Submitted:
17 March 2025
Posted:
19 March 2025
You are already at the latest version
Abstract
Influenza A virus (IAV) infections continue to threaten public health. Current strategies, such as vaccines and antiviral drugs, are limited due to time-consuming development and drug-resistant strains. Therefore, new effective treatments are needed. Here, virus-supportive cellular factors are promising drug targets, and encapsulation of candidate substances in PLGA nanoparticles (NPs) is intended to improve their bioavailability. This study investigates the potential of the indirubin derivative 6BIGOE, a GSK-3β inhibitor, for its potential to regulate IAV replication in vitro. The effects of 6BIGOE-loaded PLGA NPs on cell metabolism were assessed by MTT and LDH assays in A549 and Calu-3 cells. Viral replication and spread were monitored in various IAV-infected cell lines in absence and presence of free and 6BIGOE-loaded PLGA NPs via plaque assays and Western blot analysis. Encapsulation of 6BIGOE in PLGA NPs resulted in reduced negative side effects on cell viability while maintaining antiviral efficacy. Both encapsulated and free 6BIGOE exhibited antiviral activity, potentially through GSK-3β inhibition and disruption of key signaling pathways required for viral replication. The data indicate 6BIGOE, particularly after encapsulation in NPs, as a potential candidate for further investigation and development as an antiviral agent to treat IAV infections.
Keywords:
influenza A virus
; PLGA
; indirubin
; nanoparticles
; antiviral
; GSK-3
1. Introduction
Seasonal influenza remains a major global health problem, with a high morbidity and mortality rate, responsible for an estimated annual burden of 3 to 5 million severe cases and 290,000 to 650,000 deaths [1,2]. In particular, elderly and immunocompromised patients are in danger of severe infection courses. Influenza A viruses (IAVs), which belong to the family of Orthomyxoviridae, are characterized by their single-stranded, negative-sensed RNA genome. Their high mutation rates, by the rapid genetic drift and shift of the virus, pose a challenge to control efforts, leading to seasonal outbreaks and occasional pandemics [3].
Although influenza vaccines are the most effective prophylactic measure, low coverage and limited efficacy against newly emerging subtypes highlight the urgent need for efficient and broadly active antivirals to limit viral replication in infected hosts [1,4].
Currently, three classes of substances are approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) for the therapeutic management of influenza virus infections: M2 ion channel inhibitors, neuraminidase inhibitors and the cap-dependent endonuclease inhibitor baloxavir marboxil [5,6]. The latter as well as neuraminidase inhibitors such as oseltamivir and peramivir are active against both, influenza A and B viruses, while M2 inhibitors, including amantadine and rimantadine, target only IAVs [6]. In any case, the medication has to be applied in the early disease course to be effective. Additionally, the rapid emergence of resistance to these antiviral agents underscores the need for alternative therapeutic strategies with a high barrier to resistance [7].
Since influenza viruses hijack the host cell machinery for replication, targeting virus-supportive cellular factors offer promising intervention options. This approach has led to the development of host-directed therapies aiming at modulating these cellular factors rather than directly attacking the virus itself [8,9,10].
One such cellular factor gaining interest is glycogen synthase kinase 3 (GSK-3). GSK-3 is a serine/threonine kinase with two isoforms, GSK-3α and GSK-3β, which are expressed in most tissues and encoded by two genes, GSK-3A and GSK-3B. Although the two isoforms share high homology in their kinase domains they differ at their C-termini [11,12]. Furthermore, the isoforms fulfill independent functions that cannot be compensated by another. Activation and deactivation of GSK-3 itself, are controlled by post-translational, site-specific phosphorylation of serine and tyrosine residues. GSK-3 regulation interferes with several key signaling pathways, including Wnt/β-catenin, nuclear factor-κB (NF-κB), and phosphatidylinositol 3-kinases (PI3K)/Akt with substrates such as the proline-rich Akt substrate of 40 kDa (PRAS40) and with no lysine 1 (WNK1). Phosphorylation of serine 9 in GSK-3β or serine 21 in GSK-3α results in the inactivation of these kinases and the subsequent reversal of their inhibitory function. In contrast, phosphorylation of tyrosine 216 in GSK-3β or tyrosine 279 in GSK-3α leads to their activation. Currently, around 100 proteins are believed to be GSK-3 substrates, however, requiring initial phosphorylation by other kinases [11]. Initially identified for its role in regulating glycogen synthase in insulin signaling [13], GSK-3 has since been implicated in a variety of cellular processes, such as cell proliferation, differentiation, apoptosis, and immune responses [14,15]. Therefore, dysregulation of GSK-3-mediated signaling is implicated in several diseases, including neurodegenerative disorders like Alzheimer's disease, metabolic disorders like type 2 diabetes, as well as in cancer progression. Concomitantly, GSK-3 inhibition shows therapeutic potential [16,17,18].
In context of viral infections GSK-3 has been shown to play crucial but diverse roles. The literature reveals contradictory results, which, however, reflect the complex interplay between GSK-3 and viral replication strategies [19,20]. In human immunodeficiency virus (HIV)-1 infection GSK-3 expression is upregulated, potentially contributing to viral persistence [21]. In coxsackievirus B3 infections, increased GSK-3β activity triggers apoptosis and enhances viral release, while GSK-3 inhibition reduces these effects [22,23]. Furthermore, GSK-3 inhibition has been shown to suppress replication in varicella zoster virus [24], and coronaviruses by disrupting nucleocapsid phosphorylation [25]. During IAV infections, GSK-3 was also shown to support viral replication, especially by supporting viral entry [26].
When developing novel antiviral strategies, the use of existing pharmaceutical substances offers a significant advantage. Reuse of drugs with proven safety and established pharmacokinetics accelerates the further investigation process, reduces costs and minimizes the risk of side effects [27]. Among those, natural products hold significant promise in drug repurposing approaches. An example is indirubin, a natural bis-indole alkaloid used in traditional Chinese medicine [28]. It has a broad spectrum of pharmacological effects, including anticancer, neuroprotective, antiviral and anti-inflammatory effects, with potential applications in chronic inflammatory diseases, cancer, Alzheimer's disease and metabolic disorders [28,29,30]. Interestingly, indirubin exhibits antiviral properties and inhibits viruses such as human adenoviruses [30], IAVs [31], HIV [32], pseudorabies virus [33] and Japanese encephalitis virus [34].
Remarkably, the indirubin derivative 6-bromoindirubin-3'-glycerol-oxime ether (6BIGOE) is a potent modulator of inflammatory cytokines and lipid mediators in human monocytes by inhibiting GSK-3β, indicating potential therapeutic applications for inflammation-related diseases [35]. Therefore, it also could be effective during an ongoing influenza virus infection and limit the symptoms. Despite its efficacy, 6BIGOE exhibits a high lipophilicity, poor water solubility, low bioavailability, and potential cytotoxicity, limiting its therapeutic use [28,36]. Conventional administration methods are less efficient to achieve controlled release rates and targeting effects. To overcome these limitations, advanced drug delivery strategies such as crystal technology, surfactants, and nanocarriers are used [37,38,39,40]. Among these, nanoparticle (NP)-based drug delivery systems have gained attention due to their ability to control drug release, increase drug bioavailability and reduce off-target effects [41]. In particular, poly(D,L-lactic-co-glycolic acid) (PLGA)-based NPs have been proven to be a highly effective drug delivery vehicle due to their excellent biocompatibility, controlled release profiles, and biodegradability [42,43]. PLGA NPs are an ideal choice to improve the bioavailability and therapeutic efficacy of hydrophobic drugs, ensuring that they are both stable and effective in clinical settings [44].
This study examined the effect of 6BIGOE on IAV replication in vitro. The data indicate that encapsulating the drug into PLGA NPs reduces its cytotoxicity while preserving its antiviral efficacy. The antiviral effect is presumably based on the 6BIGOE-mediated inhibition of GSK-3β activity that is necessary for efficient replication.
2. Materials and Methods
2.1. Cell Culture, Viruses and Substances
The human epithelial lung cancer cell line (A549) was cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Anprotec, Germany) with high glucose, stable glutamine, sodium pyruvate supplemented with 10% fetal calf serum (FCS; Anprotec, Germany) at 37 °C and 5% CO2. Madin-Darby canine kidney II (MDCK II) and the human lung-adenocarcinoma cell line (Calu-3) cells were grown in Minimum Essential Medium with Earle’s salts and L-glutamine (EMEM; Anprotec, Germany) with 10% FCS. A549 and Calu-3 cells were utilized in this study, as they closely resemble lung cells in culture, making them an excellent in vitro model for infection experiments and drug testing due to their high susceptibility to infection. The influenza virus strains used in this study were A/Puerto Rico/8/34 (PR8; H1N1), A/Jena/5258/09 (Jena5258; H1N1pdm) and A/NRW/173/09 (NRW173; H1N1pdm). 6BIGOE (Prof. Dr. Ivan Vilotijević, Friedrich Schiller University Jena, Germany) and pictilisib (Selleckchem, Germany) were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich, Germany) for biological experiments.
2.2. Preparation of PLGA NPs
6BIGOE was synthesized following a previously reported procedure [45]. The characterization data for the synthetic 6BIGOE matched the previously reported data and purity was confirmed by elemental analysis. PLGA NPs were formulated using an emulsion-diffusion-evaporation (EDE) technique. 25 mg PLGA (Resomer® RG 502; MW 7,000-17,000 g mol-1, ester terminated, 50:50 lactide:glycolide ratio; Sigma-Aldrich, Germany) and 1.25 mg 6BIGOE were dissolved in 2.5 ml dichloromethane (≥ 99.8%; Thermo Scientific™, USA) as organic phase. For the formation of an oil-in-water (O/W) emulsion, the organic phase was added dropwise at room temperature under magnetic stirring (400 rpm) to 7.5 ml of an aqueous phase consisting of 2% (w/v) poly(vinyl alcohol) (30,000-70,000 g mol-1; Sigma-Aldrich, Germany) as stabilizer in deionized water at pH 7.4. The emulsion was sonicated (Branson Sonifier Cell Disruptor B15, Branson Ultrasonic Corp., USA) for 90 s. Afterwards, the resulting nanoemulsion was diluted with deionized water to facilitate the evaporation of dichloromethane under stirring at room temperature overnight. The NP dispersion was washed thrice with deionized water by centrifugation (22,000 × g, 20 min; Beckmann Coulter Allegra 64R, USA).
2.3. Laser Light Scattering Techniques
A Zetasizer Ultra (Malvern Instruments Ltd., United Kingdom) was used for NP characterization regarding hydrodynamic diameter (HD), polydispersity index (PDI) and zeta potential (ZP) in water at 25 °C (refractive index 1.33, viscosity 0.887 mPa × s). The measurement of HD and PDI were conducted using dynamic light scattering (173°, five runs per sample) in ZEN0040 cuvettes (Brand, Germany). ZP was examined as electrophoretic mobility in deionized water with three runs per sample in a DTS 1070 folded capillary cell (Malvern Instruments). The data of three independent batches were analyzed using the Malvern Instruments Zetasizer software ZS Xplorer v1.3.1.7 and expressed as mean ± standard deviation (SD).
2.4. Transmission Electron Microscopy
For transmission electron microscopy (TEM), samples were stained with 2.0% phosphotungstic acid solution (Thermo Scientific™, USA) on formvar carbon-coated copper grids (Electron Microscopy Sciences, USA). Images were acquired on a Tecnai G2 20 (FEI, The Netherlands) at an acceleration voltage of 120 kV. Images have been recorded on a MegaView (OSIS, Olympus Soft Imaging Systems, Japan) CCD camera with 1376 x 1024 image pixels. Images have been optimized for contrast utilizing Fiji ImageJ 1.54i [46].
2.5. Quantification of 6BIGOE by UV/Vis Spectrophotometric Measurement
The amount of encapsulated 6BIGOE was determined by UV/Vis spectrophotometric measurement according to Czapka et al. [35]. Lyophilized NPs (72 h, -20 °C, 0.1 mbar; 25L Genesis SQ EL-85, SP Scientific, USA) were dissolved in DMSO (≥99.8%; Carl Roth, Germany) and analyzed by a Tecan Spark 10M plate reader (Tecan Group, Switzerland) at 520 nm. Calibration was performed with linear dilutions of 6BIGOE in DMSO between 10-150 µg ml-1 (r2=0.999). The drug load (DL; %) was calculated by the ratio of encapsulated drug (mg) per 100 mg NPs by weighting. The ratio of the determined DL to the theoretical DL corresponds to the encapsulation efficiency (EE; %). The SparkControl v3.2 software (Tecan Group, Switzerland) was used for measurements and results were presented as mean ± SD from three independent experiments.
2.6. Assessing Drug Impact on Cellular Metabolic Activity and Cytotoxicity (MTT- and LDH Assay)
To determine the impact of free and encapsulated 6BIGOE (PLGA NP [6BIGOE]) on cellular metabolic activity, MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays were performed, and lactate-dehydrogenase (LDH) release was measured to examine cytotoxic effects. Calu-3 (100,000 cells/well) or A549 cells (10,000 cells/well) were seeded into 96-well plates 24 h prior to use. The cells were treated with the indicated concentrations of 6BIGOE, PLGA NP [blank], PLGA NP [6BIGOE] or DMSO and H2O in 100 µl EMEM (Calu-3) or DMEM (A549) supplemented with 10% FCS and incubated at 37 °C with 5% CO2. After 24 h, supernatant was collected for LDH assay and stored at -20 °C until further use. For the maximum LDH release control 10 µl of lysis buffer provided by CyQUANTTM LDH Cytotoxicity Assay kit (Thermo Scientific™, USA) was added for 45 min at 37 °C, 5 % CO2 after 24 h of untreated growth. For the MTT assay 100 µl of EMEM (Calu-3) or DMEM (A549) containing MTT at a final concentration of 1 mg ml-1 was added to the cells, followed by 2 h of incubation. The medium was then removed, and cells were lysed in 30 µl of DMSO. Absorbance was measured at 562 nm using a FLUOstar Omega plate reader (BMG Labtech, Germany). LDH release was measured from the supernatants by using the CyQUANTTM LDH Cytotoxicity Assay kit according to the manufacturer’s protocol. LDH release from compound-treated cells was compared to maximum LDH release from lysis buffer-treated control cells, which were set to 100% and medium-treated samples were used as spontaneous LDH release control and represent 0%. Absorbance was measured at 490 nm and 680 nm using a FLUOstar Omega plate reader (BMG Labtech, Germany). Background absorbance at 680 nm was subtracted from that at 490 nm.
2.7. Viral Infection
A549 cells were seeded in 12-well plates (250,000 cells/well) 24 h prior to viral infection. Calu-3 cells were seeded in 12-well plates (500,000 cells/well) 36 h prior to viral infection. Cells were washed with phosphate buffered saline (PBS; Carl Roth, Germany) prior to infection and either left uninfected or infected with the indicated multiplicity of infection (MOI) of IAV in 250 µl PBS/BA (PBS supplemented with 0.2% BSA (Carl Roth, Germany), 1 mM MgCl2 (Sigma-Aldrich, Germany) and 0.9 mM CaCl2 (Sigma-Aldrich, Germany)) for 30 min at 37 °C and 5% CO2. Subsequently, cells were incubated in DMEM/BA (A549) or MEM/BA (Calu-3) containing the indicated substances until the designated time points post infection (p.i.). DMEM/BA and MEM/BA consisting of DMEM or MEM respectively were supplemented with 0.2% BSA, 1 mM MgCl2, 0.9 mM CaCl2 and 0.2 µg ml-1 TPCK trypsin (Sigma-Aldrich, Germany).
2.8. Plaque Assay
Viral titers were determined by plaque assay. MDCK II cells (2,000,000 cells/well) were seeded in 6-well plates 24 h before infection to achieve a confluent layer. Cells were washed with PBS and infected with serial dilutions of the sample in PBS/BA supplemented with 100 U ml-1/0.1 mg ml-1 penicillin/streptomycin (pen/strep; Anprotec, Germany) for 30 min at 37 °C and 5% CO2. Afterwards virus solution was discarded and replaced by 2 ml prewarmed soft agar containing MEM (Gibco™, USA) supplemented with 0.2% BSA (Carl Roth, Germany), 0.01% DEAE Dextran (Pharmacia Biotech, Germany), 0.2% NaHCO3 (Gibco™, USA), 100 U ml-1/0.1 mg ml-1 pen/strep, 0.2 µg ml-1 TPCK trypsin and 0.9% agar (Oxoid, United Kingdom). The plates were incubated at 37 °C and 5% CO2 for three days. Plaque forming units (PFU) were visualized using neutral red (Sigma-Aldrich, Germany) staining and counted.
2.9. Western Blot Analysis
To analyze protein expression Western blot analysis was performed. Triton lysis buffer (20 mM Tris-HCl (pH 7.4), 137 mM NaCl, 10% glycerol, 1% Triton-X-100, 2 mM EDTA, 50 mM β-glycerophosphate, 20 mM sodium pyrophosphate) containing protease inhibitors (0.2 mM Pefabloc® (Sigma-Aldrich, Germany), 5 µg ml-1 aprotinin (Carl Roth, Germany), 5 µg ml-1 leupeptin (Sigma-Aldrich, Germany), 1 mM sodium vanadate (Sigma-Aldrich, Germany) and 5 mM benzamidine (Sigma-Aldrich, Germany)) was used to lyse the cells for 30 min at 4 °C. Cell lysates were cleared via centrifugation (14,000 rpm, 10 min, 4 °C) and protein content was determined using Protein Assay Dye Reagent Concentrate (BioRad, USA). Lysates and protein marker (PageRuler™ Prestained Protein Ladder, Thermo Scientific™, USA) were loaded on a 7% or 10% SDS-PAGE and afterwards blotted onto a 0.2 µm nitrocellulose membrane (Amersham™ Protran®, USA). Membranes were blocked for 1 h with 2% milk. Proteins were detected using the primary antibodies (1:1,000) listed in Table 1. The secondary antibodies used were WesternSure® goat anti-rabbit HRP and WesternSure® goat anti-mouse HRP (LI-COR® Bioscience, Germany) at a dilution of 1:5,000. Membranes were incubated in Pierce® ECL Western Blotting Substrate (Thermo Scientific™, USA) and further developed with the Fusion©FX6.Edge (Vilber Lourmat, Germany). Protein levels were quantified and normalized to the α-tubulin or heat shock protein 90 (HSP90) loading control using Fiji ImageJ 1.54i [46].
3. Results
3.1. Preparation and Characterization of PLGA NP [6BIGOE]
An emulsion-diffusion-evaporation method was employed to encapsulate the small molecule 6BIGOE (430.13 g mol-1) into PLGA NPs (Figure 1a). Dynamic light scattering revealed HD of the NPs ranging from 185 to 194 nm with PDIs of 0.03 for PLGA NP [blank] and 0.12 for PLGA NP [6BIGOE], indicating monomodal particle size distributions. The ZP of the blank NPs was −22 mV, with a slight increase to approximately −14 mV observed after 6BIGOE encapsulation. Quantification of 6BIGOE via UV/Vis spectrophotometric measurement revealed a DL of approximately 1.7%, resulting in an EE of 34.8% (Figure 1b). The morphology of both, PLGA NP [blank] and PLGA NP [6BIGOE] was visualized using TEM and revealed spherical and homogenous NPs (Figure 1c).
3.2. PLGA [6BIGOE] Has Less Impact on the Cell Metabolic Activity than Free 6BIGOE
To identify non-toxic concentrations of encapsulated and free 6BIGOE suitable for in vitro treatment of A549 and Calu-3 cells, MTT assays were performed (Figure 2a,b). Only viable cells reduce the tetrazolium dye MTT to the insoluble formazan, measurable at 562 nm. For this purpose, the cells were treated with the drugs at the indicated concentrations for 24 h in comparison to solvent controls, which were arbitrarily set to 100%. In case of 6BIGOE DMSO served as acontrol, while H2O was used for the PLGA NPs. LDH release measurements complemented these assays to validate the results, as LDH is released by dying cells devoid of cell membrane integrity (Figure 2c,d). In A549 cells, exposure to 2 µM of free 6BIGOE significantly reduced metabolic activity and increased LDH release to 30% of the lysis control (Figure 2a,c). Interestingly, treatment with PLGA NP [6BIGOE] did neither affect metabolic activity nor LDH release in A549 cells, even at concentrations up to 4 µM. In Calu-3 cells, treatment with all tested concentrations of both encapsulated and free 6BIGOE (0.03 – 4 µM) had no impact on metabolic activity (Figure 2b). However, exposure to 4 µM of free 6BIGOE resulted in an 11% increase of LDH release (Figure 2d), whereas PLGA NP [6BIGOE] showed no effect on cytotoxicity at any tested concentration. Notably, PLGA NP [blank] had no impact on Calu-3 cell viability in any of the experiments.
Since 6BIGOE is known to affect GSK-3β phosphorylation, phospho-GSK-3β (pGSK-3β) levels were monitored by Western blot analysis and the impact of free and encapsulated 6BIGOE was analyzed. Pictilisib, a known inhibitor of the phosphatidylinositol 3-kinase (PI3K) signaling pathway, which plays a crucial role in regulating GSK-3 activity, served as a positive control. Treatment of A549 (Figure 2e) and Calu-3 (Figure 2f) cells with either free or PLGA-encapsulated 6BIGOE resulted in significantly reduced pGSK-3β levels within the concentration range of 0.05 to 0.5 μM for A549 cells and 0.1 to 1 µM for Calu-3 cells.
The results indicate that encapsulated 6BIGOE has lower effects on the cellular metabolic activity and cell survival compared to free 6BIGOE, while the 6BIGOE-mediated effects on GSK-3β phosphorylation are comparable. Overall, these data suggest that A549 cells are more sensitive to treatment with free 6BIGOE than Calu-3 cells, which can tolerate slightly higher concentrations. Consequently, concentrations of up to 1 µM of both free and encapsulated 6BIGOE were chosen for further experiments in epithelial cells.
3.3. 6BIGOE Treatment of IAV-Infected Cells Results in Reduced Viral Titers In Vitro and Is Associated with Inhibition of the GSK-3β-Mediated Signaling Processes
To further investigate the effects of 6BIGOE on IAV replication, multi-cycle viral growth experiments were performed in A549 and Calu-3 cells using three different IAV strains: H1N1 strain A/Puerto Rico/8/34 (PR8) and the H1N1pdm isolates A/NRW/173/09 (NRW173) and A/Jena/5258/09 (Jena5258). After the initial infection of 30 min and treatment with 0.1 and 1 µM of both free and encapsulated 6BIGOE in A549 (Figure 3a, c, e) and Calu-3 cells (Figure 3b, d, f) for 24 h, progeny infectious virus particles were quantified by plaque assay. Independent of the cell line used, treatment with both 6BIGOE and PLGA NP [6BIGOE] resulted in a concentration-dependent reduction in virus titers for all three virus strains, with the inhibitory effect being slightly more pronounced for free 6BIGOE. Thus, these data indicate that 6BIGOE inhibits the replication of IAV.To confirm the inhibitory effect of 6BIGOE on viral replication and to further examine possible cellular targets, infection experiments were carried out within the first replication cycle of IAV. Here, the expression of IAV hemagglutinin (HA) and polymerase basic protein 1 (PB1) was monitored at 4 h, 6 h, and 8 h p.i. by Western blot analysis in A549 (Figure 3g) and Calu-3 cells (Figure 3h). Furthermore, the effects of IAV infection on GSK-3β phosphorylation and 6BIGOE-mediated effects were analyzed compared to the solvent control (DMSO). In A549 cells expression of HA was already detectable at 6 h p.i. and was reduced in presence of 6BIGOE. However, this reduction was no longer apparent at 8 h p.i., while PB1 levels remained suppressed at this time point. In Calu-3 cells, viral protein expression seemed to be delayed. Nonetheless, viral protein expression was detectable at 8 h p.i., with a visible reduction in PB1 and HA levels following 6BIGOE treatment in comparison to the DMSO-treated control. In presence of 6BIGOE the IAV-induced GSK-3β phosphorylation, particularly at 8 h p.i. is visible, correlating to the reduction of viral protein expression. During IAV infection, WNK1 and PRAS40 phosphorylation occurs in both cell lines, notably at 8 h p.i., and this activation was reduced by treatment with 6BIGOE.
These findings indicate that treatment of IAV-infected epithelial cells with 6BIGOE at non-toxic concentrations effectively reduces viral load and propagation by regulation of GSK-3β-mediated signaling.
4. Discussion
Given the global health threat associated with IAV infections, effective treatment strategies are essential to mitigate the significant number of infections and associated deaths. Targeting host intracellular factors that support virus replication is of particular interest as this reduces the risk of resistance development. Furthermore, repurposing of existing therapeutics and substances with pharmaceutical effects saves both development costs and time. The recent study examines the potential use of 6BIGOE as a GSK-3β inhibitor against IAV infections.
However, due to the physicochemical properties of bioactive small molecules such as indirubin derivatives, several challenges are associated, including poor water solubility [47,48] and membrane permeability [28]. Additionally, the short half-life in blood of nearly 1 hour after oral administration in mice [49] and cytotoxic effects against different cell lines [36] are further disadvantages. These issues can be addressed by encapsulating the drug in polymeric NPs made of PLGA, which is approved by the FDA and EMA for different controlled drug release systems. A similar approach was employed by Czapka et al., where encapsulating 6BIGOE repressed the off target effects of the free drug on human monocytes [44].
In order to overcome such drug related challenges, 6BIGOE was encapsulated into spherical PLGA NP with a HD below 200 nm and narrow and homogeneous particle size distributions. HD, PDI, ZP, and EE of the formulated PLGA NPs were found to be comparable to those reported in previous studies [44,45]. The increase in ZP after encapsulation of 6BIGOE may be attributed to a part of the drug being located on or near the particle surface, since the drug is likely uncharged at neutral pH. This observation correlates with the burst-release observed in drug release studies previously conducted for PLGA NP [6BIGOE] [44].
Our findings indicate that encapsulation of 6BIGOE in PLGA NPs reduced metabolic effects and LDH release in lung epithelial cells (A549 and Calu-3 cells). This may be due to the slower release of 6BIGOE from the NPs, resulting in lower cytotoxicity. Incorporation of 6BIGOE into PLGA NPs might lower the effective local concentration; but intracellular concentrations appeared comparable, as indicated by similar effects on GSK-3β phosphorylation observed. Additionally, off-target interactions with outer membranes, linked to cytotoxicity and premature drug degradation, could potentially be reduced. PLGA is known to deliver poorly soluble drugs into tissues like tumors [50,51] or epithelial cells [52] enhancing their effect. Since a comparable antiviral effect could be observed in our study and the cytotoxic effects of 6BIGOE were attenuated, it can be concluded that a higher dose of encapsulated 6BIGOE could be administered, even for a longer period of time. In addition, encapsulating small molecules into NPs may have more significant impact in vivo by enhancing circulation time. For instance, an encapsulated indirubin derivative was 984% more bioavailable than its free form following oral administration in rats [53].
6BIGOE exhibited a greater impact on A549 cells than on Calu-3 cells, which could tolerate up to 2 µM of free 6BIGOE. While both cell lines are human lung adenocarcinoma cells, they differ significantly in characteristics [54]. Calu-3 cells form a thin mucus layer resembling the bronchial submucosal glands [55], whereas A549 cells, lacking this feature represent type II alveolar cells and may be more sensitive to external agents [56]. To further characterize the molecular effects of 6BIGOE in the encapsulated and free form and to determine the concentrations suitable for further application in vivo, more complex cell culture systems, lung organoid, ex vivo lung models as well as in vivo animal models must be used for evaluation.
Indirubins are recognized for their antiviral properties against various viruses, including adenoviruses [30] and IAVs [31]. Derivatives such as indirubin-3′-(2,3-dihydroxypropyl)-oxime ether and indirubin-3′-oxime (I3O) were already tested in terms of their anti-inflammatory and antiviral effects in endothelial cells and macrophages [31,57]. Infection with a H9N2 or H5N1 IAV strain increased cytokine and chemokine release, which was suppressed in presence of indirubin derivatives. In these studies, mitogen-activated protein kinases (MAPKs) and activator of transcription 3 (STAT3) proteins were discussed to play a role in the reduction of indirubin-mediated anti-inflammatory effects [57]. Interestingly, 6BIGOE has already been shown to counteract GSK-3β during inflammation in human monocytes, by reducing TLR4-induced release of pro-inflammatory mediators, including IL-1β, IL-6, TNFα, IL-8 und PGE2 [35]. While the present study did not assess the anti-inflammatory properties of 6BIGOE, further research is warranted to investigate its potential to counteract the IAV-induced cytokine storms in vitro, particularly in various immune cells and lung epithelial cell lines. I3O has been shown to delay viral replication by inhibiting viral gene transcription and translation [31]. In our study, treatment with 6BIGOE resulted in delayed expression of HA protein in A549 cells at 6 h p.i., but at 8 h p.i., the viral protein expression had caught up and was similar to that in the DMSO control.
GSK-3β is a regulatory factor of various signaling pathways, including PI3K/Akt-mediated signaling and GSK-3β is induced during viral replication [20,58,59]. Since PI3K/Akt signaling has supportive functions during IAV replication, it is not surprising that inhibition of GSK-3β with 6BIGOE also results in reduced viral load. The use of non-toxic concentrations of 6BIGOE suggests that it selectively inhibits viral replication rather than disrupting cellular processes. RNA interference screening identified 295 cellular host factors that play a crucial role in influenza virus replication, including GSK-3β, though its exact mechanistic role remains unclear [10]. Previous studies reported that GSK-3 phosphorylates a key residue on the GTPase dynamin I which is involved in clathrin-mediated endocytosis [60,61]. By inhibiting GSK-3β the formation and maturation of clathrin-coated pits are promoted [60,62]. Considering the importance of receptor-mediated endocytosis in IAV entry, further research is necessary to evaluate whether inhibiting GSK-3β with 6BIGOE could disrupt endocytic processes and ultimately limit viral entry into host cells. This may occur because the enhanced formation of clathrin-coated pits could accelerate the internalization of surface receptors, leaving fewer available for virus binding and entry.
Overall, the PI3K/Akt signaling pathway, including GSK-3β, is a promising target for influenza virus intervention [59,63,64] as its inhibition interferes with replication processes from viral entry to release. In this study, IAV infection also activated PRAS40 and WNK1, although their precise role in IAV replication is largely unknown. PRAS40, a 40 kDa substrate, has a regulatory function at the intersection of the protein kinase B/Akt and mTOR signaling pathway [65,66,67]. mTOR is a highly conserved serine/threonine kinase that is part of two distinct multi-protein complexes, mTOR complex 1 (mTORC1) and 2 (mTORC2), which are well studied in diseases such as cancer and type 2 diabtes [68,69,70]. mTORC1 itself plays a pivotal role in the regulation of cellular protein synthesis, proliferation, and metabolism, particularly in response to nutrients and growth factors [68]. Interestingly, mTORC1 activation has recently been implicated in facilitating influenza virus replication, highlighting its potential role as a host factor in viral infections. However, the role of mTORC2 in IAV infection remains poorly understood and warrants further investigation [71,72]. The relationship between the PI3K/Akt cascade and the WNK protein kinase signaling pathway, which primarily regulates ion transport across cell membranes, has been extensively studied in the context of cancer [73,74,75]. PI3K/Akt signaling can activate the WNK-oxidative stress responsive kinase 1 (OSR1)/PS/Ste20-related proline-alanine-rich kinase (SPAK)-NaCl cotransporter (NCC) pathway in a hyperinsulinemic db/db mouse model, providing critical evidence of Akt-mediated kinase activation of WNKs [76]. Although the regulation and function of PRAS40 or WNK1 during IAV infection is not yet elucidated, the interaction with die PI3K/Akt signaling pathway might explain the reduced phosphorylation of PRAS40 and WNK1 through 6BIGOE-mediated GSK-3β inhibition.
5. Conclusions
In conclusion, this study highlights 6BIGOE as a promising candidate for anti-influenza intervention. Encapsulated 6BIGOE into PLGA NPs exhibits lower cytotoxicity and impact on cellular metabolic activity compared to its free form, while maintaining effective GSK-3β inhibition. 6BIGOE and PLGA NP [6BIGOE] show antiviral properties against H1N1 IAV strains in lung epithelial cells, likely through modulation of GSK-3β signaling. To further elucidate the mechanisms mediated by GSK-3β during IAV infection, additional research is required. Moreover, investigations in more complex technical systems are essential.
Author Contributions
Conceptualization, C.E. and J.S..; methodology, J.S., J.W. and S.H..; software, J.S., J.W. and S.H; validation, J.S., J.W. and S.H.; formal analysis, J.S., J.W. and S.H.; investigation, J.S., J.W. and S.H.; resources, C.E., D.F. and B.L.; data curation, J.S..; writing—original draft preparation, J.S. and J.W.; writing—review and editing, C.E., J.W., D.F., S.H., O.W., B.L., I.V. and J.S..; visualization, J.S..; supervision, C.E. and D.F..; project administration, C.E..; funding acquisition, C.E., D.F. and B.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the German Research Foundation (SFB 1278/2 PolyTarget- 316213987—D02, C02, Z01).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original Western blot pictures were submitted with this article.
Acknowledgments
We would like to thank Jessica Lux and Heike Urban for their excellent technical assistance.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Clayville, L.R. Influenza update: a review of currently available vaccines. P T 2011, 36, 659-684.
- Iuliano, A.D.; Roguski, K.M.; Chang, H.H.; Muscatello, D.J.; Palekar, R.; Tempia, S.; Cohen, C.; Gran, J.M.; Schanzer, D.; Cowling, B.J.; et al. Estimates of global seasonal influenza-associated respiratory mortality: a modelling study. Lancet 2018, 391, 1285-1300. [CrossRef]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol Rev 1992, 56, 152-179. [CrossRef]
- Fiebach, N.; Beckett, W. Prevention of respiratory infections in adults. Influenza and pneumococcal vaccines. Arch Intern Med 1994, 154, 2545-2557.
- Bonomini, A.; Mercorelli, B.; Loregian, A. Antiviral strategies against influenza virus: an update on approved and innovative therapeutic approaches. Cell Mol Life Sci 2025, 82, 75. [CrossRef]
- Duwe, S.C.; Schmidt, B.; Gartner, B.C.; Timm, J.; Adams, O.; Fickenscher, H.; Schmidtke, M. Prophylaxis and treatment of influenza: options, antiviral susceptibility, and existing recommendations. GMS Infect Dis 2021, 9, Doc02. [CrossRef]
- Hurt, A.C. The epidemiology and spread of drug resistant human influenza viruses. Curr Opin Virol 2014, 8, 22-29. [CrossRef]
- Meineke, R.; Rimmelzwaan, G.F.; Elbahesh, H. Influenza Virus Infections and Cellular Kinases. Viruses 2019, 11. [CrossRef]
- Fichera, E.; Felnerova, D.; Mischler, R.; Viret, J.F.; Glueck, R. New strategies to overcome the drawbacks of currently available flu vaccines. Adv Exp Med Biol 2009, 655, 243-252. [CrossRef]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813-817. [CrossRef]
- Nagini, S.; Sophia, J.; Mishra, R. Glycogen synthase kinases: Moonlighting proteins with theranostic potential in cancer. Semin Cancer Biol 2019, 56, 25-36. [CrossRef]
- Hermida, M.A.; Dinesh Kumar, J.; Leslie, N.R. GSK3 and its interactions with the PI3K/AKT/mTOR signalling network. Adv Biol Regul 2017, 65, 5-15. [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxid Med Cell Longev 2017, 2017, 4629495. [CrossRef]
- Lin, J.; Song, T.; Li, C.; Mao, W. GSK-3beta in DNA repair, apoptosis, and resistance of chemotherapy, radiotherapy of cancer. Biochim Biophys Acta Mol Cell Res 2020, 1867, 118659. [CrossRef]
- Terzioglu-Usak, S.; Nalli, A.; Elibol, B.; Ozek, E.; Hatiboglu, M.A. Anvirzel(TM)regulates cell death through inhibiting GSK-3 activity in human U87 glioma cells. Neurol Res 2020, 42, 68-75. [CrossRef]
- Kramer, T.; Schmidt, B.; Lo Monte, F. Small-Molecule Inhibitors of GSK-3: Structural Insights and Their Application to Alzheimer's Disease Models. Int J Alzheimers Dis 2012, 2012, 381029. [CrossRef]
- Mathuram, T.L.; Reece, L.M.; Cherian, K.M. GSK-3 Inhibitors: A Double-Edged Sword? - An Update on Tideglusib. Drug Res (Stuttg) 2018, 68, 436-443. [CrossRef]
- Ring, D.B.; Johnson, K.W.; Henriksen, E.J.; Nuss, J.M.; Goff, D.; Kinnick, T.R.; Ma, S.T.; Reeder, J.W.; Samuels, I.; Slabiak, T.; et al. Selective glycogen synthase kinase 3 inhibitors potentiate insulin activation of glucose transport and utilization in vitro and in vivo. Diabetes 2003, 52, 588-595. [CrossRef]
- Hoffmeister, L.; Diekmann, M.; Brand, K.; Huber, R. GSK3: A Kinase Balancing Promotion and Resolution of Inflammation. Cells 2020, 9. [CrossRef]
- Alfhili, M.A.; Alsughayyir, J.; McCubrey, J.A.; Akula, S.M. GSK-3-associated signaling is crucial to virus infection of cells. Biochim Biophys Acta Mol Cell Res 2020, 1867, 118767. [CrossRef]
- Guendel, I.; Iordanskiy, S.; Van Duyne, R.; Kehn-Hall, K.; Saifuddin, M.; Das, R.; Jaworski, E.; Sampey, G.C.; Senina, S.; Shultz, L.; et al. Novel neuroprotective GSK-3beta inhibitor restricts Tat-mediated HIV-1 replication. J Virol 2014, 88, 1189-1208. [CrossRef]
- Wang, T.; Zhang, J.; Xiao, A.; Liu, W.; Shang, Y.; An, J. Melittin ameliorates CVB3-induced myocarditis via activation of the HDAC2-mediated GSK-3beta/Nrf2/ARE signaling pathway. Biochem Biophys Res Commun 2016, 480, 126-131. [CrossRef]
- Yuan, J.; Zhang, J.; Wong, B.W.; Si, X.; Wong, J.; Yang, D.; Luo, H. Inhibition of glycogen synthase kinase 3beta suppresses coxsackievirus-induced cytopathic effect and apoptosis via stabilization of beta-catenin. Cell Death Differ 2005, 12, 1097-1106. [CrossRef]
- Rahaus, M.; Desloges, N.; Wolff, M.H. Varicella-zoster virus requires a functional PI3K/Akt/GSK-3alpha/beta signaling cascade for efficient replication. Cell Signal 2007, 19, 312-320. [CrossRef]
- Wu, C.H.; Yeh, S.H.; Tsay, Y.G.; Shieh, Y.H.; Kao, C.L.; Chen, Y.S.; Wang, S.H.; Kuo, T.J.; Chen, D.S.; Chen, P.J. Glycogen synthase kinase-3 regulates the phosphorylation of severe acute respiratory syndrome coronavirus nucleocapsid protein and viral replication. J Biol Chem 2009, 284, 5229-5239. [CrossRef]
- Hirata, N.; Suizu, F.; Matsuda-Lennikov, M.; Edamura, T.; Bala, J.; Noguchi, M. Inhibition of Akt kinase activity suppresses entry and replication of influenza virus. Biochem Biophys Res Commun 2014, 450, 891-898. [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: progress, challenges and recommendations. Nat Rev Drug Discov 2019, 18, 41-58. [CrossRef]
- Gaboriaud-Kolar, N.; Vougogiannopoulou, K.; Skaltsounis, A.L. Indirubin derivatives: a patent review (2010 - present). Expert Opin Ther Pat 2015, 25, 583-593. [CrossRef]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.Z.; Mandelkow, E.M.; et al. Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer's disease. A property common to most cyclin-dependent kinase inhibitors? J Biol Chem 2001, 276, 251-260. [CrossRef]
- Wang, J.; Yang, C.; Liang, Z.; Sun, J.; Zhang, M.; Qiu, S.; Du, X.; He, X.; Pang, X.; Ma, X.; et al. Indirubin-3'-monoxime exhibits potent antiviral and anti-inflammatory effects against human adenoviruses in vitro and in vivo. Biomed Pharmacother 2024, 174, 116558. [CrossRef]
- Mok, C.K.; Kang, S.S.; Chan, R.W.; Yue, P.Y.; Mak, N.K.; Poon, L.L.; Wong, R.N.; Peiris, J.S.; Chan, M.C. Anti-inflammatory and antiviral effects of indirubin derivatives in influenza A (H5N1) virus infected primary human peripheral blood-derived macrophages and alveolar epithelial cells. Antiviral Res 2014, 106, 95-104. [CrossRef]
- Heredia, A.; Davis, C.; Bamba, D.; Le, N.; Gwarzo, M.Y.; Sadowska, M.; Gallo, R.C.; Redfield, R.R. Indirubin-3'-monoxime, a derivative of a Chinese antileukemia medicine, inhibits P-TEFb function and HIV-1 replication. AIDS 2005, 19, 2087-2095. [CrossRef]
- Hsuan, S.L.; Chang, S.C.; Wang, S.Y.; Liao, T.L.; Jong, T.T.; Chien, M.S.; Lee, W.C.; Chen, S.S.; Liao, J.W. The cytotoxicity to leukemia cells and antiviral effects of Isatis indigotica extracts on pseudorabies virus. J Ethnopharmacol 2009, 123, 61-67. [CrossRef]
- Chang, S.J.; Chang, Y.C.; Lu, K.Z.; Tsou, Y.Y.; Lin, C.W. Antiviral Activity of Isatis indigotica Extract and Its Derived Indirubin against Japanese Encephalitis Virus. Evid Based Complement Alternat Med 2012, 2012, 925830. [CrossRef]
- Czapka, A.; Konig, S.; Pergola, C.; Grune, C.; Vougogiannopoulou, K.; Skaltsounis, A.L.; Fischer, D.; Werz, O. The indirubin derivative 6-bromoindirubin-3'-glycerol-oxime ether (6BIGOE) potently modulates inflammatory cytokine and prostaglandin release from human monocytes through GSK-3 interference. Biochem Pharmacol 2020, 180, 114170. [CrossRef]
- Dan, N.T.; Quang, H.D.; Van Truong, V.; Huu Nghi, D.; Cuong, N.M.; Cuong, T.D.; Toan, T.Q.; Bach, L.G.; Anh, N.H.T.; Mai, N.T.; et al. Design, synthesis, structure, in vitro cytotoxic activity evaluation and docking studies on target enzyme GSK-3beta of new indirubin-3'-oxime derivatives. Sci Rep 2020, 10, 11429. [CrossRef]
- Savjani, K.T.; Gajjar, A.K.; Savjani, J.K. Drug solubility: importance and enhancement techniques. ISRN Pharm 2012, 2012, 195727. [CrossRef]
- Su, Y.; Zhang, B.; Sun, R.; Liu, W.; Zhu, Q.; Zhang, X.; Wang, R.; Chen, C. PLGA-based biodegradable microspheres in drug delivery: recent advances in research and application. Drug Deliv 2021, 28, 1397-1418. [CrossRef]
- Mahar, R.; Chakraborty, A.; Nainwal, N.; Bahuguna, R.; Sajwan, M.; Jakhmola, V. Application of PLGA as a Biodegradable and Biocompatible Polymer for Pulmonary Delivery of Drugs. AAPS PharmSciTech 2023, 24, 39. [CrossRef]
- Kwok, P.C.; Chan, H.K. Nanotechnology versus other techniques in improving drug dissolution. Curr Pharm Des 2014, 20, 474-482. [CrossRef]
- Dobrucki, L.W.; Pan, D.; Smith, A.M. Multiscale Imaging of Nanoparticle Drug Delivery. Curr Drug Targets 2015, 16, 560-570. [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel) 2011, 3, 1377-1397. [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Le Breton, A.; Preat, V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 2012, 161, 505-522. [CrossRef]
- Czapka, A.; Grune, C.; Schadel, P.; Bachmann, V.; Scheuer, K.; Dirauf, M.; Weber, C.; Skaltsounis, A.L.; Jandt, K.D.; Schubert, U.S.; et al. Drug delivery of 6-bromoindirubin-3'-glycerol-oxime ether employing poly(D,L-lactide-co-glycolide)-based nanoencapsulation techniques with sustainable solvents. J Nanobiotechnology 2022, 20, 5. [CrossRef]
- Bachmann, V.; Schadel, P.; Westhoff, J.; Peric, M.; Schomberg, F.; Skaltsounis, A.L.; Hoppener, S.; Pantsar, T.; Fischer, D.; Vilotijevic, I.; Werz, O. Bromo-substituted indirubins for inhibition of protein kinase-mediated signalling involved in inflammatory mediator release in human monocytes. Bioorg Chem 2024, 149, 107470. [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012, 9, 671-675. [CrossRef]
- Vougogiannopoulou, K.; Ferandin, Y.; Bettayeb, K.; Myrianthopoulos, V.; Lozach, O.; Fan, Y.; Johnson, C.H.; Magiatis, P.; Skaltsounis, A.L.; Mikros, E.; Meijer, L. Soluble 3',6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase -3 alter circadian period. J Med Chem 2008, 51, 6421-6431. [CrossRef]
- Heshmati, N.; Wagner, B.; Cheng, X.; Scholz, T.; Kansy, M.; Eisenbrand, G.; Fricker, G. Physicochemical characterization and in vitro permeation of an indirubin derivative. Eur J Pharm Sci 2013, 50, 467-475. [CrossRef]
- Tchoumtchoua, J.; Halabalaki, M.; Gikas, E.; Tsarbopoulos, A.; Fotaki, N.; Liu, L.; Nam, S.; Jove, R.; Skaltsounis, L.A. Preliminary pharmacokinetic study of the anticancer 6BIO in mice using an UHPLC-MS/MS approach. J Pharm Biomed Anal 2019, 164, 317-325. [CrossRef]
- Mundekkad, D.; Cho, W.C. Nanoparticles in Clinical Translation for Cancer Therapy. Int J Mol Sci 2022, 23. [CrossRef]
- Feng, S.S.; Mu, L.; Win, K.Y.; Huang, G. Nanoparticles of biodegradable polymers for clinical administration of paclitaxel. Curr Med Chem 2004, 11, 413-424. [CrossRef]
- Cartiera, M.S.; Johnson, K.M.; Rajendran, V.; Caplan, M.J.; Saltzman, W.M. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials 2009, 30, 2790-2798. [CrossRef]
- Heshmati, N.; Cheng, X.; Eisenbrand, G.; Fricker, G. Enhancement of oral bioavailability of E804 by self-nanoemulsifying drug delivery system (SNEDDS) in rats. J Pharm Sci 2013, 102, 3792-3799. [CrossRef]
- Wiese-Rischke, C.; Murkar, R.S.; Walles, H. Biological Models of the Lower Human Airways-Challenges and Special Requirements of Human 3D Barrier Models for Biomedical Research. Pharmaceutics 2021, 13. [CrossRef]
- Shen, B.Q.; Finkbeiner, W.E.; Wine, J.J.; Mrsny, R.J.; Widdicombe, J.H. Calu-3: a human airway epithelial cell line that shows cAMP-dependent Cl- secretion. Am J Physiol 1994, 266, L493-501. [CrossRef]
- Lieber, M.; Smith, B.; Szakal, A.; Nelson-Rees, W.; Todaro, G. A continuous tumor-cell line from a human lung carcinoma with properties of type II alveolar epithelial cells. Int J Cancer 1976, 17, 62-70. [CrossRef]
- Kwok, H.H.; Poon, P.Y.; Fok, S.P.; Ying-Kit Yue, P.; Mak, N.K.; Chan, M.C.; Peiris, J.S.; Wong, R.N. Anti-inflammatory effects of indirubin derivatives on influenza A virus-infected human pulmonary microvascular endothelial cells. Sci Rep 2016, 6, 18941. [CrossRef]
- Ehrhardt, C.; Ludwig, S. A new player in a deadly game: influenza viruses and the PI3K/Akt signalling pathway. Cell Microbiol 2009, 11, 863-871. [CrossRef]
- Ehrhardt, C.; Wolff, T.; Pleschka, S.; Planz, O.; Beermann, W.; Bode, J.G.; Schmolke, M.; Ludwig, S. Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. J Virol 2007, 81, 3058-3067. [CrossRef]
- Srinivasan, S.; Burckhardt, C.J.; Bhave, M.; Chen, Z.; Chen, P.H.; Wang, X.; Danuser, G.; Schmid, S.L. A noncanonical role for dynamin-1 in regulating early stages of clathrin-mediated endocytosis in non-neuronal cells. PLoS Biol 2018, 16, e2005377. [CrossRef]
- Clayton, E.L.; Sue, N.; Smillie, K.J.; O'Leary, T.; Bache, N.; Cheung, G.; Cole, A.R.; Wyllie, D.J.; Sutherland, C.; Robinson, P.J.; Cousin, M.A. Dynamin I phosphorylation by GSK3 controls activity-dependent bulk endocytosis of synaptic vesicles. Nat Neurosci 2010, 13, 845-851. [CrossRef]
- Reis, C.R.; Chen, P.H.; Srinivasan, S.; Aguet, F.; Mettlen, M.; Schmid, S.L. Crosstalk between Akt/GSK3beta signaling and dynamin-1 regulates clathrin-mediated endocytosis. EMBO J 2015, 34, 2132-2146. [CrossRef]
- Denisova, O.V.; Soderholm, S.; Virtanen, S.; Von Schantz, C.; Bychkov, D.; Vashchinkina, E.; Desloovere, J.; Tynell, J.; Ikonen, N.; Theisen, L.L.; et al. Akt inhibitor MK2206 prevents influenza pH1N1 virus infection in vitro. Antimicrob Agents Chemother 2014, 58, 3689-3696. [CrossRef]
- Wu, M.S.; Yen, H.R.; Chang, C.W.; Peng, T.Y.; Hsieh, C.F.; Chen, C.J.; Lin, T.Y.; Horng, J.T. Mechanism of action of the suppression of influenza virus replication by Ko-Ken Tang through inhibition of the phosphatidylinositol 3-kinase/Akt signaling pathway and viral RNP nuclear export. J Ethnopharmacol 2011, 134, 614-623. [CrossRef]
- Kovacina, K.S.; Park, G.Y.; Bae, S.S.; Guzzetta, A.W.; Schaefer, E.; Birnbaum, M.J.; Roth, R.A. Identification of a proline-rich Akt substrate as a 14-3-3 binding partner. J Biol Chem 2003, 278, 10189-10194. [CrossRef]
- Nascimento, E.B.; Snel, M.; Guigas, B.; van der Zon, G.C.; Kriek, J.; Maassen, J.A.; Jazet, I.M.; Diamant, M.; Ouwens, D.M. Phosphorylation of PRAS40 on Thr246 by PKB/AKT facilitates efficient phosphorylation of Ser183 by mTORC1. Cell Signal 2010, 22, 961-967. [CrossRef]
- Lin, J.; Fang, Y.; Zhang, M.; Wang, X.; Li, L.; He, M.; Xue, A.; Zhu, K.; Shen, Y.; Li, B. Phosphorylation of PRAS40 contributes to the activation of the PI3K/AKT/mTOR signaling pathway and the inhibition of autophagy following status epilepticus in rats. Exp Ther Med 2020, 20, 3625-3632. [CrossRef]
- Wiza, C.; Nascimento, E.B.; Ouwens, D.M. Role of PRAS40 in Akt and mTOR signaling in health and disease. Am J Physiol Endocrinol Metab 2012, 302, E1453-1460. [CrossRef]
- Lv, D.; Guo, L.; Zhang, T.; Huang, L. PRAS40 signaling in tumor. Oncotarget 2017, 8, 69076-69085. [CrossRef]
- Zhou, Q.; Tang, S.; Zhang, X.; Chen, L. Targeting PRAS40: a novel therapeutic strategy for human diseases. J Drug Target 2021, 29, 703-715. [CrossRef]
- Mata, M.A.; Satterly, N.; Versteeg, G.A.; Frantz, D.; Wei, S.; Williams, N.; Schmolke, M.; Pena-Llopis, S.; Brugarolas, J.; Forst, C.V.; et al. Chemical inhibition of RNA viruses reveals REDD1 as a host defense factor. Nat Chem Biol 2011, 7, 712-719. [CrossRef]
- Kuss-Duerkop, S.K.; Wang, J.; Mena, I.; White, K.; Metreveli, G.; Sakthivel, R.; Mata, M.A.; Munoz-Moreno, R.; Chen, X.; Krammer, F.; et al. Influenza virus differentially activates mTORC1 and mTORC2 signaling to maximize late stage replication. PLoS Pathog 2017, 13, e1006635. [CrossRef]
- Martini, M.; De Santis, M.C.; Braccini, L.; Gulluni, F.; Hirsch, E. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med 2014, 46, 372-383. [CrossRef]
- Morgensztern, D.; McLeod, H.L. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs 2005, 16, 797-803. [CrossRef]
- Gallolu Kankanamalage, S.; Karra, A.S.; Cobb, M.H. WNK pathways in cancer signaling networks. Cell Commun Signal 2018, 16, 72. [CrossRef]
- Nishida, H.; Sohara, E.; Nomura, N.; Chiga, M.; Alessi, D.R.; Rai, T.; Sasaki, S.; Uchida, S. Phosphatidylinositol 3-kinase/Akt signaling pathway activates the WNK-OSR1/SPAK-NCC phosphorylation cascade in hyperinsulinemic db/db mice. Hypertension 2012, 60, 981-990. [CrossRef]
Figure 1.
6BIGOE is successfully encapsulated into PLGA NPs. (a) PLGA NPs were prepared by an emulsion-diffusion-evaporation (EDE) method. (b) Table shows particle size (hydrodynamic diameter), polydispersity index (PDI), zeta potential (ZP), drug load (DL) and encapsulation efficiency (EE) of PLGA NP [blank] and PLGA NP [6BIGOE]. (c) Chemical structure of 6BIGOE encapsulated into a PLGA NP. Transmission electron microscopy (TEM) show the PLGA NP [blank] and PLGA NP [6BIGOE]. Scale bars represent 500 nm. Data represent the means ± SD (b) or a representation (c) of three independent batches.
Figure 1.
6BIGOE is successfully encapsulated into PLGA NPs. (a) PLGA NPs were prepared by an emulsion-diffusion-evaporation (EDE) method. (b) Table shows particle size (hydrodynamic diameter), polydispersity index (PDI), zeta potential (ZP), drug load (DL) and encapsulation efficiency (EE) of PLGA NP [blank] and PLGA NP [6BIGOE]. (c) Chemical structure of 6BIGOE encapsulated into a PLGA NP. Transmission electron microscopy (TEM) show the PLGA NP [blank] and PLGA NP [6BIGOE]. Scale bars represent 500 nm. Data represent the means ± SD (b) or a representation (c) of three independent batches.
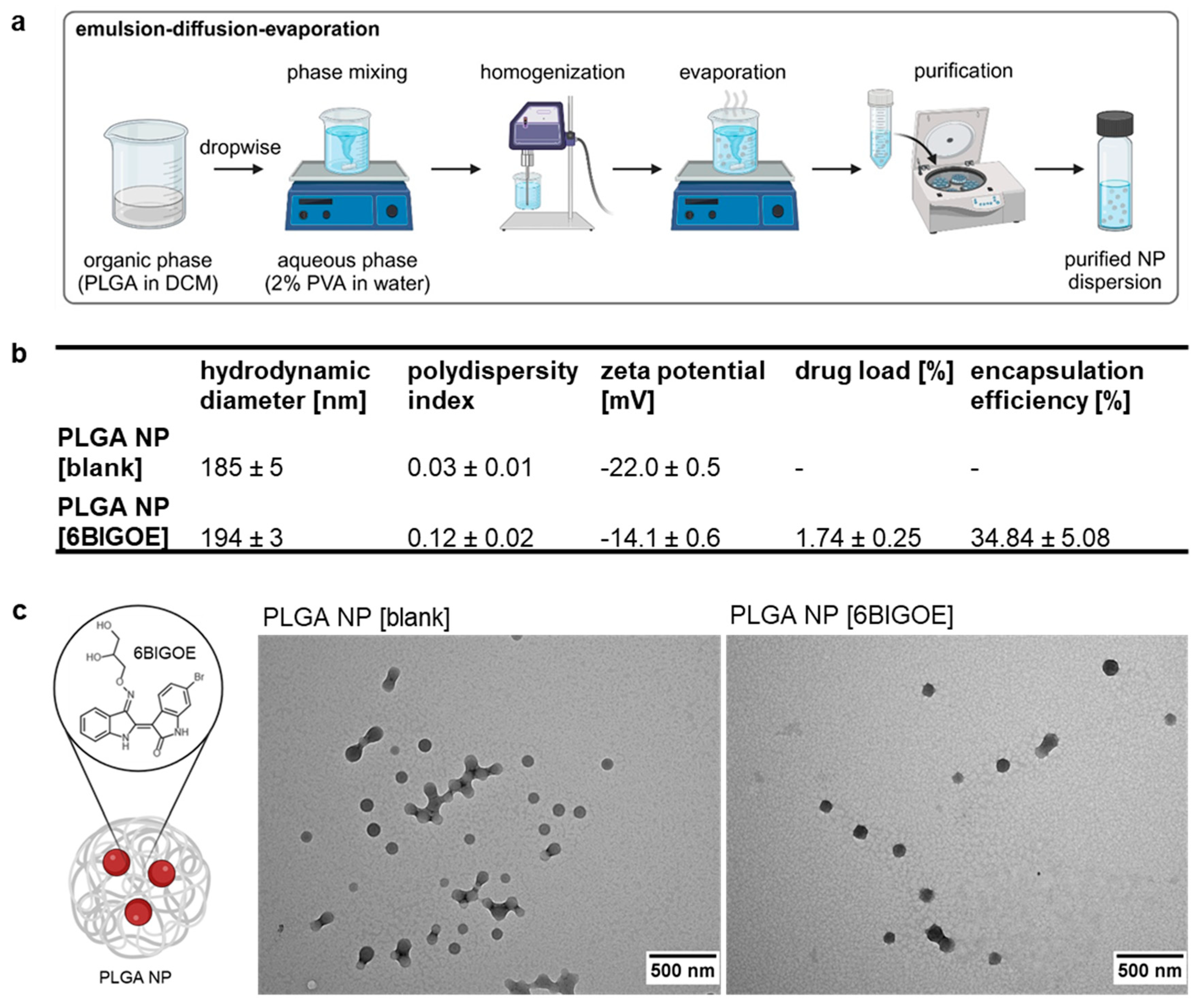
Figure 2.
PLGA NP [6BIGOE] has less impact on the cellular metabolic activity than free 6BIGOE. A549 (a ,c ,e) and Calu-3 (b ,d , f) cells were treated with the indicated concentrations of 6BIGOE, PLGA NP [6BIGOE] or PLGA NP [blank] for 24 h (a-d) or 18 h (e, f). (a, b) The metabolic activity was measured using MTT assay and normalized to the solvent control-treated samples (DMSO, H2O). The data represent the mean ± SD of four independent experiments with two technical replicates. Values are given in percent of the solvent-treated control. Statistical significance was determined by two-way ANOVA with Dunnett’s multiple comparisons test. ***p<0.001, ****p<0.0001. (c, d) LDH level was measured by using cell supernatants after 24 h treatment with the indicated substances. The lysis sample served as the maximum LDH release control and was arbitrarily set to 100%, medium-treated samples were used as spontaneous LDH release control and represent 0%. The data represent the mean ± SD of three independent experiments with two technical replicates. Values are given in percent of the lysis buffer-treated control. Statistical significance was determined by two-way ANOVA with Dunnett’s multiple comparisons test. ****p<0.0001. (e, f) Phosphorylation status of GSK-3β was measured with Western blot analysis in A549 (e) and Calu-3 (f) cells for DMSO, pictilisib and different concentrations of 6BIGOE and PLGA NP [6BIGOE]. Quantification was performed by using α tubulin as loading control via Fiji ImageJ. Data represent the mean ± SD of three independent experiments. Statistical significance was determined by one-sample t-test comparing to a hypothetical mean of 1. *p<0.05, **p<0.01.
Figure 2.
PLGA NP [6BIGOE] has less impact on the cellular metabolic activity than free 6BIGOE. A549 (a ,c ,e) and Calu-3 (b ,d , f) cells were treated with the indicated concentrations of 6BIGOE, PLGA NP [6BIGOE] or PLGA NP [blank] for 24 h (a-d) or 18 h (e, f). (a, b) The metabolic activity was measured using MTT assay and normalized to the solvent control-treated samples (DMSO, H2O). The data represent the mean ± SD of four independent experiments with two technical replicates. Values are given in percent of the solvent-treated control. Statistical significance was determined by two-way ANOVA with Dunnett’s multiple comparisons test. ***p<0.001, ****p<0.0001. (c, d) LDH level was measured by using cell supernatants after 24 h treatment with the indicated substances. The lysis sample served as the maximum LDH release control and was arbitrarily set to 100%, medium-treated samples were used as spontaneous LDH release control and represent 0%. The data represent the mean ± SD of three independent experiments with two technical replicates. Values are given in percent of the lysis buffer-treated control. Statistical significance was determined by two-way ANOVA with Dunnett’s multiple comparisons test. ****p<0.0001. (e, f) Phosphorylation status of GSK-3β was measured with Western blot analysis in A549 (e) and Calu-3 (f) cells for DMSO, pictilisib and different concentrations of 6BIGOE and PLGA NP [6BIGOE]. Quantification was performed by using α tubulin as loading control via Fiji ImageJ. Data represent the mean ± SD of three independent experiments. Statistical significance was determined by one-sample t-test comparing to a hypothetical mean of 1. *p<0.05, **p<0.01.
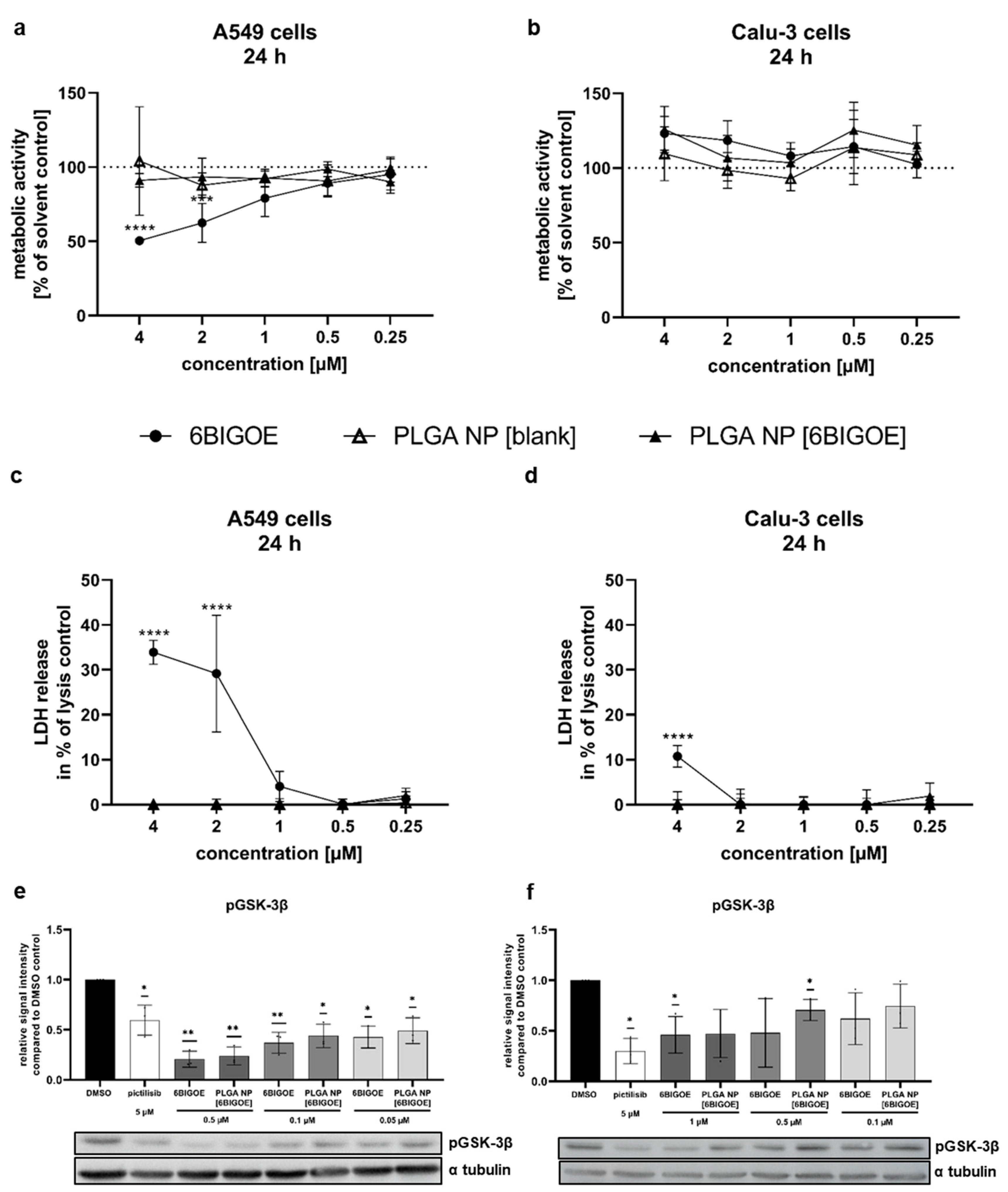
Figure 3.
PLGA NP [6BIGOE] treatment causes an inhibition of IAV replication in vitro through inhibition of GSK-3β. A549 (a, c, e, g) and Calu-3 (b, d, f, h) cells were infected with the indicated IAV strains (PR8 with 5 MOI (g) or 1 MOI (h), Jena5258 and NRW173) for 30 min and were subsequently treated with 0.1 and 1 µM (a-g), 0.5 µM (g) or 1 µM (h) of 6BIGOE, PLGA NP [6BIGOE] or controls (DMSO, PLGA NP [blank]) (a-f). After 24 h of incubation supernatants were collected and progeny virus titers (plaque forming units (PFU)/ml) were determined by plaque assay. The mean + SD of three independent experiments with two technical replicates is depicted. Statistical significance was determined using one-way ANOVA Dunnett's multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001 (a-f). Cell lysates were harvested after 4 h, 6 h and 8 h p.i. for SDS-PAGE and Western blot analysis. Effect of 6BIGOE on, PRAS40, WNK1 and GSK-3β phosphorylation, as well as relative protein expression of viral HA and PB1 is presented. A representative example of three independent experiments is depicted (g, h).
Figure 3.
PLGA NP [6BIGOE] treatment causes an inhibition of IAV replication in vitro through inhibition of GSK-3β. A549 (a, c, e, g) and Calu-3 (b, d, f, h) cells were infected with the indicated IAV strains (PR8 with 5 MOI (g) or 1 MOI (h), Jena5258 and NRW173) for 30 min and were subsequently treated with 0.1 and 1 µM (a-g), 0.5 µM (g) or 1 µM (h) of 6BIGOE, PLGA NP [6BIGOE] or controls (DMSO, PLGA NP [blank]) (a-f). After 24 h of incubation supernatants were collected and progeny virus titers (plaque forming units (PFU)/ml) were determined by plaque assay. The mean + SD of three independent experiments with two technical replicates is depicted. Statistical significance was determined using one-way ANOVA Dunnett's multiple comparisons test. *p<0.05, **p<0.01, ***p<0.001 ****p<0.0001 (a-f). Cell lysates were harvested after 4 h, 6 h and 8 h p.i. for SDS-PAGE and Western blot analysis. Effect of 6BIGOE on, PRAS40, WNK1 and GSK-3β phosphorylation, as well as relative protein expression of viral HA and PB1 is presented. A representative example of three independent experiments is depicted (g, h).

Table 1.
Antibodies for Western blot analysis.
| Target | Manufacturer |
|---|---|
| IAV (HA) | GeneTex (GTX127357) |
| IAV (PB1) | GeneTex (GTX125923) |
| α-tubulin | Cell signaling Technology (2125) |
| HSP90 | Cell signaling Technology (4877) |
| pGSK-3β (Ser9) | Cell signaling Technology (5558) |
| pPRAS40 (Thr246) | Cell signaling Technology (13175) |
| pWNK1 (Thr60) | Cell signaling Technology (4946) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



