
Submitted:
02 October 2024
Posted:
04 October 2024
You are already at the latest version
Abstract
The failure of immunotherapies in cancer patients is being widely studied due to the complexities present in the tumor microenvironment (TME), where regulatory T cells (Treg) appear to actively participate in providing an immune escape mechanism for tumors. Therefore, therapies to specifically inhibit tumor-infiltrating Treg represent a challenge because Treg are distributed throughout the body and provide physiological immune homeostasis to prevent autoimmune diseases. Characterization of immunological and functional profiles could help to identify the mechanisms that need to be inhibited or activated to ensure Treg modulation in the tumor. To address this, quantitative in-silico approaches based on mechanistic mathematical models integrating multi-scale information from immune and tumor cells and the effect of different therapies have allowed the building of computational frameworks to simulate different hypotheses, some of which have subsequently been experimentally validated. Therefore, this review presents a list of diverse computational mathematical models that examine the role of Treg as a crucial immune resistance mechanism contributing to the failure of immunotherapy. In addition, this review highlights the relevance of certain molecules expressed in Treg that are associated with the TME immunosuppression, which could be incorporated into the mathematical model for a better understanding of the contribution of Treg modulation. Finally, different pre-clinical and clinical combinations of molecules are also included to show the trend of new therapies targeting Treg.
Keywords:
Treg cells
; QSP model
; Immunotherapy
; Tumor growth dynamics
; computational immune framework
Introduction
Immunotherapy in oncology enhances the ability of the immune system to control or eliminate cancer cells, thus changing the paradigm of cancer treatment [1]. However, tumor cells can develop various resistance or immune evasion mechanisms, such as the introduction of mutations and/or upregulation of immune checkpoints, in particular programmed death ligands 1 (PD-L1). This upregulation contributes to generate an immunosuppressive tumor microenvironment (TME) that enables tumor growth and proliferation [2,3,4,5].
The relationship between tumor growth dynamics and the changes in the TME is highly complex, due to the behavior of the immune system, which is mainly responsible for this complexity. Figure 1 illustrates this relationship in a simplified way, which is based on the cancer-immunity cycle described by Mellman et al. [6]. Briefly, dendritic cells (DC) recognize and engulf neoantigens and damaged associated molecular proteins (DAMPs) produced by dead tumor cells, activating the circulating naive T cells that link the innate and adaptive immune responses. However, it is the balance between immune activation and suppression that determines whether a tumor proliferates or is eliminated. The anti-tumor response is induced by DC, M1 macrophages, CD4+ T cells, and cytotoxic CD8+ T cells [7,8,9,10]. On the other hand, tumor resistance is derived from M2 macrophages, Myeloid-Derived Suppressor Cells (MDSC), and regulatory T-cells (Treg) [11,12,13]. In this context, the design of strategies to improve antitumor efficacy requires substantial knowledge of these complex interactions between the immune system and cancer cells, and the identification of the essential mechanisms that need to be inhibited or stimulated to achieve total tumor rejection and generate immune memory. Given these unmet medical requirements in immune oncology, mathematical models stand out for their ability to describe and quantify the dynamics of different physio-pathological processes involved in tumor progression and the activity of immunotherapy.
Different types of models have been reported in the literature, including empirical and semi-mechanistic models which aim to describe and/or predict tumor growth. These models have provided a good description of tumor growth kinetics after different treatments in animals and patients. They show interesting applications for the development of new agents and propose different drug combinations or even new dose regimens to reduce the toxicities induced by chemotherapy or radiotherapy. However, in order to integrate the knowledge of the mechanisms triggered by tumors, therapies, and the immune system, the model structure requires greater complexity, which is addressed by the quantitative system pharmacology (QSP) approach.
The QSP approach in immune-oncology (IO) represents a differentiated scientific and bioinformatic tool that incorporates physiology, physiopathology, inter-individual variability, and therapeutic activity to describe and characterize the interactions between cancer cells and immune cells, in a granular way. Importantly, QSP model building is, generally, based on multi-scale information integrating relevant biomarkers and cell populations. In some of these models, the activity of immunotherapy has been adapted by incorporating the effect of activated tumor-specific cytotoxic CD8+ T cells responsible for tumor elimination together with the immune resistance response associated with the failure of many immunotherapeutic approaches, mainly due to the presence of tumor-infiltrating Treg. Although serious efforts have been made to identify up- or down-regulated biomarkers in Treg, this information has not yet been elucidated and integrated in a quantitative framework [14].
Therefore, this review summarizes a list of quantitative computational models that relate Treg activity to antitumor immunity, exploring the impact of some relevant mechanisms involved in IO therapies. Furthermore, different therapeutic strategies to counteract the immunoresistance effect of Treg are briefly discussed as a possible contribution to the development of QSP models.
1. Regulatory T Cells in TME
Treg maintain physiological immune homeostasis by balancing excessive immune responses with suppression of autoimmune responses.
Treg belong to the CD4+ T cell population and are characterized by the expression of CD4 and CD25 (or IL-2 receptor) on their surface, and the intracellular nuclear transcription factor Forkhead box P3 (Foxp3), which controls Treg proliferation and activity. However, both biomarkers, Foxp3 and CD25, are also transiently expressed on activated effector T cells (cytotoxic CD8+ T cells). Therefore, it is relevant to selectively identify Treg to study their role in IO. A summary of such biomarkers is provided in Supplementary Table S1.
It is known that Treg can migrate to specific tissues in response to chemokine signalling and differentiate into CD25high Foxp3high, a characteristic of immunosuppressive Treg (iTreg), which represents one of the immune escape mechanisms developed by the tumors [15,16]. These chemokines, which are able to bind to their receptors, CCR4 and CCR8, are particularly involved in Treg modulation and immunotherapy efficacy. CCL17 and CCL22 are endogenous ligands that bind to CCR4, promoting Th2 response and thereby, facilitating Treg tumor infiltration [17,18]. Similarly, CCL18 or CCL1 bound to CCR8 which is strongly expressed in tumor-infiltrated Treg, lead to Treg proliferation and activation through Foxp3 upregulation [19]. These Treg release immunosuppressive cytokines such as IL-10, TGFβ, and IL-35 within TME, competing with the effector CD8+ T cells for IL-2 by upregulating the expression of CD25. This activity promotes the depletion of NK cells and disrupts the activation and proliferation of effector CD8+ T cells, which become exhausted, inducing an immunotolerant and immunosuppressive TME. As a consequence of this process, iTreg infiltration in the TME is associated with a poor prognosis and the failure of immunotherapies [20].
As the efficacy of immunotherapies lies in the enhancement of immune surveillance, appropriate modulation of functional Treg activity may improve this mechanism and thus clinical response. To address this, mathematical modeling provides a useful tool to better explore these complexities using an in-silico approach to identify mechanisms that could explain the observed clinical or preclinical results [21].
2. Relevance of Quantitative Mathematical Models to Explore Treg Activity in Tumor Growth
Interest in characterising tumour growth using quantitative approaches, including empirical, semi-mechanistic and QSP models, has grown over the years. One of the pivotal mathematical models is the data-driven pharmacokinetic/pharmacodynamic (PKPD) model, which is capable of describing tumor growth and identifying significant covariates affecting some model parameters under different scenarios through in-silico simulations [22]. The main goal of such PKPD model is the screening of tumor sensitivity to drugs and drug distribution in the tumor, relying on data from immunosuppressed animal models (i.e., xenograft mouse), and thus not considering immunological aspects.
Notably, the inclusion of immunoresistance mechanisms and the antitumor effects exerted by effector CD8+ T cells were later accounted for by using data from syngenic murine tumor models. This animal model allows the identification of inter-subject variability in the immune response and the stratification of individuals into responders and non-responders [23]. In addition, these models have provided a framework for testing the effects of different therapeutic agents including vaccines, toll-like receptor (TLR) agonists, and immune checkpoint inhibitors, among others. However, the role of pro-tumor factors such as infiltrating Treg, MDSC, and PD-L1 upregulation were implicitly modeled due to the lack of specific information on lymphocyte populations and biomarker levels. This highlights the critical need for experimental data to elucidate the underlying pro-tumor mechanisms.
In recent years, new experimental methodologies using functional and phenotypic assays have greatly improved the ability 1) to study the temporal characteristics of tumor and immune cell progression, 2) to capture the heterogeneity of tumors and TME [24], and 3) to provide more comprehensive in-vitro and in-vivo information on novel therapies. These advances challenge the incorporation of the obtained data into mechanistic computational models, as occurs in the QSP model structure.
Understanding the TME, its elements, and the immune framework, is essential to determine the impact of effector CD8+ T cells alongside Treg. Figure 2 depicts the main processes involved in either i) tumor elimination (represented in red) where CD8+ T cells are essential, or ii) tumor progression (represented in blue) where Treg are the main responsible elements. In addition, coordination across lymphocytes CD8+, CD4+, Treg, NK cells, macrophages M1 and M2, MDSC and DC is required to regulate the development and progression of the disease. Integrating these processes with their corresponding biomarkers into a mathematical framework that replicates the most complex immunological state triggered by the presence of a tumor is the most challenging exercise in predicting the final immune response.
Several promising biological markers have been identified to determine the balance between antitumor immune response and resistance mechanisms. In particular, an increased intratumoral CD8+:Treg ratio has been identified as a predictor of tumor shrinkage in preclinical studies and has also been translated into clinical trials [24]. An increase in activated CD8+ T cells shifts the balance in favour of the immune system to achieve an antitumor response. However, the presence of Treg and their balance with CD8+ T cells has not fully explained clinical outcomes. This has led to a great interest in preclinical studies to investigate the role of Treg inhibition, stimulation, or depletion within the TME, which ultimately determine the efficacy of immunotherapeutic approaches in cancer [25]. It should be noted that cytokines and chemokines are also relevant players in Treg modulation, and therefore, their integration into these computational QSP models is necessary. On the other hand, the role of other immune cells present in the TME, such as the T helper cells and macrophage subsets that control the Th1/Th2 and M1/M2 ratios, respectively, in tumor clearance is not yet fully understood [26].
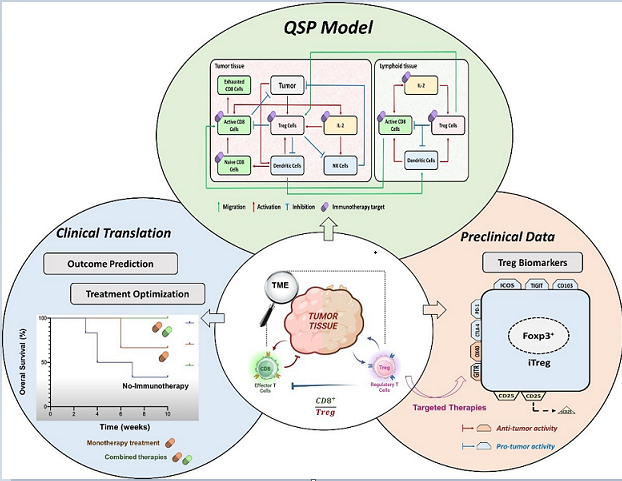
Different QSP quantitative frameworks have been published in the literature partially covering the pro and antitumor response elements described in Figure 2 [27]. In order to explore and consider the role of Treg in cancer in a comprehensive manner, we focused our review on those that included the antitumoral effect of Treg in immunotherapy and applied the following inclusion criteria:1) preclinical and clinical studies, 2) time-evolution dynamics of cell populations (described by ordinary differential equations, ODEs), and 3) tumor dynamic profiles as the main outcome. We also included articles studying longitudinal clinical data on cytokines and relevant lymphocyte populations. A total of six articles combining preclinical and clinical data were selected, numbered and presented in Table 1, according to increasing complexity in terms of mechanistic aspects, cell populations, assay complexity, spatial components (tumor vasculature, tumor compartment), and tested therapies.
The first article, reported by Kronik et al., presents a mathematical nonspatial model developed to describe cellular immunotherapy for melanoma using clinical trial data integrating CD8+ T cells and cytokines [28]. The model includes melanoma cells that express immunogenic antigens in the context of the major histocompatibility complex I (MHC I) molecules, but also secrete the pro-tumor factor TGF-β, which inhibits T-cell activity, implicitly representing the effects of Treg. The tested treatment consists of adoptive T-cell therapy, based on ex-vivo expanded tumor-specific T-cell infusion that lyses their target and secretes the antitumor factor IFN-ϒ, which has a positive feedback on MHC I molecules, increasing cytotoxic T lymphocytes (CTL)-mediated antitumor effect. In addition, a treatment with IL-2 infusions prolongs the persistence of infused CTL. The combination of both therapies promotes a higher antitumor efficacy than adoptive T-cell therapy alone, with assumable adverse effects. Tumor growth rate and tumor size are essential in predicting the outcome of therapy, and this mathematical model supports the decision of the most favorable schedule for each patient.
Other studies have reported models focusing on one (Wilson et al.) or on parallel (Robertson-Tessi et al.) pathways of inhibitory factors induced by Treg, TGF-β and/or IL-10 that promote tumor growth and reduce the cytotoxicity of effector cells (i.e., CD8+ cells and others such as DC, CD4+ helper T cells, and IL-2). In the model reported by Wilson et al., using data from a TC1 murine tumor model, the authors considered two types of interactions between tumor cells and T cells: 1) the interaction with Treg through TGF-β release to investigate the anti-TGF-β treatment, and 2) the interaction with CD8+ cells, by using cancer vaccines as effector cell stimulators [29]. The authors conclude that tumor elimination requires combined immunotherapy treatments capable of working through different mechanisms since vaccine monotherapy is not sufficient to eradicate the tumor.
Increasing the complexity of the model, Robertson-Tessi et al. considered different simultaneous pathways of pro-tumor effects [30]. The model, based on in silico multiscale data from tumors with different growth rates and antigenicities, estimates and simulates typical time profiles of tumor growth by determining the relevance of various immunosuppressive mechanisms at the different stages of tumor growth. In addition to TGF-β-induced immunosuppression, this model accounts for the conversion of CD4+ helper T cells into Treg, and the evaluation of dendritic cell therapy. The model structure allows for tumor escape, which is mainly supported by the immunosuppression conferred by TGF-β (greater effects with larger tumor sizes), the presence of Treg at all tumor growth stages, and the limited access of immune cells to the TME at large tumor sizes (sufficient tumor vascularization). Interestingly, for a given tumor growth rate, there is a specific antigenicity and optimal dose of transfused dendritic cells that leads to an adequate response of the immune system that shall be large enough to affect the tumor, but small enough to avoid excessive Treg promotion and suppressive effects.
In addition to different cytokines and the most representative immune cell populations involved in immunotherapy, the role of M1 and M2 macrophages in tumor growth, and the use of the M2/M1 ratio as a pro-tumor biomarker, has garnered considerable attention in recent years. Den Breems et al. reported a mathematical model that analyzes the stimulation of immunity by Th1 and M1 cells, and their complementary immunosuppressive pro-tumor response derived from M2 and Th2 cells, to describe data from a murine melanoma model [26]. However, type I and type II cytokines, which are required to mediate the interaction between M1 and Th1, and between M2 and Th2 cells, respectively, are not included in order to keep the model structure relatively simple. The authors conclude that an M2/M1 macrophage ratio greater than 1 can explain the tumor size, but caution that experimental studies with longitudinal data on M2/M1 ratio biomarker are too limited to confront these findings. Furthermore, data from human trials are also required to evaluate/validate the results.
The model proposed by Coletti et al. for prostate cancer successfully added compartmentalization into the prostate gland and lymphoid tissue to the structure using data from pre-clinical experiments, given the paucity of clinical studies [31]. The prostate gland compartment contains two types of prostate cancer cells, androgen-dependent (representing tumor cell sensitivity) and independent (corresponding to tumor cell resistant) cells. Other players involved, connecting both compartments, are mature DC, which in lymphoid tissues act as functional DC (Df) or regulatory DC (Dr) activating CTL or Treg, respectively. Finally, the pro-tumor immunosuppression exerted by MDSC, an essential contributor to prostate cancer along with NK, androgens, and IL-2, have been included to explore different immunotherapies. Historically, prostate cancer has not responded well to immunotherapy possibly due to a strong immunosuppressive TME [32]. Thus, this model provides the framework to test in silico a variety of seven combinatorial immunotherapies: 1) androgen deprivation therapy, 2) cancer vaccines targeting mature dendritic cells, 3) anti-IL-2 for Treg inhibition, 4) anti-CD25 for Treg depletion, 5) administration of NK cells, 6) combination of anti-CTLA4 with anti-PD1 antibodies, and 7) cabozantinib (anti-MDSC). A possible limitation of this model is the lack of effect of the suppressive molecules TGF-β and IL-10 secreted by Treg, which may exert a pro-tumoral effect. Given that, these biomarkers are easy to obtain from peripheral blood, suggesting their inclusion in future models. In addition, although beyond the scope of the current review focused on Treg function in the TME, the cell mutation from androgen-dependent to independent prostate cancer cells may provide a novel aspect in QSP modeling in IO.
Finally, the most complex QSP model is reported by Ji et al. [33]. This model was developed for DTA-1.mIgG2a (DTA-1), a mAb agonist of GITR (glucocorticoid-induced tumor necrosis factor receptor-related protein), which is constitutively overexpressed on the surface of Treg. DTA-1, tested in two syngenic murine tumor models (CT26 and A20), has previously been shown to inhibit Treg and activate cytotoxic CD8+ T and NK cells. This is due to a dual activity: 1) inhibition of Treg by the binding of DTA-1 to GITR, and 2) a direct depletion of Treg by antibody-dependent cellular phagocytosis (ADPC). This depletion, mediated mainly by macrophages, downregulates IL-10 and allows the activation of effector T cells influenced by IL-2 expression. To address this hypothesis, data consisting of serum concentrations of DTA-1, soluble GITR (sGITR), and anti-drug antibodies (ADAs) for pharmacokinetic (PK) assessment, in addition to the major T cell subsets including intratumoral levels of Treg (distinguishing high and low GITR expression in Treg), effector CD8+ T cells (accounted as the sum of inactivated and activated effector T cells), as well as macrophages (FcgRIV) for pharmacodynamic (PD) assessment, were collected from different compartments and analyzed. DTA-1 PK was described by a two-compartment model with a non-linear elimination, due to the presence of ADAs capable of binding to sGITR. DTA-1 plasma concentrations were directly connected with the tumor compartment for drug trafficking. Interestingly, the authors explain the tumor shrinkage as a result of the balance of the CD8+/Treg ratio, which first requires depletion of Treg capable of causing a reduction in the immunosuppressive effects of IL-10, then promoting the proliferation and differentiation of CD8+ T cells that eliminate cancer cells. Note that several complexities were addressed in this study, such as the different antitumor responses depending on the tumor model, and the optimal dose. To describe tumor elimination, the authors hypothesized that treatment responses were associated with the presence of Treg with high GITR expression which can induce a greater ADCP effect. Although the model, under certain assumptions, successfully described the PK and PD of DTA-1 in the two preclinical tumor models, verification/validation using more complex experimental designs and the inclusion of other immune pathways will be necessary to understand/elucidate and quantify the mechanisms hypothesized here.
Remarkably, the dual activity assumed for DTA-1 is also shared by other mAbs against certain biomarkers expressed on Treg. This is the case of anti-CTLA-4, which can inhibit Treg activity and also induce Treg depletion via the ADCC mechanism, leading to an increase in antitumor efficacy. However, as activated effector T cells and DC also express CTLA-4, this strategy could also cause a reduction of these immune cell populations, thereby hampering the activity of effector CD8+ T cells [34].
All the models reviewed have some characteristics that need to be highlighted. A shared limitation to models 1 to 4 is that they are all non-spatial tumor-immune models, confined to the TME. Although the model proposed by Robertson-Tessi et al was developed without spatial elements, it considers the number of tumor cells accessible to the immune system, linked with tumor size and vascularization. The spatial localization of immune cells may contribute to tumor growth and invasion, thus affecting the performance of the models and therefore, the efficacy of immunotherapy, as mentioned by Ji et al [33]. This highlights the need to compartmentalize the mathematical framework to take into account spatial heterogeneity. Tumor and TME heterogeneity may determine different responses to treatments as IO therapy does not achieve similar antitumor responses in all treated patients. Several internal and external factors are considered to explain the inter- and intratumor heterogeneity. New technologies, in particular, the spatial transcriptomic applied to cancer research, offer a great opportunity to understand the localization of the different components of TME and tumor cells, which combined with the characterization of the diversity of gene expression and mutations, can explain the different phenotypes and thus, the different therapeutic responses of tumors. In this context, the QSP approach is a valuable tool to integrate all information to explore diverse hypotheses and simulate potential combinations based on each immune patient profile. However, longitudinal preclinical and clinical data on subpopulations of pro- and antitumor cells are essential to fully assess the trade-off between Treg and CD8+ cells, responsible for IO efficacy.
In this work, all models are based on CD25+ and Foxp3+ Treg, except for the model by Ji et al, which describes Treg subpopulations according to GITR expression levels. The relevance of the mentioned subpopulations was demonstrated by a transcriptomic analysis of 99 solid tumors from cancer patients enrolled in a clinical trial [35]. The authors analyzed the correlation between the expression of GITR and its endogenous ligand and the efficacy of several GITR agonists tested in preclinical and clinical trials. The heterogeneous expression of these proteins between and within cancer types evidenced the necessity to incorporate these data to achieve patient stratification to establish the most appropriate IO treatment. For example, tumor responders were those associated with high GITR and low-moderate GITR ligand expression, a phenotype found in approximately 30% of breast and lung cancer patients, respectively. However, this high GITR expression was independent of other immune modulators (PD1, CTLA4, OX40), suggesting that a more detailed analysis may be critical to providing rational combinations targeting different Treg biomarkers. The authors also concluded that other characteristics such as concomitant therapies, and the disease progression state might be investigated in each patient as these may also influence the antitumor response. Overall, further studies are warranted to identify the most complete immune profile of patients to achieve efficacy.
For all mentioned, these QSP frameworks are useful tools in decoding cancerous processes, providing an understanding of the disease, and evaluating the mechanism of action of molecular entities. The ultimate goal of these QSP models is to use virtual animal/patient populations to inform the design of clinical studies and to test in silico the efficacy of new treatments and combinatorial therapies [31]. The common conclusion of all the models described here, based mainly on preclinical data, is related to the extrapolation of the models to the clinical setting, highlighting the urgent need for the availability of clinical data to confront model predictions, as well as the inclusion of human parameters in QSP models.
3. New Molecules for Treg Modulation
Based on the QSP model proposed for Treg expressing GITR, other biomarkers such as CD25, CTLA-4, PD-1, ICOS, OX40, CCR4, and CCR8 (Figure 3) are useful targets that can also be exploited and integrated into similar QSP frameworks. Interesting mAbs that bind to OX-40 (BAT6026, BGB-A445) or CCR8 (Nb-Fc1B) are being tested in preclinical models, as shown in Table 2. The expression of biomarkers in Treg therefore offers a wide range of possibilities for the development of therapies against immunosuppressive activity.
Notably, Treg targeting does not always guarantee the efficacy of immunotherapies. Therefore, a comprehensive integration of the mechanisms triggered by many of these therapeutic approaches to deal with Treg activity into a computational mathematical model can enrich the knowledge of immune activity and exquisitely improve the understanding of clinical benefit by predicting the most likely response according to different therapeutic combinations. Currently, some of these interesting advances in Treg modulation have reached clinical translation and are involved in several clinical trials (Table 3).
On the other hand, Foxp3, the transcription factor considered the master regulator of the Treg immunosuppressive phenotype, is an interesting biomarker that represents a challenge for the development of targeted therapies due to its intranuclear location. In this sense, intensive research has led to the spread of several peptides that reproduce parts of the Foxp3 amino acid sequences and compete for the binding partners and cofactors of Foxp3, or non-specific peptides with the ability to bind and inhibit Foxp3 activity [36]. Specifically, P60 is a linear peptide capable of selectively binding to Foxp3 preventing its nuclear translocation and inducing antitumor response in preclinical tumor models. However, its very short systemic half-life requires a high daily dose administration, limiting its clinical translation.
To overcome this shortcoming various modifications have been tested, such as the cycling of the linear structure or by the conjugation with the CD28 aptamer. Although, these strategies provided a slight increase in the half-life of the peptide and an acceptable antitumor effect, other more efficient approaches are being sought. Serrano et al [37] have reported a new nanosystem for P60 delivery. The authors have successfully encapsulated P60 in advanced nanoliposomes, which were decorated with monovalent variable fragments of anti-CD25 (Fab’-CD25) to selectively target Treg. This novel formulation was assayed in the MC38 mouse tumor model, and induced complete tumor remission in 40% of animals after monotherapy and in 100% after combination with anti-PD1. All mice developed immune memory. This result contrasts with the modest 10% of animals that showed tumor elimination after free P60 administration at a dose 50 times higher than that of the targeted liposome. Therefore, this nanosystem constitutes an interesting platform to exploit the transport and delivery of not only peptides but also other new molecules or treatments including different types of genes, i.e. small interfering-RNA (siRNA), which is capable of inhibiting specific gene expression [38]. Patisiran (Onpattro™), a lipid nanoparticle encapsulating siRNA, approved by the US Food and Drug Administration (FDA) in 2018 for the treatment of a rare metabolic disease, is a good example of the nanosystem potential [39].
Selective therapies to modulate Treg activity, by depletion and/or inhibition are therefore being actively investigated, with a particular focus on several biomarkers (Table 2 and Table 3). Interestingly, many clinical trials are using inhibitors against OX40, GITR, or LAG3 with promising results. However, the availability of longitudinal data from these trials could help to propose and develop more complete QSP model structures that integrate pro- and antitumor mechanisms allowing the validation of model predictions, accelerating the approval of new therapies, and proposing the most rational combination depending on the individual immune profile. Computational models can also guide the design of clinical trials to improve the benefit of IO.
Conclusions
Model-based quantitative pharmacology has demonstrated an important role in the development of new therapeutic molecules, providing information on doses and regimens and also exploring different mechanisms involved in tumor progression and elimination. QSP models applied to IO allow the elucidation of specific immune mechanisms to better understand the biology of the disease, therapies, and individual immune profile with the aim of identifying different cancer types, and patients to propose particular combinations to achieve efficient antitumor response.
This review presents a list of diverse computational mathematical models that examine the role of regulatory T cells as a crucial immune resistance mechanism contributing to the failure of immunotherapy. In all models, both Treg and cytotoxic CD8+ T cells are included, either explicitly or implicitly, in the quantitative immune frameworks proposed for evaluating tumour growth based on the immune therapies tested in preclinical models. While QSP models have demonstrated their capacity to explore intriguing hypotheses, longitudinal preclinical and clinical data on pro- and antitumour cell subpopulations are indispensable for identifying the most plausible mechanisms involved in IO. Furthermore, new strategies for targeting Treg based on specific biomarkers involved in their activation status are emerging as promising therapies to enhance the antitumour immune response, thereby generating considerable interest in many of them.
Finally, tumor heterogenicity, spatial localization of immune cells within the TME, and other factors including disease progression, concomitant treatments, and pro- and anti-inflammatory cytokine levels, among others, which contribute to tumor growth need to be incorporated into the model in order to propose individualized treatments and improve the efficacy of immunotherapies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualisation, I.F.T, J.J.L., N.R. and M.J.G.; resources, A.S., S.Z., N.R. and M.J.G.; writing—original draft preparation, A.S., N.R. and M.J.G.; writing—review and editing, J.J.L., I.F.T., N.R. and M.J.G. All authors have read and agreed to the published version of the manuscript.
Funding
Alejandro Serrano was supported by ADA scholarship (from University of Navarra) and FPU program from Ministerio de Universidades (Spanish Government).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data can be made available on request to corresponding authors.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- J.C. Crispin, G.C. Tsokos, Cancer immunosurveillance by CD8 T cells, F100 Res. 9 (2020) 1–7. [CrossRef]
- T.K. Kim, E.N. Vandsemb, R.S. Herbst, L. Chen, Adaptive immune resistance at the tumour site: mechanisms and therapeutic opportunities, Nat. Rev. Drug Discov. 21 (2022) 529–540. [CrossRef]
- S.A. Rosenberg, Progress in human tumour immunology and immunotherapy, Nature. 411 (2001) 380–384. [CrossRef]
- A. Ribas, L.H. Butterfield, J.A. Glaspy, J.S. Economou, Current developments in cancer vaccines and cellular immunotherapy, J. Clin. Oncol. 21 (2003) 2415–2432. [CrossRef]
- N. Mason, S. Dow, Cancer immunotherapy, Ther. Strateg. Vet. Oncol. 305 (2023) 121–152. [CrossRef]
- I. Mellman, D.S. Chen, T. Powles, S.J. Turley, The cancer-immunity cycle: Indication, genotype, and immunotype, Immunity. 56 (2023) 2188–2205. [CrossRef]
- S.T. Ferris, V. Durai, R. Wu, D.J. Theisen, J.P. Ward, M.D. Bern, J.T.D. Iv, P. Bagadia, T. Liu, G. Carlos, L. Li, W.E. Gillanders, G.F. Wu, W.M. Yokoyama, T.L. Murphy, R.D. Schreiber, K.M. Murphy, cDC1 prime and are licensed by CD4 T cells to induce antitumour immunity, 584 (2021) 624–629. [CrossRef]
- Y. Pan, Y. Yu, X. Wang, T. Zhang, Tumor-Associated Macrophages in Tumor Immunity, Front. Immunol. 11 (2020). [CrossRef]
- F. Castellino, R.N. Germain, Cooperation between CD4+ and CD8+ T cells: When, where, and how, Annu. Rev. Immunol. 24 (2006) 519–540. [CrossRef]
- S. Tang, Q. Ning, L. Yang, Z. Mo, S. Tang, Mechanisms of immune escape in the cancer immune cycle, Int. Immunopharmacol. 86 (2020) 106700. [CrossRef]
- M. Sengupta, R. Pal, A. Nath, B. Chakraborty, L.M. Singh, B. Das, S.K. Ghosh, Anticancer efficacy of noble metal nanoparticles relies on reprogramming tumor-associated macrophages through redox pathways and pro-inflammatory cytokine cascades, Cell. Mol. Immunol. 15 (2018) 1088–1090. [CrossRef]
- D.I. Gabrilovich, Myeloid-derived suppressor cells, Cancer Immunol Res. 5 (2018) 3–8. [CrossRef]
- A. Tanaka, S. Sakaguchi, Regulatory T cells in cancer immunotherapy, Cell Res. 27 (2017) 109–118. [CrossRef]
- M. V. Goldberg, C.G. Drake, LAG-3 in Cancer Immunotherapy, Curr Top Microbiol Immunol. 344 (2011) 269–278. [CrossRef]
- V. Gouirand, I. Habrylo, M.D. Rosenblum, Regulatory T Cells and Inflammatory Mediators in Autoimmune Disease, J. Invest. Dermatol. 142 (2022) 774–780. [CrossRef]
- E.M. Shevach, A.M. Thornton, tTregs, pTregs, and iTregs: similarities and differences, Immunol. Rev. 259 (2014) 88–102. [CrossRef]
- O. Yoshie, Ccr4 as a therapeutic target for cancer immunotherapy, Cancers (Basel). 13 (2021) 1–28. [CrossRef]
- O. Yoshie, K. Matsushima, CCR4 and its ligands: From bench to bedside, Int. Immunol. 27 (2015) 11–20. [CrossRef]
- S.K. Whiteside, F.M. Grant, D.S. Gyori, A.G. Conti, C.J. Imianowski, P. Kuo, R. Nasrallah, F. Sadiyah, S.A. Lira, F. Tacke, R.L. Eil, O.T. Burton, J. Dooley, A. Liston, K. Okkenhaug, J. Yang, R. Roychoudhuri, CCR8 marks highly suppressive Treg cells within tumours but is dispensable for their accumulation and suppressive function, Immunology. 163 (2021) 512–520. [CrossRef]
- D. Noyes, A. Bag, S. Oseni, J. Semidey-Hurtado, L. Cen, A.A. Sarnaik, V.K. Sondak, D. Adeegbe, Tumor-associated Tregs obstruct antitumor immunity by promoting T cell dysfunction and restricting clonal diversity in tumor-infiltrating CD8+ T cells, J. Immunother. Cancer. 10 (2022) e004605. [CrossRef]
- K. Peskov, I. Azarov, L. Chu, V. Voronova, Y. Kosinsky, G. Helmlinger, Quantitative mechanistic modeling in support of pharmacological therapeutics development in immuno-oncology, Front. Immunol. 10 (2019) 1–11. [CrossRef]
- M. Simeoni, P. Magni, C. Cammia, G. De Nicolao, V. Croci, E. Pesenti, M. Germani, I. Poggesi, M. Rocchetti, Predictive Pharmacokinetic-Pharmacodynamic Modeling of Tumor Growth Kinetics in Xenograft Models after Administration of Anticancer Agents, Cancer Res. 64 (2004) 1094–1101. [CrossRef]
- Z.P. Parra-Guillen, P. Berraondo, E. Grenier, B. Ribba, I.F. Troconiz, Mathematical model approach to describe tumour response in mice after vaccine administration and its applicability to immune-stimulatory cytokine-based strategies, AAPS J. 15 (2013) 797–807. [CrossRef]
- Y. Bulliard, R. Jolicoeur, M. Windman, S.M. Rue, S. Ettenberg, D.A. Knee, N.S. Wilson, G. Dranoff, J.L. Brogdon, Activating fc γ receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies, J. Exp. Med. 210 (2013) 1685–1693. [CrossRef]
- B. Chaudhary, E. Elkord, Regulatory T cells in the tumor microenvironment and cancer progression: Role and therapeutic targeting, Vaccines. 4 (2016) 1–25. [CrossRef]
- N.Y. den Breems, R. Eftimie, The re-polarisation of M2 and M1 macrophages and its role on cancer outcomes, J. Theor. Biol. 390 (2016) 23–39. [CrossRef]
- A. Arabameri, D. Asemani, J. Hadjati, A structural methodology for modeling immune-tumor interactions including pro- and anti-tumor factors for clinical applications, Math. Biosci. 304 (2018) 48–61. [CrossRef]
- N. Kronik, Y. Kogan, P.G. Schlegel, M. Wölflz, Erratum: Improving T-cell immunotherapy for melanoma through a mathematically motivated strategy: Efficacy in numbers (Journal of Immunotherapy (2012) (116)), J. Immunother. 35 (2012) 292. [CrossRef]
- S. Wilson, D. Levy, A Mathematical Model of the Enhancement of Tumor Vaccine Efficacy by Immunotherapy, Bull. Math. Biol. 74 (2012) 1485–1500. [CrossRef]
- M. Robertson-Tessi, A. El-Kareh, A. Goriely, A mathematical model of tumor-immune interactions, J. Theor. Biol. 294 (2012) 56–73. [CrossRef]
- R. Coletti, L. Leonardelli, S. Parolo, L. Marchetti, A QSP model of prostate cancer immunotherapy to identify effective combination therapies, Sci. Rep. 10 (2020) 1–18. [CrossRef]
- M. Movassaghi, R. Chung, C.B. Anderson, M. Stein, Y. Saenger, I. Faiena, Overcoming immune resistance in prostate cancer: Challenges and advances, Cancers (Basel). 13 (2021) 1–16. [CrossRef]
- Y. Ji, K. Madrasi, D.A. Knee, L. Gruenbaum, J.F. Apgar, J.M. Burke, B. Gomes, Quantitative systems pharmacology model of GITR-mediated T cell dynamics in tumor microenvironment, CPT Pharmacometrics Syst. Pharmacol. 12 (2023) 413–424. [CrossRef]
- D. Qin, Y. Zhang, P. Shu, Y. Lei, X. Li, Y. Wang, Targeting tumor-infiltrating tregs for improved antitumor responses, Front. Immunol. 15 (2024) 1–15. [CrossRef]
- P. Moussa, Transcriptomic analysis of GITR and GITR ligand reveals cancer immune heterogeneity with implications for GITR targeting, Am. J. Cancer Res. 14 (2024) 1634–1648. [CrossRef]
- Y. Dong, C. Yang, F. Pan, Post-Translational Regulations of Foxp3 in Treg Cells and Their Therapeutic Applications, Front. Immunol. 12 (2021) 1–16. [CrossRef]
- A. Serrano, N. Casares, I.F. Trocóniz, T. Lozano, J.J. Lasarte, S. Zalba, M.J. Garrido, Foxp3 inhibitory peptide encapsulated in a novel CD25-targeted nanoliposome promotes efficient tumor regression in mice, Acta Pharmacol. Sin. (2024) 1–13. [CrossRef]
- S. Mirzaei, M.H. Gholami, H.L. Ang, F. Hashemi, A. Zarrabi, A. Zabolian, K. Hushmandi, M. Delfi, H. Khan, M. Ashrafizadeh, G. Sethi, A.P. Kumar, Pre-clinical and clinical applications of small interfering rnas (Sirna) and co-delivery systems for pancreatic cancer therapy, Cells. 10 (2021). [CrossRef]
- S. Yonezawa, H. Koide, T. Asai, Recent advances in siRNA delivery mediated by lipid-based nanoparticles, Adv. Drug Deliv. Rev. 154–155 (2020) 64–78. [CrossRef]
- U. Jarry, S. Donnou, M. Vincent, P. Jeannin, L. Pineau, I. Fremaux, Y. Delneste, D. Couez, Treg depletion followed by intracerebral CpG-ODN injection induce brain tumor rejection, J. Neuroimmunol. 267 (2014) 35–42. [CrossRef]
- S.A. Quezada, K.S. Peggs, T.R. Simpson, Y. Shen, D.R. Littman, J.P. Allison, Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T cell depletion against established melanoma, J. Exp. Med. 205 (2008) 2125–2138. [CrossRef]
- M. Semmrich, J.B. Marchand, L. Fend, M. Rehn, C. Remy, P. Holmkvist, N. Silvestre, C. Svensson, P. Kleinpeter, J. Deforges, F. Junghus, K.L. Cleary, M. Bodén, L. Mårtensson, J. Foloppe, I. Teige, E. Quéméneur, B. Frendéus, Vectorized Treg-depleting αcTLA-4 elicits antigen cross-presentation and CD8 + T cell immunity to reject cold’ tumors, J. Immunother. Cancer. 10 (2022) 1–14. [CrossRef]
- L.X. Zhu, M. Davoodi, M.K. Srivastava, P. Kachroo, J.M. Lee, M.S. John, M. Harris-White, M. Huang, R.M. Strieter, S. Dubinett, S. Sharma, GITR agonist enhances vaccination responses in lung cancer, Oncoimmunology. 4 (2015) 1–12. [CrossRef]
- A.D. Cohen, D.A. Schaer, C. Liu, Y. Li, D. Hirschhorn-Cymmerman, S.C. Kim, A. Diab, G. Rizzuto, F. Duan, M.A. Perales, T. Merghoub, A.N. Houghton, J.D. Wolchok, Agonist anti-GITR monoclonal antibody induces melanoma tumor immunity in mice by altering regulatory T cell stability and intra-tumor accumulation, PLoS One. 5 (2010). [CrossRef]
- D. Coe, S. Begom, C. Addey, M. White, J. Dyson, J.G. Chai, Depletion of regulatory T cells by anti-GITR mAb as a novel mechanism for cancer immunotherapy, Cancer Immunol. Immunother. 59 (2010) 1367–1377. [CrossRef]
- M.D. Oberst, C. Augé, C. Morris, S. Kentner, K. Mulgrew, K. McGlinchey, J. Hair, S. Hanabuchi, Q. Du, M. Damschroder, H. Feng, S. Eck, N. Buss, L. de Haan, A.J. Pierce, H. Park, A. Sylwester, M.K. Axthelm, L. Picker, N.P. Morris, A. Weinberg, S.A. Hammond, Potent Immune Modulation by MEDI6383, an Engineered Human OX40 Ligand IgG4P Fc Fusion Protein, Mol. Cancer Ther. 17 (2018) 1024–1038. [CrossRef]
- S. Liang, D. Zheng, X. Liu, X. Mei, C. Zhou, C. Xiao, C. Qin, H. Yue, J. Lin, C. Liu, S. Li, J.C. Yu, BAT6026, a novel anti-OX40 antibody with enhanced antibody dependent cellular cytotoxicity effect for cancer immunotherapy, Front. Oncol. 13 (2023) 1–11. [CrossRef]
- B. Jiang, T. Zhang, M. Deng, W. Jin, Y. Hong, X. Chen, X. Chen, J. Wang, H. Hou, Y. Gao, W. Gong, X. Wang, H. Li, X. Zhou, Y. Feng, B. Zhang, B. Jiang, X. Lu, L. Zhang, Y. Li, W. Song, H. Sun, Z. Wang, X. Song, Z. Shen, X. Liu, K. Li, L. Wang, Y. Liu, BGB-A445, a novel non-ligand-blocking agonistic anti-OX40 antibody, exhibits superior immune activation and antitumor effects in preclinical models, Front. Med. 17 (2023) 1170–1185. [CrossRef]
- H. Van Damme, B. Dombrecht, M. Kiss, H. Roose, E. Allen, E. Van Overmeire, D. Kancheva, L. Martens, A. Murgaski, P.M.R. Bardet, G. Blancke, M. Jans, E. Bolli, M.S. Martins, Y. Elkrim, J. Dooley, L. Boon, J.K. Schwarze, F. Tacke, K. Movahedi, N. Vandamme, B. Neyns, S. Ocak, I. Scheyltjens, L. Vereecke, F.A. Nana, P. Merchiers, D. Laoui, J.A. Van Ginderachter, Therapeutic depletion of CCR8 + tumor-infiltrating regulatory T cells elicits antitumor immunity and synergizes with anti-PD-1 therapy, J. Immunother. Cancer. 9 (2021) 1–16. [CrossRef]
- J.R. Campbell, B.R. McDonald, P.B. Mesko, N.O. Siemers, P.B. Singh, M. Selby, T.W. Sproul, A.J. Korman, L.M. Vlach, J. Houser, S. Sambanthamoorthy, K. Lu, S. V. Hatcher, J. Lohre, R. Jain, R.Y. Lan, Fc-optimized anti-CCR8 antibody depletes regulatory t cells in human tumor models, Cancer Res. 81 (2021) 2983–2994. [CrossRef]
- A.J. Rech, R.H. Vonderheide, Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells, Ann. N. Y. Acad. Sci. 1174 (2009) 99–106. [CrossRef]
- R. Okita, Y. Yamaguchi, M. Ohara, K. Hironaka, M. Okawaki, I. Nagamine, T. Ikeda, A. Emi, J. Hihara, M. Okada, Targeting of CD4+CD25high cells while preserving CD4+CD25low cells with low-dose chimeric anti-CD25 antibody in adoptive immunotherapy of cancer., Int. J. Oncol. 34 (2009) 563–72. [CrossRef]
- M.T. Litzinger, R. Fernando, T.J. Curiel, D.W. Grosenbach, J. Schlom, C. Palena, IL-2 immunotoxin denileukin diftitox reduces regulatory T cells and enhances vaccine-mediated T-cell immunity, Blood. 110 (2007) 3192–3201. [CrossRef]
- D.J. Powell, A. Felipe-Silva, M.J. Merino, M. Ahmadzadeh, T. Allen, C. Levy, D.E. White, S. Mavroukakis, R.J. Kreitman, S.A. Rosenberg, I. Pastan, Administration of a CD25-Directed Immunotoxin, LMB-2, to Patients with Metastatic Melanoma Induces a Selective Partial Reduction in Regulatory T Cells In Vivo, J. Immunol. 179 (2007) 4919–4928. [CrossRef]
- D.J. Powell, P. Attia, V. Ghetie, J. Schindlerv, E.S. Vitetta, S.A. Rosenberg, Partial reduction of human FOXP3+ CD4 T cells in vivo after CD25-directed recombinant immunotoxin administration, J. Immunother. 31 (2008) 189–198. [CrossRef]
- D.S. Hong, O. Rixe, V.K. Chiu, P.M. Forde, T. Dragovich, Y. Lou, A. Nayak-Kapoor, R. Leidner, J.N. Atkins, A. Collaku, F.E. Fox, M.A. Marshall, A.J. Olszanski, Mogamulizumab in Combination with Nivolumab in a Phase I/II Study of Patients with Locally Advanced or Metastatic Solid Tumors, Clin. Cancer Res. 28 (2022) 479–488. [CrossRef]
- J.M. Kirkwood, P. Lorigan, P. Hersey, A. Hauschild, C. Robert, D. McDermott, M.A. Marshall, J. Gomez-Navarro, J.Q. Liang, C.A. Bulanhagui, Phase II trial of tremelimumab (CP-675,206) in patients with advanced refractory or relapsed melanoma, Clin. Cancer Res. 16 (2010) 1042–1048. [CrossRef]
- W. Chi, L. Zhang, X. Wang, J. Li, F. Li, Y. Ma, Q. Zhang, Effects of Nivolumab and Ipilimumab on the suppression of cisplatin resistant small cell lung cancer cells, Invest. New Drugs. 40 (2022) 709–717. [CrossRef]
- C. Yi, L. Chen, Z. Lin, L. Liu, W. Shao, R. Zhang, J. Lin, J. Zhang, W. Zhu, H. Jia, L. Qin, L. Lu, J. Chen, Lenvatinib Targets FGF Receptor 4 to Enhance Antitumor Immune Response of Anti–Programmed Cell Death-1 in HCC, Hepatology. 74 (2021) 2544–2560. [CrossRef]
- J. Koh, J.Y. Hur, K.Y. Lee, M.S. Kim, J.Y. Heo, B.M. Ku, J.M. Sun, S.H. Lee, J.S. Ahn, K. Park, M.J. Ahn, Regulatory (FoxP3+) T cells and TGF-β predict the response to anti-PD-1 immunotherapy in patients with non-small cell lung cancer, Sci. Rep. 10 (2020) 1–10. [CrossRef]
- D. Davar, R. Zappasodi, H. Wang, G.S. Naik, T. Sato, T. Bauer, D. Bajor, O. Rixe, W. Newman, J. Qi, A. Holland, P. Wong, L. Sifferlen, D. Piper, C.A. Sirard, T. Merghoub, J.D. Wolchok, J.J. Luke, Phase IB Study of GITR Agonist Antibody TRX518 Singly and in Combination with Gemcitabine, Pembrolizumab, or Nivolumab in Patients with Advanced Solid Tumors, Clin. Cancer Res. 28 (2022) 3990–4002. [CrossRef]
- R. Geva, M. Voskoboynik, K. Dobrenkov, K. Mayawala, J. Gwo, R. Wnek, E. Chartash, G. V. Long, First-in-human phase 1 study of MK-1248, an anti–glucocorticoid-induced tumor necrosis factor receptor agonist monoclonal antibody, as monotherapy or with pembrolizumab in patients with advanced solid tumors, Cancer. 126 (2020) 4926–4935. [CrossRef]
- A.S. Balmanoukian, J.R. Infante, R. Aljumaily, A. Naing, A. V. Chintakuntlawar, N.A. Rizvi, H.J. Ross, M. Gordon, P.R. Mallinder, N. Elgeioushi, I. Gonzalez-García, N. Standifer, J. Cann, N. Durham, S. Rahimian, R. Kumar, C.S. Denlinger, Safety and clinical activity of MEDI1873, a novel GITR agonist, in advanced solid tumors, Clin. Cancer Res. 26 (2020) 6196–6203. [CrossRef]
- S.A. Piha-Paul, R. Geva, T.J. Tan, D.W.T. Lim, C. Hierro, T. Doi, O. Rahma, A. Lesokhin, J.J. Luke, J. Otero, L. Nardi, A. Singh, A. Xyrafas, X. Chen, J. Mataraza, P.L. Bedard, First-in-human phase I/Ib open-label dose-escalation study of GWN323 (anti-GITR) as a single agent and in combination with spartalizumab (anti-PD-1) in patients with advanced solid tumors and lymphomas, J. Immunother. Cancer. 9 (2021). [CrossRef]
- O. Hamid, A.A. Chiappori, J.A. Thompson, T. Doi, S. Hu-Lieskovan, F.A.L.M. Eskens, W. Ros, A. Diab, J.P. Spano, N.A. Rizvi, J.S. Wasser, E. Angevin, P.A. Ott, A. Forgie, W. Yang, C. Guo, J. Chou, A.B. El-Khoueiry, First-in-human study of an OX40 (ivuxolimab) and 4-1BB (utomilumab) agonistic antibody combination in patients with advanced solid tumors, J. Immunother. Cancer. 10 (2022) 1–12. [CrossRef]
- S. Postel-Vinay, V.K. Lam, W. Ros, T.M. Bauer, A.R. Hansen, D.C. Cho, F. Stephen Hodi, J.H.M. Schellens, J.K. Litton, S. Aspeslagh, K.A. Autio, F.L. Opdam, M. McKean, N. Somaiah, S. Champiat, M. Altan, A. Spreafico, O. Rahma, E.M. Paul, C.M. Ahlers, H. Zhou, H. Struemper, S.A. Gorman, M. Watmuff, K.M. Yablonski, N. Yanamandra, M.J. Chisamore, E. V. Schmidt, A. Hoos, A. Marabelle, J.S. Weber, J. V. Heymach, First-in-human phase I study of the OX40 agonist GSK3174998 with or without pembrolizumab in patients with selected advanced solid tumors (ENGAGE-1), J. Immunother. Cancer. 11 (2023) e005301. [CrossRef]
- M. Gutierrez, V. Moreno, K.M. Heinhuis, A.J. Olszanski, A. Spreafico, M. Ong, Q. Chu, R.D. Carvajal, J. Trigo, M.O. de Olza, M. Provencio, F.Y. de Vos, F. de Braud, S. Leong, D. Lathers, R. Wang, P. Ravindran, Y. Feng, P. Aanur, I. Melero, OX40 agonist BMS-986178 alone or in combination with nivolumab and/or ipilimumab in patients with advanced solid tumors, Clin. Cancer Res. 27 (2021) 460–472. [CrossRef]
- R. Duhen, C. Ballesteros-Merino, A.K. Frye, E. Tran, V. Rajamanickam, S.C. Chang, Y. Koguchi, C.B. Bifulco, B. Bernard, R.S. Leidner, B.D. Curti, B.A. Fox, W.J. Urba, R.B. Bell, A.D. Weinberg, Neoadjuvant anti-OX40 (MEDI6469) therapy in patients with head and neck squamous cell carcinoma activates and expands antigen-specific tumor-infiltrating T cells, Nat. Commun. 12 (2021). [CrossRef]
- T.W. Kim, H.A. Burris, M.J. de Miguel Luken, M.J. Pishvaian, Y.-J. Bang, M. Gordon, A. Awada, D.R. Camidge, F.S. Hodi, G.A. McArthur, W.H. Miller, A. Cervantes, L.Q. Chow, A.M. Lesokhin, A. Rutten, M. Sznol, D. Rishipathak, S.-C. Chen, E. Stefanich, T. Pourmohamad, M. Anderson, J. Kim, M. Huseni, I. Rhee, L.L. Siu, First-In-Human Phase I Study of the OX40 Agonist MOXR0916 in Patients with Advanced Solid Tumors., Clin. Cancer Res. 28 (2022) 3452–3463. [CrossRef]
- B.S. Glisson, R.S. Leidner, R.L. Ferris, J. Powderly, N.A. Rizvi, B. Keam, R. Schneider, S. Goel, J.P. Ohr, J. Burton, Y. Zheng, S. Eck, M. Gribbin, K. Streicher, D.M. Townsley, S.P. Patel, Safety and clinical activity of MEDI0562, a humanized OX40 agonist monoclonal antibody, in adult patients with advanced solid tumors, Clin. Cancer Res. 26 (2020) 5358–5367. [CrossRef]
Figure 1.
Scheme of cancer cycle immunity. Tumor antigens derived from cell death are engulfed by DC and presented to naive T cells, resulting in the activation of effector cytotoxic T cells (CD8+) capable of inducing tumor cell apoptosis and generating a humoral response. This process promotes the secretion of pro-inflammatory cytokines and DAMPs that enhance T-cell activation.
Figure 1.
Scheme of cancer cycle immunity. Tumor antigens derived from cell death are engulfed by DC and presented to naive T cells, resulting in the activation of effector cytotoxic T cells (CD8+) capable of inducing tumor cell apoptosis and generating a humoral response. This process promotes the secretion of pro-inflammatory cytokines and DAMPs that enhance T-cell activation.
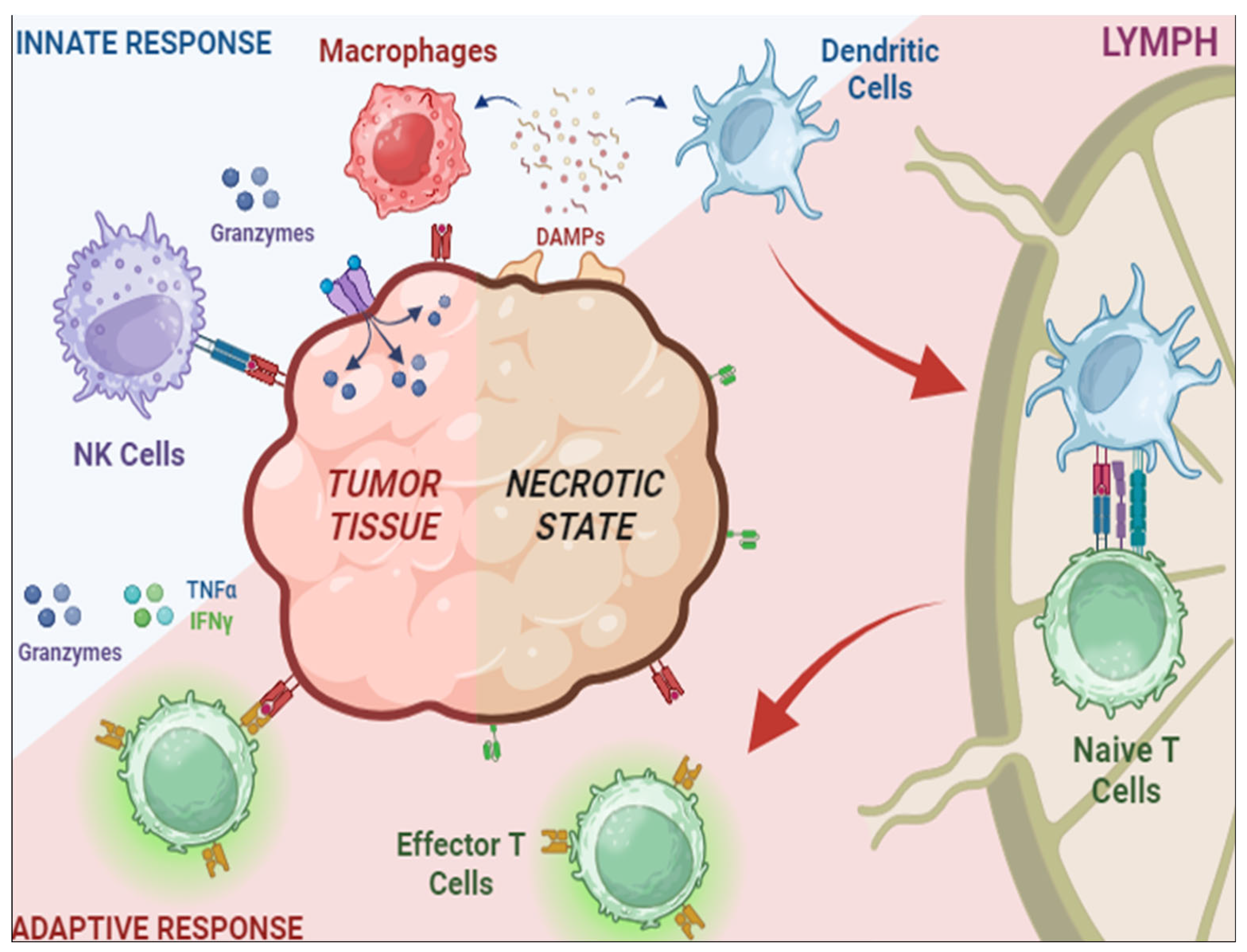
Figure 2.
Processes involved in tumor elimination (represented in red) or tumor progression (represented in blue). Stimulatory mechanisms to induce antitumor effect must overcome inhibitory activities. Intra-tumoral infiltration of activated CD8+ T cells triggers tumor cell death through the action of interferon-gamma (IFN-γ), and granzyme B. However, the tumor evades this control by inducing specific immunoresistance mechanisms, in particular, the proliferation of Treg cells and the upregulation of PD-L1 on the surface of tumor cells, leading to effector cell exhaustion.
Figure 2.
Processes involved in tumor elimination (represented in red) or tumor progression (represented in blue). Stimulatory mechanisms to induce antitumor effect must overcome inhibitory activities. Intra-tumoral infiltration of activated CD8+ T cells triggers tumor cell death through the action of interferon-gamma (IFN-γ), and granzyme B. However, the tumor evades this control by inducing specific immunoresistance mechanisms, in particular, the proliferation of Treg cells and the upregulation of PD-L1 on the surface of tumor cells, leading to effector cell exhaustion.
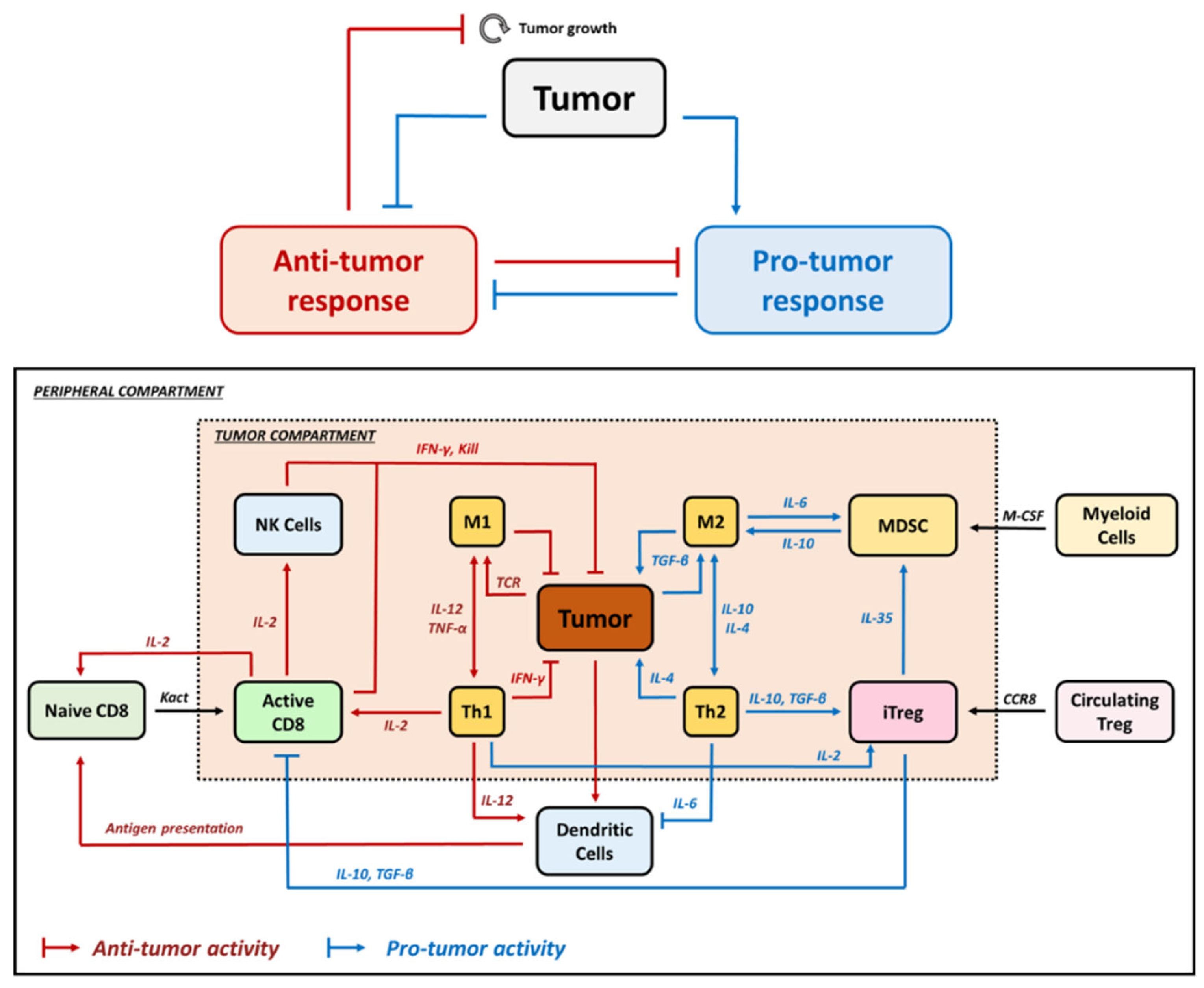
Figure 3.
Schematic representation of the different biomarkers expressed in Treg and involved in the different mechanisms of action identified for this lymphocyte subset. Note that Foxp3 is expressed intracellularly and determines Treg activation.
Figure 3.
Schematic representation of the different biomarkers expressed in Treg and involved in the different mechanisms of action identified for this lymphocyte subset. Note that Foxp3 is expressed intracellularly and determines Treg activation.
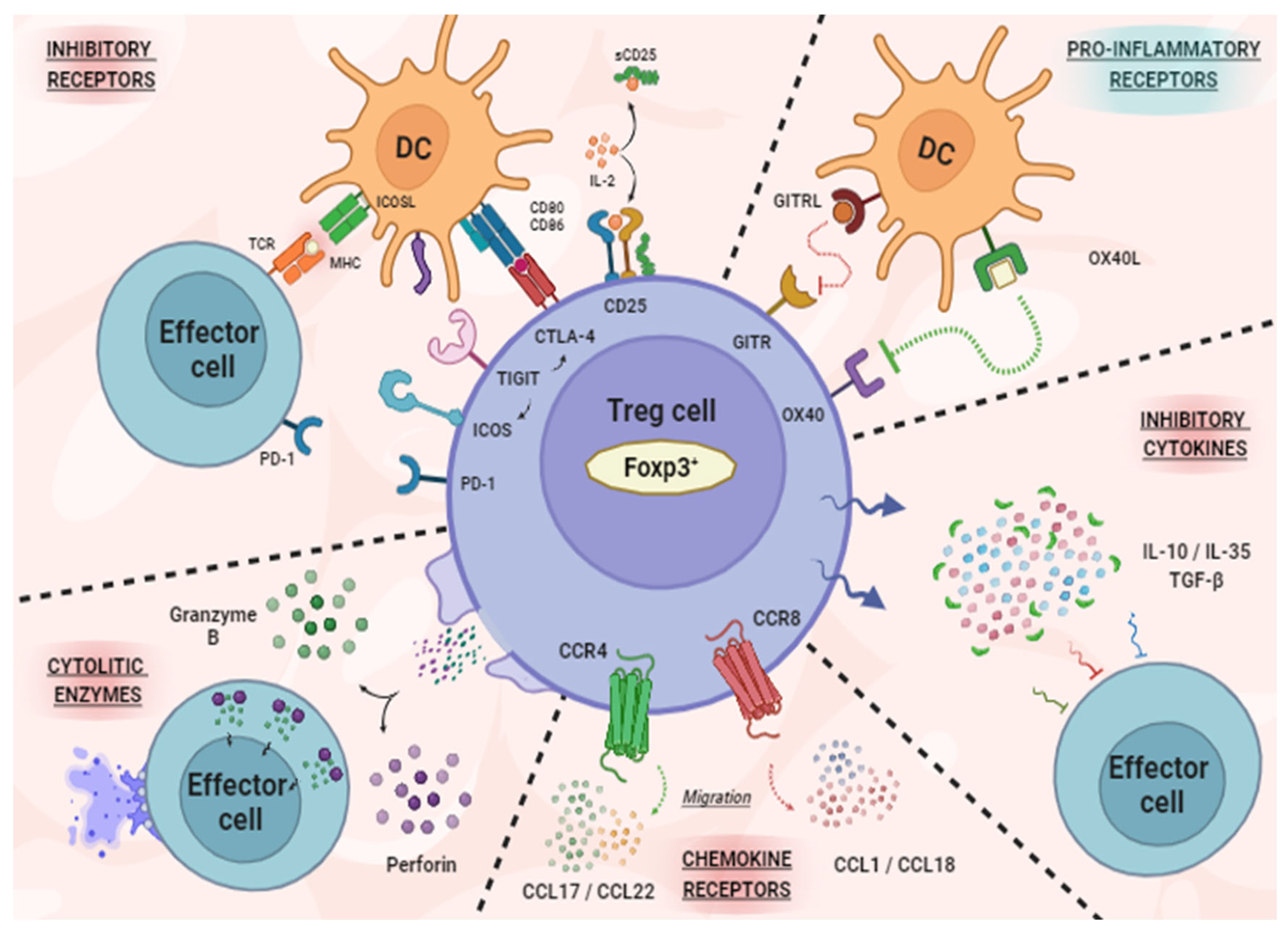

Table 1.
Summary of selected QSP models for describing the mechanisms derived from Treg in tumor growth.
Table 1.
Summary of selected QSP models for describing the mechanisms derived from Treg in tumor growth.
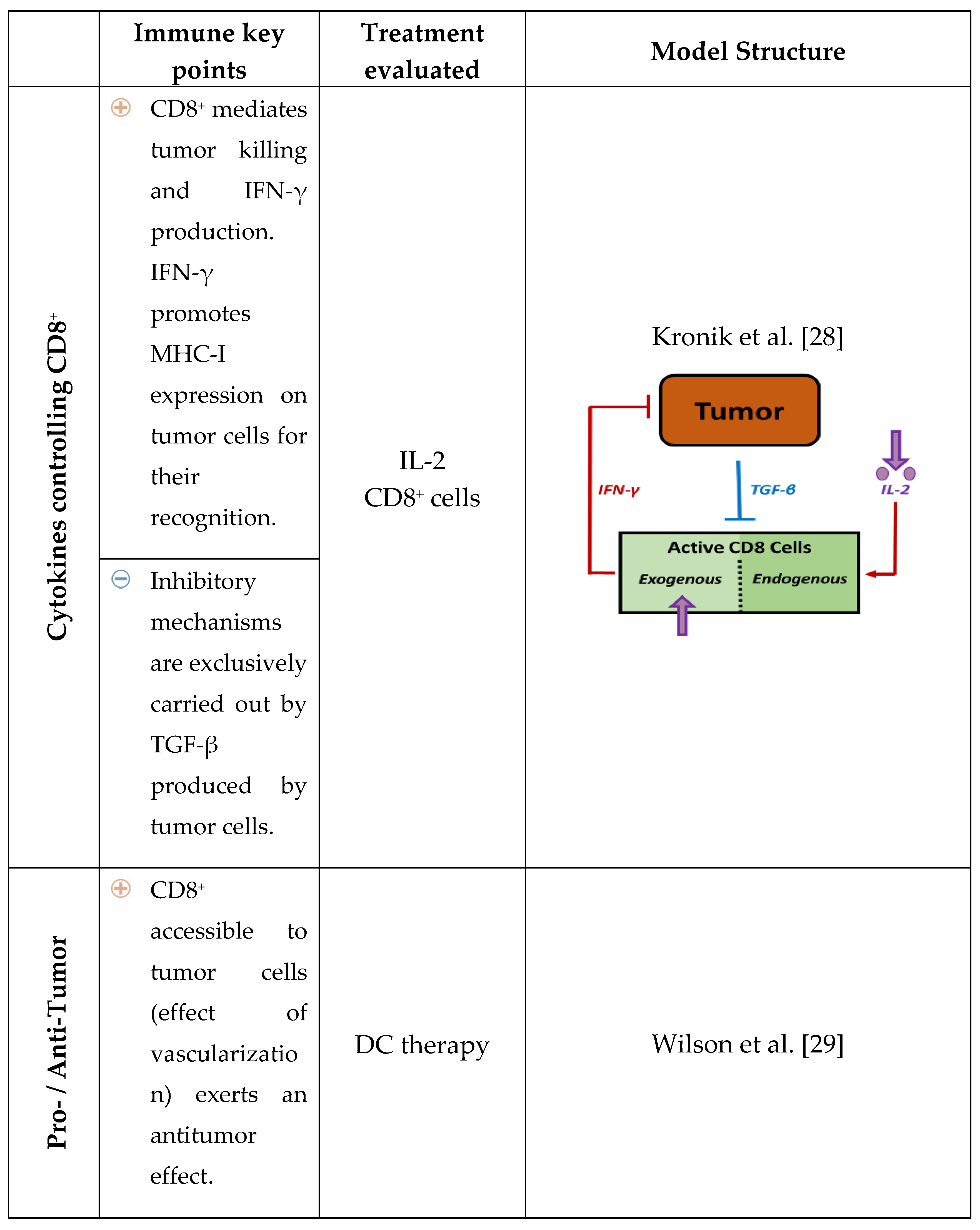
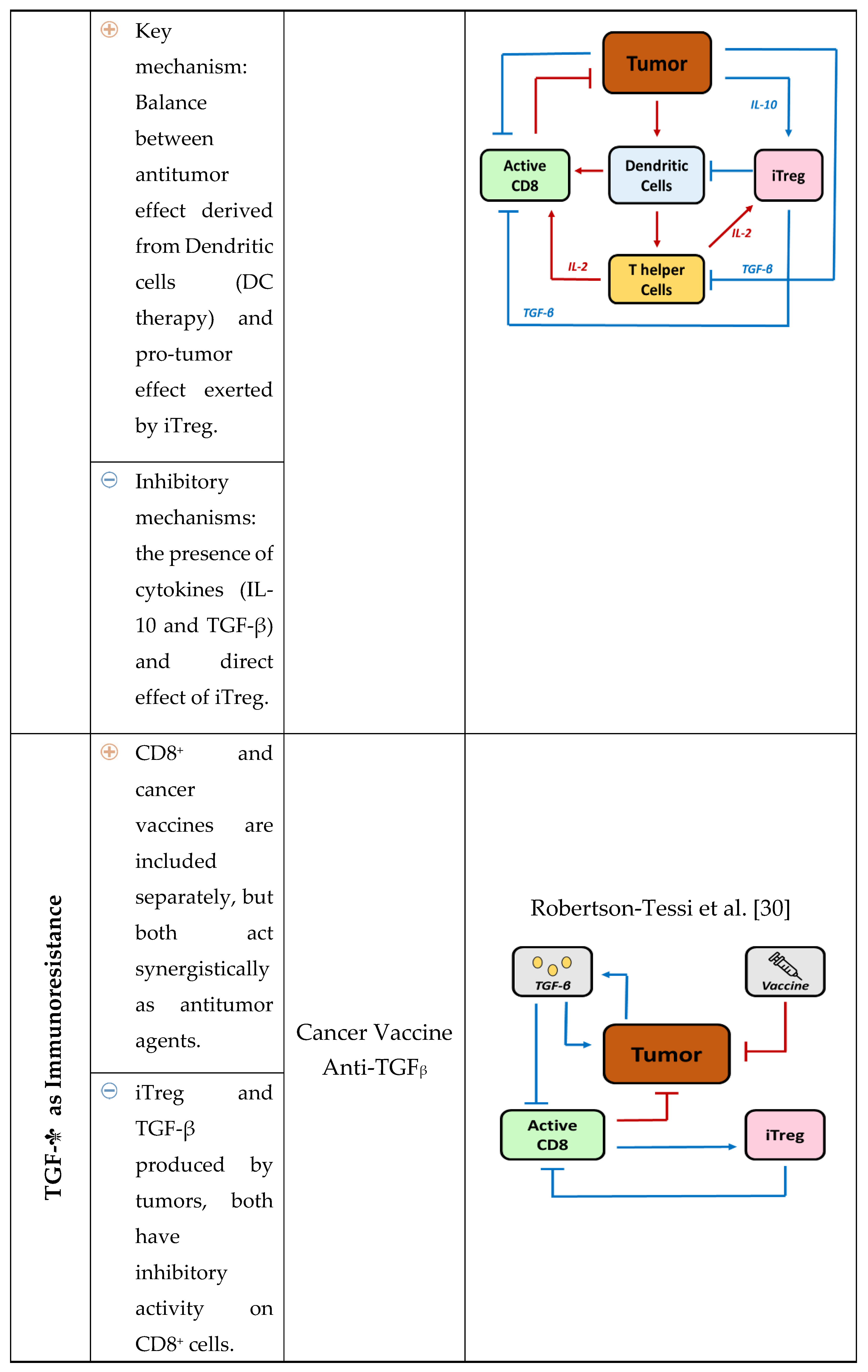
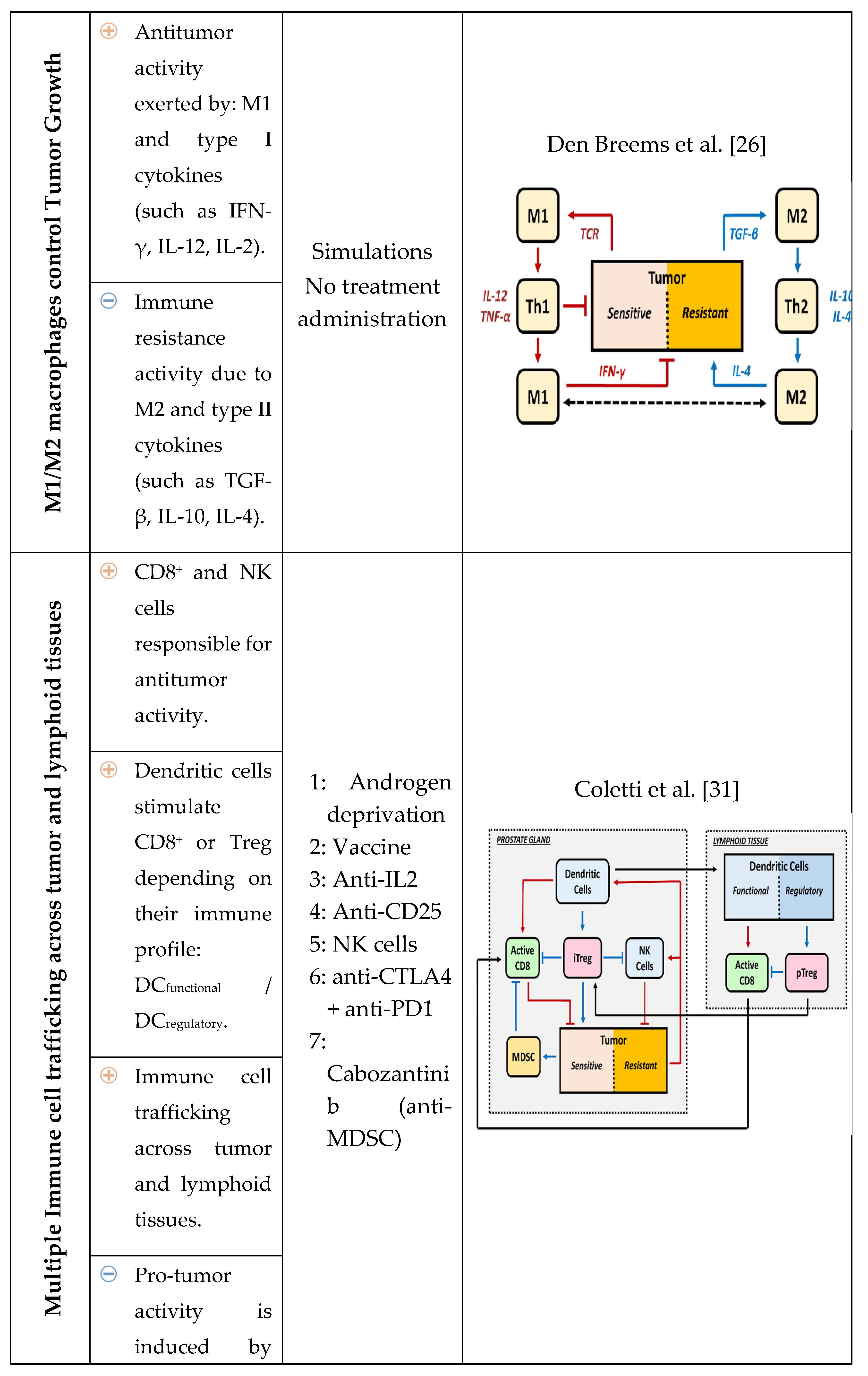
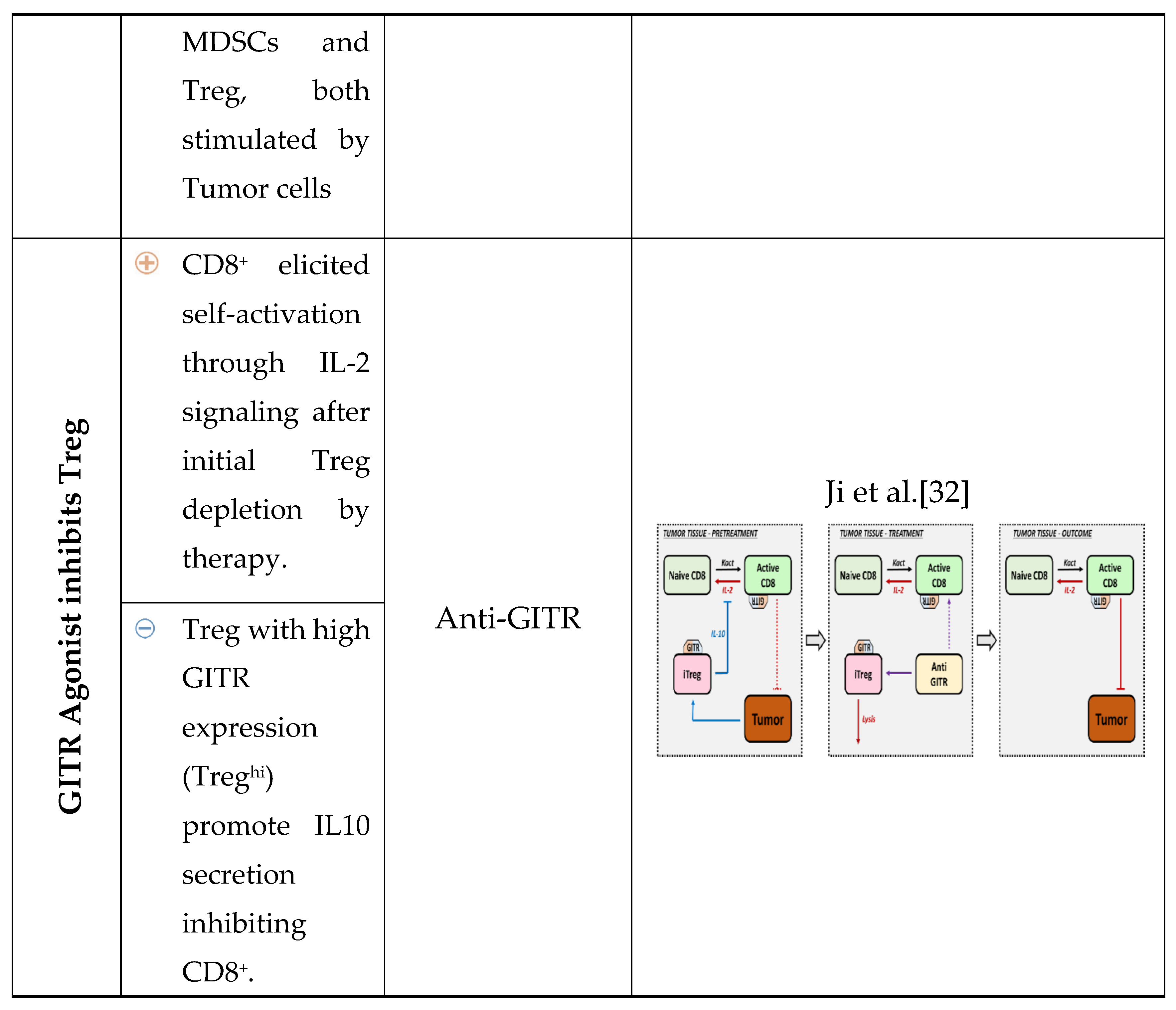
Table 2.
Treg-targeted strategies developed and tested in preclinical studies.
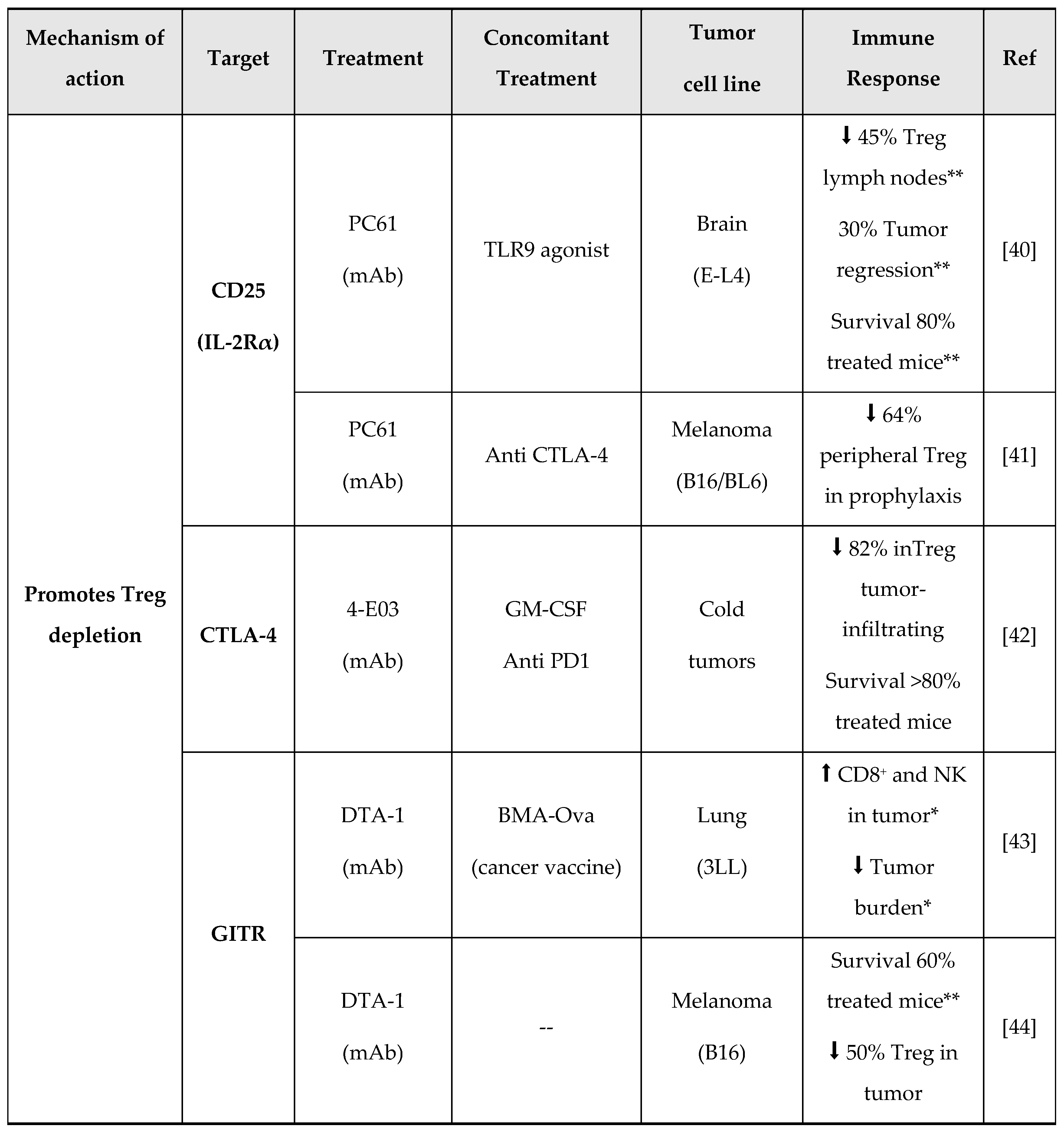
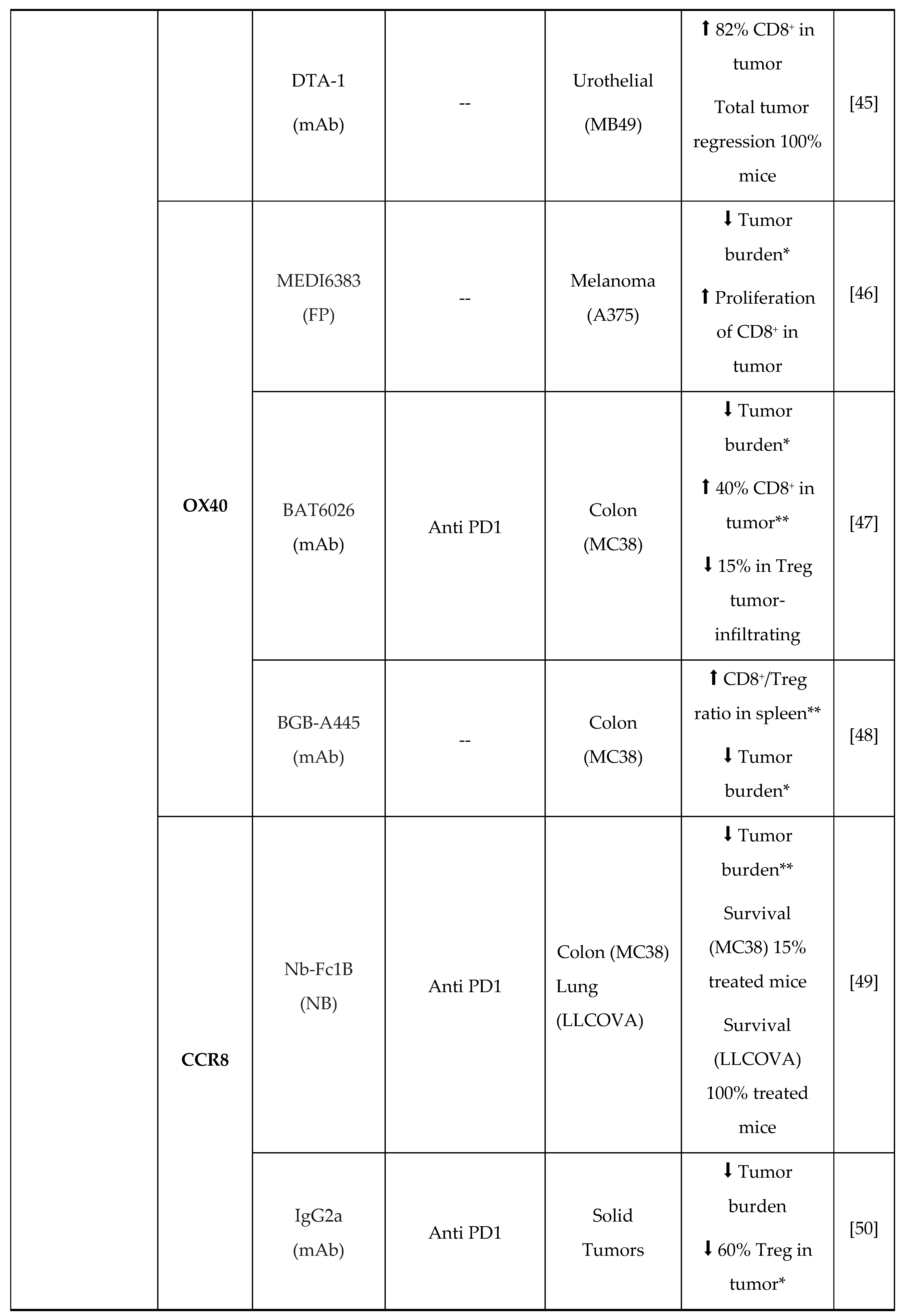
OS, Overall Survival; mAb, monoclonal Antibody; FP, Fusion Protein; NB, Nanobody. [*p<0.05; **p>0.001].
Table 3.
Treg-targeted strategies involved in clinical trials (*p<0.05; **p<0.001).
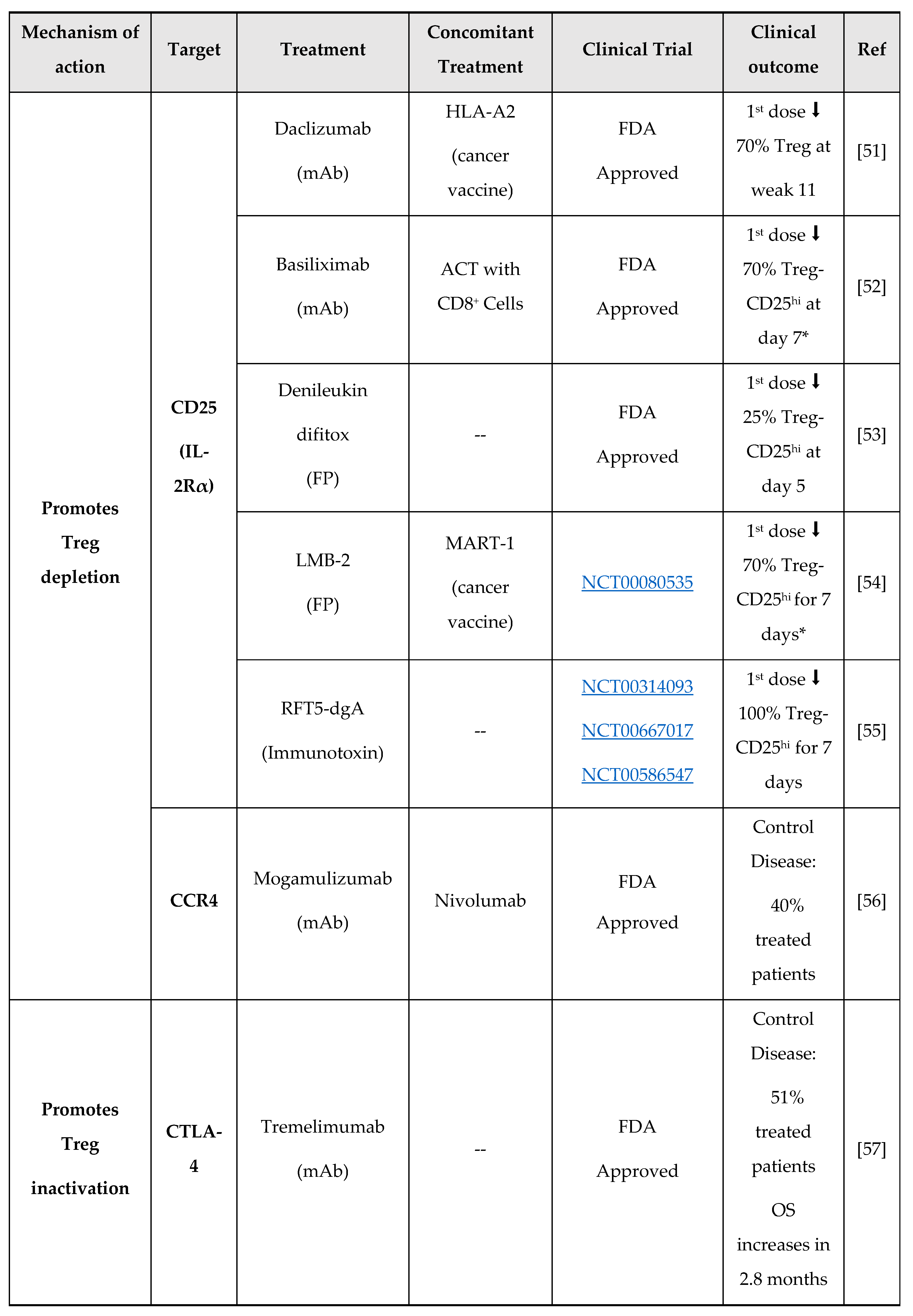
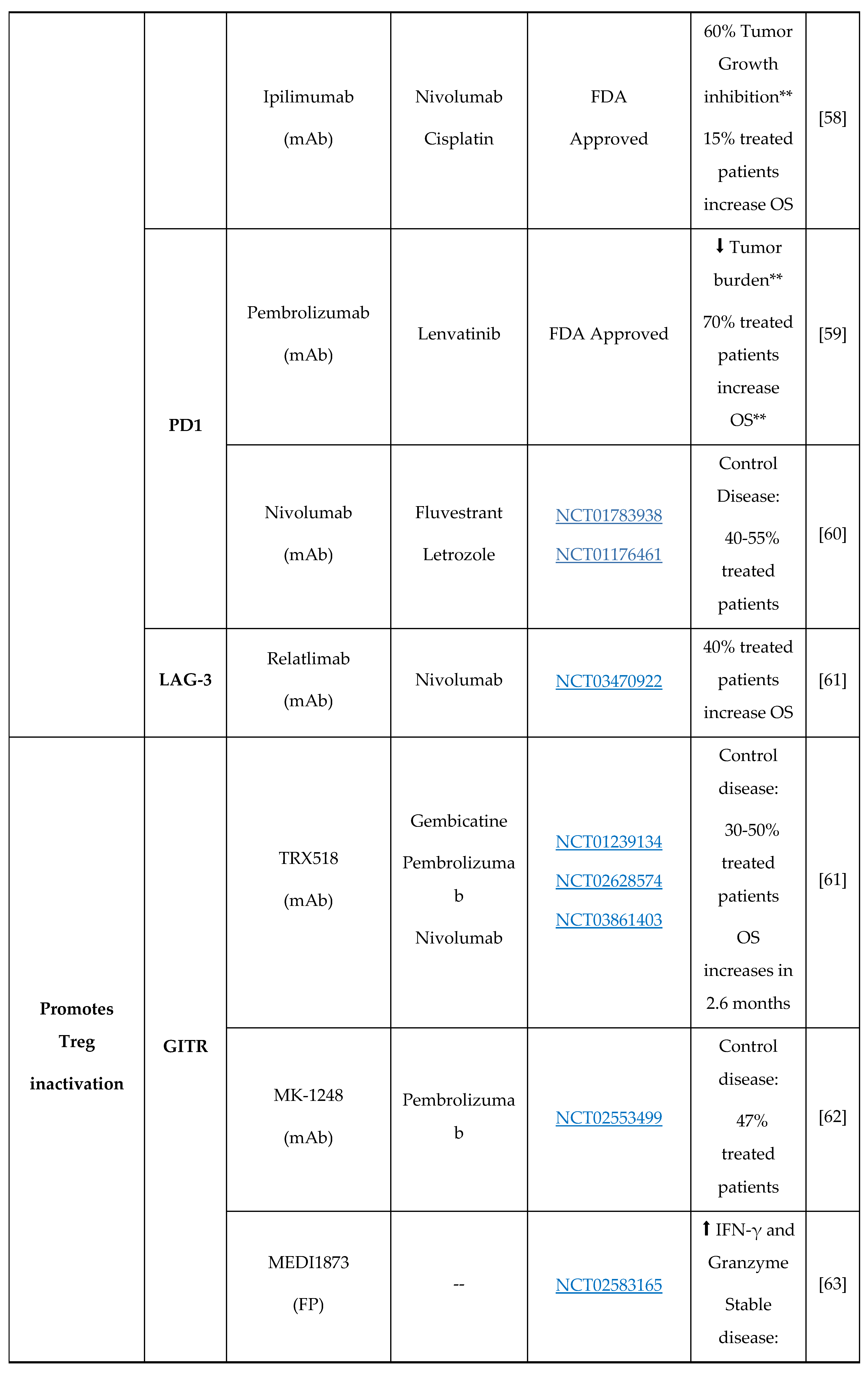
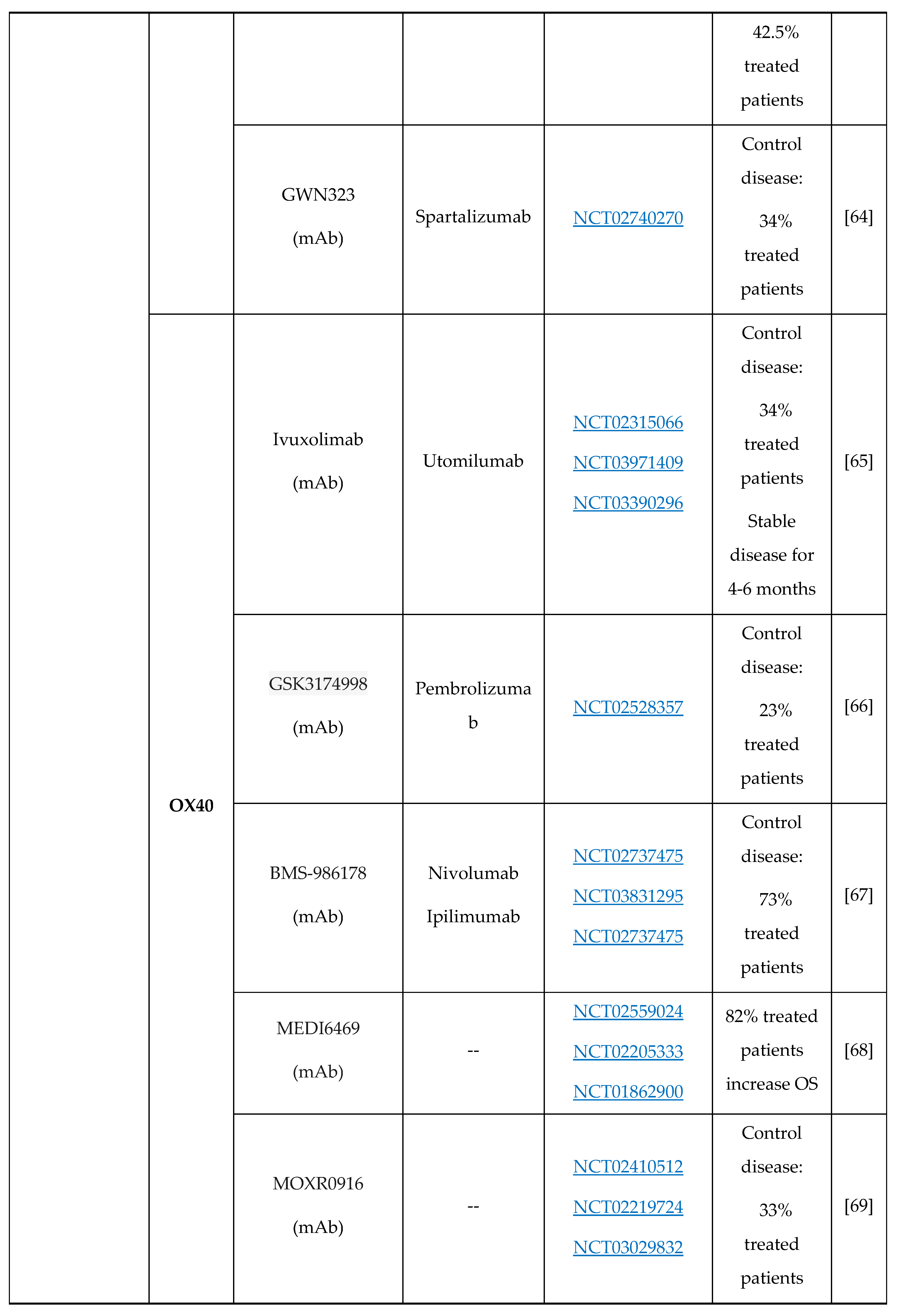
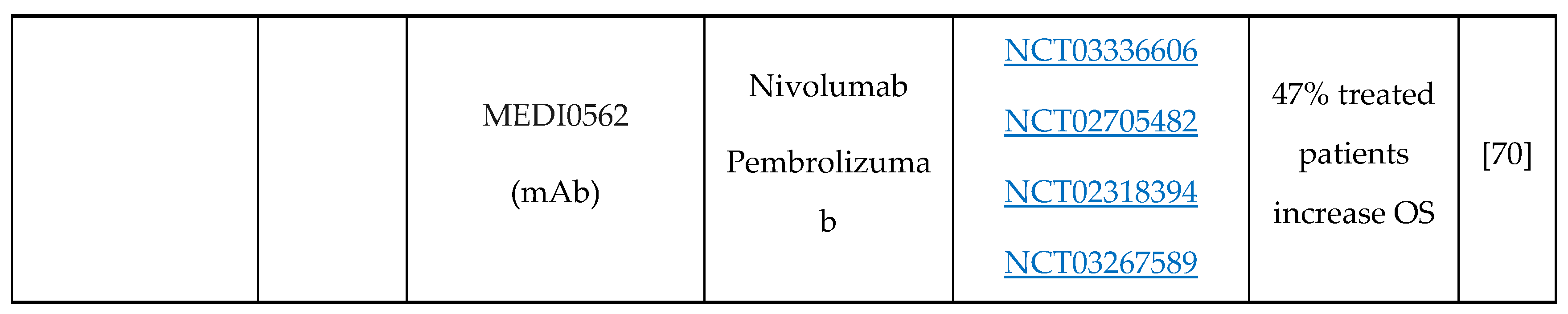
OS, Overall Survival; mAb, Monoclonal Antibody; FP, Fusion Protein; ACT, Adoptive Cell Therapy; Control Disease encompasses patients who have achieved a complete response, a partial response, or a stable disease, all of which are defined according to the RECIST criteria.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



