Submitted:
13 January 2025
Posted:
14 January 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
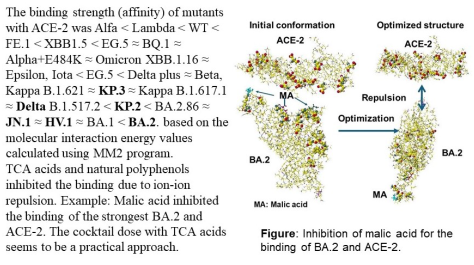
The fast mutation of COVID-19 viruses still confuses us, and the mRNA vaccines do not inhibit the infection and may protect against the heavy disease. The infection mechanism is described with the protein-protein binding stereo structure; therefore, the infection strength of variants has been estimated from the protein-protein (S-RBD binding with ACE-2) interaction energy values calculated using a molecular mechanics program. The binding strength order was Alfa < Lambda < WT < FE.1 < XBB1.5 < EG.5 ≈ BQ.1 ≈ Alpha+E484K ≈ Omicron XBB.1.16 ≈ Epsilon, Iota < EG.5 < Delta plus ≈ Beta, Kappa B.1.621 ≈ KP.3 ≈ Kappa B.1.617.1 ≈ Delta B.1.517.2 < KP.2 < BA.2.86 ≈ JN.1 ≈ HV.1 ≈ BA.1 < BA.2. The mutation from acidic amino acid to basic amino acid strength the binding. The substitute size of amino acids causes the steric hindrance for the binding. The affinity level supports the infection strength. Various proposed infection inhibitors are quantitatively analyzed. TCA acids and natural polyphenols inhibit the binding of S-RBD to ACE-2. The cocktail dose of known medicines may enhance their performance. The inhibiting multiplication may be achieved using glycated compounds that bind glycoproteins and reduce glycoprotein activities.
Keywords:
SARS-CoV S-RBD
; ACE-2
; protein-protein interaction energy values
; infection inhibitors
; multiplication inhibitors
; cocktail dose with TCA acids
; natural polyphenols
; glycosyl compounds
1. Introduction
The SARS-Cov-2 infection mechanism was described in detail, and the target approach was proposed [1]; however, the solution is far from conclusive. The infection of COVID-19 is caused by the binding of the SARS-CoV-2 protein with Angiotensin-converting enzyme 2 (ACE-2), which is an integral part of the renin-angiotensin-aldosterone system that exists to keep the body’s blood pressure. Once the death numbers soared by Omicron BA.1 and BA.2 in 2022; then, new mutants, Omicron BA.4 and BA.5, appeared, and the death numbers declined to be compatible with the influenza death numbers. Several countries announced that the disease was not to be pandemic. The pandemic’s definition is unclear, but the disease was defined as not a pandemic but a syndemic [2,3,4,5]; however, we are still fighting to control Covid-19 infection. The mutants Omicron BA.2, BA.4, and BA.5 are listed as variants of concern, and Omicron BA.2.75 and BQ.1 are considered variants of interest [6]. The mutation of viruses is too fast, and the development of new vaccines and medicines is far behind. Previous infection with an older variant such as Alpha, Beta, or Delta offers some protection against reinfection with Omicron BA.4 and BA.5 [7]. This may be a similar mutation of the Omicron BA.4 and BA.5 S-RBD binding site to that of the older variants and different from that of Omicron BA.1 and BA.2, in which various amino acids are mutated.
Furthermore, Omicron BA4 and BA.5 showed reduced neutralization by the serum from individuals vaccinated with triple doses of AstraZeneca or Pfizer vaccine compared with Omicron BA.1 and BA.2. In addition, using the serum from Omicron BA.1 vaccine breakthrough infections, there were significant reductions in the neutralization of Omicron BA.4 and BA.5 whose multiplication extend beyond that to other variants [8], raising the possibility of repeat Omicron infections. A greater escape from neutralization of Omicron BA.4 and BA.5 compared with Omicron BA.1 and BA.2 was reported [8]. Serum from triple-vaccinated donors had a 2 to 3-fold reduction in neutralization titers compared with the neutralization of Omicron BA.1 and BA.2. The prevention of transmission might become less effective as viruses evolve antigenically further from ancestral strains. While vaccination was unlikely to eliminate transmission, the combination of vaccines with boosting by natural infection will probably continue to protect the majority from severe disease. The L452R and F486V mutations both made major contributions to Omicron BA.4 and BA.5 escape [9]. However, based on previous analysis, the L452R mutation strengthens the binding with ACE-2. One emerging sublineage, Omicron BA.2.75, which might escape from antibodies due to the mutation, did not show greater antibody evasion than Omicron BA.5 [10]. Therefore, further study was carried out transmissibility of Omicron BA.4 and BA.5, BA.2.75, BQ.1, and BQ.1.1 as the difference in molecular interaction energy values. The BA.2.86 variant appeared, and the effectiveness of a new monovalent XBB.1.5 vaccine showed good levels of neutralizing antibodies [11]. The storm of new variants, HV.1 and JN.1, was rounding the world. Now, we face new variants of Omicron KP.2 and KP.3.
Numerous sublineages emerged from the continued evolution of the SARS-CoV-2 Omicron variants with different patterns of evasion from neutralizing antibodies. Various facts were observed such as human leukocyte antigen (HLA) class 1 and class 2 resented T-cell epitopes in the spiked-glycoprotein were highly conserved across the entire evolution of SARS-CoV-2 including Alpha, Beta, and Delta and Omicron sublineages, suggested that CD8+ and CD4+ T-cell recognition of Omicron, BQ.1.1, BA.2.75.2 and XBB might be largely intact. Omicron sublineages effectively evaded B-cell immunity by altering neutralizing antibody epitopes [12]. N-acetyl cysteine covalent conjugation perturbed the stereo-specific orientations of the interacting key residues of spike protein, resulting in a threefold weakening in the binding affinity of spike protein with the ACE-2 receptor [13]. Persistent SARS-CoV-2 RNA was detected in multiple anatomic sites, including throughout the brain [14]. SARS-CoV-2 non-structural protein 1 is sufficient to confer resistance to Natural killer cell killing [15]. Coldspot-guided antibody discovery reveals donor-derived neutralizing antibodies that are cross-reactive with Orthocoronavirine, including SARS-CoV-2 variants [16]. T cell epitope mapping across the SARS-CoV-2 proteome will allow us to understand better the risks presented by merging SARS-CoV-2 variants and the mechanisms that drive viral evolution [17]. Adding adjuvants or changes in doses and more mechanistic interventions may be implemented, such as using IL-7 [18].
A wide range of adverse/side effects have been reported in humans after COVID-19 vaccination [19]: thrombotic events/thrombocytopenia, myocarditis/pericarditis, cutaneous reactions, immune-mediated effects, psychiatric adverse events, systemic lupus erythematosus, reproductive toxicity, and other miscellaneous adverse effects [20], thrombotic thrombocytopenia [21], capillary leak syndrome associated with COVID-19 [22], vaccine-induced myocarditis based on the detection of free antigen in the blood of adolescents [23,24], Myocarditis and/or pericarditis were observed after SARS-CoV-2 mRNA vaccination with a predilection for adolescent and young adult males [25]. Restricted mean survival and restricted mean time lost have shown a small but significant downside for the vaccinated population [26]. mRNA COVID-19 vaccine boosters increased the prevalence of COVID-19 infection and other pathologies due to the impairing immune system response in immune-compromised individuals; therefore, it needs careful consideration for mRNA vaccine boosters [27]. mRNA vaccines for COVID-19 fail to generate secretory/mucosal immune-like IgG responses, rendering them ineffective in halting viral spread, and the mutations in the SARS-CoV-2 binding domain reduce immune recognition by vaccine-derived antibodies [28]. Adults with moderate or severe previous SARS-COV-2 infection were more likely to have a health event sufficient to impact routine activities or require medical assessment in a week following each vaccine [29]. Age-adjusted cancer mortality increases after the third mRNA-lipid nanoparticle vaccine dose during the COVID-19 pandemic [30,31,32]. This evidence may be due to weakene immune systems due to multiple vaccinations [33].
The high rate of anaphylaxis to the mRNA vaccines occurred predominantly in people who had a history of allergies [34], and cases of glomerulopathy after COVID-19 vaccination have been reported in the adult population [35]. A strong link between COVID-19 infection and an increased risk of new-onset asthma in children was considered [36]. Therefore, a wide range of adverse/side effects have been reported in humans after COVID-19 vaccination does not allow the judgment of the international scientific community, including toxicologists [37], and the significance of additional vaccination in hybrid-immunized cases is highly questionable regarding cost-effectiveness and risk-benefit [38].
Furthermore, the immune response to the vaccine is very different from that of a SARS-CoV2 infection. Vaccination induces a profound impairment in type 1 interferon signaling, which has diverse adverse consequences for human health [39]. The vulnerability of aged people may be supported by the evidence that both antigen structure and the delivery platform are crucial to efficiently prime humoral immune responses in old mice and might be relevant for designing “age-adapted” vaccine strategies [40]. People who have a history of allergies, chronic diseases, medication usage, and side effects of a strong magnitude for the BNT162b2 and ChAdOx-S vaccines should avoid vaccinations. The association of the female sex and infection with SRS-CoV-2 increased the potential of developing stronger side effects with certainanti-SARS-CoV-2 vaccines [41]. The coronavirus mortality rate in 55 African countries is almost 4.5 times lower than in the coronavirus, despite Africa having over 4.2 times more poorly vaccinated people [42]. Due to the nature of polymorphic cell-mediated immune responses may continue to contribute to prevention/limitation or severe COVID-19 manifestation [43]. Patients infected with SARS-CoV-2 to be outcomes of self-DNA driven inflammation. How inflammatory self-DNA contributes to some of the most frequent adverse events after vaccination with the Pfizer/BioNTech or Modelna mRNA vaccine can be explained the similar mechanisms to those derived by gp41. A wider application of the lessons learned from the experiences with COVID-19 and the new mRNA vaccines to combat future non-COVID-19 diseases. [44].
A new drug discovery approach was proposed and created minimal virions of wild-type (WT) and mutant SARS-CoV-2 with precise molecular composition and programmable complexity by the bottom-up assembly to study the free fatty acid binding pocket that was an allosteric regulatory site enabling adaptation of SARS-CoV-2 immunogenicity to inflammation states via binding of pro-inflammatory free fatty acids. Vitamin K and dexamethasone might work in the pocket instead of fatty acids; therefore, the approach may find a future COVID-19 therapy [45]. Drug resistance to Nirmatrelvir (Paxlovid) of SARS-Cov-2 protease mutants was extensively studied—the mutation around the binding site reduced enzyme activity [46]. The mutation prevented antibody binding and neutralization. The antibodies almost always interacted with the amino acid at position K417N or E484A [47]. The original amino acid K417 is the key mutation amino acid; the mutation to neutral amino acids reduces the S-RBD binding strength with ACE-2. The S-RBD E484 is repulsed from ACE-2; therefore, the E484A mutation erases the repulsion from ACE-2. R493 and R498 of S-RBD demonstrated their binding with ACE-2 via ion-ion interaction [48]. This fact supported the previous observation. The mutation of COVID-19 is too fast, following the analysis of transmissibility and estimation of the multiplication are delayed. Preventing infection is an urgent subject. The previous analyses suggested that the inhibitor candidates are citric acid and related Creb’s cycle organic acids, synthesized from glucose as normal metabolic processes [49].
On the other hand, monoclonal antibodies have been considered to prevent the virus from infecting human cells by binding to the virus’s spike protein; however, Omicron is totally or partially resistant to all currently available monoclonal antibodies [50]. Another report using lentivirus-based pseudo virus demonstrated that Bamlanivimab, Casirivimab, Etesevimab, Imdevimab, and Tixagevimab were less functional against BA.2 but Bebtelovimav, Cilgavimab, Imdevimab, Sotrovimab demonstrated a little effectiveness for neutralizing Omicron BA.1 and BA.2 variants [51]. Bebtelovimab showed remarkable preserved in vitro activity against all SARS-CoV-2 variants, including Omicron BA.4 and BA.5 [52]. The monoclonal antibody may bind to the S-RBD for protection; however, the binding with ACE-2 has not been described in detail. A plant-derived xanthophyll-carotenoid, Lutein, which protects from neurodegenerative diseases, protects against oxidative and nitrosative stress, both of which play a major role in long COVID and mRNA vaccination injury syndromes [53]. Glatiramer acetate made a patient clinically stable with relapsing-remitting multiple sclerosis and chronic lymphocytopenia after receiving the third dose of the Moderna mRNA vaccine [54].
Kids constantly produce TCA acids and then synthesize related amino acids and other important compounds for growing. Aging reduces such activity; therefore, adults must eat food containing essential metabolites. This known metabolism may support the idea that kids are less infected [55]. Children’s self-protection against COVID-19 was reported, even though children do not have a particular compound, and the fundamental defense mechanisms should be the same as those of adults [56,57]. It may be the activity of ordinal metabolisms and mass production of various compounds for their growth. Aged people may not have enough vital metabolisms to protect from COVID-19 infection if they do not eat balanced foods. Food habits may affect the mortality of meat-eating and vegetable-eating populations by comparison to their food habits. Before vaccination became a common practice, the USA’s total death/ and infectibility/population ratio was approximately 14 and 10 times Japan’s total death/ and infectibility/population ratio. America’s relative intake of citric acid-related metabolites [58] is lower than Japanese. The “Healthy Eating” report presented the regional food habits [59], suggesting transmissibility and mortality are very high in certain countries. Excess dairy and animal protein eating seems to relate to the urgent problem. We have to analyze the evidence continuously based on quantitative analytical chemistry.
The necessity and importance of reevaluating vaccine design and scheduling was proposed to increase oral or respiratory mucosal immunity against SARS-COV2 [60]. In addition, the intranasal recombinant protein subunit COVID-19 vaccine as a booster in adults [61]. Diagnosis is necessary to avoid the side effects of vaccination, and IgA can be considered a diagnosis [62,63] due to spiked-specific secretory IgA in mucosal secretions, highlighting the potential protective benefits of a vaccine targeting the upper respiratory tract [64]. The modified vaccine virus Ankara vector expressing the SARS-CoV-2 spike protein conferred full protection against SARS-CoV-2 cerebral infection may be a promising vaccine candidate against SARS-CoV-2 [65]. We should stop trying to prevent all symptomatic infections in healthy, young people by boosting them with vaccines containing mRNA from strains that might disappear a few months later [66]. Fragment crystallizable-mediated effector functions are critical for antiviral immunity; therefore, the choice and timing of vaccination regimens using mRNA vaccines against SARS-CoV-12 because the induction of IgG4 antibodies was not observed after homologous or heterologous SARS-CoV-2 vaccination with adenoviral vectors [67]. Carrier-free naked mRNA vaccines generated neutralizing antibodies against SARS-CoV-2 in non-human primates at levels comparable to those observed in mice; this may be a new effective approach vaccine [68]. Combined cryo-electron tomography with molecular dynamic simulation proposed pan-betacoronavirus S2-targeting antibodies as new medicines due to neutralizing arrecting pre-hear pin intermediates [69]. A new treatment is proposed to dose the spike surface protein of SARS-CoV-2 to a person’s immune cells to stimulate long-lived antibody production [70].
Despite the availability of vaccines and medicines, the infection has not been under control, and Omicron infections are as high as ever. The quantitative analysis of S-RBD mutant binding strength and inhibitor candidates was achieved in silico by calculating binding strength with ACE-2, including Hydrochloroquine, Ketoprofen, Metformin, Niacin, Nitazoxanide, Ritonavir, Simnotrelvir, Stain, and natural polyphenols. The in silico analytical method is the same as that previously described. Improving the performance of known medicines as cocktail doses and the practical inhibition of natural compounds, and glycosylation effect are described in the following sections.
2. The Fundamental Aspect of the Binding Strength of S-RBD Mutants
Acid-base (ion-ion) interaction strength is superior to hydrogen bonding, Lewis-acid-base interaction, and van der Waals (hydrophobic) interaction. Therefore, a mutation from an acidic amino acid to a basic amino acid strengthens the binding force. On the other hand, a mutation from a basic amino acid to an acidic amino acid weakens the binding force. A mutation from a small-size amino acid to a large-size amino acid weakens the binding strength due to steric hindrance. The simple quantitative analysis of these molecular interactions can be performed using model amino acids. Several examples are given in Figure 1.
The molecular interaction (MI) energy (kcal·mol-1) between Glu and an amino acid on the surface of
SARS-CoV-2 was calculated using the same approach as that used for the MI energy value calculation analyzed using the same approach represented in chromatographic molecular interaction mechanisms [71]. The MI energy value of the Final (optimized) structure is the sum of the individual molecules (one example: glutamic acid (Glu) and Alanine (Ala) minus that of the complex (Glu + Ala). The calculated MI energy values are presented in Table 1, where MIFS, MIHB, MIES, and MIVW are the MI energy values of the optimized structure, hydrogen bonding, electrostatic interactions, and van der Waals interactions, respectively. The MIHB, MIES, and MIVW indicate specific contributions to the interactions.
The main glutamic acid (Glu) and arginine (Arg) interaction is ion-ion interaction: the MIFS energy value is 84.05 kcal mol-1, and the MIES value is 64.31 kcal mol-1. The main lysine (Lys) and tyrosine (Tyr) interaction is hydrogen bonding: the MIHB energy value is 28.95 kcal mol-1, and the MIES energy value is 5.17 kcal mol-1. The contribution of van der Waals interaction is null for these amino acids. The simple calculation between two amino acids provides the binding strength by mutation of S-RBD. This fundamental molecular interaction force is applied to understand the mutant binding strength with ACE-2 protein. The mutated amino acids of various mutants are listed in Table 2, and the stereo structures of several COVID-19 A-RBD are shown in Figure 2 for easy understanding of the binding strength. Actual binding strength is calculated as molecular interaction energy values of these mutant’s S-RBD and ACE-2 docking analysis using a molecular mechanics program (MM2).
The downloaded stereo structure of SARS-CoV-2 with ACE-2 complex (RSCB PFB 7MJK) indicates that the binding site of ACE-2 contains many acidic amino acids [72]. The structure is corrected using UniProtKB-Q9BYF1(ACE2- HUMAN) 4a ACE angiotensin-converting enzyme 2 [Homo sapiens (human)], Gene ID: 59272, 24-May-2021. Figure 2A shows the downloaded COVID-19 protein 7MKK structure, which consists of a complex with saccharides. One extracted protein structure is shown in Figure 2B, and the circle indicates the S-RBD, whose structure is shown in Figure 2C. The full-size ACE-2 structure is shown in Figure 2D, where acidic amino acids at the contact site are indicated using large atom-size molecules (D: Asp; E: Glu) with the amino acid symbols and the residual numbers. Omicron KP.2 (Figure 2L) and KP.3 (Figure 2M) are mutants from Omicron JN.1 (Figure 2K); therefore, only the different mutated amino acids are clearly indicated.
3. Binding Inhibitors for S-RBD with ACE-2
The MI between an acidic amino acid and a basic amino acid strengthens the binding interactions, and the acidic amino acid repulses an acidic amino acid (Table 1). If acidic compounds bind on the S-RBD surface, the complex should be repulsed from the ACE-2 bonding site. Therefore, the inhibition effects of several acidic (medicines) were first studied. These structures are shown in Figure 3, summarising the optimized results in Table 3.
Acidic compounds did not inhibit the binding except di- or tri-carboxy acids, where a carboxy group binds with a basic amino acid of the S-RBD, and the free carboxy group is repulsed from ACE-2. The most effective compounds are TCA organic acids such as citric and malic acid. The binding inhibition effects of several compounds were performed. First, the inhibitor candidates are adsorbed on the binding surface of S-RBD, then the complexes are faced to ACE-2, and the conformations are optimized. The optimized results are visualized in Figure 4. The complexes of S-RBD with weak infection power with acidic compounds are repulsed from ACE-2.
A popular flu medicine Aspirin inhibited the binding of Omicron XBB.1.16 binding with ACE-2. The initial conformation of XBB.1.16 and three Aspirins and their complex structure are shown in Figure 4A, and the initial conformation of XBB.1.16 and Asprin complex and ACE-2 and their complex structure are shown in Figure 4A. Further analysis was performed for Delta S-RBD, which exhibited stronger binding affinity compared to that of BQ.1 and XBB,1.16. The initial location of Delta S-RBD, three Aspirin molecules, and the optimized (complex) structure are shown in Figure 4B. The initial conformation of the Delta-S-RBD and Aspirin complex and ACE-2 are shown in Figure 4B, where Aspirin is indicated as a molecular color for easy identification. The in silico analysis demonstrated that it did not inhibit the binding of Delta S-RBD with ACE-2. The binding site was extracted, and the structure was detailed, where K478 of Delta-S-RGB should be rejected from the binding with R357 of ACE-2 because both amino acids are basic. However, Aspirin connected these amino acids. This result indicated that a single carboxy group might not effectively inhibit the binding of these proteins (S-RGB and ACE-2). This in silico model analysis indicates that a flu medicine Aspirin may inhibit COVID-19 S-PDB binding with the ACE-2 if strong basic amino acids do not exist at the binding site of S-RBD. Such in silico analysis of molecular interactions should help to understand the feasibility of binding inhibitors. The actual experiment using viruses is required before studying animal tests. Overdose of medicines should be avoided, as overtaking vitamins and amino acids has caused health problems.
3.1. Lactic Acid
An inhibitor candidate lactic acid was bound on the S-RPD binding site, as shown in Figure 4C, where three molecular form lactic acids were used to cover the S-RBD binding site. The complex of Delta S-RPD and three lactic acids was located under the ACE-2 and optimized the conformation. The complex and the ACE-2 bound together. The lactic acid did not inhibit the binding. This is a pharmacological mistake. Lactic acid is ionized in physiological conditions. Therefore, ionized lactic acid should be used for the complex formation and binding inhibition study. Three ionized lactic acids were bound with Delta S-RPD, and the complex was located below the ACE-2 and optimized the conformation. Figure 4D shows that the ionized lactic acid inhibited the binding. This type of mistake was found in reports where an autodocking program was introduced.
3.2. Acidic Compounds as Inhibitor Candidates
The feasibility of these acidic compounds-binding inhibition was analyzed using the Delta variant. Acidic inhibitor candidates are ionized m-crboxyl-L-tyrosine, m-carboxyl-L-tyrosine N-mannoside, citric acid, citric acid dimethyl, citric acid O-mannoside, citric acid di-O-mannoside, Ferulic acid, Gallic acid, Ibuprofen, Glycyrrhizic acid, mefenamic acid, nalidixic acid, naproxen, probenecid, and Cyonic acid. The residual amino acid of Delta S-RBD is K478, which binds one of these carboxy groups. These ionized form compounds inhibited the binding but not their molecular form compounds. The process of these compounds is shown in Figure 4E. However, the ionized carboxy group repulsed each other when these acids were located close together. One example is Nalidixic acid, which is repulsed, and only two Nalidixic acids are bound with Delta S-RBD.
The same analysis was performed for the Omicron BA.2.86 variant. The mutated amino acids of S-RBD are D339H, R403K, V445H, G446S, N450D, L452W, N481K, E484K, and R486P, and the mutation was complicated compared to the mutation of the Delta, whose spike mutations of interest were L452R, T478K, D614G, and P681R. The key amino acids for binding of BA.2.86 are R403K, N481K, and E484K. Especially, E484K enhances the binding. However, many acidic compounds inhibited the binding of the Delta and the ACE-2 but failed to inhibit the binding of BA.2.86 and the ACE-2. The fundamental docking study demonstrated that natural multi-carboxy acids inhibit the docking of S-RBD protein with ACE-2 enzyme but not inhibit ACE-2 enzyme activity. Malic acid inhibits the docking of Delta S-RBD and ACE-2, summarized in Table 3. Further, malic acid inhibits the docking of Omicron BA.2, which exhibits the strongest docking power with ACE-2. The demonstration process follows: three malic acids are put above the Omicron BA.2 docking site, and the optimized structure is shown in Figure 4F. The complex faces ACE-2 and optimized the structure. The BA.2 malic acid complex is repulsed from ACE-2 (Figure 4F). The same result is exhibited for citric acid, which exhibits strong repulsion from ACE-2 (Table 3).
The Osertrivir amino group is bound with a carboxy group of ACE-2 D38 and E329. It also bound peptide bridge carbonyl via hydrogen bond. Cytonic acid phenolic hydroxy group forms hydrogen bonds with both D36 carboxy and K353 amino groups. One Gallic acid contributes to the binding where a hydroxy group binds with the Q38 amino group. The other two Gallic acid hydroxy groups did not involve the binding. The Furosemide amino group binds with E35 and D38 carboxy groups via hydrogen bonding, and The ferulic acid phenolic hydroxy group binds to the D38 carboxy group. The last three compounds did not inhibit the binding, but these compounds mainly bound to ACE-2 via hydrogen bonding. The results indicated that these acidic compound carboxy groups bound tightly with the BA.2.86 S-RBD via ion-ion interactions, and the binding to ACE-2 was weak hydrogen bonding. That is, the steric hindrance is a crucial factor in the affinity of the protein substitute.
The binding strength of S-RBD mutants and ACE-2 was calculated using a molecular mechanics program, and the optimized molecular interaction (MIFS) energy values demonstrated that Omicron BA. 2’s MIFS value was 1.4 times Delta MIFS and 2.7 times Alfa MIFS.The Omicron BA.2 S-RBD demonstrated the most vital transmissible strength. These mutant’s MIFS energy values increased from Alpha to Omicron BA2. 14 proposed medical treatment compounds did not show as the inhibitors to block the Omicron S-RBD and ACE-2 binding. A popular Omicron JN.1 is further mutated valiant from Omicron XBB.1.5 and exhibited stronger binding affinity with ACE-2 than that of BA.2.86. The mutation L455S increases the flexibility of JN.1, and the R454 mutation contributes strong binding affinity of JN.1. The binding strength of current popular KP.2 and KP.3 is around that of Delta. The calculated binding strength (molecular interaction energy kcal mol-1) of variants is Alfa (321.0) < Lambda (345.4) < WT (346.1) < RE.1 (350.6) < XBB1.5 (373.9) < EG.5 420.4) ≈ BQ.1 (424.6) ≈ Alpha+E484K (442.3) ≈ Omicron XBB.1.16 (443.8) ≈ Epsilon, Iota (465.4) < EG.5 (487.6) < Delta plus (511.3) ≈ Beta, Kappa B.1.621 (515.8) ≈ KP.3 (535.8) ≈ Kappa B.1.617.1 (544.6) ≈ Delta B.1.517.2 (594.2) < KP.2 (662.2) < BA.2.86 (726.1) ≈ JN.1 (737.5) ≈ HV.1 (756.0) ≈ BA.1 (761.7) < BA.2 (904.3) based on protein (S-RBD) and protein (ACE-2) interaction energy values, calculated using in silico analysis [73]. Malic acid and glycyrrhizinic acid inhibited the binding with ACE-2 except for Glycyrrhizinic and Crebs cycle acids. The analysis processes are shown in Figure 4G using JN.1 mutant, which demonstrated a similar binding affinity to ACE-2 to that of BA.2.86.
The results demonstrated that malic acid might inhibit the binding of the strongest binding capability, Omicron BA.2 with ACE-2. Citric acid was also exhibited as the binding inhibitor (Figure 4F). The possible reason why acidic compounds inhibited Delta S-RBD and ACE-2 binding did not inhibit BA.2.86 and ACE-2 binding is the conformation of R454. Delta R454 is buried inside, but BA.2.86 R454 is located in a relatively open space; therefore, the R454 guanidyl group can bind with ACE-2. Such steric hindrance contributes to the binding. Computational chemistry can analyze chemical interactions quantitatively, but it is difficult to analyze biological interactions.
In physiological conditions, the carboxy group and some polyphenol phenolic hydroxy groups are ionized, and these ionized acidic groups are effectively repulsed from the ACE-2 binding site. The ionization of a phenolic hydroxy group can be estimated from the atomic partial charge values (unit: au) calculated by the MOPAC AM1 program. The failed conformations to inhibit the binding of BA.2.86 S-RBD and ACE-2 are not due to the ion-ion interactions between the inhibitor candidates and ACE-2.
4. Practical Use and Modification of Known Medicines for Inhibiting the Infection
The analysis of binding inhibitor candidates from binding strength with ACE-2 indicates that carboxy compounds were repulsed from the ACE-2 binding site where acidic amino acids line up; therefore, further modification of medical treatment candidates may produce an effective binding inhibitor. The modified PF-07321332, whose cyano group was replaced with a carboxy group based on precursor search in organic reaction, rejected the binding from ACE-2. The chemical structure of PF07321352 and the modified compound is shown in Figure 5.
4.1. PF07321332
The complex structure of PF 07321332 and protein is PDB 7TLL [74]. The downloaded and corrected structure of 7TLL is shown in Figure 6A, where the location of PF07321332 is far from the S-RBD binding site with ACE-2. The modified PF 07321332 complex structure is also shown in Figure 6A. The residual amino acids with 2.5Å from PF 07321332 and the modified PF 07321332 are shown in Figure 6B. The PF07321332 cyano group contacted N143, G143, C145, and E166. The cyano group’s main binding residual amino acids seemed to be cysteine and asparagine. Those of the modified PF07321332 were N142, G143, C145, E166, and H172. These Figures indicate that the replacement from PF 07321332 to the modified PF 07321332 did not cause significant complex conformation changes and seemed very identical. The interaction with the protein negatively shifted both the cyano and carboxy group atomic partial charge. The cyano and carboxy groups are circled for their identification.
For the inhibition study, the feasibility of PF07321352 (PF) and the modified PF07321352 (mPF) was studied to determine whether these compounds inhibit the binding of Delta with ACE-2. The experimental protocol follows the constructed and optimized Delta and three PFs conformation (Figure 5). The optimized complex is located about 10Å below the part of ACE-2 and optimized these compounds to obtain their complex structure (Figure 7).
The same study was performed for mPF. The initial conformation of Delta S-RBD, mPF complex, and ACE-2 and their optimized structures are shown in Figure 7B.
The original PF and Delta S-RBD complex was bound with ACE-2, but the mPF and Delta S-RBD complex was repulsed from ACE-2. The modification seems to be a promised approach for the effective use of PF 07321332.
Omicron BA.2 has the strongest binding strength with ACE-2; therefore, the feasibility of mPF was studied. First, three mPF molecules were located above Omicron BA.2 S-RBD, and the structure has optimized the structure where three mPF molecules were used to cover the binding site. The optimized complex of S-RBD and three mPF molecules faced an ACE-2 binding site, and these compounds were optimized. However, the complex was not repulsed and bound with the ACE-2, as shown in Figure 7C. The result indicated that even the modification of PF by the addition of a carboxy group might not inhibit the binding to ACE-2.
Further study is carried out for Omicron BQ.1 mutant. Omicron BQ.1 bound ACE-2. Three PF 07321332 molecules were located at 10Å above BQ.1 for covering the BQ.1 binding site, and the structure was optimized (complex structure in Figure 7D). Then, the PF07321332-bound complex was located below the ACE-2, and the complex structure was optimized. As a result, PF was sandwiched between BQ.1 and ACE-2, as shown in Figure 7D. The same experiment was performed for the modified PF07321332 (Figure 7E). The in silico analysis indicated that PF07321332 could not inhibit the binding of BQ.1 S-RBD binding with the ACE-2, but the modified PF07321332 can inhibit the binding of S-RBD mutants having weak binding strength with ACE-2.
The original PF 07321332 and Delta S-RBD complex were bound with ACE-2, but the modified PF07321332 and Delta S-RBD complex were repulsed from ACE-2. Modification seems to be a promising approach for the effective use of PF07321332. The same approach was performed for modified Molnupiravir.
4.2. Molnupiravir (EIDD-2801)
Large molecular size acidic molecules may inhibit the docking of S-RBD with ACE-2, and nitrogen groups bind with ACE-2 acidic amino acids. Reported drug candidates adsorbed with ACE-2 even their docking position in SARS-CoV-2 protein is known. Therefore, the feasibility of another marketed medicine, Molnupiravir, was adsorbed on both Delta S-RBD and ACE-2. The experimental protocol was the same as that used for PF-07321332. The chemical structures of Molnupiravir and the modified compound are shown in Figure 5. Molnupiravir was modified; the hydroxy-amino group was replaced with a carbamate. The three original and modified Molnupiravir molecules are located about 10Å above the Delta S-SBD binding site, then optimized these complexes. Molnupiravir bound ACE-2 structure is shown in Figure 8A. These complexes were located about 10Å below the part of ACE-2 and optimized these complexes. The complex with the original Molnuporavir bound with ACE-2 (Figure 8B) and Molnupiravir did not inhibit the binding.
The same binding analysis was performed for modified Molnupiravir. The initial conformation of Delta S-RBD and three modified Molnupiravir molecules, their complex structure, and Delta S-RBD, modified Molnupiravir complex, and ACE-2 and their optimized structures are shown in Figure 8B. The complex with the original Molnuporavir bound with ACE-2 and Molnupiravir did not inhibit the binding, but the modified molnupiravir inhibited the binding. The compound with an ionized carboxy group was rejected to bind with ACE-2 due to ion-ion repulsing.
The modified Molunupiravir bound side of Delta S-RBD was repulsed from ACE-2 and completely turned, as shown in Figure 8B. This in silico analysis indicated that Molnupiravir could not inhibit Delta S-RBD binding with the ACE-2, but the modified compound can inhibit the binding. However, modified Molnupiravir bound BA.2 was not repulsed from ACE-2. Further analysis is carried out for Omicron BQ.1. The initial conformation of Molnupiravir and BQ.1 and their complex structures and The initial conformation of BQ.1 and Molnupiravir complex with ACE-2 and their optimized structure are shown in Figure 8C.
Molnupiravir did not inhibit the binding of weak binding affinity BQ.1 with ACE-2. Therefore, the same analysis was performed for modified Molnupiravir. The initial conformation of BQ.1 and three modified Monlunipiravir and their optimized (complex) structure are shown in Figure 8D.
The complex of BQ.1 with the modified Molnupiravir was repulsed from ACE-2. The compound with ionized carboxy compounds was rejected to bind with ACE-2 due to ion-ion repulsing. The Molnupiravir bound side was repulsed and completely turned. This in silico analysis indicated that Molnupiravir could not inhibit BQ.1 S-RBD binding with the ACE-2, but the modified compound can inhibit the binding.
4.2. a. Improving the Performance of PF 07321332, Molnupiravir, and Xocova
This demonstration indicates that multi-carboxy acids can inhibit the binding of Omicron BA.2, which exhibited the strongest binding power binding to ACE-2 (Table 3). These acids are candidates for making cocktail drugs; citric acid is the strongest docking inhibitor, but the taste is not good for a drug additive. Therefore, the feasibility of malic acid was studied for PF07321332, Molnupiravir, and Xocova. First, PF07321332, Molnupiravir, and Xocova malic acid complexes were constructed, and the complexes were put above the Delta binding site and optimized for the structures. The initial conformation of the optimized complex and ACE-2 shown in Figure 9 was optimized. Omicron BA.2 malic acid molnupiravir complex was repulsed from ACE-2 (Figure 9).
Xocova is also a medicine to treat COVID-19 patients. It is a very basic compound and easily binds to the ACE-2 binding site via ion-ion interaction. Furthermore, replacing a group with a carboxy group like PF07321332 is difficult. The basic property easily makes a complex with carboxyl acid. The structure of Xocova is shown in Figure 5, where three nitrogen (blue or dark gray) rings indicate strong basic properties and fluorine atoms support the sterically stable structure. The Xocova bound Delta S-RBD (Figure 9). The optimized structure exhibited that Xocova did not inhibit the binding. The effective use of Xocova may bind mRNA and inhibit multiplication but requires modification for cocktail use. Therefore, the feasibility of the cocktail with malic acid is studied.
Xocoba (Ensitrelvir Fumaric Acid) is like an extended β-alanine derivative and may bind RNA and/or DNA phosphate and inhibit the multiplication. Xocova was later modified as fumarate, and the drug activity improved (Ensitrelvir Fumaric Acid). It is a very basic compound and easily binds to the ACE-2 binding site via ion-ion interaction. Furthermore, replacing a group with a carboxy group like PF07321332 is difficult. The basic property easily makes a complex with carboxyl acid. The Xocova bound Omicron BA.2 S-RBD The complex faces ACE-2 and optimized the structure. The optimized structure exhibits that Xocova did not inhibit the binding.
4.2. b Further Analyses Using Omicron 2.86 and BA.2
The feasibility of the cocktail with malic acid was studied. The cocktail performances of PF07321332, Molnupiravir, and Xacova are shown in Figure 10.
PF07321332 and Molnupiravir did not inhibit the binding of BQ.1 with ACE-2; however, the modified PF07321332 and molnupiravir inhibited the binding of BQ.1 with ACE-2 but not for BA.2 binding with ACE-2, and their malic acid complexes inhibited the binding. PF07321332, Molunupiravir, and Xocova cocktail with malic acid inhibited the binding with Omicron BA.2.86 and ACE-2.; however, the PF07321332 with a malic acid cocktail was not strong enough to inhibit the binding with BA.2. Xocova did not inhibit the binding of BA.2 with ACE-2; however, the malic acid complex inhibited the binding. These visualized Figures clearly exhibit the useful dose of cocktail medicines except PF07321332 malic acid complex.
4.3. Cocktail Inhibitors
There are two types of drugs. One is trapped by proteins and is not metabolized by the protein (enzyme). This type of compound is a suicide helper, and various acidic drugs have been developed against enzyme activity. Basic compounds (drugs) bind with the phosphate of DNA and RNA and inhibit the multiplication. Molnupiravir, having a basic compound, seemed to be an effective drug for binding with the phosphate of DNA and RNA; however, it does not inhibit the S-RBD and ACE-2 binding. If these compounds are not trapped by ACE-2 and keep ACE-2 enzyme activities, they may work as active drugs. The modification is one solution, and a cocktail with another compound is another solution. Cocktail drugs consisting of a mixture of the drug candidate and acidic compounds may help the feasibility of the proposed compounds.
4.3. a Barcitinib and Malic Acid Cocktail
Barcitinib (Figure 11) is a medicine for treating COVID-19 patients but is not an officially recommended medicine for COVID-19 patients. This molecule has an electron-rich cyano group that may be modified to a carboxy group like a modified PF07321332. However, three Barcitinib molecules-bound Delta S-RBD were not repulsed from ACE-2 (Figure 12). These results were applied to a popular FE.1 mutant. The mutation of amino acids of FE.1 S-RBD is similar to those of XBB.1.5; therefore, the stereo structure of XBB.1.5 was used for further analysis. Even XBB.1.5 has no strong binding (basic) amino acid with ACE-2 because K462 is located far from the binding site.
The Baricitinib malic acid complex bound Delta S-RBD was repulsed from ACE-2. Delta has a strong binding affinity to ACE-2 and Omicron XBB.1.5, which has no basic amino acid at the binding site with ACE-2. Therefore, the binding repulsion effect of Baricitinib was studied using Omicron XBB.1.5. The Baricitinib bound XBB.1.5 (Figure 12) was partly bound to ACE-2, and Baricitinib bridged between ACE-2 A387 and XBB.1.5 Q498 (Figure 12). However, this binding is not a strong interaction like ion-ion interaction; therefore, it may be interrupted by the addition of malic acid. Their malic acid complexes inhibited the binding even though only two complexes were used for the docking instead of three complexes (Figure 12).
4.3. b Clofazimine and Malic Acid Cocktail
Clofazine is an upper-limit compound of the rule of five and is used for the treatment of COVID-19 patients. This large-size molecule, Clofazimine, did not inhibit the Delta or XBB.1.5 complexes to bind ACE-2. Clofazimine could not inhibit the binding of XBB.1.5 S-RBD Q498 and with ACE-2 A387. The location is circled (Figure 13). Their malic acid complexes inhibited the binding for the docking of Delta; however, two clofazimine and malic acid complexes on XBB.1.5 S-RBD; therefore, the optimization of the conformation was achieved using the S-RBD with two Baricitinib and malic acid complexes (Figure 13).
4.3. c Zanamivir and Malic Acid Cocktail
Zanamivir has dual ion of a basic guanidino and acidic carboxy groups. The carboxy group should be repulsed from ACE-2 acidic amino acids, and the guanidino group should bind with the ACE-2 acidic amino acids. Malic acid forms a complex with the guanidino group but not with malic acid. Therefore, the initial conformation produces different conclusions. Zanamivir is different from Baricitinib and Clofazimine; the carboxy group did not bind well to the XBB.1.5 S-RBD binding site and kick off from the binding site at the optimization process with ACE-2. The XBB.1.5 (also EG.5) exhibits weak binding power with ACE-2, but the binding cannot be inhibited using these compounds without the cocktail dose.
Three molecules are used to cover the S-RBD binding site. The results of Zanamivir were different from those compounds. The Zanamivir carboxy group inhibited the binding but could not inhibit ACE-2 K313 and D-RBD K444 binding. The location is circled in Figure 14. However, the Zanamivir does not form a complex with a malic acid due to ion-ion repulsion. The Zanamivir may not inhibit the multiplication of mRNA. Further details of other medicines will be visualized to help you understand their binding mechanisms easily. The in silico analysis provides fundamental chemistry, but the actual biochemical experiments are required as real drug candidates.
4.4. Further Study of Cocktail Applications
Omicron BA.2.86 is a current VOC. The binding strength is about the Delta but below that of Omicron BA.1 and 2. However, the BA.2.86 binding strength with ACE-2 is still stronger than previous Omicron BA.4 and 5 variants. The key mutations of Omicron BA.2.86 are N481K and E484K among I332V, D339H, R403K, V445H, G446S, N450F, L452W, and F486P. R403K reduces the binding strength, but E484K (replacing an acidic amino acid with a basic amino acid) strengthens the binding. Therefore, the property of six medicine candidates was investigated. The selected compounds were Chloroquine, Dalcetrapib, Deguetin, Dolutegravir, Ebselen, and Etravine. The in silico analytical method is the same as that previously described. Figure 15 shows their optimized structures.
The binding sites of PF07321332 and Molnupiravir are known, but these six compounds have no similarity to replace these known compounds. Ensitrelvir has two basic groups that may bind two phosphates of DNA and RNA, like derivatized β-alanine, which was once an excellent target compound in medicines. These six compounds may not have the capability of PF07321332 and Monupiravir. TCA-cycle acids can inhibit the binding of the variant and ACE-2, and medicine is required to inhibit the multiplication of the protein. If these six compounds inhibit the multiplication of COVID-19 protein, the cocktail with organic acid may improve medical activity.
These compounds did not inhibit the binding of BA.2.86 S-RBD binding with ACE-2. Considering the covering of basic amino acids, organic acid should face the ACE-2 binding site. Their malic acid complexes were prepared for their cocktail dose, and their malic acid complexes bound S-RBD were faced with an ACE-2 binding site, then optimized their conformations. The optimized structures of these six compounds are shown in Figure 16. These visualized Figures clearly exhibit the useful dose of cocktail medicines.
4.5. Ivermectin and Malic Acid Cocktail
Various drugs have been used for infected patients. Three of them are officially recommended. Many drugs have been applied, and the Pros and Cons of performance have been reported. According to Science-Doirect, 1198 scientific reports related to Ivermectin and COVID-19 have been published since 2019. The total number of research and review articles is 405 and 352, respectively. The balance indicates that the conclusion does not produce a clear answer. Therefore, the quantitative interaction of Ivermectin with COVID-19 protein was studied, and the actual conformations were visualized for the discussions.
The original structure (PDB 7mjk) consists of trimers; therefore, one protein with aspargine (N) bound saccharides that are eight 2-acetamido-2-deoxy-β-D-glucopyranose and four dimers (2-acetamide-2-deoxy-β-D-glucopyranose-(1-4)-2-acetamido-2-deoxy-β-D-glucopyranose (large size molecules) was picked up (Figure 17). Only one glucoside is located in the S-RPD, and the location indicates that the glucoside may not be directly involved in the infection. Structures of these saccharides and Ivermectin are shown in Figure 18. Ivermectin is a glycoside; therefore, the binding of Ivermectin on the surface of the protein was examined. Four Ivermectins were adsorbed at different locations in the initial study (Figure 19). Many Ivermectins seem to cover the surface and inhibit the enzyme activity. The observation is supported by glycocalyx damage COVID-19 activity.
Saccharides and Ivermectin
Ivermectin-bound Delta S-RBD was not repulsed from ACE-2; therefore, the Ivermectin-malic acid complex was used to study the binding inhibition. The structure of the Ivermectin-malic acid complex adsorbed Delta S-RBD is shown in Figure 20. ACE-2 repulsed this complex. Ivermectin is adsorbed on both ACE-2 and the glycoprotein via hydrogen bonding but not strong ion-ion interaction. Therefore, the bind can be easily cut by other molecules like water. The carboxy and ionized phenolic hydroxy group should inhibit the binding, and oligosaccharides bound with glycoproteins and reduce the glycoprotein activity. The glycocalyx acts as a buffer between the cell and extracellular matrix and plays an important role in vascular homeostasis; therefore, it contributes to treating COVID-19 [75]. Glucosylation has a profound influence on protein activity and cell biology through a variety of mechanisms, such as protein stability, receptor interactions, and signal transformation [76]. Polyphenols affect glucose-induced metabolites [77] and inhibit the binding of S-RBD with ACE-2 [78]. Therefore, the binding of Ivermectin may damage S-RND activity.
The key mutations of Omicron BA.2.86 are N481K and E484K among I332V, D339H, R403K, V445H, G446S, N450F, L452W, and F486P. R403K reduces the binding strength, but E484K (replacing an acidic amino acid with a basic amino acid) strengthens the binding. These compounds did not inhibit the binding of BA.2.86 S-RBD binding with ACE-2. Considering the covering of basic amino acids, organic acid should face the ACE-2 binding site. Their malic acid complexes were prepared for their cocktail dose, and their malic acid complexes bound S-RBD were faced with an ACE-2 binding site, then optimized their conformations.
Various compounds have been applied for Covid-19 treatment. Acidic compounds of cannabinoids did block the cellular entry of SARS-CoV-2[79]. Effectiveness of several compounds such as Ursodeoxycholic acid [80], a combination of Nirmatrelvir and Ritonavir [81], fungal metabolites [82], Bruceine A [83], Gamabufotalin [83], VITAMIC BIOSEN [84], prophylactics [85], and Azvudine [86] was reported, and Sotrovimab remained weakly reactive and broadly neutralizing antibody SA55 was still highly effective [87]. However, Nirmatrelvir, Ritonavir, Bruceine A and D, Gamabufotalin, and Azvudine were adsorbed on the ACE-2 in the above experiment. Ursodeoxycholic acid was repulsed from the ACE-2. Further study is required for their practical use.
Omicron JN.1 is further mutated valiant from Omicron XBB.1.5 and exhibited stronger binding affinity with ACE-2 than that of BA.2.86. The JN.1 mutation of amino acid from leucine to serine (L455S) reduced the steric hindrance for the S-RBD binding with ACE-2 and enhanced the binding affinity [88,89]. The mutation L455S increases the flexibility of JN.1 R454. The mutation contributed to a strong binding affinity of JN.1; therefore, further extended study for JN.1 is necessary. Sweden experienced fewer deaths per population unit during pandemic seasons than most high-income countries and was comparable to neighboring Nordic countries [90], and the modified vaccine virus Anlara vector expressing the SARS-CoV-2 spike protein conferred full protection against SARS-CoV-2 cerebral infection may be a promising vaccine candidate against SARS-CoV-2 [91]; therefore, we should study our immune system capability and further practical use of current practice medicines. Natural polyphenols are practically used to protect against inhibition.
5. Binding Inhibition of Natural Compounds
5.1. Cannabinoids
Polyphenolic compounds of cannabinoids inhibited the binding of JN.1 depending on their chemical structures in physiological conditions. The original structures of these compounds in Figure 21 are in molecular form but not ionized form [92]; therefore, these structures should be fixed in physiological conditions before autodocking. The molecular form Cannabinoids did not inhibit the S-RBD binding with ACE-2 but supported the binding. These compounds bind via hydrogen bonding between a cannabinoid carboxyl group and the residual amino acids. Ionized carboxy group binds tightly via ion-ion interaction or weak Lewis acid-base interaction. Figure 21 shows the structure of three compounds (CBCA, CBDA, and THCA-A), where ionized hydroxy groups are circled. The ionization was estimated from the atomic partial charge calculated using MOPAC AM1 [93]. The feasibility of the inhibition mechanism of these compounds is shown in Figure 22.
Cannabinoid acids exhibited the capability to inhibit COVID-19 infection; however, we should consider the side effects of cannabis components. Natural polyphenols can protect the infection. As many people suggest, colorful vegetables and fruits may keep us healthy. We should select plant products that have fewer side effects.
5.2. Curcumin
Curcumin is an inflammasome medicine that exists as an enoli-form in organic solvent and keto-form in water [94]. Therefore, the feasibility as a binding inhibitor was studied for both enol- and keto-forms. The dissociation of two phenolic hydroxy groups is not strong based on the atomic partial charge. The chemical structures are shown in Figure 23, where the ionizable hydroxy groups are circled. Their docking process is shown in Figure 24.
The keto-form inhibited the binding, but the enol-form did not. The chemistry of different mechanisms was analyzed using simple molecule propionic acid instead of amino acid and ACE-2 protein. The docked results of keto- and enol-form curcumin with propionic acid are shown in Figure 25. Figure 26 clearly shows the deformed curcumins after optimization. The change of atomic partial charge was a little, but the keto-curcumin was twisted after optimization due to the strong repulsion of high electron density keto-oxygen. The results suggested that the selection of chemical structures will guide to a wrong conclusion and the necessary of the right design of inhibitor structure.
5.3. Polyphenols of Green and Black Rea
Green and black tea components inhibited the binding of S-RBD with ACE-2 [95,96,97,98,99]. The components EGCG (Epigallocatechin gallate), TFDG (Theaflavin digallate), and Catechin are polyphenols. Phenolic compounds are classified as acidic compounds, but the dissociation constant of common phenolic compounds is more than 9; therefore, common phenolic compounds are neutral in physiological conditions. Generally, ionized acidic compounds inhibit the binding of S-RBD with ACE-2, and basic amino acids enhance the binding strength [73]. The molecular form phenolic compounds did not inhibit the binding and just adsorbed on the binding site; however, the dissociation constant (pKa) of EGCG, TFDG, and Catechin was not known. Therefore, the dissociation of these compounds was estimated from the atomic partial charge (apc) of their phenolic hydroxy groups. A pKa prediction method for acidic compounds used the apc value of the hydroxy group of oxygen and hydrogen calculated using the MOPAC AM1 program [100]. The method was applied to estimate the dissociation of green and black tea components. While the hydrogen partial atomic charge value exceeded 0.23 au, the pKa value was less than 7.5.
The calculated apc value of several hydroxy group hydrogen of these three compounds was more than 0.23 au [73], and these gallate hydroxy groups indicated the possible ionization because of their hydrogen apc values. The hydroxyl groups are circled in the Figure. Tea leaves' partly ionized acidic compounds were applied to binding inhibition analysis. First, the ionized compounds were docked at the S-RBD docking site, then the complex was faced to the ACE-2 docking site and optimized the structures. The processes are shown in the Figures.
These partly ionized phenolic compounds inhibited the binding of S-RBD with ACE-2. The Figure shows the inhibition of TFDG as an example. This in silico analysis supported that these phenolic compounds in tea indeed inhibit the binding of S-RBD with ACE-2. In addition, the binding strength of Omicron BA.2.86 was weaker than that of the Delta variant.
6. Glycisydation (Glycosylation) as an Effective Derivatization
Medicines against COVID-19 require two capabilities: inhibiting the multiplication and the binding of S-RPD with ACE-2. Designing medicine candidates for binding inhibition is not difficult due to the known binding mechanism. There are two medicines that clearly explain the multiplication inhibitors. However, many medicines and natural products have been used for the treatment. Natural phenolic compounds Noringenin, Prunin, and Narigin have been used as medicines. Naringin, Naringenin-7-rhamnoglucoside, is a disaccharide derivative that is (S)-Naringenin substituted by α-2-O-(α-L-rhamnopyranosyl)-β-D-glucopyranosyl moiety at position 7 via a glucosidic linkage. It has a role as a metabolite, an antineoplastic agent, and an anti-inflammatory agent. It is a disaccharide derivative, a dihydroxyflavanone, a member of 4'-hydroxyflavanones, α-(2S)-flavan-4-one and a neohesperidoside. The end saccharide of Naringin is α-L-rhamnopyranose. This rhamnose was replaced with an acidic saccharide, D-@@glucopyranuronic acid, or D-galactopyranuronic acid. Additionally, α-L-rhamnopyranose was replaced by acetylneuramic acid and glycolylneuraminic acid. Naringenin exhibited a potential anti-inflammatory role in COVID-19 infection [101]. Naringenin and naringin demonstrated palliative effects on various COVID sequelae exhibited various positive effects, including reducing inflammation, preventing viral infections, and providing antioxidants used potential as extended long COVID mediation [102]. Naringenin might exert therapeutic effects against COVID-19 through the inhibition of COVID-19 main protease, 3-chymotrypsin-like protease, and reduction of angiotensin-converting enzyme receptors activity [103]. Naringenin may be a promising treatment strategy against COVID-19 [101,102,103,104]. Prunin and Narigin are Naringenin glycosides. c The structures of these compounds are shown in Figure 27, where probably ionizable phenolic hydroxy groups are circled. Their binding inhibition processes are shown in Figure 28.
Narinenin has three ionized phenolic hydroxy groups inhibited strongly. Glycosylated Prunin and Naringin having two ionized phenolic hydroxy groups, also inhibited the binding. Glycosylation did not affect the binding inhibition, and the glycoside side is attractive to bind glycoproteins. Glycosylation will be one potential approach for designing new medicines.
In physiological conditions, compounds with carboxy and ionized phenolic hydroxy groups inhibit the binding of COVID-19 S-RBD with ACE-2, including Omicron BA.2, which has the strongest binding affinity with ACE-2. The simplest binding inhibitor is citric acid even the mannoside inhibited the binding. Natural polyphenols also inhibited the binding. However, compounds with a single carboxy group were not strong enough as the inhibitor, and benzoic acid could not clearly inhibit the binding. The chemistry of the inhibition mechanism is simple: one basic amino acid of S-RBD binds with one carboxy group of inhibitor candidates, and an extra carboxy group is repulsed from ACE-2 lined acidic amino acids. The mechanism can be applied for polyphenol inhibition. At least two ionized phenolic hydroxy groups are required as the inhibitor candidate molecules. The second requirement as a practical medicine against COVID-19 is inhibiting multiplication. PF07321332 and Molnupiravir are recognized as medicines to inhibit multiplication, even if these compounds do not inhibit the binding of S-RBD with ACE-2. The modified PF0731332 and Molnupiravir could inhibit the binding of Delta S-RBD [105]. In the previous study, polyphenol-glycosides inhibited the binding of S-RBD with ACE-2. These results suggested that glycated acidic compounds should have two capabilities as inhibitors of multiplication and binding with ACE-2 as medicine candidates. Therefore, the acidic group effect for the inhibition was studied using Naringin, which inhibited the binding of S-RBD with ACE-2 [106].
Sialic acids are 3-deoxy forms of monosaccharides [107] and are recognized for their bioactivities, including brain and cognition development, immune-enhancing, anti-hypertensive, anti-cancer, and skin-whitening properties [108]. Sialic acid exists at the end of oligosaccharides of membrane glycol-proteins, and many viruses engage sialylated glycan to bind to and infect cells [109]. It plays important roles in human physiology of cell-cell interaction, communication, cell-cell signaling, carbohydrate-protein interactions, cellular aggregation, development processes, immune reactions, reproduction, and in neurobiology and human diseases in enabling the infection process by bacteria and viruses, tumor growth, and metastasis, microbiome biology, and pathology. It enables molecular mimicry in pathogens that allows them to escape host immune responses. Recently, sialic acid has found a role in therapeutics. Sialic acid-acquired surfaces that can affect the host-parasite interaction. Bacteria can process them by common pathways for sialic acid metabolism and use sialic acid in different roles, such as colonization, owing to its disease-causing property. Viral sialic acid-recognizing lectins or hemagglutinins can agglutinate red blood cells. Viruses use sialic acids linked to glycoproteins and gangliosides to attach to host cells, followed by their entry, for example, coronavirus, DNA tumor viruses, hepatitis virus, influenza viruses (A, B, and C), mouse polyomavirus, mumps, Newcastle disease virus (NDV), norovirus, parainfluenza viruses, rotavirus, and Sendai virus. Sialic acid-recognizing lectins from adenoviruses and picornaviruses have not been identified. [110].
Furthermore, the fact that many of these trials are validating trials (Phase 3 and 4) clearly indicates a strong desire to confirm the efficacy and safety of Ivermectin for COVID-19. A large number of 27 meta-analyses were conducted on the clinical trial results of Ivermectin against COVID-19, with 15 concluding that Ivermectin was effective and 12 failing to demonstrate efficacy. These clinical trial results mainly focused on improving the condition of affected patients. Analysis of real-time web-delivered trial results showed Ivermectin to be effective in early treatment, late treatment, and prevention of disease onset [111]. Ivermectin also enhances the effects of chemo and radiation therapy. It has a broad impact on the immune system, increasing immune offense against cancers. It also inhibits cancer cell cycles, helping prevent the formation of new cancer cells. The drug promotes the killing of cancer cells by inducing mitochondrial stress and prevents cancer survival by preventing new blood vessels, which transport energy and fuel to cancers, from forming near cancer cells. Ivermectin can kill cancer cells in a way that drives the host immune response—immunogenic cell death [112]. The Ivermectin action was multiple antiviral and host target activation; however, the precise action was not determined [113]. In silico analysis exhibited the adsorption of Ivermectin on the surface of SARS-CoV-2 protein, but Ivermectin did not inhibit the binding of S-RBD with ACE-2, and the TCA-acid complex inhibited the binding [114]. Therefore, the sialic acid effect was studied for designing medicine candidates against COVID-19, and the structures of modified Naringin are shown in Figure 29.
These acidic glycoside-bound JN.1 S-RBDs were repulsed from ACE-2, even two glycosides were bound with S-RBD, and one glycoside was repulsed; however, the acidic glycosides enhanced the ACE-2 repulsion force. The experimental processes to analyze modified Naringenin glycosides are shown in Figure 30.
Naringenin glucopyranuronic acid, Naringenin galactopyranuronic acid, Naringenin N-acetylneuraminateglucoside, and Naringenin N-glycolylneuraminateglucoside inhibited the binding of JN.1 S-RBD with ACE-2. The next analysis is whether or not these modified Naringin glycosides bind with JN.1. These compounds were located near the 7mjk protein as performed for analyzing Ivermectin capability [115]. The locations of N-acetyl-D-glucosamine of 7mjk are indicated as full-size molecules. The locations of acidic amino acids are indicated as half-atomic-size molecules. The four acidic modified Naringins are indicated as molecular color. The optimized structures are shown in Figure 31.
The initial locations of four modified Naringins are the same as those of Naringin and Ivermectin. These modified Naringins bound at location A shown in Figure 30. The original saccharides (N-acetyl-D-glucopyranose) are shown as color molecules at additional locations B-D of protein PSB 7mjk. Acidic amino acids of 7mjk small circles in Figure 3 are indicated as 0.5A atoms. After optimization, Naringenin-glucopyranuronate-glucoside and Naringenin-galactopyranuronate-glucoside bound at four locations of 7mjk (B-E), Naringenin N-acetylnuraminate-glucoside bound only one location (C), and Naringin N-glycolylnuraminate-glucoside bound at two locations (B, C).
Acidic glycosides may improve the inhibition of the binding of S-RBD with ACE-2 and inhibit the binding at other locations of the SARS-CoV-2 protein. The modification of natural compounds may enhance them as active medicines. Oligossarides with sialic acid at their end oligomer contribute a specific action of the glycoproteins. However, whether or not the modified Naringin glycosides may act like Ivermectin inhibits COVID-19 action remains. These glycosides bound at the head of S-RBD and inhibited the binding with ACE-2; however, the inhibition effect against the enzyme multiplication is not clear.
The above docking experiment exhibited that glycosides with sialic acid can inhibit the binding of S-RBD with ACE-2; however, sialic acids repulsed from SAES-CoV-2 (7mjk) protein except in the locations of A and C. The result indicates that the SARS-CoV-2 protein may bind with glycoproteins containing sialic acids at 7mjk A location but may be repulse from 7mjk locations B and D, and ACE-2. The results suggested that the infection can be protected by covering 7mjk sites A and C using compounds that repulsed from ACE-2.
Glycosylation is an immune system-friendly modification; however, glucosylation produces Amadori compounds that produce oxygen radicals. We need skills for glycosylation reactions [116]. Glycosylation of albumin reduced the drug binding affinity significantly. Commercialized human serum albumin contains a large quantity of glycated albumin; therefore, the human serum albumin-drug binding affinity values were varied without notice of the purity [117]. Therefore, people use clean animal serum albumin instead.
7. Discussion
The mutation of COVID-19 is too fast, following the analysis of transmissibility and estimation of the multiplication are delayed. Preventing infection is an urgent subject. The previous analyses [75] suggested that the inhibitor candidates are citric acid and related Creb’s cycle organic acids, synthesized from glucose as normal metabolic processes.
Natural immunity is considered superior to vaccine-induced immunity. A growing body of literature provides evidence on natural immunity after COVID-19 infection that not only confers robust, durable, and high-level protection against COVID-19 [118]. Herbal medicine is exploring untapped therapeutic potential in neurodegenerative disease management [119], and various effective compounds have been identified in herbal medicines [120]. The in silico analysis of the binding inhibition by natural polyphenols supported the effect of food components that may inhibit the infection and help natural immunity. Several RNA vaccines are developing. mRNA vaccines produce new, toxic proteins to viruses and bacteria; however, the products are new intruders to our immune system that may cause some side effects. While we are healthy, our bodies can compete against intruders. However, aging and other incidents weaken our metabolic system, and our immune system is also weakened. Why not produce mRNA to recover our weakened metabolism? The best approach we can take now is to reconsider our eating habits and do certain exercises. Healthy lifestyle habits dramatically cut long-term CIVID-19 risks[121]. One example against COVID-19 infection is eating certain foods containing compounds that inhibit the S-RBD binding to ACE-2. Therefore, a balanced diet can protect against COVID-19 infection. Remain question is how to inhibit virus multiplication. The possible compounds are oligosaccharides that bind glycoproteins and reduce their activity. The oligosaccharide effect may support pros ivermectin. Various oligosaccharides and polyphenolic compounds are contained in natural foods. Therefore, eating colorful vegetables may protect us.
There are two types of drugs. One is trapped by proteins (enzymes) and is not metabolized by the protein. This type of compound is a suicide helper, and various acidic drugs have been developed against enzyme activity. Basic compounds (drugs) bind with the phosphate of DNA and RNA and inhibit the multiplication, but they work without specificity. Molnupiravir and Xacova have basic groups for binding with the phosphate of DNA and RNA; however, they do not inhibit the S-RBD and ACE-2 binding. PF07321332 was also trapped by ACE-2. If ACE-2 does not trap these compounds and keep the ACE-2 enzyme active, they may work as active drugs. The modification is one solution, and a cocktail with TCA acids is another solution. In physiological conditions, ionized carboxy and phenolic hydroxy groups can inhibit the S-RBS and ACE-2 binding.
New approaches have been reported; corticosteroids reduced the neuroinflammatory [122], Pemivibart remained moderately active against HN.1 sub-variants, and Sopabibart neutralized HN.1..1 but lost antiviral efficacy against KP.2.2, LB.1, and KP.3 [123]. Ibuzatrelvir robust antiviviral activity [124]. Oral administration of the GZ BL-P36 significantly improved survival and THE notable reduction in lung viral loads and lesions in the infection mouse model [125]. New proposed medicines have been available; however, the mutation is too fast to be controlled using medicines.
Therefore, a simple solution to add inhibition capability is a cocktail dose using known medicine with TCA acids. It does not take time to apply a mixture for the treatment because TCA acids are not specific medicines and are contained in everyday foods. Even. We produce them in our metabolic pathways; kids primarily produce them a lot. Such cocktail drugs consisting of a mixture of the drug candidate and acidic compounds may help the feasibility of the proposed compounds. We must consider finding effective ways to treat people who suffer from COVID-19 as an urgent and foremost priority [126]. The quality control of the mRNA vaccines has been doubted, exceeding amounts of foreign DNA [127], and 62 undeclared chemical elements have been found in the various products [128]. Improving the quality of mRNA. mRNA-based medicines have gained great interest in both immunotherapy and nonimmunogenic applications. The mRNA production technique is improved by urgent mass production of COVID-19 vaccines. mRNA medicines present a double-edged sword; therefore, further study is required [129].
8. Conclusion
The fundamental study of ion-exchange group properties was achieved using bio-mimic ion-exchange groups. The quantitative analytical method was successfully applied to the quantitative analysis of molecular interaction between a small molecule (substitute) and proteins (quantitative analysis of enzyme reactions). Furthermore, the method applied the quantitative analysis of protein-protein interaction for quantitative analysis of Covid-19 variant transmissibility.
Acidic amino acids lined up at the ACE-2 binding site, and highly transmissible valiant’s S-RBD has mutated from acidic and neutral amino acids to basic amino acids. That is, ion-ion interaction enhanced the binding. Therefore, molecular interaction energy calculation between two amino acids supported the transmissibility. This calculation does not require a high-level computational chemical system. The direct calculation of molecular interaction energy values between valiant’s S-RBD and ACE-2 improves the precision of molecular interaction energy difference due to the effect of the steric hindrance of protein stereo structure. The key mutated amino acids are L452R, T478K, N440K, L452R, T478K, E484K, Q493R, and Q498R, and these multi-mutations to basic amino acids further enhanced the transmissibility.
The number of mutated amino acids of variant’s S-RBD was Alfa → Lambda → WT → FE.1 → XBB.1.5 → EG.5 → Delta plus → Beta → Kappa B 1.620 → KP.3 → Kappa B1.617.1 → Delta B.617.4 → KP.3 → BA.2.86 → JN.1 → HV.1 → BA.1 → BA.2 = 1 → 2 → 0 → 3 → 4 → 4 → 3 → 3 → 2 → 25 → 2 → 2 → 25 → 11 → 23 → 18 → 15 → 16 and the binding strength (affinity) was Alfa < Lambda < WT < FE.1 < XBB1.5 < EG.5 ≈ BQ.1 ≈ Alpha+E484K ≈ Omicron XBB.1.16 ≈ Epsilon, Iota < EG.5 < Delta plus ≈ Beta, Kappa B.1.621 ≈ KP.3 ≈ Kappa B.1.617.1 ≈ Delta B.1.517.2 < KP.2 < BA.2.86 ≈ JN.1 ≈ HV.1 ≈ BA.1 < BA.2. Delta has two mutated basic amino acids; however, BA.5, BQ.1.1, and XBB.1.16 have only one extra basic amino acid. This result indicates that the transmissibility is not due to the binding strength but the facile multiplication. Another possibility is that our immune system does not warn us that new viruses are as dangerous as we faced at the beginning. XBB.1.16 does not increase basic amino acid at the binding site, but the binding strength is enhanced. Worry-some will appear delta-related variants that have more basic amino acids at the binding site, and a few mutations are necessary (strong binding affinity with easy multiplication).
The current three medicines are considered to inhibit virus multiplication; however, S-RBDs bound with these medicines were not repulsed from the ACE-2 binding site. The replacement of a group with a carboxy group permitted the repulsion of a modified PF07321332 and a modified Molnupiravir. A common inhibitor was tricarboxylic acid cycle acids like citric acid and natural polyphenols; therefore, the cocktail dose with TCA acids should be effective in inhibiting infection. In addition, the capability of our killer cells has limitations to erase the invented viruses; therefore, compounds that inhibit the multiplication of viruses are required. The virus multiplication of strong binding affinity can be reduced by avoiding taking excess amounts of lysine and arginine used for their mutation. A modified oral drug seemed to inhibit the S-RBD binding with the ACE-2. Understanding the fundamental molecular interaction mechanisms can support designing new drug candidates. The binding strength is based on the mutation to basic amino acids. Acidic amino acids were repulsed from ACE-2 lined-up acidic amino acids. That is, the binding can be inhibited by the compounds having ionized acidic groups. Furthermore, the 82 medicine candidates were analyzed as the binding inhibitors from binding with ACE-2. Only carboxy compounds were repulsed from the ACE-2 binding site, indicating that further modification of medical treatment candidates may convert to an effective binding inhibitor. The most effective binding inhibitors are TCA acids and natural polyphenols also inhibit the binding. The glycosides bind saccharides of glycoproteins and reduce the glycoprotein activities.
The fundamentally basic compounds bind phosphate ions of DNA and RNA via strong ion-ion interaction. Acidic compounds are selectively bound proteins (enzymes) and damage enzymatic activity. Therefore, we must carefully use basic compounds for medical treatment because of the non-selective binding, including normal DNA and RNA. TCA-cycle acids can inhibit the binding of the variant and ACE-2, and medicine is required to inhibit the multiplication of the COVID-19 protein. We must consider finding effective ways to treat people who suffer from COVID-19 as an urgent and foremost priority. We need temporary help with vaccines; however, we must maintain our daily health by exercising and eating balanced food. We should avoid continuous vaccination to maintain our immune system as our normal condition against unpredictable viruses and bacteria. Continuous vaccination is like living in a greenhouse and eating supplements. People who used to live in such conditions are vulnerable outside of the greenhouse. The unbalanced habits seem to be clear evidence for COVID-19 transmissibility and mortality like diabetes. Understanding fundamental chemistry should help individuals reconsider their lifestyle, including vaccination and drugs; therefore, the contribution of natural polyphenols as binding inhibitors was investigated.
Acknowledgments
The author appreciates Ivoyl P. Koutsaroff and his group for fruitful knowledge exchanges.
References
- Scudellari, M. How SARS-CoV-2 infects cells – and why Delta is so dangerous, Nature 2021, 595, 640-644. [CrossRef]
- Editorial, Cocid-19, a pandemic or not?, Lancet, 2020, 20, 383, www.thelancet.com/infection. [CrossRef]
- Horton, R. Offline: Covid-19 is not a pandemic, Lancet, 2020, 396, 874. Richard.horton@lancet.com.
- Shmerling, R.H. Is the Covid-19 pandemic over, or not?, Harvard Health Publishing, October 26, 2022.
- Topol, E. An optimistic outlook, Ground Truths, Daily pandemic update #6, November 7, 2022, erictioik@substack.com.
- European Centre for Disease Prevention and Control; SARS-CoV-2 variants of dashboard 10 Nov. 2022. http://www.ecdc.eu/en/covid-19/variant-concern.
- Prillaman, M. Prior omicron infection protects against BA.4 and BA.5 variants, Nature News 21 July 2022. [CrossRef]
- 8. Topol, E. The BA.5 story, The takeover by this omicron sub-variant is not pretty, Ground Truths, November 8, 2022.
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliu, S.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.R.; Das, R.; Skelly, D.; Ritter, T.G.; Amini, A.; Bibi, S. Adele, S.; Johnson, S.A.; Constantinides, A.; Webster, H.; Temperton, N.; Klenerman, P.; Barnes, E.; Dunachie, S.J.; Crook, D.; Pollard, A.H.; Lambe, T.; Goulder, P.; Paterson, N.G.; Williams, M.A.; Hall. D.R.; Fry, E.E.; Huo, J.; Mongkolsapaya, J.; Ren, J.; Stuart, D.I.; Screaton, G.T.; Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum, Cell, 2022, 185, 2422-2433.e13. [CrossRef]
- Sheward, D.J.; Kim, C.; Fischbach, J.; Muschiol, S.; Ehling, R.A.; Bjorkstrom, N.K.; Hedestam, S.T. Reddy, G.B.K.; Albert, J.; Peacock, T.P.; Murrell, B. Evasion of neutralizing antibodies by omicron sublineage BA.2.75, Lancet Infect Dis 2022, Sept 1 2022. [CrossRef]
- Topol. E. The BA2.86 variant and the new tooster, Ground Truths, Sept. 9, 2023. pp.12.
- Muik, A.; Lui, B.G.; Diao, H.; Fu, Y.; Bacher, M.; Toker, A.; Grosser, J.; Ozhelvaci, O.; Grikscheit, S. Hoehl, K.; Kohmer, N.; Lustig, Y.; Regev-Yochay, G.; Ciesek, S.; Beguir, K.; Poran, A.; Türeci, U. Sahin, Ö. Progressive loss of conserved spike protein neutralizing antibody sites in Omicron sublineages is balanced by preserved T-cell recognition epitopes, bioRxiv 2022, preprint: https://doi.org/10.1101/2022.12.15.520569, Cell Reports, 2023, 42, (8), 112888, Aug 29, 2023, pp.13. [CrossRef]
- Debnath, U.; Mitra, A.; Dewake, V.; Prabhakar, Y.S.; Tadala, R.; Krishnan, K.; Wagh, P.; Velusamy, C. Subramani, U.; Agarwal, S.; Vrati, S.; Baliyan, A.; Kurpad, A.V.; Bhattacharyya, P.; Mandal, A.K. N-acetyl cysteine: A tool to perturb SARS-CoV-2 spike protein conformation, http://chemorg/engage/api-gateway/chemrxiv/ssets/ Preprint from ChemRxiv, 06 Jan 2021, pp.21. [CrossRef]
- Stein, S.R.; Ramelli, S.C.; Grazioli, A.; Chung, J.-Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; Ko, S.H.; Platt, A.P.; Burbelo, P.D.; Quezado, M.; Pittaluga, S.; Purcell, M.; Munster, V.J.; Belinky, F.; Ramos-Benitez, M.J.; Boritz, E.A.; Lach, I.A.; Herr, D.L.; Rabin, J.; Saharia, K.K.; Madathil, R.J.; Tabatabai, A.; Soherwardi, S.; McCurdy, M.T.; Consortium, A.; Peterson, K.E.; Cohen, J.I.; de Wit, E.; Vannella, K. M.; Hewitt, S.M.; Kleiner, D.E.; Chertow, D.S. SARS-CoV-2 infection and persistence in the human body and brain at autopsy, Nature 2022, 612, 758-763.
- Lee, M.J.; Leong, M.W.; Rustagi, A.; Beck, A.; Zeng, L.; Holmes, S.; Qi, L.S.; Blish, C.A. SARS-CoV-2 escapes direct NK cell killing through Nsp1-mediated downregulation of ligands for NKG2D, Cell Reports 2022, 41, 111892, Dec. 27, 2022. [CrossRef]
- Bianchini, F.; Crivelli, V.; Abernathy, M.E.; Guerra, C.; Palus, M.; Muri, J.; Marcotte, H.; Piralla, A.; Pedotti, M.; De Gasparo, R.; Simonelli, L.; Matkovic, M.; Toscano, C.; Biggiogero, M.; Calvaruso, V.; Svoboda, P.; Rincón, T.C.; Fava, T.; Podešvová, L.; Shanbhag, AA.; Celoria, A.; Sgrignani, J.; Stefanik, M.; Hönig, V.; Pranclova, V.; Michalcikova, T.; Prochazka, J.; Guerrini, G.; Mehn, D.; Ciabattini, A.; Abolhassani, H.; Jarrossay, D.; Uguccioni, M.; Medaglini, D.; Pan-Hammarström, Q.; Calzolai, L.; Fernandez, D.; Baldanti, F.; Franzetti-Pellanda, A.; Garzoni, C.; Sedlacek, R.; Ruzek, D.; Varani, L.; Cavalli, A.; Barnes, C.O.; Robbiani, D.F. Human neutralizing antibodies to cold linear epitopes and subdomain 1 of the SARS-CoV-2 spike glycoprotein, Sci. Immunol., 2023, 8,1-17.
- Tye, E.X.C.; Jinks, E.; Haigh, T.; Kaul, B.; Patek, P.; Parry, H.M.; Newby, M.K.; Crispin, M.; Kaur, N.; Moss, P.; Drennan, S.J.; Tayloe, G.S.; Long, H.M. Mutations in SARS-CoV-2 spike protein impair epitope-specific CD4+ T cell recognition. Nat. Immunol. 2022, 23, 1726–1736. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Pawelec, G.; Cohen, A.A.; Provost, G.; Khalil, A.; Lacombe, G.; Rodrigues, S.; Desroches, M.; Hirokawa, K.; et al. Immunosenescence and Altered Vaccine Efficiency in Older Subjects: A Myth Difficult to Change. Vaccines 2022, 10, 607. [Google Scholar] [CrossRef] [PubMed]
- Taishi Kayano, Misaki Sasanami, Hiroshi Nishiura, Science-based exit from stringent countermeasures against COVID-19: Mortality prediction using immune landscape between 2021 and 2022 in Japan, Vaccine: X 20 (2024) 100547, 1-9. [CrossRef]
- Jose L. Domingo, A review of the scientifc literature on experimental toxicity studies of COVID-19 vaccines, with special attention to publications in toxicology journals, Archives of Toxicology (2024) 98:3603–3617. [CrossRef]
- Christopher D Richardson, Heterologous ChAdOx1-nCoV19–BNT162b2 vaccination provides superior immunogenicity against COVID-19, Lancet, Published Online August 12, 2021 https://doi.org/10.1016/ S2213-2600(21)00366-0, www.thelancet.com/respiratory Vol 9 November 2021, 1207-1209. [CrossRef]
- Ruggiero, R.; Balzano, N.; Di Napoli, R.; Mascolo, A.; Berrino, P.M.; Rafaniello, C.; Sportiello, L.; Rossi, F.; Capuano, A. Capillary leak syndrome following COVID-19 vaccination: Front. Immunol. 2023, 13, 956825. [Google Scholar] [CrossRef]
- Schwab, C.; Domke, L.M.; Hartmann, L.; Stenzinger, A.; Longerich, T.; Schirmacher, P. Autopsy-based histopathological characterization of myocarditis after anti-SARS-CoV-2-vaccination, Clin. Res. Cardiol. 2023, 112, 431–440. [Google Scholar] [CrossRef]
- Yonker, L.M.; Z. Swank, Z.; Bartsch, Y.C.; Burns, M.D.; Kane, A.; Boribong, B.P.; Davis, J.P.; Loiselle, M.; Novak, T.; Senussi, Y.; Cheng, C.-A.; Burgess, E.; Edlow, A.G.; Chou, J.; Dionne, A.; Balaguru, D.; Lahoud-Rahme, M.; Arditi, M.; Julg, B.; Randolph, A.G.; Alter, G.; Fasano, A.; Walt, D.R. Circulating Spike Protein Detected in Post–COVID-19 mRNA Vaccine Myocarditis, Circulation. 2023, 147, 867-876. [CrossRef]
- Barmada A.; Klein J.; Ramaswamy A.; Brodsky N.N.; Joycox J.R.; Sheikha H.; Nines K.M.; Habet V.; Campbell M.; Shimoda T.S.; Kontorovich A;. Bogunovic D.; Oliveira C.R.; Steele J.; Hall E.K.; Pena-Hernandez M.; Monteiro V.; Lucan C.; Ring A.M.; Omer S.B.; Iwasaki A.; Yildirim I.; Lucas C.L.; Cytokinopathy with aberrant cytotoxic lymphocytes and profibrotic myeloid response in SARS-CoV-2 mRNA vaccine-associated myocarditis, Additional introduction doi: 10.1126/sciimmunol.adh3455. Sci. Immunol. 2023, 8:1-18, eadh3455, Epub 2023, May 5. [CrossRef]
- Marco Alessandria 1,† , Giovanni M. Malatesta 2,†, Franco Berrino 3 and Alberto Donzelli, A Critical Analysis of All-Cause Deaths during COVID-19 Vaccination in an Italian Province, Microorganisms 2024, 12, 1343. [CrossRef]
- lberto Boretti, mRNA vaccine boosters and impaired immune system response in immune compromised individuals: a narrative review, Clinical and Experimental Medicine (2024) 24:23. [CrossRef]
- /Sunil J. WimalawansaUnlocking insights: Navigating COVID-19 challenges and Emulating future pandemic Resilience strategies with strengthening natural immunity, Heliyon 10 (2024) e34691. [CrossRef]
- Julie A. Bettinger, Michael A. Irvine, Hennady P. Shulha, Louis Valiquette, Matthew P. Muller, Otto G. Vanderkooi, James D. Kellner, Karina A. Top, Manish Sadarangani, Allison McGeer, Jennifer E. Isenor, Kimberly Marty, Phyumar Soe, and Gaston De Serres Adverse Events Following Immunization With mRNA and Viral Vector Vaccines in Individuals With Previous Severe Acute Respiratory Syndrome Coronavirus 2 Infection From the Canadian National Vaccine Safety Network, Research Network CID 2023:76 (15 March) 1088-1102.
- Gibo M, Kojima S, Fujisawa A, Kikuchi T, Fukushima M, Increased Age-Adjusted Cancer Mortality After the Third mRNA-Lipid Nanoparticle Vaccine Dose During the COVID-19 Pandemic in Japan., Cureus. 2024 Apr; 16(4): e57860. Published online 2024 Apr 8. [CrossRef]
- /Ronald Palacios Castrillo, Cancer Mortality Surges Post COVID ModRNA Vaccination Ronald Palacios Castrillo, European Journal of Clinical and Biomedical Sciences 2024, Vol. 10, No. 2, pp. [CrossRef]
- /Valdes Angues R, Perea Bustos Y (December 17, 2023) SARS-CoV-2 Vaccination and the Multi-Hit Hypothesis of Oncogenesis. Cureus 15(12): e50703.
- /Yeon-Woo Heo;Jae Joon Jeon;Min Chul Ha, ;et al,You Hyun Kim,;Solam Lee, Long-Term Risk of Autoimmune and Autoinflammatory Connective Tissue Disorders Following COVID-19, JAMA Dermatol. Published online November 6, 2024. [CrossRef]
- /Muhammad Bilal Khalid, Pamela A. Frischmeyer-Guerrerio,, The conundrum of COVID-19 mRNA vaccine– induced anaphylaxi, J ALLERGY CLIN IMMUNOL GLOBAL FEBRUARY 2023.
- /4.Gwo-Tsann Chuang, Wei-Chou Lin, Luan-Yin Chang, I-Jung Tsai, Yong-Kwei Tsau, Pediatric glomerulopathy after COVID-19 vaccination: A case series and review of the literature, Journal of the Formosan Medical Association, Volume 122, Issue 11, November 2023, Pages 1125-1131. [CrossRef]
- /Chiao-Yu Yang, · Yu-Hsiang Shih, Chia-Chi Lung, The association between COVID-19 vaccine/infection and new-onset asthma in children - based on the global TriNetX database, Infection Open access, Published: 21 June 2024, 1-13. [CrossRef]
- /Jose L. Domingo, A review of the scientifc literature on experimental toxicity studies of COVID-19 vaccines, with special attention to publications in toxicology journals, Archives of Toxicology (2024) 98:3603–3617. [CrossRef]
- /Hiroshi Kusunoki, Michiko Ohkusa, Rie Iida, Ayumi Saito, Mikio Kawahara, Kazumi Ekawa, Nozomi Kato, Masaharu Motone, Hideo Shimizu, Increase in antibody titer and change over time associated with severe acute respiratory syndrome coronavirus 2 infection after mRNA vaccination: Consideration of the significance of additional vaccination, Clin Case Rep. 2024;12:e8953. [CrossRef]
- /Stephanie Seneff, Greg Nigh, Anthony M. Kyriakopoulos, Peter A. McCullough, Innate immune suppression by SARS-CoV-2 mRNA vaccinations: The role of G-quadruplexes, exosomes, and MicroRNAs, Food and Chemical Toxicology 164 (2022) 113008,1-19. [CrossRef]
- /Dominik Pflumm , Alina Seidel , Fabrice Klein, Rüdiger Groß, Lea Krutzke, Stefan Kochanek, Joris Kroschel , Jan Münch, Katja Stifter, Reinhold Schirmbeck, Heterologous DNA-prime/ protein-boost immunization with a monomeric SARS-CoV-2 spike antigen redundantizes the trimeric receptor-binding domain structure to induce neutralizing antibodies in old mice, Frontiers in Immunology, pp.15, TYPE Original Research PUBLISHED 11 September 2023. [CrossRef]
- /27.Elias A. Said, Afnan Al-Rubkhi, Sanjay Jaju, Crystal Y. Koh, Mohammed S. Al-Balushi, Khalid Al-Naamani , Siham Al-Sinani , Juma Z. Al-Busaidi and Ali A. Al-Jabri , Association of the Magnitude of Anti-SARS-CoV-2 Vaccine Side Effects with Sex, Allergy History, Chronic Diseases, Medication Intake, and SARS-CoV-2 Infection, Vaccines 2024,12, 104. [CrossRef]
- /5.Mina Thabet Kelleni, COVID-19 mortality paradox (United States vs Africa): Mass vaccination vs early treatment, World J Exp Med. 2024 Mar 20;14(1):88674. [CrossRef]
- /Alexander Muik,Bonny Gaby Lui,Jasmin Quandt,Huitian Diao, Yunguan Fu, Maren Bacher, Jessica Gordon, Aras Toker, Jessica Grosser, Orkun Ozhelvaci, Katharina Grikscheit, Sebastian Hoehl, Niko Kohmer, Yaniv Lustig, Gili Regev-Yochay, Sandra Ciesek, Karim Beguir, Asaf Poran, Isabel Vogler, O¨ zlem Tureci,, Progressive loss of conserved spike protein neutralizing antibody sites in Omicron sublineages is balanced by preserved T cell immunity, Cell Reports 42, 112888, August 29, 2023 ª 2023 BioNTech SE. [CrossRef]
- /Martin Heil*, Self-DNA driven inflammation in COVID-19 and after mRNAbased vaccination: lessons for non-COVID-19 pathologies, Immunol. 14:1259879. [CrossRef]
- Staufer, O.; Gupta, K.; Bucher, J.E.H.; Kohler, F.; Sigl, C.; Singh, G.; Vasileiou, K.; Relimpio, A.Y.; Macher, M.; Fabritz, S.; Dietz, H.; Adam, E.A.C.; Schaffitzel, C.; Ruggieri, A.; Platzman, I.; Berger, I.; Spatz, J.P. Synthetic virions reveal fatty acid-coupled adaptive immunogenicity of SARS-CoV-2 spike glycoprotein. Nature Communications 2022, 13, 868. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Lewandowski, E.M.; Tan, H.; Zhang, X.; Morgan, R.T.; Zhang, X.; Jacobs, L.M.C.; Bitler, S.G.; Gongora, M.V. Choy, J.; Deng, X.; Chen, Y.; Wang, J. Naturally Occurring Mutations of SARS-CoV-2 Main Protease Confer Drug Resistance to Nirmatrelvir, ACS Cent. Sci. 2023, 9, 8, 1658–1669, Publication Date: July 24, 2023. [CrossRef]
- Doctrow, B. How COVID-19 variants evade the immune response, NIH Research Matter, June 8, 2021.
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Hana, P.; Li, X.; Peng, Q.; Su, C.; Huang, B.; Li, D.; Zhang, R.; Tian, M.; Fu, L.; Gao, Y.; Zhao, X.; Liu, K.; Qi, J.; Gao, G.; Wang, P. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2, Cell, 2022, 185, 630-640. [CrossRef]
- Kozlov, M. Omicron overpowers key CONID antibody treatments in early tests, Nature, 2021, Dec 21, https://doi.org/10.1038/d41586-021-03829-0 (Sotrovimab, DXP-604). [CrossRef]
- Yamasoba, D.; Kosugi, Y.; Kimura, I.; Fujita, S.; Uriu, K.; Ito, J.; Sato, K. Neutralisation sensitivity of SARS-CoV-2 omicron subvariants to therapeutic monoclonal antibodies, Lancet, www.thelancet.com/infection, July 2022, 22, 942. [CrossRef]
- Hentzien, M.; Autran, B.; Pitoh, I.; Yazdanpanah, Y.; Calmy, A. A monoclonal antibody stands out against omicron subvariants: a call to action for wider access to Bebtelovimab, Lancet, www.thelancet.com/infection 22, Sept 2022. [CrossRef]
- Hanai, T. Quantitative in silico analysis of SARS-CoV-2 S-RBD omicron mutant transmissibility, Talanta, 2022, 240, 123206. [CrossRef]
- Anthony M Kyriakopoulos 1, Greg Nigh2, Peter A McCullough3, Stephanie Seneff, Clinical rationale for dietary lutein supplementation in long COVID and mRNA vaccine injury syndromes [version 3; peer, review: 2 approved], F1000Research 2024, 13:191 Last updated: 08 NOV 2024. [CrossRef]
- Ahmad A Toubasi, Steven Allon and Francesca Bagnato, Disseminated histoplasmosis mimicking postvaccination side effects in an immunocompromised person with multiple sclerosis, Multiple Sclerosis Journal— Experimental, Translational and Clinical July–September 2024, 1–4. [CrossRef]
- Cheriyedath, S. Understanding immune responses to Sars-Cov-2 infections in children, News Med. Sci. Jan. 26, 2022, https://www.news-medical.net/news/202201125/Understanding-immune-responses-to-SARS-CoV-2-infections-inchildren.
- Mallapaty S. Kids Covid: Why young immune systems are still on top. Doi: 10.1038/d41586-02502423-8 Nature, 2021, 597, 166-168.
- Mallapaty S. Kids show mysteriously low levels of COVID-19 antibodies. Doi:10.1038/d41586-022-00681-8. Nature, News, 2022, Mar.10. [CrossRef]
- Briere, J.J.; Favier, J.; Gimenez-Roqueple, A.-P.; Rustin, P. Tricarboxylic acid cycle dysfunction as a cause of human diseases and tumor formation. Am. J. Physiol. Cell Physiol. 2006, 291, C1114–C1120. [Google Scholar]
- Smith K. (2021) Healthy eating, Nature News, Dcs. 10, 2021, (W. Willett et al. Food in the Anthropocene: the EAT-Lancet commission on healthy diets from sustainable food systems. Doi: 10.1016/s170-6736(18)31788-4; Lancet, 2019, 383:447-492, Hirvonen CK et al. Affordability of the EAT-Lancet reference diet: a global analysis, http;//doi.org/10.1038/d41586-021. Lancet Glob. Health, 2020, 8: e59-e66.
- Michinobu Yoshimura, Atsuhiko Sakamoto, Ryo Ozuru, Yusuke Kurihara, Ryota Itoh, Kazunari Ishii, Akinori Shimizu , Bin Chou, Yusuke Sechi, Aya Fujikane, Shigeki Nabeshima, Kenji Hiromatsu, Insufficient anti-spike RBD IgA responses after triple vaccination with intramuscular mRNA BNT162b2 vaccine against SARS-CoV-2, Received 4 August 2023; Received in revised form 7 Heliyon 10 (2024) e23595, 1-8. [CrossRef]
- Mohammad Hossein Fallah Mehrabadi, Monireh Hajimoradi, Ali Es-haghi, Saeed Kalantari, Mojtaba Noofeli, Ali Rezaei Mokarram, Seyed Hossein Razzaz, Maryam Taghdiri, Ladan Mokhberalsafa, Fariba Sadeghi, Vahideh Mohseni, Safdar Masoumi, Rezvan Golmoradi-Zadeh, Mohammad Hasan Rabiee, Masoud Solaymani-Dodaran, Seyed Reza Banihashemi, Safety and Immunogenicity of Intranasal Razi Cov Pars as a COVID-19 Booster Vaccine in Adults: Promising Results from a Groundbreaking Clinical Trial, Vaccines 2024, 12, 1255. [CrossRef]
- Ming Gao, Xiaomin Xing, Wenbiao Hao, Xulei Zhang, Kexin Zhong, Canhui Lu, Xilong Deng, Lei Yu, Diverse immune responses in vaccinated individuals with and without symptoms after omicron exposure during the recent outbreak in Guangzhou, China, Heliyon 10 (2024) e24030. [CrossRef]
- Khaleqsefat Esmat, Baban Jamil, Ramiar Kaml Kheder, Arnaud John Kombe Kombe, Weihong Zeng, Huan Ma, Tengchuan Jin, Immunoglobulin A response to SARS-CoV-2 infection and immunity, REVIEW ARTICLE HELYON, Volume 10, Issue 1e24031January 15, 2024. [CrossRef]
- Oscar Bladh, Katherina Aguilera, Ulrika Marking, Martha Kihlgren, Nina Greilert Norin, Anna Smed-Sörensen, Margaret Sällberg Chen, Jonas Klingström, Kim Blom, Michael W. Russell, Sebastian Havervall , Charlotte Thålin, Mikael Åberg, Comparison of SARS-CoV-2 spike-specific IgA and IgG in nasal secretions, saliva and serum, Frontiers in Immunology, PUBLISHED 15 March 2024. [CrossRef]
- Villadiego, J.; García-Arriaza, J.; Ramírez-Lorca, R.; García-Swinburn, R.; Cabello-Rivera, D.; Rosales-Nieves, A.E.; Álvarez-Vergara, M.I.; Cala-Fernández, F.; García-Roldán, E.; López-Ogáyarl, J.L.; Zamora, C.; Astorgano, D.; Albericio, G.; Pérez, P.; Muñoz-Cabello, A.M.; Pascual, A.; Esteban, M.; López-Barneo, J.; Toledo-Ara, J.J. Full protection from SARS-CoV-2 brain infection and damage in susceptible transgenic mice conferred by MVA-CoV2-S vaccine candidate, Nature Neuroscience, 2022, 26, 226-238. [CrossRef]
- Offit, P.A. Bivalent Covid-19 Vaccines — A Cautionary Tale, N. Engl. J. Med. 2023, 388, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Irrgang, P.; Gerling, J.; Kocher, K.; Lapuente, D.; Steininger, P.; Habenicht, K.; Wytopi, M.; Beileke, S.; Schäfer, S.; Zhong, J.; Ssebyatika, G.; Krey, T.; Falcone, V.; Schülein, C.; Peter, A.S.; Nganou-Makamdop, K.; Hengel, H.; Held, J.; Bogdan, C.; Überla, K.; Schober, K.; Winkler, T.H.; Tenbusch, M. Class switch towards non-inflammatory, spike-specific IgG4 antibodies after repeated SARS-CoV-2 mRNA vaccination. Sci. Immunol. 2023, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saed Abbasi,1,7 Miki Matsui-Masai,2,7 Fumihiko Yasui,3 Akimasa Hayashi,4 Theofilus A. Tockary,1 Yuki Mochida,1,5 Shiro Akinaga,2 Michinori Kohara,6 Kazunori Kataoka,1 and Satoshi Uchida1,, Carrier-free mRNA vaccine induces robust immunity against SARS-CoV-2 in mice and non-human primates without systemic reactogenicity, Molecular Therapy Vol. 32 No 5 May 2024 1267,p.1266-1283. [CrossRef]
- Michael W. Grunst, Zhuan Qin, Esteban Dodero-Rojas, Shilei Ding, Jérémie Prévost, Yaozong Chen, Yanping Hu, Michael W. Grunst, Zhuan Qin, Esteban Dodero-Rojas, Shilei Ding, Jérémie Prévost, Yaozong Chen, Yanping Hu, Marzena Pazgier, Shenping Wu, Xuping Xie, Andrés Finzi, José N. Onuchic, Paul C. Whitford, Walther Mothes, Wenwei Li, Structure and inhibition of SARS-CoV-2 spike refolding in membranes, SCIENCE, ug 2024 Vol 385, Issue 6710 pp. 757-765. [CrossRef]
- Jon Cohen, Missing immune cells may explain why COVID-19 vaccine protection quickly wanes, New insights on what stimulates long-lived antibody production could spur better vaccines, SCIENCE INSIDER, 11 OCT 2024, PP.
- Hentzien, M.; Autran, B.; Pitoh, I.; Yazdanpanah, Y.; Calmy, A. A monoclonal antibody stands out against omicron subvariants: a call to action for wider access to Bebtelovimab, Lancet, www.thelancet.com/infection 22, Sept 2022.
- Barmada A.; Klein J.; Ramaswamy A.; Brodsky N.N.; Joycox J.R.; Sheikha H.; Nines K.M.; Habet V.; Campbell M.; Shimoda T.S.; Kontorovich A; Bogunovic D.; Oliveira C.R.; Steele J.; Hall E.K.; Pena-Hernandez M.; Monteiro V.; Lucan C.; Ring A.M.; Omer S.B.; Iwasaki A.; Yildirim I.; Lucas C.L.; Cytokinopathy with aberrant cytotoxic lymphocytes and profibrotic myeloid response in SARS-CoV-2 mRNA vaccine-associated myocarditis, Additional introduction doi: 10.1126/sciimmunol.adh3455. Sci. Immunol. 2023, 8:1-18, eadh3455, Epub 2023, May 5. [CrossRef]
- Hanai, T. Further quantitative in silico analysis of SARS-CoV-2 S-RBD Omicron BA.4, BA.5, BA.2.75, BQ.1, and BQ.1.1 transmissibility, Talanta, 2023, 254, 124127. [CrossRef]
- RCSB PDB 7mjk.
- D. Zha, M. Fu, Y, Qian, Vascular Endothelial Glycocalyx Damage and Potential Targeted Therapy in COVID-19, Cells `2022, 11, 1972. [CrossRef]
- Kissel, G.T.; René, E.; Toes, M.; Thomas, W.; Huizinga1, J.; Manfred, W. Glycobiology of rheumatic, diseases, Nat Rev Rheumatol. 2023 Jan;19(1):28-43. [CrossRef]
- Williamson, G. Effects of polyphenols on glucose-induced metabolic changes in healthy human subjects and on glucose transporters, Mol. Nutr. Food Res. 2022, 66, 2101113. [Google Scholar] [CrossRef] [PubMed]
- Hanai, T. (Ed) Quantitative in Silico Analytical Chemistry, Quantitative analysis of molecular interactions from chromatography retention to enzyme selectivity, http://www.hanai-toshihiko.net/ or http://hanai-tohihiko.net/, 2024, pp.327, 43.5MB (10 Chapters).
- Van Breemen, R.B.; Muchiri, R.N.; Bates, T.A.; Weinstein, J.B.; Leier, H.C.; Farly, S.; Tafesse, F.G. Cannabinoids block cellular entry of SARS-CoV-2 and the emerging variants, J. Nat. Prod. 2022, 85, 496–184. [Google Scholar] [CrossRef] [PubMed]
- Brevini, T.; Maes, M.; Webb, G.J.; John, B.V.; Fuchs, C.D.; Buescher, G.; Wang, L.; Griffiths, C.; Brown, M.L.; Scott III, W.E.; Pereyra-Gerber, P.; Gelson, W.T.H.; Brown, S.; Dillon, S.; Muraro, D.; Sharp, J.; Neary, M.; Box, H.; Tatham, L.; Stewart, J.; Curley, P.; Pertinez, H.; Forrest, S.; Mlcochova, P. FXR inhibition may protect from SARS-CoV-2 infection by reducing ACE2, Nature, 2023,615, 134–142. [CrossRef]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A.; Abreu, P.; Bao, W.; Wisemandle, W.; Baniecki, M.- L.; Hendrick, V.M.; Damle, B.; Simón-Campos, A.; Pypstra, R.; Rusnak, J.M. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with Covid-19, N Engl J Med. 2022, 386, 1397-408. [CrossRef]
- Takahashi, J.A.; Barbosa, B.V.R.; Lima, M.T.N.S.; Cardoso, P.G.; Consigli, C.; Pimenta, L.P.S. Antiviral fungal metabolites and some insights into their contribution to the current COVID-19 pandemic, Bioorg. Med. Chem. 2021, 46, 116366. [Google Scholar] [CrossRef]
- Yang, J.; Xiao, Y.; Lidsky, P.V.; Wu, C.-T.; Bonser, L.R.; Peng, S.; Garcia-Knight, M.A.; Tassetto, M.; Chung, C.-I.; Li, X.; Nakayama, T.; Lee, I.T.; Nayak, J.V.; Ghias, K.; Hargett, K.L.; Shoichet, B.K.; Erle, D.J.; Jackson, P.K.; Andino, R.; Shu, X. Fluorogenic reporter enables identification of compounds that inhibit SARS-CoV-2, Nature Microbiology, 2023, 8, 121–134. [CrossRef]
- Cordero, F.M.M. Research study to evaluate the clinical effectiveness of the nutritional supplement VITAMIC BIOSEN®, Combination of Curcumin, Vitamin C, and Boswellia Serrata, on individuals with symptoms consistent with LONG COVID who have been vaccinated against the SARS-CoV2, Biomed J Sci & Tech Res.2022, 47, BJSTR.MS.ID.007512, 38567-38581.
- Bodie, N.M.; Hashimoto, R.; Connolly, D.; Chu, J.; Takayama, K.; Uhal, B.D. Design of a chimeric ACE-2/Tc-silent fusion protein with ultrahigh affinity and neutralizing capacity for SARS-CoV-2 variants, Antib. Ther. 2023, 20, 59-74, https://academic.oup.com/abt/advance-article/doi/10.1093/abt/tbad001/6994056 by guest on Jan. 25, 2023.
- Zhang, J.-L.; Li, Y.-H.; Wang, L.-L.; Liu, H.-Q.; Lu, S.-Y.; Liu, Y.; Li, K.; Li, B.; Li, S.-Y.; Shao, F.-M.; Wang, K.; Sheng, N.; Li, R.; Cui, J.-J.; Sun, P.-C.; Ma, C.-X.; Zhu, B.; Wang, Z.; Wan, Y.-H.; Yu, S.-S.; Che, Y.; Wang, C.-Y.; Wang, C.; Zhang, Q.; Zhao, L.-M.; Peng, X.-Z.; Cheng, Z.; Chang, Jun-B.; Jiang, J.-D. Azvudine is a thymus-homing anti-SARS-CoV-2 drug effective in treating COVID-19 patients, Signal Transduction and Targeted Therapy, 2021, 6, 414. [CrossRef]
- Yue, C.; Song, W.; Wang, L.; Jian, F.; Chen, X.; Gao, F.; Shen, Z.; Wang, Y.; Wang, X.; Cao, Y. ACE2 binding and antibody evasion in enhanced transmissibility of XBB.1.5, The Lancet Infectious Diseases, Lancet Infect Dis. 2023 Mar, 23(3):278-280. [CrossRef]
- Topol, E. The virus takes a detour in its evolution arc, Ground Truths, OCT. 26, 2023, https:/erictopod.substack.com/p/the-virus-takes-a-detour-in-its-evolution-arc/.
- 89. Kaku, Y.; Okumura, K.; Padilla-Blanco, M.; Kosugi, Y.; Uriu, K.; Hinay Jr, A.A.; Chen, L.; Plianchaisuk, A.; Kobiyama, K.; Ishii, K.J.; Zahradnik, J.; Ito, J.; Sato, K. Virological characteristics of the SARS-CoV-2 JN.1 variant Lancet Infect Dis. 2024, Jan.3. 2024.
- Bjorkman, A.; Gisslen, M.; Gullberg, M.; Ludvigsson, J. The Swedish COVID-19 approach: a scientific dialogue on mitigation policies, Frontiers in Public Health, 2023, 20, 1-4. [CrossRef]
- Villadiego, J.; García-Arriaza, J.; Ramírez-Lorca, R.; García-Swinburn, R.; Cabello-Rivera, D.; Rosales-Nieves, A.E.; Álvarez-Vergara, M.I.; Cala-Fernández, F.; García-Roldán, E.; López-Ogáyarl, J.L.; Zamora, C.; Astorgano, D. Albericio, G.; Pérez, P.; Muñoz-Cabello, A.M.; Pascual, A.; Esteban, M.; López-Barneo, J.; Toledo-Ara, J.J. Full protection from SARS-CoV-2 brain infection and damage in susceptible transgenic mice conferred by MVA-CoV2-S vaccine candidate. Nature Neuroscience, 2022; 26, 226–238. [Google Scholar]
- Van Breemen, R.B.; Muchiri, R.N.; Bates, R.A.; Weistein, J.B.; Leier, H.C.; Farly, S.; Tafesse, F.G. Cannabinoids block cellular entry of SARS-CoV-2 and the emerging variants. J. Nat. Rod. 2022, 85, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Hanai, T. (Ed) Quantitative in silico Chromatography, Computational Modeling of Molecular Interactions, 2014, RSC, Cambridge, pp.338. ISBN 978-1-84973-991-7.
- Tozser, J.; Benko, S. Natural compounds as regulators of NLRP3 inflammasome-mediated IK-1b production, Mediator of Inflammation, 2016, Article ID: 5460302, pp.16.
- T.E. Tallei, Fatimawali, N.J. Niode, R. Idroes, R.M. Zidan, S. Mitra, I. Celik, F. Nainu, D. Ağagündüz, T.B. Emran, and R. Capasso, A Comprehensive Review of the Potential Use of Green Tea Polyphenols in the Management of COVID-19, Evid Based Complement Alternat Med. 2021, 7170736. Published online 2021 Dec 3. [CrossRef]
- R. Ghosh, A. Chakraborty, A. Biswas, and S. Chowdhuri, Evaluation of green tea polyphenols, as novel coronavirus (SARS CoV-2) main protease (Mpro) inhibitors – an in silico docking and molecular dynamics simulation study, J Biomol Struct Dyn. 2020, 1–13. Published online 2020 Jun 22. [CrossRef]
- L. Henss, A. Auste, C. Schürmann, C. Schmidt, C. von Rhein, M.D. Mühlebach, B.S Schnierle, The green tea catechin epigallocatechin gallate inhibits SARS-CoV-2 infection, J Gen Virol. 102(4) (2021), 001574. [CrossRef]
- J. Park, R. Park, M. Jang, and Y-I. Park, Therapeutic Potential of EGCG, a Green Tea Polyphenol, for Treatment of Coronavirus Diseases, Life (Basel). 197, (2021) 197. Published online 2021 Mar 4. [CrossRef]
- H. Nishimura, M. Okamoto, I. Dapat, M. Katsumi, H. Oshitani, Inactivation of SARS-CoV-2 by Catechins from Green Tea, Jpn J Infect Dis. 74(5), (2021) 421-423. [CrossRef]
- T. Hanai (ed) Quantitative in silico chromatography, Computational modeling of molecular interactions, 2014, RSC, Cambridge, p.94-100. (T. Hanai, Quantitative structure-retention relationship of phenolic compounds without Hammet’s equations, J. Chromatogr. A, 985 (2003) 343-349.
- Ricardo Wesley Alberca1, Franciane Mouradian Emidio Teixeira, Danielle Rosa Beserra, Emily Araujo de Oliveira, Milena Mary de Souza Andrade, Anna Julia Pietrobon, Maria Notomi Sato, The Potential Effects of Naringenin in COVID-19, Front. Immunol., 25 September 2020, Sec. Nutritional Immunology, Volume 11 - 2020. [CrossRef]
- Siqi Liu, Mengli Zhong, Hao Wu, Weiwei Su, Yonggang Wang, Peibo Li, Potential Beneficial Effects of Naringin and Naringenin on Long COVID—A Review of the Literature, Microorganisms 2024, 12, 332. [CrossRef]
- Helda Tutunchi, Fatemeh Naeini, Alireza Ostadrahimi, Mohammad Javad Hosseinzadeh-Attar Naringenin, a flavanone with antiviral and anti-inflammatory effects: A promising treatment strategy against COVID-19, Phytotherapy Research. 2020;34:3137–3147. [CrossRef]
- Nicola Clementi, Carolina Scagnolari, Antonella D’Amore, Fioretta Palombi, Elena Criscuolo, Federica Frasca, Alessandra Pierangeli, Nicasio Mancini, Guido Antonelli, Massimo Clementi, Armando Carpaneto, Antonio Filippini, Naringenin is a powerful inhibitor of SARS-CoV-2 infection in vitro, Pharmacological Research 163 (2021) 105255. [CrossRef]
- Hanai, T. Visualization (XX) Glycosidation (Glycosylation), Effective derivatization, 2024, http://hanai-toshihiko.net/ quantitative in silico analytical chemistry/Covid-19 transmissibility and designing the binding inhibitors/.
- Lewis, A.L.; Toukach, P.; Bolton, E.; Chen, X.; Frank, M.; Lutteke, T.; Knirel, Y.; Schoenhofen, I.; Varki, A.; Vinogradov, E.; Woods, R.J.; Zachara, N.; Zhang, J.; Karmerling, J.P.; Neelamegham, S. Cataloging natural sialic and other nonulosonic acids (NulOs), and their representation using the Symbol Nomenclature for Glycans, Glycobiology. 2023, 33, 99-103. [CrossRef]
- Ling, A.J.W.; Chang, L.S.; Babji, A.S.; Latip, J. Koketsu, M.; Lim, S.J. Review of sialic acid’s biochemistry, sources, extraction and functions with special reference to edible bird’s nest Affiliations expand, Food Chem. 2022 Jan 15, 367,130755. [CrossRef]
- Stencel-Baerenwalg, J.E.; Reiss, K.; Reiter, D.K.; Stehle. T.; Dermody, T.S. The sweet spot: defining virus-sialic acid interactions, Nature Reviews Microbiology, 2014, 12, 739-749.
- Ghosh, S. Sialic acid and biology of life: An introduction, Sialic Acids and Sialoglycoconjugates in the Biology of Life, Health and Disease. 2020, 1–61. [CrossRef]
- Yagisawa, M.; Foster, P.J.; Hanaki, H.; Omura, S. Global trends in clinical trials of Ivermectin for Covid-19, Part 2. The Japanese Journal of Antibiotics, 2024, 77, 45. [Google Scholar]
- Zhang, M. Ivermectin could be a powerful drug for fighting cancer, The Epoch Times, Nar 21, 2024, 1-5.
- Zaidi, A.K.; Dehgani-Mobaraki, P. The mechanisms of action of Ivermectin against SARS-CoV-2- an extensive review. J. Antibiotics, 2022, 75, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Hanai, T. 2023-0917 Visualization (IV) of Pros and Cons of Ivermectin for S-RBDs and ACE-2 interaction and the inhibitors, http://hanai-tohihiko.net/ Quantitative in silico analytical chemistry/ COVID-19 transmissibility and designing the binding inhibitors, 2023.
- Hanai, T.; Shimada, K.; Koyama, N.; Nohara, Y.; Takayanagi, H.; (late) Kinoshita, T. Quantitative analysis of selective glycosylation of saccharides with aromatic amines, Carbohydrate Research, 2020, 498, 108171 http://doi.org/10.1016/j.carres.2020.108171 (Hanai, T.; Kato, K.; Furukoshi, E.; Hennmi, M.; Imoto, S.; Kotaka, A.; Shimada, K. Thoma, M.; Ishikawa, M.; Watanabe, T.; Ohgawara, E.; Koyama, N.; Furuhata, K.; Nagahara, K.; Takayanagi, H.; Nohara, Y.; Kinoshita, T.). [CrossRef]
- 108/Hanai, T. Evaluation of measuring methods of human serum albumin-drug binding affinity. Current Pharmaceutical Analysis, 2007, 3, 205–212. [Google Scholar]
- Pavelic, S.; Pavelic, K. Open questions over the COVID-19 pandemic, DOI: 10.5005/jp-hournals-11005-0027. Science, Art and Religion, 2023, 1, 211-220.
- Guan, Y.; Tang, G.; Li, L.; Shu, J.; Zhao, Y.; Huang, L.; Tang, J. Herbal medicine and gut microbiota: exploring untapped therapeutic potential in neurodegenerative disease management. Archives of Pharmacal Research, 2024, 47, 146–164. [Google Scholar] [CrossRef] [PubMed]
- Tozser, J.; Benko, S. Natural compounds as regulators of NLRP3 inflammasome-mediated IK-1b production, Mediator of Inflammation, 2016, Article ID: 5460302, pp.16.
- Priyanjana Pramanik, Healthy lifestyle habits dramatically cut long-term COVID-19 risks, URL: pp.5, https://www.news-medical.net/news/20240730/.
- Mathew G. Frank, Jayson B. Ball, Shelby Hopkins, Tel Kelley, Angelina J. Kuzma, Robert S. Thompson, Monika Fleshner, Steven F. Maier, SARS-CoV-2 S1 subunit produces a protracted priming of the neuroinflammatory, physiological, and behavioral responses to a remote immune challenge: A role for corticosteroids, Brain, Behavior and Immunity, 121, 2024, 87-103. [CrossRef]
- Delphine Planas, Isabelle Staropoli, Cyril Planchais, Emilie Yab, Banujaa Jeyarajah, Yannis Rahou, Matthieu Prot, Florence Guivel-Benhassine, Frederic Lemoine, Vincent Enouf , Etienne SimonLoriere, Hugo Mouquet, Marie-Anne Rameix-Welti , Olivier Schwartz., Escape of SARS-CoV-2 variants KP1.1, LB.1 and KP3.3 from approved monoclonal antibodies, bioRxiv preprint doi: https://doi.org/10.1101/2024.08.20.608835; this version posted August 21, 2024, 1-7. [CrossRef]
- Mahta Mortezavi, Abigail Sloan, Ravi Shankar P. Singh, Luke F. Chen, MBBS, Jin Hyang Kim, Negin Shojaee, Sima S. Toussi, John Prybylski, Mary Lynn Baniecki, Arthur Bergman, Anindita Banerjee, Charlotte Allerton, Negar Niki Alami, Virologic Response and Safety of Ibuzatrelvir, a Novel SARS-cov-2 Antiviral, in Adults With COVID-19, Clinical Infectious Diseases, 1-19, 2024, acspted. [CrossRef]
- Yongzhi Lu, Qi Yang, Ting Ran, Guihua Zhan, Wenqi Li, Peiqi Zhou, Jielin Tang, Minxian Dai, Jinpeng Zhong, Hua Chen, Pan He, Anqi Zhou, Bao Xue, Jiayi Chen, Jiyun Zhang, Sidi Yang, Kunzhong Wu, Xinyu Wu, Miru Tang, Wei K. Zhang, Deyin Guo, Xinwen Chen, Hongming Chen, Jinsai Shang, Discovery of orally bioavailable SARS-CoV-2 papain-like protease inhibitor as a potential treatment for COVID-19 , Nature Communications | (2024) 15:10169, 1-16. [CrossRef]
- Topol., E. Long-term Long Covid, Ground Truths, 2023, Aug. 22, pp.12.
- Stefanie Kaiser, Steffen Kaiser, Jenny Reis, and Rolf Marschalek, Quantification of objective concentrations of DNA impurities in mRNA vaccines, SSRN https://papers.ssrn.com › papers.cfm.
- Lorena Diblasi, Martín Monteverde, David Nonis, PhD, Marcela Sangorrín, At Least 55Undeclared Chemical Elements Found in COVID-19 Vaccines from AstraZeneca, CanSino, Moderna, Pfizer, Sinopharm and Sputnik V, with Precise ICP-MS, International Journal of Vaccine Theory, Practice, and Research 3(2) October 11, 2024 | Page 1367. [CrossRef]
- Yingying Shi1 , Meixing Shi1 , Yi Wang 1 and Jian You, Progress and prospects of mRNA-based drugs in pre-clinical and clinical applications, Signal Transduction and Targeted Therapy (2024) 9:322. [CrossRef]
Figure 1.
The molecular interaction between two amino acids. Yellow (light gray), red (black), blue (dark gray), and white balls: carbon, oxygen, nitrogen, and hydrogen.
Figure 1.
The molecular interaction between two amino acids. Yellow (light gray), red (black), blue (dark gray), and white balls: carbon, oxygen, nitrogen, and hydrogen.
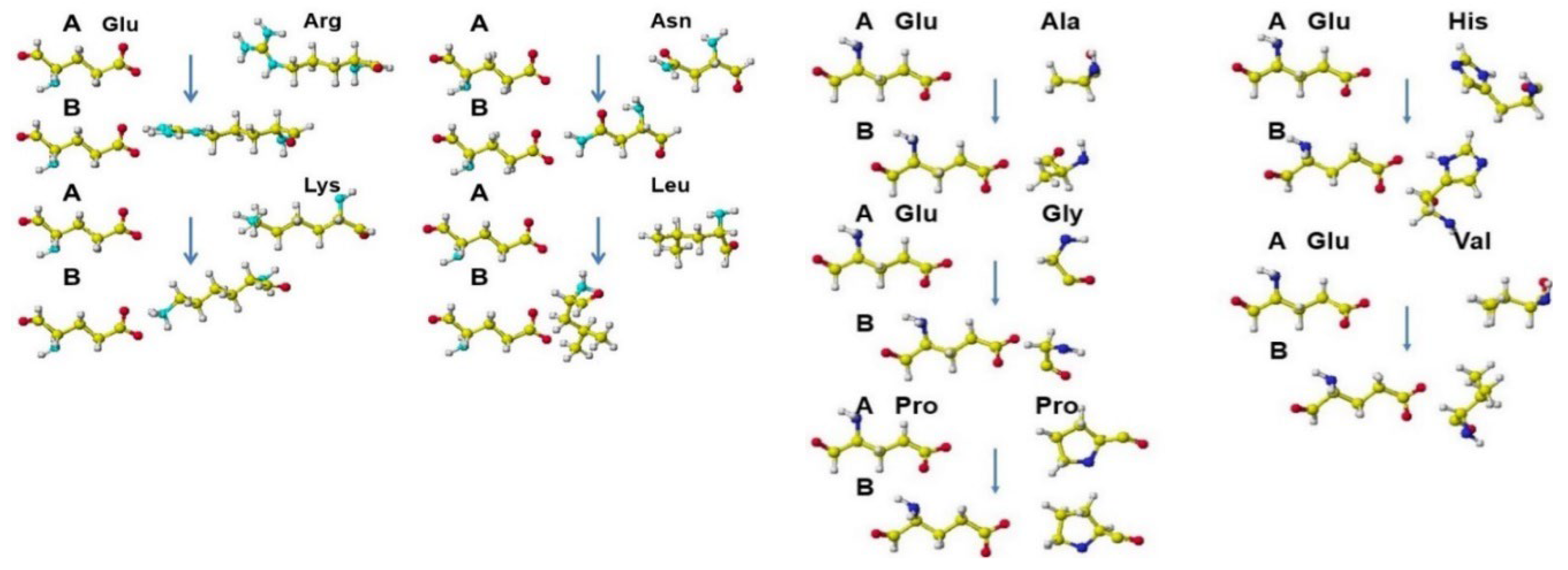
Figure 2.
A-O. Stereo structures of several SARS-CoV-2, ACE-2 and S-RBD. White, yellow (light gray), blue (dark gray), red (black) balls: hydrogen, carbon, nitrogen, oxygen.
Figure 2.
A-O. Stereo structures of several SARS-CoV-2, ACE-2 and S-RBD. White, yellow (light gray), blue (dark gray), red (black) balls: hydrogen, carbon, nitrogen, oxygen.

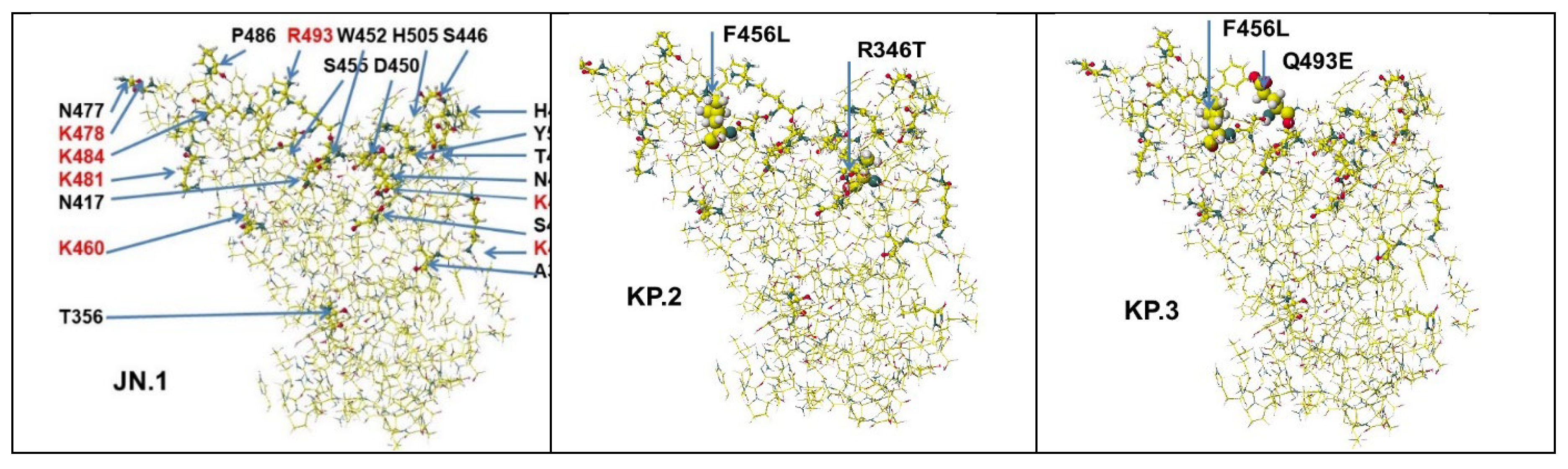
Figure 3.
Binding inhibitor candidates. White, yellow (light gray), blue (dark gray), red (black) balls: hydrogen, carbon, nitrogen, oxygen.
Figure 3.
Binding inhibitor candidates. White, yellow (light gray), blue (dark gray), red (black) balls: hydrogen, carbon, nitrogen, oxygen.
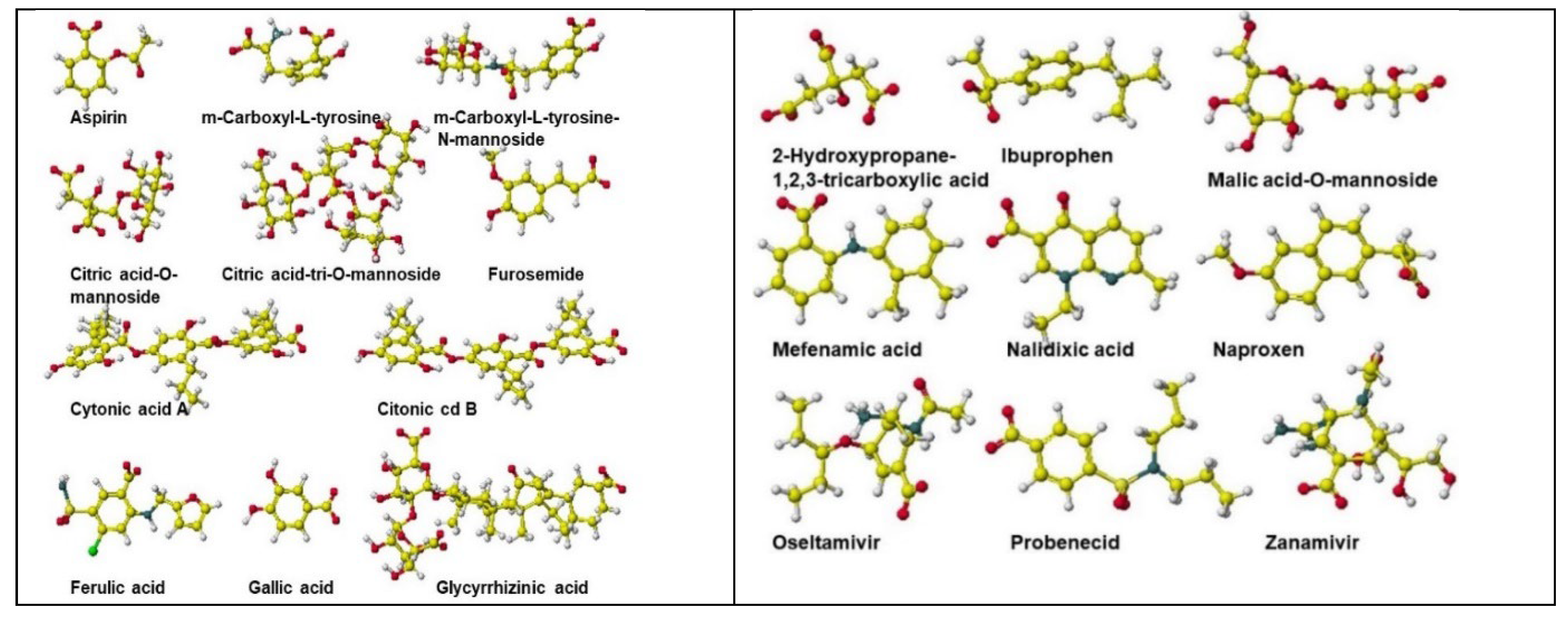
Figure 4.
(A) Initial conformation of BQ.1 S-RBD and three Aspirin, and ACE-2 and BQ.1 three Aspirin complex and their optimized (repulsed) structure. (B) Initial location of Delta S-RPD and three Aspirin and the optimized complex structure of Delta-S-RPD and three Aspirin complex, and ACE-2 and three Aspirin optimized structure. (C) Properties of docking inhibitor candidate of molecular form lactic acid. (D) Properties of docking inhibitor candidate of ionized form lactic acid. (E) Performance of acidic compounds inhibited binding Delta and ACE-2. (F) Performance of TCA acids binding inhibition; these binding inhibitors are indicated as color molecules for easy identification of their binding location with S-RPD. (G) Inhibition of glycyrrhizic acid of BA.2.86, BA.2, and JN.1 binding with ACE-2, these binding inhibitors are indicated as color molecules for easy identification of their binding location with S-RPD.
Figure 4.
(A) Initial conformation of BQ.1 S-RBD and three Aspirin, and ACE-2 and BQ.1 three Aspirin complex and their optimized (repulsed) structure. (B) Initial location of Delta S-RPD and three Aspirin and the optimized complex structure of Delta-S-RPD and three Aspirin complex, and ACE-2 and three Aspirin optimized structure. (C) Properties of docking inhibitor candidate of molecular form lactic acid. (D) Properties of docking inhibitor candidate of ionized form lactic acid. (E) Performance of acidic compounds inhibited binding Delta and ACE-2. (F) Performance of TCA acids binding inhibition; these binding inhibitors are indicated as color molecules for easy identification of their binding location with S-RPD. (G) Inhibition of glycyrrhizic acid of BA.2.86, BA.2, and JN.1 binding with ACE-2, these binding inhibitors are indicated as color molecules for easy identification of their binding location with S-RPD.

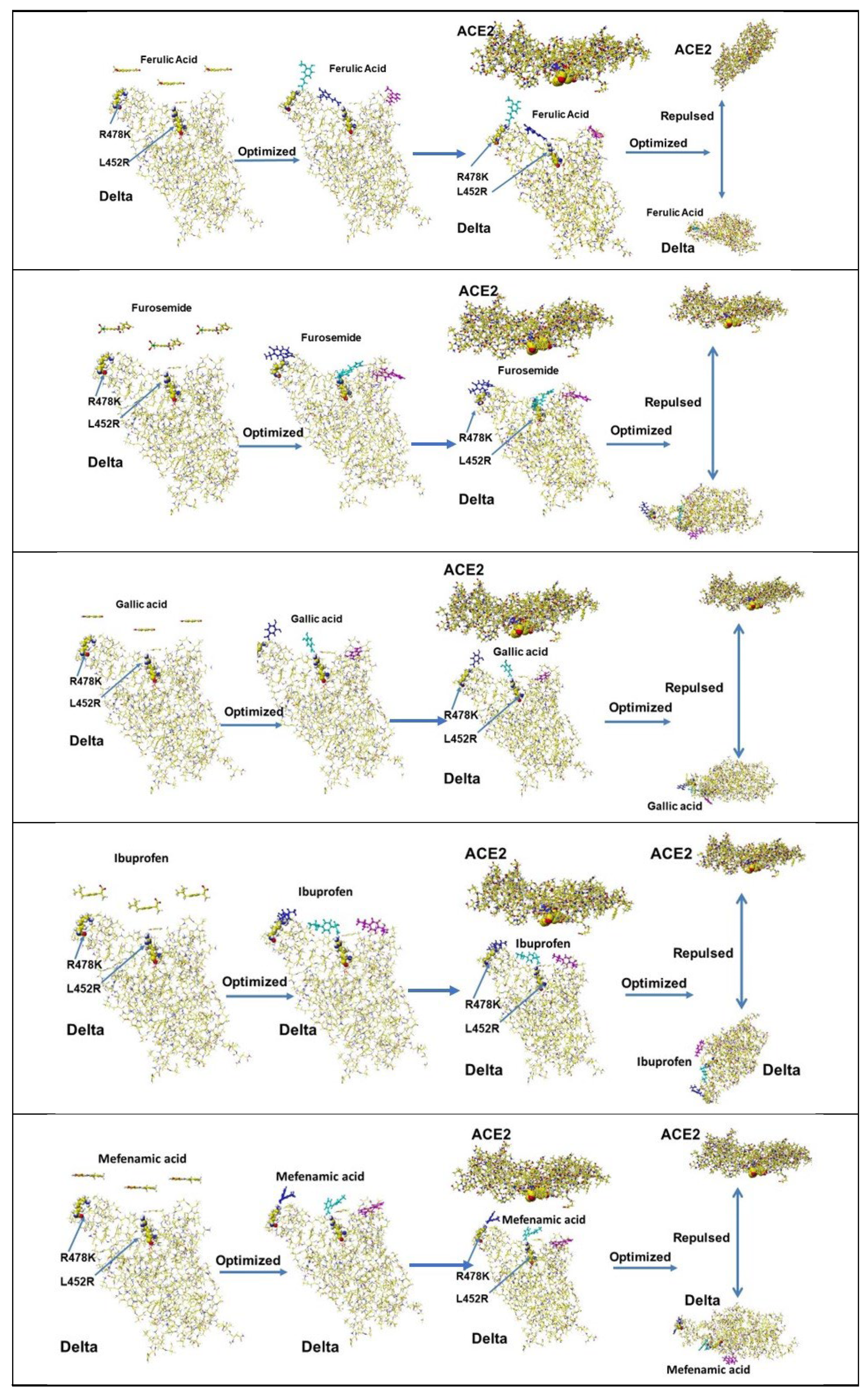
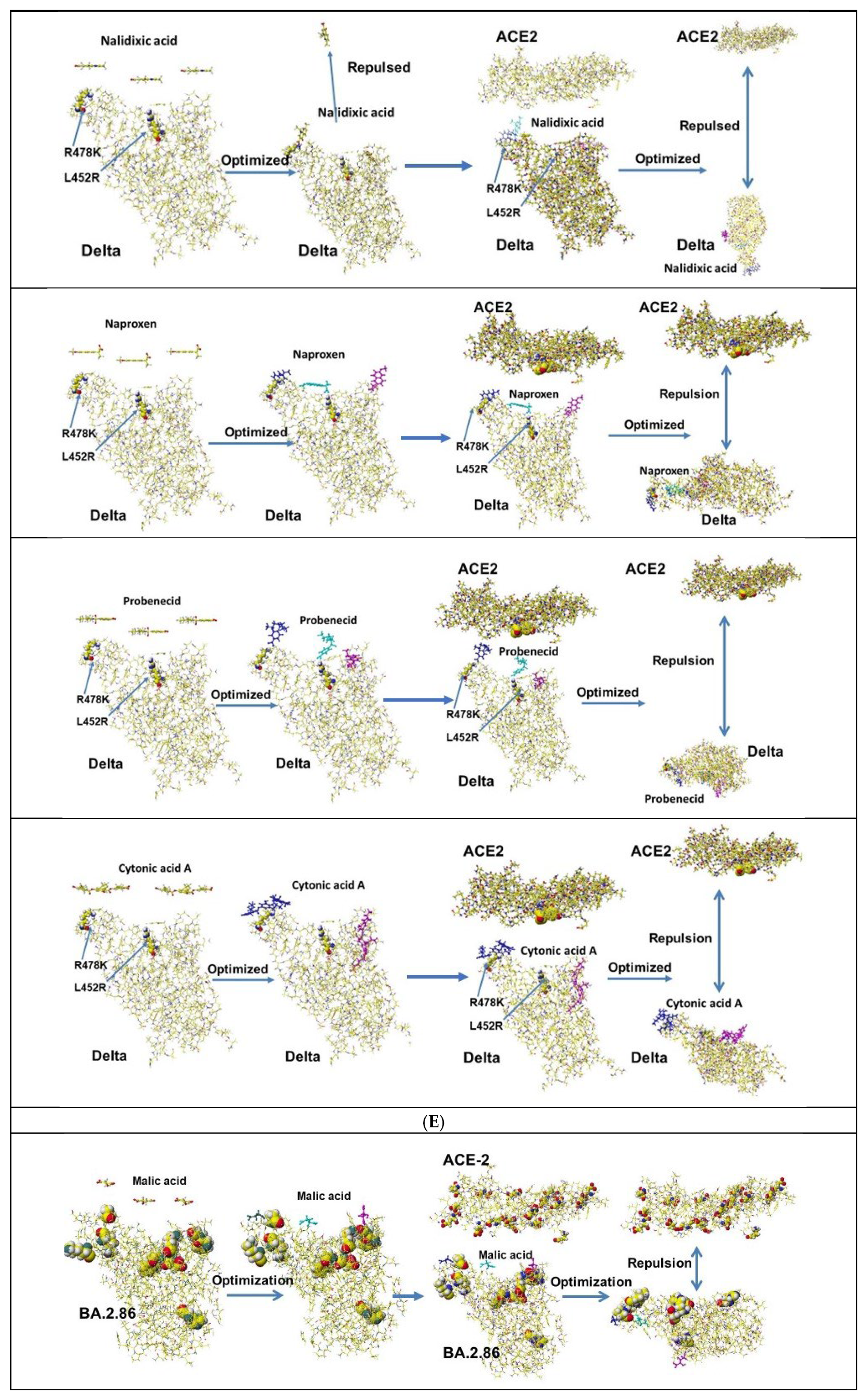
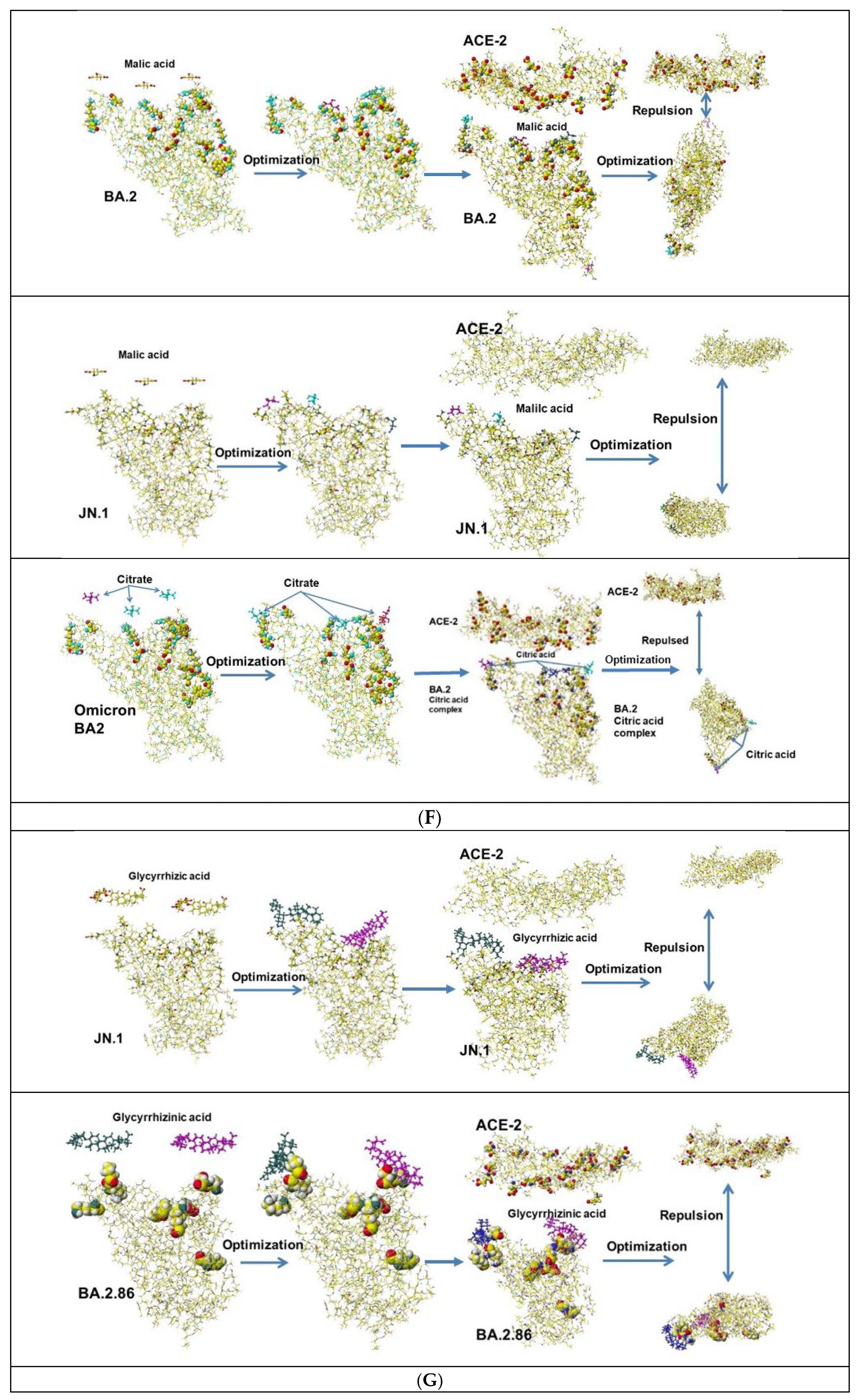
Figure 5.
Original and modified stereo structure of PF 07321332, Molnupiravir, and Xacova where atoms are indicated as color or grayscale; nitrogen (blue or dark gray balls), oxygen (red or black balls), carbon (yellow or gray balls), hydrogen (small white balls), and fluorine (light green of light gray balls). Generally, fluorone atoms support the sterically stable structure.
Figure 5.
Original and modified stereo structure of PF 07321332, Molnupiravir, and Xacova where atoms are indicated as color or grayscale; nitrogen (blue or dark gray balls), oxygen (red or black balls), carbon (yellow or gray balls), hydrogen (small white balls), and fluorine (light green of light gray balls). Generally, fluorone atoms support the sterically stable structure.
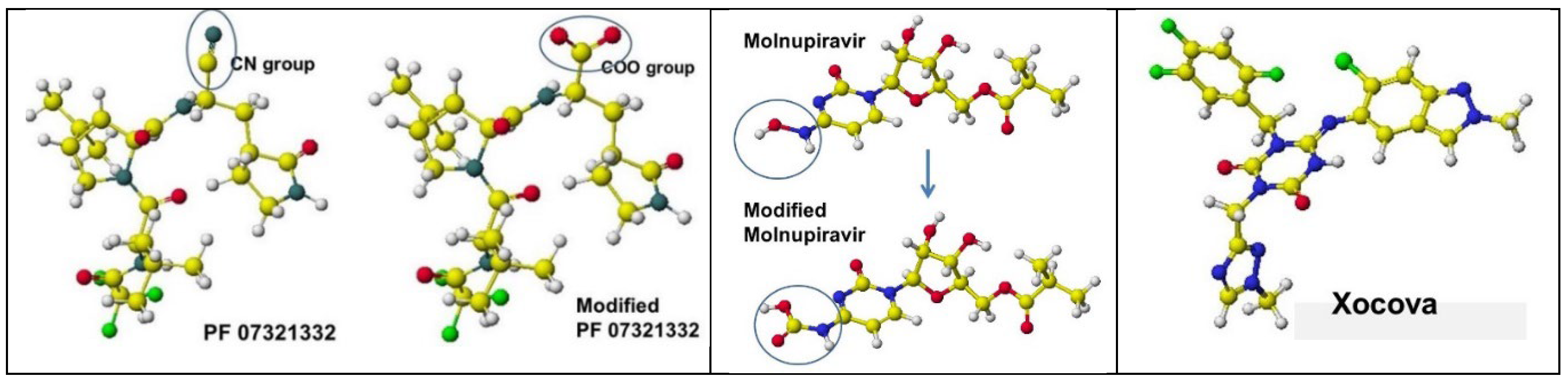
Figure 6.
(A) Conformation of PF 07321332 and modified PF 07321332 trapped protein The cyano and carboxy groups are circled for their identification. (B) Amino acid residues within 2.5 Å from PF 07321332 and modified PF 07321332.
Figure 6.
(A) Conformation of PF 07321332 and modified PF 07321332 trapped protein The cyano and carboxy groups are circled for their identification. (B) Amino acid residues within 2.5 Å from PF 07321332 and modified PF 07321332.
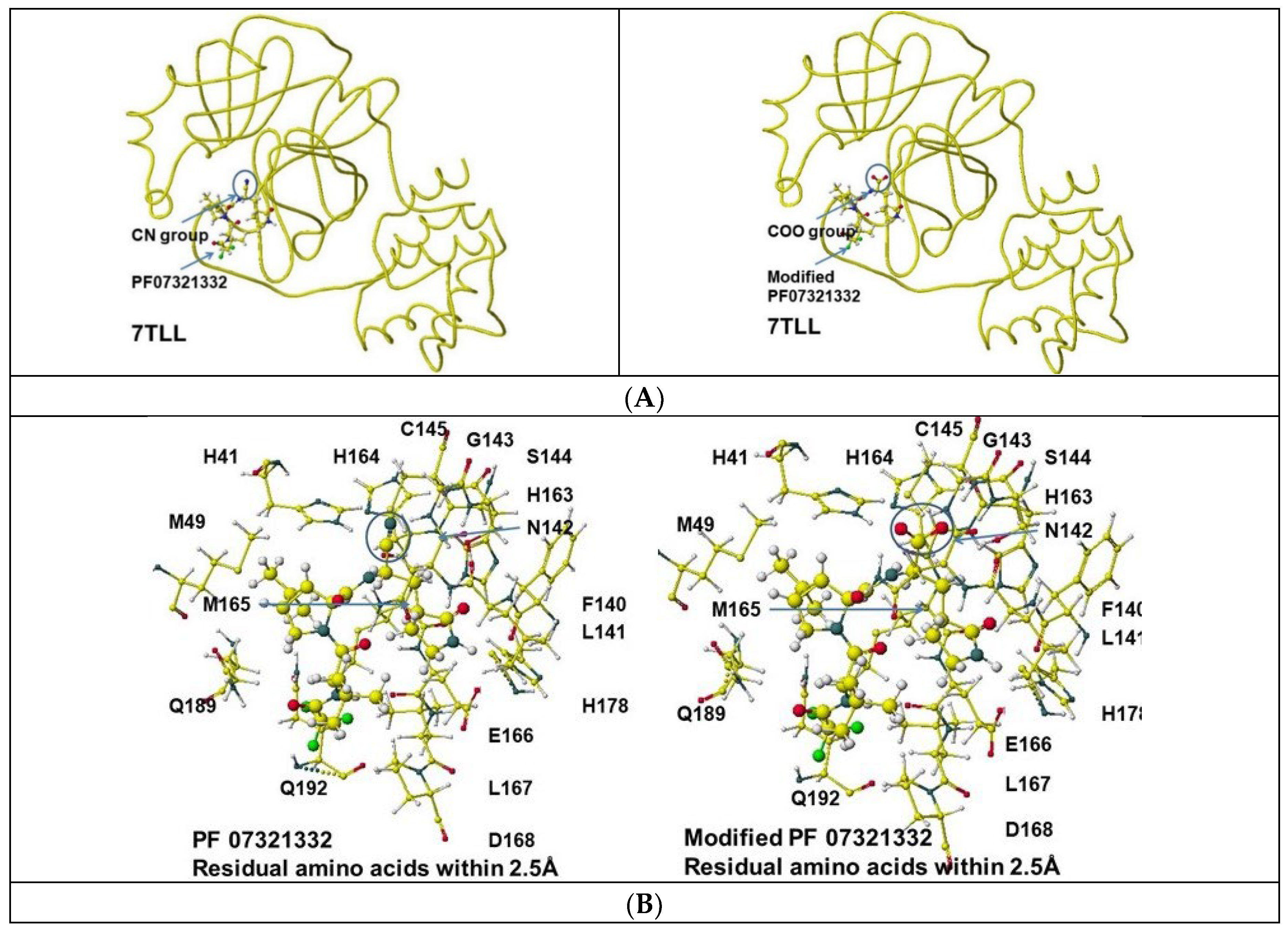
Figure 7.
(A) Initial conformation of Delta and three PF07321332 and their complex structure, Delta and three PF07321332 complex, and ACE-2 and their complex structure. (B) Initial conformation of Delta and three modified PF07321332 and their complex structure, Delta and three modified PF07321332 complex, and ACE-2 and their complex structure. (C) Initial conformations of BA.2 and three modified PF07321332 and their complex structure, BA.2 and three modified PF07321332 complex, and ACE-2 and their complex structure. (D) Initial conformations of BQ.1 S-RBD with three PF 07321332 molecules, the complex (optimized) structure of BQ.1 S-RBD with three PF 07321332 molecules complex, and ACE-2 and their optimized structure. (E) The initial position of Omicron BQ.1-S-RBD and modified PF-07321332 and Omicron BQ.1 and modified PF-07321332 complex and ACE-2 and their optimized structure.
Figure 7.
(A) Initial conformation of Delta and three PF07321332 and their complex structure, Delta and three PF07321332 complex, and ACE-2 and their complex structure. (B) Initial conformation of Delta and three modified PF07321332 and their complex structure, Delta and three modified PF07321332 complex, and ACE-2 and their complex structure. (C) Initial conformations of BA.2 and three modified PF07321332 and their complex structure, BA.2 and three modified PF07321332 complex, and ACE-2 and their complex structure. (D) Initial conformations of BQ.1 S-RBD with three PF 07321332 molecules, the complex (optimized) structure of BQ.1 S-RBD with three PF 07321332 molecules complex, and ACE-2 and their optimized structure. (E) The initial position of Omicron BQ.1-S-RBD and modified PF-07321332 and Omicron BQ.1 and modified PF-07321332 complex and ACE-2 and their optimized structure.
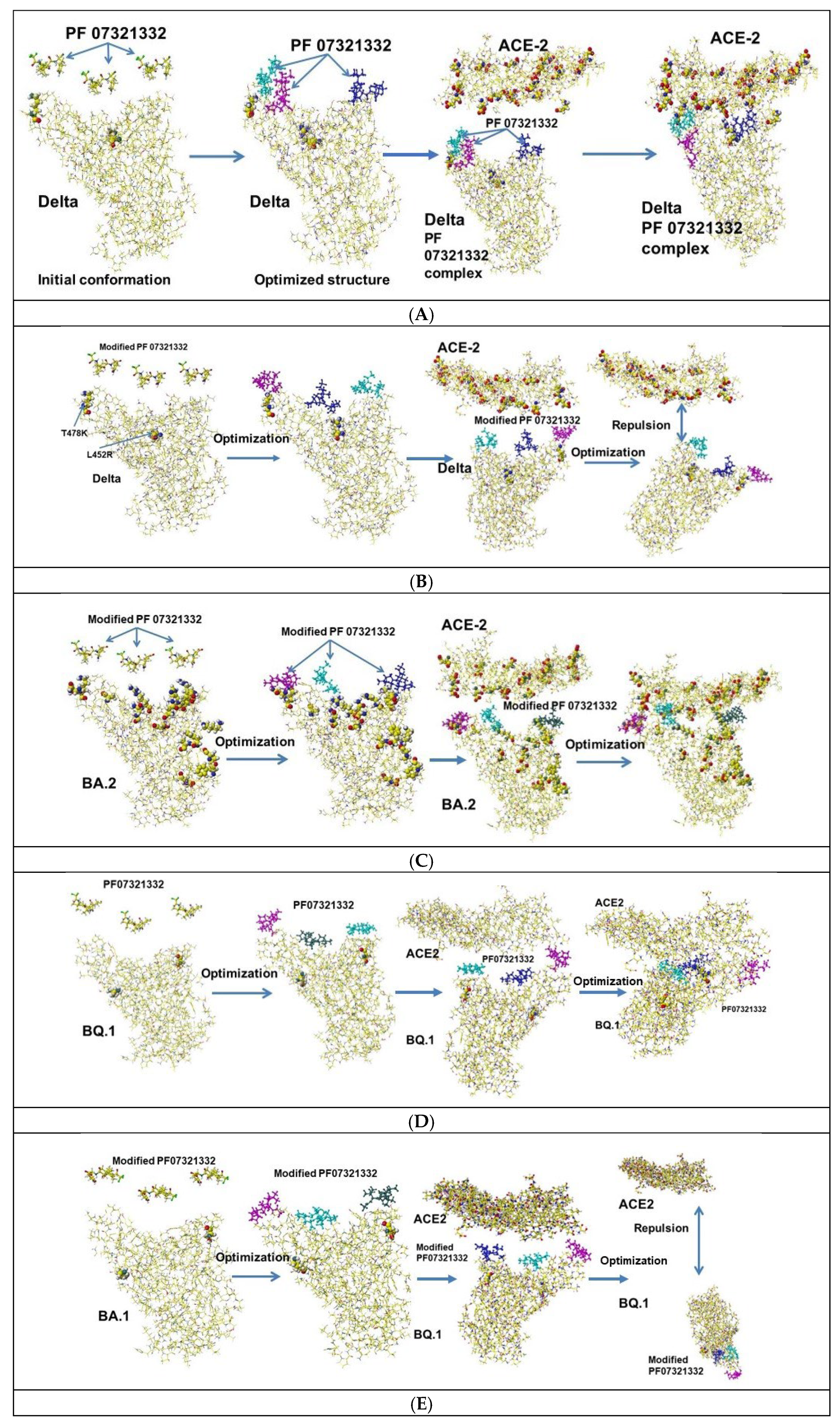
Figure 8.
(A) The initial position of Delta-S-RBD and Molnupiravir and Delta-s-RBD and Molnupiravir complex and ACE-2 and their optimized structure. (B) Initial conformations of Delta S-RBD and Omicron BA.2 with three modified Molnupiravir molecules, the complex (optimized) structure of Delta and BA.2 S-RBD with three modified Molnupiravir molecules complex and ACE-2 and their optimized structure. (C) The initial position of BQ.1-S-RBD and Molnupiravir and BQ.1-S-RPF and Molnupiravir complex and ACE-2 and their optimized structure. (D) The initial position of BQ.1-S-RBD and modified Molnupiravir and BQ.1 S-RBD and modified Molnupiravir complex and ACE-2 and their optimized structure.
Figure 8.
(A) The initial position of Delta-S-RBD and Molnupiravir and Delta-s-RBD and Molnupiravir complex and ACE-2 and their optimized structure. (B) Initial conformations of Delta S-RBD and Omicron BA.2 with three modified Molnupiravir molecules, the complex (optimized) structure of Delta and BA.2 S-RBD with three modified Molnupiravir molecules complex and ACE-2 and their optimized structure. (C) The initial position of BQ.1-S-RBD and Molnupiravir and BQ.1-S-RPF and Molnupiravir complex and ACE-2 and their optimized structure. (D) The initial position of BQ.1-S-RBD and modified Molnupiravir and BQ.1 S-RBD and modified Molnupiravir complex and ACE-2 and their optimized structure.
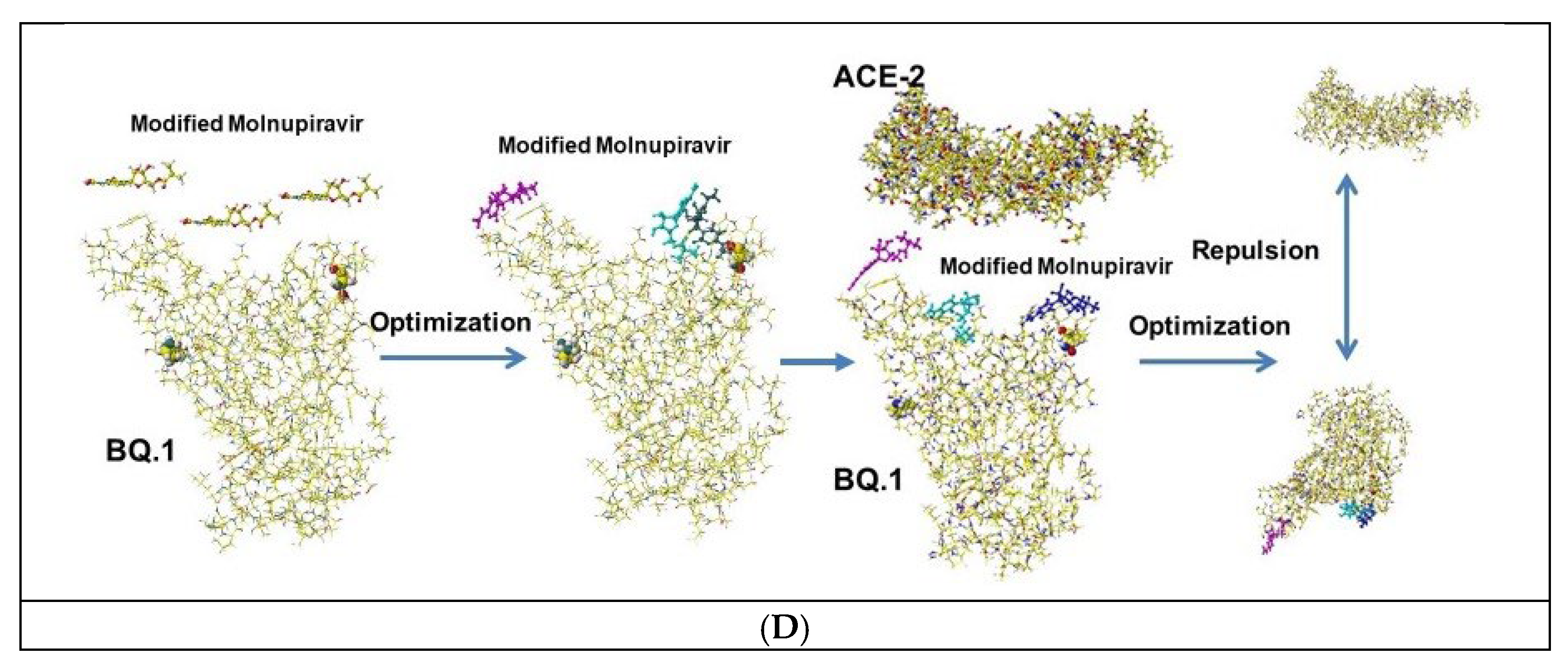
Figure 9.
Binding inhibition of PF07321332, Molnupiravir, and Xocova as malic acid cocktails.

Figure 10.
Effect of cocktail medicines with malic acid.
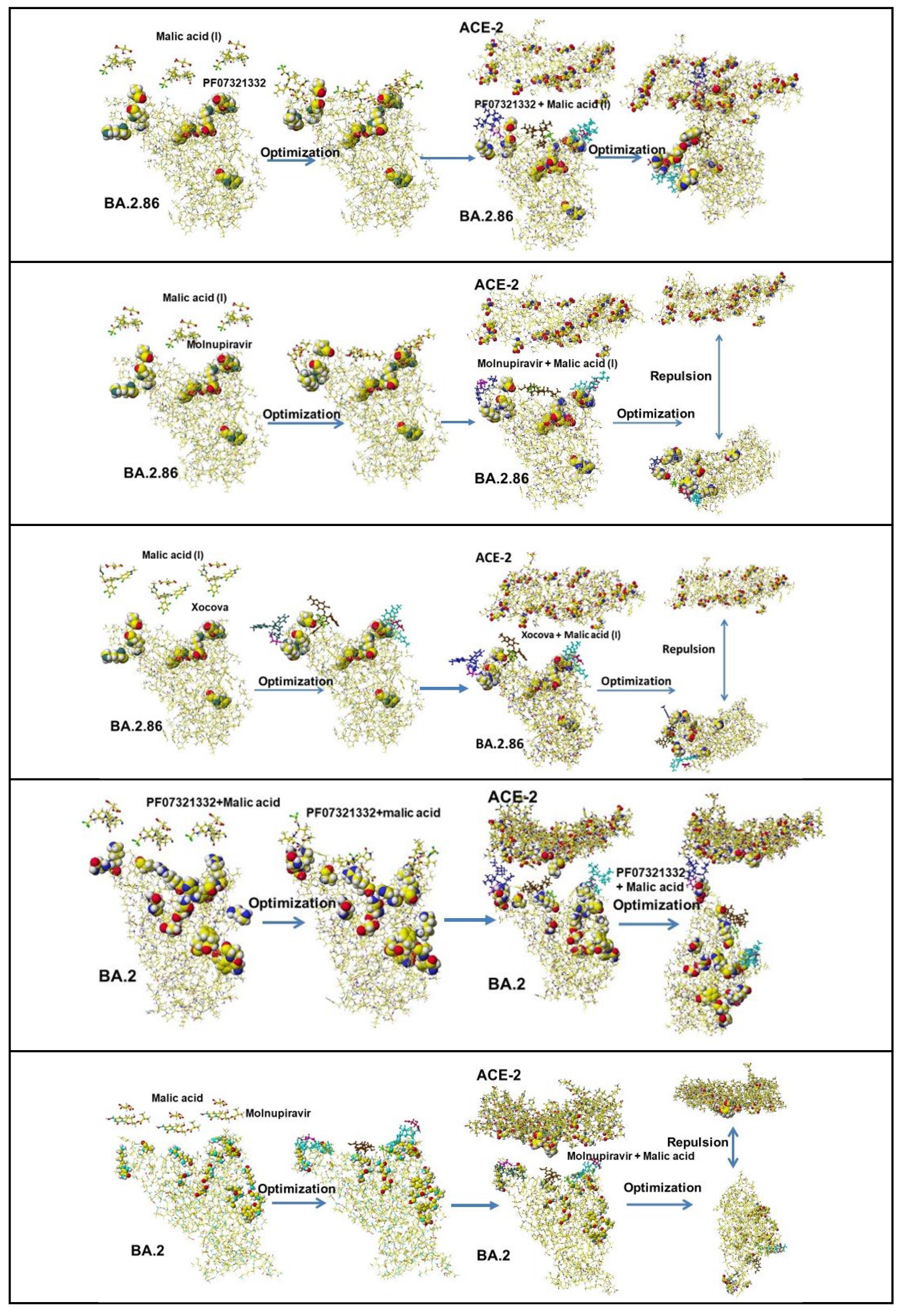
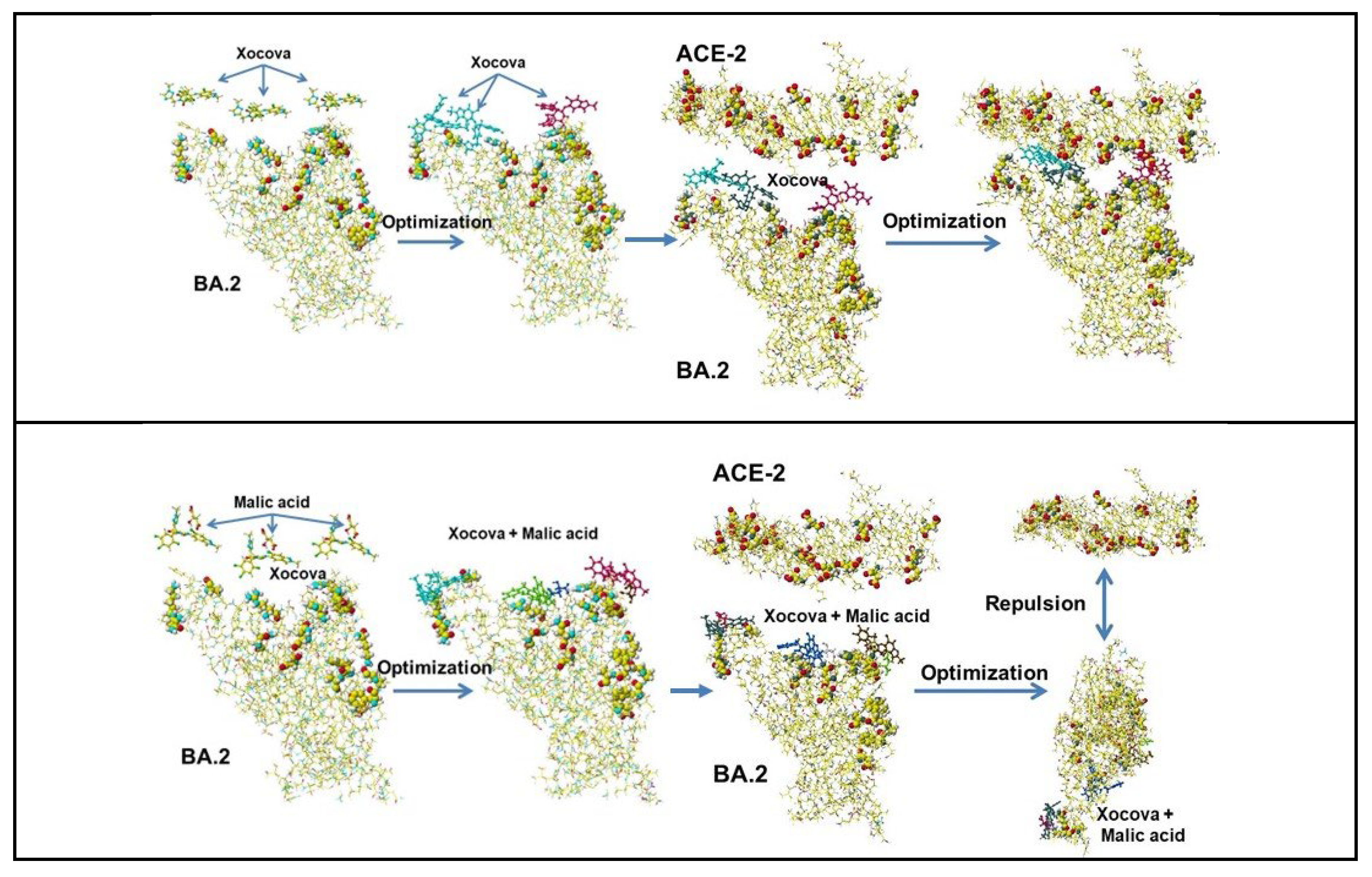
Figure 11.
Structure of Baricithinib, Clofazimine, and Zanamivir.
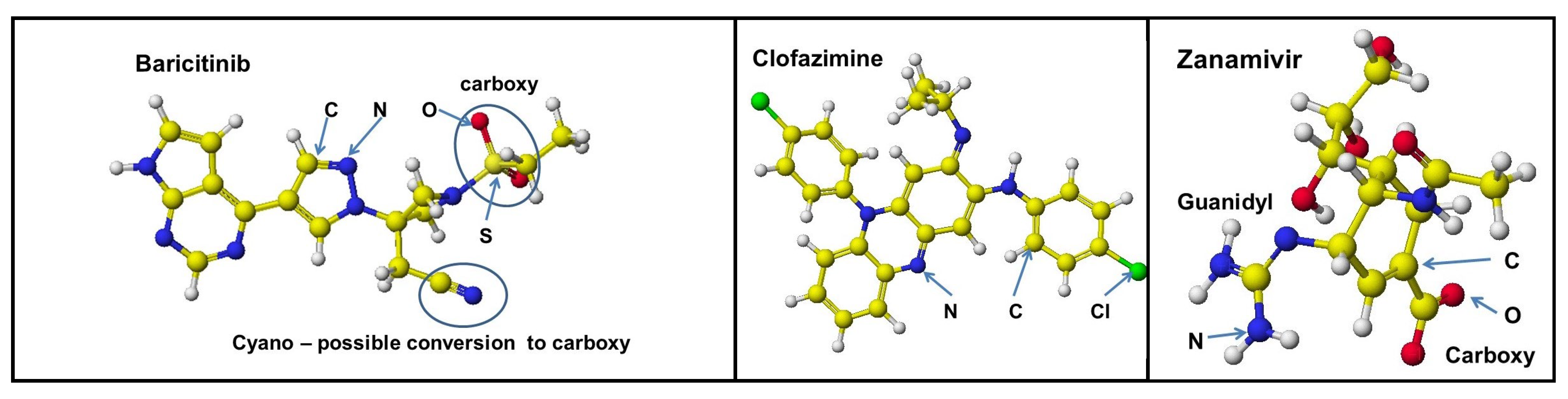
Figure 12.
Binding and repulsion process of Barcitinib.
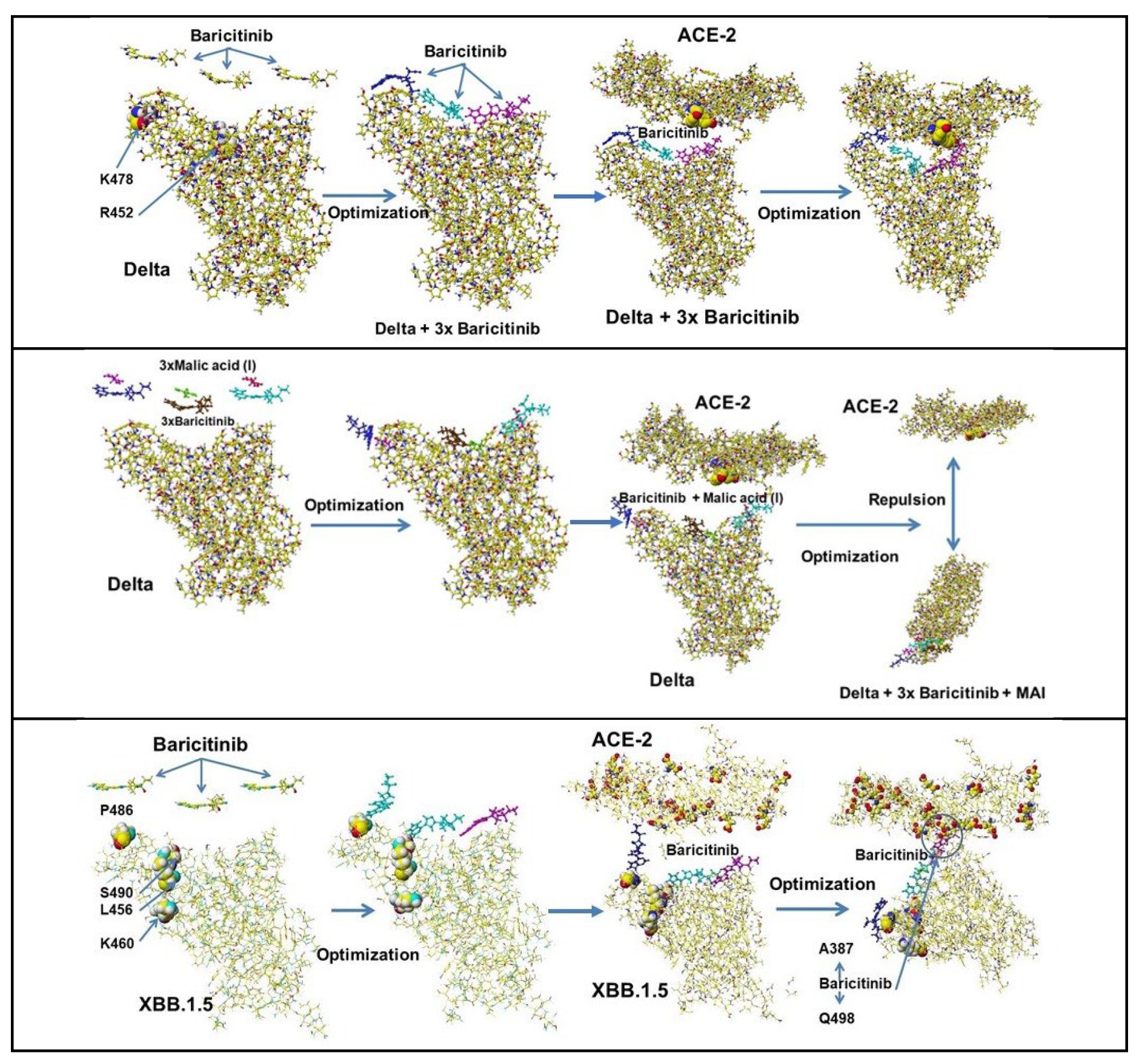

Figure 13.
Binding and repulsion process of Clofazimine.
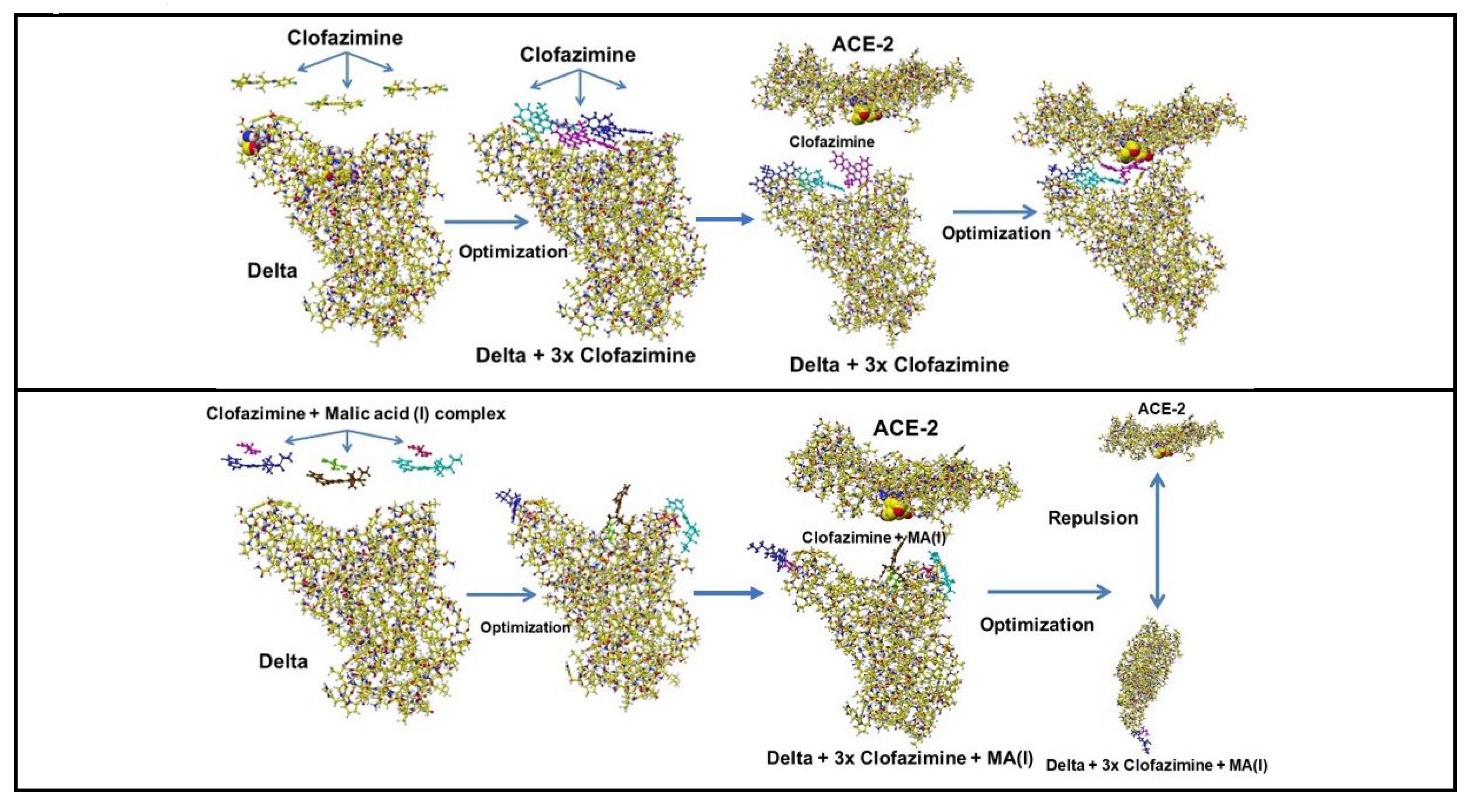
Figure 14.
Binding and repulsion process of Zanamivir.
Figure 15.
Structure of Chloroquine, Dalcetrapib, Deguetin, Dolutegravir, Ebselen, and Etravine, where atoms are indicated as color or grayscale; nitrogen (blue or dark gray balls), oxygen (red or black balls), carbon (yellow or gray balls), hydrogen (small white balls), and fluorine (light green of light gray balls). Generally, fluorone atoms support the sterically stable structure.
Figure 15.
Structure of Chloroquine, Dalcetrapib, Deguetin, Dolutegravir, Ebselen, and Etravine, where atoms are indicated as color or grayscale; nitrogen (blue or dark gray balls), oxygen (red or black balls), carbon (yellow or gray balls), hydrogen (small white balls), and fluorine (light green of light gray balls). Generally, fluorone atoms support the sterically stable structure.
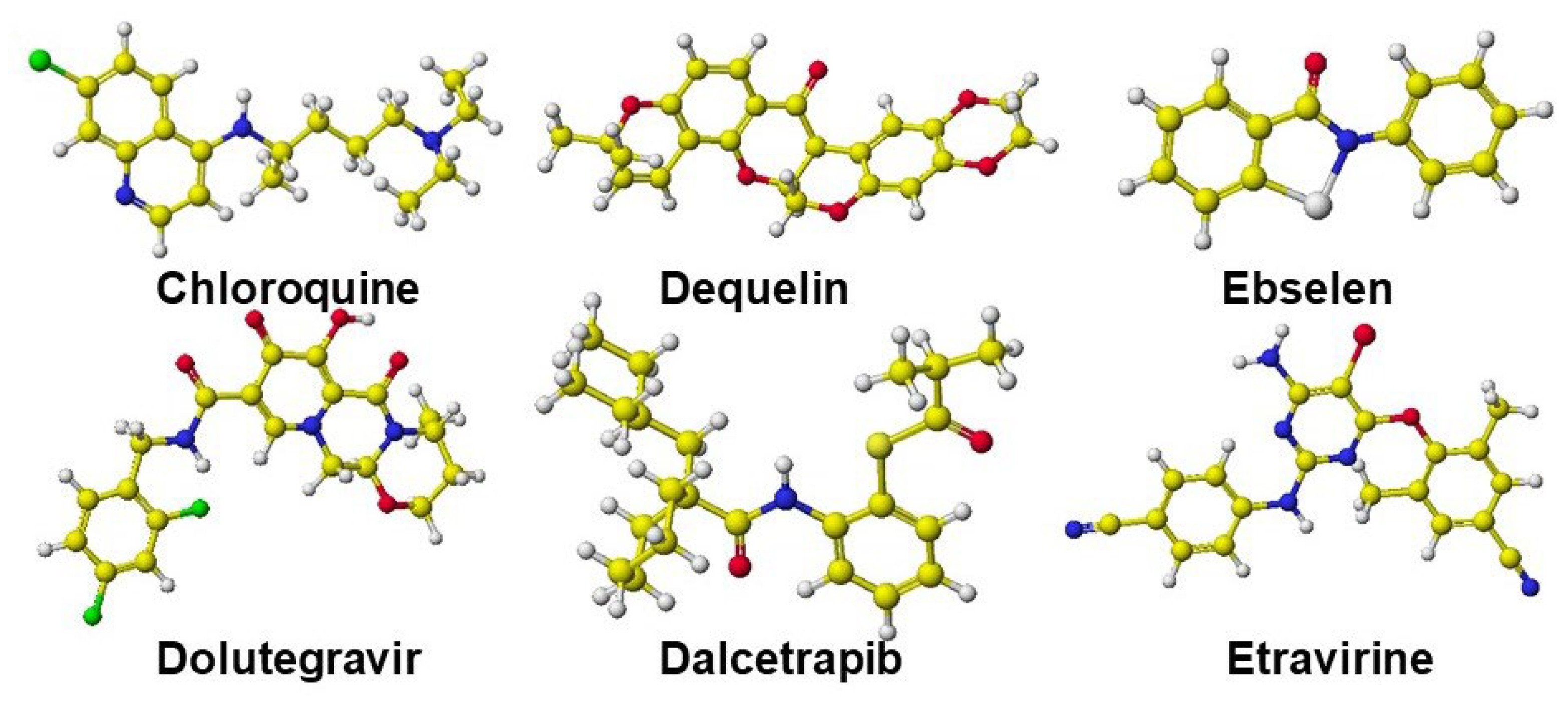
Figure 16.
Optimized bound structures of Chloroquine, Dalcetrapib, Deguetin, Dolutegravir, Ebselen, and Etravine.
Figure 16.
Optimized bound structures of Chloroquine, Dalcetrapib, Deguetin, Dolutegravir, Ebselen, and Etravine.
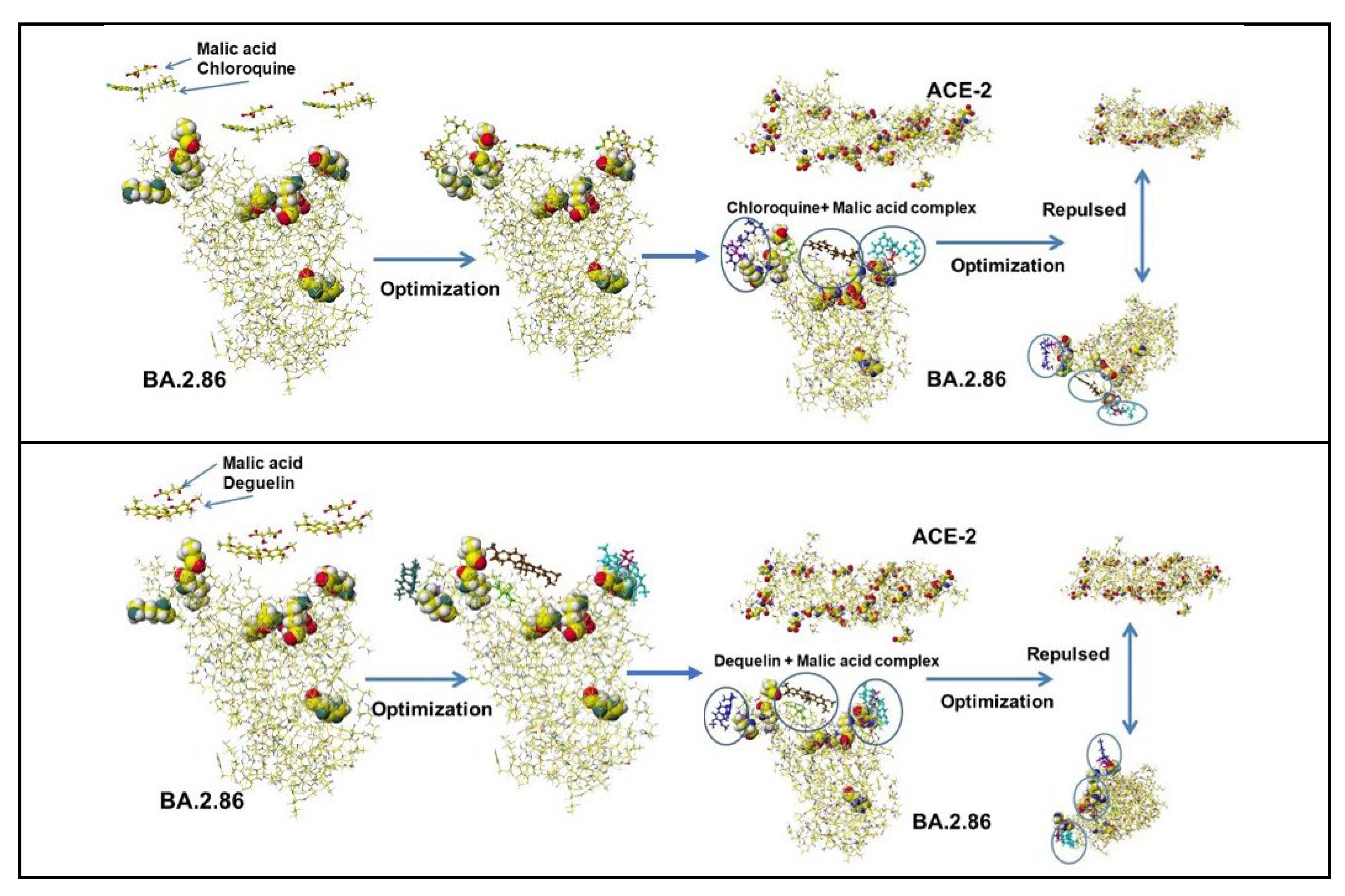
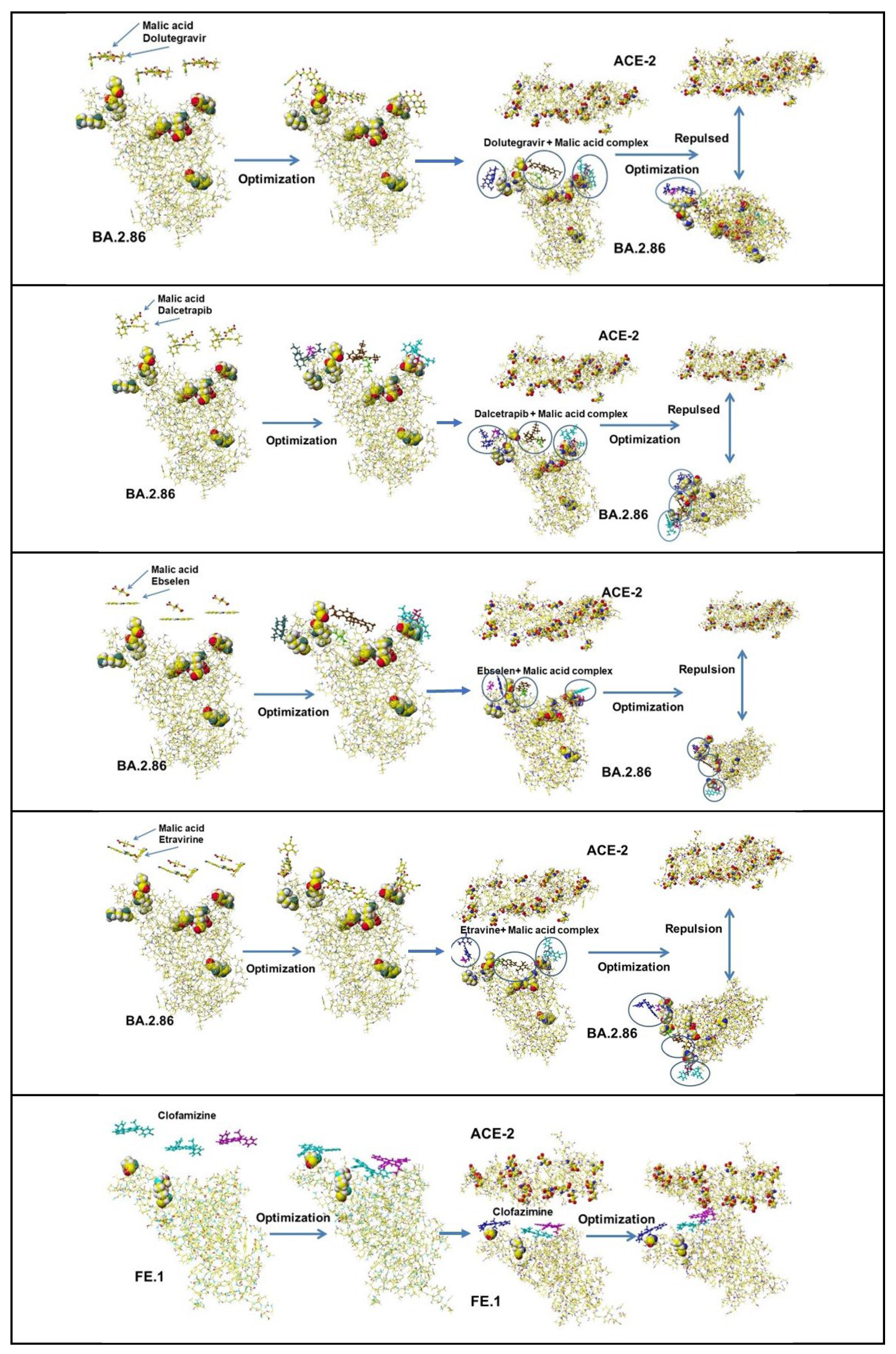
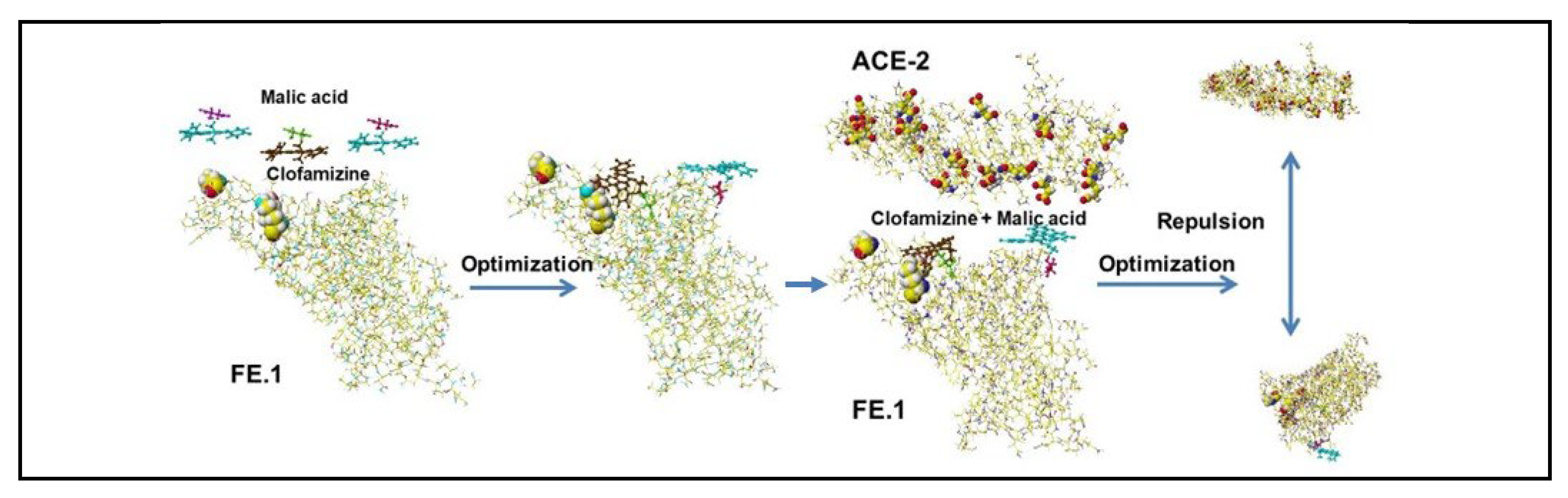
Figure 17.
Structure of WT protein.
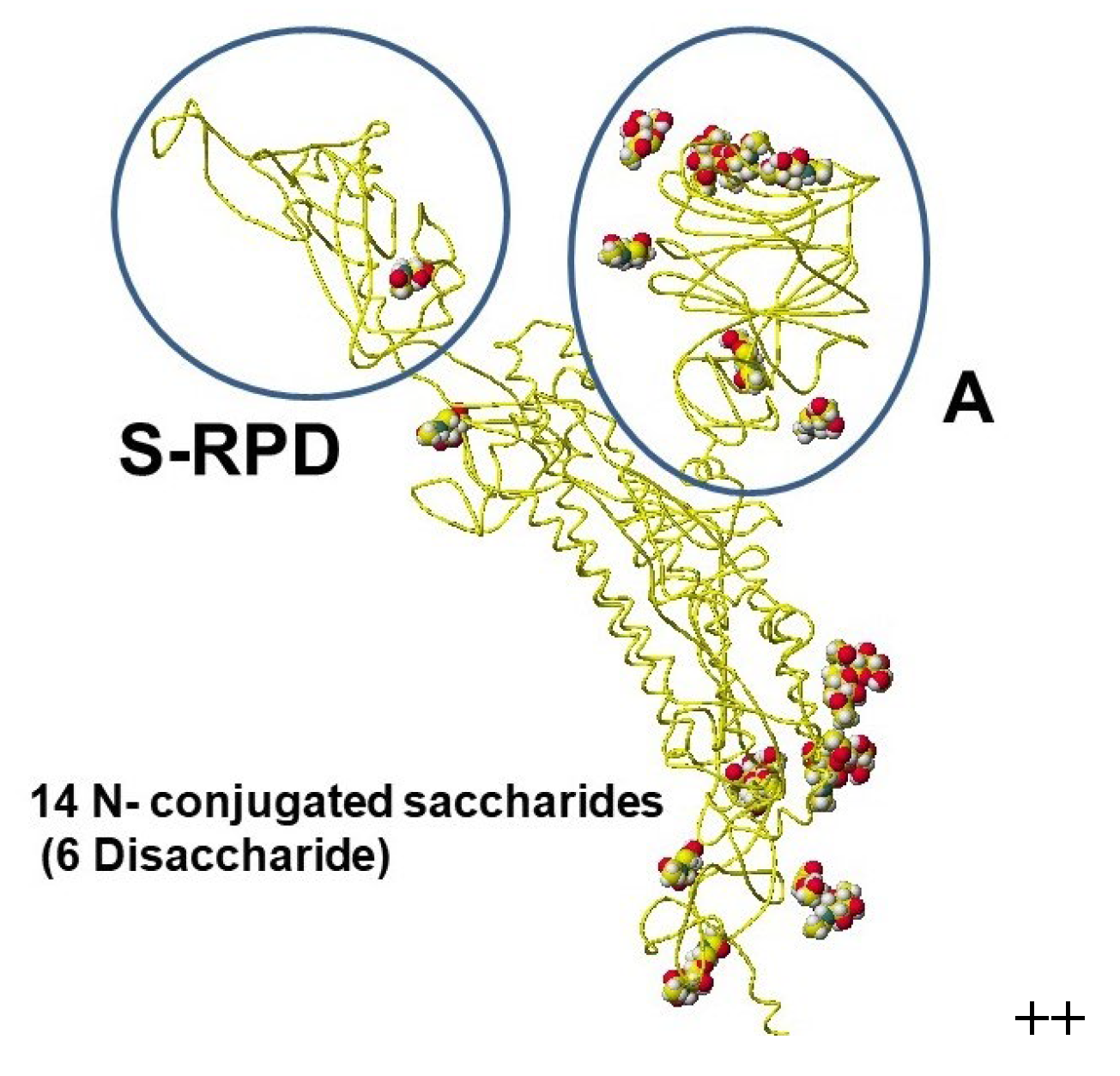
Figure 18.
Structure of conjugated.
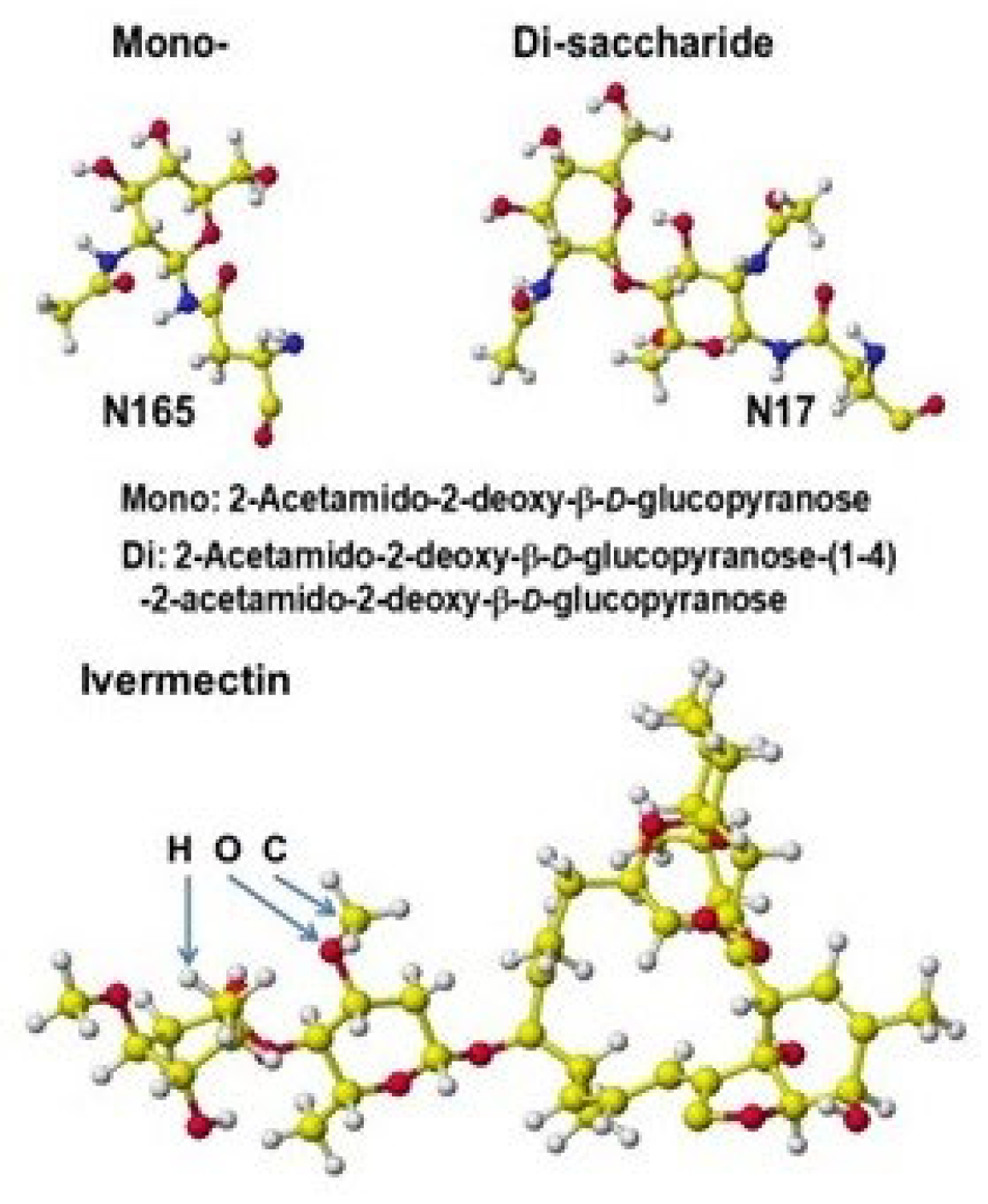
Figure 19.
Ivermectin bound WT.
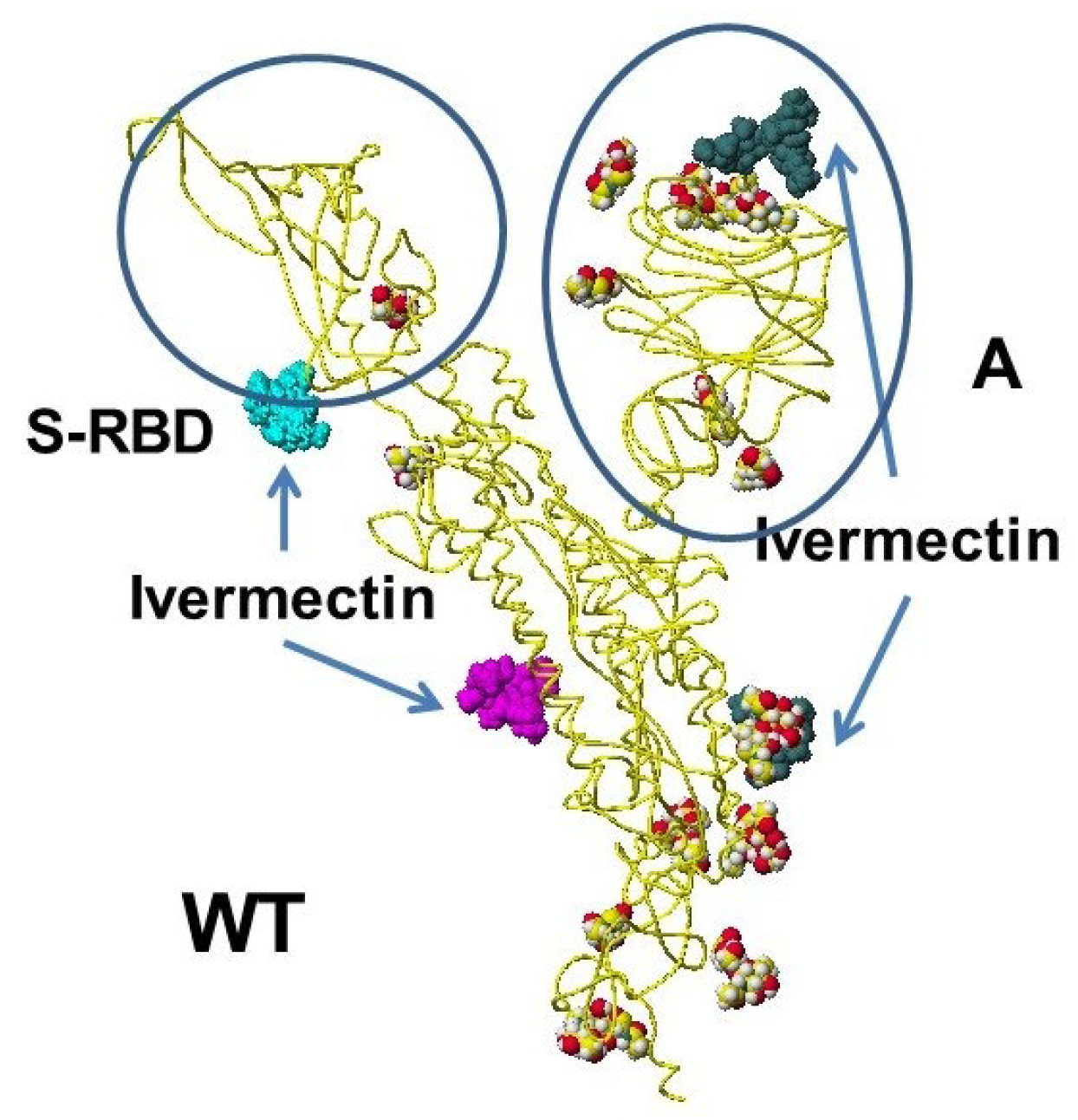
Figure 20.
Practical use of Ivermectin with malic acid as an inhibitor.
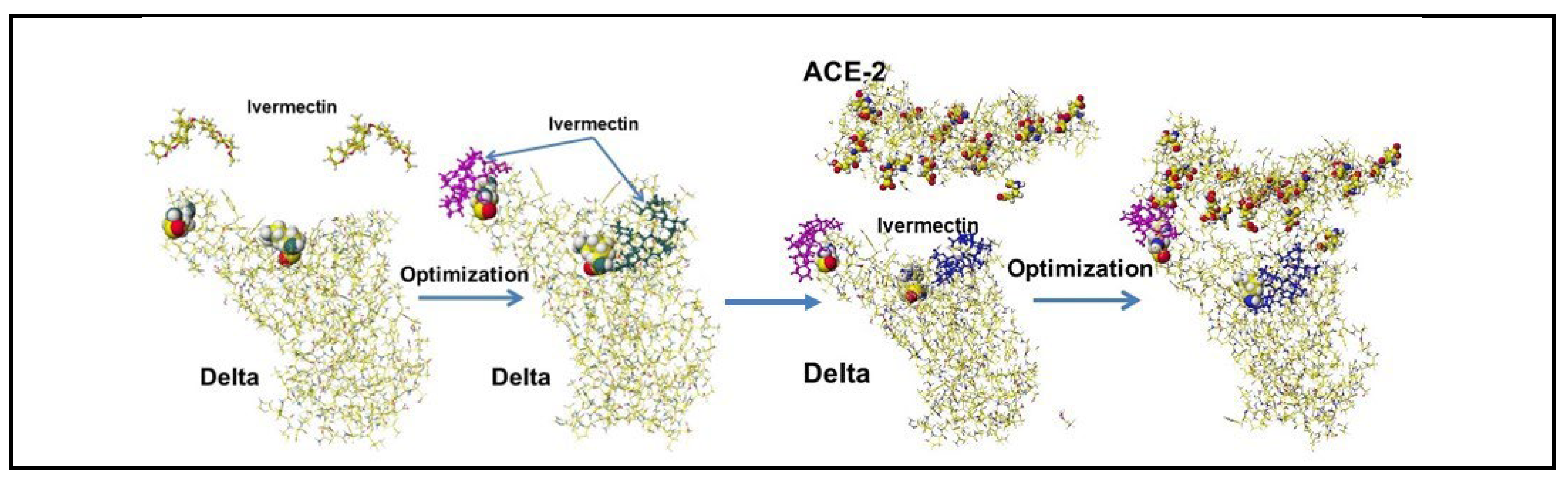
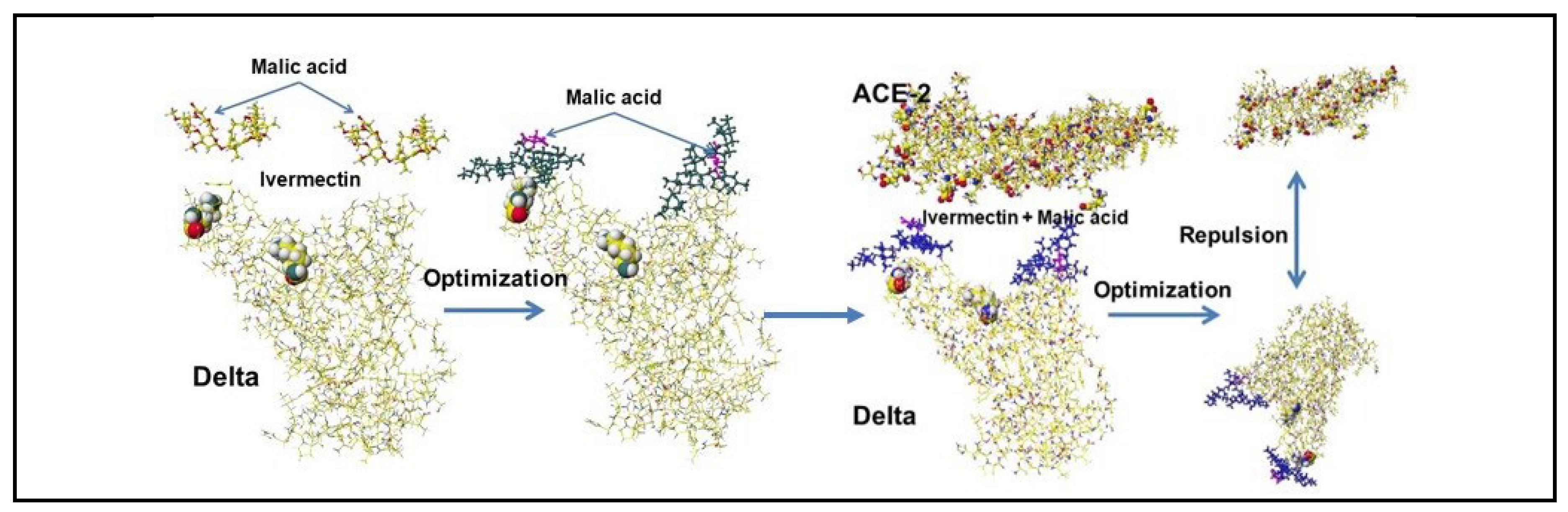
Figure 21.
Optimized structures of cannabinoid acids.
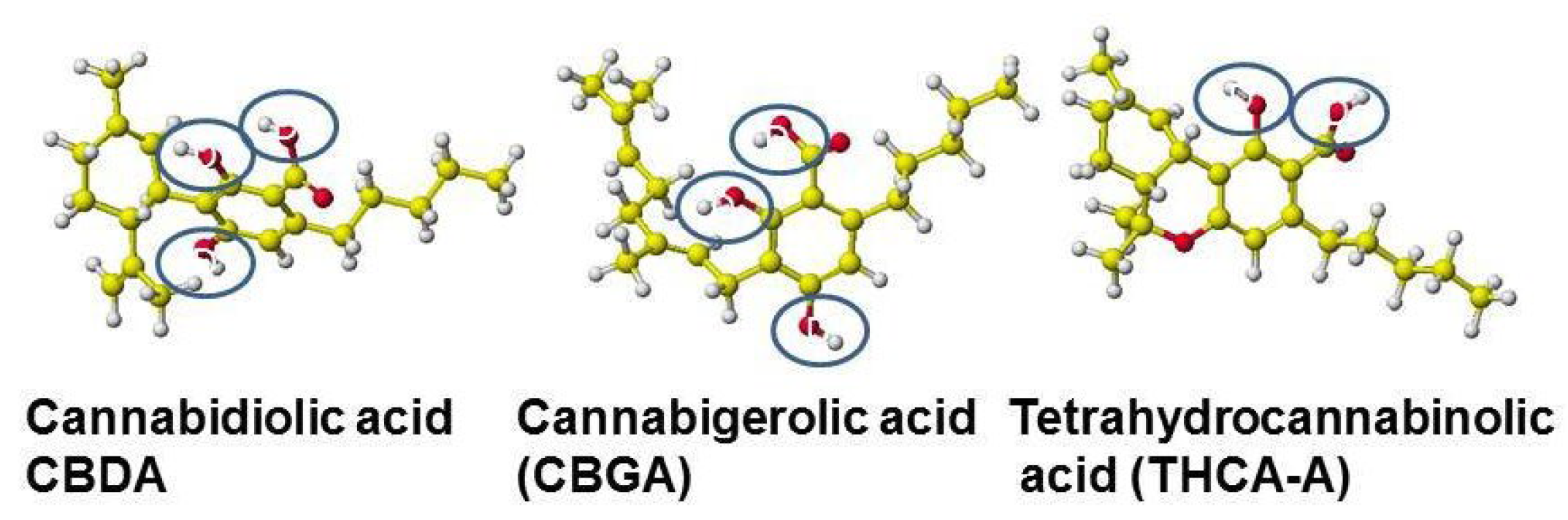
Figure 22.
Optimization process of CBDA, CBGA, RHCA-A.
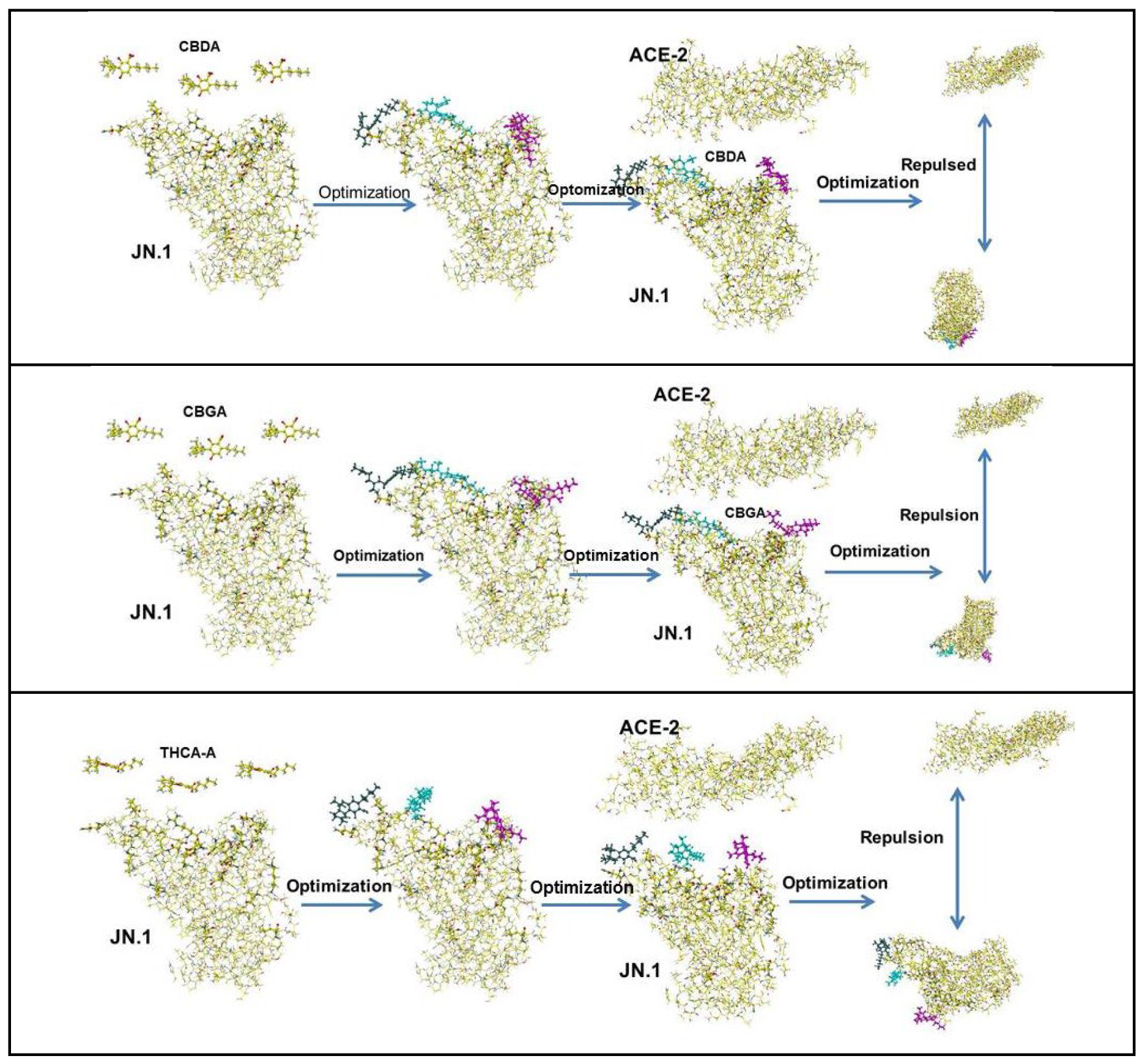
Figure 23.
keto- and enol-form curcumin.
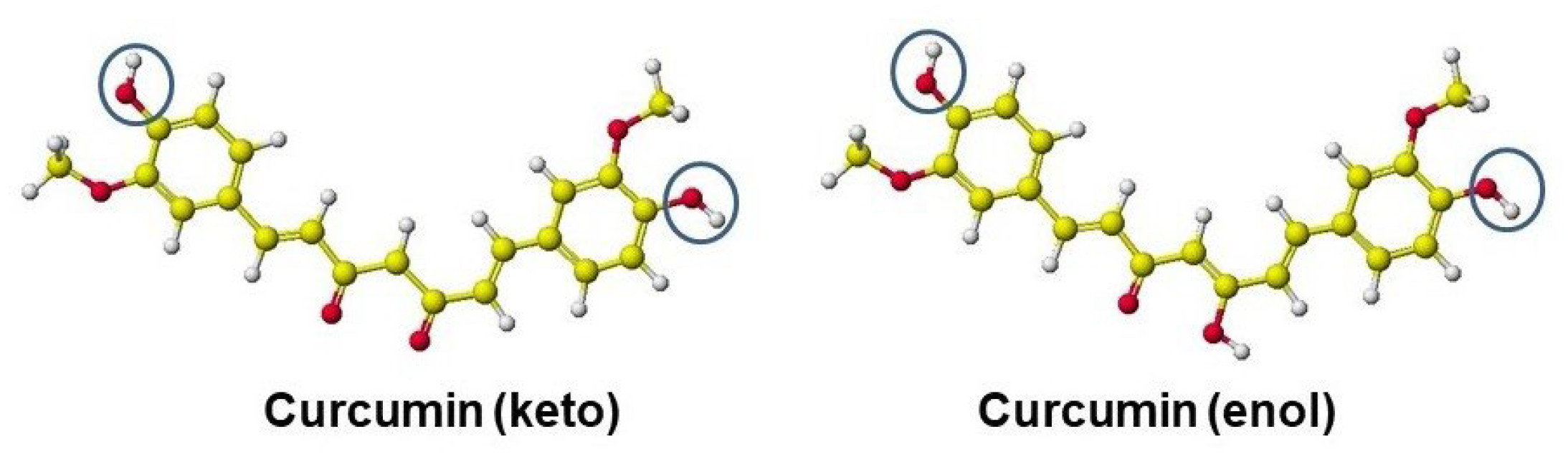
Figure 24.
Analysis of binding inhibitors of keto- and enol-form curcumin.
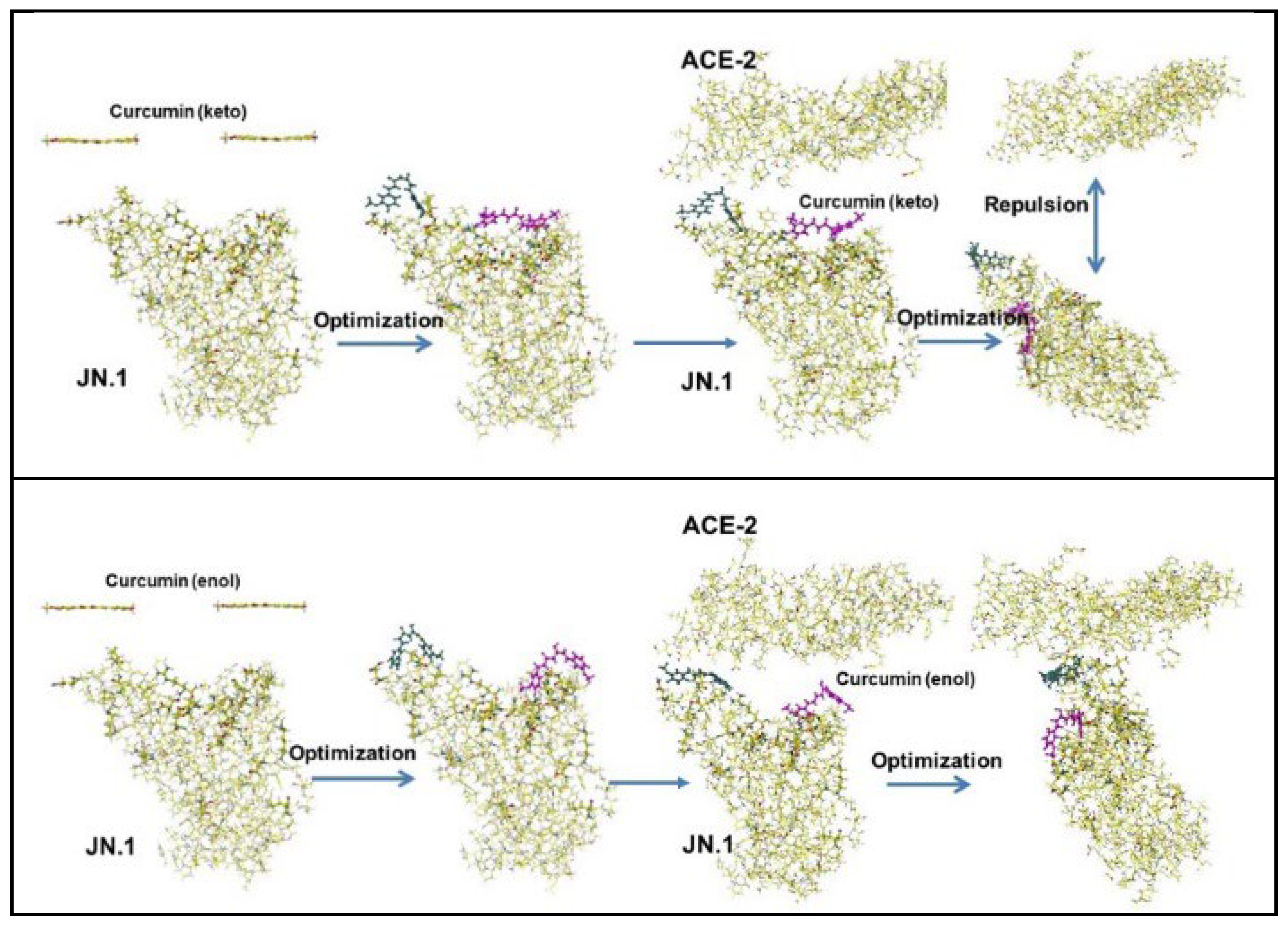
Figure 25.
Model analysis of curcumin inhibition by an acid .
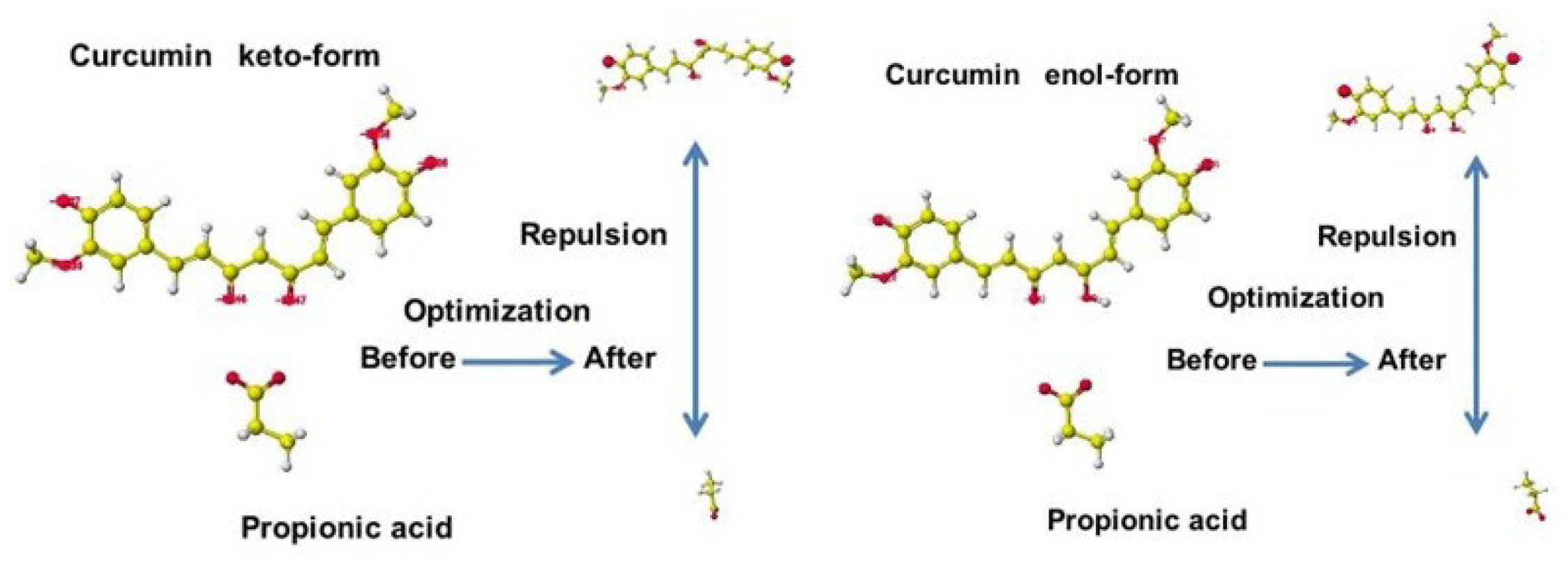
Figure 26.
Optimized structures of keto- and enol-curcumin after binding process.
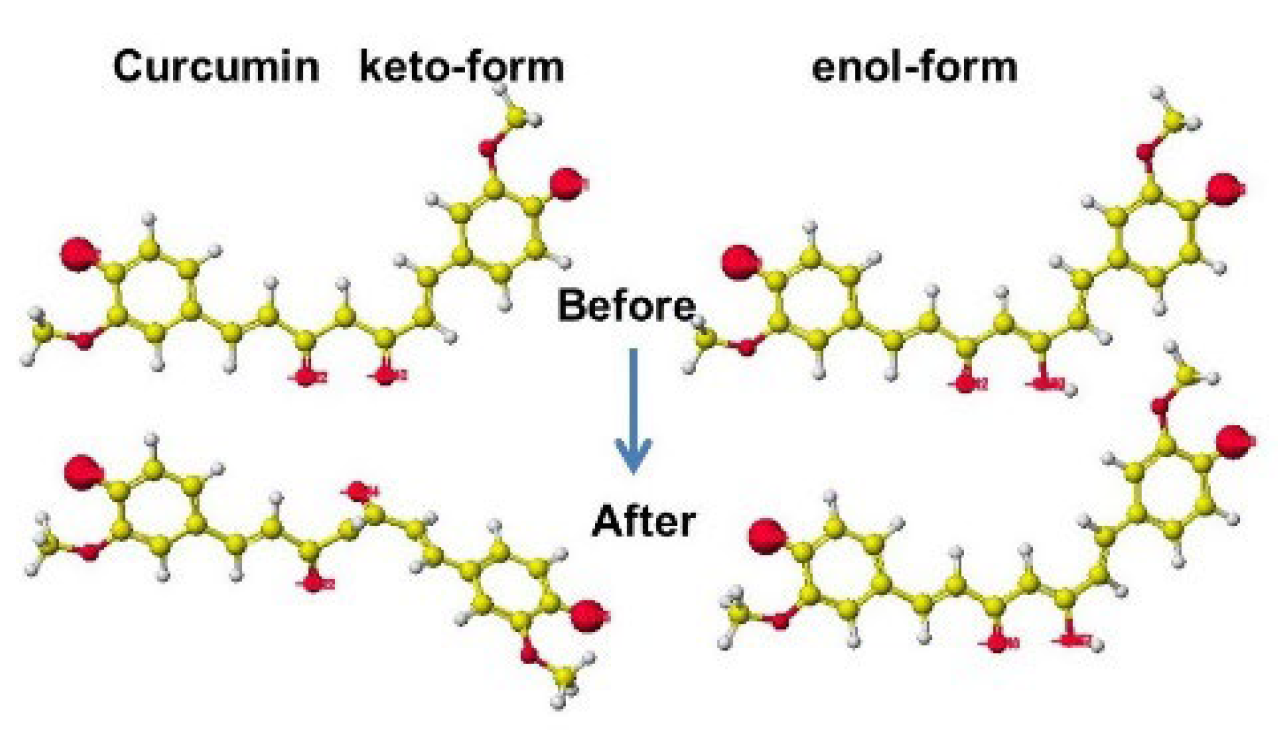
Figure 26.
Polyphenols found in tea leaves and the inhibition effects.
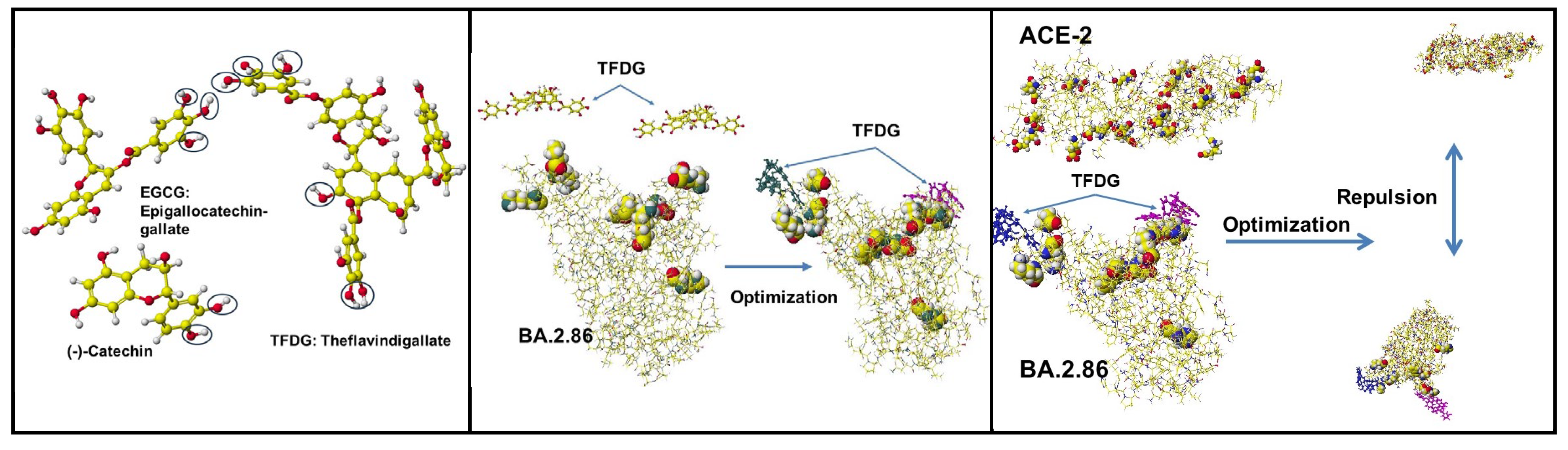
Figure 27.
Chemical structure of Noringenin, Prunin, and Narigin.
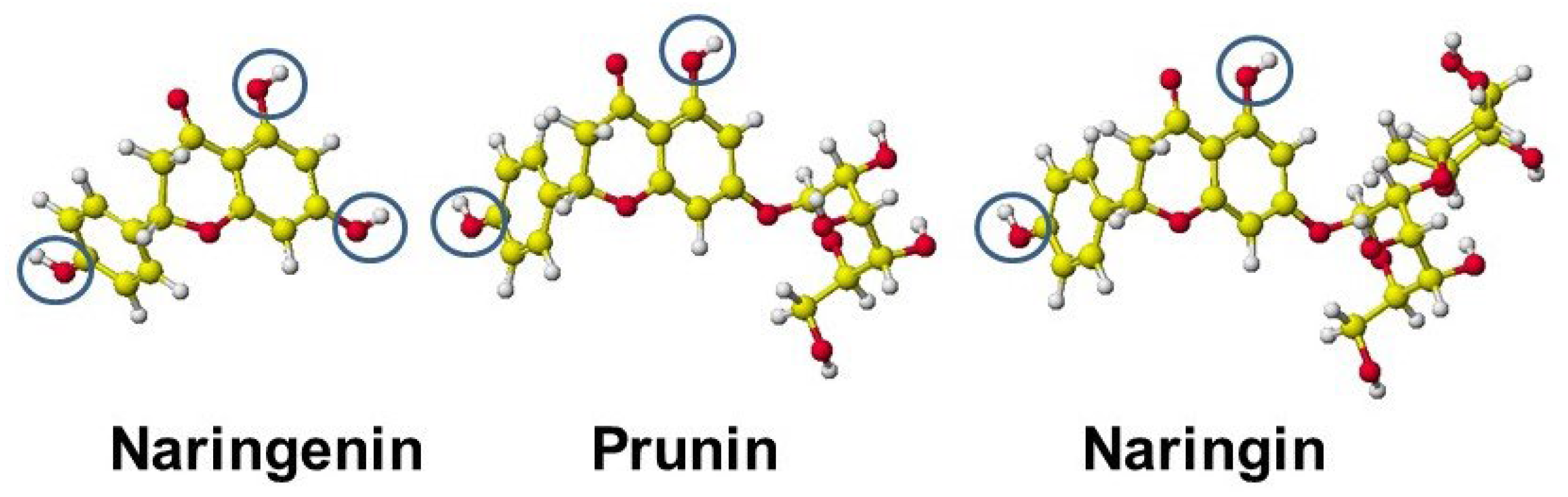
Figure 28.
Inhibition effect of Noringenin, Prunin, and Narigin.
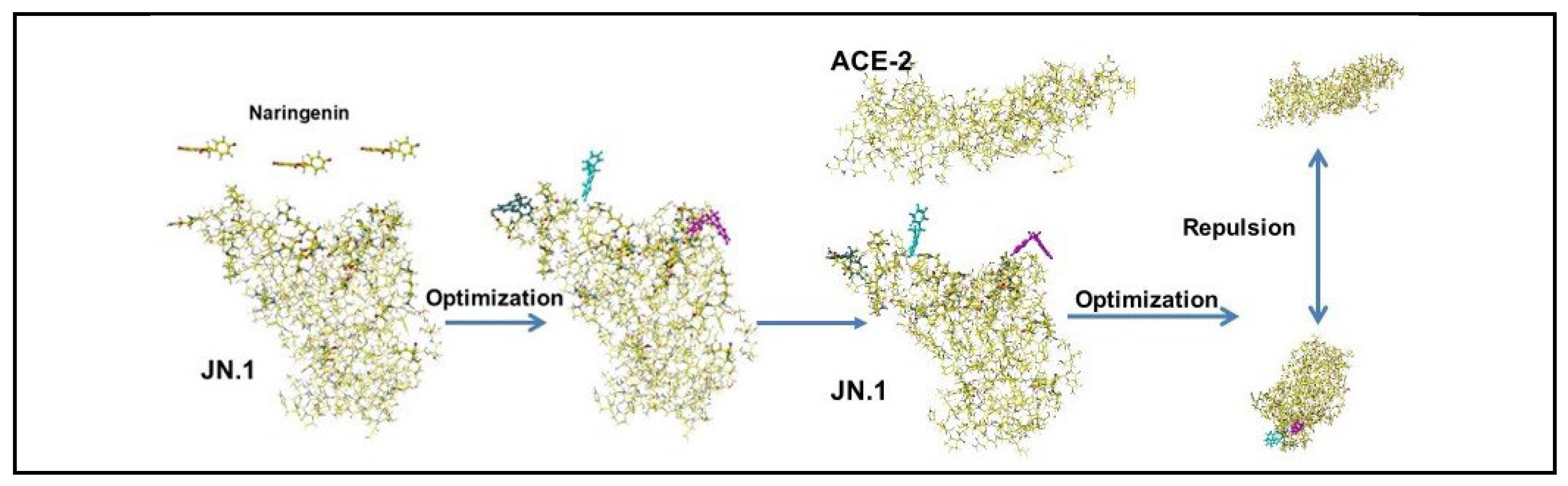
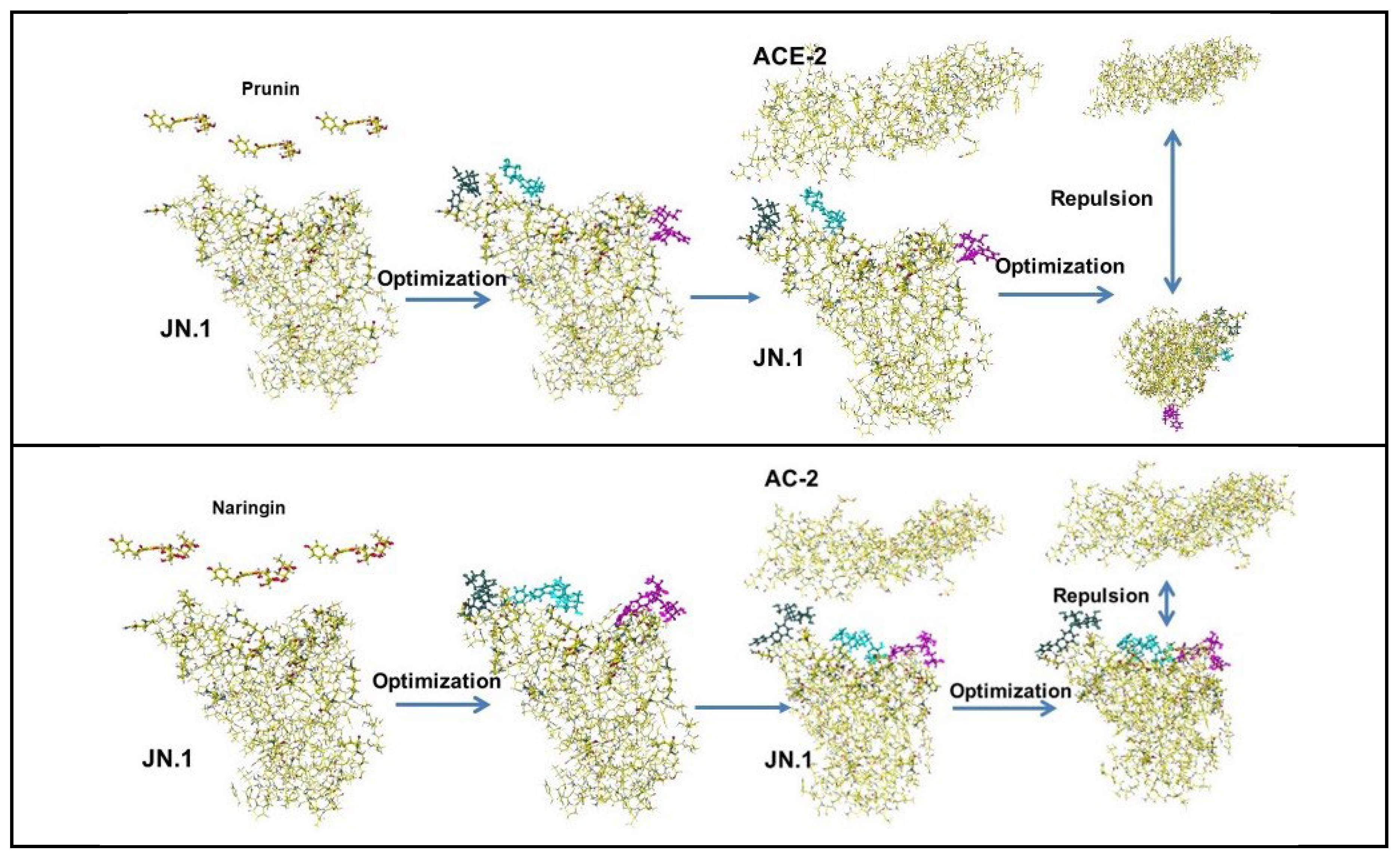
Figure 29.
Structures of modified Naringin: Methyl and carboxy groups are circled.

Figure 30.
Repulsion effect of modified Naringenin glycosides: Naringenin glycosides are shown as color molecules.
Figure 30.
Repulsion effect of modified Naringenin glycosides: Naringenin glycosides are shown as color molecules.
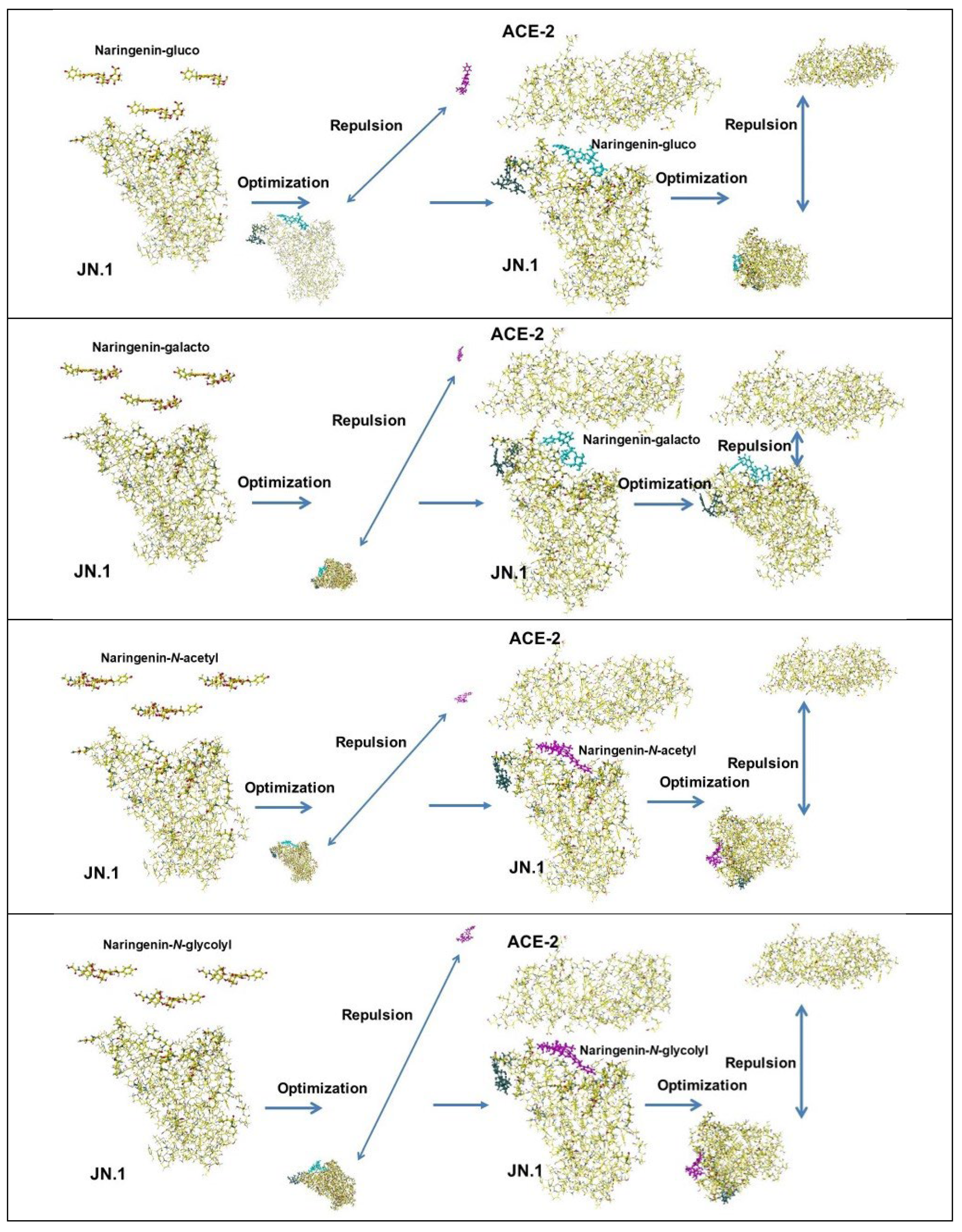
Figure 31.
Binding of modified Naringin glycosides with SARS-CoV-2 protein PDB 7mjk: modified Naringin glycosides are shown as color molecules. Ivermectin and Naringin are references.
Figure 31.
Binding of modified Naringin glycosides with SARS-CoV-2 protein PDB 7mjk: modified Naringin glycosides are shown as color molecules. Ivermectin and Naringin are references.
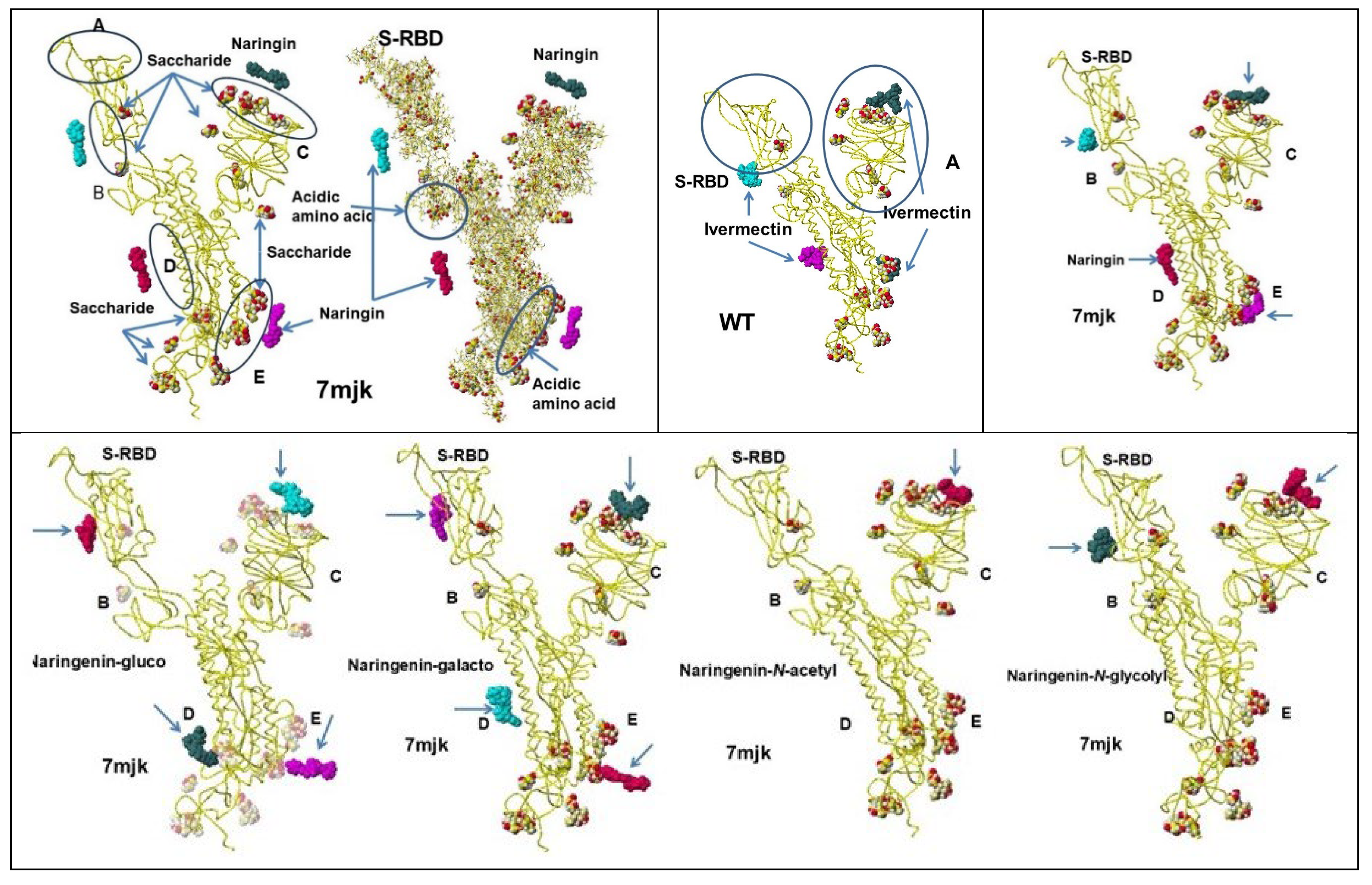

Table 1.
Molecular interaction energy values between two amino acids kcal·mol-1.
| Amino acid | MIFS | MIHB | MIES | MIVW | Amino acid | MIFS | MIHB | MIES | MIVW |
|---|---|---|---|---|---|---|---|---|---|
| Glu + Ala (A) | 10.48 | 0.07 | 11.35 | -0.92 | Glu + Pro (P) | 5.32 | -0.004 | 5.40 | 0.53 |
| Glu + Arg (E) | 84.05 | 25.29 | 64.31 | -3.30 | Glu + Thr (T) | 25.17 | 9.29 | 16.69 | 0.08 |
| Glu + Asn (N) | 33.56 | 18.74 | 16.47 | -2.17 | Glu + Tyr (Y) | 24.75 | 9.85 | 20.04 | -0.26 |
| Glu + Asp*(D) | -5.05 | -0.05 | -5.09 | 0.01 | Glu + Phe (F) | 12.61 | 0.07 | 9.41 | 2.91 |
| Glu + Glu*(E) | -5.73 | 0.02 | -5.75 | -0.01 | Glu + Ser (S) | 22.53 | 9.68 | 14.34 | -0.57 |
| Glu + Gln (Q) | 31.27 | 17.19 | 16.49 | -2.40 | Glu + Val (V) | 13.26 | 0.28 | 13.97 | -0.54 |
| Glu + Gly (G) | 16.41 | -0.11 | 15.50 | -1.06 | |||||
| Glu + His (H) | 21.20 | 4.30 | 17.04 | 0.25 | Tyr + Asn (N) | -27.71 | -14.69 | -13.24 | 5.56 |
| Glu + Leu (L) | 8.16 | 0.11 | 7.15 | 1.06 | Lys + Asn (N) | 43.92 | 27.77 | 20.73 | -4.65 |
| Glu + Lys (K) | 142.03 | 33.80 | 120.21 | -8.67 | Lys + Tyr (Y) | 31.38 | 28.95 | 5.17 | -3.52 . |
MIFS, MIHB, MIES, MIVW: Molecular interaction energy values of final structure (FS), hydrogen bonding (HB), electrostatic interaction (ES), and van der Waals force (VW). *Repulsion, no MI energy. Unit: kcal·mol-1.
Table 2.
Mutated amino acids of SARS-CoV-2 S-RBD mutants.
| WHO name | Lincage plus | Mutated amino acids |
|---|---|---|
| WT | N501 | |
| Alpha | B.1.1.7 | N501Y |
| Alpha plus | B.1.1.7+E484K | N501N, E484K*1 |
| Beta | B-1.351 | N417K, E484K, N-501Y |
| Delta | B.1.517.2 | L452R, T478K |
| Delta plus | AY.1 | K417N, L452R, T478K |
| Epsilon | B.1.427/B.1.429 | L452R |
| Eta | B.1.525 | E484K |
| Gamma | P.1 | K417N, E484K, N501Y |
| Gamma | P.1 | K417N, E484K, N501Y*2 |
| Iota | B.1.526 | E484k, (Eta B.1.525) |
| Iota | B.1.526.1 | L452R |
| Kappa | B.1.617.1 | L452R, E484Q |
| Kappa | B.1.620 | S477N, E484K |
| Lambda | C37 | L452Q, F490S |
| Mu | B.1.621 | R346K, E484K, N501Y |
| Theta | P.3 | E484K, N501Y, (Alpha B.1.7 + E484K) |
| Theta | B.1.616 | V483A |
| Zeta | P.2 | E484K, (Eta B.1.525) |
| Omicron | BA1 | G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, |
| E484A, Q493R, G496S, Q498R, N501Y, Y505H, | ||
| Omicron | BA2 | G339D, S371F, S373P, S375F, T376A, D405N, R408S, K417N, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H, |
| Omicron | B.2.75 | D339H, G446S, N460K, Q493R |
| Omicron | BA.4/5 | L452R, F486V, Q493R |
| Omicron | BQ.1 | K444T, N460K |
| Omicron | BQ.1.1 | R346T, K444T, N460K |
| Omicron | XBB | R346T, V445P, G446S, N460K, F486S, F490S, Q493R |
| Omicron | XBB.1.5 | F456L,N460K, F486P, F490S |
| Omicron | XBB.1.16 | T478R, F486P |
| Omicron | FE.1 | F456L, F486P, F490S |
| Omicron | EG.5 | F456L, N460K, S486P, FS490S |
| Omicron | BA.2.86 | I332V, D339H, R403K, V445H, G446S, N450F, L452W, N481K,483del, E484K, F486P |
| Omicron | BA.2.87.1 | G339D, S371F, S373P, S376F, T376A, D405N, R408S,K417T, N471K, E484A, Q498R, N501Y, Y506H |
| Omicron | HV.1 | R346T, L368I, T376A, D405N, K417N, N440K, V445P, G446S, L452R, V460K, S477N, T478K, E484A, F486P, F490S, Q493R, N501Y, Y505H |
| Omicron | JN.1 | T332V, K356T,T376A, R403K, D405N, K417N, N440K, V445H, G446S, N450D, L452W, L455S, V460K, S477N, T478K, N481K, 483V, E484A, F486P, F490P, Q493R, N501Y, Y505H |
| Omicron | JN.1.17 | T332V, K356T,T376A, R403K, D405N, K417N, N440K, V445H, G446S, N450D, L452W, L455S, V460K, S477N, T478K, N481K, 483V, E484A, F486P, F490P, Q493R, N501Y, Y505H, T572I |
| Omicron | JN.1.18 | T332V, R346T, K356T,T376A, R403K, D405N, K417N, N440K, V445H, G446S, N450D, L452W, L455S, V460K, S477N, T478K, N481K, 483V, E484A, F486P, F490P, Q493R, N501Y, Y505H, |
| Omicron | KP.2 | T332V, K356T, R346T,T376A, R403K, D405N, K417N, N440K, V445H, G446S, N450D, L452W, L455S, F456L, V460K, S477N, T478K, N481K, 483V, E484A, F486P, F490P, Q493R, N501Y, Y505H, T572I |
| Omicron | KP.3 | T332V, K356T,T376A, R403K, D405N, K417N, N440K, V445H, G446S, N450D, L452W, L455S, F456L, V460K, S477N, T478K, N481K, 483V, E484A, F486P, F490P, Q493E, N501Y, Y505H, T572I |
Table 3.
Inhibitor candidates for binding of SARS-CoV-2 with ACE-2.
| Compounds | S-RBD | ACE-2 | C+A* | Compounds | S-RBD | ACE-2 | C+A* |
|---|---|---|---|---|---|---|---|
| Acetaminophen | B | B | - | Furosemide | B | B | - |
| Adrenaline | B | B | - | Gallic acid | B | R | R |
| Ascorbic acid | B | B | - | Gingerol | B | B | - |
| Aspirin I | B | R | PB | Glycyrrhizinic acid | B | R | R |
| Aspirin-O-mannoside M | B | B | - | Hydroxychloroquine | B | B | - |
| Aspirin-O-mannoside I | B | R | - | Ibuprofen I | B | R | R |
| Azithromycin | B | B | - | Ibuprofen M | B | B | B |
| Benzoic acid I | B | R | PB | Isopropylantipyrine | B | B | - |
| b-Blocker | B | B | - | Lactic acid I | B | B | R |
| m-Carboxyl-L-tyrosine | B | R | R | Lactic acid M | B | B | R |
| m-Carboxyl-L-tyrosine | B | R | R | Lopinavir | B | B | - |
| N-mannoside | Malic acid D-mannoside M | B | B | - | |||
| Catechin | B | B | - | Malic acid D-mannoside I | B | R | R |
| Captopril | B | B | - | Mefenamic acid I | B | R | R |
| Clofazimine | B | B | - | Mefloquine | B | B | - |
| Chlorophenyl amine- | B | B | - | Molnupiravir | B | B | B |
| Malonate | N3 | B | B | - | |||
| Citric acid | B | R | R | Nalidixic acid I | B | R | R |
| Citric acid dimethyl | B | R | R | Naproxen I | B | R | R |
| Citric acid trimethyl | B | B | PB | Oseltamivir | B | R | B |
| Citric acid O-mannoside | B | R | R | PF-07321332 | B | B | B |
| Citric acid di-O-mannoside | B | R | R | Probenecid I | B | R | - |
| Citric acid tri-O-mannoside | B | B | - | Remdesivir | B | B | - |
| Ethionamide | B | B | - | Ritonavir | B | B | - |
| Ephedrine | B | B | - | Unifenovir | B | B | - |
| Favipiravir | B | B | - | Unifenovir I | B | B | |
| Ferulic acid I | B | R | R | Xocova | B | B | B |
| Ferulic acid M | B | B | - | Zanamivir | B | R | B |
| Flavonoid | B | B |
*: Delta-SRBD+ACE-2 complex, M: molecular form, I: ionized form, B: bound, R: rejection,. PB: partly bound, -: not calculated because of rejection by ACE-2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



