Submitted:
16 February 2025
Posted:
17 February 2025
You are already at the latest version
Abstract
Spirocalcaridines A and B are among the most challenging members of the marine invertebrate derived Leucetta alkaloids. Approaches for construction and elaboration of the highly compact spirocyclic core are described. Synthesis of tricyclic guanidine via a tandem oxidative amination dearomatizing spirocyclization (TOADS) using hypervalent iodine set the stage for total synthesis via migration of the C4/C8 double bond to C4/C5 position followed by oxidation. The undesired but not surprising propensity of the spirocyclic cyclohexadienone to undergo rearrangement to the phenol hindered the desired olefin migration. Furthermore, initial efforts to install the oxidation sequentially first, at C5 followed by C4 of the complete carbon skeleton were fraught with unforeseen challenges and unusual outcomes. In addition, the scope, and limitations of hypervalent iodine mediated tandem oxidative dearomatizing spirocyclization on various substrates were explored. Urethanes and thiourethanes undergo spirocyclization in excellent yield whereas, the reaction with allylic substrates and species lacking the p-methoxy substituent does not proceed. Attempts to prepare other guanidine precursors are briefly discussed.
Keywords:
Hypervalent iodine
; Dearomatizing spirocyclization
; Allylic hydroxylation
; dearomatization
1. Introduction
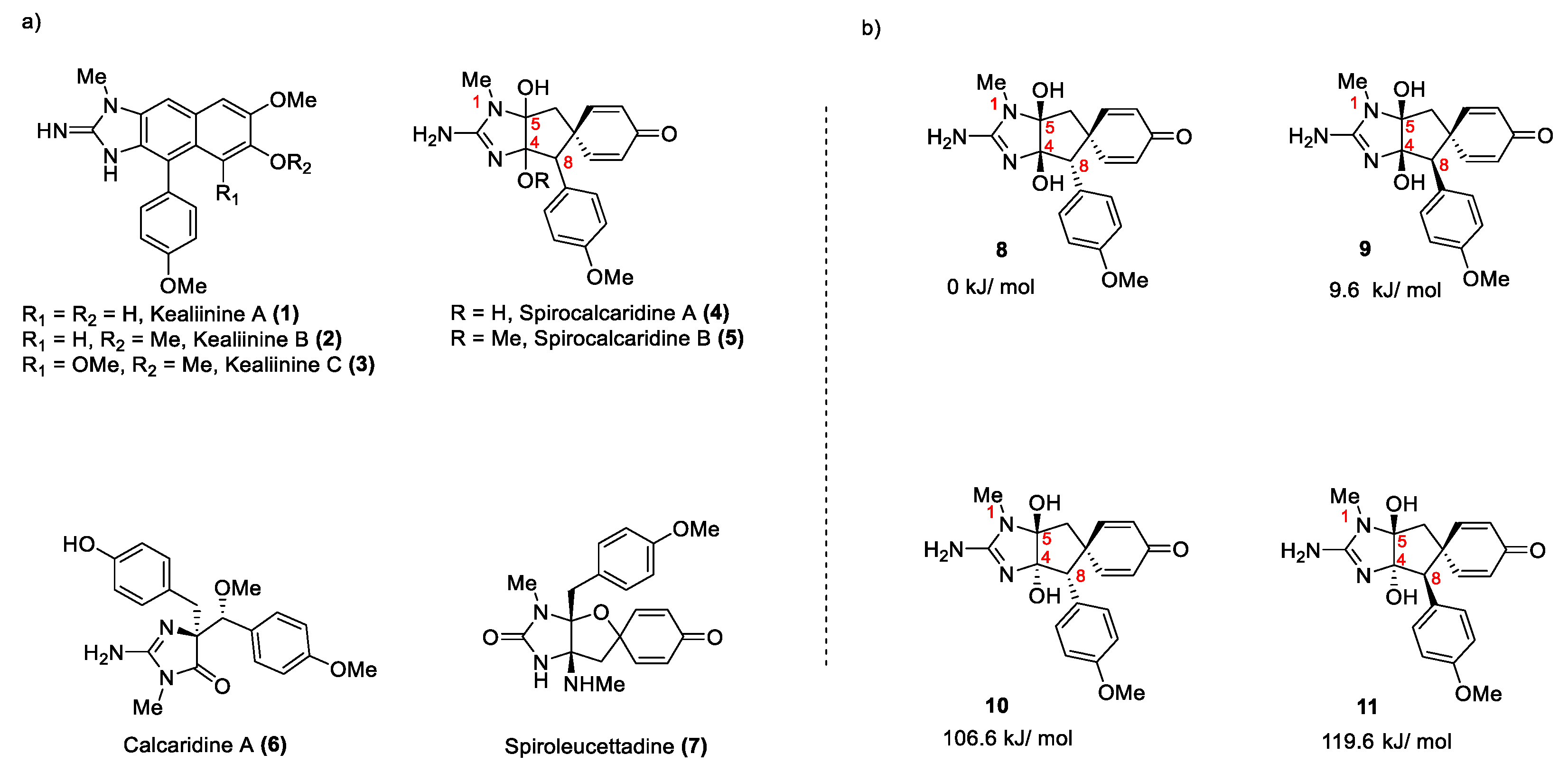
Deromatizing reactions are of substantial interest in modern synthetic organic chemistry, offering versatile and efficient approaches to the construction of complex, functionally rich molecular architectures [1] including natural products [2]. When conducted intramolecularly, this flexible methodology leads to the formation of spirocyclic compounds, where two rings share a common atom, serving as a pivotal step in the synthesis of various natural products, [3] pharmaceuticals and functional materials [4,5]. By breaking aromaticity, the resulting products possess unique reactivity profiles and three-dimensional structural motifs, facilitating access to diverse chemical space and enabling the creation of molecules with tailored properties. Leucetta alkaloids have received significant attention from medicinal chemists because of the potentially useful biological activities and have challenged the creativity and ability of the synthetic chemistry community to solve the synthetic challenges posed by the presence of unusual structural features often belying the intrinsic challenges [6,7,8,9]. Spirocalcaridines A (4) and B (5) were described by the Crews lab in 2003 [17]. These two alkaloids, along with calcaridine A (6), were isolated from the Calcareous sponge Leucetta sp. These molecules exhibit unprecedented structural features for this family of marine sponges, including the first intrinsically chiral members of this group. For example, at the time (+)-calcaridine A (6) was the only member of the family to contain a rearranged 4,4-dibenzyl-5-imidazol(on)e framework, although recently additional examples of this skeleton have appeared in the literature [10,11]. On the other hand, spirocalcaridine A (4) has a unique structure featuring a hexahydrocyclopentamidazol-2-ylidenamine ring system spiro-fused to a cyclohexadienone ring. In addition to 4, the corresponding OMe derivative (-)-spirocalcaridine B (5) was isolated from the same sample. Interestingly, spirocalcaridine B (5) does not seem to be an artifact from the isolation process as both (4) and (5) were shown to be stable in methanol solutions for extended periods by Crews [12]. The constitutions of these molecules were determined by NMR spectroscopy but the relative stereochemistry of the three chiral centers are as yet unknown. The two compounds are deficient in appropriately positioned protons for use in the NOE studies precluding this assignment, in addition, the location of the methoxy group in spirocalcaridine B is considered tentative. The spirocalcaridines are not the only member of this family in which hydrogen deficiency has resulted in ambiguity in the precise structure. Spiroleucettadine was intitially misassigned and several unsuccessful synthetic efforts [13,14] to construct the proposed framework raised questions regarding its constitution ultimately resulting in revision of the structure [15] and confirmation through a total synthesis [2]. Given this stereochemical ambiguity for 4 (and 5), there are four possible (relative) stereochemical arrangements 8-11 that can be suggested, but based on ring strain considerations of trans fusion of the two five membered rings, the two cis [3.3.0] diastereomers (8 and 9) were considered more likely. In fact, it was clear from DFT calculations at wB97XD/def2tzvp level conducted on possible spirocalcaridine A stereoisomers, that the cis-carbinolamines (8 and 9) are much more stable than trans. From optimized structures it seems they not only benefit from less strain but also may be stablized due to a feasible intramolecular H-bond between the two –OH groups, which does not exist in the trans stereoisomers (10 and 11). Out of the two cis isomers, the structure with an endo p-methoxyphenyl group is lower in energy and is likely the more correct structure.
In general, imidazole containing natural products have been synthesized via two complementary approaches; first through the de novo construction of imidazole ring [2,16] and second via functionalizing an existing imidazole ring [17,18]. In general, our group has pursued the latter approach which has provided us with significant success in the total synthesis of several other family members [17,18,19]. However, functionalizing the existing imidazole ring to access the spirocalcaridines presented significant roadblocks, prompting us to investigate a de novo approach to imdazole construction. Indeed, in our exploratory approaches to the spirocalcaridines, we investigated dearomatization chemistries developed by Larock [20] and Li [21] to establish the spiro fused 5,6 rings [22]. Furthermore, significant efforts were made towards the de novo imidazole synthesis giving access to several highly functionalized intermediates suitable for the total synthesis along with some undesired yet constructive outcomes [19,22,23,24,25,26,27,28]. We have reported an unusual one-pot spirocyclization-N-cyanation reaction assisted by cyanogen bromide, leading directly to the spiro fused derivative, a potential intermediate in synthetic approaches to several Leucetta alkaloids [29]. Hypervalent iodine-mediated dearomatizing spirocyclization represents a cutting-edge synthetic strategy that leverages the unique reactivity of hypervalent iodine reagents to facilitate the construction of spirocyclic compounds from aromatic precursors [30]. Moreover, the mild reaction conditions and broad substrate scope of hypervalent iodine-mediated reactions make it a versatile and environmentally benign strategy for accessing structurally and stereochemically diverse spirocycles, thus fueling innovation in synthetic methodology and molecular design. We have also reported the synthesis of tricyclic guanidine derivatives from propargyl guanidines via hypervalent iodine mediated tandem oxidative amination dearomatizing spirocyclization (TOADS) reaction, that set the stage for total synthesis of spirocalcaridines [16]. Specifically through migration of the C4/C8 double bond to C4/C5 position followed by oxidation and removal of protecting groups. Herein we report our efforts towards exploration of these advanced intermediates for completion of total synthesis and expand the substrate scope and limitations of hypervalent iodine mediated dearomatizing spirocyclization.
Figure 1.
a) A selection of the more highly oxidized Leucetta alkaloids. b) possible diastereomeric structures of spirocalcaridine A and their energies.
Figure 1.
a) A selection of the more highly oxidized Leucetta alkaloids. b) possible diastereomeric structures of spirocalcaridine A and their energies.
2. Results and Discussions
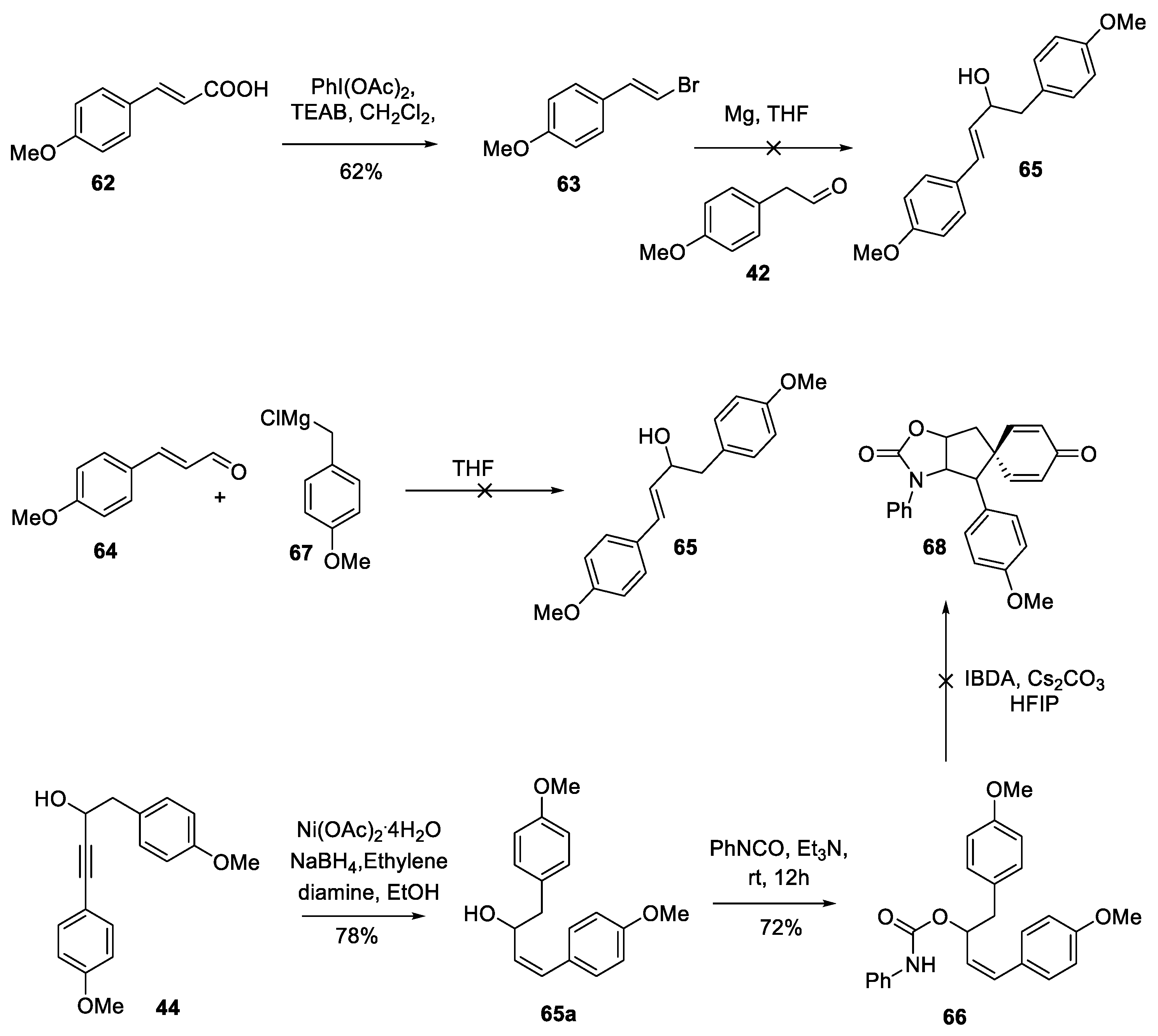
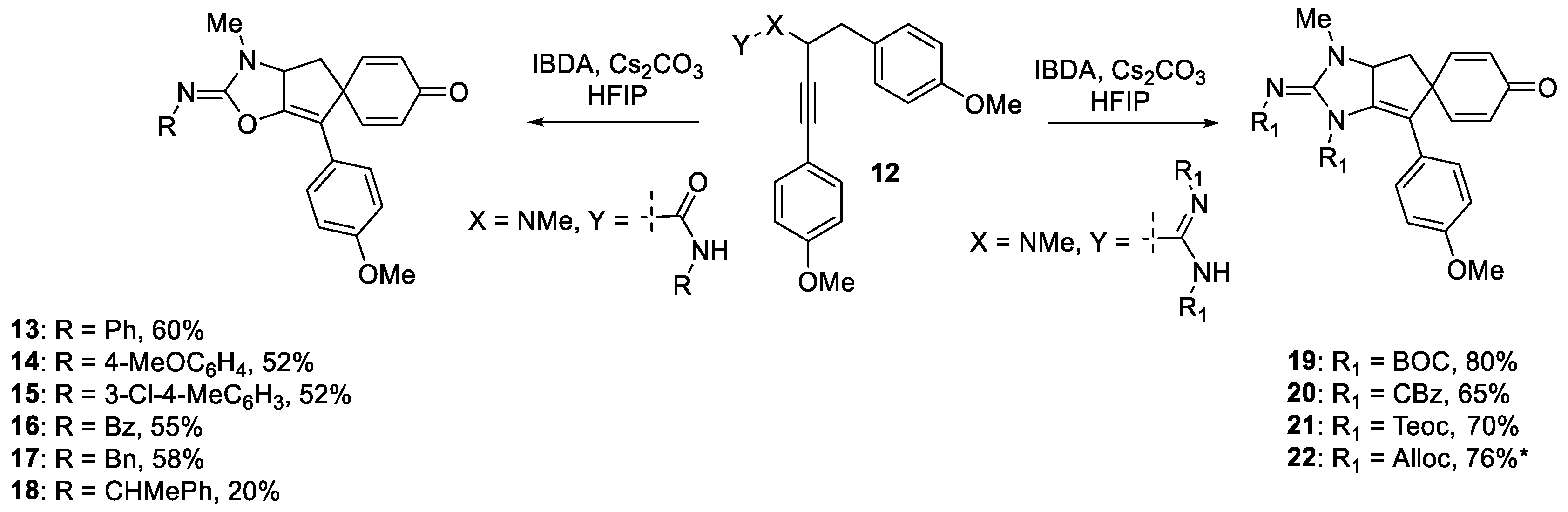
As depicted in Scheme 1, our earlier report describes a hypervalent iodine mediated oxidative dearomatizing spirocyclization of propargyl guanidines to construct the complete core present in spirocalcaridines A and B (12→19, 20 and 21) in addition to the previously unreported bis alloc analogue (22*). Apart from the propargyl guanidines, propargylic ureas also participate in this chemistry upon treatment with iodosobenzene diacetate (IBDA), providing spiro-fused iminooxazoles (12→13, 14, 15, 16, 17, and 18) [16].
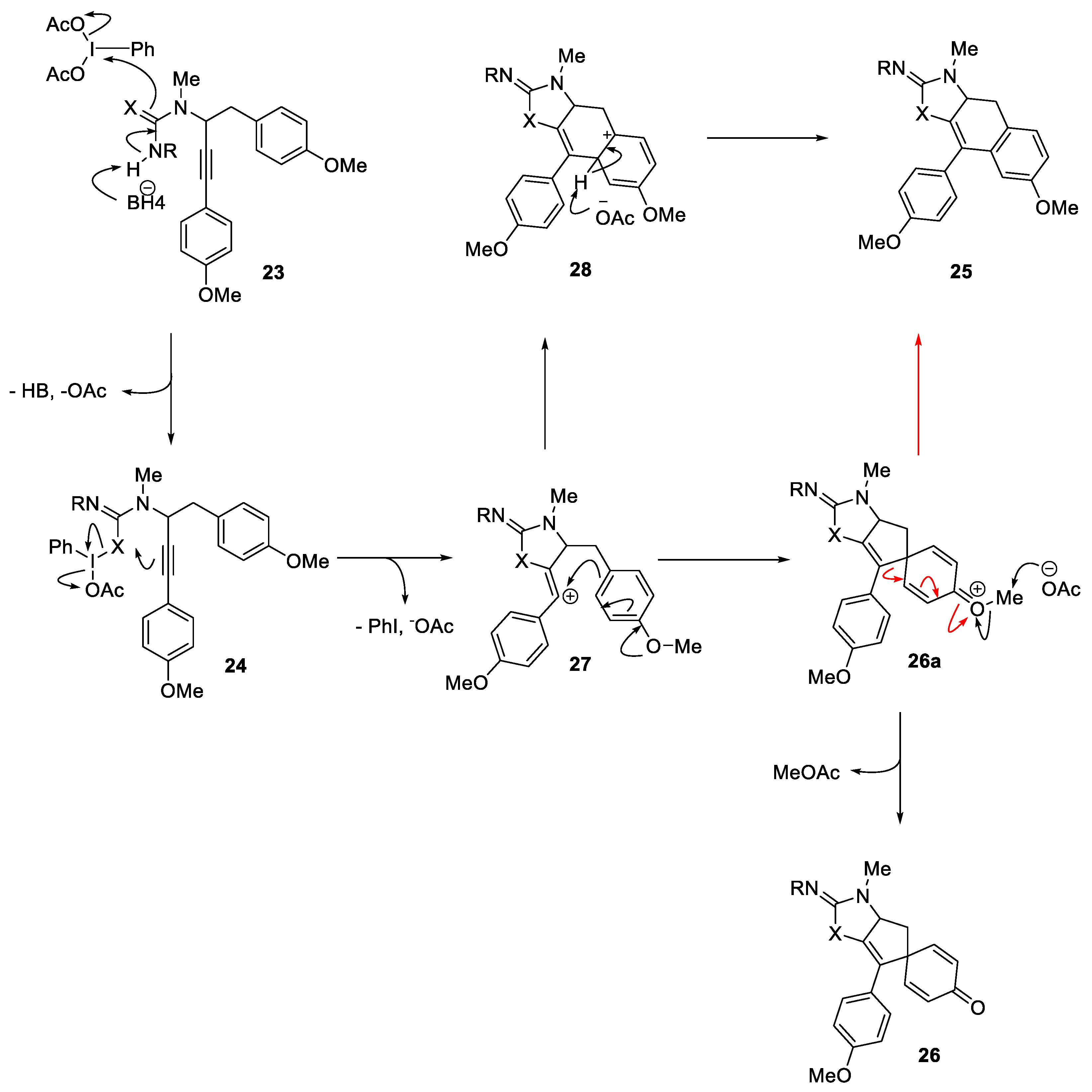
HFIP was found to be the only solvent to produce the spiro-fused iminooxazoles (13 to 18) and 2-iminoimidazoles (19 to 22). Interestingly, we observed the formation of naphthoxazole 25 (X = O, Scheme 2) upon using TFE as solvent with the urea. A putative mechanism is given in Scheme 2, specifically, the urea oxygen or the guanidine nitrogen of 23 is activated by IBDA through ligand exchange, forming the electrophilic species 24. Intramolecular addition to the electrophilic heteroatom then delivers the stablized vinylic carbocation 27, which then affords the spirocyclic product 26 upon ipso addition of the electron-rich aromatic ring followed by demethylation, presumably by acetate ion. It is postulated that in TFE, due to its relatively lower dipolarity/polarizability (0.908) compared to HFIP (1.007),31 the nucleophilicity of the acetate ion is impacted which in turn results in rearrangement of the spiro intermediate to the naphthalene derivative 28 prior to demethylation. Alternatively, the naphthalene framework 25 can be formed through the phenyl ring engaging directly with the vinyl carbocation to form a tertiary carbocation 28 which gets aromatized to produce 25. Other pathways, such as a concerted process or a radical-based pathway cannot be ruled out at this time.
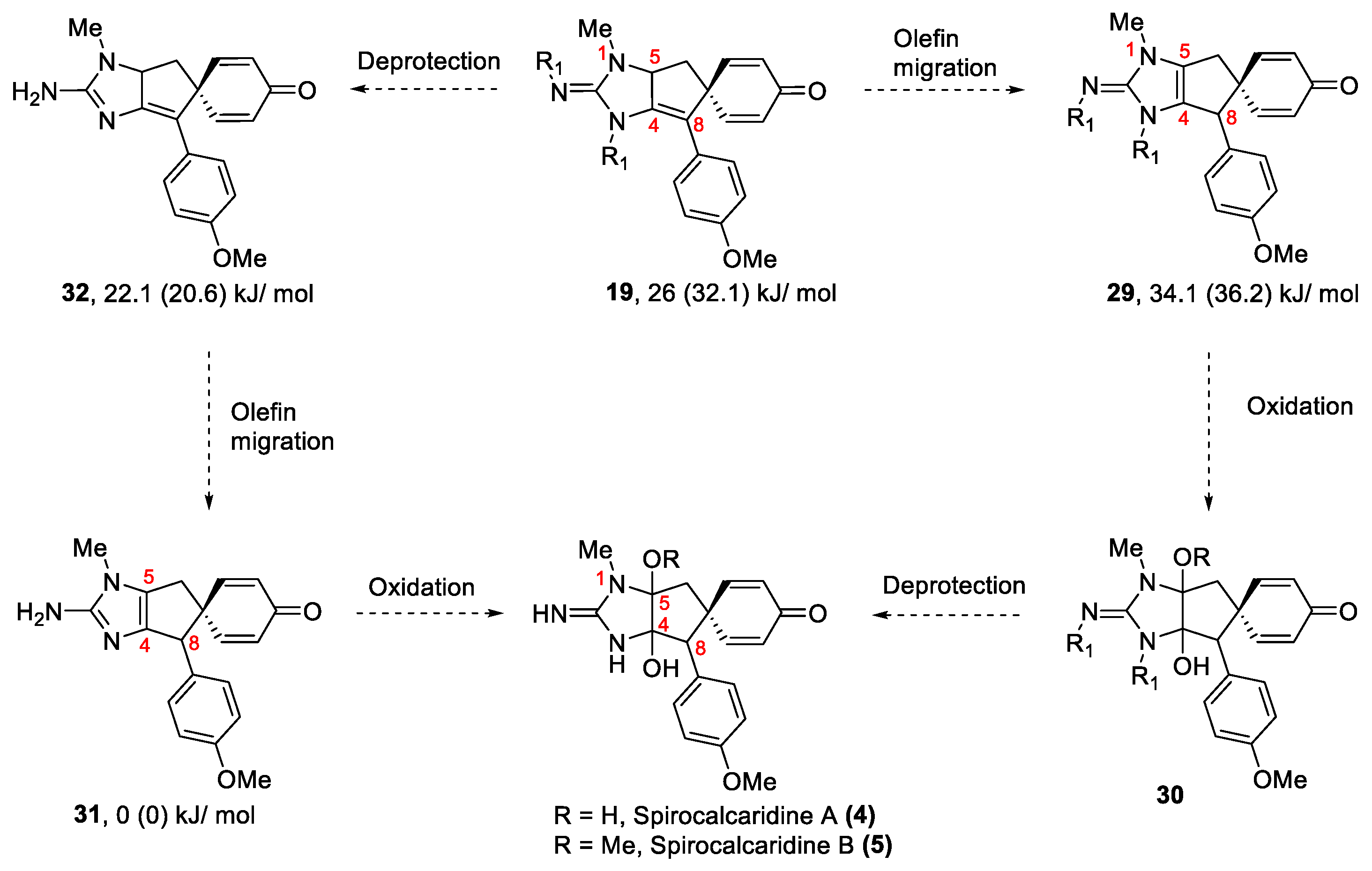
These tricylic guanidines (19 to 22) contain the complete carbon skeleton of spirocalcaridine A and requires migration of the double bond from C4/C8 to the desired position C4/C5 (19→29, Scheme 3). Our expectation was that this migration would be driven by the aromatization of the imidazole. This type of process, in a general sense, has been observed in the Diels-Alder products from vinylimidazoles where the initial adduct places a double bond exo-cyclic to the imidazole and this migrates either under thermal conditions (i.e., directly after the cycloaddition) or in the presence of electrophiles [32]. The precise repositioning of the olefin would allow us to perform oxidation across this double bond using chemistry developed in our group [33]. previously to produce 30 and finally deprotection of the imidazole would complete the total synthesis. Alternatively, it was also anticipated that deprotection of the carbamates forming 32 followed by the double bond migration would allow us the access to 31 and eventually oxidation would conclude the total synthesis.
Numerous examples of double bond migrations promoted by transition metals, [34,35] acids [36,37] and bases [38,39,40] are described in the literature. Since the bis-BOC substrate 19 is relatively easy to synthesize, attempts to effect the double bond migration on 19 using these conditions were made first. Several transition metal catalysts were tested [41,42] which include RhCl(PPh3)3, [43] RuCl3, [44] Pd(OAc)2, [45] (C6H5CN)2PdCl2, [46] PtCl2, [47] PdI2, [42] Pt(acac)2, [47] Pt(COD)Cl2, [47] to perform the double bond migration. In most cases, we observed complex mixtures including products formed via cyclohexadienone-phenol rearrangement (Scheme 4). Base-catalyzed reactions were also not effective for double bond migration; weaker bases such as DBU and pyridine were ineffective, whereas, complex reaction mixtures were observed when stronger bases e.g. NaOH and KOH were used. In one attempt to perform this reaction using sodium ethoxide (made in-situ by reacting NaH and EtOH), [40] we observed cleavage of one BOC group. This outcome was not necessarily surprising per se but what was unexpected was the deprotected product was found to be significantly less-polar on TLC. Based on this observation it was hypothesized that the BOC group on the ring nitrogen (N3) was cleaved instead of the one on the 2-imino nitrogen. Since the double bond has moved inside the ring and is now in conjugation with the C4/C8 double bond renders it more stable. The hypothesis was further supported by 1H NMR data. The 1H NMR spectrum of 19 shows two distinct signals for the t-Bu groups. The one in the vicinity of p-anisyl substituent has a signal at δH 1.0 ppm, whereas the other one on the exocyclic nitrogen appears at 1.5 ppm. After deprotection, the product with a mono BOC group shows the t-Bu signal at 1.5 ppm and the 1.0 ppm signal disappears suggesting that the ring BOC was cleaved. In addition, the removal of this BOC group allows greater rotational mobility of the p-anisyl group and there is now more space for it to conjugate with the C4-C8 double bond. The upfield shift of the methyl signals of the t-Bu group is presumably a function of lying above the shielding cone of the p-anisyl group. This outcome is in accordance with our previously reported result where a similar outcome was observed after treating the bis-BOC substrate with TFA. The process resulted in the cleavage of one of the BOC groups and cyclohexadienone-phenol rearrangement to produce 34, the order of events is not known. The dihydronaphthimidazole intermediate 34 was crystalline and X-ray crystal structure clearly shows the connectivity of the BOC group as stated above. The remaining N-BOC group appeared at δH 1.50 in the 1H NMR spectrum of 34 [16].
As we were unable to migrate the double bond, there was some concern regarding the viability of this step due to increased strain of the double bond along the ring fusion and thus DFT calculations were performed [48] to illuminate and possibly guide the next steps. The free energies of both 19 and 29 were calculated at wB97XD [49]/ def2tzvp [50,51] level. Interestingly, it was found that the rearranged product 29 was infact 12.1 kJ/mole less stable than the precursor 19 in the gas phase. Calculations performed with solvation models [52] also showed that 19 was more stable, but the difference was attenuated quite substantially. In contrast, when the deprotected congener 32 and the corresponding rearranged congener 31 were evaluated, the relative stabilities switched. In this case, the incorporation of a solvation model resulted only in a modest change in relative stability. Whether this change is a function of the imidazole ring being fully aromatic in 31 compensating for lost stabilization due to conjugation of the C4-C5 bond whereas in 29 the imidazole is not aromatic is unclear. However, this suggested that deprotection prior to rearrangement might be a constructive avenue for investigation.
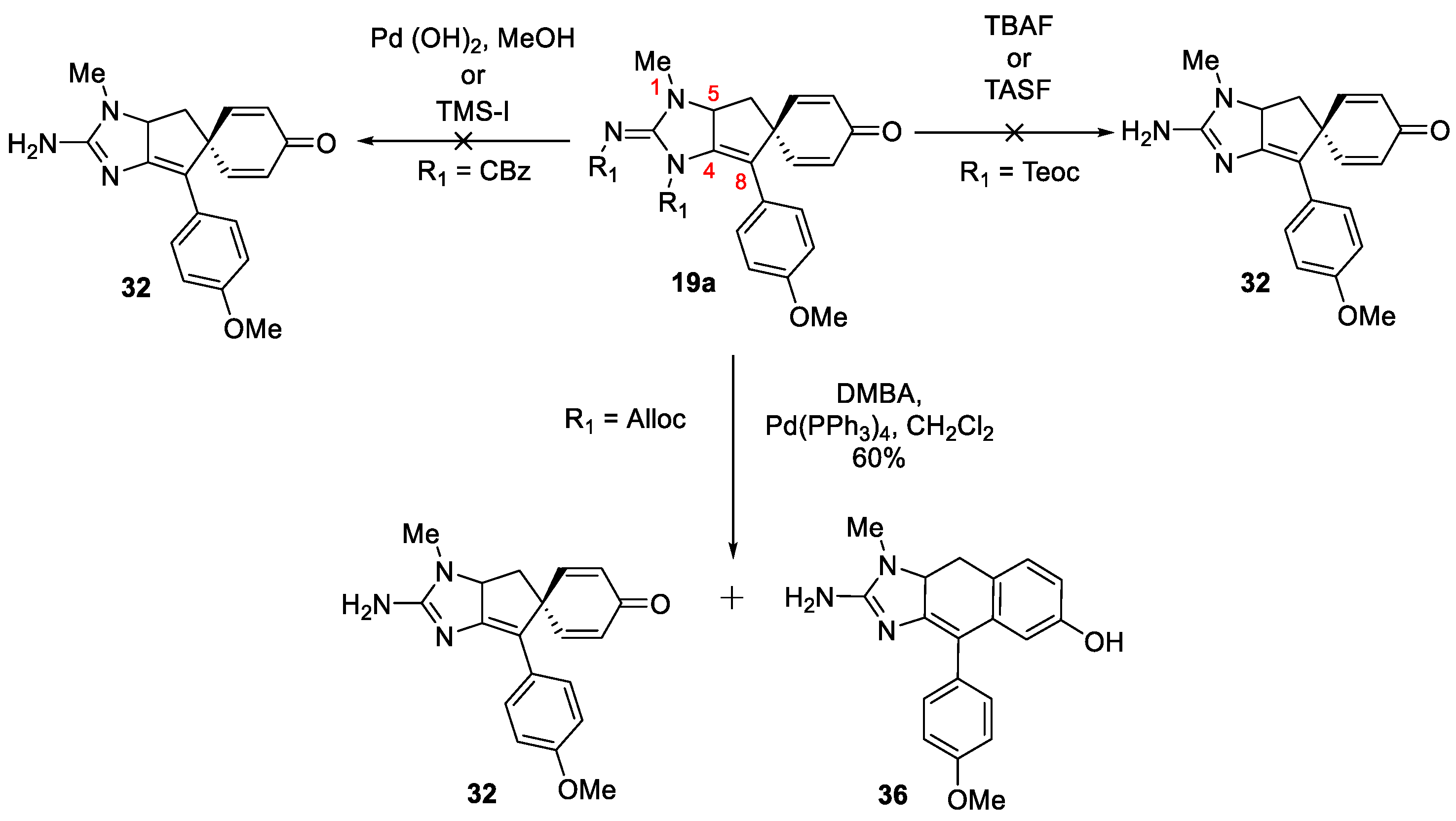
After unsuccessful attempts to effect double bond migration and deprotection with the bis-BOC substrate, focus shifted to the removal of protecting groups on the closely related analogs, specifically bis-Cbz, bis-Alloc and bis-Teoc groups. Removal of bis-Cbz 20 was investigated using Pd(OH)2 and TMSI, but in both cases starting material decomposition was observed. A similar outcome was witnessed when the bis-Teoc intermediate 21 was treated with TBAF or TASF, the starting material was found to be unstable and decomposition was observed. Deprotection of bis-Alloc derivative 22 using DMBA and Pd(PPh3)4 resulted in the formation of a mixture of two products; one formed as a result of the usual and required deprotection process (32, Scheme 6) and the other was formed as a result of deprotection followed by cyclohexadienone-phenol rearrangement (36, Scheme 6). The structures are tentatively assigned based on the NMR data of the mixture and these two products were found to be inseparable, possibily due to the continous rearrangement of 32 to 36 during silica gel chromatography.
Scheme 5.
Efforts towards removal of protecting groups.
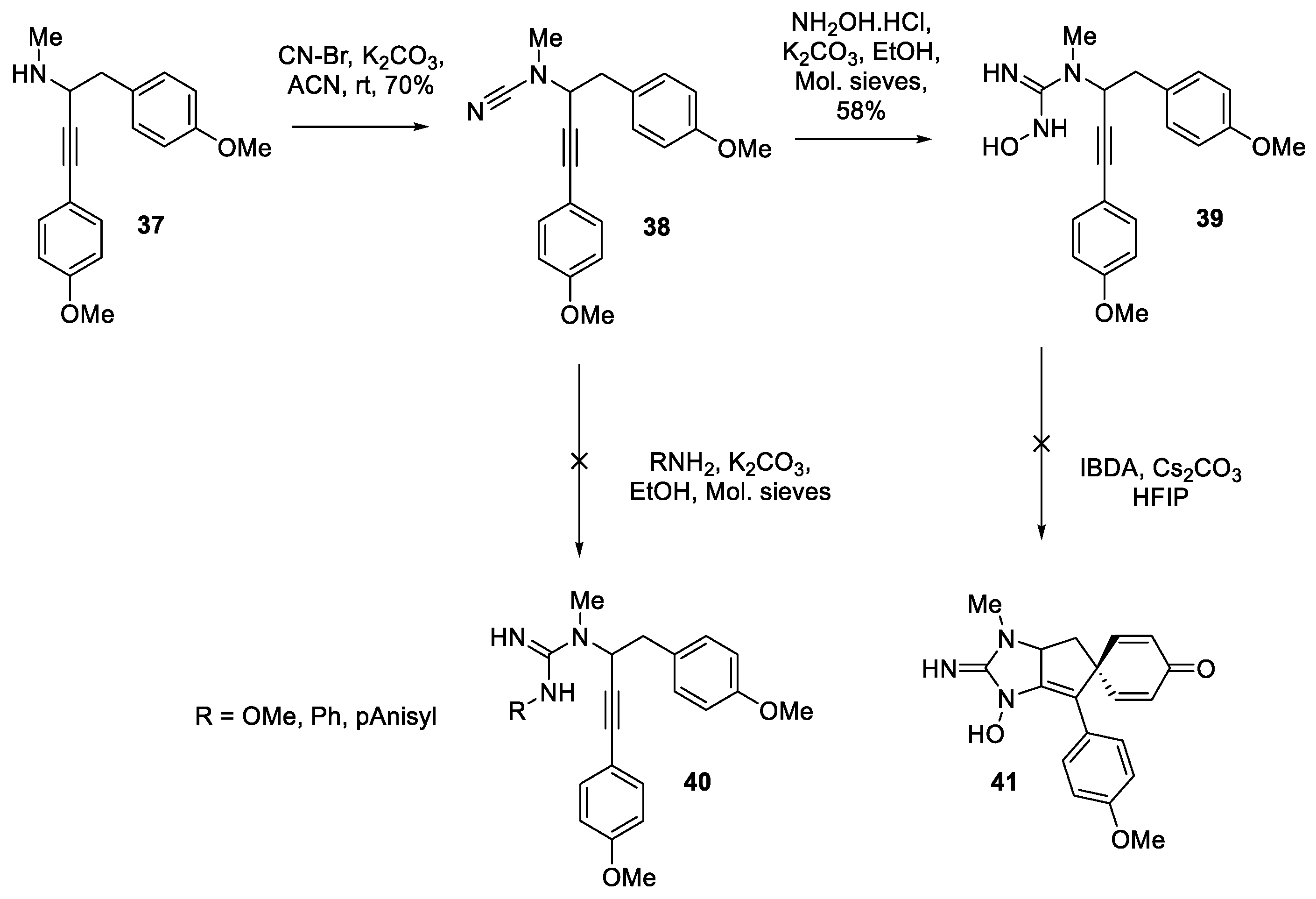
As these efforts outlined in Scheme 5 and Scheme 6 depict, the removal of the nitrogen protecting groups and the migration of double bond provided a significant roadblock in the completion of the total synthesis and thus has forced us towards exploration of alternative strategies. Accordingly, substrates which would contain a propargylic guanidine precursor lacking N-protecting groups thereby avoiding deprotection at later stage. The synthesis of these free guanidines was envisioned through amination of corresponding cyanamide 38 (Scheme 6). Accordingly, as reported, the propargyl amine 37 was treated with CN-Br in presence of K2CO3 to deliver the cyanamide via an N-cyanation (Scheme 6) [29]. Cyanamide 38 was anticipated to be a potential substrate to access the corresponding guanidine, and an attempt was made towards guanylation followed by spirocyclization. Cyanamide 38 was converted to the corresponding N-hydroxyguanidine [53] 39 by treating with hydroxylamine hydrochloride and K2CO3 in anhydrous EtOH. Other amines including anilines and methoxyamine were not effective for guanylation. The N-hydroxyguanidine 39 was subjected to the TOADS conditions but unfortunately a complex mixture formation was observed.
Scheme 6.
Efforts towards synthesis of un-protected guanidines.
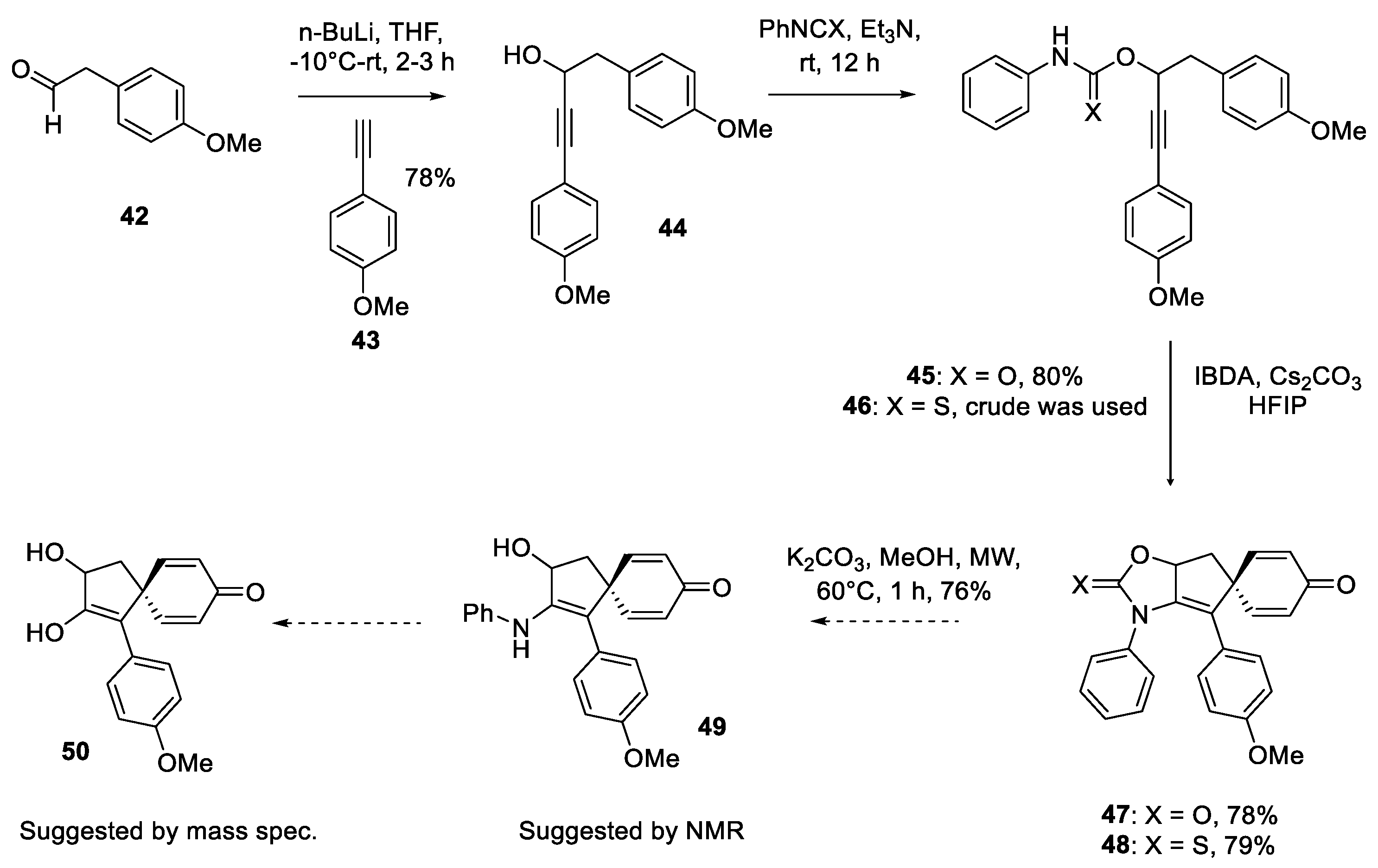
An alternative stategy was considered which involved late stage introduction of guanidine, post spirocyclization. This was the first investigation where the feasibility of the propargyl alcohol framework rather than using the corresponding amine was evaluated. Accordingly, acetaldehyde 42 was treated with lithium acetylide of 4-ethynylanisole 43 to construct the propargyl alcohol 44 which was then converted to the corresponding phenyl urethane 45 and thiourethane 46 by reacting it with phenyl isocyanate and isothiocyanate respectively. Both of these substrates were subjected to the previously established oxidative dearomatization conditions and both substrates produced the spirocyclic compounds with good yields. Initially, it was uncertain whether cyclization took place via nitrogen or X (O/S), and the structures to the spirocyclic compounds were assigned tentatively as 47 and 48. However, a hydrolysis experiment was performed upon 47 following the methods reported for the cleavage of oxazolidinones [54,55]. In particular, intermediate 47 was treated with K2CO3 in MeOH to produce a polar species. 1H and 13C NMR data showed that the cleavage was completed but the N-phenyl group was still intact suggesting 49 was the product. Interestingly, however, mass spectrometry data showed the mass of the hydrolyzed product 50. Since conversion of enamines to enols is well known [56] it is conceivable that the hydrolysis had occurred under the mass spec. conditions. This outcome certainly confirms that cyclization is taking place through nitrogen to produce the oxazolone 47. While these spirocycles hold promise for application in total synthesis of spirocalcaridines, discovery of the spirocyclization of urethane brings new opportunities for the synthesis of spirocyclic α-hydroxy ketones. At this stage guanidine substrates were revisited.
Scheme 7.
Dearomatizing spirocyclization of propargyl urethanes and thiourethanes. .
As stated above, there are a number of ways to migrate the C4/C8 double bond inside the imidazole ring [32,57,58] and it was thought that the C4/C8 double bond migration to C4/C5 position has been recalcitrant because it is both tetrasubstituted and in conjugation with the p-methoxyphenyl D-ring [59,60]. Although in the presence of N-protecting groups it is unlikely to be fully coplanar due to steric crowding. Thus, the construction of a propargyl guanidine containing a cyclohexyl D-ring instead of p-anisyl D-ring was explored as a means to mitigate conjugation with C4/C8 double bond. The synthesis of propargyl guanidine 53 was identical to its aryl congener through a three component coupling and was completed upon treatment of the propargyl amine 52 with N,N′-di-BOC-S-methylisothiourea and mercuric oxide in presence of Et3N in CH2Cl2. The platform was set for the construction of spirocyclic frameworks (54 or 55), and these intermediates would provide access to spirocalcaridines after oxidation state adjustments [61,62] and deprotection steps (Scheme 8). Although it should be noted that the cyclohexyl group was just a model in the first instance. Thus, the bis-BOC-guanidine 53 was subjected to the previously developed TOADS reaction conditions. Unfortunately, the reaction resulted in the decomposition of the substrate. This outcome is consistent with the intermediacy of a vinylic carbocation 27 (Scheme 3), and its reduced stability in this case in absence of the strongly electron donating p-methoxyphenyl group [63]. Although the result of this particular campaign was disappointing, it offers some insights into the limitations of the TOADS chemistry.
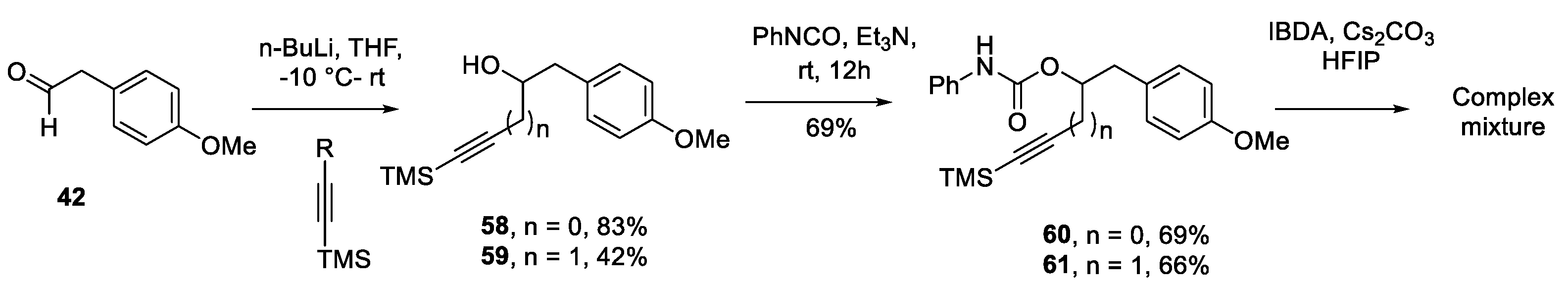
Despite some limitations, the versatility of the TOADS reaction is quite remarkable, and we wanted to test the viability of a few other alkynes as nucleophiles. Synthesis of TMS propargyl urethanes appeared simpler than the corresponding guanidines and construction of propargyl urethane 60 and homopropargyl urethane 61 was undertaken simultaneously. In particular, aldehyde 42 was treated with lithium (trimethylsilyl)acetylide and (3-(trimethylsilyl)prop-2-yn-1-yl)lithium to afford the corresponding alcohols 58 and 59, [64] which were converted to the corresponding urethanes 60 and 61 respectively by reacting with phenylisocyanate and Et3N in CH2Cl2. The two TMS-protected urethanes 60 and 61 were subjected to the TOADS conditions. However, the conversion was not clean and both substrates produced a complex mixture, providing additional insights towards the limitation of TOADS reaction.
Based on the outcome of the chemistries described in the synthetic schemes above, it seemed likely that a spirocyclic framework lacking both C4/C8 and C4/C5 double bond might serve more effectively en route to the total synthesis of spirocalcaridines A and B. In order to construct such a spirocyclic system, an allylic substrate 66 was required which would not only undergo IBDA assisted TOADS reaction to deliver a spirocyclic framework 68, but also would provide insight regarding the general feasibility of TOADS chemistry . To best of our knowledge there was no prior report for the synthesis of an analogous allylic guanidine. Again, as the construction of allylic alcohol framework appeared easier in the first instance, our efforts were directed towards its assembly. Accordingly, two parallel approaches were begun to construct the allylic alcohol 65. Having the aldehyde 42 in hand, it was planned to react this with the Gignard of styryl bromide derivative 63 to deliver the allylic alcohol 65. The styryl bromide 63 was accessed via IBDA assisted decarboxylative bromination of 4-methoxycinnamic acid 62 [65]. However, all attempts to convert the styryl bromide to the corresponding Grignard were unsuccessful. Alternatively, changing the reaction sequence and reacting trans-p-methoxycinnamaldehyde with p-methoxybenzyl magnesium chloride was evaluated, but again to no avail. Encouraged by a prior report of the efficient syn-reduction of an alkyne to alkene, [66,67] the previously synthesized propargyl alcohol 44 was reduced using NiBH4 (prepared in situ from Ni(OAc)2 and NaBH4) to afford the corresponding Z-alcohol 65a in excellent yield. The Z-allylic alcohol, thus obtained, was treated with phenylisocyanate to produce the corresponding urethane 66. Subjection of the urethane 66 to the standard TOADS conditions did not afford the desired product but a complex mixture was formed instead. It is important to mention here that after addition of HFIP to the urethane 66, the color of the solution changed from colorless to light yellow. We wanted to investigate the HFIP solution of 66, consequently an aliquot was taken and was briefly examined. The HFIP treated product was found to be relatively polar on TLC, the 1H NMR spectrum was also different. Complexity of the NMR data due to presence of other products, hindered us to reach to a conclusion, and once again, because of our focus towards completeion of total synthesis, no attempts were made to further characterize the HFIP treated product or to screen more oxidants towards TOADS reaction of allylic urethanes.
Scheme 10.
Scope of spirocyclization toward allylic urethanes.
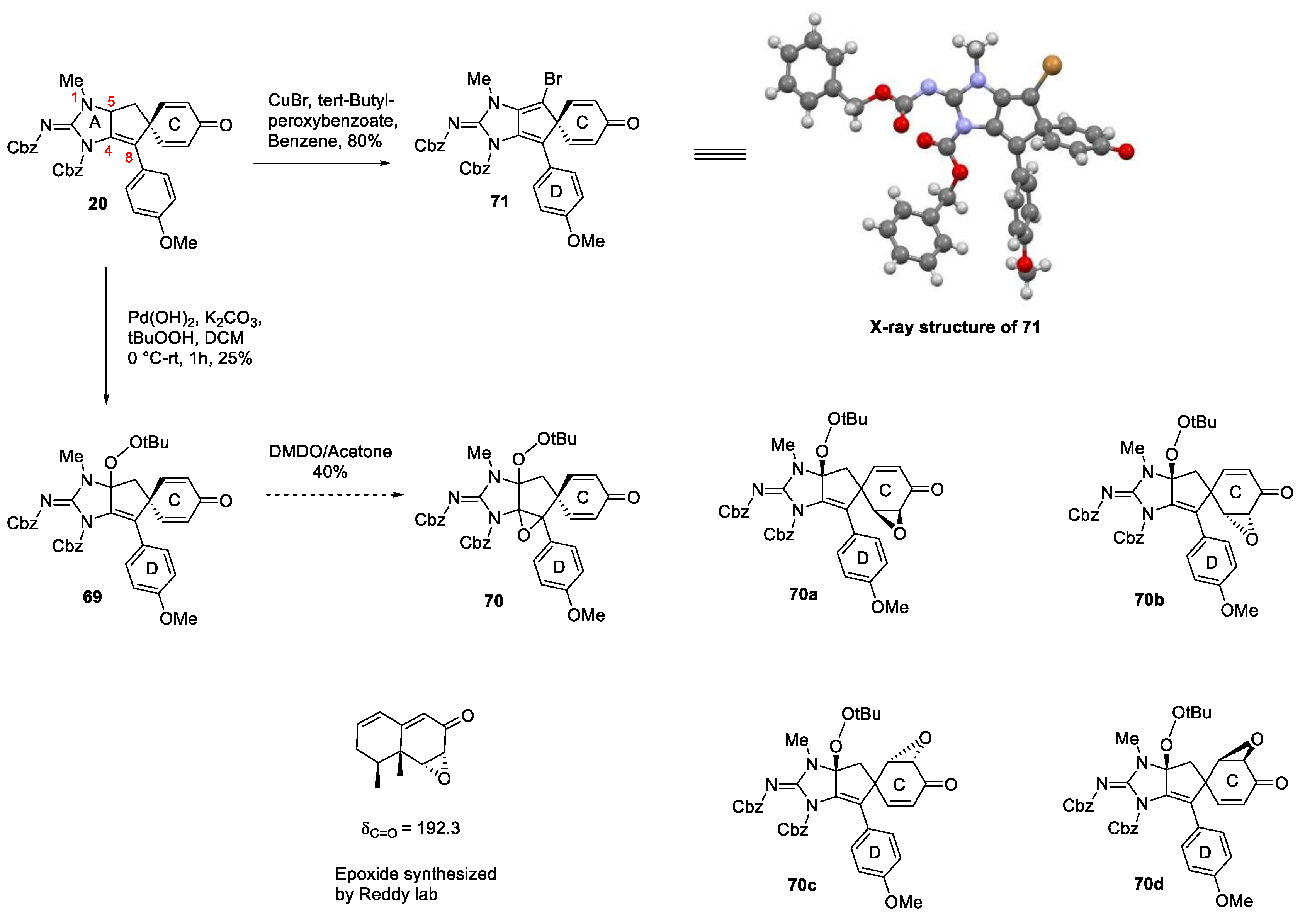
One of the advantages of synthetic chemistry is the ability to use different reagents or change the order of events to either circumvent roadblocks or to obtain different selectivities. Accordingly, we sought to investigate oxidative chemistries prior to deprotection and whether the C4-C8 double bond might engage in oxidative processes thereby obviating the need for rearrangement. In a scouting experiment the oxidation of the allylic position of the bis-Cbz substrate 20 using conditions developed by Corey and Yu were tested [68]. Although the yield was modest, installation of the required functionality at C5 was accomplished and the peroxy intermediate 69 was obtained in 25% yield, which was sufficient to carry out further scouting experiments. The initial plan was to carry out epoxidation of the C4/C8 double bond of 69 and subject it to site-selective reductive ring opening. In the event, the peroxy intermediate 69 was treated with dimethyldioxirane (DMDO) in acetone for 2 days. The epoxidation was very slow but this was somewhat expected because of the sterically hindered nature of the double bond. NMR data showed that a mixture of products was formed, which seemed reasonable by anticipating syn/anti epoxidation with respect to the peroxy moiety. Several unsuccessful attempts were made to separate the products chromatographically, eventually the mixture was subjected to conditions to effect reductive epoxide ring opening [69]. The TLC profile of this epoxide ring opening reaction was exciting as two new polar spots were formed with no starting material remaining. The two components were separated, and the NMR spectra were recorded of the two isolated components. It was clear from these data that the isolated products were in fact not single compounds. On closer examination of the NMR data of the initial epoxidation product and comparison with previously reported data [70], it became obvious that epoxidation had actually occurred on the cyclohexadienone ring (Scheme 11, 70a-70d, δC=O = 192.3 (product) vs 185.2 substrate) and not the cyclopentene. It is assumed that oxidation anti to the p-methoxybenzene group has occurred giving rise to two possible constitutional isomers. Taking into consideration the possible future hurdles during the epoxidation, no further attempt was made to take this approach forward.
One other reaction worth mentioning was uncovered during an attempt to improve the yield of the allylic hydroxylation reaction with the bis-Cbz analogue 20, it was observed that when stoichiometric CuBr was used with tert-butyl peroxybenzoate as an oxidant, a new oxidation product was formed [71]. In fact, the aim was to improve the yield of allylic oxidation, and exploration of other metals was warranted with copper serving as a viable alternative for palladium in the oxidation [72]. Initially, catalytic CuBr was used in presence of tert-butyl peroxybenzoate and benzene as a solvent. The reaction was sluggish and low yielding at room temperature, while decomposition was observed at elevated temperatures. To circumvent this observation, a small excess of the reagent and catalyst were used. With 1 equiv. of catalyst and 5 equiv. of the oxidant, one major spot was observed on TLC and starting material 20 was completely consumed. The yield of the isolated product was excellent but initially, determining the structure of this product was a challenge. However, it was clear from the 1H-NMR data that both -CH2 and -CH protons on the B-ring were missing and an assumption of ring desaturation was made. Mass spectrometry data indicated that bromine had been included into the product, based on the 1H NMR data the most obvious location was on the B-ring. Fortunately, the material was crystalline and upon obtaining an X-ray crystal structure it was revealed that the product formed was in fact the ring oxidized product 71. We did not perform any control reactions or further explore this chemistry but the origin of the observed product is consistent with net oxidation of the C5,C6 bond followed by bromine addition. It is assumed that this is via a radical pathway, but a polar mechanism cannot be fully ruled out in the absence of additional data.
3. Conclusions
In summary, we have evaluated several approaches to advance the multifunctional spirocyclic core of spirocalcaridines A and B obtained via TOADS chemistries towards completion of total synthesis. Significant roadblocks have emerged in the elaboration of the spirocyclic framework to permit the completion of total synthesis. Migration of the double bond from C4/C8 to C4/C5 position has been very challenging and DFT calculations suggest that this may in fact be fatal to the approach based on an increase in strain. In fact DFT calculations show that energy difference between reactant and product (19 and 33, R1 = BOC, Scheme 4) of the isomerization is about 12 kJ/mol with the reactant being more stable. Oxidation of C5 was shown to be partially successful and a new oxidative bromination reaction was discovered using CuBr and tert-butyl peroxybenzoate. Efforts continue to utilize these versatile intermediates towards completion of total synthesis and the results will be reported in due course.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, C.J.L. and R.P.S.; methodology, R.P.S.; software, X.M. and D.G.; validation, R.P.S., C.J.L., D.G., X.M. and P.K.; formal analysis, CJL, R.P.S., D.G. and X.M.; resources, C.J.L. and P.K.; data curation, R.P.S., D.G. and X.M.; writing—original draft preparation, R.P.S.; writing—review and editing, C.J.L., PK, X.M., R.P.S.; supervision, C.J.L.; project administration, C.J.L.; funding acquisition, C.J.L. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research was funded by the Robert A. Welch Foundation (Y-1362) and the NSF (CHEM1956328).
Data Availability Statement
Any reasonable request for original data supporting the work reported in this manuscript will be honored by the corresponding author.
Acknowledgments
Authors acknowledge the support from Shimadzu Center for Advanced Analytical Chemistry, College of Science, UTA for HRMS analysis and Texas A&M High Performance Research Computing for providing the advanced computing resources.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Li, L.; Hou, Z.-W.; Li, P.; Wang, L. Electrochemical Dearomatizing Spirocyclization of Alkynes with Dimethyl 2-Benzylmalonates to Spiro[4.5]deca-trienones. J. Org. Chem. 2022, 87, 8697–8708. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Aberle, N.S.; Lucas, N.T.; Lessene, G.; Hawkins, B.C. Total Synthesis of (−)-Spiroleucettadine. Angew. Chem. Int. Ed. 2017, 56, 14663–14666. [Google Scholar] [CrossRef] [PubMed]
- Roose, T.R.; McSorley, F.; Groenhuijzen, B.; Saya, J.M.; Maes, B.U.W.; Orrù, R.V.A.; Ruijter, E. Dearomative Spirocyclization of Tryptamine-Derived Isocyanides via Iron-Catalyzed Carbene Transfer. J. Org. Chem. 2023, 88, 17345–17355. [Google Scholar] [CrossRef]
- Inprung, N.; Ho, H.E.; Rossi-Ashton, J.A.; Epton, R.G.; Whitwood, A.C.; Lynam, J.M.; Taylor, R.J.K.; James, M.J.; Unsworth, W.P. Indole-ynones as Privileged Substrates for Radical Dearomatizing Spirocyclization Cascades. Org. Lett. 2022, 24, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Chi, Z.; Gao, Y.; Yang, L.; Zhou, C.; Zhang, M.; Cheng, P.; Li, G. Dearomative spirocyclization via visible-light-induced reductive hydroarylation of non-activated arenes. Chin. Chem. Lett. 2022, 33, 225–228. [Google Scholar] [CrossRef]
- Jin, Z. Muscarine, imidazole, oxazole, and thiazolealkaloids. Nat. Prod. Rep. 2011, 28, 1143–1191. [Google Scholar] [CrossRef]
- Koswatta, P.B.; Lovely, C.J. Structure and synthesis of 2-aminoimidazole alkaloids from Leucetta and Clathrina sponges. Nat. Prod. Rep. 2011, 28, 511–528. [Google Scholar] [CrossRef]
- Sullivan, J.D.; Giles, R.L.; Looper, R.E. 2-Aminoimidazoles from Leucetta Sponges: Synthesis and Biology of an Important Pharmacophore. Curr. Bioact. Compd. 2009, 5, 39–78. [Google Scholar] [CrossRef]
- Roué, M.; Quévrain, E.; Domart-Coulon, I.; Bourguet-Kondracki, M.-L. Assessing calcareous sponges and their associated bacteria for the discovery of new bioactive natural products. Nat. Prod. Rep. 2012, 29, 739–751. [Google Scholar] [CrossRef]
- Tang, W.-Z.; Yang, Z.-Z.; Sun, F.; Wang, S.-P.; Yang, F.; Jiao, W.-H.; Lin, H.-W. (-)-Calcaridine B, a new chiral aminoimidazole-containing alkaloid from the marine sponge Leucetta chagosensis. J. Asian Nat. Prod. Res. 2019, 21, 1123–1128. [Google Scholar] [CrossRef]
- Campos, P.-E.; Herbette, G.; Fougère, L.; Clerc, P.; Tintillier, F.; de Voogd, N.J.; Le Goff, G.; Ouazzani, J.; Gauvin-Bialecki, A. An Aminopyrimidone and Aminoimidazoles Alkaloids from the Rodrigues Calcareous Marine Sponge Ernsta naturalis Mar. Drugs [Online], 2022, p. 637.
- Edrada, R.A.; Stessman, C.C.; Crews, P. Uniquely Modified Imidazole Alkaloids from a Calcareous Leucetta Sponge. J. Nat. Prod. 2003, 66, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Aberle, N.; Ovenden, S.P.B.; Lessene, G.; Watson, K.G.; Smith, B.J. Spiroleucettadine: synthetic studies and investigations towards structural revision. Tetrahedron Lett. 2007, 48, 2199–2203. [Google Scholar] [CrossRef]
- Chang, J.J.; Chan, B.; Ciufolini, M.A. Synthetic studies toward spiroleucettadine. Tetrahedron Lett. 2006, 47, 3599–3601. [Google Scholar] [CrossRef]
- White, K.N.; Amagata, T.; Oliver, A.G.; Tenney, K.; Wenzel, P.J.; Crews, P. Structure Revision of Spiroleucettadine, a Sponge Alkaloid with a Bicyclic Core Meager in H-Atoms. J. Org. Chem. 2008, 73, 8719–8722. [Google Scholar] [CrossRef]
- Singh, R.P.; Das, J.; Yousufuddin, M.; Gout, D.; Lovely, C.J. Tandem Oxidative Dearomatizing Spirocyclizations of Propargyl Guanidines and Ureas. Org. Lett. 2017, 19, 4110–4113. [Google Scholar] [CrossRef]
- Das, J.; Koswatta, P.B.; Jones, J.D.; Yousufuddin, M.; Lovely, C.J. Total Syntheses of Kealiinines A–C. Org. Lett. 2012, 14, 6210–6213. [Google Scholar] [CrossRef] [PubMed]
- Koswatta, P.B.; Sivappa, R.; Dias, H.V.R.; Lovely, C.J. Total Synthesis of (±)-Calcaridine A and (±)-epi-Calcaridine A. Org. Lett. 2008, 10, 5055–5058. [Google Scholar] [CrossRef]
- Singh, R.P.; Bhandari, M.R.; Torres, F.M.; Doundoulakis, T.; Gout, D.; Lovely, C.J. Total Synthesis of (±)-2-Debromohymenin via Gold-Catalyzed Intramolecular Alkyne Hydroarylation. Org. Lett. 2020, 22, 3412–3417. [Google Scholar] [CrossRef]
- Zhang, X.; Larock, R.C. Synthesis of Spiro[4.5]trienones by Intramolecular ipso-Halocyclization of 4-(p-Methoxyaryl)-1-alkynes. J. Am. Chem. Soc. 2005, 127, 12230–12231. [Google Scholar] [CrossRef]
- Tang, B.-X.; Tang, D.-J.; Tang, S.; Yu, Q.-F.; Zhang, Y.-H.; Liang, Y.; Zhong, P.; Li, J.-H. Selective Synthesis of Spiro[4,5]trienyl Acetates via an Intramolecular Electrophilic ipso-Iodocyclization Process. Org. Lett. 2008, 10, 1063–1066. [Google Scholar] [CrossRef]
- Koswatta, P.B.; Das, J.; Yousufuddin, M.; Lovely, C.J. Studies towards the Leucetta-Derived Alkaloids Spirocalcaridine A and B – Possible Biosynthetic Implications. Eur. J. Org. Chem. 2015, 2015, 2603–2613. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Gout, D.; Lovely, C.J. Tandem Thioacylation-Intramolecular Hydrosulfenylation of Propargyl Amines – Rapid Access to 2-Aminothiazolidines. European Journal of Organic Chemistry 2019, 2019, 1726–1740. [Google Scholar] [CrossRef]
- Singh, R.P.; Aziz, M.N.; Gout, D.; Fayad, W.; El-Manawaty, M.A.; Lovely, C.J. Novel thiazolidines: Synthesis, antiproliferative properties and 2D-QSAR studies. Bioorg. Med. Chem. 2019, 27, 115047. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.N.; Singh, R.P.; Gout, D.; Lovely, C.J. Dearomatizing spirocyclization of thioureas, ureas and guanidines. Tetrahedron Lett. 2021, 72, 153054. [Google Scholar] [CrossRef]
- Singh, R.P.; Lovely, C.J. Chapter 3 - The Leucetta alkaloids: Synthetic aspects. In Stud. Nat. Prod. Chem., Atta ur, R. Ed. Elsevier: 2019; Vol. 63, pp 43-79.
- Yousufuddin, M.; Morales, R.; Cassis, W.; Brown, G.W.; Singh, R.P.; Lovely, C.J. [1,4-Bis(4-meth oxy phen yl)but-3-yn-2-yl](cyano) methyl amine. IUCrData 2018, 3, x180389. [Google Scholar] [CrossRef]
- Morris, L.L.; Alvarado, C.A.; Goncalves, J.M.; Singh, R.P.; Lovely, C.J.; Yousufuddin, M. [4-(4-Meth oxy phen yl)-8-oxo-3-(phenyl selan yl)spiro [4.5]deca-3,6,9-trien-2-yl]methyl cyanamide. IUCrData 2020, 5, x200078. [Google Scholar] [CrossRef]
- Singh, R.P.; Spears, J.A.; Dalipe, A.; Yousufuddin, M.; Lovely, C.J. Dearomatizing spirocyclization reactions of alkynyl cyanamides. Tetrahedron Lett. 2016, 57, 3096–3099. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef]
- Catalán, J.; López, V.; Pérez, P. Solvent dipolarity/polarizability (SPP) of alcoholic solvents. Liebigs Annalen 1995, 1995, 793–795. [Google Scholar] [CrossRef]
- Lovely, C.J.; Du, H.; Sivappa, R.; Bhandari, M.R.; He, Y.; Dias, H.V.R. Preparation and Diels−Alder Chemistry of 4-Vinylimidazoles. J. Org. Chem. 2007, 72, 3741–3749. [Google Scholar] [CrossRef]
- Sivappa, R.; Koswatta, P.; Lovely, C.J. Oxidative reactions of tetrahydrobenzimidazole derivatives with N-sulfonyloxaziridines. Tetrahedron Lett. 2007, 48, 5771–5775. [Google Scholar] [CrossRef]
- Grieco, P.A.; Nishizawa, M.; Marinovic, N.; Ehmann, W.J. Remote double bond migration via rhodium catalysis: a novel enone transposition. J. Am. Chem. Soc. 1976, 98, 7102–7104. [Google Scholar] [CrossRef]
- Murai, M.; Nishimura, K.; Takai, K. Palladium-catalyzed double-bond migration of unsaturated hydrocarbons accelerated by tantalum chloride. Chem Commun (Camb) 2019, 55, 2769–2772. [Google Scholar] [CrossRef]
- Seto, H.; Fujioka, S.; Koshino, H.; Takatsuto, S.; Yoshida, S. Stereo and chemical course of acid-catalyzed double bond migration of cholesta-5,7-dien-3β-ol to 5α-cholesta-8,14-dien-3β-ol. J. Chem. Soc. Perkin Trans. 1 2000, 1697–1703. [Google Scholar] [CrossRef]
- Noyce, D.S.; Evett, M. Mechanism of the acid-catalyzed double bond migration in 3-cyclohexen-1-one and 3-methyl-3-cyclohexen-1-one. J. Org. Chem. 1972, 37, 394–397. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, C.; Zhao, Y.; Li, D.; Zhao, J.; Wang, Z.; Qu, J. Base-Promoted Double-Bond-Migration/Hydrolysis/Isomerization of 4-Aryl-1,1,1-trifluorobut-2-en-2-yl Trifluoromethanesulfonates: A Metal-Free Approach to β-Trifluoromethyl Ketones. Eur. J. Org. Chem. 2018, 2018, 6217–6222. [Google Scholar] [CrossRef]
- Kloosterziel, H.; van Drunen, J.A.A. Stereochemistry of base-catalysed double-bond migration in allyl ethers. Recl. Trav. Chim. Pays-Bas 1970, 89, 32–36. [Google Scholar] [CrossRef]
- Walker, D.P.; Eklov, B.M.; Bedore, M.W. Practical Synthesis of 3-Oxa-6-azabicyclo[3.1.1]heptane Hydrotosylate; A Novel Morpholine-Based Building Block. Synthesis 2012, 44, 2859–2862. [Google Scholar] [CrossRef]
- Krompiec, S.; Krompiec, M.; Penczek, R.; Ignasiak, H. Double bond migration in N-allylic systems catalyzed by transition metal complexes. Coord. Chem. Rev. 2008, 252, 1819–1841. [Google Scholar] [CrossRef]
- Fiorito, D.; Scaringi, S.; Mazet, C. Transition metal-catalyzed alkene isomerization as an enabling technology in tandem, sequential and domino processes. Chem. Soc. Rev. 2021, 50, 1391–1406. [Google Scholar] [CrossRef]
- Grigg, R.; Stevenson, P.J. Rhodium(I)-Catalysed Isomerisation of N-Allylimines to 2-Aza-1,3-dienes. Synthesis 1983, 1983, 1009–1010. [Google Scholar] [CrossRef]
- Smadja, W.; Valery, J.-M.; Ville, G.; Bernassau, J.-M. RuCl3, NaOH-catalyzed isomerization of allylic alcohols to saturated ketones: Part II Regiochemistry. J. Mol. Catal. 1985, 30, 389–394. [Google Scholar] [CrossRef]
- Nishiwaki, N.; Kamimura, R.; Shono, K.; Kawakami, T.; Nakayama, K.; Nishino, K.; Nakayama, T.; Takahashi, K.; Nakamura, A.; Hosokawa, T. Efficient double bond migration of allylbenzenes catalyzed by Pd(OAc)2–HFIP system with unique substituent effect. Tetrahedron Lett. 2010, 51, 3590–3592. [Google Scholar] [CrossRef]
- Golborn, P.; Scheinmann, F. Isomerisation of allyl phenyl ethers and allylphenols with transition metal catalysts. J. Chem. Soc. Perkin Trans. 1 1973, 2870–2875. [Google Scholar] [CrossRef]
- Chen, Z.; Jia, X.; Huang, J.; Yuan, J. Platinum-Catalyzed Tandem Cycloisomerization Reaction of Benzoendiynyl Esters: Regioselective Long-Range 1,5-Acyl Migration. J. Org. Chem. 2014, 79, 10674–10681. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.V.; Bloino, J.; Janesko, B.G.; Gomperts, R.; Mennucci, B.; Hratchian, H.P.; Ortiz, J.V.; Izmaylov, A.F.; Sonnenberg, J.L.; Williams; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V.G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery Jr. J. A.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.J.; Heyd, J.J.; Brothers, E.N.; Kudin, K.N.; Staroverov, V.N.; Keith, T.A.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.P.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Millam, J.M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Farkas, O.; Foresman, J.B.; Fox, D.J. Gaussian 16 Rev. C.01, Wallingford, CT, 2016.
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Renodon-Cornière, A.; Dijols, S.; Perollier, C.; Lefevre-Groboillot, D.; Boucher, J.-L.; Attias, R.; Sari, M.-A.; Stuehr, D.; Mansuy, D. N-Aryl N‘-Hydroxyguanidines, A New Class of NO-Donors after Selective Oxidation by Nitric Oxide Synthases: Structure−Activity Relationship. J. Med. Chem. 2002, 45, 944–954. [Google Scholar] [CrossRef]
- Noshita, M.; Shimizu, Y.; Morimoto, H.; Ohshima, T. Diethylenetriamine-Mediated Direct Cleavage of Unactivated Carbamates and Ureas. Org. Lett. 2016, 18, 6062–6065. [Google Scholar] [CrossRef] [PubMed]
- Moussavi, Z.; Lesieur, D.; Lespagnol, C.; Sauzieres, J.; Olivier, P. Acyl-7 dihydro-2,3 benzoxazin-1,4 ones-3 et propriétés normolipémiantes7-Acyl-2,3-dihydro-1,4-benzoxazin-3-ones and normolipemic properties. Eur. J. Org. Chem. 1989, 24, 55–60. [Google Scholar]
- Buckle, D.R.; Cantello, B.C.C.; Smith, H.; Smith, R.J.; Spicer, B.A. Synthesis and antiallergic activity of 2-hydroxy-3-nitro-1,4-naphthoquinones. J. Med. Chem. 1977, 20, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Lovely, C.J.; Du, H.; He, Y.; Rasika Dias, H.V. Oxidative Rearrangement of Imidazoles with Dimethyldioxirane. Org. Lett. 2004, 6, 735–738. [Google Scholar] [CrossRef]
- Watson, L.J.; Harrington, R.W.; Clegg, W.; Hall, M.J. Diastereoselective intermolecular ene reactions: synthesis of 4,5,6,7-tetrahydro-1H-benzo[d]imidazoles. Org. Biomol. Chem. 2012, 10, 6649–6655. [Google Scholar] [CrossRef]
- Larionov, E.; Li, H.; Mazet, C. Well-defined transition metal hydrides in catalytic isomerizations. Chem Commun (Camb) 2014, 50, 9816–9826. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Koyama, Y.; Moro-Oka, Y.; Ikawa, T. Novel peparation of silyl enol ethers from allyl alcohols. Tetrahedron Lett. 1979, 20, 1415–1418. [Google Scholar] [CrossRef]
- Jin, X.; Taniguchi, K.; Yamaguchi, K.; Nozaki, K.; Mizuno, N. A Ni–Mg–Al layered triple hydroxide-supported Pd catalyst for heterogeneous acceptorless dehydrogenative aromatization. Chem Commun (Camb) 2017, 53, 5267–5270. [Google Scholar] [CrossRef]
- Giustra, Z.X.; Chou, L.-Y.; Tsung, C.-K.; Liu, S.-Y. Kinetics of −CH2CH2– Hydrogen Release from a BN-cyclohexene Derivative. Organometallics 2016, 35, 2425–2428. [Google Scholar] [CrossRef]
- Tian, G.; Fedoseev, P.; Van der Eycken, E.V. Hypervalent Iodine(III)-Mediated Cascade Cyclization of Propargylguanidines and Total Syntheses of Kealiinine B and C. Chem. Eur. J 2017, 23, 5224–5227. [Google Scholar] [CrossRef]
- Chenna Reddy, M.L.; Patil, V.B.; Nawaz Khan, F.R.; Saravanan, V. Synthesis of Imidazo[1,2-a]pyridines and Imidazo[2,1-b]thiazoles Attached to a Cycloalkyl or Saturated Heterocycle Containing a Tertiary Hydroxy Substitution. J. Heterocycl. Chem. 2019, 56, 1486–1497. [Google Scholar] [CrossRef]
- Fursule, R.A.; Patil, P.O.; Shewale, B.D.; Kosalge, S.B.; Deshmukh, P.K.; Patil, D.A. Novel system for decarboxylative bromination of alpha,beta-unsaturated carboxylic acids with diacetoxyiodobenzene. Chem. Pharm. Bull. 2009, 57, 1243–1245. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Ojima, I. Asymmetric synthesis of 1-vinyltetrahydroisoquinoline through Pd-catalyzed intramolecular allylic amination. Tetrahedron 2007, 63, 8563–8570. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, J.; Sun, X.; Du, Y. An efficient cis-reduction of alkyne to alkene in the presence of a vinyl iodide: stereoselective synthesis of the C22–C31 fragment of leiodolide A. Tetrahedron 2013, 69, 1553–1558. [Google Scholar] [CrossRef]
- Yu, J.-Q.; Corey, E.J. A Mild, Catalytic, and Highly Selective Method for the Oxidation of α,β-Enones to 1,4-Enediones. J. Am. Chem. Soc. 2003, 125, 3232–3233. [Google Scholar] [CrossRef] [PubMed]
- Finkielsztein, L.M.; Aguirre, J.M.; Lantaño, B.; Alesso, E.N.; Iglesias, G.Y.M. ZnI2/NaCNBH3 as an Efficient Reagent for Regioselective Ring Opening of the Benzylic Epoxide Moiety. Synth. Commun. 2004, 34, 895–901. [Google Scholar] [CrossRef]
- Handore, K.L.; Jadhav, P.D.; Hazra, B.; Basu, A.; Reddy, D.S. Total Syntheses and Biological Evaluation of (±)-Botryosphaeridione, (±)-Pleodendione, 4-epi-Periconianone B, and Analogues. ACS Med. Chem. Lett. 2015, 6, 1117–1121. [Google Scholar] [CrossRef]
- Zhu, N.; Qian, B.; Xiong, H.; Bao, H. Copper-catalyzed regioselective allylic oxidation of olefins via C–H activation. Tetrahedron Lett. 2017, 58, 4125–4128. [Google Scholar] [CrossRef]
- Kharasch, M.S.; Sosnovsky, G. THE REACTIONS OF t-BUTYL PERBENZOATE AND OLEFINS—A STEREOSPECIFIC REACTION. J. Am. Chem. Soc. 1958, 80, 756–756. [Google Scholar] [CrossRef]
Scheme 1.
Previously reported dearomatizing spirocyclization of ureas and guanidines (*compound 22 not previously described).
Scheme 1.
Previously reported dearomatizing spirocyclization of ureas and guanidines (*compound 22 not previously described).
Scheme 2.
Putative mechanism for the synthesis of spirocyclic guanidine and naphthoxazole in HFIP and TFE respectively.
Scheme 2.
Putative mechanism for the synthesis of spirocyclic guanidine and naphthoxazole in HFIP and TFE respectively.
Scheme 3.
Proposed synthetic scheme to elaborate spirocyclic intermediate to complete the total synthesis and the energies of the TOADS products and corresponding rearranged products in gas phase and (after applying the solvation model-MeOH). .
Scheme 3.
Proposed synthetic scheme to elaborate spirocyclic intermediate to complete the total synthesis and the energies of the TOADS products and corresponding rearranged products in gas phase and (after applying the solvation model-MeOH). .
Scheme 4.
Efforts towards the double bond migration. .
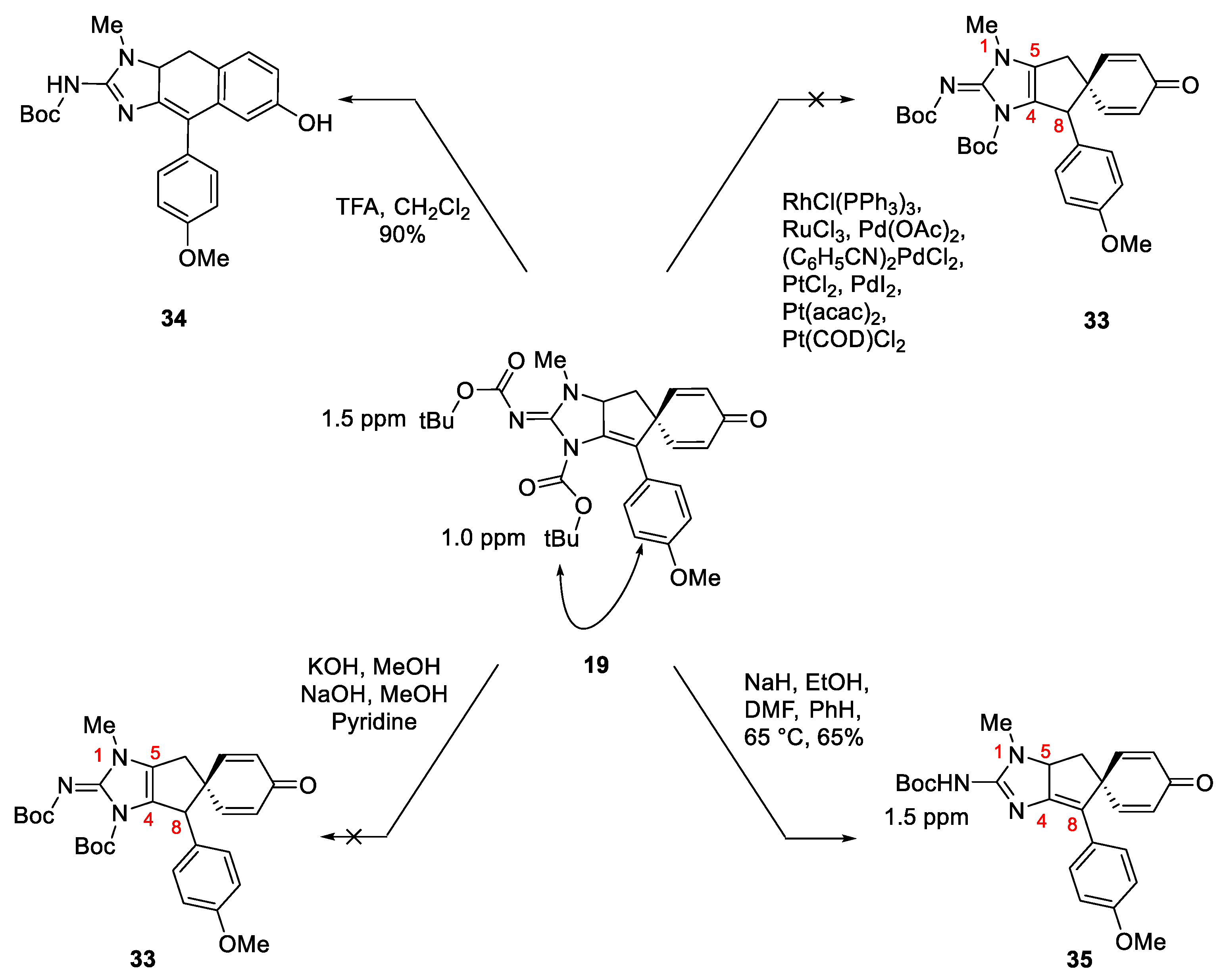
Scheme 8.
Scope of spirocyclization toward propargylic guanidines in absence of stabilizing p-anisyl group-part 1.
Scheme 8.
Scope of spirocyclization toward propargylic guanidines in absence of stabilizing p-anisyl group-part 1.
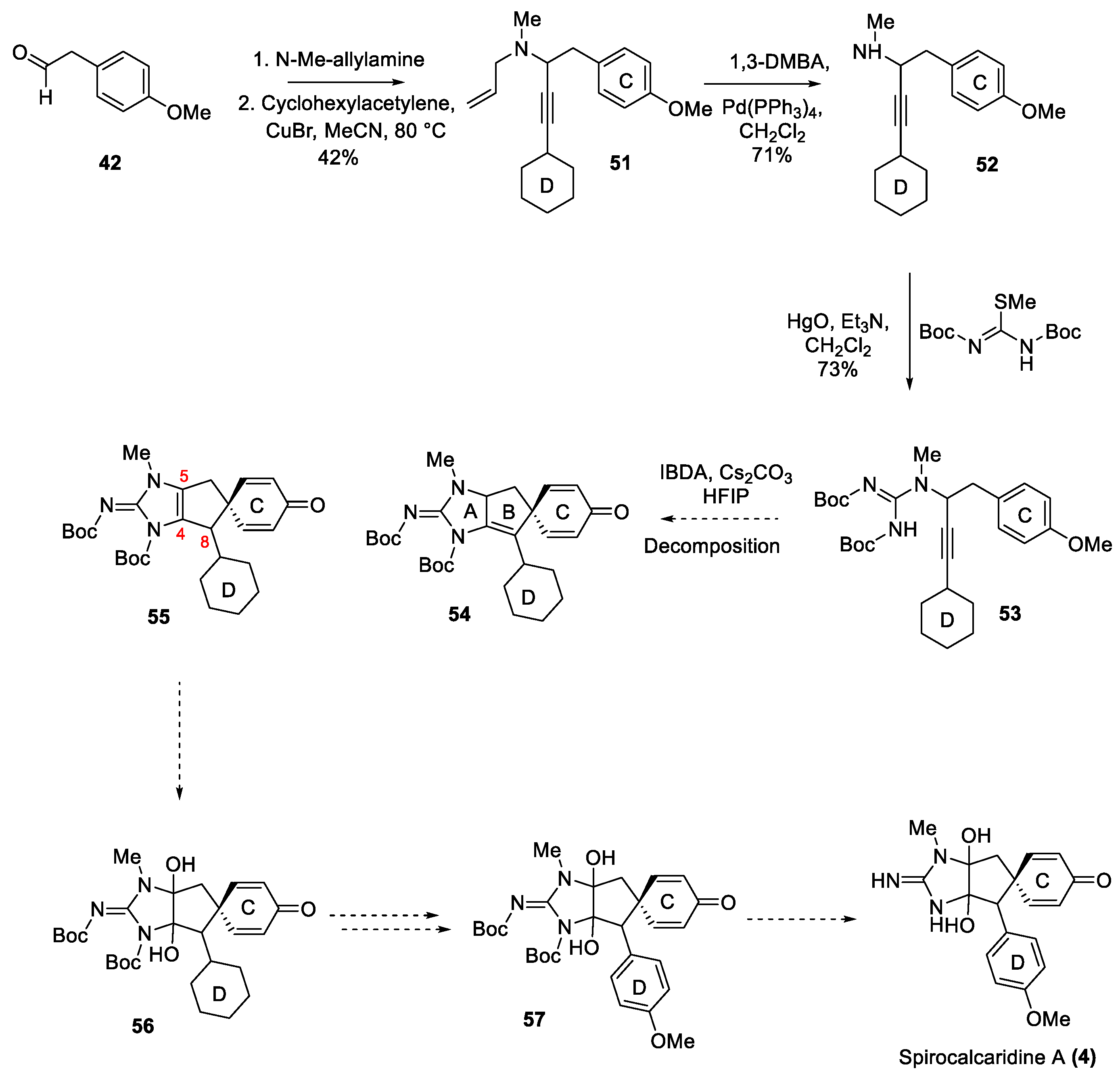
Scheme 9.
Scope of spirocyclization toward propargylic guanidines in absence of stabilizing p-anisyl group-part 2.
Scheme 9.
Scope of spirocyclization toward propargylic guanidines in absence of stabilizing p-anisyl group-part 2.
Scheme 11.
Exploration of oxidative strategies to complete the total synthesis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



