Submitted:
11 April 2025
Posted:
14 April 2025
You are already at the latest version
Abstract
When using chalcones as therapeutic agents, a potential drawback is their susceptibil-ity in solution to light-induced isomerisation around the double bond of their α,β-unsaturated carbonyl moiety giving rise to a mixture of cis- and trans-isomers. This photoisomerisation can be circumvented by transforming chalcones into indanones thus creating a conformationally constrained frame incorporating a five membered ring whilst retaining the structural key features of the parent compound. Herein, we have synthesised indanone DMU5401, i.e. 3-(benzo[d][1,3]dioxol-5-yl)-4,5,6-trimethoxy-2,3-dihydro-1H-inden-1-one, starting from chalcone DMU135, i.e. (E)-3-(benzo[d][1,3]dioxol-5-yl)-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one, via Nazarov cyclisation reaction using a green solvent, i.e. 4-methyltetrahydropyran (4-MeTHP). Although the yield was lower compared to that of previously reported methods, the green solvent procedure we adopted here did not include harmful sol-vents or hazardous chemicals, and significantly shortened the work-up stage of the Nazarov cyclisation reaction making this process more sustainable.
Keywords:
chalcone
; indanones
; Nazarov cyclisation
; 4‐methyltetrahydropyran
; green chemistry
1. Introduction
Chalcones are a privileged scaffold in medicinal chemistry endowed with many pharmacological properties [1,2]. However, in solution, this class of compounds is susceptible to light-induced isomerisation around the α,β-double bond, giving a mixture of the cis- and trans-isomers [3,4]. The few studies that have addressed this problem have shown that the rate of the isomerisation and the equilibrium ratio in solution, for some very specific compounds, is influenced by substitution patterns in the aromatic ring and the solvent used [5]. Our group previously investigated a series of in-house synthesised methoxy and/or methylenedioxy trans-chalcones, including DMU135 [6], and polyhydroxylated natural chalcones butein and isoliquiritigenin, for their cis-trans photo isomerisation properties (Figure 1) (results not published). It was found that solutions of synthetic chalcones, which were kept in dark glass vials under minimal daylight, did undergo photo isomerisation to a mix of cis-trans isomers. In contrast, under similar conditions, the natural chalcones, such as isoliquiritigenin, did not undergo photo isomerisation due to keto-enol tautomerisation via the 4-hydroxy A-ring substituent (results not published).
One solution to the photoisomerisation problem is to prepare a series of conformationally constrained chalcones incorporating a five membered ring whilst retaining the parent compounds’ structural key features. The resulting class of molecules is referred to as 1-indanones in which the β- carbon of its corresponding chalcone is attached directly to C-5 of the B-ring (Figure 2). Chalcones lacking α-substitution have a preference for the s-cis conformation over the s-trans. Chalcones are planar molecules due to carbons sp2 hybridisation and, upon cyclisation to indanones, a chiral centre at C-9(*) is introduced with removal of the alkene double bond (Figure 2). Therefore, the “constrained” indanone frame is no longer planar and mimics the s-trans conformation of chalcone that has proven to be important to its biological activity [7].
Indanones and their analogues have been reported as useful and versatile key intermediates for the synthesis of biologically active compounds such as analgesics, antihypertensive or flavouring agents in tobacco [8,9]. In spite of this, only few publications have been reported on the preparation of indanones. Indanone synthesis is usually carried out by Friedel-Crafts reaction or Fries rearrangement using acyl chloride or α, β- unsaturated carboxylic acids [10,11]. However, limitations to this methodology include low yields, use of toxic reagents and long reaction times. Indanones can also be prepared via Nazarov cyclisation reactions (Scheme 1), although this route also poses some risk due to the need of potentially hazardous reagents such as strong acids, e.g., trifluoroacetic acid (TFA), and high temperatures, which are required for reactions to proceed with high-pressure build-up in the reaction vessels [12].
One previously reported Friedel-Crafts protocol involved the reaction between p-methylphenol and α,β-unsaturated carboxylic acids in the presence of a Lewis acid (AlCl3) that was either heated at reflux or subjected to microwave solventless irradiation [9] (Scheme 1). The latter method resulted in significant reaction time reduction from several hours to 2 minutes and improved yields (30–60%) without O-acylated by-product formation.
Examples of Nazarov cyclisation reactions include the preparation of indanones by conventional heating and microwave assisted (100 W, 120–130 °C, 20min) processes starting from chalcones in neat TFA [13,14].
Although the use of microwave irradiation has arisen as a more sustainable alternative to traditional heating techniques, the use of multiple equivalents of strong Lewis or protic acids (e.g., TFA) for the previously reported indanone synthesis still poses some risks due to their corrosive and hazardous nature. Further, protic acids are often incompatible with sensitive functional groups of the substrates, thus limiting the reaction protocol scope. In our effort to improve the environmental sustainability of our chemistry research and reduce the negative impact of wet lab work on the environment, we searched for a green solvent in which to carry out the synthesis of the indanone core using the Nazarov cyclisation reaction.
It is now widespread practice to replace traditional ethereal solvents (e.g., THF, Et2O and 1,4-dioxane) with safer and greener solvents. Therefore, we directed our attention to methyltetrahydropyran (4-MeTHP), which is a hydrophobic cyclic ether that was shown to be a valid substitute of conventional ethers and harmful halogenated solvents. We were also intrigued by the fact that 4-MeTHP was previously found non-compatible with strong Lewis acids that are required for the Nazarov reaction, as they could be deactivated as a result of the coordination of the 4-MeTHP oxygen lone pairs to the boron empty orbitals [15].
Nevertheless, we sought to explore boron trifluoride etherate (BF3OEt2) as a milder source of boron trifluoride and reaction promoter for the synthesis of indanone DMU5401 via Nazarov cyclisation starting from chalcone DMU135 (Scheme 2). DMU135 was selected for indanone preparation as this chalcone is an excellent substrate for CYP1 isozymes and had shown significant anti-proliferative activity with good tumour selectivity towards MDA468 cancer cells over MCF10A non-tumour cells [16]. It is anticipated that the conformationally constrained analogue of DMU135 will undergo, in a similar fashion to the parent compound, CYP1-mediated aromatic hydroxylation or O-demethylation, leading to CYP1 metabolites that will form DNA adducts, possess antimitotic activity or inhibit tyrosine kinase activity.
2. Results and Discussion
The precursor chalcone DMU135 for the synthesis of indanone DMU5401 was readily prepared by base-catalysed Claisen-Schmidt condensation of 3′,4′,5′-trimethoxyacetophenone and piperonal (e.g., 1,3-benzodioxole-5-carboxaldehyde) using our previously published protocols [1].
Three strategies were adopted for the Nazarov cyclisation-mediated synthesis of DMU5401, including the new green solvent method (e.g., Route 3 in Scheme 2).
Initial chalcone cyclisation to indanone was attempted with DMU135 and TFA in a sealed tube by conventional heating at 120 0C as reported by Rice and co-workers [14]. This approach, however, failed to give the desired indanone after 6h, as indicated by TLC showing the presence of only chalcone DMU135 and the absence of a new spot in the reaction mixture. The technique was then modified for microwave synthesis using the established microwave irradiation reaction described by Lawrence [13] on a 100mg scale. Initially, DMU135 in neat TFA was heated at 102°C for 10 minutes at 100W power and microwave irradiated. The reaction progress was monitored by TLC that showed the appearance of a new spot with a retention factor (Rf) of 0.55 and presence of unreacted chalcone at Rf 0.6 (ethyl acetate/petroleum ether 4:6). The reaction was then repeated under the same condition except increasing the period of reaction time from 10 min to 20 min. The TLC analysis confirmed complete clean conversion of chalcone to indanone. The reaction was quenched with water, extracted with ethyl acetate, followed by purification using flash column chromatography, which afforded the desired indanone in 72% yield.
We then sought to compare the above protocols with our new method involving the use of green solvent 4-MeTHP and BF3OEt2. The reaction mixture was heated at reflux at the temperature of 110 0C overnight and the crude product purified by silica gel chromatography. Although the yield (35%) of the reaction was much lower compared to that of Route 2, the work up procedure was much more efficient and the evaporation of hazardous chemicals, such as strong acid TFA, was not required.
The major benefit associated with the use of 4-MeTHP resided in the fact the reaction mixture did not need to be concentrated or extracted during work-up, thus saving energy, time and resources. In our procedure, we quenched the reaction with deionised water, and, after separating the inorganic phase (e.g., water), 4-MeTHP was evaporated. Therefore, 4-MeTHP acted as both the reaction and extraction solvent (normally, we would have used EtOAc to extract the sample from the aqueous phase). This process greatly simplified and expedited the procedure of product recovery.
The synthetic methods reported above are not enantioselective and a mixture of enantiomers was recovered in our experiments. In regard to Route 3 experiment (Scheme 2), HPLC chiral column analysis revealed the presence of two peaks eluting at 14.6 and 15.8 min with percentage areas of 21.4 and 27.3, respectively, confirming the presence of R and S enantiomers (Figure S4 in Supplementary Material).
The analytical data obtained for the DMU5401 were consistent with the structures of the samples obtained with Route 2 and 3 methods. Mass spectrometry showed [M+ H]+ corresponding to the expected molecular ion and accurate mass analysis confirmed the expected molecular formula calculated for C19H18O6, i.e., [M+H]+ 343.1181. IR spectra showed indanone carbonyl absorption at higher wave-numbers (1650-1700cm-1) than the corresponding chalcone carbonyl absorption (1640-1650cm-1)
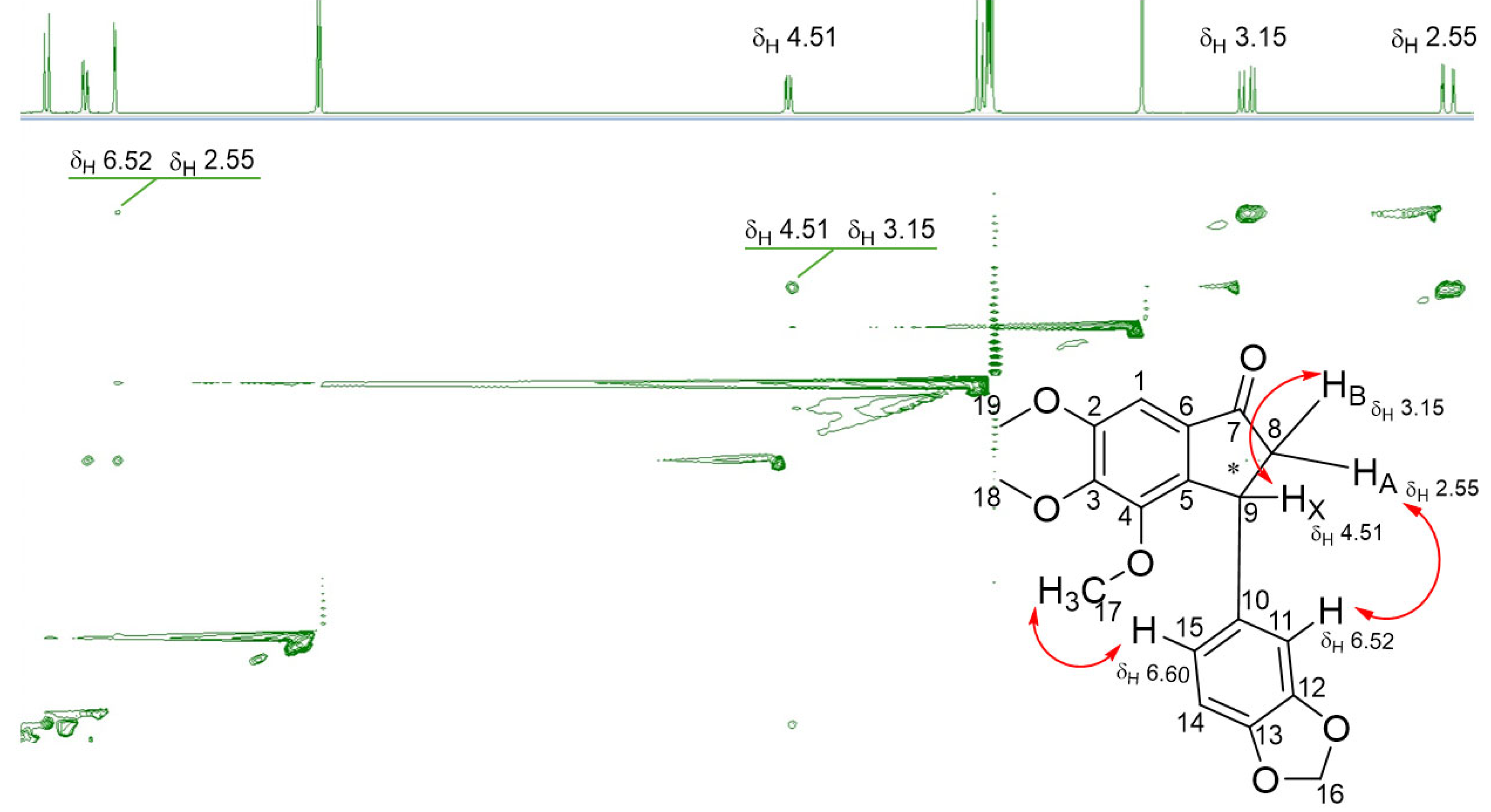
The 1H-NMR spectra of indanones showed the absence of characteristic alkene doublets, previously observed with chalcones. The chemical shift of aromatic and methoxy protons generally appeared up-field compared to the corresponding chalcone protons. In addition, 1H NMR spectra showed the presence of three distinct signals (doublet of a doublet) at δH 4.51ppm, 3.15ppm and 2.55ppm corresponding to cyclopentanone aliphatic protons H9X (chiral centre), H8B and H8A, respectively.
Further analysis of 1D and 2D 1H NMR spectra provided useful information about the cyclic structure of the indanone. It was found that the coupling constants of geminal protons HA and HB attached to C8 have a high value (e.g., 2JAB = 19.2 Hz) due to the adjacent carbonyl group at C7. Although DMU5401 indanone synthesis furnished a diastereomer mixture, some key information about the orientation of the functional groups surrounding the stereogenic centre at C9 can be gathered by analysing the NOESY spectral data and vicinal coupling constants between protons HX (C9) and geminal protons HA and HB (C8). Vicinal coupling constants are sensitive to angles between the coupling protons according to the Karplus equation. The 3JBX = 7.96 Hz shows that the dihedral angle between HB and HX is around 0-20°, thus implying a syn stereochemical arrangement. Conversely, the smaller 3JAX = 2.6 Hz indicates a larger dihedral angle and an anti conformation between HA and HX (Figure 3).
4. Materials and Methods
General Chemistry Information
Reagents were used as received from Sigma-Aldrich Chemical Company (Dorset, UK) or Alfa Aesar (Lancashire, UK).and Fisher Scientific (Loughborough, UK). 4-Methyltetrahydropyran (4-MeTHP) was supplied by IMCD Regulated Synthesis UK Ltd. (Surrey, UK). 1H and 13C Nuclear Magnetic Resonance (NMR) spectroscopy analyses were carried out using a JEOL JNM-ECZR 600 MHz (equipped with a ROYAL probe). Tetramethylsilane (TMS) was used as an internal standard. Chemical shifts are reported in units relative to the TMS signal and coupling constants (J) expressed in Hertz (Hz). 1H-NMR information is provided in the following format: number of protons, multiplicity, coupling constant (where necessary) and assignment. Multiplicities are reported as follow; s=singlet, d=doublet, t=triplet, q=quartet, dd=doublets of doublet, m=multiplet. Infrared spectra (IR) were recorded on a Perkin-Elmer 298 Spectrophotometer with absorption expressed in cm-1. High-Resolution Mass Spectrometry analysis was performed on a Waters UPLC coupled with a Xevo G2-XS QTOF (Waters, Wilmslow, UK). Chromatographic analysis was achieved using an Acquity BEH C18 UPLC column (1.7 µm, 2.1 × 50 mm) at 40°C. The mobile phases consisted of 0.1% formic acid in LCMS grade water (A) and 0.1% formic acid in LCMS grade acetonitrile (B). The flow rate was 0.5 mL min-1, with the following gradient program: 0min 5% B, 0-0.5 min 5% B, 0.5-1.5 min 5-25% B, 1.5-2 min 25% B, 2-3.5 min 25-70% B, 3.5-3.9 min 70% B, 3.9-5min 5% B. The total run time was 5 min. The injection volume was 0.5 µL. Chiral HPLC analysis was performed using a Daicel Chiralcel OD analytical column (250 × 4.6 mm, 10 μm) and UV detection was monitored at 254 nm. The injection volume was 20 µL with an isocratic phase of 2% isopropanol in hexane, flow rate = 1 mL min−1, λ = 254 nm. Melting points (uncorrected) were determined on a Gallenkamp melting point apparatus in open glass capillary tubes. Thin layer chromatography (TLC) was performed on Merck Aluminium Sheet- Silica Gel 60 F254 coated plates. The TLC plates visualised under Multiband UVGL-58 UV-254/366 nm UV light and stained with 2,4-dinitrophenylhydrazine (DNP to stain for the carbonyl group) or iodine absorbed on sand or phosphomolybdic acid (PMA). Silica gel (Fluka Silica 60; standard 30-45 μm / fine grade 20-45 μm) was used for Flash Column Chromatography. CEM Discover S-ClassTM microwave synthesizer was used for the synthesis of DMU5401 according to Route 2.
General Procedure to the Preparation of Chalcone DMU135
Sodium hydroxide solution (50 % w/v, 66.7 mmoles, 5.3 mL) was added to a stirred solution of the acetophenone (6.67 mmoles) and aldehyde (6.67 mmoles) in methanol (30 mL). The resulting mixture was stirred at room temperature and sequentially monitored by TLC (ethyl acetate/petroleum ether (3:7) until the reaction was complete. The reaction was quenched with distilled water (30 mL) and extracted with ethyl acetate (3x30 mL). The combined organic extracts were washed with brine (50 mL), dried with anhydrous magnesium sulphate and the solvent removed in vacuo. The crude product was recrystalised from ethanol to afford yellow solid.
(E)-3-(3,4-methylenedioxyphenyl)-1-(3,4,5-trimethoxyphenyl) prop-1-en-3-one (DMU 135) Yellow crystals (1.75 g, 77%), TLC: Rf 0.72 (ethyl acetate/petroleum ether 2:8); m.p. 135-136 °C, m/z [FAB] (343 [M+H]+, 100%), υmax (KBr) /cm-1 1650 (C=O), δH (CDCl3) 3.92 (3H, s, OMe), 3.93 (6H, s, OMe), 6.01 (2H, s, OCH2O), 6.83 (1H, s, ArH), 7.11 (1H, d, ArH), 7.15 (1H, d, ArH), 7.25 (2H, s, ArH), 7.30 (1H, d, J=16Hz, CH=CH), 7.72 (1H, d, J=16Hz, CH=CH); δC (CDCl3), 56.4, 61.0, 101.7, 106.1, 106.7, 108.7, 119.8, 125.3, 133.7, 142.4, 144.6, 148.4, 150.0, 153.2, 189.1 (C=O); HRMS found [M+H]+ 343.1176, C19H19O6 requires [M+H]+ 343.1176; Anal Calcd for C19H18O6 : C,66.66; H,5.30, Found, C, 66.49; H, 5.44.
Procedures to the Preparation of Indanone DMU5401
Route 2 (microwave irradiation)
The precursor chalcone DMU135 (0.1 g, 0.29 mmol) was dissolved in anhydrous trifluroacetic acid (TFA, 0.3 mL) and sealed in a pressure-rated reaction tube (10 mL). The reaction vial was irradiated in a self-tuning single-mode CEM DiscoverTM Focused Synthesiser. The reaction was maintained at 120 °C (power: 100W) for 20 min. The mixture was allowed to cool to room temperature and quenched with water (10 mL) and extracted with ethyl acetate (3x10 mL). The combined organic extract was washed saturated NaHCO3 (2x10 mL), brine (2x10 mL) and dried over anhydrous magnesium sulphate and concentrated in vacuo. The crude product was purified by flash column chromatography, e.g., SiO2, petroleum ether 40-60 with an increasing gradient of ethyl acetate (20-50%), to afford the product indanone as a pale brown solid. TLC: Rf 0.60 (ethyl acetate/petroleum ether 4:6). Yield 0.072 g (72%).
Route 3 (using 4-Methyltetrahydropyran)
To a solution of DMU135 (0.1 g, 0.29 mmol) in 4-MeTHP (2 mL) was added boron trifluoride diethyl etherate (38 µL, 0.29 mmol). The reaction mixture was refluxed at the temperature 115 0C overnight. Progression of the reaction was monitored by TLC and absence of the starting material indicated end of the reaction. The reaction mixture was then cooled to the room temperature and quenched with water. The green solvent was evaporated in vacuo. The crude product was purified via column chromatography, e.g., SiO2, hexane with an increasing gradient of ethyl acetate (20-50%), to afford the product indanone as a pale brown solid. TLC: Rf 0.37 (ethyl acetate/hexane 3:7). Yield 0.035 g (34%).
4,5,6-trimethoxy-3-(3,4-methylendioxyphenyl)indan-1-one (DMU 5401) m.p. 105- 106 °C; νmax (KBr) /cm-1 1690 (C=O), m/z [FAB] 343 [M+H]+ 50%; 1H-NMR (600 MHz, CHLOROFORM-D) δ 7.07 (s, 1H), 6.72 (d, J = 7.9 Hz, 1H), 6.60 (dd, J = 8.0, 1.8 Hz, 1H), 6.52 (d, J = 1.7 Hz, 1H), 5.91 (dd, J = 5.7, 1.4 Hz, 2H), 4.51 (dd, J = 8.0, 2.5 Hz, 1H), 3.91 (s, 3H), 3.90 (s, 3H), 3.46 (s, 3H), 3.15 (dd, J = 19.2, 7.9 Hz, 1H), 2.55 (dd, J = 19.2, 2.6 Hz, 1H)
13C-NMR (151 MHz, CHLOROFORM-D) δ 205.2, 154.9, 150.4, 148.8, 147.8, 146.2, 144.4, 138.3, 132.2, 120.3, 108.2, 107.5, 106.0, 105.9, 101.0, 100.3, 60.9, 60.4, 60.2, 56.4, 56.3, 56.2, 47.3, 41.3
HRMS found [M+H]+ 343.1181, C19H19O6 requires [M+H]+ 343.1182.
5. Conclusions
The greater rigidity of indanones compared to chalcones may result in increased prodrug activity and/or increased CYP1 isozyme selectivity. Further, the 1- indanone locked ring system should eliminate the possibility of cis –trans isomerisation, a problem which exists with chalcones. Herein, we have adopted a green chemistry approach for the convenient and safe conversion of chalcones DMU135 to indanone DMU5401 via Nazarov cyclisation reaction using an eco-friendly solvent (4-MeTHP) in the presence of Lewis acid BF3OEt2. The product recovery process was greatly simplified by the use of the green solvent and a yield of 35% was achieved with our method, which did not require extraction step with polar aprotic solvents and improved the sustainability aspects of the experiment.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1. 1H NMR spectrum of 4,5,6-trimethoxy-3-(3,4-methylendioxyphenyl)indan-1-one. Figure S2. 13C NMR spectrum of 4,5,6-trimethoxy-3-(3,4-methylendioxyphenyl)indan-1-one. Figure S3. 1H-13C HSQC NMR spectrum of 4,5,6-trimethoxy-3-(3,4-methylendioxyphenyl)indan-1-one. Figure S4. Chromatogram of of 4,5,6-trimethoxy-3-(3,4-methylendioxyphenyl)indan-1-one using chiral chromatography column Daicel Chiracel OD showing the presence of two enantiomers. Figure S4-6. High-Resolution Mass Spectrometry analytical data including HPLC/MS chromatogram, mass spectrum and single mass analysis calculated for molecular formula C19H18O6.
Author Contributions
Conceptualization, F.B.; methodology, K.R. and F.B.; investigation, P.P., K.R. and R.A.; writing—original draft preparation, K.R. and F.B; writing—review and editing, F.B.; supervision, F.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Data supporting reported results can be found in the Supplementary Materials.
Acknowledgments
Brandon Moulds and the De Montfort University technical team are thanked for the support in collecting Accurate Mass spectrometry data.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Aziafor, K.; Ruparelia, K.; Moulds, B.; Zloh, M.; Parish, T.; Brucoli, F. Design and Synthesis of Pyridyl and 2-Hydroxyphenyl Chalcones with Antitubercular Activity. Molecules. 2024, 29, 4539. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C., Zhang, W., Sheng, C., Zhang, W., Xing, C.,; Miao. Chalcone: A Privileged Structure in Medicinal Chemistry. Chemical Reviews 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, D.; Nikolic, D.; Zhu, D.; Pezzuto, J.M.; van Breemen, R.B. In vitro metabolism of isoliquiritigenin by human liver microsomes. Drug metabolism and disposition 2008, 36, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Nishino, T.; Inoue, H.; Nagata, N.; Satomi, Y.; Nishino, H.; Shibata, S. Antitumorigenic activities of chalcones (II). Photo-isomerization of chalcones and the correlation with their biological activities. Biological and Pharmaceutical Bulletin 1997, 20, 1266–1270. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-L.; Kuo, P.L.; Lin, C.C. Isoliquiritigenin induces apoptosis and cell cycle arrest through p53-dependent pathway in Hep G2 cells. Life Sciences 2005, 77, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Sale, S.; Tunstall, R.G.; Ruparelia, K.C.; Butler, P.C.; Potter, G.A.; Steward, W.P.; Gescher, A.J. Effects of the potential chemopreventive agent DMU-135 on adenoma development in the Apc Min+ mouse. Investigational new drugs. 2006, 459–64. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S.; Forrest, R.; Hadfield, J.A.; Kendall, A.; Lawrence, N.J.; McGown, A.T.; Rennison, D. Potent antimitotic and cell growth inhibitory properties of substituted chalcones. Bioorganic & Medicinal Chemistry Letters 1998, 1051–1056. [Google Scholar] [CrossRef]
- Patil, S.A.; Patil, R.; Patil, S.A. Recent developments in biological activities of indanones. European Journal of Medicinal Chemistry 2017, 138, 182–198. [Google Scholar] [CrossRef] [PubMed]
- da Camera e Silva, E.; Figueroa-Villar, J.D.; de Aguiar, A.P. Preparation of 1-indanones with conventional heating versus microwaves. Synthetic Communications, 2002, 32, 3193–3198. [Google Scholar] [CrossRef]
- Muckensturm, B.; Diyani, F. An improved preparation of 7-hydroxyindan-1-ones. Journal of Chemical Research-S 1995, 442–443. [Google Scholar] [CrossRef]
- Turek, M.; Szczęsna, D.; Koprowski, M.; Bałczewski, P. Synthesis of 1-indanones with a broad range of biological activity. Beilstein Journal of Organic Chemistry 2017, 13, 451–494. [Google Scholar] [CrossRef] [PubMed]
- Prakasham, A.P.; Saxena, A.K.; Luqman, S.; Chanda, D.; Kaur, T.; Gupta, A.; Yadav, D.K.; Chanotiya, C.S.; Shanker, K.; Khan, F.; Negi, A.S. Synthesis and anticancer activity of 2-benzylidene indanones through inhibiting tubulin polymerization. Bioorganic & Medicinal Chemistry 2012, 20, 3049–3057. [Google Scholar] [CrossRef]
- Lawrence N.J., Armitage E.S.M., Greedy B., Cook D., Ducki S., McGown A.T. The synthesis of indanones related to combretastatin A-4 via microwave-assisted Nazarov cyclization of chalcones. Tetrahedron letters 2006, 47, 1637–1640. [Google Scholar] [CrossRef]
- Gu, X.H.; Yu, H.; Jacobson, A.E.; Rothman, R.B.; Dersch, C.M.; George, C.; Flippen-Anderson, J.L.; Rice, K.C. Design, synthesis, and monoamine transporter binding site affinities of methoxy derivatives of indatraline. Journal of medicinal chemistry. 2000, 43, 4868–4876. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi S., Tamura T., Yoshimoto S., Kawakami T., Masuyama A. 4-Methyltetrahydropyran (4-MeTHP): Application as an Organic Reaction Solvent. Chemistry–An Asian Journal 2019, 14, 3921–3937. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, K.C.; Zeka, K.; Ijaz, T.; Ankrett, D.N.; Wilsher, N.E.; Butler, P.C.; Tan, H.L.; Lodhi, S.; Bhambra, A.S.; Potter, G.A.; Arroo, R.R. The synthesis of chalcones as anticancer prodrugs and their bioactivation in CYP1 expressing breast cancer cells. Medicinal Chemistry 2018, 14, 322–332. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Photoisomerisation of synthetic chalcones and keto-enol tautomerization of isoliquiritigenin via 4-hydroxy group preventing isomerisation.
Figure 1.
Photoisomerisation of synthetic chalcones and keto-enol tautomerization of isoliquiritigenin via 4-hydroxy group preventing isomerisation.
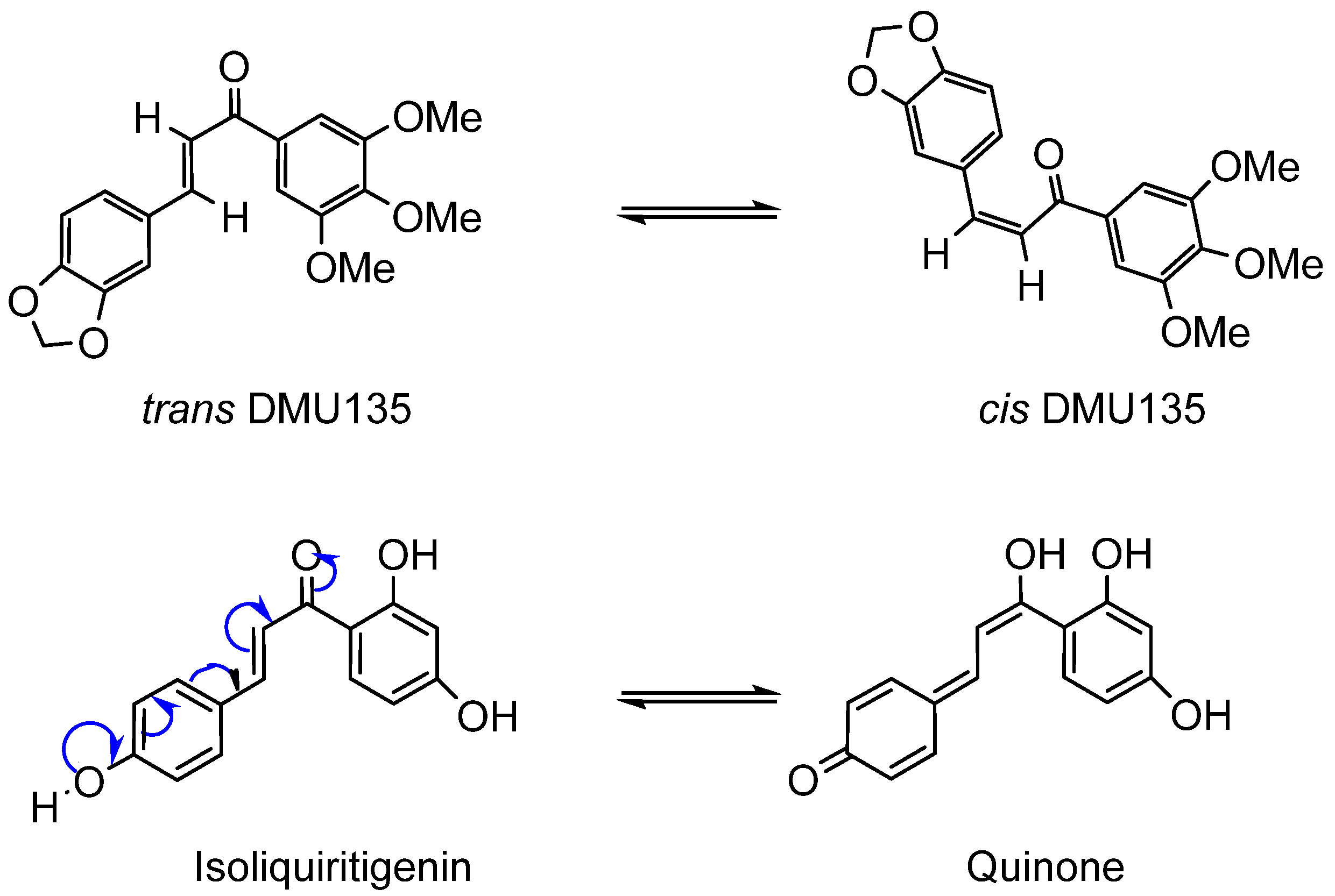
Figure 2.
Overlay of chalcone and indanone structures.
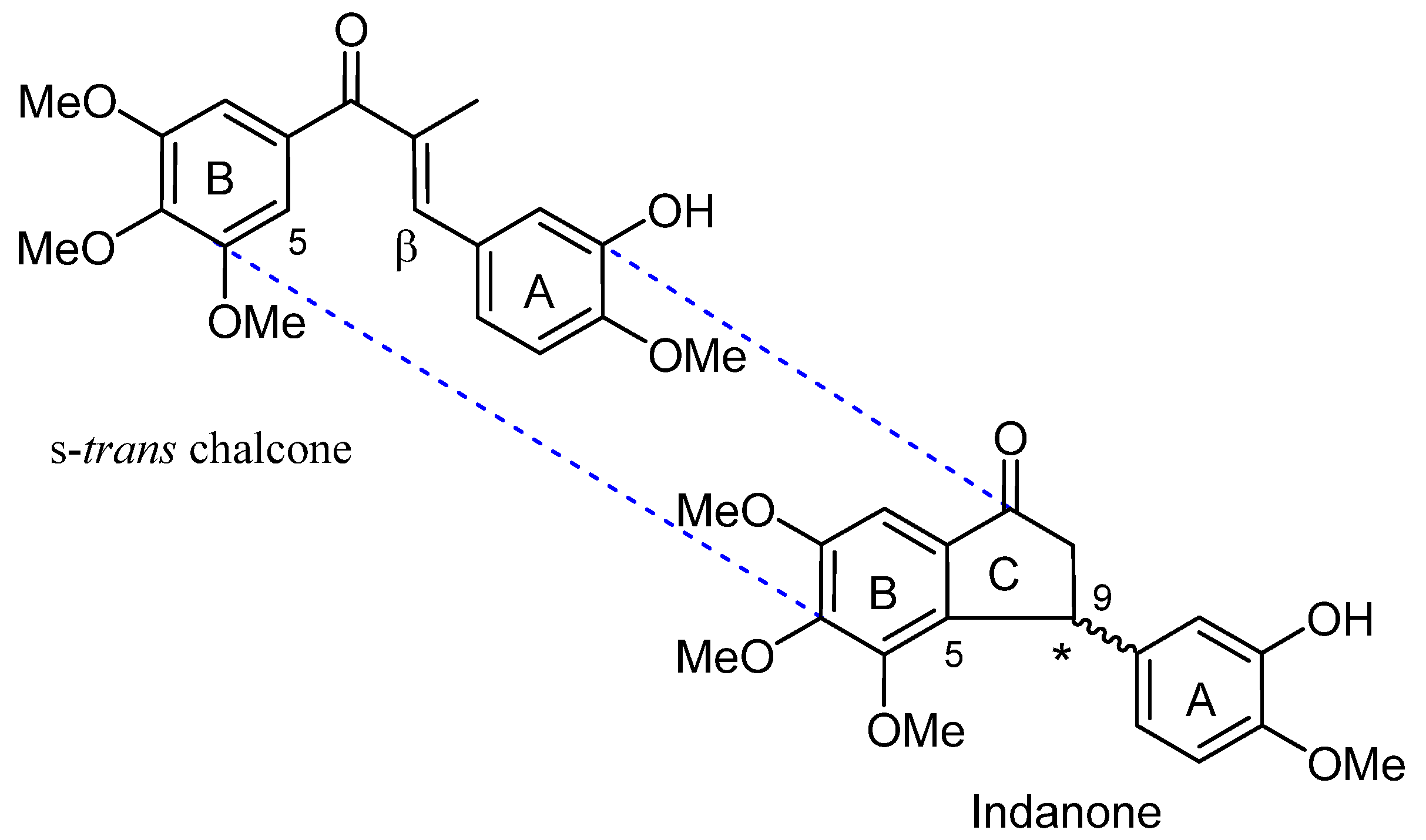
Scheme 1.
Examples of synthesis of 1-indanones via Friedel-Crafts and Nazarov cyclisation reactions using microwave irradiation and conventional heating. (E)-3-(4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one was used as the starting material in the Nazarov cyclisation reaction below to furnish 4,5,6-trimethoxy-3-(4-methoxyphenyl)-2,3-dihydro-1H-inden-1-one.
Scheme 1.
Examples of synthesis of 1-indanones via Friedel-Crafts and Nazarov cyclisation reactions using microwave irradiation and conventional heating. (E)-3-(4-methoxyphenyl)-1-(3,4,5-trimethoxyphenyl)prop-2-en-1-one was used as the starting material in the Nazarov cyclisation reaction below to furnish 4,5,6-trimethoxy-3-(4-methoxyphenyl)-2,3-dihydro-1H-inden-1-one.
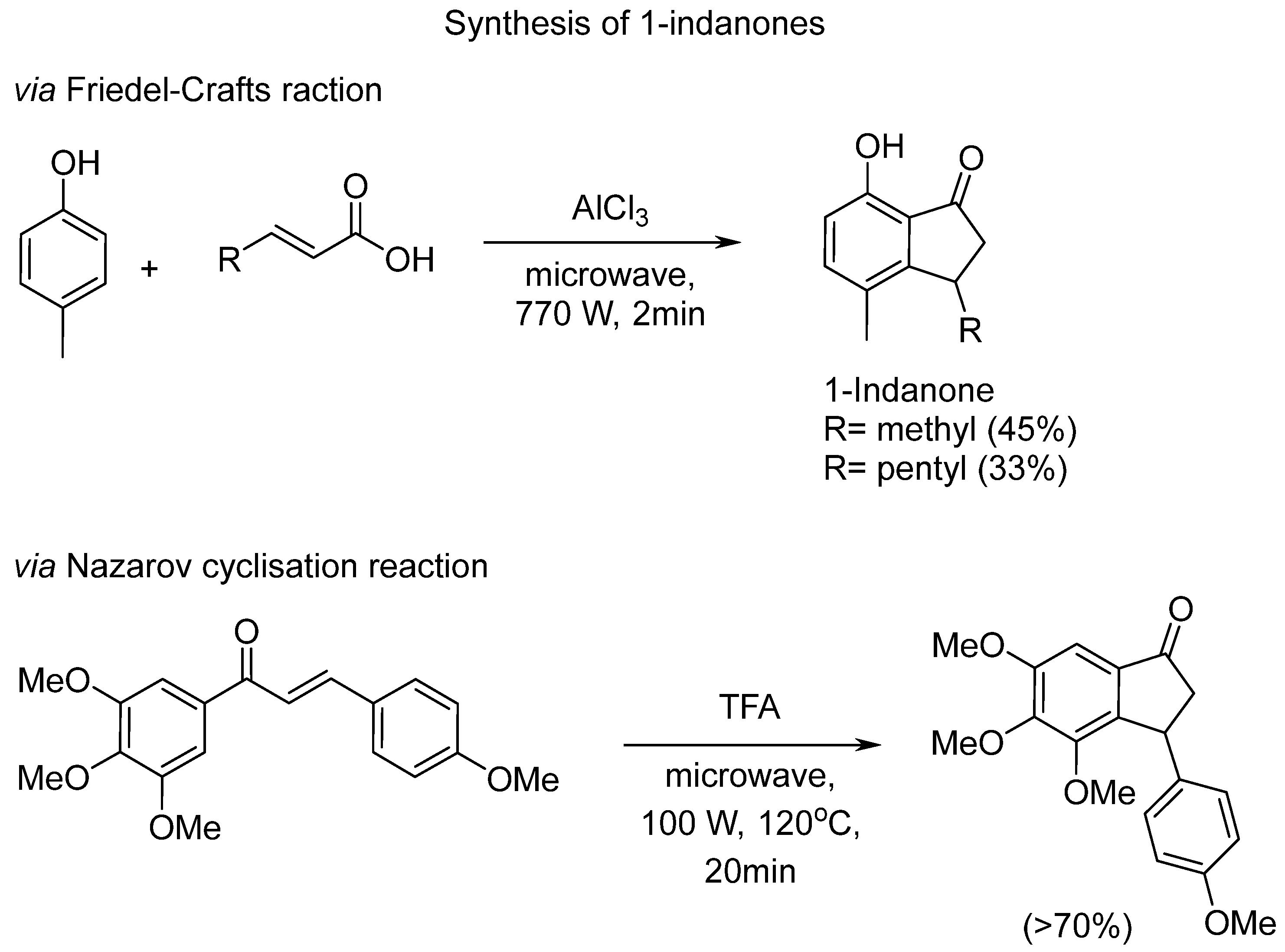
Scheme 2.
The Nazarov cyclisation reaction of chalcone DMU135 to yield indanone DMU5401 was conducted using three methods, e.g., in the presence of neat TFA at either reflux (Route 1) or under microwave irradiation (Route 2), and employing Lewis acid BF3OEt2 and 4-MeTHP, as the solvent (Route 3).
Scheme 2.
The Nazarov cyclisation reaction of chalcone DMU135 to yield indanone DMU5401 was conducted using three methods, e.g., in the presence of neat TFA at either reflux (Route 1) or under microwave irradiation (Route 2), and employing Lewis acid BF3OEt2 and 4-MeTHP, as the solvent (Route 3).
Figure 3.
NOESY spectrum of DMU5401 showing key 2D 1H-1H chemical shift correlations recorded on a Jeol JNM-ECZR 600 MHz in MeOD.
Figure 3.
NOESY spectrum of DMU5401 showing key 2D 1H-1H chemical shift correlations recorded on a Jeol JNM-ECZR 600 MHz in MeOD.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



