Submitted:
17 April 2025
Posted:
17 April 2025
You are already at the latest version
Abstract
Peripheral artery disease (PAD) is a chronic progressive accumulation of atherosclerotic lesions with varying degrees of arterial obstruction determining ischemic symptoms of the involved extremities. PAD is associated with decreased bioavailable nitric oxide due to endothelial cell dysfunction and the development and progression of vascular arterial stiffening (VAS). Atherosclerosis also plays an essential role in the development and progression of vascular arterial stiffening (VAS), which associates with endothelial cell activation and dysfunction that results in a proinflammatory endothelium with a decreased ability to produce bioavailable nitric oxide (NO). NO is one of three gasotransmitters along with carbon monoxide and hydrogen sulfide that promotes vasodilation. NO plays a crucial role in the regulation of PAD and a deficiency in its bioavailability is strongly linked to the development of atherosclerosis, VAS, and PAD. A decreased arterial patency may also occur due to a reduction in the elasticity or diameter of the vessel wall due to the progressive nature of VAS and atherosclerosis in PAD. Progressive atherosclerosis and VAS promote narrowing over time, which lead to impairment of vasorelaxation and extremity blood flow. This narrative review examines how atherosclerosis, aging and hypertension, metabolic syndrome and type 2 diabetes, tobacco smoking, endothelial cell activation and dysfunction with decreased NO, VAS with its increased damaging pulsatile pulse pressure results in microvessel remodeling. Further, the role of ischemia and ischemia reperfusion injury is discussed and how it contributes to ischemic skeletal muscle remodeling, ischemic neuropathy, and pain perception in PAD.
Keywords:
Atherosclerosis
; Brain endothelial cell activation and dysfunction
; Ischemic myopathy and myalgia
; Ischemic neuropathy
; Microvessel disease
; Nitric oxide
; Peripheral artery disease
; Pulse pressure
; Vascular arterial stiffening
“Man is as old as his arteries”
Thomas Sydenham (1624-1689) an English physician.
1. Introduction
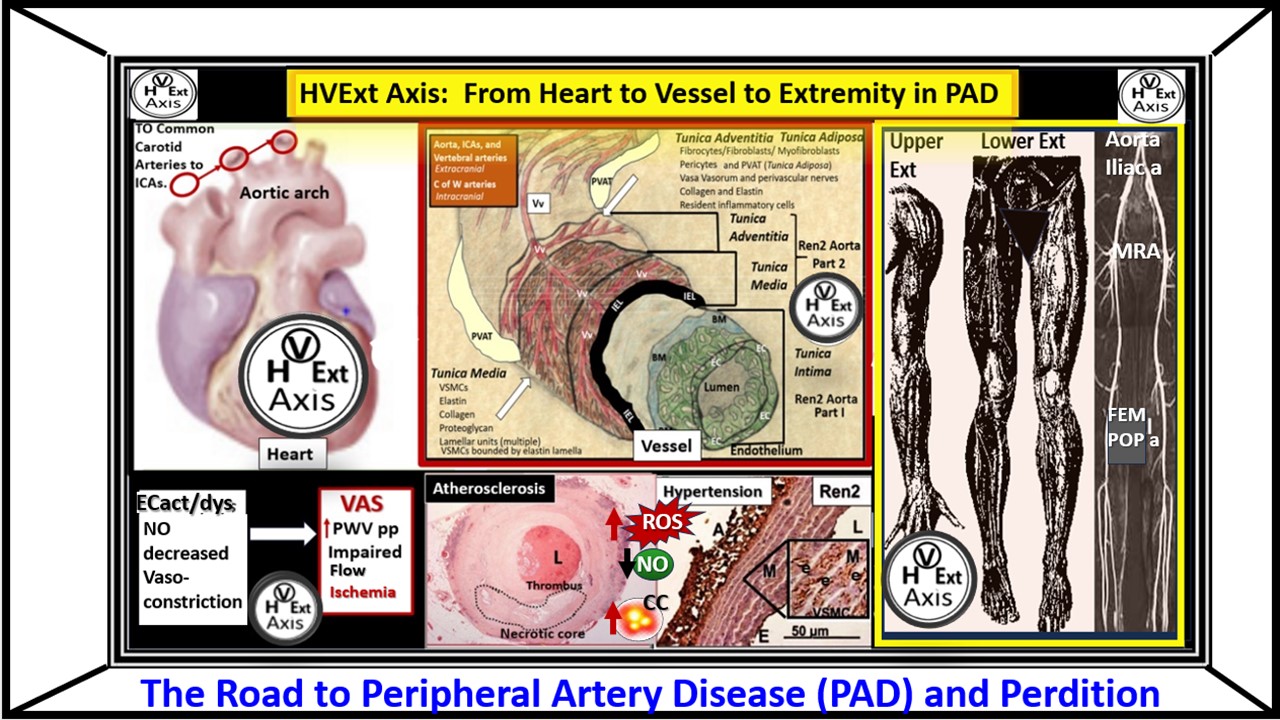
Age-related peripheral artery disease (PAD) and atherosclerosis with vascular ossification-calcification (VOC) have been around since the times of ancient Egyptians that were discovered by the studies of mummies via computed tomography scanning, which reveled vascular calcifications [1,2,3]. Globally, PAD affects 200-250 million people [4,5]. Currently, we are living in one of the oldest living populations in history [6,7,8] and therefore, we can expect to experience an even higher occurance of symptomatic PAD, atherosclerosis, VOC, hypertension (specifically systolic hypertension) with increased injurious pulsatile pulse pressure (pp) and cardiovascular disease (CVD) [4,5,9,10].
The presence of PAD is associated with a marked increase in the prediction of cerebrocardiovascular disease (CCVD) events such as myocardial infarction and stroke with increased morbidity and mortality [11]. Further, Grenon et al. were able to demonstrate that coronary artery disease (CAD) plus PAD had greater risk for cerebrocardiovascular disease (CCVD) events such as myocardial infarction and stroke with increased morbidity and mortality than those individuals with just CAD alone [12]. Indeed, PAD should be considered a red flag warning in that it serves as a biomarker for the increased risk, morbidity and mortality of CCVD events [11,12].
The vascular endothelial cell(s) (ECs) with their protruding cell surface protective endothelial glycocalyx (ecGCx) are the first cells and surface layer that are exposed to the circulating blood plasma, red blood cells, leukocytes, and multiple injurious species that are capable of triggering EC activation and dysfunction (ECact/dys) (Figure 1 and Figure 2) [13,14,15,16,17,18,19,20,21].
ECact/dys results in a proinflammatory surface with attenuation and/or loss of the ecGCx (activation), while ECdys results in a decrease in bioavailable nitric oxide (NO) [9]. Indeed, EC dysfunction with decreased bioavailability of NO may sound an alarm within the cells of the vessel wall (Figure 3) [13,14,15,16,17,18,19,20,21,22].
Interestingly, the decrease in the bioavailability of NO may not only sound an alarm but also may act to activate an alarmin system also referred to as damage-associated molecular patterns (DAMPs), wherein multiple response to injury inflammatory immune signals interact with pattern recognition receptors such as Toll-like receptors (TLRs) [23,24]. A brief list of EC alarmins involved with atherosclerosis include: high mobility group box 1(HMGB1), Fas ligand, heat shock proteins (HSP70-90), S100AB, and S100A9 [23,24].
PAD results primarily from chronic and progressive atherosclerotic disease characterized by narrowing of the peripheral arteries and reduced blood flow to the lower extremities [25]. Vascular arterial stiffening (VAS) of the extremities is closely related to the continued development of atherosclerosis and its progression. As atherosclerotic plaque burden progresses and extracellular matrix accumulate, the arteries and particularly the arterioles will have less lumen diameter and stiffening will continue to progess with less ability to dilate and carry blood oxygen and nutrients resulting in ischemia, ischemic pain and neuropathy in the extremities with intermittent claudication (IC) that may progress to resting pain with ischemic ulceration and or limb loss with critical limb ischemia (CLI) [26].
The primary aim of this narrative review is to increase the database of knowledge regarding the development and progression of PAD in order to decrease its increasing morbidity and mortality. In the following sections, author will discuss PAD and the role of atherosclerosis, aging and HTN, metabolic syndrome and type 2 diabetes mellitus. Also addressed, is the importance of bioavailable NO and its pathologic injurious structural remodeling roles and functional abnormalities in PAD that associate with decreased bioavailability in the presence of ECact/dys as in Figure 3. Additionally, VAS and vascular ossification calcification (VOC), microvessel remodeling in skeletal muscle, and ischemic neuropathy with neuronal remodeling in the lower extremities and the perception of pain will be discussed along with concluding remarks and future directions.
2. Peripheral Arterial Disease (PAD)
PAD results from a chronic progressive accumulation of atherosclerotic lesions that is associated with increased VAS, which is associated with increasing injurious pulsatile pulse pressure (pp) to the endothelium and vessel wall. PAD also associates with decreasing NO bioavailability (as a result of ECact/dys), which results in varying degrees of vascular obstruction and determines the clinical ischemic symptoms of the involved extremities. Further, PAD is often described as a partial or complete occlusion of circulating blood flow to the extremities due to a decrease in patency as a result of not only a reduction in elasticity associated with VAS but also a reduction in the diameter (due to atherosclerosis with enlarging atherosclerotic plaques with or without thrombosis) of the extremity vessels [11,12].
2.1. Atherosclerosis in PAD
Atherosclerosis is a systemic, focal occurring, chronic, progressive, fibroprolifertive, multifactorial, and dysfunctional endothelial-intimal disease that is also a primary underlying and significant cause of VAS and PAD. Further, atherosclerosis is caused by the retention of modified low density lipoprotein-cholesterol (modLDL-C), hemodynamic, and oxidative redox stress (OxRS), which is a term that is inclusive of reactive oxygen species (ROS), reactive nitrogen species (RNS), reactive oxygen, nitrogen and sulfur species (RONSS) and the reactive species interactome (RSI) that results in remodeling of the arterial vessel wall (Figure 4) [1,21,25,27,28,29,30]
Atherosclerosis is known to be the leading cause of vascular disease (cardiovascular, cerebrovascular, and peripheral artery disease) globally and is significantly associated with dementia when utilizing carotid intima-media thickness to assess the degree of atherosclerosis [31,32]. It initially occurs at areas of turbulent blood flow and low shear stress such as bifurcations and side branches of arteries such as iliac; femoral bifurcations distally and progresses in a cephalad manner to involve the coronaries, carotids and extracranial arteries [1,27,33,34]. While atherosclerosis is a systemic disease it is also focal occurring in regions where there is low shear stress and turbulent blood flow such as bifurcations and side branches (known as atherosclerotic prone regions of the arterial tree). Additionally, atherosclerosis is usually absent in arterial segments with high shear stress and laminar blood flow such as the internal mammary arteries (referred to as atherosclerotic resistant regions) [34]. Importantly, atherosclerosis is not only a fibroproliferative, angiogenic, prothrombotic, multifactorial disease but also a chronic inflammatory disease that begins in the intima. This intimal disease is facilitated by the multiple injurious species as in Figure 1 and Figure 2 (the dirty dozen) and especially the penetration and retention of atherogenic modified-oxidized (modLDL-C/small dense LDL-C or the atherogenic lipid triad phenotype (high triglycerides, low high-density lipoprotein (HDL) and increased small dense lipoprotein-cholesterol (sdLDL-C [35]. Further, this intimal retention of modified and atherogenic lipids are associated with the monocyte to macrophage transformation and the formation of the macrophage foam cell due to the engorgement of modified intimal atherogenic lipids [36,37,38].
Atherosclerosis has been defined by eight major types of plaque lesions by light microscopy histopathology and more recently by magnetic resonance imaging (MRI) (Box 1) [34,39].
Box 1. Comparison of plaque types (I-VIII) by histopath microscopic images to the emerging modified American Heart Association classification for MRI. Note the importance dashed outlined type VI lesions that are known to be vulnerable and prone to rupture or undergo erosion with major and dire clinical consequences. Panel A depicts thrombo-erosion in a type 2 diabetic individual and panel B depicts plaque rupture with thrombosis. Both panels A and B were originally stained with hematoxylin and eosin; however, panel B was pseudo-colored with blue coloring in Microsoft paint to emphasize rupture as compared to erosion (see Figure 5).
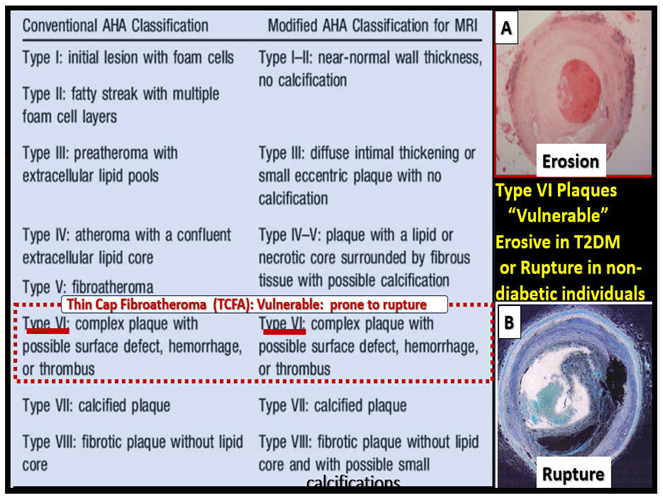
Importantly, the American Heart Association classification of type VI atherosclerotic plaques is considered the most prone to rupture because they are defined as "complicated lesions" with features like surface defects, hematoma, hemorrhage, and thrombus formation, indicating a high risk of plaque instability and potential for rupture or even erosion [40]. Additionally, note that type VII plaques are also clinically important, because they allow clinicians to diagnose subclinical coronary artery disease and also aid in predicting those individuals at high risk for future cardiovascular clinical events. Type VII calcified plaques provide the substrate for the indentification of the calcification score, which assist in the evaluation of the severity of atherosclerotic cardiovascular disease [41].
Evidence that T2DM, MetS, and obesity are associated with greater arterial stiffness as compared to non-diabetic individuals is convincing according to Stehouwer et al. and he adds at least 16 supportive references [42]. This increased VAS also helps to explain the increased CVD risk in individuals with diabetes and the MetS (Figure 5) [42,43].
Figure 5.
Comparison of type 2 diabetes mellitus (T2DM) culprit thrombo-erosive type VI lesion and sudden death due to arrhythmia to non-diabetic culprit type VI plaque rupture with thrombosis and death from drowning and acute coronary syndrome. Panel A depicts a thrombo-erosion type VI plaque with complete occlusion of lumen. Note that this plaque type is associated with a marked monocytic infiltrate in the adventitia (asterisks) and vasa vasorum (Vv) microvessels. While each of these type VI vulnerable plaques associate with increased vascular arterial stiffness they may result in similar clinical outcomes, i.e. sudden death due to atherothrombosis via different vulnerable plaque morphology. Non-diabetic plaques are more prone to rupture while T2DM individuals are more prone to develop thrombo-erosion and result in sudden death. Additionally, T2DM individuals are known to have increased vascular stiffness as compared to non-diabetic individuals. Panels A and A’ are hematoxylin and eosin (H&E) stained and Panel B is H&E stained that was pseudo-colored blue in Microsoft paint color program in order to provide contrasting colors with A and A’. Modified images provided with permission by CC 4.0 [43]. Phox 22, underlined in red is the light chain component of the reduced nicotinamide adenine dinucleotide phosphate oxidase (NADPHOx) enzyme; its positive staining strongly suggests increased oxidative stress within the plaque and the adventitia that may contribute to plaque instability and erosion in T2DM.
Figure 5.
Comparison of type 2 diabetes mellitus (T2DM) culprit thrombo-erosive type VI lesion and sudden death due to arrhythmia to non-diabetic culprit type VI plaque rupture with thrombosis and death from drowning and acute coronary syndrome. Panel A depicts a thrombo-erosion type VI plaque with complete occlusion of lumen. Note that this plaque type is associated with a marked monocytic infiltrate in the adventitia (asterisks) and vasa vasorum (Vv) microvessels. While each of these type VI vulnerable plaques associate with increased vascular arterial stiffness they may result in similar clinical outcomes, i.e. sudden death due to atherothrombosis via different vulnerable plaque morphology. Non-diabetic plaques are more prone to rupture while T2DM individuals are more prone to develop thrombo-erosion and result in sudden death. Additionally, T2DM individuals are known to have increased vascular stiffness as compared to non-diabetic individuals. Panels A and A’ are hematoxylin and eosin (H&E) stained and Panel B is H&E stained that was pseudo-colored blue in Microsoft paint color program in order to provide contrasting colors with A and A’. Modified images provided with permission by CC 4.0 [43]. Phox 22, underlined in red is the light chain component of the reduced nicotinamide adenine dinucleotide phosphate oxidase (NADPHOx) enzyme; its positive staining strongly suggests increased oxidative stress within the plaque and the adventitia that may contribute to plaque instability and erosion in T2DM.
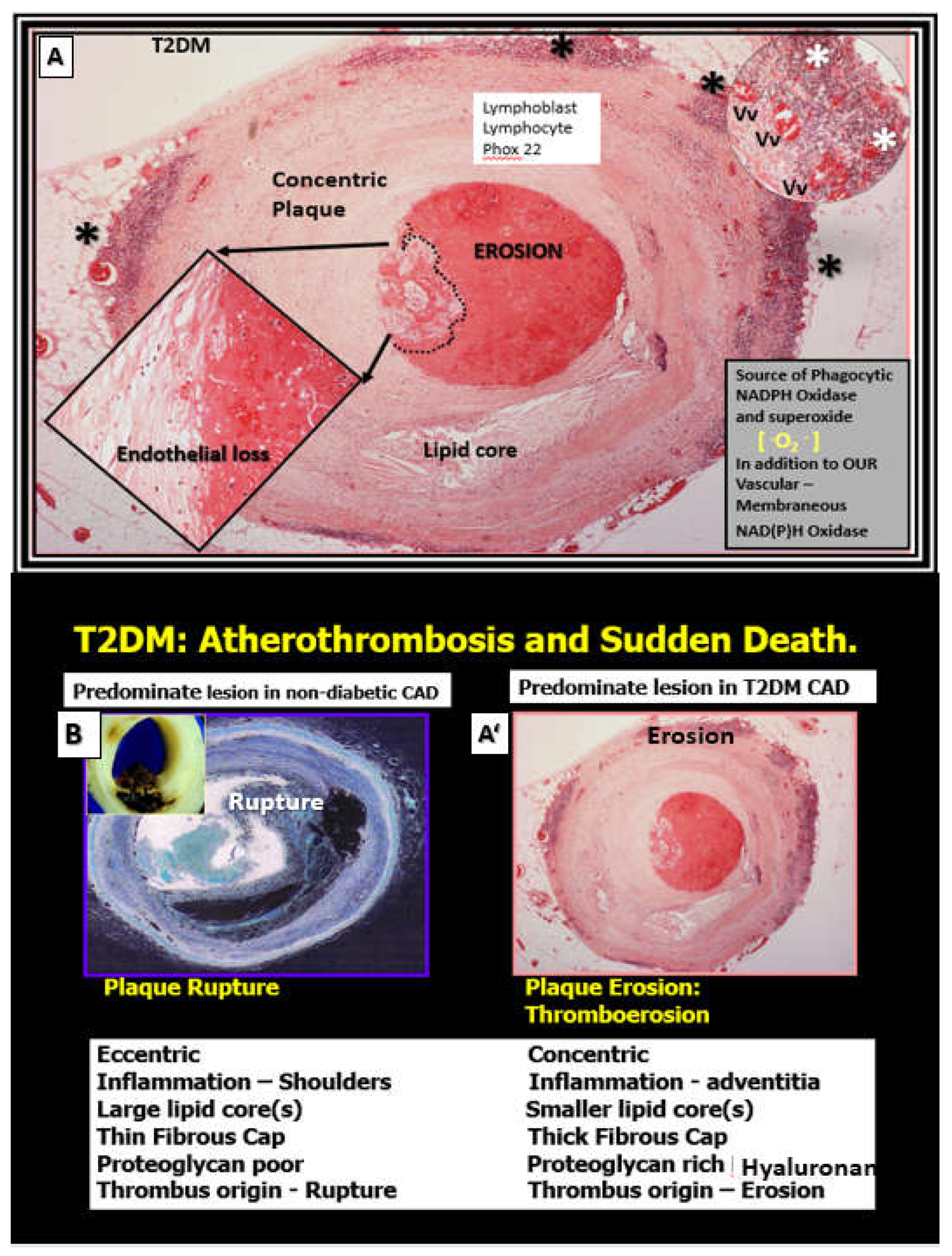
Glucose elevation in T2DM causes an increase in advanced glycation products (AGE) over time, which then promotes collagen crosslinking and interacts with its receptor RAGE and this interaction of AGE/RAGE gives rise to increased OxRS that activate matrix metalloproteinases (MMPs), which result in increased fragmentation of elastin within the media to promote stiffening of the vessel wall. Concurrently, this increase in glucose and AGE instigate a ‘phenotypic switch’ in vascular smooth muscle cell(s) (VSMCs) of the media to form a synthetic type of cell as compared to the normal contractile type that increases the synthesis of stiffer collagen and also the stiff ECM hyaluronan-glycosaminoglycan components as in Figure 4 and Figure 5 to result in increased VAS [44].
While atherosclerosis is defined as being a chronic progressive intimal disease, it is important to note that even when there is continued atheroma lipid, PGN, hyaluronan expansion that these lesions do not usually become clinically symptomatic unless there are superimposed progressive remodeling changes. These changes include the deposition of thrombotic material (intraplaque hemorrhage), inflammation, calcification, and malignant-like invasion via angiogenesis of the atheromatous plaque with plaque hemorrhage and rupture [27,34,43]. The malignant-like invasion of the plaque by the adventitial-derived vasa vasorum (Vv) vessels [27,45] allow for the direct delivery of inflammatory cells. Also, these thin walled Vv vessels are prone to rupture and result in intraplaque hemorrhage with the subsequent release of red blood cels into the lipid core and supply a rich source of membranous lipid material to increase free cholesterol into the plaque. Additionally, these changes are associated necrosis/apoptosis of macrophage foam cells and VSMCs to further increase inflammation and thinning of the luminal surface plaque cap to create a thin-cap atheroma that is prone to rupture [46]. At the shoulders of these thin-cap atheroma, there is an accumulation of inflammatory macrophages resulting in hot plaques [42]. These macrophages are known to result in the activation of the proteolytic matrix metalloproteinases (MMPs) to result in thinning and eventual rupture of these vulnerable plaques with devastating acute coronary syndromes, which present clinically as luminal thrombosis and myocardial infarction in the heart or stroke in the brain (Figure 6) [46].
Notably, these hot plaques with increased temperature represent calor or heat of the four cardinal signs of inflammation: rubor (increased blood flow), dolar (pain), calor (heat), and tumor (swelling) were determined to be an independent predictor in regards to the clinical outcomes of individuals undergoing coronary interventions [47].
When thoughts turn to the realm of PAD and its transition to chronic limb-threatening ischemia (CLTI) or CLI that is considered to be the end-stage of PAD, one has to think about how differently the role of atherosclerotic plaques may differ from the coronary regional beds and the iliac, femoral-popliteal, and the infra-popliteal regional arterial beds (Figure 7).
For example, Narula et al. were able to demonstrate that luminal occlusion by thrombosis associated with insignificant atherosclerotic plaques in CLI, especially in the infra-popliteal (INFRA-POP) regions as in Figure 7 [48]. Also, they were able to show that pathological remodeling of PAD resulted from thrombosis regardless of the extent of atherosclerotic plaques and further that 75% of distal infra-popliteal arteries with stenosis had thrombi even in the presence of insignificant atherosclerotic plaques [49]. These findings by Narula et al. in 2018 and 2020 strongly suggest the need for both a preventive and therapeutic course of dual antiplatelet therapeutic regimens in order to decrease thrombosis, especially in the INFRA-POP regions of the extremities [48,49].
2.2. Advancing Age and essential hypertension (eHTN-HTN) in PAD
eHTN-HTN (also known as primary or idiopathic HTN) is the chronic elevation of blood pressure without a definite identifiable cause such as occurs in secondary reno-vascular hypertension with a known cause. Advancing chronological age (aging) is strongly associated with PAD and HTN, plus they are well-known risk factors for atherosclerosis that is the most common cause of PAD (as discussed in section 2.1.), VAS, and eHTN [50]. Also, many feel that VAS may precede the development and progression of atherosclerosis, PAD, and importantly HTN [50,51,52]. Importantly, aging is thought to be the strongest risk factor for the development of VAS [53]. Indeed, the fatigue and failure of the vessel wall due to the increased injurious pp with chronic wall stress that also associates with stiffening of endothelial cells, internal elastic lamina fragmentation or loss, and dysfunction, loss of elastin lamellar units, and collagen cross-linking with stiffening contribute to the development and progression of VAS and PAD. Also, there is VSMC stiffening and accumulation of advanced glycation end product(s) (AGEs) that bind to its receptor (RAGE) known to increase OxRS with increased MMP production [54]. Notably, HTN is the second most common cause of VAS following aging [54].
HTN is known to associate with and contribute to the development and progression of atherosclerosis, PAD, and VAS [55] as well as acting as one of the leading causes of atherosclerotic plaque formation (Figure 8) [11,56,57].
Despite the enormous efforts put forth in the treatment of HTN, only 53% of those with documented HTN have their blood pressure controlled according to current guidelines [58] and this is precisely why the wheel icon in Figure 8 was chosen as an icon since HTN just keeps on turning and allows for the negative interaction between HTN and the multiple clinical diseases including PAD.
Many decades of research have implicated HTN and atherosclerosis as potential modifiable risk factors in the development of VAS, cerebrocardiovascular disease and PAD [57,59]. Indeed, both atherosclerosis and HTN are implicated in the development and progression of VAS, CCVD, and small vessel disease (SVD); however, they frequently co-exist and may be present simultaneously especially in older individuals [55]. Also, multiple risk factors for the development of atherosclerosis and eHTN including the MetS play an important role (Figure 9) [59,60,61].
2.3. The Metabolic Syndrome (MetS) and Type 2 Diabetes Mellitus (T2DM) in PAD
The MetS is a cluster of multiple risk factors including the four “Hs” (horsemen): I. Hyperlipidemia (obesity); II. Hyperinsulinemia (IR); III. HTN-isolated systolic hypertension (ISH). IV. Hyperglycemia (impaired glucose tolerance (IGT) with or without T2DM and associated variables [21,57]. In addition to paralleling the obesity epidemic, the MetS associates with an increased risk for the development of T2DM and cerebrocardiovascular disease [57]. Each of the previous four Hs (horsemen) that forms the risk factor clustering, in addition to the MetS itself and the other variables, play an important role in the development of both VAS and PAD [62,63,64]. MetS is a strong predictor of CCVD disease, VAS, and PAD, which is increasingly being recognized as a CCVD risk factor. Additionally, other components/variables of the MetS are capable of interacting synergistically to impact carotid intimal media thickness, extracranial VAS, and PAD (Figure 10) [57,62,63,64,65].
Importantly, VAS has been determined to be independently associated with PAD even when adjusting for several atherosclerotic risk factors including HTN [26]. Also, MetS has a 1.76-fold increased prevalence of PAD as compared to non-MetS individuals [66].
Diabetes (specificallyT2DM) has been increasing globally in the past decade with the International Diabetes Federation indicating that 537 million people worldwide had diabetes in 2021 with a projection of in excess of one billion by 2050 with approximately 90-95% having T2DM [67]. These numbers are huge and with T2DM playing a major role in the development of atherosclerosis and a two-fold increased prevalence of PAD [66] it is important to better understand the relation between T2DM and the development of progression of PAD since T2DM is known to be associated with accelerated atherosclerosis (Figure 11) [1,67,68,69].
2.4. Tobacco smoking, oxidative redox stress (OxRS), and accelerated atherosclerosis in PAD
Smoking is considered to be one the four most common risk factors (smoking, HTN, hyperlipidemia/hypercholesterolemia, and T2DM) for the development of cardiovascular diseases and PAD [70]. Importantly, smoking is one of the most preventable causes of PAD. Indeed, cigarette smoking has a very high risk of developing PAD primarily due to increased OxRS, which induces macrovascular atherosclerosis as a result of at least 4,000 chemicals found in cigarette smoke (highly addictive nicotine, thiocyanates, aldehydes, benzenes, nitrosamines, and polycyclic aromatic hydrocarbons to name a few), and a very large number of highly unstable and damaging free radicals that promote OxRS with depletion of antioxidant reserves. Further, smoking with its known increased OxRS is capable of inducing ECact/dys with increased vascular inflammation and decreased nitric oxide (NO) due to eNOS enzyme uncoupling. This promotes monocyte to macrophage transformation, which instigates VSMC remodeling and a phenotypic switch from a contractile phenotype to a synthetic phenotype that predisposes to migration and the development of vascular ossification-calcification (VOC), and the initiation of thrombo-inflammation [71,72]. Importantly, smoking also associates with impaired folate one-carbon metabolism that is associated with elevated levels of homocysteine (Hcy) and decreased levels of folate (vitamin B9), vitamin B6, and B12 [73]. Elevations of Hcy are a known biomarker of impaired folate one-carbon metabolism, which is associated with increased OxRS that contributes to ECact/dys to increase endothelial inflammation and decrease endothelial-derived bioavailable NO, which results in accelerated atherosclerosis and increased cardiovascular diseases including VAS and PAD [74,75]. Unfortunately, smoking/nicotine still remains extremely addictive and remains difficult for many individuals to quit even with its known perceived damaging effects to the vessels. As a result, many individuals are unable to quit smoking even with the supportive assistance and encouragement by health care providers.
3. Decreased Bioavailability of Endothelial Cell-Derived Nitric Oxide in PAD
When the endothelium is exposed to the multiple injurious species as presented in figures one and two in the introduction, it becomes activated and dysfunctional. ECact/dys associates with a proinflammatory EC (activation) and decreased NO bioavailability (dysfunction). This ECact/dys plays an important role in the development of CCVDs, which includes PAD. Indeed, decreased NO bioavailability plays an important role in the development of atherosclerosis, HTN, VAS and PAD [76,77,78].
In health, vascular EC-derived NO (one of three EC gasotransmitters along with carbon monoxide and hydrogen sulfide) is known to be responsible for numerous pleiotropic and vasculoprotective functions. These protective functions include its anti-inflammatory, antioxidant, antithrombotic, antiatherosclerotic, antiplatelet activation (decreasing adherence and aggregation) with potent vasodilation properties [75,79,80,81,82]. NO synthesis is dependent on its substrate L-arginine oxidation with NADPH and oxygen serving as co-substrates and the endothelial nitric oxide synthase enzyme (eNOS) with its essential cofactor tetrahydrobiopterin (BH4) that must be in its totally reduced form in order to run the eNOS reaction to produce NO and L-citrulline [83]. Unfortunately, when there is increased OxRS, BH4 will be oxidized to BH2 and the eNOS reaction will not run to produce NO. The eNOS reaction is very sensitive to detrimental effects of OxRS since this not only interferes with the total reduction of BH4 but also associates with increased asymmetrical dimethyl arginine (ADMA) that competes with L-arginine for NO production [77]. Also, the known decrease in NO due to the excessive and rapid oxidation of NO to nitrite (NO2−), peroxynitrite, and nitrate (NO3−) via increased OxRS also contributes to decreased NO bioavailability [84]. Unfortunately, when there develops a decreased in NO bioavailability due to ECact/dys as in PAD in Figure 3, a vascular alarm is sounded and this results in at least seven detrimental cellular effects of the arterial vessel wall that promotes not only atherogenesis but also VAS and PAD.
Nuclear factor-kappa B (NF-κB) is an important mediator between the vicious cycle of inflammation and OxRS wherein one instigates the other bidirectionally. Additionally, homeostatic levels of NO are known to directly inhibit NF-κB activation by preventing the phosphorylation and degradation of IκB-α, a protein that keeps NF-κB inactive in the cytoplasm and this relationship results in the findings that when NO is decreased it associates with increased activity of NF-κB [85]. Interestingly, the other gasotransmitter hydrogen sulfide (H2S) is capable of NO production via a cascade of phosphorylation events that starts with p38 mitogen activated protein kinase (MAPK) and Akt to eNOS, to form NO [86,87]. Indeed, EC-derived NO via the eNOS enzyme plays a critical role in the maintenance of vascular homeostasis (Figure 12) [88].
4. Vascular Arterial Stiffening (VAS) in PAD
VAS and PAD are clinically determined by the careful examination of individuals for decreased or asymmetrical pulsations of distal lower extremity arteries and elevated systolic blood pressure-isolated systolic hypertension (ISH) with an increase in pulse pressure. Additional evaluation for VAS includes the more precise and quantitative measurement of the ankle-brachial index (ABI) or the gold standard, carotid-femoral pulse wave velocity (cfPWV) [89]. Importantly, the finding of VAS is well known to be an independent predictor of CVD morbidity and mortality [90,91]. Multiple risk factors for the development of VAS include: aging [62,92,93,94]; MetS [62]; T2DM and insulin resistance [95,96,97]; obesity [98,99]. Also, lifestyle risks are involved such as: smoking [100], physical inactivity-sedentary lifestyle [101], and high salt, high caloric intake as occurs with the diet-induced obesity Western style diets [102,103]. Risk factors for the development of VAS and PAD closely parallel those of CVD [104,105] and importantly, VAS may even begin in youth or middle age with some reports that VAS may precede the development of HTN and metabolic risks [106].
The pathobiology mechanisms for the development and progression of VAS are currently thought to be initiated by injuries to the endothelium by a number of injurious stimuli as previously listed in Figure 2 as the dirty dozen [21]. Once injured, ECact/dys develops, which results in a proinflammatory endothelium with increased OxRS and decreased NO bioavailability. This functional change in the activated and dysfunctional endothelium results in a response to injury wound healing mechanism that becomes chronically activated along with the renin-angiotensin-aldosterone system (RAAS) and the direct damaging effects of AngII and aldosterone induced OxRS and transforming growth factor-beta one (TGF-β1). Following the activation and dysfunction of the endothelium there is an activation of MMPs and various elastases to result in the degradation, fragmentation, and splitting of elastin and loss of compliance and recoil upon stretching. These remodeling changes of elastin give rise to elastin-derived fragments (EDFs) that are a rich source of elastin-derived peptides (EDPs) and along with OxRS are known to result in a vascular smooth muscle cell(s) (VSMC(s) “phenotype switch” (from a contractile phenotype to a synthetic phenotype) in the media. Additionally, in the proliferative phases of the wound healing mechanism the cytokine growth factor TGF-β1 is known to be increased and also stimulates these reactive VSMCs to produce stiffer organized collagen, i.e. fibrosis. In turn, these reactive, dedifferentiated, synthetic, proliferative VSMCs are known to be highly synthetic (with increased production of stiffer collagen (types I) and proteoglycans (PGNs) within the media, and a heightened capability for migration to the intima of the vessel wall to result in and contribute to the neogenesis of the intimal space. These structural and functional remodeling changes to the ECM proteins elastin and collagen result in a decreased elastin/collagen ratio with ensuing increased vascular stiffness (Figure 13 and Figure 14) [21,98,105,107,108,109,110].
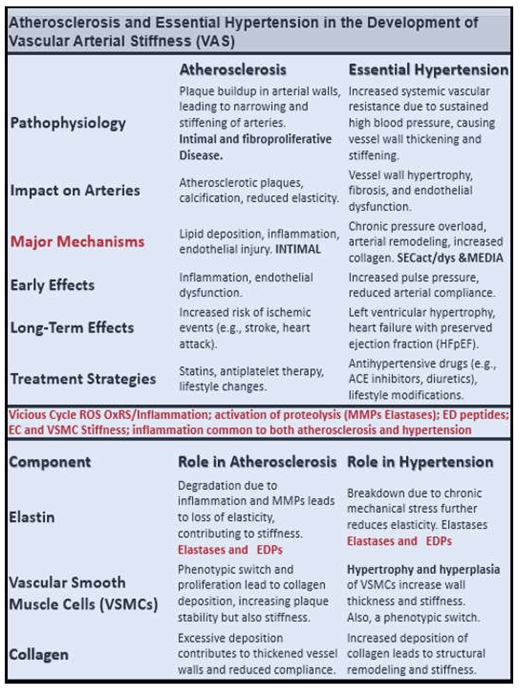
T2DM with its accelerated atherosclerosis and HTN frequently co-occur in aging [111,112]. Therefore, it is important to compare some of the effects of atherosclerosis and HTN in the development of VAS in PAD (Box 2).
Box 2. Comparison of atherosclerosis and hypertension (HTN) in the development of vascular arterial stiffening (VAS) and peripheral artery disease (PAD). Notably, atherosclerosis is an intimal disease that associates primarily with PAD and that HTN is a disease that associates with the media of the arterial vessel wall that associates primarily with VAS. However, note that many of the pathologic mechanisms of HTN and atherosclerosis may be associated with either VAS or PAD. Reproduced image provided with permission by CC 4.O [21]. ACE, angiotensin converting enzyme; EC(s), endothelial cells; EDPs, elastin-derived peptides; MMP(s), matrix metalloproteinases; SECact/dys, systemic endothelial cell activation/dysfunction; VSMC(s), vascular smooth muscle cells.
4.1. Bioactive Elastin-Derived Peptides (EDPs) and Transglutaminase 2 (TG2)
The degradation of the internal elastic lamina (IEL) and elastin lamella result in the formation/production of elastin-derived fragments (EDFs) that are a rich source of elastin-derived peptides as visualized in Figure 14 in panels A-C; however, these similar remodeling changes are also found to be present in hypertension (not shown). These bioactive EDPs (also termed matrikines or elastokines) interact with their receptors the elastin receptor complex and are capable of instigating multiple interactions that contribute to the development and progression of VAS in PAD. EDPs are known to promote inflammation and react with RAGE to increase OxRS, NFkappa B, MMP activation, cytokine activation including TNFa and IL-6, increased crosslinking of collagen and elastin to collagen, and importantly to promote phenotypic switching from contractile VSMCs to a synthetic, proliferative, migratory phenotype, that increases the synthesis of collagen and proteoglycans. Indeed, EDPs contribute to VSMC remodeling and further amplify elastin degradation to promote VAS in both atherosclerosis and hypertension that may be present in the development of PAD [113,114,115].
Importantly, transglutaminase 2 (TG2) is an enzyme that is ubiquitously expressed in the vasculature, which promotes the synthesis and crosslinking of extracellular matrix collagen that is also important in the development of VAS. Increased TG2 is a known collagen crosslinker that associates with VAS and affects the extracellular matrix by increasing collagen deposition, and crosslinking (in a calcium-dependent manner) that has been long recognized as critical player in vascular stiffening [102,116]. Herein, it is notable that Ramirez-Perez et al. were able to demonstrate that cystamine (a nonspecific TG2 inhibitor) reduced vascular stiffness in Western diet-fed female mice; however, they did not investigate cysteamines’ role as also being an antioxidant that could additionally contribute to the vascular destiffening effects [102].
The multiple functional and structural remodeling changes discussed in this section are multifactorial and complicated to say the least. However, it is possible to create a more simplified possible sequence of events that help to understand the development and progression of VAS in the development and progression of PAD, which can supplement the recent findings by the multiorganizational bodies for the recommended reading of the management of lower extremity PAD (Figure 15) [117].
Importantly, it has been demonstrated that VAS with its damaging increased pulsatile pulse pressure (an injurious stimuli to the endothelium) is independently associated with PAD even after adjusting for several atherosclerotic risk factors and that there is a significant association between VAS and the onset of PAD regardless of blood pressure status [26,118].
4.2. Vascular Ossification Calcification (VOC) in Type VII Plaques
VOC is the term utilized to define the process of calcium phosphate complexes deposition in the form of hydroxyapatite (Ca10(PO4)6(OH)2) in the media of the vasculature, which also results in increased VAS [21,118,119,120,121,122]. Arterial VOC is a definite characteristic of vascular aging [123,124] and is also increased in clinical diseases such as diabetes, HTN, chronic kidney disease (CKD), and hereditary disorders (Figure 16) [21,107,125,126].
VOC occurs at different locations, e.g. in HTN VOC occurs primarily in the tunica media regions and in diabetes it occurs primarily in the tunica intima; however, in certain situations VOC may co-occur in both regions since these two clinical diseases often co-occur [119,127]. Notably, VOC is no longer thought to be a passive process that associates with aging, but involves a reprogramming of tunica media VSMC from its contractile phenotype to a synthetic and proliferative, migratory phenotype that is capable of becoming an osteogenic cell [108,128]. Indeed, local VSMC cues are different for medial calcification that associates with aging, HTN, senescence, and uremia of renal failure with multiple elevations of uremic toxins and specifically elevated calcium and phosphate levels in contrast to atherosclerosis-related aortic tunica intima calcification, which involves inflammation, OxRS, and apoptosis [99,128].
Also, elastin degradation/fragmentation as depicted in Figure 14C results in the release of bioactive EDPs, which can readily induce an osteogenic phenotype switch along with OxRS from an activated endothelium to induce a contractile VSMC to a synthetic phenotype. Once this phenotype switch occurs these VSMC are capable of producing calcification similar to osteogenic cells in bone in the media that have a predilection to adhere to the damaged elastin lamella within the media or to the internal elastic lamella of the intima [129]. While VOC was not included in the six-steps regarding the development of PAD in Figure 15, it is nevertheless a very important step in its progression.
4.3. PAD involves arterial vessels large and small: Microvessel (Mv) remodeling in skeletal muscle (SkM)
Lower extremity PAD is caused by the initial progression and expansion of atherosclerosis of the large conduit arteries that develop VAS and PAD with ischemia due to progressive luminal narrowing and or thrombosis. However, it is the microvessels (arterioles, capillaries, and venules with lumen diameters ranging from 5 to 500 micrometers) that undergo structural remodeling and functional changes that contribute to the horrendous and horrifying pain associated with claudication, nocturnal leg cramps (lower extremity myalgias and myopathy), and the stabbing-like pain of ischemic peripheral neuropathy [130,131].
Mv remodeling is extensive in PAD and consists of i) basement membrane thickening (primarily due to increased type IV collagen; ii) perivascular collagen I and type III fibrosis due to the initial response to injury to acute or early critical ischemia with an initial increased pericyte coverage of Mv capillary ECs and their capability to transition to myofibroblasts as a result of increased transforming growth factor-beta 1 (TGF-β1) chronically; iii) endothelial cell remodeling, which includes endothelial activation that is proinflammatory and endothelial dysfunction that associates with decreased protective bioavailable NO primarily due to eNOS uncoupling; iv) increased OxRS that is central to the peripheral neuronal unit (PNU), neuromuscular junction (NMJ), and the skeletal muscle myopathy aberrant remodeling of PAD [130,131]. Importantly, there is evidence that decreased lower extremity skeletal muscle microvascularization is primarily responsible for exercise intolerance in PAD [132] and that microvascular disease potentiates the risk of amputation in PAD [133]. Further, the initial response to regional hypoxia due to lower extremity PAD would be increased angiogenesis due to ischemia-induced hypoxia inducible factor-1alpha (HIF-1α) and activation of vascular endothelial growth factor (VEGF) to result in angiogenesis and capillary expansion and density. However, over time this initial angiogenesis may be complicated by capillary loss with the development of string-like microvessels and capillary rarefaction due to the ongoing chronic ischemia induced ECact/dys, OxRS, inflammation, and decreased NO (Figure 17) [131,132,133].
Lower extremity PAD impacts skeletal muscle by increased OxRS damage, inflammation, aberrant mitochondria dysfunction/loss, and decreased adenosine triphosphate (ATP) production, loss of number and function of myofibers, dysfunction and loss of capillary density, increased proteolysis and apoptosis. Although ischemia/ischemia reperfusion injury cycles are considered the principal cause of skeletal muscle pathophysiology in PAD, the impact of other comorbidities such as hyperlipidemia on endothelial dysfunction and skeletal muscle pathophysiology should not be overlooked [135].
In addition to microvessel remodeling there is a co-occurance of skeletal muscle (SkM) remodeling that includes skeletal muscle loss (atrophy/ischemia-induced sarcopenia), endomysial fibrosis, myocyte lipid infiltration, and aberrant mitochondria with loss of pericapillary skeletal muscle mitochondrial mounding and loss of subsarcolemmal mitochondria with reduced ATP production in PAD [136].
Thus, as PAD progresses from chronic atherosclerotic macrovessel disease it results in the development of hypoperfusion and hypometabolism to the lower extremity with microvessel remodeling that transition from the early symptoms of IC, which often progresses to CLI. This results, in structural remodeling in three critical regional tissue beds that are also involved with aberrant functional changes, which include microvessel (Mv), peripheral neuronal unit (PNU), neuromuscular junction (NMJ), and lower extremity SkM regions (Figure 18).
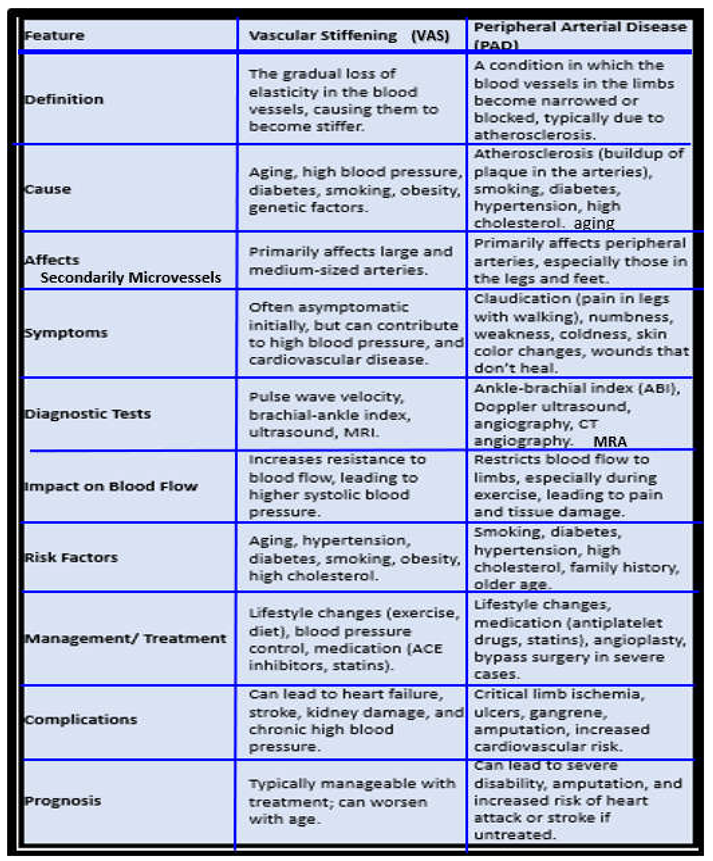
While VAS describes a condition or a process of stiffening of the arterial vessel wall due to a loss of elastin and an increase in stiffer collagen, PAD describes a specific vascular disease associated with the accumulation of intimal atherosclerotic plaques that eventually result in a narrowing of the arterial vessel lumen resulting in ischemia to peripheral arteries as well as an increased incidence of thrombosis and embolism to the distal extremities. Indeed, while VAS and PAD are each important in the development and progression of cerebrocardiovascular diseases, when compared side by side what stands out is how they co-develop, co-occur and share so many commonalities (Box 3) [26,51,70,90,91,137].
Box 3. A simplistic comparison of vascular arterial stiffening (VAS) and peripheral arterial disease (PAD). ACE, angiotensin converting enzyme; CT, computed tomography; MRA, magnetic resonance angiography; MRI, magnetic resonance imaging.
At this juncture, it is noteworthy to point out that PAD may be considered a spectrum disorder/disease as portrayed in following image (Figure 19) [131,133,138].
The clinical identification of PAD is important and ankle brachial index (ABI) and/or toe brachial index (TBI) measurements will allow for its diagnosis that is more sensitive than the clinical findings of intermittent claudication since only 50% of individuals with PAD have symptoms of IC, while the other half with PAD still remain asymptomatic. Additionally, microvessel remodeling is associated with elevations of vascular endothelial growth factor (VEGF) due to hypoxia induced factor alpha (HIF-1α) that induces VEGF.
5. Ischemia and Ischemia/Reperfusion (IR) Injury, Microvessel (Mv) Remodeling, Ischemic Neuropathy, Ischemic Skeletal Muscle (SkM) Remodeling with Myopathy and Myalgias, and Pain Perception
Progressive atherosclerotic macrovascular remodeling changes result in luminal narrowing and additionally contribute to Mv remodeling with ECact/dys and decreased NO that contribute to neuronal and SkM ischemia and ischemia/reperfusion injury. Initially ischemia results in hypoperfusion with hypometabolism and hypoxia to tissues. This combined energy and oxygen deficiency or deficit contribute to both Mt dysfunction and inflammation in SkM. Notably, this brings into play the ROS-inflammation ongoing damaging vicious cycle. Mt dysfunction results in increased OxRS–ROS, which overtime depletes the natural antioxidant reserves. Anerobic metabolism contributes to increased lactic acid, H+ proton accumulation, and the initial decrease in ATP to supply cellular energy; however, over time increased ATP leakage via the opening of Mt transition pores and liberation of ATP upon cellular death allow for the signaling of afferent sensory neurons. These abnormalities along with increased cytokines and chemokines of inflammation also contribute to cellular damage and death via apoptosis-necrosis. Thus, IR injury is largely due to mitochondrial dysfunction and increased mtROS production with increased OxRS and inflammation to result in SkM remodeling (Figure 20) [13,139,140].
Also, Mv remodeling as previously discussed in subsection 4.3. and Figure 18 and Figure 19 is accompanied with a co-occurance of peripheral neuronal unit remodeling in the lower extremities in regards to the development and progression of ischemic neuropathy that occurs in intermittent claudication as it progresses to CLI in lower extremity PAD (Figure 21) [141,142,143].
Ischemic neuropathy is tightly associated with Mv remodeling and subsequent ischemia to the PNU and associated remodeling as in Figure 19, Figure 20 and Figure 21. The perception of pain in ischemic neuropathy of PAD is initially instigated via the ischemic injury, which results in the accumulation of toxic metabolic stimuli such as increased lactic acid, decreased pH, increased hydrogen protons, and ATP. Additionally, this ischemic injury is capable of instigating an activation of mast cells since they serve as the first responders to injury to create a cellular inflammatory response to injury and pain perception. In turn, these activated mast cells are capable of forming a neuroimmune axis with the sensory afferent neurons within the SkM interstitial or endomysial spaces between SkM myocytes (Figure 22) [144].
SkM remodeling in PAD was specifically discussed in section 4.3. Figure 17 and this section Figure 21 and consisted primarily of skeletal muscle loss (atrophy/ischemic sarcopenia), endomysial fibrosis, myocyte lipid infiltration, and aberrant mitochondria with loss of pericapillary skeletal muscle mitochondrial mounding and loss of subsarcolemmal mitochondria with reduced ATP production [136]. Once the lower extremity SkM is injured and remodeled due to chronic atherosclerotic macrovascular disease progression with continued luminal narrowing and Mv remodeling with ischemia occur or acute peripheral embolic events occur, there develops a sequence of signaling events, which result in the perception of pain (Figure 23) [145].
Incidently, these same SkM groups III and IV sensory afferents as depicted in Figure 22 also form the sensory components of the exercise pressor reflex that involves the increase in heart rate and blood pressure following muscle contraction during casual walking and exercise [146,147,148].
The two most common forms of neuropathy are diabetic peripheral neuropathy (DPN) of diabetes (specifically T2DM) and ischemic neuropathy of PAD as discussed in subsection 2.3. and illustrated in Figure 11. Therefore, it is appropriate to compare these two most common forms of neuropathy, especially since diabetes has a 2-fold increase in the prevalence of PAD (Figure 24) [67].
6. Conclusions and Future Directions
PAD is the third most common manifestation of atherosclerosis after coronary artery (CAD) and cerebrovascular disease (CVD) that currently affects between 200-250 million individuals globally, and it is considered to be an aging-associated disease [4,5]. Since we are now living in one of the oldest living populations in history, we can only expect to experience an even greater number of individuals to develop PAD as the global aging population expands along with the major risk factors associated with the development and progression of PAD. Further, numerous authors have pointed out that PAD is underappreciated, understudied, underdiagnosed, and undertreated [150,151,152,153,154] and in the past some have even pointed out that PAD may be the last major pandemic of cardiovascular disease [4,5,150]. Indeed, the diagnosis of PAD should be considered a ‘red flag’ in that it serves as a biomarker for the increased risk, morbidity, and mortality of cardiovascular and cerebrovascular disease that should merit earlier detection and a more aggressive and sustained treatment plan [150].
This narrative review has examined the background of PAD and how it is associated with ECact/dys due to multiple injurious species, atherosclerosis development and progression, aging and HTN, MetS and T2DM, and tobacco smoking. Also discussed, was how ECact/dys with eNOS uncoupling associated with the decrease in vasculoprotective bioavailable nitric oxide in the development and progression of PAD. Further, the role of VAS with elastin loss and associated increase in collagen and proteoglycans, the role of transglutaminase 2, and vascular ossification and calcification and how they are involved with the development and progression of PAD was presented. Then, PAD progression via progressive macrovascular atherosclerosis accumulation with lumen encroachment to limit blood flow to the lower extremities was presented. Also, Mv remodeling as a result of increased VAS and increased pulsatile pp and impaired vasodilation with the persistence of ECact/dys and decreased NO bioavailability, which resulted in ischemia, ischemia/reperfusion (IR) injury was discussed. Finally, IR injury and its effects not only on Mv remodeling but also its effects on the development of ischemic neuropathy with remodeling of the PNU and NMJ, ischemic SkM remodeling. and pain perception were presented.
In regards to future directions, we may need to focus more on the microvessel disease associated with VAS and PAD in regards to the development ischemic neuropathy and the skeletal muscle remodeling in the lower extremities. Especially, since these remodeling changes along with macrovascular remodeling changes are responsible for the horrific debilitating pain that control individual lives every day if they are not eligible for surgical revascularization.
Indeed, future directions should include methods to increase the awareness of PAD not only to individuals but also to their health care providers, especially since the study by Hirsch et al. reported that only 49% of caring physicians were aware that their patients had PAD that were identified by ankle-brachial index (ABI) studies in their large reported study with 6,979 individuals [150]. Therefore, creative planning by primary care provider offices may be necessary in order to overcome the findings that limited time by staff and limited reimbursement were thought to be roadblocks to perform ABI testing [155]. In the future, clinicians might consider annual or new patient vascular health questioners for their patients to fill out prior to examination. Also, regional hospitals that serve multiple out-patient clinics might consider having outpatient vascular health screening clinics that include BP recordings, blood tests to access for hyperlipidemia, and ABI measurements to overcome these concerns regarding underappreciation and underdiagnosis concerns by multiple authors in this field of study.
Because PAD is associated with increased morbidity and mortality and its association with increased prevalence of risk for morbid, mortal CVDs, newer treatment strategies for the prevention and delaying the progression in order to extend life expectancy and prevention of disability, pain, and limb loss are needed. Further, Arabzadeh et al. have discussed the possibility of utilizing strategies like regenerative medicine, cell-based therapies, nano-therapy, gene therapy, and targeted therapy, besides other traditional drug combination therapy for PAD that may be considered promising novel therapies [156]. Also, novel or innovative treatments might be considered regarding the treatment of PAD in addition to aggressive therapy with antihypertensive medications to lower blood pressure (specifically angiotensin converting enzyme inhibitors and angiotensin receptor blockers), lipid lowering strategies with the use of statins and/or monoclonal antibodies along with diet to slow the progression of atherosclerosis, dual antiplatelet therapies to prevent thromboembolism, emphasizing smoking cessation, and if possible regular exercise that are already recommended.
In this regard, author has recently proposed utilizing intravenous sodium thiosulfate (STS) as an innovative, multi-targeting, and repurposed therapeutic approach for the treatment of late-onset Alzheimer’s disease [156] and has also previously suggested STS treatment for its novel antioxidant potential in the treatment of diabetic peripheral neuropathy [142]. Because STS has been successfully and widely utilized in the global treatment of calciphylaxis [127,157], it is felt that STS might also be utilized as an innovative treatment modality for PAD. PAD like late-onset Alzheimer’s disease is also multifactorial in its development and progression, which involves oxidant stress, inflammation, excess calcium deposition in the development of VOC, and ECact/dys with decreased bioavailability of NO due to excessive oxidation and dysfunction of the essential coenzyme tetrahydrobiopterin (BH4) that allows the eNOS enzyme to produce NO may also benefit with the use of STS treatment (Figure 25) [157].
While STS has not yet been approved for the treatment of calciphylaxis in humans, it has now been sucessfully used off-label to treat calciphylaxis for just over two decades and results have been reported in the literature primarily as case reports, small case series, and reviews of calciphylaxis with discussions of STS embedded within these reviews [127,158,159,160,161,162,163,164,165,166,167]. Additionally, STS has been utilized to prevent the excessive oxidative stress associated with cisplatin induced hearing loss for solid tumors and head and neck cancers [168,169] and also recently approved for the treatment in pediatric age populations [170]. Also, STS has been shown to improve cardiac dysfunction and the extracellular matrix remodeling in a heart failure induced arterial venous fistula mouse model, in part, by increasing ventricular H2S generation [171]. Additionally, Wen et al. found that STS attenuated the progression of vascular calcification and importantly vascular arterial stiffness (VAS) in hemodialysis treated individuals [172]. These findings by Wen et al. are pertinent in that VAS plays an important role in the development and progression of PAD. Further, ischemia and IR injury to skeletal muscle has been shown to be associated with increased OxRS and decreased H2S with SkM dysfunction and weakness. Recently, Du et al were able to demonstrate in rat models that H2S treatment was able to significantly protect rat SkM against ischemia and IR injury [173]. These findings are important since we now know that STS serves as an H2S donor/mimetic as presented in Figure 25 and may be able to restore SkM dysfunction and possibly weakness.
Notably, STS treatment has been consistently associated with a rapid improvement of the neuritic/ischemic pain associated with the vascular calcification of calciphylaxis in human individuals and this may be due to a restoration of eNOS enzyme function and NO production [126,174]. Indeed, STS has U.S. Food and Drug Administration (FDA) approved medical uses, off-label uses, side effects, and possible future uses in clinical diseases (Figure 26) [157].
Of great concern to suffering individuals and their health care providers is that a full understanding of the pathobiology for the development of nocturnal leg cramping and rest pain as occur in critical limb ischemia (CLI) or chronic limb-threatening ischemia (CLTI) still remain uncertain and therefore poses a treatment problem [175]. While Figure 19 provides somewhat of a spectrum and a roadmap for the development and progression of PAD, this image also points to the importance of microvessel remodeling and its relation to the development of ischemic neuropathy and ischemia-related myopathy and myalgias. Author’s personal observations have concluded that a personalized approach of utilizing a walker to prevent falling and stretching the involved cramping muscle(s) by standing upright from the supine nocturnal sleeping position to literally walk away from this horrendous cramping pain of the lower extremity myalgias. While this may not prevent future nocturnal cramping that may occur later during the night, it at least helps to prevent the active cramping at that moment in time. Additionally, author highly recommends the algorithm created by Allen and Kirby for the evaluation and treatment of nocturnal leg cramps [175].
Over the years we have been taught that PAD-associated atherosclerosis begins distally in the abdominal aorta at the iliac artery bifurcations while concurrently developing VAS that progresses cephalad initially to the coronary arteries resulting in CVD, CAD, and congestive heart failure and then to the extracranial carotid arteries to involve the brain’s microvessels with brain ECact/dys of the neurovascular unit to result in the co-occurance of vascular dementia and VCID that co-occurs with neurodegeneration resulting in mixed dementias (cerebrovascular disease) and stroke [21]. In reality, while atherosclerosis is progressing cephalad it is concurrently progressing caudad to the distal lower extremities via the femoral, popliteal, peroneal, tibial, and dorsalis pedis arteries on the road to PAD and perdition. PAD frequently presents clinically with asymptomatic stenosis; however, it is known to progress from intermittent claudication to the most severe form of critical limb ischemia that leads to severe and debilitating pain due to ischemic neuropathy and SkM myopathy with the development of ischemic skin ulcerations, gangrene, and eventual limb amputation that also associates with a high risk of morbidity and mortality [176]. Indeed, we are as old as our arteries [177].
Funding
Author has not received grants from any funding agency in the public, commercial, or not-for-profit sectors in regards to this review.
Institutional Review Board Statement
The tissues provided for the representative electron microscopic images utilized in this manuscript were all approved in advance by the University of Missouri Institutional Animal Care and Use Committee (No. 190; July 7, 2017). The animals were cared for in accordance with National Institutes of Health guidelines and by the Institutional Animal Care and Use Committees at the Harry S. Truman Memorial Veterans Hospital and the University of Missouri, Columbia, MO, USA, which conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH).
Informed Consent Statement
Not applicable.
Data Availability Statement
The data and materials can be provided upon reasonable request.
Acknowledgments
The author would like to acknowledge DeAna Grant Research Specialist of the Electron Microscopy Core Facility at the Roy Blunt NextGen Precision Health Research Center, University of Missouri, Columbia, Missouri.
Conflicts of Interest
The author declares no conflict of interest.
Abbreviations
ABI, ankle brachial index; AGE, advanced glycation endproducts; AGE/RAGE, advanced glycation endproducts and it receptor for AGE ; ATP, adenosine triphosphate; BH4, tetrahydrobiopterin; BH2, dihydrobiopterin; CAD, coronary artery disease; CLI, critical limb ischemia; CLTI, chronic limb-threating ischemia; CVD, cerebrovascular disease-cardiovascular disease; CCVD, cerebrocardiovascular disease; Ca2+ and Ca++, calcium cation;; CBF, cerebral blood flow; CL, capillary lumen; ECact/dys, endothelial cell activation/dysfunction; ecGCx, endothelial cell glycocalyx; EDFs, elastin-derived fragments; EDPs, elastin-derived peptides eHTN, essential hypertension; eNOS, endothelial nitric oxide synthase; FDA, U.S. Food and Drug Administration; H2S, hydrogen sulfide; Hif-1α, hypoxia inducible factor 1-alpha; HTN, hypertension; IC, intermittent claudication; IGT, impaired glucose tolerance; IR, ischemia reperfusion; LOAD, late-onset Alzheimer’s disease; MMP(s), matrix metalloproteinases; MMP-2,-9, matrix metalloproteinase-2,-9; MRI, magnetic resonance imaging; mtROS, mitochondrial ROS; mod-LDL-C, modified low density lipoprotein-cholesterol; Mv, microvessel; NF-kB, nuclear factor-kappa B; NO, nitric oxide; OxRS, oxidative redox stress; NMJ, neuromuscular junction; pCC, peripheral cytokines chemokines; PGNs, proteoglycans; PNU, peripheral neuronal unit; pp, pulsatile pulse pressure; pWV, pulse wave velocity; RAGE, receptor for AGE; ROS, reactive oxygen species, RONS, reactive oxygen, nitrogen species; RONSS, reactive oxygen, nitrogen, sulfur species; rPVACfp/ef, reactive perivascular astrocyte foot processes/endfeet; rPVMΦ, reactive resident perivascular macrophages; RSI, reactive species interactome; SkM, skeletal muscle; STS, sodium thiosulfate; SVD, cerebral small vessel disease; T2DM, type 2 diabetes mellitus; TEM, transmission electron microscopy; TG2, transglutaminase 2; TGFβ, transforming growth factor beta; TNFα, tumor necrosis factor alpha; VaD, vascular dementia; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; VOC, vascular ossification calcification; VSMC(s), vascular smooth muscle cells; WMH, white matter hyperintensities.
References
- Hayden, M.R.; Tyagi, S.C. Intimal redox stress: Accelerated atherosclerosis in metabolic syndrome and type 2 diabetes mellitus. Atheroscleropathy. Cardiovasc Diabetol. 2002, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Abdelfattah, A.; Allam, A.H.; Wann, S.; Thompson, R.C.; Abdel-Maksoud, G.; Badr, I.; et al. Int J Cardiol. 2013, 167, 570-574. [CrossRef]
- Allam, A.H.; Thompson, R.C.; Wann, L.S.; Miyamoto, M.I.; Nur El-Din, A.e.l.-H.; El-Maksoud, G.A.; et al. Atherosclerosis in ancient Egyptian mummies: the Horus study. Cardiovasc Imaging. 2011, 4, 315–327. [Google Scholar] [CrossRef]
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010, a systematic review and analysis. Lancet. 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Song, P.; Rudan, D.; Zhu, Y.; Fowkes, F.J.I.; Rahimi, K.; Fowkes, F.G.R.; Rudan, I. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015, an updated systematic review and analysis. Lancet Glob Health. 2019, 7, e1020–e1030. [Google Scholar] [CrossRef] [PubMed]
- The Diabetes Prevention Program Research Group; Crandall, J. Schade D. Ma Y. Fujimoto WY. Barrett-Connor E. Fowler S. Dagogo-Jack S. Andres R. The Influence of Age on the Effects of Lifestyle Modification and Metformin in Prevention of Diabetes. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2006, 61, 1075–1081, United Nations Department of Economics and Social Affairs Populations Division. Population Ageing and Sustainable Development. No2017/1. June 2017. Available online: https://www.un.org/development/desa/publications/world-population-prospects-the-2017-revision.htmlm (accessed on 14 January 2025).
- United Nations Department of Economics and Social Affairs Populations Division. Population Ageing and Sustainable Development. Available online: https://www.un.org/en/development/desa/population/publications/pdf/popfacts/PopFacts_2017-1.pdf (accessed on 14 January 2025).
- Hayden, M.R. Type 2 Diabetes Mellitus Increases the Risk of Late-Onset Alzheimer’s Disease: Ultrastructural Remodeling of the Neurovascular Unit and Diabetic Gliopathy. Brain Sci. 2019, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Macabrey, D.; Longchamp, A.; MacArthur, M.R.; Lambelet, M.; Urfer, S.; Deglise, S.; Allagnat, F. Sodium thiosulfate, a source of hydrogen sulfide, stimulates endothelial cell proliferation and neovascularization. EBioMedicine. 2022, 78, 103954. [Google Scholar] [CrossRef]
- Eraso, L.H.; Fukaya, E.; Mohler, E.R.; Xie, D.; Sha, D.; Berger, J.S. Peripheral arterial disease, prevalence and cumulative risk factor profile analysis. Eur J Prev Cardiol. 2014, 21, 704–711. [Google Scholar] [CrossRef]
- Parwani, D.; Ahmed, M.A.; Mahawar, A.; Gorantla, V.R. Peripheral Arterial Disease: A Narrative Review. Cureus. 2023, 15, e40267. [Google Scholar] [CrossRef]
- Grenon, S.M.; Vittinghoff, E.; Owens, C.D.; Conte, M.S.; Whooley, M.; Cohen, B.E. Peripheral artery disease and risk of cardiovascular events in patients with coronary artery disease: Insights from the Heart and Soul Study. Vasc Med. 2013, 18, 176–184. [Google Scholar] [CrossRef]
- Liao, J.K.; Shin, W.S.; Lee, W.Y.; Clark, S.L. Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J Biol Chem. 1995, 270, 319–324. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone MAJr Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest. 1995, 96, 60–68. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Hayden, M.R. Endothelial activation and dysfunction in metabolic syndrome, type 2 diabetes mellitus, and coronavirus disease 2019. J Int Med Res. 2020, 48, 300060520939746. [Google Scholar] [CrossRef]
- Hayden, M.R. Brain Endothelial Cells Play a Central Role in the Development of Enlarged Perivascular Spaces in the Metabolic Syndrome. Medicina. 2023, 59, 1124. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K. Linking endothelial dysfunction with endothelial cell activation. J Clin Invest. 2013, 123, 540–541. [Google Scholar] [CrossRef]
- Hayden, M.R. The Brain Endothelial Cell Glycocalyx Plays a Crucial Role in the Development of Enlarged Perivascular Spaces in Obesity, Metabolic Syndrome, and Type 2 Diabetes Mellitus. Life (Basel). 2023, 13, 1955. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Brain endothelial cell activation and dysfunction associate with and contribute to the development of enlarged perivascular spaces and cerebral small vessel disease. Histol Histopathol. 2024, 18792. [Google Scholar] [CrossRef]
- Hayden, M. R.; Tyagi, N. Extracranial Vascular Arterial Stiffness Contributes to Cerebral Small Vessel Disease, Stroke, and Late-Onset Alzheimer’s Disease. Preprints. 2025, 2025010993. [Google Scholar] [CrossRef]
- Refaat, A.; Abdou, M.; Ismael, A.; Alhelali, I. Aortic stiffness and microalbuminuria in patients with chronic obstructive pulmonary disease. Egyptian Journal of Chest Diseases and Tuberculosis. 2015, 64, 541–549. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Yang, D. Alarmins: chemotactic activators of immune responses. Curr Opin Immunol. 2005, 17, 359–365. [Google Scholar] [CrossRef]
- Yang, D.; Han, Z.; Oppenheim, J.J. ALARMINS AND IMMUNITY. Immunol Rev. 2017, 280, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. 2000, 407, 233-41. [CrossRef]
- Allen, M.F.; Pekas, E.J.; Park, S.Y. Arterial Stiffness as a Prognostic Marker for Peripheral Artery Disease Risk: Clinical Relevance and Considerations. JACC Asia. 2023, 3, 298–300. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Vasa vasorum in plaque angiogenesis, metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: a malignant transformation. Cardiovasc Diabetol. 22;1, 3. [CrossRef]
- Falk, E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. C: Suppl). [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature. 2021, 592, 524–533. [Google Scholar] [CrossRef]
- Hayden, M.R.; Reidy, M. Many roads lead to atheroma. Nat Med. 1995, 1, 22–23. [Google Scholar] [CrossRef] [PubMed]
- Herrrington W, Lacey B, Sherliker P, Armitage J, Lewington S Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ Res. [CrossRef]
- Xie, B.; Shi, X.; Xing, Y.; Tang, Y. Association between atherosclerosis and Alzheimer's disease: A systematic review and meta-analysis. Brain Behav. 2020, 10, e01601. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C. Composition and classification of human atherosclerotic lesions. Virchows Arch A Pathology Anat Histopathol. 1992, 421, 277–290. [Google Scholar] [CrossRef]
- Stary, H.C.; Blankenhorn, D.H.; Chandler, A.B.; Glagov, S.; Insull Jr, W.; Richardson, M.; et al. A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association, Circulation. 1992, 85, 391–405. [Google Scholar] [CrossRef]
- Rizzo, M.; Berneis, K.J. Lipid triad or atherogenic lipoprotein phenotype: a role in cardiovascular prevention? Atheroscler Thromb. 2005, 12, 237–239. [Google Scholar] [CrossRef]
- Gui, Y.; Zheng, H.; Cao, R.Y. Foam Cells in Atherosclerosis: Novel Insights Into Its Origins, Consequences, and Molecular Mechanisms. Font Cardiovasc Med. 2022, 9, 845942. [Google Scholar] [CrossRef]
- Yang, K.; Zhang, X.J.; Cao, L.J.; Liu, X.H.; Liu, Z.H.; Wang, X.Q.; et al. Toll-like receptor 4 mediates inflammatory cytokine secretion in smooth muscle cells induced by oxidized low-density lipoprotein. PLoS One. 2014, 9, e95935. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xue, Q.; Cao, L.; Wang, Y.; Chen, Y.; Zhang, X.; Xiao, F.; Yang, Y.; Hayden, M.R.; Liu, Y.; Yang, K. Toll-Like Receptor 4 Mediated Oxidized Low-Density Lipoprotein-Induced Foam Cell Formation in Vascular Smooth Muscle Cells via Src and Sirt1/3 Pathway. Mediators Inflamm. 2021, 2021, 6639252. [Google Scholar] [CrossRef]
- Cai JM, Hatsukami TS, Ferguson MS, Small R, Polissar NL, Yuan C. Classification of Human Carotid Atherosclerotic Lesions With In Vivo Multicontrast Magnetic Resonance Imaging. Circulation. 2002, 106, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Costopoulos, C.; Huang, Y.; Brown, A.J.; Calvert, P.A.; Hoole, S.P.; West, N.E.J. Plaque Rupture in Coronary Atherosclerosis Is Associated With Increased Plaque Structural Stress. JACC Cardiovasc Imaging. 2017, 10, 1472–1483. [Google Scholar] [CrossRef] [PubMed]
- Nasir, K.; Cainzos-Achirica, M. Role of coronary artery calcium score in the primary prevention of cardiovascular disease. BMJ. 2021, 373, n776. [Google Scholar] [CrossRef]
- Stehouwer, C.D.; Henry, R.M.; Ferreira, I. Arterial stiffness in diabetes and the metabolic syndrome: a pathway to cardiovascular disease. Diabetologia. 2008, 51, 527–539. [Google Scholar] [CrossRef]
- Hayden, M.R.; Karuparthi, P.R.; Chaudhury, N.A.; Gorindarajan, G.; Habibi, J.; Ortman, R.A. Autoimmune Vasculitis and Plaque Erosion in the Cardiometabolic Syndrome and Type 2 Diabetes mellitus. J Cardiometabolic Syn. 2007, 1, 228–232. [Google Scholar] [CrossRef]
- Lorentzen, K.A.; Chai, S.; Chen, H.; Danielsen, C.C.; Simonsen, U.; Wogensen, L. Mechanisms involved in extracellular matrix remodeling and arterial stiffness induced by hyaluronan accumulation. Atherosclerosis. 2016, 244, 195–203. [Google Scholar] [CrossRef]
- Barger, A.C.; Beeuwkes 3rd, R.; Lainey, L.L.; Silverman, K.J. Hypothesis: vasa vasorum and neovascularization of human coronary arteries. A possible role in the pathophysiology of atherosclerosis. N Eng J Med. 1984, 310, 175–177. [Google Scholar] [CrossRef]
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Virmani, R. Intraplaque Hemorrhage and Progression of Coronary Atheroma. N Engl J Med. 2003, 349, 2316–1325. [Google Scholar] [CrossRef]
- Stefanadis, C.; Toutouzas, K.; Tsiamis, E.; Stratos, C.; Vavuranakis, M.; Kallikazaros, I.; Panagiotakos, D.; Toutouzas, P. Increased local temperature in human coronary atherosclerotic plaques: an independent predictor of clinical outcome in patients undergoing a percutaneous coronary intervention. J Am Coll Cardiol. 2001, 37, 1277–1283. [Google Scholar] [CrossRef] [PubMed]
- Narula, N.; Dannenberg, A.J.; Olin, J.W.; Bhatt, D.L.; Johnson, K.W.; Nadkarni, G.; et al. Pathology of Peripheral Artery Disease in Patients With Critical Limb Ischemia. J Am Coll Cardiol. 2152; ;72. [Google Scholar] [CrossRef]
- Narula, N.; Olin, J.W.; Narula, N. Pathologic Disparities Between Peripheral Artery Disease and Coronary Artery Disease. Arter Thromb Vascul Biology. 2020, 40, 1982–1989. [Google Scholar] [CrossRef]
- Savji, N.; Rockman, C.B.; Skolnick, A.H.; Guo, Y.; Adelman, M.A.; Riles, T.; Berger, J.S. Association Between Advanced Age and Vascular Disease in Different Arterial Territories: A Population Database of Over 3.6 Million Subjects. J Am Coll Cardiol. 2013, 61, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Hooglugt, A.; Klatt, O.; Huveneers, S. Vascular stiffening and endothelial dysfunction in atherosclerosis/ Curr Opin Lipidol. 2022, 33, 353-363. 33. [CrossRef]
- Chirinos, J.A.; Segers, P.; Hughes, T.; Townsend, R. Large Artery Stiffness in Health and Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019 Sep 3;74, 1237–1263. [CrossRef]
- Mitchell, G.F. Arterial Stiffness and Hypertension: Chicken or Egg? Hypertension. 2014, 64, 210–214. [Google Scholar] [CrossRef]
- Greenwald, S.E. Ageing of the conduit arteries. J Pathol. 2007, 211, 157–172. [Google Scholar] [CrossRef]
- Abraham AT, Mojaddedi S, Loseke IH, Bray C. Hypertension in Patients With Peripheral Artery Disease: An Updated Literature Review. Cureus. 2024, 16, e62246. [Google Scholar] [CrossRef]
- Hurtubise, J.; McLellan, K.; Durr, K.; Onasanya, O.; Nwabuko, D.; Ndisang, J.F. The different facets of dyslipidemia and hypertension in atherosclerosis. Curr Atheroscler Rep. 2016, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Overview and New Insights into the Metabolic Syndrome: Risk Factors and Emerging Variables in the Development of Type 2 Diabetes and Cerebrocardiovascular Disease. Medicina (Kaunas). 2023, 59, 561. [Google Scholar] [CrossRef]
- Go, A.S.; Mozaffarian, D.; Roger, V.L.; Benjamin, E.J.; Berry, J.D.; Blaha, M.J.; et al. Heart disease and stroke statistics--2014 update: a report from the american heart association. Circulation. 2014, 129, e28–e292. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Melnichenko, A.A.; Grechko AVOrekhov, A.N. Hypertension as a risk factor for atherosclerosis: Cardiovascular risk assessment. Front Cardiovasc Med. 2022, 9, 959285. [Google Scholar] [CrossRef]
- Scuteri, A.; Najjar, S.S.; Muller, D.C.; Andres, R.; Hougaku, H.; Metter, J.; Lakatta, E.G. Metabolic syndrome amplifies the age-associated increases in vascular thickness and stiffness. Am Coll Cardiol. 2004, 43, 1388–1395. [Google Scholar] [CrossRef]
- Schillaci, G.; Pirro, M.; Vaudo, G.; Mannarino, M.R.; Savarese, G.; Pucci, G.; Franklin, S.S.; Mannarino, E. Metabolic Syndrome Is Associated With Aortic Stiffness in Untreated Essential Hypertension. Hypertension. 2005, 45, 1078–82. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Vicente, W.R.P.; Rodrigues, S.; Cepeda, F.X.; Jordão, C.P.; Costa-Hong, V.; Dutra-Marques, A.C.B.; et al. Arterial stiffness and its association with clustering of metabolic syndrome risk factors. Diabetol Metabol Syndr. 2017, 9, 87. [Google Scholar] [CrossRef]
- Scuteri, A.; Najjar, S.S.; Muller, D.C.; Andres, R.; Hougaku, H.; Metter, J.; Lakatta, E.G. Metabolic syndrome amplifies the age-associated increases in vascular thickness and stiffness. Am Coll Cardiol. 2004, 43, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhao, W.; Liu, O.; Qin, M. Association Between Metabolic Syndrome and Peripheral Artery Disease in Elderly Patients with Type 2 Diabetes Mellitus. 2021, 14, 4783-4789. [CrossRef]
- Schillaci, G.; Pirro, M.; Vaudo, G.; Mannarino, M.R.; Savarese, G.; Pucci, G.; Franklin, S.S.; Mannarino, E. Metabolic Syndrome Is Associated With Aortic Stiffness in Untreated Essential Hypertens ion. Hypertension. 2005, 45, 1078–82. [Google Scholar] [CrossRef]
- Soyoye, D.O.; Abiodun, O.O.; Ikem, R.T.; Kolawole, B.A.; Akintomide, A.O. Diabetes and peripheral artery disease: A review. World J Diabetes. 2021, 12, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050, a systematic analysis for the Global Burden of Disease Study 2021. GBD 2021 Diabetes Collaborators. Lancet. 2023, 402, 203–234. [Google Scholar] [CrossRef]
- Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri V, Orekhov AN. The Diabetes Mellitus–Atherosclerosis Connection: The Role of Lipid and Glucose Metabolism and Chronic Inflammation. Int J Mol Sci. 2020, 21, 1835. [Google Scholar] [CrossRef]
- Ye J, Li L, Wang M, Ma Q, Tian Y, Zhang Q, Liu J, Li B, Zhang B, Liu H, Sun G Diabetes Mellitus Promotes the Development of Atherosclerosis: The Role of NLRP3. Front Immunol. 2022, 13, 900254. [CrossRef]
- Joosten, M.M.; Pai, J.K.; Bertoia, M.L.; Rimm, F.B.; Spiegelman, D.; Mittleman, M.A.; Mukamal, K.J. Associations between conventional cardiovascular risk factors and risk of peripheral artery disease in men. JAMA. 2012, 308, 1660–7. [Google Scholar] [CrossRef]
- Alnima, T.; Meijer, R.I.; Spronk, H.M.H.; Warle, M.; ten Cate, H. Diabetes- versus smoking-related thrombo-inflammation in peripheral artery disease. Cardiovasc Diabet. 2023, 22, 257. [Google Scholar] [CrossRef]
- Wang W, Zhao T, Geng K, Yuan G, Chen Y, Smoking and the Pathophysiology of Peripheral Artery Disease. Front Cardiovasc Med. 7041. [CrossRef]
- O'Callaghan P, , Meleady R, Fitzgerald T, Graham I; European COMAC group. Smoking and plasma homocysteine. 2002, 23, 1580–1586. [CrossRef]
- Hayden, M.R.; Tyagi, S.C. Homocysteine and reactive oxygen species in metabolic syndrome, type 2 diabetes mellitus, and atheroscleropathy: the pleiotropic effects of folate supplementation. Nutr J. 2004, 10, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C. Impaired folate one-carbon metabolism in Type 2 Diabetes, Late-Onset Alzheimer's Disease and Long COVID. Medicina (Kaunas). 2021, 58, 16. [Google Scholar] [CrossRef] [PubMed]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef]
- Ismaeel, A.; Brumberg, R.S.; Kirk, J.S.; Papoutsi, E.; Farmer, P.J.; Bohannon, W.T.; Smith, R.S.; Eidson, J.L.; Sawicki, I.; Koutakis, P. Oxidative Stress and Arterial Dysfunction in Peripheral Artery Disease. Antioxidants (Basel). 2018, 7, 145. [Google Scholar] [CrossRef]
- Chen, J.Y.; Ye, Z.X.; Wang, X.F.; Chang, J.; Yang, M.W.; Zhong, H.H.; et al. Nitric oxide bioavailability dysfunction involves in atherosclerosis. Biomed Pharmacother. 2018, 97, 423–428. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N Engl J Med. 1993, 329, 2002–2012. [Google Scholar] [CrossRef]
- Cooke, J.P.; Dzau, V.J. Nitric oxide synthase: role in the genesis of vascular disease. Annu Rev Med. 1997, 48, 489–509. [Google Scholar] [CrossRef]
- Jin, R.C.; Loscalzo, J. Vascular nitric oxide: formation and function. J Blood Med. 2010, 1, 147–162. [Google Scholar] [CrossRef]
- Cooke, J.P. The pivotal role of nitric oxide for vascular health. Can J Cardiol. 2004, 20 Suppl B:7B-15B.
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001, 357, 593–615. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.D.; Giordano, T.; Kevil, C.G. Nitrite and Nitric Oxide Metabolism in Peripheral Artery Disease. Nitric Oxide. 2012, 26, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.; Giaginis, C.; Tsigris, C.; Patsouris, E.; Theocharis, S. NF-κB signaling at the crossroads of inflammation and atherogenesis: searching for new therapeutic links. Expert Opin Ther Targets. 2014 Sep;18, 1089-101. [CrossRef]
- Altaany, Z.; Yang, G.; Wang, R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J Cell Mol Med. 2013, 17, 879–888. [Google Scholar] [CrossRef]
- Lee, K.S.; Kim, J.; Kwak, S.N.; Lee, K.S.; Lee, D.K.; Ha, K.S. Functional role of NF-κB in expression of human endothelial nitric oxide synthase. Biochem Biophys Res Commun. 2014, 448, 101–107. [Google Scholar] [CrossRef]
- Barbato, J.E.; Tzeng, E. Nitric oxide and arterial disease. Vasc Surg. 2004, 40, 187–193. [Google Scholar] [CrossRef]
- Kamieńska, A.; Danieluk, A.; Niwinska, M.M.; Chlabicz, S. Arterial Stiffness and Ankle-Brachial Index – Cross-Sectional Study of 259 Primary Care Patients ≥50 Year-Old. Med Sci Monit. 2024, 30, e942718–1. [Google Scholar] [CrossRef]
- Cecelja, M.; Chowienczyk, P. Role of arterial stiffness in cardiovascular disease. JRSM Cardiovasc Dis. 2012, 1, cvd2012012016. [Google Scholar] [CrossRef]
- Mitchell, G.F.; Parise, H.; Benjamin, E.J.; et al. "Changes in arterial stiffness and wave reflection with advancing age in the Framingham Heart Study. " Hypertension. 2004, 43, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Andrés, V.; Castiglioni, S.; Giudici, A.; Lau, E.S.; Nemcsik, J.; et al. Aging and Vascular Disease: A Multidisciplinary Overview. J Clin Med. 2023, 12, 5512. [Google Scholar] [CrossRef]
- Laurent, S.; Boutouyrie, P.; Asmar, R.; Gautier, I.; Laloux, B.; Guize, L.; Ducimetiere, P.; Benetos, A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001, 37, 1236–41. [Google Scholar] [CrossRef]
- Safar, M.E.; Asmar, R.; Benetos, A.; Blacher, J.; Boutouyrie, P.; Lacolley, P.; et al. Interaction Between Hypertension and Arterial Stiffness. Hypertension. 2018, 72, 796–805. [Google Scholar] [CrossRef]
- Cohen, J.B.; Mitchell, G.F.; Gill, D.; Burgess, S.; Rahman, M.; Hanff, T.C.; Ramachandran, V.S.; Mutalik, K.M.; Townsend, R.R.; Chirinos, J.A. Arterial Stiffness and Diabetes Risk in Framingham Heart Study and UK Biobank. Circ Res. 2022, 131, 545–554. [Google Scholar] [CrossRef]
- Wang M, Huang J, Wu T, Qi L Arterial Stiffness, Genetic Risk, and Type 2 Diabetes: A Prospective Cohort Study. Diabetes Care. 2022, 45, 957–964. [CrossRef] [PubMed]
- Urbina, E.M.; Gao, Z.; Khoury, P.R.; Martin, L.J.; Dolan, L.M. Insulin resistance and arterial stiffness in healthy adolescents and young adults. Diabetologia. 2012, 55, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Safar, M.E.; Czernichow, S.; Blacher, J. Obesity, arterial stiffness, and cardiovascular risk. J Am Soc Nephrol. 2006, 17(4 Suppl 2):S109-11. [CrossRef]
- Aroor, A.R.; Jia, G.; Sowers, J.R. Cellular mechanisms underlying obesity-induced arterial stiffness. Am J Physiol Regul Integr Comp Physiol. 2017, 314, R387–R398. [Google Scholar] [CrossRef] [PubMed]
- Camplain, R.; Meyer, M.L.; Takana, H.; Palta, P.; Agarwal, S.K.; Aguila, D.; Butler, K.R.; Heiss, G. Smoking Behaviors and Arterial Stiffness Measured by Pulse Wave Velocity in Older Adults: The Atherosclerosis Risk in Communities (ARIC) Study. Am J Hypertens. 2016, 29, 1268–1275. [Google Scholar] [CrossRef]
- Saladini, F. Effects of Different Kinds of Physical Activity on Vascular Function. J Clin Med. 2023, 13, 152. [Google Scholar] [CrossRef]
- Ramirez-Perez, F.I.; Cabral-Amador, F.J.; Whaley-Connell, A.T.; Aroor, A.R.; Morales-Quinones, M.; Woodford, M.L.; et al. Cystamine reduces vascular stiffness in Western diet-fed female mice. Am J Physiol Heart Circ Physiol. [CrossRef]
- Padilla, J.; Ramirez-Perez, F.I.; Habibi, J.; Bostick, B.; Aroor, A.R.; Hayden, M.R.; et al. Regular Exercise Reduces Endothelial Cortical Stiffness in Western Diet-Fed Female Mice. Hypertension. 2016 Nov;68, 1236-1244. [CrossRef]
- van Sloten TT, Stehouwer CDA. Carotid Stiffness: A Novel Cerebrovascular Disease Risk Factor. Pulse (Basel). Pulse (Basel). 2016, 4, 24–27. [CrossRef]
- Zieman, S.J.; Melenovsky, V.; Kass, D.A. Mechanisms, pathophysiology, and therapy of arterial stiffness. Arterioscler Thromb Vasc Biol. 2005, 25, 932–43. [Google Scholar] [CrossRef]
- Agbaje, A.O. Arterial stiffness precedes hypertension and metabolic risks in youth: a review. Journal of Hypertension. 2022, 49, 1887–1896. [Google Scholar] [CrossRef]
- Shirwany, N.A.; Zou, M.H. Arterial stiffness: a brief review. Acta Pharmacol Sin. 2010, 31, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Sowers, K.M.; Hayden, M.R. Calcific uremic arteriolopathy: pathophysiology, reactive oxygen species and therapeutic approaches. Oxid Med Cell Longev. 2010, 3, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R. Empagliflozin Ameliorates Tinica Adiposa Expansion and Vascular Stiffening of the Descending Aorta in Female db/db Mice. Adipobiology. 2020, 10(2019).1. [CrossRef]
- Hayden, M.R.; Sowers, J.R.; DeMarco, V.G. Ultrastructure study of the transgenic REN2 rat aorta – part 2, media, external elastic lamina, and adventitia. Biomed Reviews. 2019, 30, 111–123. [Google Scholar] [CrossRef]
- Sowers, J.R.; Whaley-Connell, A.; Hayden, M. The role of overweight and obesity in the cardiorenal syndrome. Cardiorenal Med. 2011, 1, 5–12. [Google Scholar] [CrossRef]
- Petrie, J.R.; Guzik, T.J.; Touyz, R.M. Diabetes, Hypertension, and Cardiovascular Disease: Clinical Insights and Vascular Mechanisms. Can J Cardiol. 2018, 34, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Duca, L.; Blaise, S.; Romier, B.; Laffargue, M.; Gayral, S.; El Btaouri, G.; et al. Matrix ageing and vascular impacts: focus on elastin fragmentation. Cardiovasc Res. 2016, 110, 298–308. [Google Scholar] [CrossRef]
- Le Page, A.; Khalil, A.; Vermette, P.; Frost, E.H.; Larbi, A.; Witkowski, J.M.; Fulop, T. The role of elastin-derived peptides in human physiology and diseases. Matrix Biol. 2019, 84, 81–96. [Google Scholar] [CrossRef]
- Wahart, A.; Hocine, T.; Albrecht, C.; Henry, A.; Sarazin, T.; Martiny, L.; et al. Role of elastin peptides and elastin receptor complex in metabolic and cardiovascular diseases. FEBS J. 2019, 286, 2980–2993. [Google Scholar] [CrossRef]
- Babici, D.; Kudej, R.K.; McNulty, T.; Zhang, J.; Oydanich, M.; Berkman, T.; et al. Mechanisms of increased vascular stiffness down the aortic tree in aging, premenopausal female monkeys. Am J Physiol Heart Circ Physiol. 2020, 319, H222–H234. [Google Scholar] [CrossRef]
- Gornik, H.L.; Aronow, H.D.; Goodney, P.P.; Arya, S.; Brewster, L.P.; Byrd, L.; Peer Review Committee Members et, a.l. Circulation. 2024ACC/AHA/AACVPR/APMA/ABC/SCAI/SVM/SVN/SVS/SIR/VESS Guideline for the Management of Lower Extremity Peripheral Artery Disease: A Report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. 2024, 149, e1313-e1410. [CrossRef]
- Wu, Z.; Jiang, Y.; Zhu, Q.; Zhang, H.; Li, Z.; Wang, J.; et al. Combined Evaluation of Arterial Stiffness and Blood Pressure Promotes Risk Stratification of Peripheral Arterial Disease. JACC Asia. 2023, 3, 287–297. [Google Scholar] [CrossRef]
- Wang, X.; Chen, X.; Chen, Z.; Zhang, M. Arterial Calcification and Its Association With Stroke: Implication of Risk, Prognosis, Treatment Response, and Prevention. Front Cell Neurosci. 2022, 16, 845215. [Google Scholar] [CrossRef] [PubMed]
- Demer, L.L.; Tintut, Y. Mineral exploration: search for the mechanism of vascular calcification and beyond: the 2003 Jeffrey M. Hoeg Award lecture. Arterioscler Thromb Vasc Biol. 2003, 23, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Chen, N.X. Pathophysiology of vascular calcification in chronic kidney disease. Circ Res. 2004, 95, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, K.; Xiong, Y.; He, Y.; Zhou, Y.; Hayden, M.R. Phosphate Toxicity and Vascular Calcification in Chronic Kidney Disease: A Closer Look Utilizing Transmission Electron Microscopy. Curr Protein Pept Sci. 2023, 24, 621–639. [Google Scholar] [CrossRef]
- Wu, X.H.; Chen, X.Y.; Wang, L.J.; Wong, K.S. Intracranial Artery Calcification and Its Clinical Significance. J Clin Neurol. 2016, 12, 253–261. [Google Scholar] [CrossRef]
- Tesauro, M.; Mauriello, A.; Rovella, V.; Annicchiarico-Petruzzelli, M.; Cardillo, C.; Melino, G.; Daniele, N.D. Arterial ageing: from endothelial dysfunction to vascular calcification. J Intern Med. 2017, 281, 471–482. [Google Scholar] [CrossRef]
- Wu, M.; Rementer, C.; Giachelli, C.M. Vascular Calcification: an Update on Mechanisms and Challenges in Treatment. Calcif Tissue Int. 2013, 93, 365–373. [Google Scholar] [CrossRef]
- Hayden MRTyagi, S.C.; Kolb, L.; Sowers, J.R.; Khanna, R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: the emerging role of sodium thiosulfate. Cardiovasc Diabetol. 2005, 4, 4. [Google Scholar] [CrossRef]
- Vos, A.; Van Hecke, W.; Spliet, W.G.M.; Goldschmeding, R.; Isgum, I.; Kockelkoren, R.; et al. Predominance of Nonatherosclerotic Internal Elastic Lamina Calcification in the Intracranial Internal Carotid Artery. Stroke. 2016, 47, 221–223. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. 2018, 114, 590–600. [Google Scholar] [CrossRef]
- McEniery, C.M.; McDonnell, B.J.; So, A.; Aitken, S.; Bolton, C.E.; Munnery, M.; et al. Aortic calcification is associated with aortic stiffness and isolated systolic hypertension in healthy individuals. Hypertension. 2009, 53, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Bethel, M.; Annex, B.H. Peripheral arterial disease: A small and large vessel problem. Am Heart J Plus. 2023, 28, 100291. [Google Scholar] [CrossRef]
- Mietus, C.J.; Lackner, T.J.; Karvelis, P.S.; Willcockson, G.T.; Shields, C.M.; Lambert, N.G. Abnormal Microvascular Architecture, Fibrosis, and Pericyte in the Calf Muscle of Peripheral Artery Disease Patients with Claudication and Critical Limb Ischemia. J Clin Med. 2020, 9, 2575. [Google Scholar] [CrossRef]
- Duscha, B.D.; Kraus, W.E.; Jones, W.S.; Robbins, J.L.; Piner, L.W.; Huffman, K.M.; Allen, J.D.; Annex, B.H. Skeletal muscle capillary density is related to anaerobic threshold and claudication in peripheral artery disease. Vasc Med. 2020, 25, 411–418. [Google Scholar] [CrossRef]
- Behroozian, A.; Beckman, J.A. Microvascular Disease Increases Amputation in Patients With Peripheral Artery Disease. Arterioscler Thromb Vasc Biol. 2020 Mar;40, 534-540. [CrossRef]
- Hayden, M.R.; Yang, Y.; Habibi, J.; Bagree, S.V.; Sowers, J.R. Pericytopathy: Oxidative Stress and Impaired Cellular Longevity in the Pancreas and Skeletal Muscle in Metabolic Syndrome and Type 2 Diabetes. Oxidative Medicine and Cellular Longevity. 2010, 3, 290–303. [Google Scholar] [CrossRef]
- Sfyri, P.; Matsakas, A. Crossroads between peripheral atherosclerosis, western-type diet and skeletal muscle pathophysiology: emphasis on apolipoprotein E deficiency and peripheral arterial disease. Biomed Sci. 2017, 24, 42. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, M.; Lee, N.H.; Moon, J.Y.; Han, W.M.; Anerson, S.E.; Choi, J.J.; et al. Critical Limb Ischemia Induces Remodeling of Skeletal Muscle Motor Unit, Myonuclear-, and Mitochondrial-Domains. Sci Rep. 2019, 9, 9551. [Google Scholar] [CrossRef]
- Wu, Z.; Jiang, Y.; Zhu, Q.; Zhang, H.; Li, Z.; Wang, J.; et al. Combined Evaluation of Arterial Stiffness and Blood Pressure Promotes Risk Stratification of Peripheral Arterial Disease. JACC: Asia. 2023, 3, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Shu, J.; Santulli, G. Update on peripheral artery disease: Epidemiology and evidence-based facts. Atherosclerosis. 2018, 275, 379–381. [Google Scholar] [CrossRef]
- Paradis, S.; Charles, A.L.; Meyer, A.; Lejay, A.; Scholey, J.W.; Chakfé, N.; Zoll, J.; Geny, B. Chronology of mitochondrial and cellular events during skeletal muscle ischemia-reperfusion. Am J Physiol Cell Physiol. [CrossRef]
- Liang, Z.; Zhang, W.; Zhu, T.; Li, Y.; Cao, P.; Wu, Y.; Zhang, Y. Ischemia-Reperfusion Injury in Peripheral Artery Disease and Traditional Chinese Medicine Treatment. Evid Based Complement Alternat Med. 2021, 2021, 4954070. [Google Scholar] [CrossRef]
- England, J.D.; Regensteiner, J.G.; Ringel, S.P.; Carry, M.R.; Hiatt, W.R. Muscle denervation in peripheral arterial disease. Neurology. 1992, 42, 994–9. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Salam, M.; Sowers, J.R. Reactive Oxygen Species and Diabetic Peripheral Neuropathy - A Closer Look. In Systems Biology of Free Radicals and Antioxidants. Laher, ED.: Springer-Verlag Berlin Heidelberg, 2014, 14, pp3375-3403. [CrossRef]
- Lastra, G.; Habibi, J.; Whaley-Connell, A.T.; Manrique, C. Hayden MR, Rehmer J, et al. Direct Renin Inhibition Improves Systemic Insulin Resistance and Skeletal Muscle. Endocrinology. 2009, 150, 2561–2568. [Google Scholar] [CrossRef]
- Gupta, K.; Harvima, I.T. Mast cell-neural interactions contribute to pain and itch. Immunol Rev. 2018, 282, 168–187. [Google Scholar] [CrossRef]
- Queme, L.F.; Ross, J.L.; Jankowski, M.P. Peripheral Mechanisms of Ischemic Myalgia. Front Cell Neurosci. 2017, 11, 419. [Google Scholar] [CrossRef]
- Smolderen KG, Alabi O, Collins TC, Dennis B, Goodney BP, Mena-Hurtado C, Spertus JA, Decker C. American Heart Association Council on Peripheral Vascular Disease and Council on Lifestyle and Cardiometabolic Health. Advancing Peripheral Artery Disease Quality of Care and Outcomes Through Patient-Reported Health Status Assessment: A Scientific Statement From the American Heart Association. Circulation. 2022, 146, e286-e297. [CrossRef]
- Hayes, S.G.; McCord, J.L.; Kaufman, M.P. Role played by P2X and P2Y receptors in evoking the muscle chemoreflex. J Appl Physiol (1985). 2008, 104, 538–41. [Google Scholar] [CrossRef] [PubMed]
- Ives, S.J.; McDaniel, J.; Witman, M.A.; Richardson, R.S. Passive limb movement: evidence of mechanoreflex sex specificity. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H154–H161. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Moris, D.; Karaolanis, G.; Kakkos, S.K.; Filis, K.; Sigals, F. Rivaroxaban versus Clopidogrel for Peripheral Artery Disease: A Clinico-Economic Approach of the COMPASS Trial. Curr Pharm Des. 2018, 24, 4516–4517. [Google Scholar] [CrossRef]
- Hirsch, A.T.; Criqui, M.H.; Treat-Jacobson, D.; Regensteiner, J.G.; Creager, M.A.; Olin, J.W.; et, a.l. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA 2001, 286, 1317–1324. [Google Scholar] [CrossRef]
- Jansen, S.; de Borst, G.J.; Hinchliffe, R.; Teraa, M. Peripheral Artery Disease: Underappreciated Impact and Residual Cardiovascular Risk Despite Revascularization. Clin Ther. 2023, 45, 1019–1022. [Google Scholar] [CrossRef]
- Olin, J.W.; Sealove, B.A. Peripheral Artery Disease: Current insight Into the Disease and Its Diagnosis and Management. Mayo Clin Proc. 2010, 85, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Mastracci, T.M.; Anand, S.S.; Aday, A.W. Peripheral Artery Disease: A High-Risk Yet Understudied, Underdiagnosed, and Undertreated Condition-A Call to Action. Can J Cardiol. 2022, 38, 553–554. [Google Scholar] [CrossRef]
- McDermott, M.M.; Kerwin, D.R.; Liu, K.; Martin, G.J.; O'Brien, E.; Kaplan, H.; Greenland, P. Prevalence and significance of unrecognized lower extremity peripheral arterial disease in general medicine practice. J Gen Intern Med. 2001, 16, 384–390. [Google Scholar] [CrossRef] [PubMed]
- Mohler ER3rd Treat-Jacobson, D.; Reilly, M.P.; Cunningham, K.E.; Miani, M.; Criqui, M.H.; Hiatt, W.R.; Hirsch, A.T. Utility and barriers to performance of the ankle-brachial index in primary care practice. Vasc Med. 2004, 9, 253–260. [Google Scholar] [CrossRef]
- Arabzadeh, A.A.; Faghfuri, E.; Soofiyani, S.R.; Addolahinia, E.R.; Siapush, S.; Nejati-Koshki, K.; et al. Current and Novel Emerging Medical Therapies for Peripheral Artery Disease: A Literature Review. Adv Pharm Bull. 2022, 13, 259–268. [Google Scholar] [CrossRef]
- Hayden, M.R.; Tyagi, N. Sodium Thiosulfate: An Innovative Multi-Target Repurposed Treatment Strategy for Late-Onset Alzheimer’s Disease. Pharmaceuticals (Basel). 2024, 17, 1741. [Google Scholar] [CrossRef]
- Cicone, J.S.; Petronis, J.B.; Embert, C.D.; Spector, D.A. Successful treatment of calciphylaxis with intravenous sodium thiosulfate. Am. J. Kidney Dis. 2004, 43, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Tyagi, S.C.; Kolb, L.; Sowers, J.R.; Khanna, R. Vascular ossification-calcification in metabolic syndrome, type 2 diabetes mellitus, chronic kidney disease, and calciphylaxis-calcific uremic arteriolopathy: The emerging role of sodium thiosulfate. Cardiovasc Diabetol. 2005, 4, 4. [Google Scholar] [CrossRef]
- Hayden, M.R.; Kolb, L.G.; Khanna, R. Calciphylaxis and the cardiometabolic syndrome. J Cardiometab Syndr. 2006, 1, 76–79. [Google Scholar] [CrossRef]
- Yerram, P.; Saab, G.; Karuparthi, P.R.; Hayden, M.R.; Khanna, R. Nephrogenic systemic fibrosis: A mysterious disease in patients with renal failure--role of gadolinium-based contrast media in causation and the beneficial effect of intravenous sodium thiosulfate. Clin. J. Am. Soc. Nephrol. 2007, 2, 258–263. [Google Scholar] [CrossRef]
- Rogers, N.M.; Coates, P.T. Calcific uraemic arteriolopathy: An update. Curr. Opin. Nephrol. Hypertens. 2008, 17, 629–634. [Google Scholar] [CrossRef]
- Araya, C.E.; Fennell, R.S.; Neiberger, R.E.; Dharnidharka, V.R. Sodium thiosulfate treatment for calcific uremic arteriolopathy in children and young adults. Clin J Am Soc Nephrol. 2006, 1, 1161–1166. [Google Scholar] [CrossRef]
- Hayden, M.R.; Goldsmith, D.; Sowers, J.R. Khanna R. Calciphylaxis: calcific uremic arteriolopathy and the emerging role of sodium thiosulfate. Int Urol Nephrol. 2008, 40, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.R.; Goldsmith, D.J. New hope for the treatment of calciphylaxis. Semin. Dial. 2010, 23, 258–262. [Google Scholar] [CrossRef]
- Nigwekar SU, Brunelli SM, Meade D, Wang W, Hymes J, Lacson E Jr. Sodium thiosulfate therapy for calcific uremic arteriolopathy. Clin J Am Soc Nephrol. 2013, 8, 1162-1170. [CrossRef]
- Generali JA, Cada DJ. Sodium Thiosulfate: Calciphylaxis. Hosp Pharm. 2015, 50, 975–977. [CrossRef]
- Pfeifle, C.E.; Howell, S.B.; Felthouse, R.D.; Woliver, T.B.; Andrews, P.A.; Markman, M.; Murphy, M.P. High-dose cisplatin with sodium thiosulfate protection. J. Clin. Oncol. 1985, 3, 237–244. [Google Scholar] [CrossRef]
- Brock, P.R. Maibach R, Childs M, Rajput K, Roebuck D, Sullivan MJ, et al. Sodium Thiosulfate for Protection from Cisplatin-Induced Hearing Loss. N. Engl. J. Med. 2018, 378, 2376–2385. [Google Scholar] [CrossRef]
- Dhillon, S. Sodium Thiosulfate: Pediatric First Approval. Paediatr Drugs. 2023, 25, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Sen, U.; Vacek, T.P.; Hughes, W.M.; Kumar, M.; Moshal, K.S.; Tyagi, N.; Metreveli, N.; Hayden, M.R.; Tyagi, S.C. Cardioprotective role of sodium thiosulfate on chronic heart failure by modulating endogenous H2S generation. Pharmacology. 2008, 82, 201–213. [Google Scholar] [CrossRef]
- Wen, W.; Portales-Castillo, I.; Seethapathy, R.; Krinsky, S.; Kroshinsky, D.; Kalim, S.; et al. Intravenous sodium thiosulphate for vascular calcification of hemodialysis patients-a systematic review and meta-analysis. Nephrol Dial Transplant. 2023, 38, 733–745. [Google Scholar] [CrossRef]
- Du, J.T.; Li, W.; Yang, J.Y.; Tang, C.S.; Li, Q.; Jin, H.F. Hydrogen sulfide is endogenously generated in rat skeletal muscle and exerts a protective effect against oxidative stress, Chin Med J (Engl). 2013, 126, 930-936.
- Tokashiki, K.; Ishida, A.; Kouchi, M.; Ishihara, S.; Tomiyama, N.; Kohagura, K.; Iseki, K.; Takishita, S. Successful management of critical limb ischemia with intravenous sodium thiosulfate in a.
- chronic hemodialysis patient. 2006, 66, 140-143. [CrossRef]
- Allen, R.E.; Kirby, K.A. Nocturnal leg cramps. Am Fam Physician. 2012, 86, 350–355. [Google Scholar] [PubMed]
- Criqui, M.H.; Matsushita, K.; Aboyans, V.; Hess, C.N.; Hicks, C.W.; Kwan, T.W.; McDermott, M.M.; Misra, S.; Ujueta, F. Lower Extremity Peripheral Artery Disease: Contemporary Epidemiology, Management Gaps, and Future Directions: A Scientific Statement From the American Heart Association. Circulation. 2021, 144, e171–e191. [Google Scholar] [CrossRef] [PubMed]
- Weber, T.; Meyer, C.C. “Man Is as OLD as His Arteries” Taken Literally: In Search of the Best Metric. Hypertension. 2020, 76, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Multiple injurious species in the plasma result in systemic endothelial cell (SEC) activation and dysfunction (ECact/dys) resulting in a proinflammatory endothelium with decreased nitric oxide (NO). Note that the initial endothelial cell glycocalyx surface layer (a gel-like layer of proteoglycans, glycoproteins and extracellular matrix components) is absent from his image due to the size constraint of the figure. Further, note that tobacco smoke was left out due to the large numbers of chemicals that are found within smoke; nevertheless, a large number of highly unstable reactive and damaging free radicals are found in smoke that instigates injury to the endothelium. Image reproduced with permission by CC4.0 [17,19,21]. Ang II, angiotensin 2; AGE/RAGE, advanced glycation endproducts and its receptor RAGE; AJ, adherens junction; aMMPs, activated matrix metalloproteinases; CCL2, monocyte chemoattractant protein-1; downward arrows, decrease; ecGCx, endothelial cell glycocalyx; ICAM-1, intercellular adhesion molecule-1; LPS, lipopolysaccharide; modLDL-C, modified low density lipoprotein-cholesterol; N, nucleus; O2̇̄ , superoxide anion; ONOO−, OxRS, oxidative redox stress; peroxinitrite; pCC, peripheral cytokines, chemokines; pp, pulsatile pulse pressure; RBC, red blood cell; RONSS, reactive oxygen, nitrogen, sulfur species; ROS, reactive oxygen species; RSI, reactive species interactome; upward arrows, increase; SEC, systemic endothelial cell(s); T, transcytosis; VAS, vascular arterial stiffness; VCAM-1, vascular cellular adhesion molecule -1; VE-cadherins, vascular endothelial cell cadherins; WBC, white blood cell; ZO-1 zona occludins 1.
Figure 1.
Multiple injurious species in the plasma result in systemic endothelial cell (SEC) activation and dysfunction (ECact/dys) resulting in a proinflammatory endothelium with decreased nitric oxide (NO). Note that the initial endothelial cell glycocalyx surface layer (a gel-like layer of proteoglycans, glycoproteins and extracellular matrix components) is absent from his image due to the size constraint of the figure. Further, note that tobacco smoke was left out due to the large numbers of chemicals that are found within smoke; nevertheless, a large number of highly unstable reactive and damaging free radicals are found in smoke that instigates injury to the endothelium. Image reproduced with permission by CC4.0 [17,19,21]. Ang II, angiotensin 2; AGE/RAGE, advanced glycation endproducts and its receptor RAGE; AJ, adherens junction; aMMPs, activated matrix metalloproteinases; CCL2, monocyte chemoattractant protein-1; downward arrows, decrease; ecGCx, endothelial cell glycocalyx; ICAM-1, intercellular adhesion molecule-1; LPS, lipopolysaccharide; modLDL-C, modified low density lipoprotein-cholesterol; N, nucleus; O2̇̄ , superoxide anion; ONOO−, OxRS, oxidative redox stress; peroxinitrite; pCC, peripheral cytokines, chemokines; pp, pulsatile pulse pressure; RBC, red blood cell; RONSS, reactive oxygen, nitrogen, sulfur species; ROS, reactive oxygen species; RSI, reactive species interactome; upward arrows, increase; SEC, systemic endothelial cell(s); T, transcytosis; VAS, vascular arterial stiffness; VCAM-1, vascular cellular adhesion molecule -1; VE-cadherins, vascular endothelial cell cadherins; WBC, white blood cell; ZO-1 zona occludins 1.
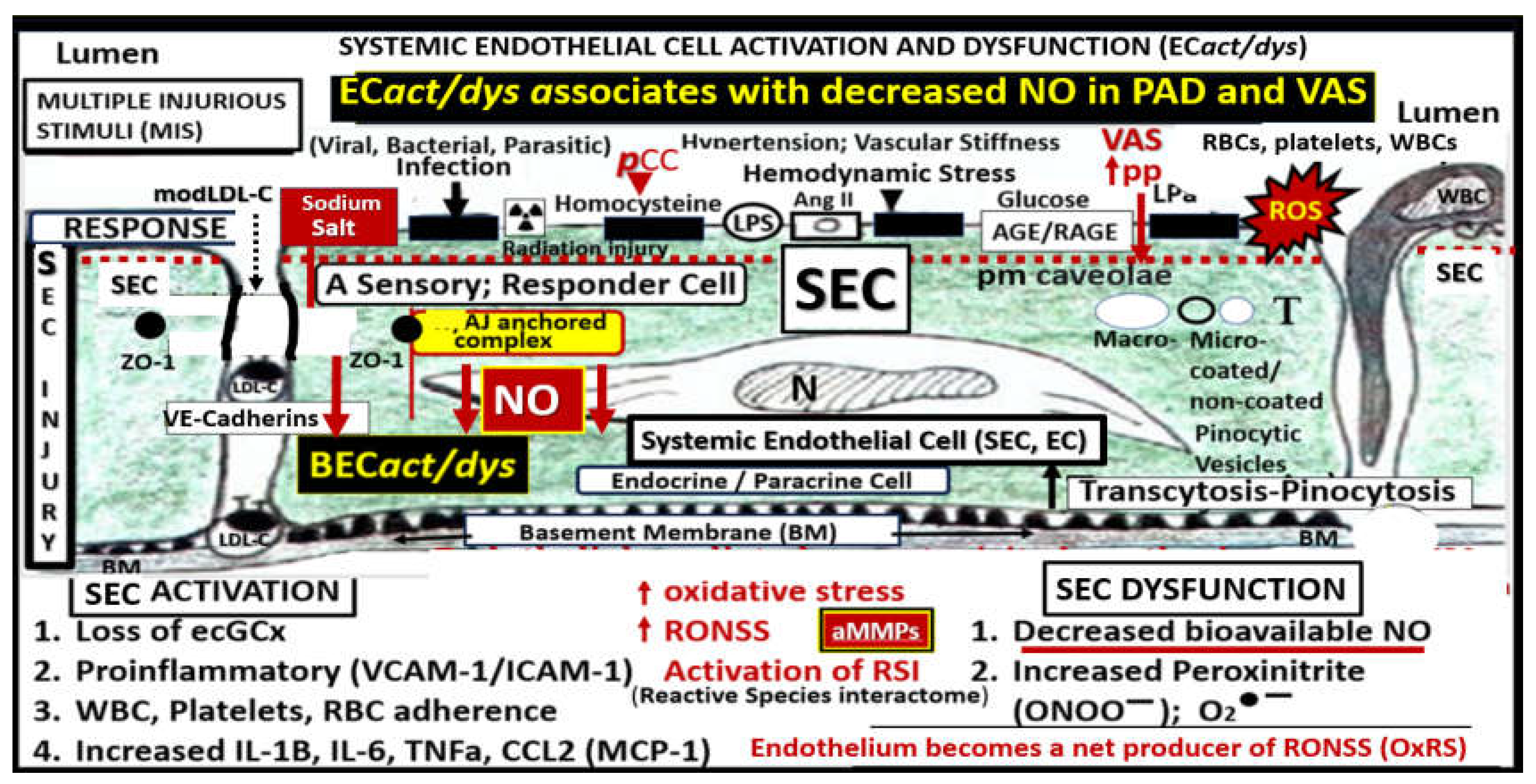
Figure 2.
Multiple injurious species: “The dirty dozen” affecting the systemic endothelium. Regarding number eight, the ultimate bacterial endotoxin is lipopolysaccharide (LPS) and that number 12, sodium acts as an injurious stimulus via the endothelial sodium channel (eNAC). CCL2, chemokine (C-C motif) ligand 2; CCL-5, chemokine (C-C motif) ligand 5 (RANTES); ECs, endothelial cells; Fractalkine(s), chemokine (C-X3-C motif) ligand 1; ROS, reactive oxygen species; RONS, reactive oxygen, nitrogen species; RONSS, reactive oxygen, nitrogen, sulfur species; RSI, reactive species interactome; VAS, vascular arterial stiffening.
Figure 2.
Multiple injurious species: “The dirty dozen” affecting the systemic endothelium. Regarding number eight, the ultimate bacterial endotoxin is lipopolysaccharide (LPS) and that number 12, sodium acts as an injurious stimulus via the endothelial sodium channel (eNAC). CCL2, chemokine (C-C motif) ligand 2; CCL-5, chemokine (C-C motif) ligand 5 (RANTES); ECs, endothelial cells; Fractalkine(s), chemokine (C-X3-C motif) ligand 1; ROS, reactive oxygen species; RONS, reactive oxygen, nitrogen species; RONSS, reactive oxygen, nitrogen, sulfur species; RSI, reactive species interactome; VAS, vascular arterial stiffening.

Figure 3.
ECact/dys results in a decrease of bioavailable endothelial nitric oxide (NO), which sounds an alarm to ECs and the cells of the vessel wall. In addition to the seven detrimental effects of decreased nitric oxide also note the important role of increased oxidative redox stress (OxRS) and its important role in uncoupling the NO generating eNOS enzyme reaction that requires its essential co-enzyme tetrahydrobiopterin (BH4) to be totally reduced. If BH4 is oxidized to dihydrobiopterin (BH2) via excessive oxidative redox stress (OxRS) the endothelial nitric oxide synthase (eNOS) enzyme reaction will not produce NO. Also, note the pulse wave velocity wave forms on the left-hand side of the image that illustrates the premature arrival of the reflected pulse wave contributing to the increased pulsatile pulse pressure (pp) to further result in vascular injury and remodeling with increased vascular arterial stiffening of peripheral artery disease. Pulse wave velocity image lower left-hand side reproduced with permission by CC 4.0 [22]. AFM, atomic force microscopy; Asterisk, indicates emphasis; BP, blood pressure; ECact/dys, endothelial cell activation and dysfunction; EC(s), endothelial cells; Ext, extremities; H, heart; OxRS, oxidative redox stress; PAD, peripheral artery disease; Pc, pericyte; V, vessel-vascular; PGN, proteoglycan; VSMC, vascular smooth muscle cell.
Figure 3.
ECact/dys results in a decrease of bioavailable endothelial nitric oxide (NO), which sounds an alarm to ECs and the cells of the vessel wall. In addition to the seven detrimental effects of decreased nitric oxide also note the important role of increased oxidative redox stress (OxRS) and its important role in uncoupling the NO generating eNOS enzyme reaction that requires its essential co-enzyme tetrahydrobiopterin (BH4) to be totally reduced. If BH4 is oxidized to dihydrobiopterin (BH2) via excessive oxidative redox stress (OxRS) the endothelial nitric oxide synthase (eNOS) enzyme reaction will not produce NO. Also, note the pulse wave velocity wave forms on the left-hand side of the image that illustrates the premature arrival of the reflected pulse wave contributing to the increased pulsatile pulse pressure (pp) to further result in vascular injury and remodeling with increased vascular arterial stiffening of peripheral artery disease. Pulse wave velocity image lower left-hand side reproduced with permission by CC 4.0 [22]. AFM, atomic force microscopy; Asterisk, indicates emphasis; BP, blood pressure; ECact/dys, endothelial cell activation and dysfunction; EC(s), endothelial cells; Ext, extremities; H, heart; OxRS, oxidative redox stress; PAD, peripheral artery disease; Pc, pericyte; V, vessel-vascular; PGN, proteoglycan; VSMC, vascular smooth muscle cell.

Figure 4.
A pull-out vessel model demonstrating the three major layers of arteries and how they are structurally interrelated. This figure illustrates the lumen that is surrounded by systemic endothelial cells (SECs pseudo-colored green) that have a basement membrane (BM) pseudo-colored blue, and the proteoglycan (PGN)-rich intima-subintimal space (SiS noted by double arrows). These three tunica layers begin most luminally with the tunica intima composed of the endothelium and its basement membrane the subendothelial intimal space where atherosclerosis is known to originate and expand. Next the thickened black line forms the internal elastic lamina (IEL), which is demarcated by the bold white line, which separates the intima from the tunica media that encircles the artery and serves to allow elastic recoil of the artery during contraction and relaxation. Once the vessel is stretched along with its multiple layers of the elastin lamella that bound the lamellar units containing the vascular smooth muscle cell(s) (VSMCs) pseudo-colored red and denoted by open white arrows that comprise the second tunica that is bound abluminally by the external elastic lamina (yellow dashed line). The most abluminal and third tunica is the tunica adventitia, which is demarcated by the yellow dashed line luminally and the perivascular adipose tissue (PVAT) of the tunica adventitia that contains loose connective tissue with multiple cells including pericytes, leukocytes, and occasional mast cells through which, the vasa vasorum (Vv) flows as it penetrates the media to be referred to as the vessel within the vessel. The perivascular adipose tissue (PVAT underlined and pseudo-colored yellow) contains visceral adipocytes and extracellular matrix) is an extension of the tunica adventitia; however, some feel that it merits the distinction to be called the tunica adiposa due to its proinflammatory-metainflammation potential to result in aberrant arterial remodeling from the outside in. Modified image provided with permission by CC 4.0 [1,21,27]. EC, endothelial cell; EL, elastin lamina; HVExt, heart-vessel-extremity axis; PGN, proteoglycan; Ren2 rat part I and II, ultrastructure study of the young male transgenic heterozygous (mRen2)27 (Ren2) rat hypertensive model: Aorta intima Part I and media-adventitia Part II with elevated renin, angiotensin II, angiotensin type-1 (AT-1) receptors, and aldosterone; Vv, vasa vasorum.
Figure 4.
A pull-out vessel model demonstrating the three major layers of arteries and how they are structurally interrelated. This figure illustrates the lumen that is surrounded by systemic endothelial cells (SECs pseudo-colored green) that have a basement membrane (BM) pseudo-colored blue, and the proteoglycan (PGN)-rich intima-subintimal space (SiS noted by double arrows). These three tunica layers begin most luminally with the tunica intima composed of the endothelium and its basement membrane the subendothelial intimal space where atherosclerosis is known to originate and expand. Next the thickened black line forms the internal elastic lamina (IEL), which is demarcated by the bold white line, which separates the intima from the tunica media that encircles the artery and serves to allow elastic recoil of the artery during contraction and relaxation. Once the vessel is stretched along with its multiple layers of the elastin lamella that bound the lamellar units containing the vascular smooth muscle cell(s) (VSMCs) pseudo-colored red and denoted by open white arrows that comprise the second tunica that is bound abluminally by the external elastic lamina (yellow dashed line). The most abluminal and third tunica is the tunica adventitia, which is demarcated by the yellow dashed line luminally and the perivascular adipose tissue (PVAT) of the tunica adventitia that contains loose connective tissue with multiple cells including pericytes, leukocytes, and occasional mast cells through which, the vasa vasorum (Vv) flows as it penetrates the media to be referred to as the vessel within the vessel. The perivascular adipose tissue (PVAT underlined and pseudo-colored yellow) contains visceral adipocytes and extracellular matrix) is an extension of the tunica adventitia; however, some feel that it merits the distinction to be called the tunica adiposa due to its proinflammatory-metainflammation potential to result in aberrant arterial remodeling from the outside in. Modified image provided with permission by CC 4.0 [1,21,27]. EC, endothelial cell; EL, elastin lamina; HVExt, heart-vessel-extremity axis; PGN, proteoglycan; Ren2 rat part I and II, ultrastructure study of the young male transgenic heterozygous (mRen2)27 (Ren2) rat hypertensive model: Aorta intima Part I and media-adventitia Part II with elevated renin, angiotensin II, angiotensin type-1 (AT-1) receptors, and aldosterone; Vv, vasa vasorum.
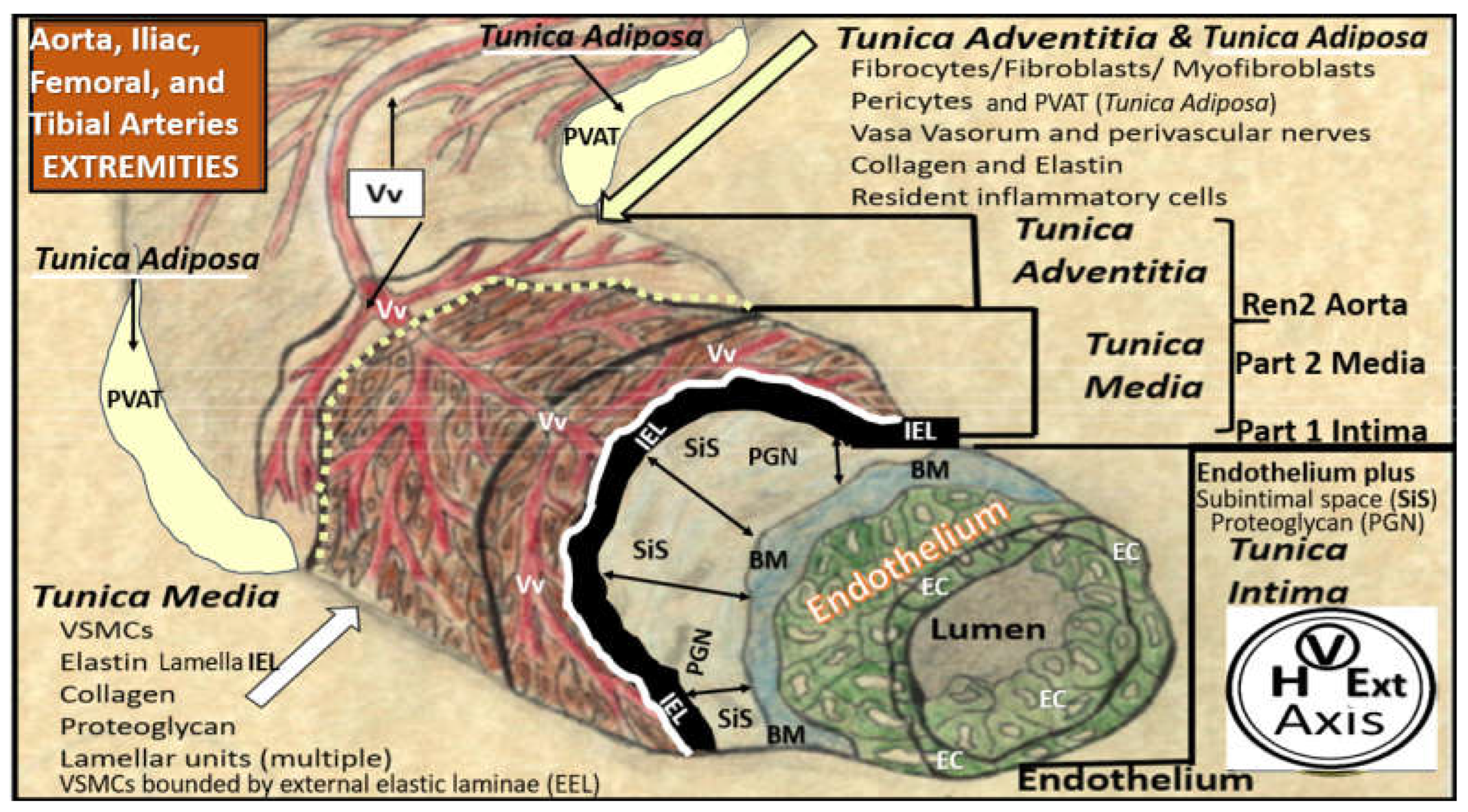
Figure 6.
Malignant-like invasion of vasa vasorum (Vv) to result in intraplaque hemorrhage (IPH) of a type VI plaque to result in thrombosis and distal ischemia. Note the increased angiogenesis of the hand-drawn vessel on the left and how it invades the type VI plaque on the right to result in a type VI thin cap atheroma prone to rupture resulting in thrombosis. This process of Vv invasion results in a lipid laden, prothrombotic, acidic, angiogenic, inflamed, ‘hot plaque’ due to IPH, Revised figures provided with permission by CC 4.0 [27]. ECact/dys, endothelial activation and dysfunction; eNOS, endothelial nitric oxide synthase; ph, pH or potential of hydrogen and a measure of acidity; PVAT, perivascular adipose tissue; NO, nitric oxide; perivascular adipose tissue; OxRS, oxidative redox stress.
Figure 6.
Malignant-like invasion of vasa vasorum (Vv) to result in intraplaque hemorrhage (IPH) of a type VI plaque to result in thrombosis and distal ischemia. Note the increased angiogenesis of the hand-drawn vessel on the left and how it invades the type VI plaque on the right to result in a type VI thin cap atheroma prone to rupture resulting in thrombosis. This process of Vv invasion results in a lipid laden, prothrombotic, acidic, angiogenic, inflamed, ‘hot plaque’ due to IPH, Revised figures provided with permission by CC 4.0 [27]. ECact/dys, endothelial activation and dysfunction; eNOS, endothelial nitric oxide synthase; ph, pH or potential of hydrogen and a measure of acidity; PVAT, perivascular adipose tissue; NO, nitric oxide; perivascular adipose tissue; OxRS, oxidative redox stress.
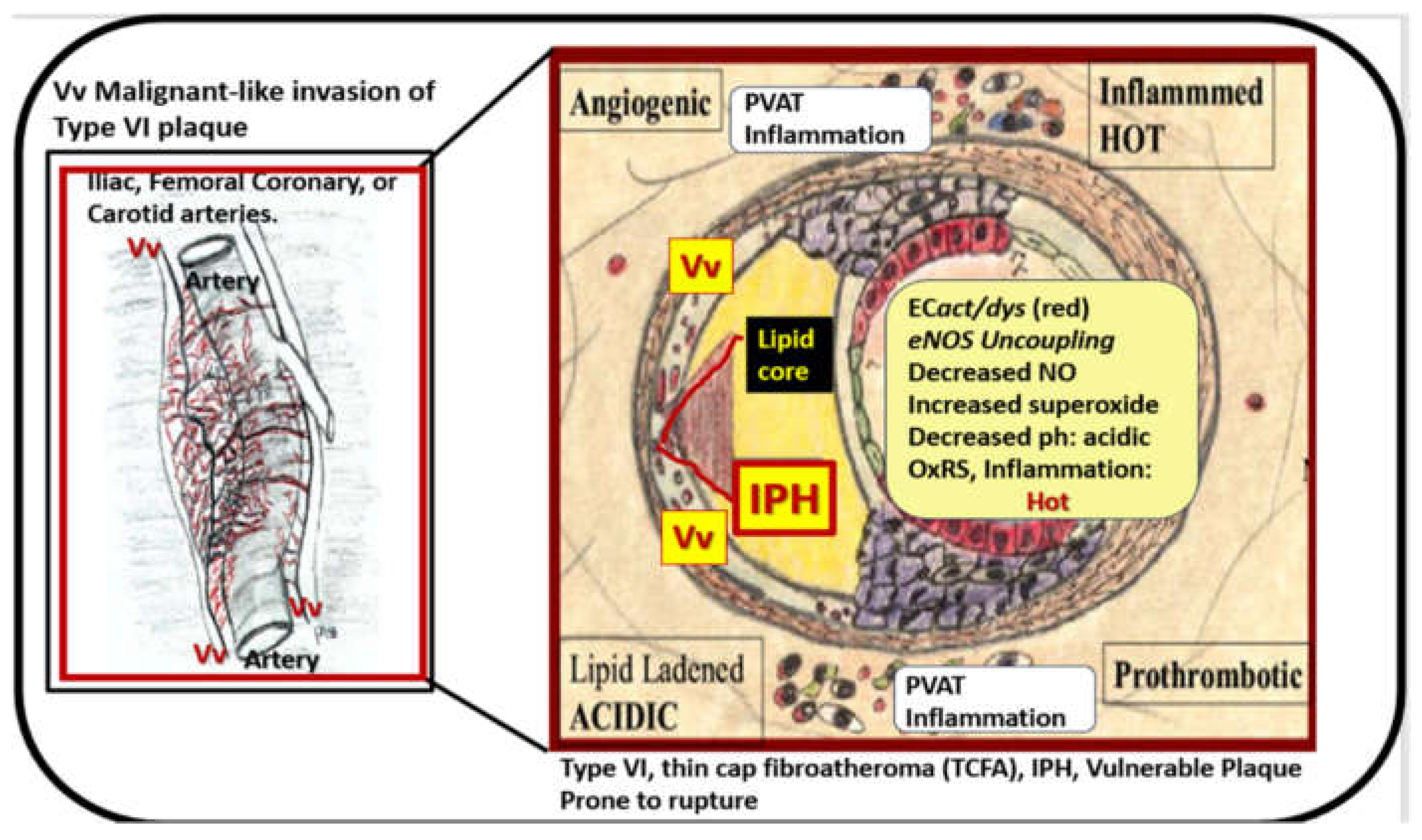
Figure 7.
In femoral-popliteal arteries acute thrombi and atherosclerotic plaques are more common in contrast to infra-popliteal arteries (anterior tibial, posterior tibial, peroneal, and dorsalis pedis arteries) where chronic thrombosis and emboli predominate with medial calcifications and fewer atherosclerotic plaques. Note circular insert (upper far-left) that depicts the abdominal aorta, the iliac bifurcations and the femoral arteries. In the regular magnetic resonance angiography (MRA) (far-left) note the yellow horizontal line that separates the superior femoral-popliteal (FEM-POP) regions from the more distal infra-popliteal (INFRA-POP) regions. a, artery; ATA, anterior tibial artery’ ECact/dys, endothelial cell activation and dysfunction; eNOS, endothelial nitric oxide synthase; FEM, femoral artery; H2S, hydrogen sulfide; NO, nitric oxide; PA, peroneal artery; PTA, posterior tibial artery; TPT, tibia peroneal trunk; STS, sodium thiosulfate.
Figure 7.
In femoral-popliteal arteries acute thrombi and atherosclerotic plaques are more common in contrast to infra-popliteal arteries (anterior tibial, posterior tibial, peroneal, and dorsalis pedis arteries) where chronic thrombosis and emboli predominate with medial calcifications and fewer atherosclerotic plaques. Note circular insert (upper far-left) that depicts the abdominal aorta, the iliac bifurcations and the femoral arteries. In the regular magnetic resonance angiography (MRA) (far-left) note the yellow horizontal line that separates the superior femoral-popliteal (FEM-POP) regions from the more distal infra-popliteal (INFRA-POP) regions. a, artery; ATA, anterior tibial artery’ ECact/dys, endothelial cell activation and dysfunction; eNOS, endothelial nitric oxide synthase; FEM, femoral artery; H2S, hydrogen sulfide; NO, nitric oxide; PA, peroneal artery; PTA, posterior tibial artery; TPT, tibia peroneal trunk; STS, sodium thiosulfate.
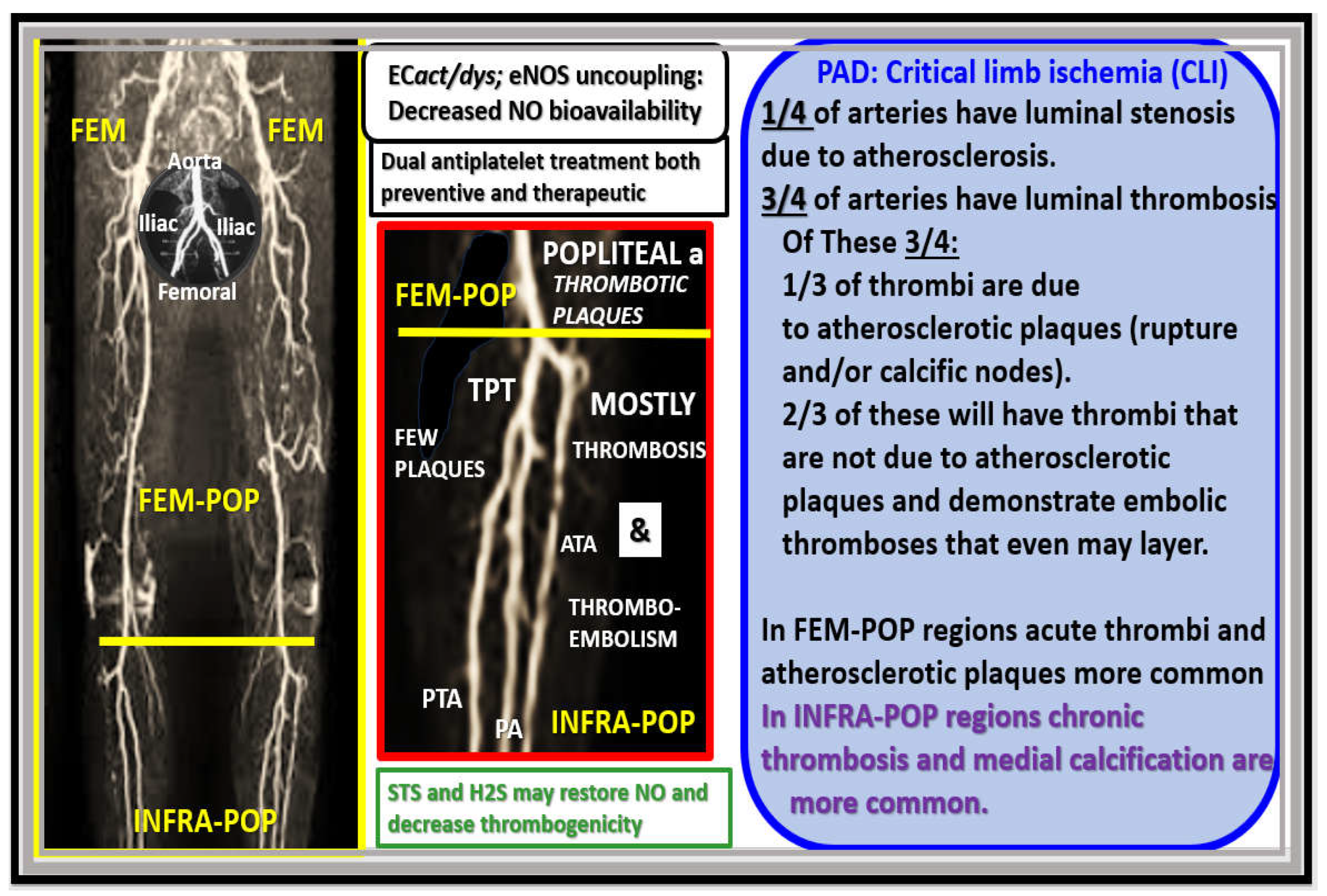
Figure 8.
The essential hypertension (eHTN-HTN) wheel. Insulin resistance (IR) presents as a central core feature of the HTN wheel as it is also the central core feature in the metabolic syndrome (MetS). IR is a central mediator for the development of HTN and impaired fasting glucose (IFG)-impaired glucose tolerance (IGT)–type two diabetes mellitus (T2DM), along with multiple metabolic and clinical boxed-in clinical conditions that surround the outer positions of this wheel. Extracranial vascular arterial stiffening (VAS) and PAD are placed at the top of the wheel because they are believed to be a driving force behind the subsequent clinical end-organ remodeling and disease in addition to the development of HTN. Endothelial cell activation and dysfunction (ECact/dys) at the bottom of the wheel results in increased peroxynitrite (ONOO−) and decreased nitric oxide (NO) bioavailability (asterisk). Peroxynitrite is generated by the reaction between superoxide and NO. Note the multiple clinical diseases that are associated with HTN, and the wheel depicts the interconnectedness between HTN and the multiple clinical disease states, including vascular stiffening and PAD. Thus, obesity, IR, HTN, and T2DM are not to be underestimated. The wheel background icon was chosen because it goes round and round, and over time it just keeps on turning and results in vascular arterial stiffening (VAS), PAD, and end-organ damage in the heart-vessel-extremity axis. Modified image provided with permission by CC 4.0 [21]. Asterisk, indicates emphasis; CAD, coronary artery disease; CCVD, cerebrocardiovascular disease; CHF, congestive heart failure; CKD, chronic kidney disease; Dd, diastolic dysfunction; eNOS, endothelial nitric oxide synthase; ESRD, end-stage renal disease; LOAD, late-onset Alzheimer’s disease; MASLD, metabolic dysfunction-associated liver disease; MI, myocardial infarction; mtROS, mitochondrial reactive oxygen species; O2•–, superoxide; OxRS, oxidative redox stress; PAD, peripheral artery disease;.
Figure 8.
The essential hypertension (eHTN-HTN) wheel. Insulin resistance (IR) presents as a central core feature of the HTN wheel as it is also the central core feature in the metabolic syndrome (MetS). IR is a central mediator for the development of HTN and impaired fasting glucose (IFG)-impaired glucose tolerance (IGT)–type two diabetes mellitus (T2DM), along with multiple metabolic and clinical boxed-in clinical conditions that surround the outer positions of this wheel. Extracranial vascular arterial stiffening (VAS) and PAD are placed at the top of the wheel because they are believed to be a driving force behind the subsequent clinical end-organ remodeling and disease in addition to the development of HTN. Endothelial cell activation and dysfunction (ECact/dys) at the bottom of the wheel results in increased peroxynitrite (ONOO−) and decreased nitric oxide (NO) bioavailability (asterisk). Peroxynitrite is generated by the reaction between superoxide and NO. Note the multiple clinical diseases that are associated with HTN, and the wheel depicts the interconnectedness between HTN and the multiple clinical disease states, including vascular stiffening and PAD. Thus, obesity, IR, HTN, and T2DM are not to be underestimated. The wheel background icon was chosen because it goes round and round, and over time it just keeps on turning and results in vascular arterial stiffening (VAS), PAD, and end-organ damage in the heart-vessel-extremity axis. Modified image provided with permission by CC 4.0 [21]. Asterisk, indicates emphasis; CAD, coronary artery disease; CCVD, cerebrocardiovascular disease; CHF, congestive heart failure; CKD, chronic kidney disease; Dd, diastolic dysfunction; eNOS, endothelial nitric oxide synthase; ESRD, end-stage renal disease; LOAD, late-onset Alzheimer’s disease; MASLD, metabolic dysfunction-associated liver disease; MI, myocardial infarction; mtROS, mitochondrial reactive oxygen species; O2•–, superoxide; OxRS, oxidative redox stress; PAD, peripheral artery disease;.
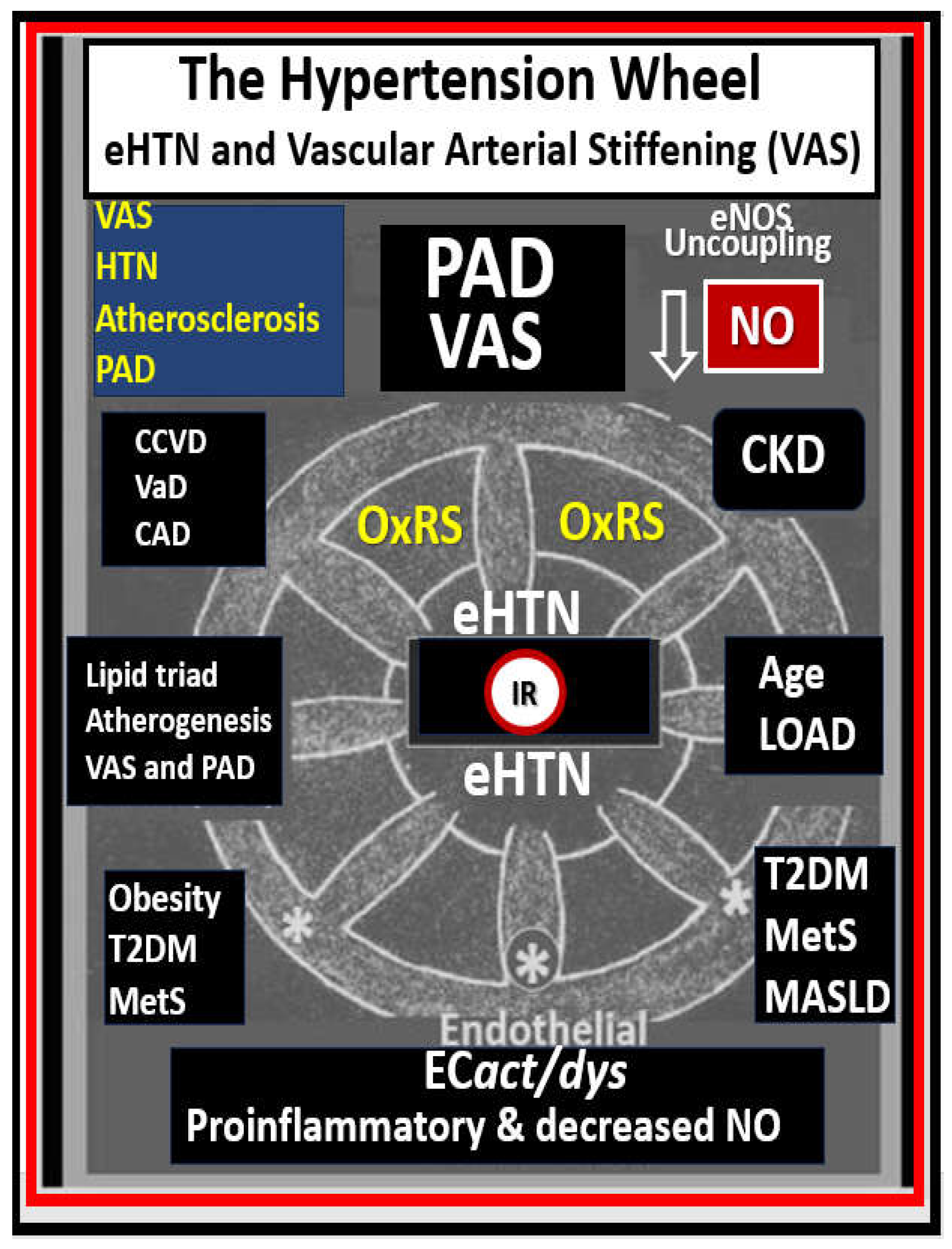
Figure 9.
Multiple risk factors for the development of peripheral artery disease (PAD) include both hypertension (HTN), atherosclerosis, and the metabolic syndrome (MetS). Note how the metabolic syndrome (MetS) upper right is a major supplier for the modifiable risk factors for the development cerebrocardiovascular diseases (CCVDs) including PAD of the extremities. Also note the non-modifiable risks lower-left and the lifestyle and social risks lower-right. Importantly, note the importance of endothelial cell activation and dysfunction (ECact/dys) and how the uncoupling of the eNOS reaction leads to a decrease in the bioavailability of nitric oxide (NO), which contributes to a prothrombotic state and thrombotic emboli due to decreased NO. Also, note the importance of the vicious cycle upper left between hypertension, atherosclerosis, and vascular stiffness (VAS). Modified image supplied with permission by 4.0 [21]. CCVD, cerebrocardiovascular disease; CHF, congestive heart failure; CVD(s), cardiovascular diseases; ECact/dys, endothelial cell activation and dysfunction; FFA, free fatty acids; SVD, small vessel disease; T2DM, type 2 diabetes mellitus.
Figure 9.
Multiple risk factors for the development of peripheral artery disease (PAD) include both hypertension (HTN), atherosclerosis, and the metabolic syndrome (MetS). Note how the metabolic syndrome (MetS) upper right is a major supplier for the modifiable risk factors for the development cerebrocardiovascular diseases (CCVDs) including PAD of the extremities. Also note the non-modifiable risks lower-left and the lifestyle and social risks lower-right. Importantly, note the importance of endothelial cell activation and dysfunction (ECact/dys) and how the uncoupling of the eNOS reaction leads to a decrease in the bioavailability of nitric oxide (NO), which contributes to a prothrombotic state and thrombotic emboli due to decreased NO. Also, note the importance of the vicious cycle upper left between hypertension, atherosclerosis, and vascular stiffness (VAS). Modified image supplied with permission by 4.0 [21]. CCVD, cerebrocardiovascular disease; CHF, congestive heart failure; CVD(s), cardiovascular diseases; ECact/dys, endothelial cell activation and dysfunction; FFA, free fatty acids; SVD, small vessel disease; T2DM, type 2 diabetes mellitus.
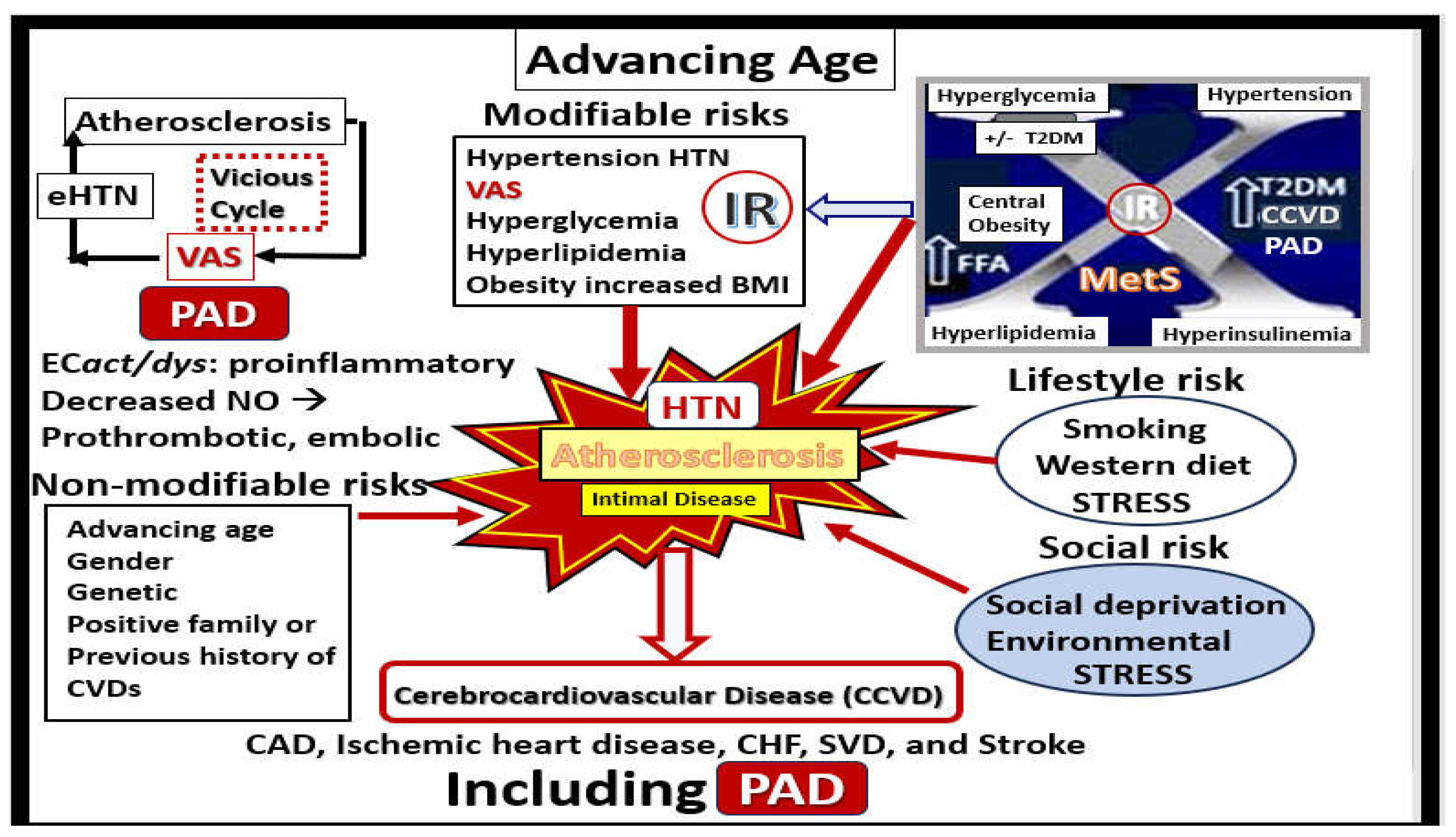
Figure 10.
Metabolic syndrome (MetS). The “H” phenomenon (four horsemen) of the MetS includes: I. Hyperlipidemia (obesity and the atherogenic lipid triad); II. Hyperinsulinemia (insulin resistance-(IR)), III. Hypertension (HTN); IV. Hyperglycemia (impaired glucose tolerance (IGT) +/- type 2 diabetes mellitus (T2DM), as well as, the clustering effect of each of the multiple variables of the MetS itself and contribute to extracranial vascular arterial stiffening (VAS) and peripheral artery disease (PAD). Additionally, IR and LR contribute to vascular stiffening primarily through the actions of obesity on the development of VAS. Modified graphic abstract image provided with permission by CC 4.0 [57]. AAA, abdominal aortic aneurysm; aMt, aberrant mitochondria; CCVD, cerebrocardiovascular disease; CAD, coronary artery disease; CKD, chronic kidney disease; CL, capillary lumen; CVD, cardiovascular disease; BHK, brain-heart-kidney axis; CHF, congestive heart failure; Dd, diastolic dysfunction; EC, endothelial cell; FFA, free fatty acids; H, hyperlipidemia, hyperinsulinemia, hypertension, and hyperglycemia; HCY, homocysteine; HHCY, hyperhomocyteinemia; HVExt, heart-vessel-extremity axis; HVB, heart-vessel-brain axis; IR, insulin resistance; LR, leptin resistance; lncRNAs, long non-coding ribonucleic acids MI, myocardial infarction; miRNAs, micro ribonucleic acids; MΦ, macrophage; NO, nitric oxide; ROS, reactive oxygen species; RONS, reactive oxygen, nitrogen species; RONSS, reactive oxygen, nitrogen, sulfur species; RSI, reactive species interactome.
Figure 10.
Metabolic syndrome (MetS). The “H” phenomenon (four horsemen) of the MetS includes: I. Hyperlipidemia (obesity and the atherogenic lipid triad); II. Hyperinsulinemia (insulin resistance-(IR)), III. Hypertension (HTN); IV. Hyperglycemia (impaired glucose tolerance (IGT) +/- type 2 diabetes mellitus (T2DM), as well as, the clustering effect of each of the multiple variables of the MetS itself and contribute to extracranial vascular arterial stiffening (VAS) and peripheral artery disease (PAD). Additionally, IR and LR contribute to vascular stiffening primarily through the actions of obesity on the development of VAS. Modified graphic abstract image provided with permission by CC 4.0 [57]. AAA, abdominal aortic aneurysm; aMt, aberrant mitochondria; CCVD, cerebrocardiovascular disease; CAD, coronary artery disease; CKD, chronic kidney disease; CL, capillary lumen; CVD, cardiovascular disease; BHK, brain-heart-kidney axis; CHF, congestive heart failure; Dd, diastolic dysfunction; EC, endothelial cell; FFA, free fatty acids; H, hyperlipidemia, hyperinsulinemia, hypertension, and hyperglycemia; HCY, homocysteine; HHCY, hyperhomocyteinemia; HVExt, heart-vessel-extremity axis; HVB, heart-vessel-brain axis; IR, insulin resistance; LR, leptin resistance; lncRNAs, long non-coding ribonucleic acids MI, myocardial infarction; miRNAs, micro ribonucleic acids; MΦ, macrophage; NO, nitric oxide; ROS, reactive oxygen species; RONS, reactive oxygen, nitrogen species; RONSS, reactive oxygen, nitrogen, sulfur species; RSI, reactive species interactome.

Figure 11.
Schematic for how type 2 diabetes mellitus (T2DM) associates with accelerated atherosclerosis and a two-fold increase in prevalence of peripheral artery disease (PAD). Note that ECact/dys is outlined in red color to highlight its importance because EC activation (proinflammatory endothelium) and dysfunction (decrease of protective bioavailable nitric oxide (NO) of the endothelium) of the vascular endothelium is the hallmark of atherosclerosis in T2DM. ECs, endothelial cells; ECact/dys, endothelial cell activation and dysfunction; HDL-C, high density lipoprotein cholesterol; IR, insulin resistance; ISH, isolated systolic hypertension; LDL, low-density lipoprotein cholesterol; MetS, metabolic syndrome; NO, nitric oxide; OxRS, oxidative redox stress; NFkappaB, nuclear factor kappa B; PAI-1, plasminogen activator inhibitor-1; RAGE, receptor for advanced glycation end products. .
Figure 11.
Schematic for how type 2 diabetes mellitus (T2DM) associates with accelerated atherosclerosis and a two-fold increase in prevalence of peripheral artery disease (PAD). Note that ECact/dys is outlined in red color to highlight its importance because EC activation (proinflammatory endothelium) and dysfunction (decrease of protective bioavailable nitric oxide (NO) of the endothelium) of the vascular endothelium is the hallmark of atherosclerosis in T2DM. ECs, endothelial cells; ECact/dys, endothelial cell activation and dysfunction; HDL-C, high density lipoprotein cholesterol; IR, insulin resistance; ISH, isolated systolic hypertension; LDL, low-density lipoprotein cholesterol; MetS, metabolic syndrome; NO, nitric oxide; OxRS, oxidative redox stress; NFkappaB, nuclear factor kappa B; PAI-1, plasminogen activator inhibitor-1; RAGE, receptor for advanced glycation end products. .
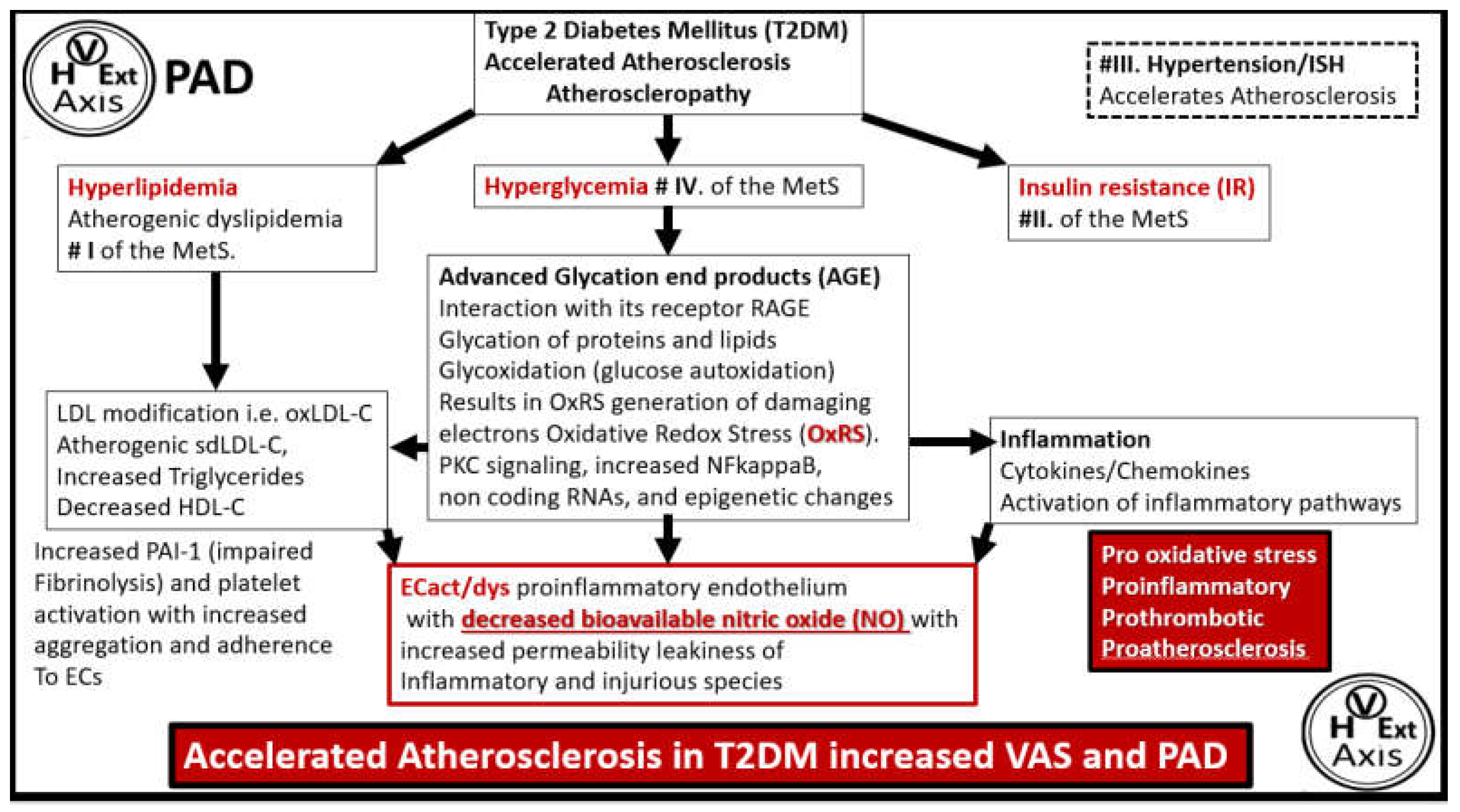
Figure 12.
Nitric oxide has at least five positive and protective effects on the arterial vessel wall. Antithrombotic, anti-inflammatory, anti-atherosclerotic, vasodilatory, and proangiogenic effects. BH4. Tetrahydrobiopterin necessary coenzyme for eNOS enzyme; EC, endothelial cell; eNOS, endothelial nitroxide synthase; VSMC, vascular smooth muscle cell. .
Figure 12.
Nitric oxide has at least five positive and protective effects on the arterial vessel wall. Antithrombotic, anti-inflammatory, anti-atherosclerotic, vasodilatory, and proangiogenic effects. BH4. Tetrahydrobiopterin necessary coenzyme for eNOS enzyme; EC, endothelial cell; eNOS, endothelial nitroxide synthase; VSMC, vascular smooth muscle cell. .
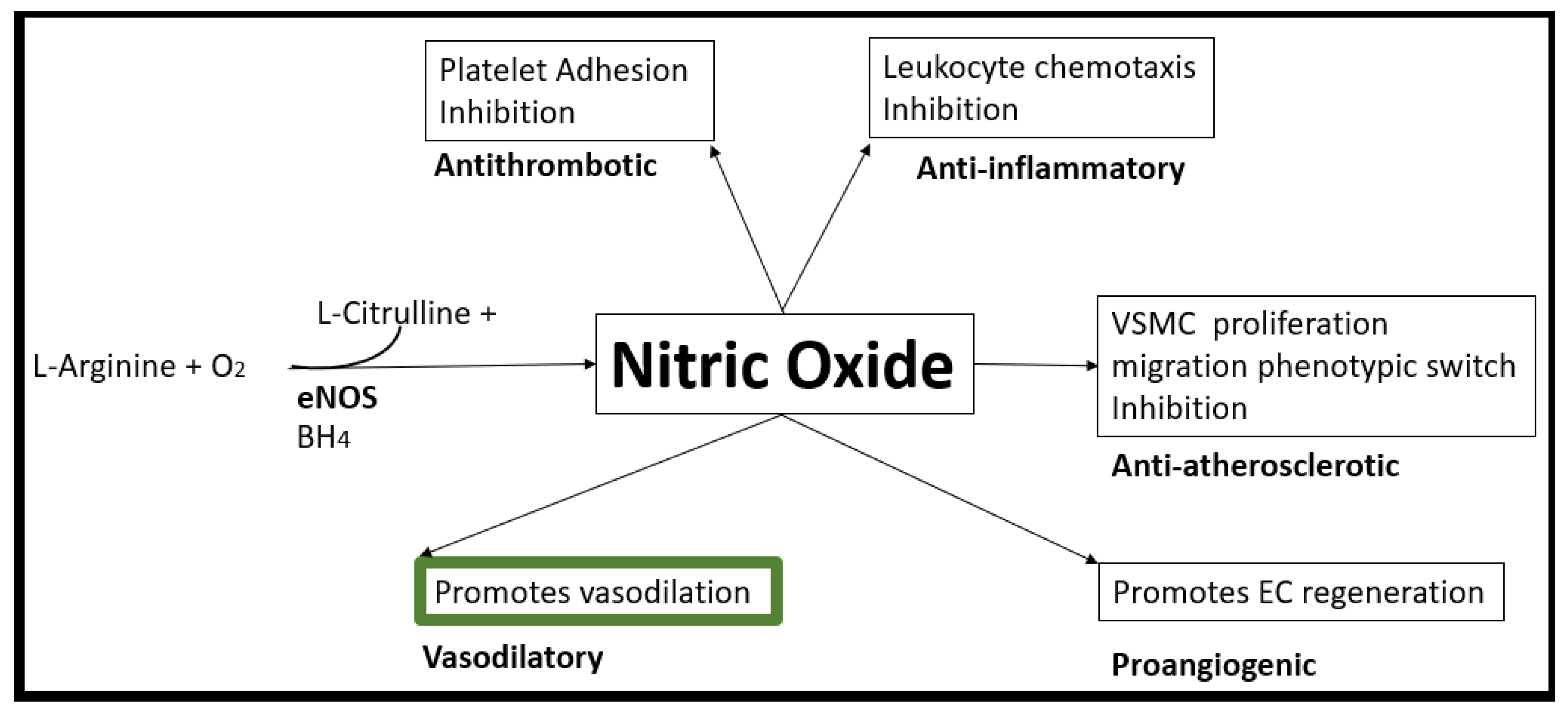
Figure 13.
Development of vascular arterial stiffening (VAS) in peripheral artery disease (PAD). The process of VAS development begins with the presence of injurious species within the lumen and note the pulsatile pulse pressure (pp) boxed-in red along with the dirty dozen from Figure 2 along with neurohormonal and sympathetic stimuli with increased shear stress (step 1). Next, the endothelium becomes activated and dysfunctional (ECact/dys) with increased permeability (step 2), which allows these injurious species to penetrate the vessel wall with subsequent structural remodeling and dysfunctional steps that follow (steps 3-5) in the development of VAS. Note the increased pulsatile pp (red arrows) and pulse wave velocity (pWV) upper right insert that play an important role in both macro-and microvessel aberrant remodeling with dysfunction and damage in VAS. Incidently, it is important to note that both hypertension and atherosclerosis also contribute to the development of VAS. Modified background image provided with permission by 4.0 [108]. AGE(s), advanced glycation end-products; CaPO4, calcium phosphate; EDFs, elastin-derived fragments; EDPs, elastin-derived peptides; I-CAM, intercellular adhesion molecule; NO, MΦ, macrophage; nitric oxide; RAAS, renin-angiotensin-aldosterone system; ROS, reactive oxygen species; TGF-β1, transforming growth factor beta-one; VOC, vascular ossification-calcification; VSMCs, vascular smooth muscle cells. .
Figure 13.
Development of vascular arterial stiffening (VAS) in peripheral artery disease (PAD). The process of VAS development begins with the presence of injurious species within the lumen and note the pulsatile pulse pressure (pp) boxed-in red along with the dirty dozen from Figure 2 along with neurohormonal and sympathetic stimuli with increased shear stress (step 1). Next, the endothelium becomes activated and dysfunctional (ECact/dys) with increased permeability (step 2), which allows these injurious species to penetrate the vessel wall with subsequent structural remodeling and dysfunctional steps that follow (steps 3-5) in the development of VAS. Note the increased pulsatile pp (red arrows) and pulse wave velocity (pWV) upper right insert that play an important role in both macro-and microvessel aberrant remodeling with dysfunction and damage in VAS. Incidently, it is important to note that both hypertension and atherosclerosis also contribute to the development of VAS. Modified background image provided with permission by 4.0 [108]. AGE(s), advanced glycation end-products; CaPO4, calcium phosphate; EDFs, elastin-derived fragments; EDPs, elastin-derived peptides; I-CAM, intercellular adhesion molecule; NO, MΦ, macrophage; nitric oxide; RAAS, renin-angiotensin-aldosterone system; ROS, reactive oxygen species; TGF-β1, transforming growth factor beta-one; VOC, vascular ossification-calcification; VSMCs, vascular smooth muscle cells. .
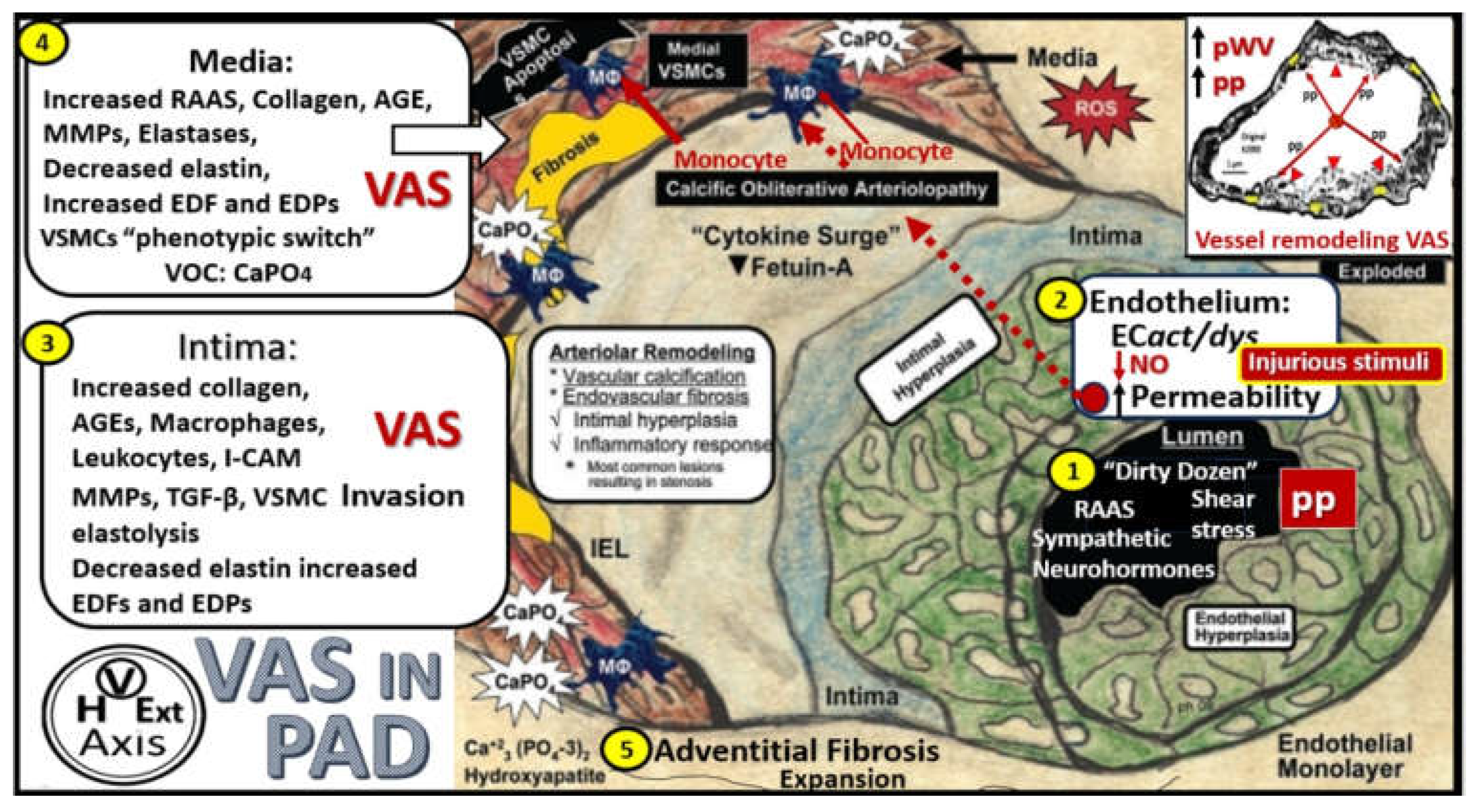
Figure 14.
A collection of representitive ultrastructural images that are only meant to illustrate the following: Endothelial cell (EC) lifting and separation in panel A, elastin separation and degradation in panel B, the formation of elastin-derived fragments (EDFs) via elastolysis that are a rich source of elastin-derived peptides (EDPs) in panel C, which represent remodeling changes at the endothelium and intima regions, which involves both the internal elastic lamina (IEL) and the elastin that forms the boundaries of the elastin lamellar units containing vascular smooth muscle cell(s) (VSMCs) sandwiched between the elastin lamella (EL). Then the phenotypic shift of VSMCs is illustrated in panel C, phenotype shift of VSMCs in panel D and the migratory “red dragon” VSMC representing the migratory potential and the creation of neointima just abluminally to the EC cell in panel E, and the migratory VSMCs creating and invading the neointimal space in panel F. Images A-C are from the obese, insulin resistant female diabetic db/db mouse models at 20 weeks of age without hypertension and images D-G are from a male genetic-induced hypertensive Ren2 rat at 14 weeks of age. Modified images panels A-C and panels D-F are provided with permission respectfully by CC4.0 [21,109,110]. Asterisks, elastin splitting and degradation; DBC, female, obese, insulin resistant diabetic db/db models at 20-weeks of age; Mt, mitochondria; N, nucleus; PGN(s), proteoglycans; Ren2, male transgenic hypertensive models TGR(mRen2)27 rat at 10-weeks of age; ROS, reactive oxygen species; V, vacuole; v, vesicle; WP, Weible-Palade bodies.
Figure 14.
A collection of representitive ultrastructural images that are only meant to illustrate the following: Endothelial cell (EC) lifting and separation in panel A, elastin separation and degradation in panel B, the formation of elastin-derived fragments (EDFs) via elastolysis that are a rich source of elastin-derived peptides (EDPs) in panel C, which represent remodeling changes at the endothelium and intima regions, which involves both the internal elastic lamina (IEL) and the elastin that forms the boundaries of the elastin lamellar units containing vascular smooth muscle cell(s) (VSMCs) sandwiched between the elastin lamella (EL). Then the phenotypic shift of VSMCs is illustrated in panel C, phenotype shift of VSMCs in panel D and the migratory “red dragon” VSMC representing the migratory potential and the creation of neointima just abluminally to the EC cell in panel E, and the migratory VSMCs creating and invading the neointimal space in panel F. Images A-C are from the obese, insulin resistant female diabetic db/db mouse models at 20 weeks of age without hypertension and images D-G are from a male genetic-induced hypertensive Ren2 rat at 14 weeks of age. Modified images panels A-C and panels D-F are provided with permission respectfully by CC4.0 [21,109,110]. Asterisks, elastin splitting and degradation; DBC, female, obese, insulin resistant diabetic db/db models at 20-weeks of age; Mt, mitochondria; N, nucleus; PGN(s), proteoglycans; Ren2, male transgenic hypertensive models TGR(mRen2)27 rat at 10-weeks of age; ROS, reactive oxygen species; V, vacuole; v, vesicle; WP, Weible-Palade bodies.

Figure 15.
Possible sequence of events leading to the development and progression of peripheral artery disease (PAD). The sequence of events begins with the injury to the endothelium at the top of the figure that was previously presented in Figure 1 and Figure 2 and is followed sequentially by the six steps and representative images (1-6) with explanation to the left-hand side to arrive at the magnetic resonance angiography (MRA) images of the aorta, iliac, femoral, popliteal, and the distal anterior and posterior tibial, and peroneal arteries of the lower extremity images on the right-hand side of the Figure (7, 8) in regards to clinical assessment of PAD. Note that steps and figures in Figure 3 and Figure 4 are from diabetic models), and that step 5 is from hypertensive models and denote the beginning of the vessel wall cellular remodeling that is associated with both atherosclerosis and hypertension, which associate with vascular arterial stiffening (VAS) and the development and progression of PAD. Further, note that steps 3, 4, and 5 are associated with vascular calcification that is discussed in greater detail in the following section. As can be observed, the development and progression PAD involves the multifactorial interactions and remodeling changes, which culminate in hypertensive changes and excessive atherosclerosis that associate with stiffening of the vascular wall to result in the development and progression of PAD. Importantly, note the two vicious cycles on the road to develop PAD. Once PAD is established it is the formation of progressive vascular narrowing with thrombosis and emboli that result in ischemia and multiple painful clinical stages from intermittent claudication to critical limb ischemia and potential skin ulceration. Notably, most of the clinical symptoms in the in Infra-POP regions result from thrombosis and embolic phenomenon and not atherosclerosis remodeling changes. While these sequential six steps are presented, it is important to note that each of these steps are in a dynamic interaction with one another to result in the multifactorial development and progression of PAD. Notably, vascular ossification and calcification was not included in this six-step process that would occur between step-5 and step-6, it is nevertheless very important in some individuals in regards to the development and progression of PAD and vascular stiffening that is discussed in the following section 4.2. EC, endothelial cell; ECact/dys, endothelial cell activation and dysfunction; ecGCx, endothelial glycocalyx; EDF, elastin-derived fragments; EDP, elastin-derived products; eHTN, essential hypertension; FEM-POP, femoral-popliteal arteries; HPA, hypothalamus pituitary axis; INFRA-POP, inferior popliteal arteries; MMPs, matrix metalloproteinases; NFkappaB, nuclear factor kappa B; NO, nitric oxide; OxRS, oxidative redox stress; PTSD, post-traumatic stress disorder; VSMC, vascular smooth muscle cell.
Figure 15.
Possible sequence of events leading to the development and progression of peripheral artery disease (PAD). The sequence of events begins with the injury to the endothelium at the top of the figure that was previously presented in Figure 1 and Figure 2 and is followed sequentially by the six steps and representative images (1-6) with explanation to the left-hand side to arrive at the magnetic resonance angiography (MRA) images of the aorta, iliac, femoral, popliteal, and the distal anterior and posterior tibial, and peroneal arteries of the lower extremity images on the right-hand side of the Figure (7, 8) in regards to clinical assessment of PAD. Note that steps and figures in Figure 3 and Figure 4 are from diabetic models), and that step 5 is from hypertensive models and denote the beginning of the vessel wall cellular remodeling that is associated with both atherosclerosis and hypertension, which associate with vascular arterial stiffening (VAS) and the development and progression of PAD. Further, note that steps 3, 4, and 5 are associated with vascular calcification that is discussed in greater detail in the following section. As can be observed, the development and progression PAD involves the multifactorial interactions and remodeling changes, which culminate in hypertensive changes and excessive atherosclerosis that associate with stiffening of the vascular wall to result in the development and progression of PAD. Importantly, note the two vicious cycles on the road to develop PAD. Once PAD is established it is the formation of progressive vascular narrowing with thrombosis and emboli that result in ischemia and multiple painful clinical stages from intermittent claudication to critical limb ischemia and potential skin ulceration. Notably, most of the clinical symptoms in the in Infra-POP regions result from thrombosis and embolic phenomenon and not atherosclerosis remodeling changes. While these sequential six steps are presented, it is important to note that each of these steps are in a dynamic interaction with one another to result in the multifactorial development and progression of PAD. Notably, vascular ossification and calcification was not included in this six-step process that would occur between step-5 and step-6, it is nevertheless very important in some individuals in regards to the development and progression of PAD and vascular stiffening that is discussed in the following section 4.2. EC, endothelial cell; ECact/dys, endothelial cell activation and dysfunction; ecGCx, endothelial glycocalyx; EDF, elastin-derived fragments; EDP, elastin-derived products; eHTN, essential hypertension; FEM-POP, femoral-popliteal arteries; HPA, hypothalamus pituitary axis; INFRA-POP, inferior popliteal arteries; MMPs, matrix metalloproteinases; NFkappaB, nuclear factor kappa B; NO, nitric oxide; OxRS, oxidative redox stress; PTSD, post-traumatic stress disorder; VSMC, vascular smooth muscle cell.
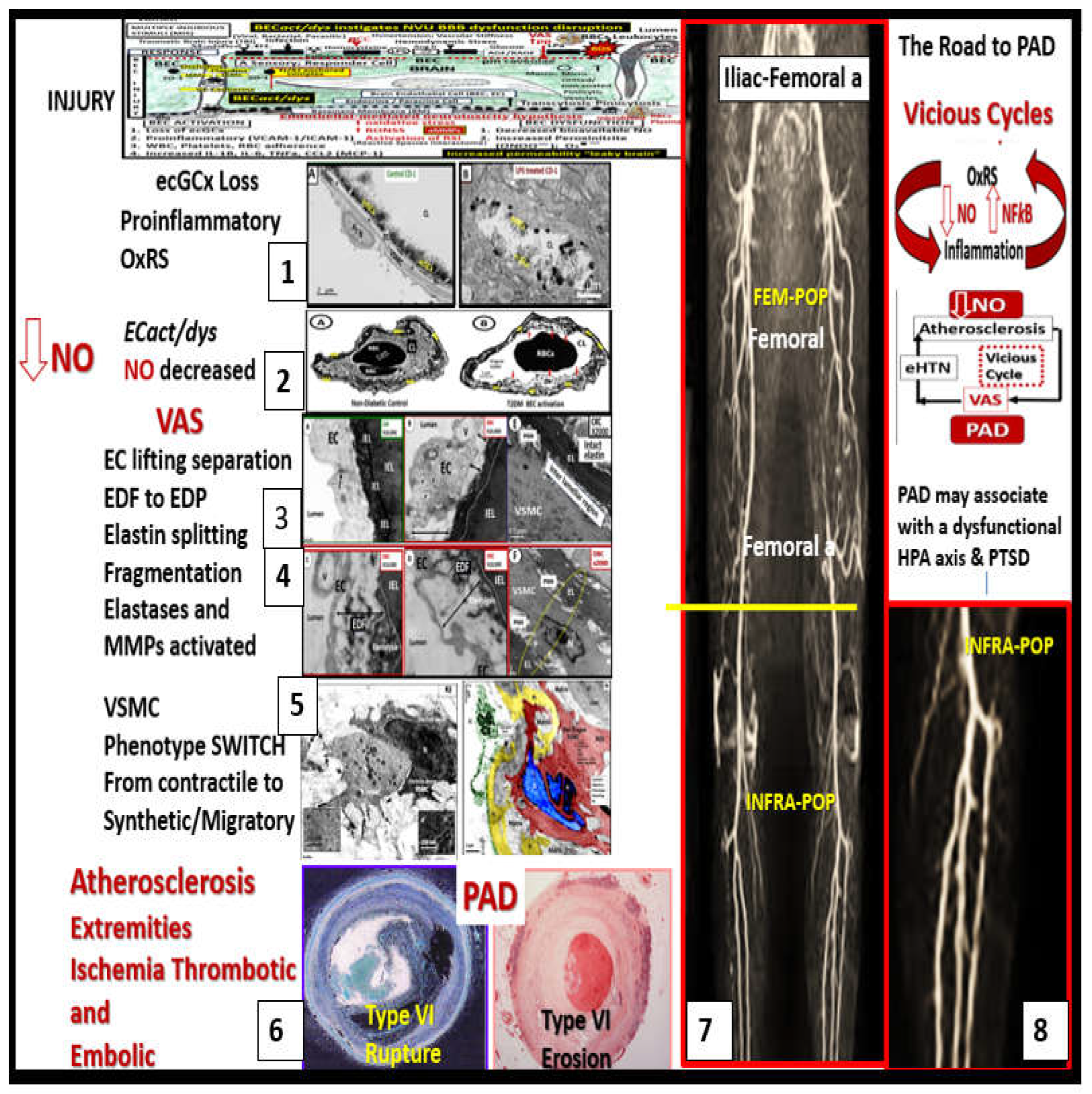
Figure 16.
Phenotype Switching of vascular smooth muscle cells (VSMCs) predisposes to vascular ossification, calcification (VOC) in the media of arteries and arterioles. This image depicts the possible involved mechanisms that involve oxidative redox stress (OxRS -reactive oxygen species) and various uremic toxins that are responsible for VOC. VOC represents a pathologic secondary ossification site or dystrophic calcification in a response to injury to endothelial cells and the subsequent VSMC phenotypic switch phenomenon from contractile to synthetic phenotype. Multiple injurious species as in the dirty dozen plus specific uremic toxins (increased parathyroid hormone (PTH), phosphorus (Pi), phosphate (P043-), calcium, calcium x phosphorus product, vitamin D3 trigger this mechanism of inducing the osteoblast-like phenotype that occurs during the phenotype shift in VSMCs. Phosphate absorption into these cells is facilitated by the sodium phosphate cotransporter (Pit-1) resulting in an osteogenic switch due to activation of transcription factors: osteoblast-specific cis-acting element (Osf2)—core binding factor alpha1 (Cbfa-1/Runx2). Osteocalcin, osteonectin, bone morphogenic protein-2alpha and alkaline phosphatase (ALP) are also known inducers of calcification. In contrast, the systemic and local inhibitors of calcification fetuin-A (alpha2-Heremans-Schmid glycoprotein (AHSG) and matrix Gla protein (MGP) are decreased in uremia and calciphylaxis. ROS and associated inflammatory cytokine surges may contribute to decreased hepatic synthesis of fetuin-A (insert a). Uremic toxins—ROS promote uncoupling of endothelial nitric oxide synthase (eNOS) enzyme via the oxidation of the requisite tetrahydrobiopterin (BH4) cofactor and results in the endothelium becoming a net producer of superoxide—ROS (insert b). Importantly, decreased bioavailable NO due to eNOS enzyme uncoupling promotes a proinflammatory, proconstrictive, prothrombotic vascular endothelium and note the constricted, collapsed arteriole (insert b). ROS are also capable of promoting VSMC apoptosis in the arterial vascular wall and when this occurs the matrix vesicles and apoptotic bodies serve as nucleating sites for further calcium deposition in the extracellular matrix of the arteriole media (inserts c–e). Image reproduced with permission by CC 4.0 [21,108,126].
Figure 16.
Phenotype Switching of vascular smooth muscle cells (VSMCs) predisposes to vascular ossification, calcification (VOC) in the media of arteries and arterioles. This image depicts the possible involved mechanisms that involve oxidative redox stress (OxRS -reactive oxygen species) and various uremic toxins that are responsible for VOC. VOC represents a pathologic secondary ossification site or dystrophic calcification in a response to injury to endothelial cells and the subsequent VSMC phenotypic switch phenomenon from contractile to synthetic phenotype. Multiple injurious species as in the dirty dozen plus specific uremic toxins (increased parathyroid hormone (PTH), phosphorus (Pi), phosphate (P043-), calcium, calcium x phosphorus product, vitamin D3 trigger this mechanism of inducing the osteoblast-like phenotype that occurs during the phenotype shift in VSMCs. Phosphate absorption into these cells is facilitated by the sodium phosphate cotransporter (Pit-1) resulting in an osteogenic switch due to activation of transcription factors: osteoblast-specific cis-acting element (Osf2)—core binding factor alpha1 (Cbfa-1/Runx2). Osteocalcin, osteonectin, bone morphogenic protein-2alpha and alkaline phosphatase (ALP) are also known inducers of calcification. In contrast, the systemic and local inhibitors of calcification fetuin-A (alpha2-Heremans-Schmid glycoprotein (AHSG) and matrix Gla protein (MGP) are decreased in uremia and calciphylaxis. ROS and associated inflammatory cytokine surges may contribute to decreased hepatic synthesis of fetuin-A (insert a). Uremic toxins—ROS promote uncoupling of endothelial nitric oxide synthase (eNOS) enzyme via the oxidation of the requisite tetrahydrobiopterin (BH4) cofactor and results in the endothelium becoming a net producer of superoxide—ROS (insert b). Importantly, decreased bioavailable NO due to eNOS enzyme uncoupling promotes a proinflammatory, proconstrictive, prothrombotic vascular endothelium and note the constricted, collapsed arteriole (insert b). ROS are also capable of promoting VSMC apoptosis in the arterial vascular wall and when this occurs the matrix vesicles and apoptotic bodies serve as nucleating sites for further calcium deposition in the extracellular matrix of the arteriole media (inserts c–e). Image reproduced with permission by CC 4.0 [21,108,126].
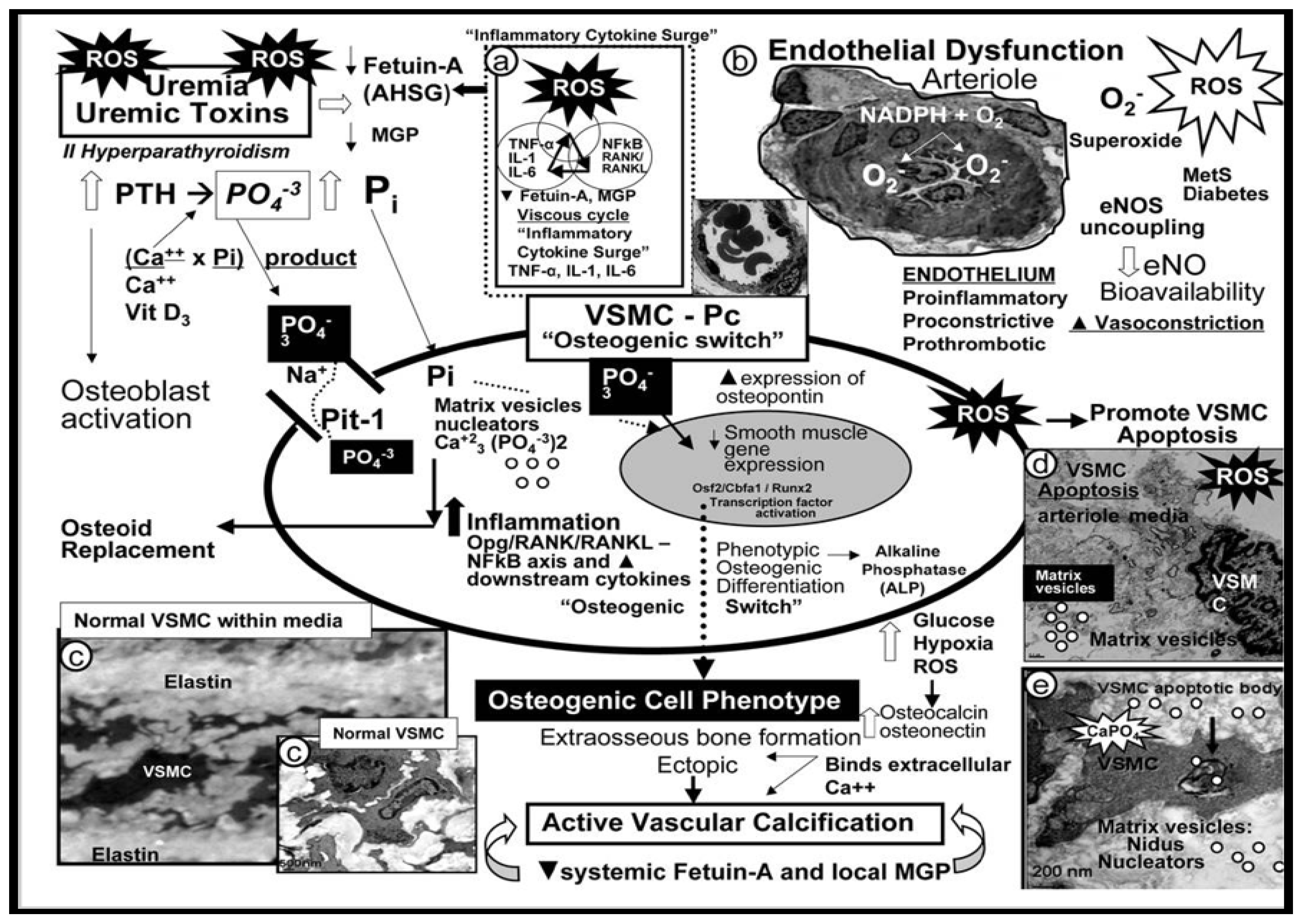
Figure 17.
Skeletal muscle (SkM) microvessel (Mv) remodeling in peripheral artery disease (PAD). Panel A demonstrates a normal image of the control gastrocnemius skeletal muscle (pseudo- colored rust). Note the membraneous location of nuclei (pseudo-colored blue) and the capillary pericyte (pseudo-colored green). Additionally, note that the depicted capillary is serving 3 myocytes and that the myocyte mitochondria are both subsarcolemmal and perinuclear (pseudo-colored purple). The encircled numbers 1, 2, and 3 depict the pericyte, subsarcolemmal and perinuclear mitochondria, and SkM microvessel capillary respectively. This trinity (1-3) is important since they are known to become aberrant or lost as occurs with capillary rarefaction as PAD progresses. Panel B specifically depicts the lifting and separation of the pericyte from the capillary that is undergoing apoptosis in this soleus skeletal muscle from the hypertensive male Ren2 rat model. Modified images provided with permission by CC 4.0 [134]. Cl, capillary lumen; Mt, mitochondria; N, myocyte nucleus; Pc, pericyte; PcP, pericyte process; RBC, red blood cell; X, loss of pericapillary mitochondria mounding.
Figure 17.
Skeletal muscle (SkM) microvessel (Mv) remodeling in peripheral artery disease (PAD). Panel A demonstrates a normal image of the control gastrocnemius skeletal muscle (pseudo- colored rust). Note the membraneous location of nuclei (pseudo-colored blue) and the capillary pericyte (pseudo-colored green). Additionally, note that the depicted capillary is serving 3 myocytes and that the myocyte mitochondria are both subsarcolemmal and perinuclear (pseudo-colored purple). The encircled numbers 1, 2, and 3 depict the pericyte, subsarcolemmal and perinuclear mitochondria, and SkM microvessel capillary respectively. This trinity (1-3) is important since they are known to become aberrant or lost as occurs with capillary rarefaction as PAD progresses. Panel B specifically depicts the lifting and separation of the pericyte from the capillary that is undergoing apoptosis in this soleus skeletal muscle from the hypertensive male Ren2 rat model. Modified images provided with permission by CC 4.0 [134]. Cl, capillary lumen; Mt, mitochondria; N, myocyte nucleus; Pc, pericyte; PcP, pericyte process; RBC, red blood cell; X, loss of pericapillary mitochondria mounding.
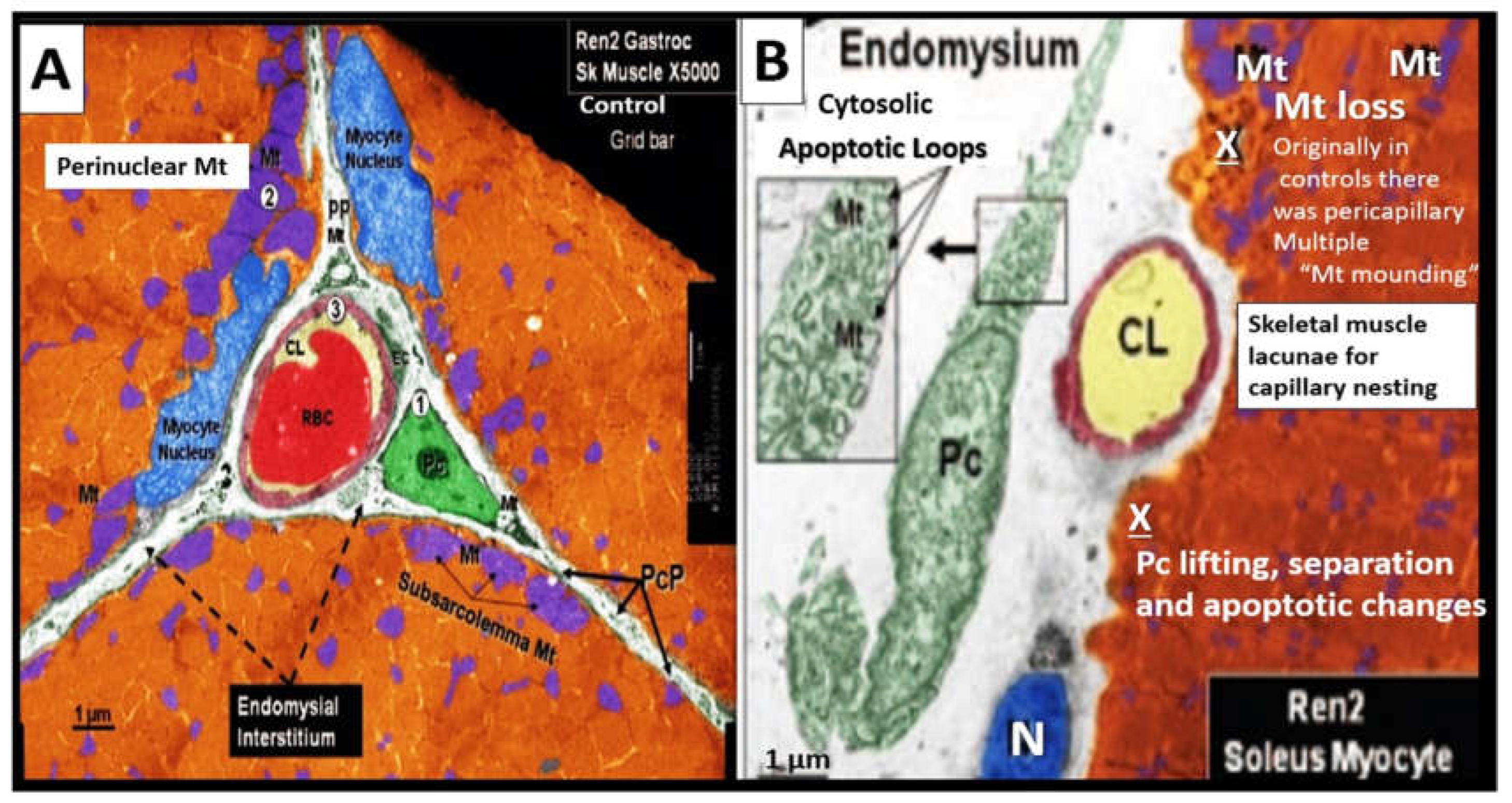
Figure 18.
Three major tissue beds affected by microvessel remodeling and ischemia in intermittent claudication (IC) as it transitions to critical limb ischemia (CLI). aMt, aberrant mitochondria; ATP, adenosine triphosphate; BM, basement membrane; ECact/dys, endothelial cell activation and dysfunction; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; Mt, mitochondria; mtROS, mitochondria-derived reactive oxygen species; NO, nitric oxide; Pc, pericyte; VEGF, vascular endothelial growth factor.
Figure 18.
Three major tissue beds affected by microvessel remodeling and ischemia in intermittent claudication (IC) as it transitions to critical limb ischemia (CLI). aMt, aberrant mitochondria; ATP, adenosine triphosphate; BM, basement membrane; ECact/dys, endothelial cell activation and dysfunction; ECM, extracellular matrix; eNOS, endothelial nitric oxide synthase; Mt, mitochondria; mtROS, mitochondria-derived reactive oxygen species; NO, nitric oxide; Pc, pericyte; VEGF, vascular endothelial growth factor.
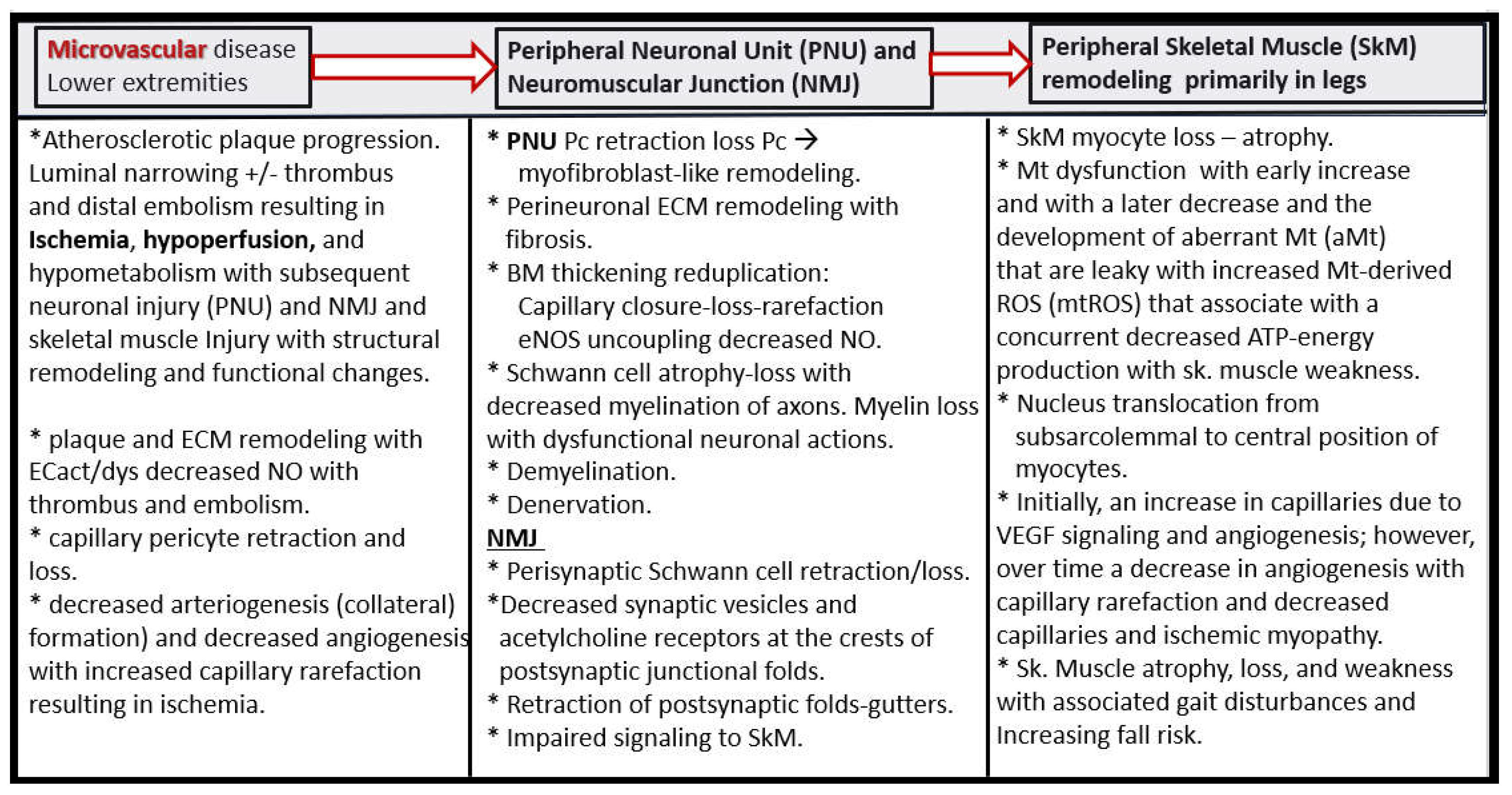
Figure 19.
Peripheral artery disease (PAD) may be considered a spectrum disorder-disease. This image illustrates PAD as a spectrum disease that begins to develop on the left-hand side of the image and progresses to critical limb ischemia on the right-hand side of the image that provides somewhat of roadmap to the development of PAD. Note that in the red zone (after the development of intermittent claudication (IC) (vertical red line) the importance of microvessel remodeling and emboli, which are primarily responsible for the progression to ischemic skeletal muscle myalgia and myopathy and ischemic neuropathy. Thrombi that occur in the macrovessels (more cephalad) may embolize to the more distal microvessels to result in ischemic injury that induces more aberrant remodeling in microvessels as occurs in the inferior popliteal arteries as in previous Figure 15. Also, it should be noted that on-going ischemia/ischemia reperfusion injury contributes to microvessel remodeling. CCVD, cerebrocardiovascular disease; FFA, free fatty acids; HVExt, heart, vessel extremity logo; IC, intermittent claudication; IGT, impaired glucose tolerance; IR, insulin resistance; MetS, metabolic syndrome; T2DM, type 2 diabetes mellitus.
Figure 19.
Peripheral artery disease (PAD) may be considered a spectrum disorder-disease. This image illustrates PAD as a spectrum disease that begins to develop on the left-hand side of the image and progresses to critical limb ischemia on the right-hand side of the image that provides somewhat of roadmap to the development of PAD. Note that in the red zone (after the development of intermittent claudication (IC) (vertical red line) the importance of microvessel remodeling and emboli, which are primarily responsible for the progression to ischemic skeletal muscle myalgia and myopathy and ischemic neuropathy. Thrombi that occur in the macrovessels (more cephalad) may embolize to the more distal microvessels to result in ischemic injury that induces more aberrant remodeling in microvessels as occurs in the inferior popliteal arteries as in previous Figure 15. Also, it should be noted that on-going ischemia/ischemia reperfusion injury contributes to microvessel remodeling. CCVD, cerebrocardiovascular disease; FFA, free fatty acids; HVExt, heart, vessel extremity logo; IC, intermittent claudication; IGT, impaired glucose tolerance; IR, insulin resistance; MetS, metabolic syndrome; T2DM, type 2 diabetes mellitus.
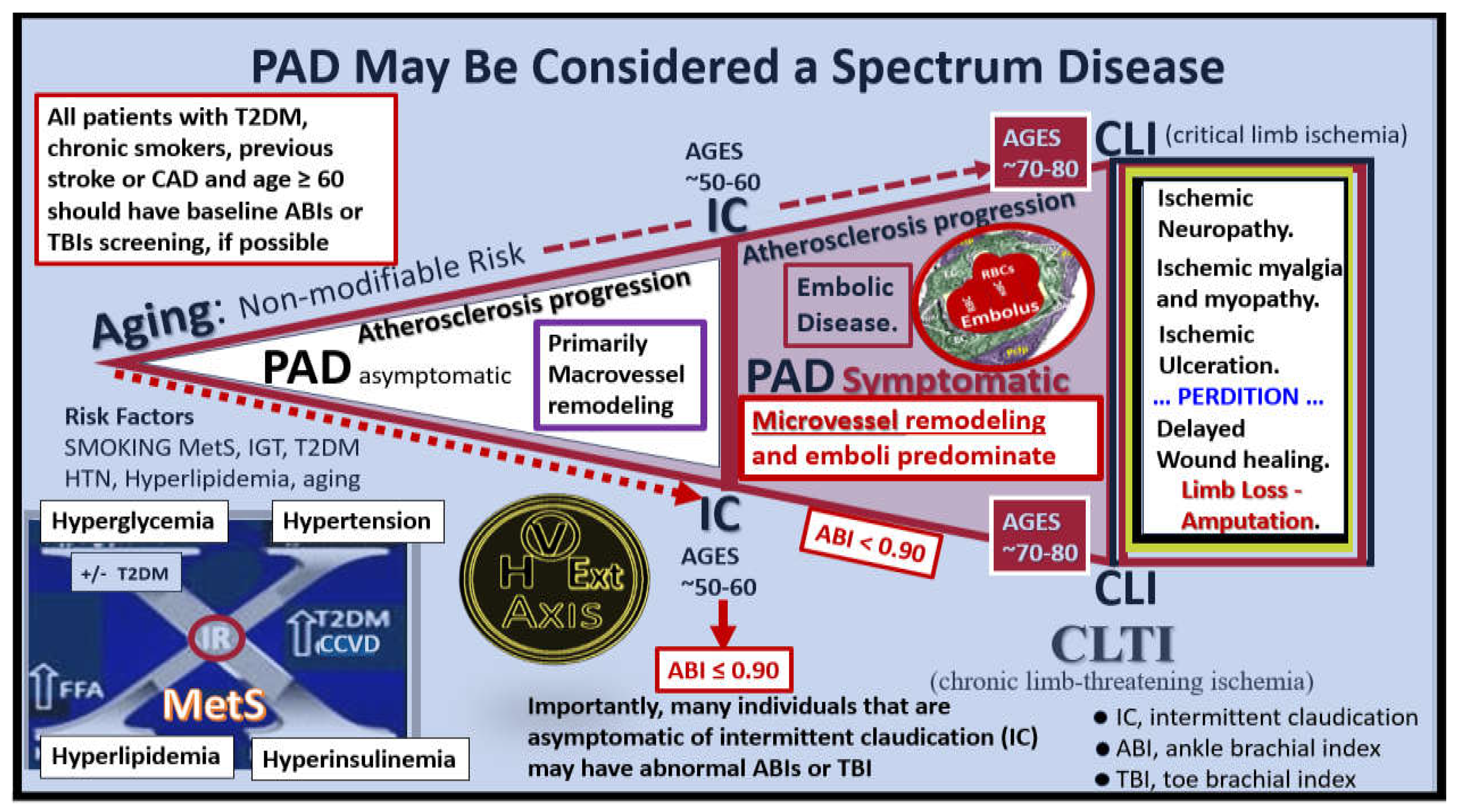
Figure 20.
Possible sequence of events in the remodeling of ischemic neuropathy, skeletal muscle (SkM), and ischemia/reperfusion (IR) injury in peripheral artery disease (PAD). Note the sarcopenia of skeletal muscle (loss of mass, strength, and function) is related specifically to ischemia or ischemia-induced sarcopenia. Asterisk, denotes emphasis; ATP, adenosine triphosphate; Mt, mitochondria; mtROS, mitochondria-derived reactive oxygen species; Mv, microvessel; NMJ, neuromuscular junction; pH, potential of hydrogen, PNU, peripheral neuronal unit; ROS, reactive oxygen species.
Figure 20.
Possible sequence of events in the remodeling of ischemic neuropathy, skeletal muscle (SkM), and ischemia/reperfusion (IR) injury in peripheral artery disease (PAD). Note the sarcopenia of skeletal muscle (loss of mass, strength, and function) is related specifically to ischemia or ischemia-induced sarcopenia. Asterisk, denotes emphasis; ATP, adenosine triphosphate; Mt, mitochondria; mtROS, mitochondria-derived reactive oxygen species; Mv, microvessel; NMJ, neuromuscular junction; pH, potential of hydrogen, PNU, peripheral neuronal unit; ROS, reactive oxygen species.
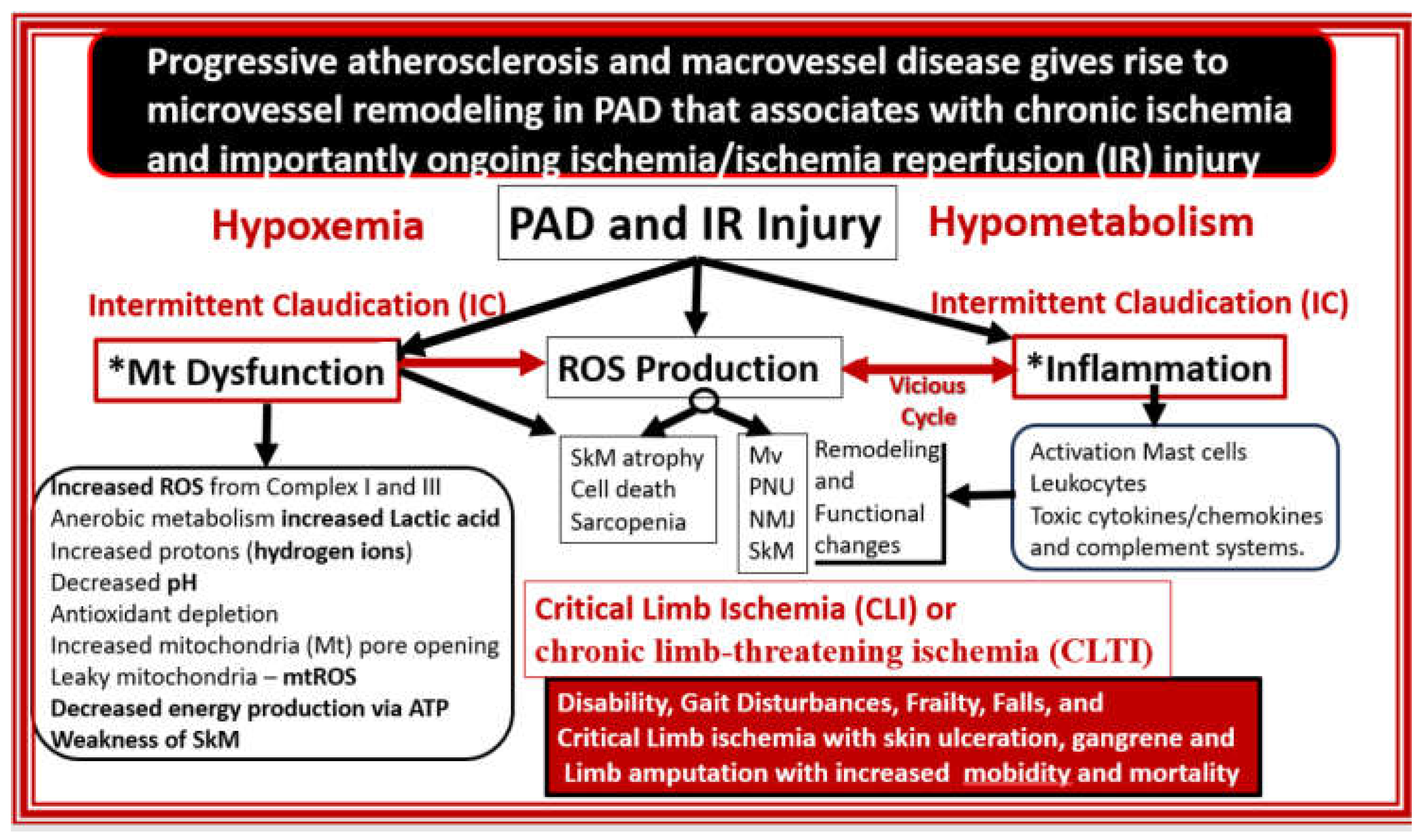
Figure 21.
Microvessel (Mv) remodeling and ischemia leads to peripheral neuronal unit (PNU) and skeletal muscle (SkM) innervation remodeling. Panel A depicts a PNU with dysfunction and loss (rarefaction) of the Mv capillaries of the vasa nervorum primarily due to endothelial cell activation and dysfunction (ECact/dys) with increased inflammation and decreased nitric oxide (NO) bioavailability. Note the Mv capillary involvement with the development of ghost-like capillaries and capillary rarefaction of the vasa nervorum within the endoneurium that associate with myelin dysfunction or loss that results in impaired signaling of the neuromuscular junction (NMJ) in panel B. Mvs exibit basement membrane thickening and reduplication with increased capillary closures along with initial aberrant pericyte (Pc) function contributing to fibrosis and eventually Pc detachment and loss. Note the three major divisions of the PNU (epineurium, perineurium, and endoneurium). Extracellular matrix (ECM) remodeling within the PNU consists of fibrosis due to hypoxia and excessive oxidative redox stress (OxRS). This ECM fibrosis also has the potential to result in a compressive type of ischemic neuropathy. Further, capillary remodeling changes contribute to Schwan cell dysfunction with impaired myelin synthesis and protection to innervating neurons with axonal degeneration resulting in denervation. The initial remodeling changes of the Mv capillaries result in increased permeability and over time a loss of function of blood-nerve barrier, which result in an acceleration of all the above remodeling changes contributing to ischemic neuropathy. Panel B depicts the background gastrocnemius SkM from the male Ren2 control model with inserts b and b’ illustrating the innervation by the efferent PNU from panel A with its presynaptic bouton (b’) with white circles encircled in red acetylcholine neurotransmitters and the postsynaptic junctional folds with acetylcholine receptors and insert b’’ illustrating an afferent sensory neuron responsible for the transfer of pain signals to provide neuronal, inflammatory, and nociception pain signals from ischemic SkM. These remodeling changes in the PNU and neuromuscular junction (NMJ) are co-occurring with the microvessel remodeling discussed previously. Modified images in panels A and B provided with permission by CC 4.0 [142,143]. BM, basement membrane; DPN, diabetic peripheral neuropathy; eNOS, endothelial nitric oxide synthase; Pc, pericyte; Pc N, pericyte nucleus; ROS, reactive oxygen species; SC, Schwann cell; UM, unmyelinated axon.
Figure 21.
Microvessel (Mv) remodeling and ischemia leads to peripheral neuronal unit (PNU) and skeletal muscle (SkM) innervation remodeling. Panel A depicts a PNU with dysfunction and loss (rarefaction) of the Mv capillaries of the vasa nervorum primarily due to endothelial cell activation and dysfunction (ECact/dys) with increased inflammation and decreased nitric oxide (NO) bioavailability. Note the Mv capillary involvement with the development of ghost-like capillaries and capillary rarefaction of the vasa nervorum within the endoneurium that associate with myelin dysfunction or loss that results in impaired signaling of the neuromuscular junction (NMJ) in panel B. Mvs exibit basement membrane thickening and reduplication with increased capillary closures along with initial aberrant pericyte (Pc) function contributing to fibrosis and eventually Pc detachment and loss. Note the three major divisions of the PNU (epineurium, perineurium, and endoneurium). Extracellular matrix (ECM) remodeling within the PNU consists of fibrosis due to hypoxia and excessive oxidative redox stress (OxRS). This ECM fibrosis also has the potential to result in a compressive type of ischemic neuropathy. Further, capillary remodeling changes contribute to Schwan cell dysfunction with impaired myelin synthesis and protection to innervating neurons with axonal degeneration resulting in denervation. The initial remodeling changes of the Mv capillaries result in increased permeability and over time a loss of function of blood-nerve barrier, which result in an acceleration of all the above remodeling changes contributing to ischemic neuropathy. Panel B depicts the background gastrocnemius SkM from the male Ren2 control model with inserts b and b’ illustrating the innervation by the efferent PNU from panel A with its presynaptic bouton (b’) with white circles encircled in red acetylcholine neurotransmitters and the postsynaptic junctional folds with acetylcholine receptors and insert b’’ illustrating an afferent sensory neuron responsible for the transfer of pain signals to provide neuronal, inflammatory, and nociception pain signals from ischemic SkM. These remodeling changes in the PNU and neuromuscular junction (NMJ) are co-occurring with the microvessel remodeling discussed previously. Modified images in panels A and B provided with permission by CC 4.0 [142,143]. BM, basement membrane; DPN, diabetic peripheral neuropathy; eNOS, endothelial nitric oxide synthase; Pc, pericyte; Pc N, pericyte nucleus; ROS, reactive oxygen species; SC, Schwann cell; UM, unmyelinated axon.
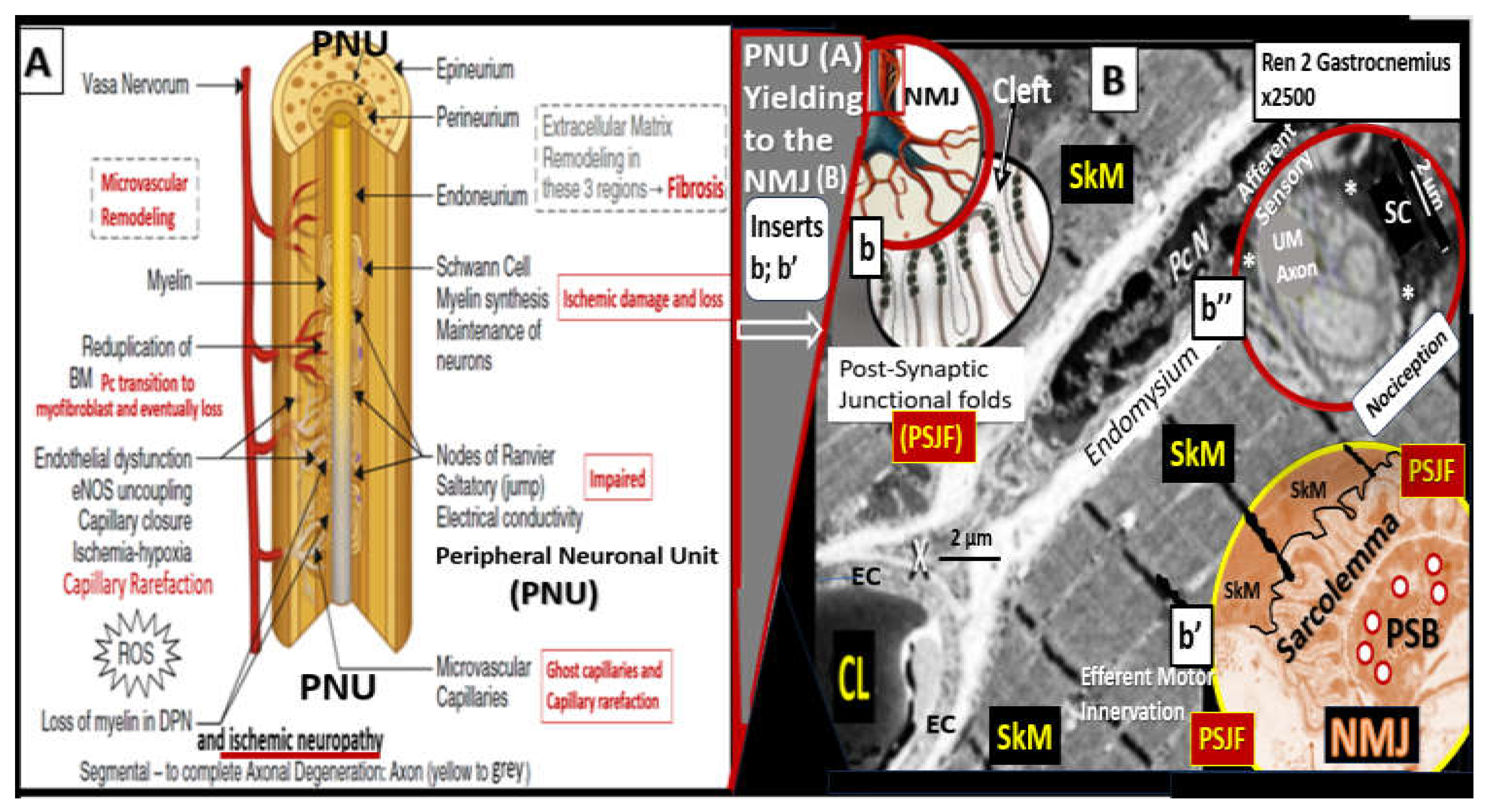
Figure 22.
Skeletal muscle (SkM) sensory afferent neurons interact with mast cell(s) to create a SkM neuroimmune axis within the endomysium that contributes to neuroimmune pain perception associated with ischemic neuropathy. ECM, extracellular matrix; MC, mast cell; N, nucleus; NT, neuron terminal; UN, unmyelinated sensory afferent neurons-axons. This representative image of gastrocnemius SkM was obtained from transgenic male Ren2 hypertensive rat at 16-weeks of age and previously unpublished.
Figure 22.
Skeletal muscle (SkM) sensory afferent neurons interact with mast cell(s) to create a SkM neuroimmune axis within the endomysium that contributes to neuroimmune pain perception associated with ischemic neuropathy. ECM, extracellular matrix; MC, mast cell; N, nucleus; NT, neuron terminal; UN, unmyelinated sensory afferent neurons-axons. This representative image of gastrocnemius SkM was obtained from transgenic male Ren2 hypertensive rat at 16-weeks of age and previously unpublished.
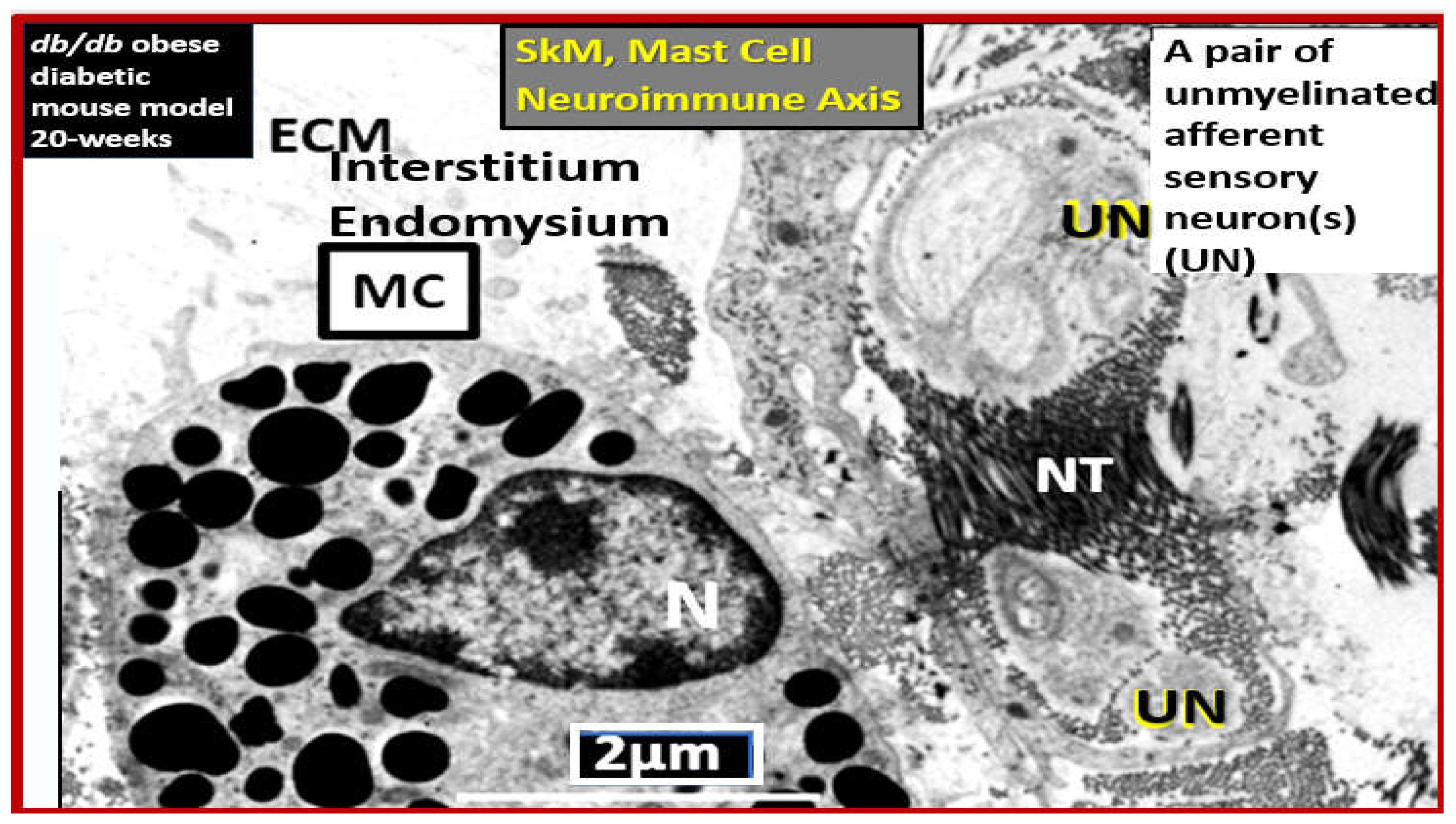
Figure 23.
Macrovessel and microvessel (Mv) disease results in skeletal muscle (SkM) ischemia that is perceived as pain via a 5-step signaling and transference mechanisms. Step 1 represents the injury due to macrovessel or Mv ischemia due to chronic disease progression or acute ischemia due to an embolic thrombus as depicted in the near-complete occlusion of the Mv capillary lumen (pseudo-colored yellow adjacent to the red blood cells (RBCs)). Step 2 depicts the signaling of groups III and IV unmyelinated (UN) sensory afferent neurons as a result of increased lactic acid, adenosine triphosphate (ATP), hydrogen protons (H+), and decreased pH. Step 3 depicts the transference of pain information as a result of increased receptors and channels of sensory afferents such as acid sensing ion channels (ASICs), purinergic receptors (P2X and P2Y), transient receptor potential vanilloid type 1(TRPV1), interleukin 1 receptor, type I (IL1R1). Step 4 illustrates the secondary transference of pain information from the dorsal root ganglia (DRG) to the thalamus via the spinal thalamic tracts that are then transferred to the cortex to realize pain perception, Notably, nociceptive, inflammatory, and neuropathic mechanisms all play a role in the generation and perception of pain in individuals with peripheral artery disease (PAD) [145]. Background gastrocnemius SkM provided with permission by CC 4.0 [142]. Hif-1α, hypoxia inducible factor alpha; IL-1β, interleukin one beta; IL-6, interleukin 6; Mt, mitochondria; mtROS, mitochondrial-derived reactive oxygen species; NGF, nerve growth factor; RBCs, red blood cells; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor.
Figure 23.
Macrovessel and microvessel (Mv) disease results in skeletal muscle (SkM) ischemia that is perceived as pain via a 5-step signaling and transference mechanisms. Step 1 represents the injury due to macrovessel or Mv ischemia due to chronic disease progression or acute ischemia due to an embolic thrombus as depicted in the near-complete occlusion of the Mv capillary lumen (pseudo-colored yellow adjacent to the red blood cells (RBCs)). Step 2 depicts the signaling of groups III and IV unmyelinated (UN) sensory afferent neurons as a result of increased lactic acid, adenosine triphosphate (ATP), hydrogen protons (H+), and decreased pH. Step 3 depicts the transference of pain information as a result of increased receptors and channels of sensory afferents such as acid sensing ion channels (ASICs), purinergic receptors (P2X and P2Y), transient receptor potential vanilloid type 1(TRPV1), interleukin 1 receptor, type I (IL1R1). Step 4 illustrates the secondary transference of pain information from the dorsal root ganglia (DRG) to the thalamus via the spinal thalamic tracts that are then transferred to the cortex to realize pain perception, Notably, nociceptive, inflammatory, and neuropathic mechanisms all play a role in the generation and perception of pain in individuals with peripheral artery disease (PAD) [145]. Background gastrocnemius SkM provided with permission by CC 4.0 [142]. Hif-1α, hypoxia inducible factor alpha; IL-1β, interleukin one beta; IL-6, interleukin 6; Mt, mitochondria; mtROS, mitochondrial-derived reactive oxygen species; NGF, nerve growth factor; RBCs, red blood cells; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor.
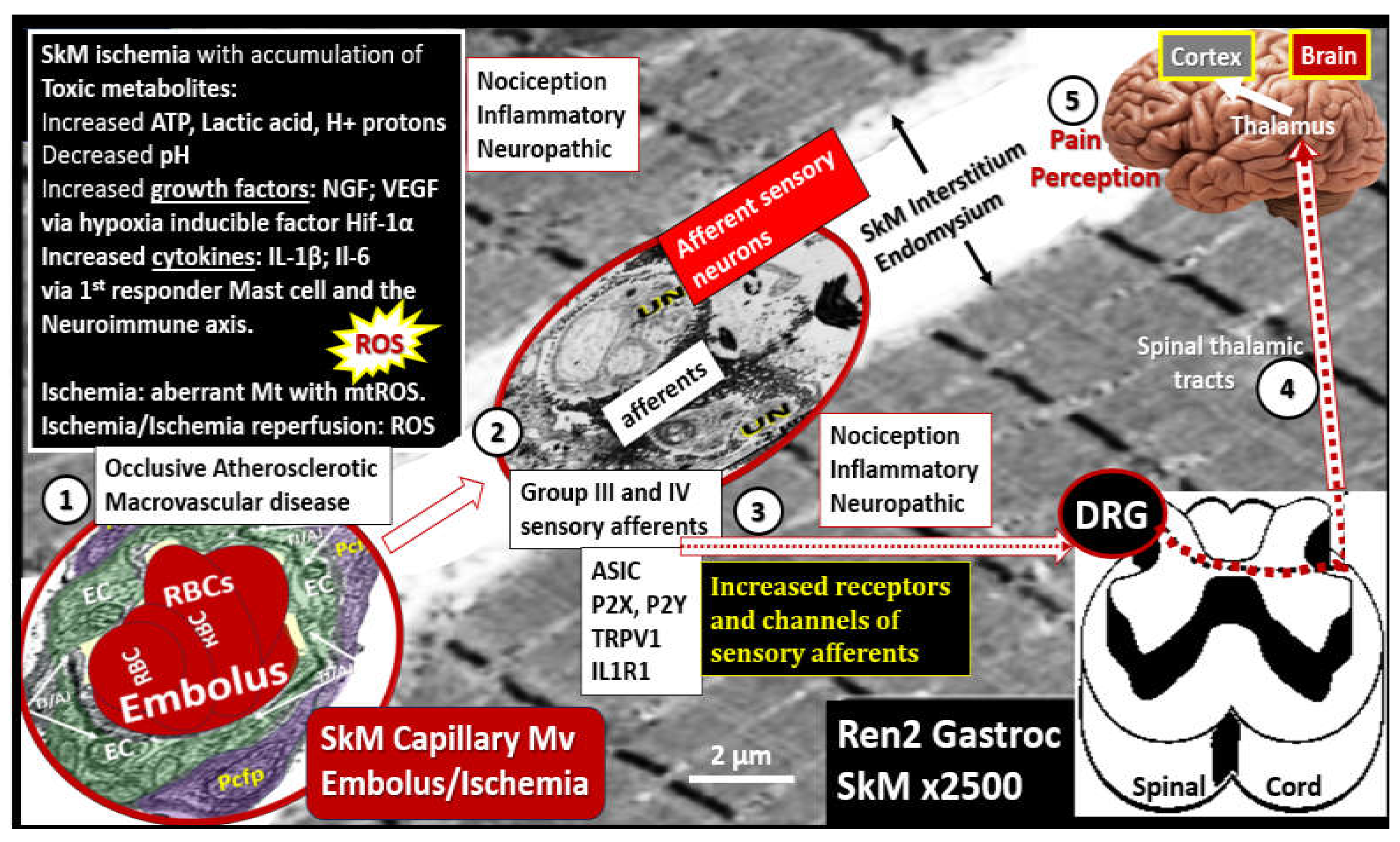
Figure 24.
Comparison of ischemic neuropathy in peripheral artery disease to diabetic peripheral neuropathy. Note that there are many overlaps between these two clinically distinct neuropathies and they frequently co-occur. Modified image of Harriet’s nerves provided with permission by CC 4.0 [142].
Figure 24.
Comparison of ischemic neuropathy in peripheral artery disease to diabetic peripheral neuropathy. Note that there are many overlaps between these two clinically distinct neuropathies and they frequently co-occur. Modified image of Harriet’s nerves provided with permission by CC 4.0 [142].
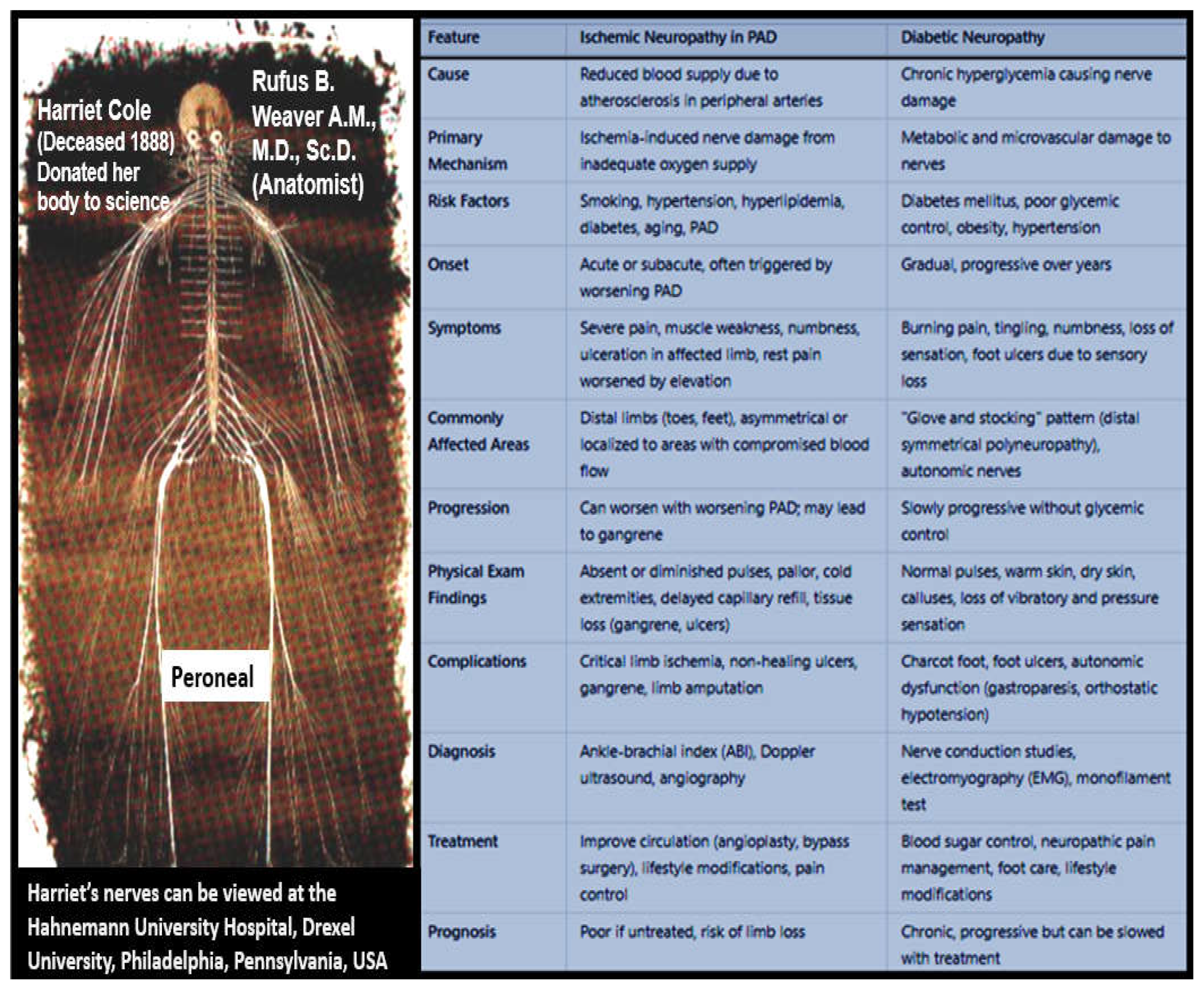
Figure 25.
Sodium thiosulfate (STS) posesses antioxidant, anti-inflammatory, chelation of cations, and the possibility to restore endothelial nitric oxide synthase (eNOS) enzyme production of nitric oxide (NO) capabilities to improve peripheral artery disease (PAD). Importantly, STS is a hydrogen sulfide (H2S) donor/mimetic. Notably, brain endothelial cells also include systemic, peripheral endothelial cells in PAD. Modified graphic abstract image provided by CC 4.0 [156]. Athero, atherosclerosis; BH4, tetrahydrobiopterin an essential cofactor for the eNOS enzyme; Ca++, calcium cation; Cu++, copper cation; Fe++, iron cation; GSH, glutathione; NF-kB, nuclear factor kappa B; R, various amino acid, peptide groups; VAS, vascular arterial stiffness; VOC, vascular ossification and calcification.
Figure 25.
Sodium thiosulfate (STS) posesses antioxidant, anti-inflammatory, chelation of cations, and the possibility to restore endothelial nitric oxide synthase (eNOS) enzyme production of nitric oxide (NO) capabilities to improve peripheral artery disease (PAD). Importantly, STS is a hydrogen sulfide (H2S) donor/mimetic. Notably, brain endothelial cells also include systemic, peripheral endothelial cells in PAD. Modified graphic abstract image provided by CC 4.0 [156]. Athero, atherosclerosis; BH4, tetrahydrobiopterin an essential cofactor for the eNOS enzyme; Ca++, calcium cation; Cu++, copper cation; Fe++, iron cation; GSH, glutathione; NF-kB, nuclear factor kappa B; R, various amino acid, peptide groups; VAS, vascular arterial stiffness; VOC, vascular ossification and calcification.
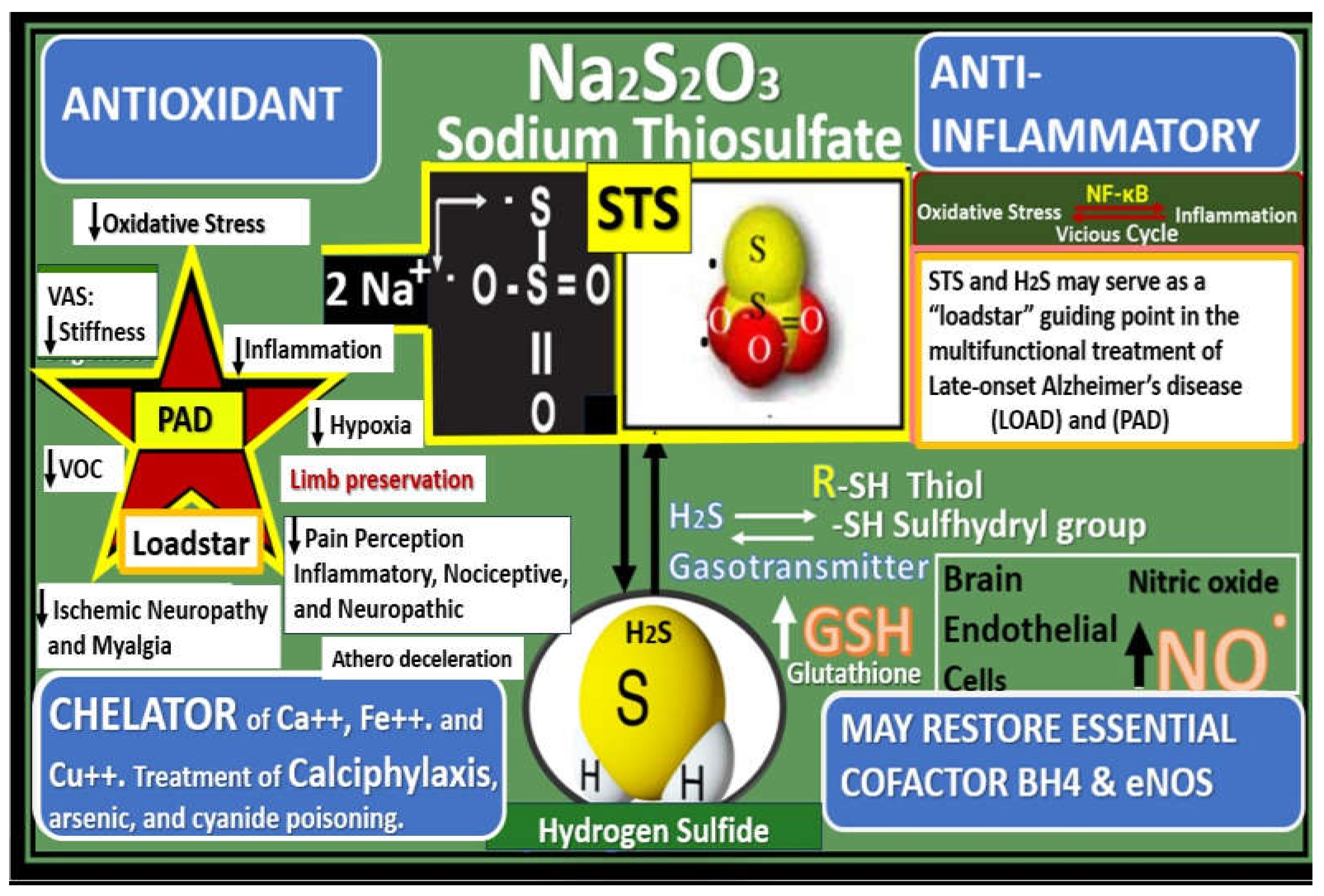
Figure 26.
Sodium thiosulfate (STS) and its medical uses: U.S. Food and Drug Administration (FDA) approved, off-label uses, side effects, and possible future uses in clinical diseases. Modified image provided with permission by CC 4.0 [157]. BH2, dihydrobiopterin; BH4, tetrahydrobiopterin; NO, nitric oxide. .
Figure 26.
Sodium thiosulfate (STS) and its medical uses: U.S. Food and Drug Administration (FDA) approved, off-label uses, side effects, and possible future uses in clinical diseases. Modified image provided with permission by CC 4.0 [157]. BH2, dihydrobiopterin; BH4, tetrahydrobiopterin; NO, nitric oxide. .
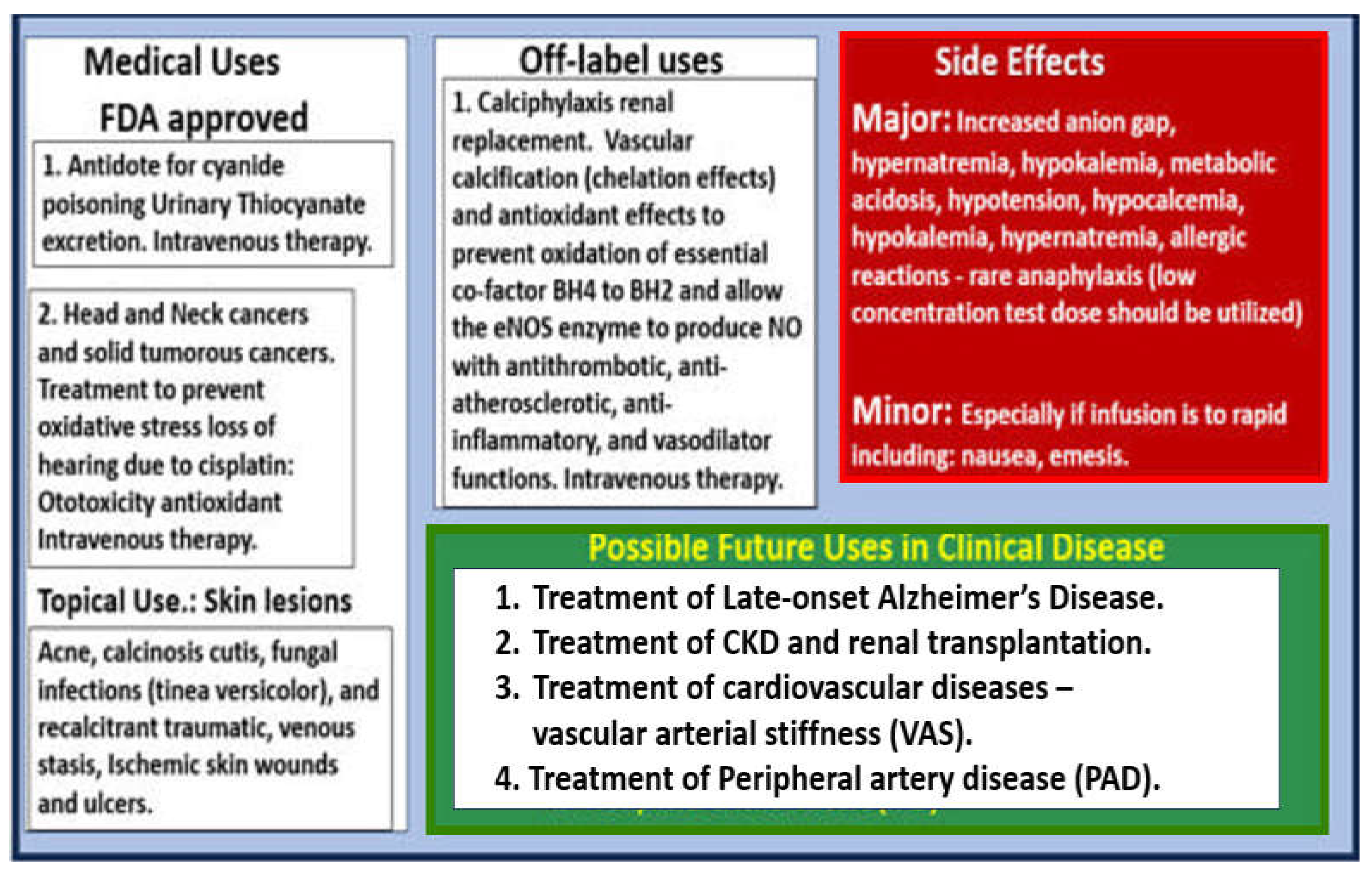
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



