
Submitted:
19 April 2025
Posted:
21 April 2025
You are already at the latest version
Abstract
Myelodysplastic syndromes (MDS) are a heterogeneous group of hematological malignancies characterized by ineffective hematopoiesis, resulting in cytopenias, morphologic dysplasia in hematopoietic lineages, and a variable risk of progression to acute myeloid leukemia. Significant advances in the understanding of MDS have been made in recent years, largely due to the implementation of molecular tools. Latin America is a highly diverse region, both ethnically and racially, and often faces resource limitations that challenge the broad applicability of recent advances in MDS. In this review, we discuss the key genes implicated in the pathogenesis and classification of MDS, including those related to germline predisposition syndromes, and their relevance to diagnosis, prognosis, and therapeutic decision-making. We also highlight research efforts from Latin America and present the results of a survey assessing the availability of molecular tools in Latin American centers.
Keywords:
myelodysplastic syndrome
; NGS
; mutations
; prognostic
; germline
1. Introduction
Myelodysplastic syndromes (MDS) are a heterogeneous group of hematological malignancies characterized by ineffective hematopoiesis, leading to cytopenia, morphologic dysplasia in the hematopoietic lineages, and a variable progression rate to acute myeloid leukemia [1]. MDS comprise multiple neoplastic clones derived from hematopoietic stem and progenitor cells [2]. Based on this concept, the most recent World Health Organization (WHO) classification renamed MDS as myelodysplastic neoplasms, while retaining the traditional abbreviation [3]. Classifications in MDS have evolved following the advances in cytogenetic and molecular findings, with implications for diagnosis, prognosis, and therapeutic algorithms [1,4]. As MDS are associated with aging, it predominantly affects the elderly, and the global burden of MDS appears to be increasing, particularly in Latin America (LA) [5].
2. Myelodysplastic Syndromes in Latin America
LA is a region comprising several countries, covering an area of approximately 19,197,000 km2. As of 2024, the population of LA and the Caribbean reached 663 million people [6,7]. The predominant languages are Spanish and Portuguese [7]. The impact of race and ethnicity on MDS outcomes has been studied in the United States using data from the Surveillance, Epidemiology, and End Results (SEER) program, revealing significant differences in age at diagnosis, disease risk, and survival outcomes based on racial/ethnic backgrounds [8]. In the molecular setting, different polymorphisms in the nucleotide excision repair pathway were studied in 173 Brazilian and 96 Argentinean MDS patients [9]. This study reinforces the heterogeneity of MDS and highlights the importance of ethnic differences and regional influences in pathogenesis and prognosis of MDS. Accordingly, it has been suggested that incorporating ethnic diversity, through the evaluation of genetic ancestry, into prognostic models is essential to ensure their universal applicability [10]. In the clinical context, the first multicenter study from LA which included 1,080 patients with MDS (median age 69 years, with 75% over the age of 60 at diagnosis), reported differences in clinical features and outcomes among countries [11]. Despite regional variability, the most widely used clinical prognostic scores for MDS were validated LA patients [11,12,13], and the efficacy of specific treatments [14,15,16,17] was also confirmed.
3. Molecular Data
In recent years, the use of high-throughput/next-generation sequencing (NGS) has significantly advanced the understanding of MDS [18]. The recognition of a high frequency of mutations in genes involved in RNA splicing [19] expanded the number of known affected pathways in MDS, which now include epigenetic regulation, signal transduction, DNA damage, transcription factors, cohesins complexes, among others [20]. Also, mutations can be classified in early or late events in the progression of MDS [20,21]. At least one gene mutation can be found in over 90% of the cases when analyzing a panel of approximately 50 recurrently mutated genes [22]. In agreement with the findings of Papaemmanuil et al., the series by Haferlach et al., identified 47 significantly mutated genes, with TET2, SF3B1, ASXL1, SRSF2, DNMT3A, and RUNX1 mutated in over 10% of cases [21,22]. Within this molecular, 110 Brazilian patients were included among 2,957 patients profiled for mutation 152 genes [23]. Bernard et al., confirmed that at least one oncogenic genomic alteration was present in 94% of patients with MDS. They also reported 3,186 cytogenetic alterations in 41% of patients and 9,254 oncogenic mutations across 121 genes in 90% of patients [23].
The first multicenter study from LA included 145 patients with MDS and 37 patients with chronic myelomonocytic leukemia (CMML) (median age 65 years, male-to-female ratio 1:3) from Argentina and Uruguay [24]. This retrospective real-world study used six different commercially available myeloid-focused NGS based panels, covering 30-63 genes per panel, with molecular data generated at each institution. The results showed that 81% of patients had at least one detectable variant, and that individuals with CMML exhibited a significantly higher mutational burden compared to those with MDS (3.5 variants vs. 2 variants, respectively). The five most frequently mutated genes were TET2, ASXL1, SRSF2, SF3B1, and DNMT3A [24]. Similarly, a recent study conducted in Uruguay analyzed 52 patients with MDS or CMML (median age 67 years) and found that 75% harbored at least one somatic mutation. The most frequently mutated genes were DNMT3A, TP53, TET2, ASXL1, and SF3B1. Copy number variations (CNV) were detected in 38% of cases, including abnormalities not identifiable by conventional cytogenetics. The presence of three or more somatic mutations was significantly associated with adverse clinical features and outcomes. Patients with ≥3 mutations had higher bone marrow blast percentages (median 7% vs. 1%, p = 0.005), elevated LDH levels (p = 0.0027), and increased International Prognostic Scoring System–Revised (IPSS-R) scores (p = 0.013) compared to those with fewer mutations. Moreover, this group demonstrated significantly reduced overall survival (OS) (p = 0.0032) and leukemia-free survival (LFS) (p = 0.001). These findings underscore the prognostic relevance of mutational burden in MDS and support its integration into contemporary risk stratification models [25].
4. Key Genetic Affected Pathways
4.1. RNA Splicing
Mutations in splicing factors have been shown to occur early in tumor evolution [21]. These proteins play key roles in assembling major U-2 type spliceosomes or minor U-12 type spliceosomes, which catalyze pre-mRNA splicing. Mutations in these splicing factors can lead to aberrant alternative splicing events [26]. They have been observed in approximately 50% of the patients with MDS and are frequent in cases progressing to AML [19,27,28,29,30]. These mutations were often mutually exclusive, with the most frequent involving splicing factor 3b subunit 1 (SF3B1), serine and arginine rich splicing factor 2 (SRSF2), U2 small nuclear RNA auxiliary factor 1 (U2AF1) and Zinc finger CCCH-type, RNA binding motif, and serine/arginine rich 2 (ZRSR2) [19].
SF3B1 mutations in MDS result in distinct splicing alterations, defining a disease characterized by ring sideroblasts, ineffective erythropoiesis, a lower risk of leukemic transformation, and longer OS, provided that other poor prognostic markers are absent [23,31,32]. In a bioinformatic analysis of an RNAseq data set (GSE114922) of CD34+ cells of MDS patients with SF3B1 variants, Lincango et al., identified 1,342 differentially expressed genes when compared with patients lacking other splicing factor mutations and healthy controls. Functional enrichment analysis (gseGo and enrichGo) revealed dysregulated pathways involving ribosomes, oxidative phosphorylation, mitochondrial gene expression and translation [33]. Among them, NDUFA8, RBM25, MRRF and AC, related to mitochondrial pathways and telomeric maintenance, were confirmed to be significantly dysregulated. This novel gene Hub explained 77.9% of the variance associated with SF3B1 mutations in Argentine MDS patients [34]. MDS-SF3B1 is now recognized as a distinct entity, comprising over 90% of MDS with ≥5% ring sideroblasts [3,32,35]. Lincango et al., reported SFR3B1 mutations in 16% of patients from Argentina and Uruguay [24], doubling up the reported frequency in Brazil [36]. Catalán AI et al. found a 13% mutation rate among Uruguayan patients with MDS, associated with older age (median age 76 years), elevated ferritin levels and lower bone marrow blast counts compared to those without SF3B1 mutations [25]. MDS can also occur alongside systemic mastocytosis; in such complex clinical scenarios, treatment is guided by the prevailing clinical features. Lenalidomide has shown efficacy in a patient with SF3B1-mutant MDS and systemic mastocytosis, achieving transfusion independence after failure of a hypomethylating agent (HMA) and serving as a bridge to transplant, based on the complexity of accompanying mutational profile [37].
U2AF1 mutations predominantly occur at two hotspots (S34F, Q157) within zinc finger domains [19]. Patients with the S34F mutations appear to have better survival than those with Q157 variant, including those undergoing allogeneic stem cell transplantation [38]. U2AF1 mutations have been associated with del(20q), chromosome 7 alterations, higher rates of transfusion-dependent anemia, and excess of bone marrow blasts [39]. These mutations are typically acquired later in life and are linked with rapid progression to MDS and AML [38].
Mutations in SRSF2, usually affecting the Pro95 residue and associated with aberrant exon-skipping alternative splicing patterns, were found in 15–20% of patients with MDS. They are associated with advanced age, male sex, normal cytogenetics and CMML subtype [19,28,40,41]. A South American series reported a 19% frequency, likely reflecting the inclusion of CMML patients [24]. Bersanelli et al., described two prognostic clusters enriched for SRSF2 variants: one associated with TET2, and another with ASXL1, RUNX1, IDH2 or EZH2. Both are linked to trisomy 8 and monocytosis; however, the second cluster showed more cytopenias, multilineage dysplasia, and higher blast percentages, indicating a worse prognosis [39].
ZRSR2 mutations are less frequent (2-10%) in MDS but are significantly enriched in high-risk cases compared to low-risk MDS [19,21]. Although their impact on AML progression and OS is less pronounced [42], ZRSR2 mutations are classified among MDS-related genes mutations by both the ICC and the WHO AML classifications [3,35].
Having multiple spliceosome mutations does not appear to worsen prognosis compared to single mutation [43]. However, concurrent mutations in ASXL1, RUNX1, and KRAS may negatively affect outcomes, varying across different morphological diagnostic categories, suggesting complex gene–gene interactions [44].
4.2. Chromatin Modification
Mutations in the Additional Sex Combs Like 1 (ASXL1) gene occur in 20.7% of MDS cases, with approximately 70% being frameshift mutations and 30% heterozygous point mutations that result in translational alterations [45]. ASXL1 mutations are common in clonal hematopoiesis and are considered relatively early events in leukemogenesis, with increased dominant of mutant clones observed in secondary AML following MDS [46]. ASXL1 mutations are associated with an adverse prognostic outcome in patients with MDS [47].
Enhancer of Zeste Homolog 2 (EZH2) mutations in myeloid neoplasms correspond to loss-of-function alterations and are found in approximately 5% of patients with MDS [48]. Patients with EZH2-mutated MDS are typically older and show lower response rates to HMAs compared to those with wild type EZH2. Among patients with EZH2-mutated MDS, co-occurrence of ASXL1 or RUNX1 mutations is associated with shorter OS compared to those without such co-mutations [49].
BCOR (BCL6 Corepressor) and BCORL1 (BCL6 Corepressor like 1) are two homologous genes located on chromosome X [50]. The BCOR gene product is primarily involved in the suppression of myeloid regulatory genes and the promotion of lymphopoiesis [51]. BCOR mutations are most commonly detected in patients in their sixth to seventh decades of life and are associated with low white blood cell counts at diagnosis. BCOR mutations occur in 3–8% of the patients with MDS, with RUNX1 and U2AF1 being among the most frequently co-mutated genes, while TP53 mutations were less commonly observed. Mutations in the BCOR gene are distributed throughout the exon sequence, with no identifiable hotspot [52]. A complex karyotype adversely impacted survival among mutated BCOR-mutated AML/MDS cases, whereas allogeneic stem cell transplant has been shown to improve survival [52]. The BCORL1 mutations are present in approximately 1–5% of patients with MDS and are more frequent in patients with prior therapy [53].
The human KMT2A gene, located on chromosome 11q23, is recurrently rearranged in acute leukemias [54]. In contrast, KMT2A partial tandem duplication (PTD) involves intragenic duplications and has been described in approximately 2% of patients with MDS, where it is associated with poor response to HMAs and adverse outcome [55,56].
PHF6 is thought to regulate gene expression via chromatin modification, acting as a tumor suppressor and influencing hematopoietic lineage differentiation. PHF6 mutations have been reported in 0.7% to 5% of myeloid malignancies, including MDS [57]. In patients with MDS, a variant allele fraction (VAF) > 20% for PHF6 mutations has been significantly associated with inferior OS and disease progression [57,58].
The cohesin complex, comprising SMC1, SMC3, RAD21, and either STAG1 or STAG2, is a recurrent mutational target, with STAG2 (stromal antigen 2) mutations accounting for more than half of cohesin mutations in myeloid malignancies [59]. These mutations were mostly mutually exclusive and occur in 10% of MDS cases [60]. STAG2, located on chromosome Xq25, is considered one of eight secondary-type mutations specific to secondary AML (including SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, and BCOR) [30]. STAG2 mutations are associated with high-risk disease and adverse outcomes in MDS, despite sensitivity to HMA. Katamesh et al., found that 18% of MDS cases progressed to AML, with an increased co-mutational burden on progression, suggesting that STAG2 mutations promote leukemic transformation by inducing genetic instability and facilitating the acquisition of additional mutations [61]. Morphologically, a striking megakaryocytic dysplasia has been described in patients with STAG2 mutation [62].
4.3. DNA Methylation
DNA methyltransferase 3 A (DNMT3A) mutations are the most common aberrations in de novo AML (20–25%), MDS (about 10%), and clonal hematopoiesis of indeterminate potential (CHIP), and are mostly heterozygous [63]. Reduced DNMT3A function results in global genomic hypermethylation, particularly enriched at promoters near the transcription start sites of genes involved in cellular differentiation [64,65]. Canonical mutations in DNMT3A, along with TET2 and ASXL1 mutations, are key drivers of age-related clonal hematopoiesis [66]. DNMT3A variants are found in 17% of patients with MDS and are enriched in patients with MDS-SF3B1 cases, particularly in women [67,68]. The DNMT3A R882 mutation is a known hotspot, and MDS patients harboring this mutation exhibit more severe leukopenia, frequent co-mutations in SRSF2 and IDH2 mutations, a higher frequency of excess blasts, a markedly increased risk of transformation to AML, and worse progression-free-survival (PFS) compared to non-R882 mutant MDS cases [69].
Ten-eleven translocation (TET) family proteins (TETs), specifically, TET1, TET2 and TET3, can modify DNA by oxidizing 5-methylcytosine (5mC). TET2, in particular, is a key regulator of DNA methylation whose primary function is to catalyze the conversion of 5-methylcytosine to 5-hydroxymethylcytosine during DNA demethylation [70]. The loss or attenuation of TET function is implicated in genomic hypermethylation and transcriptional reprogramming that promotes oncogenesis [71]. Smith et al., identified 71 TET2 mutations in 12% of 355 patients with MDS. Mutant clones were present in T cells, as well as CD34(+) and total bone marrow cells. There was no significant prognostic association between TET2 mutations and WHO subtypes, IPSS score, cytogenetic abnormalities, or transformation to AML [72]. However, biallelic TET2 mutations may be associated with adverse outcomes [73], and may drive monocytic differentiation, consistent with their prevalence in CMML [74].
Isocitrate dehydrogenase 1 or 2 (IDH1 or IDH2) mutations are detected in about 12% of patients with MDS and are enriched in high-risk disease and severe neutropenia [75,76]. These recurrent mutations in key metabolic enzymes result in the production of the oncometabolite 2-hydroxyglutarate (2-HG), which promotes leukemogenesis through a block in normal myeloid differentiation [76]. Mutation frequencies vary by MDS morphological subtype: 4% in refractory anemia with ring sideroblasts, 12% in refractory cytopenia with multilineage dysplasia, 14% in MDS-unclassifiable, 14% in refractory anemia with excess blasts RAEB-1, and 23% in RAEB-2. IDH1 mutations were associated with inferior survival, whereas IDH2 mutations did not affect survival [77,78].
4.4. Transcriptional Regulation
The transcription factor RUNX1 is mutated in familial platelet disorder with associated myeloid malignancy, as well as in sporadic MDS and leukemia [79,80]. Approximately 10% of patients with MDS harbor RUNX1 mutations, which are associated with AML transformation [2,81] and poor OS [18,82]. In patients with low-risk MDS, RUNX1 mutations are linked to disease progression, even in the absence of overt AML transformation [79]. RUNX1 expression levels may further refine prognostication, with higher RUNX1 expression correlating with shorter LFS and OS [83]. RUNX1 has also been implicated with autoinflammatory features observed in patients with MDS [84,85].
The CUT-like homeobox 1 (CUX1) gene, located on chr7q22.1, encodes multiple CUX1 isoforms, including the full-length p200 CUX1 protein [87]. CUX1 transcription factors regulate a large number of genes and microRNAs involved in multiple cellular processes [86], and exerts the tumor suppressor activity in part by regulating cell cycle and proliferative genes and DNA damage repair [86,87]. Most patients with MDS and AML with -7/del(7q), and up to 15% of MDS patients and 5% of AML patients diploid for the CUX1 locus, exhibited down modulated CUX1 expression. In 75% of mutant cases, CUX1 mutations were heterozygous, whereas microdeletions and homozygous and compound-heterozygous mutations were less common. CUX1 mutated or deleted were associated with worse survival compared with CUX1 wild-type [88]. A high association with dysplastic changes has been reported [87].
The ETV6 gene (formerly TEL) belongs to the ETS (E26 transformation specific) family of transcription factors [89]. ETV6 mutations are rare but recurrent somatic events in myeloid neoplasms, and are negatively prognostic in MDS [18]. Point mutations in the ETV6 gene have been reported in 2.7% of patients with MDS, were frequently subclonal, and never occurred in isolation, suggesting a role as a late event in disease progression [18,90]. Higher rates of ASXL1, SETBP1, RUNX1 and U2AF1 co-mutations were observed in ETV6-mutated cases, compared to those with wild-type ETV6 [90].
4.5. DNA Damage Response
TP53, also known as the “guardian of the genome”, is a tumor suppressor gene. It is the most commonly mutated gene in human malignancies [91]. In MDS, TP53 mutations are identified in 12% of cases [23]. Patients with multiple TP53 mutations or with both mutation and loss of heterozygosity (LOH; either due to chromosomal deletion or copy-neutral LOH, cnLOH as a result of an acquired uniparental disomy) are classified as TP53 multi-hit. This category comprises nearly 70% of patients of TP53-mutated MDS cases, and is associated with complex karyotypes in 90% of them [23]. Alterations affecting TP53 can be detected by conventional cytogenetics, typically by evidencing loss of chromosome 17 or its short arm (17p), including deletions or unbalanced translocations involving TP53 locus 17p13.3. Other techniques, such as FISH or arrays can identify deletions and, depending on the design of the array, cnLOH. Targeted NGS not only detects mutations but also provides the variant allele frequency (VAF), with VAFs greater than 50% suggesting additional alterations in the remaining locus. Bernard et al., reported that 22% of cases showed >1 mutation, 21% had accompanying deletions, and 19% exhibited 19% cnLOH. These multi-hits events are among one the strongest predictors of adverse outcomes [23], and are recognized as a distinct entity in the WHO 5th edition classification [3]. Similarly, the ICC defines TP53-mutated MDS based on the presence of two mutations with a VAF >10%, or one mutation associated with a complex karyotype (typically involving 17p-), VAF>50%, or cnLOH, in patients with bone marrow blast below 10%. For those with higher percentages, one alteration with VAF >10% is sufficient for classification [35]. In a recently published South-American series, 18/182 (10%) of patients harbored TP53 mutations, and a third of them were classified as bi-TP53 based on either the presence of two mutations or a VAF>50%. Limited access to advanced complementary technologies may hinder the detection of other alterations [24]. A retrospective cohort study of LA patients with TP53-mutated MDS reported a mean age was 68 years; 65% had de novo MDS, and 58% were classified as high or very high risk according to the IPSS-R, with a median OS of 16 months. The mean VAF was 33%, with 59% of patients harboring multi-hit mutations, and 30% showing complex karyotype [92]. In Uruguay, Catalán et al. reported a TP53 mutation prevalence of 21.2%, higher than typically reported at diagnosis. Notably, 70% of TP53-mutated patients exhibited multi-hit alterations, accounting for 14.9% of the overall cohort. These patients displayed significantly worse clinical features, including lower hemoglobin levels, increased transfusion dependence, elevated ferritin, thrombocytopenia, and had significantly poorer OS and LFS [25].
PPM1D, is a phosphatase that plays as a central role in the DNA damage response as part of a regulatory feedback loop with TP53: activated TP53 induces PPM1D expression, which then both directly and indirectly dephosphorylates TP53, leading to downregulation of TP53-mediated apoptosis. Mutations in PPM1D, typically nonsense or frameshift variants in exon 6, result in a gain-of-function that constitutively inhibits TP53 activation in response to DNA damage. The increased and stabilized expression of these truncated variants provides a survival advantage to hematopoietic clones by making them resistant to DNA-damaging agents such as cisplatin or the topoisomerase inhibitor etoposide [93]. PPM1D mutations are frequently seen in therapy-related clonal hematopoiesis, as well as therapy-related AML and MDS. These mutations may occur in the founding clone, with 81% expanding under exposure to alkylating agents [94]. In the aforementioned South-American series, only a minority of patients were tested for PPM1D mutation, as this gene was only recently incorporated into clinical diagnostic panels [24].
4.6. Signal Transduction
The genes CBL, FLT3, KIT, KRAS, NF1, NRAS, PTPN11, and RIT1 are key components of cellular signaling pathways in MDS [20]. Somatic mutations involved in signaling appeared to be a precondition for AML transformation from MDS, particularly NRAS/KRAS, FLT3, CBL, and PTPN11 [95,96]. In a cohort of 72 patients with MDS, 15 (20.8 %) tested positive for FLT3-ITD and exhibited significantly worse OS and PFS compared to those without the mutation [97]. Single-gene mutations in the RAS pathway are relatively rare in MDS and their clinical significance remains unclear. Both NRAS and KRAS have been associated with disease progression in this neoplasm [98]. Ren et al. studied 370 patients with MDS and identified RAS pathway mutations in 57 (15.4%) of them. Patients with RAS mutations had a higher median percentage of marrow blasts, a greater number of co-mutated genes, were more frequently classified as higher-risk according to IPSS-R, and showed a higher AML transformation compared to those with wild-type RAS MDS patients. Although RAS mutations did not significantly affect the response to disease-modifying treatments, OS was significantly shorter in patients harboring these mutations compared to those with wild-type RAS [99].
4.7. Others
One of the hallmarks of MDS is aberrant DNA hypermethylation, particularly affecting genes involved in DNA repair. In a cohort of Brazilian MDS patients, the Ataxia-telangiectasia mutated (ATM) gene exhibited higher methylation levels among individuals who progressed to AML. Furthermore, ATM expression progressively declined from low-risk MDS subtypes to high-risk MDS and AML. These findings suggest that ATM is frequently silenced or downregulated in MDS, likely a due to promoter hypermethylation or gene mutations [100].
Among the enzymes involved in histone modification and gene expression, histone deacetylases play a critical role [101]. Within this group, the Sirtuins (SIRTs) family of enzymes has shown diverse functions in the transcriptional regulation, metabolic control, and genome maintenance [102]. Goes et al., in a study of 80 bone marrow samples from Brazilian patients with MDS, reported altered SIRT expression pattern associated with specific clinical and prognostic features of MDS [103].
5. Molecular Data Integrated in Clinical Scores
Molecular information in MDS has been integrated into a comprehensive model combining clinical parameters and cytogenetics findings: the IPSS-R for Molecular data (IPSS-M) [23] based on the earlier IPSS-R [104]. This model, developed by the International Working Group for prognosis in MDS, incorporates molecular information on 31 genes and classifies patients into six risk categories with distinct prognostic implications. The IPSS-M outperforms the IPSS-R, and allows for re-stratification of 46% of the patients [23]. Nevertheless, emerging data on specific entities, such as MDS with DDX41 mutations, suggest that the IPSS-M may not be applicable in all cases [105].
Artificial intelligence is playing an increasing role in the prognostication of hematological malignancies, including MDS [106]. The Spanish Group of Myelodysplastic Syndromes developed an Artificial Intelligence Prognostic Scoring System for MDS (AIPSS-MDS), using data from 7,202 patients. The optimal model was based on eight variables: age, gender, hemoglobin, leukocyte count, platelet count, neutrophil percentage, bone marrow blast percentage, and cytogenetic risk group. This model accurately predicted OS and PFS, and showed superiority over both the IPSS-R and the age-adjusted IPSS-R [107]. Data from LA comes from Lincango et al., who compared the performance of the IPSS-M with various other prognostic systems, including molecular scores like the European MDS (EuroMDS) clustering system and the Munich Leukemia Laboratory (MLL) models, as well as non-molecular scores such as the AIPSS-MDS and IPSS-R. This retrospective study included 182 adult patients (>18 years old) diagnosed with myelodysplastic neoplasms between 2009 and 2022 at five centers in Argentina and one in Uruguay [39,108]. The IPSS-M model predicted OS more accurately than other molecular-based models. Interestingly, the AIPSS-MDS performed similarly to IPSS-M, with slight differences depending on whether the scores were analyzed as continuous or grouped into risk categories. AIPSS-MDS demonstrated superior prognostic power compared to the IPSS-R. When simplifying classification into low- and high-risk groups for clinical management, 51% of patients categorized as low-risk (<3.5) by the IPSS-R would have been reclassified as high-risk according to the AIPSS-MDS, with no cases being downstaged. In contrast, only 13% of patients were upstaged from low-risk by IPSS-R to high-risk by IPSS-M, while 5% were downstaged and might have been considered for lower-intensity treatment. Using the median risk score from the original training set, AIPSS-MDS appears to better identify low risk patients with longer OS [24,107]. This model may represent a more suitable prognostic tool than the IPSS-R, particularly in the resource-limited settings in our region, where molecular testing is not routinely available due to infrastructure and reimbursement constraints [24].
6. Molecular Data Incorporated in Current Classifications and New Proposals
The fifth edition of the WHO classification of hematolymphoid tumors introduces significant advancements in the understanding and diagnosis of myeloid neoplasms. Particularly in the context of MDS, the WHO classification now recognizes novel entities such as clonal hematopoiesis, and broadly divides MDS into two categories (1) MDS with defining genetic abnormalities and (2) MDS defined morphologically. The category of MDS with defining genetic abnormalities includes the following entities: MDS with biallelic TP53 inactivation, MDS with low blasts and SF3B1 mutation (MDS-SF3B1), and MDS with low blasts and del(5q) [109]. In turn, the ICC of myeloid neoplasms highlights TP53 and SF3B1 mutations, establishing them as distinct entities [35]. The latter classification also remain MDS-related molecular findings for those patients with bone marrow blasts between 10 and 19% described by Lindsley et al., including mutations in splicing factors, STAG2, EZH2, ASXL1, BCOR [30], and additionally RUNX1 [30,35]. The remaining MDS cases are defined by the blast count. In this regard, mutational profile, gene associations and co-mutation patterns may lead to the identification of even more new entities. Since blast count remains as a key parameter distinguishing MDS and AML, while softened in the presence of AML-related abnormalities, it is subjective measure, and the boundary between low and elevated blasts can be controversial. Huber et al. identified nine genetically defined, non-overlapping hierarchical subgroups: (1) biallelic TP53 mutations (biTP53), (2) complex karyotype, (3) mutated RUNX1, (4) mutated ASXL1, (5) deletion of 5q [del(5q)], (6) mutated SF3B1, (7) mutations in U2AF1, SRSF2, and/or ZRSR2 (splicing factor-positive, SP+), (8) presence of at least one mutation in DNMT3A, TET2, or other genes recurrently mutated in MDS (splicing factor-negative with ≥1 mutation, SP-/≥1), and (9) complete absence of any of the above genetic markers (SP-/0) [108].
More recently, Bernard et al., investigated how genetic alterations define specific disease subtypes using 3,233 representative MDS patient samples (median age, 72 years) from the IPSS-M cohort, and a 152-gene targeted NGS panel in conjunction with cytogenetics [4]. Diagnostic subtypes defined by genetic biomarkers (MDS-5q, MDS-SF3B1, or MDS-biTP53) represented 23% and 22% of cases according to the WHO 2022 and ICC classifications, respectively. A total of 10,564 oncogenic mutations across 126 genes (in 91% of patients), 3,558 chromosomal alterations (in 43% of patients), and 375 cnLOH events (in 11% of patients), were identified. The analysis enabled the identification of 16 molecular subgroups (comprising 86% of the patients) and two residual groups with distinct phenotypes (Table 1), further refining the molecular taxonomy of the MDS [4,110].
7. Molecular Data to Tailor Treatment Strategies
Despite the emergence of new therapies, no significant improvement in outcomes has been observed in LA over the last two-three decades [111,112]. Data from the U.S. National Cancer Data Base showed that OS has not improved over time, except in younger patients (<40 years old) [113]. These findings may not yet reflect the impact of molecular profiling on treatment decisions. Indeed, molecular data provide not only prognostic information but are also essential for guiding targeted therapies. For example, SF3B1-positive variants responds to luspartercept in the low risk MDS [114]; IDH1-positive patients may benefit from ivosidenib [115]; and IDH2-positive patients from enasidenib [116]. TP53 mutations may serve as markers of response to lenalidomide and predictors of relapse after transplantation. Somatic mutations in splicing factor genes, NPM1 and IDH1/2 are aslo likely to predict response to venetoclax [117,118,119]. Additionally, patients carrying DDX41 mutations have shown exceptional responses to venetoclax combined with HMA [120]. Although data remain limited and sometimes conflicting, TET2 and/or DNMT3A mutation and the ASXL1 wild-type status were predictor of better responses to HMA [121,122,123].
8. Germline Predispositions: Implications and Challenges in Latin America
Recent advances in genomic research have revealed that a significant proportion of patients diagnosed with MDS carry germline pathogenic variants in genes associated with predisposition to hematologic malignancies. This discovery has markedly shifted the traditional paradigm, as MDS was previously considered solely an acquired disorder. Today, it is estimated that between 4% and 10% of MDS cases are linked to germline mutations, with higher frequencies observed in younger individuals and those with a family history indicative of inherited susceptibility. In pediatric populations, familial or inherited forms of AML and MDS may account for up to 50% of cases, while in young adults the figure ranges from 10% to 20%, dropping to around 5–10% in older adults [124,125,126,127,128,129]. In one cohort, 4% of 588 unselected pediatric patients with acute leukemias carried deleterious germline variants [130]. Among adults with AML, 6% were found to harbor clinically significant germline mutations in cancer-predisposing genes [131]. Furthermore, in a cohort of 404 patients with MDS undergoing allogeneic stem cell transplantation, at least one pathogenic germline variant was detected in 7% of cases, with the highest detection rate among adolescents [132]. Importantly, these numbers may underestimate the true prevalence, as many studies are limited by incomplete gene panels, lack of evaluation for copy number or noncoding variants, and exclusion of patients without confirmed germline status.
The clinical and molecular landscape of germline predisposition to myeloid malignancies is highly heterogeneous. These conditions may result from variants inherited from a parent or arise de novo during gametogenesis or early embryonic development. Modes of inheritance vary depending on the gene involved, and include autosomal dominant, autosomal recessive, and X-linked patterns [133]. The penetrance, age at onset, and spectrum of hematologic or systemic manifestations differ markedly between syndromes. The significance of these germline syndromes was acknowledged with the inclusion of a dedicated chapter in the 2016 revision of the WHO classification of hematopoietic neoplasms [134], reflecting growing clinical awareness and increased availability of NGS. The WHO 2022 and ICC 2022 classifications further expanded the list of recognized syndromes [3,35]. Table 2 shows 2022 WHO and ICC classification of myeloid neoplasms with germline predisposition.
Given the overlap between sporadic and inherited cases in terms of clinical presentation, comprehensive molecular profiling and detailed family history are essential for identifying germline cases (Table S1). Timely recognition of these conditions has clinical implications for clinical management, including treatment, optimization transplant donor selection, and also genetic familial counseling and cancer surveillance in at-risk relatives. As research progresses, refining the criteria for germline testing and integrating them into diagnostic pathways will be crucial for improving outcomes in MDS and related myeloid neoplasms.
Among the most recognized syndromes is familial AML with germline CEBPA mutations. These patients typically develop leukemia in early to middle adulthood, often without preceding hematologic abnormalities [135,136,137]. Although the precise prevalence is unknown, familial cases may represent up to 1% of all AML, particularly in the context of a normal karyotype [138]. CEBPA mutations usually affect the N-terminal domain and follow an autosomal dominant inheritance pattern, with nearly complete penetrance. A second somatic mutation in the alternate allele is often acquired during leukemogenesis [135,136]. These leukemias generally respond well to standard therapy, but careful donor selection is essential, as using related donors with the same germline variant has resulted in donor-derived leukemia [137].
Another well-characterized syndrome is associated with germline mutations in DDX41, which encodes a DEAD-box helicase involved in RNA processing. Studies have found DDX41 mutations in 1.5–2.5% of adults with MDS or AML, making it one of the most common hereditary myeloid neoplasia syndromes [139]. This condition predominantly affects older adults and is often asymptomatic until the onset of MDS or AML. Although the penetrance appears high, many affected individuals lack a family history of hematologic malignancies, likely due to age-related expression [140]. Germline mutations of DDX41, located at 5q35.3, most commonly include, missense and frameshift variants [139], although large deletions have also been reported [141]. Most cases involve an inherited truncating variant with a second somatic hit in the opposite allele. Interestingly, cytogenetic analysis has shown that acquisition of a del(5q) may represent the second hit in these patients [142]. Ongoing clinical trials are evaluating the potential benefits of monitoring unaffected DDX41 mutation carriers, focusing on detecting low-frequency secondary somatic mutations in DDX41 [143]. Treatment of hematologic malignancies associated with germline variants of DDX41 is similar to that of others AML and MDS [139]. Importantly, these patients may experience more severe graft-versus-host disease after transplantation, particularly when receiving grafts from wild-type donors [144].
Mutations in RUNX1 cause familial platelet disorder with a predisposition to myeloid malignancy. This autosomal dominant syndrome is characterized by lifelong mild-to-moderate thrombocytopenia and impaired platelet function, frequently accompanied by an elevated risk of MDS, AML, or T-acute lymphoblastic leukemia (T-ALL) [145,146]. The lifetime risk of hematologic malignancy in affected individuals ranges from 35–40%, with wide variability in age at diagnosis. The phenotypes of MDS, AML, and T-ALL in familial platelet disorder vary widely [145,147,148,149,150]. Leukemic transformation commonly involves a second somatic hit in RUNX1 or cooperating mutations (e.g., in GATA2), and cytogenetic abnormalities such as del(7q), 3q aberrations plus or gain of an additional chromosome 21 bearing the mutations [142].
Other hereditary syndromes involve genes such as ANKRD26 and ETV6, both associated with autosomal dominant thrombocytopenia and an increased predisposition to myeloid malignancies, including MDS and acute leukemia. Patients typically present with lifelong mild to moderate thrombocytopenia and normal platelet size, often without significant bleeding complications. Despite the usually mild hemorrhagic phenotype, the underlying clonal instability confers a substantial long-term risk of malignant transformation [151]. ANKRD26-related thrombocytopenia (THC2) is a rare disorder, with fewer than 100 pedigrees described in the literature to date. Pathogenic variants typically occur in the 5’ untranslated region (5’UTR) of the gene, leading to abnormal upregulation of ANKRD26 in megakaryocytes, defective platelet production, and a lifetime risk of MDS or AML estimated at 8–20% [152,153,154]. Germline ETV6 mutations are similarly rare, with estimated prevalence below 1% among individuals with inherited thrombocytopenia. They may also be associated with a broader cancer predisposition, including lymphoid malignancies and, less frequently, solid tumors. The lifetime risk of hematologic malignancy in ETV6 mutation carriers ranges from 30% and 40%, though penetrance and age of onset are highly variable [155,156,157].
The syndrome of GATA2 deficiency has a heterogeneous presentation and may be associated with MDS, acute leukemia (AL), bone marrow failure, congenital neutropenia, or other hematologic abnormalities. It is among the more common inherited AL/MDS disorders, especially in children and young adults. GATA2 deficiency encompasses a wide range of clinical phenotypes, including immunodeficiency, lymphedema, and myeloid neoplasia [158,159]. Many patients develop MDS or AML by their third decade, often presenting with monosomy 7/del(7q), trisomy 8 or other cytogenetic abnormalities such as der(1;7)(q10;p10), although 35% of cases show a normal karyotype [142,160]. Germline mutations in GATA2 (chromosome band 3q21.3) may involve missense, truncating frameshift, nonsense, large genomic rearrangements, or mutations within a conserved enhancer element in intron 5 [161]. Commercial panels not designed to detect intronic sequences or whole-exome approaches may miss theses alterations. Surveillance and timely transplantation are critical in these patients due to progressive cytopenia and risk of severe infections.
SAMD9 and SAMD9L germline mutations are a significant cause of pediatric MDS, especially in the context of monosomy 7 and inherited bone marrow failure where it is detected in up to 20% of patients [162,163]. These gain-of-function mutations inhibit hematopoiesis, sometimes leading to spontaneous loss of the affected chromosome, which may restore hematopoietic function but increase the risk of MDS. Clinical presentations can include MIRAGE syndrome and ataxia-pancytopenia phenotypes. Mutations in these genes are identified in up to 20% of pediatric MDS cases with monosomy 7. Management requires a multidisciplinary approach due to complex clinical manifestations [164].
Although TP53 mutations are more frequently associated with solid tumors, germline alterations in this gene also confer a rare but significant predisposition to hematologic malignancies, particularly in the context of Li-Fraumeni syndrome. This autosomal dominant disorder is classically associated with early-onset breast cancer, sarcomas, brain tumors, and adrenocortical carcinomas; however, MDS and AL can occur in a subset of carriers without prior solid tumors, with a median age of first cancer diagnosis between 20 and 30 years [165]. It has been suggested that young individuals with biallelic TP53 mutations should be tested to rule out a germline variant. Therapeutic decisions and follow-up require a multidisciplinary approach.
These examples illustrate the complexity of inherited predisposition to myeloid neoplasms and highlight the importance of integrating germline evaluation into routine hematologic care, especially for younger patients and those with atypical clinical presentations or suggestive family histories.
Diagnostic genetic testing is increasingly recognized as a fundamental component in the evaluation of patients with AML or MDS when germline predisposition is suspected. While no single global consensus exists, published guidelines from the Nordic MDS Group and the NCCN provide key clinical criteria to guide testing [166,167]. The Spanish group has also published guidelines online, covering a broad aspect from laboratory testing to genetic counseling and clinical management [168]. These guidelines emphasize the importance of germline evaluation in patients diagnosed with MDS/AML before the age of 50, especially in the presence of a personal or family history suggestive of inherited predisposition. According to the Nordic group, testing is recommended in patients with: (1) two first- or second-degree relatives with MDS/AML or chronic thrombocytopenia, with at least one diagnosed before age 50; (2) a proband with MDS/AML and two relatives with solid tumors, one of them under 50; or (3) a diagnosis before age 50 associated with features of bone marrow failure syndromes or cytogenetic alterations, particularly involving chromosome 7 [166]. The NCCN guidelines reinforce these and expand the indications to include: (1) monosomy 7 or other chromosome 7 abnormalities in patients under 50; (2) hypocellular MDS or aplastic bone marrow at diagnosis; (3) a clinical suspicion of predisposition at any age; and (4) family donors in the context of allogeneic transplantation or candidates for transplantation with clinical signs of inherited marrow failure syndromes [167]. Both guidelines also recommend confirmatory germline testing from non-hematopoietic tissue when somatic mutations with VAF suggestive of germline origin (typically >40%) are detected. Germline testing is particularly essential when selecting related donors for hematopoietic stem cell transplantation, to avoid the risk of donor-derived leukemia [166,167].
Genetic testing should ideally be coordinated by clinicians or genetic counselors with expertise in hereditary cancer syndromes and genomic data interpretation. Testing strategies can be either targeted or broad. Broader approaches, such as whole-genome sequencing (WGS) or whole-exome sequencing (WES), may be particularly useful when no familial diagnosis has been established or when clinical features suggest multiple possible conditions. Targeted testing through gene panels is appropriate when a specific mutation has been identified in a relative or when molecular features strongly suggest a known syndrome. Depending on the method, single nucleotide variants (SNV) as well as small insertions and deletions, called Indels can be identified. To identify larger genomic rearrangements and CNV, complementary techniques such as microarrays, multiplex ligation–dependent probe amplification (MLPA), or optical genome mapping (OGM) may be necessary. Gene selection and methodology vary by laboratory, underscoring the importance of choosing appropriate strategies based on clinical context [169]. In urgent scenarios, such as a pending stem cell transplant, testing may need to be expedited or done in parallel.
In patients with hematologic malignancy, the preferred specimen for germline testing is typically cultured skin fibroblasts, as blood or bone marrow may be contaminated by malignant or clonal hematopoietic cells. Hair follicle is also recommended, particularly by the Spanish guidelines [168]. Saliva, buccal swabs, and nails may be used in some cases, but have limitations due to DNA yield or potential contamination. Blood may be used in remission or from patients not previously transplanted, but clonal hematopoiesis must be considered [166,167,170].
In LA, despite limited resources and infrastructure, early efforts are underway to better characterize germline predisposition in MDS, though data remain limited. Most countries face significant barriers to routine germline testing, including limited availability of NGS panels validated for constitutional DNA, high costs, and a shortage expertise in variant interpretation. Consequently, underdiagnosis of hereditary syndromes is likely. However, some centers, such as Uruguay, Brazil and Argentina have begun to implement protocols for analyzing germline tissue, and are developing targeted panels or applying WES to detect germline alterations. Notably, collaborations between the University of Chicago and Hospital das Clínicas (São Paulo, Brazil), on ANKRD26 and ETV6 [171], and the development of a machine learning algorithm that helps with the diagnosis of bone marrow failure in collaboration of centers from Brazil with the National Institute of Health (NIH) [172] have been established. Further collaborations include studies on RUNX1 between Argentina and Italy [173], and participation in international consortia on familial platelet disorder with associated myeloid malignancy [174]. Reports from Brazil and Argentina have described individual cases or small series involving germline mutations in RUNX1, GATA2, DDX41, and telomere biology genes. In Uruguay, additional reports described complex hereditary backgrounds, such as dual germline mutations in RUNX1 and DDX41 [175], and familial cases with CEBPA mutations affecting donor selection for transplantation [176,177]. These examples underscore the feasibility and clinical relevance of integrating germline analysis even in resource-limited settings.
Improving recognition of suggestive phenotypes and increasing access to genetic counseling and molecular testing are critical for equitable implementation of personalized medicine across LA. However, a survey conducted by Grupo Latinoamericano de Mielodisplasia (GLAM) across 21 centers in 12 countries revealed that only 28% of laboratories (6/21) had the capacity to confirm the germline origin of variants [178], mainly due to a lack of access to confirmatory testing using non-hematopoietic tissues (e.g., fibroblasts culture). This gap has serious implications, particularly for genetic counseling, familial risk assessment, and transplant donor selection. In a region where related donors are frequently used, failure to detect germline mutations can negatively impact outcomes.
To address this gap, it is essential to establish regional or national reference centers, define standardized criteria for germline testing, and promote clinician and laboratory training in variant interpretation. Strengthening referral networks and fostering collaborative frameworks will ensure timely and accurate diagnosis. Initiatives such as GLAM play a key role in raising awareness, building technical capacity, and promoting equitable access to molecular diagnostics across the region.
9. Molecular Tools in Latin America
The diagnosis and treatment of MDS remain difficult challenging due to significant economic and technological disparities within and among LA countries [7,179]. In 2015, the GLAM was established as a multidisciplinary group of healthcare professionals dedicated to the study of MDS, working in close collaboration with regional hematology societies [7]. In alignment with its goals, GLAM conducted an initial survey involving 458 respondents from nine LA countries (Argentine, Bolivia, Chile, Colombia, Dominican Republic, Ecuador, Paraguay, Peru and Uruguay). The results revealed heterogeneous access to the conventional cytogenetic analysis (ranging to 53% to 100%) and flow cytometry (42% to 100%), despite efforts toward standardized implementation [7,179,180]. Subsequently, the GLAM MDS registry, known as Re-GLAM, showed that NGS panels were used in only 15.2% of the registered patients [181]. To evaluate the real-world laboratory practice regarding NGS technology, a 33-question survey was distributed and responses collected between March 2022 and January 2023. The survey addressed various aspects of NGS implementation across LA institutions. In total, 37 centers in 12 countries were contacted, with and 21 of 26 providing nearly complete responses.
Responding centers were fairly evenly distributed between private and publicly funded institutions, including universities, health or research centers, and clinical laboratories (Figure 1). Among them, 11 centers focused on adult patients, while 8 had mixed-age patient population. Initial funding sources included governmental support (n=12), the American Society of Hematology (ASH) (n=2), private investment (n=11), and combined funding mechanisms (n=5), including International Consortium on Acute Leukemia (ICAL) projects (n=4). Most laboratories reported using Illumina platforms (73%) and commercial NGS panels (69%), employing either amplicon- or captured-based technologies, primarily developed by Illumina (n=9) or Sophia Genetics (n=7). In terms of expertise, 9 out of 21 centers reported having less than one year of experience or being in the early stages of NGS implementation. The majority (57%) adapted several guidelines to report their findings, despite six having no one defined. Others results are shown in Figure 1. We consider this effort a foundational step toward optimizing laboratory capacities, promoting the share of best practices, and facilitating the transfer of scientific and technical expertise across LA [178].
10. Conclusions and Perspectives
In the era of a molecular revolution with the widespread use of NGS technology, comprehensive analyses of the genetic heterogeneity in MDS have guided advances in the diagnostic workup over the last fifteen years. These advances have enabled the identification of disease subgroups associated with disease phenotypes and outcomes, as well as prognostic information that impacts the clinical treatment. New genetically defined entities, such as MDS with del(5q), MDS with SF3B1 mutation, or and the important role of TP53 inactivation, have been included in both the WHO5 and the ICC 2022 classification. Both systems have eliminated the ≥20% blast requirement in the presence of AML-defining abnormalities and, in turn, incorporated the concept of clonal hematopoiesis. The recognition of the importance of germline predisposition gives insights for outlining disease models and their therapeutic management.
Risk stratification and prognostication are crucial for the appropriate management of patients who are commonly assessed using the IPSS-R. Mutational information is also being incorporated into prognostic scoring systems, with IPSS-M as a valuable tool for personalized risk assessment and treatment decisions. Among other molecular-based proposals for classification, the recently developed molecular taxonomy of MDS will likely facilitate the implementation of precision medicine approaches.
The main challenge related to these latest advances is to grant access of MDS patients to their genomic profiling. In the past few years, new laboratories adopting the NGS technology have been opening and collaborative projects are underway; however, only a minority of our patients get access to this, which clearly indicates the needs of LA patients with MDS are unmet. Ideally, access to new technologies would extend across all of LA and not remain confined to specialized centers. As emphasized by Dr Cazola and Dr Malcovati concluded [182], achieving this goal will require the development of specialized infrastructures within healthcare systems, driven by close collaboration among healthcare providers, academia, and the life sciences industry throughout LA.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1: Hereditary Syndromes Associated with Myeloid Malignancies.
Author Contributions
Conceptualization, A.L.B., C.B.; methodology, A.L.B., V.A., S.G., C.B.; resources, A.L.B., C.B. and A.W.; writing—original draft preparation, A.L.B., S.G.; writing—review and editing, V.A., S.G., C.B.; supervision, A.L.B., C.B.; project administration, A.L.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank all members of GLAM and Andrés Posse for their assistance with the language revision of the manuscript.
Conflicts of Interest
A.L.B. received honoraria for conference from Abbvie. The other authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| MDS | Myelodysplastic syndromes |
| AML | Acute myeloid leukemia |
| WHO | World Health Organization |
| LA | Latin America |
| SEER | Surveillance, Epidemiology, and End Results |
| CMML | Chronic myelomonocitic leukemia |
| NGS | Next-generation sequencing |
| CNV | Copy number variations |
| IPSS-R | International Prognostic Scoring System Revised |
| IPSS-M | International Prognostic Scoring System Molecular |
| OS | Overall Survival |
| LFS | Leukemia Free Survival |
| VAF | Variant allele frequency |
| HMA | Hypomethylating agents |
| CHIP | Clonal hematopoiesis of indeterminate potential |
| PFS | Progression free survival |
| AIPSS | Artificial Intelligence Prognostic Scoring System |
References
- Hasserjian, R.P., U. Germing, and L. Malcovati, Diagnosis and classification of myelodysplastic syndromes. Blood, 2023. 142(26): p. 2247-2257.
- Luo, B., et al., Myelodysplastic syndromes are multiclonal diseases derived from hematopoietic stem and progenitor cells. Exp Hematol Oncol, 2022. 11(1): p. 28.
- Khoury, J.D., et al., The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia, 2022. 36(7): p. 1703-1719.
- Bernard, E., et al., Molecular taxonomy of myelodysplastic syndromes and its clinical implications. Blood, 2024. 144(15): p. 1617-1632.
- Gou, X., Z. Chen, and Y. Shangguan, Global, regional, and national burden of myelodysplastic syndromes and myeloproliferative neoplasms, 1990-2021: an analysis from the global burden of disease study 2021. Front Oncol, 2025. 15: p. 1559382.
- Economic Commission for Latin America and the Caribbean (ECLAC). Demographic Observatory, 2024 (LC/PUB.2024/22-P), Santiago, 2024.. 2024. Available online: https://repositorio.cepal.org/server/api/core/bitstreams/dc566b9e-b3ef-44e9-a167-995614696404/content.
- Crisp, R., et al., Myelodysplastic syndromes in Latin America: state of the art. Blood Adv, 2018. 2(Suppl 1): p. 60-62.
- Goksu, S.Y., et al., The impact of race and ethnicity on outcomes of patients with myelodysplastic syndromes: a population-based analysis. Leuk Lymphoma, 2022. 63(7): p. 1651-1659.
- Borges, D.P., et al., Functional polymorphisms of DNA repair genes in Latin America reinforces the heterogeneity of Myelodysplastic Syndrome. Hematol Transfus Cell Ther, 2023. 45(2): p. 147-153.
- Ribeiro Junior, H.L., Molecular monitoring of myelodysplastic neoplasm: Don’t just watch this space, consider the patient’s ancestry. Br J Haematol, 2024. 205(3): p. 759-760.
- Belli, C.B., et al., Myelodysplastic syndromes in South America: a multinational study of 1080 patients. Am J Hematol, 2015. 90(10): p. 851-8.
- Belli, C.B., et al., Application of the revised International Prognostic Scoring System for myelodysplastic syndromes in Argentinean patients. Ann Hematol, 2014. 93(4): p. 705-7.
- Gonzalez, J.S., et al., Prognostic assessment for chronic myelomonocytic leukemia in the context of the World Health Organization 2016 proposal: a multicenter study of 280 patients. Ann Hematol, 2021. 100(6): p. 1439-1449.
- Azevedo, R.S., et al., Age, Blasts, Performance Status and Lenalidomide Therapy Influence the Outcome of Myelodysplastic Syndrome With Isolated Del(5q): A Study of 58 South American Patients. Clin Lymphoma Myeloma Leuk, 2022. 22(1): p. e1-e6.
- Lazzarino, C., et al., Severe thrombocytopenia as a predictor of survival and response to hypomethylating agents in myelodysplastic syndromes: A Latin-American cohort of 212 patients. Am J Hematol, 2020. 95(12): p. E323-E326.
- Castelo, L., et al., Alternative Dosing Schedules of Azacitidine: A Real-World Study Across South American Centers. Clin Lymphoma Myeloma Leuk, 2024. 24(6): p. 407-411.
- Moura, A.T.G., et al., Prolonged response to recombinant human erythropoietin treatment in patients with myelodysplastic syndrome at a single referral centre in Brazil. Clinics (Sao Paulo), 2019. 74: p. e771.
- Bejar, R., et al., Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med, 2011. 364(26): p. 2496-506.
- Yoshida, K., et al., Frequent pathway mutations of splicing machinery in myelodysplasia. Nature, 2011. 478(7367): p. 64-9.
- Duployez, N. and C. Preudhomme, Monitoring molecular changes in the management of myelodysplastic syndromes. Br J Haematol, 2024. 205(3): p. 772-779.
- Papaemmanuil, E., et al., Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood, 2013. 122(22): p. 3616-27; quiz 3699.
- Haferlach, T., et al., Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia, 2014. 28(2): p. 241-7.
- Bernard, E., et al., Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid, 2022. 1(7): p. EVIDoa2200008.
- Lincango, M., et al., Assessing the Relevance of Non-molecular Prognostic Systems for Myelodysplastic Syndrome in the Era of Next-Generation Sequencing. Ann Lab Med, 2025. 45(1): p. 44-52.
- Catalán, A.I., et al., Integration of NGS and CNV Analysis in Prognostic Evaluation of MDS in a Developing Country: Insights from Uruguay. Blood Global Hematology, 2025. Accepted for publication, 2025.
- Tseng, C.C. and E.A. Obeng, RNA splicing as a therapeutic target in myelodysplastic syndromes. Semin Hematol, 2024. 61(6): p. 431-441.
- Reinig, E., et al., Targeted Next-Generation Sequencing in Myelodysplastic Syndrome and Chronic Myelomonocytic Leukemia Aids Diagnosis in Challenging Cases and Identifies Frequent Spliceosome Mutations in Transformed Acute Myeloid Leukemia. Am J Clin Pathol, 2016. 145(4): p. 497-506.
- Thol, F., et al., Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood, 2012. 119(15): p. 3578-84.
- Tefferi, A., et al., Targeted next-generation sequencing in myelodysplastic syndromes and prognostic interaction between mutations and IPSS-R. Am J Hematol, 2017. 92(12): p. 1311-1317.
- Lindsley, R.C., et al., Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood, 2015. 125(9): p. 1367-76.
- Jiang, M., et al., SF3B1 mutations in myelodysplastic syndromes: A potential therapeutic target for modulating the entire disease process. Front Oncol, 2023. 13: p. 1116438.
- Malcovati, L., et al., SF3B1-mutant MDS as a distinct disease subtype: a proposal from the International Working Group for the Prognosis of MDS. Blood, 2020. 136(2): p. 157-170.
- Lincango, M., I. Larripa, and C. Belli, Bioinformatic Evaluation of Differentially Expressed Genes in Myelodysplastic Syndromes with SF3B1 Variants. Medicina (B Aires), 2022. 82(Supl. V): p. 98.
- Lincango, M., et al., Validation of Novel Hub Genes Expression in Patients with Myelodysplastic Syndrome and SF3B1 Mutations. Medicina (B Aires), 2024. 84(Suppl. V).
- Arber, D.A., et al., International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood, 2022. 140(11): p. 1200-1228.
- Donaires, F.S., et al., Splicing factor SF3B1 mutations and ring sideroblasts in myelodysplastic syndromes: a Brazilian cohort screening study. Rev Bras Hematol Hemoter, 2016. 38(4): p. 320-324.
- Sarmiento, M., et al., Efficacy of lenalidomide in a patient with systemic mastocytosis associated with SF3B1-mutant myelodysplastic syndrome. Leuk Lymphoma, 2021. 62(12): p. 3027-3030.
- Badar, T., et al., U2AF1 pathogenic variants in myeloid neoplasms and precursor states: distribution of co-mutations and prognostic heterogeneity. Blood Cancer J, 2023. 13(1): p. 149.
- Bersanelli, M., et al., Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J Clin Oncol, 2021. 39(11): p. 1223-1233.
- Kuendgen, A., et al., Efficacy of azacitidine is independent of molecular and clinical characteristics - an analysis of 128 patients with myelodysplastic syndromes or acute myeloid leukemia and a review of the literature. Oncotarget, 2018. 9(45): p. 27882-27894.
- Kim, E., et al., SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell, 2015. 27(5): p. 617-30.
- Makishima, H., et al., Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet, 2017. 49(2): p. 204-212.
- Nagehan, P., et al., Impact of single versus multiple spliceosome mutations on myelodysplastic syndrome. J Clin Exp Hematop, 2023. 63(3): p. 173-176.
- Cockey, S.G., et al., Molecular landscape and clinical outcome of SRSF2/TET2 Co-mutated myeloid neoplasms. Leuk Lymphoma, 2025. 66(3): p. 469-478.
- Thol, F., et al., Prognostic significance of ASXL1 mutations in patients with myelodysplastic syndromes. J Clin Oncol, 2011. 29(18): p. 2499-506.
- Yang, L., X. Wei, and Y. Gong, Prognosis and risk factors for ASXL1 mutations in patients with newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Cancer Med, 2024. 13(1): p. e6871.
- Lin, Y., et al., Prognostic significance of ASXL1 mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia: A meta-analysis. Hematology, 2016. 21(8): p. 454-61.
- Rinke, J., et al., EZH2 in Myeloid Malignancies. Cells, 2020. 9(7).
- Ball, S., et al., Clinical characteristics and outcomes of EZH2-mutant myelodysplastic syndrome: A large single institution analysis of 1774 patients. Leuk Res, 2023. 124: p. 106999.
- Damm, F., et al., BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood, 2013. 122(18): p. 3169-77.
- Sportoletti, P., D. Sorcini, and B. Falini, BCOR gene alterations in hematologic diseases. Blood, 2021. 138(24): p. 2455-2468.
- Baranwal, A., et al., Genetic landscape and clinical outcomes of patients with BCOR mutated myeloid neoplasms. Haematologica, 2024. 109(6): p. 1779-1791.
- Liu, Y., et al., Next generation sequencing reveals the mutation landscape of Chinese MDS patients and the association between mutations and AML transformations. Hematology, 2024. 29(1): p. 2392469.
- Meyer, C., et al., The KMT2A recombinome of acute leukemias in 2023. Leukemia, 2023. 37(5): p. 988-1005.
- Wei, Q., et al., Detection of KMT2A Partial Tandem Duplication by Optical Genome Mapping in Myeloid Neoplasms: Associated Cytogenetics, Gene Mutations, Treatment Responses, and Patient Outcomes. Cancers (Basel), 2024. 16(24).
- Lee, W.H., et al., Validation of the molecular international prognostic scoring system in patients with myelodysplastic syndromes defined by international consensus classification. Blood Cancer J, 2023. 13(1): p. 120.
- Fathima, S., et al., Myeloid neoplasms with PHF6 mutations: context-dependent genomic and prognostic characterization in 176 informative cases. Blood Cancer J, 2025. 15(1): p. 28.
- Zuo, Z., et al., Concurrent Mutations in SF3B1 and PHF6 in Myeloid Neoplasms. Biology (Basel), 2022. 12(1).
- Sudunagunta, V.S. and A.D. Viny, Untangling the loops of STAG2 mutations in myelodysplastic syndrome. Leuk Lymphoma, 2025. 66(1): p. 6-15.
- Kon, A., et al., Recurrent mutations in multiple components of the cohesin complex in myeloid neoplasms. Nat Genet, 2013. 45(10): p. 1232-7.
- Katamesh, B., et al., Clinical and prognostic impact of STAG2 mutations in myeloid neoplasms: the Mayo Clinic experience. Blood Adv, 2023. 7(8): p. 1351-1355.
- Deb, P.Q. and W. Xiao, “Ring-form” megakaryocytic dysplasia in STAG2-mutated myelodysplastic neoplasm. Blood, 2024. 143(21): p. 2218.
- Kawashima, N., et al., Landscape of biallelic DNMT3A mutant myeloid neoplasms. J Hematol Oncol, 2024. 17(1): p. 87.
- Yang, L., R. Rau, and M.A. Goodell, DNMT3A in haematological malignancies. Nat Rev Cancer, 2015. 15(3): p. 152-65.
- Challen, G.A., et al., Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet, 2011. 44(1): p. 23-31.
- Jaiswal, S., et al., Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med, 2014. 371(26): p. 2488-98.
- Lee, W.H., et al., Clinico-genetic and prognostic analyses of 716 patients with primary myelodysplastic syndrome and myelodysplastic syndrome/acute myeloid leukemia based on the 2022 International Consensus Classification. Am J Hematol, 2023. 98(3): p. 398-407.
- GenoMed4All, c., A sex-informed approach to improve the personalised decision making process in myelodysplastic syndromes: a multicentre, observational cohort study. Lancet Haematol, 2023. 10(2): p. e117-e128.
- Jawad, M., et al., DNMT3A R882 Mutations Confer Unique Clinicopathologic Features in MDS Including a High Risk of AML Transformation. Front Oncol, 2022. 12: p. 849376.
- Zhang, X., et al., TET (Ten-eleven translocation) family proteins: structure, biological functions and applications. Signal Transduct Target Ther, 2023. 8(1): p. 297.
- Hawking, Z.L. and J.M. Allan, Landscape of TET2 Mutations: From Hematological Malignancies to Solid Tumors. Cancer Med, 2025. 14(6): p. e70792.
- Smith, A.E., et al., Next-generation sequencing of the TET2 gene in 355 MDS and CMML patients reveals low-abundance mutant clones with early origins, but indicates no definite prognostic value. Blood, 2010. 116(19): p. 3923-32.
- Danishevich, A., et al., Myelodysplastic Syndrome: Clinical Characteristics and Significance of Preclinically Detecting Biallelic Mutations in the TET2 Gene. Life (Basel), 2024. 14(5).
- Awada, H., et al., Invariant phenotype and molecular association of biallelic TET2 mutant myeloid neoplasia. Blood Adv, 2019. 3(3): p. 339-349.
- Bezerra, M.F., et al., Screening for myeloid mutations in patients with myelodysplastic syndromes and AML with myelodysplasia-related changes. Hematol Transfus Cell Ther, 2022. 44(3): p. 328-331.
- Komrokji, R., et al., IDH mutations are enriched in myelodysplastic syndrome patients with severe neutropenia and can be a potential for targeted therapy. Haematologica, 2023. 108(4): p. 1168-1172.
- Patnaik, M.M., et al., Differential prognostic effect of IDH1 versus IDH2 mutations in myelodysplastic syndromes: a Mayo Clinic study of 277 patients. Leukemia, 2012. 26(1): p. 101-5.
- Wang, N., et al., IDH1 Mutation Is an Independent Inferior Prognostic Indicator for Patients with Myelodysplastic Syndromes. Acta Haematol, 2017. 138(3): p. 143-151.
- Jain, A.G., et al., Patterns of lower risk myelodysplastic syndrome progression: factors predicting progression to high-risk myelodysplastic syndrome and acute myeloid leukemia. Haematologica, 2024. 109(7): p. 2157-2164.
- Bellissimo, D.C. and N.A. Speck, RUNX1 Mutations in Inherited and Sporadic Leukemia. Front Cell Dev Biol, 2017. 5: p. 111.
- Sutandyo, N., et al., Association of Somatic Gene Mutations with Risk of Transformation into Acute Myeloid Leukemia in Patients with Myelodysplastic Syndrome: A Systematic Review and Meta-Analysis. Asian Pac J Cancer Prev, 2022. 23(4): p. 1107-1116.
- He, W., C. Zhao, and H. Hu, Prognostic effect of RUNX1 mutations in myelodysplastic syndromes: a meta-analysis. Hematology, 2020. 25(1): p. 494-501.
- Wang, Y.H., et al., Higher RUNX1 expression levels are associated with worse overall and leukaemia-free survival in myelodysplastic syndrome patients. EJHaem, 2022. 3(4): p. 1209-1219.
- Watad, A., et al., Somatic Mutations and the Risk of Undifferentiated Autoinflammatory Disease in MDS: An Under-Recognized but Prognostically Important Complication. Front Immunol, 2021. 12: p. 610019.
- Okano, T., et al., Somatic mutation in RUNX1 underlies mucocutaneus inflammatory manifestations. Rheumatology (Oxford), 2021. 60(12): p. e429-e431.
- Hulea, L. and A. Nepveu, CUX1 transcription factors: from biochemical activities and cell-based assays to mouse models and human diseases. Gene, 2012. 497(1): p. 18-26.
- Dermawan, J.K., et al., Clinically Significant CUX1 Mutations Are Frequently Subclonal and Common in Myeloid Disorders With a High Number of Co-mutated Genes and Dysplastic Features. Am J Clin Pathol, 2022. 157(4): p. 586-594.
- Aly, M., et al., Distinct clinical and biological implications of CUX1 in myeloid neoplasms. Blood Adv, 2019. 3(14): p. 2164-2178.
- Wang, Q., et al., ETV6 mutation in a cohort of 970 patients with hematologic malignancies. Haematologica, 2014. 99(10): p. e176-8.
- Gurney, M., et al., The clinical and molecular spectrum of ETV6 mutated myeloid neoplasms. Br J Haematol, 2023. 202(2): p. 279-283.
- Siddon, A.J. and O.K. Weinberg, Diagnosis and Classification of Myelodysplastic Syndromes with Mutated TP53. Clin Lab Med, 2023. 43(4): p. 607-614.
- Boada, M., et al., TP53 Myelodysplastic Syndromes in Latin America. Real World Data from Latin American MDS Group (GLAM). Leukemia Research, 2023. 128S: p. 107158.
- Hsu, J.I., et al., PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell, 2018. 23(5): p. 700-713 e6.
- Fandrei, D., et al., Clonal Evolution of PPM1D Mutations in the Spectrum of Myeloid Disorders. Clin Cancer Res, 2025.
- Xu, F., et al., Somatic mutations of activating signalling, transcription factor, and tumour suppressor are a precondition for leukaemia transformation in myelodysplastic syndromes. J Cell Mol Med, 2022. 26(23): p. 5901-5916.
- Menssen, A.J., et al., Convergent Clonal Evolution of Signaling Gene Mutations Is a Hallmark of Myelodysplastic Syndrome Progression. Blood Cancer Discov, 2022. 3(4): p. 330-345.
- Sumiyoshi, R., et al., The FLT3 internal tandem duplication mutation at disease diagnosis is a negative prognostic factor in myelodysplastic syndrome patients. Leuk Res, 2022. 113: p. 106790.
- da Silva-Coelho, P., et al., Clonal evolution in myelodysplastic syndromes. Nat Commun, 2017. 8: p. 15099.
- Ren, Y., et al., Co-mutation landscape and clinical significance of RAS pathway related gene mutations in patients with myelodysplastic syndrome. Hematol Oncol, 2023. 41(1): p. 159-166.
- Pinheiro, R.F., et al., The Ataxia-telangiectasia mutated (ATM) is the most important gene for repairing the DNA in Myelodysplastic Neoplasm. DNA Repair (Amst), 2025. 146: p. 103803.
- Yang, X.J. and E. Seto, HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene, 2007. 26(37): p. 5310-8.
- Dai, Y. and D.V. Faller, Transcription Regulation by Class III Histone Deacetylases (HDACs)-Sirtuins. Transl Oncogenomics, 2008. 3: p. 53-65.
- Goes, J.V.C., et al., Gene expression patterns of Sirtuin family members (SIRT1 TO SIRT7): Insights into pathogenesis and prognostic of Myelodysplastic neoplasm. Gene, 2024. 915: p. 148428.
- Greenberg, P.L., et al., Revised international prognostic scoring system for myelodysplastic syndromes. Blood, 2012. 120(12): p. 2454-65.
- Baliakas, P., et al., How to manage patients with germline DDX41 variants: Recommendations from the Nordic working group on germline predisposition for myeloid neoplasms. Hemasphere, 2024. 8(8): p. e145.
- Nazha, A., et al., Personalized Prediction Model to Risk Stratify Patients With Myelodysplastic Syndromes. J Clin Oncol, 2021. 39(33): p. 3737-3746.
- Mosquera Orgueira, A., et al., Machine Learning Improves Risk Stratification in Myelodysplastic Neoplasms: An Analysis of the Spanish Group of Myelodysplastic Syndromes. Hemasphere, 2023. 7(10): p. e961.
- Huber, S., et al., MDS subclassification-do we still have to count blasts? Leukemia, 2023. 37(4): p. 942-945.
- Loghavi, S., et al., Fifth Edition of the World Health Classification of Tumors of the Hematopoietic and Lymphoid Tissue: Myeloid Neoplasms. Mod Pathol, 2024. 37(2): p. 100397.
- Vaughan, L. and J.E. Pimanda, Seeing MDS through the lens of genomics. Blood, 2024. 144(15): p. 1552-1554.
- Mendonca, P.D.S., R.F. Pinheiro, and S.M.M. Magalhaes, Myelodysplastic Syndrome Over Time: A Comparative Analysis of Overall Outcome. Mayo Clin Proc, 2019. 94(12): p. 2593-2594.
- Basquiera, A., et al., Myelodysplasia-Related Mortality Remains the Main Cause of Death Along Different Groups of Risks: An Analysis from MDS Argentinean Study Group. Haematologica, 2017. 102(s2): p. 485. Abstract n. 1182.
- Al-Kali, A., et al., Outcome of Myelodysplastic Syndromes Over Time in the United States: A National Cancer Data Base Study From 2004-2013. Mayo Clin Proc, 2019. 94(8): p. 1467-1474.
- Burke, S., O. Chowdhury, and K. Rouault-Pierre, Low-risk MDS-A spotlight on precision medicine for SF3B1-mutated patients. Hemasphere, 2025. 9(3): p. e70103.
- DiNardo, C.D., et al., Final phase 1 substudy results of ivosidenib for patients with mutant IDH1 relapsed/refractory myelodysplastic syndrome. Blood Adv, 2024. 8(15): p. 4209-4220.
- DiNardo, C.D., et al., Targeted therapy with the mutant IDH2 inhibitor enasidenib for high-risk IDH2-mutant myelodysplastic syndrome. Blood Adv, 2023. 7(11): p. 2378-2387.
- Othman, J., et al., Molecular MRD is strongly prognostic in patients with NPM1-mutated AML receiving venetoclax-based nonintensive therapy. Blood, 2024. 143(4): p. 336-341.
- Senapati, J., et al., Venetoclax abrogates the prognostic impact of splicing factor gene mutations in newly diagnosed acute myeloid leukemia. Blood, 2023. 142(19): p. 1647-1657.
- Lachowiez, C.A., et al., Contemporary outcomes in IDH-mutated acute myeloid leukemia: The impact of co-occurring NPM1 mutations and venetoclax-based treatment. Am J Hematol, 2022. 97(11): p. 1443-1452.
- Nanaa, A., et al., Venetoclax plus hypomethylating agents in DDX41-mutated acute myeloid leukaemia and myelodysplastic syndrome: Mayo Clinic series on 12 patients. Br J Haematol, 2024. 204(1): p. 171-176.
- Itzykson, R., et al., Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia, 2011. 25(7): p. 1147-52.
- Traina, F., et al., Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia, 2014. 28(1): p. 78-87.
- Hu, C. and X. Wang, Predictive and prognostic value of gene mutations in myelodysplastic syndrome treated with hypomethylating agents: a meta-analysis. Leuk Lymphoma, 2022. 63(10): p. 2336-2351.
- Zhang, J., et al., Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med, 2015. 373(24): p. 2336-2346.
- Schwartz, J.R., et al., The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun, 2017. 8(1): p. 1557.
- Feurstein, S., et al., Germline variants drive myelodysplastic syndrome in young adults. Leukemia, 2021. 35(8): p. 2439-2444.
- Sebert, M., et al., Germline DDX41 mutations define a significant entity within adult MDS/AML patients. Blood, 2019. 134(17): p. 1441-1444.
- Churpek, J.E., et al., Genomic analysis of germ line and somatic variants in familial myelodysplasia/acute myeloid leukemia. Blood, 2015. 126(22): p. 2484-90.
- DiNardo, C.D., et al., Evaluation of Patients and Families With Concern for Predispositions to Hematologic Malignancies Within the Hereditary Hematologic Malignancy Clinic (HHMC). Clin Lymphoma Myeloma Leuk, 2016. 16(7): p. 417-428 e2.
- Zhang, J., K.E. Nichols, and J.R. Downing, Germline Mutations in Predisposition Genes in Pediatric Cancer. N Engl J Med, 2016. 374(14): p. 1391.
- Yang, F., et al., Identification and prioritization of myeloid malignancy germline variants in a large cohort of adult patients with AML. Blood, 2022. 139(8): p. 1208-1221.
- Feurstein, S., et al., Germ line predisposition variants occur in myelodysplastic syndrome patients of all ages. Blood, 2022. 140(24): p. 2533-2548.
- Trottier, A.M., S. Feurstein, and L.A. Godley, Germline predisposition to myeloid neoplasms: Characteristics and management of high versus variable penetrance disorders. Best Pract Res Clin Haematol, 2024. 37(1): p. 101537.
- Arber, D.A., et al., The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood, 2016. 127(20): p. 2391-405.
- Owen, C., M. Barnett, and J. Fitzgibbon, Familial myelodysplasia and acute myeloid leukaemia--a review. Br J Haematol, 2008. 140(2): p. 123-32.
- Smith, M.L., et al., Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med, 2004. 351(23): p. 2403-7.
- Tawana, K., et al., Disease evolution and outcomes in familial AML with germline CEBPA mutations. Blood, 2015. 126(10): p. 1214-23.
- Taskesen, E., et al., Prognostic impact, concurrent genetic mutations, and gene expression features of AML with CEBPA mutations in a cohort of 1182 cytogenetically normal AML patients: further evidence for CEBPA double mutant AML as a distinctive disease entity. Blood, 2011. 117(8): p. 2469-75.
- Polprasert, C., et al., Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell, 2015. 27(5): p. 658-70.
- Quesada, A.E., et al., DDX41 mutations in myeloid neoplasms are associated with male gender, TP53 mutations and high-risk disease. Am J Hematol, 2019. 94(7): p. 757-766.
- Makishima, H., et al., Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood, 2023. 141(5): p. 534-549.
- Auger, N., et al., Cytogenetics in the management of myelodysplastic neoplasms (myelodysplastic syndromes, MDS): Guidelines from the groupe francophone de cytogenetique hematologique (GFCH). Curr Res Transl Med, 2023. 71(4): p. 103409.
- Homan, C.C., et al., Somatic mutational landscape of hereditary hematopoietic malignancies caused by germline variants in RUNX1, GATA2, and DDX41. Blood Adv, 2023. 7(20): p. 6092-6107.
- Saygin, C., et al., Allogeneic hematopoietic stem cell transplant outcomes in adults with inherited myeloid malignancies. Blood Adv, 2023. 7(4): p. 549-554.
- Owen, C.J., et al., Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood, 2008. 112(12): p. 4639-45.
- Song, W.J., et al., Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet, 1999. 23(2): p. 166-75.
- Arepally, G., et al., Evidence for genetic homogeneity in a familial platelet disorder with predisposition to acute myelogenous leukemia (FPD/AML). Blood, 1998. 92(7): p. 2600-2.
- Beri-Dexheimer, M., et al., Clinical phenotype of germline RUNX1 haploinsufficiency: from point mutations to large genomic deletions. Eur J Hum Genet, 2008. 16(8): p. 1014-8.
- Churpek, J.E., et al., Identification and molecular characterization of a novel 3′ mutation in RUNX1 in a family with familial platelet disorder. Leuk Lymphoma, 2010. 51(10): p. 1931-5.
- Michaud, J., et al., In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood, 2002. 99(4): p. 1364-72.
- Noris, P. and A. Pecci, Hereditary thrombocytopenias: a growing list of disorders. Hematology Am Soc Hematol Educ Program, 2017. 2017(1): p. 385-399.
- Noris, P., et al., Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood, 2011. 117(24): p. 6673-80.
- Noris, P., et al., ANKRD26-related thrombocytopenia and myeloid malignancies. Blood, 2013. 122(11): p. 1987-9.
- Marconi, C., et al., 5’UTR point substitutions and N-terminal truncating mutations of ANKRD26 in acute myeloid leukemia. J Hematol Oncol, 2017. 10(1): p. 18.
- Noetzli, L., et al., Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet, 2015. 47(5): p. 535-538.
- Rampersaud, E., et al., Germline deletion of ETV6 in familial acute lymphoblastic leukemia. Blood Adv, 2019. 3(7): p. 1039-1046.
- Zhang, M.Y., et al., Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet, 2015. 47(2): p. 180-5.
- Wlodarski, M.W., et al., Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood, 2016. 127(11): p. 1387-97; quiz 1518.
- Hahn, C.N., et al., Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet, 2011. 43(10): p. 1012-7.
- Donadieu, J., et al., Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica, 2018. 103(8): p. 1278-1287.
- Spinner, M.A., et al., GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood, 2014. 123(6): p. 809-21.
- Sahoo, S.S., E.J. Kozyra, and M.W. Wlodarski, Germline predisposition in myeloid neoplasms: Unique genetic and clinical features of GATA2 deficiency and SAMD9/SAMD9L syndromes. Best Pract Res Clin Haematol, 2020. 33(3): p. 101197.
- Bluteau, O., et al., A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood, 2018. 131(7): p. 717-732.
- Narumi, S., et al., SAMD9 mutations cause a novel multisystem disorder, MIRAGE syndrome, and are associated with loss of chromosome 7. Nat Genet, 2016. 48(7): p. 792-7.
- Gonzalez, K.D., et al., Beyond Li Fraumeni Syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol, 2009. 27(8): p. 1250-6.
- Baliakas, P., et al., Nordic Guidelines for Germline Predisposition to Myeloid Neoplasms in Adults: Recommendations for Genetic Diagnosis, Clinical Management and Follow-up. Hemasphere, 2019. 3(6): p. e321.
- National Comprehensive Cancer Network. Myelodysplastic Syndromes (Version 2.2025). 2025 April 13, 2025]. Available online: https://www.nccn.org/professionals/physician_gls/pdf/mds.pdf.
- Hemoterapia, S.E.d.H.y. Manual para el Diagnóstico y Atención Clínica en la Predisposición Germinal a Neoplasias Hematológicas. 2024. Available online: https://sehh.es/publicaciones/manuales-publicaciones/126044-manual-para-el-diagnostico-y-atencion-clinica-en-la-predisposicion-germinal-a-neoplasias-hematologicas.
- Roloff, G.W., L.A. Godley, and M.W. Drazer, Assessment of technical heterogeneity among diagnostic tests to detect germline risk variants for hematopoietic malignancies. Genet Med, 2021. 23(1): p. 211-214.
- DeRoin, L., et al., Feasibility and limitations of cultured skin fibroblasts for germline genetic testing in hematologic disorders. Hum Mutat, 2022. 43(7): p. 950-962.
- Drazer, M.W., et al., Clonal hematopoiesis in patients with ANKRD26 or ETV6 germline mutations. Blood Adv, 2022. 6(15): p. 4357-4359.
- Gutierrez-Rodrigues, F., et al., Differential diagnosis of bone marrow failure syndromes guided by machine learning. Blood, 2023. 141(17): p. 2100-2113.
- Kamiya, L.J., et al., Two novel families with RUNX1 variants indicate glycine 168 as a new mutational hotspot: Implications for FPD/AML diagnosis. Br J Haematol, 2024. 205(6): p. 2315-2320.
- Cunningham, L., et al., Natural history study of patients with familial platelet disorder with associated myeloid malignancy. Blood, 2023. 142(25): p. 2146-2158.
- Bove, V., et al., Myelodysplastic syndrome with dual germline RUNX1 and DDX41 variants: a rare genetic predisposition case. Fam Cancer, 2025. 24(1): p. 20.
- Spangenberg, M.N., et al., Unusual Co-Occurrence of Multiple Myeloma and AML in a Patient With Germline CEBPA Variant. Expanding the Spectrum of Hereditary Hematologic Malignancies. Clin Genet, 2025. 107(5): p. 576-578.
- Boada, M., et al., Germline CEBPA mutation in familial acute myeloid leukemia. Hematol Rep, 2021. 13(3): p. 9114.
- Belli, C., et al., P008 - Topic: AS01-Diagnosis/AS01c-Molecular aberrations (cytogenetic, genetic, gene expression): REAL-WORLD LABORATORY PRACTICE PATTERNS ON NEXT-GENERATION-SEQUENCING (NGS) TECHNOLOGY IN LATIN AMERICA. Leukemia Research, 2023. 128: p. 107143.
- Crisp, R., et al., [Preferences and limitations of hematologists to address the complexity of myelodysplastic syndromes]. Medicina (B Aires), 2019. 79(3): p. 174-184.
- Grille Montauban, S., et al., Flow cytometry “Ogata score” for the diagnosis of myelodysplastic syndromes in a real-life setting. A Latin American experience. Int J Lab Hematol, 2019. 41(4): p. 536-541.
- Grille, S., et al., Myelodysplastic Syndromes in Latin-America - Results from a Novel International Registry: Re-Glam. Blood, 2022. 140(Supplement 1): p. 4096-4097.
- Cazzola, M. and L. Malcovati, Genome sequencing in the management of myelodysplastic syndromes and related disorders. Haematologica, 2025. 110(2): p. 312-329.
Figure 1.
Data from a survey conducted by GLAM in LA to know about laboratory practices regarding NGS technology.
Figure 1.
Data from a survey conducted by GLAM in LA to know about laboratory practices regarding NGS technology.
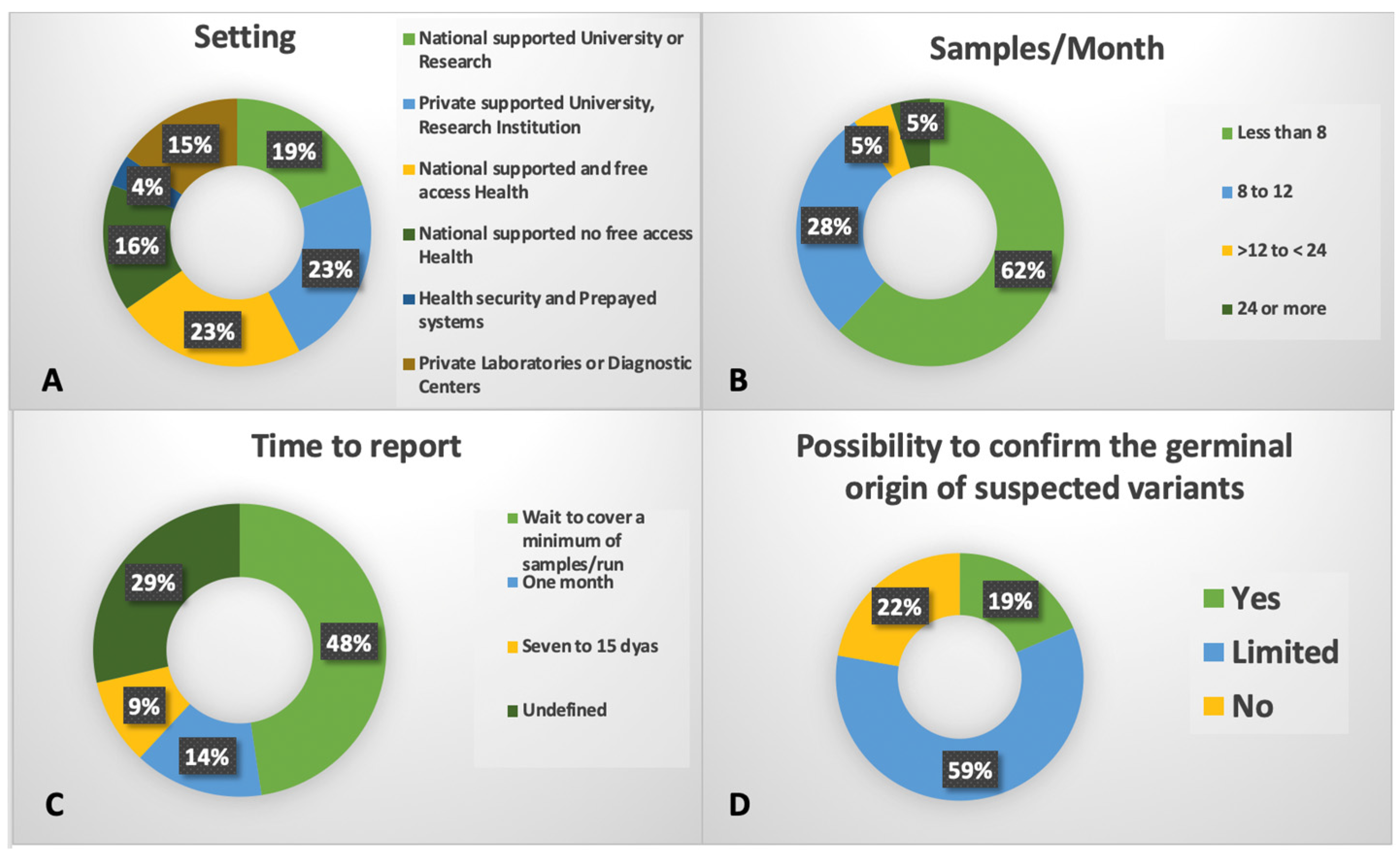

Table 1.
Molecular classifications of myelodysplastic syndromes.*.
| Group | Entity | % | Comments |
|---|---|---|---|
| Validated 5 established subgroups | DDX41 | 3.3 | 56% of patients with mutated DDX41 had both a putative germ line DDX41 variant (defined here as >30% VAF) and a somatic DDX41 mutation, 37% only a putative germ line DDX41 variant, and 7% only somatic DDX41 mutations. |
| AML-like | 2 | NPM1 mutations or at least 2 events from WT1, FLT3, MLLPTD, or MYC mutations. | |
| TP53 complex | 10 | Multihit TP53 mutations were present in 74% cases, of which 91% had complex karyotype. | |
| del/5q) | 6.9 | Presence of del(5q) as the sole cytogenetic abnormality or with 1 additional abnormality excluding −7/7q. Monoallelic TP53 mutations were significantly enriched in this group | |
| SF3B1 | 14 | Indolent clinical course. | |
| Confirmed 3 previously reported subgroups | Bi-allelic TET2 | 13 | Early biallelic TET2 mutations with splicing factor mutations in 80% of patients, most commonly affecting SRSF2, SF3B1, or ZRSR2. Modulation of phenotype by ASXL1 and RAS mutations driving monocytosis and JAK2 driving thrombocytosis. |
| der(1;7) | 0.5 | ETNK1 mutations were enriched in this group. | |
| CCUS-like | 6.9 | 46% had a single mutated gene (TET2, or DNMT3A), 8% had loss of Y without gene mutations, and 6% only had ≥2 DTA mutations. | |
| Eight novel subgroups | -7/SETBP1 | 4.9 | SETBP1 mutations and/or −7 in the absence of complex karyotype. GATA2 variants were prevalent. |
| EZH2-ASXL1 | 4 | ASXL1 and EZH2 mutation co-occurrence. High molecular complexity (75% of patients with ≥5 mutated genes). | |
| IDH-STAG2 | 8.9 | Mutations at the IDH2 R140 hot spot, IDH1, and/or STAG2 co-occurring with either SRSF2 or ASXL1 mutations | |
| BCOR/L1 | 3.5 | 83% of patients had mutations in BCOR, 33% in BCORL1, and 17% in both genes. | |
| U2AF1 | 4.3 | 58% had a Q157 mutation, 41% had a S34 mutation, and 1% had both. | |
| SRSF2 | 2.2 | Aggressive disease. | |
| ZRSR2 | 1.3 | Indolent clinical course. | |
| Two subgroups without defining genetic events | Not otherwise specified | 7.9 | Presence of other cytogenetic abnormalities and/or mutations in 51 other recurrently mutated genes |
| No event | 6.5 | Absence of any recurrent drivers evaluated. |
* Abbreviations: AML: acute myeloid leukemia; CCUS: clonal cytopenia of undetermined significance.
Table 2.
Classifications of Myeloid Neoplasms Associated with Germline Predisposition.*.
| World Health Organization (2022) fifth edition | International Consensus Classification (2022) |
|---|---|
|
Myeloid neoplasms with germline predisposition without a preexisting platelet disorder or organ dysfunction: Germline CEBPA P/LP variant Germline DDX41 P/LP variant Germline TP53 P/LP variant |
Hematologic neoplasms with germline predisposition without a constitutional disorder affecting multiple organ systems: Myeloid neoplasms with germline CEBPA variant Myeloid or lymphoid neoplasms with germline DDX41 variant Myeloid or lymphoid neoplasms with germline TP53 variant |
|
Myeloid neoplasms with germline predisposition and pre-existing platelet disorder: Germline RUNX1 P/LP variant Germline ANKRD26 P/LP variant Germline ETV6 P/LP variant |
Hematologic neoplasms with germline predisposition associated with a constitutional platelet disorder: Myeloid or lymphoid neoplasms with germline RUNX1 variant Myeloid neoplasms with germline ANKRD26 variant Myeloid or lymphoid neoplasms with germline ETV6 variant |
|
Myeloid neoplasms with germline predisposition and potential organ dysfunction: Germline GATA2 P/LP variant Bone marrow failure syndromes (Severe congenital neutropenia Shwachman, Diamond syndrome, Fanconi anemia) Telomere biology disorders RASopathies (Neurofibromatosis type 1, CBL syndrome Noonan syndrome or Noonan syndrome–like disorders) Down syndrome Germline SAMD9 P/LP variant (MIRAGE Syndrome) Germline SAMD9L P/LP variant (SAMD9L-related Ataxia Pancytopenia Syndrome) Biallelic germline BLM P/LP variant (Bloom syndrome) |
Hematologic neoplasms with germline predisposition associated with a constitutional disorder affecting multiple organ systems: Myeloid neoplasms with germline GATA2 variant Myeloid neoplasms with germline SAMD9 variant Myeloid neoplasms with germline SAMD9L variant Myeloid neoplasms associated with bone marrow failure syndromes (Fanconi anemia, Shwachman-Diamond syndrome, Telomere biology disorders, including dyskeratosis congenita Severe congenital neutropenia, Diamond-Blackfan anemia) JMML associated with neurofibromatosis JMML associated with Noonan syndrome and Noonan syndrome–like disorder Myeloid or lymphoid neoplasms associated with Down syndrome |
* Abbreviations: JMML: juvenile myelomonocytic leukemia.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



