Submitted:
02 July 2024
Posted:
03 July 2024
You are already at the latest version
Abstract
Sickle cell disease (SCD) is a hereditary blood disorder characterized by abnormal hemoglobin, leading sickle shape of red blood cells. It has several vascular complications and the cerebrovascular ones are among the most frequent and severe both in children and in adults. This review summarizes the main neurovascular manifestations of SCD, including acute stroke, silent cerebral infarction, large vessel diseases (moyamoya arteriopathy and aneurysms) and brain bleeding. Both epidemiology, pathophysiology and treatment issues are addressed and prevention of cerebrovascular events, including silent cerebral infarctions, is particularly relevant in SCD patients, being associated to poor functional outcome and cognitive complaints. Transfusions and hydroxyurea are the main available therapy at the moment, but contraindications, availability and complications might prevent their long term use, particularly in low income countries. The role of Transcranial Doppler in monitoring the patients (mainly children) is analyzed and a practical approach has been selected in order to give the main messages from the actual knowledge for a better management of the SCD patients.
Keywords:
sickle cell disease
; SCD
; anemia
; ischemic stroke
; bleeding
; moyamoya arteriopathy
; intracranial aneurysm
; subarachnoid hemorrhage
; fibromuscular dysplasia
; MRI
; CT
; angiography
1. Introduction
Sickle cell disease (SCD) is a hereditary blood disorder characterized by the production of abnormal hemoglobin molecules, leading to red blood cells adopting a crescent or sickle shape [1]. The primary cause of SCD is a mutation in the beta-globin gene (HBB), which produces abnormal hemoglobin known as hemoglobin S (HbS) [2]. Unlike normal adult hemoglobin (HbA), HbS causes red blood cells to become stiff and sticky, resulting in their deformation and impaired ability to flow through small blood vessels [3]. This altered shape contributes to various severe health complications associated with SCD.
The clinical manifestations of SCD are diverse and can vary significantly in severity among individuals. The hallmark symptom is recurrent episodes of severe pain, known as vaso-occlusive crises, caused by the obstruction of blood flow in small vessels by sickled red blood cells. Additionally, individuals with SCD often experience fatigue, anemia, and an increased susceptibility to infections. These symptoms can significantly impact the daily lives of those with SCD, affecting their physical and psychosocial well-being [4,5]. SCD presents a significant clinical challenge due to its associated hematological and neurological complications, including stroke. The link between SCD and stroke is well-established, with approximately 11% of SCD patients experiencing a stroke by age 20. This elevated risk is attributed to the pathophysiology of SCD, which encompasses hemolysis, vaso-occlusion, and inflammation. These factors collectively disrupt cerebral blood flow and promote vasculopathy, often resulting in ischemic or hemorrhagic strokes [5,6,7,8]. Since Platt et al.‘s seminal study, research has increasingly elucidated the mechanisms connecting SCD to stroke. Hemolysis plays a crucial role in vascular dysfunction [5,9], while inflammation exacerbates cerebrovascular complications [10]. Accurate classification of strokes in SCD patients as either ischemic or hemorrhagic is critical. Adams et al. highlighted the predictive value of transcranial Doppler for pediatric ischemic stroke [11]. DeBaun et al. reviewed management strategies for both stroke types, emphasizing the importance of transfusion and anticoagulation therapies [12].
The aim of this review is to address the link between SCD and cerebrovascular manifestations, highlighting lights and shadows of the current knowledge on this topic in a multidisciplinary approach involving vascular neurologists, neuroradiologists and hematologists.
2. Sickle Cell Disease
2.1. Definition and Diagnostic Criteria
SCD is a genetic disease characterized by the synthesis of falciform hemoglobin (HbS), which polymerizes by deoxygenation damaging erythrocytes and leading to multiorgan damage and premature death.The cause is a genetic defect, transmitted as an autosomal recessive character, which consists in a point mutation on the gene that encodes the beta chain of hemoglobin (β-globin) and involves the replacement of glutamic acid with the amino acid valine in position 6 (β6Glu Val or βS) [13,14,15]. Recent studies, however, document that the basis of drepanocytic disease exists a more complex multifactorial process, involving plasma factors and other blood cells, in addition to falcemic red blood cells, including reticulocytes, white blood cells, platelets, as well as endothelial cells lining the inner walls of blood vessels. Because of a chronic pro-inflammatory state, in fact, a generalized cell hyperactivation occurs and a multicellular adhesion process is triggered that leads to vasculopathy and vessel-occlusion.Several adhesion proteins are chronically overexpressed in patients with sickle cell anemia and, among them, P-selectin plays a central role in multicellular interactions that cause formation of cell aggregates and vessel-occlusion [16,17].These mechanisms play a key role in the pathogenesis of acute and chronic clinical manifestations typical of sickle cell anemia such as chronic hemolytic anemia, due to increased removal of damaged sickle cell erythrocytes from the peripheral circulation, both by extravascolar hemolysis (70%, through the macrophage system) and intravascular (30%), and sickle-painful crises, caused by acute vascular-occlusive phenomena with hypoxic tissue damage of ischemic-reperfusion type. The pathogenesis of these vase-occlusive crises (VOC) is helped by a series of events that act in a proinflammatory and prothrombotic sense (endothelial and platelet activation, neutrophil recall, release of soluble factors such as inflammatory cytokines and clotting factors), as well as with vasoconstrictor effect, following reduced district bioavailability of nitric oxide (NO, which decreases as it is avidly bound by the free Hb present in excess circulation, as well as heme, due to the effect of chronic hemolysis, leading to saturation of the physiological binding systems by haptoglobin and hemopexin) [18,19].
The most common form of severe SCD is related to the homozygous genotype for hemoglobin S (hbss) by inheritance of βS abnormal alleles from both parents
2.2. Epidemiology and Main Clinical Features
Sickle cell anemia is the most common hereditary monogenic hemoglobinopathy in the world. The prevalence of the disease is particularly high in large areas of sub-Saharan Africa, the Mediterranean basin, the Middle East and India, ie in the areas of maximum spread of malaria, as the sickle-shaped tract (i.e., heterozygous for the mutation of hbs) provides some degree of protection from Plasmodium falciparum infection, although the exact mechanisms are still under discussion, favoring host survival and, consequently, the transmission of the abnormal hemoglobin gene [20,21]. Epidemiological data collected by the World Health Organization (WHO) indicate that about 5% of the world’s population carries a severe hemoglobin variant (homozygous) and 2% is heterozygous for hemoglobin S [21]. Globally, there are over 300,000 new cases per year of children born with severe sickle cell disease, of which the majority (about 200,000 cases/ year) concerns the African continent (in particular Nigeria and Congo) and India, and it is estimated that this number is expected to grow further, reaching 400,000 new cases/year by 2050; the prevalence of the disease among newborns ranges from 0.1/1000 in countries not endemic to 20/1000 in some parts of Africa (as in Nigeria). Migratory phenomena have increased the frequency of the gene in the American continent and today there are almost 100,000 people with the disease diagnosed in the United States [20,21,22]. Although in most endemic countries precise data are not available, the prevalence of the sickle tract (the frequency of heterozygous carriers who inherited the mutant gene from a single parent) varies between 10% and 40% in equatorial Africa, decreasing to 1-2% on the North African coast and standing below 1% in South Africa [21,23]. Since the second half of the twentieth century, from developing countries, that is, from areas where sickle cell anemia is endemic, important migratory flows to Europe have begun, dramatically increased in the last 10-1015 years, which led to the increase in the prevalence of this disease also in the “Old Continent”, where currently there are about 25,000 patients suffering from SCD [14,19,23]. Although at European level it is considered a rare disease because of its overall frequency in EU countries, SCD is the most common genetic disease in France and the UK, and its prevalence is constantly increasing in many other countries of central and southern Europe [22]. In countries with high health standards, this trend also reflects the increase in life expectancy of patients, following therapeutic interventions that improve survival to acute complications [20]. The United Nations and the WHO recognise hereditary haemoglobin disorders as a global public health problem, affecting both developing and developed Western nations, whereas sickle cell anemia is one of the emerging diseases of the new millennium that require adaptation of haematological care by the National Health Systems and the implementation of comprehensive programmes for disease prevention and management [21,22]. In Italy, the SCD is historically present in the South, in particular in Sicily and Calabria, with a frequency between 2% and 13%.Since the 2000s, the distribution in our territory has radically changed, both for the displacement of immigrant populations in the South to the industrialized regions of the North and the Centre, and, above all, for new massive migratory flows from North and Central Africa, from Balkan countries such as Albania and Montenegro or from areas of Central and South America, and directed mainly towards the northern area of our country, which led to the spread of the disease in all Italian regions [17,19]. Moreover, the immigrant population resident in Italy consists mainly of young people of childbearing age with a fertility rate higher than that of the native population and this has contributed on the one hand to the increase in the birth rate observed in some regions Italians and, on the other hand, the spread of inherited diseases such as haemoglobinopathies [20]. As a result, in the last 15 years the number of children with SCDs in Italy has progressively increased, with an increase in new diagnoses especially in the northern part of the country, making this disease a widespread public health problem and posing new clinical challenges to pediatricians and haematologists. Data from Regional Reference Centre registers and screening programmes allow to estimate about 1000-1500 children and 2500-3000 total patients with SCDs currently resident in Italy [19]. As a result of immigration, sickle cell anemia has become an emerging disease even in our national health care reality, representing the new haematological challenge for both clinicians who must learn to recognize and manage the disease, both for the NHS, which must respond to the specific needs of this type of patient by ensuring an adaptation of care services [18,23].
2.3. Management Issues and Treatment Options
SCD is a chronic disease that involves global care and specific follow-up by dedicated centres and requires a holistic and multidisciplinary care approach. Dedicated centres are the reference structures for management of patients, for the training of the patient and his family members and for collaboration with peripheral centers, pediatricians and family doctor. Management involves prevention of infections and cerebrovascular and pulmonary complications, treatment of VOCs, self-management education, psychological support, and intensive medical treatment. Pain is the most characteristic symptom of sickle cell anemia and is associated with acute VOCs, also known as sickle cell crises, which represent the most typical clinical manifestation of the disease. Painful seizures are responsible for over 90% of admissions in adult patients with SCD. The severity, location and duration of VOCs are subject to significant variability between different patients; moreover, the frequency and severity of painful seizures can also change in the same patient in relation to age, worsening from childhood to adulthood, with a turning point in late adolescence. The frequency of VOCs and associated admissions has been identified as the factors that most seriously affect the quality of life of patients. Some scientific evidence has shown a positive correlation between painful VOC frequency and premature death, also because they can precede serious and potentially fatal complications such as acute splenic seizure (ASSC) and acute thoracic syndrome (ACS) [15,24]. Risk factors for painful crises include increased hematocrit, the presence of thalassemia, a reduced saturation of oxygen in the inhaled air, heart and lung disorders, dehydration, infections, acidosis and hypothermia [24]. Acute pain, distinctive sign of VOCs, can affect any part of the body, most frequently the distal extremities (hands and feet), the back, abdomen and chest, and can vary in intensity from mild to severe. Acute pain in sickle cell anemia is supported by the presence of at least three components: neuropathic pain, nociceptive pain and idiopathic pain of unknown origin [18]. Acute pain with musculoskeletal involvement is the main clinical presentation of VOCs. The bone districts most frequently involved are the long bones of the upper and lower limbs, but also the vertebral bodies, especially in the dorso-lumbar area, which can mimic the osteoarticular pain of acute arthritis or osteomyelitis [15,24]. Most sickle-cell crises are a medical emergency requiring patient access emergency department. VOCs are acute “time dependent” events that need timely and intensive medical interventions to prevent their development under severe and life-threatening conditions. Patients with VOC accessing PS must receive a correct Triage identifying the presence of SCD All patients with VOC must undergo pain assessment with VAS scale and receive intensive analgesic treatment within 30 minutes of PS access, including Triage time. Pain treatment involves opioid analgesics (tramadol or morphine) in association with NSAIDs or paracetamol and has a minimum initial goal of a 50% reduction in the VAS score (re-evaluated every 10 min). Other acute complications include acute thoracic syndrome (ACS) stroke and intracranial bleeding, acute anemic syndrome, aplastic crisis, infections, medullary hyperplasia, osteomyelitis, bone infarction, bone fractures (with vertebral collapse), septic arthritis, priapism. ACS is an event characterized by chest pain, fever, tachypnea, wheezing or coughing associated with the detection of a “new lung infiltrate detected by chest X-ray”. ACS is a frequent cause of hospitalization in young adult patients and is characterized by high mortality. It is often associated with acute VOC responsible for sterno-costal bone infarction, bone marrow gas embolism, pulmonary infarction or infectious pneumonia, mimicking the symptoms of an acute pulmonary thromboembolism [13,14,18]. Episodes of acute anemia may appear in SCD patients due to acute splenic seizure or other causes such as hemolytic transfusion reactions or aplastic seizures, which may occur when reduced circulatory survival of falcemic red blood cells is associated with the suppression of medullary erythropoiesis during infection (typical of Parvovirus B-19) or inflammatory processes responsible for transient red blood cell aplasia with reticulocytopenia (reticulocyte count <50.000/L). Clinically, acute anemization syndrome is manifested by hypotension, dyspnea and tachycardia [13,14,18].
Chronic complications and organ damage usually appear within 30 years in the target districts (kidney, lungs, brain, bones) as a result of repeated ischemic damage and chronic anemia. Cerebrovascular disease with neurological and cognitive deficits, pulmonary hypertension and cardiomyopathy, kidney failure, hyposplenism, retinopathy, Hepatopathy, painful skin ulcers in the limbs and recurrent priapism are important causes of low quality of life and premature mortality.
In patients with severe and frequent painful crises (>3 per year) involving hospitalization or multiple episodes of ACS (>2) intensive medical treatment with hydroxyurea (HU) or transfusion strategy is indicated. The benefits of hydroxyurea are largely linked to the increased synthesis of hbf, which increases the intra-erythrocyte share of this by reducing the polymerization of hbs, but also the reduction of the number and inactivation of leukocytes, increased production of nitric oxide (NO) and reduced expression of adhesion receptors on the endothelial wall of microcirculation vessels. HU treatment effectively reduces mortality and SCD-related morbidity. Treatment with HU effectively reduces mortality and SCD-related morbidity and is generally well tolerated, but requires regular monitoring of blood chemistry and should be discontinued in anticipation of pregnancy, at least 3 months before conception, for its teratogenic effect, while it can cause male aspermity and infertility. Moreover, hydroxyurea must be taken in a chronic way and poses an important problem of adhesion, associated with poor acceptance, as well as a considerable share of ineffective clinical responses [14,15,19]. Hemotransfusion therapy has the dual objective of correcting anemia and reducing levels of hbs ( 30% or about 30-50%) to prevent SCD complications and organ damage. It is used both for the treatment of acute events (aplastic crises, cardiopulmonary complications, etc.), as a chronic prophylaxis (typically for the prevention of stroke or its recurrence), or in patients where therapy with HU is not tolerated or accepted, or has not proven effective (HU not responder). However, blood transfusion support depends on blood availability and is associated with important complications such as alloimmunization, hyperviscosity, hemolysis from immunogenic reaction and, in the case of traditional transfusions, iron overload with risk of progressive organ damage. Manual or automated erythrocyte exchange procedures are preferable to traditional hemotransfusion strategy as they avoid the risk of hyperviscosity or martial overload (requiring the initiation of ferrochelating treatment) [24,25]. Finally, the transplantation of hematopoietic stem cells from the HLA-identical donor cord represents an important curative and healing option of sickle cell anemia, indicated in patients who have developed a stroke, with history of recurrent hospitalizations and need for chronic ACS transfusion therapy or severe recurrent acute painful seizures (>3 per year). However, it is a treatment option limited to the availability of a compatible HLA donor, the risk-benefit ratio of which should be carefully assessed, and reserved for younger subjects (<16 years) and have not yet developed serious organ damage related to the underlying disease [14,18,26]. Innovative gene therapy approaches for the treatment of sickle cell anemia are currently being studied, with encouraging preliminary results. Other interesting molecules for therapeutic effectiveness are waiting for response from regulatory bodies.
2.4. Children vs. Adult Patients
In SCD patients, adolescence is also a critical step from a clinical point of view, as well as from a psychological point of view. In adolescence the frequency of VOC increases both for the greater susceptibility of the target organs, consequent to the functional damage for the previous repeated VOC, and for the psycho-affective stress related to the adolescent crisis, worsened by the presence of the disease. The transition from pediatric to adult care requires a planned and coordinated management step, in close multidisciplinary collaboration between the two settings and with psychological service, aimed at gradually bringing the young patient to the center of the treatment process.
3. Neurovascular Manifestations
Cerebrovascular disease is a relevant complication of SCD, with a notable prevalence of 4% and an incidence rate of 0.61 per 100 patient years [27]. This condition is broadly defined in studies as including both acute ischemic and hemorrhagic strokes, but also covert cerebrovascular disease has gained attention and intracranial large vessel involvement may not be neglected. Stroke, silent cerebral infarcts (SCI), and cognitive impairments are the most common long-term complications of SCD in both children and adults. A significant cohort study in the United States before 1990 [8] revealed that by age 40, about 20% of adults with HbSS and 10% with HbSC had experienced a cerebrovascular accident. In low- to middle-income settings, approximately 11% of children with HbSS will have a stroke by age 18.
However, the leading cause of permanent neurological damage in individuals with HbSS or HbSb0 thalassemia is SCI, affecting around 39% of children and more than 50% of adults [28]. Detecting these infarcts requires MRI and a formal neurological examination to rule out overt strokes. Both stroke and silent cerebral infarcts pose a high risk of recurrence and significant cognitive impairments, potentially affecting employment and quality of life. In 2013, the American Heart Association/American Stroke Association (AHA/ASA) expanded the definition of stroke to include silent cerebral infarctions and hemorrhages, typically identified by brain MRI [29]. This change emphasizes radiological evidence of infarction or hemorrhage, acknowledging that permanent neurological damage can occur even if symptoms resolve within 24 hours. For patients with cerebral ischemia, the AHA/ASA recommends treatment based on the cause of the ischemic event rather than the presence or size of an infarction.
The pathophysiology of cerebrovascular manifestations in SCD is complex, involving vessel occlusion, hemolysis, inflammation and vessel wall changes as main phenomena. Red blood cells in SCD become rigid and sickle-shaped, being lodged in tiny blood vessels, obstructing blood circulation and depriving brain tissue of oxygen, causing ischemic stroke [3,30,31]. Sickle-shaped red blood cells are more likely to adhere to blood vessel walls, increasing blood viscosity and slowing blood flow, which raises the risk of clotting [31,32]. Furthermore, sickle cells have a shorter lifespan than normal red blood cells, resulting in increased cell debris and turnover [32,33]. This heightened turnover leads to inflammation and damages or weakens blood vessel walls. Weakened vessels are prone to rupture, which can block blood flow and cause a hemorrhagic or ischemic stroke [34]. Chronic anemia associated with SCD increases cardiac output, raising shear forces on blood vessel walls and contributing to their damage [35,36]. In SCD, there is also increased platelet activation, which promotes clot formation [37]. Children with SCD have a very high risk of stroke, estimated at 10% to 15%, and without intervention, approximately 11% of patients experience clinical ischemic stroke by age 20 [38]. Strokes often recur in these patients.
Ischemic strokes in SCD are often due to stenosis or occlusion of major cerebral arteries such as the internal carotid, middle cerebral, and anterior cerebral arteries [34]. Small vessel disease caused by sickling in capillaries and venules also increases the risk of ischemic stroke [12]. Vulnerable areas of the brain include regions with end arteries, such as the subcortical white matter, deep gray matter, and watershed zones [39,40]. SCD also causes intimal hyperplasia and the proliferation of smooth muscle cells in arteries, thickening vessel walls, narrowing their diameter, and reducing blood flow [41,42]. Endothelial dysfunction is common, reducing nitric oxide levels and promoting vascular inflammation and thrombosis [42]. Abnormal interactions between sickle cells, endothelial cells, platelets, and coagulation factors further encourage abnormal clotting [37].
The compensatory high-velocity blood flow in sickle cell anemia increases shear stress on blood vessels over time, damaging the endothelium [40]. Impaired vascular autoregulation makes it harder for the brain to adjust blood flow with changes in pressure and oxygen levels [43]. Both significant artery stenosis and Moyamoya syndrome are observed; the latter causes fragile collateral vessels prone to rupture and hemorrhage. Overall, both ischemic and hemorrhagic stroke are increased in SCD due to these numerous vascular effects. Blood transfusions can significantly reduce stroke risk by diluting the concentration of sickle cells, but they are not associated to a restoration of normal hemodynamic parameters in the brain and a biological model of vascular instability has been recently propose to explain the pathophysiology of stroke in SCD [44].
A recent systematic review [45] provided interesting information about the association between SCD and stroke. A preliminary issue is the association between stroke risk and genotype. As previously said, SCD includes several genotypes, with hemoglobin SS (homozygous for hemoglobin S), hemoglobin SC (compound heterozygous for hemoglobin S and hemoglobin C), and hemoglobin S beta-thalassemia being the most common. Hemoglobin SS is the most prevalent and carries the highest risk for both ischemic and hemorrhagic strokes [46]. The SENET study [8] reported an incidence rate of 0.61 first strokes per 100 patient-years in children with SS disease, compared to 0.17 in SC disease and 0.09 in S-beta thalassemia. Although some studies suggest that the stroke risk in SC disease approaches that of SS disease when adjusting for factors like socioeconomic status and haplotype, data on other genotypes like sickle-hemoglobin C disease are limited due to their rarity [47]. Research typically groups all sickle cell genotypes together or focuses on SS disease, leading to a lack of specific data on stroke risks across different genotypes. Current evidence indicates that SS disease poses the greatest stroke risk, while SC disease and S-beta thalassemia also have a significant but lower risk. All SCD patients should undergo continuous screening and monitoring for stroke.
Additionally, patients with SCD may present acute coma, seizures, and headaches, often attributed to posterior reversible encephalopathy syndrome (PRES) [48,49,50]. These symptoms can occur following acute chest syndrome (ACS) or in situations involving hypertension and/or immunosuppression. The differential diagnosis for these presentations in SCD includes a broad range of focal and generalized vascular and non-vascular pathologies. These may include intracranial hemorrhage, extensive infarction, central nervous system infections, and autoimmune encephalopathies. Differentiating among these conditions is crucial for appropriate management, with Magnetic Resonance Imaging (MRI) playing a pivotal role in diagnosis. Diffusion-weighted imaging (DWI) is particularly useful in distinguishing between infarction and reversible conditions such as edema, penumbral ischemia, PRES. These last manifestations were not the focus of the present review.
3.1. Ischemic Stroke
As previously written, without screening and prophylactic treatment, approximately 11% of patients with SCD will experience an overt stroke by age 20, and this number increases to 24% by age 45 [8]. Ischemic strokes are the most prevalent, making up about 75% of SCD-related strokes [51,52]. However, SCD patients are at high risk for both ischemic and hemorrhagic strokes. Ischemic strokes are more common in children, while hemorrhagic strokes are more frequent in young adults [53,54,55].
A recent cohort study [56] found that overt strokes accounted for 10% of deaths among SCD patients. Although overt ischemic strokes are rarely fatal, 26% of hemorrhagic stroke cases result in death [8]. Recurrence rates for overt ischemic strokes can reach up to 70%, with the highest risk occurring within 36 months of the initial stroke [57]. Both ischemic and hemorrhagic strokes lead to significant long-term morbidity, including seizures, physical disabilities, and cognitive impairments [12].
Then, stroke is one of the most severe complications of SCD, commonly occurring in childhood [8,58]. Major risk factors for ischemic stroke include prior transient ischemic attack (TIA), low hemoglobin levels, high leukocyte counts, and hypertension [59,60]. In studies focusing on ischemic stroke in sickle cell disease SCD, the most consistently identified risk factors include prior TIA [46,60], emphasizing the significance of low hemoglobin levels and high leukocyte counts. Research indicates that individuals with a history of TIA in SCD face a tenfold higher risk of subsequent IS compared to those without such a history [61]. Long-term transfusion programs following TIA have notably reduced stroke incidence, underscoring the role of TIA as a critical warning sign.
The clinical features of ischemic stroke in SCD patients are similar to those in non-SCD pediatric strokes, with common symptoms including hemiparesis, speech difficulties, and seizures [58,62]. However, SCD stroke patients often exhibit a higher frequency of bilateral motor deficits due to multilobar infarction. Additionally, cranial nerve palsies and ataxia are reported more frequently in SCD stroke patients.
Ischemic strokes in children and adults with SCD represent significant and common medical emergencies, especially in regions where primary stroke prevention is not a standard practice. When a patient with SCD presents with an ischemic stroke or TIA, a timely response is crucial to minimize further ischemic damage. While the optimal timing for intervention with blood transfusion therapy and brain imaging has not been rigorously studied, principles of acute ischemic stroke and TIA management, along with observational studies, offer evidence for best practices. In the absence of randomized clinical trials specific to SCD, comprehensive guidelines that cover the entire spectrum of brain imaging and interventions for reducing ischemic brain injury are not currently available. However, practical approaches can be derived from understanding the principles of cerebral hemodynamics unique to SCD, combined with the collective experiences of multidisciplinary panels. Table 1 summarizes the main features of ischemic stroke in patients with SCD.
Effective management of acute ischemic stroke and TIA in individuals with SCD hinges on rapid evaluation, multidisciplinary collaboration, and adapting to evolving clinical insights. While definitive randomized clinical trials are lacking, practical approaches rooted in cerebral hemodynamic principles and clinical experiences are crucial for optimizing outcomes in this patient population. Continued research and shared experiences will further enhance our understanding and management of acute ischemic events in SCD. The main steps of acute management of SCD patients with nacute neurological deficit are summarized in Table 2.
The final diagnosis of ischemic stroke or TIA in individuals with SCD relies heavily on a thorough neurological history and examination. While brain MRI, especially DWI, can aid in diagnosing acute cerebral ischemia, it is crucial to note that a normal DWI does not definitively rule out an ischemic stroke and a TIA [64]. This underscores the importance of clinical judgment and comprehensive assessment in making accurate diagnoses. When a patient with SCD presents with acute-onset focal neurological deficits suggestive of ischemic stroke or TIA, prompt intervention is essential to mitigate the risk of ongoing ischemic brain injury. This approach is crucial regardless of the immediate availability of diagnostic imaging confirmation. Regarding management strategies, there is limited empirical evidence specific to SCD. One retrospective observational study [65] focused on children with HbSS or HbSβ0 thalassemia provides data supporting the use of exchange transfusion (either apheresis or manual) over simple transfusion for acute management of ischemic strokes. The study reported that children who received exchange transfusion had a lower rate of recurrent strokes compared to those managed with simple transfusion (relative risk [RR] 5.0, 95% confidence interval [CI] 1.3-18.6). This finding suggests that exchange transfusion may offer advantages in reducing the risk of recurrent ischemic events in this population.
In summary, while MRI can support the diagnosis of acute cerebral ischemia in SCD, clinical evaluation remains paramount. The choice between exchange transfusion and simple transfusion for acute management of ischemic strokes should consider available empirical evidence, clinical judgment, and the multidisciplinary management approach tailored to individual patient needs.
The management of acute ischemic stroke in individuals with SCD involves considerations specific to the altered cerebral hemodynamics and unique pathophysiology of SCD [66,67] (Table 3).
In summary, optimizing hemoglobin levels through transfusion therapy plays a crucial role in managing acute ischemic strokes in individuals with SCD, aiming to improve cerebral oxygen delivery and minimize further ischemic brain injury. The choice between simple transfusion and apheresis depends on the severity of anemia and availability of resources, emphasizing the need for a multidisciplinary approach to ensure timely and effective care.
The main considerations due to the underlying pathophysiology of ischemic damage in SCD are summarized in Table 4.
In summary, the management of acute ischemic stroke or TIA in individuals with SCD requires a tailored approach that considers optimizing hemoglobin levels while mitigating the risk of viscosity-related complications. Close collaboration between hematologists, neurologists, and acute-care providers is essential to ensure timely and effective treatment, aligned with the unique aspects of cerebral hemodynamics in SCD.
The decision to use either simple blood transfusion or exchange transfusion (apheresis) in the treatment of acute ischemic injury in SCD must weigh the potential benefits against the risks associated with each type of transfusion. The main complications associated with these treatments are summarized in Table 5.
Despite these potential complications, the benefits of timely treatment with transfusion therapy for acute ischemic injury in the brain are generally considered to outweigh the risks. This decision is based on the urgent need to improve oxygen delivery to the brain, mitigate ongoing ischemic damage, and reduce the risk of recurrent strokes in individuals with SCD. The choice between simple transfusion and exchange transfusion depends on factors such as the patient’s hemoglobin level, percentage of sickle hemoglobin (HbS), and the presence of complications like ACS. The main recommendations for acute ischemic stroke management in SCD are ilustrated in Table 6.
Some remarks may be advanced according with the guidelines [88]. First, the optimal timing for therapy is to provide a transfusion within 2 hours of presentation to medical care for children and adults with SCD who present within 72 hours of the onset of suspected acute stroke, new neurological deficit, or TIA symptoms. Second, for children and adults presenting to medical care more than 72 hours after the onset of new neurological deficit or TIA symptoms, without recent worsening, it is suggested to assess for anemia and the percentage of sickle hemoglobin and to consider transfusion on a case-by-case basis. Third, for individuals with hemoglobin levels above 8.5 g/dL presenting with focal neurological deficits or TIA, it is suggested to perform exchange transfusion therapy to reduce the risk of hyperviscosity syndrome.
To date, only one study has examined the use of tPA in adults with SCD [89]. Overall, the SCD population experiences a first stroke at a younger age and has a higher proportion of hemorrhagic strokes compared to the general adult population with strokes. There is no existing evidence to determine the optimal initial treatment—tPA or blood transfusion. Conceptually, treatment prioritization should be guided by the underlying stroke etiology (SCD vs non-SCD), but this distinction is often unclear in the hyperacute setting. Given the increased overall survival of adults with SCD into middle and old age and the cumulative effect of traditional cardiovascular risk factors leading to stroke, administering tPA to older adults with SCD presenting with acute ischemic strokes within 4.5 hours of symptom onset is advisable. However, no absolute age cutoff has been defined. In some cases, tPA treatment may occur before recognizing the patient has SCD. In such instances, blood transfusion should be considered as soon as possible after SCD identification. A systematic review on endovascular thrombectomy was not conducted because there are no interventional studies specific to SCD. However, multiple large clinical trials support thrombectomy for stroke in selected patients with acute large vessel occlusion outside of SCD. Evidence supports endovascular thrombectomy for stroke with acute large vessel occlusion in the general population. However, we lack data on the risks and benefits in adults with SCD. The utility of endovascular thrombectomy in adults with SCD should be carefully evaluated due to the prevalence of cerebral vasculopathy and moyamoya syndrome and the absence of data describing the benefits and risks.
For children with HbSS or HbSb0 thalassemia and a past ischemic stroke, some issues should be clarified. First, hydroxyurea therapy is an inferior alternative to regular blood transfusion for secondary stroke prevention in children who cannot receive or refuse blood transfusion. However, it is noted that hydroxyurea therapy is still superior to no therapy at all in terms of reducing the risk of secondary stroke. Second, adolescents who experienced a stroke during childhood are recommended to continue receiving regular blood transfusions into adulthood for secondary stroke prevention. This approach aims to reduce the risk of recurrent strokes in individuals with SCD. Third, adults who experience their first stroke as adults should undergo a thorough evaluation for stroke-modifiable risk factors. Secondary stroke prevention measures should include regular blood transfusion therapy as well as other recommended interventions to manage and mitigate the risk of subsequent strokes.
The evidence from large multicenter studies underscores the ongoing risk of stroke recurrence in children and adults with HbSS or HbSb0 thalassemia, despite receiving regular blood transfusion therapy for secondary prevention. The key findings from the available studies are summarized in Table 7.
Regular blood transfusion therapy is shown to provide partial effectiveness in reducing the risk of secondary strokes in children and adults with HbSS or HbSb0 thalassemia. However, the studies highlight that despite this treatment, the risk of recurrent strokes remains significant. Progressive vasculopathy observed in these patients underscores the ongoing challenges in managing stroke prevention effectively. These findings emphasize the need for continued research into optimizing secondary stroke prevention strategies, including exploring adjunctive therapies or interventions to further mitigate the risk of recurrent strokes in this high-risk population.
Hydroxyurea therapy has been shown to be less effective than regular blood transfusion in preventing secondary strokes in children with HbSS or HbSb0 thalassemia. There is insufficient data regarding other sickle cell disease SCD phenotypes. In a recent single-center study [92], 35 children previously receiving regular transfusions for stroke prevention were switched to hydroxyurea at the highest tolerated dose, alongside serial phlebotomy to manage iron overload. The study reported a stroke recurrence rate of 5.7 events per 100 patient-years. Although phlebotomy effectively reduced iron levels and normalized hepatic iron, the analysis did not adjust for the critical initial 2-year period with the highest stroke recurrence rates. Based on these findings, a phase 3 non-inferiority randomized controlled trial, Stroke With Transfusions Changing to Hydroxyurea (SWiTCH) [93], was conducted. Results indicated that hydroxyurea, combined with phlebotomy, was inferior to continued transfusions and ongoing iron chelation therapy due to excessive iron accumulation. The secondary stroke rate was 5.6 events per 100 patient-years with hydroxyurea, compared to zero events in those continuing transfusion (standard care). Consequently, the trial was halted after interim analysis demonstrated equivalent liver iron content, suggesting futility in reducing the composite primary endpoint of stroke increase, while requiring superiority for iron removal. In settings where transfusion is unavailable, hydroxyurea therapy offers superior secondary stroke prevention compared to no treatment at all. A prospective study in Nigeria [94] supported this, with 13 children receiving hydroxyurea showing a significantly lower secondary stroke incidence (7 events per 100 person-years) compared to those who did not receive treatment (28 per 100 person-years, P < 0.001; odds ratio 3.80, 95% CI 1.55-9.31).
These findings underscore the importance of tailored treatment strategies in managing SCD-related stroke risk, balancing the benefits and limitations of different therapeutic approaches.
For children and adults with strokes and severe cerebral vasculopathy like moyamoya syndrome, additional treatments such as revascularization surgery can be considered alongside regular blood transfusion therapy for preventing secondary strokes. Hematopoietic stem cell transplantation (HSCT) [95,96] is recognized as a curative and definitive option for secondary stroke prevention in children, especially those with matched related donors or undergoing haploidentical bone marrow transplantation with post-transplantation cyclophosphamide [97]. Recent studies have identified new cerebral infarcts through centralized neuroimaging and neurology assessments conducted as part of HSCT protocols [98,99]. It is crucial for individuals and their families to understand both the potential long-term benefits and risks associated with these therapies for preventing secondary strokes.
These statements reflect the importance of tailored management strategies for stroke prevention in individuals with sickle cell disease, emphasizing the role of regular blood transfusion where indicated and the consideration of alternative therapies when transfusion is not feasible or accepted. Each approach aims to reduce the incidence of recurrent strokes and improve long-term outcomes for patients with sickle cell disease who are at risk for ischemic events.
3.2. Silent Cerebral Infarction
Silent cerebral infarction (SCI) stands as the most common cause of permanent neurological damage in children and adults with SCD [100], affecting around 39% of children by the age of 18 and more than 50% of adults by the age of 30 [28]. Despite being considered a small vessel disease, there is no direct evidence supporting this classification. A systematic review found no guidance for managing new cases of SCI in adults [101]. The risk factors for SCI include low hemoglobin levels, elevated systolic blood pressure, and signs of cerebrovascular disease on both intracranial and extracranial MR angiography. The exact mechanisms causing these infarctions are unclear, though their frequent occurrence in border-zone brain areas suggests hemodynamic factors may be involved [70,100,102,103,104,105].
Once SCI is detected, preventing its progression is crucial due to its significant impact on cognitive function and its role as a biomarker for recurrent infarction in children and adults with HbSS or HbSβ0 thalassemia [106,107,108]. The Silent Cerebral Infarct Transfusion (SIT) trial [106] investigated whether regular exchange blood transfusions could prevent cerebrovascular diseases, including symptomatic strokes and the progression of SCI, in children with pre-existing silent infarctions. The trial showed a significant reduction in the relative risk of cerebrovascular disease with regular transfusions, with a number needed to treat (NNT) of 13.
Additionally, a recent systematic review indicated that long-term exchange blood transfusion might reduce the incidence of SCI in children with abnormal transcranial Doppler velocities, though it may have little to no effect on children with normal velocities [101]. For adults who have an incidental finding of SCI on an MRI, it may be appropriate to offer periodic MR scans and consider interventions if there is evidence of progressive ischemia. The main features of SCI in SCD are summarized in Table 8.
The prevalence and consiquences of SCI in SCD impact on the need of screening this cerebrovascular manifestation in children and adults. The main recommendations about screening are summarized in Table 9.
After identifying an infarct-like lesion, the following plan of action is recommended:
1. Neurological Evaluation: Ensure that the infarcts are classified as silent cerebral infarcts rather than overt strokes.
2. Discussion on Management:
- Secondary Prevention Options: Consider regular blood transfusions and hematopoietic stem cell transplantation (HSCT).
- Cognitive Screening Assessment: Conduct cognitive screening.
3. MRI Surveillance:
- Conduct MRI scans every 12 to 24 months to monitor for cerebral infarct progression.
- If new infarcts are detected, discuss with the patient and family the pros and cons of increasing therapy intensity to prevent recurrence.
Given the link between silent cerebral infarcts and the risk of progressive brain damage, MRI screening for silent cerebral infarcts is recommended at least once in children with HbSS or HbSb0 thalassemia. Adults with these conditions should also be screened, even though data on secondary prevention in adults is lacking. Identifying SCI allows for close monitoring and consideration of preventive measures such as regular blood transfusions or experimental treatments, including HSCT and gene therapy.
The reasons to justify screening for SCI in children and adults with HbSS or HbSb0 thalassemia are summarized in the Table 10:
3.3. Intracranial Bleeding
Hemorrhagic stroke is less common than ischemic stroke in SCD but has higher mortality rates. The prevalence of HS in SCD patients varies widely across studies, from 1.3% to 11%, due to differences in diagnostic methods, patient populations, and follow-up periods [110,111]. Intracerebral hemorrhage (ICH) in SCD patients occurs most frequently between ages 20 and 29 [112,113]. Hypertension is a significant risk factor, present in over half of SCD-ICH cases in some studies. Other risk factors include low hemoglobin, high reticulocyte count, prior ICH, and smoking [114,115].
The pathophysiology of ICH in SCD is not fully understood. Potential mechanisms include vessel wall damage due to chronic anemia and sickling, increased vessel fragility from collagen depletion and smooth muscle hypertrophy, and aneurysm formation [115,116]. Neuroimaging studies often locate hemorrhages at the corticomedullary junction and lenticulostriate arteries, suggesting that fragile collateral vessels may be the origin.
As previously pointed out, in individuals with SCD, intracranial hemorrhage becomes notably more prevalent between the ages of 20 and 29 years [8]. Historically, from the 1970s to 2010, approximately 75% of reported strokes in children and adults with SCD were ischemic, with the remaining 25% categorized as hemorrhagic [57,59]. Among adults, intracranial hemorrhage often correlates with the formation of aneurysms [117], as highlighted by studies indicating prevalence rates of 1.2% in children and significantly higher rates of 10.8% in adults with SCD [118]. These aneurysms, when ruptured, typically manifest as relatively small (2–9 mm) lesions situated at major vessel bifurcations [117,118]. The clinical features of hemorrhagic stroke in SCD patients are similar to those observed in other populations, including acute severe headache, nausea, seizures, and focal neurological deficits depending on the hemorrhage location. However, SCD patients often present with smaller hematoma volumes compared to hypertensive ICH patients [119].
The management of acute intracranial hemorrhage in adults with SCD poses a substantial challenge due to the scarcity of evidence-based guidelines specific to this population. In the absence of dedicated protocols, it is advisable to adhere to established guidelines from the European Stroke Organization [120] that govern the general management of acute intracranial hemorrhage in adults without SCD. This approach underscores the critical need for a multidisciplinary team comprising specialists in neurosurgery, interventional neuroradiology, hematology, and neurology/stroke to collaboratively address the complex clinical needs of affected patients. Regarding the role of exchange blood transfusion in managing acute intracranial hemorrhage in SCD, its efficacy and safety profile remain inadequately researched. Therefore, decisions regarding transfusion strategies should be made on a case-by-case basis, considering the specific clinical context and in consultation with the multidisciplinary team. In summary, the management of intracranial hemorrhage in adults with SCD necessitates a coordinated and interdisciplinary approach, guided by established stroke management principles tailored to the unique challenges posed by this hematologic disorder.
There is limited evidence on the management of acute ICH specific to SCD patients beyond controlling blood pressure and considering surgery. The role of blood transfusions is debated due to concerns about increasing blood viscosity and exacerbating the hemorrhage. Many aspects of HS in SCD remain unknown, including precise risk factors and optimal treatment strategies. Further research through multicenter prospective studies is needed to better characterize ICH incidence, establish screening protocols, and explore novel therapies to improve prognosis. Standardized management protocols are also essential.
3.4. Intracranial Arteriopathy
Patients with SCD are susceptible to progressive narrowing of intracranial arteries over time, which can lead to the development of ill-defined collateral vessels resembling moyamoya vasculopathy. The term “moyamoya” originates from the characteristic “puff of smoke” appearance observed in cerebral vasculature on digital subtraction angiography (DSA). This delicate network of collateral vessels is exposed to presumed flow-related stress, resulting in fragmentation of the elastic lamina, thinning of the vessel media, and the formation of microaneurysms, thereby increasing the risk of intracranial hemorrhage [121]. Conversely, moyamoya-related vessels may also collapse or thrombose, contributing to ischemic stroke [122]. In approximately 30% to 40% of individuals with SCD and stroke, moyamoya neovascularization can develop. This condition particularly manifests in children and young adults with symptoms such as cognitive decline, silent infarcts, or border zone ischemic stroke, all stemming from steno-occlusive changes in the primary cerebral arteries [123,124]. Reticulocytosis is commonly associated with vasculopathy in SCD through chronic oxygen desaturation as well as hemolysis [125,126], sometimes non responding to regular blood transfusion [127].Other associatioted factors are mean prior values for hemoglobin, oxygen content, reticulocytes, and indirect bilirubin [126].
Surgical revascularization options, when used adjunctively with long-term exchange blood transfusion, aim to mitigate these risks. Techniques such as pial synangiosis or encephaloduroarteriosynangiosis involve dissecting the superficial temporal artery from the scalp, passing it into the skull, and suturing it to the brain surface beyond the stenosis, thereby enhancing cerebral circulation [128,129,130]. While surgical revascularization is not universally endorsed as a definitive solution, when combined with exchange blood transfusion, it has demonstrated efficacy in reducing both initial and recurrent strokes in patients with moyamoya syndrome associated with SCD [131]. However, it’s important to note that the curative treatment for SCD remains allogeneic hematopoietic stem cell transplantation, although its specific impact on neurovascular outcomes requires further clarification.
Progression of cerebral infarcts and cerebral vasculopathy remains common despite regular blood transfusion therapy for secondary stroke prevention. Many patients advance to moyamoya vasculopathy, which significantly raises the risk of TIA, ischemic and hemorrhagic strokes [124], and cognitive decline [132]. Outcome studies on children and adults with sickle cell disease (SCD) who develop moyamoya and have experienced strokes are generally limited to small or single-center series, with little follow-up beyond five years [130,133,134,135,136,137,138,139]. No rigorous, prospective controlled trials have compared the benefits and risks of combining revascularization surgery with regular blood transfusion therapy against blood transfusion therapy alone for secondary stroke prevention in children and adults with SCD and moyamoya syndrome. Several studies have examined stroke incidence before and after revascularization surgery; however, these studies have intrinsic limitations. The highest recurrence rate for cerebral infarcts occurs within two years of the initial stroke, regardless of preventive treatment. Adverse outcomes primarily occurred within the first month after these procedures. Due to the heterogeneity of the five different neurosurgical procedures (pial synangiosis, encephalo-duro-arterio-myosynangiosis [EDAMS], encephalo-duro-arterio-synangiosis [EDAS], encephalo-myo-arterio-synangiosis, multiple burr holes) and the lack of uniform neurological assessments (surveillance MRI of the brain and neurological evaluation for infarct recurrence) during the follow-up period, no pooled analysis of these studies could be completed. Future studies aimed at assessing the additional benefits of surgical revascularization in SCD-related moyamoya syndrome for secondary stroke prevention should incorporate brain MRIs to detect silent cerebral infarcts, formal neurological assessments by neurologists to identify subtle deficits associated with overt strokes, and adjustment for the high-risk period of stroke recurrence. To date, none of the published studies on cerebral revascularization in SCD have included these strategies to enhance their scientific rigor, limiting the inferences that can be drawn about the potential benefits.
For individuals, both adults, and children, with SCD and moyamoya syndrome who have a history of stroke or TIA, the American Society of Hematology (ASH) guideline panel [88] suggests considering evaluation for revascularization surgery in addition to continuing regular blood transfusions, but this recommendation is conditional due to the very low certainty in the evidence regarding its effects. In this context the key points of the management are highlighted in Table 11.
In summary, the ASH guideline [88] provides a framework for considering revascularization surgery alongside regular blood transfusions in individuals with SCD, moyamoya syndrome, and a history of stroke or TIA. It advocates for careful evaluation by a multidisciplinary team and emphasizes the importance of long-term outcome monitoring to guide future treatment recommendations. One limitation is the different definitions used in different studies for intracranial vasculopathy, as outlined by Guilliams et al. [140], making extremely variable the prevalence estimates and hard the comparisons between studies.
3.5. Intracranial Aneurysms
Stroke is a prevalent complication in patients with SCD, affecting 30% to 40% of this population. These patients frequently visit hospitals due to vaso-occlusive headaches. However, when headaches are severe and unusual, they often signal hemorrhagic conditions. Additionally, in patients with hemoglobin levels below 8 to 10 g/dL, the reduced sensitivity of computed tomography (CT) scans can lead to the misdiagnosis of subarachnoid hemorrhage (SAH) caused by aneurysms [141]. Due to the rarity of the condition, intracranial aneurysms (IAs) in SCD patients have not been thoroughly studied, althout being often present some characteristics, such as their multiplicity and tendency to affect posterior circulation [117,142]. However, definitive diagnosis and treatment protocols remain underdeveloped. Improved detection technologies have revealed that the incidence of IAs in SCD patients is higher than previously thought. The presence of sickled cells complicates these aneurysms, resulting in clinical patterns that differ from those in the general population. Intracranial aneurysms in SCD patients, a specific type of aneurysm that poses a significant hemorrhagic stroke risk, exhibit unique characteristics and require tailored treatment approaches. A recent systematic review [143] addressed these issues, allowing to summarize the main features od SCD-associated aneurysms through collecting data on 111 patients (31 children and 80 adults) and 218 aneurysms. The average number of aneurysms per person was 1.9 in children and 2.0 in adults. The mean age of the study population was 27 years (range 5-54 years) with a male-to-female ratio of 0.96 (52:54). In the child subgroup, the male-to-female ratio was 1.9 (20:11), while in the adult subgroup it was 0.7 (32:43). Among the recorded hemoglobin types, 86.2% (75 out of 87 cases) were the SS type, known as sickle cell anemia. The most common presenting symptom was SAH, followed by incidental discovery. In the child cohort, 13 out of 31 (41.9%) aneurysms ruptured, whereas 62 out of 79 (78.5%) ruptured in the adult cohort. Regarding multiplicity, 50 out of 111 patients (45.0%) had multiple intracranial aneurysms, with 159 (73.0%) of the total 218 aneurysms occurring in these patients, averaging 3.2 aneurysms per person. Fifty-four of these 159 multiple aneurysms were located in the internal carotid artery. In both child and adult groups, most aneurysms were small (85.6%) and saccular (90.8%). Of the 218 aneurysms, 149 (68.3%) were in the anterior circulation and 69 (31.7%) were in the posterior circulation. The internal carotid artery was the most common location, with the posterior cerebral artery being the most involved site in the posterior circulation. Table 12 sumarizes thr main features of aneurysms in SCD.
4. Main neuroimaging Issues
Since the mid-1980s, MRI has played a crucial role in investigating brain pathology in SCD patients, particularly those presenting with acute neurological symptoms [145]. MRI techniques commonly used in clinical settings include T1-weighted, T2-weighted fluid attenuated inversion recovery (T2-FLAIR), T2*-weighted, susceptibility-weighted imaging, diffusion-weighted MRI, and time-of-flight angiography (MRA) and venography (MRV). In research settings, there has been a shift towards structural and hemodynamic quantitative approaches in MRI analysis. Techniques such as T1 mapping, diffusion imaging, perfusion imaging, quantitative susceptibility mapping (QSM), and functional MRI have been employed. The findings provided by conventional MRI and MRA techniques, together with other vascular imaging techniques, are the main focus of the present review [146].
4.1. Ischemic Stroke
In patients with SCD, infarctions typically occur in the territory of large intracranial vessels, particularly affecting the watershed regions of the deep white matter [55,147,148]. This vulnerability exists regardless of the presence of concurrent intracranial cerebral vasculopathy [149]. Silent cerebral infarction has a similar distribution pattern, with up to 90% of SCIs occurring in a small deep watershed white matter region that makes up only 5.6% of brain volume [72]. On MRI, SCI and overt ischemic stroke often appear indistinguishable. Several studies suggest that the differences in lesion size and location, rather than the underlying physiological mechanisms, may determine whether an ischemic event results in overt symptoms (ischemic stroke) or remains clinically silent (SCI) [149,150].
The role of MRI techniques in Acute Ischemic Events is not different in SCD as compared with non-SCD patients:
- T2-fluid attenuated inversion recovery (T2-FLAIR) MRI typically shows abnormalities hours to days after the event.
- DWI reveals hyperintense ischemic lesions within minutes, depicting early signs of ischemia.
- Apparent diffusion coefficient (ADC) maps show corresponding hypointense areas indicative of ischemic injury.
4.2. Silent Brain Infarctions and Small Vessel Disease
Silent cerebral infarction is more prevalent than overt stroke in individuals with SCD [109]. SCI is characterized by hyperintensities on brain MRI indicating infarction or ischemia without focal neurological symptoms and can occur as early as six months of age [151,152]. Research indicates that the prevalence of SCI reaches 25% by age six [153], 39% by age 18 [100], and 53% by young adulthood [28], with no evidence of a plateau. Despite being clinically “silent,” the co-operative study of SCD (CSSD) [154] first demonstrated that SCI significantly increases the risk of overt ischemic stroke by 14 times and found that 25% of children with SCI had new or enlarged lesions upon follow-up. SCI has also been linked to cognitive decline [155]. Subsequent studies have confirmed these findings, showing that SCI in children under five is associated with progressive ischemia, vasculopathy, academic difficulties, and a higher risk of overt ischemic stroke later in life [151]. Supporting the notion of progressive ischemia, a recent clinical review of 60 unselected adult cases revealed that 37% of SCI patients had multiple lesions [28].
The definition of a silent cerebral infarct-like lesion is an MRI signal abnormality of at least 3 mm in one dimension, visible in two planes on FLAIR or T2-weighted images (or similar images with 3-dimensional [3D] imaging), with no correlative neurological findings. Silent cerebral infarcts are common in children and adults with HbSS or HbSb0 thalassemia, with a prevalence of approximately 39% and 50% [18,100], respectively. An individual with SCD is diagnosed with a silent cerebral infarct if the following three features are present:
(1) no history of focal neurologic deficits;
(2) an MRI of the brain showing a T2-weighted image with FLAIR signal abnormality that is at least 3 mm in one dimension and visible in two planes (or similar image with 3D imaging); and
(3) a normal neurological examination, preferably conducted by a neurologist, or an abnormality on examination that cannot be explained by the location of the brain lesion or lesions [156].
Some considerations can be advanced:
1. This definition of silent cerebral infarct in children has been validated. Using a definition that requires a 5-mm size with corresponding T1-weighted hypointensity on MRI instead of 3-mm only would lead to significant misclassification, with fewer children being identified with silent cerebral infarcts [157]. A minimum size of 3 mm has been used in adult SCD studies and is predictive of infarct recurrence [158].
2. Diagnosing a silent cerebral infarct can be challenging if the radiologist is unfamiliar with the definition in SCD. This definition should not be extrapolated to include the common definition of lacunar strokes in the general population [159], which includes a T1 hypointensity in addition to a 5-mm FLAIR hyperintensity [157].
3. When available, imaging should be performed on a 3.0 T magnet instead of a 1.5 T to improve the detection of silent cerebral infarcts. As new FLAIR sequences are acquired via whole-brain 3D imaging with no gaps between slices, the requirement for imaging in two planes to confirm a silent infarct may not be necessary.
4. Imaging examples of silent cerebral infarcts and mimics, such as Virchow-Robin spaces and periventricular leukomalacia.
5. The Silent Cerebral Infarct Transfusion (SIT) Trial [156] demonstrated that blood transfusion therapy is superior to observation for secondary stroke prevention, showing a 56% relative risk reduction in cerebral infarct recurrence. However, the number needed to treat to prevent infarct recurrence with regular blood transfusions is 13. There are no controlled trial data demonstrating the noninferiority of hydroxyurea therapy to regular blood transfusion therapy for children or adults with silent cerebral infarcts.
Figure 1 shows an example of SCI and accompanying extracranial arteriopathy.
Quantitative MRI studies suggest that cerebral tissue injury in SCD extends beyond overt stroke, SCI, and large vessel disease. Reports indicate reduced cortical and subcortical gray matter volumes [160,161,162,163], as well as diminished subcortical white matter volumes [164,165,166]. Abnormal brain maturation patterns have also been observed [167,168,169]. Diffusion imaging studies show significant reductions in white matter integrity, particularly in the watershed regions of the centrum semiovale, affecting both SCI patients and those without MRI-defined lesions [170,171,172,173,174]. Research indicates that alterations in volumetric and structural integrity contribute to cognitive impairment in SCD patients, regardless of the presence of SCI [175].
4.3. Hemorrhagic Stroke
Approximately 25% of strokes in SCD are hemorrhagic, although sudden death events likely underestimate this statistic. Types of hemorrhages observed include intraparenchymal, intraventricular, subarachnoid, and occasionally subdural, detectable using T1, T2, T2*, and susceptibility-weighted MR sequences, as well as CT imaging. In older SCD patients, cerebral hemorrhage is commonly associated with aneurysm formation, which can be identified on MRA [176]. Aneurysms that rupture typically occur at vessel bifurcations, particularly in the vertebrobasilar circulation. Intraparenchymal bleeding may correlate with large vessel vasculopathy, especially in the presence of moyamoya formation. Other associated conditions include venous sinus thrombosis and reversible posterior leukoencephalopathy syndrome, which may also lead to hemorrhage. There are documented instances of epidural hematomas occurring in SCD patients without significant head trauma, likely due to hypervascular bone areas [177,178].
Acute blood appears hyperattenuating on CT scans, but this hyperattenuation decreases over time. MRI, particularly with FLAIR, gradient echo imaging, and susceptibility-weighted imaging (SWI), is at least as sensitive as CT in detecting acute blood [179,180,181]. However, MRI is significantly more sensitive than CT in detecting subacute and chronic blood products [182]. Local susceptibility-induced dephasing results in T2-weighted hypointensity within a parenchymal hematoma, except during the late subacute period when hemoglobin becomes extracellular. The development of methemoglobin in subacute blood causes T1 shortening. DWI interpretation can be challenging due to T2-shine through and T2-blackout effects, but true diffusion restriction may occur in acute hematoma where red blood cells are densely packed [183]. In the absence of significant mass effect, hematoma treatment is generally supportive and does not differ significantly from the management of intracranial hemorrhage in the general population [118].
4.4. Intracranial Arteriopathy
Vasculopathy is common in SCD patients, both with and without overt stroke and SCI. MRA frequently shows intra- and extra-cranial steno-occlusive arteriopathy, particularly involving the distal ICA and the proximal ACA and MCA [186]. This is often reported in patients with overt ischemic stroke and SCI [100]. The incidence of progressive stenosis with compensatory collateral vessel formation is as high as 30–40% among SCD patients with vasculopathy [124,187]. A multi-center pediatric study found that 38% of chronically transfused patients presented with new vessel stenosis or occlusion on follow-up MRA [91]. Despite aggressive hematological management, children with vasculopathy progression were 12 times more likely to develop new SCI or overt ischemic stroke compared to those without progression.
A sequential moyamoya-like model was proposed for SCD vasculopathy and stroke [188], where early ischemic events are linked to stenosis and later hemorrhagic events to the development and rupture of fragile collateral vessels. However, most SCD-related intracerebral and subarachnoid hemorrhages are associated with aneurysm rupture rather than collateral vessel rupture [143,189]. Beyond intracerebral aneurysms, tortuosity and ectasia are documented in both humans and animal models of SCD [190,191,192,193]. While aneurysms are not significantly linked to collateral vessel formation, they do appear to form in the context of progressive vasculopathy [176], with many patients having multiple aneurysms [117]. The simultaneous presence of overt ischemic stroke and/or SCI in most children with SCD-related ICH and/or aneurysm [194] suggests a concurrent development of pathologies underlying both ischemia and hemorrhage, potentially with shared mechanisms [195,196,197].
In patients with SCD, anterior cerebral circulation vasculopathy is well-documented, often visualized using DSA [198], which provides detailed vessel wall anatomy. MRA also plays a role in identifying abnormal vessels [102,126,200,201,202,203,204,205], though its effectiveness can be affected by flow disturbances or turbulence in the vessels. Old studies from non-SCD pediatric stroke cases suggests that MRA is effective in detecting large vessel disease, comparable to DSA [206]. However, in SCD patients, flow disturbances can complicate MRA assessments of vascular disease severity. These limitations come mainly from very old studies, as one [207] involving 21 children with SCD (14 with prior stroke, 7 asymptomatic), where MRA showed varying sensitivity in detecting occlusive lesions: 81% in the anterior circulation and 50% in the posterior circulation]. Specificity for detecting stenosis in ICA and MCA was high (86-100%). In addition, it is expected that the improving in technical issues over years might have increased the accuracy of MRA in comparison with old technologies. Time-of-flight MRA, commonly used in children, may sometimes overestimate stenosis due to intravoxel dephasing (turbulent flow), particularly problematic in SCD patients [208]. This dephasing has been observed even in patients initially showing normal cerebral angiography, potentially indicating early signs of vascular disease progression [207]. However, there’s a lack of large-scale studies examining whether intravoxel dephasing predicts future large vessel vascular disease development.
The prevalence of MRA abnormalities varies widely, influenced by study populations (including asymptomatic individuals and stroke patients) and varying definitions of abnormality [186]. Studies often define abnormality based on degrees of arterial stenosis (>50%), or use grading scales incorporating multiple vessel segments to assess severity [199]. Despite its non-invasive nature, MRA remains a crucial tool in screening asymptomatic SCD patients, detecting typical signal dropouts in the ICA, MCA, and ACA, and potentially revealing conditions like moyamoya syndrome.
In patients with SCD who have experienced at least one ischemic stroke, the prevalence of moyamoya syndrome is approximately 43% [102,124]. Additionally, 10.4% of asymptomatic children with SCD show evidence of large vessel vasculopathy on magnetic resonance MRA. The presence of moyamoya collaterals increases the risk of recurrent ischemic stroke by 2.4 times [102,124].
On T2-weighted imaging, serpiginous collateral vessels appear as thread-like flow-related signal voids and should be specifically sought out. Dedicated angiography, such as computed tomography angiography (CTA), is essential to accurately assess the degree of stenosis and the extent of collateral vessels. The “ivy sign,” indicative of proximal stenosis, can be observed as hyperintensity on FLAIR imaging and post-gadolinium enhancement, reflecting slow flow and compensatory dilation of pial vessels [209,210]. Figure 2 shows an example of intracranial occlusive arteriopathy in a SCD patient.
Recent findings also highlight extra-cranial vasculopathy in SCD, including dissections, narrowing, or occlusions in neck imaging, which are associated with SCI [100,211]. Tortuosity and ectasia in basilar and intracranial circulations are also common and linked to low hematocrit levels [190,192].
Aneurysm rupture is the most frequent cause of intracranial hemorrhage in SCD patients. The increased prevalence of aneurysms in SCD is attributed to sustained endothelial injury, which weakens vessel walls. Although similar pathophysiology is suggested for moyamoya vasculopathy [176], studies indicate little correlation between these two conditions, hinting at different underlying mechanisms. Saccular aneurysms are detected in approximately 6% of adults [176] and 4% of children [194] with SCD during routine imaging. SCD patients are more likely to have multiple aneurysms [144,176,194], with a higher propensity for these to occur in the posterior circulation (30% in SCD patients vs. 5-14% in the general population) [117,212]. The ICA is the most commonly affected, followed by the PCA. Most aneurysms in SCD patients are saccular, similar to the general population [143]. DSA is the gold standard for detecting intracranial aneurysms. However, due to the practical advantages of noninvasive imaging, time-of-flight magnetic resonance angiography (TOF MRA) and computed tomography angiography (CTA) are generally preferred, both offering over 95% accuracy for detecting unruptured intracranial aneurysms [213]. Close monitoring is crucial in SCD patients, as their aneurysms are more likely to rupture at smaller sizes compared to the general population. Women aged 30-39 with SCD are at the highest risk for subarachnoid hemorrhage due to aneurysm rupture [176].
In addition to saccular aneurysms, SCD often results in vertebrobasilar system dilatation, inversely related to hematocrit levels [193]. Vessel dilatation and tortuosity, responses to the hyperdynamic circulation caused by chronic anemia, are associated with an increased stroke risk, even in the absence of other brain abnormalities on MRI [193].
Given the higher risk of rupture at smaller sizes, treatment is recommended for aneurysms larger than 5 mm [214]. Traditional treatment involves surgical clipping, but endovascular coiling and stenting are becoming more common [215]. Aneurysm treatment in SCD patients is complicated by the risk of intraoperative sickling and hypercoagulability, increasing the risk of embolization after the insertion of permanent devices. To reduce periprocedural complications, it is essential to maintain preoperative hemoglobin S (HbS) levels below 30%, avoid hypoxia, and use nonionic contrast media [144]. An example is illustrated in Figure 3.
4.5. Cerebral Venous Drainage
The venous drainage anatomy in individuals with SCD differs from that of healthy controls, as observed in susceptibility-weighted imaging. SCD patients exhibit reduced cortical venous visibility [215], venular rarefaction, and a distinct distribution with fewer long venules and more short venules [217]. Additionally, they have a greater dural venous sinus diameter [218]. Although these anatomical differences are not linked to a history of stroke, venular rarefaction may be associated with memory impairment [217].
There have been several case reports of cerebral venous thrombosis (CVT) in SCD patients, often related to vaso-occlusive crises, transfusions, or acute respiratory illnesses [219,220,221]. CVT can lead to infarction or hemorrhage and is likely underdiagnosed in SCD, despite the potential for secondary edema to cause death [222]. Anticoagulation has been shown to reduce mortality and morbidity in the general adult population with CST. For acute neurological symptoms in SCD patients, neuroimaging should include CT and/or MR venography.
5. Transcranial Doppler and Stroke Prevention Strategie
The cornerstone of preventing ischemic strokes in children with SCD hinges on insights drawn from pediatric studies [11,106,223,224]. A critical part of this strategy is the use of transcranial Doppler (TCD) ultrasound screening, which pinpoints children at high risk for future strokes. The transformative Stroke Prevention (STOP) trial revealed that regular exchange blood transfusions could reduce the risk of symptomatic stroke by an impressive 92% in children with TCD velocities above 200 cm/s, compared to standard treatment [11]. When exchange transfusions aren’t feasible, hydroxycarbamide, administered at the highest tolerable dose, steps in as a formidable ally, lowering TCD velocities and stroke risk. Recent research even suggests that starting with a low-dose of hydroxycarbamide is beneficial for primary stroke prevention, mirroring the effectiveness of monthly transfusions seen in the STOP trial, all without added toxicity [225,226].
Currently, there is no validated tool to systematically assess stroke risk in adults with SCD. Research has shown that in adults with acute ischemic stroke, TCD values are not elevated, and no specific cut-off points have been established to aid in risk stratification. Additionally, TCD measurements have not proven beneficial in individuals with SCD over the age of 16 [227]. There is a significant gap in research regarding the efficacy of transfusions for primary ischemic stroke prevention in defined adult SCD populations. Furthermore, there are no studies addressing the management of adults who have transitioned from childhood to adulthood while receiving long-term transfusions for primary stroke prevention. Managing such patients requires a coordinated effort by a multidisciplinary team. This team should engage in thorough discussions with young adults and their caregivers to evaluate the risks and benefits of continuing transfusions, increasing the frequency of transfusion cycles, or switching to hydroxycarbamide.
According to the guidelines from the British [228] and American [88] Societies for Hematology, children and adults presenting with signs or symptoms indicative of an acute ischemic stroke should receive an immediate blood transfusion to maintain HbS levels below 30% and enhance tissue oxygen delivery via non-sickling red blood cells [229]. Exchange blood transfusion is the preferred initial treatment for acute ischemic stroke. However, in cases of severe anemia (defined as a hemoglobin level of 80 g/L) or when there are logistical challenges in arranging central line access and an apheresis team for exchange transfusion, a simple transfusion may be used as an immediate measure. Despite this, achieving and maintaining HbS levels below 30% is challenging with simple transfusions unless the patient is significantly anemic [230,231]. Simple transfusions, or top-up transfusions, involve adding blood without removing any, unlike exchange transfusions, which use more units of donor red cells and thus carry a higher risk of transfusion reactions and antibody formation. However, exchange transfusions also help mitigate the risk of iron overload by removing native red blood cells during the process. The main recommendations of the American Society for Hematology [88] are summarized in Table 13.
The Transcranial Doppler with Transfusions Changing to Hydroxyurea trial demonstrated that children with SCD and high transcranial Doppler velocities, but without MR angiography-defined vasculopathy, who had been on regular exchange transfusions for at least a year, could transition to hydroxyurea or hydroxycarbamide therapy at the maximum tolerated dose over approximately six months [232]. Hydroxycarbamide serves as an alternative to transfusions, particularly when the cost, availability, or burden of long-term exchange transfusions poses a challenge. It has been shown to be more effective than no exchange transfusion for secondary stroke prevention. However, the optimal dosage and timing for initiating hydroxycarbamide therapy in adults with SCD who have experienced ischemic or hemorrhagic strokes, silent cerebral ischemic lesions, or cerebral vasculopathy remain unclear. However, it should be pointed out that:
1. For children with abnormal TCD results, but without MRA-defined vasculopathy or SCI, who have received at least one year of transfusions, hydroxyurea therapy at the maximum tolerated dose should be considered as an alternative to regular blood transfusion therapy. This recommendation is based on the entry criteria of the TCD With Transfusions Changing to Hydroxyurea (TWiTCH) Trial [232].
2. For children with abnormal TCD results, MRA-defined vasculopathy, or SCI, regular blood transfusions should be continued indefinitely. This recommendation aligns with the exclusion criteria of the TWiTCH Trial [232] with a conditional recommendation. The suggested threshold for treatment is two TCD measurements with a time-averaged mean of the maximum velocity (TAMMV) of ≥200 cm/s or a single measurement >220 cm/s in the distal internal carotid artery (ICA) or proximal middle cerebral artery (MCA). Two measurements are required for values between 200 cm/s and 220 cm/s due to the ultrasonographer’s large coefficient of variation, which can be up to 12% within the same child measured only three hours apart [233]. Additionally, a significant intrasubject standard deviation of 14.9 cm/s was observed in a study of 812 children with HbSS and HbSb0 thalassemia who had at least two TCD examinations within six months without any intervention [234]. If Transcranial Color Coded Sonography (TCCS) technique is used for assessment, then two measurements with a time-averaged mean maximum (TAMX) of ≥185 cm/s or a single measurement >205 cm/s are required in the distal ICA or proximal MCA. Predictive values of the TCD measurements in other intracranial arteries have not been rigorously addressed and should not be used to classify children into high- and low-risk groups for future strokes.
Other considerations are that the threshold for hemolysis requiring regular TCD surveillance should be determined based on individual patient characteristics, considering factors such as hemoglobin level, reticulocyte count, and degree of hemolysis in relation to HbSS. Then, we could not define a laboratory threshold to determine who should undergo TCD. Additionally, there is no evidence available to demonstrate that children with HbSC should undergo TCD screening for primary stroke prevention.
However, the application of results from the Optimizing Primary Stroke Prevention in Sickle Cell Anemia (STOP) Trial [11] has significantly advanced the management of SCD. TCD screening, combined with regular blood transfusion therapy for those with abnormal TAMMV TCD measurements, is associated with a 92% reduction in stroke incidence compared with observation alone [11].
STOP 2 [223] demonstrated that for STOP participants who received transfusions for 30 months or longer and whose TCD measurements normalized, continued regular blood transfusions were necessary to prevent strokes or reversion to abnormal TCD measurements. Therefore, children with abnormal TCD measurements are presumed to have an indefinite risk of strokes. The results from STOP and STOP 2 trials underscore the clear benefit of regular blood transfusion compared with no transfusion (observation). STOP 2 excluded children with severe stenotic lesions on cerebral MRA. Given the extremely low incidence rate of strokes in the transfusion arm of the STOP Trial [11] (<1 stroke per 100 patient-years), no formal assessment of stroke risk factors in the treatment arm can be used to determine the subgroup of children likely to have a stroke while receiving regular blood transfusion therapy. In STOP 2 [223], among those randomly allocated to regular blood transfusion therapy, 21.6% had persistent abnormal TCD measurements with no stroke occurrences over a mean follow-up of 2.4 years.
The optimal interval for reassessment of children with conditional TCD measurements (170-199 cm/s) has not been determined, but reassessment is commonly done within six months, and often sooner. Both HbSS and HbSb0 thalassemia phenotypes were eligible for the STOP trials due to the clinical challenges of distinguishing HbSS from HbSb0 thalassemia using clinical laboratory values [235] and their inclusion in primary stroke prevention trials [11,223].
Children evaluated for abnormal TCD measurements should not have had a recent blood transfusion, as TCD velocities are associated with transfusions. Typically, TCD measurement should be done at least three months after the last transfusion and when the child is at their baseline state of health [236]. Regular blood transfusion therapy commonly requires iron chelation therapy to manage excessive iron stores from transfusions. If a child who meets the criteria for transitioning to hydroxyurea after one year of regular blood transfusion therapy, a discussion with the family should include whether hydroxyurea is preferable. Prior to transitioning from transfusion therapy to hydroxyurea, MRI of the brain should be performed to exclude silent cerebral ischemic lesions, and intracranial MRA should be completed to assess cerebral vasculopathy, as per the TWiTCH protocol [232].
Compound heterozygotes for HbS (excluding HbSC) deserve a separate consideration, in particular among children, because a small proportion are at high risk of stroke. The utility of TCD screening in these children is not well defined. Given the relationship between TCD values and hemoglobin levels in children with HbSS and the association between low hemoglobin levels and strokes, evidence suggests that TCD screening and treatment will likely prevent strokes in children with compound heterozygous SCD with evidence of hemolysis similar to children with HbSS [236]. Decisions regarding TCD screening and subsequent treatment for children with compound heterozygous SCD should be made on an individual basis. For children with HbSC, the risk of abnormal TCD measurements and stroke is lower than for those with HbSS.
In well-implemented primary stroke programs in high-income settings, less than 1% of children who receive TCD screening coupled with regular blood transfusion therapy for abnormal TCD measurements will have strokes [237]. For children with an abnormal TCD measurement, the risk of ischemic strokes is exceptionally high at 10.7 strokes per 100 patient-years [11]. This is much higher than the stroke incidence rate of 4.4 events per 100 patient-years in untreated adults with atrial fibrillation [238], highlighting the significant public health impact of preventing strokes in children with HbSS or HbSb0 thalassemia in both low- and high-income settings. This is of paramount relevance for involving the stakeholders in allowing a dedicated treatment for SCD patients in low-income countries [239,240,241].
There is currently limited evidence supporting the use of antiplatelet or anticoagulant therapy in SCD for the prevention of thrombotic events. Further research is needed to establish clear guidelines for these treatments in the context of SCD.
In cases where children or adults with SCD present with acute neurological deficits, including transient ischemic attacks, exchange blood transfusion is strongly recommended. The primary goal of this intervention is to decrease the percentage of sickled red blood cells to less than 30%, particularly if the neurological symptoms are related to sickle cell disease. This approach aims to improve oxygen delivery to tissues by increasing the proportion of normal, non-sickling red blood cells circulating in the bloodstream. The rationale behind using exchange blood transfusion lies in its ability to rapidly lower the concentration of sickle hemoglobin (HbS) and reduce the risk of further sickling-related complications, including ischemic events in the brain. This method involves removing and replacing a significant volume of the patient’s blood with donor red blood cells. By doing so, it not only dilutes the sickled cells but also reduces the overall burden of HbS in circulation. Exchange transfusion is preferred over simple transfusion in these acute scenarios because it allows for more effective control of HbS levels below the critical threshold of 30%. This targeted reduction is crucial in preventing ongoing sickle-related vascular occlusions that can lead to progressive neurological damage or stroke. Overall, in the management of acute neurological events in individuals with SCD, prompt initiation of exchange blood transfusion is advocated to mitigate the immediate risks associated with sickle cell-related ischemic complications and to optimize neurological outcomes. The main measures to prevent and manage ischemic stroke in SCD are summarized in Table 14.
5. Conclusions
Cerebrovascular disease in SCD patients is complex and requires a nuanced understanding of its unique characteristics, risk factors, and management strategies. Comprehensive, multidisciplinary approaches and further research are essential to improve outcomes for this vulnerable patient population. It is likely that different measures may be needed in children and adults, but in this last subgroup, some information is still lacking.
Author Contributions
Conceptualization, M.Z. and R.P.; methodology, M.Z.; data curation, all the autjors; writing—original draft preparation, M.Z., M.Q., I.C., and R.P.; writing—review and editing, all the authors.; supervision, F.M., F.V. and R.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–31.
- Thein SL. The molecular basis of β-thalassemia. Cold Spring Harb Perspect Med. 2013;3:a011700.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–9.
- Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21:37–47.
- Platt OS, Brambilla DJ, Rosse WF, et al.. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44. 10.1056/NEJM199406093302303.
- Ballas SK, Lusardi M: Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol. 2005, 79:17-25. 10.1002/ajh.20336.
- Platt OS, Thorington BD, Brambilla DJ, Milner PF, Rosse WF, Vichinsky E, Kinney TR: Pain in sickle cell disease. Rates and risk factors. N Engl J Med. 1991, 325:11-16. 10.1056/NEJM199107043250103.
- Ohene-Frempong K, Weiner SJ, Sleeper LA, et al.: Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998, 91:288-294.
- Hebbel RP, Osarogiagbon R, Kaul D: The endothelial biology of sickle cell disease: Inflammation and a chronic vasculopathy. Microcirculation. 2004, 11:129-151. 10.1080/10739680490278402.
- Ataga KI, Orringer EP: Hypercoagulability in sickle cell disease: a curious paradox. Am J Med. 2003, 115:721-728. 10.1016/j.amjmed.2003.07.011.
- Adams RJ, McKie VC, Hsu L, et al.: Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N Engl J Med. 1998, 339:5-11. 10.1056/NEJM199807023390102.
- DeBaun MR, Kirkham FJ: Central nervous system complications and management in sickle cell disease. Blood. 2016, 127:829-838. 10.1182/blood-2015-09-618579.
- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017 Jul 15;390(10091):311-323.
- Perrotta S, Russo G, et al.; Gruppo di Lavoro “Patologia del globulo rosso” Associazione Italiana Ematologia Oncologia Pediatrica. Linee-Guida per la gestione della Malattia Drepanocitica in età pediatrica in Italia-versione 3 (30 gennaio 2018). https://www.aieop.org › web › uploads › 2018/03P3.
- Kutlar A, Kanter J, Liles DK, et al. Effect of crizanlizumab on pain crises in subgroups of patients with sickle cell disease: A SUSTAIN study analysis. Am J Hematol. 2019 Jan;94(1):55-61.
- Matte A, Zorzi F, Mazzi F, et al. New Therapeutic Options for the Treatment of Sickle Cell Disease. Mediterr J Hematol Infect Dis. 2019;11(1):e2019002.
- Russo G, De Franceschi L, Colombatti R, et al. Current challenges in the management of patients with sickle cell disease - A report of the Italian experience. Orphanet J Rare Dis. 2019 May 30;14(1):120.
- De Franceschi L. La drepanocitosi: un problema emergente di salute pubblica. Prospettive in pediatria. Società Italiana di Pediatria. 2014; 44(174): 88-95.
- De Franceschi L, Graziadei G, Rigano P, et al. Raccomandazioni per la gestione del paziente adulto affetto da anemia falciforme. Società Italiana Talassemie ed Emoglobinopatie-SITE. Collana Scientifica S.I.T.E. n.2 2014.
- Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med. 2017 Jul 20;377(3):305.
- World Health Organization (WHO). Report by the Secretariat of the Fifty-ninth World Health Assembly A59/9, 2006.
- Colombatti R, Perrotta S, Samperi P et al. Italian Association of Pediatric Hematology-Oncology (AIEOP) Sickle Cell Disease Working Group. Organizing national responses for rare blood disorders: the Italian experience with sickle cell disease in childhood. Orphanet J Rare Dis 2013;8:169.
- Roberts I, de Montalembert M. Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe. Haematologica. 2007 Jul;92(7):865-71.
- Colombatti R, Casale M, Russo G. Disease burden and quality of life of in children with sickle cell disease in Italy: time to be considered a priority. Ital J Pediatr. 2021 Jul 29;47(1):163.
- De Franceschi L, Russo G, Sainati L. Raccomandazioni per lo screening neonatale nelle sindromi falciformi. Società Italiana Talassemie ed Emoglobinopatie- SITE. Collana Scientifica S.I.T.E. n. 5 2017.
- Niscola P, Sorrentino F, Scaramucci L, et al. Pain syndromes in sickle cell disease: an update. Pain Med. 2009 Apr;10(3):470-80.
- Dormandy E, James J, Inusa B, et al. How many people have sickle cell disease in the UK? J Public Health 2018;40:e291–5.
- Kassim AA, Pruthi S, Day M, et al. Silent cerebral infarcts and cerebral aneurysms are prevalent in adults with sickle cell anemia. Blood. 2016;127(16):2038-2040.
- Sacco RL, Kasner SE, Broderick JP, et al.; Council on Nutrition, Physical Activity and Metabolism. An updated definition of stroke for the 21st century: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44(7):2064-2089.
- Veluswamy S, Shah P, Denton CC, Chalacheva P, Khoo MC, Coates TD. Vaso-occlusion in sickle cell disease: is autonomic dysregulation of the microvasculature the trigger? J Clin Med. 2019;8:1690.
- Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH. Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med. 1980;302:992–995.
- Kaul DK, Fabry ME, Nagel RL. Vaso-occlusion by sickle cells: evidence for selective trapping of dense red cells. Blood. 1986;68:1162–1166.
- Weigand M, Gomez-Pastora J, Palmer A, Zborowski M, Desai P, Chalmers J. Continuous-flow magnetic fractionation of red blood cells based on hemoglobin content and oxygen saturation— clinical blood supply implications and sickle cell anemia treatment. Processes. 2022;10:927.
- Hirtz D, Kirkham FJ. Sickle cell disease and stroke. Pediatr Neurol. 2019;95:34–41.
- Gladwin MT, Sachdev V. Cardiovascular abnormalities in sickle cell disease. J Am Coll Cardiol. 2012;59:1123–1133.
- Chennupati R, Solga I, Wischmann P, et al. Chronic anemia is associated with systemic endothelial dysfunction. Front Cardiovasc Med. 2023;10.
- Ataga KI, Key NS. Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematology Am Soc Hematol Educ Program. 2007:91–96.
- Parikh T, Goti A, Yashi K, Ravikumar NPG, Parmar N, Dankhara N, Satodiya V. Pediatric sickle cell disease and stroke: a literature review. Cureus. 2023;15:0.
- Pavlakis SG, Bello J, Prohovnik I, et al. Brain infarction in sickle cell anemia: magnetic resonance imaging correlates. Ann Neurol. 1988;23:125–130.
- Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14:263–292.
- Kassim AA, DeBaun MR. Sickle cell disease, vasculopathy, and therapeutics. Annu Rev Med. 2013;64:451–466.
- Elsharawy MA, Moghazy KM, Shawarby MA. Atherosclerosis in sickle cell disease — a review. Int J Angiol. 2009;18:62–66.
- Prohovnik I, Pavlakis SG, Piomelli S, Bello J, Mohr JP, Hilal S, De Vivo DC. Cerebral hyperemia, stroke, and transfusion in sickle cell disease. Neurology. 1989;39:344–348.
- Stotesbury H, Kawadler JM, Hales PW, Saunders DE, Clark CA and Kirkham FJ (2019) Vascular Instability and Neurological Morbidity in Sickle Cell Disease: An Integrative Framework. Front. Neurol. 10:871. [CrossRef]
- Hakami F, Alhazmi E, Busayli WM, Althurwi S, Darraj AM, Alamir MA, Hakami A, Othman RA, Moafa AI, Mahasi HA, Madkhali MA. Overview of the Association Between the Pathophysiology, Types, and Management of Sickle Cell Disease and Stroke. Cureus. 2023 Dec 15;15(12):e50577. [CrossRef]
- Balkaran B, Char G, Morris JS, Thomas PW, Serjeant BE, Serjeant GR. Stroke in a cohort of patients with homozygous sickle cell disease. J Pediatr. 1992;120:360–366.
- DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012;119:4587–4596.
- Dowling MM, Noetzel MJ, Rodeghier MJ, et al. Headache and migraine in children with sickle cell disease are associated with lower hemoglobin and higher pain event rates but not silent cerebral infarction. J Pedatr. 2014;164:1175–1180.
- Solh Z, Taccone MS, Marin S, et al. Neurological presentations in sickle cell patients are not always stroke: a review of posterior reversible encephalopathy syndrome in sickle cell disease. Pediatr Blood Cancer. 2016;63:983–989.
- Vargas A, Testai FD. Posterior reversible encephalopathy syndrome in adult sickle-cell patients: case series and literature review. J Clin Neurosci. 2019;70:249–250.
- Adams RJ, McKie VC, Brambilla D, Carl E, Gallagher D, Nichols FT, et al. Stroke prevention trial in sickle cell anemia. Control Clin Trials. (1998) 19:110–29. [CrossRef]
- Hart RG, Kanter MC. Hematologic disorders and ischemic stroke. A selective review. Stroke. (1990) 21:1111–21. [CrossRef]
- Jabbarli R, Dinger TF, Pierscianek D, Oppong MD, Chen B, Dammann P, et al. Intracranial aneurysms in sickle cell disease. Curr Neurovasc Res. (2019) 16:63–76. [CrossRef]
- Strouse JJ, Hulbert ML, DeBaun MR, Jordan LC, Casella JF. Primary hemorrhagic stroke in children with sickle cell disease is associated with recent transfusion and use of corticosteroids. Pediatrics. (2006) 118:1916–24. [CrossRef]
- Switzer JA, Hess DC, Nichols FT, Adams RJ. Pathophysiology and treatment of stroke in sickle-cell disease: present and future. Lancet Neurol. (2006) 5:501–12. [CrossRef]
- de Araujo OMR, Ivo ML, Ferreira Júnior MA, Pontes ERJC, Bispo IMGP, Oliveira, et al. Survival and mortality among users and non-users of hydroxyurea with sickle cell disease. Rev Lat Am Enfermagem. (2015) 23:67– 73. [CrossRef]
- Powars D, Wilson B, Imbus C, Pegelow C, Allen J. The natural history of stroke in sickle cell disease. Am J Med. (1978) 65:461–71. [CrossRef]
- Ataga KI, Gordeuk VR, Agodoa I, Colby JA, Gittings K, Allen IE. Low hemoglobin increases risk for cerebrovascular disease, kidney disease, pulmonary vasculopathy, and mortality in sickle cell disease: a systematic literature review and meta-analysis. PLoS One. 2020;15:0.
- Strouse JJ, Lanzkron S, Urrutia V. The epidemiology, evaluation and treatment of stroke in adults with sickle cell disease. Expert Rev Hematol. 2011;4:597–606.
- Lopez-Vicente M, Ortega-Gutierrez S, Amlie-Lefond C, Torbey MT. Diagnosis and management of pediatric arterial ischemic stroke. J Stroke Cerebrovasc Dis. 2010;19:175–183.
- Belisário AR, Silva CM, Velloso-Rodrigues C, Viana MB. Genetic, laboratory and clinical risk factors in the development of overt ischemic stroke in children with sickle cell disease. Hematol Transfus Cell Ther. 2018;40:166–181.
- Kirkham FJ, Lagunju IA. Epidemiology of stroke in sickle cell disease. J Clin Med. 2021;10.
- Alakbarzade V, Maduakor C, Khan U, Khandanpour N, Rhodes E, Pereira AC. Cerebrovascular disease in sickle cell disease. Pract Neurol. 2023 Apr;23(2):131-138. [CrossRef]
- Makin SD, Doubal FN, Dennis MS, Wardlaw JM. Clinically confirmed stroke with negative diffusion-weighted imaging magnetic resonance imaging: longitudinal study of clinical outcomes, stroke recurrence, and systematic review. Stroke. 2015;46(11):3142-3148.
- Hulbert ML, Scothorn DJ, Panepinto JA, et al. Exchange blood transfusion compared with simple transfusion for first overt stroke is associated with a lower risk of subsequent stroke: a retrospective cohort study of 137 children with sickle cell anemia. J Pediatr. 2006;149(5):710-712.
- Guilliams KP, Fields ME, Ragan DK, et al. Red cell exchange transfusions lower cerebral blood flow and oxygen extraction fraction in pediatric sickle cell anemia. Blood. 2018;131(9):1012-1021.
- Juttukonda MR, Lee CA, Patel NJ, et al. Differential cerebral hemometabolic responses to blood transfusions in adults and children with sickle cell anemia. J Magn Reson Imaging. 2019;49(2):466-477.
- Prohovnik I, Hurlet-Jensen A, Adams R, De Vivo D, Pavlakis SG. Hemodynamic etiology of elevated flow velocity and stroke in sickle-cell disease. J Cereb Blood Flow Metab. 2009;29(4):803-810.
- Bush AM, Borzage MT, Choi S, et al. Determinants of resting cerebral blood flow in sickle cell disease. Am J Hematol. 2016;91(9):912-917.
- Scothorn DJ, Price C, Schwartz D, et al. Risk of recurrent stroke in children with sickle cell disease receiving blood transfusion therapy for at least five years after initial stroke. J Pediatr. 2002;140(3):348-354.
- Silva GS, Vicari P, Figueiredo MS, Carrete H Jr, Idagawa MH, Massaro AR. Brain magnetic resonance imaging abnormalities in adult patients with sickle cell disease: correlation with transcranial Doppler findings. Stroke. 2009;40(7):2408-2412.
- Ford AL, Ragan DK, Fellah S, et al. Silent infarcts in sickle cell disease occur in the border zone region and are associated with low cerebral blood flow. Blood. 2018;132(16):1714-1723.
- Kim HC. Red cell exchange: special focus on sickle cell disease. Hematology Am Soc Hematol Educ Program. 2014;2014:450-456.
- Swerdlow PS. Red cell exchange in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2006;2006:48-53.
- Faye BF, Sow D, Seck M, et al. Efficacy and safety of manual partial red cell exchange in the management of severe complications of sickle cell disease in a developing country. Adv Hematol. 2017;2017:3518402.
- Schmalzer EA, Lee JO, Brown AK, Usami S, Chien S. Viscosity of mixtures of sickle and normal red cells at varying hematocrit levels. Implications for transfusion. Transfusion. 1987;27(3):228-233.
- Alexy T, Pais E, Armstrong JK, Meiselman HJ, Johnson CS, Fisher TC. Rheologic behavior of sickle and normal red blood cell mixtures in sickle plasma: implications for transfusion therapy. Transfusion. 2006;46(6):912-918.
- Hurlet-Jensen AM, Prohovnik I, Pavlakis SG, Piomelli S. Effects of total hemoglobin and hemoglobin S concentration on cerebral blood flow during transfusion therapy to prevent stroke in sickle cell disease. Stroke. 1994;25(8):1688-1692.
- Saver JL. Time is brain--quantified. Stroke. 2006;37(1):263-266.
- Ferriero DM, Fullerton HJ, Bernard TJ, et al.; American Heart Association Stroke Council and Council on Cardiovascular and Stroke Nursing. Management of stroke in neonates and children: a scientific statement from the American Heart Association/American Stroke Association. Stroke. 2019;50(3): e51-e96.
- Brush LN, Monagle PT, Mackay MT, Gordon AL. Hypertension at time of diagnosis and long-term outcome after childhood ischemic stroke. Neurology. 2013;80(13):1225-1230.
- Rivkin MJ, Bernard TJ, Dowling MM, Amlie-Lefond C. Guidelines for urgent management of stroke in children [published correction appears in Pediatr Neurol. 2016;64:105]. Pediatr Neurol. 2016;56:8-17.
- Pegelow CH, Colangelo L, Steinberg M, et al. Natural history of blood pressure in sickle cell disease: risks for stroke and death associated with relative hypertension in sickle cell anemia. Am J Med. 1997;102(2):171-177.
- Aygun B, Wruck LM, Schultz WH, et al.; TCD With Transfusions Changing to Hydroxyurea (TWiTCH) Trial Investigators. Chronic transfusion practices for prevention of primary stroke in children with sickle cell anemia and abnormal TCD velocities. Am J Hematol. 2012;87(4):428-430.
- Vichinsky EP, Luban NL, Wright E, et al.; Stroke Prevention Trail in Sickle Cell Anemia. Prospective RBC phenotype matching in a stroke-prevention trial in sickle cell anemia: a multicenter transfusion trial. Transfusion. 2001;41(9):1086-1092.
- Files B, Brambilla D, Kutlar A, et al. Longitudinal changes in ferritin during chronic transfusion: a report from the Stroke Prevention Trial in Sickle Cell Anemia (STOP). J Pediatr Hematol Oncol. 2002;24(4):284-290.
- Adamkiewicz TV, Abboud MR, Paley C, et al. Serum ferritin level changes in children with sickle cell disease on chronic blood transfusion are nonlinear and are associated with iron load and liver injury. Blood. 2009;114(21):4632-4638.
- DeBaun MR, Jordan LC, King AA, et al. American Society of hematology 2020 guidelines for sickle cell disease: prevention, diagnosis, and treatment of cerebrovascular disease in children and adults. Blood Adv 2020;4:1554–88.
- Adams RJ, Cox M, Ozark SD, et al. Coexistent sickle cell disease has no impact on the safety or outcome of lytic therapy in acute ischemic stroke: findings from Get With The Guidelines-Stroke. Stroke. 2017;48(3):686-691.
- Pegelow CH, Adams RJ, McKie V, et al. Risk of recurrent stroke in patients with sickle cell disease treated with erythrocyte transfusions. J Pediatr. 1995; 126(6):896-899.
- Hulbert ML, McKinstry RC, Lacey JL, et al. Silent cerebral infarcts occur despite regular blood transfusion therapy after first strokes in children with sickle cell disease. Blood. 2011;117(3):772-779.
- Aygun B, Mortier NA, Kesler K, et al.; Stroke With Transfusions Changing to Hydroxyurea (SWiTCH) Trial Investigators. Therapeutic phlebotomy is safe in children with sickle cell anaemia and can be effective treatment for transfusional iron overload. Br J Haematol. 2015;169(2):262-266.
- Ware RE, Helms RW; SWiTCH Investigators. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood. 2012;119(17):3925-3932.
- Lagunju IA, Brown BJ, Sodeinde OO. Stroke recurrence in Nigerian children with sickle cell disease treated with hydroxyurea. Niger Postgrad Med J. 2013;20(3):181-187.
- Bernaudin F, Dalle JH, Bories D, et al.; Soci ’et ´e Française de Greffe de Moelle et de Therapie Cellulaire. Long-term event-free survival, chimerism and fertility outcomes in 234 patients with sickle-cell anemia younger than 30 years after myeloablative conditioning and matched-sibling transplantation in France. Haematologica. 2020;105(1):91-101.
- de la Fuente J, Dhedin N, Koyama T, et al. Haploidentical bone marrow transplantation with post-transplantation cyclophosphamide plus thiotepa improves donor engraftment in patients with sickle cell anemia: results of an international learning collaborative. Biol Blood Marrow Transplant. 2019;25(6):1197-1209.
- Fitzhugh CD, Hsieh MM, Taylor T, et al. Cyclophosphamide improves engraftment in patients with SCD and severe organ damage who undergo haploidentical PBSCT. Blood Adv. 2017;1(11):652-661.
- Jordan LC, Juttukonda MR, Kassim AA, et al. Haploidentical bone marrow transplantation improves cerebral hemodynamics in adults with sickle cell disease. Am J Hematol. 2019;94(6):E155-E158.
- King AA, McKinstry RC, Wu J, et al. Functional and radiologic assessment of the brain after reduced-intensity unrelated donor transplantation for severe sickle cell disease: Blood and Marrow Transplant Clinical Trials Network Study 0601. Biol Blood Marrow Transplant. 2019;25(5):e174-e178.
- Bernaudin F, Verlhac S, Arnaud C, et al. Chronic and acute anemia and extracranial internal carotid stenosis are risk factors for silent cerebral infarcts in sickle cell anemia. Blood 2015;125:1653–61.
- Estcourt LJ, Fortin PM, Hopewell S, et al. Interventions for preventing silent cerebral infarcts in people with sickle cell disease. Cochrane Database Syst Rev 2017;5:CD012389.
- Thangarajh M, Yang G, Fuchs D, et al. Magnetic resonance angiography-defined intracranial vasculopathy is associated with silent cerebral infarcts and glucose-6-phosphate dehydrogenase mutation in children with sickle cell anaemia. Br J Haematol 2012;159:352–9.
- DeBaun MR, Sarnaik SA, Rodeghier MJ, et al. Associated risk factors for silent cerebral infarcts in sickle cell anemia: low baseline hemoglobin, sex, and relative high systolic blood pressure. Blood 2012;119:3684–90.
- Dowling MM, Quinn CT, Plumb P, et al. Acute silent cerebral ischemia and infarction during acute anemia in children with and without sickle cell disease. Blood 2012;120:3891–7.
- Arkuszewski M, Krejza J, Chen R, et al. Sickle cell anemia: intracranial stenosis and silent cerebral infarcts in children with low risk of stroke. Adv Med Sci 2014;59:108–13.
- DeBaun MR, Gordon M, McKinstry RC, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med Overseas Ed 2014;371:699–710.
- Jordan LC, Kassim AA, Donahue MJ, et al. Silent infarct is a risk factor for infarct recurrence in adults with sickle cell anemia. Neurology 2018;91:e781–4.
- Rigano P, De Franceschi L, Sainati L, et al. Real-Life experience with hydroxyurea in sickle cell disease: a multicenter study in a cohort of patients with heterogeneous descent. Blood Cells Mol Dis 2018;69:82–9.
- DeBaun MR, Armstrong FD, McKinstry RC, Ware RE, Vichinsky E, Kirkham FJ. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012;119(20):4587-4596.
- Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118:19–27.
- Allali S, Taylor M, Brice J, de Montalembert M. Chronic organ injuries in children with sickle cell disease. Haematologica. 2021;106:1535–1544.
- Kamath SD, Pai MG. A case series of hemorrhagic neurological complications of sickle cell disease: multiple faces of an underestimated problem! Asian J Transfus Sci. 2021;15:241–246.
- Hariharan N, Brunson A, Mahajan A, Keegan TH, Wun T. Bleeding in patients with sickle cell disease: a population-based study. Blood Adv. 2020;4:793–802.
- Jordan LC, Hillis AE. Hemorrhagic stroke in children. Pediatr Neurol. 2007;36:73–80.
- An SJ, Kim TJ, Yoon BW. Epidemiology, risk factors, and clinical features of intracerebral hemorrhage: an update. J Stroke. 2017;19:3–10.
- Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 2017;127:750–760.
- Preul MC, Cendes F, Just N, et al. Intracranial aneurysms and sickle cell anemia: multiplicity and propensity for the vertebrobasilar Territory. Neurosurgery 1998;42:971–7.
- Nabavizadeh SA, Vossough A, Ichord RN, et al. Intracranial aneurysms in sickle cell anemia: clinical and imaging findings. J Neurointerv Surg 2016;8:434–40.
- Brandow AM, Liem RI. Advances in the diagnosis and treatment of sickle cell disease. J Hematol Oncol. 2022;15:20.
- Steiner T, Salman RA-S, Beer R, et al. European stroke organisation (ESO) guidelines for the management of spontaneous intracerebral hemorrhage. Int J Stroke 2014;9:840–55.
- Yamashita M, Tanaka K, Matsuo T, et al. Cerebral dissecting aneurysms in patients with moyamoya disease. Report of two cases. J Neurosurg 1983;58:120–5.
- Oka K, Yamashita M, Sadoshima S, et al. Cerebral haemorrhage in moyamoya disease at autopsy. Virchows Arch A Pathol Anat Histol 1981;392:247–61.
- Russell MO, Goldberg HI, Hodson A, et al. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood 1984;63:162–9.
- Dobson SR, Holden KR, Nietert PJ, et al. Moyamoya syndrome in childhood sickle cell disease: a predictive factor for recurrent cerebrovascular events. Blood 2002;99:3144–50.
- Dlamini N, Saunders DE, Bynevelt M, et al. Nocturnal oxyhemoglobin desaturation and arteriopathy in a pediatric sickle cell disease cohort. Neurology. 2017;89:2406–2412.
- Jacob M, Saunders DE, Sangeda RZ, et al. Cerebral infarcts and vasculopathy in Tanzanian children with sickle cell anemia. Pediatr Neurol. 2020;107:64–70.
- Kaushal M, Byrnes C, Khademian Z, et al. Examination of reticulocytosis among chronically transfused children with sickle cell anemia. PLoS One. 2016;11:e0153244.
- Griessenauer CJ, Lebensburger JD, Chua MH, et al. Encephaloduroarteriosynangiosis and encephalomyoarteriosynangiosis for treatment of moyamoya syndrome in pediatric patients with sickle cell disease. J Neurosurg Pediatr 2015;16:64–73.
- Smith ER, McClain CD, Heeney M, et al. Pial synangiosis in patients with moyamoya syndrome and sickle cell anemia: perioperative management and surgical outcome. Neurosurg Focus 2009;26:E10.
- Fryer RH, Anderson RC, Chiriboga CA, et al. Sickle cell anemia with moyamoya disease: outcomes after EDAS procedure. Pediatr Neurol 2003;29:124–30.
- Lawrence C, Webb J. Sickle cell disease and stroke: diagnosis and management. Curr Neurol Neurosci Rep 2016;16:27.
- Hogan AM, Kirkham FJ, Isaacs EB, Wade AM, Vargha-Khadem F. Intellectual decline in children with moyamoya and sickle cell anaemia. Dev Med Child Neurol. 2005;47(12):824-829.
- Alamri A, Hever P, Cheserem J, Gradil C, Bassi S, Tolias CM. Encephaloduroateriosynangiosis (EDAS) in the management of moyamoya syndrome in children with sickle cell disease. Br J Neurosurg. 2019;33(2):161-164;
- Hall EM, Leonard J, Smith JL, et al. Reduction in overt and silent stroke recurrence rate following cerebral revascularization surgery in children with sickle cell disease and severe cerebral vasculopathy. Pediatr Blood Cancer. 2016;63(8):1431-1437.
- Ng J, Thompson D, Lumley JP, Saunders DE, Ganesan V. Surgical revascularisation for childhood moyamoya. Childs Nerv Syst. 2012;28(7):1041-1048.
- Smith ER, McClain CD, Heeney M, Scott RM. Pial synangiosis in patients with moyamoya syndrome and sickle cell anemia: perioperative management and surgical outcome. Neurosurg Focus. 2009;26(4):E10.
- Winstead M, Sun PP, Martin K, et al. Encephaloduroarteriosynangiosis (EDAS) in young patients with cerebrovascular complications of sickle cell disease: Single-institution experience. Pediatr Hematol Oncol. 2017;34(2):100-106.
- Yang W, Xu R, Porras JL, et al. Effectiveness of surgical revascularization for stroke prevention in pediatric patients with sickle cell disease and moyamoya syndrome. J Neurosurg Pediatr. 2017;20(3):232-238.
- Hankinson TC, Bohman LE, Heyer G, et al. Surgical treatment of moyamoya syndrome in patients with sickle cell anemia: outcome following encephaloduroarteriosynangiosis. J Neurosurg Pediatr. 2008;1(3):211-216.
- Guilliams KP, Fields ME, Dowling MM. Advances in understanding ischemic stroke physiology and the impact of vasculopathy in children with sickle cell disease. Stroke. (2019) 50:266–73. [CrossRef]
- Lyon M, Jeter J, Lottenberg R. Approach to the diagnosis and treatment of acute subarachnoid hemorrhage in a patient with sickle cell disease. Am J Emerg Med. 2015;33:481.e3-481.e4.
- Brandao RA, de Carvalho GT, Reis BL, Bahia E, de Souza AA. Intracranial aneurysms in sickle cell patients: report of 2 cases and review of the literature. Surg Neurol. 2009;72:296-299.
- Yao Z, Li J, He M, You C. Intracranial Aneurysm in Patients with Sickle Cell Disease: A Systematic Review. World Neurosurg. 2017 Sep;105:302-313. [CrossRef]
- Saini S, Speller-Brown B, Wyse E, Meier ER, Carpenter J, Fasano RM, et al. Unruptured intracranial aneurysms in children with sickle cell disease: analysis of 18 aneurysms in 5 patients. Neurosurgery. 2015;76:531-538.
- Kirkham FJ. Therapy insight: stroke risk and its management in patients with sickle cell disease. Nat Clin Pract Neurol. 2007;3:264–278.
- Stotesbury H, Kawadler JM, Saunders DE, Kirkham FJ. MRI detection of brain abnormality in sickle cell disease. Expert Rev Hematol. 2021 May;14(5):473-491. [CrossRef]
- Adams RJ, Nichols FT, McKie V, McKie K, Milner P, Gammal TE. Cerebral infarction in sickle cell anemia: mechanism based on CT and MRI. Neurology. (1988) 38:1012–7. [CrossRef]
- Pavlakis SG, Bello J, Prohovnik I, Sutton M, Ince C, Mohr JP, et al. Brain infarction in sickle cell anemia: magnetic resonance imaging correlates. Ann Neurol. (1988) 23:125–30. [CrossRef]
- Guilliams KP, Fields ME, Ragan DK, Chen Y, Eldeniz C, Hulbert ML, et al. Large-vessel vasculopathy in children with sickle cell disease: a magnetic resonance imaging study of infarct topography and focal atrophy. Pediatr Neurol. (2017) 69:49–57. [CrossRef]
- Dowling MM, Kirkham FJ. Stroke in sickle cell anaemia is more than stenosis and thrombosis: the role of anaemia and hyperemia in ischaemia. Br J Haematol. (2017) 176:151–3. [CrossRef]
- Cancio MI, Helton KJ, Schreiber JE, Smeltzer MP, Kang G, Wang WC. Silent cerebral infarcts in very young children with sickle cell anaemia are associated with a higher risk of stroke. Br J Haematol. (2015) 171:120–9. [CrossRef]
- Wang WC, Langston JW, Steen RG, Wynn LW, Mulhern RK, Wilimas JA, et al. Abnormalities of the central nervous system in very young children with sickle cell anemia. J Pediatr. (1998) 132:994–8. [CrossRef]
- Kwiatkowski JL, Zimmerman RA, Pollock AN, Seto W, Smith-Whitley K, Shults J, et al. Silent infarcts in young children with sickle cell disease. Br J Haematol. (2009) 146:300–5. [CrossRef]
- Pegelow CH, Macklin EA, Moser FG, Wang WC, Bello JA, Miller ST, et al. Longitudinal changes in brain magnetic resonance imaging findings in children with sickle cell disease. Blood. (2002) 99:3014–8. [CrossRef]
- Wang W, Enos L, Gallagher D, Thompson R, Guarini L, Vichinsky E, et al. Neuropsychologic performance in school-aged children with sickle cell disease: a report from the cooperative study of sickle cell disease. J Pediatr. (2001) 139:391–7. [CrossRef]
- DeBaun MR, Gordon M, McKinstry RC, et al. Controlled trial of transfusions for silent cerebral infarcts in sickle cell anemia. N Engl J Med. 2014;371(8):699-710.
- Choudhury NA, DeBaun MR, Rodeghier M, King AA, Strouse JJ, McKinstry RC. Silent cerebral infarct definitions and full-scale IQ loss in children with sickle cell anemia. Neurology. 2018;90(3):e239-e246.
- Jordan LC, Kassim AA, Donahue MJ, et al. Silent infarct is a risk factor for infarct recurrence in adults with sickle cell anemia. Neurology. 2018;91(8):e781-e784.
- Vichinsky EP, Neumayr LD, Gold JI, et al.; Neuropsychological Dysfunction and Neuroimaging Adult Sickle Cell Anemia Study Group. Neuropsychological dysfunction and neuroimaging abnormalities in neurologically intact adults with sickle cell anemia. JAMA. 2010;303(18):1823-1831.
- Kawadler JM, Clayden JD, Kirkham FJ, Cox TC, Saunders DE, Clark CA. Subcortical and cerebellar volumetric deficits in paediatric sickle cell anaemia. Br J Haematol. (2013) 163:373–6. [CrossRef]
- Kim JA, Leung J, Lerch JP, Kassner A. Reduced cerebrovascular reserve is regionally associated with cortical thickness reductions in children with sickle cell disease. Brain Res. (2016) 1642:263–9. [CrossRef]
- Kirk GR, Haynes MR, Palasis S, Brown C, Burns TG, McCormick M, et al. Regionally specific cortical thinning in children with sickle cell disease. Cereb Cortex. (2009) 19:1549–56. [CrossRef]
- Mackin RS, Insel P, Truran D, Vichinsky EP, Neumayr LD, Armstrong FD, et al. Neuroimaging abnormalities in adults with sickle cell anemia: associations with cognition. Neurology. (2014) 82:835–41. [CrossRef]
- Baldeweg T, Hogan AM, Saunders DE, Telfer P, Gadian DG, Vargha-Khadem F, et al. Detecting white matter injury in sickle cell disease using voxel-based morphometry. Ann Neurol. (2006) 59:662–72. [CrossRef]
- Choi S, Bush AM, Borzage MT, Joshi AA, Mack WJ, Coates TD, et al. Hemoglobin and mean platelet volume predicts diffuse T1-MRI white matter volume decrease in sickle cell disease patients. NeuroImage Clin. (2017) 15:239–46. [CrossRef]
- Schatz J, Buzan R. Decreased corpus callosum size in sickle cell disease: relationship with cerebral infarcts and cognitive functioning. J Int Neuropsychol Soc. (2006) 12:24–33. [CrossRef]
- Chen R, Arkuszewski M, Krejza J, Zimmerman RA, Herskovits EH, Melhem ER. A prospective longitudinal brain morphometry study of children with sickle cell disease. AJNR Am J Neuroradiol. (2015) 36:403–10. [CrossRef]
- Darbari DS, Eigbire-Molen O, Ponisio MR, Milchenko MV, Rodeghier MJ, Casella JF, et al. Progressive loss of brain volume in children with sickle cell anemia and silent cerebral infarct: a report from the silent cerebral infarct transfusion trial. Am J Hematol. (2018) 93:E406–8. [CrossRef]
- Kawadler JM, Clark CA, McKinstry RC, Kirkham FJ. Brain atrophy in paediatric sickle cell anaemia: findings from the silent infarct transfusion (SIT) trial. Br J Haematol. (2017) 177:151–3. [CrossRef]
- Balci A, Karazincir S, Beyoglu Y, Cingiz C, Davran R, Gali E, et al. Quantitative brain diffusion-tensor MRI findings in patients with sickle cell disease. AJR Am J Roentgenol. (2012) 198:1167–74. [CrossRef]
- Chai Y, Coloigner J, Qu X, Choi S, Bush A, Borzage M, et al. Tract specific analysis in patients with sickle cell disease. Proc SPIE Int Soc Opt Eng. (2015) 9681:968108. [CrossRef]
- Choi S, Bush AM, Borzage M, Joshi A, Coates TD, Leahy R, et al. Regional susceptibility to chronic anemia in WM microstructure using diffusion tensor imaging. Blood. (2016) 128:3640.
- Kawadler JM, Kirkham FJ, Clayden JD, Hollocks MJ, Seymour EL, Edey R, et al. White matter damage relates to oxygen saturation in children with sickle cell anemia without silent cerebral infarcts. Stroke. (2015) 46:1793–9. [CrossRef]
- Sun B, Brown RC, Hayes L, Burns TG, Huamani J, Bearden DJ, et al. White matter damage in asymptomatic patients with sickle cell anemia: screening with diffusion tensor imaging. AJNR Am J Neuroradiol. (2012) 33:2043–9. [CrossRef]
- Stotesbury H, Kirkham FJ, Kölbel M, Balfour P, Clayden JD, Sahota S, et al. White matter integrity and processing speed in sickle cell anemia. Neurology. (2018) 90:e2042–50. [CrossRef]
- Birkeland P, Gardner K, Kesse-Adu R, et al. Intracranial aneurysms in sickle-cell disease are associated with the hemoglobin SS genotype but not with moyamoya syndrome. Stroke. 2016;47:1710–1713.
- Hamm J, Rathore N, Lee P, et al. Cranial epidural hematomas: a case series and literature review of this rare complication associated with sickle cell disease. Pediatr Blood Cancer. 2017;64:64.
- Saha B, Saha A. spontaneous epidural hemorrhage in sickle cell disease, Are they all the same? A case report and comprehensive review of the literature. Case Rep Hematol. 2019:8974580.
- Mitchell P, Wilkinson I, Hoggard N, et al. Detection of subarachnoid haemorrhage with magnetic resonance imaging. J Neurol Neurosurg Psychiatry 2001;70:205-11.
- Chalela JA, Kidwell CS, Nentwich LM, et al. Magnetic resonance imaging and computed tomography in emergency assessment of patients with suspected acute stroke: a prospective comparison. Lancet (London, England) 2007;369:293-8.
- Verma RK, Kottke R, Andereggen L, et al. Detecting subarachnoid hemorrhage: comparison of combined FLAIR/SWI versus CT. Eur J Radiol 2013;82:1539-45.
- Kidwell CS, Chalela JA, Saver JL, et al. Comparison of MRI and CT for detection of acute intracerebral hemorrhage. JAMA 2004;292:1823-30.
- Kang BK, Na DG, Ryoo JW, et al. Diffusion-weighted MR imaging of intracerebral hemorrhage. Korean J Radiol 2001:2;183-91.
- Kija EN, Saunders DE, Munubhi E, et al. Transcranial Doppler and magnetic resonance in Tanzanian children with sickle cell disease. Stroke 2019;50:1719-26.
- Novelli EM, Elizabeth Sarles C, Jay Aizenstein H, et al. Brain venular pattern by 7T MRI correlates with memory and haemoglobin in sickle cell anaemia. Psychiatry Res 2015;233:18-22.
- Guilliams KP, Fields ME, Dowling MM. Advances in understanding ischemic stroke physiology and the impact of vasculopathy in children with sickle cell disease. Stroke. (2019) 50:266–73. [CrossRef]
- Seeler RA, Royal JE, Powe L, Goldberg HR. Moyamoya in children with sickle cell anemia and cerebrovascular occlusion. J Pediatr. (1978) 93:808–10. [CrossRef]
- Powars D, Adams RJ, Nichols FT, Milner P, Charache S, Sarnaik S. Delayed intracranial hemorrhage following cerebral infarction in sickle cell anemia. J Assoc Acad Minor Phys. (1990) 1:79–82.
- Kassim AA, Galadanci NA, Pruthi S, DeBaun MR. How I treat and manage strokes in sickle cell disease. Blood. (2015) 125:3401–10. [CrossRef]
- Buch K, Arya R, Shah B, Nadgir RN, Saito N, Qureshi MM, et al. Quantitative analysis of extracranial arterial tortuosity in patients with sickle cell disease. J Neuroimaging. (2017) 27:421–27. [CrossRef]
- Hyacinth HI, Sugihara CL, Spencer TL, Archer DR, Shih AY. Higher prevalence of spontaneous cerebral vasculopathy and cerebral infarcts in a mouse model of sickle cell disease. J Cereb Blood Flow Metab. (2019) 39:342–51. [CrossRef]
- Steen RG, Langston JW, Ogg RJ, Manci E, Mulhern RK, Wang W. Ectasia of the basilar artery in children with sickle cell disease: relationship to hematocrit and psychometric measures. J Stroke Cerebrovasc Dis. (1998) 7:32–43. [CrossRef]
- Steen RG, Reddick WE, Glass JO, Wang WC. Evidence of cranial artery ectasia in sickle cell disease patients with ectasia of the basilar artery. J Stroke Cerebrovasc Dis. (1998) 7:330–8. [CrossRef]
- Kossorotoff M, Brousse V, Grevent D, Naggara O, Brunelle F, Blauwblomme T, et al. Cerebral haemorrhagic risk in children with sickle-cell disease. Dev Med Child Neurol. (2015) 57:187–93. [CrossRef]
- Koshy M, Thomas C, Goodwin J. Vascular lesions in the central nervous system in sickle cell disease (neuropathology). J Assoc Acad Minor Phys. (1990) 1:71–8. 50.
- Oyesiku NM, Barrow DL, Eckman JR, Tindall SC, Colohan AR. Intracranial aneurysms in sickle-cell anemia: clinical features and pathogenesis. J Neurosurg. (1991) 75:356–63. [CrossRef]
- Connes P, Verlhac S, Bernaudin F. Advances in understanding the pathogenesis of cerebrovascular vasculopathy in sickle cell anaemia. Br J Haematol. (2013) 161:484–98. [CrossRef]
- Russell MO, Goldberg HI, Hodson A, et al. Effect of transfusion therapy on arteriographic abnormalities and on recurrence of stroke in sickle cell disease. Blood. 1984;63:162–169.
- Helton KJ, Adams RJ, Kesler KL, et al. Magnetic resonance imaging/angiography and transcranial Doppler velocities in sickle cell anemia: results from the SWiTCH trial. Blood. 2014;124:891–898.
- Dlamini N, Saunders DE, Bynevelt M, et al. Nocturnal oxyhemoglobin desaturation and arteriopathy in a pediatric sickle cell disease cohort. Neurology. 2017;89:2406–2412.
- Guilliams KP, Fields ME, Ragan DK, et al. Large-vessel vasculopathy in children with sickle cell disease: a magnetic resonance imaging study of infarct topography and focal atrophy. Pediatric Neurology. 2017;69:49–57.
- Montanaro M, Colombatti R, Pugliese M, et al. Intellectual function evaluation of first generation immigrant children with sickle cell disease: the role of language and sociodemographic factors. Ital J Pediatr. 2013;39:36.
- Nottage KA, Ware RE, Aygun B, et al. Hydroxycarbamide treatment and brain MRI/MRA findings in children with sickle cell anaemia. Br J Haematol. 2016;175:331–338.
- Green NS, Munube D, Bangirana P, et al. Burden of neurological and neurocognitive impairment in pediatric sickle cell anemia in Uganda (BRAIN SAFE): a cross-sectional study. BMC Pediatr. 2019;19:381.
- Kija EN, Saunders DE, Munubhi E, et al. Transcranial doppler and magnetic resonance in Tanzanian children with sickle cell disease. Stroke. 2019;50:1719–1726.
- Husson B, Rodesch G, Lasjaunias P, et al. Magnetic resonance angiography in childhood arterial brain infarcts: a comparative study with contrast angiography. Stroke. 2002;33:1280–1285.
- Kandeei AY, Zimmerman RA, Ohcnc-Frempong K, et al. Comparison of magnetic resonance angiography and conventional angiography in sickle cell disease: clinical significance and reliability. Diagnostic Neuroradiol. 199638:409-16.
- Husson B, Lasjaunias P. Radiological approach to disorders of arterial brain vessels associated with childhood arterial stroke - A comparison between MRA and contrast angiography. Pediatr Radiol. 2004;34:10–15.
- Mori N, Mugikura S, Higano S, et al. The leptomeningeal “ivy sign” on fluid-attenuated inversion recovery MR imaging in moyamoya disease: a sign of decreased cerebral vascular reserve? Am J Neuroradiol 2009;30:930-5.
- Maeda M, Tsuchida C. “Ivy sign” on fluid-attenuated inversion recovery images in childhood moyamoya disease. AJNR Am J Neuroradiol 1999;20:1836-8.
- Telfer PT, Evanson J, Butler P, et al. Cervical carotid artery disease in sickle cell anemia: clinical and radiological features. Blood. 2011;118:6192–6199.
- Rinne J, Hernesniemi J, Puranen M, et al. Multiple intracranial aneurysms in a defined population: prospective angiographic and clinical study. Neurosurgery 1994;35:803-8.
- Sailer AMH, Wagemans BAJM, Nelemans PJ, et al. Diagnosing intracranial aneurysms with MR angiography: systematic review and meta-analysis. Stroke 2014;45:119-26.
- Buonanno FS, Schmahmann JD, Romero JM, et al. Case records of the Massachusetts General Hospital. Case 10–2016. A 22-year-old man with sickle cell disease, headache, and difficulty speaking. N Engl J Med 2016;374:1265-7.
- Gallas S, Tuilier T, Ebrahiminia V, et al. Intracranial aneurysms in sickle cell disease: aneurysms characteristics and modalities of endovascular approach to treat these patients. J Neuroradiol 2019;47:221-6.
- Winchell AM, Taylor BA, Song R, et al. Evaluation of SWI in children with sickle cell disease. Am J Neuroradiol. 2014;39:1016–1021.
- Novelli EM, Sarles C, Aizenstein J, et al. Brain venular pattern by 7T MRI correlates with memory and haemoglobin in sickle cell anaemia. Psychiatry Res – Neuroimaging. 2015;233:18–22.
- Adler K, Reghunathan A, Hutchison LH, et al. Dural venous sinus diameters in children with sickle cell disease: correlation with history of stroke in a case-control study. South Med J. 2016;109:511–515.
- Ciurea SO, Thulborn KR, Gowhari M. Dural venous sinus thrombosis in a patient with sickle cell disease: case report and literature review. Am J Hematol 2006;81:290–293.
- Sidani CA, Ballourah W, El Dassouki M, et al. Venous sinus thrombosis leading to stroke in a patient with sickle cell disease on hydroxyurea and high hemoglobin levels: treatment with thrombolysis. Am J Hematol. 2008;83:818–820.
- Wang MK, Shergill R, Jefkins M, et al. A sickle cell disease patient with dural venous sinus thrombosis: a case report and literature review. Hemoglobin. 2019;43:193–197.
- Sébire G, Tabarki B, Saunders DE, et al. Cerebral venous sinus thrombosis in children: risk factors, presentation, diagnosis and outcome. Brain. 2005;128:477–489.
- Adams RJ, Brambilla D. Optimizing primary stroke prevention in sickle cell anemia trial I. discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med 2005;353:2769–78.
- Abboud MR, Yim E, Musallam KM, et al. Discontinuing prophylactic transfusions increases the risk of silent brain infarction in children with sickle cell disease: data from stop II. Blood 2011;118:894–8.
- Rankine-Mullings A, Reid M, Soares D, et al. Hydroxycarbamide treatment reduces transcranial Doppler velocity in the absence of transfusion support in children with sickle cell anaemia, elevated transcranial Doppler velocity, and cerebral vasculopathy: the EXTEND trial. Br J Haematol 2021;195:612–20.
- Abdullahi SU, Jibir BW, Bello-Manga H, et al. Hydroxyurea for primary stroke prevention in children with sickle cell anaemia in Nigeria (spring): a double-blind, multicentre, randomised, phase 3 trial. Lancet Haematol 2022;9:e26–37.
- Valadi N, Silva GS, Bowman LS, et al. Transcranial Doppler ultrasonography in adults with sickle cell disease. Neurology 2006;67:572–4.
- Haematology GBSf. An audit of compliance with the British Society for haematology (BSH) guideline on red cell transfusion in sickle cell disease (SCD). Part II: indications for transfusion; 2016.
- Lawrence C, Webb J. Sickle cell disease and stroke: diagnosis and management. Curr Neurol Neurosci Rep 2016;16:27.
- Mack AK, Thompson AA. Primary and secondary stroke prevention in children with sickle cell disease. J Pediatr Health Care 2017;31:145–54.
- Chou ST, Fasano RM. Management of patients with sickle cell disease using transfusion therapy: guidelines and complications. Hematol Oncol Clin North Am 2016;30:591–608.
- Ware RE, Davis BR, Schultz WH, et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial Doppler flow velocities in children with sickle cell anaemia—TCD with transfusions changing to hydroxyurea (twitch): a multicentre, open-label, phase 3, non-inferiority trial. The Lancet 2016;387:661–70.
- Galadanci NA, Umar Abdullahi S, Vance LD, et al. Feasibility trial for primary stroke prevention in children with sickle cell anemia in Nigeria (SPIN trial). Am J Hematol. 2017;92(8):780-788.
- Brambilla DJ, Miller ST, Adams RJ. Intra-individual variation in blood flow velocities in cerebral arteries of children with sickle cell disease. Pediatr Blood Cancer. 2007;49(3):318-322.
- Day ME, Rodeghier M, Driggers J, Bean CJ, Volanakis EJ, DeBaun MR. A significant proportion of children of African descent with HbSb0 thalassaemia are inaccurately diagnosed based on phenotypic analyses alone. Br J Haematol. 2019;185(1):153-156.
- Venketasubramanian N, Prohovnik I, Hurlet A, Mohr JP, Piomelli S. Middle cerebral artery velocity changes during transfusion in sickle cell anemia. Stroke. 1994;25(11):2153-2158.
- Enninful-Eghan H, Moore RH, Ichord R, Smith-Whitley K, Kwiatkowski JL. Transcranial Doppler ultrasonography and prophylactic transfusion program is effective in preventing overt stroke in children with sickle cell disease. J Pediatr. 2010;157(3):479-484.
- Gage BF, Waterman AD, Shannon W, Boechler M, Rich MW, Radford MJ. Validation of clinical classification schemes for predicting stroke: results from the National Registry of Atrial Fibrillation. JAMA. 2001;285(22):2864-2870.
- Kilonzi, M.; Mlyuka, H.J.; Felician, F.F.; Mwakawanga, D.L.; Chirande, L.; Myemba, D.T.; Sambayi, G.; Mutagonda, R.F.; Mikomangwa, W.P.; Ndunguru, J.; et al. Barriers and Facilitators of Use of Hydroxyurea among Children with Sickle Cell Disease: Experiences of Stakeholders in Tanzania. Hemato 2021, 2, 713–726. [CrossRef]
- Fome, A.D.; Sangeda, R.Z.; Balandya, E.; Mgaya, J.; Soka, D.; Tluway, F.; Masamu, U.; Nkya, S.; Makani, J.; Mmbando, B.P. Hematological and Biochemical Reference Ranges for the Population with Sickle Cell Disease at Steady State in Tanzania. Hemato 2022, 3, 82-97. [CrossRef]
- Sharma, A.; Dahiya, A.; Alavi, A.; Woldie, I.; Sharma, A.; Karson, J.; Singh, V. Patterns of Blood Transfusion in Sickle Cell Disease Hospitalizations. Hemato 2024, 5, 26-34. [CrossRef]
Figure 1.
The brain MRI (panel a) and extracranial CTA (panel b and c) of a patient with heterozygous HbS is proposed. In the axial FLAIR slices (panel a) white matter hypertntensities are present with a dotted pattern. In the MIP/MPT reconstructed CTA of the right (panel b) and left (panel c) internal carotid artery an irregular, dysplasic pattern is well evident.
Figure 1.
The brain MRI (panel a) and extracranial CTA (panel b and c) of a patient with heterozygous HbS is proposed. In the axial FLAIR slices (panel a) white matter hypertntensities are present with a dotted pattern. In the MIP/MPT reconstructed CTA of the right (panel b) and left (panel c) internal carotid artery an irregular, dysplasic pattern is well evident.
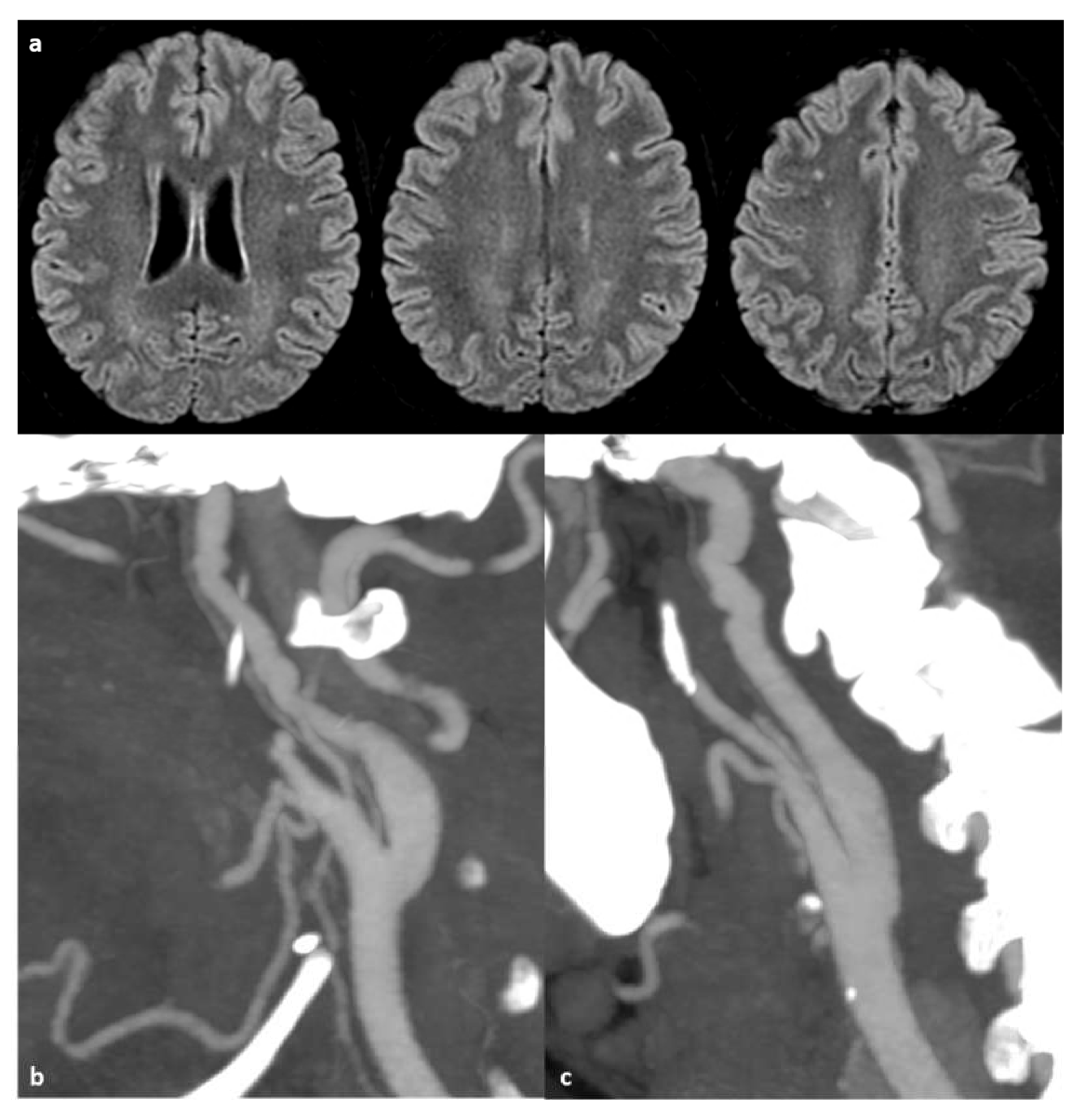
Figure 2.
Intracranial and extracranial arteriopathy in a 65 years old woman with a compound heterozygosis (HbSC), investigated using several techniques: - a: brain MRI with axial FLAIR slices and the association of ivy sign and spaghetti sign on the left hemisphere. - b: TOF-MRA in coronal view reconstructed with MIP/MPR showing the flow gap in the left MCA and a network of tiny tortuous vessel around the ICA terminus on the right side. - c: left carotid angiogram with a severe atheromasic stenosis on the internal carotid artery. - d and e: left common carotid artery angiogram in lateral view (d) and in posterior-anterior view (e), showing the known intracranial arteriopathy of M1 MCA with the development of collaterals coming from the external carotid artery with an artero-venous shunt feeded by the petrosquamous branch of the left middle meningeal artery. - f and g: right internal carotid artery angiogram in posterior-anterior view in early (f) and late (g) arterial phase with the collateralization through the anterior cerebral artery feeding a portion of the left MCA territory.
Figure 2.
Intracranial and extracranial arteriopathy in a 65 years old woman with a compound heterozygosis (HbSC), investigated using several techniques: - a: brain MRI with axial FLAIR slices and the association of ivy sign and spaghetti sign on the left hemisphere. - b: TOF-MRA in coronal view reconstructed with MIP/MPR showing the flow gap in the left MCA and a network of tiny tortuous vessel around the ICA terminus on the right side. - c: left carotid angiogram with a severe atheromasic stenosis on the internal carotid artery. - d and e: left common carotid artery angiogram in lateral view (d) and in posterior-anterior view (e), showing the known intracranial arteriopathy of M1 MCA with the development of collaterals coming from the external carotid artery with an artero-venous shunt feeded by the petrosquamous branch of the left middle meningeal artery. - f and g: right internal carotid artery angiogram in posterior-anterior view in early (f) and late (g) arterial phase with the collateralization through the anterior cerebral artery feeding a portion of the left MCA territory.
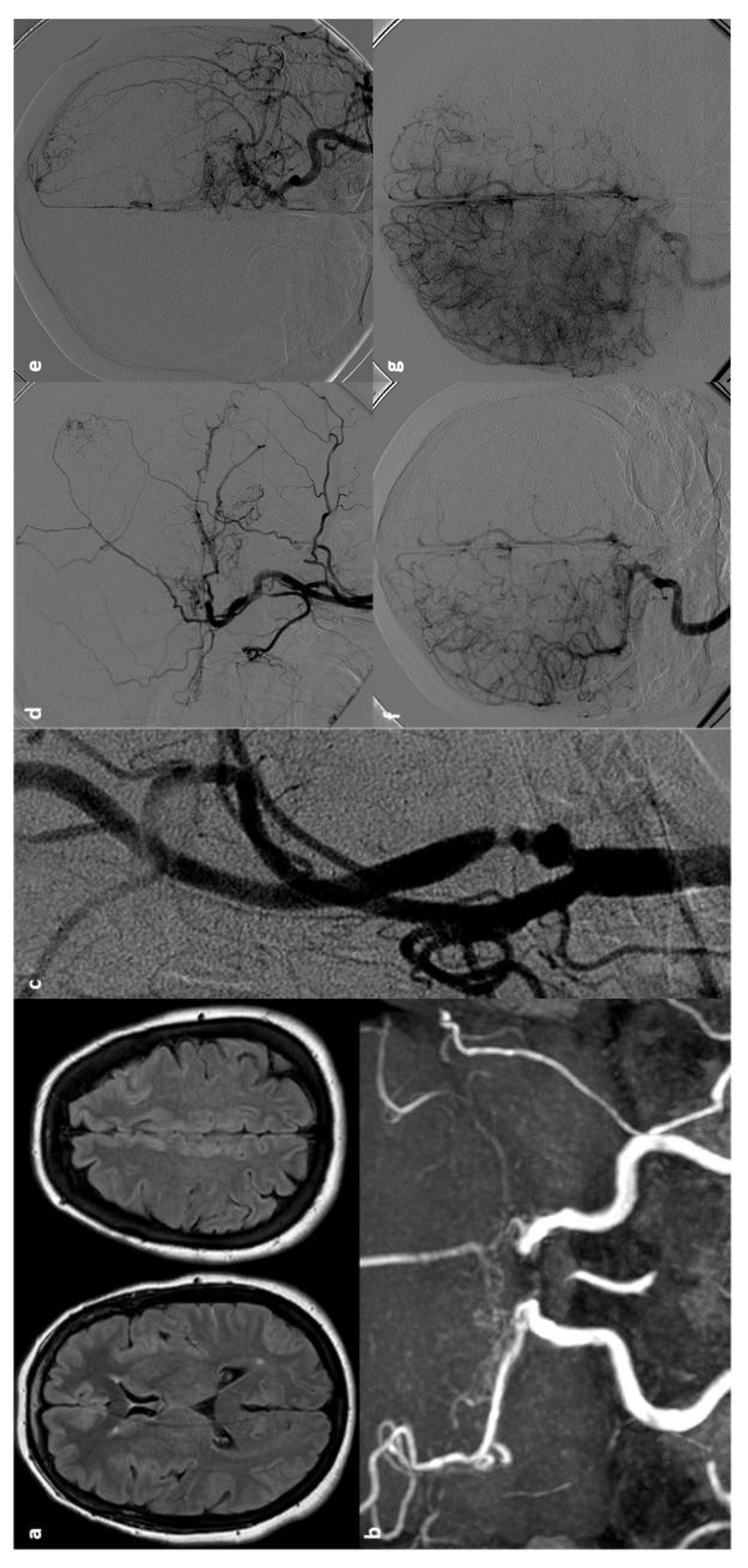
Figure 3.
Left internal carotid angiogram in posterior-anterior view (a) and 3D-RA reconstructed image, showing a saccular internal carotid artery aneurysm of choroidal subtype.
Figure 3.
Left internal carotid angiogram in posterior-anterior view (a) and 3D-RA reconstructed image, showing a saccular internal carotid artery aneurysm of choroidal subtype.
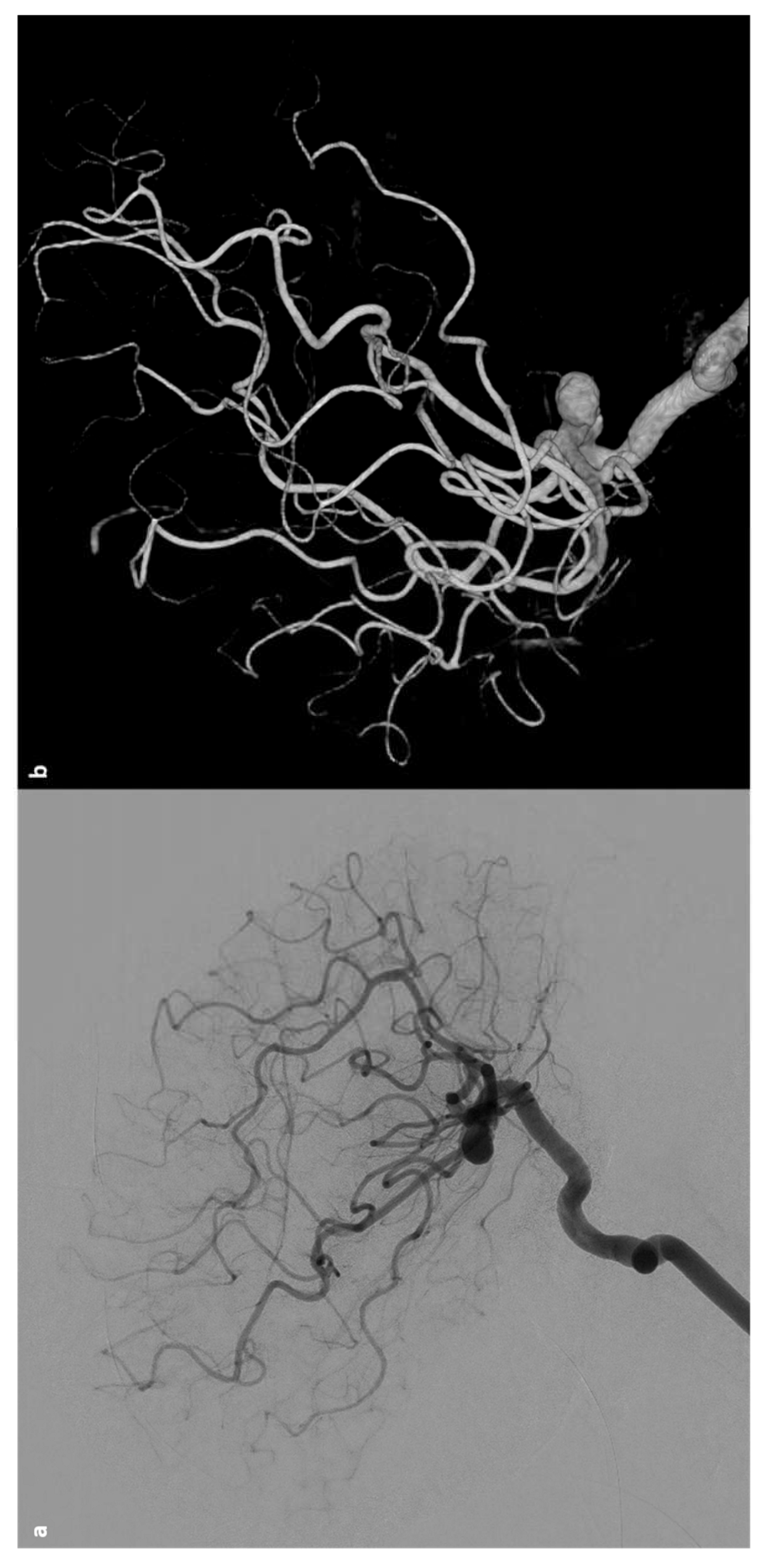

Table 1.
Main features of ischemic stroke in SCD [63].


Table 2.
Main steps of management of acute ischemic stroke in SCD [63].
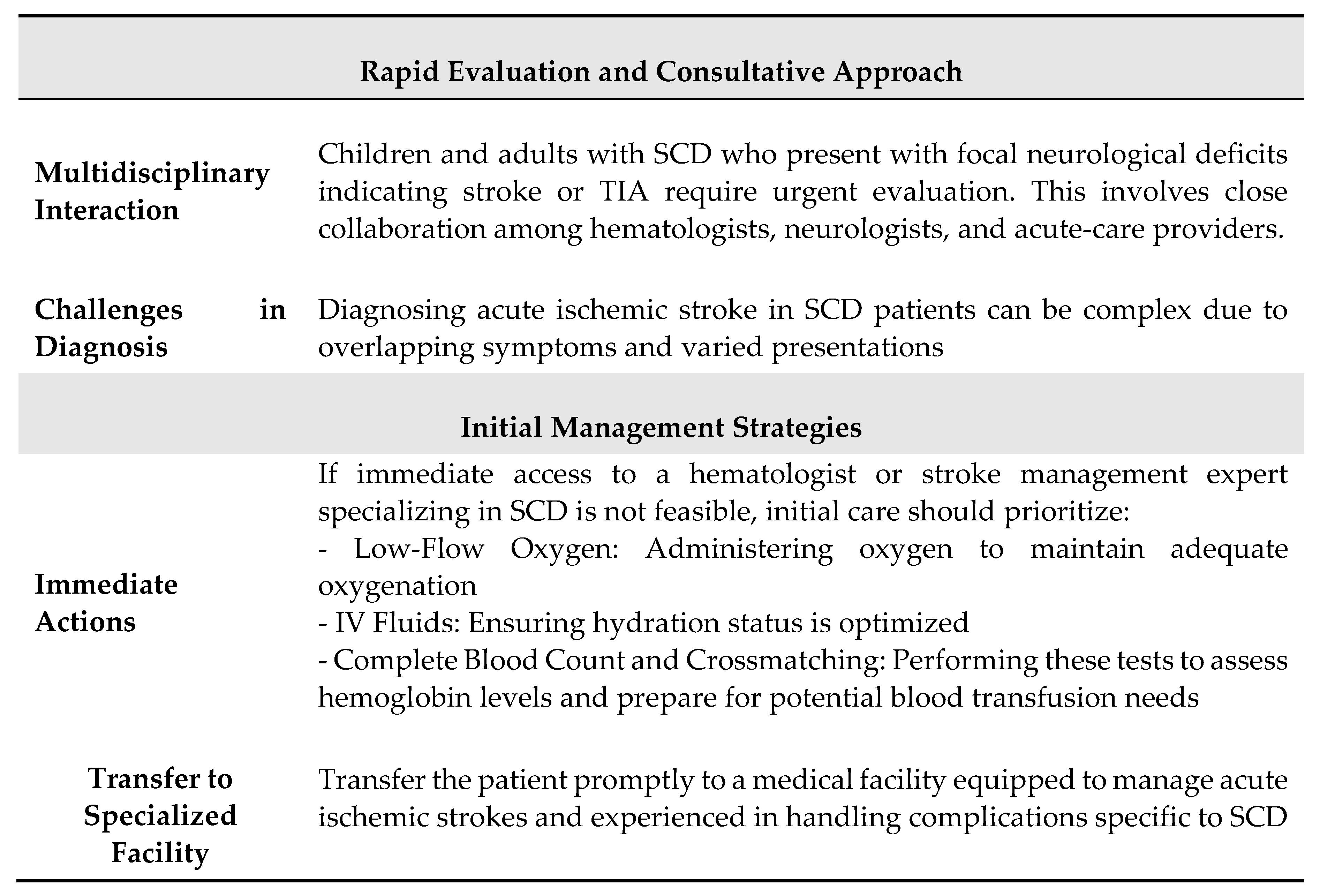
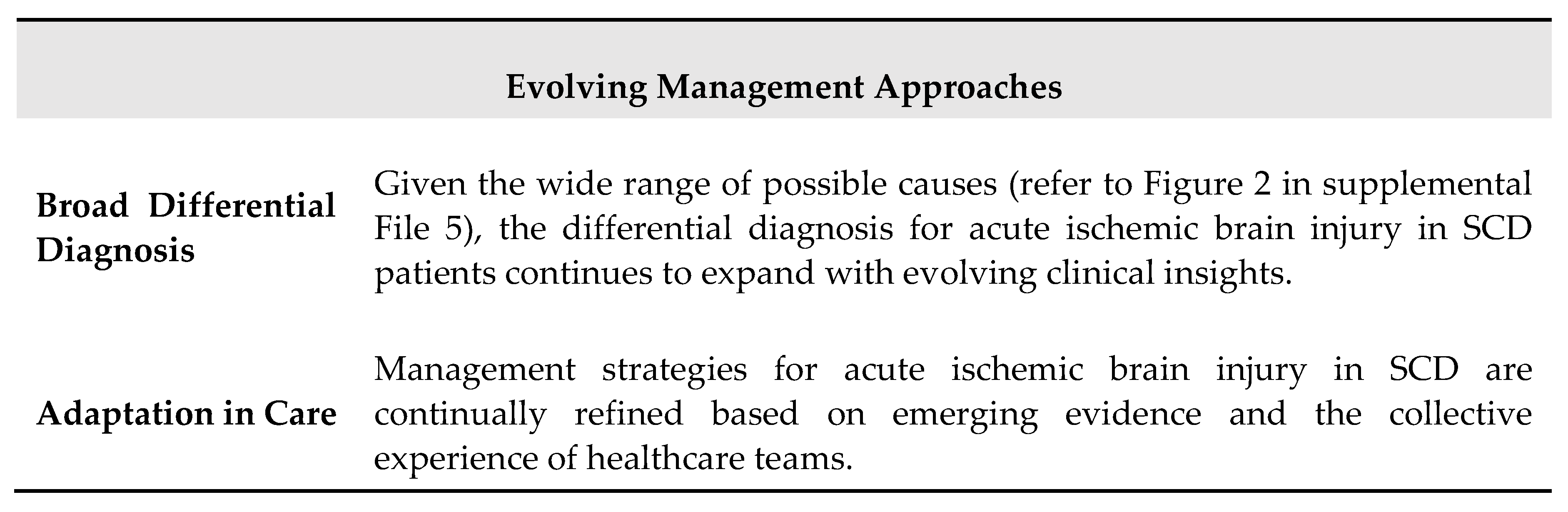
Table 3.
Main specific features of acute stroke management in SCD [63].


Table 4.
Summary of pathophysiological issues in SCD-related acute stroke.
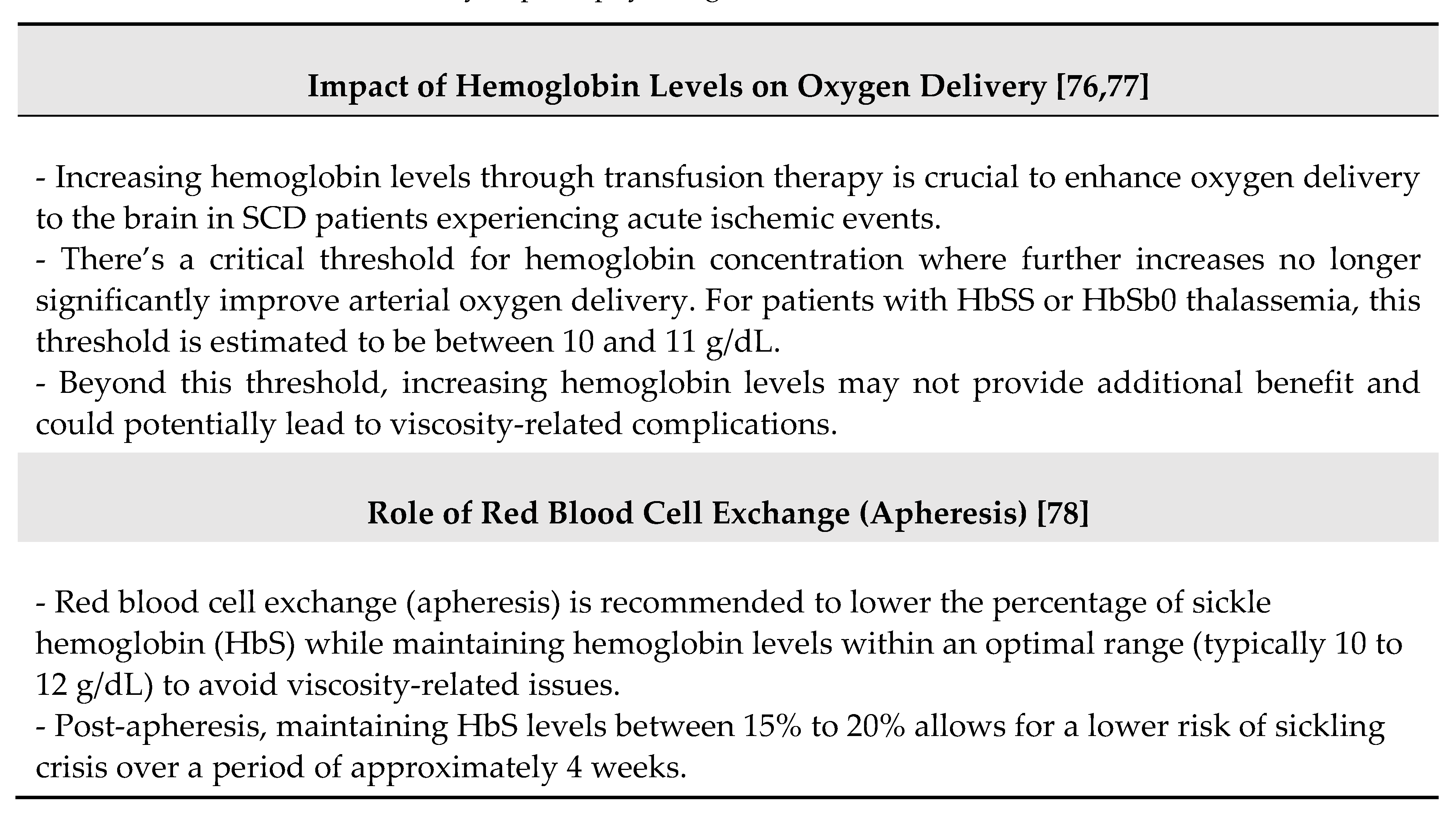
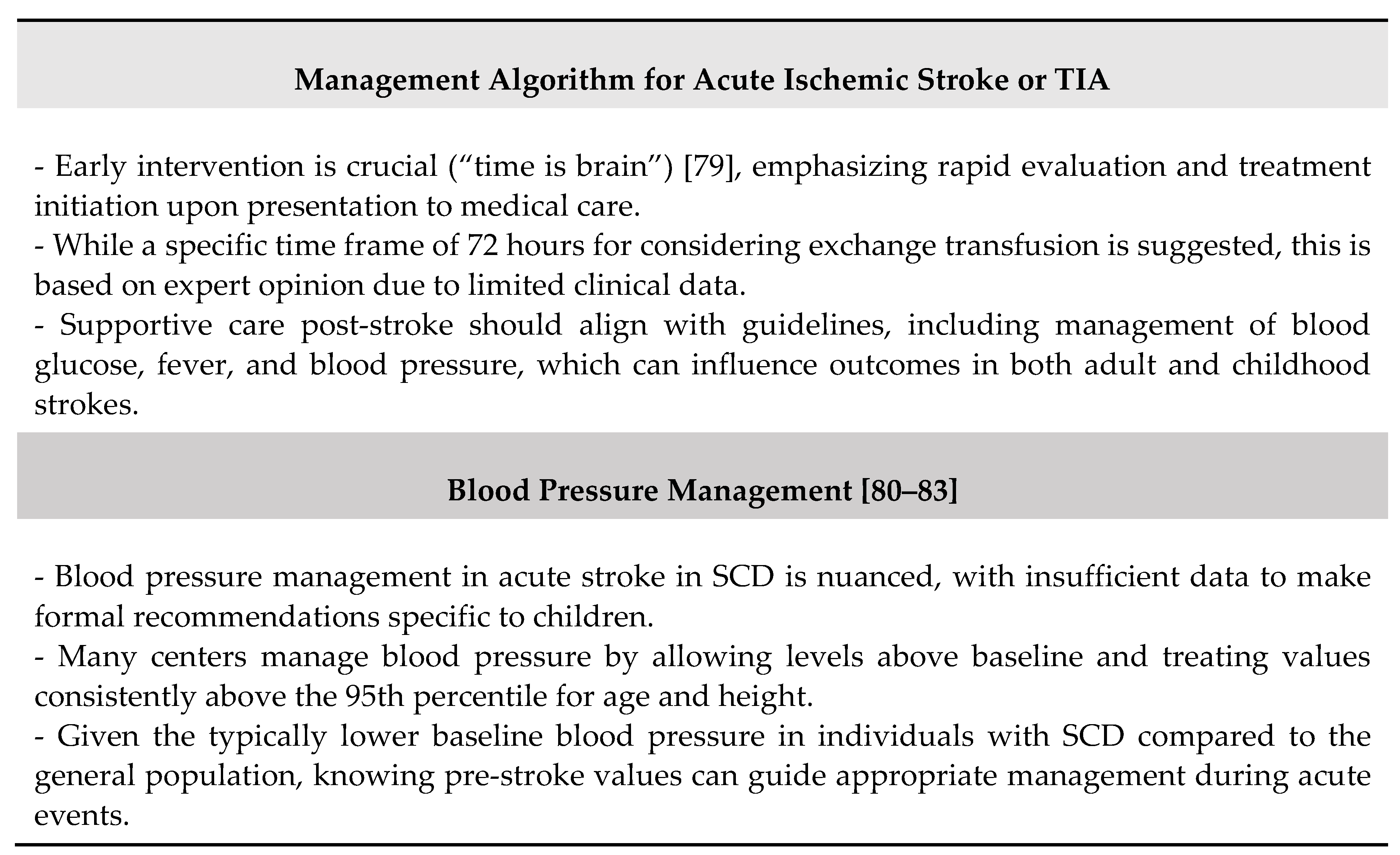
Table 5.
Potential complications of treatments in SCD-related acute stroke.
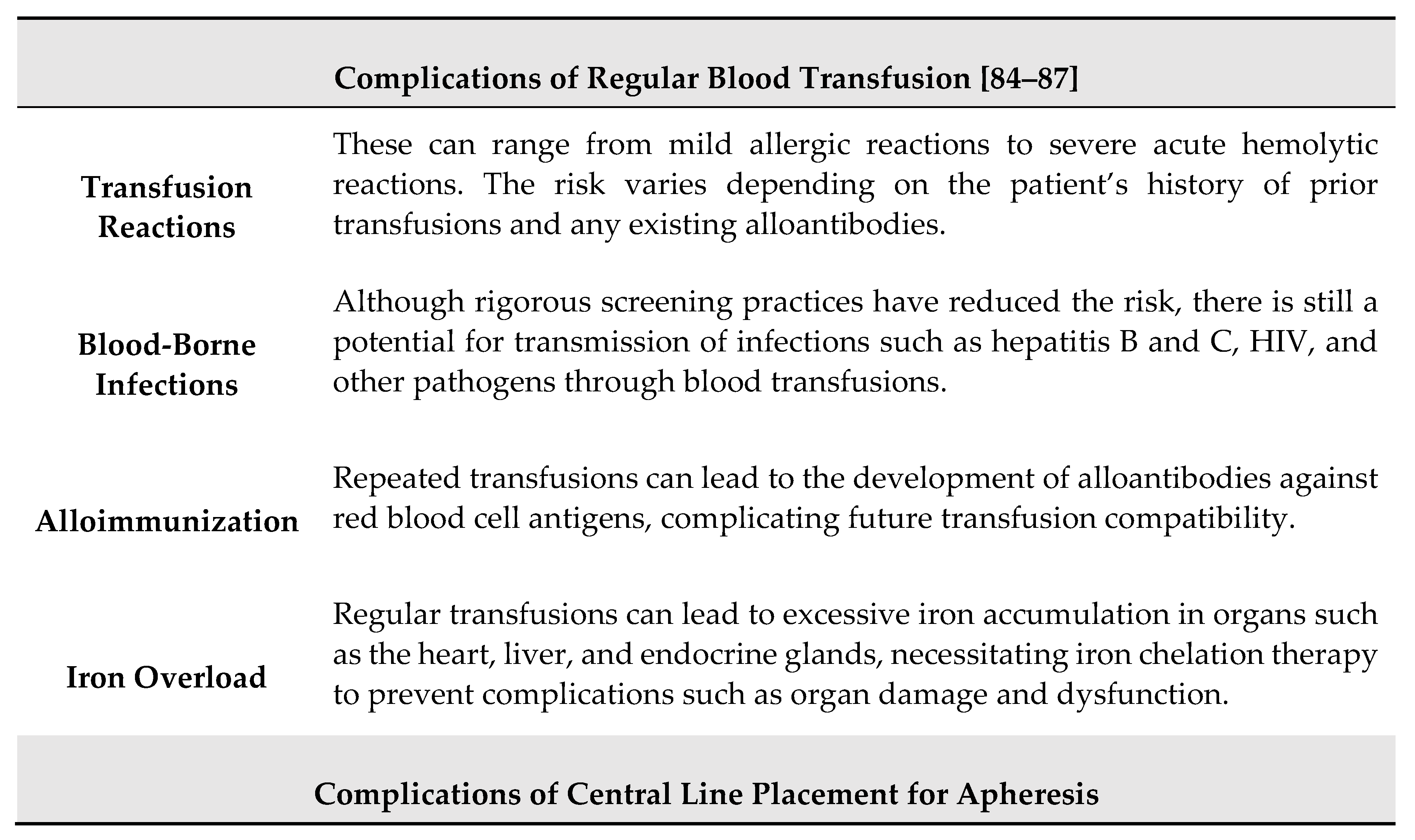

Table 6.
Main recommendations for acute ischemic stroke treatment in children and adults with SCD [88].
Table 6.
Main recommendations for acute ischemic stroke treatment in children and adults with SCD [88].

* <18 years.
Table 7.
Summary of findings of the main studies about risk of stroke recurrence in SCD.
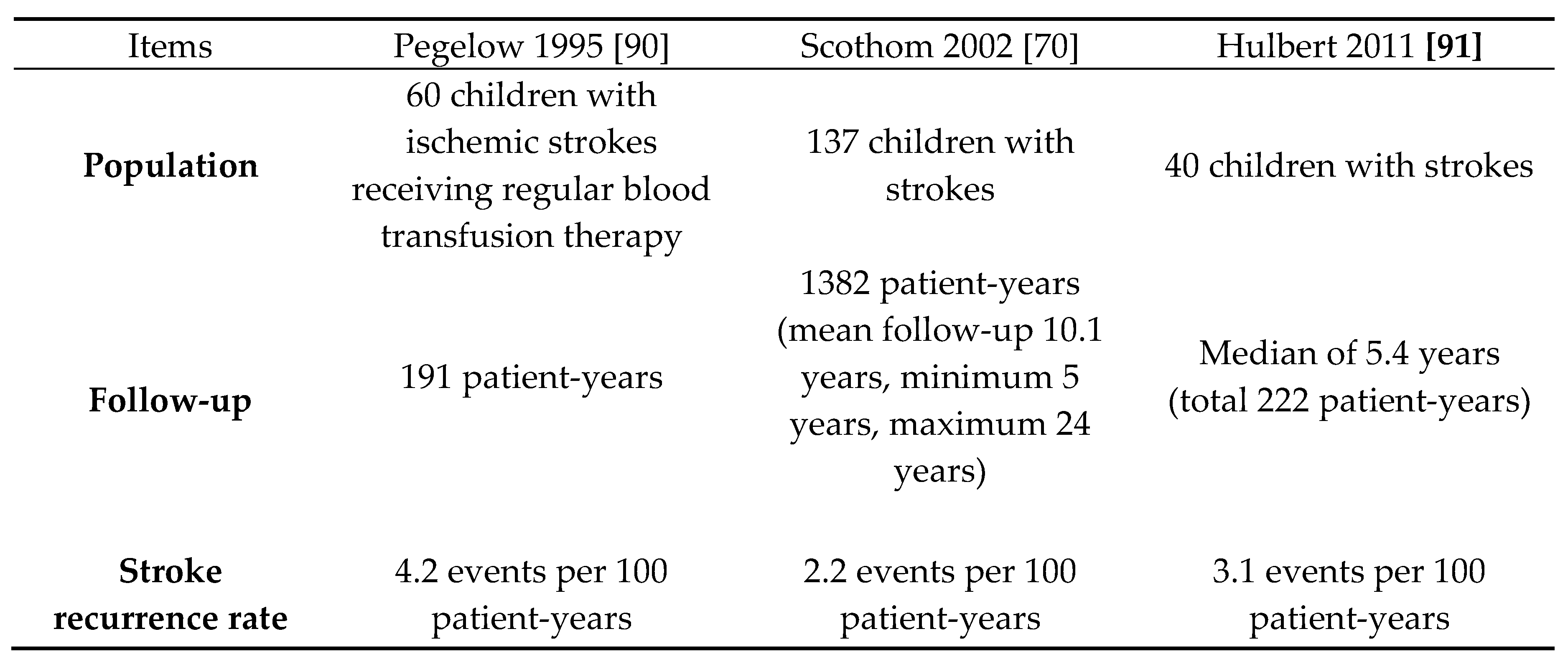

* Tables may have a footer.
Table 8.
Main features of SCI in SCD.

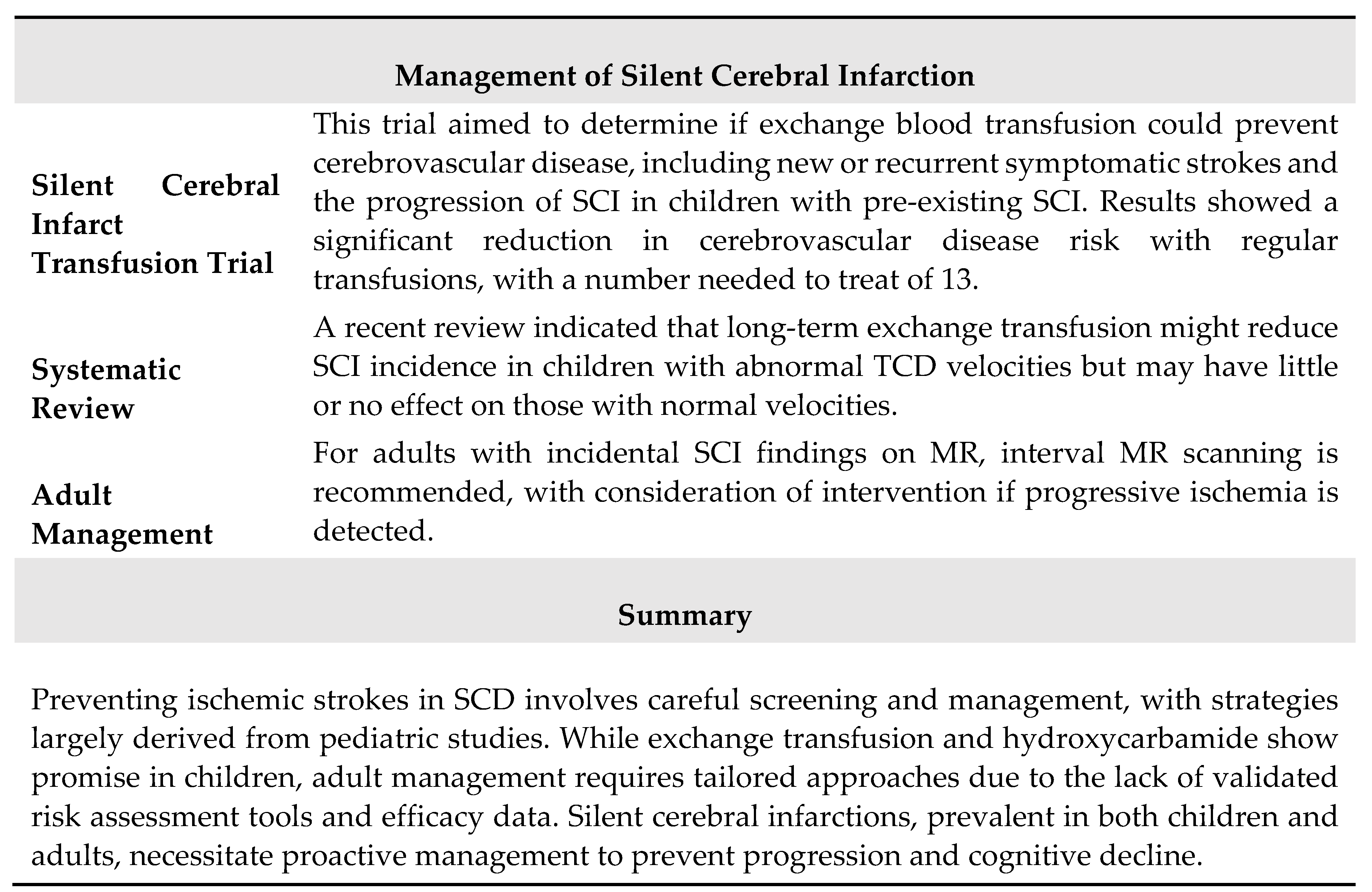
Table 9.
Main recommendations for screening for silent cerebral infarcts in children and adults with HbSS or HbSb0 thalassemia [88].
Table 9.
Main recommendations for screening for silent cerebral infarcts in children and adults with HbSS or HbSb0 thalassemia [88].

Table 10.
Main reasons for screening for silent cerebral infarcts in children and adults with HbSS or HbSb0 thalassemia [88].
Table 10.
Main reasons for screening for silent cerebral infarcts in children and adults with HbSS or HbSb0 thalassemia [88].
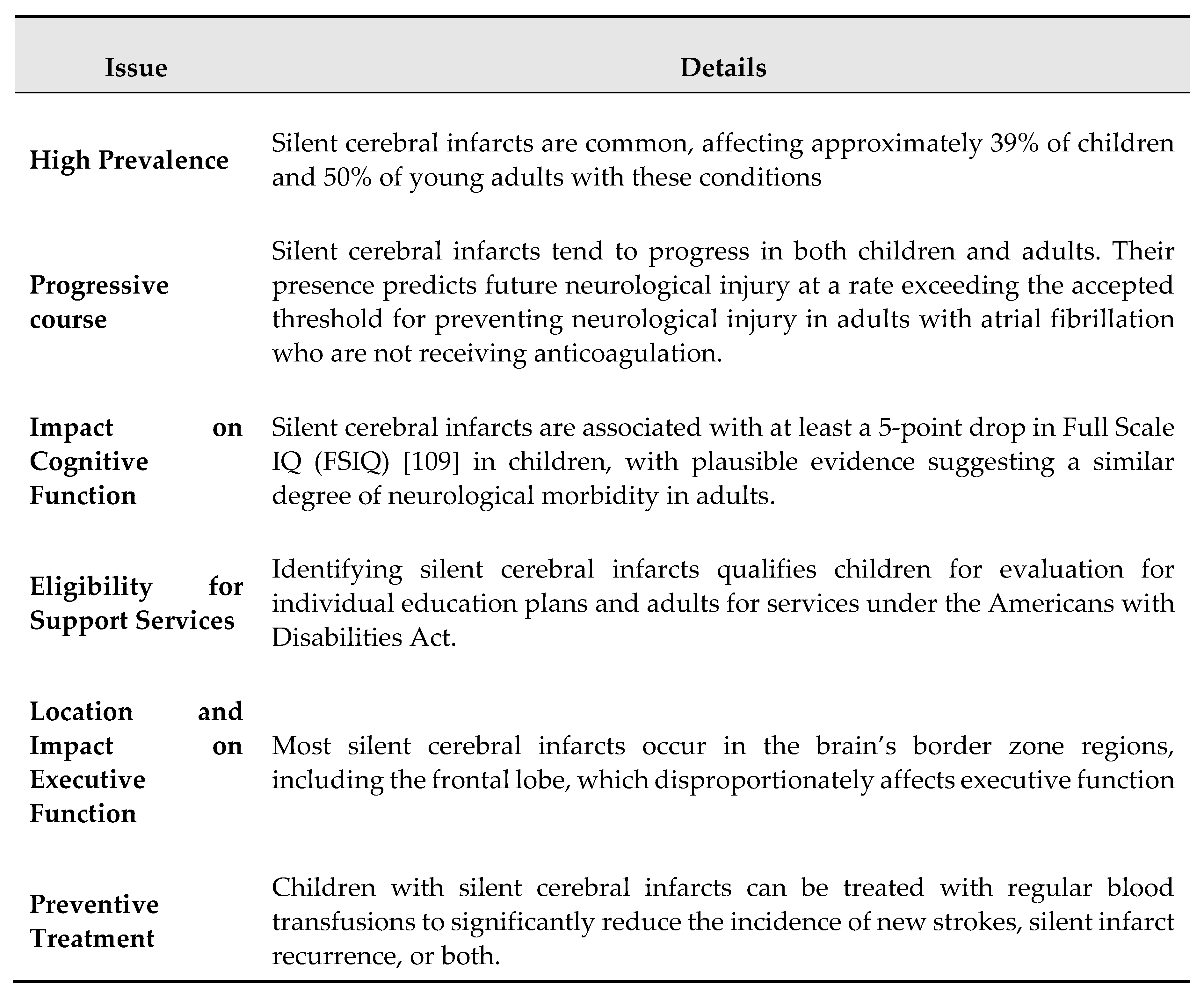
Table 11.
Main key points in patients with SCD and intracranial progressive arteriopathy [88].


Table 12.
Main features of aneurysmal disease in SCD acconding with [143].

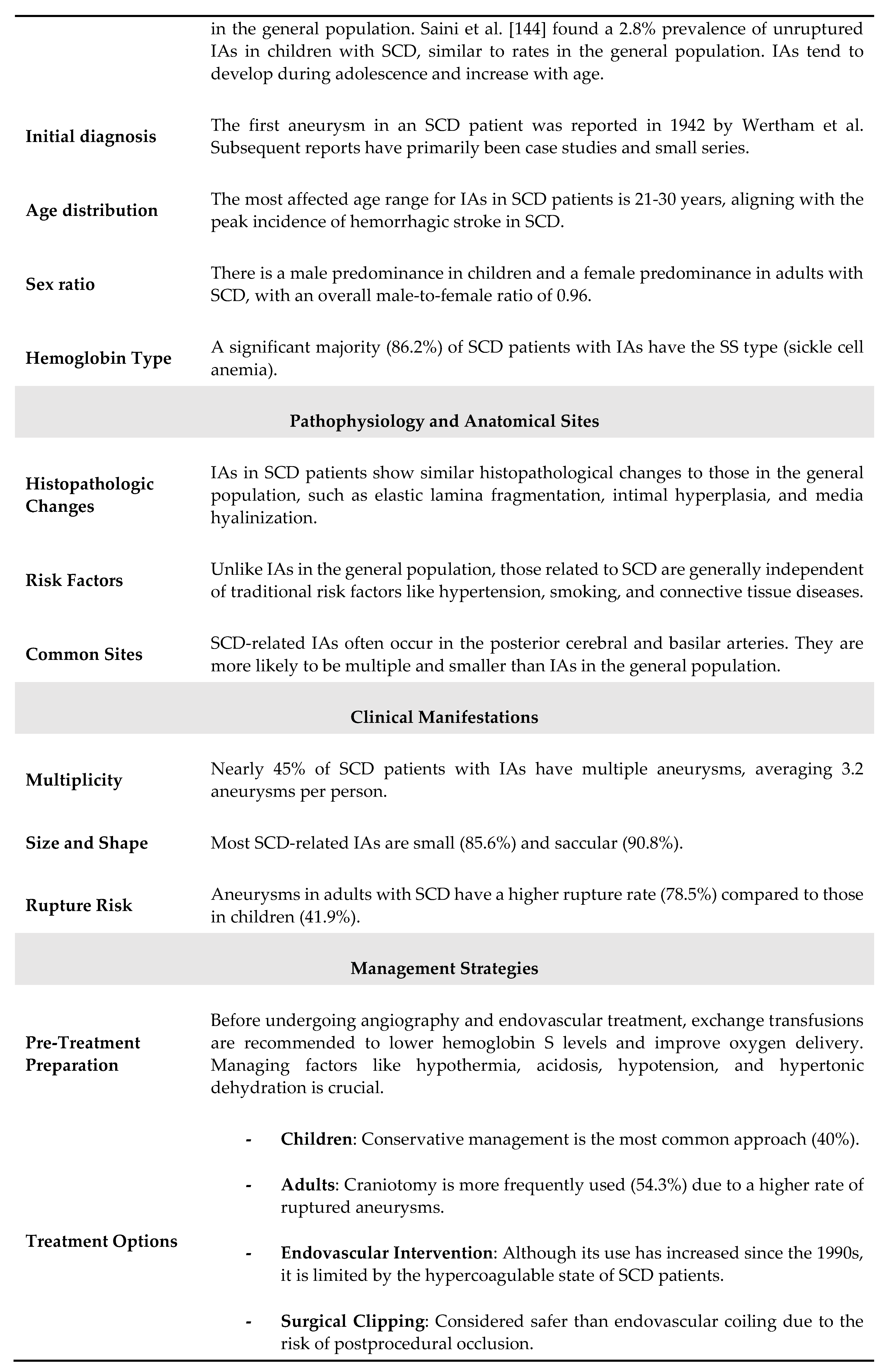

Table 13.
Main recommendations for prevention, diagnosis, and treatment of cerebrovascular disease in children and adults with SCD [88].
Table 13.
Main recommendations for prevention, diagnosis, and treatment of cerebrovascular disease in children and adults with SCD [88].


* ages 2-16 years.
Table 14.
Prevention and Management of Ischemic Stroke in SCD.
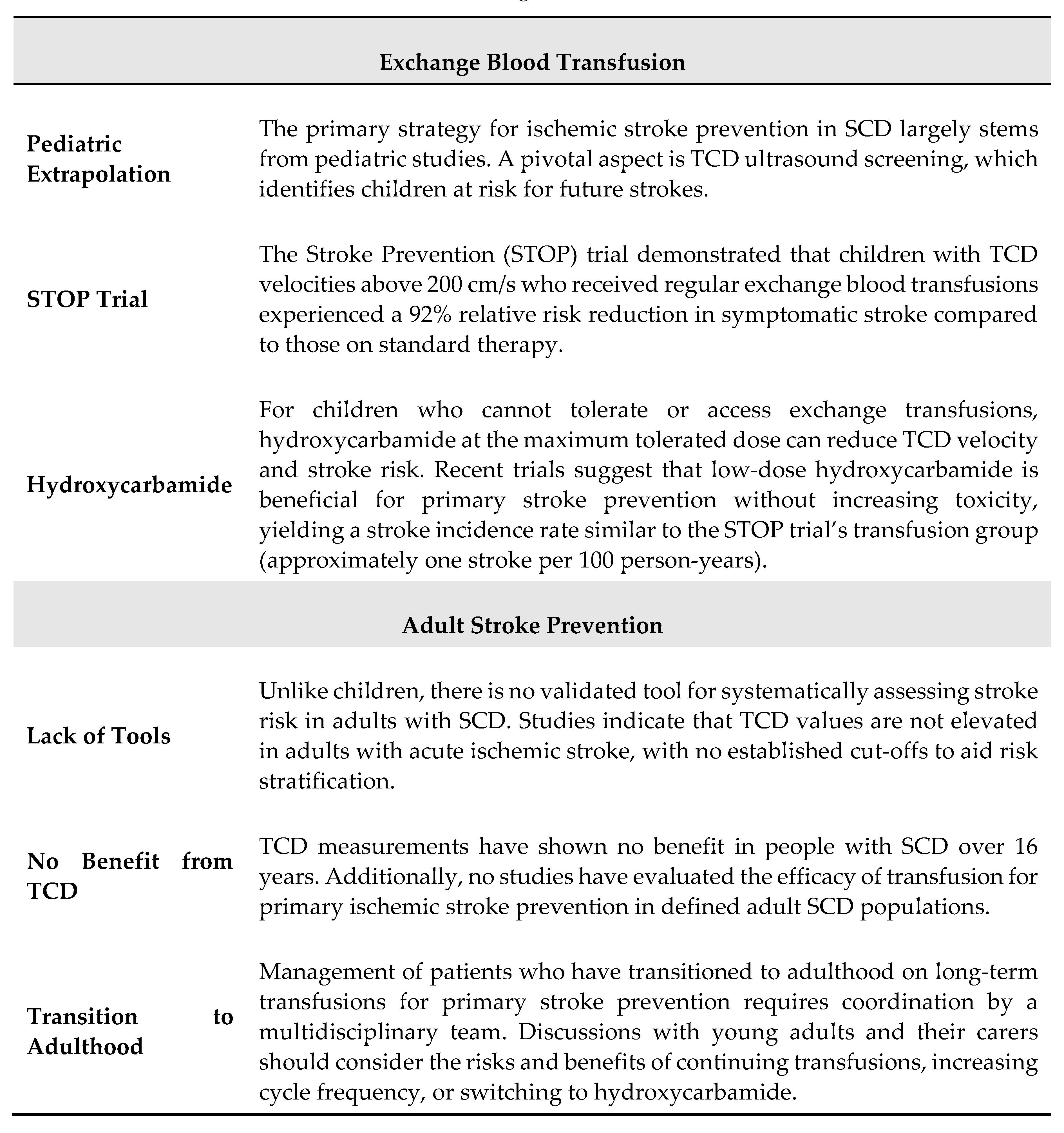
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



