Submitted:
08 March 2025
Posted:
11 March 2025
You are already at the latest version
Abstract
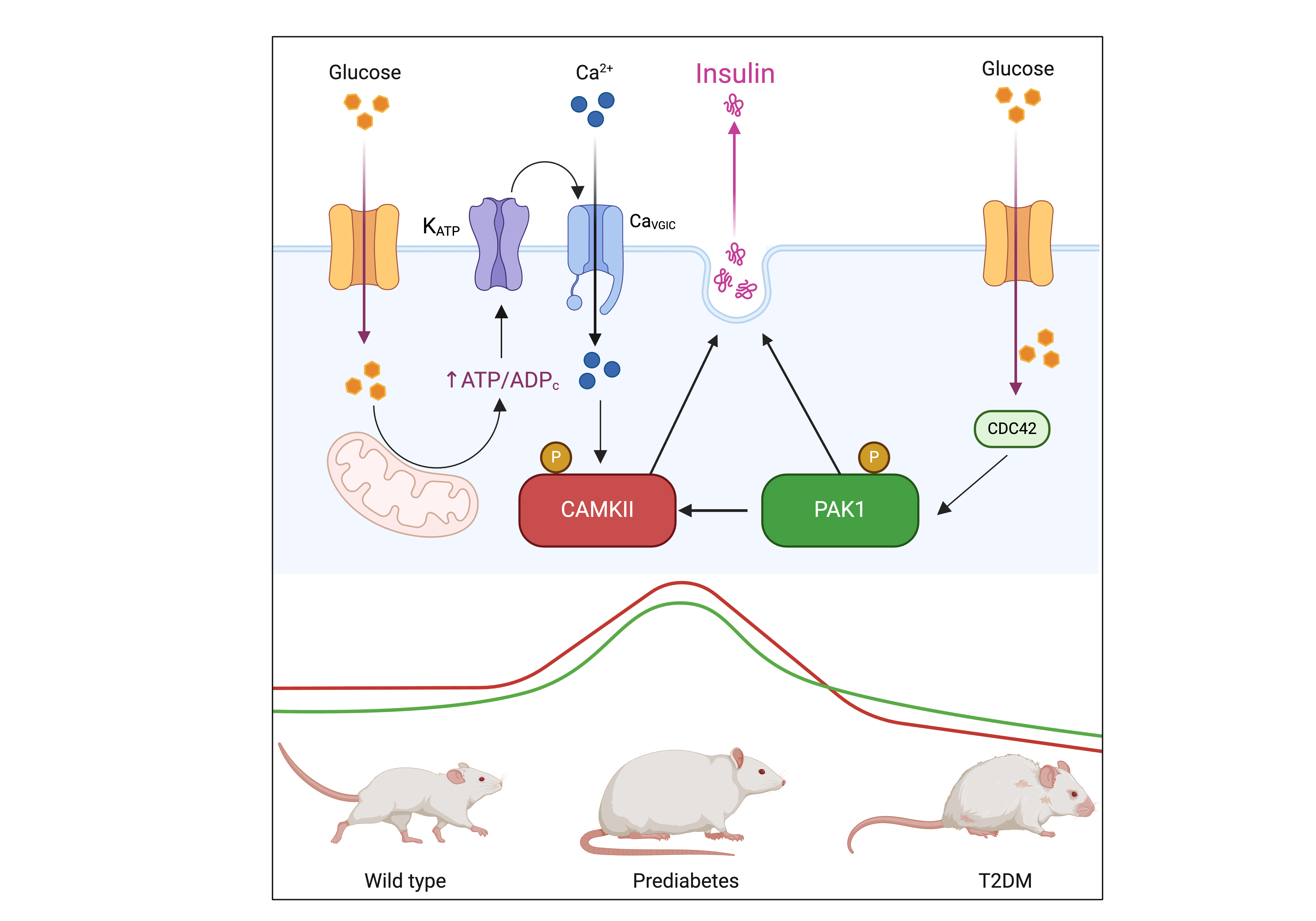
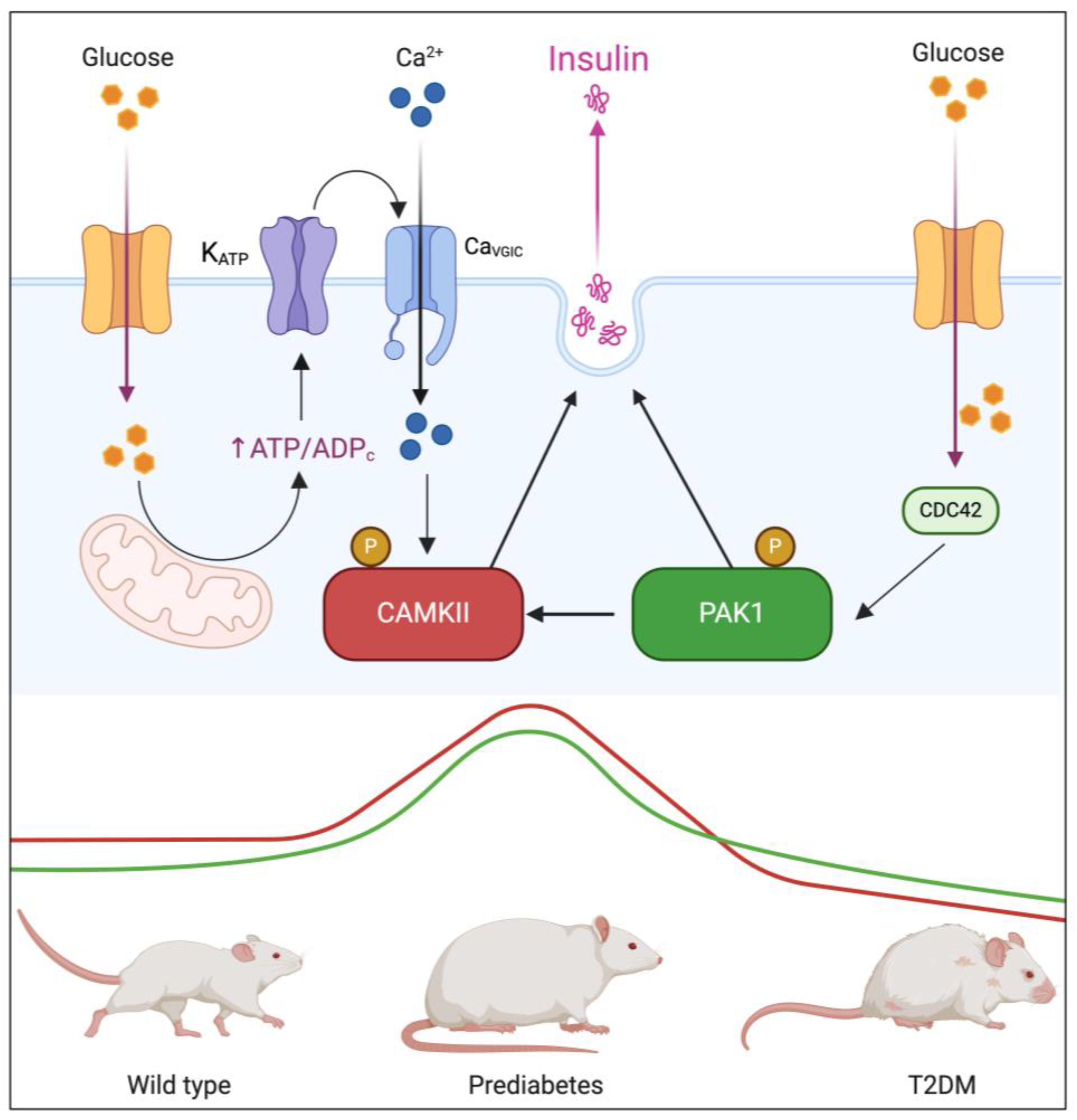
Background/Objectives: Type 2 diabetes mellitus (T2DM) is a major global health challenge, primarily driven by insulin resistance and beta-cell dysfunction. This study investigated the roles of p21-activated kinase 1 (PAK1) and calci-um/calmodulin-dependent protein kinase II (CAMKII) in insulin secretion, aiming to elucidate their involvement in this process and their implications in T2DM pathophysi-ology. Methods: Using the Beta-TC-6 insulinoma cell line, we assessed colocalization and interaction of PAK1 and CAMKII under glucose stimulation through indirect immuno-fluorescence (IFI) and proximity ligation assays (PLA). To examine their expression dynamics in a physiological context, we performed immunohistochemistry (IHC) on pancreatic sections from wild-type (WT), prediabetic, and T2DM murine models. Addi-tionally, bioinformatic analysis of publicly available RNA sequencing (RNA-Seq) data from human islets of healthy donors, prediabetic individuals, and T2DM patients pro-vided translational validation. Results: High glucose conditions significantly increased PAK1-CAMKII colocalization, correlating with enhanced insulin secretion. Pharmaco-logical inhibition of these kinases reduced insulin release, confirming their regulatory roles. Murine and human islet analyses showed a progressive increase in kinase ex-pression from prediabetes to T2DM, highlighting their relevance in disease progression. Conclusions: The coordinated function of PAK1 and CaMKII in insulin secretion suggests their potential as biomarkers and therapeutic targets in T2DM. Further studies are warranted to explore their mechanistic roles and therapeutic applications in preserving beta-cell function.
Keywords:
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Glucose-Stimulated Insulin Secretion Assay (GSIS)
2.2. Immunofluorescence and Confocal Microscopy
2.3. Proximity Ligation Assay (PLA)
2.4. Enzyme-Linked ImmunoSorbent Assay
2.5. Western Blotting
2.6. Animal Model of T2DM and Immunohistochemistry
2.7. Immunohistochemistry.
2.8. Bioinformatic Analysis of RNA-Sequencing Data from Human Pancreatic Islets
2.9. Statistical Analysis
3. Results
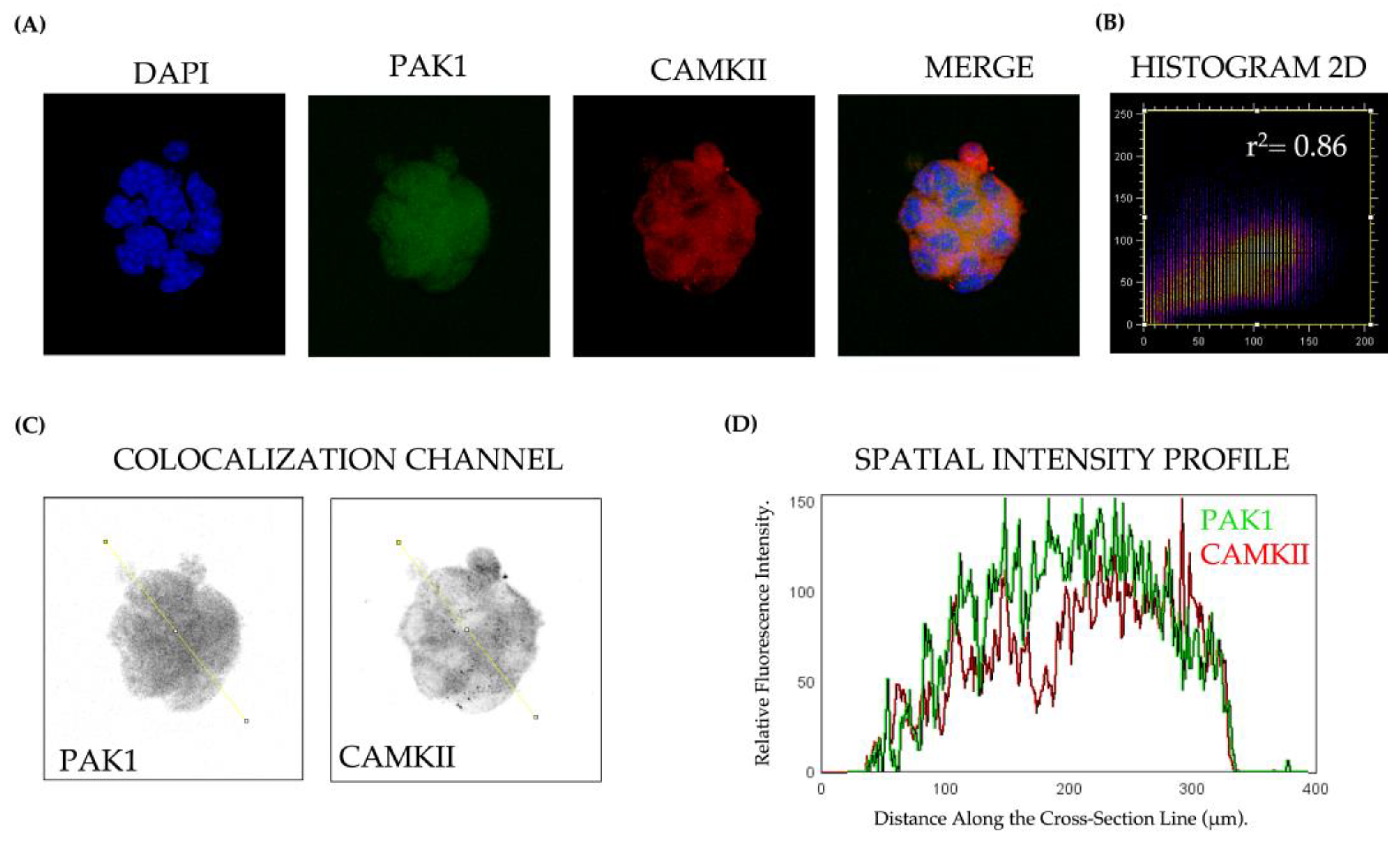
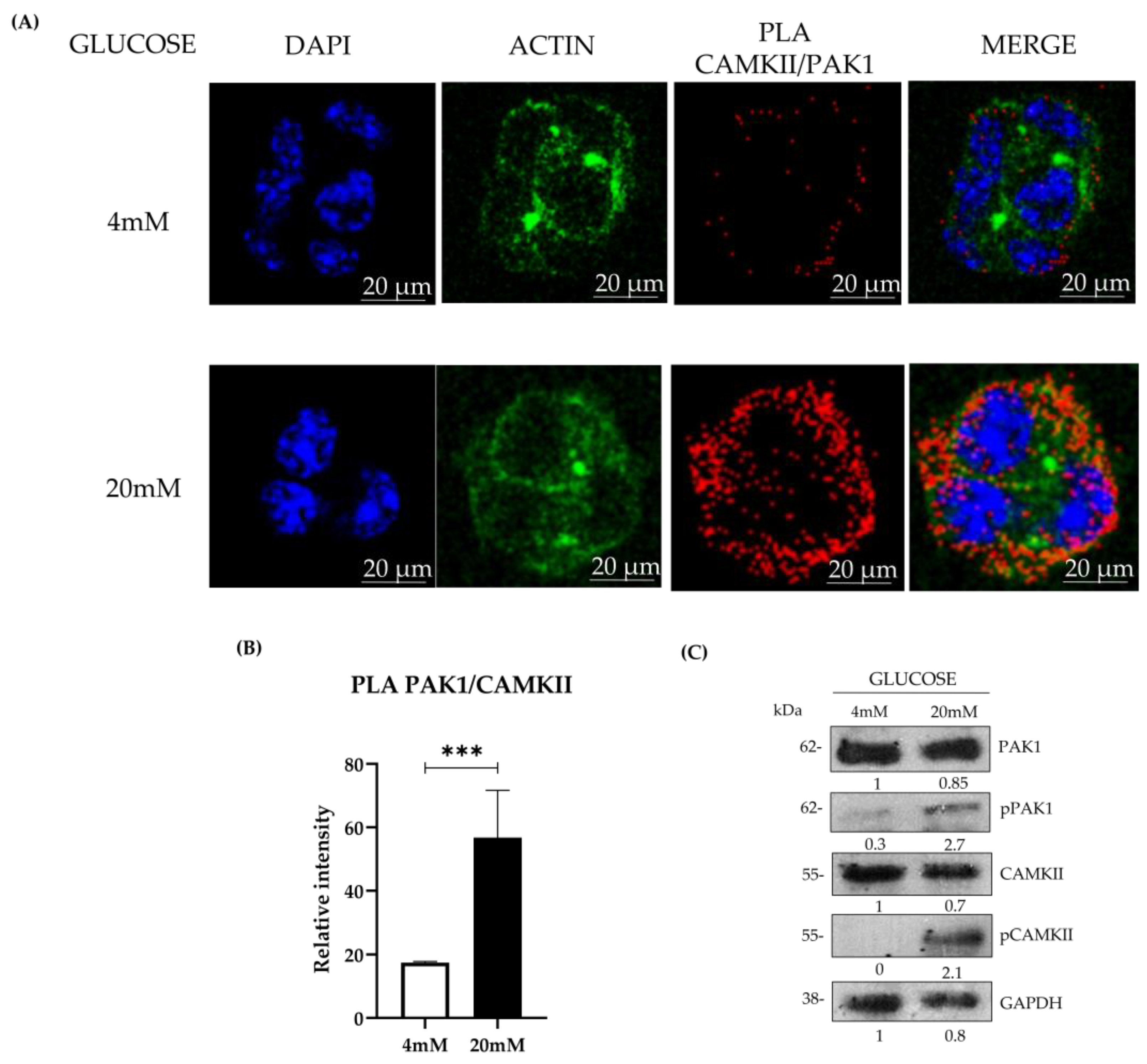
3.1. PAK1 and CAMKII Colocalization and Interaction in Response to Glucose Stimulation
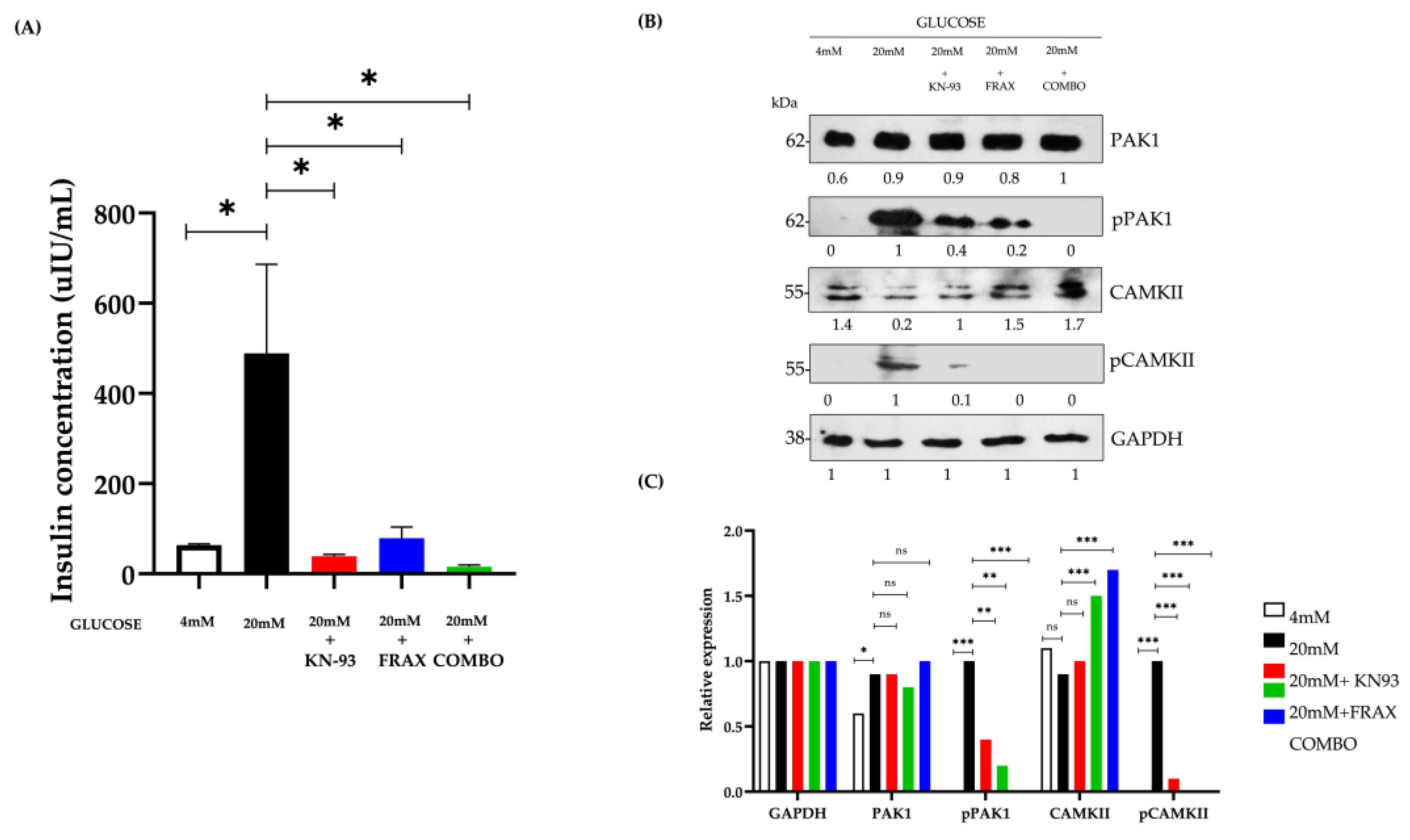
3.2. Dual Kinase Inhibition Disrupts Glucose-Stimulated Insulin Secretion
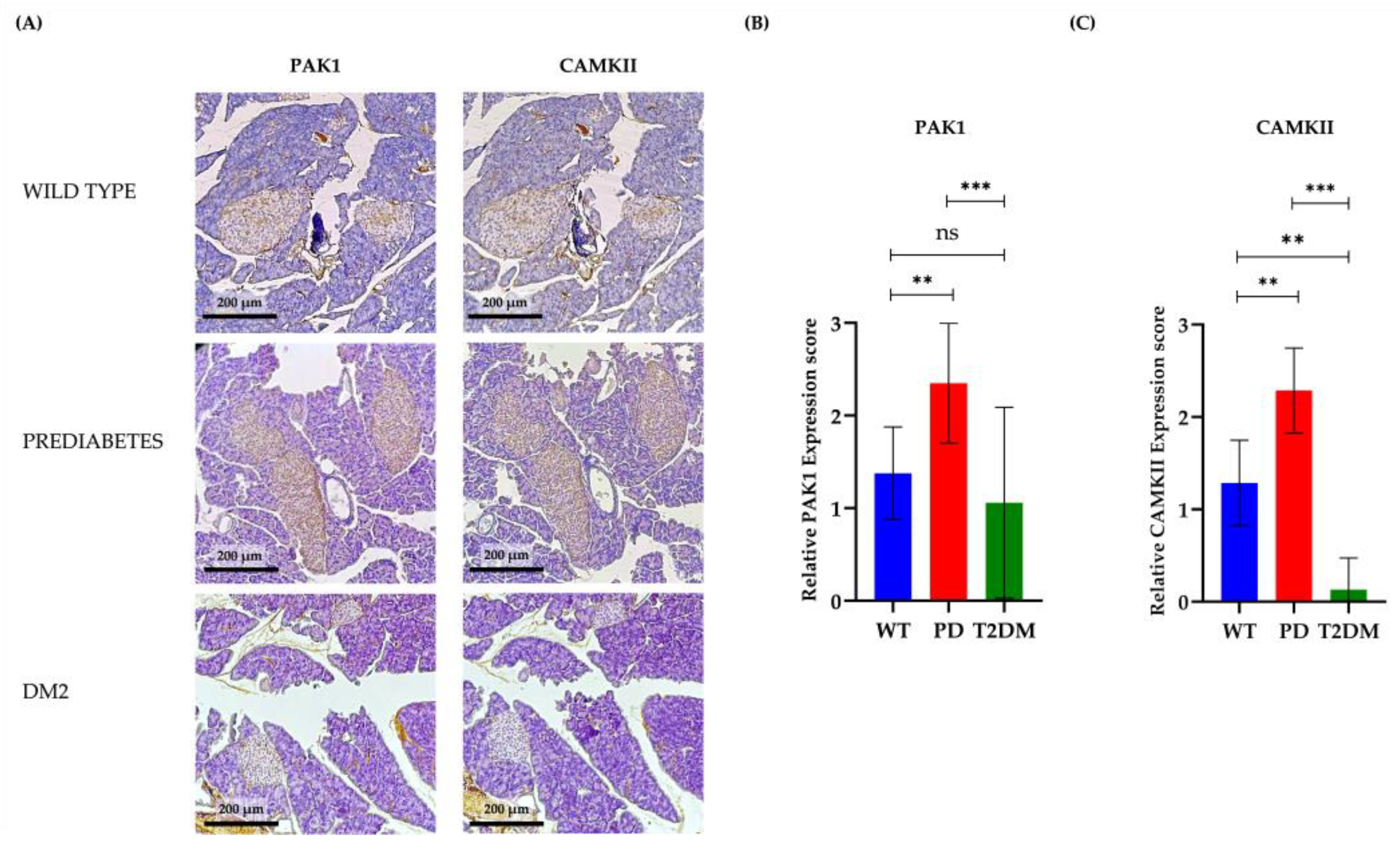
3.3. PAK1 and CAMKII Expression in Pancreatic Islets During Diabetes Progression
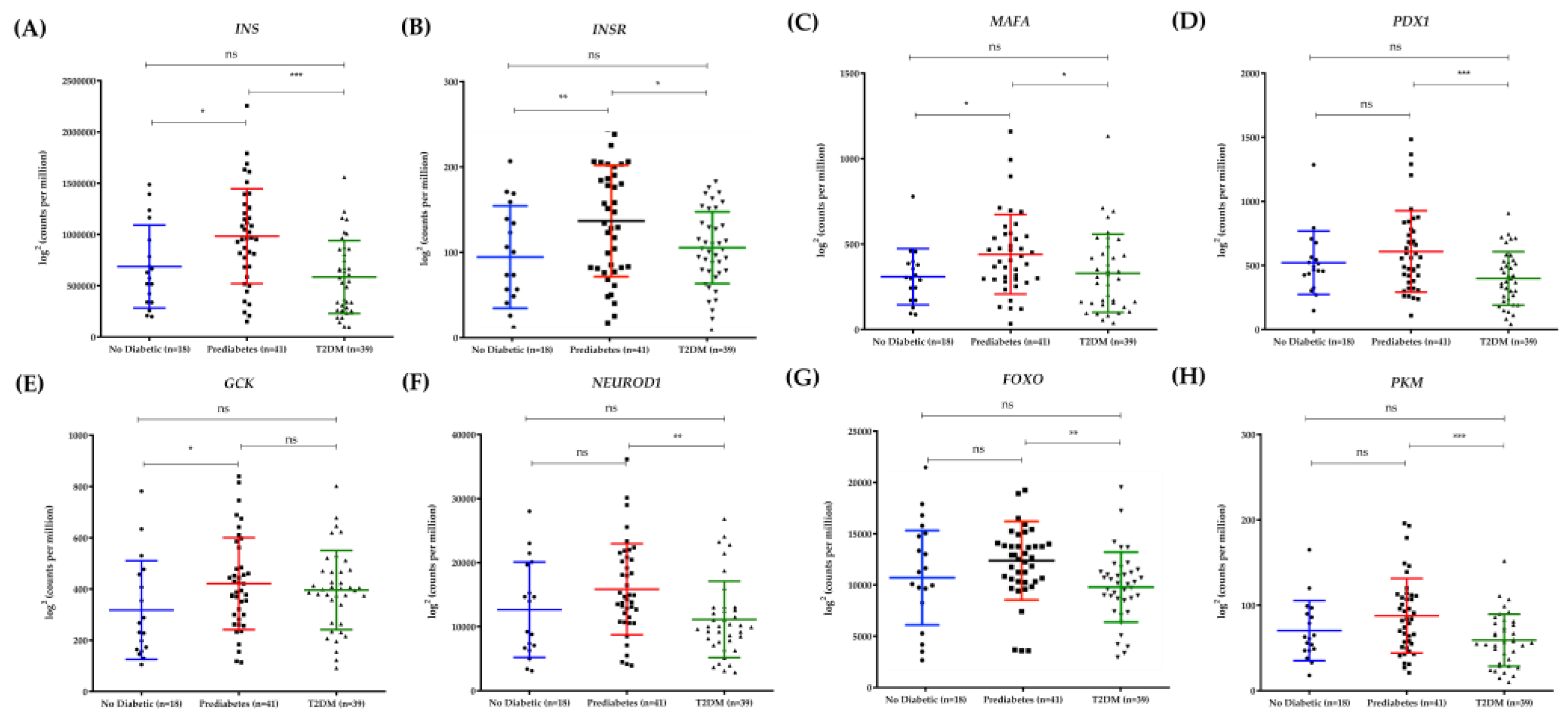
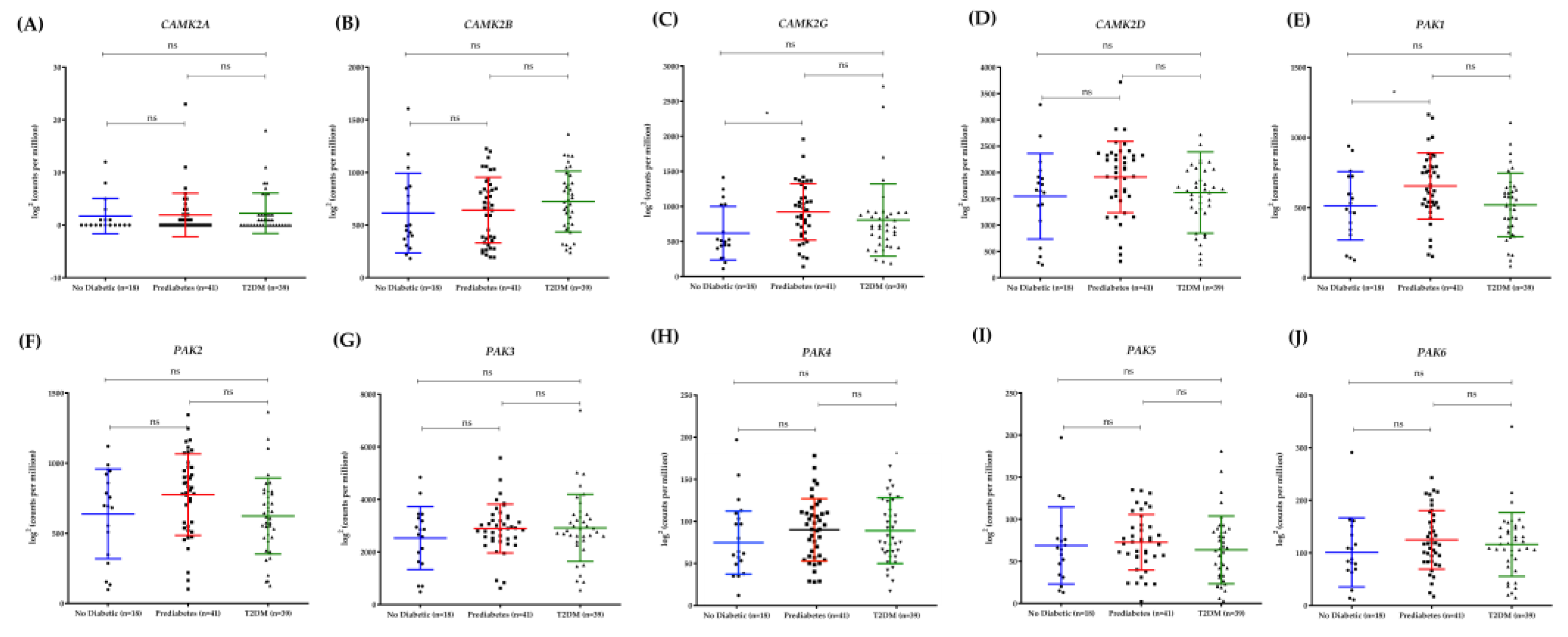
3.4. Differential Expression of PAK1 and CAMK2G in Human Pancreatic Islets Reflects Diabetes Progression
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| T2DM | Type 2 Diabetes Mellitus |
| PAK1 | P21-activated kinase 1 |
| CAMKII | Calcium/calmodulin-dependent protein kinase II |
| GSIS | Glucose-stimulated insulin secretion |
| PLA | Proximity ligation assay |
| PD | Prediabetes |
| INS | Insulin gene |
| INSR | Insulin receptor gene |
| MAFA | Maf bZIP transcription factor A gene |
| PDX1 | Pancreatic and duodenal homeobox 1 gene |
| GCK | Glucokinase gene |
| NEUROD1 | Neurogenic differentiation factor 1 gene |
| FOXO | Forkhead box O gene |
| PKM | Pyruvate kinase muscle isoform gene |
References
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nature reviews. Disease primers 2015, 1, 15019. [CrossRef]
- Rutter, G.A.; Hodson, D.J. Beta cell connectivity in pancreatic islets: a type 2 diabetes target? Cellular and molecular life sciences : CMLS 2015, 72, 453-467. [CrossRef]
- Nauck, M.A.; Meier, J.J. The incretin effect in healthy individuals and those with type 2 diabetes: physiology, pathophysiology, and response to therapeutic interventions. The lancet. Diabetes & endocrinology 2016, 4, 525-536. [CrossRef]
- Rorsman, P.; Eliasson, L.; Renström, E.; Gromada, J.; Barg, S.; Göpel, S. The Cell Physiology of Biphasic Insulin Secretion. News in physiological sciences : an international journal of physiology produced jointly by the International Union of Physiological Sciences and the American Physiological Society 2000, 15, 72-77. [CrossRef]
- Wang, Z.; Thurmond, D.C. Mechanisms of biphasic insulin-granule exocytosis - roles of the cytoskeleton, small GTPases and SNARE proteins. Journal of cell science 2009, 122, 893-903. [CrossRef]
- Nie, J.; Sun, C.; Faruque, O.; Ye, G.; Li, J.; Liang, Q.; Chang, Z.; Yang, W.; Han, X.; Shi, Y. Synapses of amphids defective (SAD-A) kinase promotes glucose-stimulated insulin secretion through activation of p21-activated kinase (PAK1) in pancreatic β-Cells. The Journal of biological chemistry 2012, 287, 26435-26444. [CrossRef]
- Kalwat, M.A.; Yoder, S.M.; Wang, Z.; Thurmond, D.C. A p21-activated kinase (PAK1) signaling cascade coordinately regulates F-actin remodeling and insulin granule exocytosis in pancreatic β cells. Biochemical pharmacology 2013, 85, 808-816. [CrossRef]
- Kumar, R.; Sanawar, R.; Li, X.; Li, F. Structure, biochemistry, and biology of PAK kinases. Gene 2017, 605, 20-31. [CrossRef]
- Semenova, G.; Chernoff, J. Targeting PAK1. Biochemical Society transactions 2017, 45, 79-88. [CrossRef]
- Radu, M.; Semenova, G.; Kosoff, R.; Chernoff, J. PAK signalling during the development and progression of cancer. Nature reviews. Cancer 2014, 14, 13-25. [CrossRef]
- Kichina, J.V.; Goc, A.; Al-Husein, B.; Somanath, P.R.; Kandel, E.S. PAK1 as a therapeutic target. Expert opinion on therapeutic targets 2010, 14, 703-725. [CrossRef]
- Wang, Y.; Wang, S.; Lei, M.; Boyett, M.; Tsui, H.; Liu, W.; Wang, X. The p21-activated kinase 1 (Pak1) signalling pathway in cardiac disease: from mechanistic study to therapeutic exploration. British journal of pharmacology 2018, 175, 1362-1374. [CrossRef]
- Kreis, P.; Barnier, J.V. PAK signalling in neuronal physiology. Cellular signalling 2009, 21, 384-393. [CrossRef]
- Ramos-Alvarez, I.; Jensen, R.T. The Important Role of p21-Activated Kinases in Pancreatic Exocrine Function. Biology 2025, 14. [CrossRef]
- Wang, Z.; Oh, E.; Thurmond, D.C. Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. The Journal of biological chemistry 2007, 282, 9536-9546. [CrossRef]
- Sylow, L.; Kleinert, M.; Pehmøller, C.; Prats, C.; Chiu, T.T.; Klip, A.; Richter, E.A.; Jensen, T.E. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cellular signalling 2014, 26, 323-331. [CrossRef]
- Sylow, L.; Jensen, T.E.; Kleinert, M.; Højlund, K.; Kiens, B.; Wojtaszewski, J.; Prats, C.; Schjerling, P.; Richter, E.A. Rac1 signaling is required for insulin-stimulated glucose uptake and is dysregulated in insulin-resistant murine and human skeletal muscle. Diabetes 2013, 62, 1865-1875. [CrossRef]
- Ahn, M.; Yoder, S.M.; Wang, Z.; Oh, E.; Ramalingam, L.; Tunduguru, R.; Thurmond, D.C. The p21-activated kinase (PAK1) is involved in diet-induced beta cell mass expansion and survival in mice and human islets. Diabetologia 2016, 59, 2145-2155. [CrossRef]
- Kořánová, T.; Dvořáček, L.; Grebeňová, D.; Röselová, P.; Obr, A.; Kuželová, K. PAK1 and PAK2 in cell metabolism regulation. Journal of cellular biochemistry 2022, 123, 375-389. [CrossRef]
- Yasuda, R.; Hayashi, Y.; Hell, J.W. CaMKII: a central molecular organizer of synaptic plasticity, learning and memory. Nature reviews. Neuroscience 2022, 23, 666-682. [CrossRef]
- Reyes Gaido, O.E.; Nkashama, L.J.; Schole, K.L.; Wang, Q.; Umapathi, P.; Mesubi, O.O.; Konstantinidis, K.; Luczak, E.D.; Anderson, M.E. CaMKII as a Therapeutic Target in Cardiovascular Disease. Annual review of pharmacology and toxicology 2023, 63, 249-272. [CrossRef]
- Richter, E.A.; Hargreaves, M. Exercise, GLUT4, and skeletal muscle glucose uptake. Physiological reviews 2013, 93, 993-1017. [CrossRef]
- Dadi, P.K.; Vierra, N.C.; Ustione, A.; Piston, D.W.; Colbran, R.J.; Jacobson, D.A. Inhibition of pancreatic β-cell Ca2+/calmodulin-dependent protein kinase II reduces glucose-stimulated calcium influx and insulin secretion, impairing glucose tolerance. The Journal of biological chemistry 2014, 289, 12435-12445. [CrossRef]
- Santos, G.J.; Ferreira, S.M.; Ortis, F.; Rezende, L.F.; Li, C.; Naji, A.; Carneiro, E.M.; Kaestner, K.H.; Boschero, A.C. Metabolic memory of ß-cells controls insulin secretion and is mediated by CaMKII. Molecular metabolism 2014, 3, 484-489. [CrossRef]
- Hegyi, B.; Bers, D.M.; Bossuyt, J. CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. Journal of molecular and cellular cardiology 2019, 127, 246-259. [CrossRef]
- Saldivar-Cerón, H.I.; Villamar-Cruz, O.; Wells, C.M.; Oguz, I.; Spaggiari, F.; Chernoff, J.; Patiño-López, G.; Huerta-Yepez, S.; Montecillo-Aguado, M.; Rivera-Pazos, C.M.; et al. p21-Activated Kinase 1 Promotes Breast Tumorigenesis via Phosphorylation and Activation of the Calcium/Calmodulin-Dependent Protein Kinase II. Frontiers in cell and developmental biology 2021, 9, 759259. [CrossRef]
- Wigger, L.; Barovic, M.; Brunner, A.D.; Marzetta, F.; Schöniger, E.; Mehl, F.; Kipke, N.; Friedland, D.; Burdet, F.; Kessler, C.; et al. Multi-omics profiling of living human pancreatic islet donors reveals heterogeneous beta cell trajectories towards type 2 diabetes. Nature metabolism 2021, 3, 1017-1031. [CrossRef]
- Seino, S.; Shibasaki, T.; Minami, K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. The Journal of clinical investigation 2011, 121, 2118-2125. [CrossRef]
- Illario, M.; Monaco, S.; Cavallo, A.L.; Esposito, I.; Formisano, P.; D'Andrea, L.; Cipolletta, E.; Trimarco, B.; Fenzi, G.; Rossi, G.; et al. Calcium-calmodulin-dependent kinase II (CaMKII) mediates insulin-stimulated proliferation and glucose uptake. Cellular signalling 2009, 21, 786-792. [CrossRef]
- Li, G.; Hidaka, H.; Wollheim, C.B. Inhibition of voltage-gated Ca2+ channels and insulin secretion in HIT cells by the Ca2+/calmodulin-dependent protein kinase II inhibitor KN-62: comparison with antagonists of calmodulin and L-type Ca2+ channels. Molecular pharmacology 1992, 42, 489-488. [CrossRef]
- Keane, K.N.; Cruzat, V.F.; Carlessi, R.; de Bittencourt, P.I., Jr.; Newsholme, P. Molecular Events Linking Oxidative Stress and Inflammation to Insulin Resistance and β-Cell Dysfunction. Oxidative medicine and cellular longevity 2015, 2015, 181643. [CrossRef]
- Luc, K.; Schramm-Luc, A.; Guzik, T.J.; Mikolajczyk, T.P. Oxidative stress and inflammatory markers in prediabetes and diabetes. Journal of physiology and pharmacology : an official journal of the Polish Physiological Society 2019, 70. [CrossRef]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41-46. [CrossRef]
- Weir, G.C.; Bonner-Weir, S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 2004, 53 Suppl 3, S16-21. [CrossRef]
- Khalid, M.; Alkaabi, J.; Khan, M.A.B.; Adem, A. Insulin Signal Transduction Perturbations in Insulin Resistance. International journal of molecular sciences 2021, 22. [CrossRef]
- Kaihara, K.A.; Dickson, L.M.; Jacobson, D.A.; Tamarina, N.; Roe, M.W.; Philipson, L.H.; Wicksteed, B. β-Cell-specific protein kinase A activation enhances the efficiency of glucose control by increasing acute-phase insulin secretion. Diabetes 2013, 62, 1527-1536. [CrossRef]
- Oakie, A.; Wang, R. β-Cell Receptor Tyrosine Kinases in Controlling Insulin Secretion and Exocytotic Machinery: c-Kit and Insulin Receptor. Endocrinology 2018, 159, 3813-3821. [CrossRef]
- Dixit, S.S.; Wang, T.; Manzano, E.J.; Yoo, S.; Lee, J.; Chiang, D.Y.; Ryan, N.; Respress, J.L.; Yechoor, V.K.; Wehrens, X.H. Effects of CaMKII-mediated phosphorylation of ryanodine receptor type 2 on islet calcium handling, insulin secretion, and glucose tolerance. PloS one 2013, 8, e58655. [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).



