myth regarding the pathogenesis of weight gain and obesity outlined here, with its inherent blame and focus on human vices, has contributed towards a widespread dispassionate perspective on obesity, and its associated stigma [12]. Furthermore, this obesity myth also likely contributes towards a relative lack of funding for effective therapies for obesity. This is particularly evident in recent times, with a gaping disconnect between the unprecedented interest and development of novel incretin-based pharmacotherapies for obesity [with ‘big pharma’ and >200 biotechnology industries globally interested in the obesity space] and the relative lack of funding and reimbursement to support the widespread administration and adoption of such therapies at a community-focused level [both within NHS and healthcare settings globally]. Whilst focus on treatment strategies for obesity is important, we argue that the development of a clear understanding and insight into the pathogenesis of obesity is also important to guide future preventive and therapeutic strategies, but also to help to address the widespread misconceptions of obesity within our society, to reduce and eliminate stigma, and ultimately to foster a more compassionate approach towards people living with obesity.
Whilst much published work highlights the obesogenic environment [13], in this concise narrative review we focus on the heritability of Body Mass Index [BMI] to illustrate that obesity has an important genetic component, an insight that is often neglected by and omitted from the popularized myth of obesity pathogenesis outlined above. We discuss the complex interlinks between the gut microbiota and the central hypothalamic control of appetite and metabolism. We look beyond the human genome to explore the hypothesis that the missing heritability of BMI from reported ‘Genome Wide Association Studies’ [GWAS] in obesity, stems in part from the heritable gut microbiome and its associated vast metagenome [Figure 1]. Finally, we consider the implications of the dynamicity and mutability of the gut microbiome as a promising therapeutic target for the prevention and management of obesity, and even as a possible future maternal pre-conception strategy to help protect offspring from the vicissitude of future weight gain and obesity development.
2. Methodology
We performed a narrative review of the current literature, using PubMed for this purpose. The search terms were as follows: ‘Body Mass Index; genetics; heritability; gut microbiota; appetite; vaginal delivery; breast feeding’. We only considered articles written in English, and there was no restriction on the date of publication.
3. The Heritability of BMI
Heritability refers to the quality of a characteristic being transmissible from parent to offspring. Technically the term ‘heredity’ refers to the specifics of genetic transmission between generations. Conversely, the term ‘heritability’ has a broader meaning, and encompasses the transmission of a biologically interesting phenotype from parent to offspring, and to quantify this level of predictability. Any biological phenotype that has multiple contributing factors to its expression [including genetic and environmental factors] tends to assume a normal distribution, common examples being height and BMI. The heritability of such traits refers numerically to the variation within that normal distribution that is due to biological transmission from parent to offspring. Given the obvious importance of genetics as a transmissible biological trait, and the relatively recent emergence of the field of epigenetics and metagenomics, traditionally the terms ‘heritability’ and ‘heredity’ have tended to be used synonymously, and heritability to refer only to genetic factors. However, it is important for our discussion within this review to understand and appreciate that heritability can include other biologically transmissible traits that are separate from the human genome.
To gain a deeper understanding and insight into the pathogenesis of obesity, it is essential to explore the heritability of BMI. Identification of genetic and other biological contributors to weight gain and obesity would provide therapeutic targets for the prevention and management of obesity. Furthermore, such insights would help to address the widely believed mythical explanation for obesity outlined above, in which lifestyle choices and human vices account solely for weight gain and obesity [and independent from any genetic or other biological effects]. Any heritability of BMI would argue strongly against such a naïve and simplistic model of weight gain but rather support obesity as a biological disease that is hard-wired in our genetics, and perhaps in our other heritable biological traits. Some of our best evidence for the heritability of BMI stem from twin studies. In one such study from the ‘Chinese National Twin Registry’, 1421 twin pairs were included, and the heritability of BMI was calculated at 72% [14]. Interestingly, in this study the heritability of both BMI and cardiometabolic traits declined with age, with environmental factors assuming a relatively greater role than genetics in older people [14]. Regarding the phenotypic correlations between BMI and cardiometabolic traits, genetic factors played a consistently greater role than environmental factors [14]. In another population study using data from medical screening of military personnel in Israel, including >447,000 offspring, there was a calculated heritability of 39% between mid-parental and offspring BMI [15]. The clear conclusion from these and other population-wide genetics studies reported in the literature is that BMI, and by implication obesity, is highly heritable. Overall, the heritability of BMI is estimated to be some 40-50% [16]. However, the heritability of BMI varies according to the BMI subgroup within the population. For normal weight individuals, the heritability of BMI is around 30% [16]. However, in the subgroup of people with obesity, heritability of BMI is 60-80% [16]. Furthermore, fat distribution and the presence of ectopic fat are also heritable traits [around 30-55%] [16].
A detailed discussion of the genetic architecture of obesity is beyond the scope of this review and has been covered elsewhere [17,18]. Here, we provide a brief overview. There are two main elements to this discussion. Monogenic forms of obesity are rare and stem, by definition, from a mutation in a single gene that often have major adverse effects on the control of appetite and metabolism and often manifest with substantial weight gain and obesity from an early age. We have identified mutations in at least 15 genes, mainly those that result in deficiencies in the leptin-melanocortin signalling pathway within the hypothalamic appetite centre [16]. The study of the genetics of monogenic obesity has provided much insight into the neurobiological control of appetite and metabolism. In this review, we focus on the second element of obesity genetics: polygenic forms of obesity that are common and underlie the high prevalence of obesity within the populace. Within the realm of polygenic obesity, there is heterogeneity with distinct phenotypic subtypes based on measures of body composition, insulin sensitivity, physical fitness, glycaemia, and cardiovascular risk [19]. However, for this review we consider polygenic obesity as a single entity.
GWAS studies of obesity have identified >1,000 gene variants that impact on BMI, with most alleles that influence BMI only contributing a few grams more, or less, to body weight [16,18]. Interestingly, alleles that promote obesity tend to have a greater effect in those individuals with a propensity for weight gain and obesity and exert minimal effects in individuals of normal weight [16]. Therefore, the penetrance of BMI-influencing alleles seems variable according to BMI although it is not known whether the effect size of BMI-increasing alleles precedes the onset of weight gain and obesity, or is enhanced by the obese state [16]. Indeed, gene-environment interaction analyses reveal that our obesogenic environment could be amplifying genetic risk for obesity [17]. Our current genetic model of polygenic obesity promotes heritability of BMI being influenced by thousands of gene variants [16], each of which individually has a relatively small effect on body weight, but their overall cumulative effect underlies the phenotypic expression of BMI in each of us. For most GWAS-identified loci that influence BMI, we lack understanding of causality [translation from variant to function] [18]. However, insights from GWAS reveal that gene variants that impact on total body mass are expressed primarily within the Central Nervous System [CNS], particularly the hypothalamic centres that regulate the control of appetite and metabolism [17]. Conversely, gene variants that impact on fat distribution are enriched within the adipose tissue [17].
In recent times, data from GWAS have transformed our understanding of polygenic obesity and contributed towards our insight into the central neurobiological control of appetite and metabolism. However, despite the power of recently reported GWAS on tens of thousands of participants, the gene variants implicated in BMI control only account for a small proportion of the overall heritability of BMI. The ‘polygenic score’ [PGS] is used to assess the genetic susceptibility to complex diseases such as obesity. Including data on >2M gene variants identified from GWAS, the PGS only explains 8.4% of the overall variation of BMI [18]. The low PGS for obesity is redolent of that for other complex diseases based on GWAS data, including for example Polycystic Ovary Syndrome [PCOS] [4,20,21], and T2D [22]. The question then is: where is all the missing heritability of obesity hiding? It is possible that the reported GWAS in obesity simply lack power and that some of the missing heritability is lurking within many gene variants with very small effect sizes that are only detectable with GWAS of much greater power [that would require hundreds of thousands or even millions of participants to detect]. Alternatively, epigenetic effects may contribute to the missing heritability. However, given that heritability refers the generational transmission of biological traits [not limited to genetic factors], it is important for us to consider non-genetic factors that may mediate part of the missing heritability of BMI. The gut microbiome is an excellent candidate.
4. The Gut Microbiome and the Metagenome
Human cellular function within well-defined organs and systems forms a firm foundation for our traditional model of human physiology. In recent times, our understanding of physiology has morphed from this traditional human-centric perspective to a vista that accommodates a complex interaction and symbiotic co-evolution over eons of hominid evolution, between human cells and 100 trillion foreign microbes, the latter of which vastly outnumber our own cells. These foreign microbes are referred to collectively as the ‘microbiome’. Although terms like ‘virome’ and ‘mycobiome’ refer to the collection of viruses and fungi, respectively, found in or on an organism, the microbiome is an umbrella term that includes all prokaryotic cells and viruses that associate with an organism, and from our perspective, the human body [23,24]. The microbiota accumulate on any epithelial or endothelial surface that is exposed to the environment, including the genitourinary tract, respiratory epithelia and skin [25]. However, the vast majority of the human microbiota reside within the gut, and the majority of these [around 70%] exist within the colon [26]. The gut microbiome play a central role in the regulation of the immune and inflammatory systems, and as such play a central pathogenic role in the development of many modern-day chronic illnesses. These include chronic inflammatory and auto-immune conditions, atopies, food intolerances, and possibly cardio-metabolic conditions [27], and neuro-psychiatric disorders such as Parkinson’s Disease [28], Autism Spectrum Disorder [29], chronic pain [30] and disorders of mood and affect [31]. A healthy gut microbiome [eubiosis], outside of the neonatal period, is typified by a diverse and rich array of microbes, achieved and maintained through a high-fibre plant-based diet [26]. Conversely, an unhealthy gut microbiome [dysbiosis] is impoverished and imbalanced, and may also associate with ‘leakiness’ of the gut epithelium, with the propensity for gut microbes [or components of gut microbes] to translocate across the gut wall into the vascular system [endotoxinaemia] [32]. It is important to note that the term, ‘leaky gut’, is poorly defined and should not be confused with actual defects in the gut barrier that occur in the context of chronic inflammatory bowel diseases. Furthermore, there is much public misinformation and advertising that promote the use of various prebiotic, postbiotic and symbiotic supplements to ‘heal leaky gut’ with little supportive evidence. A challenge for the future will be to better define ‘leaky gut’ and to determine its effective and evidence-based management.
The metagenome refers to the collective genome of the human microbiome. Given that the microbiome vastly outnumbers our own cells, it follows that the metagenome is orders of magnitude larger than the human genome [that contains around 20,000 genes]. Metagenomics refers to the techniques [including massive sequencing platforms] used to identify a microbial population through analysis of its collective genetic material [33]. When the human genome project was in its infancy, our understanding of the gut microbiome was also in its infancy, and we had incomplete understanding of the role of the gut microbiome for health and chronic illness. Future genetic studies should include both the human genome and metagenome, although the latter will bring many challenges, including the sheer number of microbiota and their combined genomes, but also the dynamic nature of the gut microbiome, as opposed to the fixed and immutable nature of the human genome [excepting acquired somatic mutations]. Future genetic studies should also focus on epigenetic changes within our genome [including epigenetic programming during fetal development influenced by the maternal environment and metabolic status], that likely play an important role in the development and heritability of obesity.
5. Interlinks Between the Gut Microbiome and Central Appetitive and Metabolic Control
As outlined, BMI is heritable. Our genetic architecture is a key mediator of such heritability, particularly regarding the expression of gene variants that influence the central hypothalamic control of appetite and metabolism. In recent times there has been a renaissance in our understanding and insight into the control of appetite and metabolism [stemming mainly from rodent-based studies], going beyond human genetics, with the gut microbiome looming as a cynosure [34]. Broadly, the gut microbiota [and/or their metabolic by-products] communicate with the brain both indirectly [via entero-endocrine mechanisms and autonomic afferents within the gut wall itself], and directly via the bloodstream. This latter mechanism requires the translocation of components of the gut microbiota [endotoxins] and/or their metabolic by-products across the gut wall and into the bloodstream, enabling the exertion of both peripheral and central effects. As such, the gut wall forms a boundary between the gut microbiota and human cells [23]. Gut leakiness enables translocation of microbial wall components such as lipopolysaccharides [LPS] or microbes directly. Microbial metabolic components including amino acids, lipid species and many more which may be transported by intestinal transporters. These metabolites probably contribute to beneficial metabolic effects even though some are unhealthy. To defend and protect the gut wall, there is a layer of mucus [mucin] [23]. In one fascinating human-based study, there was an inverse correlation between the thickness of the colonic mucus [measured as the distance between the gut epithelial lining and the gut microbiota on colonic biopsies] and metabolic measures of BMI and glycaemic indices [HbA1c and fasting glucose levels] [35]. These data are consistent with the gut wall mucus layer having a protective effect on gut wall leakiness and reducing propensity for weight gain and obesity [and dysmetabolic effects] through optimized central hypothalamic control of appetite and metabolism.
Our gut microbiome [and its impact on the central control of appetite and metabolism] are influenced by our diet, particularly our intake of dietary fibre [36,37]. Through its effects on our gut microbiota, our intake of dietary fibre may impact on propensity for weight gain, BMI and insulin sensitivity. Indeed, there is a correlation between body weight and composition of the gut microbiota, with lean and obese people having distinct gut microbiota compositional phenotypes [38]. In obese rodent models, weight loss associates with concurrent changes in gut microbiota composition towards a lean phenotype [38,39]. Furthermore, rodent-based studies involving fecal transplantation from obese or lean mice to germ-free mice reveal that only the recipients of gut microbiota from obese mice exhibited a subsequent increase in fat mass under isoenergetic conditions [39]. These data provide proof of concept that the gut microbiota influence fat mass, at least in rodents. In a human-based study, a high-fibre [oligofructose] diet reduced the content of gram-negative gut bacteria and body weight, whereas a high-fat diet increased the content of gram-negative bacterial LPS within the gut [40]. In the same study, continuous subcutaneous infusion of LPS for 4-weeks increased body weight, hepatic fat content, and markers of both insulin resistance and inflammation, comparable to that following a high-fat diet [40]. These data reveal that dietary fibre consumption influences the composition of the gut microbiota [and the relative proportions of gut microbiota species], thereby linking the gut microbiota with insulin sensitivity, inflammation and the central control of appetite and metabolism [38].
Recently, we published an overview of the complex and bi-directional interlinks between the gut microbiome and the brain [23]. We have also published discussion of the bi-directional interlinks of the gut microbiome with the liver [5] and the peripheral immune-inflammatory systems [13,23]. In this subsection, we provide a key summary of the evidence that links the gut microbiome [and/or their metabolic by-products] with central hypothalamic control of appetite and metabolism, implicating entero-endocrine, autonomic, and neuro-humeral pathways [34,41].
5.1. Entero-Endocrine Pathway
The gut microbiome impacts on the hypothalamic control of appetite and metabolism via effects on the modulation of hormonal signals from entero-endocrine cells within the gut wall [23]. Some of these effects are indirect and stem from metabolic by-products of the gut microbiome [released during their utilization of food for energy]. These metabolic by-products include tryptophan metabolites, secondary bile acids, and Short Chain Fatty Acids [SCFAs] [34,42]. SCFAs are precursors of odd-chain fatty acids [bioactive compounds], and as such these lipid species can be used as biomarkers for fibre-rich diets [43,44,45]. SCFAs are produced by caecal anaerobic microbes such as Enterococcus in the process of fermentation of dietary fibre [23]. SCFAs stimulate G protein-coupled receptors [GPRs] including GPR41 and GPR43 expressed on entero-endocrine cells [46]. In a rodent-based study that compared wildtype with GPR41 knockout mice, SCFA stimulated GPR41 receptors resulting in enhanced secretion of Peptide YY [PYY, a potent appetite-suppressant gut-derived incretin-like hormone] only in the wildtype mice [47].
To complement the effects on GPR41, SCFAs also stimulate GPR43 resulting in the release of glucagon-like peptide-1 [GLP-1] that induces satiety and thereby supports hypothalamic appetite control [48]. Finally, SCFAs may translocate across the gut wall into the systemic circulation, and from there cross the blood-brain barrier into the brain parenchyma to influence the hypothalamic control of appetite and metabolism [49,50]. Through indirect effects on the entero-endocrine release of key incretin hormones like GLP1, and through direct central effects, SCFAs may impact meaningfully on the hypothalamic control of appetite and metabolism and thereby represent a biological contributor to BMI. To corroborate this assertion, in a study on obese women undergoing bariatric surgery, colonic levels of Enterococcus were associated with appetitive inhibition [51]. Furthermore, in a randomised-controlled crossover study on overweight human adults, compared with inulin ingestion [control group], the ingestion of propionate [a common SCFA released by human gut microbiota] resulted in early postprandial release of the intestinal hormone PYY, and the incretin hormone GLP-1 from colonic entero-endocrine cells and reduced caloric intake. Metabolic benefits of regular propionate ingestion occurred over 24-weeks, with reduced hepatic lipid content and intra-abdominal adipose tissue volume, preserved insulin sensitivity, and significant weight loss [52]. However, the role of PYY in appetite regulation and its association with human body weight is controversial [53,54]. Furthermore, inulin [as a prebiotic] is fermentable and may also elicit metabolic effects of SCFAs over a longer time-period [55]. Therefore, inulin is not an ideal control comparator for propionate ingestion.
Despite the suggested metabolic benefits of SCFAs demonstrated in some rodent-and human-based studies, there is controversy in the literature. SCFAs rely upon fermentation of soluble dietary fibre [23]. In the short-term, the ingestion of soluble [and highly fermentable] fibre improves insulin sensitivity and has some short-term glycometabolic benefits with anti-inflammatory effects [56]. However, the longer-term metabolic effects of dietary soluble fibre are less clear [56]. Conversely, there is much evidence to support the metabolic benefits of ingested insoluble cereal fibres [little-fermentable oat extracts, wheat and whole grain products [57]], including improved insulin sensitivity and reduced risk for the development of T2D [38,58,59,60,61,62]. However, it should be noted that the beneficial long-term effects of insoluble cereal fibres are only observed in reported observational studies. Furthermore, interventional studies are sparse, particularly those that compare the metabolic effects of ingested insoluble with soluble fibre, such as one of the longest duration rodent-based studies reported on to date, a 45-week study of an isoenergetic diet that compared the metabolic effects of additional dietary soluble fibre versus insoluble cereal fibre, with a matched high-fat diet in obesity-prone mice [63]. In this study, compared with the insoluble fibre mice, those fed soluble fibre had a greater production of SCFA via colonic fermentation, a significant reduction in energy loss via the faeces, a significant increase in body weight and elevated markers of insulin resistance [63]. Furthermore, gene expression analysis in white adipose tissue revealed a significant increase in the levels of the fatty acid target G-protein coupled receptor-40 in the mice that were fed soluble fibre [63]. Conversely, liver gene expression in the insoluble fibre group showed a pattern consistent with increased fatty acid oxidation [63]. Therefore, there were clear differences in the metabolic effects [including body weight and measures of insulin resistance] of soluble versus insoluble dietary fibre added to a high-fat Western-style diet in obesity-prone mice [63].
Therefore, although dietary intake of soluble fibre results in significantly increased production of SCFAs that may have beneficial short-term metabolic benefits, these may be outweighed, at least in rodents, by adverse metabolic outcomes over the longer-term. Arguably, in the rodent-based study outlined above [63], there was a relative overdose of dietary fibre and this may have explained the rather atypical weight-gain for the mice assigned to the soluble fibre group mice [the dosage of fibre was 100g per kg of food ingested, and in humans this would represent around 200g of soluble dietary fibre per day]. Interestingly, a somewhat similar study from China assessed the metabolic effects of dietary ingestion of 40g of resistant starch in humans with low baseline dietary fibre intake, revealing body weight loss under these conditions [64]. Finally, in a 67-week duration rodent-based study, adolescent rats fed guar gum or guar by-product diets gained less weight than those fed cellulose, and only those fed guar gum had improved carbohydrate tolerance [65].
In addition to the stimulated release of GLP1 [via SCFAs], the gut microbiota may also influence the release of another incretin hormone, glucose dependent insulinotropic polypeptide [GIP] [66]. In recent times, there has been much interest in the pharmacotherapeutic potential of GIP [in combination with GLP1 agonism] in the context of obesity and T2D [67]. GIP may exert some of its metabolic effects indirectly via the suppression of ghrelin release [68]. However, unlike GLP1, insights from rodent-based models reveal that the direct effects of GIP may be less desirable metabolically, including the mediation of energy intake and markers of insulin resistance in response to carbohydrate ingestion [69]. Indeed, such metabolic benefits of suppressed GIP release provides a rational for the ‘Endo-barrier’ as a weight-loss intervention [through creating a physical barrier between nutrients and the duodenal wall] [70]. In a rodent-based model, C57BL/6 mice and GIP-receptor knockout mice were exposed to ovariectomy [simulating menopause] or sham operation and observed for 26-weeks for changes in metabolic regulation and body weight [71]. The ovariectomized wild-type mice gained weight with increased fat mass and insulin resistance as expected, through effects of reduced estrogen on the regulation of body weight, mediated in part by GIP signalling [71]. Conversely, the ovariectomized GIP-receptor knockout mice were protected from these adverse metabolic outcomes with significant reduction in food intake, stemming from changes in the neurobiological regulation of the hypothalamic appetite centre [71]. This may provide one explanation for the weight neutrality of dipeptidyl peptidase 4 [DPP4] inhibitors, that inhibit the breakdown of endogenous GLP1 and GIP.
5.2. Autonomic Pathway
Bi-directional signalling via the vagus nerve connects the gut microbiota with the brain. Such signalling may impact on the hypothalamic control of appetite and metabolism indirectly via effects on mood and the functioning of the hypothalamo-pituitary adrenal [HPA] axis. In a rodent-based study, chronic ingestion of a lactobacillus strain resulted in a reduction of depression- and anxiety-related behaviours, reduced stress-induced activity of the HPA axis, and regional changes in the expression of Gamma-Amino Butyric Acid [GABA] neurones within the brain. The absence of such effects in vagotomised mice supports an important role for the vagus nerve in the mediation of signals between the gut microbiota and the brain [72]. The vagus nerve also links the gut microbiota with the liver, that in turn communicates with the hypothalamus to control appetite, feeding behaviour and metabolism through myriad ways that include the release of hepatokines [73].
5.3. Neuro-Humeral Pathway
In addition to changes in entero-endocrine and autonomic nervous pathways, the gut microbiota may also influence the levels of important neurotransmitters and receptivity of neuroreceptors within the brain, that regulate key physiological and psychological processes and emotional behaviours [including depression and anxiety] [72,74,75]. GABA is one such major inhibitory neurotransmitter [27,72]. In a rodent-based study, chronic ingestion of Lactobacillus rhamnosus [JB-1] compared with control-fed mice, resulted in regionally dependent changes in the expression of GABA receptors within the brain that in turn associated with reduced depression- and anxiety-related behaviour [72]. In addition to GABA, the gut microbiota influence the expression of other neuroreceptors within the brain such as those for N-methyl-D-aspartate [NMDA, mediating effects of the excitatory neurotransmitter glutamate] [75], serotonin receptor 1A [74] and tryptophan [as a precursor to the neurotransmitter serotonin] [76]. Given the complexities of the neurobiology of central control of appetite and metabolism, including the impact of mood and emotions [77,78], it is likely that such neuro-humeral changes in response to the gut microbiota mediate changes in body weight and BMI.
6. Heritability of the Gut Microbiome
The gut microbiome can only contribute towards the heritability of BMI if the gut microbiome is itself heritable. In this section, we explore the available evidence to support the heritability of the gut microbiome, and its transmissibility between generations. This discussion will include a role for vaginal delivery in the establishment of the gut microbiota in the newborn, and evidence that breast milk contains maternal gut microbiota as a means of seeding the gut microbiota of early infancy. We also consider the shared food environment of offspring and parents that influences the infant gut microbiota. Although such a shared environment would not strictly constitute a ‘heritable’ mechanism, it may nonetheless help to explain some of the familial concordance of BMI.
6.1. Vaginal Delivery in the Establishment of the Gut Microbiome of the Newborn
Recently, there has been much interest in the impact of mode of delivery on the initial establishment of the gut microbiota and future health implications. Caesarean section [C-section] eliminates contact of the newborn with maternal microbes, and in this scenario the newborn gut microbiota are derived from the environment [including possible pathogens], with potential negative implications for long-term metabolic and immunological functioning [79]. [Intrapartum antibiotic exposure can also diminish the transfer of maternal gut microbiota to the offspring during vaginal delivery [79]]. Several large cohort studies and meta-analyses have revealed an association between C-section birth and long-term health problems, including obesity and chronic inflammatory and immune diseases that may extend into adulthood [79,80]. Although confounding factors exist, given the important effects of the gut microbiota on physiological and immunological development, these data are consistent with a lack of protection from maternally derived gut microbiota [during vaginal delivery] for offspring born through C-section [79].
In contrast to C-section, during vaginal delivery there is oral exposure of the newborn to the maternal microbiome. In addition to vaginal microbes and skin commensals, this also includes oral exposure to the maternal gut microbiome [81]. In a study that used quantitative PCR, there were significant differences in the composition of the gut microbiota in young adults based on their mode of delivery [vaginal vs C-section] [79,82]. In a systematic review that explored mode of delivery on the gut microbiota of newborn infants, it was shown that compared with C-section, vaginal delivery resulted in a greater overall diversity and colonization pattern of the infant gut microbiota [including significantly more Bifidobacterium and Bacteroides genera] during the first 3-months of life [83]. Bifidobacteria are important for early gut health and have widespread immunological effects that facilitate the normal development of the immune system [79]. Low gut microbial abundance of Bifidobacteria [especially B. longum] early in life, associates with an increased risk of allergic diseases later in childhood [84], and is predictive of adiposity later in life [79,85]. During vaginal delivery, maternally derived microbes colonize the infant gut permanently, whereas non-maternal gut microbes are transient typically [79,86]. The permanence of maternally derived microbes within the gut suggests compatibility between maternal and infant gut microbes that may be mediated genetically through immune factors and gut mucus composition [79].
The published data outlined here support a role for vaginal delivery on the early establishment [and permanence] of the infant gut microbiota with future health implications. Furthermore, vaginal delivery enables the transmission of maternal gut microbiota to her offspring, therefore supporting the heritability of gut microbiota in vaginally delivered infants.
6.2. Breast Feeding as a Means of Seeding the Gut Microbiome of Early Infancy
Although traditionally considered as sterile, over the last two decades our understanding of human breast milk [HBM] constituents has been transformed by the identification of commensal bacteria [87], including lactic acid bacteria [LAB] genetically distinct from the LAB isolated from maternal breast skin and mammary areola [87,88]. Within healthy HBM, Staphylococcus and Streptococcus appear as universally predominant genera [87,89]. However, there is a rich diversity of the HBM microbiome including Firmicutes and Proteobacteria [87,90]. It is now widely accepted that HBM contains an abundance of diverse microorganisms [87,90], and that these play a key role in the colonization of the infant gut, and the establishment of the infant gut microbiome [87,91]. As such, the transmission of maternal microbiota to the offspring via HBM may represent a heritable biological trait. However, this would depend on the translocation of maternal gut microbiota via a ‘entero-mammary pathway’ [EMP].
The origin of the HBM microbiome is incompletely understood [87]. In addition to the EMP, another theory is that the HBM microbiome develops secondary to contamination from maternal surface skin and the infant’s oral cavity [87]. Early studies seemed to favour the latter hypothesis, based on similarities between the HBM microbiome and skin micro-organisms [such as Staphylococcus and Corynebacterium] [87,92,93]. However, there are marked differences between the microbiome of the skin and HBM [87,94,95]. Furthermore, anaerobic bacteria such as Bacteroides, Clostridium, Bifidobacterium and Parabacteroides are shared between HBM and infant faeces but are absent from adult skin [87,88,96]. Therefore, it seems unlikely that the HBM microbiome originates from the maternal breast skin and mammary areola. An EMP would implicate the translocation of maternal gut microbiota across the gut wall, followed by endogenous transfer to the mammary glands [87]. Consistent with this hypothesis is the presence of an entero-mammary circulation of IgA-producing cells [87,97], and the observation that dendritic cells can penetrate the gut wall and open tight junctions that then allow the translocation of the gut microbiota across the gut wall [87,98]. Furthermore, some microbiota species such as Bifidobacterium and Lactobacillus are shared between the maternal HBM and the infant feces [87,88,93,99]. There is also evidence for the presence of extracellular vesicles [EVs] derived from bacteria [especially Bifidobacterium and Lactobacillus] within HBM [87,100,101], that likely facilitate the microbiota colonization of the infant gut and may act as receptors for bioactive molecules in the host cell [87,100,102]. These observations are all consistent with the existence of a EMP that enables the transmission of maternal gut microbiota to her offspring, thereby supporting the heritability of gut microbiota in breast-fed infants. Future research should explore the exact mechanisms that underlie the EMP.
6.3. Shared Food Environment of Offspring and Parents and the Gut Microbiome
Heritability refers to the transmission of biological traits between generations. Heritability does not include environmental factors. However, it is known that environmental and lifestyle factors, particularly diet, influences the composition of the gut microbiome. Therefore, shared food environments between parents and offspring may account for some familial traits of the gut microbiota [103]. Indeed, in a three-generational cohort study based on a Dutch population, it was shown that around 48.6% of taxa within the gut microbiota were significantly explained by co-habitation [with only 6.6% of taxa being heritable] [104]. We reviewed the impact of diet on the gut microbiome in detail previously [23]. Here, we provide a summary.
In the industrialized era, western diets have changed radically, with diminished dietary fibre [from unprocessed plant-based foods], coupled with an abundance of ultra-highly processed foods, laden with additional fats and carbohydrates [37]. Evidence from rodent-based studies reveal that even within a single day, the gut microbiota alters in response to changes in dietary macronutrient intake [105]. Human-based studies show variable timeframes for diet to impact on the composition of the gut microbiota, including the absence of any significant changes in the gut microbiota in response to dietary changes [106], limited diet-related changes to the composition of the gut microbiota occurring over weeks or months [107,108,109], and changes to the gut microbiota manifesting over the short-term in response to changes in the diet [110]. In a well-phenotyped study using metagenomic sequencing, the ingestion of healthy and diverse plant-based foods was a key factor that promoted a significant association between gut microbes and specific nutrients and food groups [111]. Furthermore, the composition of the gut microbiota was predictive for multiple cardio-metabolic blood markers [111]. There is clear evidence from the literature that dietary fibre [including both fermentable [mostly soluble] and little-fermentable [often insoluble] fibres], derived from plant-based foods, play a key role in the establishment and nurture of a healthy gut microbiome and the promotion of health and wellbeing [37]. [soluble] fibres associate with Bacteroides species, and butyrate-producing bacteria [Clostridium leptum and Eubacterium rectale] [112,113]. Furthermore, the ingestion of complex carbohydrates promotes the growth of Bifidobacteria species [Bifidobacterium longum, Bifidobacterium breve, and Bacteroides thetaiotaomicron], each of which are favourable for health [114].
One caveat to a shared food environment between parents and offspring is that offspring [particularly older children and adolescents] often have diets and food preferences that differ from their parents [115]. However, co-habiting families do share their food and meal environments, and children model their own eating behaviours, food choices and taste preferences on that of their parents [116]. Therefore, even with some generational differences in food preferences, co-habitation likely results in at least a partial confluence of the gut microbiota between parents and offspring.
7. Conclusions and Future Directions
To summarise, common polygenic obesity, unlike monogenic conditions, has a complex and incompletely understood underlying pathogenesis, that reflects an elaborate interplay between our genetic architecture and modern-day obesogenic environment. BMI is a heritable trait, but GWAS studies have only identified a small proportion of the overall heritability of BMI. Given that heritability incorporates any inherited biological trait, it is important to search beyond the human genome to identify some of this missing heritability. It seems likely that the gut microbiome contributes towards the heritability of BMI [117,118] given its impact on the control of hypothalamic appetite and metabolic control through entero-endocrine, autonomic and neuro-humeral effects, and the maternal-offspring inter-generational transmissibility of the gut microbiome, at least in the context of vaginal delivery and/or breast feeding.
There are limitations to our outlined hypothesis. Our evidence for the impact of the gut microbiota on the hypothalamic control of appetite and metabolism stems primarily from rodent-based studies, with a relative lack of human-based studies. It is important not to over-extrapolate the implications of data between species, and it remains possible that the gut microbiota play less of a role in appetite and metabolic control in humans than in rodents, although we need to await further data from human-based studies to make further conclusions on this point. Furthermore, there are confounders that are implicit in the data outlined, including for example associations between mode of delivery and future risk of chronic illness and BMI. Our hypothesis is based on observational data, with obvious difficulties, hurdles, and challenges for any future randomised controlled trials in this field.
This review has focused on the gut microbiome as a heritable trait. Unlike the human genome, the gut microbiome is highly mutable and has inherent dynamicity that makes its study challenging. However, such dynamicity does not preclude an important role for the gut microbiome in the heritability of BMI, based on the evidence outlined that support a notion of the gut microbiome as a familial trait, particularly a tendency for permanence of maternally derived gut microbiota. Furthermore, the dynamicity and modifiability of the gut microbiome presents an opportunity for the future prevention of weight gain and management of obesity. Numerous lifestyle factors including physical activity, sleep, and stress influence the gut microbiome in important ways [23]. As outlined, dietary factors [particularly the ingestion of diverse and unprocessed plant-based foods] are also important for the nurturing of a healthy gut microbiome [23]. Beyond dietary and lifestyle optimization, it is interesting to speculate on potential future preventive and management strategies for obesity that focus on modifying the gut microbiome in some way. Currently, faecal transplantation [FT] is used within the NHS only for the management of patients with intractable colonic colonisation with Clostridium difficile [23,119]. There is compelling evidence from rodent-based studies for important effects of FT on both metabolic status and body weight [120]. Human-based studies have shown some favourable effects of FT on glucose tolerance, but disappointing effects on weight loss in recipients with obesity [120,121]. There is a need for further human-based studies to explore the potential metabolic benefits of FT [or other therapeutic strategies to transform the gut microbiota], including as a future weight-loss strategy. Furthermore, given the inter-generational transmissibility of the maternal gut microbiota to offspring during vaginal delivery and breast feeding, there is a case for optimization of the maternal gut microbiome pre-conception, analogous to our current approach to optimise maternal body weight and glycaemic control pre-conception.
To conclude, it is useful to provide learning points from our review. Our gut microbes influence our health and wellbeing through myriad mechanisms. Aspects of our gut microbiome manifest mutability, with influence from all aspects of lifestyle, particularly our diet. We should all strive to nurture our gut microbiome through healthy lifestyles and dietary intake of diverse, unprocessed plant-based foods. This should include the ingestion of fermented foods such as kefir, kimchi, sauerkraut and natto [122]. Other aspects of our gut microbiome manifest a degree of permanence, particularly our maternally derived microbiota from infancy. Given the heritability of our gut microbiome, at least in the context of vaginal delivery and/or breast-fed infants, it is important for reproductive age women to strive for optimisation of their gut microbiome through healthy lifestyle and dietary strategies [as outlined above] throughout pre-conception and extending into antenatal and postnatal periods. Finally, it is important that insights from our review are disseminated broadly throughout society, to address the widespread misconceptions and myths regarding the development of weight gain and obesity that are often promulgated and stigmatised by the mass media. Improved public understanding of the heritability of BMI, including the role of genetic factors and other biological and heritable traits such as our gut microbiome, would help to address societal myths and misconceptions regarding the origins of obesity. Such insights would also help to encourage and promote the widespread adoption of healthy lifestyles and diet. Finally, we live in a cruel and unfair society in which human myths abound [to a greater extent than what most of us appreciate], in which weight gain and obesity are often incorrectly attributed to human vices, redolent of a medieval mindset in which plague and other poorly understood diseases were attributed to the wrath of God as a punishment for human misdemeanors [with associated stigmatisation of such diseases]. Improved public understanding of obesity pathogenesis, including an appreciation of our genetic misalignment to our modern-day obesogenic environment, and that our BMI is largely biologically inherited through both our genetics and our gut microbiome, would help to foster cultural change apropos of societal attitudes towards people living with obesity. A cultural change in which there is vanquishment of obesity-related stigma and abuse, and a new dawn of understanding, support, and compassion.
Funding
This research received no funding.
Conflicts of Interest
None of the authors has any conflicts of interest.
References
- World Health Organization. Obesity and Overweight Factsheet 2024. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 15 December 2024).
- Fruh, S.M. Obesity: Risk factors, complications, and strategies for sustainable long-term weight management. Journal of the American Association of Nurse Practitioners. 2017, 29, S3–S14. [Google Scholar] [CrossRef]
- Barber, T.M.; Franks, S. Obesity and polycystic ovary syndrome. Clin Endocrinol [Oxf]. 2021, 95, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.M. Why are women with polycystic ovary syndrome obese? Br Med Bull. 2022, 143, 4–15. [Google Scholar] [CrossRef]
- Barber, T.M.; Kabisch, S.; Pfeiffer, A.F.H.; Weickert, M.O. Metabolic-Associated Fatty Liver Disease and Insulin Resistance: A Review of Complex Interlinks. Metabolites. 2023, 13, 757. [Google Scholar] [CrossRef]
- Barber, T.M.; Franks, S. The link between polycystic ovary syndrome and both Type 1 and Type 2 diabetes mellitus: what do we know today? Womens Health [Lond]. 2012, 8, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, X. The medical risks of obesity. Postgraduate medicine. 2009, 121, 21–33. [Google Scholar] [CrossRef]
- Peeters, A.; Barendregt, J.; Willekens, F.; Mackenbach, J.; Al Mamun, A.; Bonneux, L. Obesity in adulthood and its consequences for life expectancy: a life-table analysis. Annals of internal medicine. 2003, 138, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Cecchini, M. Use of healthcare services and expenditure in the US in 2025: The effect of obesity and morbid obesity. PLoS One. 2018, 13, e0206703. [Google Scholar] [CrossRef]
- Nigatu, Y.T.; van de Ven, H.A.; van der Klink, J.J.; Brouwer, S.; Reijneveld, S.A.; Bultmann, U. Overweight, obesity and work functioning: the role of working-time arrangements. Appl Ergon. 2016, 52, 128–134. [Google Scholar] [CrossRef]
- Sarwer, D.B.; Polonsky, H.M. The Psychosocial Burden of Obesity. Endocrinol Metab Clin North Am. 2016, 45, 677–688. [Google Scholar] [CrossRef]
- Westbury, S.; Oyebode, O.; van Rens, T.; Barber, T.M. Obesity Stigma: Causes, Consequences, and Potential Solutions. Curr Obes Rep. 2023, 12, 10–23. [Google Scholar] [CrossRef]
- Barber, T.M.; Kabisch, S.; Pfeiffer, A.F.H.; Weickert, M.O. Dietary and Lifestyle Strategies for Obesity. Nutrients. 2024, 16, 2714. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Wu, Z.; Cao, W.; Lv, J.; Yu, C.; Huang, T.; et al. Cardiometabolic Traits in Adult Twins: Heritability and BMI Impact with Age. Nutrients. 2022, 15, 164. [Google Scholar] [CrossRef]
- Chodick, G.; Simchoni, M.; Jensen, B.W.; Derazne, E.; Pinhas-Hamiel, O.; Landau, R.; et al. Heritability of Body Mass Index Among Familial Generations. JAMA Netw Open. 2024, 7, e2419029. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C. Genetics of Obesity: What We Have Learned Over Decades of Research. Obesity [Silver Spring]. 2021, 29, 802–820. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O. Genetics of obesity: what genetic association studies have taught us about the biology of obesity and its complications. Lancet Diabetes Endocrinol. 2018, 6, 223–236. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: from discovery to biology. Nat Rev Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef]
- Abraham, A.; Yaghootkar, H. Identifying obesity subtypes: A review of studies utilising clinical biomarkers and genetic data. Diabet Med. 2023, 40, e15226. [Google Scholar] [CrossRef]
- Barber, T.M.; Franks, S. Genetics of polycystic ovary syndrome. Front Horm Res. 2013, 40, 28–39. [Google Scholar]
- Hayes, M.G.; Urbanek, M.; Ehrmann, D.A.; Armstrong, L.L.; Lee, J.Y.; Sisk, R.; et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun. 2015, 6, 7502. [Google Scholar] [CrossRef]
- Dong, S.S.; Guo, Y.; Yang, T.L. Addressing the Missing Heritability Problem With the Help of Regulatory Features. Evol Bioinform Online. 2019, 15, 1176934319860861. [Google Scholar] [CrossRef]
- Barber, T.M.; Valsamakis, G.; Mastorakos, G.; Hanson, P.; Kyrou, I.; Randeva, H.S.; et al. Dietary Influences on the Microbiota-Gut-Brain Axis. Int J Mol Sci. 2021, 22, 3502. [Google Scholar] [CrossRef]
- Barber, T.M.; Hanson, P.; Weickert, M.O. Metabolic-Associated Fatty Liver Disease and the Gut Microbiota. Endocrinol Metab Clin North Am. 2023, 52, 485–496. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.T.; Bakacs, E.; Combellick, J.; Grigoryan, Z.; Dominguez-Bello, M.G. The infant microbiome development: mom matters. Trends in molecular medicine. 2015, 21, 109–117. [Google Scholar] [CrossRef]
- Mayer, E.A.; Tillisch, K.; Gupta, A. Gut/brain axis and the microbiota. J Clin Invest. 2015, 125, 926–938. [Google Scholar] [CrossRef] [PubMed]
- de Vos, W.M.; de, V.o.s.EA. Role of the intestinal microbiome in health and disease: from correlation to causation. Nutr Rev. 2012, 70 (Suppl 1), S45–S56. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Padua, D.; Tillisch, K. Altered brain-gut axis in autism: comorbidity or causative mechanisms? Bioessays. 2014, 36, 933–939. [Google Scholar] [CrossRef]
- Amaral, F.A.; Sachs, D.; Costa, V.V.; Fagundes, C.T.; Cisalpino, D.; Cunha, T.M.; et al. Commensal microbiota is fundamental for the development of inflammatory pain. Proc Natl Acad Sci U S A. 2008, 105, 2193–2197. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Oduro-Donkor, D.; Turner, M.C.; Farnaud, S.; Renshaw, D.; Kyrou, I.; Hanson, P.; et al. Modification of fecal microbiota as a mediator of effective weight loss and metabolic benefits following bariatric surgery. Expert Rev Endocrinol Metab. 2020, 15, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Cardenas, J.A.; Manzano-Agugliaro, F. The metagenomics worldwide research. Curr Genet. 2017, 63, 819–829. [Google Scholar] [CrossRef]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proceedings of the National Academy of Sciences. 2011, 108, 16050–16055. [Google Scholar] [CrossRef]
- Chassaing, B.; Raja, S.M.; Lewis, J.D.; Srinivasan, S.; Gewirtz, A.T. Colonic microbiota encroachment correlates with dysglycemia in humans. Cellular and molecular gastroenterology and hepatology. 2017, 4, 205–221. [Google Scholar] [CrossRef]
- Barber, T.M.; Kabisch, S.; Pfeiffer, A.F.H.; Weickert, M.O. The Effects of the Mediterranean Diet on Health and Gut Microbiota. Nutrients. 2023, 15, 2150. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.M.; Kabisch, S.; Pfeiffer, A.F.H.; Weickert, M.O. The Health Benefits of Dietary Fibre. Nutrients. 2020, 12, 3209. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Pfeiffer, A.F. Metabolic effects of dietary fiber consumption and prevention of diabetes. J Nutr. 2008, 138, 439–442. [Google Scholar] [CrossRef]
- Backhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A. 2007, 104, 979–984. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Goehler, L.E.; Gaykema, R.P.; Opitz, N.; Reddaway, R.; Badr, N.; Lyte, M. Activation in vagal afferents and central autonomic pathways: early responses to intestinal infection with Campylobacter jejuni. Brain, behavior, and immunity. 2005, 19, 334–344. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Anfora, A.T.; Liu, J.; Schultz, P.G.; Lesley, S.A.; Peters, E.C.; et al. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proceedings of the national academy of sciences. 2009, 106, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Weitkunat, K.; Bishop, C.A.; Wittmuss, M.; Machate, T.; Schifelbein, T.; Schulze, M.B.; et al. Effect of Microbial Status on Hepatic Odd-Chain Fatty Acids Is Diet-Dependent. Nutrients. 2021, 13, 1546. [Google Scholar] [CrossRef]
- Weitkunat, K.; Schumann, S.; Nickel, D.; Hornemann, S.; Petzke, K.J.; Schulze, M.B.; et al. Odd-chain fatty acids as a biomarker for dietary fiber intake: a novel pathway for endogenous production from propionate. Am J Clin Nutr. 2017, 105, 1544–1551. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, B.J.; Seyssel, K.; Chiu, S.; Pan, P.H.; Lin, S.Y.; Stanley, E.; et al. Odd Chain Fatty Acids; New Insights of the Relationship Between the Gut Microbiota, Dietary Intake, Biosynthesis and Glucose Intolerance. Sci Rep. 2017, 7, 44845. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.-J.; Bakker, BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. Journal of lipid research. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proceedings of the National Academy of Sciences. 2008, 105, 16767–16772. [Google Scholar] [CrossRef]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; et al. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes. 2012, 61, 364–371. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; et al. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Haghikia, A.; Jörg, S.; Duscha, A.; Berg, J.; Manzel, A.; Waschbisch, A.; et al. Dietary fatty acids directly impact central nervous system autoimmunity via the small intestine. Immunity. 2015, 43, 817–829. [Google Scholar] [CrossRef]
- Sanmiguel, C.P.; Jacobs, J.; Gupta, A.; Ju, T.; Stains, J.; Coveleskie, K.; et al. Surgically induced changes in gut microbiome and hedonic eating as related to weight loss: preliminary findings in obese women undergoing bariatric surgery. Psychosomatic medicine. 2017, 79, 880. [Google Scholar] [CrossRef]
- Chambers, E.S.; Viardot, A.; Psichas, A.; Morrison, D.J.; Murphy, K.G.; Zac-Varghese, S.E.; et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut. 2015, 64, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Pfluger, P.T.; Kampe, J.; Castaneda, T.R.; Vahl, T.; D'Alessio, D.A.; Kruthaupt, T.; et al. Effect of human body weight changes on circulating levels of peptide YY and peptide YY3-36. J Clin Endocrinol Metab. 2007, 92, 583–588. [Google Scholar] [CrossRef]
- Weickert, M.O.; Spranger, J.; Holst, J.J.; Otto, B.; Koebnick, C.; Mohlig, M.; et al. Wheat-fibre-induced changes of postprandial peptide YY and ghrelin responses are not associated with acute alterations of satiety. Br J Nutr. 2006, 96, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Han, X.; Ruan, M.; Huang, F.; Yang, L.; Xu, T.; et al. Prebiotic inulin controls Th17 cells mediated central nervous system autoimmunity through modulating the gut microbiota and short chain fatty acids. Gut Microbes. 2024, 16, 2402547. [Google Scholar] [CrossRef]
- Kabisch, S.; Weickert, M.O.; Pfeiffer, A.F.H. The role of cereal soluble fiber in the beneficial modulation of glycometabolic gastrointestinal hormones. Crit Rev Food Sci Nutr. 2024, 64, 4331–4347. [Google Scholar] [CrossRef] [PubMed]
- Weickert, M.O.; Arafat, A.M.; Blaut, M.; Alpert, C.; Becker, N.; Leupelt, V.; et al. Changes in dominant groups of the gut microbiota do not explain cereal-fiber induced improvement of whole-body insulin sensitivity. Nutr Metab [Lond]. 2011, 8, 90. [Google Scholar] [CrossRef]
- Weickert, M.O. High fiber intake, dietary protein, and prevention of type 2 diabetes. Expert Rev Endocrinol Metab. 2018, 13, 223–224. [Google Scholar] [CrossRef]
- Weickert, M.O.; Pfeiffer, A.F.H. Impact of Dietary Fiber Consumption on Insulin Resistance and the Prevention of Type 2 Diabetes. J Nutr. 2018, 148, 7–12. [Google Scholar] [CrossRef]
- Weickert, M.O.; Roden, M.; Isken, F.; Hoffmann, D.; Nowotny, P.; Osterhoff, M.; et al. Effects of supplemented isoenergetic diets differing in cereal fiber and protein content on insulin sensitivity in overweight humans. Am J Clin Nutr. 2011, 94, 459–471. [Google Scholar] [CrossRef]
- Schulze, M.B.; Schulz, M.; Heidemann, C.; Schienkiewitz, A.; Hoffmann, K.; Boeing, H. Fiber and magnesium intake and incidence of type 2 diabetes: a prospective study and meta-analysis. Arch Intern Med. 2007, 167, 956–965. [Google Scholar] [CrossRef]
- de Munter, J.S.; Hu, F.B.; Spiegelman, D.; Franz, M.; van, D.a.m.RM. Whole grain, bran, and germ intake and risk of type 2 diabetes: a prospective cohort study and systematic review. PLoS Med. 2007, 4, e261. [Google Scholar] [CrossRef]
- Isken, F.; Klaus, S.; Osterhoff, M.; Pfeiffer, A.F.; Weickert, M.O. Effects of long-term soluble vs. insoluble dietary fiber intake on high-fat diet-induced obesity in C57BL/6J mice. J Nutr Biochem. 2010, 21, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, L.; Li, J.; Wu, Q.; Qian, L.; He, J.; et al. Resistant starch intake facilitates weight loss in humans by reshaping the gut microbiota. Nat Metab. 2024, 6, 578–597. [Google Scholar] [CrossRef] [PubMed]
- Track, N.S.; Cawkwell, M.E.; Chin, B.C.; Chiu, S.S.; Haberer, S.A.; Honey, C.R. Guar gum consumption in adolescent and adult rats: short- and long-term metabolic effects. Can J Physiol Pharmacol. 1985, 63, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Russo, S.; Mingrone, G. Incretin hormones, obesity and gut microbiota. Peptides. 2024, 178, 171216. [Google Scholar] [CrossRef]
- Statham, L.; Pelling, M.; Hanson, P.; Kyrou, I.; Randeva, H.; Barber, T.M. Designer GLP1 poly-agonist peptides in the management of diabesity. Expert Rev Endocrinol Metab. 2023, 18, 231–240. [Google Scholar] [CrossRef]
- Rudovich, N.N.; Nikiforova, V.J.; Otto, B.; Pivovarova, O.; Gogebakan, O.; Erban, A.; et al. Metabolomic linkage reveals functional interaction between glucose-dependent insulinotropic polypeptide and ghrelin in humans. Am J Physiol Endocrinol Metab. 2011, 301, E608–17. [Google Scholar] [CrossRef]
- Isken, F.; Weickert, M.O.; Tschop, M.H.; Nogueiras, R.; Mohlig, M.; Abdelrahman, A.; et al. Metabolic effects of diets differing in glycaemic index depend on age and endogenous glucose-dependent insulinotrophic polypeptide in mice. Diabetologia. 2009, 52, 2159–2168. [Google Scholar] [CrossRef]
- Ruban, A.; Ashrafian, H.; Teare, J.P. The EndoBarrier: Duodenal-Jejunal Bypass Liner for Diabetes and Weight Loss. Gastroenterol Res Pract. 2018, 2018, 7823182. [Google Scholar] [CrossRef]
- Isken, F.; Pfeiffer, A.F.; Nogueiras, R.; Osterhoff, M.A.; Ristow, M.; Thorens, B.; et al. Deficiency of glucose-dependent insulinotropic polypeptide receptor prevents ovariectomy-induced obesity in mice. Am J Physiol Endocrinol Metab. 2008, 295, E350–5. [Google Scholar] [CrossRef]
- Bravo, J.A.; Forsythe, P.; Chew, M.V.; Escaravage, E.; Savignac, H.M.; Dinan, T.G.; et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. Proc Natl Acad Sci U S A. 2011, 108, 16050–16055. [Google Scholar] [CrossRef]
- Ringseis, R.; Gessner, D.K.; Eder, K. The Gut-Liver Axis in the Control of Energy Metabolism and Food Intake in Animals. Annu Rev Anim Biosci. 2020, 8, 295–319. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, K.M.; Kang, N.; Bienenstock, J.; Foster, J.A. Reduced anxiety-like behavior and central neurochemical change in germ-free mice. Neurogastroenterol Motil. 2011, 23, 255–264, e119. [Google Scholar] [CrossRef]
- Neufeld, K.A.; Kang, N.; Bienenstock, J.; Foster, J.A. Effects of intestinal microbiota on anxiety-like behavior. Commun Integr Biol. 2011, 4, 492–494. [Google Scholar] [CrossRef]
- Clarke, G.; Grenham, S.; Scully, P.; Fitzgerald, P.; Moloney, R.D.; Shanahan, F.; et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry. 2013, 18, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Shaban, H.; O'Connor, R.; Ovsepian, S.V.; Dinan, T.G.; Cryan, J.F.; Schellekens, H. Electrophysiological approaches to unravel the neurobiological basis of appetite and satiety: use of the multielectrode array as a screening strategy. Drug Discov Today. 2017, 22, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Berthoud, H.R. Metabolic and hedonic drives in the neural control of appetite: who is the boss? Curr Opin Neurobiol. 2011, 21, 888–896. [Google Scholar] [CrossRef]
- Korpela, K. Impact of Delivery Mode on Infant Gut Microbiota. Ann Nutr Metab. 2021, 77, 11–19. [Google Scholar] [CrossRef]
- Andersen, V.; Moller, S.; Jensen, P.B.; Moller, F.T.; Green, A. Caesarean Delivery and Risk of Chronic Inflammatory Diseases [Inflammatory Bowel Disease, Rheumatoid Arthritis, Coeliac Disease, and Diabetes Mellitus]: A Population Based Registry Study of 2,699,479 Births in Denmark During 1973-2016. Clin Epidemiol. 2020, 12, 287–293. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010, 107, 11971–11975. [Google Scholar] [CrossRef]
- Nagpal, R.; Yamashiro, Y. Gut Microbiota Composition in Healthy Japanese Infants and Young Adults Born by C-Section. Ann Nutr Metab. 2018, 73 (Suppl 3), 4–11. [Google Scholar] [CrossRef] [PubMed]
- Rutayisire, E.; Huang, K.; Liu, Y.; Tao, F. The mode of delivery affects the diversity and colonization pattern of the gut microbiota during the first year of infants' life: a systematic review. BMC Gastroenterol. 2016, 16, 86. [Google Scholar] [CrossRef] [PubMed]
- Low, J.S.Y.; Soh, S.E.; Lee, Y.K.; Kwek, K.Y.C.; Holbrook, J.D.; Van der Beek, E.M.; et al. Ratio of Klebsiella/Bifidobacterium in early life correlates with later development of paediatric allergy. Benef Microbes. 2017, 8, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Kalliomaki, M.; Collado, M.C.; Salminen, S.; Isolauri, E. Early differences in fecal microbiota composition in children may predict overweight. Am J Clin Nutr. 2008, 87, 534–538. [Google Scholar] [CrossRef]
- Korpela, K.; Costea, P.; Coelho, L.P.; Kandels-Lewis, S.; Willemsen, G.; Boomsma, D.I.; et al. Selective maternal seeding and environment shape the human gut microbiome. Genome Res. 2018, 28, 561–568. [Google Scholar] [CrossRef]
- Yi, D.Y.; Kim, S.Y. Human Breast Milk Composition and Function in Human Health: From Nutritional Components to Microbiome and MicroRNAs. Nutrients. 2021, 13, 3094. [Google Scholar] [CrossRef]
- Martin, R.; Langa, S.; Reviriego, C.; Jiminez, E.; Marin, M.L.; Xaus, J.; et al. Human milk is a source of lactic acid bacteria for the infant gut. J Pediatr. 2003, 143, 754–758. [Google Scholar] [CrossRef]
- Fitzstevens, J.L.; Smith, K.C.; Hagadorn, J.I.; Caimano, M.J.; Matson, A.P.; Brownell, E.A. Systematic Review of the Human Milk Microbiota. Nutr Clin Pract. 2017, 32, 354–364. [Google Scholar] [CrossRef]
- Togo, A.; Dufour, J.C.; Lagier, J.C.; Dubourg, G.; Raoult, D.; Million, M. Repertoire of human breast and milk microbiota: a systematic review. Future Microbiol. 2019, 14, 623–641. [Google Scholar] [CrossRef]
- Asnicar, F.; Manara, S.; Zolfo, M.; Truong, D.T.; Scholz, M.; Armanini, F.; et al. Studying Vertical Microbiome Transmission from Mothers to Infants by Strain-Level Metagenomic Profiling. mSystems. 2017, 2, e00164–16. [Google Scholar] [CrossRef]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr. 2017, 171, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Kordy, K.; Gaufin, T.; Mwangi, M.; Li, F.; Cerini, C.; Lee, D.J.; et al. Contributions to human breast milk microbiome and enteromammary transfer of Bifidobacterium breve. PLoS One. 2020, 15, e0219633. [Google Scholar] [CrossRef]
- Hunt, K.M.; Foster, J.A.; Forney, L.J.; Schutte, U.M.; Beck, D.L.; Abdo, Z.; et al. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS One. 2011, 6, e21313. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rubio, R.; Collado, M.C.; Laitinen, K.; Salminen, S.; Isolauri, E.; Mira, A. The human milk microbiome changes over lactation and is shaped by maternal weight and mode of delivery. Am J Clin Nutr. 2012, 96, 544–551. [Google Scholar] [CrossRef]
- Murphy, K.; Curley, D.; O'Callaghan, T.F.; O'Shea, C.A.; Dempsey, E.M.; O'Toole, P.W.; et al. The Composition of Human Milk and Infant Faecal Microbiota Over the First Three Months of Life: A Pilot Study. Sci Rep. 2017, 7, 40597. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Uhr, T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004, 303, 1662–1665. [Google Scholar] [CrossRef]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; et al. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef]
- Milani, C.; Mancabelli, L.; Lugli, G.A.; Duranti, S.; Turroni, F.; Ferrario, C.; et al. Exploring Vertical Transmission of Bifidobacteria from Mother to Child. Appl Environ Microbiol. 2015, 81, 7078–7087. [Google Scholar] [CrossRef]
- Kim, K.U.; Kim, W.H.; Jeong, C.H.; Yi, D.Y.; Min, H. More than Nutrition: Therapeutic Potential of Breast Milk-Derived Exosomes in Cancer. Int J Mol Sci. 2020, 21, 7327. [Google Scholar] [CrossRef]
- Kim, S.Y.; Yi, D.Y. Analysis of the human breast milk microbiome and bacterial extracellular vesicles in healthy mothers. Exp Mol Med. 2020, 52, 1288–1297. [Google Scholar] [CrossRef]
- Bryant, W.A.; Stentz, R.; Le Gall, G.; Sternberg, M.J.E.; Carding, S.R.; Wilhelm, T. In Silico Analysis of the Small Molecule Content of Outer Membrane Vesicles Produced by Bacteroides thetaiotaomicron Indicates an Extensive Metabolic Link between Microbe and Host. Front Microbiol. 2017, 8, 2440. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Gacesa, R.; Kurilshikov, A.; Vich Vila, A.; Sinha, T.; Klaassen, M.A.Y.; Bolte, L.A.; et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature. 2022, 604, 732–739. [Google Scholar] [CrossRef]
- Faith, J.J.; McNulty, N.P.; Rey, F.E.; Gordon, J.I. Predicting a human gut microbiota's response to diet in gnotobiotic mice. Science. 2011, 333, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011, 334, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: human gut microbes associated with obesity. Nature. 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Duncan, S.H.; Belenguer, A.; Holtrop, G.; Johnstone, A.M.; Flint, H.J.; Lobley, G.E. Reduced dietary intake of carbohydrates by obese subjects results in decreased concentrations of butyrate and butyrate-producing bacteria in feces. Appl Environ Microbiol. 2007, 73, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Asnicar, F.; Berry, S.E.; Valdes, A.M.; Nguyen, L.H.; Piccinno, G.; Drew, D.A.; et al. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat Med. 2021, 27, 321–332. [Google Scholar] [CrossRef]
- Louis, P.; Scott, K.P.; Duncan, S.H.; Flint, H.J. Understanding the effects of diet on bacterial metabolism in the large intestine. J Appl Microbiol. 2007, 102, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Simoes, C.D.; Maukonen, J.; Kaprio, J.; Rissanen, A.; Pietilainen, K.H.; Saarela, M. Habitual dietary intake is associated with stool microbiota composition in monozygotic twins. J Nutr. 2013, 143, 417–423. [Google Scholar] [CrossRef]
- Pokusaeva, K.; Fitzgerald, G.F.; van Sinderen, D. Carbohydrate metabolism in Bifidobacteria. Genes Nutr. 2011, 6, 285–306. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.G.; Worsley, A.; Liem, D.G. Parents' food choice motives and their associations with children's food preferences. Public Health Nutr. 2015, 18, 1018–1027. [Google Scholar] [CrossRef]
- Kral, T.V.; Rauh, E.M. Eating behaviors of children in the context of their family environment. Physiol Behav. 2010, 100, 567–573. [Google Scholar] [CrossRef]
- Smoczek, M.; Vital, M.; Wedekind, D.; Basic, M.; Zschemisch, N.H.; Pieper, D.H.; et al. A combination of genetics and microbiota influences the severity of the obesity phenotype in diet-induced obesity. Sci Rep. 2020, 10, 6118. [Google Scholar] [CrossRef] [PubMed]
- Pigeyre, M.; Yazdi, F.T.; Kaur, Y.; Meyre, D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin Sci [Lond]. 2016, 130, 943–986. [Google Scholar] [CrossRef]
- Kim, K.O.; Gluck, M. Fecal Microbiota Transplantation: An Update on Clinical Practice. Clinical endoscopy. 2019, 52, 137. [Google Scholar] [CrossRef]
- Lee, P.; Yacyshyn, B.R.; Yacyshyn, M.B. Gut microbiota and obesity: An opportunity to alter obesity through faecal microbiota transplant [FMT]. Diabetes Obes Metab. 2019, 21, 479–490. [Google Scholar] [CrossRef]
- Kootte, R.S.; Levin, E.; Salojarvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; et al. Improvement of Insulin Sensitivity after Lean Donor Feces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 26, 611–619 e6. [Google Scholar] [CrossRef]
- Leeuwendaal, N.K.; Stanton, C.; O'Toole, P.W.; Beresford, T.P. Fermented Foods, Health and the Gut Microbiome. Nutrients. 2022, 14, 1527. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Impact of the gut microbiota on BMI via hypothalamic control of appetite and metabolism, and mechanisms that underlie the trans-generational effects of the gut microbiota on the heritability of BMI.
Figure 1.
Impact of the gut microbiota on BMI via hypothalamic control of appetite and metabolism, and mechanisms that underlie the trans-generational effects of the gut microbiota on the heritability of BMI.
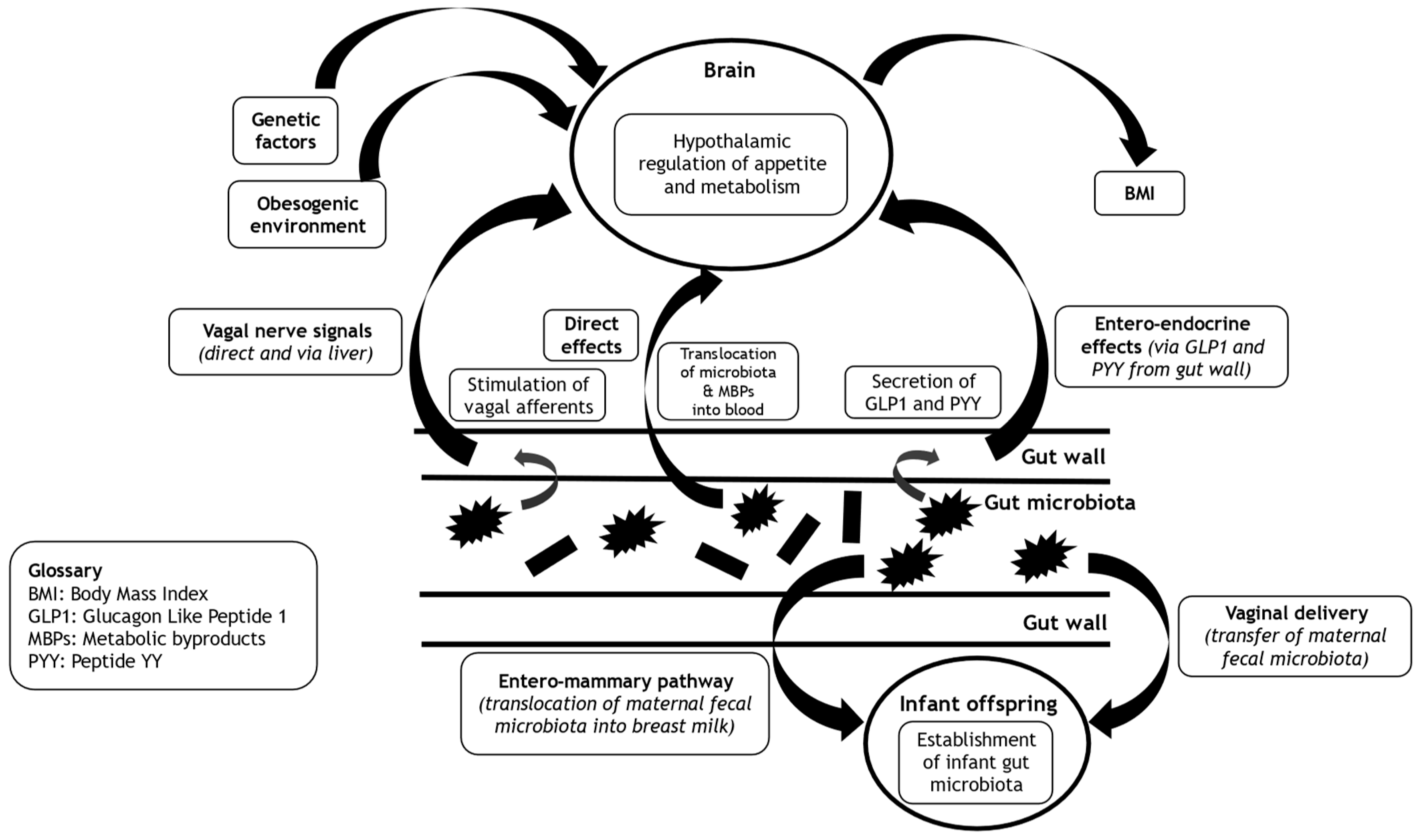
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



