Submitted:
19 February 2025
Posted:
19 February 2025
You are already at the latest version
Abstract
DNA damage response (DDR) is crucial for maintaining genomic stability and preventing the accumulation of mutations that can lead to various diseases, including cancer. DDR is a complex cellular regulatory network that involves DNA damage sensing, signal transduction, repair, and cell cycle arrest. Modifications in histone phosphorylation play important roles in these processes, facilitating DNA repair factor recruitment, damage signal transduction, chromatin remodeling, and cell cycle regulation. The precise regulation of histone phosphorylation is critical for the effective repair of DNA damage, genomic integrity maintenance, and prevention of diseases such as cancer, where DNA repair mechanisms are often compromised. Thus, understanding histone phosphorylation in DDR provides insights into DDR mechanisms and offers potential therapeutic targets for diseases associated with genomic instability, including cancers.
Keywords:
histone phosphorylation
; DNA damage response
; γH2AX
; kinase
; DNA repair
1. Introduction
Eukaryotic cells are frequently subjected to endogenous and exogenous DNA damage, which threatens genome stability and may lead to cellular and systemic imbalances, contributing to the onset of diseases such as cancer [1]. DNA damage arises from a variety of endogenous or exogenous sources, including replication errors, reactive oxygen species, abnormal metabolites, chemical agents, ultraviolet (UV) radiation and ionizing radiation (IR). To counter these inevitable threats, cells have evolved various DNA damage response (DDR) pathways, which are responsible for detecting, signaling, and repairing DNA damage [2,3]. As DDR plays a crucial role in maintaining genomic stability, DDR pathway defects can lead to diseases such as premature aging, neurodegenerative disorders, immunodeficiencies, and cancer [1,3,4,5].
The DDR network encompasses a series of intricate signaling and repair mechanisms. The core DDR components include DNA damage recognition, signal transduction, cell cycle regulation, and DNA repair. Initially, cells employ specific sensor proteins to recognize aberrant DNA structures. For example, excessive long-strectch of single-stranded DNA (ssDNA) is accumulated due to DNA unwinding and syntheis uncoupling, which is promptly bound by replication protein A (RPA). Next, these initial signals and their readers recruit and activate apical kinases such as ataxia telangiectasia-mutated (ATM) and ATM- and Rad3-related (ATR) kinases, which trigger the phosphorelay reactions through the mediators to downstream effector kinases CHK1 and CHK2. These pathways regulate various effector proteins that coordinate DNA repair and induce cell cycle arrest, ensuring sufficient time for repair [2,3].
Eukaryotic cells utilize multiple conserved repair mechanisms depending on the type of DNA damage and cell cycle state. For example, nucleotide excision repair is used to address UV-induced DNA damage [6], whereas mismatch repair (MMR) corrects base-pairing mismatches or insertion/deletion loops formed during DNA replication [7,8,9,10]. Double-strand breaks (DSBs) are among the most severe and lethal forms of DNA damage, often leading to genomic instability. In mammalian cells, DSBs are predominantly repaired via error-prone nonhomologous end-joining (NHEJ) and complementary via error-free homologous recombination (HR) or other pathways in certain circumstances [11,12,13]. NHEJ operates throughout the cell cycle via a template-independent rejoining mechanism with minimal end processing [14]. In contrast, HR relies on homologous DNA sequences as templates so it occurs strictly during S and G2 phases [15,16,17]. In addition, single-strand annealing (SSA) also contributes to DSB repair [12,18]. Furthermore, DDR is closely linked to apoptosis and senescence pathways. When DNA damage is irreparable, cells undergo programmed cell death to prevent the propagation of the damaged genome. These DDR mechanisms form a complex interconnected network that maintains genomic stability [11,19,20].
Increasing evidence has suggested that posttranslational modifications (PTMs) in histones play a pivotal role in DDR. In eukaryotic cells, chromatin comprises DNA and nucleosomal protein complexes. Each nucleosome core particle consists of an octamer of four core histones (H2A, H2B, H3, and H4), with two molecules of each wrapped in 147 bp of DNA. The linker histone H1 stabilizes the chromatin structure by connecting nucleosomes [21]. Histones are essential not only for maintaining DNA structure and genomic stability but also for regulating gene expression. Accordingly, histone PTMs are involved in various biological processes, including gene transcription, DNA replication, chromatin condensation, and DNA damage repair [22,23,24]. Following DNA damage, histones undergo multiple types of PTMs such as phosphorylation, acetylation, methylation, and ubiquitination. These modifications are triggered at damaged sites and facilitate DNA repair through diverse mechanisms [25,26,27].
This review primarily highlights the roles of histone phosphorylation in DDR (Figure 1), as well as its implications in cancer research and therapy. This modification is known to aid in the recruitment of repair factors to damaged sites, transmission of damage signals, modulation of chromatin openness and closure, transcription, regulation of cell cycle progression and apoptosis, ultimately ensuring effective DNA damage repair and genome stability.
2. Histone Phosphorylation in the DNA Damage Response
2.1. H2A Phosphorylation in the DNA Damage Response
2.1.1. γH2AX
One of the most well-characterized histone PTMs involved in the DDR is the phosphorylation of the histone variant H2AX. Upon DNA damage, H2AX is phosphorylated at Ser139 by DDR kinases, including ATM and ATR, and DNA-dependent protein kinase (DNA-PK) in mammalian cells. This phosphorylated form is known as γH2AX [28,29,30]. In yeast, where H2AX is absent, a corresponding phosphorylation occurs at S129 of H2A (γH2A). ATR is the primary kinase responsible for γH2AX phosphorylation during single-strand damage and replication stress, whereas DNA-PKcs mediate this modification during apoptosis [31,32]. In contrast, DSB-induced H2AX phosphorylation is primarily mediated by ATM or the yTel1 and yMec1 homologs in budding yeast [30,33]. Furthermore, VRK1, a chromatin kinase, is critically involved in the phosphorylation of H2AX at Ser139 in response to DNA damage induced by IR [34].
γH2AX is majorly involved in DDR pathways, including damage signal transduction, NHEJ and HR [35,36]. This histone modification serves as a biomarker of DNA damage in cancer cells, marking DSBs and facilitating repair protein recruitment, making it one of the most extensively studied histone modifications in DDR [29]. The importance of γH2AX in DNA repair is highlighted by studies showing that mice lacking H2AX or cells unable to phosphorylate S139 exhibit heightened sensitivity to DNA damage and increased genomic instability [37]. In yeast, mutation of H2AS129 to a nonphosphorylatable alanine results in hypersensitivity to DNA-damaging agents such as phleomycin and methyl methane-sulphonate (MMS), confirming the critical role of γH2AX in DSB repair. In addition, as γH2AX facilitates sister chromatid recombination, its absence increases reliance on the error-prone SSA repair pathway [35,38].
Following DSB induction by exogenous or endogenous factors, DDR is promptly activated, recruiting numerous signaling molecules and repair proteins to the damaged site to maintain genomic integrity and cellular functions [39,40]. Initially, γH2AX foci are formed, which provide binding sites for other repair proteins. More specifically, upon DSB occurrence, the Mre11-Rad50-Nbs1 (MRN) complex recognizes the damage and recruits ATM to the site. ATM then phosphorylates H2AX at Ser139, generating γH2AX foci [41]. These foci spread bidirectionally, covering approximately 50 kb in yeast, and several Mb in mammals. This extensive γH2AX distribution establishes a signaling platform that facilitates the recruitment and retention of key DDR proteins such as MDC1, p53-binding protein 1 (53BP1), breast cancer 1 (BRCA1) , and the MRN complex [35,42,43,44]. MDC1, a critical mediator protein, binds γH2AX via its tandem C-terminal BRCT domains, while interacts with the FHA domain of P95, a subunit of the MRN complex, and recruits the latter to the DSB site. This interaction amplifies ATM activity, enhancing H2AX phosphorylation and the DDR signal, which facilitates the recruitment of other repair proteins, such as 53BP1 and BRCA1, thus initiating DNA repair [45,46,47].
Notably, γH2AX can also coordinate the DDR by regulating other types of histone modifications. For instance, γH2AX promotes the recruitment of E3 ubiquitin ligases, including RNF8 and RNF168, to damaged sites, regulating ubiquitylation signaling in DSBs. Upon the occurrence of DNA double-strand breaks, γH2AX is extensively formed at the damage sites, facilitating the recruitment of MDC1. MDC1 subsequently recruits RNF8 to the damaged regions. The mono-ubiquitination of γH2AX, catalyzed by RNF8, provides a binding platform for RNF168, thereby amplifying the ubiquitination signaling cascade and enhancing the recruitment of downstream repair factors [48,49,50]. Moreover, the adaptor protein Rad9, related to 53BP1/Crb2, interacts with γH2AX via its BRCT domain and with methylated H3K79 through its Tudor domain. This specificity enables Rad9 recruitment to the DSB site, where it is phosphorylated by Mec1, triggering a DNA damage checkpoint that delays G1/S progression and allows repair [51,52,53]. In yeast, γH2AX recruits the NuA4 acetyltransferase complex to DSBs. NuA4 mediates H4 hyperacetylation and promotes chromatin relaxation [54,55]. Furthermore, γH2AX assists in recruiting chromatin-remodeling complexes such as INO80 and SWR1, which enhance DNA repair accessibility [56,57].
Following DNA repair, γH2AX removal is essential for preventing persistent repair protein recruitment, DNA damage-induced cell cycle arrest recovery, and chromatin integrity restoration. Two primary mechanisms have been proposed for γH2AX clearance. First, γH2AX can be replaced by unphosphorylated H2A or removed from DSB sites by chromatin remodelers [58,59]. Second, γH2AX is dephosphorylated by various protein phosphatases, regenerating H2AX. In yeast, the HTP-C phosphatase complex regulates H2AS129 dephosphorylation in vivo, enabling DNA damage checkpoint recovery [60]. Similarly, in mammals, phosphatases such as PP2A, Wip1, PP6, and PP4 dephosphorylate γH2AX, allowing effective DNA repair and cell cycle arrest recovery. Among them, PP2A primarily dephosphorylates γH2AX during DSB repair. Comprising a structural subunit A, a regulatory subunit B, and a catalytic subunit C, PP2A directly binds γH2AX at DSB sites, mediating dephosphorylation through its catalytic subunit C. PP2A deficiency results in repair defects and persistent γH2AX accumulation, highlighting the importance of dephosphorylation in postrepair chromatin processing [61,62,63]. Other phosphatases are also involved in γH2AX dephosphorylation. PP6 interacts with the catalytic subunit of DNA-PK to mediate γH2AX dephosphorylation, whereas Wip1 directly induces γH2AX dephosphorylation. PP4 primarily dephosphorylates γH2AX mediated by ATR, enabling DNA damage checkpoint and cell cycle recovery after DNA damage [64,65,66,67]. In summary, the orderly subsequent γH2AX dephosphorylation is essential for maintaining genomic stability following DNA damage repair.
2.1.2. Other H2A sites
Although γH2AX is widely used as a DNA damage marker, H2A contains multiple phosphorylation sites that contribute to the DDR apart from serine 139 (S139) phosphorylation.
For instance, tyrosine 142 (Y142) phosphorylation, which is regulated in a DNA-damage-dependent manner, is catalyzed by the WSTF kinase. Unlike γH2AX, Y142 phosphorylation is ubiquitously present in cells but decreases significantly upon DNA damage. This inverse relationship is critical for maintaining γH2AX stability at DNA repair foci, as the EYA1/3 phosphatase-mediated Y142 dephosphorylation is essential for recruiting repair factors to damaged sites. Failure to dephosphorylate Y142 impairs the accumulation of repair factors and disrupts DDR [68,69].
In yeast, serine 122 (S122) and serine 129 (S129) on histone H2A are dynamically phosphorylated during DNA damage, contributing to DDR processes. Studies have shown that S122 is critical for cell survival under DNA damage induced by camptothecin, MMS, hydroxyurea (HU), or ultraviolet light. The phosphorylation of S129 in the DDR is dependent on the Tel1 and Mec1 kinases, while the phosphorylation of S122 in Schizosaccharomyces pombe and S. cerevisiae is mediated by the Bub1 kinase. Both modifications may facilitate interactions with the DDR machinery without altering global chromatin structure. The concurrent phosphorylation of S122 and S129 during DNA damage suggests that they may play synergistic roles in the recruitment or retention of repair factors [70,71,72]. Moreover, recent study has revealed that the phosphorylation of H2A S122, mediated by the Bub1 kinase, plays a critical role in regulating chromosome segregation [73].
Recently, DNA damage-induced H2A phosphorylation at S15, catalyzed by Mec1, was found to be linked to DNA end resection in yeast. DNA end resection provides the single-stranded DNA required for HR, thereby potentially assisting in the repair of breaks [74]. Threonine 101 (T101), which is also phosphorylated after DNA damage, is another phosphorylation site. Mutations at this site render cells sensitive to IR, indicating its pivotal role in H2AX-dependent DDR function [75]. Furthermore, the phosphorylation of the threonine 126 (Thr126) residue in H2A.1 is linked to the stability and repair of fragile DNA regions, particularly CAG repeat sequences [76].
These findings collectively highlight the importance of phosphorylation at other H2A sites in DDR regulation. However, the specific regulatory mechanisms of these phosphorylation events in DDR remain unclear, highlighting the need for further studies to elucidate the mechanisms by which these modifications cooperate with γH2AX to maintain genomic stability following DNA damage.
2.2. H3
Histone H3 phosphorylation also plays an important role in DDR. Studies of key phosphorylation sites such as serine 10 (S10), threonine 11 (T11), and serine 28 (S28), together with their respective kinases, have demonstrated their significance in genomic stability maintenance. These residues are phosphorylated during mitosis to facilitate chromatin compaction [77,78,79,80].
The Aurora kinase family, particularly Aurora B kinase, mediates H3S10 and S28 phosphorylation in DDR. As a serine/threonine kinase, Aurora B participates in chromosome segregation, cell cycle regulation, and chromatin remodeling. Direct phosphorylation Ser10 in H3 by VRK1 both in vitro and in vivo was observed [34,81]. G1-phase cells exhibit specific reductions in H3S10 phosphorylation following DNA damage [79]. Concurrent decreases have been demonstrated in additional histone modifications, such as acetylations, accompanied by chromatin condensation. Studies have also suggested the potential crosstalk between H3S10 phosphorylation and other modifications, such as H3K9 acetylation or methylation, collectively affecting chromatin compaction and DNA repair protein recruitment [82,83,84].These researches suggest dynamic changes of chromatin structure and/or transcriptional repression during DNA damage response.
Histone H3 threonine 11 (H3T11) phosphorylation also participates in DDR by regulating chromatin relaxation and DNA repair factor recruitment. Protein kinase C (PKC) or Chk1 kinases typically catalyze H3T11 phosphorylation. Following DNA damage, activated PKC phosphorylates H3T11 directly. This modification occurs primarily during the S or G2/M phases, when chromatin structure undergoes significant changes, to facilitate repair protein recruitment Moreover, under environmental stress conditions such as radiation-induced damage, the DDR core kinases ATM and ATR may enhance H3T11 phosphorylation indirectly through Chk1 activity modulation, thus influencing chromatin dynamics and DNA repair [78,84]. Moreover, H3T11 phosphorylation mediated by Casein kinase II (CKII) is a key modification for the formation and maintenance of heterochromatin in Neurospora, contributing to genomic stability and the regulation of gene expression [85]. Additionaly, AKT phosphorylates H3-threonine 45 to facilitate termination of gene transcription in response to DNA damage [86].
Collectively, these H3 phosphorylation events mainly modulate chromatin structure and regulate the recruitment of DNA repair factors, which are essential for effective DNA damage repair. Furthermore, since H3 phosphorylation is crosslinked with other epigenetic modifications, elucidating the mechanisms underlying histone H3 phosphorylation will enhance our understanding of the intricate DDR regulatory networks.
2.3. H4
During DNA damage, histone H4 undergoes site-specific phosphorylation by kinases that regulate chromatin structure, repair, and checkpoint regulation.
CKII catalyzes H4 serine 1 phosphorylation (H4S1ph) in yeast in response to UV light-, MMS-, or phleomycin-induced genotoxic stress. This modification contributes to NHEJ [87,88]. H4S1ph accumulates at DSBs, supporting its DNA repair role in humans as well [89]. Interestingly, H4S1ph demonstrates inverse correlation with H4 acetylation, with its levels decreasing as repair concludes. H4S1ph inhibits the histone acetyltransferase activity of the NuA4 complex in vitro. The association of CKII with the Rpd3S deacetylase complex in vivo, suggests that H4S1ph stabilizes newly assembled nucleosomes through acetylation prevention, thereby promoting chromatin restoration [88]. These findings demonstrate the cooperation of histone phosphorylation and deacetylation in mediating NHEJ.
H4Y51, another H4 phosphorylation site, was the first tyrosine phosphorylation modification identified in this histone. This modification, which is catalyzed by the TIE2 kinase, has been linked to NHEJ [90]. Another phosphorylation site, H4T80, also participates in DDR. H4T80 is phosphorylated by the kinase Cla4 and is recognized by the histone-binding scaffold protein RTT107. The interaction between RTT107 and H4T80p prevents chromatin binding by Rad9, facilitating checkpoint recovery following DNA damage [91].
2.4. H2B and H1
In budding yeast, DSBs trigger extensive Tel1 (ATM)- and Mec1 (ATR)-mediated H2A phosphorylation near break sites, leading to γ-H2AX formation. Similarly, DNA damage triggers Tel1- and Mec1-mediated H2B phosphorylation at T129. The distribution of H2BT129p mirrors that of γ-H2AX in yeast, forming large domains around break sites. Notably, the absence of γ-H2AX impaired γ-H2B formation [92], suggesting a potential cooperation between these modifications in DDR. In mammalian cells, DSBs induce H2B phosphorylation at serine 14 by MST1 kinase [93]. In addition, H2BS14p is a hallmark histone modification closely associated with chromatin remodeling and apoptosis [94]. However, the regulatory mechanisms and functions of this modification remain incompletely understood. Current understanding of DSB-induced H2B phosphorylation remains limited, particularly regarding specific enzymes and recognition mechanisms.
Phosphorylation of the linker histone H1 has also been found to be associated with the DNA damage response. Studies show that a H1 subtype, H1.2, is phosphorylated at threonine 145 (H1.2T145p) in the p53-dependent DDR. Under normal conditions, unphosphorylated H1.2 interacts with p53 to keep its target genes repressed. Following DNA damage, DNA-PK phosphorylates H1.2 at T145, disrupting its interaction with p53. This promotes the recruitment of chromatin remodeling complexes and transcription factors to p53 target promoters, ultimately activating the p53 transcriptional program to maintain genome stability [95,96].
In summary, apart from γH2AX, the phosphorylation of other sites also plays crucial roles in DDR, including facilitating DNA damage repair, regulating the cell cycle, modulating chromatin dynamics, and promoting apoptosis. Unraveling these mechanisms will enhance our understanding of the complex DDR regulatory networks and offer new avenues for the diagnosis and treatment of related diseases.
3. Histone Phosphorylation in Cancer Research and Therapy
Histone phosphorylation is crucial for DDR and genome stability maintenance, holding significant therapeutic implications. Studies have demonstrated a strong correlation between abnormal histone phosphorylation and cancer development. Research on histone phosphorylation in human cancers has revealed its roles beyond DDR pathways.
For example, colorectal cancer tissues exhibit elevated mRNA levels of H2AX and increased γH2AX expression compared with those in normal tissues, correlating with aggressive tumor behavior and poor patient survival [97,98,99]. Notably, H2AX phosphorylation levels increase significantly during DNA fragmentation and apoptosis [35]. The relationship between H2AX expression and microsatellite instability, a carcinogenic mechanism driven by mismatch repair defects, further emphasizes the connection between γH2AX and cancer progression. In colorectal cancer, reduced H3 Ser10 (H3S10) and Y74 and Y272 phosphorylation levels mediated by T-LAK cell-originated protein kinases (TOPK) promote tumor development [100]. Aurora B, which is critical for H3 phosphorylation and chromosome segregation, is overexpressed in various cancers, including colorectal and breast cancers [101]. In prostate cancer cells, androgen stimulation activates kinase PKCβ and PRK1, which phosphorylate H3Thr6 and H3Thr11, respectively [102,103] . In addition, Mst1 kinase phosphorylates H2AX, and its overexpression induces apoptosis in HELA cells via H2AXSer139p [104].
Histone phosphorylation is intricately linked to transcriptional regulation, particularly that of genes involved in cell cycle control and proliferation [105]. For instance, Janus kinase 2 (JAK2) phosphorylates H3Tyr41, disrupting the interaction between heterochromatin protein 1α (HP1α) and chromatin. This loss of HP1α binding leads to constitutive activation of the JAK2 signaling pathway, including the proto-oncogene imo2, thereby driving oncogenesis. JAK2-mediated H3Y41 phosphorylation facilitates the transcriptional activation of diverse gene sets in a cancer patient-specific manner [106,107]. Furthermore, phosphorylation of H3 at Ser10 and Ser28 and H2B at Ser32 is associated with epidermal growth factor (EGF)-mediated gene transcription. UVB radiation exposure increases H3Ser10p and H2BSer32p levels, upregulating the expression of proto-oncogenes such as c-myc, c-fos, and c-jun, whereas H3Ser28p specifically regulates c-fos and α-globin activation [108,109,110]. The levels of H2BSer32 phosphorylation, which is mediated by RSK2, are significantly elevated in skin cancer cells [111].
In addition to its role as a DNA damage marker, γH2AX has gained importance in cancer research and treatment. It has been widely used to evaluate radiotherapy and chemotherapy efficacy, predict tumor cell sensitivity to treatment, and serve as a potential therapeutic target [35,98,112,113]. Cancer cells, characterized by genomic instability, exhibit alterations in γH2AX levels that closely correlate with therapy response. Some cancer cells evade treatment by enhancing their DNA repair capabilities, with increased γH2AX levels facilitating effective repair of chemotherapy- or radiotherapy-induced DNA damage, thus contributing to treatment resistance [98,114,115].
γH2AX demonstrates utility in auxiliary diagnosis and prognosis monitoring across multiple diseases. High-throughput mass spectrometry quantification of γH2AX changes has been used to detect DNA damage in human peripheral blood cells exposed to low-dose environmental IR [116]. In addition, γH2AX levels in circulating tumor cells of chemotherapy patients serve as prognostic markers [117]. In reproductive cell research, γH2AX has been used for assessing DNA damage and repair capacity in sperm and oocytes and is involved in maintaining embryonic stem cell self-renewal [118,119]. Glycolytic metabolite pyruvate has been shown to promote FACT complex-mediated γH2AX loading onto chromatin, enhancing DNA damage signaling and repair, thereby supporting glioblastoma cell survival after DNA damage. These findings provide new strategies for improving the efficacy of Glioblastoma Multiforme treatment [120].
H2AX has emerged as a crucial target for therapeutic strategy development, with drugs that modulate its function undergoing investigation from preclinical studies to clinical trials. Synthetic lethality approaches, particularly in combination with chemotherapy or radiotherapy, have been explored by targeting key enzymes in the DDR pathways, such as ATM, ATR, and DNA-PK. These strategies aim to exploit vulnerabilities in cancer cells with defective DNA repair mechanisms, thereby enhancing treatment specificity and efficacy [121,122].
The integration of histone phosphorylation research with therapeutic development reflects the evolving landscape of oncology and offers innovative treatment paradigms. This approach promises significant breakthroughs in cancer therapy through targeted and personalized interventions.
4. Conclusions and Perspectives
This review summarizes the research progress on histone phosphorylation in DDR (Table 1) and its application significance in cancer research and therapy.
Histone phosphorylation plays a crucial role in DDR by facilitating chromatin remodeling, recruiting damage repair proteins, mediating signal transduction, and regulating cell cycle checkpoints. Notably, H2AX phosphorylation at Ser139 (γH2AX) represents a hallmark of DDR following DNA DSBs. γH2AX serves as an early marker of DDR and plays a pivotal role in detecting and repairing DNA damage. γH2AX formation and dephosphorylation are the most extensively studied histone phosphorylation events [35,37,115]. γH2AX participates in repair pathways such as NHEJ and HR and provides a critical tool for cancer diagnosis, treatment evaluation, and prognosis monitoring. In addition, histone phosphorylation interacts with other epigenetic modifications, such as methylation and acetylation, to coordinate DDR regulation.
Despite significant progress in elucidating the relationship between histone phosphorylation and DDR, many critical questions remain unanswered. For instance, first, the precise molecular mechanisms underlying histone phosphorylation in DDR require clarification, particularly regarding the specific roles of different phosphorylated histones in DNA damage recognition, signaling, and repair. Second, the mechanisms by which histone phosphorylation interacts with other epigenetic modifications to orchestrate DDR require further investigation. Third, the reversible histone phosphorylation/dephosphorylation cycle is intimately embedded throughout the full round of DDR process, i.e., activation/deactivation (recovery). Therefore, a challenging task is to resolve the histone phosphorylation and its related PTM levels in a precise quantitative spatio-temporal manner. Furthermore, the association between aberrant histone phosphorylation and cancer initiation and progression, as well as its potential as a therapeutic target, require extensive clinical and fundamental research.
Future research directions should focus on several key areas:employing advanced proteomics and genomics technologies to systematically identify and elucidate the roles and regulatory networks of histone phosphorylation in DDR, developing cell and animal models to investigate the mechanism by which aberrant histone phosphorylation affects genomic stability and tumorigenesis, developing drugs targeting histone phosphorylation modifications, and evaluating their potential efficacy and safety in cancer therapy. These efforts are expected to uncover the intricate roles of histone phosphorylation in DDR and cancer, paving the way for novel diagnostic, preventive, and therapeutic strategies for tumor management.
Author Contributions
Conceptualization and writing, P.G. and Q.C.; all authors proofread the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (32100069) to P.G. and Beijing Municipal Natural Science Foundation (5212010) to Q.C.
Acknowledgments
The authors thank Prof. Huiqiang Lou for the critical comments, discussion and proofreading of this manuscript. The figure was created using Biorender.com under a granted license.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| DDR | DNA damage response |
| 53BP1 | p53-binding protein 1 |
| ATM | ataxia telangiectasia-mutated |
| ATR | ATM- and Rad3-related |
| BRCA1 | breast cancer 1 |
| CKII | casein kinase II |
| DNA-PK | DNA-dependent protein kinase |
| DSB | double-strand break |
| EGF | epidermal growth factor |
| HR | homologous recombination |
| HU | hydroxyurea |
| IR | ionizing radiation |
| JAK2 | janus kinase 2 |
| MMR | mismatch repair |
| MMS | methyl methane-sulphonate |
| MRN | Mre11-Rad50-Nbs1 |
| NHEJ | nonhomologous end-joining |
| PTM | posttranslational modification |
| RPA | replication protein A |
| SSA | single-strand annealing |
| ssDNA | single-stranded DNA |
| TOPK | T-LAK cell-originated protein kinases |
| UV | ultraviolet |
| DDR | DNA damage response |
| 53BP1 | p53-binding protein 1 |
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Molecular cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—an evolving hallmark of cancer. Nature reviews Molecular cell biology 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Delint-Ramirez, I.; Madabhushi, R. DNA damage and its links to neuronal aging and degeneration. Neuron 2025, 113, 7–28. [Google Scholar] [CrossRef]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nature reviews Molecular cell biology 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nature reviews Molecular cell biology 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Mullenders, L.H. Solar UV damage to cellular DNA: from mechanisms to biological effects. Photochemical & Photobiological Sciences 2018, 17, 1842–1852. [Google Scholar]
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Research 2020, 48, 11227–11243. [Google Scholar] [CrossRef]
- Ijsselsteijn, R.; Jansen, J.G.; de Wind, N. DNA mismatch repair-dependent DNA damage responses and cancer. DNA repair 2020, 93, 102923. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Ünsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annual review of biochemistry 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair pathway choices and consequences at the double-strand break. Trends in cell biology 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Molecular cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual review of biochemistry 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Heyer, W.-D.; Ehmsen, K.T.; Liu, J. Regulation of homologous recombination in eukaryotes. Annual review of genetics 2010, 44, 113–139. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Critical reviews in biochemistry and molecular biology 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nature cell biology 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Ui, A.; Chiba, N.; Yasui, A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer science 2020, 111, 1443–1451. [Google Scholar] [CrossRef]
- Katsuki, Y.; Jeggo, P.A.; Uchihara, Y.; Takata, M.; Shibata, A. DNA double-strand break end resection: a critical relay point for determining the pathway of repair and signaling. Genome Instability & Disease 2020, 1, 155–171. [Google Scholar]
- Wang, K.; Li, L.; Zhang, Y.; Gao, D. Crosstalk between signaling pathways and DNA damage response. Genome Instability & Disease 2020, 1, 81–91. [Google Scholar]
- Hammond, C.M.; Strømme, C.B.; Huang, H.; Patel, D.J.; Groth, A. Histone chaperone networks shaping chromatin function. Nature reviews Molecular cell biology 2017, 18, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of histone modification. Histone Mutations and Cancer 2021, 1–16. [Google Scholar]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harbor perspectives in biology 2016, 8, a019521. [Google Scholar] [CrossRef]
- Lawrence, M.; Daujat, S.; Schneider, R. Lateral thinking: how histone modifications regulate gene expression. Trends in Genetics 2016, 32, 42–56. [Google Scholar] [CrossRef]
- Van, H.T.; Santos, M.A. Histone modifications and the DNA double-strand break response. Cell Cycle 2018, 17, 2399–2410. [Google Scholar] [CrossRef] [PubMed]
- Arnaudo, A.M.; Garcia, B.A. Proteomic characterization of novel histone post-translational modifications. Epigenetics & chromatin 2013, 6, 1–7. [Google Scholar]
- Campos, E.I.; Reinberg, D. Histones: annotating chromatin. Annual review of genetics 2009, 43, 559–599. [Google Scholar] [CrossRef]
- Lai, P.M.; Chan, K.M. Roles of Histone H2A Variants in Cancer Development, Prognosis, and Treatment. International Journal of Molecular Sciences 2024, 25, 3144. [Google Scholar] [CrossRef]
- Stope, M.B. Phosphorylation of histone H2A. X as a DNA-associated biomarker. World Academy of Sciences Journal 2021, 3, 1–5. [Google Scholar] [CrossRef]
- Yao, S.; Feng, Y.; Zhang, Y.; Feng, J. DNA damage checkpoint and repair: From the budding yeast Saccharomyces cerevisiae to the pathogenic fungus Candida albicans. Computational and Structural Biotechnology Journal 2021, 19, 6343–6354. [Google Scholar] [CrossRef]
- Merighi, A.; Gionchiglia, N.; Granato, A.; Lossi, L. The phosphorylated form of the histone H2AX (γH2AX) in the brain from embryonic life to old age. Molecules 2021, 26, 7198. [Google Scholar] [CrossRef] [PubMed]
- Rahmanian, N.; Shokrzadeh, M.; Eskandani, M. Recent advances in γH2AX biomarker-based genotoxicity assays: A marker of DNA damage and repair. DNA repair 2021, 108, 103243. [Google Scholar] [CrossRef]
- Zhao, S.; Allis, C.D.; Wang, G.G. The language of chromatin modification in human cancers. Nature Reviews Cancer 2021, 21, 413–430. [Google Scholar] [CrossRef] [PubMed]
- Salzano, M.; Sanz-García, M.; Monsalve, D.M.; Moura, D.S.; Lazo, P.A. VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics 2015, 10, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, K.S.; Kuttikrishnan, S.; Ahmad, N.; Habeeba, U.; Mariyam, Z.; Suleman, M.; Bhat, A.A.; Uddin, S. H2AX: A key player in DNA damage response and a promising target for cancer therapy. Biomedicine & Pharmacotherapy 2024, 175, 116663. [Google Scholar]
- Song, H.; Shen, R.; Liu, X.; Yang, X.; Xie, K.; Guo, Z.; Wang, D. Histone post-translational modification and the DNA damage response. Genes & Diseases 2023, 10, 1429–1444. [Google Scholar]
- Oberdoerffer, P.; Miller, K.M. Histone H2A variants: Diversifying chromatin to ensure genome integrity. Seminars in cell & developmental biology 2023, 135, 59–72. [Google Scholar]
- Xie, A.; Puget, N.; Shim, I.; Odate, S.; Jarzyna, I.; Bassing, C.H.; Alt, F.W.; Scully, R. Control of sister chromatid recombination by histone H2AX. Molecular cell 2004, 16, 1017–1025. [Google Scholar] [CrossRef]
- Oh, J.-M.; Myung, K. Crosstalk between different DNA repair pathways for DNA double strand break repairs. Mutation Research/Genetic Toxicology and Environmental Mutagenesis 2022, 873, 503438. [Google Scholar] [CrossRef]
- Guha, S.; Bhaumik, S.R. Transcription-coupled DNA double-strand break repair. DNA repair 2022, 109, 103211. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paull, T.T. Cellular functions of the protein kinase ATM and their relevance to human disease. Nature Reviews Molecular Cell Biology 2021, 22, 796–814. [Google Scholar] [CrossRef] [PubMed]
- Rass, E.; Willaume, S.; Bertrand, P. 53BP1: keeping it under control, even at a distance from DNA damage. Genes 2022, 13, 2390. [Google Scholar] [CrossRef]
- Kciuk, M.; Gielecińska, A.; Mujwar, S.; Mojzych, M.; Kontek, R. Cyclin-dependent kinases in DNA damage response. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 2022, 1877, 188716. [Google Scholar] [CrossRef]
- Shibata, A.; Jeggo, P.A. ATM’s role in the repair of DNA double-strand breaks. Genes 2021, 12, 1370. [Google Scholar] [CrossRef]
- Danovski, G.; Panova, G.; Keister, B.; Georgiev, G.; Atemin, A.; Uzunova, S.; Stamatov, R.; Kanev, P.-B.; Aleksandrov, R.; Blagoev, K.B. Diffusion of activated ATM explains γH2AX and MDC1 spread beyond the DNA damage site. Iscience 2024, 27. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Ho, T.L.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R. Rapid recruitment of p53 to DNA damage sites directs DNA repair choice and integrity. Proceedings of the National Academy of Sciences 2022, 119, e2113233119. [Google Scholar] [CrossRef]
- Salguero, I.; Belotserkovskaya, R.; Coates, J.; Sczaniecka-Clift, M.; Demir, M.; Jhujh, S.; Wilson, M.D.; Jackson, S.P. MDC1 PST-repeat region promotes histone H2AX-independent chromatin association and DNA damage tolerance. Nature communications 2019, 10, 5191. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Zhu, Q.; Wani, G.; He, J.; Wang, Q.-e.; Wani, A.A. USP3 counteracts RNF168 via deubiquitinating H2A and γH2AX at lysine 13 and 15. Cell cycle 2014, 13, 106–114. [Google Scholar] [CrossRef]
- Kocyłowski, M.K.; Rey, A.J.; Stewart, G.S.; Halazonetis, T.D. Ubiquitin-H2AX fusions render 53BP1 recruitment to DNA damage sites independent of RNF8 or RNF168. Cell Cycle 2015, 14, 1748–1758. [Google Scholar] [CrossRef]
- Krishnan, R.; Lapierre, M.; Gautreau, B.; Nixon, K.C.; El Ghamrasni, S.; Patel, P.S.; Hao, J.; Yerlici, V.T.; Guturi, K.K.N.; St-Germain, J. RNF8 ubiquitylation of XRN2 facilitates R-loop resolution and restrains genomic instability in BRCA1 mutant cells. Nucleic Acids Research 2023, 51, 10484–10505. [Google Scholar] [CrossRef]
- Sadoughi, F.; Hallajzadeh, J.; Asemi, Z.; Mansournia, M.A.; Alemi, F.; Yousefi, B. Signaling pathways involved in cell cycle arrest during the DNA breaks. DNA repair 2021, 98, 103047. [Google Scholar] [CrossRef] [PubMed]
- Siler, J.; Guo, N.; Liu, Z.; Qin, Y.; Bi, X. γH2A/γH2AX Mediates DNA Damage-Specific Control of Checkpoint Signaling in Saccharomyces cerevisiae. International Journal of Molecular Sciences 2024, 25, 2462. [Google Scholar] [CrossRef] [PubMed]
- Hammet, A.; Magill, C.; Heierhorst, J.; Jackson, S.P. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO reports 2007, 8, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Aricthota, S.; Rana, P.P.; Haldar, D. Histone acetylation dynamics in repair of DNA double-strand breaks. Frontiers in Genetics 2022, 13, 926577. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.W.; Yu, D.Y.; Pray-Grant, M.G.; Qiu, Q.; Harmon, K.E.; Megee, P.C.; Grant, P.A.; Smith, M.M.; Christman, M.F. Acetylation of histone H4 by Esa1 is required for DNA double-strand break repair. Nature 2002, 419, 411–415. [Google Scholar] [CrossRef]
- van Attikum, H.; Fritsch, O.; Hohn, B.; Gasser, S.M. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004, 119, 777–788. [Google Scholar] [CrossRef]
- Horigome, C.; Oma, Y.; Konishi, T.; Schmid, R.; Marcomini, I.; Hauer, M.H.; Dion, V.; Harata, M.; Gasser, S.M. SWR1 and INO80 chromatin remodelers contribute to DNA double-strand break perinuclear anchorage site choice. Molecular cell 2014, 55, 626–639. [Google Scholar] [CrossRef]
- Papamichos-Chronakis, M.; Krebs, J.E.; Peterson, C.L. Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptationin response to DNA damage. Genes & development 2006, 20, 2437–2449. [Google Scholar]
- Gerhold, C.B.; Gasser, S.M. INO80 and SWR complexes: relating structure to function in chromatin remodeling. Trends in cell biology 2014, 24, 619–631. [Google Scholar] [CrossRef]
- Keogh, M.-C.; Kim, J.-A.; Downey, M.; Fillingham, J.; Chowdhury, D.; Harrison, J.C.; Onishi, M.; Datta, N.; Galicia, S.; Emili, A. A phosphatase complex that dephosphorylates γH2AX regulates DNA damage checkpoint recovery. Nature 2006, 439, 497–501. [Google Scholar] [CrossRef]
- Chen, L.; Lai, Y.; Zhu, X.; Ma, L.; Bai, Q.; Vazquez, I.; Xiao, Y.; Liu, C.; Li, D.; Gao, C. The role of specific PP2A complexes in the dephosphorylation of γ-H2AX. Journal of Cell Science 2015, 128, 421. [Google Scholar] [CrossRef]
- Li, X.; Nan, A.; Xiao, Y.; Chen, Y.; Lai, Y. PP2A–B56ϵ complex is involved in dephosphorylation of γ-H2AX in the repair process of CPT-induced DNA double-strand breaks. Toxicology 2015, 331, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Cao, P.; Greer, P.; Nagengast, E.; Kolb, R.; Mumby, M.; Cowan, K. Protein phosphatase 2A has an essential role in the activation of γ-irradiation-induced G2/M checkpoint response. Oncogene 2010, 29, 4317–4329. [Google Scholar] [CrossRef] [PubMed]
- Nakada, S.; Chen, G.I.; Gingras, A.C.; Durocher, D. PP4 is a γH2AX phosphatase required for recovery from the DNA damage checkpoint. The EMBO Reports 2008, 9, 1019–1026. [Google Scholar] [CrossRef]
- Moon, S.-H.; Nguyen, T.-A.; Darlington, Y.; Lu, X.; Donehower, L.A. Dephosphorylation of γ-H2AX by WIP1: an important homeostatic regulatory event in DNA repair and cell cycle control. Cell cycle 2010, 9, 2092–2096. [Google Scholar] [CrossRef]
- Zhong, J.; Liao, J.; Liu, X.; Wang, P.; Liu, J.; Hou, W.; Zhu, B.; Yao, L.; Wang, J.; Li, J. Protein phosphatase PP6 is required for homology-directed repair of DNA double-strand breaks. Cell cycle 2011, 10, 1411–1419. [Google Scholar] [CrossRef]
- Dziegielewski, J.; Bońkowska, M.A.; Poniecka, E.A.; Heo, J.; Du, K.; Crittenden, R.B.; Bender, T.P.; Brautigan, D.L.; Larner, J.M. Deletion of the SAPS1 subunit of protein phosphatase 6 in mice increases radiosensitivity and impairs the cellular DNA damage response. DNA repair 2020, 85, 102737. [Google Scholar] [CrossRef]
- Xiao, A.; Li, H.; Shechter, D.; Ahn, S.H.; Fabrizio, L.A.; Erdjument-Bromage, H.; Ishibe-Murakami, S.; Wang, B.; Tempst, P.; Hofmann, K. WSTF regulates the H2A. X DNA damage response via a novel tyrosine kinase activity. Nature 2009, 457, 57–62. [Google Scholar] [CrossRef]
- Cook, P.J.; Ju, B.G.; Telese, F.; Wang, X.; Glass, C.K.; Rosenfeld, M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature 2009, 458, 591–596. [Google Scholar] [CrossRef]
- Harvey, A.C.; Jackson, S.P.; Downs, J.A. Saccharomyces cerevisiae histone H2A Ser122 facilitates DNA repair. Genetics 2005, 170, 543–553. [Google Scholar] [CrossRef]
- Rossetto, D.; Avvakumov, N.; Côté, J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef]
- Moore, J.D.; Yazgan, O.; Ataian, Y.; Krebs, J.E. Diverse roles for histone H2A modifications in DNA damage response pathways in yeast. Genetics 2007, 176, 15–25. [Google Scholar] [CrossRef]
- Kozmin, S.G.; Dominska, M.; Kokoska, R.J.; Petes, T.D. A tale of two serines: the effects of histone H2A mutations S122A and S129A on chromosome nondisjunction in Saccharomyces cerevisiae. Genetics 2025, 229, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Côté, V.; Côté, J. DNA damage-induced phosphorylation of histone H2A at serine 15 is linked to DNA end resection. Molecular and Cellular Biology 2021, 41, e00056–00021. [Google Scholar] [CrossRef]
- Xie, A.; Odate, S.; Chandramouly, G.; Scully, R.A. H2AX post-translational modifications in the IR response and homologous recombination. Cell cycle 2010, 9, 3602–3610. [Google Scholar] [CrossRef] [PubMed]
- House, N.C.; Polleys, E.J.; Quasem, I.; De la Rosa Mejia, M.; Joyce, C.E.; Takacsi-Nagy, O.; Krebs, J.E.; Fuchs, S.M.; Freudenreich, C.H. Distinct roles for S. cerevisiae H2A copies in recombination and repeat stability, with a role for H2A. 1 threonine 126. Elife 2019, 8, e53362. [Google Scholar] [CrossRef]
- Sawicka, A.; Seiser, C. Histone H3 phosphorylation–a versatile chromatin modification for different occasions. Biochimie 2012, 94, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Shimada, M.; Niida, H.; Zineldeen, D.H.; Tagami, H.; Tanaka, M.; Saito, H.; Nakanishi, M. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell 2008, 132, 221–232. [Google Scholar] [CrossRef]
- Sharma, A.K.; Bhattacharya, S.; Khan, S.A.; Khade, B.; Gupta, S. Dynamic alteration in H3 serine 10 phosphorylation is G1-phase specific during ionization radiation induced DNA damage response in human cells. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 2015, 773, 83–91. [Google Scholar] [CrossRef]
- Ozawa, K. Reduction of phosphorylated histone H3 serine 10 and serine 28 cell cycle marker intensities after DNA damage. Cytometry Part A: The Journal of the International Society for Analytical Cytology 2008, 73, 517–527. [Google Scholar] [CrossRef]
- Monte-Serrano, E.; Morejón-García, P.; Campillo-Marcos, I.; Campos-Díaz, A.; Navarro-Carrasco, E.; Lazo, P.A. The pattern of histone H3 epigenetic PTMs is regulated by the VRK1 chromatin kinase. Epigenetics & Chromatin 2023, 16, 18. [Google Scholar]
- Monaco, L.; Kolthur-Seetharam, U.; Loury, R.; Murcia, J.M.-d.; de Murcia, G.; Sassone-Corsi, P. Inhibition of Aurora-B kinase activity by poly (ADP-ribosyl) ation in response to DNA damage. Proceedings of the National Academy of Sciences 2005, 102, 14244–14248. [Google Scholar] [CrossRef] [PubMed]
- Tjeertes, J.V.; Miller, K.M.; Jackson, S.P. Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. The EMBO journal 2009, 28, 1878–1889. [Google Scholar] [CrossRef]
- Lan, J.; Lepikhov, K.; Giehr, P.; Walter, J. Histone and DNA methylation control by H3 serine 10/threonine 11 phosphorylation in the mouse zygote. Epigenetics & chromatin 2017, 10, 1–19. [Google Scholar]
- Tian, Y.; Zhang, C.; Tian, X.; Zhang, L.; Yin, T.; Dang, Y.; Liu, Y.; Lou, H.; He, Q. H3T11 phosphorylation by CKII is required for heterochromatin formation in Neurospora. Nucleic Acids Research 2024, 52, 9536–9550. [Google Scholar] [CrossRef]
- Lee, J.-H.; Kang, B.-H.; Jang, H.; Kim, T.W.; Choi, J.; Kwak, S.; Han, J.; Cho, E.-J.; Youn, H.-D. AKT phosphorylates H3-threonine 45 to facilitate termination of gene transcription in response to DNA damage. Nucleic acids research 2015, 43, 4505–4516. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.L.; Turner, F.B.; Krishnamoorthy, T.; Wolner, B.; Ahn, S.-H.; Foley, M.; Dorsey, J.A.; Peterson, C.L.; Berger, S.L.; Allis, C.D. Phosphorylation of histone H4 serine 1 during DNA damage requires casein kinase II in S. cerevisiae. Current biology 2005, 15, 656–660. [Google Scholar] [CrossRef]
- Utley, R.T.; Lacoste, N.; Jobin-Robitaille, O.; Allard, S.; Côté, J. Regulation of NuA4 histone acetyltransferase activity in transcription and DNA repair by phosphorylation of histone H4. Molecular and cellular biology 2005. [Google Scholar] [CrossRef]
- Clouaire, T.; Legube, G. A snapshot on the cis chromatin response to DNA double-strand breaks. Trends in Genetics 2019, 35, 330–345. [Google Scholar] [CrossRef]
- Hossain, M.B.; Shifat, R.; Johnson, D.G.; Bedford, M.T.; Gabrusiewicz, K.R.; Cortes-Santiago, N.; Luo, X.; Lu, Z.; Ezhilarasan, R.; Sulman, E.P. TIE2-mediated tyrosine phosphorylation of H4 regulates DNA damage response by recruiting ABL1. Science advances 2016, 2, e1501290. [Google Scholar] [CrossRef]
- Millan-Zambrano, G.; Santos-Rosa, H.; Puddu, F.; Robson, S.C.; Jackson, S.P.; Kouzarides, T. Phosphorylation of histone H4T80 triggers DNA damage checkpoint recovery. Molecular cell 2018, 72, 625–635. [Google Scholar] [CrossRef]
- Lee, C.-S.; Lee, K.; Legube, G.; Haber, J.E. Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nature structural & molecular biology 2014, 21, 103–109. [Google Scholar]
- Fernandez-Capetillo, O.; Allis, C.D.; Nussenzweig, A. Phosphorylation of histone H2B at DNA double-strand breaks. The Journal of experimental medicine 2004, 199, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.L.; Ajiro, K.; Samejima, K.; Kloc, M.; Cheung, P.; Mizzen, C.A.; Beeser, A.; Etkin, L.D.; Chernoff, J.; Earnshaw, W.C. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 2003, 113, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Andrés, M.; García-Gomis, D.; Ponte, I.; Suau, P.; Roque, A. Histone H1 post-translational modifications: update and future perspectives. International journal of molecular sciences 2020, 21, 5941. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Jeong, K.W.; Kim, H.; Choi, J.; Lu, W.; Stallcup, M.R.; An, W. Functional interplay between p53 acetylation and H1. 2 phosphorylation in p53-regulated transcription. Oncogene 2012, 31, 4290–4301. [Google Scholar] [CrossRef]
- Bonner, W.M.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Sedelnikova, O.A.; Solier, S.; Pommier, Y. γH2AX and cancer. Nature Reviews Cancer 2008, 8, 957–967. [Google Scholar] [CrossRef]
- Kawashima, S.; Kawaguchi, N.; Taniguchi, K.; Tashiro, K.; Komura, K.; Tanaka, T.; Inomata, Y.; Imai, Y.; Tanaka, R.; Yamamoto, M. γ-H2AX as a potential indicator of radiosensitivity in colorectal cancer cells. Oncology Letters 2020, 20, 2331–2337. [Google Scholar] [CrossRef]
- Lee, Y.-C.; Yin, T.C.; Chen, Y.-T.; Chai, C.-Y.; Wang, J.Y.; Liu, M.-C.; Lin, Y.-C.; Kan, J.Y. High expression of phospho-H2AX predicts a poor prognosis in colorectal cancer. Anticancer Research 2015, 35, 2447–2453. [Google Scholar]
- Xiao, J.; Duan, Q.; Wang, Z.; Yan, W.; Sun, H.; Xue, P.; Fan, X.; Zeng, X.; Chen, J.; Shao, C. Phosphorylation of TOPK at Y74, Y272 by Src increases the stability of TOPK and promotes tumorigenesis of colon. Oncotarget 2016, 7, 24483. [Google Scholar] [CrossRef]
- Hsu, J.-Y.; Sun, Z.-W.; Li, X.; Reuben, M.; Tatchell, K.; Bishop, D.K.; Grushcow, J.M.; Brame, C.J.; Caldwell, J.A.; Hunt, D.F. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell 2000, 102, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Imhof, A.; Patel, D.; Kahl, P.; Hoffmeyer, K.; Friedrichs, N.; Müller, J.M.; Greschik, H.; Kirfel, J.; Ji, S. Phosphorylation of histone H3T6 by PKCβI controls demethylation at histone H3K4. Nature 2010, 464, 792–796. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Wen, W.; Zhu, F.; Zhang, J.; Keum, Y.-S.; Zykova, T.; Yao, K.; Peng, C.; Zheng, D.; Cho, Y.-Y.; Ma, W.-y. MST1 promotes apoptosis through phosphorylation of histone H2AX. Journal of Biological Chemistry 2010, 285, 39108–39116. [Google Scholar] [CrossRef]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications—cause and consequence of genome function. Nature Reviews Genetics 2022, 23, 563–580. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1α from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, M.K.; Sinha, S.; Mendez, M.T.; Sanyal, T.; Mahmud, H.; Kay, N.E.; Gupta, M.; Xu, C.; Vesely, S.K.; Mukherjee, P. Aberrantly Expressed Mitochondrial Lipid Kinase, AGK, Activates JAK2–Histone H3 Axis and BCR Signal: A Mechanistic Study with Implication in CLL Therapy. Clinical Cancer Research 2024, OF1–OF15. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Choi, B.Y.; Cho, Y.-Y.; Mizuno, H.; Kang, B.S.; Bode, A.M.; Dong, Z. Phosphorylation of histone H3 at serine 10 is indispensable for neoplastic cell transformation. Cancer research 2005, 65, 5818–5827. [Google Scholar] [CrossRef]
- Komar, D.; Juszczynski, P. Rebelled epigenome: histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clinical Epigenetics 2020, 12, 1–14. [Google Scholar] [CrossRef]
- Lau, P.N.I.; Cheung, P. Histone code pathway involving H3 S28 phosphorylation and K27 acetylation activates transcription and antagonizes polycomb silencing. Proceedings of the National Academy of Sciences 2011, 108, 2801–2806. [Google Scholar] [CrossRef]
- Cho, Y.-Y. RSK2 and its binding partners in cell proliferation, transformation and cancer development. Archives of pharmacal research 2017, 40, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Widjaja, L.; Werner, R.A.; Krischke, E.; Christiansen, H.; Bengel, F.M.; Bogdanova, N.; Derlin, T. Individual radiosensitivity reflected by γ-H2AX and 53BP1 foci predicts outcome in PSMA-targeted radioligand therapy. European Journal of Nuclear Medicine and Molecular Imaging 2023, 50, 602–612. [Google Scholar] [CrossRef]
- Banjarnahor, C.T.U.; Hardiany, N.S.; Wahjoepramono, E.J.; Hariyanto, A.D.; Sadikin, M. High concentration of γ-H2AX correlates with a marker of apoptotic suppression and PI3K/Akt pathway upregulation in glioblastoma multiforme. Oncology Letters 2023, 25, 1–7. [Google Scholar] [CrossRef]
- Hosking, H.; Pederick, W.; Neilsen, P.; Fenning, A. Considerations for the Use of the DNA Damage Marker γ-H2AX in Disease Modeling, Detection, Diagnosis, and Prognosis. Aging and Cancer 2024, 5, 62–69. [Google Scholar] [CrossRef]
- Aitmagambetova, M.; Smagulova, G.; Sakhanova, S.; Kereyeva, N.; Koishybaev, A.; Amanzholkyzy, A.; Tulyaeva, A.; Zholmukhamedova, D.; Kandygulova, G.; Imanbaev, N. The γ-H2AX foci as an indicator for double-stranded DNA breaks and response to ongoing chemotherapy in breast cancer women: a pilot study. European Review for Medical & Pharmacological Sciences 2023, 27. [Google Scholar]
- Zhao, H.; Qu, M.; Li, Y.; Wen, K.; Xu, H.; Song, M.; Xie, D.; Ao, X.; Gong, Y.; Sui, L. An estimate assay for low-level exposure to IR based on mass spectrometry quantification of γ-H2AX in human peripheral blood lymphocytes. Frontiers in Public Health 2022, 10, 1031743. [Google Scholar] [CrossRef]
- Zhu, H.; Chen, K.; Chen, Y.; Liu, J.; Zhang, X.; Zhou, Y.; Liu, Q.; Wang, B.; Chen, T.; Cao, X. RNA-binding protein ZCCHC4 promotes human cancer chemoresistance by disrupting DNA-damage-induced apoptosis. Signal transduction and targeted therapy 2022, 7, 240. [Google Scholar] [CrossRef] [PubMed]
- Llavanera, M.; Delgado-Bermudez, A.; Ribas-Maynou, J.; Salas-Huetos, A.; Yeste, M. A systematic review identifying fertility biomarkers in semen: a clinical approach through omics to diagnose male infertility. Fertility and Sterility 2022, 118, 291–313. [Google Scholar] [CrossRef]
- Zorzompokou, C.; Ipeirotis, M.; Martzoukos, M.K.; Marangos, P. Detection of DNA Double-Stranded Breaks in Mouse Oocytes. J. Vis. Exp 2023, 196, e65494. [Google Scholar] [CrossRef]
- Wu, S.; Cao, R.; Tao, B.; Wu, P.; Peng, C.; Gao, H.; Liang, J.; Yang, W. Pyruvate Facilitates FACT-Mediated γH2AX Loading to Chromatin and Promotes the Radiation Resistance of Glioblastoma. Advanced Science 2022, 9, 2104055. [Google Scholar] [CrossRef]
- Kono, T.; Ozawa, H. A comprehensive review of current therapeutic strategies in cancers targeting DNA damage response mechanisms in head and neck squamous cell cancer. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer 2024, 189255. [Google Scholar]
- Moon, J.; Kitty, I.; Renata, K.; Qin, S.; Zhao, F.; Kim, W. DNA damage and its role in cancer therapeutics. International Journal of Molecular Sciences 2023, 24, 4741. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A schematic representation of histone phosphorylation and its roles in the DNA damage response (DDR).When DNA damage occurs, kinases transfer phosphate groups to specific target sites on histones, leading to the accumulation of phosphorylated histones. These phosphorylated histones recruit and coordinate with other proteins involved in the DDR to collectively carry out DDR processes. The main functions of histone phosphorylation in DDR include facilitating DNA repair, inducing cell cycle arrest, regulating transcription, and promoting apoptosis. Once DNA repair is completed, phosphatases catalyze the removal of phosphate groups, restoring chromatin to its normal state. Created with Biorender.com.
Figure 1.
A schematic representation of histone phosphorylation and its roles in the DNA damage response (DDR).When DNA damage occurs, kinases transfer phosphate groups to specific target sites on histones, leading to the accumulation of phosphorylated histones. These phosphorylated histones recruit and coordinate with other proteins involved in the DDR to collectively carry out DDR processes. The main functions of histone phosphorylation in DDR include facilitating DNA repair, inducing cell cycle arrest, regulating transcription, and promoting apoptosis. Once DNA repair is completed, phosphatases catalyze the removal of phosphate groups, restoring chromatin to its normal state. Created with Biorender.com.
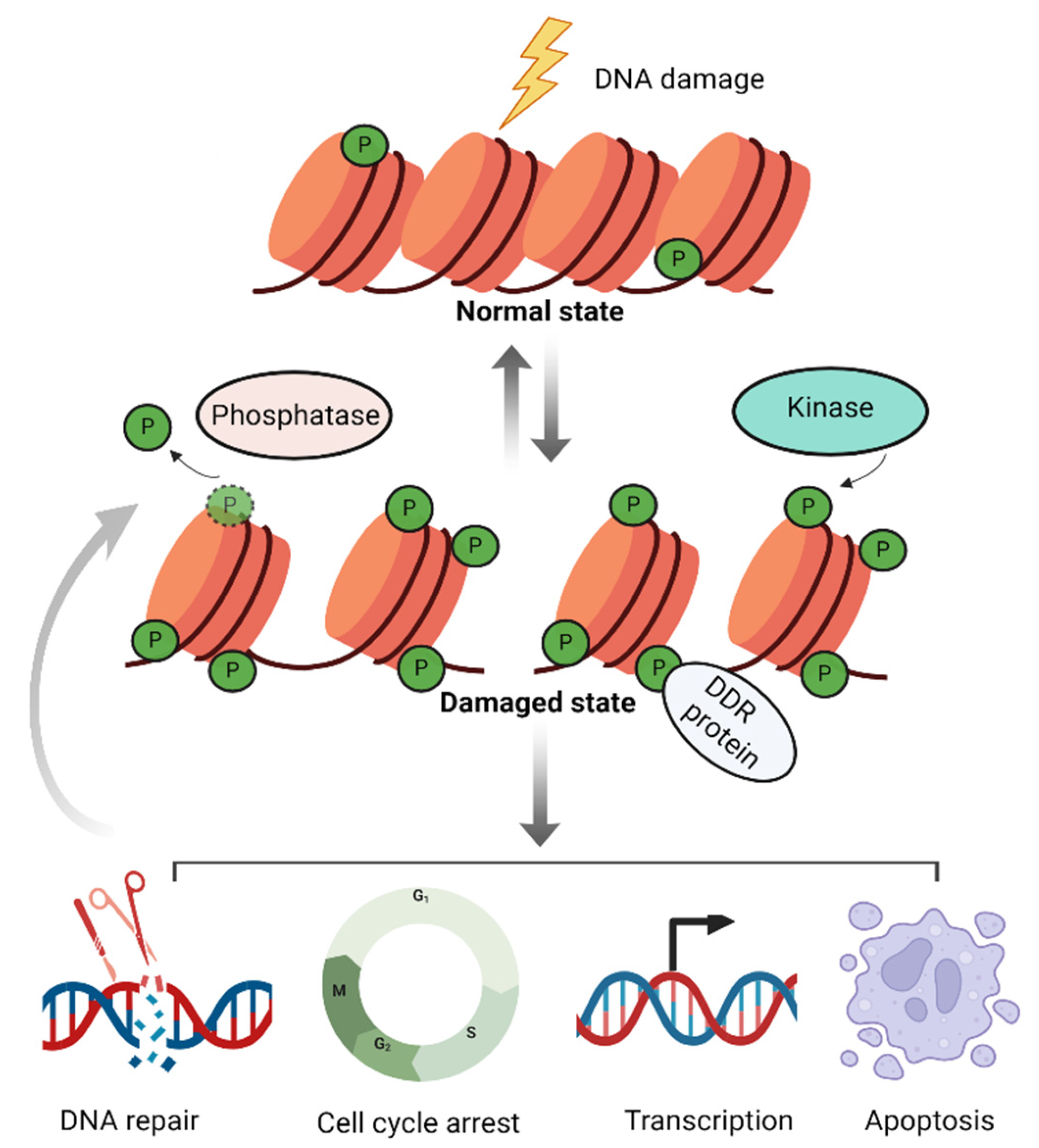

Table 1.
Summary of histone phosphorylation in the DDR discussed in this review.
| Histone phosphorylation sites | Kinases | Function | Ref. |
|---|---|---|---|
| H1.2-T145 | DNA-PK | chromatin remodel; p53 transcription | [95,96] |
| H2A.1-T126 | unknown | affects the stability and repair of fragile DNA regions | [76] |
| H2A-S122 | Bub1 | DNA repair; chromosome segregation | [70,73] |
| H2A-S15 | Mec1 | influencing chromatin dynamics and DNA end resection | [74] |
| H2AX-S139 (H2A-S129 in yeast) | ATM, ATR, DNA-PK | DNA repair; damage signal transduction; transcription; checkpoint regulation; apoptosis | [35,39,40,47,53,56,57,104] |
| H2AX-T101 | unknown | reduce cells’ sensitivity to IR | [75] |
| H2AX-Y142 | WSTF | DNA repair | [68,69] |
| H2B-S14 | MST1 | chromatin remodeling and apoptosis | [93,94] |
| H2B-T129 | Mec1/Tel1 | unclear, possibly coordinated with the function of γH2AX | [92] |
| H3-S10 | Aurora-B | transcription; modulate chromatin structure | [79,80,84,108] |
| H3-S28 | MSK1 | modulate chromatin structure; transcription | [80,110] |
| H3-T11 | CHK1, CKII | DNA repair; transcription; maintenance of heterochromatin | [78,84,85] |
| H3-T45 | AKT | transcription | [86] |
| H4-S1 | CKII | DNA repair | [87,88] |
| H4-T80 | Cla4 | checkpoint regulation | [91] |
| H4-Y51 | TIE2 | DNA repair | [90] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



