Submitted:
04 March 2025
Posted:
05 March 2025
You are already at the latest version
Abstract
Cystic fibrosis (CF) is a life-threatening disorder caused by mutations in the CFTR gene, leading to defective chloride ion transport and thickened mucus in the respiratory and gastrointestinal systems. CFTR modulators, including ivacaftor, lumacaftor, tezacaftor, and elexacaftor, have improved patient outcomes, but interindividual pharmacokinetic variability and potential drug-drug interactions require therapeutic drug monitoring (TDM) for optimal efficacy and safety. In this context, a liquid chromatography-tandem mass spectrometry (LC-MS/MS) method has been developed and validated for the simul-taneous quantification of CFTR modulators and their major active metabolites in human plasma to support pharmacokinetic studies and routine TDM. The multiplex LC-MS/MS assay was established using plasma protein precipitation, followed by chromatographic separation on an Xselect HSS T3 column and positive electrospray ionization mode detec-tion. The method was validated based on FDA and EMA guidelines for specificity, linear-ity, accuracy, precision, matrix effects, and stability, demonstrating robust performance with excellent precision and accuracy. International interlaboratory comparisons con-firmed the reliability of the assay. The developed method has been applied for monitoring caftors plasma levels for patient TDM and also to research studies in alternative matrices such as breast milk. Future studies will help at characterizing caftors pharmacokinetic variability in patients and drug-drug interactions to further refine personalized dosing strategies.
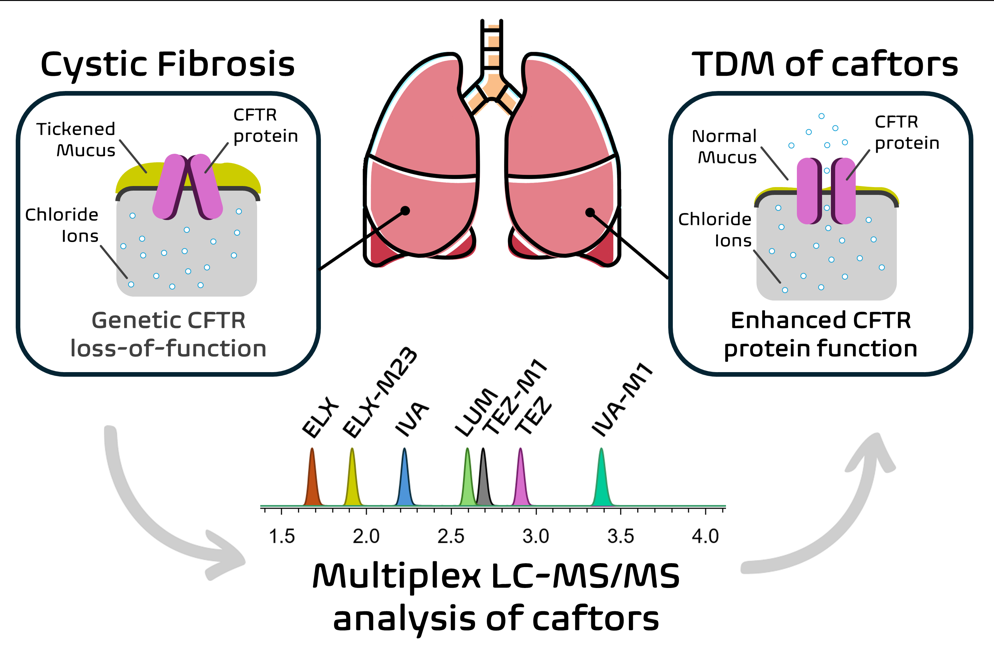
Keywords:
1. Introduction
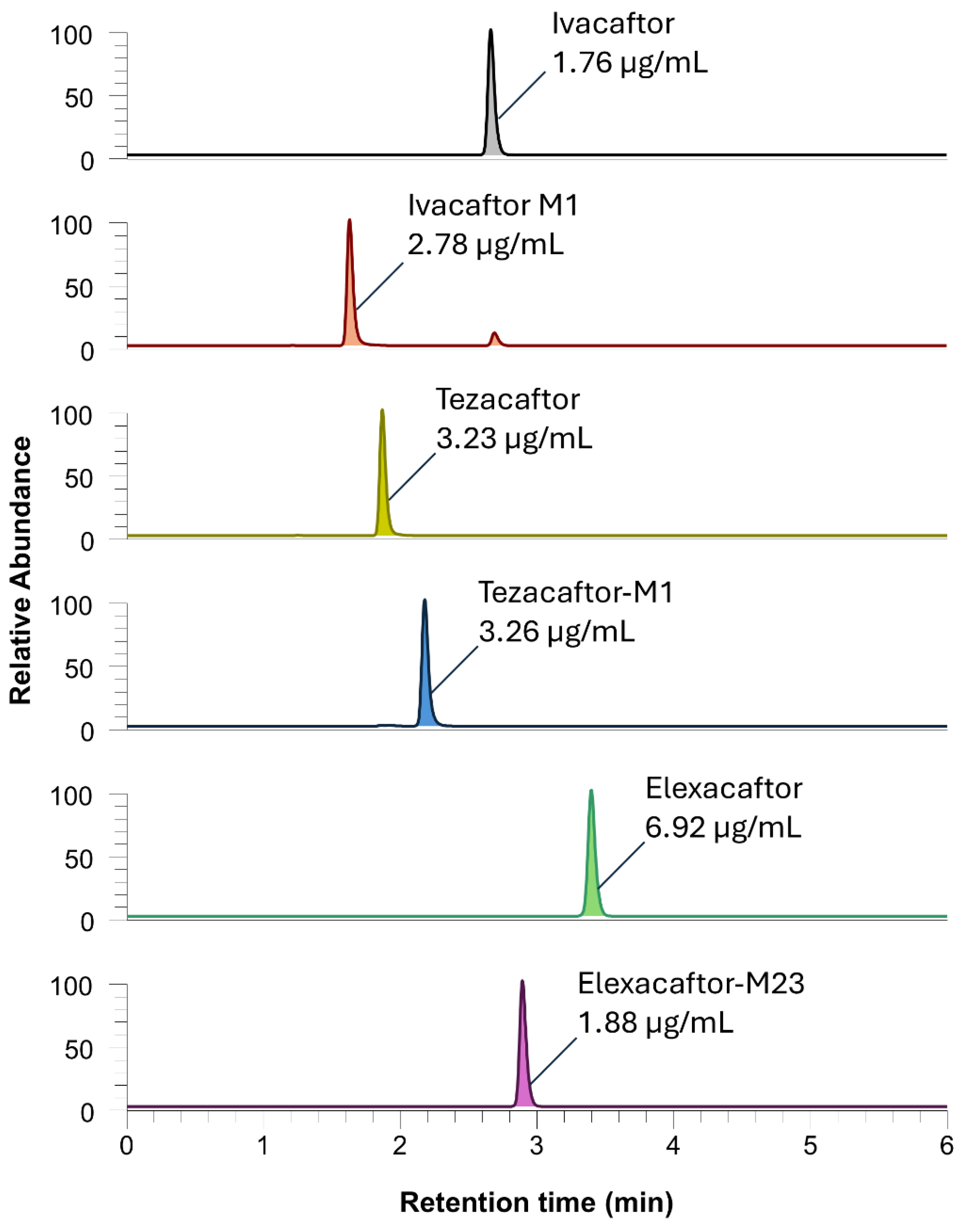
2. Results and Discussion
2.1. Analytical Method Development
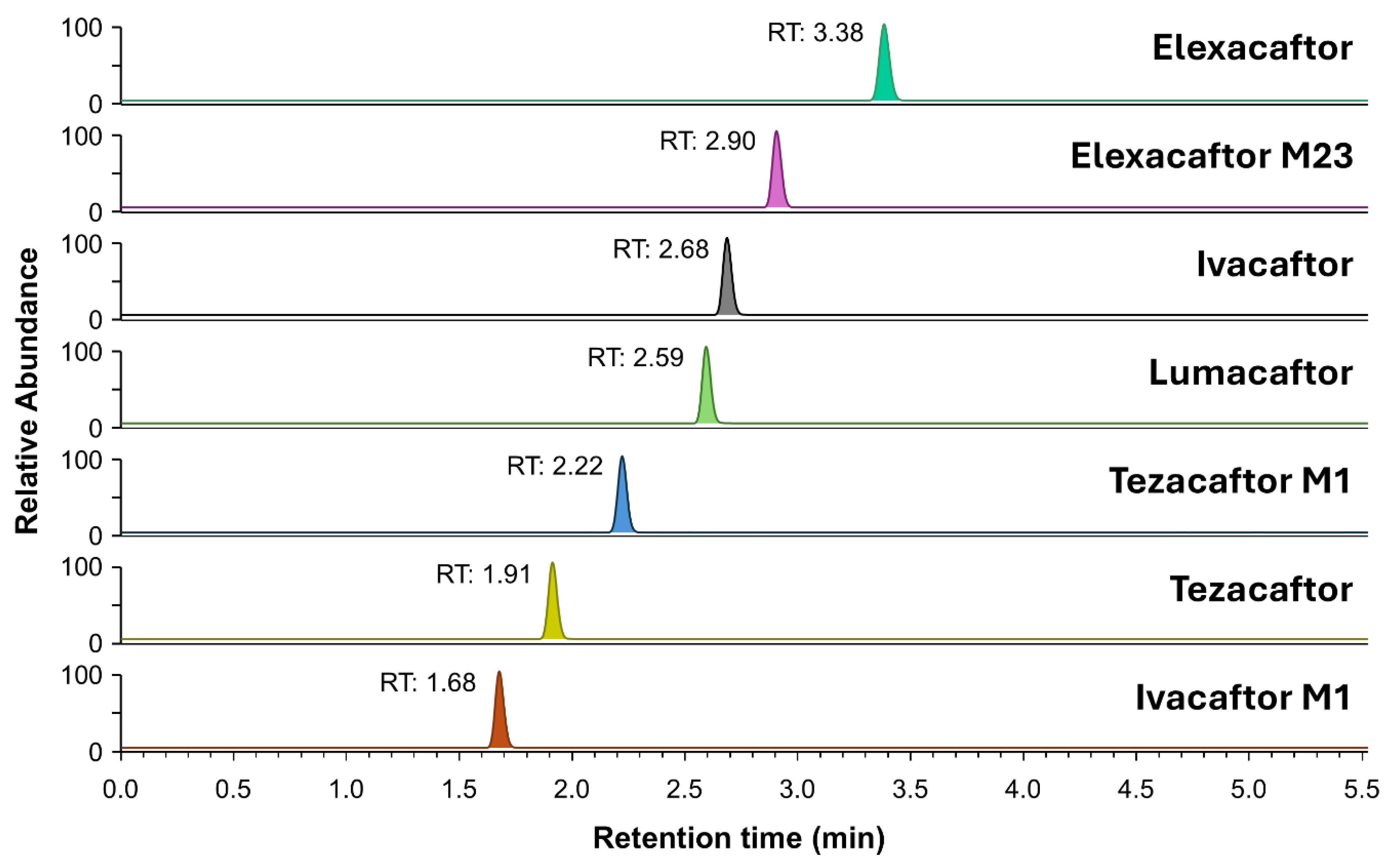
2.2. Validation of the Method
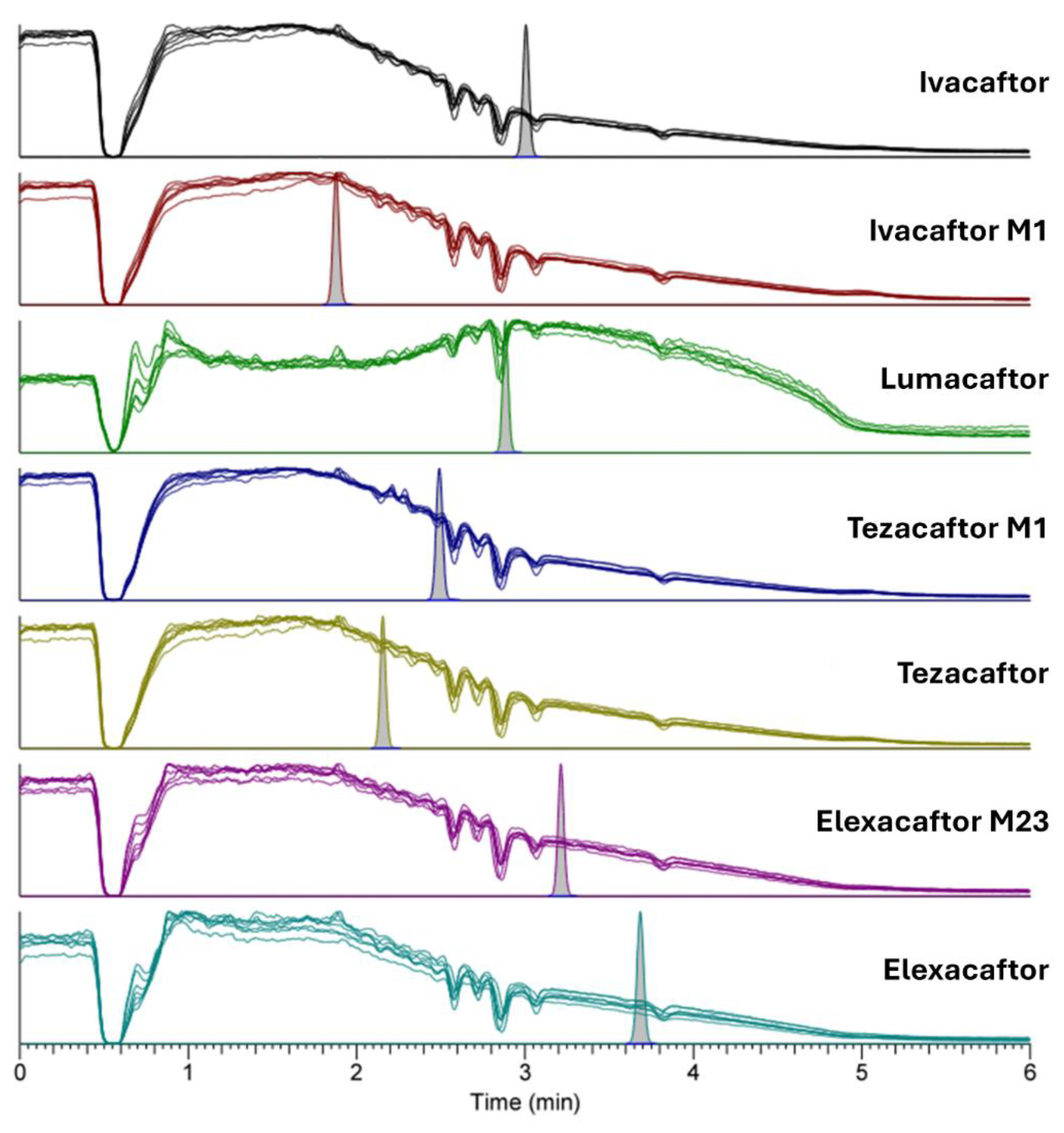
2.2.1. Selectivity, Specificity, Crosstalk, and Carryover
2.2.2. Limit of Detection, and Linearity
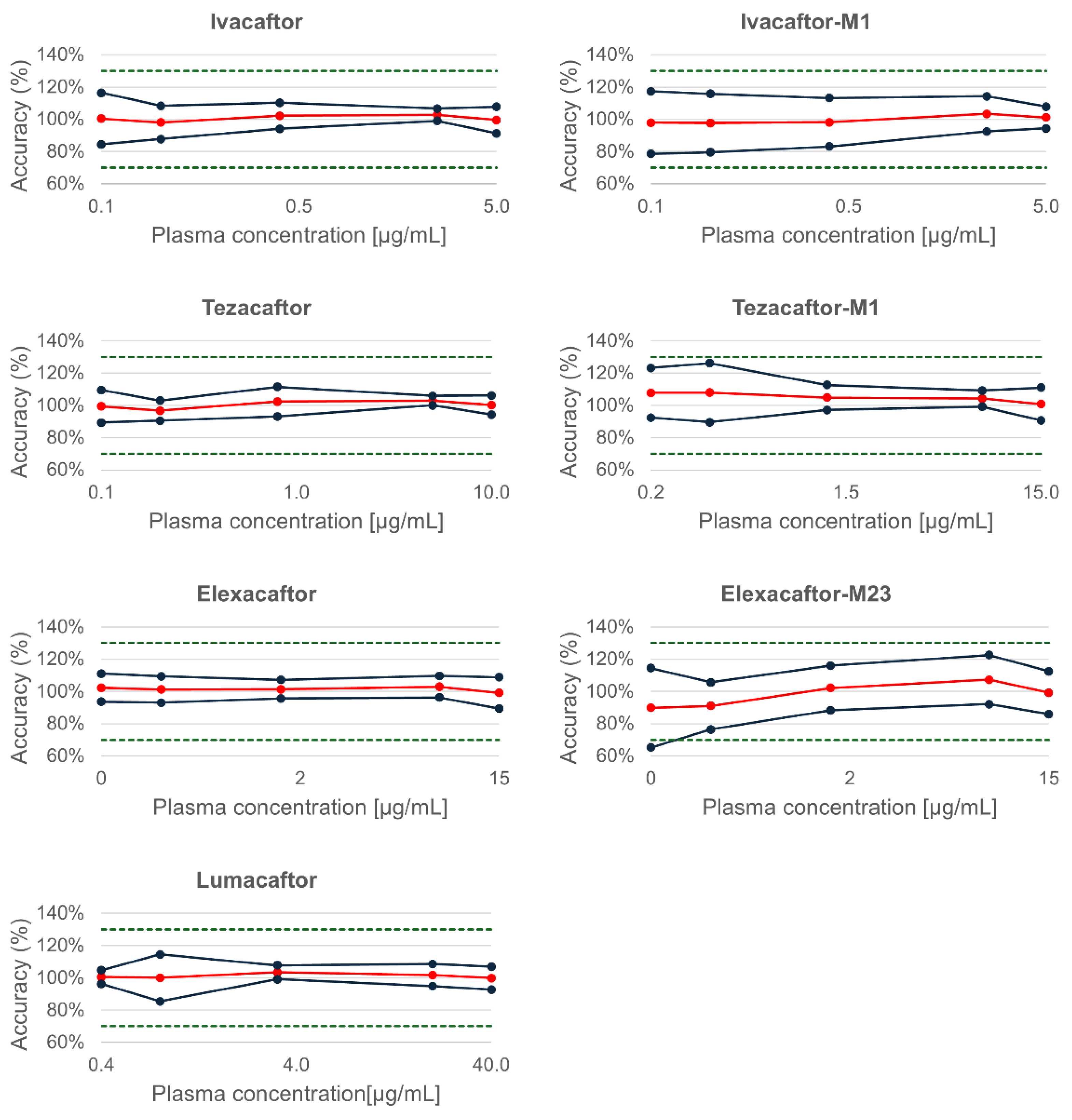
2.2.3. Trueness and Precision
2.2.4. Evaluation of Matrix Effect
2.2.5. Stability Studies
2.3. Inter-Laboratory Comparisons
2.4. Clinical Application: Exploratory Analyses
3. Materials and Methods
3.1. Chemical and Reagents
3.2. Stock Solutions, Calibration and Validation Standards Preparation
3.3. Plasma Sample Extraction Procedure
3.4. Instrumentation and Experimental Conditions
3.5. Validation Procedure
3.5.1. Selectivity, Specificity, Crosstalk, and Carryover
3.5.2. Evaluation of Matrix Effect
3.5.3. Limit of Quantification, and Linearity
3.5.4. Trueness and Precision
3.5.5. Stability Studies
3.5.6. Inter-Laboratory Comparisons
3.6. Clinical Application of the Method
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garg, V.; Shen, J.; Li, C.; Agarwal, S.; Gebre, A.; Robertson, S.; Huang, J.; Han, L.; Jiang, L.; Stephan, K.; et al. Pharmacokinetic and Drug-Drug Interaction Profiles of the Combination of Tezacaftor/Ivacaftor. Clin Transl Sci 2019, 12, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Choong, E.; Sauty, A.; Koutsokera, A.; Blanchon, S.; Andre, P.; Decosterd, L. Therapeutic Drug Monitoring of Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor in Cystic Fibrosis: Where Are We Now? Pharmaceutics 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Noetzli, M.; Choong, E.; Ansermot, N.; Eap, C.B. Simultaneous determination of antidementia drugs by HPLC-MS: validation data of the method and plasma level determinations in 300 patients. In Proceedings of the Therapeutic Drug Monitoring, Aug; 2011; pp. 501–501. [Google Scholar]
- Courlet, P.; Alves Saldanha, S.; Cavassini, M.; Marzolini, C.; Choong, E.; Csajka, C.; Gunthard, H.F.; Andre, P.; Buclin, T.; Desfontaine, V.; et al. Development and validation of a multiplex UHPLC-MS/MS assay with stable isotopic internal standards for the monitoring of the plasma concentrations of the antiretroviral drugs bictegravir, cabotegravir, doravirine, and rilpivirine in people living with HIV. J Mass Spectrom 2020, 55, e4506. [Google Scholar] [CrossRef] [PubMed]
- Hansson, A.B.; Wadstrom, H.; Eliasson, E.; Al Shakirchi, M.; de Monestrol, I.; Barclay, V. Development and clinical implementation of an LC-HRMS method for ivacaftor, lumacaftor, tezacaftor and elexacaftor in human plasma and breast milk. Anal Bioanal Chem 2024, 416, 5565–5577. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Rouillon, S.; Khemakhem, M.; Balakirouchenane, D.; Lui, G.; Abdalla, S.; Sanoufi, M.R.; Sauvaitre, L.; Thebault, L.; Hirt, D.; et al. A rapid LC-MS/MS method for the simultaneous quantification of ivacaftor, lumacaftor, elexacaftor, tezacaftor, hexyl-methyl ivacaftor and ivacaftor carboxylate in human plasma. J Pharm Biomed Anal 2024, 248, 116322. [Google Scholar] [CrossRef] [PubMed]
- Vonk, S.E.M.; van der Meer-Vos, M.; Kos, R.; Neerincx, A.H.; Terheggen-Lagro, S.W.J.; Altenburg, J.; Maitland-van der Zee, A.H.; Mathot, R.A.A.; Kemper, E.M.; Amsterdam Mucociliary Clearance Disease research, g. Dried Blood Spot Method Development and Clinical Validation for the Analysis of Elexacaftor, Elexacaftor-M23, Tezacaftor, Tezacaftor-M1, Ivacaftor, Ivacaftor Carboxylate, and Hydroxymethyl Ivacaftor Using LC-MS/MS. Ther Drug Monit 2024, 46, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, S.; Erdogan Uzunoglu, U.; Levent, S.; Can, N.O. Liquid chromatographic determination of lumacaftor in the presence of ivacaftor and identification of five novel degradation products using high-performance liquid chromatography ion trap time-of-flight mass spectrometry. J Sep Sci 2023, 46, e2300228. [Google Scholar] [CrossRef] [PubMed]
- Pigliasco, F.; Cafaro, A.; Stella, M.; Baiardi, G.; Barco, S.; Pedemonte, N.; D'Orsi, C.; Cresta, F.; Casciaro, R.; Castellani, C.; et al. Simultaneous Quantification of Ivacaftor, Tezacaftor, and Elexacaftor in Cystic Fibrosis Patients' Plasma by a Novel LC-MS/MS Method. Biomedicines 2023, 11. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.J.; Guimbellot, J.S.; Dowell, A.E.; Reed-Walker, K.D.; Kerstner-Wood, C.D.; Anderson, J.D.; Liu, Z.; Acosta, E.P. Quantitation of cystic fibrosis triple combination therapy, elexacaftor/tezacaftor/ivacaftor, in human plasma and cellular lysate. J Chromatogr B Analyt Technol Biomed Life Sci 2022, 1213, 123518. [Google Scholar] [CrossRef] [PubMed]
- European public assessment report. Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/variation-report/kalydeco-h-c-2494-x-83-g-epar-assessment-report-variation_en.pdf (accessed on June 3, 2022).
- Vonk, S.E.M.; van der Meer-Vos, M.; Bos, L.D.J.; Neerincx, A.H.; Majoor, C.J.; Maitland-van der Zee, A.H.; Mathot, R.A.A.; Kemper, E.M.; Amsterdam Mucociliary Clearance Disease research, g. Quantitative Method for the Analysis of Ivacaftor, Hydroxymethyl Ivacaftor, Ivacaftor Carboxylate, Lumacaftor, and Tezacaftor in Plasma and Sputum Using Liquid Chromatography With Tandem Mass Spectrometry and Its Clinical Applicability. Ther Drug Monit 2021, 43, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.K. Cytochrome P450 3A4 Induction: Lumacaftor versus Ivacaftor Potentially Resulting in Significantly Reduced Plasma Concentration of Ivacaftor. Drug Metab Lett 2018, 12, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Habler, K.; Kalla, A.S.; Rychlik, M.; Bruegel, M.; Teupser, D.; Nahrig, S.; Vogeser, M.; Paal, M. Isotope dilution LC-MS/MS quantification of the cystic fibrosis transmembrane conductance regulator (CFTR) modulators ivacaftor, lumacaftor, tezacaftor, elexacaftor, and their major metabolites in human serum. Clin Chem Lab Med 2022, 60, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.K.; Reyes-Ortega, F.; Wilson, J.W.; Kotsimbos, T.; Keating, D.; Li, J.; Velkov, T. Development of HPLC and LC-MS/MS methods for the analysis of ivacaftor, its major metabolites and lumacaftor in plasma and sputum of cystic fibrosis patients treated with ORKAMBI or KALYDECO. J Chromatogr B Analyt Technol Biomed Life Sci 2016, 1038, 57–62. [Google Scholar] [CrossRef] [PubMed]
- European public assessment report. Guideline on Bioanalytical Method Validation and Study Sample Analysis. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m10-bioanalytical-method-validation-step-5_en.pdf (accessed on June 3, 2023).
- Feinberg, M. Validation of analytical methods based on accuracy profiles. J Chromatogr A 2007, 1158, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.; Kringle, R. A total error approach for the validation of quantitative analytical methods. Pharm Res 2007, 24, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Rozet, E.; Hubert, C.; Ceccato, A.; Dewe, W.; Ziemons, E.; Moonen, F.; Michail, K.; Wintersteiger, R.; Streel, B.; Boulanger, B.; et al. Using tolerance intervals in pre-study validation of analytical methods to predict in-study results. The fit-for-future-purpose concept. J Chromatogr A 2007, 1158, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry: Bioanalytical Method Validation. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf. (accessed on June 3, 2022).
- Rozet, E.; Ceccato, A.; Hubert, C.; Ziemons, E.; Oprean, R.; Rudaz, S.; Boulanger, B.; Hubert, P. Analysis of recent pharmaceutical regulatory documents on analytical method validation. J Chromatogr A 2007, 1158, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Choong, E.; Uppugunduri, C.R.S.; Marino, D.; Kuntzinger, M.; Doffey-Lazeyras, F.; Lo Piccolo, R.; Chalandon, Y.; Peters, C.; Daali, Y.; Ansari, M. Therapeutic Drug Monitoring of Busulfan for the Management of Pediatric Patients: Cross-Validation of Methods and Long-Term Performance. Ther Drug Monit 2018, 40, 84–92. [Google Scholar] [CrossRef] [PubMed]
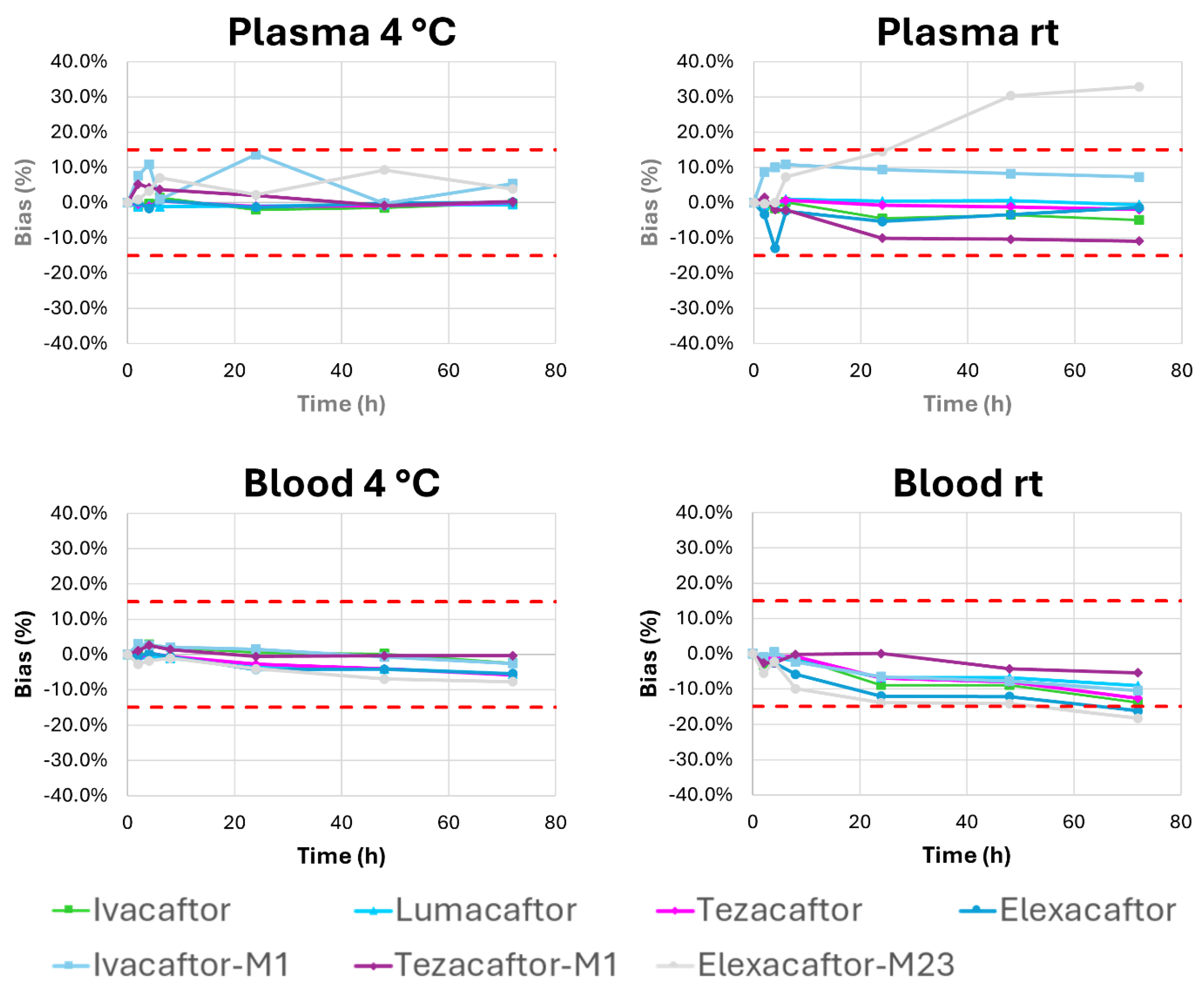
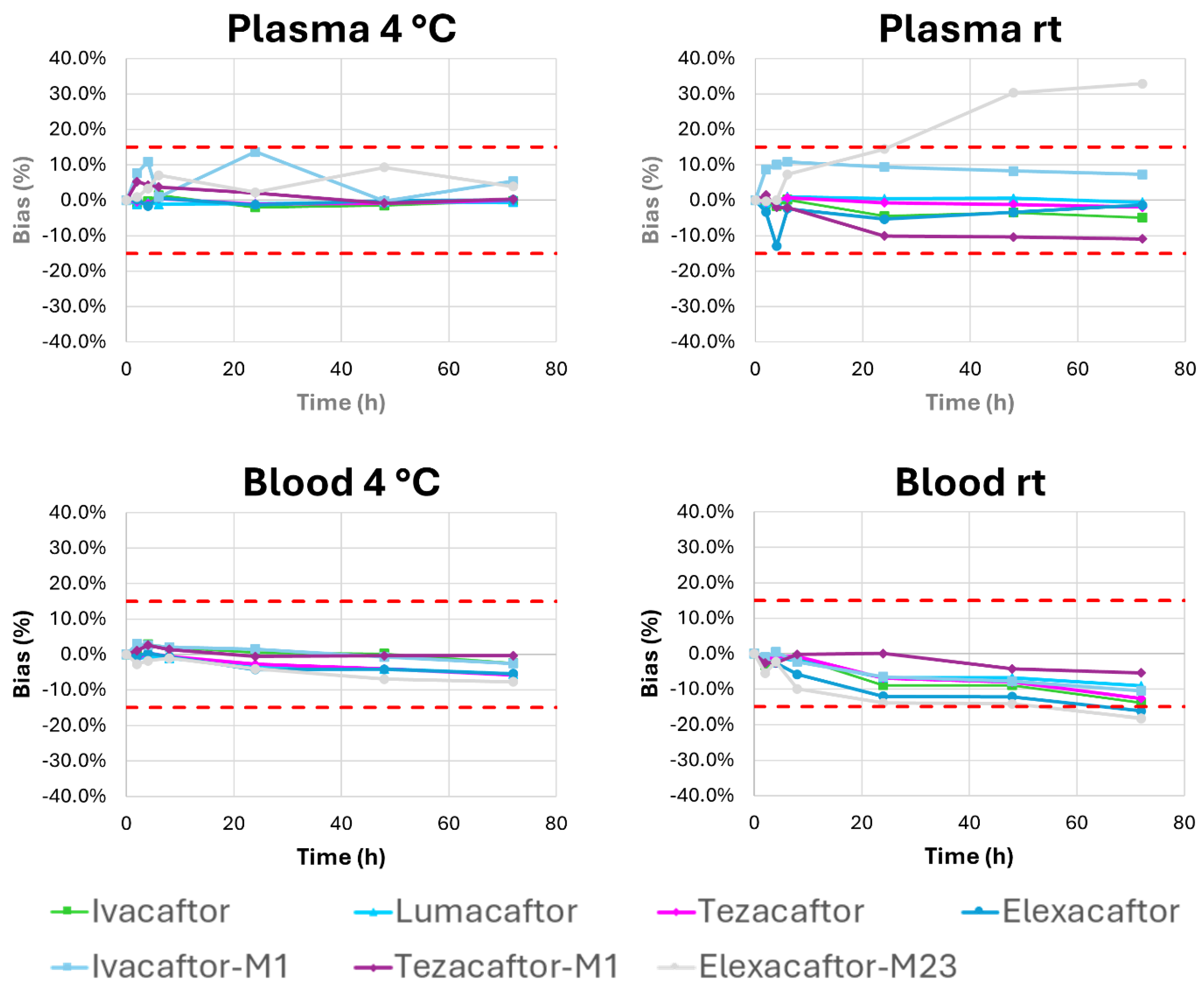
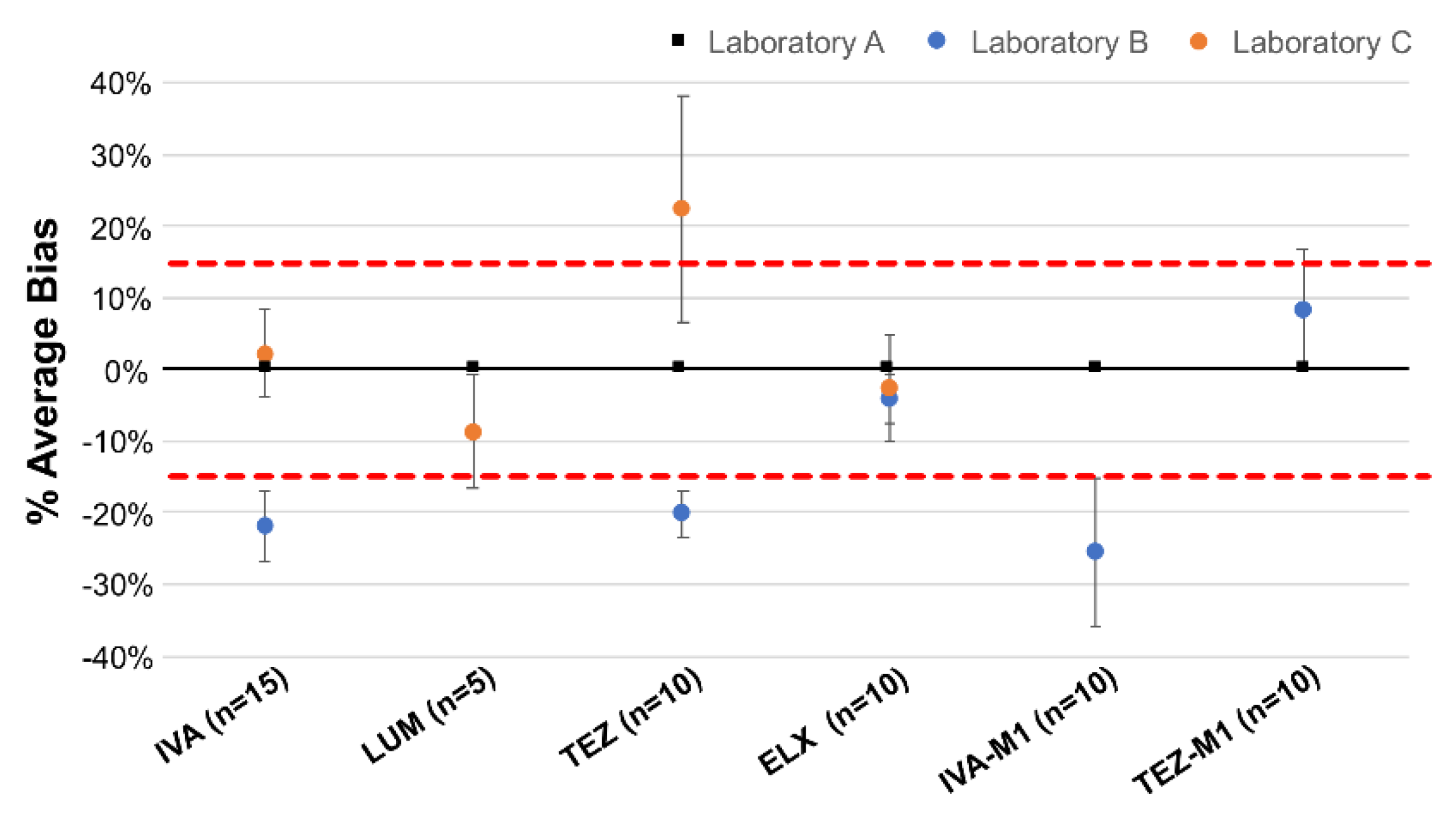
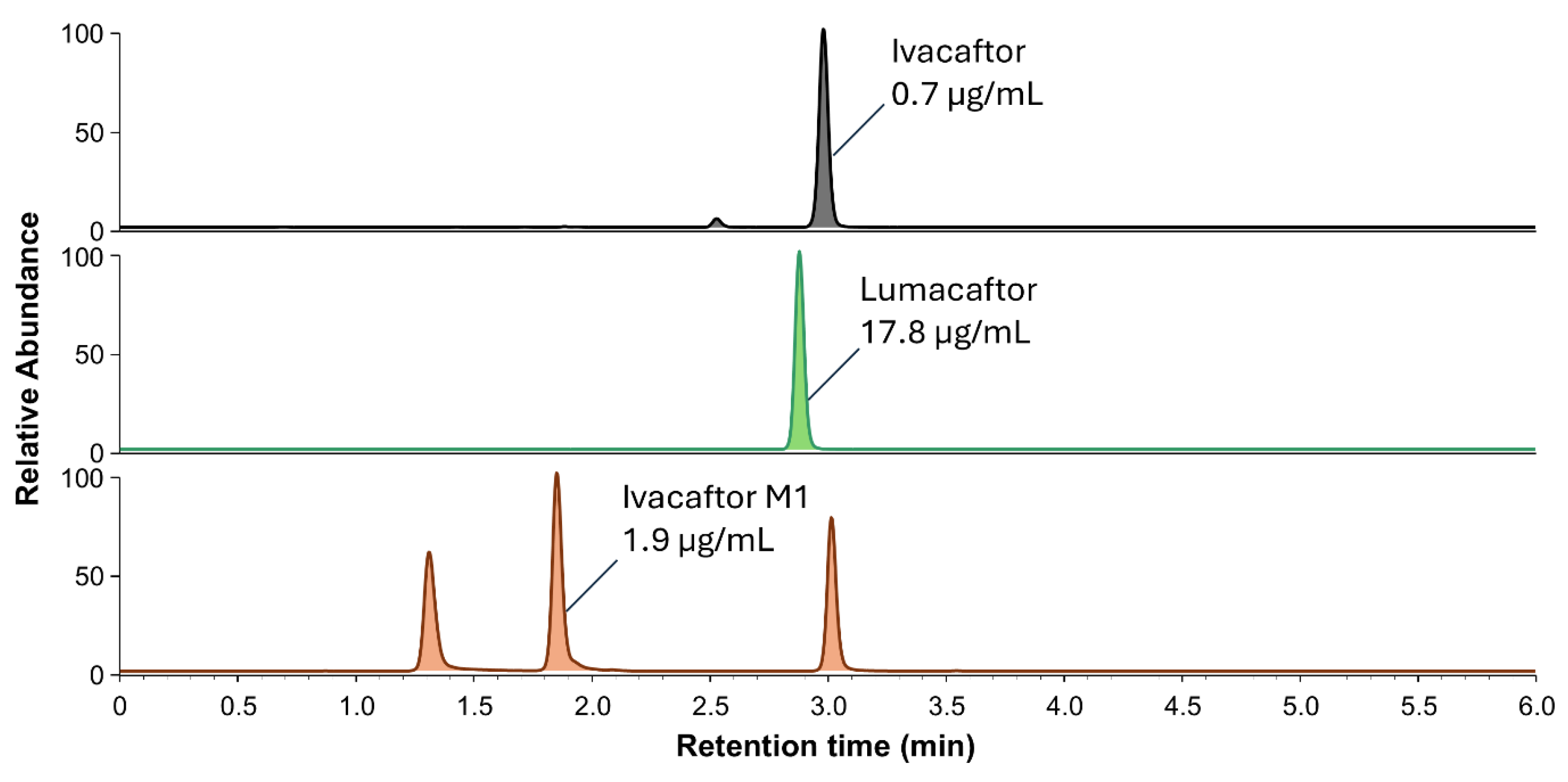

| Compound | Precursor ion (m/z) |
Product ion (m/z) |
CE [V] | RT [min] | Internal standard |
|---|---|---|---|---|---|
| Ivacaftor | 393.3 | 337.167 | 14 | 2.66 | Ivacaftor-D4 |
| Lumacaftor | 453.3 | 413 | 26 | 2.57 | Lumacaftor-D4 |
| Tezacaftor | 521.3 | 449.083 | 21 | 1.90 | Tezacaftor-D4 |
| Elexacaftor | 598.3 | 422.333 | 26 | 3.36 | Elexacaftor-D3 |
| Ivacaftor-M1 | 409.3 | 353.083 | 16 | 1.66 | Voriconazole-D3 |
| Tezacaftor-M1 | 519.3 | 501.25 | 17 | 2.19 | Tezacaftor-D4 |
| Elexacaftor-M23 | 584.3 | 422.583 | 25 | 2.89 | Elexacaftor-D3 |
| Ivacaftor-D4 | 397.3 | 341.167 | 14 | 2.66 | |
| Lumacaftor-D4 | 457.3 | 417 | 26 | 2.57 | |
| Tezacaftor-D4 | 525.3 | 453.25 | 22 | 1.90 | |
| Elexacaftor-D3 | 601.3 | 422.25 | 26 | 3.36 | |
| Voriconazole-D3 | 353.1 | 284.2 | 15 | 1.40 |
| Precision | ||||
|---|---|---|---|---|
| Compound | Concentration [µg/mL] | Truness (%) | Repeatability (%) | Intermediate precision (%) |
| Ivacaftor | 0.05 | 100.4 | 1.9 | 7.1 |
| 0.1 | 98.0 | 2.7 | 4.6 | |
| 0.4 | 102.2 | 2.2 | 3.5 | |
| 2.5 | 102.8 | 1.4 | 1.7 | |
| 5 | 99.5 | 2.0 | 3.7 | |
| Lumacaftor | 0.4 | 100.5 | 1.9 | 1.9 |
| 0.8 | 100.0 | 6.4 | 6.4 | |
| 3.2 | 103.5 | 1.5 | 1.9 | |
| 20 | 101.8 | 1.8 | 3.1 | |
| 40 | 99.9 | 3.1 | 3.1 | |
| Tezacaftor | 0.1 | 99.3 | 2.4 | 4.4 |
| 0.2 | 96.7 | 1.8 | 2.8 | |
| 0.8 | 100.3 | 3.7 | 4.1 | |
| 5 | 102.9 | 1.3 | 1.3 | |
| 10 | 100.2 | 1.4 | 2.6 | |
| Elexacaftor | 0.15 | 100.4 | 2.4 | 5.1 |
| 0.3 | 98.6 | 3.3 | 4.0 | |
| 1.2 | 103.5 | 5.0 | 5.6 | |
| 7.5 | 103.0 | 2.5 | 2.8 | |
| 15 | 99.0 | 1.8 | 2.6 | |
| Ivacaftor-M1 | 0.05 | 98.0 | 6.7 | 8.5 |
| 0.1 | 97.7 | 8.0 | 8.0 | |
| 0.4 | 98.2 | 6.6 | 6.6 | |
| 2.5 | 103.3 | 2.8 | 4.8 | |
| 5 | 101.1 | 2.0 | 3.0 | |
| Tezacaftor-M1 | 0.15 | 107.8 | 3.6 | 6.8 |
| 0.3 | 107.9 | 8.1 | 8.1 | |
| 1.2 | 104.9 | 3.4 | 3.4 | |
| 7.5 | 104.2 | 1.6 | 2.2 | |
| 15 | 100.8 | 2.9 | 4.5 | |
| Elexacaftor-M23 | 0.15 | 89.8 | 4.0 | 10.9 |
| 0.3 | 91.0 | 4.9 | 6.4 | |
| 1.2 | 102.2 | 4.9 | 6.1 | |
| 7.5 | 107.3 | 5.6 | 6.7 | |
| 15 | 99.2 | 4.0 | 5.9 | |
| Molecule | CAL1 | CAL2 | CAL 3 | CAL4 | CAL5 | CAL6 | QC 1 | QC 2 | QC 3 |
|---|---|---|---|---|---|---|---|---|---|
| Ivacaftor | 0.05 | 0.10 | 0.20 | 0.50 | 1.25 | 5.00 | 0.15 | 0.75 | 3.75 |
| Lumacaftor | 0.40 | 0.80 | 1.60 | 4.00 | 10.00 | 40.00 | 1.20 | 6.00 | 30.00 |
| Tezacaftor | 0.10 | 0.20 | 0.40 | 1.00 | 2.50 | 10.00 | 0.30 | 1.50 | 7.50 |
| Elexacaftor | 0.15 | 0.30 | 0.60 | 3.00 | 7.50 | 15.00 | 0.45 | 2.25 | 11.25 |
| Ivacaftor-M1 | 0.05 | 0.10 | 0.20 | 1.00 | 2.50 | 5.00 | 0.15 | 0.75 | 3.75 |
| Tezacaftor-M1 | 0.15 | 0.30 | 0.60 | 3.00 | 7.50 | 15.00 | 0.45 | 2.25 | 11.25 |
| Elexacaftor-M23 | 0.15 | 0.30 | 0.60 | 3.00 | 7.50 | 15.00 | 0.45 | 2.25 | 11.25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).



