Submitted:
02 April 2025
Posted:
03 April 2025
You are already at the latest version
Abstract
Electroconvulsive therapy (ECT) remains one of the most effective interventions for treatment-resistant psychiatric disorders, particularly major depressive disorder and bipolar disorder. Despite extensive clinical and preclinical investigations the precise neurobiological mechanisms underlying ECT’s therapeutic effects are not fully understood. This review explores the molecular and cellular pathways involved in ECT, emphasizing its impact on neurotrophic signaling, oxidative stress, apoptosis, and neuroplasticity. Evidence suggests that ECT modulates brain-derived neurotrophic factor (BDNF) and other neurotrophic factors, promoting synaptic plasticity and neuronal survival. Additionally, ECT influences the hypothalamic-pituitary-adrenal (HPA) axis, reduces neuroinflammation, and alters neurotransmitter systems, contributing to its antidepressant effects. Recent findings also highlight the role of mitochondrial function and oxidative stress regulation in ECT-induced neural adaptation. By synthesizing current molecular insights, this review provides a comprehensive perspective on the neurobiological mechanisms of ECT, offering potential directions for future research and therapeutic advancements in brain stimulation.
Keywords:
electroconvulsive therapy
; neurobiological mechanisms
; neurotrophic factors
; neuroplasticity
; oxidative stress
1. Introduction
Major depressive disorder (MDD) is a complex psychiatric condition defined by at least one depressive episode lasting a minimum of two weeks. The primary clinical features of MDD include a persistently depressive mood or anhedonia, accompanied by various neurocognitive and neurovegetative symptoms, such as impaired concentration, changes in sleep patterns, and other disturbances in physiological functioning [1]. Globally, it is estimated that approximately 280 million individuals are affected by depression [2], with a higher prevalence observed in women compared to men [3]. In 2008, the World Health Organization (WHO) recognized severe depression as the third leading cause of global disease burden, based on factors including financial costs, mortality, morbidity, and associated health consequences. Projections indicate that by 2030, severe depression is expected to emerge as the leading cause of global disease burden [4].
First-line treatments for MDD, encompassing both psychopharmacological and psychological interventions, do not provide sufficient efficacy for all patients, with approximately one-third remaining unresponsive to these approaches [5]. For individuals with treatment-resistance depression (TRD), the electroconvulsive therapy (ECT) has emerged as a preferred intervention [6] demonstrating both rapid antidepressant effects [7] and a reduction in suicidal ideation [8]. ECT is a medical procedure in which precisely controlled electrical currents are administered to the brain under general anesthesia, intentionally inducing a generalized seizure for therapeutic purposes [9]. Since its discovery in the early 20th century, ECT has undergone significant advancements and remains a cornerstone treatment for severe mood disorders, particularly in cases of treatment-resistant MDD [10,11]. The evolution of ECT has led to substantial advancements in anesthesia techniques, electrode placement, and dosage optimization [12]. These improvements have not only enhanced the safety profile of ECT but have also significantly reduced the cognitive side effects that historically contributed to its controversial reputation. In comparison to alternative therapeutic options, ECT is the most effective treatment for symptom remission in MDD patients [13]. Response rates for ECT are notably high, ranging from 60% to 80%, with clinical improvement occurring more rapidly than with standard pharmacological treatments. Therefore, ECT is considered as one of the most potent and swift-acting therapies for affective disorders [15,16]. Moreover, research indicates that ECT can significantly reduce the duration of hospital stays and decrease the frequency of hospitalizations over a three-year period for patients undergoing maintenance ECT sessions [17]. The efficacy of ECT is strongly supported by robust clinical evidence, consistently showing superior outcomes in managing depression and other mood disorders, including bipolar depression, mania, and certain subtypes of schizophrenia [10,18,19].
The exact mechanism of action of ECT remains unclear, though significant scientific progress has been made in recent years. Several theories have been previously proposed, categorized into neurophysiological, neurobiochemical, and neuroplastic processes, which include effects on neurotransmitters, neurotrophic factors, the immune system, the hypothalamic–pituitary–adrenal (HPA) axis, neuroplasticity, epigenetic changes, brain neurophysiology, circuitry, and structure [20]. Despite extensive clinical and preclinical investigations conducted up to 2025 and its established utilization for over 80 years, the precise molecular mechanisms driving its efficacy remain incompletely understood. Consequently, a deeper comprehension of how ECT operates is essential for illuminating the underlying causes of severe MDD and advancing personalized treatment strategies for these patients. Hence, the aim of our review is to present the most discussed neurobiological mechanisms and associated signaling pathways involved in ECT’s mechanism of action.
2. Understanding the Mechanisms of ECT: Key Theories
In previous decades, several theories has been proposed in order to elucidate the precise mechanism of action during ECT. The amnesia hypothesis suggested that ECT's therapeutic effect stemmed from memory loss of events that triggered symptom onset [21], which led to unsupported multiple ECT administrations per session to enhance amnesia [22]. Studies show ECT releases endogenous opioids like beta-endorphin and Met-enkephalin, linked to memory loss, while naloxone counteracts these effects, supporting opioid involvement [23]. Neuroimaging and electrophysiology studies reveal that ECT temporarily disrupts memory-related brain regions, though these effects are typically reversible [24]. However, this hypothesis was abandoned when research showed right unilateral or bifrontal placements with ultrabrief pulses caused less amnesia than bitemporal placements while maintaining efficacy [25,26]. On the other hand, the anticonvulsant theory emerged from the observation that during ECT both seizure threshold increases and seizure duration decreases. This led to the hypothesis that the inhibitory brain processes linked to the rising seizure threshold also contribute to depression relief. Supporting evidence from electroencephalogram (EEG) and cerebral blood flow studies shows a suppression of neural activity, particularly in the frontal lobes, after ECT, which correlates with its antidepressant effects [27]. However, later studies have failed to replicate the correlation between increase in seizure threshold and antidepressant outcomes [28] and magnetic resonance spectroscopy (MRS) has shown no significant gamma-aminobutyric acid (GABA) changes related to ECT’s efficacy [29]. The neurogenesis hypothesis suggests that the therapeutic effects of ECT are driven by an increase in the number of neurons or the strengthening of connections between neurons [30]. The theory is based on neurotrophic effects occurring after electroconvulsive seizures [31], with additional studies reporting amplified signaling of brain derived growth factor (BDNF) in numerous brain areas and vascular endothelial growth factor (VEGF) in the hippocampus after exposure to electroconvulsive seizures [31,32], as well as increased precursor cell proliferation in the subgranular zone of the hippocampal dentate gyrus (DG) in the monkey hippocampus [33] The neuroendocrine hypothesis of ECT suggests that seizures activate the HPA axis, as evidenced by a postictal surge in blood levels of adrenocorticotropic hormone, cortisol, and prolactin [34]. It has been reported that ECT induces rapid increase in serum concentrations of these hormones, suggesting a significant stimulation of the HPA axis [35]. Additionally, research indicates that ECT decreases serum levels of cortisol, acting as a regulator of HPA axis activity [36]. These findings support the notion that neuroendocrine responses play an important role in the antidepressant efficacy of ECT. To date, four main hypotheses have survived in an attempt to explain the potential mechanisms of action of ECT, including neuroplasticity hypothesis, neurotransmitter hypothesis, receptor hypothesis, and cytokine hypothesis (Figure 1).
The neuroplasticity (or neurotrophic) hypothesis posits that morphological changes - such as neurogenesis, gliogenesis, or alterations in dendritic or axonal arborization of existing neurons - are critical for antidepressant effects achieved with ECT [37]. This theory is supported by preclinical animal studies indicating that electroconvulsive stimulation (ECS) leads to a dose-dependent increase in neurogenesis within the DG of the hippocampus [38]. Shahin and colleagues reported increased levels of plasma BDNF in patients with treatment-resistant schizophrenia after ECT [39]. ECT beneficial effect can arise from induction of BDNF production, which in turn affect neuronal proliferation in the DG and the sprouting of its efferent fibers [40]. The neurotransmitter theory is based on the impact of ECT on monoamine neurotransmitter functioning, such as the enhancement of serotoninergic transmission [41]. Preclinical studies have demonstrated that ECT increases serotonergic neurotransmission, with enhanced expression and activity in the hippocampus and prefrontal cortex of both postsynaptic serotonin 1A receptor (5-HT1A) and serotonin 2A receptor (5-HT2A) receptors. In human studies, it has been demonstrated that the binding of both 5-HT1A and 5-HT2A receptors is generally reduced after ECT [42]. Additionally, ECT has been found to affect the GABA system, the primary inhibitory neurotransmitter in the brain, by increasing GABAergic tone and enhancing GABA transmission, thus contributing to its anticonvulsant and anxiolytic effects. Furthermore, same study showed that ECT-induced activation of the dopamine system likely contributes to the alleviation of depressive and anxious symptoms, accompanied by improvements in motivation, concentration, and attention [43]. Collectively, these findings underscore the multifaceted impact of ECT on neurotransmitter systems, which is central to its efficacy in treating depressive disorders. Receptor hypothesis proposed that an increased affinity of ɑ2 adrenergic receptors is present in the frontal cortex (FC) and hippocampus (CA) in depressive patients [44,45] while this affinity decreases following ECT [45]. At the same time, ECT can influence the expression of genes encoding dopamine receptors, leading to an upregulation of dopamine D1 receptors in the hippocampal CA3 region, which contributes to the treatment of severe mental disorders [46]. Finally, the cytokine hypothesis explains mechanisms of ECT to be related with alterations in cytokine levels after ECT sessions, specifically the levels of interleukin (IL)-6 and tumor necrosis factor-α (TNF-α), while these markers significantly decrease after ECT [47].
These hypothesis suggest that ECT promotes neurogenesis, modulates monoamine and GABAergic neurotransmission, alters receptor affinity, and reduces pro-inflammatory cytokines, all contributing to its therapeutic effects. While each hypothesis provides valuable insight, further research is needed to fully integrate these mechanisms into a comprehensive understanding of ECT’s efficacy.
3. Neurotransmitter Modulation by ECT
Previous research on depression and other psychiatric diseases has focused on exploring the relationship between various neurotransmitter systems and the pathophysiology of these conditions. There is a well-established consensus that at least three neurotransmitter systems - serotonin, noradrenaline, and dopamine - are crucial in the pathogenesis of MDD. This is supported by extensive evidence, including studies utilizing animal models, neuroimaging techniques, genetic analyses, and the pharmacological effects of antidepressant medications, which specifically target one or more components of these neurotransmitter systems. Furthermore, a meta-analysis of monoamine depletion studies has demonstrated an indirect correlation between monoamine levels and mood regulation [48].
Regarding the serotonergic system, early preclinical studies have demonstrated enhanced serotonergic neurotransmission due to the upregulation of postsynaptic 5-HT1A receptors in specific brain regions, alongside temporally and anatomically distinct alterations in 5-HT1A and 5-HT2A receptor expression [49,50]. Other studies have reported increased 5-HT2A receptor binding without significant changes in 5-HT1A receptor binding or 5-HT1A messenger RNA (mRNA) expression [51]. Furthermore, Chaput and colleagues found that, in addition to postsynaptic 5-HT1A receptor sensitization, there was no suppression of serotonin production via negative feedback from presynaptic 5-HT2A receptors in rats, a process typically observed during antidepressant treatment with paroxetine [52]. On the other hand, Strome and colleagues demonstrated a significant reduction in 5-HT2A receptor binding 24 hours and 7 days after ECT in non-human primates, with receptor levels returning to baseline after six weeks [53]. In contrast, study on patients resistant to antidepressant therapy have reported reduced 5-HT1A receptor binding after ECT, specifically in brain regions implicated in emotional regulation and MDD pathology, including the amygdala (AM), anterior cingulate cortex (ACC), orbitofrontal cortex (OFC), and insula (IN). Notably, these reductions were not observed in two separate pre-ECT measurements, nor were they correlated with baseline 5-HT1A receptor levels in relation to the Hamilton Depression Rating Scale (HDRS) scores at the end of therapy [54]. Meanwhile, Saijo and colleagues found no significant difference in 5-HT1A receptor binding before and after ECT in MDD patients [55]. Additionally, another study reported a post-ECT reduction in 5-HT2A receptor expression, with changes in the right lingual gyrus, right medial frontal gyrus, and right parahippocampal gyrus correlating with improvements in depressive symptoms [42]. These findings align with studies conducted on non-human primates and research on antidepressant treatments [53,56,57], highlighting the potential role of 5-HT2A receptor modulation as a key mechanism underlying ECT's therapeutic effects.
In contrast to serotonin, where discrepancies exist between rodent and human studies, research on the effects of ECT on the dopaminergic system has demonstrated a relatively high degree of consistency across both animal and human models. Nikisch and colleagues reported a significant post-ECT increase in dopamine and serotonin metabolites - homovanillic acid (HVA) and 5-hydroxyindoleacetic acid (5-HIAA) - as well as elevated levels of neuropeptide Y (NPY)-like immunoreactivity (NPY-LI) in the cerebrospinal fluid of depressive patients (n=6) [58]. Another study found that patients who responded to ECT exhibited higher baseline HVA levels before treatment, followed by a significant reduction in HVA concentrations five weeks after ECT initiation, which correlated with clinical improvement based on the HDRS [59]. Saijo and colleagues observed a decrease in dopamine D2 receptor binding in the rostral ACC of MDD patients who responded to ECT [60], while others reported an increase in D1 receptor levels in the DG following ECT [46]. In non-human primates, Landau and colleagues found a transient increase in dopamine transporter binding after six ECT sessions, whereas D2 receptor binding remained unchanged [61]. Additionally, Lammers and colleagues demonstrated that ten days of ECT in rats led to a significant increase in D3 receptor mRNA expression and D3 receptor binding in the nucleus accumbens shell [62]. Furthermore, results from one study indicated that ECT induces an increase in prolactin levels, mediated by dopaminergic but not serotonergic neuronal activity [48]. Huuhka and colleagues investigated the polymorphisms of the dopamine D2 receptor (DRD2) gene C957T (rs6277) and the catechol-O-methyltransferase (COMT) gene Val158Met (rs4680) in relation to ECT response in 118 MDD patients and 383 healthy controls. Their findings suggest that interactions between these genetic variants may be associated with ECT responsiveness [63]
NPY, a key regulator of feeding, circadian rhythms, and memory, has also been implicated in the etiopathogenesis of MDD [64]. A study examining the effects of antidepressants on NPY reported a significant increase in serum NPY concentration in depressed patients, with the most pronounced elevation observed after six months of treatment [65]. Similarly, Altar and colleagues demonstrated that ECS increases the expression of NPY pathway genes, followed by elevated NPY levels in the hippocampus and DG two weeks post-stimulation [31]. Regarding norepinephrine (NE), preclinical studies in rats have shown reduced α2-adrenoceptor binding in the FC, hippocampus, and AM following ECT, suggesting that ECT may enhance NE release via α2-adrenoceptor downregulation [66]. Early studies in patients with major depressive episodes also reported significant increases in plasma norepinephrine levels following ECT [67]. However, one study found a post-ECT decrease in plasma NE concentrations, with significant differences between pre-ECT levels in depressed patients and controls. These changes did not correlate with HDRS scores [68], a finding which is also showed by Kelly and colleagues in certain patients [69]. On the other hand, Pollak and colleagues further reported a significant reduction in epinephrine levels in ECT responders, though no significant differences were observed in NE or cortisol levels between responders and non-responders [70]. It is essential to recognize that monoaminergic systems do not function in isolation but rather interact dynamically. NE modulates dopamine release in the ventral tegmental area (VTA) via α1- and α2-adrenoceptors, while dopamine inhibits NE release from the locus coeruleus. Additionally, both neurotransmitters facilitate serotonin release via α1 (NE) and D2 (dopamine) receptor activation [71]
Glutamate is another neurotransmitter implicated in mood regulation and the therapeutic effects of ECT. Dong and colleagues demonstrated that depressed rats exhibit elevated glutamate levels, which decreased in the hippocampus following ECT [72]. Additionally, an increased glutamate-to-GABA ratio has been observed in the hippocampus and prefrontal cortex in rodent models of depression [73]. In human studies, alterations in glutamate levels have also been reported. Postmortem analyses of patients with affective disorders revealed increased glutamate concentrations in the FC [74], while reductions were noted in the AM, dorsolateral prefrontal cortex, and ACC [75]. Notably, ECT has been shown to normalize glutamate concentrations in the ACC in MDD patients, which was in correlation with therapeutic response [76]. Another study reported an increase in glutamate levels in the ACC and a decrease in the hippocampus after ECT in MDD patients [77]. Pfleiderer and colleagues previously demonstrated that ECT induces a significant increase in glutamate levels in the left ACC specifically in responders, whereas non-responders showed no statistically significant change [78] However, some studies have failed to detect significant glutamate alterations following ECT [79].
ECT exerts its therapeutic effects through complex interactions within serotonergic, dopaminergic, noradrenergic, and glutamatergic systems, leading to neurotransmitter modulation and receptor alterations. Overall, the available evidence underscores the multifaceted neurochemical effects of ECT, highlighting its capacity to restore balance across multiple neurotransmitter systems. While these findings provide valuable insights into the biological underpinnings of ECT, further research is required to fully elucidate its mechanisms of action and optimize its clinical application in MDD and other psychiatric conditions.
4. The Role of Neuroplasticity, Functional Network Reorganization, and Neuroanatomical Changes in the Therapeutic Effects of ECT
An increasing amount of evidence suggests that neuroplasticity - the brain’s ability to adapt structurally and functionally in response to stimuli - plays a crucial role in therapeutic effects of ECT. Neuroplasticity encompasses various processes, including synaptic remodeling, neurogenesis, dendritic growth, and changes in neural connectivity, which are essential for mood regulation and cognitive function [80]. It has been shown that application of ECT induces extensive neuroplastic changes across neocortical, limbic, and paralimbic areas, with these alterations closely linked to the degree of the antidepressant response. Various studies showed that ECT induced neuroplasticity in the hippocampus and AM, which was associated with improved clinical response and pronounced in regions with prominent connections to ventromedial prefrontal cortex and other limbic structures. Both hippocampal and AM volumes increased following ECT and correlated with evident improvement of symptoms [81,82,83].A study by Bouckaert and colleagues enrolling 66 depressed patients who predominantly received right unilateral ECT also found a significant bilateral increase in hippocampal volume one week after treatment. However, these changes were no longer detectable at a six-month follow-up [84]. Sartorius and colleagues further reported post-ECT increases in hippocampal and AM gray matter volume, particularly in the right hemisphere, but these changes did not correlate with improvements in depression or cognitive function in patients primarily treated with right unilateral ECT [85]. Camilleri and colleagues found increased gray matter volume in the right hippocampus and AM in patients with unipolar depression after ECT compared to healthy controls. However, they did not assess the relationship between these changes and clinical outcomes [86]. While most studies indicate no clear link between hippocampal volume increases and antidepressant efficacy, some research suggests a connection to cognitive impairment. For example, Oostrom and collegues reported that larger increases in hippocampal volume after ECT correlated with poorer cognitive performance [87]. Overall, changes in hippocampal volume and function induced by ECT may indicate neuroplasticity; however, these effects are often temporary and do not consistently correlate with clinical outcomes in depression or cognitive side effects.
Beyond the hippocampus, there is a smaller body of research on ECT-induced neuroplasticity in other brain regions and white matter. Volumetric increases have been also observed in the ACC, postcentral gyrus, fusiform gyrus, medial prefrontal cortex, supplementary motor cortex, insula, and striatum [88]. Moreover, variations in ACC thickness, which can early distinguish treatment responders and non-responders, may serve as a biomarker for overall clinical outcomes [89]. Lyden and colleagues studied white matter changes using diffusion tensor imaging (DTI) in 20 patients with MDD who received right unilateral or bitemporal ECT. Authors demonstrated increased fractional anisotropy in the bilateral anterior cingulum, forceps minor, and left superior longitudinal fasciculus, which were associated with reductions in depressive symptoms. This suggests that ECT may enhance the integrity of fronto-limbic pathways involved in mood regulation [90].
The neuroplasticity and neurogenesis hypothesis suggests that the therapeutic effects of ECT are driven by an increase in the number of neurons or the strengthening of neural connections [30]. Preclinical research has demonstrated that ECS, the animal model equivalent of ECT, increases the proliferation of neural progenitor cells in the DG of the hippocampus, a region crucial for memory processing and emotional regulation as well as and bromodeoxyuridine (BrdU)-positive cells in the same region [38,91,92]. When extended to adult non-human primates, ECS was found to increase precursor cell proliferation in the subgranular zone of the DG, with most of these cells differentiating into either neurons or endothelial cells [33]. Also, ECT has been shown to modulate synaptic plasticity by increasing the expression of BDNF, a key molecule involved in neuronal survival, synaptic strength, and adaptive responses to stress and VEGF, specifically in the hippocampus [32]. BDNF levels are often reduced in MDD patients, and their restoration following ECT has been associated with symptom improvement [93]. Furthermore, ECT alters the expression of genes and proteins associated with synaptic function, including glutamatergic and gamma-aminobutyric acid (GABA)-ergic signaling, which are critical for maintaining excitatory-inhibitory balance in the brain. Various studies indicate that ECS enhances neurogenesis by increasing the volume of certain brain regions, which correlates with improved behavioral outcomes and neuroplasticity [94,95]. The protein Homer-1, primarily found in two forms - short (Homer1a) and long (Homer1b/c) - is found to be crucial for postsynaptic density, connecting metabotropic glutamate receptors (mGluRs), and regulating their signaling pathways [96]. Homer1a, a rapidly produced variant in response to neuronal activity, competes with the more stable Homer1b/c for mGluR binding. This balance is of particular importance for neuronal plasticity; Homer1a dominance promotes homeostatic plasticity, while Homer1b/c is associated with heightened activation [96,97]. Homer1a, mainly located in the CA1 hippocampus, is activated by neuronal stimulation, such as seizure activity [96,98]. It increases Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor clustering, enhancing synaptic transmission and excitatory postsynaptic potential (EPSC), without changing presynaptic glutamate release. Additionally, Homer1a modulates the mGluR-IP3 signaling pathway, reducing excitability in pyramidal neurons and acting as a negative feedback mechanism to prevent excessive excitation. Research shows that increased Homer1a in the medial prefrontal cortex has antidepressant effects, while lower levels are linked to depression [99]. In the hippocampus, high Homer1a may increase stress vulnerability [98]. Homer1 also regulates the HPA axis independently of mGluR1/5. By interacting with mGluR1/5 and NMDA receptors, Homer1a can induce rapid antidepressant responses [100]. Thus, Homer1a is essential for mediating antidepressant effects, with its splice variants, Homer1b/c, having distinct regulatory roles. ECS remodels neuroplasticity by balancing mGluR1/5 and AMPA receptors, leading to rapid antidepressant effects. It activates presynaptic glutamatergic neurons and inhibits GABAergic neurons, resulting in increased glutamate release and AMPA receptor activation while inhibiting NMDA receptors. This process promotes the release of BDNF, which activates the TrkB receptor and subsequently signals Akt to mTORC1, encouraging neurogenesis. Additionally, Homer1 disrupts dysfunctional complexes with mGluR1/5 and partially opens the BK channel, contributing to the hyperpolarization of the postsynaptic neuron and enhancing the antidepressant effect [101].
Additionally, neuroplasticity induced by ECT goes beyond just molecular and cellular changes; it also affects the functional connectivity within large-scale brain networks. Depression is often associated with dysregulation in the default mode network (DMN), which is linked to self-referential thinking and rumination. Functional neuroimaging studies indicate that ECT decreases hyperconnectivity within the DMN while enhancing connectivity in cognitive control networks, such as the central executive network (CEN). These connectivity changes, such as altered communication between the medial prefrontal cortex and ventrolateral prefrontal cortex, as well as between the dorsomedial prefrontal cortex and posterior cingulate cortex, have been associated with clinical improvement and contributes to mood stabilization and cognitive recovery [102,103].
In conclusion, the proposed model of ECT’s neurobiological effects suggests that individuals with depression have low plastic potential before treatment, contributing to severe symptoms. Each ECT session induces temporary brain disruption, which can cause postictal confusion but also triggers physiological changes like reduced N-acetylaspartate levels, altered connectivity, and changes in white matter integrity. This disruption, leads to a heightened state of neuroplasticity, promoting the reorganization of neural circuits related to depression. It has been also suggested that excessive ECT dosing may result in significant structural and functional changes, providing both antidepressant and cognitive side effects. Conversely, insufficient dosing may not yield an adequate antidepressant response but could minimize side effects. Understanding these dynamics can help optimize ECT protocols to balance benefits and risks [88,104].
4. Molecular Pathways Related to ECT
4.1. Neurotrophins and ECT
One of the most extensively studied systems implicated in the pathogenesis of MDD is the neurotrophic system, while it has been shown that antidepressants influence the concentrations of neurotrophins and neurotrophic factors [105]. BDNF is a crucial neurotrophic factor that plays a key role in neuronal development, maintenance, and plasticity in the central nervous system (CNS), particularly by promoting neurogenesis, enhancing synaptic plasticity, and supporting neuronal survival [106]. During ECT, alterations occur in various neurobiochemical molecules, including BDNF, but also other neurotrophic factors, which further triggers neuroplastic changes within the brain. These modifications, alongside enhanced neuronal proliferation, also have a neuroprotective function. Notably, a single application of ECT has been shown to induce neuronal proliferation in the hippocampal DG of rats, with these new neurons surviving for several months [38,107].
Studies investigating the relationship between baseline BDNF levels and response to drug therapy, as assessed by depression scales, have yielded conflicting results [108,109,110]. Various studies have highlighted a strong association between the response to antidepressant treatment and early increases in serum or plasma BDNF levels during the first few weeks of therapy [111,112,113,114]. Karege and colleagues found that individuals with depression exhibit significantly lower baseline serum BDNF levels, which increase in response to ketamine treatment [115,116]. Also, some studies have shown that patients suffering from TRD have lower BDNF levels before therapy compared to healthy controls and a significant increase in these levels after ECT. It was also found that there is a temporal correlation between clinical response to ECT and increases in BDNF levels, suggesting that BDNF levels could represent a biological marker of remission during ECT sessions [117]. However, some studies which examined the predictive power of baseline BDNF as an indicator of potential response to ECT did not show positive results [93,118,119,120]. Ryan and colleagues assessed baseline BDNF levels in medicated patients with depression and revealed no significant differences in BDNF concentrations between these patients and controls. Additionally, the study showed no notable change in BDNF levels in depressed patients before and after ECT administration [119]. However, some less frequent studies, such as the one conducted by Sorri and colleagues, reported a decrease in BDNF levels during ECT. These contrasting findings have led some researchers to question the reliability of BDNF as a determinant of ECT efficacy [93,119,121,122].
The discrepancies in BDNF levels observed in studies involving depression and ECT may arise due to several factors. First, methodological differences between studies, such as variations in sample size, measurement techniques (e.g., serum vs. plasma BDNF levels), and the timing of BDNF measurements (e.g., pre- and post-treatment time points), could contribute to conflicting results. Sorri and colleagues reported that serum BDNF levels were not affected by ECT, while BDNF plasma levels decreased during the fifth ECT session [93]. Second, variations in treatment protocols can differ in terms of the type, dosage, and frequency of antidepressant medications or ECT, which could lead to varying effects on BDNF expression. Some treatments might stimulate BDNF production more robustly than others, while some may not induce noticeable changes in BDNF levels at all. Finally, individual variability and genetic differences in patients, particularly variations in genes associated with BDNF production or its receptor or in the pathophysiology of depression can influence how BDNF levels are regulated in different patients. Depression is a heterogeneous condition, meaning that varying biological mechanisms may contribute to its development in different individuals, which could lead to inconsistent BDNF responses across studies. Overall, in a recent review by Zelada and colleagues authors reported lower BDNF serum levels in patients with MDD compared with those in healthy controls, and pharmacological treatments usually lead to an increase in these levels, which correlated with an improvement in the clinical presention [123]. However, there is more controversy in the literature regarding non-pharmacological treatments and BDNF levels in this population of patients [123].
Another growth factor hypothesized to play a role in the pathogenesis of MDD is VEGF. VEGF promotes vasculogenesis, angiogenesis, and neurogenesis in the hippocampus, with hypoxia increasing its expression in both the hippocampus and peripheral organs [124]. VEGF also regulates glutamatergic synaptic function, suggesting its role in the pathophysiology of psychiatric disorders [124]. However, studies on VEGF levels in MDD patients have yielded mixed results compared to controls [125]. Minelli and colleagues found that VEGF levels increased in patients undergoing ECT, with a temporal correlation between VEGF levels and symptom improvement, supporting its potential role in ECT's mechanism of action [126]. In another study which enrolled 67 MDD patients, authors observed a significant correlation between reduced depressive symptoms and VEGF levels before ECT, suggesting that pre-treatment VEGF levels may predict treatment response [127].
The role of neurotrophic factors, such as nerve growth factor (NGF), neurotrophin-3 (NT3), neurotrophin-3 (NT4), glial cell-derived neurotrophic factor (GDNF), NPY, and their receptors (TrkA, TrkB, TrkC, p75), in relation to ECT therapy remains insufficiently explored. Grønli and colleagues studied NT3, NGF, and NPY levels in patients with affective disorders before and after ECT, finding no significant change in NT3 and NGF, but a notable increase in NPY levels [128]. However, the study had limitations, including a small sample size and broader diagnostic criteria, and authors warranted further research to confirm these findings. Zhang and colleagues found that patients with TRD who responded to ECT (based on a >50% reduction in HDRS scores) had a significant increase in serum GDNF, while non-responders did not [129]. Another study showed decreased GDNF levels in the hippocampus and striatum of rats treated with ECT, but increased NGF in the FC and BDNF in the hippocampus, FC, and striatum [130].
There is a lack of studies examining other molecules in the BDNF signaling pathway and the impact of ECT on BDNF and neurotrophic factor receptors. Enomoto and colleagues found that ECT treatment in rats for 10 days led to downregulation of full-length TrkB receptors, potentially in response to elevated mBDNF concentrations, but also an increase in phosphorylated TrkB expression in the dorsal and ventral hippocampus, suggesting enhanced BDNF/TrkB signaling [131]. Schurgers and colleagues found that ECT significantly increased the concentration of molecules involved in the BDNF/TrkB signaling cascade, which negatively correlated with depression scores in TRD patients [132].
Collectively, while several studies have explored the role of neurotrophic factors and their receptors in relation to ECT therapy, the findings remain mixed and it is hard to bring strong conclusions. Some research suggests that ECT may influence the expression of neurotrophic factors like NPY, GDNF, and BDNF, which could be linked to therapeutic outcomes in patients with TRD. However, the limited sample sizes, varied diagnostic criteria, and insufficient exploration of other molecules in the BDNF signaling cascade indicate that more comprehensive studies are needed to fully understand the underlying mechanisms. Ultimately, future research should aim to clarify the precise role of these factors and their receptors in ECT's therapeutic effects, potentially offering valuable insights for optimizing depression treatment strategies.
4.2. ECT and Immunological Alterations
The involvement of immune system cells in the pathogenesis of depressive and other psychiatric disorders has gained increasing attention in recent years. In this context, it has been hypothesized that ECT may exert its effects, at least in part, through modulation of the immune system, particularly cytokines. It has been shown that in MDD patients, the release of cytokines in response to external stress is enhanced both peripherally and centrally [133], with pro-inflammatory cytokines such as TNF-α, C-reactive protein (CRP), and IL-6 being particularly significant and often recorded in increased levels [133,134]. Previous studies have demonstrated that modulating the activity of the innate immune system, by antagonizing the action of certain cytokines, can improve depressive symptoms in patients with inflammatory diseases, even without treating the underlying disease. Furthermore, studies in mice have shown that knocking out genes for the TNF-α receptors resulted in decreased anxiety during infections and the development of an antidepressant-like phenotype [135,136,137].
Microglia, the resident macrophages of the CNS, originate from the mesoderm and migrate to the CNS during development, where they play a crucial role in maintaining homeostasis and protecting against injury [138]. They support neuronal survival, differentiation, and circuit formation through BDNF signaling and are involved in synaptic formation related to learning. However, persistently activated microglia can contribute to neurodegenerative diseases by producing high levels of proinflammatory cytokines and chemokines, leading to neuronal dysfunction [138]. In animal models of depression, microglia were shown to be activated, and modulating their activity led to an improvement in the clinical signs of the disease [139]. It has been shown that electrical neuronal stimulation regulates microglia and controls their activation in response to immune challenges. Although electroconvulsive seizures did not affect resting microglia, they induced transcriptomic changes in the retinoic acid receptor α response pathway, which modulated microglial response to immune stimulation [140]. Further research related to exploring the effects of ECS on brain microglia, as well as on astrocytes would be crucial, as neuronal excitation and glutamate release are known to induce calcium waves and gliotransmission, with ECS promoting increased glial fibrillary acidic protein (GFAP) expression and astrocyte proliferation [140].
Research investigating cytokine levels during ECT treatment has shown that the concentrations of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 increase, particularly in the first hour following the current application. These early changes in cytokine levels remaind consistent throughout the course of therapy, regardless of whether it is an initial or later ECT session, with no reduction of this acute effect through repeated treatments [141,142]. However, Hestad and colleagues observed that after multiple ECT sessions, there was a gradual reduction of TNF-α levels, most notably 24 hours and one week after the final treatment. This decrease was not observed in control patients receiving only pharmacotherapy [143]. Additionally, a study by Järventausta and colleagues found that IL-6 levels decreased towards the end of ECT treatment, which correlated with patient response scores as measured by the Montgomery-Åsberg Depression Rating Scale (MADRS) [144]. Kranaster and colleagues investigated the effect of ECT on the activity of innate immune system cells and concluded that ECT likely exerts its therapeutic effects, at least in part, through changes in neuroinflammation. Their study showed a reduction in the concentration of macrophage migration inhibitory factor (MIF) in the cerebrospinal fluid of depressed patients, which is a pro-inflammatory protein implicated in the regulation of the innate immune system and neurogenesis. Additionally, there was a reduction in the serum concentration of the CD14 molecule [145]. Fluitman and colleagues also noted increased concentrations of TNF-α and IL-6, along with an increase in IL-10 production by monocytes after lipopolysaccharide stimulation, and a decrease in IFN-γ production by T cells following CD2/CD28 stimulation after ECT application. They further observed a temporary increase in leukocytes, granulocytes, natural killer (NK) cells, and monocytes immediately after ECT, with these levels returning to baseline approximately 30 minutes post-treatment. Additionaly, repeated ECT sessions showed no cumulative effects on these acute changes. However, sample size and patient heterogeneity, prevent clear correlations between these immune changes and depression symptom severity [146]. Similarly, the study by Kronfoll and colleagues showed an increase in NK cells immediately after ECT application, with this effect observed in repeated sessions, though long-term effects on NK cells were not monitored [147]. Long-term studies of ECT's impact on immune cells have indicated that after multiple sessions, certain NK cell subtypes, particularly CD56dimCD16+ cells (which are responsible for cytotoxic activity), decrease in number, while CD56highCD16–/dim cells increase. This shift suggests a reduction in NK cell cytotoxicity, although opposite results are observed within the first 15 minutes post-treatment. Notably, a correlation was found between the ratio of these NK cell subtypes before ECT and the treatment outcomes [148]. S100B, a calcium-binding protein produced by astrocytes, plays a key role in protecting neurons from oxidative stress, promoting neuron growth, and regulating cell motility, proliferation, and metabolism [149,150,151]. Arts and colleagues studied the concentration of S100B following ECT and found that its serum levels increased 1 and 3 hours post-treatment. However, similar to some other studies, they observed no long-term changes in S100B levels after multiple ECT sessions [152].
Emerging evidence suggests that immune system modulation, particularly through cytokine changes, plays an important role in the therapeutic effects of ECT in patients with depression. Despite these findings, significant gaps remain in understanding the precise mechanisms through which ECT interacts with the immune system, and further research is necessary to explore these connections more thoroughly. Future studies could explore how ECT modulates immune cell signaling pathways, cytokine receptor expression, and intracellular processes within immune cells. Specifically, how ECT influences the balance between pro-inflammatory and anti-inflammatory responses could provide insight into its therapeutic effects.
4.3. Mitochondrial Function and Energy Metabolism During ECT
Mitochondria are dynamic organelles that create an interconnected network within the cytosol. Their morphology is regulated by the processes of fusion and fission, both of which are essential for maintaining optimal mitochondrial function. Fission, mediated by the dynamin-1-like protein (Drp1), contributes significantly to quality control, while fusion, facilitated by mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy-1 (OPA1), promotes the exchange of mitochondrial components, including proteins, lipids, metabolites, and mitochondrial DNA (mtDNA) [153]. Mitochondria are vital for adenosine triphosphate (ATP) production via the electron transport chain and ATP synthase. The electron transport chain, consisting of complexes I, II, III, and IV in the inner mitochondrial membrane (IMM), creates a proton gradient used by ATP synthase. This process, known as oxidative phosphorylation (OXPHOS), generates reactive oxygen species (ROS) as byproducts [154]. While ROS can act as signaling molecules, excessive amounts may cause protein and lipid oxidation, triggering autophagy, apoptosis, necrosis, and inflammation [155]. Mitochondria produce ATP for Na+-K+-ATPase, crucial for maintaining neuronal membrane potential and regulating Ca2+ during synaptic transmission [156].
Mitochondrial dysfunction can lead to neurodegenerative and neuropsychiatric disorders [153], while the relationship between depression and mitochondrial dysfunction has been already established. Initially, depression was supposed to be among the first symptoms of mitochondrial diseases or mutations of mitochondrial or mitochondrion-related genes associated with MDD [157]. Previous research suggests that abnormalities in mitochondrial morphology and function are deeply associated with neuronal function and mood disorders [158]. In a study by Gebara and colleagues, high-anxious rats had more severe depression-like behavior, along with larger mitochondria area and mitochondria tissue coverage and higher number of mitochondria-mitochondria contacts in the medium spiny neurons from the nucleus accumbens [159]. Wu and colleagues demonstrated that the prenatal exposure to dexamethasone leads to depression-like behavior and mitochondrial damage in hippocampus [160]. Furthermore, in an animal model of depression, depressive-like symptoms in mice were accompanied with reduced mitochondrial respiratory rates and a dissipated mitochondrial membrane potential in the hippocampus, cortex, and hypothalamus [161]. This suggests that depression may be associated with a disruption in brain energy metabolism due to mitochondrial genetic vulnerability and environmental influence [162]. One recent meta-analysis reported higher mtDNA concentration in circulating blood samples and skin fibroblasts in depressive patients in comparison to healthy individuals, suggesting a potential association between depression and amount of mtDNA [163]. In patients with confirmed mitochnodrial diseases due to mitochondrial gene mutations, the prevalence of depression was estimated to be 54% [164]. More than 20 years ago, Gardner and colleagues reported that 68% of depressive patients have mtDNA deletions, in comparison to 36% of non-depressive individuals [165], which could be at least partially explained by activation of inflammatory processes resulting from damaged mtDNA [166,167]. Cases of MDD exhibited rare homoplasmic mutations that may have functional implications in the ATP synthase 8 (ATP8), ATP synthase 6 (ATP6), ND5, and cytochrome b (CYTB) genes [168]. Additionally, patient with depression displayed a subthreshold heteroplasmy rate at a variant located in the displacement loop (D-loop) region of mtDNA [168].
ECS in animal models has been associated with changes in mitochondrial morphology, including fission and fusion dynamics, highlighting the intricate effects of ECT on mitochondrial regulation [169]. Collectively, these findings suggest that ECT may modulate brain metabolism through its effects on mitochondrial enzymes [170]. The interplay between ECT and mitochondrial function is complex and not yet fully elucidated. A deeper understanding of how ECT influences mitochondrial function could lead to optimized treatment protocols and the development of novel therapeutic strategies targeting mitochondrial pathways in patients with mood disorders.
4.4. Oxidative Stress and ECT
The brain, which accounts for more than 20% of the total oxygen consumption in the body, relies on oxygen for neuronal function and survival. However, while oxygen is s essential for cellular metabolism, its byproducts, including ROS and reactive nitrogen species (RNS), can exert neurotoxic effects when produced in excess [171]. Oxidative stress (OS) is defined as an imbalance between the generation of ROS/RNS and the capacity of the antioxidant defense system, leading to cellular damage, particularly in proteins, lipids, and DNA. Although OS plays a crucial role in maintaining physiological homeostasis, disruptions in redox signaling have been implicated in the onset and progression of various disorders [172]. Excessive ROS accumulation can interfere with neuronal signaling, disrupt cellular integrity, and impair brain function [173]. Furthermore, oxidative injury exposes molecular patterns known as danger-associated molecular patterns (DAMPs), which activate innate immune responses and sterile inflammation in the brain. This inflammatory cascade amplifies the production of proinflammatory cytokines, linking OS and neuroinflammation to the pathophysiology of depression [173].
The brain is particularly vulnerable to OS due to its high metabolic demand, the presence of highly peroxidizable substrates, and relatively low levels of endogenous antioxidants [174]. Increasing evidence suggests that neuroinflammation and OS, known for their roles in neurodegenerative diseases and aging, also contribute to the development of MDD, which is characterized by its multifactorial nature, neuroprogressive aspects, accelerated cellular aging, and a heightened risk of related age-related illnesses [175]. Elevated levels of OS biomarkers, such as 8-hydroxydeoxyguanosine and malondialdehyde - a byproduct of the peroxidation of polyunsaturated fatty acids and arachidonic acid - indicate oxidative DNA damage [176]. These biomarkers, together with a reduction in antioxidant enzyme activities, are commonly observed in MDD patients [176]. The "OS hypothesis of depressive disorders" posits that excessive ROS generation and the depletion of antioxidant defenses contribute to structural alterations in the brain [177]. A review by Ait Tayeb and colleagues demonstrated that increased plasma hydrogen peroxide (H2O2) levels were associated with MDD, while nitric oxide (NO) concentrations showed more variability, with both elevated serum levels and decreased erythrocyte levels observed in MDD patients [178]. Additionally, studies investigating superoxide dismutase (SOD) expression and its activity in serum, plasma, and erythrocytes have yielded inconsistent results, with some studies reporting increases in SOD activity and others showing reductions or no significant differences [178]. One recent meta-analysis indicated increased catalase (CAT) activity in MDD patients [179], which may reflect a compensatory mechanism to mitigate ROS accumulation [178].
Inconsistent results have also been reported regarding lipid oxidative damage, with some studies identifying increased lipid peroxidation in the serum and erythrocytes of MDD patients, while others found no significant differences [180]. Nevertheless, recent research by Bader and colleagues has highlighted the potential of integrating OS biomarkers with clinical and sociodemographic features to improve depression detection and severity assessment using machine learning techniques [181]. This underscores the importance of OS in understanding the pathophysiology of MDD and its potential as a reliable biomarker for personalized treatment strategies.
Alterations in redox balance are increasingly recognized as central to neuroplasticity and neuronal health, which are thought to underlie the therapeutic effects of ECT (REF). The impact of ECT on OS markers has been explored in both human and animal models, though findings remain variable [180,182,183,184]. A systematic review of 11 human studies and 9 animal studies found inconsistent results regarding the influence of ECT on OS markers in circulating blood samples, suggesting that no clear association exists between ECT and OS in psychiatric disorders [180]. For instance, Şahin and colleagues reported lower total antioxidant levels in MDD patients prior to ECT, with a significant increase in antioxidant levels following ECT treatment [183]. Some studies suggest that while ECT may reduce nitrosative stress, it could concurrently induce oxidative DNA damage, highlighting the complex interplay between oxidative and nitrosative stress in ECT’s mechanisms of action [185].
Research in rodent models has produced mixed findings as well. Barichello and colleagues reported a decrease in oxidative damage markers, such as thiobarbituric acid-reactive substances (TBARS) and protein carbonyls, in the hippocampus following single or repeated electroconvulsive shock (ECS) [182]. The same group also observed reduced oxidative damage in the hippocampus, striatum, and cerebellum, but an increase in oxidative damage in the cortex after ECS [186]. Conversely, Zupan and colleagues reported increases in hippocampal and cerebellar SOD and glutathione peroxidase (GPX) activities following single ECS-induced seizures [187]. However, other studies demonstrated decreases in SOD and GPX activity across various brain regions, with the reduction in antioxidant enzyme activity persisting for up to 48 hours post-stimulation [188]. More recently, a decrease in mitochondrial respiration and an increase in RNA oxidation were observed in rat brain tissue after chronic ECS [189], suggesting that this treatment induces an increased OS, which may drive both therapeutic and potentially neurotoxic effects of ECT.
The variability in these findings highlights the complexity of the relationship between ECT and OS. Factors such as differences in experimental designs, sample sizes, methodologies for assessing OS markers, and patient populations likely contribute to these inconsistencies. Further research, employing standardized methodologies and larger sample sizes, is needed to clarify the precise relationship between ECT and OS. Understanding this relationship may offer critical insights into the mechanisms underlying ECT’s therapeutic effects and inform strategies to mitigate OS-related side effects.
4.5. Apoptosis and ECT
Apoptosis is a highly regulated form of programmed cell death that plays a crucial role in development, cellular homeostasis, and the response to various forms of cellular stress. Unlike most other organs, the nervous system exhibits limited neuronal cell division and proliferation following embryonic development. During early stages, an overproduction of neural precursor cells (NPCs) occurs, and excess cells are subsequently eliminated through apoptosis, which is essential for refining neural connectivity and establishing proper brain function. As the nervous system matures, apoptosis thresholds increase significantly, reducing the rate of neuronal cell death and promoting the long-term survival of neurons within a stable, fully integrated neural network [190].
Disruption of apoptotic regulation is associated with various neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and Huntington's disease [191], as well as psychiatric disorders, including depression [192]. Kondratyev and colleagues previously reported that exposure to minimal ECS markedly reduced vulnerability to the neuronal cell death triggered by status epilepticus in rats due to reduced internucleosomal DNA fragmentation and decrease of apoptosis-like neuronal morphology in hippocampus and rhinal cortex [193]. On contrary, Zarubenco and colleagues demonstrated significant level of neuron death in particular parts of the mouse hippocampus following ECS. However, authors could not interprete does the neuronal loss occurs due to apoptotic or necrotic processes [194].
One clinical study investigated the levels of serum biomarkers of neuronal injury and astrocytic reactivity in patients with major depressive episode undergone to acute ECT [195]. Although authors confirmed the a temporary increase in serum GFAP suggesting astrocytic reactivity, no evidence of neuronal injury has been observed, while biomarkers such as NfL and t-tau remained unchanged during ECT [195]. On the other hand, ECT was not associated with alterations in E2F transcription factors, a group of proteins involved in different cell functions including apoptosis and cell proliferation [196]. Although E2F1 mRNA levels were significantly lower in peripherial blood of depressed patients in comparison to healthy individuals, ECT did not affect the baseline values [196]. Ito and colleagues investigated the effects of different number of ECS (single, 10, or 20 applications) on cell proliferation and apoptosis in the subgranular zone of the DG [107]. While the application of a single or 10 ECSs increased cell proliferation in the observed region, no difference in cell proliferation has been observed after 20 ECSs in comparison to control animals [107]. One of the proposed mechanisms for potential antiapoptotic actions of repeated ECT could be related to c-Myc down-regulation via ubiquitination-proteasomal degradation and Bad inactivation in the rat FC [197].
5. Future Directions
From a molecular perspective, ECT exerts profound and multifaceted effects on the brain, modulating key neurobiological systems, as neurotransmitter regulation, synaptic plasticity, neurogenesis, inflammation, oxidative stress, and apoptosis. These changes contribute to the therapeutic effects of ECT, particularly in mood disorders like MDD, by promoting neuronal survival, enhancing synaptic connectivity, and fostering neuroplasticity. Although the precise mechanisms remain to be fully elucidated, accumulating scientific evidence strongly supports the notion that ECT induces a coordinated molecular response that not only restores neurochemical balance but also fosters neural regeneration and reorganization, thereby alleviating psychiatric symptoms. This underscores the potential of ECT as a powerful therapeutic intervention, with molecular pathways playing a critical role in its antidepressant effects.
Author Contributions
E.F., N.M. and M.M. performed formal analysis and data curation; writing review and editing were performed by E.F., N.M. and M.M.; visualization was provided by V.J.; B.R.; M.F.; N.J. and D.S. V.J. and G.R. performed conceptualization, managed resources, and supervised. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
Figure 1 was created using Biorender.com.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marx, W.; Penninx, B.W.; Solmi, M.; Furukawa, T.A.; Firth, J.; Carvalho, A.F.; Berk, M. Major depressive disorder. Nat. Rev. Dis. Primers 2023., 9(1), p.44.
- Institute for Health Metrics and Evaluation. Global Burden of Disease Study 2017 (GBD 2017) Data Resources [Internet]. Seattle, WA: IHME; 2019.
- Collaborators; G.B.D.; 2019. Mental Disorders, et al.(2022) Global, regional, and national burden of 12 mental disorders in 204 countries and territories, 1990–2019: A systematic analysis for the global burden of disease study 2019. The Lancet Psychiatry, 9(2), pp.137-150.
- Mathers, C. The global burden of disease: 2004 update. World Health Organization: Geneva, Switzerland. 2008.; pp. 7-49.
- Abe, Y.; Erchinger, V.J.; Ousdal, O.T.; Oltedal, L.; Tanaka, K.F.; Takamiya, A. Neurobiological mechanisms of electroconvulsive therapy for depression: Insights into hippocampal volumetric increases from clinical and preclinical studies. J. Neurochem. 2024., 168(9), pp.1738-1750. [CrossRef]
- Espinoza, R.T.; Kellner, C.H. Electroconvulsive therapy. NEJM. 2022., 386(7), pp.667-672.
- Spaans, H.P.; Sienaert, P.; Bouckaert, F.; van den Berg, J.F.; Verwijk, E.; Kho, K.H.; Stek, M.L.; Kok, R.M. Speed of remission in elderly patients with depression: electroconvulsive therapy v. medication. Br. J. Psychiatry 2015., 206(1), pp.67-71.
- Kellner, C.H.; Fink, M.; Knapp, R.; Petrides, G.; Husain, M.; Rummans, T.; Mueller, M.; Bernstein, H.; Rasmussen, K.; O’Connor, K.;Smith, G. Relief of expressed suicidal intent by ECT: a consortium for research in ECT study. Am. J. Psychiatry 2005., 162(5), pp.977-982. [CrossRef]
- Trifu, S.; Sevcenco, A.; Stănescu, M.; Drăgoi, A.M.; Cristea, M.B. Efficacy of electroconvulsive therapy as a potential first-choice treatment in treatment-resistant depression. Exp. Ther. Med. 2021., 22(5), p.1281.
- UK Ect Review Group. Efficacy and safety of electroconvulsive therapy in depressive disorders: a systematic review and meta-analysis. Lancet 2003., 361(9360), pp.799-808. [CrossRef]
- Kritzer, M.D.; Peterchev, A.V.; Camprodon, J.A. Electroconvulsive therapy: mechanisms of action, clinical considerations, and future directions. Harv. Rev. Psychiatry 2023., 31(3), pp.101-113. [CrossRef]
- McClintock, S.M.; Brandon, A.R.; Husain, M.M.; Jarrett, R.B. A systematic review of the combined use of electroconvulsive therapy and psychotherapy for depression.J. ECT. 2011., 27(3), pp.236-243. [CrossRef]
- Chen, J.J.; Zhao, L.B.; Liu, Y.Y.; Fan, S.H.; and Xie, P. Comparative efficacy and acceptability of electroconvulsive therapy versus repetitive transcranial magnetic stimulation for major depression: a systematic review and multiple-treatments meta-analysis. Behav. Brain Res. 2017., 320, pp.30-36. [CrossRef]
- Kumar, S.; Mulsant, B.H.; Liu, A.Y.; Blumberger, D.M.; Daskalakis, Z.J.; Rajji, T.K. Systematic review of cognitive effects of electroconvulsive therapy in late-life depression. Am. J. Geriatr. Psychiatry 2016., 24(7), pp.547-565. [CrossRef]
- Karabatsiakis, A.; Schönfeldt-Lecuona, C. Depression, mitochondrial bioenergetics, and electroconvulsive therapy: a new approach towards personalized medicine in psychiatric treatment-a short review and current perspective. Transl. Psychiatry 2020., 10(1), p.226.
- Kellner, C.H.; Husain, M.M.; Knapp, R.G.; McCall, W.V.; Petrides, G.; Rudorfer, M.V.; Young, R.C.; Sampson, S.; McClintock, S.M.; Mueller, M.; Prudic, J. Right unilateral ultrabrief pulse ECT in geriatric depression: phase 1 of the PRIDE study. Am. J. Psychiatry 2016., 173(11), pp.1101-1109. [CrossRef]
- McCall, W.V.; Lisanby, S.H.; Rosenquist, P.B.; Dooley, M.; Husain, M.M.; Knapp, R.G.; Petrides, G.; Rudorfer, M.V.; Young, R.C.; McClintock, S.M.; Mueller, M.; Effects of continuation electroconvulsive therapy on quality of life in elderly depressed patients: A randomized clinical trial. J. Psychiatr. Res 2018., 97, pp.65-69. [CrossRef]
- Liang, C.S.; Chung, C.H.; Tsai, C.K.; Chien, W.C. In-hospital mortality among electroconvulsive therapy recipients: a 17-year nationwide population-based retrospective study. Eur. psychiatr. 2017., 42, pp.29-35. [CrossRef]
- Liang, C.S.; Chung, C.H.; Ho, P.S.; Tsai, C.K.; Chien, W.C. Superior anti-suicidal effects of electroconvulsive therapy in unipolar disorder and bipolar depression. Bipolar Disord. 2018., 20(6), pp.539-546. [CrossRef]
- Ryan, K.M.; McLoughlin, D.M. From Molecules to Mind: Mechanisms of Action of Electroconvulsive Therapy. FOCUS, Am. J. Psychiatry 2019., 17(1), pp.73-75. [CrossRef]
- Miller, E. Psychological theories of ECT: a review. Br. J. Psychiatry 1967., 113(496), pp.301-311.
- Cameron, D.E. Production of differential amnesia as a factor in the treatment of schizophrenia. Compr. Psychiatry 1960., 1(1), pp.26-34.
- Netto, C.A.; Izquierdo, I. Amnesia as a major side effect of electroconvulsive shock: the possible involvement of hypothalamic opioid systems. Braz. J. Med. Biol. Res. 1984., 17(3-4), pp.349-351.
- Bassa, A.; Sagués, T.; Porta-Casteràs, D.; Serra, P.; Martínez-Amorós, E.; Palao, D.J.; Cano, M.; Cardoner, N. The neurobiological basis of cognitive side effects of electroconvulsive therapy: a systematic review. Brain Sci. 2021., 11(10), p.1273. [CrossRef]
- Sackeim, H.A.; Prudic, J.; Devanand, D.P.; Kiersky, J.E.; Fitzsimons, L.; Moody, B.J.; McElhiney, M.C.; Coleman, E.A.; Settembrino, J.M. Effects of stimulus intensity and electrode placement on the efficacy and cognitive effects of electroconvulsive therapy. N. Engl. J. Med. 1993., 328(12), pp.839-846. [CrossRef]
- Sienaert, P.; Vansteelandt, K.; Demyttenaere, K.; Peuskens, J. Randomized comparison of ultra-brief bifrontal and unilateral electroconvulsive therapy for major depression: cognitive side-effects. J. Affect. Disord. 2010., 122(1-2), pp.60-67. [CrossRef]
- Farzan, F.; Boutros, N.N.; Blumberger, D.M.; Daskalakis, Z.J. What does the electroencephalogram tell us about the mechanisms of action of ECT in major depressive disorders?. J. ECT. 2014., 30(2), pp.98-106. [CrossRef]
- Duthie, A.C.; Perrin, J.S.; Bennett, D.M.; Currie, J.; Reid, I.C. Anticonvulsant mechanisms of electroconvulsive therapy and relation to therapeutic efficacy. J ECT. 2015, 31(3), pp.173-178. [CrossRef]
- Knudsen, M.K.; Near, J.; Blicher, A.B.; Videbech, P.; Blicher, J.U. Magnetic resonance (MR) spectroscopic measurement of γ-aminobutyric acid (GABA) in major depression before and after electroconvulsive therapy. Acta Neuropsychiatr. 2019, 31(1), pp.17-26. [CrossRef]
- Deng, Z.D.; Robins, P.L.; Regenold, W.; Rohde, P.; Dannhauer, M.; Lisanby, S.H. How electroconvulsive therapy works in the treatment of depression: is it the seizure, the electricity, or both?. Neuropsychopharmacol. 2024., 49(1), pp.150-162. [CrossRef]
- Altar, C.A.; Laeng, P.; Jurata, L.W.; Brockman, J.A.; Lemire, A.; Bullard, J.; Bukhman, Y.V.; Young, T.A.; Charles, V.; Palfreyman, M.G. Electroconvulsive seizures regulate gene expression of distinct neurotrophic signaling pathways. J. Neurosci. 2004., 24(11), pp.2667-2677. [CrossRef]
- Newton, S.S.; Collier, E.F.; Hunsberger, J.; Adams, D.; Terwilliger, R.; Selvanayagam, E.; Duman, R.S. Gene profile of electroconvulsive seizures: induction of neurotrophic and angiogenic factors. J. Neurosci. 2003., 23(34), pp.10841-10851. [CrossRef]
- Perera, T.D.; Coplan, J.D.; Lisanby, S.H.; Lipira, C.M.; Arif, M.; Carpio, C.; Spitzer, G.; Santarelli, L.; Scharf, B.; Hen, R.; Rosoklija, G. Antidepressant-induced neurogenesis in the hippocampus of adult nonhuman primates. J. Neurosci. 2007., 27(18), pp.4894-4901. [CrossRef]
- Haskett, R.F. Electroconvulsive therapy’s mechanism of action: neuroendocrine hypotheses. J. ECT.2014., 30(2), pp.107-110.
- Eşel, E.; Baştürk, M.; Kula, M.; Reyhancan, M.; Turan, M.T.; Sofuoğlu, S. Effects of electroconvulsive therapy on pituitary hormones in depressed patients. Klin. Psikofarmakol. B. 2003., 13, pp.109-117.
- Burgese, D.F.; Bassitt, D.P. Variation of plasma cortisol levels in patients with depression after treatment with bilateral electroconvulsive therapy. Trends Psychiatry Psychother. 2015., 37(1), pp.27-36. [CrossRef]
- Bouckaert, F.; Sienaert, P.; Obbels, J.; Dols, A.; Vandenbulcke, M.; Stek, M.; Bolwig, T. ECT: its brain enabling effects: a review of electroconvulsive therapy–induced structural brain plasticity. J. ECT. 2014., 30(2), pp.143-151.
- Madsen, T.M.; Treschow, A.; Bengzon, J.; Bolwig, T.G.; Lindvall, O.; Tingström, A. Increased neurogenesis in a model of electroconvulsive therapy. Biol. Psychiatry 2000., 47(12), pp.1043-1049. [CrossRef]
- Shahin, O.; Gohar, S.M.; Ibrahim, W.; El-Makawi, S.M.; Fakher, W.; Taher, D.B.; Abdel Samie, M.; Khalil, M.A.; Saleh, A.A. Brain-Derived neurotrophic factor (BDNF) plasma level increases in patients with resistant schizophrenia treated with electroconvulsive therapy (ECT). Int J Psychiat Clin. 2022., 26(4), pp.370-375. [CrossRef]
- Kato, N. Neurophysiological mechanisms of electroconvulsive therapy for depression. Neurosci. Res. 2009., 64(1), pp.3-11. [CrossRef]
- Baldinger, P.; Lotan, A.; Frey, R.; Kasper, S.; Lerer, B.; Lanzenberger, R. Neurotransmitters and electroconvulsive therapy. J. ECT. 2014., 30(2), pp.116-121.
- Yatham, L.N.; Liddle, P.F.; Lam, R.W.; Zis, A.P.; Stoessl, A.J.; Sossi, V.; Adam, M.J.; Ruth, T.J. Effect of electroconvulsive therapy on brain 5-HT2 receptors in major depression. Br. J. Psychiatry 2010., 196(6), pp.474-479. [CrossRef]
- Cojocaru, A.M.; Vasile, A.I.; Trifu, S.C. Neurobiological mechanisms and therapeutic impact of electroconvulsive therapy (ECT). Rom. J. Morphol. Embryology 2024., 65(1), p.13. [CrossRef]
- Landau, A.M.; Phan, J.A.; Iversen, P.; Lillethorup, T.P.; Simonsen, M.; Wegener, G.; Jakobsen, S.; Doudet, D.J. Decreased in vivo α2 adrenoceptor binding in the Flinders Sensitive Line rat model of depression. Neuropharmacol. 2015., 91, pp.97-102. [CrossRef]
- Lillethorup, T.P.; Iversen, P.; Wegener, G.; Doudet, D.J.M.; Landau, A.M. α2-adrenoceptor binding in Flinders-sensitive line compared with Flinders-resistant line and Sprague-Dawley rats. Acta Neuropsyciatr. 2015., 27(6), pp.345-352. [CrossRef]
- Kobayashi, K.; Imoto, Y.; Yamamoto, F.; Kawasaki, M.; Ueno, M.; Segi-Nishida, E.;Suzuki, H. Rapid and lasting enhancement of dopaminergic modulation at the hippocampal mossy fiber synapse by electroconvulsive treatment. J. Neurophysiol. 2017., 117(1), pp.284-289. [CrossRef]
- Dai, X.; Zhang, R.; Deng, N.; Tang, L.; Zhao, B. Anesthetic Influence on Electroconvulsive Therapy: A Comprehensive Review. Neuropsychiatr. Dis. Treat. 2024., pp.1491-1502. [CrossRef]
- Markianos, M.; Hatzimanolis, J.; Lykouras, L. Relationship between prolactin responses to ECT and dopaminergic and serotonergic responsivity in depressed patients.Eur. Arch. Psychiatry Clin. Neurosci. 2002., 252, pp.166-171. [CrossRef]
- Burnet, P.W.; Sharp, T.; LeCorre, S.M.; Harrison, P.J. Expression of 5-HT receptors and the 5-HT transporter in rat brain after electroconvulsive shock. Neurosci. Lett. 1999., 277(2), pp.79-82. [CrossRef]
- Newman, M.E.; Gur, E.; Shapira, B.; Lerer, B. Neurochemical mechanisms of action of ECS: evidence from in vivo studies. J. ECT. 1998., 14(3), pp.153-171.
- Burnet, P.W.J.; Mead, A.; Eastwood, S.L.; Lacey, K.; Harrison, P.J.; Sharp, T. Repeated ECS differentially affects rat brain 5-HT1A and 5-HT2A receptor expression. Neuroreport 1995., 6(6), pp.901-904. [CrossRef]
- Chaput, Y.; de Montigny, C.; Blier, P. Presynaptic and postsynaptic modifications of the serotonin system by long-term administration of antidepressant treatments. An in vivo electrophysiologic study in the rat. Neuropsychopharmacol. 1991., 5(4), pp.219-229.
- Strome, E.M.; Clark, C.M.; Zis, A.P.; Doudet, D.J. Electroconvulsive shock decreases binding to 5-HT2 receptors in nonhuman primates: an in vivo positron emission tomography study with [18F] setoperone. Biol. Psychiatry. 2005., 57(9), pp.1004-1010. [CrossRef]
- Lanzenberger, R.; Baldinger, P.; Hahn, A.; Ungersboeck, J.; Mitterhauser, M.; Winkler, D.; Micskei, Z.; Stein, P.; Karanikas, G.; Wadsak, W.; Kasper, S. Global decrease of serotonin-1A receptor binding after electroconvulsive therapy in major depression measured by PET. Mol. Psychiatry 2013., 18(1), pp.93-100. [CrossRef]
- Saijo, T.; Takano, A.; Suhara, T.; Arakawa, R.; Okumura, M.; Ichimiya, T.; Ito, H.; Okubo, Y. Effect of electroconvulsive therapy on 5-HT1A receptor binding in patients with depression: a PET study with [11C] WAY 100635. Int. J. Neuropsychopharmacol. 2010., 13(6), pp.785-791. [CrossRef]
- Gray, J.A.; Roth, B.L. Paradoxical trafficking and regulation of 5-HT2A receptors by agonists and antagonists. Brain Res. Bull. 2001., 56(5), pp.441-451. [CrossRef]
- Meyer, J.H.; Kapur, S.; Eisfeld, B.; Brown, G.M.; Houle, S.; DaSilva, J.; Wilson, A.A.; Rafi-Tari, S.; Mayberg, H.S.; Kennedy, S.H. The effect of paroxetine on 5-HT2A receptors in depression: an [18F] setoperone PET imaging study. Am. J. Psychiatry 2001., 158(1), pp.78-85.
- Nikisch, G.; Mathé, A.A. CSF monoamine metabolites and neuropeptides in depressed patients before and after electroconvulsive therapy. Eur. psychiatr. 2008., 23(5), pp.356-359. [CrossRef]
- Okamoto, T.; Yoshimura, R.; Ikenouchi-Sugita, A.; Hori, H.; Umene-Nakano, W.; Inoue, Y.; Ueda, N.; Nakamura, J. Efficacy of electroconvulsive therapy is associated with changing blood levels of homovanillic acid and brain-derived neurotrophic factor (BDNF) in refractory depressed patients: a pilot study. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008., 32(5), pp.1185-1190. [CrossRef]
- Saijo, T.; Takano, A.; Suhara, T.; Arakawa, R.; Okumura, M.; Ichimiya, T.; Ito, H.; Okubo, Y. Electroconvulsive therapy decreases dopamine D2 receptor binding in the anterior cingulate in patients with depression: a controlled study using positron emission tomography with radioligand [11C] FLB 457. J. Clin. Psychiatry 2010., 71(6), p.793.
- Landau, A.M.; Chakravarty, M.M.; Clark, C.M.; Zis, A.P.; Doudet, D.J. Electroconvulsive therapy alters dopamine signaling in the striatum of non-human primates. Neuropsychopharmacol. 2011., 36(2), pp.511-518. [CrossRef]
- Lammers, C.H.; Diaz, J.; Schwartz, J.C.; Sokoloff, P. Selective increase of dopamine D3 receptor gene expression as a common effect of chronic antidepressant treatments. Mol. Psychiatry 2000., 5(4), pp.378-388. [CrossRef]
- Huuhka, K.; Anttila, S.; Huuhka, M.; Hietala, J.; Huhtala, H.; Mononen, N.; Lehtimäki, T.;Leinonen, E. Dopamine 2 receptor C957T and catechol-o-methyltransferase Val158Met polymorphisms are associated with treatment response in electroconvulsive therapy. Neurosci. Lett. 2008., 448(1), pp.79-83. [CrossRef]
- Kask, A.; Harro, J.; von Hörsten, S.; Redrobe, J.P.; Dumont, Y.; Quirion, R. The neurocircuitry and receptor subtypes mediating anxiolytic-like effects of neuropeptide Y. Neurosci. Biobehav. Rev. 2002., 26(3), pp.259-283. [CrossRef]
- Ozsoy, S.; Eker, O.O.; Abdulrezzak, U. The effects of antidepressants on neuropeptide Y in patients with depression and anxiety. Pharmacopsychiatry 2016., 49(01), pp.26-31. [CrossRef]
- Lillethorup, T.P.; Iversen, P.; Fontain, J.; Wegener, G.; Doudet, D.J.; Landau, A.M. Electroconvulsive shocks decrease α2-adrenoceptor binding in the Flinders rat model of depression. Eur. Neuropsychopharmacol. 2015., 25(3), pp.404-412. [CrossRef]
- Mann, J.J.; Manevitz, A.Z.; Chen, J.S.; Johnson, K.S.; Adelsheimer, E.F.; Azima-Heller, R.; Massina, A.; Wilner, P.J. Acute effects of single and repeated electroconvulsive therapy on plasma catecholamines and blood pressure in major depressive disorder. Psychiatry Res. 1990., 34(2), pp.127-137. [CrossRef]
- Ambade, V.; Arora, M.M.; Singh, P.; Somani, B.L.; Basannar, D. Adrenaline, noradrenaline and dopamine level estimation in depression: does it help?. Med. J. Armed Forces India 2009., 65(3), pp.216-220. [CrossRef]
- Kelly, C.B.; Cooper, S.J. Plasma noradrenaline response to electroconvulsive therapy in depressive illness. Br. J. Psychiatry 1997., 171(2), pp.182-186. [CrossRef]
- Pollak, C.; Maier, H.B.; Moschny, N.; Jahn, K.; Bleich, S.; Frieling, H.; Neyazi, A. Epinephrine levels decrease in responders after electroconvulsive therapy. J. Neural. Transm. 2021., 128, pp.1917-1921. [CrossRef]
- El Mansari, M.; Guiard, B.P.; Chernoloz, O.; Ghanbari, R.; Katz, N.; Blier, P. Relevance of norepinephrine–dopamine interactions in the treatment of major depressive disorder. CNS Neurosci. Ther. 2010., 16(3), pp.e1-e17. [CrossRef]
- Dong, J.; Min, S.; Wei, K.; Li, P.; Cao, J.; Li, Y. Effects of electroconvulsive therapy and propofol on spatial memory and glutamatergic system in hippocampus of depressed rats.J. ECT. 2010., 26(2), pp.126-130. [CrossRef]
- Sartorius, A.; Mahlstedt, M.M.; Vollmayr, B.; Henn, F.A.; Ende, G.; Elevated spectroscopic glutamate/γ-amino butyric acid in rats bred for learned helplessness. Neuroreport 2007., 18(14), pp.1469-1473. [CrossRef]
- Hashimoto, K.; Sawa, A.; Iyo, M.; Increased levels of glutamate in brains from patients with mood disorders. Biol. Psychiatry. 2007., 62(11), pp.1310-1316. [CrossRef]
- Hasler, G.; van der Veen, J.W.; Tumonis, T.; Meyers, N.; Shen, J.; Drevets, W.C. Reduced prefrontal glutamate/glutamine and γ-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry. 2007., 64(2), pp.193-200. [CrossRef]
- Zhang, J.; Narr, K.L.; Woods, R.P.; Phillips, O.R.; Alger, J.R.; Espinoza, R.T. Glutamate normalization with ECT treatment response in major depression. Mol. Psychiatry 2013., 18(3), pp.268-270. [CrossRef]
- Njau, S.; Joshi, S.H.; Espinoza, R.; Leaver, A.M.; Vasavada, M.; Marquina, A.; Woods, R.P.; Narr, K.L. Neurochemical correlates of rapid treatment response to electroconvulsive therapy in patients with major depression. J. Psychiatry Neurosci. 2017., 42(1), pp.6-16. [CrossRef]
- Pfleiderer, B.; Michael, N.; Erfurth, A.; Ohrmann, P.; Hohmann, U.; Wolgast, M.; Fiebich, M.; Arolt, V.; Heindel, W. Effective electroconvulsive therapy reverses glutamate/glutamine deficit in the left anterior cingulum of unipolar depressed patients. Psychiatry Res. Neuroimaging 2003., 122(3), pp.185-192. [CrossRef]
- Erchinger, V.J.; Craven, A.R.; Ersland, L.; Oedegaard, K.J.; Bartz-Johannessen, C.A.; Hammar, Å.; Haavik, J.; Riemer, F.; Kessler, U.; Oltedal, L. Electroconvulsive therapy triggers a reversible decrease in brain N-acetylaspartate. Front. Psychiatry 2023., 14, p.1155689. [CrossRef]
- Puderbaugh, M.; Emmady, P.D. Neuroplasticity. StatPearls Publishing: Treasure Island, Florida, 2023., pp 3-9.
- Joshi, S.H.; Espinoza, R.T.; Pirnia, T.; Shi, J.; Wang, Y.; Ayers, B.; Leaver, A.; Woods, R.P.; Narr, K.L. 2016. Structural plasticity of the hippocampus and amygdala induced by electroconvulsive therapy in major depression. Biol. Psychiatry.2016., 79(4), pp.282-292. [CrossRef]
- Nordanskog, P.; Dahlstrand, U.; Larsson, M.R.; Larsson, E.M.; Knutsson, L.; Johanson, A. Increase in hippocampal volume after electroconvulsive therapy in patients with depression: a volumetric magnetic resonance imaging study. J. ECT. 2010., 26(1), pp.62-67.
- Jorgensen, A.; Magnusson, P.; Hanson, L.G.; Kirkegaard, T.; Benveniste, H.; Lee, H.; Svarer, C.; Mikkelsen, J.D.; Fink-Jensen, A.; Knudsen, G.M.; Paulson, O.B. Regional brain volumes, diffusivity, and metabolite changes after electroconvulsive therapy for severe depression. Acta Psychiatr. Scand. 2016., 133(2), pp.154-164. [CrossRef]
- Bouckaert, F.; Dols, A.; Emsell, L.; De Winter, F.L.; Vansteelandt, K.; Claes, L.; Sunaert, S.; Stek, M.; Sienaert, P.; Vandenbulcke, M. Relationship between hippocampal volume, serum BDNF, and depression severity following electroconvulsive therapy in late-life depression. Neuropsychopharmacol. 2016., 41(11), pp.2741-2748. [CrossRef]
- Sartorius, A.; Demirakca, T.; Böhringer, A.; von Hohenberg, C.C.; Aksay, S.S.; Bumb, J.M.; Kranaster, L.; Nickl-Jockschat, T.; Grözinger, M.; Thomann, P.A.; Wolf, R.C.; Electroconvulsive therapy induced gray matter increase is not necessarily correlated with clinical data in depressed patients. Brain Stimul. 2019., 12(2), pp.335-343. [CrossRef]
- Camilleri, J.A.; Hoffstaedter, F.; Zavorotny, M.; Zöllner, R.; Wolf, R.C.; Thomann, P.; Redlich, R.; Opel, N.; Dannlowski, U.; Groezinger, M.; Demirakca, T. Electroconvulsive therapy modulates grey matter increase in a hub of an affect processing network. Neuroimage Clin. 2020., 25, p.102114. [CrossRef]
- van Oostrom, I.; van Eijndhoven, P.; Butterbrod, E.; van Beek, M.H.; Janzing, J.; Donders, R.; Schene, A.; Tendolkar, I. Decreased cognitive functioning after electroconvulsive therapy is related to increased hippocampal volume: exploring the role of brain plasticity. J. ECT.2018., 34(2), pp.117-123.
- Ousdal, O.T.; Brancati, G.E.; Kessler, U.; Erchinger, V.; Dale, A.M.; Abbott, C.; Oltedal, L. The neurobiological effects of electroconvulsive therapy studied through magnetic resonance: what have we learned, and where do we go?.Biol. Psychiatry 2022., 91(6), pp.540-549. [CrossRef]
- Pirnia, T.; Joshi, S.H.; Leaver, A.M.; Vasavada, M.; Njau, S.; Woods, R.P.; Espinoza, R.; Narr, K.L. Electroconvulsive therapy and structural neuroplasticity in neocortical, limbic and paralimbic cortex. Transl. Psychiatry 2016., 6(6), pp.e832-e832. [CrossRef]
- Lyden, H.; Espinoza, R.T.; Pirnia, T.; Clark, K.; Joshi, S.H.; Leaver, A.M.; Woods, R.P.; Narr, K.L. Electroconvulsive therapy mediates neuroplasticity of white matter microstructure in major depression. Transl. Psychiatry 2014., 4(4), pp.e380-e380. [CrossRef]
- Malberg, J.E.; Eisch, A.J.; Nestler, E.J.; Duman, R.S. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000., 20(24), pp.9104-9110. [CrossRef]
- Scott, B.W.; Wojtowicz, J.M.; Burnham, W.M. Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp. Neurol. 2000., 165(2), pp.231-236. [CrossRef]
- Sorri, A.; Järventausta, K.; Kampman, O.; Lehtimäki, K.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E. Effect of electroconvulsive therapy on brain-derived neurotrophic factor levels in patients with major depressive disorder.Brain Behav. 2018., 8(11), p.e01101. [CrossRef]
- Kyeremanteng, C.; MacKay, J.C.; James, J.S.; Kent, P.; Cayer, C.; Anisman, H.; Merali, Z. Effects of electroconvulsive seizures on depression-related behavior, memory and neurochemical changes in Wistar and Wistar–Kyoto rats. Prog Neuropsychopharmacol. Biol. Psychiatry 2014., 54, pp.170-178. [CrossRef]
- Luo, J.; Min, S.; Wei, K.; Cao, J.; Wang, B.; Li, P.; Dong, J.; Liu, Y. Behavioral and molecular responses to electroconvulsive shock differ between genetic and environmental rat models of depression. Psychiatry Res. 2015., 226(2-3), pp.451-460. [CrossRef]
- Shiraishi-Yamaguchi, Y.; Furuichi, T. The Homer family proteins. Genome Biol. 2007., 8, pp.1-12. [CrossRef]
- Hu, J.H.; Park, J.M.; Park, S.; Xiao, B.; Dehoff, M.H.; Kim, S.; Hayashi, T.; Schwarz, M.K.; Huganir, R.L.; Seeburg, P.H.; Linden, D.J. Homeostatic scaling requires group I mGluR activation mediated by Homer1a. Neuron 2010., 68(6), pp.1128-1142. [CrossRef]
- Müller, H.K.; Orlowski, D.; Bjarkam, C.R.; Wegener, G.; Elfving, B. Potential roles for Homer1 and Spinophilin in the preventive effect of electroconvulsive seizures on stress-induced CA3c dendritic retraction in the hippocampus. Eur. Neuropsychopharmacol.2015., 25(8), pp.1324-1331. [CrossRef]
- Serchov, T.; Clement, H.W.; Schwarz, M.K.; Iasevoli, F.; Tosh, D.K.; Idzko, M.; Jacobson, K.A.; de Bartolomeis, A.; Normann, C.; Biber, K.; van Calker, D. Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of Homer1a. Neuron 2015., 87(3), pp.549-562. [CrossRef]
- Wagner, K.V.; Hartmann, J.; Labermaier, C.; Häusl, A.S.; Zhao, G.; Harbich, D.; Schmid, B.; Wang, X.D.; Santarelli, S.; Kohl, C.; Gassen, N.C. Homer1/mGluR5 activity moderates vulnerability to chronic social stress. Neuropsychopharmacol. 2015., 40(5), pp.1222-1233. [CrossRef]
- Li, M.; Yao, X.; Sun, L.; Zhao, L.; Xu, W.; Zhao, H.; Zhao, F.; Zou, X.; Cheng, Z.; Li, B.; Yang, W. Effects of electroconvulsive therapy on depression and its potential mechanism. Front Psychol. 2020., 11, p.80. [CrossRef]
- Pang, Y.; Wei, Q.; Zhao, S.; Li, N.; Li, Z.; Lu, F.; Pang, J.; Zhang, R.; Wang, K.; Chu, C.; Tian, Y. Enhanced default mode network functional connectivity links with electroconvulsive therapy response in major depressive disorder. J. Affect. Disord. 2022., 306, pp.47-54. [CrossRef]
- Abbott, C.C.; Lemke, N.T.; Gopal, S.; Thoma, R.J.; Bustillo, J.; Calhoun, V.D.; Turner, J.A. Electroconvulsive therapy response in major depressive disorder: a pilot functional network connectivity resting state FMRI investigation. Front. Psychiatry. 2013., 4, p.10. [CrossRef]
- Deng, Z.D.; Robins, P.L.; Regenold, W.; Rohde, P.; Dannhauer, M.; Lisanby, S.H. How electroconvulsive therapy works in the treatment of depression: is it the seizure, the electricity, or both?. Neuropsychopharmacol. 2024., 49(1), pp.150-162. [CrossRef]
- Levy, M.J.; Boulle, F.; Steinbusch, H.W.; van den Hove, D.L.; Kenis, G.; Lanfumey, L. Neurotrophic factors and neuroplasticity pathways in the pathophysiology and treatment of depression. Psychopharmacol. 2018., 235, pp.2195-2220. [CrossRef]
- Pardossi, S.; Fagiolini, A.; Cuomo, A. Variations in BDNF and Their Role in the Neurotrophic Antidepressant Mechanisms of Ketamine and Esketamine: A Review. Int. J. Mol. Sci. 2024., 25(23), p.13098. [CrossRef]
- Ito, M.; Seki, T.; Liu, J.; Nakamura, K.; Namba, T.; Matsubara, Y.; Suzuki, T.; Arai, H. Effects of repeated electroconvulsive seizure on cell proliferation in the rat hippocampus. Synapse 2010., 64(11), pp.814-821. [CrossRef]
- Ricken, R.; Adli, M.; Lange, C.; Krusche, E.; Stamm, T.J.; Gaus, S.; Koehler, S.; Nase, S.; Bschor, T.; Richter, C.; Steinacher, B. Brain-derived neurotrophic factor serum concentrations in acute depressive patients increase during lithium augmentation of antidepressants. J. Clin. Psychopharmacol. 2013., 33(6), pp.806-809. [CrossRef]
- Mikoteit, T.; Beck, J.; Eckert, A.; Hemmeter, U.; Brand, S.; Bischof, R.; Holsboer-Trachsler, E.; Delini-Stula, A. High baseline BDNF serum levels and early psychopathological improvement are predictive of treatment outcome in major depression. Psychopharmacology 2014., 231, pp.2955-2965. [CrossRef]
- Nase, S.; Köhler, S.; Jennebach, J.; Eckert, A.; Schweinfurth, N.; Gallinat, J.; Lang, U.E.; Kühn, S. Role of serum brain derived neurotrophic factor and central n-acetylaspartate for clinical response under antidepressive pharmacotherapy. Neurosignals 2018., 24(1), pp.1-14. [CrossRef]
- Yoshimura, R.; Mitoma, M.; Sugita, A.; Hori, H.; Okamoto, T.; Umene, W.; Ueda, N.; Nakamura, J. Effects of paroxetine or milnacipran on serum brain-derived neurotrophic factor in depressed patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2007., 31(5), pp.1034-1037. [CrossRef]
- Yoshimura, R.; Kishi, T.; Hori, H.; Katsuki, A.; Sugita-Ikenouchi, A.; Umene-Nakano, W.; Atake, K.; Iwata, N.; Nakamura, J. Serum levels of brain-derived neurotrophic factor at 4 weeks and response to treatment with SSRIs. Psychiatry Investig. 2014., 11(1), p.84. [CrossRef]
- Tadić, A.; Wagner, S.; Schlicht, K.F.; Peetz, D.; Borysenko, L.; Dreimüller, N.; Hiemke, C.; Lieb, K. The early non-increase of serum BDNF predicts failure of antidepressant treatment in patients with major depression: a pilot study. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011., 35(2), pp.415-420. [CrossRef]
- Dreimüller, N.; Schlicht, K.F.; Wagner, S.; Peetz, D.; Borysenko, L.; Hiemke, C.; Lieb, K.; Tadić, A. Early reactions of brain-derived neurotrophic factor in plasma (pBDNF) and outcome to acute antidepressant treatment in patients with Major Depression. Neuropharmacol. 2012., 62(1), pp.264-269. [CrossRef]
- Karege, F.; Perret, G.; Bondolfi, G.; Schwald, M.; Bertschy, G.; Aubry, J.M. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 2002., 109(2), pp.143-148. [CrossRef]
- Haile, C.N.; Murrough, J.W.; Iosifescu, D.V.; Chang, L.C.; Al Jurdi, R.K.; Foulkes, A.; Iqbal, S.; Mahoney III, J.J.; De La Garza, R.; Charney, D.S.; Newton, T.F. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int. J. Neuropsychopharmacol. 2014., 17(2), pp.331-336.
- Piccinni, A.; Del Debbio, A.; Medda, P.; Bianchi, C.; Roncaglia, I.; Veltri, A.; Zanello, S.; Massimetti, E.; Origlia, N.; Domenici, L.; Marazziti, D. Plasma Brain-Derived Neurotrophic Factor in treatment-resistant depressed patients receiving electroconvulsive therapy. Eur. Neuropsychopharmacol. 2009., 19(5), pp.349-355. [CrossRef]
- Maffioletti, E.; Gennarelli, M.; Gainelli, G.; Bocchio-Chiavetto, L.; Bortolomasi, M.; Minelli, A. BDNF genotype and baseline serum levels in relation to electroconvulsive therapy effectiveness in treatment-resistant depressed patients. J. ECT. 2019., 35(3), pp.189-194. [CrossRef]
- Ryan, K.M.; Dunne, R.; McLoughlin, D.M. BDNF plasma levels and genotype in depression and the response to electroconvulsive therapy. Brain Stimul. 2018., 11(5), pp.1123-1131. [CrossRef]
- Fernandes, B.; Gama, C.S.; Massuda, R.; Torres, M.; Camargo, D.; Kunz, M.; Belmonte-de-Abreu, P.S.; Kapczinski, F.; de Almeida Fleck, M.P.; Lobato, M.I. Serum brain-derived neurotrophic factor (BDNF) is not associated with response to electroconvulsive therapy (ECT): a pilot study in drug resistant depressed patients. Neurosci. Lett. 2009., 453(3), pp.195-198. [CrossRef]
- van Zutphen, E.M.; Rhebergen, D.; van Exel, E.; Oudega, M.L.; Bouckaert, F.; Sienaert, P.; Vandenbulcke, M.; Stek, M.; Dols, A. Brain-derived neurotrophic factor as a possible predictor of electroconvulsive therapy outcome. Transl. Psychiatry 2019., 9(1), p.155. [CrossRef]
- Vanicek, T.; Kranz, G.S.; Vyssoki, B.; Fugger, G.; Komorowski, A.; Höflich, A.; Saumer, G.; Milovic, S.; Lanzenberger, R.; Eckert, A.; Kasper, S. Acute and subsequent continuation electroconvulsive therapy elevates serum BDNF levels in patients with major depression. Brain Stimul. 2019., 12(4), pp.1041-1050. [CrossRef]
- Zelada, M.I.; Garrido, V.; Liberona, A.; Jones, N.; Zúñiga, K.; Silva, H.; Nieto, R.R. Brain-derived neurotrophic factor (BDNF) as a predictor of treatment response in major depressive disorder (MDD): a systematic review. Int. J. Mol. Sci. 2023., 24(19), p.14810. [CrossRef]
- Pu, J.; Liu, Y.; Gui, S.; Tian, L.; Xu, S.; Song, X.; Zhong, X.; Chen, Y.; Chen, X.; Yu, Y.; Liu, L. Vascular endothelial growth factor in major depressive disorder, schizophrenia, and bipolar disorder: A network meta-analysis. Psychiatry Res. 2020., 292, p.113319. [CrossRef]
- Sharma, A.N.; Soares, J.C.; Carvalho, A.F.; Quevedo, J. Role of trophic factors GDNF, IGF-1 and VEGF in major depressive disorder: A comprehensive review of human studies. J. Affect. Disord. 2016., 197, pp.9-20. [CrossRef]
- Minelli, A.; Zanardini, R.; Abate, M.; Bortolomasi, M.; Gennarelli, M.; Bocchio-Chiavetto, L. Vascular Endothelial Growth Factor (VEGF) serum concentration during electroconvulsive therapy (ECT) in treatment resistant depressed patients. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2011., 35(5), pp.1322-1325. [CrossRef]
- Minelli, A.; Maffioletti, E.; Bortolomasi, M.; Conca, A.; Zanardini, R.; Rillosi, L.; Abate, M.; Giacopuzzi, M.; Maina, G.; Gennarelli, M.; Bocchio-Chiavetto, L. Association between baseline serum vascular endothelial growth factor levels and response to electroconvulsive therapy. Acta Psychiatr. Scand. 2014., 129(6), pp.461-466. [CrossRef]
- Grønli, O.; Stensland, G.Ø.; Wynn, R.; Olstad, R. Neurotrophic factors in serum following ECT: a pilot study. World J. Biol. Psychiatry 2009., 10(4), pp.295-301. [CrossRef]
- Zhang, X.; Zhang, Z.; Sha, W.; Xie, C.; Xi, G.; Zhou, H.; Zhang, Y. Electroconvulsive therapy increases glial cell-line derived neurotrophic factor (GDNF) serum levels in patients with drug-resistant depression. Psychiatry Res. 2009., 170(2-3), pp.273-275. [CrossRef]
- Angelucci, F.; Aloe, L.; Jiménez-Vasquez, P.; Mathé, A.A. Electroconvulsive stimuli alter the regional concentrations of nerve growth factor, brain-derived neurotrophic factor, and glial cell line-derived neurotrophic factor in adult rat brain. J. ECT. 2002., 18(3), pp.138-143. [CrossRef]
- Enomoto, S.; Shimizu, K.; Nibuya, M.; Suzuki, E.; Nagata, K.;Kondo, T. Activated brain-derived neurotrophic factor/TrkB signaling in rat dorsal and ventral hippocampi following 10-day electroconvulsive seizure treatment. Neurosci. Lett. 2017., 660, pp.45-50. [CrossRef]
- Schurgers, G.; Walter, S.; Pishva, E.; Guloksuz, S.; Peerbooms, O.; Incio, L.R.; Arts, B.M.; Kenis, G.; Rutten, B.P. Longitudinal alterations in mRNA expression of the BDNF neurotrophin signaling cascade in blood correlate with changes in depression scores in patients undergoing electroconvulsive therapy.Eur. Neuropsychopharmacol. 2022., 63, pp.60-70. [CrossRef]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009., 65(9), pp.732-741.
- Mössner, R.; Mikova, O.; Koutsilieri, E.; Saoud, M.; Ehlis, A.C.; Müller, N.; Fallgatter, A.J.; Riederer, P. Consensus paper of the WFSBP Task Force on Biological Markers: biological markers in depression. World J. Biol. Psychiatry 2007., 8(3), pp.141-174. [CrossRef]
- Silverman, M.N.; Macdougall, M.G.; Hu, F.; Pace, T.W.W.; Raison, C.L.; Miller, A.H. Endogenous glucocorticoids protect against TNF-alpha-induced increases in anxiety-like behavior in virally infected mice. Mol. Psychiatry 2007., 12(4), pp.408-417. [CrossRef]
- Simen, B.B.; Duman, C.H.; Simen, A.A.; Duman, R.S. TNFα signaling in depression and anxiety: behavioral consequences of individual receptor targeting. Biol. Psychiatry. 2006, 59(9), pp.775-785. [CrossRef]
- Tyring, S.; Gottlieb, A.; Papp, K.; Gordon, K.; Leonardi, C.; Wang, A.; Lalla, D.; Woolley, M.; Jahreis, A.; Zitnik, R.; Cella, D. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. The Lancet. 2006, 367(9504), pp.29-35. [CrossRef]
- Wang, C.; Zong, S.; Cui, X.; Wang, X.; Wu, S.; Wang, L.; Liu, Y.; Lu, Z.; The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Front. immunol. 2023, 14, p.1117172. [CrossRef]
- Yirmiya, R.; Rimmerman, N.; Reshef, R. Depression as a microglial disease. Trends Neurosci. 2015, 38(10), pp.637-658.
- Goldfarb, S.; Fainstein, N.; Ganz, T.; Vershkov, D.; Lachish, M.; Ben-Hur, T. Electric neurostimulation regulates microglial activation via retinoic acid receptor α signaling. Brain Behav Immun. 2021, 96, pp.40-53. [CrossRef]
- Lehtimäki, K.; Keränen, T.; Huuhka, M.; Palmio, J.; Hurme, M.; Leinonen, E.;Peltola, J. Increase in plasma proinflammatory cytokines after electroconvulsive therapy in patients with depressive disorder. J ECT. 2008, 24(1), pp.88-91. [CrossRef]
- Kronfol, Z.A.; Lemay, L.; Nair, M.P.; Kluger, M.J. Electroconvulsive Therapy Increases Plasma Levels of Interleukin-6 a. 1990. [CrossRef]
- Hestad, K.A.; Tønseth, S.; Støen, C.D.; Ueland, T.; Aukrust, P. Raised plasma levels of tumor necrosis factor α in patients with depression: normalization during electroconvulsive therapy. J ECT. 2003, 19(4), pp.183-188.
- Järventausta, K.; Sorri, A.; Kampman, O.; Björkqvist, M.; Tuohimaa, K.; Hämäläinen, M.; Moilanen, E.; Leinonen, E.; Peltola, J.; Lehtimäki, K. Changes in interleukin-6 levels during electroconvulsive therapy may reflect the therapeutic response in major depression. Acta Psychiatr. Scand. 2017, 135(1), pp.87-92. [CrossRef]
- Kranaster, L.; Hoyer, C.; Aksay, S.S.; Bumb, J.M.; Müller, N.; Zill, P.; Schwarz, M.J.; Sartorius, A. Antidepressant efficacy of electroconvulsive therapy is associated with a reduction of the innate cellular immune activity in the cerebrospinal fluid in patients with depression. World J. Biol. Psychiatry. 2018, 19(5), pp.379-389. [CrossRef]
- Fluitman, S.B.; Heijnen, C.J.; Denys, D.A.; Nolen, W.A.; Balk, F.J.; Westenberg, H.G. Electroconvulsive therapy has acute immunological and neuroendocrine effects in patients with major depressive disorder. J. Affect. Disord. 2011, 131(1-3), pp.388-392. [CrossRef]
- Kronfol, Z.; Nair, M.P.; Weinberg, V.; Young, E.A.; Aziz, M. Acute effects of electroconvulsive therapy on lymphocyte natural killer cell activity in patients with major depression. J. Affect. Disord. 2002, 71(1-3), pp.211-215. [CrossRef]
- Moschny, N.; Jahn, K.; Maier, H.B.; Khan, A.Q.; Ballmaier, M.; Liepach, K.; Sack, M.; Skripuletz, T.; Bleich, S.; Frieling, H.; Neyazi, A.. Electroconvulsive therapy, changes in immune cell ratios, and their association with seizure quality and clinical outcome in depressed patients. Eur. Neuropsychopharmacol. 2020, 36, pp.18-28. [CrossRef]
- Donato, R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33(7), pp.637-668. [CrossRef]
- Marenholz, I.; Heizmann, C.W.; Fritz, G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem. Biophys. Res. Commun. 2004, 322(4), pp.1111-1122. [CrossRef]
- F. Winningham-Major, J.L.; Staecker, S.W.; Barger, S. Coats,; L.J. Van Eldik. Neurite extension and neuronal survival activities of recombinant S100 beta proteins that differ in the content and position of cysteine residues, J. Cell Biol. 109. 1989, 3063–3071. [CrossRef]
- Arts, B.; Peters, M.; Ponds, R.; Honig, A.; Menheere, P.; van Os, J. S100 and impact of ECT on depression and cognition. J ECT. 2006, 22:206–212. [CrossRef]
- Khan, M.; Baussan, Y.; Hebert-Chatelain, E. Connecting Dots between Mitochondrial Dysfunction and Depression. Biomolecules. 2023, Apr 20;13(4):695. [CrossRef]
- Mailloux, R.J. An update on mitochondrial reactive oxygen species production. Antioxidants. 2020, 9(6), p.472. [CrossRef]
- Andrés Juan, C.; Pérez de Lastra, J.M.; Plou Gasca, F.J; Pérez-Lebeña, E. The chemistry of reactive oxygen species (ROS) revisited: outlining their role in biological macromolecules (DNA, lipids and proteins) and induced pathologies. Int J Mol Sci. 2021, 28;22(9):4642.
- Mattson, MP.; Gleichmann, M.; Cheng, A.; Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008, 10;60(5):748-66. [CrossRef]
- Petschner, P.; Gonda, X.; Baksa, D.; Eszlari, N.; Trivaks, M.; Juhasz, G.; Bagdy, G. Genes linking mitochondrial function, cognitive impairment and depression are associated with endophenotypes serving precision medicine. Neuroscience. 2018, 370, pp.207-217. [CrossRef]
- Jiang, M.; Wang, L.; Sheng, H.; Mitochondria in depression: The dysfunction of mitochondrial energy metabolism and quality control systems. CNS Neuroscience & Therapeutics. 2024, 30(2), p.e14576. [CrossRef]
- Gebara, E.; Zanoletti, O.; Ghosal, S.; Grosse, J.; Schneider, B.L.; Knott, G.; Astori, S.;Sandi, C. Mitofusin-2 in the nucleus accumbens regulates anxiety and depression-like behaviors through mitochondrial and neuronal actions. Biol. Psychiatry. 2021, 89(11), pp.1033-1044. [CrossRef]
- Wu, T.; Huang, Y.; Gong, Y.; Xu, Y.; Lu, J.; Sheng, H; Ni, X.; Treadmill exercise ameliorates depression-like behavior in the rats with prenatal dexamethasone exposure: the role of hippocampal mitochondria. Front. Neurosci. 2019, 13, p.264. [CrossRef]
- Gong, Y.; Chai, Y.; Ding, J.H.; Sun, X.L; Hu, G. Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci. Lett. 2011, 488(1), pp.76-80. [CrossRef]
- Caruso, G.; Benatti, C.; Blom, J.M.; Caraci, F.; Tascedda, F. The many faces of mitochondrial dysfunction in depression: From pathology to treatment. Front. Pharmacol. 2019, 10, p.995. [CrossRef]
- Li, W.; Zhu, L.; Chen, Y.; Zhuo, Y.; Wan, S.; Guo, R. Association between mitochondrial DNA levels and depression: a systematic review and meta-analysis. BMC psychiatry. 2023, 23(1), p.866. [CrossRef]
- Fattal, O.; Link, J.; Quinn, K.; Cohen, B.H;Franco, K. Psychiatric comorbidity in 36 adults with mitochondrial cytopathies. CNS spectrums. 2007, 12(6), pp.429-438. [CrossRef]
- Gardner, A.; Johansson, A.; Wibom, R.; Nennesmo, I.; von Döbeln, U.; Hagenfeldt, L.; Hällström, T. Alterations of mitochondrial function and correlations with personality traits in selected major depressive disorder patients. J. Affect. Disord. 2003, 76(1-3), pp.55-68.
- Brymer, K.J.; Fenton, E.Y.; Kalynchuk, L.E.; Caruncho, H.J. Peripheral etanercept administration normalizes behavior, hippocampal neurogenesis, and hippocampal reelin and GABAA receptor expression in a preclinical model of depression. Front. Pharmacol. 2018, 9, p.121. [CrossRef]
- Wang, J.; Hodes, G.E.; Zhang, H.; Zhang, S.; Zhao, W.; Golden, S.A.; Bi, W.; Menard, C.; Kana, V.; Leboeuf, M.; Xie, M. Epigenetic modulation of inflammation and synaptic plasticity promotes resilience against stress in mice. Nat. Commun. 2018, 9(1), p.477. [CrossRef]
- Sequeira, A.; Rollins, B.; Magnan, C.; van Oven, M.; Baldi, P.; Myers, R.M.; Barchas, J.D.; Schatzberg, A.F.; Watson, S.J.; Akil, H.; Bunney, W.E. Mitochondrial mutations in subjects with psychiatric disorders. PloS one. 2015, 10(5), p.e0127280. [CrossRef]
- Kasahara, T.; Kubota, M.; Miyauchi, T.; Ishiwata, M.; Kato, T. A marked effect of electroconvulsive stimulation on behavioral aberration of mice with neuron-specific mitochondrial DNA defects. PLoS One. 2008, 3(3), p.e1877. [CrossRef]
- Búrigo, M.; Roza, C.A.; Bassani, C.; Fagundes, D.A.; Rezin, G.T.; Feier, G.; Dal-Pizzol, F.; Quevedo, J.; Streck, E.L. Effect of electroconvulsive shock on mitochondrial respiratory chain in rat brain. Neurochem. Res. 2006, 31, pp.1375-1379. [CrossRef]
- Gandhi, S.;Abramov, A.Y. Mechanism of oxidative stress in neurodegeneration. Oxid. Med. Cell. Longev. 2012, (1), p.428010.
- Yoshikawa, T.; You, F. Oxidative stress and bio-regulation. Int. J. Mol. Sci. 2024, 25(6), p.3360.
- Jazvinšćak Jembrek, M.; Oršolić, N.; Karlović, D.; Peitl, V. Flavonols in action: targeting oxidative stress and neuroinflammation in major depressive disorder. Int. J. Mol. Sci. 2023, 24(8), p.6888. [CrossRef]
- Gadoth, N.; Göbel, HH. Oxidative Stress in Applied Basic Research and Clinical Practice. Totowa, NJ: Humana Press, 2011; pp. 19–27.
- Bakunina, N.; Pariante, C.M.; Zunszain, P.A.. Immune mechanisms linked to depression via oxidative stress and neuroprogression. Immunology. 2015, 144(3), pp.365-373. [CrossRef]
- Maes, M.; Galecki, P.; Chang, Y.S.; Berk, M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro) degenerative processes in that illness. Prog. Neuro-Psychopharmacol. Biol. Psychiatry. 2011, 35(3), pp.676-692. [CrossRef]
- Bhatt, S.; Nagappa, A.N.; Patil, C.R. Role of oxidative stress in depression. Drug Discov. Today. 2020, 25(7), pp.1270-1276.
- Ait Tayeb, A.E.K.; Poinsignon, V.; Chappell, K.; Bouligand, J.; Becquemont, L.;Verstuyft, C. Major depressive disorder and oxidative stress: a review of peripheral and genetic biomarkers according to clinical characteristics and disease stages. Antioxidants. 2023, 12(4), p.942. [CrossRef]
- Jiménez-Fernández, S.; Gurpegui, M.; Garrote-Rojas, D.; Gutiérrez-Rojas, L.; Carretero, M.D.; Correll, C.U. Oxidative stress parameters and antioxidants in adults with unipolar or bipolar depression versus healthy controls: Systematic review and meta-analysis. J. Affect. Disord. 2022, 314, pp.211-221. [CrossRef]
- Atagün, M.İ.; Canbek, Ö.A. A systematic review of the literature regarding the relationship between oxidative stress and electroconvulsive therapy. Alpha Psychiatry. 2022, 23(2), p.47. [CrossRef]
- Bader, M.; Abdelwanis, M.; Maalouf, M.; Jelinek, H.F. Detecting depression severity using weighted random forest and oxidative stress biomarkers. Sci. Rep. 2024, 14(1), p.16328. [CrossRef]
- Barichello, T.; Bonatto, F.; Feier, G.; Martins, M.R.; Moreira, J.C.F.; Dal-Pizzol, F.; Izquierdo, I.; Quevedo, J. No evidence for oxidative damage in the hippocampus after acute and chronic electroshock in rats. Brain Res. 2004, 1014(1-2), pp.177-183. [CrossRef]
- Şahin, Ş.; Aybastı, Ö.; Elboğa, G.; Altındağ, A.; Tamam, L. Major depresyonda elektrokonvulsif terapinin oksidatif metabolizma üzerine etkisi. Çukurova med. j. 2017, 42(3), pp.513-517.
- Lv, Q.; Hu, Q.; Zhang, W.; Huang, X.; Zhu, M.; Geng, R.; Cheng, X.; Bao, C.; Wang, Y.; Zhang, C.; He, Y. Disturbance of oxidative stress parameters in treatment-resistant bipolar disorder and their association with electroconvulsive therapy response. International Journal of Neuropsychopharmacology. 2020, 23(4), pp.207-216. [CrossRef]
- Karayağmurlu, E.; Elboğa, G.; Şahin, Ş.K.; Karayağmurlu, A.; Taysı, S.; Ulusal, H.;Altındağ, A. Effects of electroconvulsive therapy on nitrosative stress and oxidative DNA damage parameters in patients with a depressive episode. Int. J. Psychiatry. 2022, 26(3), pp.259-268. [CrossRef]
- Barichello, T.; Bonatto, F.; Agostinho, F.R.; Reinke, A.; Moreira, J.C.F.; Dal-Pizzol, F.; Izquierdo, I.; Quevedo, J. Structure-related oxidative damage in rat brain after acute and chronic electroshock. Neurochem. Res. 2004, 29, pp.1749-1753. [CrossRef]
- Župan, G.; Pilipović, K.; Hrelja, A.; Peternel, S. Oxidative stress parameters in different rat brain structures after electroconvulsive shock-induced seizures. Prog.Neuro-Psychopharmacol. Biol. Psychiatry. 2008, 32(3), pp.771-777. [CrossRef]
- Eraković, V.; Župan, G.; Varljen, J.; Radošević, S.; Simonić, A. Electroconvulsive shock in rats: changes in superoxide dismutase and glutathione peroxidase activity. Brain Res. Mol. Brain Res. 2000, 76(2), pp.266-274. [CrossRef]
- Nielsen, B.; Cejvanovic, V.; Wörtwein, G.; Hansen, A.R.; Marstal, K.K.; Weimann, A.; Bjerring, P.N.; Dela, F.; Poulsen, H.E.; Jørgensen, M.B. Increased oxidation of RNA despite reduced mitochondrial respiration after chronic electroconvulsive stimulation of rat brain tissue. Neurosci. Lett. 2019, 690, pp.1-5. [CrossRef]
- Hollville, E.; Romero, S.E.; Deshmukh, M. Apoptotic cell death regulation in neurons. FEBS Lett. 2019, 286(17), pp.3276-3298. [CrossRef]
- Erekat, N.S. Apoptosis and its therapeutic implications in neurodegenerative diseases. Clin. Anat. 2022, 35(1), pp.65-78. [CrossRef]
- Lucassen, P.J.; Heine, V.M.; Muller, M.B.; van der Beek, E.M.; Wiegant, V.M.; Ron De Kloet, E.; Joels, M.; Fuchs, E.; Swaab, D.F.; Czeh, B. Stress, depression and hippocampal apoptosis. CNS Neurol Disord Drug Targets. 2006, 5(5), pp.531-546.
- Kondratyev, A.; Sahibzada, N.; Gale, K. Electroconvulsive shock exposure prevents neuronal apoptosis after kainic acid-evoked status epilepticus. Brain Res. Mol. Brain Res. 2001, 91(1-2), pp.1-13. [CrossRef]
- Zarubenko, I.I.; Yakovlev, A.A.; Stepanichev, M.Y.; Gulyaeva, N.V. Electroconvulsive shock induces neuron death in the mouse hippocampus: correlation of neurodegeneration with convulsive activity. Neurosci. Behav. Physiol. 2005, 35, pp.715-721. [CrossRef]
- Sigström, R.; Göteson, A.; Joas, E.; Pålsson, E.; Liberg, B.; Nordenskjöld, A.; Blennow, K.; Zetterberg, H.; Landén, M. Blood biomarkers of neuronal injury and astrocytic reactivity in electroconvulsive therapy. Mol. Psychiatry. 2024, pp.1-9. [CrossRef]
- McGrory, C.L.; Ryan, K.M.; Kolshus, E.; McLoughlin, D.M. Peripheral blood E2F1 mRNA in depression and following electroconvulsive therapy. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2019, 89, pp.380-385. [CrossRef]
- Jeon, W.J.; Kim, S.H.; Seo, M.S.; Kim, Y.; Kang, U.G.; Juhnn, Y.S.; Kim, Y.S. Repeated electroconvulsive seizure induces c-Myc down-regulation and Bad inactivation in the rat frontal cortex. Exp Mol Med. 2008, 40(4), pp.435-444. [CrossRef]
Figure 1.
Key theories of potential mechanisms of ECT. The neurotransmitter theory (top-left). Cytokine theory (top-right). The receptors theory (bottom-left). The neurotrophic theory (bottom-right).
Figure 1.
Key theories of potential mechanisms of ECT. The neurotransmitter theory (top-left). Cytokine theory (top-right). The receptors theory (bottom-left). The neurotrophic theory (bottom-right).
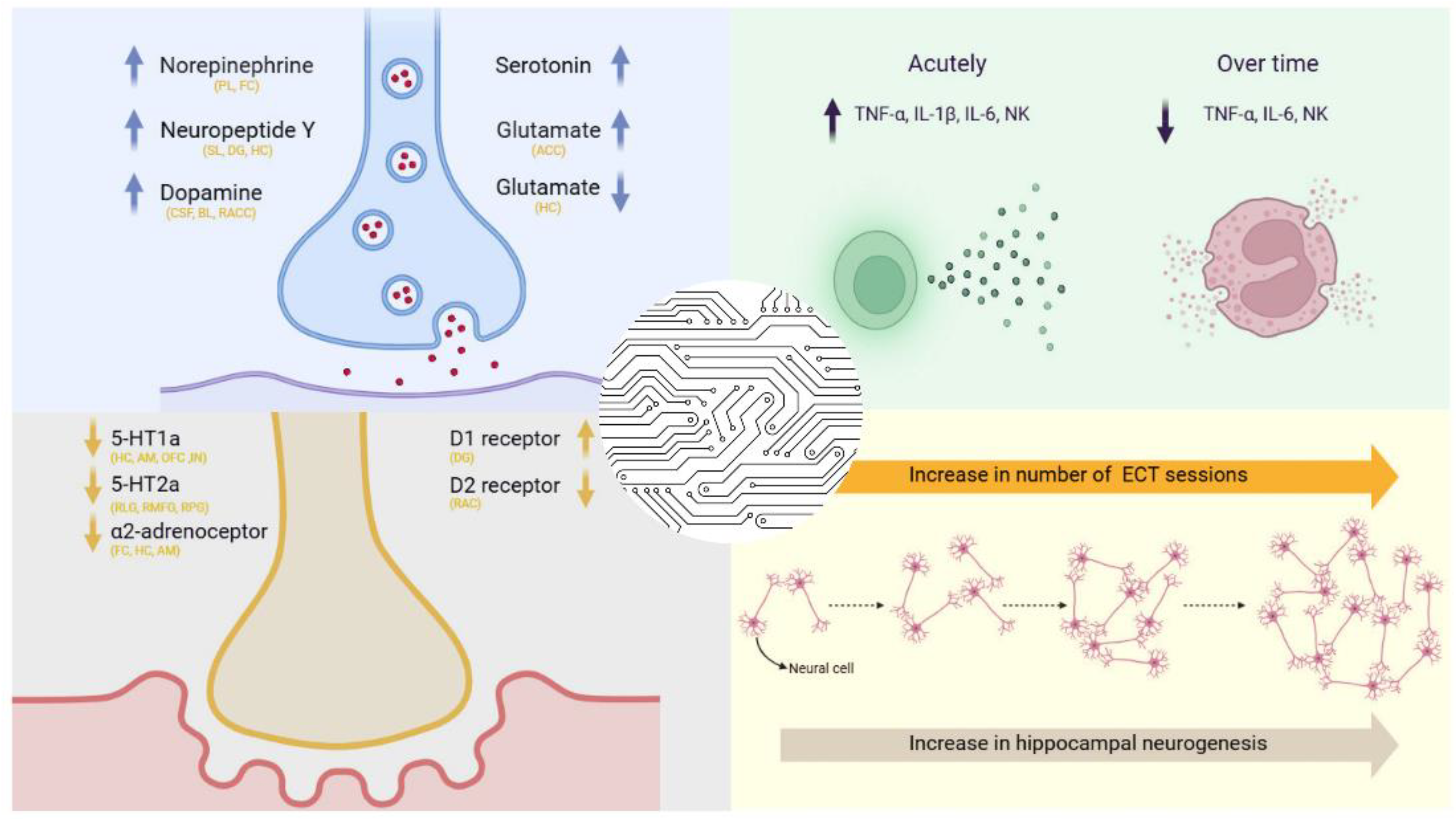
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



