
Submitted:
28 October 2024
Posted:
29 October 2024
You are already at the latest version
Abstract
Human CYP1-CYP4 families play crucial roles in the biosynthesis, bioactivation, and detoxification of a diverse range of chemicals including xenobiotics, steroid hormones, vitamins, bile acids, procarcinogens, and drugs. The same CYP isoform can be involved in the conversion of different substrates via both bioactivation and detoxification reactions depending on a substrate type. Furthermore, the same CYP isoform can convert the same substrate to different products depending on a tissue type. On the contrary, different CYP isoforms can convert the same substrate to a variety of products, either more toxic or less toxic than their parent compound. The metabolic diversity and dual character of biological effects dictate limited number of cancer types, in which CYPs are implicated. In our review, we highlight the latest advancements and current understanding of molecular mechanisms of the development of hormone-sensitive cancers tumors based on dysregulated CYP expression and mutagenic adduct formation. We discuss the variations in CYP activities including CYP gene polymorphisms, which affect interindividual differences in cancer and drug susceptibilities. We show that there is only limited data indicating the statistically significant association between CYP gene polymorphisms and cancer among different populations. The involvement of CYPs in anticancer drug metabolism associated with drug-drug interactions and drug resistance is also discussed.
Keywords:
cancer
; metabolism
; cytochrome P450
; gene polymorphisms
; drugs
1. Introduction
The superfamily of cytochromes P450 (CYPs) is composed of heme-thiolate-containing monooxygenase enzymes, which are found in all kingdoms of life to be involved in a variety of vital processes [1]. In humans, CYP superfamily contains 57 currently known functionally active genes and 46 pseudogenes, which constitute 18 families and 44 subfamilies. Their nomenclature is based on a number following the abbreviation CYP indicating a gene family (for example CYP1 and CYP2), a capital letter referring to subfamily (for example, CYP3A and CYP11B), and another numeral pointing at an individual gene (for example, CYP3A4 and CYP105D5) [2].
Human CYPs are membrane-bound proteins located either in the mitochondrial inner membrane or in the ER membranes [3]. Mostly, they are expressed in the liver; however, some isoforms can be found extrahepatically, in the lung, prostate, adrenal gland, placenta, and kidney [4]. Hepatic expression and abundance of different CYP isoforms have been shown to change during human developmental stages and life-time [5]. Additionally, the variations in the organ-, sex-, and ethnicity-specific expression of human CYPs have been also observed [6,7,8].
CYPs demonstrate a high degree of functional diversity being involved in huge amounts of metabolic processes including biosynthesis of a variety of biologically active compounds, detoxification of xenobiotics, and bioactivation of procarcinogens into highly reactive chemicals [9,10]. The most importantly, human microsomal CYP1-CYP4 families serve as major players in drug metabolism including 70-80% drugs used in clinical practice [11]. CYP3A4, CYP2C8, CYP2C9, CYP2D6, and CYP1A2 are key contributors to drug metabolism due to their broad spectrum of substrate specificity and high expression levels. Additionally, CYPs are implicated in biosynthesis and oxidation of a variety of steroid hormones including corticosteroids, androgens, and estrogens with the involvement of various types of reactions and intermediates [12,13]. CYP11A1, CYP11B1, CYP11B2, CYP17A1, and CYP19A1 are the most prominent enzymes engaged in steroid biosynthesis [14,15].
Reactions catalyzed by CYP enzymes belong to phase I reactions and can be extremely diverse toward a vast variety of substrates including reactions of hydroxylation, peroxidation, epoxidations, N-, O- and S-dealkylation, and deamination [16]. Products of the phase I reactions can further undergo conjugation, mostly, with glucuronic acid, sulfuric acid, acetic acid, and glutathione (GSH) via the phase II reactions [17]. The conjugation with the above-mentioned substances can occur with the involvement of UDP-glucanosyltransferases (UGTs), sulfotransferases (SULTs), N-acetyltransferases (NATs), and glutathione S-transferases (GSTs), respectively [18]. The both phase I and II reactions are parts of the biotransformation process, which yields the polar compounds possessing the increased water solubility, yet often preserving their activity. Such a biotransformation serves to facilitate the excretion of waste and toxic compounds from an organism by kidney and intestine.
CYP enzymes involved in detoxification of xenobiotics, and bioactivation of drugs demonstrate extensive genetic diversity, which contributes to survival in the conditions of changing environment and dietary restrictions [19]. Several hundreds of variant alleles have been identified for the genes encoding detoxifying and drug-metabolizing CYPs in humans [20,21]. However, substantial interindividual variations in CYP gene expression can exert only a limited or almost no effect on metabolic activities of CYP enzymes. Indeed, Gao et al. have found 26 gene polymorphism sites in human liver microsomes, only half of them including those for CYP2A6, CYP2B6, CYP2C9, CYP2D6 and CYP3A4/5 influence to some extent the enzyme catalytic activities [22]. Liu et al. also have identified that there was greater interindividual variation in CYP mRNA expression than in the enzyme activities, except for CYP2C19 [23]. This phenomenon can be ethnicity-dependent; for example, as shown in the latter study, Hispanics had higher CYP2C8 activity and higher CYP2B6, CYP2C9, and CYP2C19 mRNA expression as compared to Caucasians, whereas African Americans had lower CYP2D6 mRNA expression. Such variations in CYP gene expression underlie differences in the enzyme activities, thereby affecting the disease and drug susceptibilities [24].
Cancer is a complex, heterogeneous, and multifactorial disease and a major cause of death and decreasing life expectancy worldwide. According to the International Agency for Research on Cancer, nearly 19.3 million new cancer cases and approximately 10 million cancer-related deaths have been accounted in 2020 [25]. The burden of cancer incidence and mortality is rapidly growing worldwide, reflecting both aging and growth of the population as well as changes in the prevalence and distribution of the main risk factors for cancer [26]. The majority of new cases and cancer-related mortalities in both sexes have been attributed to following cancer types: breast cancer (11.7 and 6.9%, respectively), lung cancer (11.4 and 18%, respectively), prostate cancer (7.3 and 3.8%, respectively), colorectal cancer (10.0 and 9.4%, respectively), and liver cancer (4.7 and 8.3%, respectively) [27].
Cancer cells are strongly dependent on their microenvironment and are regulated by a variety of endogenous and exogenous stimuli [28,29,30]. Both reprogrammed and oxidative metabolism are maintained by cancer cells through metabolic plasticity [31,32]. A variety of metabolites have been implicated in cancer initiation and progression due to the capability to interact with macromolecules including DNA, proteins, and lipids [33,34,35]. This yields in the formation of toxic adducts, which cause dysfunction in cell signaling pathways, impairment in cell functioning and cell death.
The capabilities of CYP enzymes to cause procarcinogen and xenobiotic activation, thereby causing production of toxic adducts provide their implication in cancer initiation, progression, and metastasis [36]. Due to the ability to activate prodrugs, they are involved in chemoprevention whereas due to the involvement in anticancer drug metabolism, CYPs can serve as targets in anticancer therapy [37]. Furthermore, various types of CYP gene polymorphisms such as single nucleotide polymorphisms (SNPs), premature stop codon, variable number tandem repeats (VNTRs), gene deletions, and copy number variations (CNVs) have been implicated in differences in individual’s enzyme activity and anticancer drug response [38].
In our review, we discuss the latest advancements in elucidating the roles of human CYP metabolic activities and gene polymorphisms in cancer initiation, progression, and metastasis. CYPs are implicated, mostly, in hormone-dependent cancers and liver injury and hepatocarcinogenesis. Therefore, we discuss the roles of human CYP1-CYP4 families in steroid hormone biosynthesis, xenobiotic detoxification, procarcinogen activation, and anti-cancer drug metabolism. In this context, CYPs exert both detoxifying and bioactivation capabilities, which reflect diversity of human CYP metabolic activities. Such metabolic diversity and dual character of biological effects dictate limited number of cancer types, in which CYPs are implicated. We highlighted that only limited CYP gene polymorphisms have been implicated in various cancer types.
2. Metabolic Activities of Human CYPs
2.1. Biosynthesis of Steroid Hormones
The first studies on monooxygenase activity of bovine adrenocortical microsomes via C21 hydroxylation of 17α-hydroxyprogesterone in the pathway of steroid biosynthesis from cholesterol was performed by Cooper et al. at the beginning of 1960s [39,40]. Later, between the mid of 1970s and early 1980s, the presence of 17α-hydroxylase and 17,20-lyase activities of cytochrome P450 from rat testicular microsomes was reported [41,42]. Afterwards, the involvement of a variety redox partners in CYP-catalyzed reactions has been elucidated. In particular, Nakajin and Hall have shown that CYP-catalyzed reactions require NADPH and a flavoprotein P-450 reductase for glucocorticoid and androgen biosynthesis in neonatal pig testis [43].
The side chain of cholesterol, a first steroid in the pathway of biosynthesis of all steroid hormones, is initially oxidized and cleaved by CYP11A1 (P450scc), thereby yielding the C21-steroid, pregnenolone (Figure 1) [44]. This reaction comprises three sequential steps: hydroxylation of cholesterol at C22 followed by the product hydroxylation at C20, and a final cleavage of the C20-C22 bond. The reaction requires the involvement of steroidogenic acute regulatory (StAR) protein, which promotes transfer of cholesterol into the inner mitochondrial membrane [45]. Additionally, CYP11A1-catalyzed monooxygenase reactions involve the iron-sulfur [2Fe-2S] cluster protein, ferredoxin (Fdx), and ferredoxin NADP(+)-reductase (FdR) flavoprotein as the CYP redox partners [46].
The central roles of CYP17A1 in further conversions of pregnenolone in mammals have been well-established. Pregnenolone either can be hydroxylated at C17 to produce 17α-hydroxypregnenolone (17α-OHP5) by mitochondrial CYP17A1 or can undergo oxidation at its C3-hydroxyl group to keto-group [47]. The latter reaction is catalyzed by microsomal 3β-hydroxysteroid dehydrogenase 2 (3βHSD2) to be accompanied by a switch of a double bond from B ring to A ring to yield progesterone in the ∆4 pathways. The 17α-OHP5 is further converted to dehydroepiandrostenedione (DHEA) due to the C17-C20 side chain cleavage. To catalyze this reaction, CYP17A1 interacts with its redox partners, cytochrome P450 oxidoreductase (POR) and cytochrome b5 (cyt b5), for electron transportation [48]. The both intermediates of the ∆5 pathways of androgen and estrogen biosynthesis serve as drug targets for treatment of hormone-sensitive cancers such as prostate cancer (PCa) and breast cancer (BC) [49].
Androstenedione (∆4-dione), a precursor of both estrogens and androgens, is produced from DHEA due to the oxidation of hydroxyl group to keto-group at C3-position associated with a switch of a double bond from B to A ring by 3βHSD2 [50]. Another way of androstenedione production is from 17α-hydroxyprogesterone (17α-OHP), which is in turn produced by hydroxylation of progesterone with the involvement of CYP17A1 and POR as a redox partner in the ∆4 pathways [51]. Further, aldo-keto reductase 1C3 (AKR1C3) reversibly reduces keto-group at C17 in androstenedione to hydroxyl group to yield testosterone [52]. CYP19A1 is also known as aromatase and an enzyme playing critical roles in sex steroid hormone biosynthesis. It is expressed in the granulosa cells and lutea corpora of the ovary, Leydig and Sertoli cells of the testis, and other non-gonadal tissues such as placenta, brain, liver, vascular smooth muscle [53]. CYP19A also requires POR as a redox partner to produce estrone from androstenedione, estriol from 16-hydroxytestosterone, and 17β-estradiol from testosterone [46].
Other steroid hormones are glucocorticoids synthesized in zona fasciculata of adrenal gland with the involvement of CYP11B1 (11β-hydroxylase) and mineralocorticoids produced in the outer zona glomerulosa, which expresses CYP11B2 (aldosterone synthase) [54.]. CYP11B1 hydroxylates the 17α-OHP at C11 to produce cortisol, which is further converted to cortisone via oxidation of 11-hydroxyl group to keto-group by 11βHSD1. CYP11B1 requires Fdx and FdR for glucocorticoid biosynthesis [55]. CYP11B2 involves the soluble [2Fe-2S] cluster protein adrenodoxin (Adx), which belongs to ferredoxins, and the mitochondrial membrane-associated NADPH-dependent flavoprotein adrenodoxin reductase (AdR) [56,57].
2.2. Detoxification vs Bioactivation of Xenobiotics
Mammalian CYP1-CYP4 families are known to be involved in both detoxification and bioactivation of procarcinogens, drugs, and xenobiotics [58]. Rendic and Guengerich have found that these CYPs dominate over the other xenobiotic-metabolizing enzymes such as aldo-keto reductases (AKRs), flavin-containing monooxygenases (FMOs), and the monoamine oxidase (MAO) [59]. The authors have assessed reactions of metabolism of all chemicals, which they divide into drugs, physiological compounds (hormones, vitamins, and bile acids), and general chemicals (environmental and industrial pollutants). This has allowed identifying that 20% of reactions of xenobiotic metabolism can be attributed to the activities of CYP3A4 and 10% - CYP1A2 and CYP2D6 each, whereas 9% can be assigned to CYP2C9, 8% - CYP2C8, 7% - CYP1A1, and 5% - to CYP2B6 and CYP2E1 each. They also found that FMOs, MAO and AKRs are involved collectively in only 5% of detoxification reactions.
CYP-metabolized general chemicals comprise hundreds of compounds; among them are alkyl dimethyl benzyl ammonium chlorides known also as benzalkonium chlorides (BACs) [60]. The BACs belong to common quaternary ammonium compounds (QACs), which exert cytotoxic effects and are hazardous to human health [61]. Recombinant CYP2D6, CYP4F2, and CYP4F12 have been shown to metabolize substantial part of BACs in human liver microsomes [62]. For example, the conversions of C10 BAC occur via the ω-oxidation reactions and their major products including ω-hydroxy-, (ω−1)-hydroxy-, (ω, ω−1)-diol-, (ω−1)-ketone-, and ω-carboxylic acid metabolites (Figure 2A) possess cytotoxicity [63].
Other groups of general chemicals comprise organophosphates and carbamates, which are used as pesticides and cause acute toxicity resulted from inhibition of serine esterase enzymes including acetylcholinesterase (AChE) [64]. The AChE inhibition causes excessive nicotinic and muscarinic stimulation in the central and autonomic nervous systems and the neuromuscular junction. Chemically, organophosphates are modified esters of phosphoric acid containing P=O phosphoryl group. They can include less toxic phosphorothioates containing P=S group and phosphorodiamidates containing P-N group, which are oxidatively activated via production of corresponding oxons with P=O group [65]. Indeed, parathion, chlorpyrifos, malathion, and disulfoton are known to become more toxic than their parent compounds via the CYP-catalyzed metabolic activation (Figure 2B). For example, parathion is metabolized to diethyl 4-nitrophenyl phosphate (paraoxon), which is a highly poisonous nerve agent and a potent AChE inhibitor [66]. However, a potency of menadione (methyl-1,4-naphthoquinone, vitamin K3) to inhibit the conversion of parathion to paraoxon by recombinant CYP1A2, CYP2B6, and CYP3A4 and human liver microsomes has been reported [67].
Nevertheless, depending on a CYP isoform involved in metabolism, the both parathion and chlorpyrifos can be converted to less toxic p-nitrophenol and 3,5,6-trichloro-2-pyridinol derivatives, respectively (Figure 2B). For example, CYP2B6 has been recognized as the main CYP enzyme responsible for bioactivation of parathion and chlorpyrifos with the formation of oxon derivatives whereas CYP2C19 is primarily responsible for chlorpyrifos detoxification with the formation of 3,4,5-tricholorpyrindinol [69]. To do that, CYP2B6 catalyzes the oxidative desulfuration whereas CYP2C19 activity causes the oxidative dearylation, both reactions have low K(m) and high V(max) values. Additionally, metabolic pathways of organophosphorus pesticides yielding either more or less toxic derivatives depend on CYP gene polymorphisms as shown for CYP2B6-catalyzed conversions of chlorpyrifos [70].
Commonly used carbamate pesticides include carbaryl, carbofuran, and aminocarb, which unlike organophosphates reversibly inhibit AChE [64]. They can be activated via hydroxylation by human liver microsome-derived CYPs to form more toxic products than their parent compounds. Indeed, human CYP1A1 and CYP1A2 have been shown to catalyze the conversion of carbaryl to 5-hydroxycarbaryl whereas CYP1A2 and CYP3A4 have great potential to metabolize carbaryl to 4-hydroxtycarbaryl (Figure 2C) [71]. In this, chlorpyrifos and cimetidine compete with carbaryl and decrease its metabolism by CYPs. Moreover, both organophosphates and carbamates such as chlorpyrifos, fonofos, carbaryl, and naphthalene influence the steroid biosynthesis by inhibiting the 17β-estradiol conversion to 2-hydroxyestradiol by CYP1A2 and CYP3A4 [72]. Moreover, the inhibition of 17β-estradiol metabolism by chlorpyrifos is irreversible; therefore, this can lead to the estrogen accumulation observed in BC.
2.3. Bioactivation vs Inactivation of Procarcinogens
CYPs have been shown to catalyze the reactions of about 66% of all procarcinogen activation pathways, and among them, six microsomal CYPs (CYP1A1, CYP1A2, CYP1B1, CYP2E1, CYP3A4, and CYP2A6) have been shown to account for >90% of all procarcinogen-activating reactions [73]. In addition to CYPs, other enzymes such as SULTs (13%), NATs (7%), AKR (8%), cyclooxygenases (2%), and FMOs (1%) have been assigned to procarcinogen metabolism. CYP-catalyzed procarcinogen activation occurs, predominantly, due to the reactions of C- and N-hydroxylation, epoxidation, nitroreduction, and conjugation via O-acetylation and O-sulfonation. Products of the bioactivation reactions can interact with macromolecules such as DNA and proteins, causing their irreversible modifications, which lead to the macromolecular damage and cell dysfunction [74,75].
Shimada et al. have investigated the human CYPs-catalyzed bioactivation of a diverse range of carcinogenic chemicals including polycyclic aromatic hydrocarbons (PAHs) such as dibenzopyrenes, chrysene, phenanthrene, and anthracene and their dihydroxy- and dihydro-derivatives as well as heterocyclic and aromatic amines, aminoazo dyes, mycotoxins, insecticides, N-nitrosodimethylamine, vinyl carbamate, and acrylonitrile [76]. They have shown that CYP1A1 and CYP1B1 activate a variety of procarcinogens to produce strong carcinogenic metabolites, epoxides, both genotoxic and non-genotoxic. High-performance liquid chromatographic analysis showed that recombinant CYP1B1 and CYP1A1 catalyze the conversion of benzo[a]pyrene (B[a]P) to B[a]P epoxide, which is further converted to trans-7,8-dihydroxy-7,8-dihydrobenzo[a]pyrene (B[a]P-7,8-dihydrodiol) in the presence of liver epoxide hydrolase [77]. B[a]P-7,8-dihydrodiol gives rise to other epoxy-derivatives such as 7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydro-benzo[a]pyrene (Figure 3A). Kinetic analysis showed that ratio of V(max) to K(m) for the product formation in this system was 3.2-fold higher for CYP1B1 than for CYP1A1. The epoxy-derivatives can interact with DNA to yield toxic adducts.
Phenanthrene is not considered to be highly toxic and, therefore, it is of less importance regarding cytotoxicity and carcinogenicity as compared to B[a]P and 7,12-dimethylbenz[a]anthracene (DMBA) [78]. However, phenanthrene more readily produces metabolites excreted in higher amounts than other PAHs, and this dictates their diagnostic value in epidemiological and occupational exposure studies. Baum et al. have studied conversions of PAH benzo[c]phenanthrene (B[c]PH) to show that microsomal CYP1A2 produces, predominantly, B[c]PH-3,4-dihydrodiol in human liver and B[c]PH-5,6-dihydrodiol in the lung [79]. Both human CYP1A1 and CYP1A2 have been shown to oxidize the B[c]PH at 5,6- and 3,4-positions with similar efficiency; however, the CYPs displayed stereoselectivity in genetically engineered V79 Chinese hamster cells [80].
A variety of heterocyclic carcinogens originated from high-temperature cooking of meat and tobacco smoke can also be activated/deactivated by human liver microsomes and CYPs. An example is 3-aminodibenzofuran, carcinogenic aromatic amine with a stronger mutagenic activity than B[a]P. It undergoes metabolic activation via N-hydroxylation and further conjugation with either sulfuric acid by SULT or GSH with the involvement of GSTs [81]. The activation is followed by the interaction with Cys residues of a protein to form a hepatotoxic adduct (Figure 3B). Another example is dietary-derived coumarin, which itself has hepatotoxic potential [82]; it is oxidized by human liver cytosolic fractions to yield 2-hydroxyphenylacetic acid and 7-hydroxycoumarin, non-toxic and non-mutagenic metabolites [83]. The furafylline/α-naphthoflavone and 8-methoxypsoralen, which inhibit CYP1A2 and CYP2A6, respectively, blocked the conversions, thereby evidencing the involvement of the above-mentioned CYPs.
CYP3A4 has occurred to be more efficient than CYP1A2 in a reaction of activation of hepatocarcinogen aflatoxin B1 (AFB1) in human liver [84]. The CYP3A4-catalyzed reaction leads to the formation of aflatoxin Q1 (AFQ1) and exo-8,9-epoxide whereas CYP1A2 forms aflatoxin M1, a small amount of AFQ1, and exo- and endo-8,9-epoxides (Figure 3C). The AFQ1 formation requires 3-hydroxylation and causes detoxification whereas the AFB1 exo-8, 9-epoxide formation is an activation reaction. The AFB1 exo-8, 9-epoxide is highly reactive and interacts with DNA to give cytotoxic adducts in high yield (>98%); however, GSTs catalyze AFB1 exo-8,9-epoxide conjugation with GSH [85]. Similarly, the 2,3-dihydro-2-(N7-guanyl)-3-hydroxyaflatoxin B1 formed in DNA by CYPNF involved in the activation of AFB1 has been observed [86].
Formation of multiple DNA adducts from 3-aminobenzanthrone (3-ABA), a major metabolite formed from 3-nitrobenzanthrone (3-NBA), a carcinogen found in diesel exhaust and an ambient air pollution, has been reported [87]. Human and rat liver microsomes and recombinant CYP1A1 and CYP1A2 have been shown to metabolically activate 3-ABA via N-hydroxylation reaction, which were inhibited by α-naphthoflavone and furafylline, inhibitors of CYP1A1 and CYP1A2. One more procarcinogen and industrial pollutant, 2-nitroanisole, can be metabolized by human recombinant CYP2E1, CYP1A1, and CYP2B6, which are the most efficient enzymes causing its oxidative detoxification [88]. The detoxification products include O-demethylated metabolite, 2-nitrophenol, and two oxidation products, 2,5-dihydroxynitrobenzene and 2,6-dihydroxynitrobenzene [89].
2.4. Metabolism of Drugs
2.4.1. Drugs as CYP Substrates
Liver is a major organ, where drug metabolism takes place; however, the expression of each CYP isoform is influenced by a variety of factors including induction by xenobiotics, regulation by cytokines and hormones as well as sex, age, ethnicity, etc. [5,6,7]. Additionally, CYPs expressed by liver microsomes differ from each other by their roles in reactions of drug conversions. The most highly expressed in the liver CYP isoforms include CYP3A4, CYP2C9, CYP2C8, CYP2E1, and CYP1A2 whereas CYP2A6, CYP2D6, CYP2B6, CYP2C19, and CYP3A5 are less abundant and CYP2J2, CYP1A1, and CYP1B1 are mainly expressed extrahepatically [90]. Additionally, CYP-catalyzed metabolism of drugs depends on activities of CYP redox partners, the involvement of transporters, drug-drug interactions, pathophysiological conditions in the liver, etc. [91]. Despite CYPs have the broad and overlapping substrate specificities, at clinically relevant concentrations, many drugs are metabolized only by one or few CYPs.
About 3/4 of all CYP-catalyzed reactions in humans have been attributed to drug metabolism with the involvement of, predominantly, the following five enzymes: CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 [92]. It has been reported that 27% of drug metabolism reactions can be assigned to CYP3A4 followed by CYP2D6 (13%), CYP2C9 (10%), CYP1A2 and CYP2C19 (both 9%), CYP3A5 (6%), and CYP1A1 (5%). A meta-analysis undertaken by Achour et al. to assess the expression levels of various CYPs has shown that CYP3A4 is the most abundant drug-metabolizing CYP in liver microsomes, followed by CYP2E1, CYP2C9, and CYP1A2 [93]. However, the interindividual variability due to CYP gene polymorphisms and interlaboratory heterogeneity because of variations in measurement methodologies have been found in the latter study.
A number of important drugs can serve as CYP substrates, inhibitors, and inducers. In this, various CYPs can metabolize the same substrate to different products, thereby causing either their detoxification or bioactivation. An example is antitumor drug ellipticine, a CYP substrate and a cell-permeable lipophilic antineoplastic pyrido [3,4-b]carbazole derivative alkaloid, causing DNA intercalation and topoisomerase II inhibition [94,95]. CYP1A1 and CYP1A2 have been shown to catalyze ellipticine detoxification via the formation of 7-hydroxy- and 9-hydroxyellipticine, whereas CYP3A4 produces 13-hydroxyellipticine and catalyzes a reaction of N-hydroxylation to yield ellipticine N2-oxide (Figure 4A), the toxic metabolites responsible for the formation of DNA adducts [96]. However, the presence of cyt b5 induces the ability of CYP1A1 and CYP1A2 to efficiently produce 12-hydroxy- and 13-hydroxyellipticine, thereby switching the detoxification effect to activation one. The change in metabolite ratio results in an increased formation of covalent ellipticine-DNA adducts [97].
Another pyrido[3,4-b]carbazole derivative alkaloid, antineoplastic drug olivacine (1,5-dimethyl-6H-pyrido[4,3-b]carbazole), can be metabolized by CYP1A1, CYP1A2, and FMO3 to generate detectable amounts of products in primary hepatocytes [98]. Structure-activity relationship analyses have revealed that hydroxylation reactions with the formation of hydroxy-, hydroxymethyl- and methoxy-derivatives at positions C1, N2, C9, and C11 significantly increase cytotoxic activity of olivacine toward tumor cells [99]. Indeed, in vitro study on colorectal cancer cells has shown that hydroxylation of olivacine at C9 position with the production of 9-methoxyderivative increases its cytostatic activity greater than of ellipticine derivatives and doxorubicin [100].
Anticancer drug irinotecan [7-ethyl-10[4-(1-piperidino)-1-piperidino]-carbonyloxy camptothecin] (CPT-11), a semisynthetic analogue of DNA topoisomerase I inhibitor camphothecin, [101], undergoes conversions via several metabolic pathways catalyzed by CYP3A4 and CYP3A5 [102]. CYP3A4 has been shown to preferentially metabolize irinotecan via the oxidation of piperidinylpiperidine side chain, thereby producing 4-N-5-aminopentanoic acid and 4-aminopiperidino metabolite whereas CYP3A5 catalyzes de-ethylation of camphothecin moiety to produce M4 metabolite [103].
Tamoxifen is a commonly used antiestrogen, which is able to interact with estrogen receptor-α (ERα) to cause competitive inhibition of estrogen action, and this dictates its usage in the endocrine treatment of choice in both premenopausal and postmenopausal women with ER positive (ER+) BC [104]. Tamoxifen is a prodrug that is converted to its active metabolites by CYP2D6, CYP3A4, CYP3A5, CYP2B6, and CYP2C19, and no statistical differences due to CYP gene polymorphisms in the efficacy of tamoxifen have been observed by Singh et al. [105]. However, interindividual differences in postmenopausal women with early BC receiving tamoxifen have been observed by Goetz et al. [106]. The authors have found that patients with decreased tamoxifen metabolism identified as having CYP2D6*4 genotype exerted significantly shorter recurrence-free survival (RFS) than patients with extensive metabolism. However, Tan et al. have found that five-year RFS and overall survival (OS) were slightly better, but statistically non-significant, in women with the extensive or ultra metabolizer CYP2D6 phenotype compared to those with the intermediate phenotype [107]. Additionally, no significant difference in progression free survival (PFS) between the CYP2D6*4 genotype group and the overall study cohort has been found by Stingl et al. in the Austrian TIGER study [108].
92% of the tamoxifen metabolism is attributed to the production of quantitatively major primary metabolite N-desmethyltamoxifen, the conversion catalyzed by CYP2D6 with the highest activity and to a lesser extent by CYP3A4 and CYP3A5 [109]. CYP2C19 has an order of magnitude higher activity than CYP2D6 in formation of 4-hydroxytamoxifen, which is ~30-100-fold more potent than tamoxifen, although this pathway constitutes only 7% of tamoxifen metabolism [110]. Further, the both N-desmethyltamoxifen and 4-hydroxytamoxifen can be converted to endoxifen (4-hydroxy-N-desmethyltamoxifen) by CYP2D6 and CYP3A4/CYP5, respectively (Figure 4B). Endoxifen is the most potent antiestrogen among a variety of other metabolites produced in these pathways and significantly contributes to the tamoxifen therapeutic effects [111].
Hydroxylation of tamoxifen at ethyl group causes its conversion to α-hydroxytamoxifen, a genotoxic compound produced, predominantly, by CYP3A4 in both humans and rats [112]. The α-hydroxytamoxifen produces DNA adducts, which have been identified in the endometrium of women taken tamoxifen [113]. Alternatively, the hydroxylated metabolites of tamoxifen can undergo conjugation with sulfuric acid by SULT1A1 and glucuronic acid by UGTs for further excretion [114]. The CYP-catalyzed metabolism contributes to an intrinsic resistance to tamoxifen fueled by cross-talks with growth factor signaling pathways and hormone-binding transport proteins, which interfere with ERα-hormone interactions [115,116,117].
Stereospecific CYP activities in hydroxylation at the same positions of the same substrates have been observed. For example, anticancer drug thalidomide is converted to 5-hydroxythalidomide and diastereomeric 5'-hydroxythalidomide by liver microsomes; however, human CYP2C19 is a major enzyme catalyzing this reaction whereas in rats, thalidomide is hydroxylated extensively by CYP2C6 isoform [118]. Additionally, CYP2A6 and to lesser extent CYP1A2 and CYP2C8 have been shown to convert tegafur [5-fluoro-1-(2-tetrahydrofuryl)-2, 4 (1H, 3H)-pyrimidinedione], a component of anticancer drug S-1 used in treating breast, pancreatic, lung, and other carcinomas, to cytotoxic compound 5-fluorouracil [119]. The conversion takes place via hydroxylation of tetrahydrofuran ring at its 5’-position and catalytic activity of CYP2A6 in catalyzing this reaction is stereospecific [120].
2.4.2. Drugs as CYP Inducers: the Roles of Nuclear Receptors and Drug Transporters
Rifampin, phenytoin, and ritonavir are examples of drugs, which induce human CYP activity, the process in which human pregnane X receptor (PXR) plays a critical role by causing unfavorable and long-lasting drug-drug interactions and cytotoxicity effects [121,122]. PXR affects drug metabolism and efflux and drug-drug interactions via controlling the expression of multiple drug resistance protein-1 (MDR1), also known as P-glycoprotein (P-gp), which serves as a transmembrane pump for the elimination and disposition of drugs [123]. P-gp is a 170 kDa phosphorylated and glycosylated protein encoded by MDR1 gene and belonging to the ATP binding cassette superfamily of transport proteins [124]. In humans, two genes, MDR1 and MDR2, have been described, among which only MDR1 has been implicated in drug transport and drug resistance [125].
The PXR belongs to the evolutionarily related superfamily of nuclear receptors (NRs), which function as DNA-binding transcription factors regulating a broad spectrum of physiological processes [126]. Despite a high diversity of functions and modes of action, NRs exhibit conserved structural organization, which include six domains and among them, DNA-binding domain, ligand-binding domain, and activation function 2 (AF-2) domain are the most important ones [127]. Binding of a NR ligand causes the receptor activation and nuclear translocation followed by its homo- or heterodimerization with retinoic X receptor (RXR) for DNA binding. The role of AF-2 domain is a proper repositioning of NR structural elements upon the ligand-binding to provide the interaction with DNA and transcriptional regulation as well as co-activator and co-repressor binding [128].
Numerous CYP inducers including pregnane, steroid hormones, drugs, and environmental pollutants can activate PXR [129]. For example, PXR is a target for many drugs that induce CYP3A4 and can be activated by high concentrations of flucloxacillin with consequences for metabolism of other drugs [130]. Flucloxacillin, a β-lactam antibiotic of the isoxazolyl-penicillin group with high incidence rate of drug-induced liver injury, can be metabolized by liver microsomes and recombinant CYP3A4 to produce 5'-hydroxymethylflucloxacillin. The latter metabolite causes cytotoxic effects in human hepatocytes and gallbladder-derived biliary epithelial cells. Indeed, the incubation of primary human hepatocytes and LS180 colorectal adenocarcinoma cells with flucloxacillin led to a dose-dependent induction of MDR1 [131].
Etoposide, a semisynthetic derivative of podophyllotoxin that exhibits antitumor activity to be used in BC treatment, undergoes O-demethylation to etoposide catechol with the involvement of CYP3A4 and to lesser extent of CYP1A2 and CYP2E1 [132]. The etoposide is a substrate for P-gp, which determines its pharmacokinetics restricting its oral uptake and mediating its intestinal excretion across the gut wall [133]. In rats with DMBA-induced mammary tumors, increased absorption of etoposide from the gastrointestinal tract via the inhibition of intestinal P-gp and decreased intestinal metabolism of etoposide via the inhibition of intestinal CYP3A by morin have been observed [134].
Hofman et al. have shown that the overexpression of CYP3A4 and CYP2C8 causes the decrease in sensitivity of hepatocellular carcinoma (HCC) HepG2 cells to anticancer drugs of the taxane group, docetaxel and paclitaxel [135]. CYP3A4-mediated docetaxel resistance due to impaired apoptosis has been observed, and the drug resistance can be reversed by ketoconazole. CYP3A4 and CYP3A5 have been shown to catalyze the formation of hydroxy- and dihydroxy-derivatives of paclitaxel and docetaxel, thereby causing their inactivation in cancer cells [136,137]. Consequently, CYP3A4 and CYP3A5 gene polymorphisms contribute to interindividual variations in the drug clearance and their toxicity in BC patients [138].
In cancer cells, PXR ligands enhance PXR-mediated transcription in a ligand- and promoter-dependent fashion, thereby leading to differential regulation of individual PXR targets, especially CYP3A4 and MDR1. Indeed, in epithelial ovarian cancer cells, PXR agonists, phthalate and pregnenolone, have been shown to induce CYP3A4 expression and PXR-mediated transcription through the CYP3A4 promoter, whereas paclitaxel and cisplatin increased MDR1 expression and PXR-mediated transcription through the MDR1 promoter [139]. The PXR downregulation by small interfering RNA inhibited endometrial and ovarian cancer cell growth and enhanced apoptosis caused by the anticancer drugs and PXR ligands [139,140].
A variety of drugs such as phenobarbital, rifampicin, and hyperforin have been shown to induce the transcriptional expression of CYP2C isoforms in primary human hepatocytes and to increase the metabolism of CYP2C substrates in vivo [141]. This induction can result in drug-drug interactions, drug tolerance, and therapeutic failure. In human chronic myelogenous leukemia K-562 cells, treatment with cisplatin and two ruthenium-based coordinated complexes [cisCRu(III) and cisDRu(III)] has been shown to cause apoptosis associated with MDR1 amplification and CYP2C9 and CYP2C19 overexpression [142]. Several drug-activated nuclear receptors including PXR, constitutive androstane receptor (CAR), and vitamin D receptor (VDR) recognize drug responsive elements within the 5'-flanking promoter region of CYP2C genes to mediate the transcriptional upregulation of these genes in response to xenobiotics and steroids [143,144,145].
CAR similarly to PXR can be activated by a variety of endogenous and exogenous CYP ligands including drugs, insecticides, pesticides, and nutritional compounds and functions as a xenobiotic receptor for detoxification and clearance of toxic substances from the liver [146]. Wang et al. have observed that CAR downregulation by RNA interference causes significant ovarian cancer cell growth inhibition and apoptosis in the presence of anticancer agents [147]. CAR ligands and anticancer drugs cisplatin, paclitaxel, and arsenic trioxide cause the upregulation of MDR1 and UGT1A1 in the presence of 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehydeO-(3,4-dichlorobenzyl)oxime (CITCO), the CAR agonist.
Also, CAR has been implicated in the regulation of CYP2B1, CYP2B6, CYP3A1, and CYP3A4 in human hepatocytes [148,149]. CAR has been shown to induce the expression of CYP2B6 and CYP3A4 in human primary hepatocytes and leukemia cells, and this enhances the antitumor activity of an alkylating agent cyclophosphamide (CPA) used in the treatment of a variety of tumors. CPA undergoes oxidation and conversion to 4-hydroxy-CPA, a rate limiting reaction in CPA bioactivation [150]. The involvement of human microsomal CYP2C9 in the CPA bioactivation to 4-hydroxy-CPA has been also observed [151].
2.4.3. CYP Inhibition: Drug-Drug Interactions
Clinically important CYP3A4 inhibitors include macrolide antibiotics (clarithromycin, erythromycin, etc.), anti-HIV agents (ritonavir and delavirdine), antidepressants (fluoxetine and fluvoxamine), calcium channel blockers (verapamil and diltiazem), steroids and their modulators (gestodene and mifepristone) [152]. Ivermectin is a macrocyclic lactone with antiparasitic and antiviral activities, a derivative of avermectins, the 16-membered macrolide lactone compounds produced by the fungus Streptomyces avermitilis, is metabolized both in vivo and in vitro by CYP3A4 as the major enzyme, with a contribution of CYP3A5 and CYP2C9 [153]. The major metabolites are produced by 3″-O-demethylation, C4-hydroxylation, C25 isobutyl-hydroxylation, and 3″-O-demethyl-4-hydroxymethyl metabolite formation reactions. Co-administration of drugs that are potent inhibitors or inducers of CYP3A4 activity and CYP gene polymorphisms can provoke pronounced drug-drug interactions [154]. Additionally, many substrates of CYP3A4, which considerably overlap with those of P-gp and ivermectin can also inhibit the CYP3A4 functions [155].
CYP2C8 has been shown to increase anticancer activity of multi-kinase inhibitor (MKI) sorafenib due to the conversion to sorafenib N-oxide, thereby inhibiting proliferation, clonality, migration, invasion, and cell cycle progression in HCC cells [156]. Sorafenib is frequently co-administered with a range of other drugs such as paclitaxel to improve the efficacy of anticancer therapy and to treat comorbidities. In this, sorafenib N-oxide, a major pharmacologically active metabolite, has the potential to contribute to pharmacokinetic drug-drug interactions. Indeed, along with N-hydroxymethyl and N-desmethyl metabolites, it can be formed by CYP3A4 (Figure 4C) to inhibit the 6α-hydroxylation of paclitaxel by CYP2C8 [157]. In human liver microsomes, the metabolite formation was correlated with CYP3A4-mediated midazolam 1'-hydroxylation, but not with other CYP-specific substrate oxidations whereas CYP3A4 inhibitor ketoconazole selectively inhibited microsomal sorafenib oxidation pathways [158]. Sorafenib and its metabolites have been shown to accumulate in patients’ blood serum during therapy, which may increase toxicity of co-administered drugs due to drug-drug interactions [159].
Sorafenib has been shown to suppress the Bcl-2 family member, induced myeloid leukemia cell differentiation protein MCL1, to regulate apoptosis in THP-1, U937 and Granta519 cancer cells; however, no increased apoptosis was observed in combination with vinblastine [160]. Vinblastine is metabolized by CYP3A4, and this pathway is inhibited by vincristine and navelbine, other members of the Vinca alkaloid family, and other anticancer drugs currently associated with vinblastine in cancer chemotherapy (etoposide, adriamycin, lomustine, and teniposide) [161,162].
CYP3A4 has been implicated in metabolism of another MKI imatinib via its N-demethylation and N-desmethylimatinib formation, correlated with microsomal oxidation of the CYP3A4 substrates testosterone and midazolam, and the CYP2C8 substrate paclitaxel [163]. Recombinant CYP2C8 and CYP3A4 metabolize imatinib extensively, whereas other isoforms have minor effect on imatinib concentrations. However, during the long-term treatment with imatinib, dose- and time-dependent auto-inactivation of CYP3A4 by imatinib has been observed [164]. Imatinib increased the bleeding risks of rivaroxaban, widely used direct oral anticoagulant, whereas sunitinib had a risk of reducing therapy efficiency due to drug-drug interactions. Imatinib and gefitinib significantly inhibited CYP2J2- and CYP3A4-mediated metabolism of rivaroxaban and efflux transportation mediated by breast cancer resistance protein (BCRP)- and P-gp drug transporters in MDR1-MDCK and ABCG2-MDCK cells [165].
Filppula et al. have undertaken a screening of inhibitory effects of a variety of MKIs on the activities of CYP3A and CYP2C8 and showed that a wide range of drugs including bosutinib isomer 1, crizotinib, dasatinib, erlotinib, gefitinib, lestaurtinib, nilotinib, pazopanib, saracatinib, sorafenib, and sunitinib exhibited an increased inhibition of CYP3A after a 30-min preincubation with NADPH, as compared with no preincubation [166]. Bosutinib was the only inhibitor causing time-dependent inhibition of CYP2C8. The authors have found that midostaurin and nintedanib irreversibly inhibited the CYP3A4 and predicted drug-drug interactions between vatalanib and CYP2C8 substrates as well as between masitinib, midostaurin, and vatalanib and CYP3A4 substrates [167]. Despite CYP3A4 and CYP2C8 are major contributors in masitinib metabolic activation, CYP3A5 and CYP2D6 also contribute to the formation of N-desmethyl masitinib (M485), and several oxygenated metabolites can be detected along with four reactive metabolite cyano adducts (MCN510, MCN524, MCN526, and MCN538) [168]. Table 1 summarizes data on the involvement of various human CYP1-CYP4 families in drug metabolism.
3. Roles of CYPs in Cancer
3.1. Breast, Endometrial, and Ovarian Cancer
3.1.1. Effects of Estrogen Metabolites
Estrogen production and metabolism play critical roles in the development and pathogenesis of hormone-sensitive cancers in women – breast, endometrial, and ovarian carcinomas. BC is the first most common cancer type and the fifth cause of cancer-related deaths worldwide as estimated in 2020 [27]. BC is a genetically and clinically highly heterogeneous disease with multiple subtypes classified based on the immunohistochemical expression of hormone receptors: ER+, progesterone receptor positive (PR+), human epidermal growth factor receptor positive (HER2+), and triple-negative breast cancer (TNBC) characterized by the lack of expression of any of the above-mentioned receptors [169]. Approximately 70–75% of invasive BCs are characterized by significantly high ERα expression, whereas PR is expressed in more than 50% of ER-positive patients [170].
Endometrial cancer (EC) is the sixth most common cancer type in women worldwide [171]. EC is also heterogenous disease and is classified on the basis of ERα and PR expression into endometrioid type I (hormone receptor-positive type) and non-endometroid type II (hormone-receptor-negative type) [172]. The endometrioid type I EC risk has been linked to increased circulating levels of estrogens and persistent ERα activation [173]. Ovarian cancer (OC) is the eight most common cause of death in female population worldwide and comprises 1.2% of all the cases of cancer as estimated in 2020 [174]. About 90% of primary malignant ovarian tumors are epithelial carcinomas, and critical roles of hormones in OC development and progression have been reported [175]. Studies have evidenced that the ER+ and PR+ statuses were higher among patients with serous and endometrioid carcinomas than among mucinous and clear-cell carcinomas of malignant epithelial OC and that ER+ and PR+ OC patients have better clinical outcome [176].
Current understanding of molecular mechanisms of hormone-sensitive cancers is based on key roles of CYPs in biosynthesis and metabolism of steroids hormones, androgens and estrogens [177]. Studies have shown that in BC, OC, and EC tissues, levels of circulating estrogens and their metabolites, and genotoxic DNA adducts correlate with the activities of CYPs and conjugating enzymes. Indeed, circulating estrogen levels have been shown to influence ERα activation and DNA adduct formation in BC tissues with the involvement of CYP1B1, SULT1A1, SULT1A2, and GSTP1 [178]. Additionally, the increased level of CYP1B1 in SKOV-3 and A2780 OC cell lines [179] and overexpression of CYP1A1 in various types of OC compared with benign epithelia [180] have been reported.
CYP1A1, CYP1B1, and CYP1A2 are the most important CYPs involved in the irreversible hydroxylation of 17β-estradiol and estrone to produce catechol estrogens, 2-hydroxy- and 4-hydroxy-derivatives, whereas CYP2C19, mostly, forms 16α-hydroxy-derivative [181]. Additionally, in human liver microsomes, the 2-hydroxylation and 4-hydroxylation of 17β-estradiol have been shown to correlate with monooxygenase activities of CYP3A4 and CYP3A5 [182]. The 2-hydroxylation yields 2,3-catechols whereas 4-hydroxylation gives rise to 3,4-catechols, the both catechol types are further converted to corresponding 2,3- and 3.4-quinones involved in protein and DNA adducts formation (Figure 5A). Among them, mutagenic adduct formation with adenine and guanine, which cause DNA depurination, can serve as biomarkers of estrogen metabolites formation in BC [183].
The hydroxy-metabolites can inhibit BC cell proliferation, and therefore, they lack tumorigenic activities. Indeed, 16α-hydroxy-metabolites of 17β-estradiol and estrone have been shown to demonstrate ER agonist properties in ER+ MCF-7 and T47D human BC cells [184]. On the contrary, 2-hydroxy-metabolites of both 17β-estradiol and estrone demonstrated weak mitogenic activity and did not show ERα agonist properties in the both cell lines; instead, they exhibited protective effects. Moreover, 17β-estradiol and its metabolite 4-hydroxyestradiol have been reported to protect from oxidative stress via increasing superoxide dismutase (SOD) activity, whereas 16α-hydroxy-estrone increased GST activity [185].
The tumor protective activities of estrogen hydroxy-metabolites have been attributed to the formation of methoxy-derivatives and conjugated estrogens [186,187]. Indeed, the catechol estrogens can be either methylated by catechol-O-methyltransferase (COMT) or conjugated, for example, with glucuronic acid [188]. The O-methylation of catechol estrogens by COMT can serve as a protective pathway via minimization of depurinating DNA adduct formation and cancer prevention [189,190]. A case-control study of serum concentrations of 15 estrogens and their metabolites in both conjugated and unconjugated form has shown that almost all estrogens and estrogen metabolites in unconjugated form are associated with an increased BC risk in postmenopausal women [191]. Exception was 2-hydroxylation, which was associated with lower risk, whereas less extensive methylation of potentially genotoxic 3,4-catechols was associated with higher risk of postmenopausal BC.
The COMT inhibition has been shown to decrease the formation of 2-methoxy-derivative from 2-hydroxy-17β-estradiol and to increase the oxidative DNA damage via 8-hydroxy-2'-deoxyguanosine (8-oxo-dG) formation in MCF-7 cells exposed to 0.1 microM 17β-estradiol [192]. Additionally, the increased levels of albumin and hemoglobin protein adducts formed by 17β-estradiol 2,3- and 3,4- quinones have been shown to correlate with worse 5-year survival in BC patients [193]. The authors proposed that BC treatment with the use of aromatase inhibitors and tamoxifen may dramatically reduce burden of estrogen quinones.
The increased levels of unconjugated 17β-estradiol and estriol as well as estrone metabolites, 2-methoxyestrone and 4-methoxyestrone, in women with type I as compared to type II EC have been reported [173]. Additionally, the increased progesterone-to-estradiol ratio and level of 17α-hydroxypregnenolone have been shown to inversely associate with EC risk and positively associate with OC risk [194]. Women with OC have been shown to possess two low-activity alleles of COMT and one or two high-activity alleles of CYP1B1 associated with higher levels of estrogen-DNA adducts [195].
3.1.2. CYP Gene Polymorphisms
Various SNPs in genes encoding CYPs involved in estrogen biosynthesis have been implicated in increased risk of BC. Indeed, multiple SNPs in CYP1A1 gene have been studied and some investigators have identified positive correlations between the SNPs and ER+ BC [196]. For example, Martínez-Ramírez et al. have found that polymorphisms in genes encoding ERα, CYP1A1, CYP1B1, COMT, O-6-methylguanine DNA-methyltransferase (MGMT), and X-ray repair cross-complementing protein 1 (XRCC1) are positively associated with the BC risk in Mexican women [197]. However, other studies have found no statistically significant correlations between SNPs in genes encoding CYP1A1, CYP1B1, and GST and breast BC in both premenopausal and postmenopausal women among Indian and Nigerian population [198,199].
Zhang et al. have found that BC risk in postmenopausal Chinese women is associated with the CYP17 TC genotype compared with TT genotype [200]. The CYP19 TC+TT genotypes were associated with both overall cancer risk and premenopausal cancer risk, particularly for ER+/PR+ tumors. In multivariate analysis, the SULT1A1 genotype has been shown to be the most significant BC determinant, followed by CYP19, whereas the (TTTA) 10 allele of CYP19 was associated with tumor size, and the His allele of SULT1A1 was associated with status of lymph node metastasis [201]. Combined analyses of the two genes encoding CYP17 and CYP19 has shown that the increased frequency of high-risk alleles as compared to individual studies undertaken in BC patients [202].
A case-control study of the associations between the CYP1B1 and COMT polymorphisms and invasive EC risk has shown that carriers of the CYP1B1 N453S allele had a statistically significant decreased cancer risk; however, no significant association between CYP1B1 L432V allele and EC risk has been found [203]. Similarly, a study of three CYP1A1 genotypes (Msp1, I462V, and T461N) has shown no additional risk of EC in Caucasian women [204]. Nevertheless, the association between the rs2470893 polymorphism of the CYP1A1 gene and EC and OC in Mediterranean women has been identified [205].
The individuals with minor variant genotype of L432V polymorphism in CYP1B1 gene have been found to show high risk of EC in Caucasians [206]. However, no significant association between L432V polymorphism in CYP1B1 gene and OC risk has been found [207]. A meta-analysis has shown that the rs1056836 polymorphism of the CYP1B1 gene is associated with OC risk in Caucasians and Asians, but no associations have been observed in African-Americans [208].
3.2. Prostate Cancer
3.2.1. Effects of Androgen Metabolism
PCa is the fourth most common cancer worldwide and the eight cause of cancer-related deaths worldwide as estimated in 2020 [209]. Many early-stage PCa cases are androgen-dependent or castration-sensitive; therefore, androgen production, metabolism, and transport influence greatly the PCa progression [210]. Consequently, decreasing the androgen levels in the body or blocking their action may be an effective type of the anticancer therapy [211].
CYP17A1 is a single enzyme that catalyzes the sequential 17α-hydroxylase and 17,20-lyase steps that are required for conversion of C21 pregnenolone and progesterone to DHEA and androstenedione, the C19 androgen and C18 estrogen precursors [212]. CYP17A1 gene is expressed in the gonads, adrenal glands, and prostate cancer cells. The balance of enzyme activities and substrate preferences vary among species; in humans, 17α-hydroxylase activity is required to produce cortisol and to maintain glucocorticoid and mineralocorticoid homeostasis whereas the both 17α-hydroxylase and 17,20-lyase activities are required for sex hormone biosynthesis [213]. CYP17A1 is expressed in the ER of testicular Leydig cells and the adrenal cortex, the major sites of testosterone biosynthesis, whereas POR and cyt b5 become present in cells with high 17,20-lyase activity during human development [214].
Usage of the in vivo LNCaP PCa mouse xenograft model has enabled demonstrating that circulating cholesterol level is significantly associated with tumor size and intra-tumoral levels of testosterone and directly correlate with tumoral expression of CYP17A [215]. Moreover, the expression of CYP17A1 and nuclear androgen receptor (AR) at the both protein and mRNA levels has been observed in 50% of the hormone-dependent 22Rv1 cell line, in contrast to hormone-independent PC3 and DU145 cell lines [216]. Along with these data, about 91% of benign prostate hyperplasia (BPH) and 83% of PCa specimens have been shown to express CYP17A1 whereas and CYP11A1 and translocator protein (TSPO) transporting cholesterol into mitochondria have been expressed in all BPH and PCa specimens [217].
For the advanced PCa, androgen deprivation therapy (ADT) by surgical or medical castration is a standard frontline treatment and is initially effective in most patients; however, ADT can lead to the development of uncurable metastatic castration-resistant prostate cancer (CRPC) [218]. Several mechanisms have been proposed to explain the progression of ADT-responsive PCa to CRPC including AR overexpression and mutations, despite metastatic CRPC can exist as a mixture of cells displaying a range of AR expression levels [219]. Sustained intra-tumoral synthesis of 5α-dihydrotestosterone (DHT), more potent AR agonist than testosterone, also contributes to CRPC [220,221]. Testosterone can be either reversibly converted to androstenedione by 17βHSD or reduced by SRD5A1/2 isoenzymes to DHT, which can be, alternatively, produced from androstenedione due to sequential catalysis by 17βHSD to 5α-androstanedione (5α-dione) and by SRD5A1/2 isoenzymes to DHT (Figure 5B).
Since persistent androgen signaling has been implicated in CRPC progression and metastasis, studies are carried out to develop AR antagonists [222]. However, PCa cells can develop mechanisms to resist AR blockade due to overexpression, mutations, alterations in coregulators of AR. Therefore, studies have been undertaken to develop various potential steroidal and non-steroidal derivatives with the capabilities of reversible small-molecule CYP inhibitors [223,224]. Metastatic CRPC has been shown to display the upregulation of genes encoding steroidogenic CYP enzymes including CYP17A1 and CYP19A1 [225]. Various potential steroidal and non-steroidal CYP17A1 inhibitors have been proposed as candidates for PCa treatment [226,227,228,229,230,231]. However, the only FDA approved drug for prostate cancer treatment is a steroidal irreversible selective CYP17A1 inhibitor abiraterone [232]. Abiraterone acetate, an orally administered small molecule derived from the structure of pregnenolone, selectively and irreversibly inhibits both the 17α-hydroxylase and 17,20-lyase activities of CYP17A with approximately 10–30-fold greater potency as compared to ketoconazole, non-selective inhibitor of CYP17A1 [233].
In a randomized phase 3 clinical trial (NCT00638690), 1195 patients with metastatic CRPC who had previously received docetaxel were assigned to combination of prednisone with abiraterone acetate and showed the increase in OS (14.8 months) as compared to placebo group (10.9 months) [234]. Another clinical trial performed with the use of 57 patients with bone-metastatic CRPC have evidenced that abiraterone acetate causes sustained suppression of testosterone in both blood and bone marrow [235]. Limitation in the use of abiraterone has been found due to its ability to inhibit other AR pathway targets due to its steroidal structure.
Recent advancements in bioinformatics and computational approaches have enabled discovery of novel CYP17A1 inhibitors to replace abiraterone and elucidating mechanisms underlying the CYP-inhibitor interactions. For example, CYP17A1 inhibitors, seviteronel and abiraterone, have been demonstrated to function as competitive AR antagonists [236]. Molecular dynamics (MD) simulation study has revealed the importance of the conserved H-bond for acquiring the proper position by abiraterone in the binding site [237]. MD simulation has also demonstrated the extended residence time of abiraterone within the CYP17A1 active site, even when most abiraterone has been eliminated systemically [238]. This has been experimentally confirmed by prolonged suppression of DHEA concentrations observed in VCaP cells after abiraterone washout. The authors have concluded that their findings can provide a basis for re-evaluating the current dosing paradigm of abiraterone while mitigating its dose-dependent adverse effects.
Additionally, studies have been undertaken to discover novel molecular targets in PCa treatment. For example, Zhang et el. have shown that genetic or pharmacological activation of PXR decreases androgenic activity by inducing the expression of CYP3A4 and SULT2A1, which are enzymes important for the metabolic deactivation of androgens [239]. In human prostate cancer, the treatment with the PXR agonist rifampicin inhibited androgen-dependent proliferation of LAPC-4 cells, but had little effect on the growth of the androgen-independent isogenic LA99 cells. PXR has been shown to be expressed at the mRNA and protein level in both rat primary Leydig cells and mouse Leydig tumor MA-10 cells, and the incubation of these cells with pregnenolone 16α-carbonitrile resulted in a significant decrease in testosterone secretion [240]. This was associated with decreased protein expression of key steroidogenic enzymes such as CYP17A1 and 3βHSD.
3.2.2. CYP Gene Polymorphisms
Polymorphisms in genes involved in the testosterone biosynthetic pathways such as CYP17A1, CYP3A4, and SRD5A2 represent strong candidates for affecting androgen-dependent cancers. Indeed, genetic polymorphisms in genes encoding AR, the prostate-specific antigen (PSA), SRD5A2, CYP17, CYP3A4, and a putative hereditary PC susceptibility 2 protein (HPC2/ELAC2) have been suggested to play the roles in the PCa etiology [241]. For example, the HPC2/ELAC2 has been found to feature S217L and A541T polymorphisms whereas the SRD5A2 features A49T and V89L polymorphisms. The statistically significant correlation between A49T mutation in SRD5A2 and BPH in Turkish men has been reported [242]. Additionally, the SRD5A2 V89L variant has been suggested to influence risk of PCa among younger-aged men with diagnosis of more aggressive disease [243].
Some studies have shown that there is no evidence of associations between CYP17 and SRD5A2 polymorphisms and PCa risk [244,245]. Lindström et al. have found that PCa risk is significantly associated with multiple SNPs in the AR and CYP17 genes, as well as one SNP in the SRD5A2 gene. [246]. Mutations in individual genes encoding ERα and CYP17 showed a 2- and a 3.5-fold increased risk of PCa as compared to BPH and healthy controls, respectively; however, the combination of mutant alleles of the two genes increased the cancer risk by more than 2-fold in the North Indian population [247]. Another study has found no significant association between SRD5A2 gene polymorphism and PCa risk, but a significant association between the CYP17 gene polymorphism and PCa risk in the Japanese population has been identified [248].
Additionally, a case-control study of genes encoding CYP1A1, CYP1A2, CYP2E1, GSTM1, and GSTT1 has shown that only a combination of polymorphisms in CYP1A1 and GSTM1 is associated with PCa risk in the Japanese population [249]. The increased risk of PCa in the Japanese population was observed among individuals with the NAT2 slow acetylator, CYP1A1 GA+GG, and CYP1A2 CA+AA genotypes [250]. In the latter study, CYP1A1 GA+GG genotypes were associated with intake of heterocyclic aromatic amines, the CYP substrates.
Genetic polymorphisms in the CYP1B1 gene have also been studied to show that combinations of some of them are associated with an increased PCa risk, whereas one frequent haplotype (T-A-T-G-T) is associated with the decreased cancer risk [251]. Some CYP1B1 haplotypes were positively associated with PCa among men with the highly aggressive disease [252]. A meta-analysis undertaken for various cancer types has shown that A119S polymorphism in CYP1B1 gene is associated with PCa risk in the Caucasians [253]. Such genetic variants within CYP1B1 have been proposed to provide susceptibility to PCa by altering telomere length [254]. Importantly, the inherited genetic variations in CYP1B1 gene can serve as predictors of clinical outcomes for patients with clinically localized PCa [255].
In a meta-analysis undertaken for the Asian population, the rs1048943 and rs4646903 polymorphisms were associated with a significant increase in PCa risk; however, no association between rs10012, rs162549, rs1800440, and rs2551188 polymorphisms in the CYP1B1 gene and PCa risk has been identified [256]. When compared to Caucasians, a significant association between the rs1048943 polymorphism in the CYP1A1 gene and PCa risk has been found in the overall population, but not in Caucasians. Another case-control study revealed no significant association between PCa risk and CYP1A2 rs2472299 and rs11072508 polymorphisms [257].
The CYP3A4*1B haplotype has been found to positively associate with PCa risk in Caucasians with more aggressive disease, and a negative association has been identified between the CYP3A4*1B/CYP3A5*1 haplotype and PCa risk in Caucasians with less aggressive disease [258]. Data obtained by a meta-analysis undertaken by Liang et al. have shown that the CYP3A5*3 gene polymorphism may be associated with increased PCa risk, particularly in African populations [259]. The use of bioinformatics tools has also enabled the identification that CYP3A4*1B and CYP3A5*3 haplotypes are significantly associated with an increased PCa risk and the disease progression [260]. The interaction between CYP3A5*3/*3 and SRD5A2 A49T GG genotypes was associated with the clinical tumor stage T2-T4 whereas CYP3A5*3/*3 and KLK3 I179T CC/TC genotypes had increased OS in patients with metastatic PCa [261]. Additionally, family-based analyses have made it possible to identify associations between PCa risk or aggressiveness and several CYP3A4 SNPs and two SRD5A2 SNPs in strong linkage disequilibrium [262].
3.3. CYP Hepatotoxicity and Liver Cancer
3.3.1. Anticancer Drug-Induced Liver Injury
The expression and activities of CYP enzymes as well as drug efficacy have been shown to be altered during liver injury including chronic alcoholic liver disease and nonalcoholic fatty liver disease (NAFLD) [263]. For example, the mouse model of acetaminophen (APAP)-induced liver injury showed a decreased expression and activity of CYP3A11, CYP1A2, CYP2B10, CYP2C29, and CYP2E1 [264]. Adult male mice were more susceptible to APAP-induced hepatotoxicity and had more decreased expression of CYPs than female mice. The expression and activity of CYP1A, CYP2C19, and CYP3A appear to be particularly vulnerable to the liver diseases whereas CYP2D6, CYP2C9, and CYP2E1 were less affected [265].
On the other hand, it is well-documented that CYP-activated drugs and procarcinogens produce metabolites, which induce hepatotoxicity. For example, induction of hepatic cytochrome CYP2E1 increases the generation of N-acetyl-p-benzoquinone imine (NAPQI), a toxic metabolite of APAP [266]. APAP-induced hepatotoxicity can be potentiated by isoniazid and ethanol, which also induce CYP2E1, thereby increasing the formation of NAPQI by 2- to 3-fold [267]. However, the elevated expression of inflammatory mediators in the liver caused by excessive APAP activity can be dose-dependently reversed by natural chemicals such as 8-methoxypsoralen [268]. The latter compound binds to CYP2E1 and occupies an active site of the enzyme, competitively inhibiting the oxidative metabolism of APAP and, thereby reducing the generation of toxic products. The expression and activity of CYP2E1 can be inhibited by another natural chemical, silymarin, as shown with the use of human and rat liver microsomes and HepG2 cells [269].
Sunitinib, a MKI inhibitor, undergoes metabolic activation via aromatic and aliphatic hydroxylation, N-oxidation, and oxidative defluorination [270]. Sunitinib malate is an orally administered drug approved for treatment of metastatic tumors; however, its usage is ineffective in certain cancers such as BC [271]. Moreover, the oxidative defluorination of sunitinib gives rise to the formation of chemically reactive metabolite, quinoneimine, which causes idiosyncratic hepatotoxicity (Figure 5C). The latter metabolite undergoes the conjugation with GSH to form quinoneimine-GSH conjugate of sunitinib. Recombinant CYP1A2 and CYP3A4 have been shown to generate the highest levels of toxic metabolites of sunitinib, with less contribution of CYP2D6 [272]. In human hepatocytes, the CYP3A4 inducer rifampicin has been shown to significantly increase the sunitinib N-dealkylation to the primary active metabolite N-desethyl-sunitinib. CYP1A induced by omeprazole markedly increases the generation of a quinoneimine-cysteine conjugate, a downstream metabolite of quinoneimine-GSH conjugate [273].
Metabolic activation of a dual-tyrosine kinase inhibitor lapatinib by CYPs has also been implicated in idiosyncratic hepatotoxicity [174]. CYP3A4 and CYP3A5 are the primary enzymes responsible for quinonimine-GSH adduct formation from lapatinib and O-dealkylated lapatinib as the CYP substrates [275]. The production of the following three primary metabolites of lapatinib: O-dealkylated lapatinib, N-dealkylated lapatinib, and N-hydroxy-lapatinib in HepG2 cells, which overexpress CYP1A1, CYP3A4, CYP3A5, and CYP3A7 enzymes has been shown [276]. The latter study showed that that the N-dealkylated lapatinib is the most toxic metabolite and lapatinib-induced cytotoxicity involves multiple mechanisms including apoptosis and DNA damage.
One of the mechanisms underlying the hepatotoxic effects of anticancer drugs is their photobiological properties, which contribute to CYP-induced photo(geno)toxicity as shown under in vitro conditions. Indeed, both lapatinib itself and its chemically reactive N-dealkylated derivative produced by CYP3A4 and CYP3A5 have been shown to exert photo- and genotoxicity, which cause the photosensitized protein and DNA damage [277]. Another anticancer drug gefitinib, a selective epidermal growth factor receptor inhibitor, undergoes metabolic bioactivation by CYP3A4, CYP3A5, CYP1A1, and CYP2D6 [278,279]. This is resulted in the formation of O-demethyl-, 4-defluoro-4-hydroxy-, and O-demorpholinopropyl-derivatives. Among them, the O-demethyl gefitinib has the highest photogenotoxic potential to cause cellular DNA damage, which is hardly repaired [180].
3.3.2. CYP Gene Polymorphisms and Liver Cancer
HCC is the sixth most common primary malignant tumor of the liver with increasing incidence rate worldwide and the third most frequent cause of cancer-related deaths worldwide as estimated in 2020 [281]. Liver cirrhosis is a main cause of HCC and together with inflammation associated with hepatitis B virus (HBV) or hepatitis C virus (HCV) accompanies early stages of HCC [282]. The exposure to procarcinogen and genotoxic agent AFB1 is a crucial factor in promoting HCC in individuals infected with the HBV [283], the AFB1 has been shown to trigger nuclear translocation of aryl hydrocarbon receptor (AHR), which functions as a transcription factor mediating CYP expression induced by AFB1 [284].
Silvestri et al. have studied polymorphisms in genes encoding five enzymes of the CYP superfamily, CYP1A1, CYP1A2, CYP2D6, CYP2E1, and CY3A4 as risk factors for liver cancer progression in HCV-infected patients [285]. They found that CYP2D6 poor metabolizer genotype was significantly less frequent in hepatitis/cirrhosis and HCC patients as compared to healthy subjects. The presence of two risk alleles in CYP2D6 gene in the same rapid-metabolizer subjects demonstrates the increased risk of HCC [286]. In the latter study, the relationship between the slow acetylator NAT2 genotype and HCC risk was more pronounced in patients lacking serum HBV and HCV markers.
The CYP2D6*10 mutant homozygote has been shown to be 2.8-fold less frequent in HCC than in controls whereas the CYP2C9*3 (42614A>C), and CYP3A5*3 (6986A>G) polymorphisms exerted definite effects on enzyme activities [287]. Statistically significant difference in genetic mutant alleles has been identified for the genotype of CYP2A6*4A homozygous in the HCC patients, which was significantly higher than that in healthy subjects of the Japanese population [288]. The CYP2D6 TT genotype has also been shown to associate with decreased risk of HCC due to decreased activity of the enzyme [289].
The CYP24A1 rs6013897 genotype has been shown to associate with cirrhosis and HCC as a predictor [290], whereas the c1/c1 genotype of CYP2E1 was found in 83.3% of patients with HCC and in 63.3% of controls [291]. SNPs in CYP1A1, CYP2E1, GSTM1, GSTT1, and SULT1A1 influence susceptibility towards HCC, considering their interaction with cigarette smoking [292]. In the latter study, the presence of any CYP2E1*5B variant allele and CYP2E1*6 variant allele were inversely associated with HCC risk. There was a borderline increased risk among carriers of combined CYP1A1*2A and SULT1A1 variant alleles. A significant biological interaction was observed between GSTT1 and smoking and borderline significant interaction was observed for SULT1A1 and smoking.
The CYP2E1-mediated constitutive metabolic activation has been shown to strongly correlate with incidence and severity of liver tumorigenesis [293]. The inhibition of CYP2E1 activity decreased hepatocyte proliferation, liver injury, and hepatocarcinogenesis in the presence of diethylnitrosamine. The lower activity of CYP2E1 accompanied by POR rs10954732 (G > A) polymorphism has been shown to decrease the susceptibility to HCC with prolonged to 199% OS [294]. POR downregulation in HCC was significantly correlated with better OS whereas POR overexpression causes the pentose phosphate pathway enzyme, glucose-6-phosphate dehydrogenase (G6PD), deficiency and suppression of HCC cell growth and metastasis [295]. The G6PH overexpression suppresses ferroptosis via CYP-catalyzed metabolic pathways and increases cancer cell viability and metastasis associated with poor OS of HCC patients.
4. Conclusions and Challenges
Human CYP enzymes demonstrate the broad and overlapping substrate specificities, and a balance of enzyme activities and substrate preferences vary depending on a variety of factors. These include CYP isoforms, CYP gene polymorphisms, tissue type, developmental stage and age, sex, and ethnicity. Indeed, CYP17A1 possesses both 17α-hydroxylase and C17,20-lyase activities to be involved in the conversions of several substrates in steroid biosynthesis pathways. CYP17A1 is required for conversion of C21 pregnenolone and progesterone to DHEA and androstenedione, both C19 androgen and C18 estrogen precursors. Moreover, the same CYP isoform can catalyze both bioactivation and detoxification reactions depending on a substrate type. For example, human CYP1A1 activates a diverse range of procarcinogens to produce strong carcinogenic metabolites, epoxides. Additionally, CYP1A1 has been shown to catalyze the activation of carbaryl to more toxic 5-hydroxycarbaryl; however, it catalyzes the detoxification of 2-nitoanisole to O-demethylated metabolite, 2-nitrophenol. Furthermore, the same CYP isoform can convert the same substrate to different products depending on a tissue type. For example, microsomal CYP1A2 converts B[c]PH, predominantly, to B[c]PH-3,4-dihydrodiol in human liver and B[c]PH-5,6-dihydrodiol in the lung.
On the contrary, different CYP isoforms can convert the same substrate to a variety of products, which are either more toxic or less toxic than their parent compound. Indeed, CYP2B6 is a main CYP enzyme responsible for the bioactivation of chlorpyrifos with the formation of more toxic oxon derivatives, whereas CYP2C19 is primarily responsible for chlorpyrifos detoxification with the formation of 3,4,5-tricholorpyrindinol. Additionally, CYP3A4-catalyzed metabolism of AFB1 causes its conversion less toxic AFQ1, whereas CYP1A2 converts it to more toxic exo- and endo-8,9-epoxides. Another example is antitumor drug ellipticine, which is detoxified by CYP1A1 and CYP1A2 via the formation of 7-hydroxy- and 9-hydroxyellipticine, whereas CYP3A4 produces 13-hydroxyellipticine via the reaction of N-hydroxylation to yield toxic ellipticine N2-oxide, the metabolites responsible for the formation of DNA adducts. The anticancer drug tamoxifen is metabolized to 4-hydroxytamoxifen by CYP2C19, whereas CYP3A4/CYP3A5 convert tamoxifen to N-desmethyltamoxifen, which is further metabolized to endoxifen by CYP2D6. All these conversions increase therapeutic effects of tamoxifen; however, hydroxylation of tamoxifen at ethyl group by CYP3A4 causes its conversion to α-hydroxytamoxifen, a genotoxic compound, which produces DNA adducts.
The involvement of CYPs in drug metabolism underlies drug-drug-interactions and drug resistance, the processes, which are under control of nuclear receptors such as PXR, CAR, and VDR that regulate the expression of MDR1 (P-gp), which serves as a transmembrane pump for drug efflux. The PXR and CAR can be activated by numerous CYP inducers including pregnane, steroid hormones, drugs, and environmental pollutants. Various PXR and CAR ligands and anticancer drugs including cisplatin, docetaxel, paclitaxel, and arsenic trioxide cause the upregulation of MDR1, whereas the nuclear receptors recognize drug responsive elements within the 5'-flanking promoter region of CYP genes to mediate the transcriptional upregulation of these genes in response to xenobiotics and drugs. Anticancer drugs and their metabolites accumulate in patients’ blood serum during therapy, which may increase toxicity of co-administered drugs due to drug-drug interactions. For example, imatinib increases the bleeding risks of rivaroxaban, widely used direct oral anticoagulant. Imatinib and gefitinib significantly inhibited CYP2J2- and CYP3A4-mediated metabolism of rivaroxaban and efflux transportation mediated by breast cancer resistance protein (BCRP)- and P-gp drug transporters.
CYP-catalyzed steroid hormone production and metabolism play critical roles in the development and pathogenesis of hormone-sensitive cancers in men women – prostate, breast, endometrial, and ovarian cancers. Current understanding of molecular mechanisms of the development of hormone-sensitive tumors is based on dysregulated CYP expression and mutagenic adduct formation, which cause cellular accumulation of toxic protein and DNA adducts to lead to cell damage and dysfunction. CYP1A1, CYP1A2, CYP1B1, CYP3A4/CYP3A5 are the most important CYPs involved in irreversible hydroxylation of 17β-estradiol and estrone to produce catechol estrogens, 2-hydroxy- and 4-hydroxy-derivatives, whereas CYP2C19, mostly, forms 16α-hydroxy-derivative. However, the 16α-hydroxy-derivatives of estrogens serve as ERα agonist properties whereas their 2-hydroxy- and 4-hydroxy-derivatives along with methoxy-derivatives and conjugated estrogens exhibit tumor protective activities. In prostate cancer, the key roles have been attributed to CYP17A1 and sustained intra-tumoral synthesis of 5α-dihydrotestosterone, more potent AR agonist than testosterone. This dictates the importance of anticancer drug development based on steroidal and non-steroidal CYP17A1 and AR inhibitors.
The variations in CYP activities are provided by diverse factors including CYP gene polymorphisms, which affect interindividual differences in cancer and drug susceptibilities. Various SNPs in genes encoding CYPs involved in estrogen and androgen biosynthesis and metabolism have been implicated in increased risk of hormone-sensitive cancers. However, polymorphisms in only some CYP isoforms have been identified to show statistically significant association with breast, endometrial, ovarian, prostate, and liver cancers in different ethnic groups. Therefore, more data are needed to clearly elucidate the associations between various types of gene polymorphisms in genes encoding CYPs and other detoxifying and drug-metabolizing enzymes. Additionally, many questions on the involvement of CYPs in drug metabolism and adverse drug reactions, which underlie the patients’ response to treatment are still challenging.
References
- D.R. Nelson, A world of cytochrome P450s, Philos Trans R Soc Lond B Biol Sci., 368(1612) (2013), p. 20120430. [CrossRef]
- D.R. Nelson, Cytochrome P450 diversity in the tree of life, Biochim. Biophys. Acta Proteins Proteom., 1866(1) (2018), p. 141-154. [CrossRef]
- M. Šrejber, V. Navrátilová, M. Paloncýová, V. Bazgier, K. Berka, P. Anzenbacher, M. Otyepka, Membrane-attached mammalian cytochromes P450: An overview of the membrane's effects on structure, drug binding, and interactions with redox partners, J. Inorg. Biochem., 183 (2018), p. 117-136. [CrossRef]
- M. Nishimura, H. Yaguti, H. Yoshitsugu, S. Naito, T. Satoh, Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high-sensitivity real-time reverse transcription PCR. Yakugaku Zasshi, 123(5) (2003), p. 369-375. [CrossRef]
- N.C. Sadler, P. Nandhikonda, B.J. Webb-Robertson, C. Ansong, L.N. Anderson, J.N. Smith, R.A. Corley, A.T. Wright, Hepatic cytochrome P450 activity, abundance, and expression throughout human development, Drug Metab. Dispos., 44(7) (2016), p. 984-991. [CrossRef]
- J. McGraw, D. Waller, Cytochrome P450 variations in different ethnic populations, Expert Opin. Drug Metab. Toxicol. 8(3) (2012), p. 371-382. [CrossRef]
- S.H. Gerges, A.O.S. El-Kadi, Sexual dimorphism in the expression of cytochrome P450 enzymes in rat heart, liver, kidney, lung, brain, and small intestine, Drug Metab. Dispos. 51(1) (2023), p. 81-94. [CrossRef]
- J.C. Fuscoe, V. Vijay, J.P. Hanig, T. Han, L. Ren, J.J. Greenhaw, R.D. Beger, L.M. Pence, Q. Shi, Hepatic transcript profiles of cytochrome P450 genes predict sex differences in drug metabolism, Drug Metab. Dispos. 48(6) (2020), p. 447-458. [CrossRef]
- M.F. Hoffmann, S.C. Preissner, J. Nickel, M. Dunkel, R. Preissner, S. Preissner, The Transformer database: biotransformation of xenobiotics, Nucleic Acids Res., 42(Database issue) (2014) D1113-D1117. [CrossRef]
- S.C. Khojasteh, N.N. Bumpus, J.P. Driscoll, G.P. Miller, K. Mitra, I.M.C.M. Rietjens, D. Zhang, Biotransformation and bioactivation reactions - 2018 literature highlights, Drug Metab. Rev., 51(2) (2019), p. 121-161. [CrossRef]
- M. Zhao, J. Ma, M. Li, Y. Zhang, B. Jiang, X. Zhao, C. Huai, L. Shen, N. Zhang, L. He, S. Qin, Cytochrome P450 enzymes and drug metabolism in humans, Int. J. Mol. Sci., 22(23) (2021), p. 12808. [CrossRef]
- F.P. Guengerich, Y. Tateishi, K.D. McCarty, F.K. Yoshimoto, Updates on mechanisms of cytochrome P450 catalysis of complex steroid oxidations, Int. J. Mol. Sci., 25(16) (2024), p. 9020. [CrossRef]
- A.R. Modi, J.H. Dawson, Oxidizing intermediates in P450 catalysis: a case for multiple oxidants, Adv. Exp. Med. Biol., 851 (2015), p. 63-81. [CrossRef]
- K. Fujiyama, T. Hino, S. Nagano, Diverse reactions catalyzed by cytochrome P450 and biosynthesis of steroid hormone, Biophys. Physicobiol., (19) (2022), p. e190021. [CrossRef]
- D.W. Nebert, K. Wikvall, W.L. Miller, Human cytochromes P450 in health and disease, Philos Trans R Soc Lond B Biol Sci., 368(1612) (2013), p. 20120431. [CrossRef]
- Y. Yan, J. Wu, G. Hu, C. Gao, L. Guo, X. Chen, L. Liu, W. Song, Current state and future perspectives of cytochrome P450 enzymes for C-H and C=C oxygenation, Synth. Syst. Biotechnol., 7(3) (2022), p. 887-899. [CrossRef]
- J. Ducharme, K. Auclair, Use of bioconjugation with cytochrome P450 enzymes, Biochim. Biophys. Acta Proteins Proteom., 1866(1) (2018), p. 32-51. [CrossRef]
- M. Silva, M.D.G. Carvalho, Detoxification enzymes: cellular metabolism and susceptibility to various diseases, Rev. Assoc. Med. Bras., 64(4) (2018), p. 307-310. [CrossRef]
- I.M. Mokhosoev, D.V. Astakhov, A.A. Terentiev, N.T. Moldogazieva, Cytochrome P450 monooxygenase systems: Diversity and plasticity for adaptive stress response, Prog. Biophys. Mol. Biol., (193) (2024), p. 19-34. [CrossRef]
- M. Ingelman-Sundberg, Cytochrome P450 polymorphism: From evolution to clinical use, Adv. Pharmacol., 95 (2022), p.393-416. [CrossRef]
- I.S. Lee, D. Kim, Polymorphic metabolism by functional alterations of human cytochrome P450 enzymes, Arch. Pharm. Res., 34(110) (2011), p. 1799-1816. [CrossRef]
- N. Gao, X. Tian, Y. Fang, J. Zhou, H. Zhang, Q. Wen, L. Jia, J. Gao, B. Sun, J. Wei, Y. Zhang, M. Cui, H. Qiao, Gene polymorphisms and contents of cytochrome P450s have only limited effects on metabolic activities in human liver microsomes, Eur. J. Pharm. Sci., (92) (2016), p. 86-97. [CrossRef]
- J. Liu, Y.F. Lu, J.C. Corton, C.D. Klaassen, Expression of cytochrome P450 isozyme transcripts and activities in human livers, Xenobiotica, 51(3) (2021), p. 279-286. [CrossRef]
- S.F. Zhou, J.P. Liu, B. Chowbay, Polymorphism of human cytochrome P450 enzymes and its clinical impact, Drug Metab. Rev., 41(2) (2009), p. 89-295. [CrossRef]
- F. Bray, M. Laversanne, E. Weiderpass, I. Soerjomataram, The ever-increasing importance of cancer as a leading cause of premature death worldwide, Cancer, 127(16) (2021), p. 3029-3030. [CrossRef]
- C. Mattiuzzi, G. Lippi, Current cancer epidemiology, J. Epidemiol. Glob. Health., (9)4 (2019), p. 217-222. [CrossRef]
- H. Sung, J. Ferlay, R.L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal, F. Bray, Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J. Clin., 71(3), p. 209-249. [CrossRef]
- N.M. Anderson, M.C. Simon, The tumor microenvironment, Curr. Biol., 30(16) (2020), p. R921-R925. [CrossRef]
- C.M. Neophytou, M. Panagi, T. Stylianopoulos, P. Papageorgis, The role of tumor microenvironment in cancer metastasis: Molecular mchanisms and therapeutic opportunities, Cancers (Basel), 13(9) (2021), p. 2053. [CrossRef]
- M. Benvenuto, C. Focaccetti, Tumor microenvironment: Cellular interaction and metabolic adaptations, Int. J. Mol. Sci., 25(7) (2024), p. 3642. [CrossRef]
- S. Arfin, N.K. Jha, S.K. Jha, K.K. Kesari, J. Ruokolainen, S. Roychoudhury, B. Rathi, D. Kumar, Oxidative stress in cancer cell metabolism, Antioxidants (Basel), 10(5) (2021), p. 642. [CrossRef]
- N.T. Moldogazieva, I.M. Mokhosoev, A.A. Terentiev, Metabolic heterogeneity of cancer cells: An interplay between HIF-1, GLUTs, and AMPK, Cancers (Basel) 12(4) (2020). p. 862. [CrossRef]
- N.T. Moldogazieva, S.P. Zavadskiy, D.V. Astakhov, A.A. Terentiev, Lipid peroxidation: Reactive carbonyl species, protein/DNA adducts, and signaling switches in oxidative stress and cancer, Biochem. Biophys. Res. Commun., 687 (2023), p. 149167. [CrossRef]
- S. Zavadskiy, S. Sologova, N. Moldogazieva, Oxidative distress in aging and age-related diseases: Spatiotemporal dysregulation of protein oxidation and degradation, Biochimie, 195 (2022), p. 114-134. [CrossRef]
- A.I. Archakov, I.M. Mokhosoev, Modification of proteins by active oxygen and their degradation, Biokhimiia, 54(2) (1989), p. 179-186.
- A.M. Alzahrani, P. Rajendran, The multifarious link between cytochrome P450s and cancer, Oxid. Med. Cell Longev., 2020 (2020), p. 3028387. [CrossRef]
- F.P. Guengerich, Cytochrome P450 enzymes as drug targets in human disease, Drug Metab. Dispos., 52(6) (2024), p. 493-497. [CrossRef]
- B. Hossam Abdelmonem, N.M. Abdelaal, E.K.E. Anwer, A.A. Rashwan, M.A. Hussein, Y.F. Ahmed, R. Khashana, M.M. Hanna, A. Abdelnaser, Decoding the role of CYP450 enzymes in metabolism and disease: A comprehensive review, Biomedicines, 12(7) (2024), p. 1467. [CrossRef]
- D.Y. Cooper, R.W. Estabrook, O. Rosenthal, The stoichiometry of C21 hydroxylation of steroids by adrenocortical microsomes, J. Biol. Chem., 238 (1963), p. 1320-1323.
- D.Y. Cooper, S. Levin, S. Narasimhulu, O. Rosenthal, Photochemical action spectrum of the terminal oxidase of the mixed function oxidase systems, Science, 147(3556) (1965), p. 400-402. [CrossRef]
- R.H. Menard, S.A. Latif, J.L. Purvis, The intratesticular localization of cytochrome P-450 and cytochrome P-450-dependent enzymes in the rat testis, Endocrinology, 97(6) (1975) p. 1587-1592. [CrossRef]
- J.L. Purvis, R.H. Menard, Compartmentation of microsomal cytochrome P-450 and 17 alpha-hydroxylase activity in the rat testis, Curr. Top Mol. Endocrinol., 2 (1975), p. 65-84. [CrossRef]
- S. Nakajin, P.F. Hall, Microsomal cytochrome P-450 from neonatal pig testis. Purification and properties of A C21 steroid side-chain cleavage system (17 alpha-hydroxylase-C17,20 lyase), J. Biol. Chem., 256(8) (1981), p. 3871-3876.
- L. Schiffer, L. Barnard, E.S. Baranowski, L.C. Gilligan, A.E. Taylor, W. Arlt, C.H.L. Shackleton, K.H. Storbeck, Human steroid biosynthesis, metabolism and excretion are differentially reflected by serum and urine steroid metabolomes: A comprehensive review, J. Steroid Biochem. Mol. Biol., 194 (2019), p. 105439. [CrossRef]
- Y. Chien, K. Rosal, B.C. Chung, Function of CYP11A1 in the mitochondria, Mol. Cell. Endocrinol., 441 (2017), p. 55-61. [CrossRef]
- K. Fujiyama, T. Hino, S. Nagano, Diverse reactions catalyzed by cytochrome P450 and biosynthesis of steroid hormone, Biophys. Physicobiol., 19 (2022), p. e190021. [CrossRef]
- A.J. Conley, I.M. Bird, The role of cytochrome P450 17 alpha-hydroxylase and 3 beta-hydroxysteroid dehydrogenase in the integration of gonadal and adrenal steroidogenesis via the delta 5 and delta 4 pathways of steroidogenesis in mammals, Biol. Reprod., 56(4) (1997), p. 789-799. [CrossRef]
- D.H. Geller, R.J. Auchus, W.L. Miller, P450c17 mutations R347H and R358Q selectively disrupt 17,20-lyase activity by disrupting interactions with P450 oxidoreductase and cytochrome b5, Mol. Endocrinol., 13(1) (1999), p. 167-175. [CrossRef]
- W.L. Miller, Androgen biosynthesis from cholesterol to DHEA, Mol. Cell. Endocrinol., 198(1-2) (2002), p. 7-14. [CrossRef]
- C.M. Klinge, B.J. Clark, R.A. Prough, Dehydroepiandrosterone research: Past, current, and future, Vitam. Horm., 108 (2018), p. 1-28. [CrossRef]
- E.M. Petrunak, N.M. DeVore, P.R. Porubsky, E.E. Scott, Structures of human steroidogenic cytochrome P450 17A1 with substrates, J. Biol. Chem., 289(47) (2014), p. 32952-32964. [CrossRef]
- T.M. Penning, The aldo-keto reductases (AKRs): Overview, Chem. Biol. Interact., 234 (2015), p. 236-246. [CrossRef]
- C. Stocco, Tissue physiology and pathology of aromatase, Steroids, 77(1-2) (2012), p. 27-35. [CrossRef]
- Y. Takeda, M. Demura, M. Kometani, S. Karashima, T. Yoneda, Y. Takeda, Molecular and epigenetic control of aldosterone synthase, CYP11B2 and 11-hydroxylase, CYP11B1, Int. J. Mol. Sci., 24(6) (2023), p. 5782. [CrossRef]
- A. Midzak, V. Papadopoulos, Adrenal mitochondria and steroidogenesis: From individual proteins to functional protein assemblies, Front. Endocrinol. (Lausanne), 7 (2016), p. 106. [CrossRef]
- S. Brixius-Anderko, E.E. Scott, Structural and functional insights into aldosterone synthase interaction with its redox partner protein adrenodoxin, J. Biol. Chem., 296 (2021), p. 100794. [CrossRef]
- G.A. Ziegler, G.E. Schulz, Crystal structures of adrenodoxin reductase in complex with NADP+ and NADPH suggesting a mechanism for the electron transfer of an enzyme family, Biochemistry 39(36) (2000), p. 10986-10995. [CrossRef]
- S. Rendic, Summary of information on human CYP enzymes: human P450 metabolism data, Drug Metab. Rev., 34(1-2) (2002), p. 83-448. [CrossRef]
- S. Rendic, F.P. Guengerich, Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicals, Chem. Res. Toxicol., 28(1) (2015), p. 38-42. [CrossRef]
- B. Merchel Piovesan Pereira, I. Tagkopoulos, Benzalkonium chlorides: Uses, regulatory status, and microbial resistance, Appl. Environ. Microbiol., 85(13) (2019), p. e00377-19. [CrossRef]
- W.A. Arnold, A. Blum, J. Branyan, T.A. Bruton, C.C. Carignan, G. Cortopassi, S. Datta, J. DeWitt, A.C. Doherty, R.U. Halden, H. Harari, E.M. Hartmann, T.C. Hrubec, S. Iyer, C.F. Kwiatkowski, J. LaPier, D. Li, L. Li, J.G. Muñiz Ortiz, A. Salamova, T. Schettler, R.P. Seguin, A. Soehl, R. Sutton, L. Xu, G. Zheng, Quaternary ammonium compounds: A chemical class of emerging concern, Environ. Sci. Technol., 57(20) (2023), p. 7645-7665. [CrossRef]
- R.P. Seguin, J.M. Herron, V.A. Lopez, J.L. Dempsey, L. Xu, Metabolism of benzalkonium chlorides by human hepatic cytochromes P450, Chem. Res. Toxicol., 32(12) (2019), p. 2466-2478. [CrossRef]
- S.H. Kim, D. Kwon, S. Lee, S.W. Son, J.T. Kwon, P.-J. Kim, Y.-H. Lee, Y.-S. Jung, Concentration- and time-dependent effects of benzalkonium chloride in human lung epithelial cells: Necrosis, apoptosis, or epithelial mesenchymal transition, Toxics, 8 (2020), p. 17. doi: 10.3390/toxics8010017.
- A.M. King, C.K. Aaron, Organophosphate and carbamate poisoning, Emerg. Med. Clin. North Am., 33(1) (2015), p. 133-151. [CrossRef]
- D.A. Jett, J.R. Richardson, Neurotoxic pesticides. In: Dobbs MR, editor. Clinical Neurotoxicology: Syndromes, Substances, and Environments. San Diego, CA: Elsevier; 2009. pp. 491–499.
- Y.H. Jan, J.R. Richardson, A.A. Baker, V. Mishin, D.E. Heck, D.L. Laskin, J.D. Laskin, Novel approaches to mitigating parathion toxicity: targeting cytochrome P450-mediated metabolism with menadione, Ann. N. Y. Acad. Sci., 1378(1) (2016), p. 80-86. [CrossRef]
- Y.H. Yan, J.R. Richardson, A.A. Baker, V. Mishin, D.E. Heck, D.L. Laskin, J.D. Laskin, Vitamin K3 (menadione) redox cycling inhibits cytochrome P450-mediated metabolism and inhibits parathion intoxication, Toxicol. Appl. Pharmacol., 288(1) (2015), p. 114-120. [CrossRef]
- R.J. Foxenberg, B.P. McGarrigle, J.B. Knaak, P.J. Kostyniak, J.R. Olson, Human hepatic cytochrome p450-specific metabolism of parathion and chlorpyrifos, Drug Metab. Dispos., 35(2) (2007), p. 189-193. [CrossRef]
- H. Imaishi, T. Goto, Effect of genetic polymorphism of human CYP2B6 on the metabolic activation of chlorpyrifos, Pestic. Biochem. Physiol., 144 (2018), p. 42-48. [CrossRef]
- A.L. Crane, K. Klein, J.R. Olson, Bioactivation of chlorpyrifos by CYP2B6 variants, Xenobiotica, 42(12) (2012), p. 1255-1262. [CrossRef]
- J. Tang, Y. Cao, R.L. Rose, E. Hodgson, In vitro metabolism of carbaryl by human cytochrome P450 and its inhibition by chlorpyrifos, Chem. Biol. Interact., 141(3) (2002), p. 229-241. [CrossRef]
- K.A. Usmani, T.M. Cho, R.L. Rose, E. Hodgson, Inhibition of the human liver microsomal and human cytochrome P450 1A2 and 3A4 metabolism of estradiol by deployment-related and other chemicals, Drug Metab. Dispos., 34(9) (2006), p. 1606-1614. [CrossRef]
- S. Rendic, F.P. Guengerich, Contributions of human enzymes in carcinogen metabolism, Chem. Res. Toxicol., 25(7) (2012), p. 1316-1383. [CrossRef]
- Y.W. Sun, F.P. Guengerich, A.K. Sharma, T. Boyiri, S. Amin, K. el-Bayoumy, Human cytochromes P450 1A1 and 1B1 catalyze ring oxidation but not nitroreduction of environmental pollutant mononitropyrene isomers in primary cultures of human breast cells and cultured MCF-10A and MCF-7 cell lines, Chem. Res. Toxicol., 17(8) (2004), p. 1077-1085. [CrossRef]
- L. Reed, V.M. Arlt, D.H. Phillips, The role of cytochrome P450 enzymes in carcinogen activation and detoxication: an in vivo-in vitro paradox, Carcinogenesis, 39(7) (2018), p. 851-859. [CrossRef]
- T. Shimada, C.L. Hayes, H. Yamazaki, S. Amin, S.S. Hecht, F.P. Guengerich, T.R. Sutter, Activation of chemically diverse procarcinogens by human cytochrome P-450 1B1, Cancer Res., 56(13) (1996), p. 2979-2984.
- T. Shimada, E.M. Gillam, Y. Oda, F. Tsumura, T.R. Sutter, F.P. Guengerich, K. Inoue, Metabolism of benzo[a]pyrene to trans-7,8-dihydroxy-7, 8-dihydrobenzo[a]pyrene by recombinant human cytochrome P450 1B1 and purified liver epoxide hydrolase, Chem. Res. Toxicol., 12(7) (1999), p. 623-629. [CrossRef]
- J. Jacob, G. Raab, V. Soballa, W.A. Schmalix, G. Grimmer, H. Greim, J. Doehmer, A. Seidel, Cytochrome P450-mediated activation of phenanthrene in genetically engineered V79 Chinese hamster cells, Environ. Toxicol. Pharmacol., 1(1) (1996), p. 1-11. [CrossRef]
- M. Baum, S. Amin, F.P. Guengerich, S.S. Hecht, W. Köhl, G. Eisenbrand, Metabolic activation of benzo[c]phenanthrene by cytochrome P450 enzymes in human liver and lung, Chem. Res. Toxicol., 14(6) (2001), p. 686-693. [CrossRef]
- A. Seidel, V.J. Soballa, G. Raab, H. Frank, H. Greim, G. Grimmer, J. Jacob, J. Doehmer, Regio- and stereoselectivity in the metabolism of benzo[c]phenanthrene mediated by genetically engineered V79 Chinese hamster cells expressing rat and human cytochromes P450, Environ. Toxicol. Pharmacol., 5(3) (1998), p. 179-196. [CrossRef]
- Wang Y, Li K, Zou Y, Zhou M, Li J, Wu C, Tan R, Liao Y, Li W, Zheng J. Metabolic activation of 3-aminodibenzofuran mediated by P450 enzymes and sulfotransferases. Toxicol Lett., 360 (2022), p. 44-52. [CrossRef]
- M. Pitaro, N. Croce, V. Gallo, A. Arienzo, G. Salvatore, G. Antonini, Coumarin-induced hepatotoxicity: A narrative review, Molecules, 27(24) (2022), p. 9063. [CrossRef]
- N. Murayama, H. Yamazaki, Metabolic activation and deactivation of dietary-derived coumarin mediated by cytochrome P450 enzymes in rat and human liver preparations, J. Toxicol. Sci., 46(8) (2021), p. 371-378. [CrossRef]
- Y.F. Ueng, T. Shimada, H. Yamazaki, F.P. Guengerich, Oxidation of aflatoxin B1 by bacterial recombinant human cytochrome P450 enzymes, Chem. Res. Toxicol., 8(2) (1995), p. 218-225. [CrossRef]
- F.P. Guengerich, W.W. Johnson, T. Shimada, Y.F. Ueng, H. Yamazaki, S. Langouët, Activation and detoxication of aflatoxin B1, Mutat. Res., 402(1-2) (1998), p. 121-128. [CrossRef]
- T. Shimada, F.P. Guengerich, Evidence for cytochrome P-450NF, the nifedipine oxidase, being the principal enzyme involved in the bioactivation of aflatoxins in human liver, Proc. Natl. Acad. Sci. U S A, 86(2) (1989), p. 462-465. [CrossRef]
- V.M. Arlt, A. Hewer, B.L. Sorg, H.H. Schmeiser, D.H. Phillips, M. Stiborova, 3-aminobenzanthrone, a human metabolite of the environmental pollutant 3-nitrobenzanthrone, forms DNA adducts after metabolic activation by human and rat liver microsomes: evidence for activation by cytochrome P450 1A1 and P450 1A2, Chem. Res. Toxicol., 17(8) (2004), p. 1092-1101. [CrossRef]
- M. Miksanová, M. Sulc, H. Rýdlová, H.H. Schmeiser, E. Frei, M. Stiborová, Enzymes involved in the metabolism of the carcinogen 2-nitroanisole: evidence for its oxidative detoxication by human cytochromes P450, Chem. Res. Toxicol., 17(5) (2004), p. 663-671. [CrossRef]
- H. Dracínska, M. Miksanová, M. Svobodová, S. Smrcek, E. Frei, H.H. Schmeiser, M. Stiborová, Oxidative detoxication of carcinogenic 2-nitroanisole by human, rat and rabbit cytochrome P450, Neuro Endocrinol. Lett., 27 (Suppl 2) (2006), p. 9-13.
- U.M. Zanger, M. Schwab, Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation, Pharmacol. Ther., 138(1) (2013), p. 103-141. [CrossRef]
- Almazroo, M.K. Miah, R. Venkataramanan, Drug metabolism in the liver, Clin. Liver Dis., 21(1) (2017), p. 1-20. [CrossRef]
- S.P. Rendic, F.P. Guengerich, Human family 1-4 cytochrome P450 enzymes involved in the metabolic activation of xenobiotic and physiological chemicals: an update, Arch. Toxicol., 95(2) (2021), p. 395-472. [CrossRef]
- B. Achour, J. Barber, A. Rostami-Hodjegan, Expression of hepatic drug-metabolizing cytochrome p450 enzymes and their intercorrelations: a meta-analysis, Drug Metab. Dispos., 42(8) (2014), p. 1349-1356. [CrossRef]
- M. Samadi-Baboli, G. Favre, E. Blancy, G. Soula, Preparation of low density lipoprotein-9-methoxy-ellipticin complex and its cytotoxic effect against L1210 and P 388 leukemic cells in vitro, Eur. J. Cancer Clin. Oncol., 25(2) (1989), p. 233-241. [CrossRef]
- S.J. Froelich-Ammon, M.W. Patchan, N. Osheroff, R.B. Thompson, Topoisomerase II binds to ellipticine in the absence or presence of DNA. Characterization of enzyme-drug interactions by fluorescence spectroscopy, J. Biol. Chem., 270(25) (1995), p. 14998-5004. [CrossRef]
- M. Stiborová, J. Sejbal, L. Borek-Dohalská, D. Aimová, J. Poljaková, K. Forsterová, M. Rupertová, J. Wiesner, J. Hudecek, M. Wiessler, E. Frei, The anticancer drug ellipticine forms covalent DNA adducts, mediated by human cytochromes P450, through metabolism to 13-hydroxyellipticine and ellipticine N2-oxide, Cancer Res., 64(22) (2004), p. 8374-8380. [CrossRef]
- V. Kotrbová, B. Mrázová, M. Moserová, V. Martínek, P. Hodek, J. Hudeček, E. Frei, M. Stiborová, Cytochrome b(5) shifts oxidation of the anticancer drug ellipticine by cytochromes P450 1A1 and 1A2 from its detoxication to activation, thereby modulating its pharmacological efficacy, Biochem. Pharmacol., 82(6) (2011), p. 669-680. [CrossRef]
- L. Pichard-Garcia, R.J. Weaver, N. Eckett, G. Scarfe, J.M. Fabre, C. Lucas, P. Maurel, The olivacine derivative s 16020 (9-hydroxy-5,6-dimethyl-N-[2-(dimethylamino)ethyl)-6H-pyrido(4,3-B)-carbazole-1-carboxamide) induces CYP1A and its own metabolism in human hepatocytes in primary culture, Drug Metab. Dispos., 32(1) (2004), p. 80-88. [CrossRef]
- B. Tylińska, B. Wiatrak, Bioactive olivacine derivatives-potential application in cancer therapy, Biology (Basel), 10(6) (2021), p. 564. [CrossRef]
- J. Piasny, B. Wiatrak, A. Dobosz, B. Tylińska, T. Gębarowski, Antitumor activity of new olivacine derivatives, Molecules, 25(11) (2020), p. 2512. [CrossRef]
- Y.H. Hsiang, R. Hertzberg, S. Hecht, L.F. Liu, Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I, J. Biol. Chem., 260(27) (1985), p. 14873-14878.
- M.C. Haaz, C. Riché, L.P. Rivory, J. Robert, Biosynthesis of an aminopiperidino metabolite of irinotecan [7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecine] by human hepatic microsomes, Drug Metab. Dispos., 26(8) (1998), p. 769-774.
- A. Santos, S. Zanetta, T. Cresteil, A. Deroussent, F. Pein, E. Raymond, L. Vernillet, M.L. Risse, V. Boige, A. Gouyette, G. Vassal, Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans, Clin. Cancer Res., 6(5) (2000), p. 2012-2020.
- Early Breast Cancer Trialists' Collaborative Group (EBCTCG); C. Davies, J. Godwin, R. Gray, M. Clarke, D. Cutter, S. Darby, P. McGale, H.C. Pan, C. Taylor, Y.C. Wang, M. Dowsett, J. Ingle, R. Peto, Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials, Lancet, 378(9793) (2011), p. 771-784. [CrossRef]
- M.S. Singh, P.A. Francis, M. Michael, Tamoxifen, cytochrome P450 genes and breast cancer clinical outcomes, Breast, 20(2) (2011), p. 111-118. [CrossRef]
- M.P. Goetz, S.K. Knox, V.J. Suman, J.M. Rae, S.L. Safgren, M.M. Ames, D.W. Visscher, C. Reynolds, F.J. Couch, W.L. Lingle, R.M. Weinshilboum, E.G. Fritcher, A.M. Nibbe, Z. Desta, A. Nguyen, D.A. Flockhart, E.A. Perez, J.N. Ingle, The impact of cytochrome P450 2D6 metabolism in women receiving adjuvant tamoxifen, Breast Cancer Res. Treat., 101(1) (2007), p. 113-121. [CrossRef]
- E.Y. Tan, L. Bharwani, Y.H. Chia, R.C.T. Soong, S.S.Y. Lee, J.J.C. Chen, P.M.Y Chan, Impact of cytochrome P450 2D6 polymorphisms on decision-making and clinical outcomes in adjuvant hormonal therapy for breast cancer, World J. Clin. Oncol., 13(8) (2022), p. 712-724. [CrossRef]
- J.C. Stingl, S. Parmar, A. Huber-Wechselberger, A. Kainz, W. Renner, A. Seeringer, J. Brockmöller, U. Langsenlehner, P. Krippl, E. Haschke-Becher, Impact of CYP2D6*4 genotype on progression free survival in tamoxifen breast cancer treatment, Curr. Med. Res. Opin., 26(11) (2010), p. 2535-2542. [CrossRef]
- Z. Desta, B.A. Ward, N.V. Soukhova, D.A. Flockhart, Comprehensive evaluation of tamoxifen sequential biotransformation by the human cytochrome P450 system in vitro: prominent roles for CYP3A and CYP2D6, J. Pharmacol. Exp. Ther., 310(3) (2004), p. 1062-1075. [CrossRef]
- D.J. Boocock, K. Brown, A.H. Gibbs, E. Sanchez, K.W. Turteltaub, I.N. White, Identification of human CYP forms involved in the activation of tamoxifen and irreversible binding to DNA, Carcinogenesis, 23(11) (2002), p. 1897-901. [CrossRef]
- Y.C. Lim, Z. Desta, D.A. Flockhart, T.C. Skaar, Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen, Cancer Chemother. Pharmacol., 55(5) (2005), p. 471-478. [CrossRef]
- D.J. Boocock, J.L. Maggs, I.N. White, B.K. Park, Alpha-hydroxytamoxifen, a genotoxic metabolite of tamoxifen in the rat: identification and quantification in vivo and in vitro, Carcinogenesis, 20(1) (1999), p. 153-160. [CrossRef]
- Y. Okamoto, S. Shibutani, Development of novel and safer anti-breast cancer agents, SS1020 and SS5020, based on a fundamental carcinogenic research, Genes Environ., 41 (2019), p. 9. [CrossRef]
- D.J. Klein, C.F. Thorn, Z. Desta, D.A. Flockhart, R.B. Altman, T.E. Klein, PharmGKB summary: tamoxifen pathway, pharmacokinetics, Pharmacogenet. Genomics, 23(11) (2013), p. 643-647. [CrossRef]
- N.T. Moldogazieva, D.S. Ostroverkhova, N.N. Kuzmich, V.V. Kadochnikov, A.A. Terentiev, Y.B. Porozov, Elucidating binding sites and affinities of ERα agonists and antagonists to human alpha-fetoprotein by in silico modeling and point mutagenesis, Int. J. Mol. Sci., 21(3) (2020), p. 893. [CrossRef]
- J.N. Mills, A.C. Rutkovsky, A. Giordano, Mechanisms of resistance in estrogen receptor positive breast cancer: overcoming resistance to tamoxifen/aromatase inhibitors, Curr. Opin. Pharmacol., (41) (2018), p. 59-65. [CrossRef]
- K. Kiyotani, T. Mushiroda, Y. Nakamura, H. Zembutsu, Pharmacogenomics of tamoxifen: roles of drug metabolizing enzymes and transporters, Drug Metab. Pharmacokinet., 27(1) (2012), p. 122-131. [CrossRef]
- Y. Ando, E. Fuse, W.D. Figg, Thalidomide metabolism by the CYP2C subfamily, Clin. Cancer Res., 8(6) (2002), p. 1964-1973.
- P. Chhetri, A. Giri, S. Shakya, S. Shakya, B. Sapkota, K.C. Pramod, Current development of anti-cancer drug S-1, J. Clin. Diagn. Res., 10(11) (2016), p.XE01-XE05. [CrossRef]
- Yamamiya, K. Yoshisue, Y. Ishii, H. Yamada, K. Yoshida, Enantioselectivity in the cytochrome P450-dependent conversion of tegafur to 5-fluorouracil in human liver microsomes, Pharmacol. Res. Perspect., 1(1) (2013), p. e00009. [CrossRef]
- J.D. Tebbens, M. Azar, E. Friedmann, M. Lanzendörfer, P. Pávek, Mathematical models in the description of pregnane X receptor (PXR)-regulated cytochrome P450 enzyme induction, Int. J. Mol. Sci., 19(6) (2018), p. 1785. [CrossRef]
- T. Smutny, S. Mani, P. Pavek, Post-translational and post-transcriptional modifications of pregnane X receptor (PXR) in regulation of the cytochrome P450 superfamily, Curr. Drug Metab., 14(10) (2013), p. 1059-1069. [CrossRef]
- G. Bradley, P.F. Juranka, V. Ling, Mechanism of multidrug resistance, Biochim. Biophys. Acta, 948(1) (1988), p. 87-128. [CrossRef]
- R.L. Juliano, V. Ling, A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants, Biochim. Biophys. Acta, 455(1) (1976), p. 152-162. [CrossRef]
- Fardel, L. Payen, A. Courtois, L. Vernhet, V. Lecureur, Regulation of biliary drug efflux pump expression by hormones and xenobiotics, Toxicology, 167(1) (2001), p. 37-46. [CrossRef]
- Y. Lv, Y.Y. Luo, H.W. Ren, C.J. Li, Z.X. Xiang, Z.L. Luan, The role of pregnane X receptor (PXR) in substance metabolism, Front. Endocrinol. (Lausanne), 13 (2022), p. 959902. [CrossRef]
- P. Germain, B. Staels, C. Dacquet, M. Spedding, V. Laudet, Overview of nomenclature of nuclear receptors, Pharmacol. Rev., 58(4) (2006), p.685-704. [CrossRef]
- Y. Li, M.H. Lambert, H.E. Xu, Activation of nuclear receptors: a perspective from structural genomics, Structure, 11(7) (2003), p. 741-746. [CrossRef]
- X. Ma, J.R. Idle, F.J. Gonzalez, The pregnane X receptor: from bench to bedside, Expert Opin. Drug Metab. Toxicol., 4(7) (2008), p. 895-908. [CrossRef]
- J. Huwyler, M.B. Wright, H. Gutmann, J. Drewe, Induction of cytochrome P450 3A4 and P-glycoprotein by the isoxazolyl-penicillin antibiotic flucloxacillin, Curr. Drug Metab., 7(2) (2006), p. 119-126. [CrossRef]
- F. Lakehal, P.M. Dansette, L. Becquemont, E. Lasnier, R. Delelo, P. Balladur, R. Poupon, P.H. Beaune, C. Housset, Indirect cytotoxicity of flucloxacillin toward human biliary epithelium via metabolite formation in hepatocytes, Chem. Res. Toxicol., 14(6) (2001), p. 694-701. [CrossRef]
- T. Kawashiro, K. Yamashita, X.J. Zhao, E. Koyama, M. Tani, K. Chiba, T. Ishizaki, A study on the metabolism of etoposide and possible interactions with antitumor or supporting agents by human liver microsomes, J. Pharmacol. Exp. Ther., 286(3) (1998), p. 1294-1300.
- J.S. Lagas, L. Fan, E. Wagenaar, M.L. Vlaming, O. van Tellingen, J.H. Beijnen, A.H. Schinkel, P-glycoprotein (P-gp/Abcb1), Abcc2, and Abcc3 determine the pharmacokinetics of etoposide, Clin. Cancer Res., 16(1) (2010), p. 130-140. [CrossRef]
- S.H. Yang, H.G. Choi, S.J. Lim, M.G. Lee, S.H. Kim, Effects of morin on the pharmacokinetics of etoposide in 7,12-dimethylbenz[a]anthracene-induced mammary tumors in female Sprague-Dawley rats, Oncol. Rep., 29(3) (2013), p. 1215-1223. [CrossRef]
- J. Hofman, D. Vagiannis, S. Chen, L. Guo, Roles of CYP3A4, CYP3A5 and CYP2C8 drug-metabolizing enzymes in cellular cytostatic resistance, Chem. Biol. Interact., 340 (2021), p. 109448. [CrossRef]
- C. Martínez, E. García-Martín, R.M. Pizarro, F.J. García-Gamito, J.A. Agúndez, Expression of paclitaxel-inactivating CYP3A activity in human colorectal cancer: implications for drug therapy, Br. J. Cancer, 87(6) (2002), p. 681-686. [CrossRef]
- N.R. Powell, T. Shugg, R.C. Ly, C. Albany, M. Radovich, B.P. Schneider, T.C. Skaar, Life-threatening docetaxel toxicity in a patient with reduced-function CYP3A variants: A case report, Front. Oncol., 11 (2022), p. 809527. [CrossRef]
- S. Sim, J. Bergh, M. Hellström, T. Hatschek, H. Xie, Pharmacogenetic impact of docetaxel on neoadjuvant treatment of breast cancer patients, Pharmacogenomics, 19(16) (2018), p. 1259-1268. [CrossRef]
- H. Masuyama, K. Nakamura, E. Nobumoto, Y. Hiramatsu, Inhibition of pregnane X receptor pathway contributes to the cell growth inhibition and apoptosis of anticancer agents in ovarian cancer cells, Int. J. Oncol., 49(3) (2016), p. 1211-1220. [CrossRef]
- H. Masuyama, H. Nakatsukasa, N. Takamoto, Y. Hiramatsu, Down-regulation of pregnane X receptor contributes to cell growth inhibition and apoptosis by anticancer agents in endometrial cancer cells, Mol. Pharmacol., 72(4) (2007), p. 1045-1053. [CrossRef]
- Y. Chen, J.A. Goldstein, The transcriptional regulation of the human CYP2C genes, Curr. Drug Metab., 10(6) (2009), p. 567-578. [CrossRef]
- C.A. Vilanova-Costa, H.K. Porto, L.C. Pereira, B.P. Carvalho, W.B. Dos Santos, P. Silveira-Lacerda Ede, MDR1 and cytochrome P450 gene-expression profiles as markers of chemosensitivity in human chronic myelogenous leukemia cells treated with cisplatin and Ru(III) metallocomplexes, Biol. Trace Elem. Res., 163(1-2) (2015), p. 39-47. [CrossRef]
- J. Hakkola, C. Bernasconi, S. Coecke, L. Richert, T.B. Andersson, O. Pelkonen, Cytochrome P450 induction and xeno-sensing receptors pregnane X receptor, constitutive androstane receptor, aryl hydrocarbon receptor and peroxisome proliferator-activated receptor α at the crossroads of toxicokinetics and toxicodynamics, Basic Clin. Pharmacol. Toxicol., 123 (Suppl.5) (2018), p. 42-50. [CrossRef]
- S.S. Ferguson, E.L. LeCluyse, M. Negishi, J.A. Goldstein, Regulation of human CYP2C9 by the constitutive androstane receptor: discovery of a new distal binding site, Mol. Pharmacol., 62(3) (2002), p. 737-746. [CrossRef]
- L.C. Preiss, R. Liu, P. Hewitt, D. Thompson, K. Georgi, L. Badolo, V.M. Lauschke, C. Petersson, Deconvolution of cytochrome P450 induction mechanisms in HepaRG nuclear hormone receptor knockout cells, Drug Metab. Dispos., 49(8) (2021), p. 668-678. [CrossRef]
- P. Wei, J. Zhang, D.H. Dowhan, Y. Han, D.D. Moore, Specific and overlapping functions of the nuclear hormone receptors CAR and PXR in xenobiotic response, Pharmacogenomics J., 2(2) (2002), p. 117-126. [CrossRef]
- Y. Wang, H. Masuyama, E. Nobumoto, G. Zhang, Y. Hiramatsu, The inhibition of constitutive androstane receptor-mediated pathway enhances the effects of anticancer agents in ovarian cancer cells, Biochem. Pharmacol., 90(4) (2014), p. 356-366. [CrossRef]
- B. Goodwin, E. Hodgson, D.J. D'Costa, G.R. Robertson, C. Liddle, Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor, Mol. Pharmacol., 62(2) (2002), p. 359-365. [CrossRef]
- D. Smirlis, R. Muangmoonchai, M. Edwards, I.R. Phillips, E.A. Shephard, Orphan receptor promiscuity in the induction of cytochromes p450 by xenobiotics, J. Biol. Chem., 276(16) (2001), p. 12822-12826. [CrossRef]
- D. Wang, L. Li, H. Yang, S.S. Ferguson, M.R. Baer, R.B. Gartenhaus, H. Wang, The constitutive androstane receptor is a novel therapeutic target facilitating cyclophosphamide-based treatment of hematopoietic malignancies, Blood, 121(2) (2013), p. 329-338. [CrossRef]
- S. Ren, J.S. Yang, T.F. Kalhorn, J.T. Slattery, Oxidation of cyclophosphamide to 4-hydroxycyclophosphamide and deschloroethylcyclophosphamide in human liver microsomes, Cancer Res., 57(19) (1997), p. 4229-4235.
- S.F. Zhou, Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4, Curr. Drug Metab., 9(4) (2008), p. 310-322. [CrossRef]
- S.P. Rendic, Metabolism and interactions of ivermectin with human cytochrome P450 enzymes and drug transporters, possible adverse and toxic effects. Arch Toxicol. 2021 May;95(5):1535-1546. [CrossRef]
- Y.L. Teo, H.K. Ho, A. Chan, Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: current understanding, challenges and recommendations, Br. J. Clin. Pharmacol., 79(2) (2015), p. 241-253. [CrossRef]
- A. Didier, F. Loor, The abamectin derivative ivermectin is a potent P-glycoprotein inhibitor, Anticancer Drugs, 7(7) (1996), p. 745-751. [CrossRef]
- X. Zhou, T.M. Li, J.Z. Luo, C.L. Lan, Z.L. Wei, T.H. Fu, X.W. Liao, G.Z. Zhu, X.P. Ye, T. Peng, CYP2C8 suppress proliferation, migration, invasion and sorafenib resistance of hepatocellular carcinoma via PI3K/Akt/p27kip1 axis, J. Hepatocell. Carcinoma, 8 (2021), p. 1323-1338. [CrossRef]
- P.C. Nair, T.B. Gillani, T. Rawling, M. Murray, Differential inhibition of human CYP2C8 and molecular docking interactions elicited by sorafenib and its major N-oxide metabolite, Chem. Biol. Interact., 338 (2021), p. 109401. [CrossRef]
- S. Ghassabian, T. Rawling, F. Zhou, M.R. Doddareddy, B.N. Tattam, D.E. Hibbs, R.J. Edwards, P.H. Cui, M. Murray, Role of human CYP3A4 in the biotransformation of sorafenib to its major oxidized metabolites, Biochem. Pharmacol., 84(2) (2012), p. 215-223. [CrossRef]
- F.P. Guengerich, A history of the roles of cytochrome P450 enzymes in the toxicity of drugs, Toxicol. Res., 37(1) (2020), p. 1-23. [CrossRef]
- B.L. Salerni, D.J. Bates, T.C. Albershardt, C.H. Lowrey, A. Eastman, Vinblastine induces acute, cell cycle phase-independent apoptosis in some leukemias and lymphomas and can induce acute apoptosis in others when Mcl-1 is suppressed, Mol. Cancer Ther., 9(4) (2010), p. 791-802. [CrossRef]
- X.R. Zhou-Pan, E. Sérée, X.J. Zhou, M. Placidi, P. Maurel, Y. Barra, R. Rahmani, Involvement of human liver cytochrome P450 3A in vinblastine metabolism: drug interactions, Cancer Res., 53(21) (1993), p. 5121-5126.
- J.B. Dennison, P. Kulanthaivel, R.J. Barbuch, J.L. Renbarger, W.J. Ehlhardt, S.D. Hall, Selective metabolism of vincristine in vitro by CYP3A5. Drug Metab. Dispos., 34(8) (2006), p. 1317-1327. [CrossRef]
- N. Nebot, S. Crettol, F. d'Esposito, B. Tattam, D.E. Hibbs, M. Murray, Participation of CYP2C8 and CYP3A4 in the N-demethylation of imatinib in human hepatic microsomes, Br. J. Pharmacol., 161(5) (2010), p. 1059-1069. [CrossRef]
- A.M. Filppula, M. Neuvonen, J. Laitila, P.J. Neuvonen, J.T. Backman, Autoinhibition of CYP3A4 leads to important role of CYP2C8 in imatinib metabolism: variability in CYP2C8 activity may alter plasma concentrations and response, Drug Metab. Dispos., 41(1) (2013), p. 50-59. [CrossRef]
- T. Zhao, X. Li, Y. Chen, J. Du, X. Chen, D. Wang, L. Wang, S. Zhao, C. Wang, Q. Meng, H. Sun, K. Liu, J. Wu, Risk assessment and molecular mechanism study of drug-drug interactions between rivaroxaban and tyrosine kinase inhibitors mediated by CYP2J2/3A4 and BCRP/P-gp, Front. Pharmacol., 13 (2022), p. 914842. [CrossRef]
- A,M, Filppula, P.J. Neuvonen, J.T. Backman, In vitro assessment of time-dependent inhibitory effects on CYP2C8 and CYP3A activity by fourteen protein kinase inhibitors, Drug Metab. Dispos., 42(7) (2014), p. 1202-1209. [CrossRef]
- A.M. Filppula, T.M. Mustonen, J.T. Backman, In vitro screening of six protein kinase inhibitors for time-dependent inhibition of CYP2C8 and CYP3A4: Possible implications with regard to drug-drug interactions, Basic Clin. Pharmacol. Toxicol., 123(6) (2018), p. 739-748. [CrossRef]
- B.D. Latham, D.S. Oskin, R.D. Crouch, M.J. Vergne, K.D. Jackson, Cytochromes P450 2C8 and 3A catalyze the metabolic activation of the tyrosine kinase inhibitor masitinib, Chem. Res. Toxicol., 35(9) (2022), p. 1467-1481. [CrossRef]
- N. Eliyatkın, E. Yalçın, B. Zengel, S. Aktaş, E. Vardar, Molecular classification of breast carcinoma: From traditional, old-fashioned way to a new age, and a new way, J. Breast Health, 11(2) (2015), p. 59-66. [CrossRef]
- M.H. Zhang, H.T. Man, X.D. Zhao, N. Dong, S.L. Ma, Estrogen receptor-positive breast cancer molecular signatures and therapeutic potentials (Review), Biomed. Rep., 2(1) (2014), p. 41-52. [CrossRef]
- V. Makker, H. MacKay, I. Ray-Coquard, D.A. Levine, S.N. Westin, D. Aoki, A. Oaknin, Endometrial cancer, Nat. Rev. Dis. Primers, 7(1) (2021), p. 88. [CrossRef]
- J.L. Hecht, G.L. Mutter, Molecular and pathologic aspects of endometrial carcinogenesis, J. Clin. Oncol., 24(29) (2006), 4783-4791. [CrossRef]
- L.A. Brinton, B. Trabert, G.L. Anderson, R.T. Falk, A.S. Felix, B.J. Fuhrman, M.L. Gass, L.H. Kuller, R.M. Pfeiffer, T.E. Rohan, H.D. Strickler, X. Xu, N. Wentzensen, Serum estrogens and estrogen metabolites and endometrial cancer risk among postmenopausal women, Cancer Epidemiol. Biomarkers Prev., 25(7) (2016), p. 1081-1089. [CrossRef]
- U.M. Zamwar, A.P. Anjankar, Aetiology, epidemiology, histopathology, classification, detailed evaluation, and treatment of ovarian cancer, Cureus, 14(10) (2022), e30561. [CrossRef]
- F. Modugno, R. Laskey, A.L. Smith, C.L. Andersen, P. Haluska, S. Oesterreich, Hormone response in ovarian cancer: time to reconsider as a clinical target? Endocr. Relat. Cancer, 19(6) (2012), R255-R279. [CrossRef]
- F. Shen, X. Zhang, Y. Zhang, J. Ding, Q. Chen, Hormone receptors expression in ovarian cancer taking into account menopausal status: a retrospective study in Chinese population, Oncotarget, 8(48) (2017), p. 84019-84027. [CrossRef]
- B. Luo, D. Yan, H. Yan, J. Yuan, Cytochrome P450: Implications for human breast cancer, Oncol. Lett., 22(1) (2021), p. 548. [CrossRef]
- J. Wunder, D. Pemp, A. Cecil, M. Mahdiani, R. Hauptstein, K. Schmalbach, L.N. Geppert, K. Ickstadt, H.L. Esch, T. Dandekar, L. Lehmann, Influence of breast cancer risk factors on proliferation and DNA damage in human breast glandular tissues: role of intracellular estrogen levels, oxidative stress and estrogen biotransformation, Arch. Toxicol., 96(2) (2022), p. 673-687. [CrossRef]
- H. Piotrowska, M. Kucinska, M. Murias, Expression of CYP1A1, CYP1B1 and MnSOD in a panel of human cancer cell lines, Mol. Cell. Biochem., 383(1-2) (2013), p. 95-102. [CrossRef]
- Y.K. Leung, K.M. Lau, J. Mobley, Z. Jiang, S.M. Ho, Overexpression of cytochrome P450 1A1 and its novel spliced variant in ovarian cancer cells: alternative subcellular enzyme compartmentation may contribute to carcinogenesis, Cancer Res., 65(9) (2005), p. 3726-3734. [CrossRef]
- A.E. Cribb, M.J. Knight, D. Dryer, J. Guernsey, K. Hender, M. Tesch, T.M. Saleh, Role of polymorphic human cytochrome P450 enzymes in estrone oxidation, Cancer Epidemiol. Biomarkers Prev., 15(3) (2006), p. 551-558. [CrossRef]
- V. Kerlan, Y. Dreano, J.P. Bercovici, P.H. Beaune, H.H. Floch, F. Berthou, Nature of cytochromes P450 involved in the 2-/4-hydroxylations of estradiol in human liver microsomes, Biochem. Pharmacol., 44(9) (1992), p. 1745-1756. [CrossRef]
- E. Cavalieri, E. Rogan, The 3,4-quinones of estrone and estradiol are the initiators of cancer whereas resveratrol and N-acetylcysteine are the preventers, Int. J. Mol. Sci., 22(15) (2021), p. 8238. [CrossRef]
- M. Gupta, A. McDougal, S. Safe, Estrogenic and antiestrogenic activities of 16alpha- and 2-hydroxy metabolites of 17beta-estradiol in MCF-7 and T47D human breast cancer cells, J. Steroid Biochem. Mol. Biol., 67(5-6) (1998), p. 413-419. [CrossRef]
- E. Sawicka, A. Długosz, The role of 17β-estradiol metabolites in chromium-induced oxidative stress, Adv. Clin. Exp. Med., 26(2) (2017), p. 215-221. [CrossRef]
- H. Samavat, M.S. Kurzer, Estrogen metabolism and breast cancer, Cancer Lett., 356(Pt A) (2015), p. 231-243. [CrossRef]
- S. Verenich, P.M. Gerk, Therapeutic promises of 2-methoxyestradiol and its drug disposition challenges, Mol. Pharm., 7(6) (2010), p. 2030-2039. [CrossRef]
- C. Guillemette, a. Bélanger, J. Lépine, Metabolic inactivation of estrogens in breast tissue by UDP-glucuronosyltransferase enzymes: an overview, Breast Cancer Res., 6(6) (2004), p. 246-254. [CrossRef]
- J.D. Yager, Mechanisms of estrogen carcinogenesis: The role of E2/E1-quinone metabolites suggests new approaches to preventive intervention--A review, Steroids, 99(Pt A) (2015), p. 56-60. [CrossRef]
- F. Lu, M. Zahid, M. Saeed, E.L. Cavalieri, E.G. Rogan, Estrogen metabolism and formation of estrogen-DNA adducts in estradiol-treated MCF-10F cells. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin induction and catechol-O-methyltransferase inhibition, J. Steroid Biochem. Mol. Biol., 105(1-5) (2007), p. 150-158. [CrossRef]
- B.J. Fuhrman, C. Schairer, M.H. Gail, J. Boyd-Morin, X. Xu, L.Y. Sue, S.S. Buys, C. Isaacs, L.K. Keefer, T.D. Veenstra, C.D. Berg, R.N. Hoover, R.G. Ziegler, Estrogen metabolism and risk of breast cancer in postmenopausal women, J. Natl. Cancer Inst., 104(4) (2012), p. 326-339. [CrossRef]
- J.A. Lavigne, J.E. Goodman, T. Fonong, S. Odwin, P. He, D.W. Roberts, J.D. Yager, The effects of catechol-O-methyltransferase inhibition on estrogen metabolite and oxidative DNA damage levels in estradiol-treated MCF-7 cells, Cancer Res., 61(20) (2001), p. 7488-7494.
- C. Lin, D.R. Chen, S.J. Kuo, C.Y. Feng, D.R. Chen, W.C. Hsieh, P.H. Lin, Profiling of protein adducts of estrogen quinones in 5-year survivors of breast cancer without recurrence, Cancer Control., 29 (2022), p. 10732748221084196. [CrossRef]
- B. Trabert, A.M. Geczik, D.C. Bauer, D.S.M. Buist, J.A. Cauley, R.T. Falk, G.L. Gierach, T.F. Hue, J.V. Lacey Jr, A.Z. LaCroix, K.A. Michels, J.A. Tice, X. Xu, L.A. Brinton, C.M. Dallal, Association of endogenous pregnenolone, progesterone, and related metabolites with risk of endometrial and ovarian cancers in postmenopausal women: The B∼FIT cohort, Cancer Epidemiol. Biomarkers Prev., 30(11) (2021), p. 2030-2037. [CrossRef]
- M. Zahid, C.L. Beseler, J.B. Hall, T. LeVan, E.L. Cavalieri, E.G. Rogan, Unbalanced estrogen metabolism in ovarian cancer, Int. J. Cancer, 134(10) (2014), p. 2414-2423. [CrossRef]
- E. Taioli, H.L. Bradlow, S.V. Garbers, D.W. Sepkovic, M.P. Osborne, J. Trachman, S. Ganguly, S.J. Garte, Role of estradiol metabolism and CYP1A1 polymorphisms in breast cancer risk, Cancer Detect. Prev., 23(3) (1999), p. 232-237. [CrossRef]
- Martínez-Ramírez, C. Castro-Hernández, R. Pérez-Morales, L. Casas-Ávila, R.M. de Lorena, A. Salazar-Piña, J. Rubio, Pathological characteristics, survival, and risk of breast cancer associated with estrogen and xenobiotic metabolism polymorphisms in Mexican women with breast cancer, Cancer Causes Control., 32(4) (2021), p. 369-378. [CrossRef]
- V. Singh, N. Rastogi, A. Sinha, A. Kumar, N. Mathur, M.P. Singh, A study on the association of cytochrome-P450 1A1 polymorphism and breast cancer risk in north Indian women, Breast Cancer Res. Treat., 101(7) (2007), p. 73-81. [CrossRef]
- M. Okobia, C. Bunker, J. Zmuda, C. Kammerer, V. Vogel, E. Uche, S. Anyanwu, E. Ezeome, R. Ferrell, L. Kuller, Cytochrome P4501A1 genetic polymorphisms and breast cancer risk in Nigerian women, Breast Cancer Res. Treat. 94(3) (2005), p. 285-293. [CrossRef]
- L. Zhang, L. Gu, B. Qian, X. Hao, W. Zhang, Q. Wei, K. Chen, Association of genetic polymorphisms of ER-alpha and the estradiol-synthesizing enzyme genes CYP17 and CYP19 with breast cancer risk in Chinese women, Breast Cancer Res. Treat., 114(2) (2009), p. 327-338. [CrossRef]
- D.F. Han, X. Zhou, M.B. Hu, W. Xie, Z.F. Mao, D.E. Chen, F. Liu, F. Zheng, Polymorphisms of estrogen-metabolizing genes and breast cancer risk: a multigenic study, Chin. Med. J. (Engl), 118(18) (2005), p. 1507-1516.
- B.M. Tüzüner, T. Oztürk, H.I. Kisakesen, S. Ilvan, C. Zerrin, O. Oztürk, T. Isbir, CYP17 (T-34C) and CYP19 (Trp39Arg) polymorphisms and their cooperative effects on breast cancer susceptibility, In Vivo, 24(1) (2010), p. 71-74.
- M. McGrath, S.E. Hankinson, L. Arbeitman, G.A. Colditz, D.J. Hunter, I. De Vivo, Cytochrome P450 1B1 and catechol-O-methyltransferase polymorphisms and endometrial cancer susceptibility, Carcinogenesis, 25(4) (2004), p. 559-565. [CrossRef]
- T.N. Sergentanis, K.P. Economopoulos, S. Choussein, N.F. Vlahos, Cytochrome P450 1A1 gene polymorphisms and endometrial cancer risk: a meta-analysis. Int J Gynecol Cancer. 2011 Feb;21(2):323-31. [CrossRef]
- J.B. Ramirez Sabio, J.V. Sorli, R. Barragan, F.-C. Rebeca, E.M. Asensio, D. Cjrella, Effect of the polymorphism rs2470893 of the CYP1A1 gene on ovarian and endometrial cancer in Mediterranean women, Ann. Oncol., 29(Suppl. 8) (2018), viii6.
- J.Y Liu, Y. Yang, Z.Z. Liu, J.J. Xie, Y.P. Du, W. Wang, Association between the CYP1B1 polymorphisms and risk of cancer: a meta-analysis, Mol. Genet. Genomics, 290(2) (2015), p. 739-765. [CrossRef]
- K. Gajjar, G. Owens, M. Sperrin, P.L. Martin-Hirsch, F.L. Martin, Cytochrome P1B1 (CYP1B1) polymorphisms and ovarian cancer risk: a meta-analysis, Toxicology, 302(2-3) (2012), p. 157-162. [CrossRef]
- L. Zhang, L. Feng, M. Lou, X. Deng, C. Liu, L. Li, The ovarian carcinoma risk with the polymorphisms of CYP1B1 come from the positive selection, Am. J. Transl. Res., 13(5) (2021), p. 4322-4341.
- M.B. Culp, I. Soerjomataram, J.A. Efstathiou, F. Bray, A. Jemal, Recent global patterns in prostate cancer incidence and mortality rates, Eur. Urol., 77(1) (2020), p. 38-52. [CrossRef]
- H. Zhang, Y. Zhou, Z. Xing, R.K. Sah, J. Hu, H. Hu, Androgen metabolism and response in prostate cancer anti-androgen therapy resistance, Int. J. Mol. Sci., 23(21) (2022), p. 13521. [CrossRef]
- Y. Chen, P.H. Lin, S.J. Freedland, J.T. Chi, Metabolic response to androgen deprivation therapy of prostate cancer, Cancers (Basel), 16(11) (2024), p. 1991. [CrossRef]
- R.J. Auchus, Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic, J. Steroid Biochem. Mol. Biol., 165(Pt A) (2017), p. 71-78. [CrossRef]
- B.C. Chung, J. Picado-Leonard, M. Haniu, m. Bienkowski, P.F. Hall, J.E. Shively, W.L. Miller, Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues, Proc. Natl. Acad. Sci. U S A, 84(2) (1987), p. 407-411. [CrossRef]
- T. Suzuki, H. Sasano, J. Takeyama, C. Kaneko, W.A. Freije, B.R. Carr, W.E. Rainey, Developmental changes in steroidogenic enzymes in human postnatal adrenal cortex: immunohistochemical studies, Clin. Endocrinol (Oxf), 53(6) (2000), p. 739-747. [CrossRef]
- E.A. Mostaghel, K.R. Solomon, K. Pelton, M.R. Freeman, R.B. Montgomery, Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors, PLoS One 7(1) (2012), p. e30062. [CrossRef]
- A. Giatromanolaki, V. Fasoulaki, D. Kalamida, A. Mitrakas, C. Kakouratos, T. Lialiaris, M.I. Koukourakis, CYP17A1 and androgen-receptor expression in prostate carcinoma tissues and cancer cell lines, Curr. Urol., 13(3) (2019), p. 157-165. [CrossRef]
- M. Sakai, D.B. Martinez-Arguelles, A.G. Aprikian, A.M. Magliocco, V. Papadopoulos, De novo steroid biosynthesis in human prostate cell lines and biopsies, Prostate, 76(6) (2016), p. 575-587. [CrossRef]
- K.H. Chang, C.E. Ercole, N. Sharifi, Androgen metabolism in prostate cancer: from molecular mechanisms to clinical consequences, Br. J. Cancer, 111(7) (2014), p. 1249-1254. [CrossRef]
- S. Crnalic, E. Hörnberg, P. Wikström, U.H. Lerner, A. Tieva, O. Svensson, A. Widmark, A. Bergh, Nuclear androgen receptor staining in bone metastases is related to a poor outcome in prostate cancer patients, Endocr. Relat. Cancer, 17(4) (2010), p. 885-895. [CrossRef]
- N. Sharifi. Mechanisms of androgen receptor activation in castration-resistant prostate cancer, Endocrinology, 154(11) (2013), p. 4010-4017. [CrossRef]
- K.H.Chang, R. Li, M. Papari-Zareei, L. Watumull, Y.D. Zhao, R.J. Auchus, N. Sharifi, Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer, Proc. Natl. Acad. Sci. U S A, 108(33) (2011), p. 13728-13733. [CrossRef]
- G.L. Varaprasad, V.K. Gupta, K. Prasad, E. Kim, M.B. Tej, P. Mohanty, H.K. Verma, G.S.R. Raju, L. Bhaskar, Y.S. Huh, Recent advances and future perspectives in the therapeutics of prostate cancer, Exp. Hematol. Oncol., 12(1) (2023), p. 80. [CrossRef]
- H. Bolek, S.C. Yazgan, E. Yekedüz, M.D. Kaymakcalan, R.R. McKay, S. Gillessen, Y. Ürün, Androgen receptor pathway inhibitors and drug-drug interactions in prostate cancer, ESMO Open, 9(11) (2024), p. 103736. [CrossRef]
- T.K. Le, Q.H. Duong, V. Baylot, C. Fargette, M. Baboudjian, L. Colleaux, D. Taïeb, P. Rocchi, Castration-resistant prostate cancer: From uncovered resistance mechanisms to current treatments, Cancers (Basel), 15(20) (2023), p. 5047. [CrossRef]
- R.B. Montgomery, E.A. Mostaghel, R. Vessella, D.L. Hess, T.F. Kalhorn, C.S. Higano, L.D. True, P.S. Nelson, Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth, Cancer Res., 68(11) (2008), p. 4447-4454. [CrossRef]
- C. Padmakar Darne, U. Velaparthi, M. Saulnier, D. Frennesson, P. Liu, A. Huang, J. Tokarski, A. Fura, T. Spires, J. Newitt, V.M. Spires, M.T. Obermeier, P.A. Elzinga, M.M. Gottardis, L. Jayaraman, G.D. Vite, A. Balog, The discovery of BMS-737 as a potent, CYP17 lyase-selective inhibitor for the treatment of castration-resistant prostate cancer, Bioorg. Med. Chem. Lett., 75 (2022), p. 128951. [CrossRef]
- T.M. Wróbel, O. Rogova, K. Sharma, M.N. Rojas Velazquez, A.V. Pandey, F.S. Jørgensen, F.S. Arendrup, K.L. Andersen, F. Björkling, Synthesis and structure-activity relationships of novel non-steroidal CYP17A1 inhibitors as potential prostate cancer agents, Biomolecules, 12(2) (2022), p. 165. [CrossRef]
- S.A.H. Abdi, A. Ali, S.F. Sayed, M.J. Ahsan, A. Tahir, W. Ahmad, S. Shukla, A. Ali, Morusflavone, a new therapeutic candidate for prostate cancer by CYP17A1 inhibition: Exhibited by molecular docking and dynamics simulation, Plants (Basel), 10(9) (2021), p. 1912. [CrossRef]
- A.S. Latysheva, V.A. Zolottsev, V.S. Pokrovsky, I.I. Khan, A.Y. Misharin, Novel nitrogen containing steroid derivatives for prostate cancer treatment, Curr. Med. Chem., 28(40) (2021), p. 8416-8432. [CrossRef]
- T.M. Wróbel, O. Rogova, K.L. Andersen, R. Yadav, S. Brixius-Anderko, E.E. Scott, L. Olsen, F.S. Jørgensen, F. Björkling, Discovery of novel non-steroidal cytochrome P450 17A1 inhibitors as potential prostate cancer agents, Int. J. Mol. Sci., 21(14) (2020), p. 4868. [CrossRef]
- T.M. Wróbel, F.S. Jørgensen, A.V. Pandey, A. Grudzińska, K. Sharma, J. Yakubu, F. Björkling, Non-steroidal CYP17A1 inhibitors: Discovery and assessment, J. Med. Chem., 66(10) (2023), p. 6542-6566. [CrossRef]
- L. Yin, Q. Hu, CYP17 inhibitors--abiraterone, C17,20-lyase inhibitors and multi-targeting agents, Nat Rev Urol. 2014 Jan;11(1):32-42. [CrossRef]
- M.G. Rowlands, S.E. Barrie, F. Chan, J. Houghton, M. Jarman, R. McCague, G.A. Potter, Esters of 3-pyridylacetic acid that combine potent inhibition of 17 alpha-hydroxylase/C17,20-lyase (cytochrome P45017 alpha) with resistance to esterase hydrolysis, J. Med. Chem., 38(21) (1995), 4191-4197. [CrossRef]
- J.S. de Bono, C.J. Logothetis, A. Molina, K. Fizazi, S. North, L. Chu, K.N. Chi, R.J. Jones, O.B. Goodman, Jr, F. Saad, J.N. Staffurth, P. Mainwaring, S. Harland, T.W. Flaig, T.E. Hutson, T. Cheng, H. Patterson, J.D. Hainsworth, C.J. Ryan, C.N. Sternberg, S.L. Ellard, A. Fléchon, M. Saleh, M. Scholz, E. Efstathiou, A. Zivi, D. Bianchini, Y. Loriot, N. Chieffo, T. Kheoh, C.M. Haqq, H.I. Scher; COU-AA-301 Investigators, Abiraterone and increased survival in metastatic prostate cancer, N. Engl. J. Med., 364(21) (2011), p. 1995-2005. [CrossRef]
- E. Efstathiou, M. Titus, D. Tsavachidou, V. Tzelepi, S. Wen, A. Hoang, A. Molina, N. Chieffo, L.A. Smith, M. Karlou, P. Troncoso, C.J. Logothetis, Effects of abiraterone acetate on androgen signaling in castrate-resistant prostate cancer in bone, J. Clin. Oncol., 30(6) (2012), p. 637-643. [CrossRef]
- J.D. Norris, S.J. Ellison, J.G. Baker, D.B. Stagg, S.E. Wardell, S. Park, H.M. Alley, R.M. Baldi, A. Yllanes, K.J. Andreano, J.P. Stice, S.A. Lawrence, J.R. Eisner, D.K. Price, W.R. Moore, W.D. Figg, D.P. McDonnell, Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer, J. Clin. Invest., 127(6) (2017), p. 2326-2338. [CrossRef]
- H. Mehralitabar, A.S. Ghasemi, J. Gholizadeh, Abiraterone and D4, 3-keto Abiraterone binding to CYP17A1, a structural comparison study by molecular dynamic simulation, Steroids, 167 (2021), p. 108799. [CrossRef]
- E.J.Y. Cheong, P.C. Nair, R.W.Y. Neo, H.T. Tu, F. Lin, E. Chiong, K. Esuvaranathan, H. Fan, R.Z. Szmulewitz, C.J. Peer, W.D. Figg, C.L.L. Chai, J.O. Miners, E.C.Y. Chan, Slow-, tight-binding inhibition of CYP17A1 by abiraterone redefines its kinetic selectivity and dosing regimen, J. Pharmacol. Exp. Ther., 374(3) (2020), p. 438-451. [CrossRef]
- B. Zhang, Q. Cheng, Z. Ou, J.H. Lee, M. Xu, U. Kochhar, S. Ren, M. Huang, B.R. Pflug, W. Xie, Pregnane X receptor as a therapeutic target to inhibit androgen activity, Endocrinology, 151(12) (2010), p. 5721-5729. [CrossRef]
- J.M. Salamat, E.M. Ayala, C.J. Huang, F.S. Wilbanks, R.C. Knight, B.T. Akingbemi, S.R. Pondugula, Pregnenolone 16-alpha carbonitrile, an agonist of rodent pregnane X receptor, regulates testosterone biosynthesis in rodent Leydig cells, J. Xenobiot., 14(3) (2024), p. 1256-1267. [CrossRef]
- A. Gsur, E. Feik, S. Madersbacher, Genetic polymorphisms and prostate cancer risk, World J. Urol., 21(6) (2004), p. 414-423. [CrossRef]
- M. Izmirli, B. Arikan, Y. Bayazit, D. Alptekin, Associations of polymorphisms in HPC2/ELAC2 and SRD5A2 genes with benign prostate hyperplasia in Turkish men, Asian Pac. J. Cancer Prev., 12 (2011), p. 731-733.
- M.S. Cicek, D.V. Conti, A. Curran, P.J. Neville, P.L. Paris, G. Casey, J.S. Witte, Association of prostate cancer risk and aggressiveness to androgen pathway genes: SRD5A2, CYP17, and the AR, Prostate, 59(1) (2004), p. 69-76. [CrossRef]
- I.H. Onen, A. Ekmekci, M. Eroglu, F. Polat, H. Biri, The association of 5alpha-reductase II (SRD5A2) and 17 hydroxylase (CYP17) gene polymorphisms with prostate cancer patients in the Turkish population, DNA Cell Biol., 26(2) (2007), p. 100-107. [CrossRef]
- N.E. Allen, M.S. Forrest, T.J. Key, The association between polymorphisms in the CYP17 and 5alpha-reductase (SRD5A2) genes and serum androgen concentrations in men, Cancer Epidemiol. Biomarkers Prev., 10(3) (2001), p. 185-189.
- S. Lindström, F. Wiklund, H.O. Adami, K.A. Bälter, J. Adolfsson, H. Grönberg, Germ-line genetic variation in the key androgen-regulating genes androgen receptor, cytochrome P450, and steroid-5-alpha-reductase type 2 is important for prostate cancer development, Cancer Res., 66(22) (2006), p. 11077-11083. [CrossRef]
- R.C. Sobti, L. Gupta, S.K. Singh, A. Seth, P. Kaur, H. Thakur, Role of hormonal genes and risk of prostate cancer: gene-gene interactions in a North Indian population, Cancer Genet. Cytogenet., 185(2) (2008), p. 78-85. [CrossRef]
- Y. Yamada, M. Watanabe, M. Murata, M. Yamanaka, Y. Kubota, H. Ito, T. Katoh, J. Kawamura, R. Yatani, T. Shiraishi, Impact of genetic polymorphisms of 17-hydroxylase cytochrome P-450 (CYP17) and steroid 5alpha-reductase type II (SRD5A2) genes on prostate-cancer risk among the Japanese population, Int. J. Cancer, 92(5) (2001), p. 683-686. [CrossRef]
- M. Murata, M. Watanabe, M. Yamanaka, Y. Kubota, H. Ito, M. Nagao, T. Katoh, T. Kamataki, J. Kawamura, R. Yatani, T. Shiraishi, Genetic polymorphisms in cytochrome P450 (CYP) 1A1, CYP1A2, CYP2E1, glutathione S-transferase (GST) M1 and GSTT1 and susceptibility to prostate cancer in the Japanese population, Cancer Lett., 165(2) (2001), p. 171-177. [CrossRef]
- M. Koda, M. Iwasaki, Y. Yamano, X. Lu, T. Katoh, Association between NAT2, CYP1A1, and CYP1A2 genotypes, heterocyclic aromatic amines, and prostate cancer risk: a case control study in Japan, Environ. Health Prev. Med., 22(1) (2017), p. 72. [CrossRef]
- B.L. Chang, S.L. Zheng, S.D. Isaacs, A. Turner, G.A. Hawkins, K.E. Wiley, E.R. Bleecker, P.C. Walsh , D.A. Meyers, W.B. Isaacs, J. Xu, Polymorphisms in the CYP1B1 gene are associated with increased risk of prostate cancer, Br. J. Cancer, 89(8) (2003), p. 1524-1529. [CrossRef]
- M.S. Cicek, X. Liu, G. Casey, J.S. Witte, Role of androgen metabolism genes CYP1B1, PSA/KLK3, and CYP11alpha in prostate cancer risk and aggressiveness, Cancer Epidemiol. Biomarkers Prev., 14(9) (2005), p. 2173-2177. [CrossRef]
- J.Y. Liu, Y. Yang, Z.Z. Liu, J.J. Xie, Y.P. Du, W. Wang, Association between the CYP1B1 polymorphisms and risk of cancer: a meta-analysis, Mol. Genet. Genomics, 290(2) (2015), p. 739-765. [CrossRef]
- C.Y. Gu, G.X. Li, Y. Zhu, H. Xu, Y. Zhu, X.J. Qin, D. Bo, D.W. Ye, A single nucleotide polymorphism in CYP1B1 leads to differential prostate cancer risk and telomere length, J. Cancer, 9(2) (2018), p. 269-274. [CrossRef]
- C.Y. Gu, X.J. Qin, Y.Y. Qu, Y. Zhu, F.N. Wan, G.M. Zhang, L.J. Sun, Y. Zhu, D.W. Ye, Genetic variants of the CYP1B1 gene as predictors of biochemical recurrence after radical prostatectomy in localized prostate cancer patients, Medicine (Baltimore), 95(27) (2016), p. e4066. [CrossRef]
- W. Zhu, H. Liu, X. Wang, J. Lu, H. Zhang, S. Wang, W. Yang, Associations of CYP1 polymorphisms with risk of prostate cancer: an updated meta-analysis, Biosci. Rep., 39(3) (2019), p.BSR20181876. [CrossRef]
- M. Vilčková, M. Škereňová, D. Dobrota, P. Kaplán, J. Jurečeková, J. Kliment, M. Híveš, R. Dušenka, D. Evin, M. Knoško Brožová, M. Kmeťová Sivoňová, Polymorphisms in the gene encoding CYP1A2 influence prostate cancer risk and progression, Oncol. Lett. 25(2) (2023), p. 85. [CrossRef]
- S.J. Plummer, D.V. Conti, P.L. Paris, A.P. Curran, G. Casey, J.S. Witte, CYP3A4 and CYP3A5 genotypes, haplotypes, and risk of prostate cancer, Cancer Epidemiol. Biomarkers Prev., 12((9) (2003), p. 928-932.
- Y. Liang, W. Han, H. Yan, Q. Mao, Association of CYP3A5*3 polymorphisms and prostate cancer risk: A meta-analysis, J. Cancer Res. Ther., 14(Suppl) (2018), p. S463-S467. [CrossRef]
- S.F. Bellah, M.A. Salam, S.M.S. Billah, M.R. Karim, Genetic association in CYP3A4 and CYP3A5 genes elevate the risk of prostate cancer, Ann. Hum. Biol., 50(1) (2023), p. 63-74. [CrossRef]
- M.H. Vaarala, H. Mattila, P. Ohtonen, T.L. Tammela, T.K. Paavonen, J. Schleutker, The interaction of CYP3A5 polymorphisms along the androgen metabolism pathway in prostate cancer, Int. J. Cancer, 122(11) (2008), p. 2511-2516. [CrossRef]
- A. Loukola, M. Chadha, S.G. Penn, D. Rank, D.V. Conti, D. Thompson, M. Cicek, B. Love, V. Bivolarevic, Q. Yang, Y. Jiang, D.K. Hanzel, K. Dains, P.L. Paris, G. Casey, J.S. Witte, Comprehensive evaluation of the association between prostate cancer and genotypes/haplotypes in CYP17A1, CYP3A4, and SRD5A2, Eur. J. Hum. Genet., 12(4) (2004), p. 321-332. [CrossRef]
- J.P. Villeneuve, V, Pichette, Cytochrome P450 and liver diseases, Curr. Drug Metab., 5(3) (2004), p. 273-282. [CrossRef]
- Y. Bao, M. Phan, J. Zhu, X. Ma, J.E. Manautou, X.B. Zhong, Alterations of cytochrome P450-mediated drug metabolism during liver repair and regeneration after acetaminophen-induced liver injury in mice, Drug Metab. Dispos., 50(5) (2022), p. 694-703. [CrossRef]
- Y. Bao, P. Wang, X. Shao, J. Zhu, J. Xiao, J. Shi, L. Zhang, H.J. Zhu, X. Ma, J.E. Manautou, X.B. Zhong, Acetaminophen-induced liver injury alters expression and activities of cytochrome P450 enzymes in an age-dependent manner in mouse liver, Drug Metab. Dispos., 48(5) (2020), p. 326-336. [CrossRef]
- A. Michaut, C. Moreau, M.A. Robin, B. Fromenty, Acetaminophen-induced liver injury in obesity and nonalcoholic fatty liver disease, Liver Int., 34(7) (2014), p. e171-e179. [CrossRef]
- K.E. Thummel, J.T. Slattery, H. Ro, J.Y. Chien, S.D. Nelson, K.E. Lown, P.B. Watkins, Ethanol and production of the hepatotoxic metabolite of acetaminophen in healthy adults, Clin. Pharmacol. Ther., 67(6) (2000), p. 591-599. [CrossRef]
- S. Liu, R. Cheng, H. He, K. Ding, R. Zhang, Y. Chai, Q. Yu, X. Huang, L. Zhang, Z. Jiang, 8-methoxypsoralen protects against acetaminophen-induced liver injury by antagonising Cyp2e1 in mice, Arch. Biochem. Biophys., 741 (2023), p. 109617. [CrossRef]
- W. Yang, Z. Liang, C. Wen, X. Jiang, L. Wang, Silymarin protects against acute liver injury induced by acetaminophen by downregulating the expression and activity of the CYP2E1 enzyme, Molecules, 27(24) (2022), p. 8855. [CrossRef]
- B. Speed, H.Z. Bu, W.F. Pool, G.W. Peng, E.Y. Wu, S. Patyna, C. Bello, P. Kang, Pharmacokinetics, distribution, and metabolism of [14C]sunitinib in rats, monkeys, and humans, Drug Metab. Dispos., 40(3) (2012), p. 539-555. [CrossRef]
- D. Wang, F. Xiao, Z. Feng, M. Li, L. Kong, L. Huang, Y. Wei, H. Li, F. Liu, H. Zhang, W. Zhang, Sunitinib facilitates metastatic breast cancer spreading by inducing endothelial cell senescence, Breast Cancer Res., 22(1) (2020), p. 103. [CrossRef]
- G.M. Amaya, R. Durandis, D.S. Bourgeois, J.A. Perkins, A.A. Abouda, K.J. Wines, M. Mohamud, S.A. Starks, R.N. Daniels, K.D. Jackson, Cytochromes P450 1A2 and 3A4 catalyze the metabolic activation of sunitinib, Chem. Res. Toxicol., 31(7) (2018), p. 570-584. [CrossRef]
- E.A. Burnham, A.A. Abouda, J.E. Bissada, D.T. Nardone-White, J.L. Beers, J. Lee, M.J. Vergne, K.D. Jackson, Interindividual variability in cytochrome P450 3A and 1A activity influences sunitinib metabolism and bioactivation, Chem. Res. Toxicol., 35(5) (2022), p. 792-806. [CrossRef]
- S. Castellino, M. O'Mara, K. Koch, D.J. Borts, G.D. Bowers, C. MacLauchlin, Human metabolism of lapatinib, a dual kinase inhibitor: implications for hepatotoxicity, Drug Metab. Dispos., 40(1) (2012), p. 139-150. [CrossRef]
- J.K. Towles, R.N. Clark, M.D. Wahlin, V. Uttamsingh, A.E. Rettie, K.D. Jackson, Cytochrome P450 3A4 and CYP3A5-catalyzed bioactivation of lapatinib, Drug Metab. Dispos., 44(10) (2016), p. 1584-1597. [CrossRef]
- S. Chen, X. Li, Y. Li, X. He, M. Bryant, X. Qin, F. Li, J.E. Seo, X. Guo, N. Mei, L. Guo, The involvement of hepatic cytochrome P450s in the cytotoxicity of lapatinib, Toxicol. Sci., 197(1) (2023), p. 69-78. [CrossRef]
- G. García-Lainez, I. Vayá, M.P. Marín, M.A. Miranda, I. Andreu, In vitro assessment of the photo(geno)toxicity associated with lapatinib, a tyrosine knase inhibitor, Arch. Toxicol., 95(1) (2021), p. 169-178. [CrossRef]
- J. Li, M. Zhao, P. He, M. Hidalgo, S.D. Baker, Differential metabolism of gefitinib and erlotinib by human cytochrome P450 enzymes, Clin. Cancer Res., 13(12) (2007), p. 3731-3737. [CrossRef]
- D. McKillop, A.D. McCormick, A. Millar, G.S. Miles, P.J. Phillips, M. Hutchison, Cytochrome P450-dependent metabolism of gefitinib, Xenobiotica, 35(1) (2005), p. 39-50. [CrossRef]
- M. El Ouardi, L. Tamarit, I. Vayá, M.A. Miranda, I. Andreu, Cellular photo(geno)toxicity of gefitinib after biotransformation, Front. Pharmacol., 14 (2023), p. 1208075. [CrossRef]
- D.Y. Kim, K.H. Han, Epidemiology and surveillance of hepatocellular carcinoma, Liver Cancer 1 (2012), p. 2–14. [CrossRef]
- S. Caldwell, S.H. Park, The epidemiology of hepatocellular cancer: From the perspectives of public health problem to tumor biology, J. Gastroenterol., (2009), 44, 96–101. [CrossRef]
- Y.J. Chu, H.I. Yang, H.C. Wu, J. Liu, L.Y. Wang, S.N. Lu, M.H. Lee, C.L. Jen, S.L. You, R.M. Santella, C.J. Chen, Aflatoxin B1 exposure increases the risk of cirrhosis and hepatocellular carcinoma in chronic hepatitis B virus carriers, Int. J. Cancer, 141(4) (2017), p. 711-720. [CrossRef]
- Q. Zhu, Y. Ma, J. Liang, Z. Wei, M. Li, Y. Zhang, M. Liu, H. He, C. Qu, J. Cai, X. Wang, Y. Zeng, Y. Jiao, AHR mediates the aflatoxin B1 toxicity associated with hepatocellular carcinoma, Signal Transduct. Target Ther., 6(1) (2021), p. 299. [CrossRef]
- L. Silvestri, L. Sonzogni, A. De Silvestri, C. Gritti, L. Foti, C. Zavaglia, M. Leveri, A. Cividini, M.U. Mondelli, E. Civardi, E.M. Silini, CYP enzyme polymorphisms and susceptibility to HCV-related chronic liver disease and liver cancer, Int. J. Cancer, 104(3) (2003), p. 310-317. [CrossRef]
- J.A. Agúndez, M. Olivera, J.M. Ladero, A. Rodriguez-Lescure, M.C. Ledesma, M. Diaz-Rubio, U.A. Meyer, J. Benítez, Increased risk for hepatocellular carcinoma in NAT2-slow acetylators and CYP2D6-rapid metabolizers, Pharmacogenetics, 6(6) (1996), p. 501-12. [CrossRef]
- Z. Zhou, Q. Wen, S.F. Li, Y.F. Zhang, N. Gao, X. Tian, Y. Fang, J. Gao, M.Z. Cui, X.P. He, L.J. Jia, H. Jin, H.L. Qiao, Significant change of cytochrome P450s activities in patients with hepatocellular carcinoma, Oncotarget, 31 (2016), p. 50612-50623. [CrossRef]
- J. Mochizuki, S. Murakami, A. Sanjo, I. Takagi, S. Akizuki, A. Ohnishi, Genetic polymorphisms of cytochrome P450 in patients with hepatitis C virus-associated hepatocellular carcinoma, J. Gastroenterol. Hepatol., 20(8) (2005), p. 1191-1197. [CrossRef]
- G. Hu, F. Gao, G. Wang, Y. Fang, Y. Guo, J. Zhou, Y. Gu, C. Zhang, N. Gao, Q. Wen, H. Qiao, Use of proteomics to identify mechanisms of hepatocellular carcinoma with the CYP2D6*10 polymorphism and identification of ANGPTL6 as a new diagnostic and prognostic biomarker, J. Transl. Med., 19(1) (2021), p. 359. [CrossRef]
- B. de Jesus Brait, S.P. da Silva Lima, F.L. Aguiar, R. Fernandes-Ferreira R, C.I.F. Oliveira-Brancati, J.A.M.L. Ferraz, G.D. Tenani, M.A. de Souza Pinhel, L.C.P. Assoni, A.H. Nakahara, N. Dos Santos Jábali, O.R.F. Costa, M.E.L. Baitello, S.P. Júnior, R.F. Silva, R. de Cássia Martins Alves Silva, D.R. da Silva, Genetic polymorphisms related to the vitamin D pathway in patients with cirrhosis with or without hepatocellular carcinoma (HCC), Ecancermedicalscience, 16 (2022), p. 1383. [CrossRef]
- M.W. Yu, A. Gladek-Yarborough, S. Chiamprasert, R.M. Santella, Y.F. Liaw, C.J. Chen, Cytochrome P450 2E1 and glutathione S-transferase M1 polymorphisms and susceptibility to hepatocellular carcinoma, Gastroenterology, 109(4) (1995), 1266-1273. [CrossRef]
- S. Boccia, L. Miele, N. Panic, F. Turati, D. Arzani, C. Cefalo, R. Amore, M. Bulajic, M. Pompili, G. Rapaccini, A. Gasbarrini, C. La Vecchia, A. Grieco, The effect of CYP, GST, and SULT polymorphisms and their interaction with smoking on the risk of hepatocellular carcinoma, Biomed. Res. Int., 2015 (2015), p. 179867. [CrossRef]
- J. Gao, Z. Wang, G.J. Wang, H.X. Zhang, N. Gao, J. Wang, C.E. Wang, Z. Chang, Y. Fang, Y.F. Zhang, J. Zhou, H. Jin, H.L. Qiao, Higher CYP2E1 activity correlates with hepatocarcinogenesis induced by diethylnitrosamine, J. Pharmacol. Exp. Ther., 365(2) (2018), p. 398-407. [CrossRef]
- Y. Fang, H. Yang, G. Hu, J. Lu, J. Zhou, N. Gao, Y. Gu, C. Zhang, J. Qiu, Y. Guo, Y. Zhang, Q. Wen, H. Qiao, The POR rs10954732 polymorphism decreases susceptibility to hepatocellular carcinoma and hepsin as a prognostic biomarker correlated with immune infiltration based on proteomics, J. Transl. Med., 20(1) (2022), p. 88. [CrossRef]
- F. Cao, A. Luo, C. Yang, G6PD inhibits ferroptosis in hepatocellular carcinoma by targeting cytochrome P450 oxidoreductase, Cell Signal., 87 (2021), p. 110098. [CrossRef]
Figure 1.
The involvement of CYPs in the pathways of steroid hormone biosynthesis. The side chain of cholesterol is initially oxidized and cleaved by CYP11A1 with the involvement of the CYP redox partners, Fdx and FdR, and StAR to yield pregnenolone. The latter is either hydroxylated at C17 to produce 17α-OHP5 by mitochondrial CYP17A1 or undergoes oxidation of its hydroxyl group at C3 by 3β-HSD2 to yield progesterone. The 17α-OHP5 is further converted by CYP17A1 with the involvement of POR and cyt b5 to DHEA and androstenedione, both C19 androgen and C18 estrogen precursors. CYP19A1 plays critical roles in sex steroid hormone biosynthesis whereas CYP11B1 and CYP11B2 are essential for corticosteroid biosynthesis.
Figure 1.
The involvement of CYPs in the pathways of steroid hormone biosynthesis. The side chain of cholesterol is initially oxidized and cleaved by CYP11A1 with the involvement of the CYP redox partners, Fdx and FdR, and StAR to yield pregnenolone. The latter is either hydroxylated at C17 to produce 17α-OHP5 by mitochondrial CYP17A1 or undergoes oxidation of its hydroxyl group at C3 by 3β-HSD2 to yield progesterone. The 17α-OHP5 is further converted by CYP17A1 with the involvement of POR and cyt b5 to DHEA and androstenedione, both C19 androgen and C18 estrogen precursors. CYP19A1 plays critical roles in sex steroid hormone biosynthesis whereas CYP11B1 and CYP11B2 are essential for corticosteroid biosynthesis.
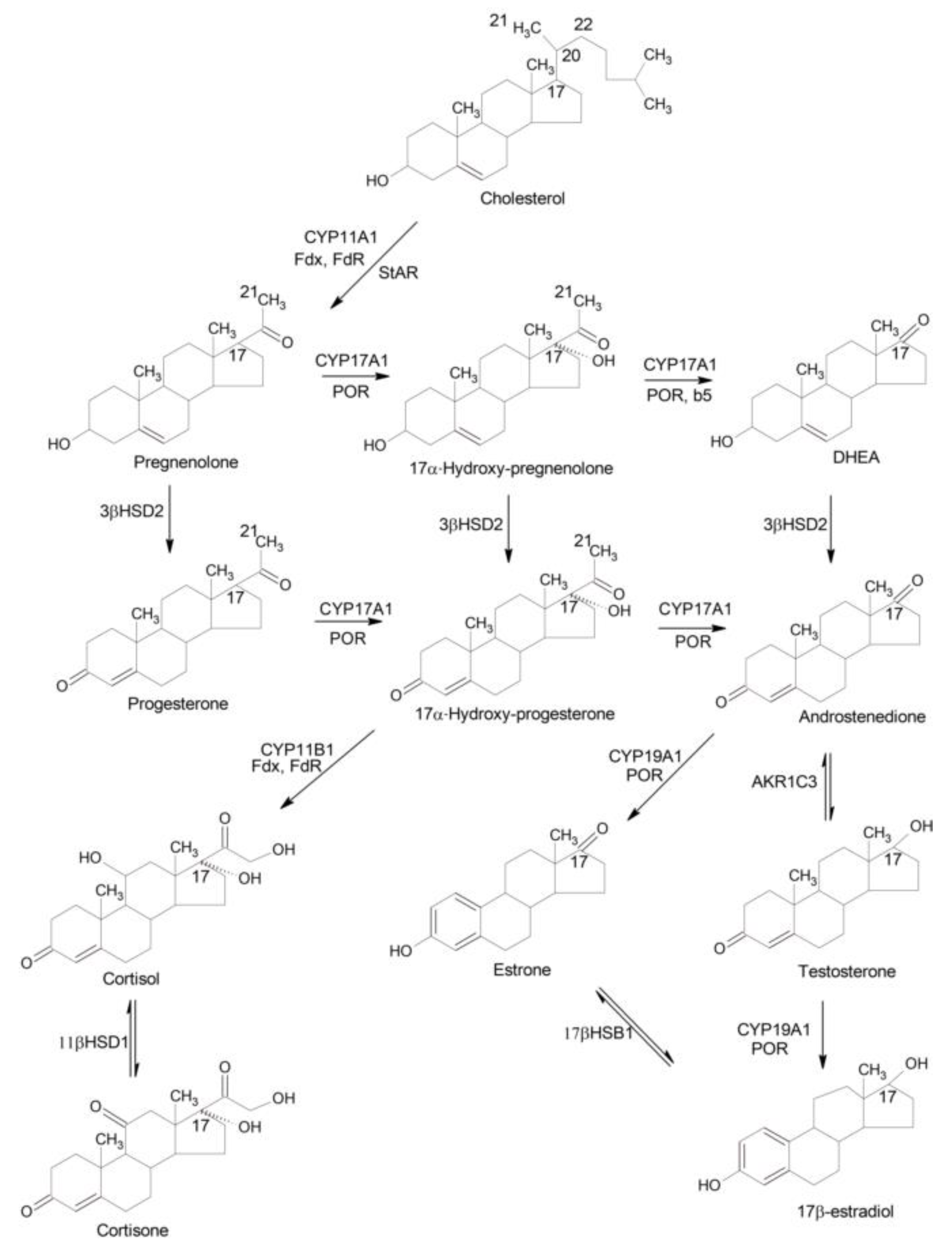
Figure 2.
The implications of CYPs in detoxification versus bioactivation of xenobiotics in the liver. (A) CYP3A4, CYP2D6, and CYP4F12 catalyze the bioactivation of C10 BACs via the ω-oxidation reactions and their major products including ω-hydroxy-, (ω−1)-hydroxy-, (ω, ω−1)-diol-, (ω−1)-ketone-, and ω-carboxylic acid metabolites. (B) Parathion is activated to diethyl 4-nitrophenyl phosphate (paraoxon) by CYP1A2 and CYP2B6; however, CYP2C19 catalyzes inactivation via oxidative cleavage of parathion to p-nitrophenol and diethyl-phosphate. (C) CYP1A1 and CYP1A2 catalyze the activation of carbaryl to 5-hydroxycarbaryl whereas CYP1A2 and CYP3A4 activate carbaryl to 4-hydroxycarbaryl.
Figure 2.
The implications of CYPs in detoxification versus bioactivation of xenobiotics in the liver. (A) CYP3A4, CYP2D6, and CYP4F12 catalyze the bioactivation of C10 BACs via the ω-oxidation reactions and their major products including ω-hydroxy-, (ω−1)-hydroxy-, (ω, ω−1)-diol-, (ω−1)-ketone-, and ω-carboxylic acid metabolites. (B) Parathion is activated to diethyl 4-nitrophenyl phosphate (paraoxon) by CYP1A2 and CYP2B6; however, CYP2C19 catalyzes inactivation via oxidative cleavage of parathion to p-nitrophenol and diethyl-phosphate. (C) CYP1A1 and CYP1A2 catalyze the activation of carbaryl to 5-hydroxycarbaryl whereas CYP1A2 and CYP3A4 activate carbaryl to 4-hydroxycarbaryl.

Figure 3.
CYP-mediated bioactivation of procarcinogens. (A) CYP1A1 and CYP1B1 catalyzes the conversion of benzo[a]pyrene (B[a]P) to 7,8-epoxy-benzo[a]pyrene that is further converted by liver epoxide hydrolase to B[a]P-7,8-dihydrodiol, which gives rise to other epoxy-derivatives with the capability to interact with DNA yielding toxic adducts. (B) Metabolic activation of 3-aminodibenzofuran by CYP1A2 and CYP2A6 via the formation of N-hydroxylated derivative and further SULT-mediated conjugation with sulfuric acid. (C) CYP3A4 and CYP1A2 are involved in inactivation of aflatoxin B1 to its Q1 and M1 derivatives as well as activation to exo- and endo-8,9-epoxides that can gives rise to toxic DNA adducts.
Figure 3.
CYP-mediated bioactivation of procarcinogens. (A) CYP1A1 and CYP1B1 catalyzes the conversion of benzo[a]pyrene (B[a]P) to 7,8-epoxy-benzo[a]pyrene that is further converted by liver epoxide hydrolase to B[a]P-7,8-dihydrodiol, which gives rise to other epoxy-derivatives with the capability to interact with DNA yielding toxic adducts. (B) Metabolic activation of 3-aminodibenzofuran by CYP1A2 and CYP2A6 via the formation of N-hydroxylated derivative and further SULT-mediated conjugation with sulfuric acid. (C) CYP3A4 and CYP1A2 are involved in inactivation of aflatoxin B1 to its Q1 and M1 derivatives as well as activation to exo- and endo-8,9-epoxides that can gives rise to toxic DNA adducts.
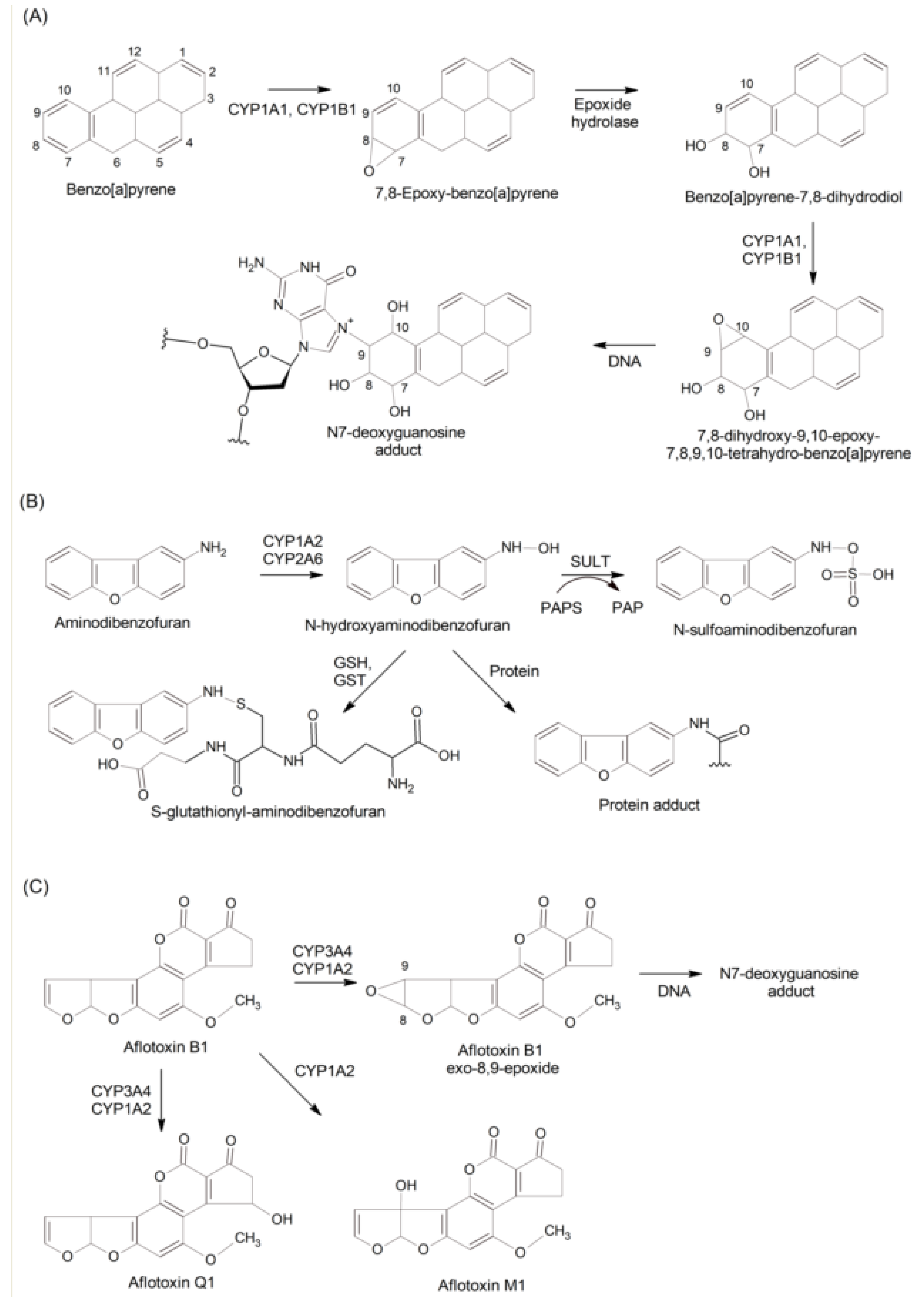
Figure 4.
CYP-catalyzed drug metabolism. (A) CYP1A1 and CYP1A2 catalyze the antitumor drug ellipticine detoxification via the formation of 7-hydroxy- and 9-hydroxyellipticine, whereas CYP3A4 catalyzes bioactivation via N-hydroxylation to yield ellipticine N2-oxide and produces 13-hydroxyellipticine that can form DNA adducts. (B) Tamoxifen is metabolized to 4-hydroxytamoxifen by CYP2C19 and CYP2D6 and to N-desmethyltamoxifen by CYP2D6, CYP3A4, and CYP3A5. The both metabolites can be converted to endoxifen (4-hydroxy-N-desmethyltamoxifen) by CYP3A4/CYP3A5 and CYP2D6, respectively. CYP3A4/CYP3A5 are also active in conversion of tamoxifen to α-hydroxytamoxifen. (C) Anticancer drug sorafenib is metabolized by CYP3A4 and CYP2C8 N-hydroxymethyl, N-desmethyl metabolites, and sorafenib N-oxide.
Figure 4.
CYP-catalyzed drug metabolism. (A) CYP1A1 and CYP1A2 catalyze the antitumor drug ellipticine detoxification via the formation of 7-hydroxy- and 9-hydroxyellipticine, whereas CYP3A4 catalyzes bioactivation via N-hydroxylation to yield ellipticine N2-oxide and produces 13-hydroxyellipticine that can form DNA adducts. (B) Tamoxifen is metabolized to 4-hydroxytamoxifen by CYP2C19 and CYP2D6 and to N-desmethyltamoxifen by CYP2D6, CYP3A4, and CYP3A5. The both metabolites can be converted to endoxifen (4-hydroxy-N-desmethyltamoxifen) by CYP3A4/CYP3A5 and CYP2D6, respectively. CYP3A4/CYP3A5 are also active in conversion of tamoxifen to α-hydroxytamoxifen. (C) Anticancer drug sorafenib is metabolized by CYP3A4 and CYP2C8 N-hydroxymethyl, N-desmethyl metabolites, and sorafenib N-oxide.
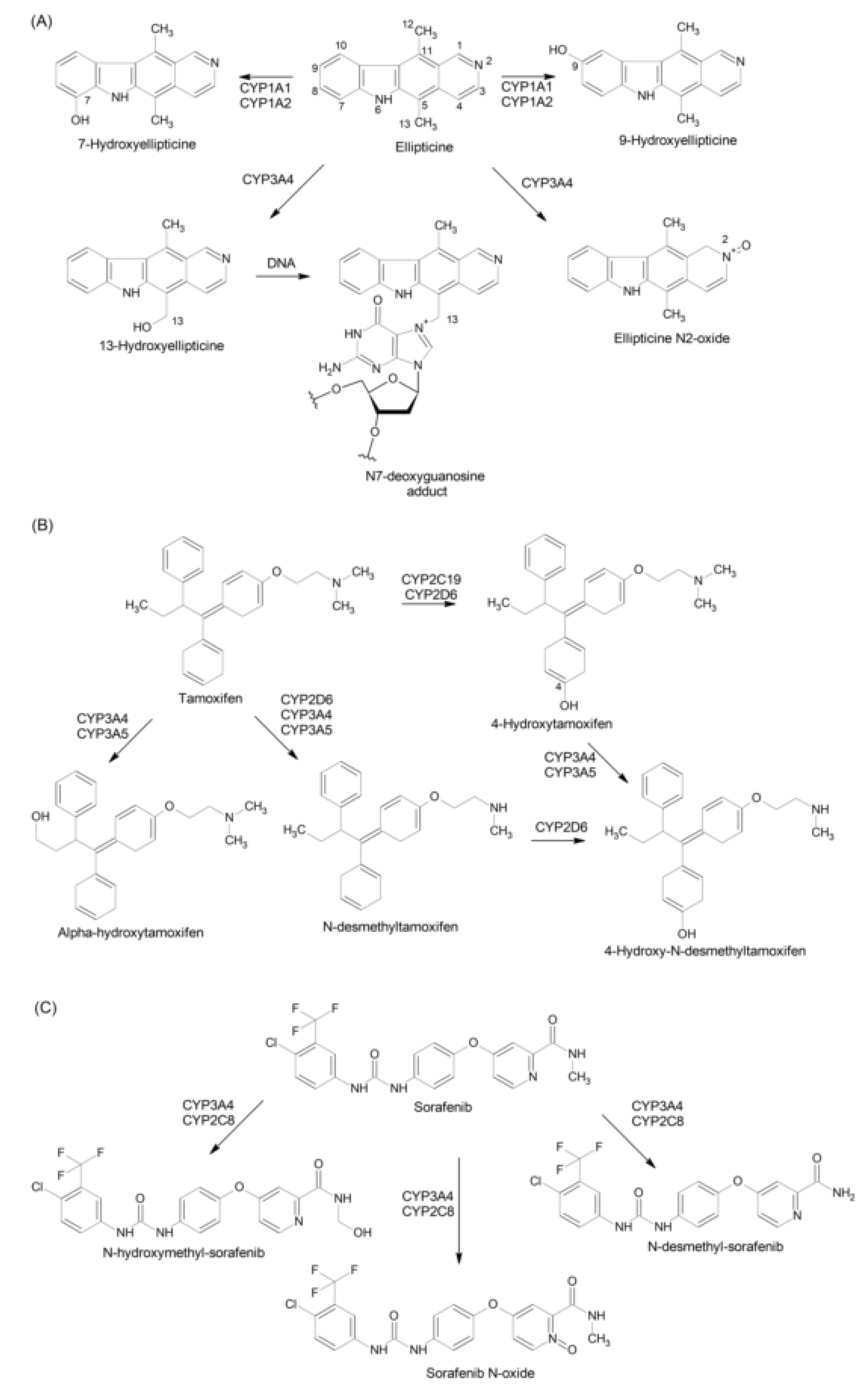
Figure 5.
Metabolism of estrogens and androgens by CYPs. (A) The pathways of 17β-estradiol conversions by CYP1A1, CYP1A2, and CYP1B1 to 2,3-cathechols and further to 2,3-quinones with subsequent either methylation by COMT or conjugation with glucuronic acid by UGT or DNA adduct formation. (B) Testosterone can be either reversibly converted to androstenedione by 17βHSD or reduced by SRD5A1/2 isoenzymes to DHT, which can be, alternatively, produced from androstenedione by 17βHSD and SRD5A1/2 isoenzymes; both testosterone and DHT are conjugated with, mostly, glucuronic acid and, at a less extent, sulfuric acid. (C) Metabolism of sunitinib by CYP1A2 and CYP3A4 via oxidative defluorination and the formation of quinoneimine; the presence of CYP3A4 inducer rifampicin causes sunitinib N-dealkylation to form N-desethyl-sunitinib.
Figure 5.
Metabolism of estrogens and androgens by CYPs. (A) The pathways of 17β-estradiol conversions by CYP1A1, CYP1A2, and CYP1B1 to 2,3-cathechols and further to 2,3-quinones with subsequent either methylation by COMT or conjugation with glucuronic acid by UGT or DNA adduct formation. (B) Testosterone can be either reversibly converted to androstenedione by 17βHSD or reduced by SRD5A1/2 isoenzymes to DHT, which can be, alternatively, produced from androstenedione by 17βHSD and SRD5A1/2 isoenzymes; both testosterone and DHT are conjugated with, mostly, glucuronic acid and, at a less extent, sulfuric acid. (C) Metabolism of sunitinib by CYP1A2 and CYP3A4 via oxidative defluorination and the formation of quinoneimine; the presence of CYP3A4 inducer rifampicin causes sunitinib N-dealkylation to form N-desethyl-sunitinib.
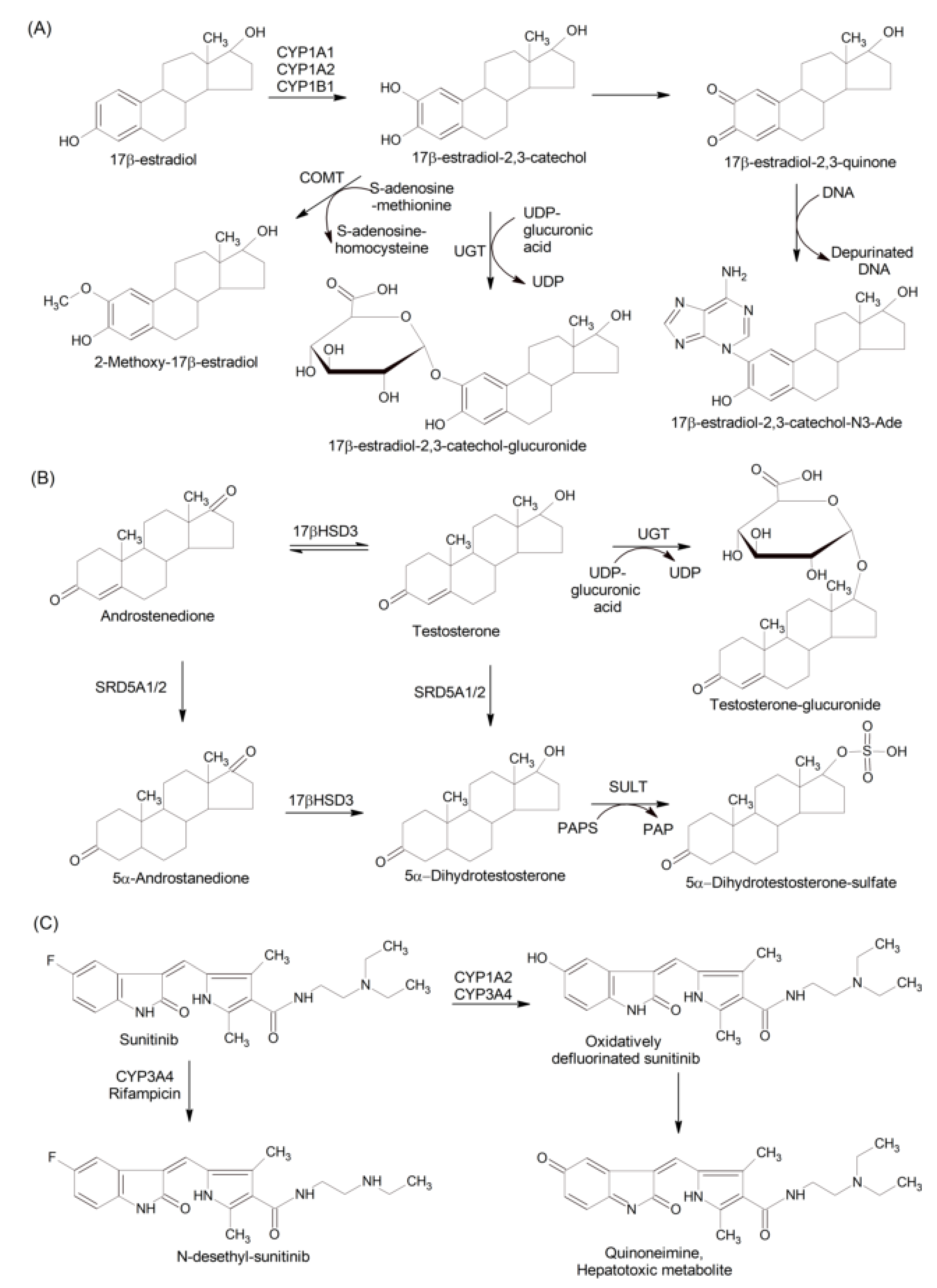
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



